Dr.ssa Paola Corti Dr.ssa Marta Verna Monza, Sabato 24 maggio 2014 EMATOLOGIA PEDIATRICA: “LEUCOPENIE A CONFRONTO” Sindromi e... Aplasie Midollari Congenite

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Dr.ssa Paola Corti Dr.ssa Marta Verna
Monza, Sabato 24 maggio 2014
EMATOLOGIA PEDIATRICA: “LEUCOPENIE A CONFRONTO”
Sindromi e...Aplasie Midollari
Congenite

Sindromi e... Aplasie Midollari Congenite
Monza, 24 maggio 2014

CASO CLINICO 1
Ragazza, 12 aa, inviata per leucopenia (riscontro occasionale):
GB 3,7*109/L; N 1,9 *109/L; PTL 118*109/L; Hb 12.9 g/dL; MCV 93 fL
E.O.: cute e mucose rosee, idratate. Non dismorfismi evidenti.
Obiettività cardio-respiratoria e addominale nei limiti di norma.
Non organomegalia.
Peso: 32 Kg (10°pct); altezza 139 cm (3°-10°pct) –secondo curva di crescita-
Accertamenti eseguiti:
• emocromo: GB 3,2*109/L; N 2,0*109/L; PTL 95*109/L; Hb 12,2 g/dL; MCV 94 fL
• autoimmunità (ANA, EMA; TTG, anticardiolipina, antifosfolipidi, LAC), dosaggio Ig A,G,M:
nella norma; C3, C4: nella norma
• B12 e folati: nella norma
• Hb F: 4,2%
• BM: midollo piuttosto povero in cellule, serie eritroide discretamente rappresentata, serie
mieloide scarsamente rappresentata con note di displasia, megacariociti poco presenti con
aspetti displastici. Presenza del 2% di blasti mieloidi scarsamente differenziati.
• campione inadeguato all’analisi citogenetica, FISH del 7 e del 8 negative.
MDSMonza, 24 maggio 2014

CASO CLINICO 1: DIAGNOSI DIFFERENZIALE
APLASIE MIDOLLARI CONGENITE:
• anemia di Fanconi
• anemia di Blackfan Diamond (DBA)
• sindrome di Shwachman Diamond (SDS)
• sindrome di Pearson
• discheratosi congenita
• trombocitopenia amegacariocitica
congenita
DEB test: analisi citogenetica delle frequenza di rottura cromosomica indotta da
diepossibutano, agente mitogeno per i linfociti T che induce rotture del DNA
(linfociti posti in coltura con l’agente mitogeno).
Anemia di Fanconi Trasmissione: autosomica recessiva
(X-LINKED)
Difetto ereditario della riparazione del DNA:
1. pancitopenia progressiva � aplasia
midollare (nella prima e seconda infanzia;
nel 90% <40 aa),
2. malformazioni congenite variabili,
3. predisposizione allo sviluppo di tumori.
Monza, 24 maggio 2014

Anomalie scheletriche: (71%)
- difetti mano ed avanbraccio (ipoplasia pollice, ipoplasia radio)
- displasia congenita dell'anca
- scoliosi, anomalie vertebrali
Cute:(64%)
- iperpigmentazione generalizzata
- macchie caffelatte
Crescita:(63%)
- ritardo pre e postnatale
- basso peso alla nascita
- bassa statura
Endocrinopatie:(81%)
- ridotta risposta al GH
- ipotiroidismo
- intolleranza glucosio
App. urinario: (34%)
- aplasia renale monolaterale
- ectopia renale
- rene “a ferro di cavallo”
Anomalie oculari: (38%)
- strabismo
- microftalmia
SNC:(24%)
- microcefalia - (ritardo mentale)
App. genitale: (20%)
- ipogonadismo ipogonadotropo-criptorchidismo, ipospadia
-- infertilità
Anomalie dell'orecchio: (11%)
- ipoacusia conduttiva
- anomalie orecchio esterno
Anomalie cardiache: (13%)
- DIV, DIA
- stenosi aortica o polmonare
- coartazione aortica
Anomalie gastrointestinali: (14%)
- atresia esofagea o duodenale o digiunale
- ano imperforato
- fistole tracheoesofagee
ANEMIA DI FANCONI: Caratteristiche cliniche
Clinicamente eterogenea, 30 % pz
fenotipicamentenormali

Le mutazioni colpiscono geni (chiamati FANC),
che codificano per proteine coinvolte nei
meccanismi di riparazione del DNA.
Instabilità cromosomica
Aumentata suscettibilità all'insorgenza di neoplasie (probabilità a 45 anni pari a 76%!):
• ematologiche (MDS, LMA, LLA);
• Ca della testa e collo, (+ cavo orale)
• Ca dello stomaco (Ca. squamocellulare),
• Ca del fegato (Ca. epatocellulare).
ANEMIA DI FANCONI
0
TERAPIA���� l'unico trattamento curativo per le manifestazioni ematologiche è
trapianto di cellule staminali ematopoietiche (HSCT), ma...
NON RIDUCE IL RISCHIO DI SVILUPPO DI NEOPLASIE!
Diagnosi precoce permette: - interventi chirurgici correttivi prima della pancitopenia,
- consulenza genetica per i genitori (25% rischio di ricorrenza),
- ev. identificazione di fratelli affetti
PREVENZIONE !!!

CASO CLINICO 2
• Nata a temine (40°sett EG) da parto eutocico, gravidanza patologica negli ultimi mesi per
IUGR. Peso alla nascita 2.530 Kg (SGA)
• Allattamento al seno, crescita staturo-ponderale regolare
• 4°mese OMA (febbre � antibioticoterapia)
• Dal 7°mese di vita:
� alvo diarroico ricorrente
� deflessione della curva di crescita (50°�10°pct).
.
.
.
.....
12 mesi, peso: 8580g (10-25°pc)
E.O.: cute rosea, ipoelastica. Mucose rosee, idratate. Pannicolo adiposo scarsamente
rappresentato. Obiettività cardio-respiratoria e addominale nei limiti di norma. Non segni di
risentimento neurologico né meningeo in atto.
Visita gastroenterologica pediatrica
Agli esami ematochimici:
• emocromo: Hb 10.9 g/dL; MCV 88 fL; GB 5,1*109/L; N
1,6 *109/L; PTL 163*109/L
• ipertransaminasemia (ALT: 121 UI; AST: 126 UI/l)
• non colestasi
Monza, 24 maggio 2014

CASO CLINICO 2: Ipotesi diagnostiche I°livello
- screening celiachia ripetutamente negativo
(HLA non indicativo);
- parassitologico/colturale feci ripetutamente
negativi;
- sospetta IPLV: idrolisato proteico pur con
RAST latte neg; challenge latte (nella norma),
reintrodotto latte vaccino
- fz tiroidea: nella norma;
- sierologie virus epatotropi negative (HAV,
HBV, HCV, CMV, EBV, Toxoplasma);
-
• CELIACHIA
• INFEZIONE INTESTINALE
•INTOLLERANZA ALIMENTARE
• IPERTIROIDISMO
• EPATITE/SINDROME
MONONUCLEOSICA
Ecografia dell’addome:
fegato di dimensioni ai
limiti superiori di norma,
ad ecostruttura
omogenea. Non
dilatiazione delle vie
biliari, colecisti
alitiasica.0
50
100
150
200
250
300
05/0
9/2
007
05/1
1/2
007
05/0
1/2
008
05/0
3/2
008
05/0
5/2
008
05/0
7/2
008
05/0
9/2
008
05/1
1/2
008
05/0
1/2
009
U/L
AST (U/L)
ALT (U/L)
Monza, 24 maggio 2014

CASO CLINICO 2: Ipotesi diagnostiche II°livello
• EPATITI AUTOIMMUNI
• FIBROSI CISTICA
• patologia del METABOLISMO
(ACIDOSI ORGANICHE E DIFETTO
DEL CICLO DELL’UREA)
• deficit α1 antiTRIPSINA
• morbo di WILSON
-pannello anticorpale (ANA, ENA, ASMA, AMA,
LKM, APCA, Ab anti-DNA nativo, anti-
cardiolipina): nella norma;
- test del sudore: negativo
- dosaggio aa plasmatici ed urinari, ammonio,
lattato nei limiti di norma, emogas venoso nella
norma.
- dosaggio alfa1 antitripsina,
- cupremia e ceruloplasmina nella norma;
• Dai 15 mesi di vita neutropenia (N 0,8-1,5 *109/L)
• Dai 16 mesi anemia normocitica (Hb 8.2 g/dL; MCV 84 fL),
• PTL border-line (110-140*109/L)
• Hb F: 7.3%
Monza, 24 maggio 2014

CASO CLINICO 2
BM: Ipoplasia di tutte le serie emopoietiche (C2 M1) ed aumento della quota istiocito-
macrofagica con presenza di immagini di emofagocitosi in assenza di elementi atipici.
Citogenetica: non valutabile; FISH 7 e 8: negativo
Saggi clonogenici in vitro: assenza di crescita della serie mieloide ed eritroide.
Aplasia Midollare... congenita ?
BOM: Midollo povero in cellule
(cellularità inferiore al 10%), rari
elementi immaturi della serie
mieloide ed eritroide e discreto
incremento della popolazione
monocito-macrofagica con aspetti di
fagocitosi; rari megacariociti; scarso
infiltrato interstiziale
linfoplasmacellulare, immagini
multiple di degenerazione fibrosa del
tessuto adiposo.
Monza, 24 maggio 2014

APLASIE MIDOLLARI CONGENITE:
DIAGNOSI DIFFERENZIALI
• anemia di Fanconi
• anemia di Blackfan Diamond
• sindrome di Shwachman Diamond
• sindrome di Pearson
• discheratosi congenita
• trombocitopenia amegacariocitica congenita
sindrome di SHWACHMAN DIAMOND
Monza, 24 maggio 2014

Sindrome di SHWACHMAN DIAMOND
Il gene SBDS localizzato sul cromosoma 7q 11.2 (Boocock et al 2003)
La proteina anomala sembra indurre apoptosi cellulare x deficit di maturazione della subunità ribosomiale 60S (Moore et al 2009)
Monza, 24 maggio 2014

1. Insufficienza del pancreas esocrino 88-100%
2. Ritardo di crescita staturo-ponderale 55-75%
3. Anomalie ematologiche 88-100%
4. Infezioni ricorrenti (+ OMA) 40-48%
5. Anomalie scheletriche: 32-52%
- coste corte
- torace carenato
- crolli vertebrali
- deficit di lunghezza tibiale
- disostosi metafisarie
6. Anomalie epatiche: ↑↑ transaminasi (+ nei primi anni di vita) 56-79%
7. Alterazioni dentarie: multiple carie (++ dentizione decidua)
8. Ritardo psicomotorio (iperattività, modesto difetto apprendimento...)
Sindrome di SHWACHMAN DIAMOND:
caratteristiche cliniche
Monza, 24 maggio 2014

• Leucopenia: pressochè sempre presente, di entità variabile;
• NEUTROPENIA: di più frequente riscontro (circa nel 50% dei casi):
- intermittente (60-65%);
- persistente (35-40%);
se severa condiziona complicanze infettive gravi e potenzialmente fatali;
• Anemia: presente 40-65% dei casi, spesso macrocitica,
• Hb F: incrementata;
• Piastrinopenia: - isolata (25-40%);
- intermittente;
- costante
raramente necessita di supporto trasfusionale di PTL.
• Pancitopenia: se presente aumenta la mortalità globale (dal 18% al 25%).
La severità della citopenia NON è predittiva della trasformazione
neoplastica (ma il rischio correla con livelli Hb F)
Sindrome di SHWACHMAN DIAMOND:
Anomalie ematologiche
Monza, 24 maggio 2014

Sindrome di SHWACHMAN DIAMOND: TERAPIA
Donadieu et al, Haematologica 2005
Registro francese delle neutropenie croniche severe:
231 pazienti con neutropenia congenita
MDS/LMA 36% a 30 aa
Monitoraggio Ematologico:
• emocromo con formula, MCV,
dosaggio HbF: ogni 6 mesi;
• aspirato midollare o biopsia
osteo-midollare:
- diagnosi;
- BM 1-2aa,
• cariotipo con citogenetica/fish
per anomalie del cromosoma 7,
8, 11, 21 (28% dei cas));
Terapia: - enzimi pancreatici (amilasi, lipasi proteasi) per via orale, prevalentemente nei primi
anni di vita;
- vitamine liposolubili A, D, E, K;
- di supporto (PTL, GRC trasfusioni)
- HCT in caso di: - citopenia persistente e refrattaria
- evoluzione in AAS (circa 20% casi);
- MDS (circa 20-30% casi con possibile � LMA 10-20% casi)

APLASIE MIDOLLARI CONGENITE:
DIAGNOSI DIFFERENZIALI
• anemia di Fanconi
• anemia di Blackfan Diamond
• sindrome di Shwachman Diamond
• sindrome di Pearson
• discheratosi congenita
• trombocitopenia amegacariocitica congenita
sindrome di BLACKFAN DIAMOND
Monza, 24 maggio 2014

Anemia di BLACKFAN DIAMOND (DBA)
RIBOSOMOPATIA: � alterazione nei geni delle proteine ribosomiali (RP) descritte nel
65% dei pazienti
� aploinsufficienza delle proteine ribosomiali
�stress ribosomiale e attivazione p53 � aumentata predisposizione ad apoptosi
GENETICA:
• sporadica, mutazione ex-novo (60%);
• familiarità (40-45%): - 2/3 dominante, - 1/3 recessiva (penetranza incompleta?)
• penetranza incompleta, espressività variabile;
• citogenetica: cariotipo normale.
�RPS19 (28%)
�RPS24 (2%)
�RPS17
�RPS7
�RPS10
�RPL35a
�RPL5 (10%)
�RPL11 (10%)
Monza, 24 maggio 2014

Anemia di BLACKFAN DIAMOND (DBA)
Aplasia pura della serie eritroide (descritta nel 1938)
• forma più comune tra ipoplasie midollari congenite (insieme Fanconi);
• rara: incidenza ≈ 1:100.000-250.000/aa nati vivi
• tutte le razze ugualmente colpite, rapporto maschi/femmine 1.1/1.
• caratterizzate da: 1) Anemia iporigenerativa
2) Malformazioni congenite
3) Predisposizione allo sviluppo di neoplasie
1) ANEMIA iporigenerativa normocitica/macrocitica, moderata/severa, trasfusione dipendente, presente � dalla nascita (25%)
� entro 1° aa di vita (>90%);
Altre linee emopoietiche normali o con � lieve leucopenia e/o � piastrinopenia e/o piastrinosi;
EPO elevata
Cellularità BM normale con ipoplasia/assenza precursori eritroidi;
Saggi clonogenici: ridotta/assente crescita in vitro BFU-E, CFU-E.
Monza, 24 maggio 2014

Anemia di BLACKFAN DIAMOND: CLINICA
2) MALFORMAZIONI: >35% pazienti(50-70%), multiple nel 25%;
distretti corporei tipici della Fanconi;
spettro di severità ↑↑, variabilità ↑↑..
•cranio-facciali (+comuni): micrognazia, microcefalia, labiopalatoschisi, palato ogivale, ipertelorismo, epicanto
• oftalmologiche: strabismo, glaucoma congenito, ptosi, anoftalmo, cataratta congenita…;
• arto superiore (10-20%): ipoplasia eminenza tenar, assenti polsi radiali, pollice trifalangeo, assente, accessorio, ipoplasico;
• difetto di crescita � < 25°pct : 45%, � < 3°pct : 30%;
• urogenitali (7-40%): monorene, rene a ferro di cavallo, ipospadia;
• cardiovascolari (7-30%): DIV, DIA, coartazione aorta, anomalie complesse;
• ritardo mentale, difficoltà apprendimento (< 2%);
Monza, 24 maggio 2014

Anemia di BLACKFAN DIAMOND: CLINICA
3) PREDISPOSIZIONE NEOPLASTICA:
in letteratura 29/700 – 3.9-4.1% (atteso <1%);
età insorgenza 15 aa, 1-43, (68 aa popol.gen.);
MDS, AML, osteosarcoma
Accertamenti diagnostici solo se sintomatologia specifica (non necessità controllo BM)
Monza, 24 maggio 2014

Anemia di BLACKFAN DIAMOND: TERAPIA
1. Terapia steroidea Prednisone 2 mg/Kg die p.o. x 4 sett
Risposta (generalmente 2-4 sett., no criteri prognostici predittivi):
• comparsa di reticolociti;
• allungamento intervallo trasfusionale;
• ↑ Hb.
steroido-responsivi (60%):
� 40% trasfusione-indipendente,
� 40% tp trasfusionale integrativa x
risposta “parziale”.
20% � remissione spontanea di malattia
(indipendenza da tp steroidea > 6 ms con
Hb accettabile).
steroido-resistenti (40%):
scalo tp dopo 4 sett, stop dopo 1-2 sett (tot
5-6 sett) � regime trasfusionale cronico;
2°ciclo dopo 18 ms (es. prima inizio tp
chelante).
solo 1-2% (casi sporadici) � remissione
spontanea.
• GRC 10-15 mL/Kg ogni 3-6 sett.
• Mantenere livelli Hb:� ben tollerati;
� buon accrescimento e sviluppo;
� adeguata qualità di vita;• Sovraccarico marziale: Deferoxamina (Desferal)
Deferasirox (Exjade)
2.Regime trasfusionale cronico
Monza, 24 maggio 2014

Anemia di BLACKFAN DIAMOND: TERAPIA
3. Trapianto di cellule emopoietiche
Non differenze significative tra MUD e MSD
Fagioli et al. 2014
Monza, 24 maggio 2014

APLASIE MIDOLLARI CONGENITE:
DIAGNOSI DIFFERENZIALI
• anemia di Fanconi
• anemia di Blackfan Diamond
• sindrome di Shwachman Diamond
• sindrome di Pearson
• discheratosi congenita
• trombocitopenia amegacariocitica congenita
sindrome di PEARSON
Monza, 24 maggio 2014

Sindrome di PEARSON
Eterogeneo disordine energetico, con coinvolgimento:
• neurologico encefalopatia, atassia, epilessia, neurodegenerazione• metabolico acidosi iperlattacidemica
• ematologico anemia sideroblastica macrocitica, piastrinopenia, leucopenia
• oculare oftalmoplegia, degenerazione retinica, ptosi etc.
• cardiovascolare cardiomiopatia ipertrofica, aritimie etc.
• gastroenterologico insuff. epatica, insuff. pancreas esocrino etc.
Mitocondriopatia, rara, sindrome midollo-pancreatica, insorgenza sporadica, caratterizzata
da:1) anemia sideroblastica refrattaria,
2) disfunzione del pancreas esocrino,
3) acidosi lattica con possibile insufficienza epatica/renale.
“… any symptom in any organ at any age”
Terapia: - di supporto;
- HCT corregge l’anemia sideroblastica iporigenerativa, ma NON l’evoluzione
neurologica progressiva (NON INDICATO)
Morte nella prima o seconda infanzia
Monza, 24 maggio 2014

GRAZIE !
WIN !

ANEMIA DI BLACKFAN-DIAMOND
• Anemia congenita per deficit selettivo eritroide
– anemia macrocitica presente nel primo anno di vita
– Reticolocitopenia
– Midollo normocellulare con deficit selettivo eritroide
– Normali o poco ridotti i neutrofili e le piastrine
Malformazioni :
� arto superiore� cuore� craniofacciali� occhio� urogenitali� ritardo mentale � bassa statura� ernia congenita� displasia congenita anca

DISCHERATOSI CONGENITA
Prevalenza stimata in 1/1.000.000.
Rara malattia multisistemica, caratterizzata da una triade di sintomi muco-cutanei:
-anomalie della pigmentazione cutanea,
-distrofia ungueale;
-leucoplachia delle mucose.
Si associa a:
• insufficienza del midollo osseo (causa principale di morte precoce);
• predisposizione alle lesioni tumorali
• complicazioni polmonari (spesso fatali).
I geni DKC1 e TERC, che sono mutati in due sottotipi di DC, codificano per componenti
del complesso delle telomerasi.
Il trapianto di cellule staminali emopoietiche rappresenta l'unica opzione per il trattamento
dei pazienti con DC che sviluppano insufficienza del midollo osseo.

APLASIE MIDOLLARI: DIAGNOSI DIFFERENZIALI
•anemia di Fanconi
•anemia di Blackfan Diamond,
•sindrome di Schwachman Diamond
•sindrome di Pearson,
-discheratosi congenita, anomalie della pigmentazione cutanea, -distrofia ungueale;-leucoplachia delle mucose.
Possono essere presenti anche altre anomalie. L'insufficienza del midollo osseo è la causa principale di morte precoce, unitamente alla predisposizione alle lesioni tumorali e alle complicazioni polmonari ad esito fatale. La DC presenta una notevole eterogeneità clinica e genetica. Sono note forme recessive, legate all'X, forme autosomiche dominanti e forme autosomiche recessive.
•trombocitopenia amegacariocitica congenita,

Leucopenia/Neutropenia
G.B. totali < al limite inferiore per l’età
Molto spesso presente, grado variabile 97-100% (92%)
Non è un problema se isolata
Esprime danno midollare per ↓ cellule staminali CD34+
Più spesso: neutropenia
PMN < 1500/mmc
• intermittente 60-65%
• persistente 36-40%
• complicanze infettive anche gravi se:
- neutropenia severa (PMN < 500/mmc)
- vie respiratorie (nei piccoli)
- talora fatali, se associato deficit di chemiotassi (test utile)
x anomalie del recettore concanavallina A o del citoscheletro
(Soll D et al 2003)

Anemia
Hb <10 gr/dl
• presente (spesso lieve) 42-65% (55%)
• isolata è rara 42%
• associata a reticolocitopenia
• spesso macrocitica
• raramente necessarie GRC trasfusioni
• spesso (77%) associata a ↑ di HbF (eritropoiesi inefficace)
attenzione ad ↑ di HbF x’ predice insorgenza di anomalie clonali e successiva evoluzione (Freedman et al)

Trombocitopenia
Piastrine < 150.000/mmc
• isolata 24-40% (36%)
• costante o intermittente
• raramente diatesi muco-cutanea
• trasfusione di concentrati piastrinici irradiati solo se
diatesi emorragica rilevante

Pancitopenia
In USA-Canada nel 19%, + frequente in Italia
Pancitopenia Sì No
Mortalità globale 24% 18%
Sopravvivenza mediana 25 aa 37aa (Alter et al)
Anticipare aspirato midollare alla comparsa

Aspirato midollare
Cellularità: - spesso ↓, ma = o ↑
- frequente ↓ cell CD34+ x
↑ dell’apoptosi Fas-mediata
- se ↓ ↓, diagnosi di AAA
Morfologia: - normale: > parte dei casi
- spostam. a sinistra/arresto maturazione: 23% dei casi
- evoluzione a MDS e/o LMA
- evoluzione a AAA
Citogenetica convenzionale: - normale
- 66% anomalie cromosoma 7: monosomia e/o i(7q)
- 16% anomalie del cromosoma 20: del (20)(q12)
- cariotipi complessi

Aspirato midollare
- Citogenetica in FISH con sonde subtelomeriche:
ricerca di monosomia del cromosoma 7
ricerca di trisomia del cromosoma 8
- Saggi clonogenici in vitro x valutaz. funzionale della capacità proliferativa di precursori mieloidi ed eritroidi:
↓ CFU-GM e BFU-E x tendenza all’apoptosi delle CD34+, doppia rispetto al normale (Freedman et al)
- Utile criopreservare MNC, DNA ed RNA, per eventuali studi futuri di approfondimento dell’etiopatogenesi

Biopsia ossea
In casi molto selezionati
Su indicazione dello specialista ematologo
► In caso di midollo ipoplasico oppure in caso di sospetta evoluzione
ematologica per approfondimento diagnostico
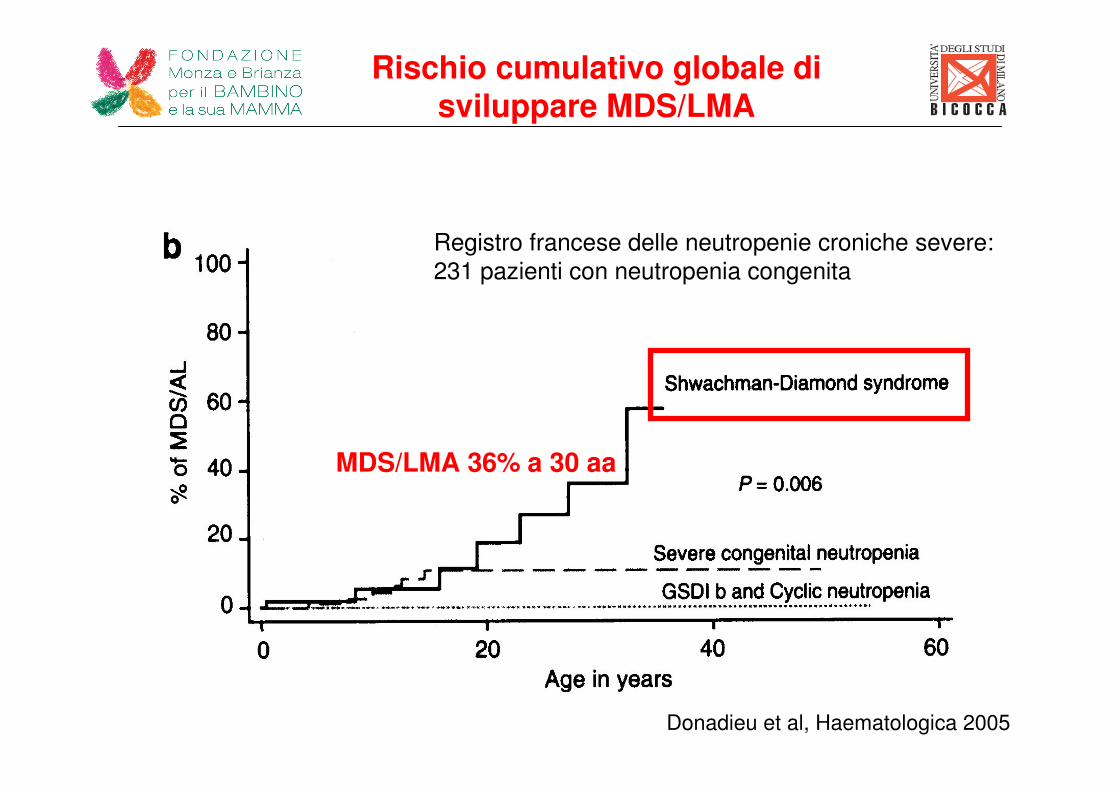
Rischio cumulativo globale di sviluppare MDS/LMA
Donadieu et al, Haematologica 2005
Registro francese delle neutropenie croniche severe:
231 pazienti con neutropenia congenita
MDS/LMA 36% a 30 aa

Conclusioni
- 10 % evoluzione ematologica
- Anomalie clonali non sono sinonimo di evoluzione rapida
- Necessità di criteri diagnostici dinamici per le MDS
- Tutti i pazienti evoluti avevano riarrangiamento SBDS
- Confermata l’importanza della sorveglianza ematologica
- Confermata l’importanza dei saggi clonogenici
- Analisi di SBDS a tutti i pazienti con citopenia primitiva?
Az. Ospedaliera di Padova

Evoluzione ematologica
Sfavorevole nel 10-33% Età mediana 14 aa (1.5-43 aa)
M:F circa 9:1
anomalie citogenetiche clonali
Aplasia midollare acquisita MDS (età mediana 8aa: 2-24) (20%)
Leucemia mieloide acuta (M6)
(12-24%) (Smith et al 1996)
TMO
allogenico
1°TMO in SDS nel 1988 per AAA(Tsai et al 1990)

La discheratosi congenita classica (DC) è una rara malattia multisistemica, con una prevalenza stimata in 1/1.000.000. La DC è caratterizzata da una triade di sintomi muco-cutanei, che comprende:
- anomalie della pigmentazione cutanea, - distrofia ungueale;- leucoplachia delle mucose.
Possono essere presenti anche altre anomalie. L'insufficienza del midollo osseo è la causa principale di morte precoce, unitamente alla predisposizione alle lesioni tumorali e alle complicazioni polmonari ad esito fatale. La DC presenta una notevole eterogeneitàclinica e genetica. Sono note forme recessive, legate all'X, forme autosomiche dominanti e forme autosomiche recessive.
I geni DKC1 e TERC, che sono mutati in due sottotipi di DC, codificano per componenti del complesso delle telomerasi; per questo, si ritiene che la DC sia soprattutto causata da un difetto delle telomerasi. L'identificazione di mutazioni di DKC1 in pazienti con sindrome di Hoyeraal-Hreidarsson (HH) e di mutazioni di TERC, in alcuni pazienti con anemia aplastica (AA) e mielodisplasia (MDS), hanno ampliato il numero dei pazienti che possono essere inquadrati nella DC. Dal punto di vista clinico, l'associazione tra DC e AA e il deficit di telomerasi suggerisce che i trattamenti rivolti alla correzione dell'attività della telomerasipossono portare beneficio ai pazienti con DC/AA, che non rispondono alla terapia convenzionale. Attualmente il trapianto di cellule staminali emopoietiche, che utilizza protocolli a bassa intensità, rappresenta l'unica opzione per il trattamento dei pazienti con DC che sviluppano insufficienza del midollo osseo.

Anemia di BLACKFAN DIAMOND: DIAGNOSI
Criteri diagnostici:• anemia macrocitica/normocitica;
• età di esordio < 1 aa (90%);
• reticolocitopenia;
• cellularità midollare normale con ipoplasia precursori eritroidi;
Criteri di supporto:� maggiori - mutazioni di geni noti correlati a DBA;
- familiarità;
� minori - anomalie congenite associate;
- ↑ eADA: 80% (v.n. 0.2-0.98 U/gHb);
- ↑ Hb F;
- non altre patologie midollari associate.
DBA Classica ���� 4 criteri diagnostici
DBA Atipica ���� 3 diagnostici + storia familiare positiva;���� 2 diagnostici + 3 supporto minore; ����3 criteri di supporto+ storia familiare positiva
DBA atipica da considerare in familiari di affetti, anche in assenza di anemia (attenzione in
caso di donatori familiari per HCT o per rischio ricorrenza in secondo figlio)
Related Documents