SILMÄLÄÄKKEIDEN FORMULOINTI JA TEOLLINEN VALMISTAMINEN KOKEELLINEN OSA: RAMAN-SPEKTROSKO-PIAN HYÖDYNTÄMINEN JAUHESEOKSEN LÄÄKEAINEPITOISUUDEN MÄÄRITTÄMISESSÄ Ville Kangas Pro gradu -tutkielma Proviisorin koulutusohjelma Itä-Suomen yliopisto, farmasian laitos Maaliskuu 2014

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

SILMÄLÄÄKKEIDEN FORMULOINTI
JA TEOLLINEN VALMISTAMINEN
KOKEELLINEN OSA:
RAMAN-SPEKTROSKO-PIAN HYÖDYNTÄMINEN
JAUHESEOKSEN LÄÄKEAINEPITOISUUDEN
MÄÄRITTÄMISESSÄ
Ville Kangas Pro gradu -tutkielma Proviisorin koulutusohjelma Itä-Suomen yliopisto, farmasian laitos Maaliskuu 2014

ITÄ-SUOMEN YLIOPISTO, terveystieteiden tiedekunta Farmasian laitos Proviisorin koulutusohjelma Farmasian teknologia KANGAS VILLE T: SILMÄVALMISTEIDEN TEOLLINEN FORMULOINTI JA VALMISTAMINEN Pro gradu -tutkielma, 108 s. ohjaajat: Professori, FaT Jarkko Ketolainen, Lehtori, dosentti Tapio Nurmi Maaliskuu 2014
Avainsanat: Silmälääke, formulointi, aseptiikka, valmistaminen, teknologia
Tiivistelmä
Silmälääkintä vaatii lääkevalmisteelta useanlaisia ominaisuuksia. Silmän pinta on lääkkeellä helposti saavutettavissa, mutta yleensä vain alle viisi prosenttia lääkeaineesta pääsee imeytymään silmän sisempiin osiin, jossa sen vaikutuskohta sijaitsee. Tätä ongelmaa pyritään ratkaisemaan yleensä parantamalla lääkevalmisteen kykyä pysyä silmässä. Erilaiset ainesosat, kuten imeytymisen parantajat, viskositeetin kohottajat, biokiinnitysaineet, faasin muuttajat ja suspension muodostajat, lisäävät lääkeaineen vaikutusaikaa silmässä. Perinteisten valmisteiden rinnalle on tullut myös pitempivaikutteisia ratkaisuja, kuten silmäluomitaskuun laitettavia implantti- ja sienivalmisteita, sekä teknologisempia annostelujärjestelmiä, kuten iontoforeesi. Kaikissa järjestelmissä on omat heikkoutensa, ja silmän herkkyyden takia valmisteiden muokkaaminen optimaalisiksi on hankalaa. Parhaimmillaankin silmävalmisteet ovat eri optimitilojen kompromisseja, joissa lääkeaineen imeytyminen, valmistettavuus, siedettävyys ja käyttäjän hoitomyöntyvyys yhdessä saavuttavat korkeimman tasonsa.
Silmävalmisteiden tuotanto on puhtaudeltaan parenteraalisen lääkinnän tasolla, ja valmistusolosuhteiden täytyy täyttää niille asetetut korkeat vaatimukset. Nykyaikaisten lääkeaineiden formulaatiot voivat olla monimutkaisia, mikä aiheuttaa tuotannolle omat vaikeutensa. Sterilointi ei useinkaan onnistu lopputuotteelle, jolloin joudutaan menemään täysin aseptiseen valmistukseen, jossa tasaisen korkean laadun saavuttaminen vaatii tarkkaa tuotantosuunnittelua ja toimivan ympäristön. Jokainen lisätty työvaihe aseptisessa valmistuksessa on riski tuotteen kontaminoitumiseen. Tätä varten on kehitetty erilaisia tekniikoita, kuten isolaattori, HEPA-ilmansuodatus, blow-fill-seal -teknologia, sterilization in place ja sterilointitunneli –järjestelmät. Näillä pystytään yhdistämään eri tuotantovaiheita samaan ympäristöön, jolloin tuotannon turvallisuus ja tehokkuus saadaan maksimoitua.

UNIVERSITY OF EASTERN FINLAND, Faculty of health science School of Pharmacy Master of Science in Pharmacy program Pharmaceutical Technology KANGAS VILLE T: FORMULATION AND INDUSTRIAL MANUFACTURING OF OPTHALMIC PRODUCTS Master’s thesis, 108 pp, Supervisors: Professor, PhD. Jarkko Ketolainen, Lecturer, Adjunct Professor Tapio Nurmi March 2014
Keywords: Eye medicine, formulation, asepsis, manufacturing, technology
Summary
Drug administration into the eye calls for several different properties of a pharmaceutical preparation. It is relatively easy for a drug to reach the surface of the eye but usually only less than five percent of an active drug ingredient is absorbed into the inner parts of the eye, that is, into the action site. Usually formulators try to solve this problem by increasing the dwelling time of the product on the surface of the eye. Different kinds of excipients are available, such as: permeation enhancers, bioadhesive agents, phase modifiers, suspending and thickening agents. It has been developed also new kind of treatments in parallel with conventional drugs like topically administered implants and gelatin products and also more technological solutions like iontophoresis. Every system has its own downsides and because eye is so vulnerable, product optimizations are difficult to be made. Even at the best case eye medicines are compromises of different optimal modes, where the permeability of an active pharmaceutical ingredient, producibility, tolerability and users comfort for medicine are combined.
The manufacture of the eye preparations needs to be as pure as drug manufacture for parenteral applications and the facilities need to fulfill the high specifications. New drug formulations can be so complicated that that may cause problems for manufacture. Some products cannot be sterilized after manufacture, but they need to be prepared in an aseptic way. In such aseptic manufacture, spesific production planning and good facilities are needed to achieve the high and consistent quality. Every additional production step in the aseptic manufacture is a risk of contamination. To avoid the contamination risk, several techniques are developed, such as isolator, HEPA air filtration, blow-fill-seal technology, sterilization-in-place and sterilization tunnel which helps to increase manufacturing safety and efficiency.

Esipuhe
Olen tehnyt opinnäytetyöni Itä-Suomen yliopiston farmasian teknologian ja
biofarmasian osastolla. Ohjaajina opinnäytetyössä toimivat FaT Jarkko Ketolainen,
Lehtori Tapio Nurmi, FaT, dos. Ossi Korhonen ja DI Tuomas Ervasti. Haluan kiittää
heitä hyvästä ohjauksesta ja tuesta opinnäytetyöni aikana.
Haluan kiittää myös perhettäni ja ystäviäni, erityisesti Jutta ja Matti Suvantoa sekä
Katri ja Ilari Kososta tuesta ja avusta opinnäytetyössä. Lisäksi kiitokset myös
ystävilleni korppikotkille yöllisestä puhelusta opinnäytetyön teonaikana sekä Suvi
Luukolle monista hajoiluhetkistä.
Lahdessa 26.02.2014
Ville Kangas

Lyhenteet
API lääkeaine
ASA asetyylisalisyylihappo
BAK Benzalkoniumkloridi
BFS Blow-Fill-Seal
CIP Clean In Place
CW jatkuva-aaltoinen lasersäde
DCP dikalsiumfosfaatti
LAKTO laktoosimonohydraatti
MAN mannitoli
MSC multiplikatiivinen sirontakorjaus esikäsittely
OP havaitut vs. ennustetut pisteet SIMCA ohjelmassa
PARA parasetamoli
PE Polyetyleeni
PP Polypropyleeni
Q2cum Q2-summa
Q2 testijoukon vaihtelun prosenttiosuus, jonka malli voi ennustaa
RMSEE sovitetun havaintojoukon keskineliöjuuren virhe
RMSEP ennusteen keskineliöjuuren virhe ennustusjoukossa
Row rivien keskitys esikäsittely
rpm kierrosta minuutissa
SA salisyylihappo
SG Savitzky-Golay esikäsittely
SIP Sterilization In Place

SNR signaali-kohina –suhde
SNV standardi normaali variaatti esikäsittely

Sisältö
1. Johdanto ............................................................................................................... 8
2. Silmän rakenne, toiminta ja tyypillisimmät formulaatiot ................................... 9
2.1 Silmän rakenne ja toiminta ................................................................................ 9
2.2 Formulaatioiden komponentit ja niiden tehtävät .............................................. 11
2.2.1 Lääkeaine ................................................................................................. 14
2.2.2 pH:n säätö ................................................................................................. 15
2.2.3 Toonisuuden säätäjät ................................................................................ 16
2.2.4 Säilytysaineet ............................................................................................ 16
2.2.5 Kostutusaineet (humektantit) .................................................................... 17
2.2.6 Imeytymisen edistäjät ................................................................................ 17
2.2.7 Viskositeetin kohottajat ja biokiinnitysaineet ............................................. 18
2.2.8 Suspension muodostajat ........................................................................... 19
2.2.9 Faasin muuttajat........................................................................................ 19
2.3. Formulaatiotyypit silmälääkinnässä ................................................................ 20
2.3.1 Perinteiset formulaatiot ............................................................................. 20
2.3.2 Uudemmat silmälääkkeiden kuljetusmekanismit ....................................... 27
2.4 Lääkepakkaus .............................................................................................. 28
3 Valmistus .............................................................................................................. 30
3.1 Valmistuslaitteistot ........................................................................................... 32
3.1.1 Isolaattori .................................................................................................. 32
3.1.2 Tuotteiden siirtojärjestelmä ....................................................................... 35
3.1.3 Komponenttien sterilointi ........................................................................... 36
3.1.4 Pakkauksen sterilointi ja puhdistus ........................................................... 36
3.1.5 Blow-Fill-Seal -laitteisto (BFS) .................................................................. 37
3.1.6 Valmistuslinjaston puhdistus ja sterilointi .................................................. 40
4. Yhteenveto .......................................................................................................... 41
KOKEELLINEN OSA: RAMAN-SPEKTROSKO-PIAN HYÖDYNTÄMINEN
JAUHESEOKSEN LÄÄKEAINEPITOISUUDEN MÄÄRITTÄMISESSÄ .................. 43
5 JOHDANTO ....................................................................................................... 47
6 MATERIAALIT JA MENETELMÄT ................................................................... 48
6.1 Raman-spektroskopia .................................................................................. 48

6.2 Spektrien esikäsittely ................................................................................... 51
6.2.1 Spektrin derivointi ................................................................................. 52
6.2.2 Spektrin normalisointi ............................................................................ 54
6.2.3 Spektrin tasoitus ................................................................................... 54
6.2.4 Sirontaa korjaavat esikäsittelyt.............................................................. 56
6.3 Raman-spektrometri ja PhAT anturi ............................................................ 59
6.4 Seokset ja niiden valmistus ......................................................................... 61
6.5 Näytteiden mittaus ja esikäsittely................................................................. 62
6.5.1 Mittaukset paikallaan olevista näytteistä ............................................... 62
6.5.2 Mittaukset liikkeessä olevista näytteistä ................................................ 62
6.5.3 Raman-spektrin esikäsittely .................................................................. 63
6.6 Kemiallinen lääkeainepitoisuuden määrittäminen ........................................ 65
6.7 Raman-spektrikirjasto .................................................................................. 67
7 TULOKSET ........................................................................................................ 68
7.1 Lääkeainepitoisuuden kemialliset määritykset ............................................. 68
7.2 Eri esikäsittelyiden vaikutukset malliin ja ennustettavuuteen ....................... 71
7.2.1 Asetyylisalisyylihappo-dikalsiumfosfaattiseos ....................................... 71
7.2.2 Ibuprofeeni-laktoosiseos ....................................................................... 72
7.2.3 Ibuprofeeni-mannitoliseos ..................................................................... 72
7.2.4 Parasetamoli-mannitoliseos .................................................................. 72
7.2.5 Parasetamoli-laktoosiseos .................................................................... 73
7.3 Liikkeessä tehdyt mittaukset ........................................................................ 79
7.3.1 Asetyylisalisyylihappo-dikalsiumfosfaattiseokset .................................. 79
7.3.2 Ibuprofeeni-laktoosiseokset .................................................................. 81
7.3.3 Parasetamoli-laktoosiseokset ............................................................... 82
7.4 Spektrimuutokset siirryttäessä puhdasaineesta seokseen .......................... 87
7.5 Raman-spektrikirjasto .................................................................................. 90
8 POHDINTA ........................................................................................................ 92
9 KIRJALLISUUS: ................................................................................................ 96
LIITETAULUKOT ................................................................................................... 110

8
1. Johdanto
Silmä on ihon lisäksi helpoimmin saavutettavia lääkkeen annostelukohteita
paikallisessa lääkinnässä. Silmälääkkeillä pyritään saamaan vaikutus joko silmän
pinnalle tai sen sisään. Suoraan silmään laitettavilla lääkkeillä saavutetaan monia
hyötyjä: paikallinen annostelu saavuttaa kudoksia, joiden hoitaminen systemaattisesti
olisi vaikeaa, vältetään lääkkeiden ensikierron metabolia, saadaan vähennettyä
lääkkeen systeemisiä haittavaikutuksia, lääkeaine saadaan nopeammin
kohteeseensa, annostelu on helppoa ja kivutonta sekä lääkkeessä voidaan käyttää
pienempiä lääkeainepitoisuuksia (Wilson ym. 2001, Kaur ja Kanwar 2002, Gibson
2009). Silmälääkevalmisteille on hyvin tarkat laatuvaatimukset, koska silmä on hyvin
pitkälle erikoistunutta kudosta ja sillä on uniikki asema ihmiskehon toiminnassa (Al-
Achi ym. 2013). Yksinkertaisimmillaan silmälääke voisi olla veteen liuotettu
lääkeaine, jota annostellaan silmään lääkepullosta. Silmän fysiologia tekee kuitenkin
asiasta haastavamman. Silmän kerrosmainen rakenne ja kyynelnestetoiminta
heikentävät lääkeaineiden imeytymisen olemattomiin, ja siitä huolimatta puhdas
lääkeaine voisi aiheuttaa silmässä paljon epämukavuuden, ärsytyksen tai polttelun
tunnetta. Tästä syystä silmälääkkeiden formulaatioihin käytetään monia apuaineita,
jotta haittoja saadaan vähennettyä ja hyötyjä lisättyä.
Valmisteen formulaatiota mietittäessä tulee myös miettiä kuinka valmiste saadaan
valmistettua ja toimitettua turvallisesti potilaalle (Agalloco ja Akers 2008).
Silmävalmisteiden tulee olla steriilejä, ja niiden annostelun täytyy olla tarkempaa
paikallisessa lääkinnässä, mikä monimutkaistaa tuotantoa huomattavasti.
Nykyaikaisissa valmistustiloissa täytyy ottaa huomioon vallitsevat olosuhteet
ympäristössä ja suunnitella järjestelmä toimimaan niiden mukaan tai muokata
olosuhteita. Olosuhteiden muokkaamiseen valmistukselle sopiviksi on kehitetty
erilaisia järjestelmiä, kuten isolaattorit, ilmanpuhdistimet, laminaarivirtaukset sekä
erilaiset automatisoidut järjestelmät. Aseptisen valmistusjärjestelmän
monimutkaistuminen asettaa tasaiselle tuotantolaadulle haasteita. Monien laitteiden
yhteen liittäminen toimivaksi kokonaisuudeksi kustannustehokkaan tuotannon
saamiseksi ei ole helppoa.

9
2. Silmän rakenne, toiminta ja tyypillisimmät formulaatiot
2.1 Silmän rakenne ja toiminta
Silmän rakenne koostuu erilaisista kalvoista (kuva 1). Niistä uloimmat kerrokset ovat
sarveiskalvo ja sen ympärillä oleva sidekalvo. Sarveiskalvo on läpinäkyvä,
halkaisijaltaan 11,7 mm oleva kalvo, jossa ei ole verisuonitusta, mutta paljon
hermopäätteitä (Järvinen ym. 1995, Gibson 2009). Sarveiskalvo koostuu kolmesta
kerroksesta: uloin epiteeli, keskimmäinen tukiosa ja sisin endoteeli. Sarveis- ja
sidekalvo muodostavat ensimmäisen imeytymisesteen silmään annosteltaville
lääkeaineille (kuva 2). Sarveiskalvon epiteelikerros koostuu pääosin rasva-aineista,
joiden takia se estää 90 % vesiliukoisista ja 10 % rasvaliukoisista lääkeaineista
(Wilson ym 2001, Gibson 2009). Keskimmäinen tukiosa (strooma) on kerroksista
paksuin ja voimakkaasti vesipitoinen, mikä huonontaa vuorostaan vahvasti
rasvaliukoisten aineiden imeytymistä. Sarveiskalvon sisin kerros on endoteeli, joka
koostuu yksittäisestä solukerroksesta, jonka solut ovat kiinnittyneet toisiinsa
aukkoliitoksilla. Endoteelin pinta sisältää Na/K-pumppuja, jotka ohjaavat
sarveiskalvon passiivista veden kulkeutumista ja siten myös sarveiskalvon paksuutta.
Runsas veden virtaus sarveiskalvoon vähentää lääkeaineiden kulkeutumista silmään
imeytymisreitin pidentyessä, mutta kokonaisuudessaan endoteelikerros läpäisee
lääkeaineita yli 200 kertaa enemmän kuin epiteelikerros.
Kuva 1. Silmän rakenne ja yleisimmät annostelukohdat (Wilson ym. 2001)

10
Kovakalvo toimii silmän uloimpien osien huoltojärjestelmänä ja sisältää runsaasti
verisuonia, joiden avulla alueelle tuodaan happea ja ravinteita. Kovakalvon etuosaa
peittää sidekalvo, joka jatkuu silmäluomien alle. Sidekalvo ja kovakalvo toimivat
monille lääkeaineille imeytymisesteenä, ja ne lääkeaineet, jotka pääsevätkin
kulkeutumaan tätä kautta sisään, voivat päätyä kalvojen verisuonistoihin, jotka
pyrkivät kuljettamaan lääkeaineet ulos silmästä systeemiseen verenkiertoon.
Kuva 2. Silmälääkkeiden hyötyosuuteen vaikuttavat tekijät (Kaur ja Smitha 2002)
Ulkoapäin silmää huoltaa ja suojaa kyynelneste ja silmäluomet. Kyynelnestettä
tuottavat kyynelrauhaset ja Meibomin rauhaset. Kyynelfilmi muodostuu kolmesta
kerroksesta. Alin on limakerros, joka sisältää musiinia ja asettuu sarveis- ja
sidekalvon päälle tasoittaen epiteelin ja muodostaen siihen hydrofiilisen pinnan (Holly
ja Lemp 1977, Al-achi ym. 2013). Toinen kerros koostuu kyynelrauhasten erittämästä
vesikerroksesta, joka asettuu alemman hydrofiilisen pinnan päälle. Kauimmaisena
silmän pinnasta on Meibomin rauhasten tuottama lipidikerros, joka vähentää veden
haihtumista silmästä (Holly ja Lemp 1977, Gibson 2009).
Erittynyt kyynelneste muuttaa silmään annostellun valmisteen pH:ta ja laimentaa sen
pitoisuutta, mikä voi heikentää lääkeaineen imeytymistä. Silmäluomet tasoittavat joka
kerta sulkeutuessaan erittynyttä kyynelnestettä silmän pinnalle, mutta samalla ne

11
ohjaavat kerrallaan noin kaksi mikrolitraa nestettä kyyneltiehyeeseen, josta se
kulkeutuu edelleen nenän ja kurkun kautta maha-suolikanavaan. Normaalisti
kyynelnesteen määrä silmässä on noin seitsemän mikrolitraa. Silmässä voi olla
kuitenkin nestettä kerrallaan enimmillään 20–30 µl, josta suurin osa alaluomitaskussa
(Wilson ym. 2001). Tätä suuremmat nestemäärät valuvat silmästä poskille tai
ohjautuvat kyyneltiehyeeseen. Silmään laitetusta nesteestä poistuu noin 16 %
minuutissa ja silmän sisempiin osiin pääsee imeytymään alle viisi prosenttia
annoksesta (Järvinen ym. 1995, Wilson ym. 2001, Gibson 2009). Loput annoksesta
imeytyy systeemiseen verenkiertoon tai valuu pois silmästä.
2.2 Formulaatioiden komponentit ja niiden tehtävät
Suurin ongelma silmälääkinnässä on oikean pitoisuuden saavuttaminen hoidettavalla
alueella (Järvinen ym. 1995, Kaur ja Kanwar 2002, Ali ja Lehmussaari 2006).
Sopivan lääkeainepitoisuuden saavuttaminen imeytymisalueella hyödyttää lääkintää
monella tapaa: se mahdollistaa paremman hyötyosuuden lääkkeelle, tuotetta voidaan
annostella harvemmin ja tuote aiheuttaa vähemmän silmän ärsytystä. Nämä kaikki
asiat tekevät tuotteesta potilaalle käyttäjäystävällisemmän.
Lääkkeen hyötyosuuteen silmässä vaikuttavat erityisesti sen kemialliset
ominaisuudet, annoksen koko, aineiden taipumus muodostaa komplekseja
proteiinien kanssa, silmän kyynelnesteen laimentava vaikutus, hyödytön
imeytyminen, sarveiskalvon heikko läpäisykyky ja silmien räpyttely. Näihin
ominaisuuksiin pyritään vaikuttamaan erilaisilla apuaineilla (taulukko 1), joita lisätään
tarpeiden ja mahdollisuuksien mukaan valmisteisiin. Optimaalisin lääkeaine silmän
pinnalle annosteltaessa on tehokas pienillä annoksilla, pieni molekyylipainoltaan ja
sisältää sekä vesi- että rasvaliukoista luonnetta (Al-Achi ym 2013). Hyötyosuuden
lisäksi lääkkeen formuloinnissa pitää ottaa huomioon lääke- ja apuaineiden
ominaisuuksia, kuten vesiliukoisuus, stabiilisuus ja siedettävyys, mutta myös
vaaditun annoksen konsentraatio sekä hoidettavan silmän kunto. Yleensä lopullinen
valmiste muodostuu kaikkien edellä mainittujen vaatimusten kompromissiksi.

12
Taulukko 1. Silmälääkkeen yleisimmät aineosat ja niiden tarkoitukset valmisteessa.
Aineosat Tarkoitus
Imeytymisen ja läpäisyn parantajat
Parantavat aineen imeytymistä erilaisilla tavoilla, kuten löystyttämällä solujen tiiviitä liitoksia ja lisäämällä membraanin juoksevuutta
Antimikrobiset säilöntäaineet Vähentävät valmisteen biokuormaa ja estävät mikro-organismien kasvua
Antioksidantit Vähentävät lääkkeen hapettumista hapettumalla itse helpommin
Puskurit Pitävät halutun pH-alueen, jotta tuotteen stabiilisuus ja liukoisuus säilyy
Kelatointiaineet Muodostavat komplekseja metallien kanssa, jotka voivat katalysoida hapettumisreaktiota
Ioninvaihtoaineet Kontrolloivat lääkeaineen vapautumista ioninvaihdon avulla, jota esiintyy kyyneleissä
Liukoisuuden lisääjät Lisäävät lääkeaineen liukoisuutta
Suspendointi- ja sakeuttamisaineet
Kohottavat nesteen rakennetta pienentäen partikkelien laskeutumisnopeutta sekä helpottaen aineen uudelleen sekoitettavuutta. Kohottavat lääkkeen säilyvyyttä ja vaikutusaikaa silmässä sekä vähentävät emulsion vaahtoavuutta
Toonisuuden säätäjät Mahdollistavat silmälle isotoniset valmisteet
Lääkeainekuljettimet Lääkeaineelle sopiva kuljetusväline, jolla sitä suojataan tai sen imeytymistä kohdistetaan
Lähde: Al-Achi ym. 2013
Pyrittäessä parantamaan lääkeaineen hyötyosuutta pyritään samalla kiertämään
silmän suojamekanismeja, kuten kyynelnesteen huuhtelevaa ja laimentavaa
vaikutusta. Kehitystyön oleellisin osa onkin ottaa huomioon, ettei valmiste kaikkien
näiden muutosten jälkeen vahingoita silmän kudoksia. Silmän ärsyyntyminen, pistely
ja polttelu lisäävät kyynelnestetoimintaa ja silmien räpyttelyä, jotka tehostavat
lääkeaineen poistumista silmästä ja vähentävät sen imeytymistä.
Edellä mainittujen itse lääkevalmisteen aiheuttamien rajoitteiden lisäksi
tuotekehityksessä otetaan huomioon potilaan tekemä annostelu ja käyttömukavuus
(Gibson 2009). Valmiste, joka aiheuttaa potilaalle ärsytystä, polttelua, pistelyä, näön

13
sumenemista tai epämiellyttäviä systeemisiä haittavaikutuksia, voi olla hänelle syy
hoidon lopettamiseen ennenaikaisesti. Silmävalmisteille ärsytysoireet ovat yleisiä ja
ne liittyvät yleensä suuriin lääkeainekonsentraatioihin. Näitä ongelmia on saatu
vähennettyä esimerkiksi muuttamalla lääkevalmisteita aihiolääkkeiksi. Kuitenkaan
yksistään hyvä formulaatio ei riitä, vaan myös lääkkeen ja pakkauksen
käytettävyyteen on kiinnitettävä huomiota. Silmävalmisteita käyttävät eniten
vanhukset, joille hankala pakkaus voi vaikeuttaa annostelua merkittävästi. Huono
pakkaus voi lisäksi aiheuttaa annoskoon vaihtelua, jos potilaalla on vaikeuksia laittaa
silmätippoja tai hän puristaa pakkausta esimerkiksi liian lujaa tai hiljaa.
Annosteltavan tipan suositeltavana tilavuus on 8–15 µl (Chrai ym. 1973, Wilson ym.
2001, Van Santvilet ja Ludwig 2004). Tippakokoon voidaan vaikuttaa pakkauksen
suunnittelulla – jokainen markkinoilla oleva moni- tai kerta-annospakkaus tuottaa
lähtökohtaisesti sille ominaisen tippakoon. Suurin osa kaupallisista valmisteista
tuottaa 25–70 µl:n tippoja. Silmälääkkeen tilavuuden lisäämisen yli suositeltavan
tippakoon on todettu vähentävän imeytyvän lääkeaineen määrää, koska tilavuuden
lisääntyminen kasvattaa valumisen määrää silmästä. Myöskään alle kahdeksan
mikrolitran tippoja ei yleensä valmisteta, koska se hankaloittaa silmälle sopivan
konsentraation saavuttamista tippaan.
Silmän pinnalle laitettavat lääkkeet imeytyvät kahta reittiä pitkin: sarveiskalvon läpi tai
side- ja kovakalvon läpi, joista jälkimmäistä kutsutaan myös sarveiskalvottomaksi
reitiksi (kuva 3) (Wilson ym. 2001). Suoraan sarveiskalvon läpi kulkeutuvista
lääkeaineista rasvaliukoiset menevät pääosin transsellulaarisesti ja vesiliukoiset
intrasellulaarisesti. Myös viitteitä lääkeaineiden aktiivisesta kuljetuksesta on.
Sarveiskalvoton reitti on usein tehoton, koska imeytynyt lääkeaine usein kulkeutuu
reitillä oleviin verisuoniin, jotka poistavat lääkeainetta systeemiseen verenkiertoon.
Reitti on kuitenkin tärkeä isoille vesiliukoisille molekyyleille, kuten timololimaleaatille
ja gentamisiinille, sekä peptideille.

14
Kuva 3. Lääkkeiden kulkeutumisreitit paikallisessa annostelussa (Wilson ym. 2001).
2.2.1 Lääkeaine
Lääkeaineen valintaan vaikuttavat eniten sen fysikokemialliset ominaisuudet, kuten
liukoisuus, lipofiilisyys, molekyylin paino, muoto ja varaus sekä ionisoitumisaste, jotka
vaikuttavat lääkeaineen kykyyn imeytyä (Gibson 2009). Kuten aikaisemmin todettiin,
sarveiskalvo sisältää sekä lipofiiliset epiteeli- ja endoteelikerrokset että hydrofiilisen
tukikudoskerroksen (strooma), joten parhaiten imeytyvät aineet, joilla on molemman
tyyppisiä ominaisuuksia. Suotuisin jakautumiskerroin lääkeaineelle imeytymistä
ajatellen fysiologisessa pH:ssa 7,4 on kahdesta kolmeen (log P) (Singh ja Shah
1996, Gibson 2009). Sarveiskalvon läpi parhaiten imeytyvät lääkeainemolekyylit,
jotka voivat esiintyä sekä ionisoituneessa että ionittumattomassa muodossa. Näistä
ionittumattomat molekyylit läpäisevät parhaiten lipofiilisen epiteelikerroksen.
Silmälääkinnässä lääkeaineen molekyylin koko ei yleensä toimi imeytymisen
rajoittajana. Vasta yli 500 u molekyylipainoltaan olevat yhdisteet ovat huonosti
imeytyviä silmän kalvolta (Gibson 2009). Lääkeaineen kemiallinen muoto kuitenkin
vaikuttaa sen ominaisuuksiin paljon. Yleinen tapa muokata lääkeaineen kemiallisia

15
ominaisuuksia onkin vaihtaa lääkeaineen suolamuoto, jolla voidaan muuttaa
lääkeaineen liukoisuutta ja lipofiilisyyttä (Gibson 2009).
Lääkkeen kehityskaaren alkuvaiheessa seulotaan suuri määrä lääkeainemolekyylejä
läpi ja poimitaan jatkokehitykseen soveltuvimmat lääkeainemolekyylit muun muassa
niiden fysikokemiallisten ominaisuuksien perusteella (Gibson 2009). Jos tarpeeksi
hyvää lääkeainemolekyyliä ei löydy, voidaan yrittää soveltuvimmasta molekyylistä
muokata sopiva, jäljittelevä inaktiivinen aine tai aihiolääke. Tällaisten aineiden
tarkoituksena on entsymaattisella tai kemiallisella reaktiolla hajota silmässä itse
vaikuttavaksi aineeksi. Aihiolääketeknologialla pyritään parantamaan lääkeaineen
imeytymistä muuttamalla sen liukoisuutta ja lipofiilisyyttä, mutta sillä pyritään usein
myös parantamaan lääkkeen stabiilisuutta, tehokkuutta, spesifisyyttä ja
käyttäjäystävällisyyttä tai vähentämään lääkeaineen systeemisiä sivuvaikutuksia
(Järvinen ym. 2005, Gibson 2009). Lääkeaineen puutteellisten ominaisuuksien
korjaaminen tai kiertäminen myöhemmissä formuloinnin vaiheissa on hankalaa,
mutta käytännössä tämä tilanne on melko yleinen (Gibson 2009).
2.2.2 pH:n säätö
Silmävalmisteen pH on tärkeä ominaisuus tuotteen käytettävyyden kannalta, koska
se usein määrää aineiden liukoisuuden, stabiilisuuden ja sarveiskalvon
läpäisevyyden (Ali ja Lehmusvaara 2006, Al-Achi ym. 2013). Silmässä vallitseva pH
on tavallisesti 7,4, ja valmisteen pH pyritään saamaan hyvin lähelle sitä, yleensä
välille 7,14–7,8. Usein tämä ei ole kuitenkaan mahdollista, ja tältä pH-väliltä
joudutaan poikkeamaan esimerkiksi lääkeaineen säilyvyyden tai liukoisuuden takia.
Tällainen poikkeama luonnollisesta pH:sta lisää silmään kohdistuvaa ärsytystä ja
epämukavuutta. Ne lisäävät vuorostaan silmien räpyttelyä ja kyynelnesteen
muodostumista, joka tehostaa lääkkeen poistumista silmästä vähentäen sen
imeytymisaikaa. Yleensä valmisteen lopullinen pH on asetettu parhaaseen
kompromissiin stabiilisuuden ja hyötyosuuden kanssa siten, että se aiheuttaa
mahdollisimman vähän silmän ärsyyntymistä. Valmisteen puskurointiominaisuus
pitää olla kuitenkin niin pieni, että kyynelneste voi neutralisoida valmisteen pH:n.

16
Silmävalmisteissa käytetyimpiä puskuriaineita ovat boraatti-, fosfaatti- ja
sitraattipuskurit.
2.2.3 Toonisuuden säätäjät
Silmän kyynelnesteen normaali osmolaliteetti on 290–310 mOsmkg-1, joka on lähellä
suolaliuoksen osmolaliteettia (Wilson ym. 2001). Silmä sietää osmolaliteetin vaihtelua
välillä 100–640 mOsmolkg-1 hyvin, tätä suuremmat vaihtelut osmolaliteetissa
aiheuttavat silmässä ärsytystä ja johtavat kyynelnesteen ja räpyttelyn lisääntymiseen.
Normaalisti kaupalliset valmisteet asettuvat osmolaliteetiltaan välille 260–330
mOsmkg-1. Lievästi hypotonisella lääkeaineliuoksella saadaan veden virtaus
kulkemaan kohti sarveiskalvoa, jolloin lääkeaineen kulkeutuminen silmään tehostuu.
Pinnalla vallitseva hypotoninen tila kestää kuitenkin vain minuutin tai kaksi, koska
vettä virtaa voimakkaasti sarveiskalvoon.
2.2.4 Säilytysaineet
Silmävalmisteet, jotka on pakattu moniannospakkauksiin, sisältävät antimikrobisen
säilöntäaineen (Ali ja Lehmusvaara 2006). Säilöntäaineen tarkoituksena on estää
potilasta annostelemasta mikrobiologisesti kontaminoitunutta valmistetta silmäänsä.
Hyvä säilöntäaine on tehokas jo pienillä pitoisuuksilla, tehoaa useisiin bakteereihin,
liukenee hyvin valmisteeseen, kestää valmisteen steriloinnin, on yhteensopiva
valmisteen muiden aineosien ja pakkausmateriaalien kanssa sekä stabiili ja tehokas
koko tuotteen eliniän (Gibson 2009). Soveltuvimmat säilytysaineet silmävalmisteisiin
ovat kvaternääriset ammoniumyhdisteet, kuten bentsalkoniumkloridi, parabeenit,
klooributanoli ja 2-poly(etyylialkoholi). Uudempia säilytysaineita ovat peroksidia
tuottavat yhdisteet, kuten Purite®.

17
2.2.5 Kostutusaineet (humektantit)
Kostutusaineet ovat erilaisia pinta-aktiivisia aineita, jotka laskevat nesteen ja
partikkelien kontaktikulmaa parantaen partikkelien liukenemista nesteeseen (Ali ja
Lehmusvaara 2006). Näiden käytössä tulee ottaa huomioon kyynelnesteen
pintajännitys, joka on 43,6–46,6 mNm-1 (Al-Achi ym. 2013). Suuri ero tähän voi
johtaa kyynelnesteen epävakauteen sekä vaikuttaa musiinien aineita sitovaan
vaikutukseen ja sarveiskalvon mikrovilluksiin. Nämä vaikutukset voivat tulla esiin
erityisesti mukoadhesiivisiin ominaisuuksiin perustuvissa formulaatioissa.
2.2.6 Imeytymisen edistäjät
Imeytymisen edistäjien tarkoitus on muokata solun rakenteita tai toimintaa, jotta
lääkeaineiden kulkeutuminen kalvojen läpi trans- ja parasellulaarisesti olisi
helpompaa (Al-Achi 2013). Lääkeaineet kulkeutuvat molempia reittejä pitkin, mutta
yleensä toinen niistä on pääasiallinen imeytymiskanava. Samalla tavalla imeytymisen
edistäjät voivat vaikuttaa yksittäiseen solun ominaisuuteen tai moneen
ominaisuuteen yhtä aikaa (Burstein 1984, Al-achi 2013). Vaikutuskohteina voivat olla
fosfolipidien kaksoiskerros, transmembraani-proteiinien liitokset tai solujenvälisten
tiiviiden- ja vyöliitosten rakenteet.
Esimerkiksi säilytysaineenakin tunnettu bentsalkoniumkloridi kykenee pitkän
hydrofobisen hiilivetyketjunsa avulla kiinnittymään lipidikaksoiskerrokseen ja
parantamaan varautuneiden molekyylien kykyä sen läpäisyyn (Burstein 1984).
Bentsalkoniumkloridin tapaiset aineet kykenevät myös häiritsemään solujen tiiviiden
liitosten rakenteita tai niiden ympärillä toimivaa entsyymitoimintaa. Tällöin
lääkeaineet pääsevät kulkeutumaan epiteelisolujen soluvälitilaan, josta niiden
helpompi kulkeutua sisempiin osiin, kun ohitettavana on enää strooma ja
endoteelisolukerros (Burstein 1984, Hornof ja Berkop-schnurch 2002).
Toiseen solujen väliseen liitokseen, vyöliitokseen, voidaan vaikuttaa aineilla, jotka
muodostavat kelaatteja Ca2+-ionien kanssa, joiden läsnäolo tekee vyöliitoksesta tiiviin

18
(Al-achi ym. 2013). Kaikkia imeytymisen edistäjiä tulee käyttää kuitenkin harkiten,
koska osa niistä aiheuttaa erityisesti pitkäaikaisessa käytössä ärsytystä ja vahinkoa
silmien kudoksille (Hornof ja Berkop-schnurch 2002, Furrer ja Mayer 2002).
2.2.7 Viskositeetin kohottajat ja biokiinnitysaineet
Yleisin tapa parantaa imeytyneen lääkeaineen määrää on pidentää valmisteen
vaikutusaikaa silmän pinnalla. Vaikutusaikaa voidaan pidentää esimerkiksi
kohottamalla lääkkeen viskositeettia tai lisäämällä valmisteeseen musiineihin
tarttuvia polymeerejä (Ali ja Lehmussaari 2006, Gibson 2009).
Liuosten viskositeettia kohottamaan käytetään usein niihin soveltuvia polymeerejä
(Wilson ym. 2001). Yleisimmät tähän käytetyt aineet ovat polyvinyylialkoholi (PVA) ja
erilaiset selluloosajohdokset, kuten hydroksipropyylimetyyliselluloosa (HPMC).
Kaikille valmisteille on optimaalinen viskositeetti, jossa valmisteen imeytymisaika on
pidentynyt, mutta se ei kuitenkaan hankaloita lääkeaineen vapautumista
formulaatiosta, ärsytä liikaa silmän pintaa tai sumenna näköä liikaa (Nayak ym. 2012,
Al-Achi ym. 2013). Käytetyimmistä yhdisteistä PVA:n erikoisuutena on veden
pintajännityksen laskeminen, ja eri selluloosajohdokset taas kattavat laajasti
tarvittavan viskositeettialueen (Wilson ym. 2001). Molemmat aineista ovat laajassa
käytössä, ne on todettu yhteensopiviksi useiden eri lääkeaineiden kanssa, ja lisäksi
ne stabiloivat silmän kyynelfilmiä.
Musiineihin kiinnittyvät polymeerit kuuluvat biokiinnitysaineisiin (Wilson ym 2001,
Kaur ym. 2002). Näitä molekyylejä on olemassa niin syynteettisiä, puolisynteettisiä
kuin luontaisiakin (Robinson ja Mlynek 1995). Yhteistä näille polymeereille on, että ne
sisältävät paljon hydrofiilisiä ryhmiä, kuten karboksyyli-, hydroksyyli-, amidi- ja
sulfaattiryhmiä. Näiden ryhmien tarkoitus on kiinnittyä sarveiskalvon pinnalla oleviin
musiineihin elektrostaattisten, hydrofobisten interaktioiden ja vetysidoksien avulla.

19
2.2.8 Suspension muodostajat
Suspendointiaineiden tarkoituksena on muuttaa ulkoisen faasin koostumusta siten,
että kiinteiden partikkelien sedimentaatio on mahdollisimman hidasta (Ali ja
Lehmusvaara 2006). Valmisteen formulaatiossa täytyy kuitenkin ottaa huomioon, että
valmiste pysyy myös helposti annosteltavana ja muutenkin soveltuvana
silmälääkintään. Suspendointiaineen valintaan vaikuttavat halutut vehikkelin
muodostusominaisuudet, yhteensopivuus muiden komponenttien kanssa ja
myrkyttömyys soluille. Yleisesti tähän käytettyjä aineita ovat selluloosajohdokset,
kuten metyyliselluloosa, karboksyylimetyyliselluloosa,
hydroksypropyylimetyyliselluloosa sekä synteettiset polymeerit, kuten karbomeerit,
poloksameerit ja polyvinyylialkoholit.
2.2.9 Faasin muuttajat
Faasitransitiossa jokin liuoksen komponentti pystyy muuttamaan valmisteen
olomuodon silmän pinnalla nestemäisestä geelimäiseksi tai kiinteäksi. Valmisteen
faasin muutoksella pyritään pidentämään sen vaikutusaikaa silmässä (Wilson ym.
2001). Faasin vaihto perustuu muuttuvan aineen pH:n, lämpötilan tai ionivahvuuden
vaihtumiseen.
Lämpötilaperustaisiin muutoksiin käytetään yleensä poloksameerejä ja pluronic F-
127:ää. Näillä aineilla faasitransitio tapahtuu 34 °C:ssa, joka on silmälääkintään
soveltuva lämpötila (Vadnere ym. 1984). F-127 on huoneenlämmössä liuosmaisessa
tilassa, mutta annostelun jälkeen sen lämpötila kohoaa yli 34 °C:n, jolloin se vaihtaa
olomuotoaan geelimäiseksi. F-127:n faasitransitio perustuu mekanismiin, jossa
valmisteen liuotin poistuu asteittain, misellien aggregoituminen lisääntyy ja
polymeeriverkkojen kietoutuminen lisääntyy. Lämpötilaperustaisen faasitransition
heikkous on, että valmisteessa poloksameerikonsentraation täytyy olla yli 25 %
(w/w), mikä vähentää sen siedettävyyttä silmässä (Wilson ym. 2001).

20
Faasitransitio, joka perustuu komponentin pH:n vaihtumiseen, tehdään esimerkiksi
käyttäen selluloosa-asetaatti-ftalaattia (Wilson ym. 2001). Tällä aineella on hyvin
pieni viskositeetti pH:ssa viisi, mutta se muodostaa hyvin nopeasti geelin pH:n
noustessa kyynelnesteen tasolle 7,4:ään. Carpopol 934 -polymeereillä on
samantyyppinen ominaisuus: pH:ssa 4,4 sen aiheuttama viskositeetti on 25 cps ja
pH:ssa 7,4 vastaavasti 9000 cps (Nayak ym. 2012).
Ionivahvuuteen perustuvissa faasitransitioissa käytetään usein Gellan gum -
polysakkaridia. Valmisteessa nestemäisenä oleva Gellan gum muuttuu annostelun
jälkeen kyynelnesteen vaikutuksesta kirkkaaksi geeliksi (Mazuel ym.1989, Wilson
ym. 2001). Geeliytymiseen vaikuttaa erityisesti kyynelnesteessä olevat yksi- ja
kaksiarvoiset kationit. Ionivahvuuteen perustuvan faasitransition etuna on erityisesti
helpompi varastointi, koska siinä tuotteen säilyttäminen ei vaadi yhtä tarkkaa
lämpötilaseurantaa, kuten esimerkiksi lämpötilaan perustuvan transition tuotteissa.
2.3. Formulaatiotyypit silmälääkinnässä
2.3.1 Perinteiset formulaatiot
Perinteiset annostelumuodot silmälääkevalmisteissa ovat liuokset, suspensiot, geelit
ja voiteet (Ali ja Lehmussaari 2006) (Kuva 4, taulukko 2). Niitä on kritisoitu paljon
muun muassa huonosta hyötyosuudesta, mutta ne ovat edelleen käytetyimpiä
annostelumuotoja silmälääkinnässä. Perinteisten annostelumuotojen rinnalle on
kehitetty uusia innovatiivisempia lääkemuotoja, kuten silmälamellit,
aihiolääkevalmisteet sekä mikro- ja nanopartikkelit, joiden käyttöiät voivat vaihdella
tunneista vuosiin. Näiden lisäksi on erilaisia silmän sisään annosteltavia
lääkemuotoja, mutta tässä katsauksessa keskitymme silmän ulkopuolelle
annosteltaviin valmisteisiin, jotka eivät tarvitse kirurgista toimenpidettä annosteluun.

21
Kuva 4. Silmävalmisteiden käyttöaika (Amo ja Urtti 2008)
Taulukko 2 Formulaatiokomponentit, joita käytetään yleisesti moniannossilmätippa-valmisteissa. (Y) Komponentti sisältyy, (O) Komponentti voidaan lisätä, ( ) Kompo-nenttia ei käytetä
Komponentti Liuos Suspensio Geeli Voide Lamelli
Lääkeaine Y Y Y Y Y
Lääkekuljetin O O O O O
Vesi Y Y Y O
Puskurointiaine Y Y Y
Toonisuuden säätäjä O O O
Säilytysaine Y Y Y Y
Viskositeetin säätäjä O O
Liuotin O O
Suolat O O O O
Luonnollinen kiinnitysaine O O O O
Suspensionmuodostaja Y
Faasinvaihtaja Y
Imeytymisen parantaja O O O O
Kytketyt polymeerit Y
Vaha/öljy/vaseliini Y
Lähde: Lang 1995

22
2.3.1.1 Liuokset
Vesipohjaiset liuosvalmisteet ovat edelleen yleisin silmälääkemuoto (Ali ja
Lehmussaari 2006, Gibson 2009). Suurin osa lääkeaineista on vesiliukoisia, mutta
tilanteissa, jossa lääkeaine ei itse ole vesiliukoinen, yritetään lääkeaineesta muokata
vesiliukoinen suola tai tehdä rasvapohjainen valmiste. Liuosmaisen valmisteen etuja
ovat pääsääntöisesti yksinkertaisempi valmistaminen teollisessa mittakaavassa sekä
helpompi annosteltavuus. Liuoksien heikkoutena taas on nopea poistuminen
imeytymisalueelta, jolloin lääkkeen hävikki on suurempi. Liuoksissa voidaan käyttää
muun muassa lääkekuljettimia, puskurointiaineita, toonisuuden säätäjiä,
säilytysaineita, viskositeetin säätäjiä, liuottimia, biokiinnitysaineita ja imeytymisen
parantajia (taulukko 2).
2.3.1.2 Geelit
Geelimäisillä valmisteilla saavutetaan liuoksia pidempi vaikutusaika silmässä (Gibson
2009). Monia liuosvalmisteita onkin muutettu geelimäisiksi, jotta tuotteelle saadaan
pidempi annosteluväli ja siten tuote saadaan käyttäjäystävällisemmäksi.
Geelimäisten valmisteiden etuna verrattuna esimerkiksi suspensioihin on niiden
helppo silmätippamainen annostelu (Robinson ja Mlynek 1995). Geelit muodostetaan
usein faasinsäätäjillä, joista on kerrottu aikaisemmin. Niissä muutoksen perustana
toimii valmisteen lämpötila, pH tai vallitseva ionivahvuus (Gupta ym. 2007, Gibson
2009).
2.3.1.3 Suspensiot
Jos lääkeaine on huonosti veteen liukeneva, valmisteesta muodostetaan usein
suspensio (Ali ja Lehmusvaara 2006). Suspensioissa lääkeaine pyritään saamaan
alle 10 µm kokoisiksi partikkeleiksi (Al-Achi ym. 2013). Liuosmaisiin valmisteisiin
nähden suspensiolla saavutetaan pidempi vaikutusaika silmässä suuremman
viskositeetin ansiosta, mutta lääkeaineen hyötyosuuteen vaikuttaa tämän lisäksi
myös lääkeaineen kyky liueta suspensiosta kyynelnesteeseen ja imeytyminen silmän
pinnalta (Sieg ja Robinson 1975, Wilson ym. 2001, Gibson 2009, Al-Achi ym. 2013).

23
Hyvän silmäsuspension valmistaminen on monimutkaisempaa kuin liuosmaisten
valmisteiden. Lääkeaineen parantunut kemiallinen säilyvyys voi vaihtua huonoon
fysikaalisen säilyvyyteen. Formulaatiossa on otettava huomioon suspension
laskeutuminen, kakun muodostuminen, partikkelikoon kasvaminen, kidemuodon
muutokset, uudelleensekoitettavuus, säilyvyys ja helppo tuotettavuus (Kaur ja
Kanwar 2002, Ali ja Lehmusvaara 2006, Gibson 2009). Partikkelikoolla on suuri
merkitys suspension fysikaalisessa säilyvyydessä ja hyötyosuudessa (Sieg ja
Robinson 1975, Ali ja Lehmusvaara 2006). Partikkelikoko vaikuttaa tuotteen
sedimentaatioon, kasautumiseen ja uudelleensekoitettavuuteen. Partikkelikoko ja
kidemuoto eivät kuitenkaan ole suspensiossa stabiileja, vaan niitä voivat muuttaa
erityisesti varastointiolosuhteet ja varastoinnin kesto (Kaur ja Kanwar 2002).
Muutoksissa pienet partikkelit voivat liueta ja suurimmat kasvaa edelleen, ja tällainen
partikkelikoon kasvaminen voi aiheuttaa pidentyneen vaikutuksen ja hitaamman
liukenemisen (Ali ja Lehmusvaara 2006). Formuloijan on tunnettava valmisteen
ainesosat ja niiden käyttäytymiseen vaikuttavat asiat, kuten kostuminen, zeta-
potentiaali, aggregoituminen, sedimentaatio ja virtauskäsitteet, jotta hän kykenee
muodostamaan tuotteesta tehokkaan, mutta samalla hyvin muodostuneen
suspension. Lääkeaineen partikkelikoon muuttumista voidaan vähentää esimerkiksi
oikean lääkeaineen suolamuodon valinnalla, mutta samalla sen vaikutus lääkeaineen
liukenemiseen silmässä on otettava huomioon (Gibson 2009). Yksi suspension
heikkouksista on ongelmallinen sterilointi. Lääkeaine voi sterilointiprosessissa
menettää tehoaan tai siihen voi tulla morfologisia muutoksia, jotka voivat vaikuttaa
valmisteen ominaisuuksiin. Nämä muutokset täytyy tunnistaa ja ottaa huomioon jo
suunnitteluvaiheessa.
Uudemman sukupolven suspensiot perustuvat mikropalloihin ja mikrohiukkasiin, jotka
sisältävät lääkeainetta (Kaur ja Kanwar 2002). Partikkelien toiminta
imeytymisalueella voi perustua kemiallisiin reaktioihin, partikkelin hajoamiseen tai
ionienvaihtoon. Mikropartikkeleita on yhdistetty myös muihin parannuskeinoihin,
kuten biokiinnittyviin aineisiin ja nanopartikkeleihin. Näiden yhdistelmien on todettu
lisäävän lääkeaineen määrää ja pidentävän sen vaikutusta lasiaisessa (Zimmer ja
Kreuter 1995, Genta ym. 1997). Suurimmat hankaluudet mikropartikkelisovelluksissa
liittyvät stabiilisuuteen, annoksien yhdenmukaisuuteen, lääkeaineen vapautumiseen
ja teollisen mittakaavan valmistukseen. (Kauer ja Kanwar 2002).

24
2.3.1.4 Voiteet
Silmävoiteet valitaan perinteisistä annosmuodoista yleensä, kun lääkevalmiste
sisältää vedessä epävakaita lääkeaineita, joille vaikutusaika silmässä on tärkeä (Al-
Achi ym 2013). Silmävoiteiden suurimmat ongelmat ovat niiden aiheuttama näön
sumeneminen, epätarkka annostelu ja hankala sterilointi (Ali ja Lehmussaari 2006).
Vedettömien voiteiden pohjat koostuvat yleensä valkovaseliinista ja mineraaliöljystä
tai muunnellusta vaseliinipohjasta (Gibson 2009, Al-Achi ym. 2013). Voiteiden
tarkoitus on sulaa annostelualueellaan kehon lämmön vaikutuksesta. Voiteiden
etuina usein on muita vähäisempi silmän ärsytys ja hyvä levittyminen silmän pinnalle.
Vähäisempi silmän ärsytys johtuu osittain siitä, että niiden valmistuksessa käytetään
vähemmän erilaisia komponentteja (taulukko 2). Voiteiden sterilointi on suspension
tapaan ongelmallista. Niihin voidaan käyttää lämpösterilointia tai ne voidaan
valmistaa aseptisesti steriileistä lähtöaineista (Ali ja Lehmusvaara 2006, Gibson
2009). Kuitenkin yleisimpiä tapoja steriloida silmävoiteita ovat soveltuvan
membraanin läpi suodatus tai kuivailmasterilointi.
2.2.1.5 Silmälamellit ja linssit
Silmälamellit kuuluvat valmisteryhmään, jolla haetaan jatkuvaa, pitkää ja tasaista
lääkeainepitoisuutta silmään. Tässä työssä ne jaotellaan kahteen tyyppiin:
biohajoavat (soluble, bioerodible ja biodegradable) ja liukenemattomat (insoluble,
nonerodible) (Gibson 2009). Ensimmäisiä mainintoja näiden tyyppisistä valmisteista
kuvattiin Britannian farmakopeassa vuonna 1948, kyseessä oli atropiinia sisältävä
liivatekiekko (Furrer ym. 2008). Ensimmäinen kaupallinen valmiste, liukenematon
Ocusert™ tuli myyntiin Yhdysvalloissa 1974. Silmälamelli asetetaan silmäluomen ja
sarveiskalvon väliin, josta se vapauttaa lääkeainetta tasaisesti (Amo ja Urtti 2008).
Joistakin polymeereistä voidaan tehdä myös silmän sisään laitettavia valmisteita.
Liukenemattomien silmälamellien etu on tasaisempi lääkkeen vapautuminen
valmisteesta, koska vapautumista säätelee kalvo (Furrer ym. 2008, Al-Achi ym.
2013). Liukenevissa valmisteissa rungon liukenemisnopeus on pääasiallinen
lääkeaineen vapautumisen säätelijä, minkä takia tuotteella on hankalaa saavuttaa

25
nolla-asteen kinetiikkaa. Ocusert™:n tapaiset liukenemattomat varastosilmälamellit
sisältävät kolme pääosaa: lääkeaineen, lääkkeen kuljetusyksikön ja alustan.
Kuljetusyksikkö sisältää lääkevaraston, kuljetusaineen ja vapautumisen säätelijän.
Ocusert™:n tapauksessa kuljetusaineena on algiinihappo ja vapautumisen
säätelijänä toimii etyleenivinyyliasetaatti (EVA) yhteispolymeeri -membraani.
Valmiste vapauttaa lääkeainetta seitsemän vuorokauden ajan.
Liukenevat silmälamellit ovat potilaalle helppokäyttöisempiä kuin liukenemattomat,
koska niitä ei tarvitse poistaa silmästä vaikutuksen loputtua. Liukenevat valmisteet
tehdään kiinnittämällä lääkeaine liukenevista materiaaleista valmistettuun runkoon.
Yleisimpiä rungon valmistusaineita ovat hydroksipropyyliselluloosa, hyaluronihappo,
karbomeerit ja polyakryylihappo. Tuotannollisena etuna liukenevissa silmälamelleissa
on, että osa valmisteista voidaan valmistaa puristamalla, ja ne voidaan steriloida
gammasäteilyllä (Al-Achi ym. 2013). Liukenevien valmisteiden vaikutusaika on
yleensä 12–24 tuntia, mutta on myös valmisteita, kuten Posurdex®, joiden
vaikutusaika on pitempi – jopa kuusi viikkoa (Amo ja Urtti 2008).
Molempien valmistetyyppien, liukenemattomien ja liukenevien silmälamellien,
heikkoutena on lamellin mahdollinen poistuminen silmästä esimerkiksi nukkuessa
sekä erityisesti vanhuksille hankala valmisteen asettaminen ja pois ottaminen (Furrer
ym. 2008, Al-Achi ym 2013).
Lamellien liukeneminen voi tapahtua kolmella tavalla riippuen rungon polymeereistä
(kuva 5) (Merkli ym. 1995). Liukenevissa valmisteissa lääkeaineen vapautuminen
perustuu polymeerirungon hydrolyyttiseen tai entsymaattiseen pilkkoutumiseen.
Tyypissä yksi vesiliukoiset polymeerit vapautuvat, kun niitä yhdistävät hydrolyyttisesti
epävakaat sillat hajoavat. Polymeerejä yhdistävät sillat hydrolysoituvat yleensä
nopeasti eikä menetelmä siksi sovellu nopeasti liukeneville lääkeaineille. Tyypissä
kaksi liuotettava polymeeri on tehty liukenemattomaksi vaihtamalla sopivia
sivuryhmiä polymeeriin. Polymeerin ryhmien vaihtuessa kemiallisissa reaktioissa
muuttuu polymeeri taas liukoiseksi. Järjestelmä soveltuu myös nopeammin
liukeneville valmisteille, joskin rakenteen saaminen optimaaliseksi on
haasteellisempaa näissä tapauksissa johtuen rakenteen turpoamisesta hidastetussa
vapauttamisessa. Tyypissä kolme polymeerin runko on kokonaisuutena
liukenematon, mutta siihen on lisätty vesiliukoisia molekyylejä, jotka pilkkovat sen

26
pieniksi paloiksi mahdollistaen niiden liukenemisen. Kolmostyyppiä voidaan pitää
varsinaisesti biohajoavana ja, koska sen hajoamistuotteet eivät ole toksisia, voidaan
näistä tehdä myös silmän sisään laitettavia valmisteita. Eri mekanismit voivat myös
yhdistyä valmisteissa. Lääkevalmisteen hankala käytettävyys sitä eniten tarvitsevilla
on haitannut silmälamellin suosiota, mutta eläinlääkinnässä sille on löytynyt
käyttökohteita (Furrer ym. 2008). Biokiinnitysaineita on tutkittu tehostamaan
imeytymistä myös lamellien toiminnassa. Lamellit valmistetaan pursottamalla 140–
160 °C lämpötilassa ja 200–300 kPa paineessa, mikä rajoittaa mahdollisia
lääkeaineita, joita voidaan käyttää.
Kuva 5. Lamellin rungon pilkkoutumistavat (Merkli ym. 1995)
Silmälamellien lisäksi on käytössä myös liukenemattomia piilolinssejä, jotka
sisältävät lääkeainetta (Gibson 2009). Niiden käyttöä on tutkittu jo 1960-luvulta
saakka, ja erilaisia sovelluksia tutkitaan tänäkin päivänä (Peng ja Chauhan 2011).
Yleisimmin käytetään poly-2-hydroksietyylimetakrylaatista valmistettuja pehmeitä
linssejä, joita voidaan käyttää lääkeaineen annostelun lisäksi myös näön
korjaamiseen (Kaur ja Kanwar 2002). Linssin polymeerit voivat absorboida jopa 80 %
painostaan vettä (Mundapa 2011). Lääkeaineen ei ole tarkoitus reagoida linssin
rakenteiden kanssa, vaan se varastoituu absorboituneen nesteen mukana
hetkellisesti linssiin. Lääkeaine imeytetään linssiin upottamalla se lääkeliuokseen
ennen silmään asettamista, annostelemalla lääkeliuos linssin päälle asettamisen
jälkeen tai annostelemalla lääkeliuos linssin koveralle puolelle ennen linssin
asettamista silmään (Kaur ja Kanwar 2002, Souza ym 2013). Suurin osa

27
varastoituneesta lääkeaineesta kuitenkin on poistunut linssistä muutaman tunnin
kuluttua, joten se ei muodosta vielä pitkäkestoista valmistetta. Kontrolloitu
pitkäaikainen vapautuminen linssistä vaatii, että linssin kanssa käytetään jotain
muuta kuljetussysteemiä, kuten mikroemulsioita, liposomeja tai nanopartikkeleita
(Gulsen ja Chauhan 2005, Danion ym 2007, Garbwal ym. 2012). Linssien
heikkoutena on tarkan lääkeainemäärän arvioinnin vaikeus linssissä, nopea
lääkeaineen poistuminen ilman erillistä vapautusmekanismia ja kallis valmistaminen
(Mundapa 2011).
2.3.2 Uudemmat silmälääkkeiden kuljetusmekanismit
2.3.2.1 Liivatesieni Gelfoam®
Gelfoam®:ia, joka on valmistettu siannahan liivatteesta, on tutkittu uutena
mahdollisuutena parantaa erityisesti suurien vesiliukoisten lääkeainemolekyylien
kulkeutumista silmään. Se asetetaan lamellien tapaan silmäluomen alle, jossa se
imee nestettä itseensä, turpoaa ja pehmenee (Hämäläinen ym. 1998, Simamora ym.
1998). Gelatiinisieni Gelfoam® voi sisältää kuluvan rungon, joka toimii lääkeaineen
vapauttajana, tai lääkeaine voidaan imeyttää suoraan liivatesieneen (Hämäläinen
ym. 1998, Simamora ym. 1998, Negvesky ym. 2000). Suurin hyöty lamelleihin
verrattuna on kostumisen aiheuttaman liivatesienen pehmentyminen silmässä, mikä
vähentää silmän ärsyyntymistä. Gelfoam® on osoittanut lupaavia merkkejä
sopivuudesta erityisesti silmän tutkimus- ja leikkauskäytössä pidentämään ja
parantamaan pupillin laajentumista sekä kuljettamaan pilokarpiinia silmään
(Simamora ym. 1998, Negvesky ym. 2000).
2.3.2.2 Iontoforeesi
Iontoforeesi on menetelmä, jossa lääkeainetta kuljetetaan aktiivisesti sarveiskalvon
läpi käyttäen hyväksi yleensä yhden tai kahden milliampeerin sähkövirtaa (Rupenthal
ja Alany 2008). Intoforeesin ideaa on tutkittu jo 1950–luvulla, ja sitä on käytetty
lääkeainekuljetukseen monissa muissa kudoksissa. Minuutista neljään minuuttiin

28
sarveis-, side- ja kovakalvon läpäisevillä iontoforeesihoidoilla pystytään
saavuttamaan jopa monikymmenkertaisia lääkeainepitoisuuksia silmän sisempiin
osiin verrattaessa perinteiseen silmätippojen annosteluun (Eljarrat-Binstock ym.
2005). Hoitomuodon soveltumista myös kotikäyttöön on tutkittu lupaavin tuloksin
(Halhal ym. 2004). 17:stä kokeiluun osallistuneesta potilaasta 15:n hoito onnistui
täysin ja lopuilla kahdella osittain, ja lisäksi hoito osoittautui hyvin siedetyksi.
Iontoforeesin turvallisuutta ja siedettävyyttä on tutkittu myös pitempiaikaisessa
käytössä, ja tulokset osoittavat hoidon olevan hyvin siedettyä ja turvallista (Patane
ym. 2013). Laitteiston hankala käyttö ja lyhytaikainen apu sekä riski kudosvaurion
syntyyn jarruttavat tekniikan laajempaa hyödyntämistä. Hoidolla on kuitenkin
potentiaalia erityisesti lyhyissä hoidoissa, joissa se tehostaa lääkkeen kulkeutumista
silmään ja vähentää annostelukertoja päivässä. Laitteiston kehittymisen myötä myös
käyttöturvallisuus ja käytettävyys varmasti parantuvat ja järjestelmän
käyttömahdollisuudet laajenevat.
2.4 Lääkepakkaus
Pakkausmateriaalit ovat tärkeässä osassa lääkevalmisteessa, koska ne määrittävät
omalta osaltaan valmisteen säilyvyyden ja käytettävyyden. Silmälääkinnässä
pakkausmateriaalit ovat kuitenkin vielä tärkeämmässä osassa, koska kaikkien
tuotteiden tulee olla steriilejä, ja niiden käyttö pitää usein soveltua erityisesti
vanhemmille ihmisille. Lääkemuoto, siinä käytetyt komponentit ja pakkauksen
käyttötarkoitus määrittävät pitkälti, millainen pakkaus tuotteelle valitaan (Gibson
2009). Silmälääkinnässä käytetään monenlaisia pakkauksia: perinteisillä
lääkemuodoilla liuoksella, suspensiolla, geelillä ja voiteella on sekä kerta- että
moniannospakkauksia niin lasisina kuin muovisina. Teollisuus on siirtynyt tiputtimilla
varustetuista lasisista pulloista ja ampulleista pääasiassa muovisiin pakkauksiin,
joissa on integroidut annostelijat. Muovisten pakkausten etuina ovat suora
annosteltavuus pullosta sekä parempi kestävyys tuotannon ja varastoinnin aikana
(Agalloco ja Akers 2008, Gibson 2009). Lasipullojen käyttö rajoittuu nykypäivänä
lähinnä tuotteisiin, joilla on stabiilius- tai yhteensopivuusongelmia nykyisten
muovipakkausten kanssa. Valitun pakkauksen tulisi täyttää ainakin seuraavat

29
ominaisuudet: pakkauksen materiaalit ovat farmakopeoiden mukaisia ja täyttävät
säädetyt vaatimukset, pakkaus on yhteensopiva valmisteen aineosien kanssa,
pakkaus pitää tuotteen stabiilina, valmiste voidaan steriloida pakkauksessa, pakkaus
säilyttää valmisteen steriiliyden sen käyttöiän, pakkaus on turvasinetöity sekä
mahdollistaa helpon annostelun potilaalle (Löfgren ym. 2003, Gibson 2009).
Muovipulloja on monenlaisia, mutta yleisimpiä ovat perinteisellä tiputinosalla
varustetut tai blow-fill-seal (BFS) -tyyppisesti valmistetut pullot ja pipetit. Ne
valmistetaan yleensä polyetyleenistä (PE) tai polypropyleenistä (PP), joita pidetään
reagoimattomina materiaaleina. Niiden tasapainoiset ominaisuudet helpottavat
teollisuuden ja käyttäjän toimintaa, koska ne mahdollistavat laajan muokattavuuden
pakkaukselle, kirkas pakkaus mahdollistaa valmisteen silmämääräisen tarkistamisen,
ja samalla kuitenkin tuote säilyttää helpon avattavuuden ja käytettävyyden (Löfgren
ym. 2003). Muovipakkausten kiinteät tippanokat ovat myös mikrobiologisesti
parempia kuin lasipullojen pipetit, koska pullosta irroitettavat pipetit kontaminoituvat
helposti annosteltaessa, jos ne lasketaan esimerkiksi pöytätasolle. Muovien etuina
teollisesta näkökulmasta ovat myös halvempi hinta, keveys, helppo valmistaminen ja
parempi kestävyys (Löfgren ym. 2003, Gibson 2009).
Muovisten pakkausten heikkoutena on, että osa muoveista absorboi joitain
formulaatioiden aineosia, kuten lääke- ja säilytysaineita (Löfgren ym. 2003, Gibson
2009). Tätä tapahtuu erityisesti lipofiilisten lääkkeiden kanssa. Yleisin pakkauksen ja
valmisteen välinen ongelma on bentsalkoniumkloridi (BAK) -säilytysaineen
absorboituminen polyetyleenimuoviin. Absorboitumisen lisäksi polymeereistä
valmistettujen muovien heikkoutena ovat mahdollinen muovin aineosien
liukeneminen lääkeaineeseen, tulostemusteen tai etiketin liiman imeytyminen
pakkauksen läpi ja formulaation hapettumisreaktio. Näitä ongelmia voidaan kiertää
valmistamalla tuote lasiseen pakkaukseen, vaihtamalla säilytysaine tai muuttamalla
tuote säilytysaineettomaksi. Säilytysaineettomat valmisteet pakataan yleensä BFS-
tekniikalla tuotettuun kerta-annospipettiin. Säilytysaineettomien tuotteiden
valmistuksessa täytyy ottaa huomioon, että valmiste pysyy steriilinä koko tuotteen
eliniän. Kerta-annospakkauksien negatiivisina puolina ovat kalliimmat
valmistuskustannukset, isommat loppupakkaukset sekä tasaisen annoksen
hankalampi saanti.

30
3 Valmistus
Nestemäinen valmiste on erittäin altis mikrobikontaminaatiolle, koska monesti se on
ideaalinen kasvualusta bakteereille, kuten Salmonella, E. coli ja Staphylococcus
aureus (Reed 2009). Tämä tekee valmistusprosessista haastavamman, kun
lopputuotteen on oltava steriili. Steriileille tuotteille on kaksi pääasiallista
valmistustapaa, loppusterilointi ja aseptinen prosessi. Loppusterilointiin päädytään
tuotteissa, jotka ovat tarpeeksi stabiileja näihin käsittelyihin (Agalloco ja Akers 2008).
Kaikista steriileistä tuotteista 5–15 % loppusteriloidaan. Loppusteriloitavassa
valmistuksessa tuotteen osien ei tarvitse olla valmiiksi steriileitä, mutta
valmistuksessa pyritään silti välttämään ylimääräisen biokuorman kertymistä
millekään tuotteen osalle (Reed 2009). Kertyvää biokuormaa pyritään vähentämään
käyttämällä puhdastilaolosuhteita, jolloin steriloinnin työ vähenee. Lopputuotteelle
valitaan sen formulaation, ominaisuuksien ja pakkauksen perusteella sopivin
sterilointitapa (EMEA 2000). Käytetyin loppusterilointi on autoklavointi, muita yleisesti
käytettyjä ovat säteily ja kuumailma (EMEA 2000, Reed 2009).
Aseptista valmistusta käytetään, kun valmiste tai pakkaus ei kestä loppusterilointi-
prosesseja. Aseptinen valmistus perustuu pitkälti lääkeaineiden, astioiden,
materiaalien ja laitteiden erillisiin sterilointeihin sopivissa kohdissa prosessia, jonka
jälkeen osaset yhdistetään (FDA 2004, Reed 2009). Aseptisessa, kuten myös
loppusteriloitavassa valmistuksessa pakkauksen täyttö ja sulkeminen ovat prosessin
herkimpiä vaiheita, ja ne tulee suorittaa mahdollisimman puhtaissa tiloissa, koska
alkusterilointien jälkeen ei tuotteen puhtautta pystytä parantamaan.
Aseptinen valmistaminen on hankalampaa ja virheherkempää kuin loppusteriloinnin
käyttö. Tämä johtuu siitä, että aseptisessa valmistuksessa käytetään monia erilaisia
sterilointitapoja, kuten kuumailmasterilointi, autoklavointi ja steriilisuodatus (Reed
2009). Jokainen erillinen sterilointi ja työvaihe monimutkaistaa valmisteen tuotantoa.
Yhdysvaltojen ruoka- ja lääkeviranomaisen (U.S. Food and Drug Administration,
FDA) takaisinkutsumista lääkkeistä, joissa oli puutteellinen steriiliys vuosina 1980–
2000, lähes kaikki oli valmistettu aseptisella prosessilla. Yleinen käsitys onkin, että
aseptista valmistusta käytetään vain tilanteissa, joissa loppusterilointi ei ole
mahdollista (FDA 2004, Gibson 2009).

31
Aseptisen valmistuksen juuret ovat 1900-luvun alkupuolella, jolloin ensimmäiset
käsinekaapit otettiin käyttöön (kuva 6) (Agalloco ja Akers 2008). Niissä ei otettu vielä
huomioon ilmankiertoa. Suurtehohiukkassuodattimien (HEPA) kehitys toi
tilanteeseen parannusta 1950–luvulla ja mahdollisti suurempien puhdastilojen
rakentamisen. Ihmistä on huonon puhdistettavuuden ja turvallisuuden takia yritetty
poistaa valmistustiloista jo alusta alkaen erilaisten este- ja ilmanvaihtomenetelmien
avulla. Fyysiset esteet kuten verhot ja seinät hankaloittavat työskentelyä, joten
väliseiniin voidaan asentaa kiinteitä käsineitä, jotka mahdollistavat työskentelyn
puhdastila-alueella. Aktiiviset ilmasulut ja puhaltimet taas perustuvat ilmavirran
ohjailuun, joka estää partikkelien kulkeutumista valmistuksen herkkiin kohtiin.
Siirryttäessä kohti nykyaikaisempia eristysjärjestelmiä on niissä alettu kiinnittämään
entistä enemmän huomiota ilmanvaihtoon, tuotteen siirtoketjuun valmistuksen
yhteydessä, laitteiston puhdistusjärjestelmiin ja näiden kaikkien toimintaan yhdessä.
Nämä järjestelmät minimoivat sisä- ja ulkotilojen vaikutukset toisiinsa ja parantavat
sekä tuotteen että valmistushenkilökunnan turvallisuutta. Jokainen järjestelmä pitää
suunnitella sopivaksi tuotteen monimutkaisuuden, ominaisuuksien ja halutun
valmistusmittakaavaan mukaan.
Kuva 6. Aseptisen prosessoinnin sukupuu (Agalloco ja Akers 2008).

32
3.1 Valmistuslaitteistot
Aseptisessa tuotannossa pyritään estämään valmisteen kontaminoituminen
tuotannon aikana, jolloin valmistetta on turvallista käyttää potilaan silmän hoitoon.
Nykyaikaisessa aseptisessa valmistuksessa käytetään mm. isolaattoreita, rajoitetun
pääsyn esteitä (RABS), blow-fill-seal (BFS) ja form-fill-seal (FFS) -järjestelmiä sekä
näiden yhdistelmiä (Whyte 2001, Chiarello 2004, Drinkwater haettu internetistä:
8.12.2013). Näillä järjestelmillä pyritään minimoimaan ihmisen läsnäolo prosessissa
ja vähentämään aseptisen valmistuksen kustannuksia. Moni lääkeaine vaikuttaa jo
pieninä pitoisuuksina, minkä takia valmistusympäristö pyritään automatisoimaan ja
ihminen poistamaan tilasta. Nykyiset lääkkeenvalmistusjärjestelmät ovatkin entistä
automatisoituneempia.
3.1.1 Isolaattori
Isolaattorit ovat voimakkaasti kasvava aseptisen valmistuksen järjestelmä (Lysfjord ja
Porter: Haettu internetistä 9.12.2013). Isolaattorin toiminta perustuu fyysisiin ja
ilmanpaine-esteisiin, jotka pyrkivät estämään bakteerien ja hiukkasten pääsyn
valmistuksen herkimpiin osiin (Drinkwater haettu internetistä: 8.12.2013). Isolaattorit
voidaan jakaa kahteen ryhmään: avoimet ja suljetut. Avoimet järjestelmät ovat
teollisuudessa käytetympiä, koska niissä isolaattoriin voidaan tuoda ulkopuolelta
esimerkiksi uusia pakkauksia (Lysfjord ja Porter: Haettu internetistä 9.12.2013).
Suljetuissa järjestelmissä prosessin aikana ainoastaan ilma siirtyy isolaattoriin ja
sieltä pois, joten avoimet järjestelmät mahdollistavat suuremmat tuotantoerät.
Isolaattorien tarkoitus on parantaa valmistusolosuhteita ja samalla pienentää
valmistuksen kustannuksia (Agalloco 2006). Isolaattoreissa saavutetaan pienemmän
käyttökustannukset, koska koko valmistustilaa ei tarvitse muokata tuotantoketjun
herkimmän osion mukaan, vaan laitteistojen ympärille voidaan suunnitella sopivan
kokoiset isolaattorit, jolloin puhdistettava tila pienenee ja laatu paranee (Whyte 2001,
Agalloco ja Akers 2008). Isolaattoreiden toimintaan kuuluu erilaisia osia, kuten
ilmastointi, tavaran kuljetus, työskentelylaitteet, erilaiset liitännät ja ilmalukot (Kuva 7)
(Whyte 2001).

33
Kuva 7. Isolaattori ja siihen vaikuttavat reitit (Whyte 2001).
Ilmastointi on tärkeässä osassa isolaattorijärjestelmässä. Se huolehtii pitkälti siitä,
että tila täyttää valmistuksen aikana vaadittavan puhtauden. Isolaattorin
ilmanvaihdosta huolehtii HEPA-suodatettu laminaarivirtaus, joka vaihtaa tilan ilman
yleensä 20–60 kertaa tunnissa (Whyte 2001, Agalloco 2006, Agalloco ja Akers
2008). Suljetuissa isolaattorissa ja käsinekaapeissa ei ilmavirtauksen tarvitse olla
laminaarinen (Euroopan komissio 2008, Agalloco 2006). Suurin osa isolaattoreista on
sijoitettu D-luokan puhdastilaan (Lysfjord ja Porter: Haettu internetistä 9.12.2013).
Ilmastoinnilla voidaan vaikuttaa tuotteen suojaamisen lisäksi myös
työskentelyhenkilöstön turvallisuuteen (Whyte 2001). Alipaineistamalla järjestelmä
estetään toksisten tai potenttien lääkeaineiden poistuminen isolaattorien ulkopuolelle
(Whyte 2001, Chiarello 2004). Lääkkeenvalmistuksen kannalta tuotteen turvaaminen
ympäristön hiukkasilta on kuitenkin oleellisinta. Partikkelien kulkeutumista
valmistustilaan voidaan estää asettamalla tilan ilmanpaine ympäristöä
korkeammaksi. Prosesseissa tavallisesti käytettävä paine on 20–70 Pa korkeampi
kuin ympäristön.

34
Isolaattorin jakaminen eri osiin mahdollistaa ilmavirran ohjailun järjestelmässä (Kuva
8) (Chiarello 2004). Laitevalmistaja SKAN suunnittelemassa järjestelmässä
isolaattorin osat on paineistettu eri tavoilla. Kuvassa 8 näkyy (+) ja (–) -symbolein
merkittyinä kyseisen osion paineistus suhteessa ympäröivän tilan paineeseen.
Ylipaine on muodostettu sterilointi-, täyttö- ja kylmäkuivausosioihin.
Viranomaisvaatimusten mukaisesti korkein paine on puhtaimman tilan vaativassa
täyttöosiossa. Kylmäkuivaimessa (FD) vallitsee täyttöosion tasoinen tai hieman
matalampi paine. Paine-erot ohjaavat ilmavirtaa kulkemaan lopulta kohti
korkitusosiota, jossa tuote on jo paremmin suojassa astiassa. Loppuvaiheen
prosessit, kuten korkitus ja pakkauksen ulkopesu, tehdään alipaineessa, jotta
prosessista ei pääse karkaamaan lääkeaineita ympäristöön. Samalla
valmistuslaitteistolla voidaan tehdä monia tuotteita, joten laitteiston ilmanpaineita
täytyy pystyä muokkaamaan kullekin prosessille sopivaksi (Whyte 2001, Chiarello
2004). Olosuhdevalvonta paineenvaihtelukonseptissa hoidetaan tietokonepohjaisesti.
Järjestelmä mittaa ilmavirtauksen jokaisesta osiosta ja säätää tilojen paineita tarpeen
mukaan. Kylmäkuivauksen tapaiset herkät vaiheet voidaan asettaa omiin
erikoisyksikköihin, kuten käsinekaappiin. Käsinekaappi mahdollistaa tuotteen
käsittelyn prosessin aikana siten, ettei se joudu tekemisiin isolaattorin ulkopuolisen
ympäristön kanssa.

35
Kuva 8. Isolaattoriin asennettu valmistuslinjasto paineenvaihtelukonseptilla (Chiarello
2004)
3.1.2 Tuotteiden siirtojärjestelmä
Tuotteiden siirtojärjestelmä on olennainen osa lääkkeenvalmistusprosessia. Hyvin
suunnitelluissa tuotantotiloissa tuote liikkuu vain yhteen suuntaan, jolloin
ristikontaminaatio voidaan välttää (Agalloco ja Akers 2008). Tavara- tai
tuoteliikenteen, jota tapahtuu isolaattorista ulos ja sisään, täytyy tapahtua siten,
etteivät tuote tai tavarat altistu kontaminaatiolle. Siirtelyyn voidaan käyttää erilaisia
apuvälineitä, kuten isolaattorin kylkeen telakoitavia kuljetusastioita tai erilaisia
liitäntöjä ja väliastioita, joita voidaan steriloida (Whyte 2001). Suuremman
mittakaavan tuotannossa tähän käytetään liukuhihnoja, jotka kulkevat
sterilointitunnelista läpi ennen isolaattoriin tuloa (Whyte 2001, Euroopan komissio
2008).

36
3.1.3 Komponenttien sterilointi
Valmisteen komponentit voidaan steriloida useammalla tavalla (FDA 2004).
Aseptisessa valmistamisessa suodatus on yksi käytetyimmistä tavoista, etenkin
lämmölle herkille aineille. Valmisteen komponentit liuotetaan haluttuun nesteeseen,
kuten Yhdysvaltojen Farmakopean (United States Pharmacopeia, USP 36)
mukaiseen injektioihin käytettävään veteen. Liuos suodatetaan tämän jälkeen
steriloivan membraanin tai suodattimen läpi. Nestemäiset valmisteet voidaan usein
steriloida myös säteilytyksellä.
3.1.4 Pakkauksen sterilointi ja puhdistus
Aseptisen valmisteen pakkauksen täytyy olla puhdistettu, steriili ja pyrogeeniton
ennen kuin valmiste voidaan annostella siihen (Agalloco ja Akers 2008, Reed 2009).
Osassa järjestelmistä pakkauksen nämä toimenpiteet on liitetty osaksi
valmistuslinjastoa, jolloin säästytään ylimääräisiltä kuljetuksilta ja varastoinneilta.
Suuren mittakaavan järjestelmissä lasisten pakkausten depyrogenointi ja sterilointi
suoritetaan lämmön avulla jatkuvatoimisessa sterilointitunnelissa. Pakkaus kulkee
sterilointitunnelissa, jonka keskivaiheilla ilman lämpötila nousee 350 ˚C:een ja laskee
siitä asteittain normilämpötilaan ennen kuin pakkaukset saavuttavat linjaston
täyttöaseman (Reed 2009). Lasipakkaus katsotaan steriloiduksi, kun se saavuttaa
180–200 ˚C lämpötilan. Osa pakkausten valmistajista voi toimittaa pakkaukset
valmiiksi käsiteltyinä, jolloin ne voidaan täyttää heti (Agalloco ja Akers 2008). Vastuu
pakkausten steriiliydestä on silloin niiden tuottajalla, kunhan lääkkeen valmistaja
täyttää ne soveltuvassa puhdastilassa.
Muovisten pakkausten sterilointiin voidaan käyttää kaasumaisia etyleenioksidi- ja
vetyperoksidi-käsittelyjä, tai pakkaukset voidaan altistaa ionisoivalle säteilylle
(Agalloco ja Akers 2008). Elektonisäde- ja kaasusterilointiin tarvittavat laitteistot
voidaan asentaa valmistuslinjaston yhteyteen, mutta gammasädesteriloinnit on
suoritettava alihankkijoilla toiminnon terveyshaittojen takia.

37
3.1.5 Blow-Fill-Seal -laitteisto (BFS)
Blow-Fill-Seal (BFS) on automatisoitu laitteisto, joka valmistaa tuotteen pakkauksen
juuri ennen sen täyttöä ja sulkee sen heti täytön jälkeen (kuva 9). BFS-laitteistoilla
valmistetaan yleensä 1–1000 ml:n nestemäisiä pullo- tai pipettivalmisteita (Reed
2002). BFS-laitteisto kehitettiin alun perin 1930-luvulla Euroopassa, mutta vasta
1970-luvulla se otettiin farmaseuttiseen käyttöön (Reed 2002, Löfgren ym. 2003).
Kuva 9. BFS-laitteisto (Reed 2002).
BFS-laitteiston pääosat ovat polymeerin varastointi- ja syöttöjärjestelmä,
kuumennettava ruuvipuristin, jossa on putkisuutin, putken katkaisuterä, steriili
täyttökammio, muotin puolikkaat pakkauksen muodostamiseen ja sulkemiseen sekä
tarkastuslaitteet esimerkiksi vuodon havaitsemiseen.
BFS-laitteisto on vain osa aseptisesta valmistusprosessista, ja se tarvitsee
ympärilleen perinteiset aseptisen valmistusprosessin välineet, joiden avulla täytettävä
neste valmistetaan (kuva 10). BFS-laitteistot käsittelevät normaalisti vain kahta
raakamateriaalia, täytettävää tuotetta ja polymeeriä, josta pakkaus valmistetaan
(Reed 2002). Molemmille materiaaleille on omat linjastonsa, jotka mahdollistavat
erittäin suurten eräkokojen valmistamisen kerralla. Laitteisto voi valmistaa
esimerkiksi 500 000 yksikön eriä, ja sen täyttöaika voi olla 120 tuntia.

38
Kuva 10. BFS-laitteiston yhteenliittäminen valmistusprosessiin (Haettu internetistä 16.12.2013 http://www.rommelag.com)
BFS-laitteistossa polymeerirakeet syötetään ruuvipuristimeen, jossa ne
kuumennetaan 160–170 ˚C tasaiseksi massaksi ja johdetaan suuttimen läpi ontoksi
polymeeriputkeksi (Reed ym. 2002). Putken rakennetta estetään romahtamasta
tukemalla sitä steriilillä ilmavirralla (Kuva 11 a). Kun putki on saavuttanut sopivan
pituuden, alempi muotti painautuu polymeeriputken ympärille ja sulkee sen alaosan
(Kuva 11 b). Samaan aikaan putken yläosa leikataan auki sähköllä kuumennetulla
terällä. Alamuotti siirtyy tämän jälkeen täyttöasemalle, jossa putki imetään ja/tai
puhalletaan kiinni muotin kylmennettyihin seinämiin. Näin saadaan muodostettua
putkesta pakkauksen alaosa. Tämän jälkeen täyttöneula annostelee oikean määrän
nestettä pakkaukseen (Kuva 11 c). Täytön jälkeen neula nostetaan pois, ja muotin
ylempi osa puristaa putken kiinni muodostaen pakkaukselle yläosan ja samalla
sulkien sen (Kuva 11 d). Tällä tavoin on saatu yhdistettyä pakkauksen muodostus,
täyttö ja sulkeminen yhteen laitteeseen, ja koko prosessin tekemiseen menee 10–15
sekuntia.

39
Kuva 11 Blow-Fill-Seal -laitteiston toimintaperiaate: a) kuumennetun polymeeriputken syöttömuoteille, b) muottien sulkeminen ja polymeeriputken katkaisu molemmista päistä ja alapään sulkeminen, c) polymmeriputken täyttö ja muotoilu muotin mukaan, d) pakkauksen yläosan sulkeminen, e) pakkauksen vapauttaminen muoteista (Reed ym. 2002).
Ilmavirtauksia käytetään hyödyksi myös BFS-laitteistossa, jotta laitteiston
ulkopuolella olevat partikkelit eivät kulkeudu täyttöalueelle, jossa tuote on
haavoittuvimmillaan (Reed 2002, FDA 2004). Laitteiston toimintakierrossa on yleensä
kolme kohtaa, jossa pakkaus on altis kontaminaatiolle: kun putki katkaistaan
kuumalla terällä, kun pakkausta siirretään täyttöasemalle ja juuri kun täyttöneula
otettu pois pakkauksesta. Katkaistaessa polymeeriputki kuumalla terällä syntyy
näkyvää savua ja siten partikkeleita, jotka poistetaan steriilillä ilmavirralla. Katkaisuun
on kehitetty uusi systeemi, KleenKut™, joka toimii yleisimmin BFS-laitteistossa
käytetyille matalan tiheyden polymeereille vähentäen partikkeleiden muodostumista
(Reed 2002, Reed 2009). KleenKut™-operaatiossa polymeeriputki katkaistaan
kuuman terän sijasta kylmällä, mikä estää näkyvän savun synnyn ja vähentää näin
syntyviä partikkeleita.
BFS-laitteistoja koskevat samat ohjeet kuin muutakin steriiliä valmistusta (FDA
2004). Laitteiston ympärillä olevan isolaattorin täytyy estää valmisteen
kontaminoitumista. Valmisteen kanssa tekemisissä olevien pintojen täytyy olla
steriilejä ja laitteistossa täytyy olla höyrysterilointi tai vastaava järjestelmä, joka
puhdistaa sen osia. Aseptisia tuotteita valmistavien BFS-laitteistojen tulee olla
asennettuja vähintään C-luokan tilaan ja henkilökunnalta vaaditaan A/B-luokan
pukeutumista (Euroopan komissio 2008). BFS-laitteistot, jolla valmistetaan
loppusteriloitavia tuotteita, voidaan kuitenkin asentaa D-luokan tilaan.
Täysin automatisoitu laitteisto parantaa aseptisen valmistuksen varmuutta tuottaa
steriiliä tuotetta perinteiseen valmistukseen nähden (Reed 2002. Schell 2010).
Järjestelmä vähentää tilantarvetta ja kustannuksia, kun pakkausten materiaali

40
toimitetaan vähän tilaa vievänä rouheena ja erillisten pakkausten varastointi ja
käsittely poistuu. Laitteen pitkälle viety automatisointi ja uuden tavaran
syöttömahdollisuudet mahdollistavat suurten eräkokojen teon, jolla pystytään
yksittäispakkauksille saamaan kilpailukykyisemmät yksikkökustannukset kuin
muuten.
BFS-laitteistojen avulla voidaan luoda myös tuoteperhekohtaisia muotoiluja, koska ne
mahdollistavat kustannustehokkaasti hyvin monenlaisten pakkausten valmistamisen
(Löfgren 2003). Pakkausten muokattavuus turvallisuuden ja käytettävyyden
takaamiseksi onkin yksi BFS-tekniikan suurimmista vahvuuksista.
3.1.6 Valmistuslinjaston puhdistus ja sterilointi
Nykyaikaisessa teollisuudessa pyritään kustannusten minimointiin, joka tarkoittaa
myös osaltaan laitteistojen käyttöasteen maksimointia. Yksittäisellä
valmistuslinjastolla tehdään usein useampia eri tuotteita, jolloin linjaston puhdistus ja
sterilointi tulee olla helppoa, nopeaa ja luotettavaa. Nykyaikaisissa aseptisissa
järjestelmissä puhutaan paikallaan steriloinnista (SIP) ja paikallaan puhdistuksesta
(CIP). Ne ovat järjestelmiä, jotka mahdollistavat laitteiston puhdistuksen ja
steriloinnin ilman että sitä tarvitsee purkaa (Agalloco 2007, Euroopan
standardisointikomitea 2011).
Isolaattoreissa toimivista sterilointijärjestelmistä vuonna 2008 92 % perustui
hapettavaan vetyperoksidisterilointiin (H2O2) (Lysfjord ja Porter: Haettu internetistä
9.12.2013). Suurin osa vetyperoksidisteriloinneista tehdään höyrystetyllä
vetyperoksidilla. Höyrystysjärjestelmän etuja ovat laaja teho erilaisiin mikro-
organismeihin, nestemäistä vetyperoksidia tehokkaampi sterilointi,
ympäristöystävällisyys, ei myrkyllisiä jäämiä pinnoille, voi toimia normaalissa
ilmanpaineessa ja lämpötilassa sekä mahdollista käsitellä suuria ja epätasaisia
pintoja (Heckert ym. 1997, Kačer ym. 2012). Steriloiva höyry muodostetaan tilaan
käyttäen 35–59 % vetyperoksidiliuosta (Lauderback ym. 2002, Keith 2012).
Järjestelmässä on erilaisia toimintaperiaatteita, mutta ne voidaan jakaa viiteen
vaiheeseen: tilan suhteellisen kosteuden laskeminen tarvittaessa 30–50 %,

41
alipaineen muodostaminen tehon parantamiseksi, vetyperoksidin konsentraation
nosto tavoitearvoon, tilan ylläpitäminen koko sterilointivaiheen ajan ja tilan
ilmaaminen suodatetulla ilmavirralla useita kertoja. Ilmaamisessa poistetaan tilassa
oleva vetyperoksidi. Vetyperoksidin konsentraatio vaihtelee tyypillisesti välillä 150–
700 ppm, siten ettei se pääse kuitenkaan tiivistymään tilassa. Prosessin syklien
määrä ja kesto määräytyvät steriloitavan laitteen koon, tyypin ja materiaalien
mukaan. Vetyperoksidin myrkyttömien hajoamistuotteiden (vesi ja ilma) takia muuta
kuivatus- tai ilmausaikaa ei tarvita. Järjestelmällä kyetään suorittamaan
sterilointiprosessi kokonaisuudessaan parissa tunnissa, riippuen järjestelmän koosta
ja luonteesta.
4. Yhteenveto
Silmälääkinnästä haasteellista tekee silmän herkkyys ja tärkeys normaalissa
arjessamme. Silmän pinta on helppo kohde lääkkeen saavuttaa, ja tuotteesta on
melko helppo valmistaa hyvin siedetty silmässä. Samaan aikaan silmän sisemmät
osat, johon lääkeaineen usein pitäisi kulkeutua, ovat erittäin vaikeasti
saavutettavissa. Alle viiden prosentin imeytymisaste tekee silmälääkinnän
huomattavan haasteelliseksi. Silmän kerroksellisuus tekeetilanteesta hankalan,
koska parhaimmassakin tilanteessa lääkevalmisteesta saadaan aikaan vain hyvä
kompromissi. Ne ominaisuudet, joita ensimmäisen kerroksen läpäisyyn vaaditaan,
eivät sovellukaan enää seuraavan kerroksen piirteille. Pyrittäessä kasvattamaan
lääkeaineen imeytymistä erilaisilla lääkeaine- ja formulaatiomuutoksilla usein
heikennetään lääkkeen siedettävyyttä silmässä.
Yleisimmin huonoa imeytymistä yritetään korjata pyrkimällä pidentämään
lääkeaineen lyhyttä vaikutusaikaa silmässä. Erilaisilla geeliytymis-, suspensio- ja
mukoadheesivisilla aineilla lääkeaineen imeytymistä onkin saatu parannettua vähän,
mutta edelleen erilaisissa hoidoissa annostelutiheys on useita kertoja päivässä.
Potilaan ja lääkehoidon tehon kannalta pidempikestoiset ja paremmin imeytyvät
valmisteet olisivat kuitenkin tärkeitä. Erilaisilla lääkeainelamelleilla onkin saavutettu
parempaa imeytymistä, mutta niiden asettaminen ja käyttäminen etenkin ikäihmisille

42
on hankalaa. Iontoforeesin kaltaiset erilaisesta lähestymiskulmasta tulevat ratkaisut,
jotka antavat positiivisia tuloksia kokeissa, ovat tervetulleita. Etenkin tekniikan
kehittyessä hoidolle, jolla saavutetaan harvoilla annosteluilla korkeita
lääkeainepitoisuuksia silmän sisemmissä osissa, on suurta kysyntää. Lyhyt,
kohdistettu ja tehokas annostelu voisi vähentää myös systeemisten vaikutusten
ilmenemistä.
Silmälääkkeillä on tarkat tuotantovaatimukset silmän herkkyyden takia. Tuotannon
suunnittelu lähtee liikkeelle formulaation komponenttien rajoittavista tekijöistä: onko
valmistetta mahdollista loppusteriloida vai täytyykö tuotantoprosessi suunnitella
täysin aseptiseksi? Nykyaikaisilla laitteistoilla pystytään tähtäämään korkeaan
puhtausasteeseen, mutta valmisteiden monimutkaistuvat formulaatiot ja suuret
mittakaavat asettavat laitteistot ja prosessit koville. Lääketeollisuuden pyrkimys
tasaiseen ja korkeaan laatuun kilpailukykyisellä hinnalla avaa ovia uusille
tuotantotekniikoille. Laadun kannalta oleellisimmat tekniikat ovat ilmastointi ja
automatisointi. Ilmastointi on oleellisessa asemassa aseptisessa valmistuksessa,
koska jokaisessa valmistusprosessissa tuote on jossain vaiheessa alttiina
ulkoympäristölle. Isolaattoreihin yhdistettynä HEPA-suodatettu ilmastointi tekee
valmistustilan olosuhteista kuitenkin niin hyvät, että lyhytaikaiset altistukset
ympäristölle eivät aiheuta laadulle vaaraa. Automatisoinnilla saadaan taas vaikeasti
puhdistettava ihminen pois tuotantotiloista, ja saadaan usein myös kasvatettua
tuotantonopeutta. Automatisointi ei korvaa ihmistä vielä kaikessa toiminnassa, koska
tekniikka ei mahdollista vielä niin hienostunutta käsittelyä, johon ihminen kykenee.
BFS- ja SIP-järjestelmien tyyliset automatisoinnit ovat kuitenkin lähes välttämättömiä
nykyaikaisissa tuotantolaitoksissa tehokkuuden lisäämiseksi huonontamatta laatua.
Lääkevalmisteiden suunnittelu on nykypäivänä kokonaisvaltaista, ja jokaisen
formulaation aineosa täytyy miettiä aineen toiminnasta tuotannon kautta aina
asiakkaan annosteluun saakka.

43
KOKEELLINEN OSA: RAMAN-SPEKTROSKO-
PIAN HYÖDYNTÄMINEN JAUHESEOKSEN
LÄÄKEAINEPITOISUUDEN MÄÄRITTÄMISESSÄ

44
ITÄ-SUOMEN YLIOPISTO, terveystieteiden tiedekunta Farmasian laitos Proviisorin koulutusohjelma Farmasian teknologia KANGAS VILLE T: Raman-spektroskopian hyödyntäminen jauheseoksen lääkeainepitoisuuden määrittämisessä Pro gradu -tutkielma, 108 s., 3 liitettä (5 s.) ohjaajat: Tutkija, diplomi-insinööri Tuomas Ervasti ja Lehtori, FaT, dosentti Ossi Korhonen Maaliskuu 2014
Avainsanat: Raman-spektroskopia, lääkeainepitoisuus, Raman-spektrikirjasto
Tiivistelmä
Tabletit ovat edelleen käytetyin lääkemuoto sairauksien hoidossa. Niiden valmistuksessa pyritään entistä enemmän jatkuvatoimisiin prosesseihin, joiden yksi suurimmista haasteista on luotettava ja tasalaatuinen lääkkeen valmistus. Sitä varten valmistuslaitteisto vaatii rinnalleen reaaliaikaisen laadunvalvontajärjestelmän, joka kykenee riittävän nopeasti ja tarkasti analysoimaan tuotetta, ja jonka tuottaman datan avulla voidaan tehdä muutoksia valmistusasetuksiin. Raman-spektroskopia on yksi lupaavista järjestelmistä reaaliaikaiseen laadunvalvontaan, koska se kykenee mittaamaan näytteet nopeasti valmistuksen aikana, sen on osoitettu kykenevän erittäin tarkkoihin pitoisuusmittauksiin ja se soveltuu käytettäväksi niin nesteiden, kiinteiden kuin kaasumaistenkin näytteiden kanssa.
Tutkimuksen tavoitteena oli selvittää PhAT-anturilla varustetun Raman-spektrometrin soveltuvuutta jauhemaisten näytteiden lääkeainepitoisuuksien määrittämiseen sekä paikallaan että liikkeessä olevilla näytteillä. Tutkimukseen valittiin yleisesti lääkevalmistuksessa tunnettuja lääke- ja apuaineita, joista tehtiin seoksia eri pitoisuuksilla. Seokset mitattiin Raman-spektrometrillä paikallaan yhden ja kuuden sekunnin mittausajoilla sekä liikkeessä yhden sekunnin mittausajalla. Kaikki Raman-spektrometrillä mitatut lääkeaineseossarjat esikäsiteltiin SIMCA–P 12 ohjelmalla ja niistä muodostettiin ennustusmalleja, joiden kykyä ennustaa näytteiden pitoisuuksia tutkittiin käyttämällä tunnettuja koenäytteitä. Asetyylisalisyylihapon ja dikalsiumfosfaatin seoksille määritettiin lääkeainepitoisuudet myös kemiallisesti, jotta Raman-spektrometrin antamien tulosten paikkansapitävyyttä voitiin arvioida.
Soveltuvimmat esikäsittelyt antoivat sarjoista muodostetuille malleille ennustustehoiksi arvoja välillä 0,8–4 (RMSEP). Liikkeessä tehdyissä mittauksissa osassa sarjoista Raman-spektrometri pystyi erottelemaan viiden prosentin pitoisuuserot toisistaan, jota voidaan pitää vähimmäisvaatimuksena lääkkeenvalmistuksen analyysilaitteistolle.
Tutkimus osoitti järjestelmän sisältävän potentiaalia reaaliaikaiseen jauheseoksen pitoisuusmittaukseen. Etenkin liikkeessä olevien seosmittausten toistettavuutta ja tarkkuutta pitää kuitenkin tutkia vielä lisää erilaisilla näytteen liikkumisnopeuksilla.
Tutkimuksen aikana luotiin myös Raman-kirjasto, joka auttaa tuntemattomien Raman-spektrien tunnistamista. Saadut spektrit osoittautuivat suurimmalta osin vertailukelpoisiksi kirjallisuudesta saatuihin referensseihin.

45
UNIVERSITY OF EASTERN FINLAND, Faculty of Health Science School of Pharmacy Master of Science in Pharmacy program Pharmaceutical Technology KANGAS VILLE T: Utilization of Raman spectroscopy to determining the drug concentration of the powder mixture Master’s Thesis, 108 pp, 3 appendixies (5 pp.) Supervisors: Researcher, Master of Science Tuomas Ervasti and Lecturer, PhD, Adjunct Professor Ossi Korhonen March 2014
Keywords: Raman spectroscopy, drug concentration, Raman spectroscopy library
Summary Tablets are still the most commonly used form of medicines in treatment of diseases. The aim of medical manufacturers is to move from large batch processes to smaller continuous processes. In such processes one of the biggest challenges is to establish a reliable process for manufacturing uniform products. Manufacturing equipment requires a parallel real-time quality control system, which can quickly and accurately analyze the product and, with its analyzed data can be used to make necessary adjusts to the manufacturing equipment. Raman spectrometry is one of the most promising systems for that type of quality control, since it is capable of measuring samples rapidly during manufacturing process, it has shown to be very precise measuring concentrations and it is suitable to be used for liquid, solid or gaseous samples.
The aim of this study was to find out suitability of Raman spectrometer with PhAT probe for measuring drug concentrations for both stationary and mobile powdered drug mixture samples. All drugs and pharmaceutical excipients chosen to the study are commonly known in pharmaceutical manufacturing, and mixtures of various concentrations were used. Stationary mixtures were measured with Raman spectrometer with one and six second measurement times and mobile mixtures were measured with one second measurement time. All series were preprocessed with SIMCA-P 12 software and probability models were constructed for each series. Prediction efficiencies of the constructed models were measured on known exhibits. For acetylsalicylic acid and dicalciumphosphate drug mixtures were also tested in chemical assays to enable evaluation of results given by the Raman spectrometer.
The Raman spectrometer gave RMSEP values from 0.8 to 4 for the most suitable preprocessed models. For some of the series measured from mobile samples Raman spectrometry was able to distinguish five percent differences in drug concentration. This difference can be considered as minimum accuracy for analytical devices in pharmaceutical manufacturing.
Raman spectroscopy demonstrated good potential for real time concentration monitoring in the study. Further studies are needed to ensure the accuracy and repeatability of measurements with different sample speeds.

46
During the study a Raman spectroscopic library was created to aid the identification of unknown Raman spectrum. Most of the obtained spectra turned out to be comparable with the spectra from literature.

47
5 JOHDANTO
Tabletit ovat tämän hetken käytetyin lääkemuoto. Tabletti on stabiili lääkemuoto, joka
on teollisuudelle yksinkertainen ja edullinen valmistaa sekä asiakkaan helppo
käyttää. Perinteinen tablettivalmistus pohjautuu suuriin tuotantoeriin, joiden
laadunvarmistus tapahtuu valmistuksen jälkeen tuotteelle määritetyillä testeillä. Vasta
hyväksyttyjen testien jälkeen tuote on valmis vapautettavaksi jakeluun.
Lääketeollisuudessa tavoitteena olisi kuitenkin siirtyä eräpohjaisesta valmistuksesta
kohti jatkuvatoimista lääkkeenvalmistusta, jossa pyritään saavuttamaan pienemmät
valmistuslaitteistot, vähentämään tuotantokatkojen ja epäkelvon lopputuotteen
määrää sekä saamaan kattavampi reaaliaikainen laadunvarmistus.
Jatkuvatoimisen lääkkeenvalmistuksen tärkeimpiä kulmakiviä on saada laitteisto
tuottamaan tasalaatuista lopputuotetta. Tähän tarvitaan reaaliaikaista
analytiikkalaitteistoa, joka pystytään liittämään osaksi valmistuslaitteiston toimintaa
(De Beer ym. 2011). Yhden tai useamman analytiikkalaitteiston avulla pyritään
tarkkailemaan valmisteen kriittisiä ominaisuuksia reaaliajassa koko tuotantoketjun
ajan. Kriittisten ominaisuuksien määrityksellä pyritään seuraamaan eri prosesseja ja
niissä tapahtuvia muutoksia. Pyrkimyksenä on rakentaa valmistusjärjestelmä, jossa
analytiikkalaitteistojen datan perusteella voidaan valmistuslaitteistojen toimintaa
muuttaa siten, että lopputuote pysyy jatkuvasti sille määritettyjen parametrien sisällä.
Raman-spektroskopia on yksi lupaavista mittaustekniikoista, joita tutkitaan
reaaliaikaiseen analytiikkaan.
Raman-spektroskopia perustuu molekyyleihin kohdistettujen fotonien energian
epäelastiseen siroamiseen (Raman ja Krishnan 1928). Raman-spektroskopiaa
pyritään soveltamaan nykypäivänä pelkästään farmasian alalla moniin eri
valmistusprosesseihin, kuten sekoitus, rakeistus, kuivaus, päällystys, kylmäkuivaus
ja ekstruusio (Vergote ym. 2004, Wiksström ym. 2005, Aaltonen ym. 2007, De Beer
ym. 2007, Kogerman ym. 2008, Saerens ym. 2010, Wirges ym. 2013). Raman-
spektroskopia on kohteeseen kajoamaton mittausmenetelmä, jolla pystytään
mittaamaan niin kiinteitä, nestemäisiä kuin kaasumaisiakin näytteitä. Laitteistolla
voidaan mitata myös lasin läpi, jolloin näyte voidaan sijoittaa esimerkiksi
lasiampulliin, ja muun muassa hygroskooppisten näytteiden mittaaminen on

48
mahdollista. Raman-spektroskopiaa voidaan soveltaa erilaisten prosessien tarkkailun
lisäksi esimerkiksi eri yhdisteiden tunnistamiseen valmiiden spektrikirjastojen avulla
(Bugay ja Brush 2010).
Tutkimuksessa pyrittiin selvittämään Raman-spektroskopian soveltuvuutta
jauheseoksien lääkeainepitoisuuksien tarkkailuun niin paikallaan kuin liikkeessä
olevista näytteistä. Liikkuvilla näytteillä pyrittiin tutkimuksessa mallintamaan
jatkuvatoimisen prosessin reaaliaikaista mittausta. Saatuihin spektrisarjoihin
käytettiin erilaisia esikäsittelytekniikoita parantamaan datan perusteella tehtäviä
ennustuksia. Saatujen tulosten avulla arvioitiin Raman-spektroskopian soveltuvuutta
jauheiden pitoisuuden tarkkailuun. Tutkimukseen valitut aineet ovat yleisesti
lääkkeenvalmistuksessa käytettyjä materiaaleja. Pitoisuustutkimuksen yhteydessä
luotiin myös Raman-spektrikirjasto tukemaan Itä-Suomen yliopistolle valmistuneen
jatkuvatoimisen linjaston mittaustekniikoiden tutkimusta (liitetaulukko 1).
Spektrikirjaston oikeellisuutta pyrittiin varmistamaan vertaamalla tuloksia muihin
tehtyihin tutkimuksiin.
6 MATERIAALIT JA MENETELMÄT
6.1 Raman-spektroskopia
Raman-sironnaksi kutsuttua ilmiötä tutkitaan altistamalla näyte monokromaattiselle
säteilylle ja mittaamalla säteilyssä tapahtuvaa aaltolukumuutosta (Ferraro ym. 2002,
Gurtu ja Khera 2009). Säteilyn energia absorboituu näytteen molekyyleihin ja saa ne
värähtelemään. Värähtelytilan purkautuessa molekyylistä lähtevä säteily sisältää
Rayleight-säteilyksi kutsuttavan alkuperäisen taajuuden lisäksi myös matalampia
taajuuksia, joita kutsutaan Stokesin säteilyksi, ja korkeampia taajuuksia, joita
kutsutaan anti-Stokesin säteilyksi.
Raman-säteilyssä molekyylit virittyvät niin kutsutulle virtuaalitasolle, joka on
huomattavasti varsinaista elektronin virittymistilaa alhaisempi (Kuva 12) (Ferraro ym.
2002). Raman-sironta on hyvin heikkoa Rayleight-sirontaan verrattuna, sillä

49
normaalisti alle yksi sadastatuhannesta näytteeseen tulevasta fotonista aiheuttaa
Raman-sirontaa.
Kuva 12: Eri säteilyjen aiheuttamia muutoksia kaksiatomisessa molekyylissä (IR= infrapuna, R=Rayleigh, S=Stokes, A=anti-Stokes) (Ferraro ym. 2002)
Kaikki molekyylissä tapahtuva värähtely ei aiheuta Raman-sirontaa. Molekyylin
värähtely on Raman-aktiivista, jos polarisoituvan molekyylin koko, muoto tai asento
muuttuu värähtelyn aikana (kuvat 13 ja 14) (Ferraro ym 2002, Gurtu ja Khera 2009).
Raman-aktiivisessa säteilyssä värähtely tapahtuu symmetrisesti molekyylin
symmetriakeskuksen nähden, kuten kuvassa 2 värähtely V1. Värähtelyt V2 ja V3 ovat
infrapuna(IR)-aktiivisia, mutta eivät Raman-aktiivisia, koska V3-värähtely ei ole
symmetristä ja V2-värähtelyssä polaroituneiden molekyylien koko, muoto sekä asento
säilyvät muuttumattomina.

50
Raman-spektristä kullekin aineelle ominaisen tekevät niiden erilaiset
molekyylirakenteet (Ferraro ym. 2002, De Beer ym. 2011). Molekyylin värähtely
koostuu monista yksinkertaisista värähtelytiloista, joista kukin värähtelee sille
ominaisella taajuudella. Raman-spektri koostuu näiden värähtelytiloista purkautuvien
säteilyjen ja alkuperäisen säteilyn erotuksista. Kuvaan 15 on koottuna joitakin
taajuuksia, joilla ilmenee tiettyjä molekyylirakenteita. Raman-säteilyn voimakkuus, eli
intensiteetti, riippuu perustilassa olevien molekyylien määrän lisäksi värähtelevästä
sidoksesta ja värähtelytyypistä. Kovalenttiset sidokset tuottavat enemmän Raman-
säteilyä, kun taas ionisidokset tuottavat enemmän IR-säteilyä. Kovalenttisten mono-,
di- ja tri-sidosten vaikutukset Raman-spektrin intensiteettiin ovat suhteessa noin
sidoksen kerrannainen, eli esimerkiksi kolmoissidos aiheuttaa kolminkertaisen
intensiteetin yksinkertaiseen sidokseen nähden. Värähtelytyypeistä molekyyliä
venyttävä värähtely tuottaa yleensä suuremman intensiteetin kuin molekyyliä
taivuttava (Ferraro ym. 2002).
Kuva 13. CO2-molekyylissä vallitsevan polarisaation muutokset eri normaali-värähtelyissä määrittävät niiden Raman-aktiivisuuden (Ferraro ym. 2002).
Kuva 14. H2O-molekyylissä vallitsevan polarisaation muutokset eri normaali-värähtelyissä. (Ferraro ym. 2002).

51
Kuva 15. Esimerkkejä Raman-spektriä muodostavien ryhmien rakenteiden ilmenemisaaltolukualueista x-akselilla (Schwartz 2005).
6.2 Spektrien esikäsittely
Tässä tutkimuksessa mittauksissa saaduille Raman-spektreille suoritettiin erilaisia
esikäsittelyitä. Esikäsittelyt ovat erilaisia tilastollisia menetelmiä, joilla pyritään
poistamaan tutkimusdatassa esiintyviä poikkeamia, kuten mittausten olosuhteissa
tapahtuvia muutoksia tai virheitä (Gemperline ym. 2006). Näin datasta saadaan
poistettua tutkittavan muutoksen kannalta merkityksetöntä tietoa, ja todelliset
muutokset mittauksissa näkyvät selvemmin, jolloin johtopäätösten ja oikeiden
tulkintojen tekeminen on helpompaa (Naes ym. 2002, Rinnan ym. 2009).
Spektroskopiassa käytetyimmät esikäsittelytekniikat voidaan jakaa kolmeen
ryhmään: spektrin derivointi, spektriä tasoittavat esikäsittelyt ja sirontaa korjaavat
esikäsittelyt (Ferraro ym. 2002, Gemperline ym. 2006, Rinnan ym. 2009).

52
6.2.1 Spektrin derivointi
Derivointi on yksinkertainen ja usein käytetty esikäsittely spektroskopiassa. Sillä
pystytään vähentämään datan pohjatason vaihtelun lisäksi myös yhteis- ja
kerrannaisvaikutuksia (Naes ym 2002. Gemberline ym. 2006, Rinnan ym. 2009).
Derivoinnin vaikutukset havaitaan, kun tarkastellaan kahta Gaussin käyrää, joista
toiseen on lisätty kohonnut pohjataso ja toiseen sen lisäksi myös kerrannaisvaikutus
(kuva 16). Ensimmäisen asteen derivaatta on alkuperäisen spektrin jokaisen pisteen
kulmakerroin ja sillä saadaan poistettua spektriltä pohjataso. Alkuperäinen spektri
sijoittuu siten, että sen minimit ja maksimit ovat derivoidussa spektrissä x-akselin
leikkauskohdissa ja alkuperäisen spektrin suurimmat kulmakertoimet ovat
ensimmäisen asteen derivaatan maksimit ja minimit. Toinen derivaatta, joka mittaa
alkuperäisen spektrin jokaisen pisteen kaarevuutta, pystyy poistamaan
alkuperäisestä spektristä pohjatason lisäksi myös taustan kaltevuutta tai
kaareutuvuutta, joka usein johtuu fluoresenssista tai muista poikkeamista. Toisen
asteen derivoinnilla saadun spektrin rakenne on myös helpommin luettava, kuin
ensimmäisen derivoinnilla saatava. Toisen asteen derivaatalla saatavassa spektrissä
alkuperäisen spektrin positiivisella puolella ilmenevät piikit terävöityvät ja säilyttävät
maksimi kohtansa x-akselilla suhteen, vaihtaen suuntansa y-akselin suhteen
negatiiviselle puolelle (Ferraro ym. 2002, Naes ym. 2002). Piikkien terävöityminen
helpottaa eri piikkien erottamista toisistaan.

53
Kuva 16. Derivoinnin vaikutukset Gaussin käyrään, jolla on kohonnut pohjataso (vihreä) ja Gaussin käyrään, jolla on kohonnut pohjataso sekä kerrannaisvaikutus (punainen) (Rinnan ym. 2009).
Ensimmäisen asteen derivaatta saadaan kahden peräkkäisen mittauspisteen
funktioiden avulla, joista muodostetaan erotusosamäärä (kaava 1). Kaavassa 1 on
ensimmäinen mittauspiste kohdassa i ja h on mittauspisteiden välinen erotus.
Kaavassa 2 on ensimmäisen asteen derivaatta pisteessä i. Toisen asteen
derivaatta saadaan kahden peräkkäisen pisteen perusteella ensimmäisen
asteen derivaatan spektristä (kaava 3) (Rinnan ym. 2009, Etälukio 2013).
(1)
(2)
(3)
Derivoinnin heikkous on, että se hävittää datan signaalia ja huonontaa siten signaali-
kohina -suhdetta. Mitä suuremman asteen derivaatta, sitä enemmän se laskee
signaali-kohina -suhdetta (Rinnan ym. 2009 ja Gemberline ym. 2006)

54
6.2.2 Spektrin normalisointi
Normalisoinnin tarkoituksena on saada vertailtavien spektrien intensiteetit samaan
mittakaavaan ja helpottaa siten erityisesti tuntemattomien näytteiden vertailua
referenssispektreihin (Kramer 1998, Ferraro ym 2002). Normalisoinnit voidaan jakaa
karkeasti kahteen kategoriaan, globaaleihin ja paikallisiin (Listgarten ja Emili 2005).
Globaaleissa tavoissa spektrien yhteisen tekijän normalisointiin käytetään koko
spektriä, kun taas paikallisissa tavoissa siihen käytetään vain osaa spektristä.
Verrattaessa yhdisteitä, joissa on yhteinen komponentti mukana, on normalisointi
helppo suorittaa paikallisella periaatteella yhteisen piikin keskikohdan mukaan.
Tällöin normalisointi suoritetaan jakamalla kunkin spektrin yhteinen taajuus kaikkien
spektrien keskimääräisellä intensiteetillä, jolloin piikin intensiteetin vaihteluksi tulee
1,0, ja loppuosat spektreistä asettuvat sen mukaan helpommin vertailtaviksi.
Jos vertailtavilla spektreillä ei ole yhteistä rakennetta, voidaan normalisointi tehdä
spektrin alle jäävälle pinta-alalle (Ferraro ym. 2002, Meko 2013). Menetelmällä
saadaan spektrien pinta-alaksi yksi (kaava 4). Kaavassa 4 on spektrin
intensiteetti yksittäisellä taajuudella. Menetelmä mahdollistaa eri spektrien suhteiden
vertailun ja auttaa tunnistamaan tuntemattomia spektrejä. Pinta-alapohjainen
normalisointi toimii heikosti erilaisten taustojen omaavien spektrien vertailussa:
voimakkaan ja pienen taustan vertaaminen tällä normalisoinnilla voi vääristää muun
spektrin mittasuhteita siten, että spektrien tunnistaminen vaikeutuu.
√∑ (4)
6.2.3 Spektrin tasoitus
Raman-laitteisto voi kärsiä joillakin näytteillä esimerkiksi vähäisestä sironnan
määrästä, joka aiheuttaa signaali-kohina -suhteen (SNR) huonontumista spektrissä
(Ferraro ym. 2002, Gemberline ym 2006). SNR:n huononemista voidaan vähentää
erilaisilla spektrin tasoitusmenetelmillä, jotka tasoittavat spektrin taustalla olevaa
kohinaa. Spektrin tasoittamisessa on otettava huomioon, ettei tasoituksesta tehdä
liian voimakasta, sillä se voi hävittää myös tärkeää tietoa spektristä. Tasoituksella

55
voidaan kuitenkin hävittää kohinaa niin paljon, että datan tulkinta voi onnistua ilman
suurempia esikäsittelyitä. Yleensä tasoitusta käytetään kuitenkin derivoinnin
yhteydessä, jossa SNR huononee huomattavasti.
Savitzky-Golay (SG) -käsittelyssä valitaan käsittelyyn käytettävien havaintopisteiden
määrä ja polynomin aste (Ferraro ym. 2002 ja Rinnan ym. 2009). Polynomin avulla
spektrin jokaiselle havaintopisteelle muodostetaan keskiarvo (kuva 17). Esimerkiksi
laskettaessa seitsemän pisteen mallilla, jossa on kolmijakoinen painotus, niin itse
mitattavan pisteen lisäksi otetaan huomioon kolme edeltävää pistettä ja kolme
seuraavaa havaintopistettä (Ferraro ym. 2002). Kolmijakoisessa painotuksessa
pisteet painotetaan siten, että keskipiste on painoarvoltaan 1,0, sen viereiset pisteet
0,75, niistä seuraavat pisteet 0,50 ja kauimmaiset pisteet 0,25. Tulos jaetaan sitten
summalla 4,0, josta saadaan painotetun mittauspisteen arvo. Savitzky-Golay -
menetelmässä nämä yksinkertaiset painotukset on korvattu epälineaarisilla painoilla,
jotka tietokone hakee valittujen asetusten perusteella muistista ja laskee tasoituksen.
Polynomin järjestysasteella voidaan vaikuttaa siihen, kuinka monimutkaisia käyriä
tasoituksella voidaan sovittaa (Ferraro ym. 2002 ja Microsoft excel-help 6.11.2013).
Esimerkiksi toisen asteen polynomilla laskettuna käyrä voi sisältää yhden huipun tai
pohjan, kun taas kolmannen asteen polynomilla se voi sisältää kaksi huippua tai
pohjaa.

56
Kuva 17. Käsittelemättömän spektrin tasoittaminen Savitzky-Golay -käsittelyllä. Kuvassa siniset käsittelemättömän datan havaintopisteet tasoitetaan seitsemän pisteen, toisen asteen polynomifunktion avulla punaiseksi viivaksi, josta kuvassa on vielä laskettu ensimmäisen asteen derivaatta (Rinnan ym. 2009).
6.2.4 Sirontaa korjaavat esikäsittelyt
6.2.4.1 Standard Normal Variate (SNV)
SNV on käytetyimpiä esikäsittelyitä Raman-spektroskopian yhteydessä (Rinnan ym.
2009 ja Ferraro ym. 2002). Sen avulla voidaan korjata spektrin taustojen eroja (kuva
18). Toisin kuin derivoinnissa, SNV-korjauksessa säilytetään spektrin alkuperäinen
ulkoasu, jolloin signaali-kohina -suhde pysyy parempana. Käsittely ei siis poista
spektreistä taustaa, vaan pyrkii parantamaan spektrien keskinäistä korreloivuutta.
SNV-käsittelyssä jokaisesta spektristä vähennetään näytteiden keskiarvo, jolla
yksittäisten spektrien vaihtelua saadaan vähennettyä (Ferraro ym. 2002, Naes ym.
2002, Gemberline ym. 2006). Esikäsittelyn jälkeen spektri on keskittynyt y-akselilla
nollaan ja sen intensiteetin vaihtelu on muuttunut alkuperäisestä. SNV käsittelee
spektriä kaavan 5 mukaisesti, missä Xkorjattu on SNV-korjauksen jälkeinen
intensiteetti, Xorg on alkuperäinen intensiteetti samassa kohdassa spektriä, a0 on
keskiarvo korjattavista spektreistä ja n on spektrien lukumäärä.

57
Kuva 18. SNV-käsittelyn vaikutukset spektrien pohjatasoon. (a) 20 mikrokiteisen selluloosajauhenäytteen NIR-spektriä. (b) samat näytteet SNV-esikäsiteltyinä (Gemberline ym. 2006).
√∑
(5)
6.2.4.2 Multiplicative Scatter Correction (MSC)
MSC-käsittely on periaatteeltaan läheinen SNV-käsittelylle (Gemberline ym. 2006,
Rinnan ym. 2009). Molemmilla käsittelyillä pyritään poistamaan pohjatason vaihtelua
ja partikkelikoon vaihtelun aiheuttamia siroamiseroja. MSC- ja SNV-käsittelyt eroavat
siinä, että MSC:ssä spektristä ei vähennetä keskiarvoa, vaan jokaiselle spektrille
tehdään lineaarinen regressio erillisen referenssispektrin avulla. Referenssispektrinä
käytetään yleensä, kuten tässäkin tutkimuksessa, mittausjoukon keskimääräistä
spektriä (kaava 6) tai vaihtoehtoisesti erillistä kalibrointispektriä (Naes ym. 2002,
Rinnan ym. 2009, SIMCA-P 12.0.1 User Guide). Spektrit saadaan näin
käyttäytymään lineaarisemmin keskimääräisen spektrin suhteen.
∑
(6)
(7)

58
(8)
Kaavoissa 6, 7 ja 8 Xik on yksi mitatuista spektreistä ja mk on joukon keskimääräinen
spektri, jota käytetään referenssispektrinä. Edelleen Xik korjattu on korjattu spektri ja ai
ja bi ovat skalaariparametreja, jotka kuvaavat kyseisen näytteen kerrannais- (b) ja
summautumisvaikutuksia (a) ja ne ovat kullekin näytteelle erilaiset. Parametrit
saadaan kuvan 19 tyyppisestä regressiokuvaajasta. eik kuvaa muita spektrissä
esiintyviä muutoksia, jotka eivät selity a:n tai b:n perusteella. MSC-käsittely ei ole
yhtä herkkä häiriöpitoiselle spektrille kuin SNV, koska MSC-käsittely sisältää
pienimmän neliösummasovituksen (Naes ym. 2002, Rinnan ym. 2009).
Kuva 19. Skalaariparametrit saadaan piirrettäessä näytespektri referenssin suhteen, jolloin ne saadaan pienimmän neliösumman regression avulla piirretyn suoran leikkauspisteestä ja kulmakertoimesta (Rinnan ym. 2009).

59
6.2.4.3 Row center (Row)
Row center -käsittelyä ei voida pitää sirontaa korjaavana käsittelynä, vaikka se
keskittääkin näytteiden vaihtelua. Row center -käsittelyssä jokaisesta pitoisuudesta
vähennetään kaikkien pitoisuuksien keskiarvo (SIMCA-P 12.0.1 User Guide).
6.3 Raman-spektrometri ja PhAT anturi
Raman-spektrometrin tärkeimmät osat ovat säteilylähde, jollaisena toimii yleensä
jatkuva-aaltoinen (CW) laser, säteilyn näytteeseen kuljettava ja sieltä keräävä
järjestelmä, aallonpituuden valitsin sekä tunnistin ja ohjausyksikkö, eli tietokone ja
käyttöohjelmisto (Ferraro ym. 2002). Kuva 20 antaa yhdenlaisen läpileikkauksen
laitteistoon.
Tutkimuksessa käytettiin Raman-spektrometriä (Kaiser Optical systems, RXN1-785,
Michigan, Yhdysvallat) ja valokuitukaapelilla varustettua PhAT-anturia. Laitteiston
säteilylähteenä toimiva laser asetettiin 200 mW teholle ja tuottamaan 785 nm
aallonpituudella olevaa säteilyä (kuva 21 ja 22). PhAT-anturi eroaa perinteisestä
anturista suuremman mittausalan takia (Kaiser Optical systems, haettu internetistä
21.2.2013). Normaalin Raman-spektrometrin lasersäteen koko on 2–500 µm, kun
taas mittauksissa käytetyssä PhAT-anturissa mittausalue on vakiona 6 mm (kuva
21). PhAT-anturin oikea mittausetäisyys 6 mm:n lasersäteelle on 25 cm, joka on
sama kuin anturin suojaputken pituus. Suurempi mittausalue antaa näytteestä
keskimääräisemmän kuvan, joka tasoittaa eri mittausten välisiä eroja varsinkin
liikkuvassa näytteessä. Säteen koko lisää tulosten toistettavuutta, mutta ei anna yhtä
tarkkaa kuvaa vaihtelusta (Kim ym. 2006). Lisäksi PhAT-anturin suurempi
mittausalue vähentää laserin aiheuttamaa paikallista lämmöntuottoa näytteeseen ja
täten näytteessä tapahtuvia mahdollisia muutoksia. Tämä on tärkeää erityisesti
mitattaessa aineita, jotka ovat lämmölle herkkiä.
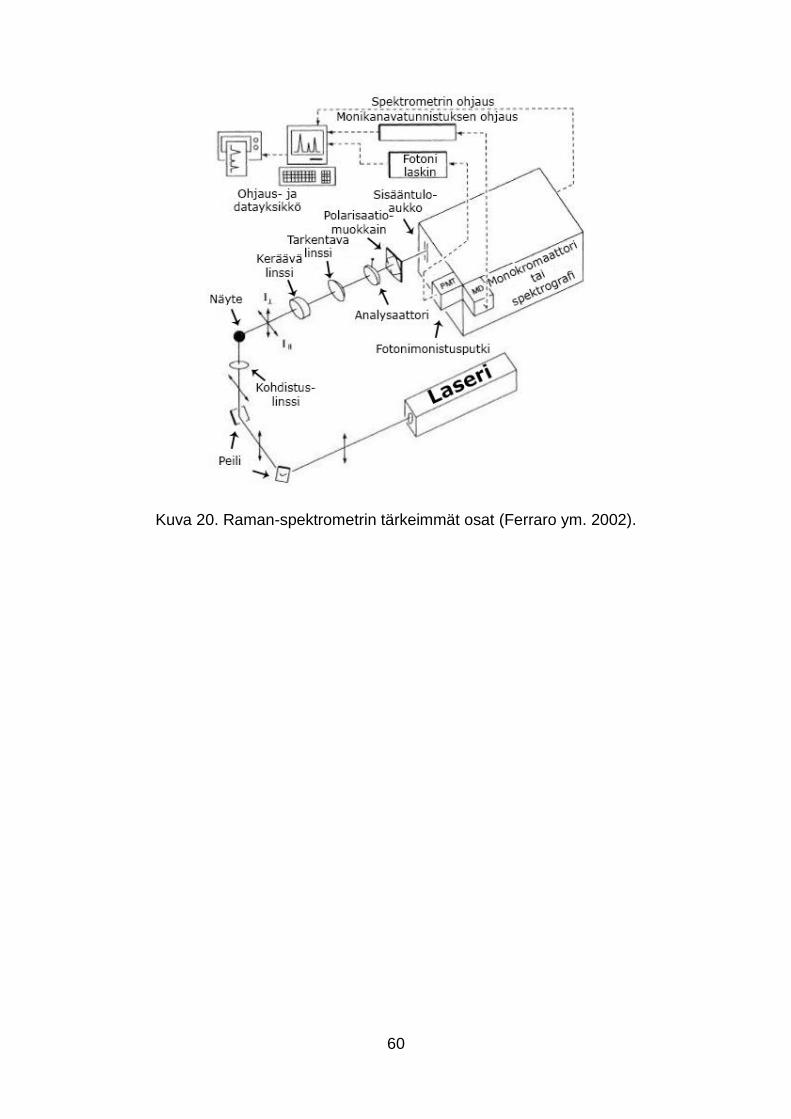
60
Kuva 20. Raman-spektrometrin tärkeimmät osat (Ferraro ym. 2002).

61
6.4 Seokset ja niiden valmistus
Tutkimukseen valittiin kolme lääkeainetta: asetyylisalisyylihappo (ASA) (Sigma-
aldrich, Steinheim, Saksa), parasetamoli (PARA) (Sigma-aldrich, Steinheim, Saksa)
ja ibuprofeeni (IBU) (BASF, Ludwigshafen, Saksa) sekä kolme apuainetta
dikalsiumfosfaatti (DCP) (Emcopress Premium, Rosenberg, Germany),
laktoosimonohydraatti 90 M (LAKTO) (DFE pharma, Veghel, Alankomaat) ja D-
mannitoli (MAN) (Merck eurolab, Briare, Ranska). Aineista tehtiin taulukon 3
mukaiset m/m-% -seossarjat punnitsemalla aineet vaa’alla (Mettler-Toledo PB3002-
S/PH Langacher Greifensee, Sveitsi) ja sekoittamalla ne geometrisessä sarjassa.
Kuva 21. PhAT-anturin lasersäteen halkaisijaksi on asetettu 6 mm ja mittausetäisyys on säädetty 25 cm:iin. Vertailuksi FT-Ramanin mikroanturin halkaisijaltaan 2–5 µm (Haettu internetistä 21.3.2013 ja muokattu www.kosi.com)
Kuva 22. Raman-spektrometri kuituoptisella PhAT-mittapäällä ja ohjausyksikkö (Haettu inter-netistä 12.2.2013 www.kosi.com)

62
Taulukko 3. Seossarjojen lääkeainepitoisuudet (m/m-%)
Mitattava seos Mitattavat lääkeainepitoisuudet (m/m-%)
IBU–MAN 0 10 15 20 30 40 50 60 70 80 85 90 100
PARA–MAN 0 10 15 20 30 40 50 60 70 80 85 90 100
IBU–LAKTO 0 5 10 15 20 30 50 45 50 55 60 70 80 85 90 95 100
PARA–LAKTO 0 5 10 15 20 30 50 45 50 55 60 70 80 85 90 95 100
ASA–DCP 0 5 10 12,5 15 17,5 20 30 40 100
6.5 Näytteiden mittaus ja esikäsittely
6.5.1 Mittaukset paikallaan olevista näytteistä
Lasinen petrimalja laitettiin täyteen näytettä ja asetettiin metalliseen kaappiin
säädettävälle tasolle. Tason avulla näyte asetettiin Raman-spektrometrin
suojaputken läheisyyteen, jolloin mittausetäisyys oli noin 25 cm. Laitteiston
asetuksina käytettiin kaksoismittausta yhden ja kuuden sekunnin mittausajoilla.
Mittausajat valittiin siten, että yhden sekunnin mittauksella liikkeessä olevista
näytteistä saadaan pistemäisempi spektri, joka kuvaa paremmin paikallista näytteen
pitoisuusvaihtelua. Kuuden sekunnin mittaus valittiin, jotta voitiin tarkastella
lyhyemmän mittausajan luotettavuutta.
6.5.2 Mittaukset liikkeessä olevista näytteistä
Liikkeessä tehdyt mittaukset suoritettiin pyörivän alustan päällä, jonka halkaisija oli
30 cm. Alusta oli jaettu kuuteen yhtä suureen osioon (kuva 23). Jauheseokset
asetettiin mittausalustalle koesuunnitelman mukaisesti. Pyörivä alustalevy ja PhAT-
anturi suljettiin pahvilaatikkoon, jotta ulkopuolinen valo ei pääsisi vaikuttamaan
mittauksiin. Alusta pyöri mittauspään alla noin 26 cm:n etäisyydellä yhden kierroksen
minuutissa, jolloin näyte liikkui nopeudella 0,016 m/s. Mittausasetuksina käytettiin
yksittäistä yhden sekunnin mittausta ja mittausten väli oli kymmenen sekuntia.
Yhdessä mittausryhmässä oli 16 mittausta. Mittaustuloksia tutkittiin kolmella tavalla:

63
omana ryhmänään, yhdistämällä pisteet sekä paikallaan otettujen näytteiden malliin
että osaan siitä.
Kuva 23. PhAT-anturi asetettuna liikkuvan alustan yläpuolelle, alustan pyörimis-nopeutena käytettiin yhtä kierrosta minuutissa.
6.5.3 Raman-spektrin esikäsittely
Raman-spektrometristä saatu data käsiteltiin Matlab (MathWorks, Yhdysvallat) -
tietojenkäsittelyohjelmalla. Mittauksissa tarkasteltiin aaltolukualuetta 250–1909.
Matlab-ohjelmalla data muutettiin Excel®-muotoon (Microsoft, Yhdysvallat), jota
SIMCA-P -mallinnusohjelma pystyy käyttämään.
Raman-spektrometrin antama data kustakin seossarjasta käsiteltiin SIMCA-P 12.0.1.
(Umetrics AB, Ruotsi) -ohjelmalla erilaisilla esikäsittelytavoilla. Esikäsitellyistä
datoista muodostettiin Partial least square (PLS) -malleja, joiden avulla on
mahdollista tunnistaa Raman-spektrometrillä mitatun näytteen lääkeaineen pitoisuus.
PLS-mallien tarkoituksena on tunnistaa X- ja Y-komponentteja yhdistäviä rakenteita
(Abdi 2003). Yhdistävillä komponenteilla pyritään selittämään mahdollisimman hyvin
halutun komponentin vaihtelua ja muodostamaan niistä malli, jonka perusteella

64
voidaan yrittää ennustaa halutun komponentin muutoksia toisen perusteella. Mitä
useampi tarkkailtava ja selittävä komponentti otetaan huomioon, sitä tarkemmaksi
malli saadaan, mutta mallin toistettavuus kärsii. Ennustusmallien monimutkaisuuden
määritysperusteeksi valittiin mallin tuottama q2-summa, joka kuvastaa mallin
ennustuskykyä uudelle datalle. Q2-summan tavoitearvoksi määriteltiin 0,93, jonka
täytyttyä mallissa oli tarpeeksi komponentteja.
Ohjelma tuottaa observed vs. predicted (OP) (kuva 24) ja ennustus (kuva 25) -
kuvaajia. OP-kuvaajat antavat Root Mean Square Error of Estimation (RMSEE) -
arvoja, jotka osoittavat, kuinka hyvin mitatut pitoisuudet (x-akseli) kuvaavat
teoreettisia pitoisuuksia (y-akseli). Pienempi arvo tarkoittaa parempaa kuvaavuutta.
Ennustuskuvaajan luonnissa mallista poistettiin joitakin mittauspisteitä ja näiden
poistettujen pisteiden avulla testattiin, kuinka hyvin jäljelle jääneen mallin avulla
pystyttiin ennustamaan poistetut mittauspisteet. Ennustus antaa tuloksen root mean
squared error of prediction (RMSEP) -arvona, jossa pienempi tulos tarkoittaa
parempaa mallin ennustustehoa eli tulokset poikkeavat nimellisarvoistaan
vähemmän. Ennen ennustuksen ja mallin tekemistä datasta poistettiin mittauspisteet,
joissa ilmeni karkeita virheitä tai jotka olivat luottamusrajojen ulkopuolella.
Kaikista seossarjoista muodostettiin taulukot, joissa eriteltiin eri esikäsittelyt ja
mittausajat. Taulukoiden avulla pystyttiin vertailemaan helpommin eri esikäsittelyiden
ja mittausaikojen vaikutusta tuloksiin.

65
Kuva 24. Asetyylisalisyylihappo-dikalsiumfosfaattiseoksen mittauspisteet ilman esikäsittelyä yhden sekunnin mittausajalla
Kuva 25. Asetyylisalisyylihappo-dikalsiumfosfaattiseoksen ennustuspisteet ilman mallin esikäsittelyä yhden sekunnin mittausajalla
6.6 Kemiallinen lääkeainepitoisuuden määrittäminen
Raman-spektroskopian tarkkuuden tarkastamiseksi päätettiin mitata yhden
seossarjan näytteiden pitoisuudet myös kemiallisesti. Mitattaviksi valittiin
asetyylisalisyylihapon ja dikalsiumfosfaatin seokset, joiden näytteillä oli pienimmät
pitoisuuserot. Menetelmäksi valittiin näytteiden liuotus ja pitoisuuksien määrittäminen
spektrofotometrillä (Shimadzu UV-1800 Kyoto, Japani) käyttäen hyödyksi
asetyylisalisyylihapon (ASA) (276 nm) ja salisyylihapon (SA) (300nm)
absorbanssiarvoja (Iwunze 2008). Mittaamalla myös salisyylihapon absorbanssi

66
haluttiin ottaa huomioon asetyylisalisyylihapon osittainen hajoaminen liuoksessa
salisyylihapoksi. Menetelmässä näytteet liuotettiin sekoituksen avulla 500 ml:aan
puhdistettua vettä, joko lämmityksessä tai ilman lämmitystä, ja liuotusaikoina
käytettiin neljää, kahdeksaa ja 24 tuntia. Liuotuksen jälkeen suodatetut ja laimennetut
liuokset mitattiin spektrofotometrin avulla. Asetyylisalisyylihapolle ja salisyylihapolle
valmistettiin pitoisuusmääritystä varten kantaliuokset puhdasaineista, joiden avulla
valmistettiin kolmet rinnakkaiset laimennossarjat (liitetaulukot 2 ja 3) standardisuorien
(kuva 26) määrittämistä varten. Kantaliuokset tehtiin punnitsemalla tarkkuusvaa’alla
(Sartorius A200S, Yhdysvallat) 0,5024 g asetyylisalisyylihappoa ja 0,5038 g
salisyylihappoa omiin 500 ml:n mittapulloihinsa ja täyttämällä mittapullot tislatulla
vedellä merkkiviivaan saakka. Kantaliuoksia sekoitettiin 24 tunnin ajan
magneettisekoittajien (Heidolph MR 3001, Saksa) päällä, ilman lämmitystä,
sekoitusnopeudella 200 kierrosta minuutissa (rpm). Valmistetut laimennokset
kantaliuoksista olivat pitoisuuksiltaan ASA: 50,2 mg/l, 101 mg/l, 201 mg/l, 301 mg/l,
402 mg/l ja 502 mg/l sekä SA 5,038 mg/l, 37,785 mg/l, 50,38 mg/l, 75,57 mg/l, 100,4
mg/l, ja yksi välilaimennos 151,14 mg/l.
ASA/DCP-näyteseoksien tutkimusliuokset valmistettiin punnitsemalla kutakin näytettä
yhden gramman verran 500 ml:n mittapulloon (liitetaulukko 4). Mittapullot täytettiin
lähelle 500 ml:n merkkiä tislatulla vedellä ja suljettiin. Tämän jälkeen mittapullot
asetettiin magneettisekoittajien (Heidolph MR 3001) päälle, jotka oli säädetty 60 ºC
lämpötilaan ja pyörimään 200 kierrosta minuutissa, jotta asetyylisalisyylihappo liukeni
nopeammin. Asetyylisalisyylihapon annettiin liueta sekoituksessa 24 tuntia.
ASA/DCP-näytteiden jauheseoksista valmistettiin myös toiset samanlaiset liuokset,
joita ei lämmitetty liuotuksen aikana, ja joiden sekoitusajat olivat lyhyemmät (neljä ja
kahdeksan tuntia). Vaihtoehtoisella liuotustavalla haluttiin vähentää ASA:n
hajoamista salisyylihapoksi liuotuksessa. Sekoituksien jälkeen kaikki mittapullot
jäähdytettiin lähelle huoneenlämpöä ja täytettiin tislatulla vedellä mittapullon
merkkiviivaan. Jokaisesta ASA/DCP-liuoksesta suodatettiin osa 20 µm (Sartorius
Minisart tai Corning Incorporated) suodattimen läpi ja laimennettiin liitetaulukon 4
mukaisesti, jotta näytteet osuisivat määritetyille kalibraatiosuoralle. Kustakin
suodatetusta ja laimennetusta ASA/DCP-liuoksesta tehtiin kolme rinnakkaista
näytettä, jotka spektrofotometri mittasi kolmeen kertaan aallonpituuksilla 276 ja 300
nm.

67
Kuva 26. Asetyylisalisyylihapon ja salisyylihapon standardisuorat
6.7 Raman-spektrikirjasto
Raman-spektrikirjastoon kerätyt aineet mitattiin käyttäen yhden ja kuuden sekunnin
mittausaikoja. Spektrit tallennettiin käsittelemättöminä aaltolukualueelta 250–1908
cm−1. Saatuja spektrejä verrattiin kirjallisuudesta löytyneisiin spektreihin, jotta
pystyttiin arvioimaan saatuja tuloksia. Spektrikirjastoon kerättiin kullekin aineelle
tunnusomaiset piikit ja kuva spektristä. Spektrit arvotettiin tunnistettavuuden mukaan
silmämääräisesti kolmeen luokkaan: huono, tyydyttävä ja hyvä. Arvotukseen vaikutti
pohjatason korkeus, piikkien korkeus pohjatasoon nähden, piikkien muoto sekä
piikkien lukumäärä.
y = 0,0056x - 0,0988 R² = 0,9995
y = 0,0244x + 0,0242 R² = 0,9997
0
0,5
1
1,5
2
2,5
3
0 100 200 300 400 500 600
Ab
so
rban
ssi
Laskennallinen pitoisuus mg/l
Asetyylisalisyylihappo
Salisyylihappo

68
7 TULOKSET
7.1 Lääkeainepitoisuuden kemialliset määritykset
Taulukossa 4 nähdään asetyylisalisyylihapon laskennalliset pitoisuudet sekä
asetyylisalisyylihappoa ja dikalsiumfosfaattia sisältävien näyteseoksien
spektrifotometrillä määritetyt pitoisuudet. Kuvissa 27 ja 28 on esitetty tulokset sekä
24 tuntia lämmitetyssä ja sekoituksessa olleille näytteille että neljä ja kahdeksan
tuntia pelkässä sekoituksessa olleille näytteille. Taulukossa 5 on esitetty
asetyylisalisyylihapon ja salisyylihapon laskennalliset ja mitatut pitoisuudet neljän ja
kahdeksan tunnin liuotuksen jälkeen. Laskennalliset pitoisuudet ylittyvät, kun
näyteseoksia on lämmitetty ja sekoitettu 24 tuntia. Pitoisuusylitykset olivat
suurimmassa osassa liuoksia 50–100 mg/l eli ne arvioitiin 2–6 % teoreettisia
pitoisuuksia vahvemmiksi. Ero korostuu, kun otetaan huomioon myös ASA:sta SA:ksi
muuttunut lääkeainemäärä. Uusituilla lyhemmillä mittauksilla näytteiden pitoisuudet
jäivät selvästi alle laskennallisten pitoisuuksien sekä neljän että kahdeksan tunnin
liuotuksilla (taulukko 5).
Taulukko 4. Asetyylisalisyylihappoa ja dikalsiumfosfaattia sisältävien seosnäytteiden teoreettiset ja kemiallisella analyysillä määritetyt pitoisuudet. Näytteitä liuotettiin 24 tuntia lämmityksessä ja sekoituksessa
ASA (m/m %)
Laskennallinen pitoisuus mg/l
Mitattu c(ASA,24h) mg/l
Mitattu ASA (m/m %)
Mitattu c(SA,24h) mg/l
5 101,67 104,43 5,14 43,354 10 204,28 280,09 13,71 128,66
12,5 250,00 345,31 17,27 157,81 15 303,54 397,13 19,62 173,47
17,5 351,05 464,65 23,16 208,44 20 404,96 454,02 22,42 175,88 30 609,00 670,94 33,05 276,75
40 808,16 818,22 40,50 321,85

69
Kuva 27. Asetyylisalisyylihapposeoksien kemiallisella analyysillä määritetyt pitoisuudet verrattuna laskennallisiin pitoisuuksiin.
Kuva 28. Salisyylihapon pitoisuus seosnäytteissä eri sekoitusolosuhteissa
0
100
200
300
400
500
600
700
800
900
0 100 200 300 400 500 600 700 800 900
Pito
isuu
s (
mg/l)
Teoreettinen pitoisuus (mg/l)
Näytteet ilmanlämmitystä 4-5hliuotuksen jälkeen
Näytteet ilmanlämmitystä 8hliuotuksen jälkeen
Teoreettinenpitoisuus
24h Lämmitetytnäytteet
0
50
100
150
200
250
300
350
0 10 20 30 40 50
Sa
lisyylih
ap
on
pito
isu
us (
mg
/l)
Asetyylisalisyylihapon pitoisuus (%)
24 tunnialämmityksessä
4-5 tuntia ilmanlämmitystä
8 tuntia ilmanlämmitystä

70
Taulukko 5. Asetyylisalisyylihappoa ja dikalsiumfosfaattia sisältävien seosten lääkeainepitoisuudet laskennallisesti ja kemiallisen analyysin avulla määritettynä. Näytteitä sekoitettiin liuotuksen aikana ilman lämmitystä neljä ja kahdeksan tuntia.
ASA (m/m %)
Laskennallinen pitoisuus (mg/l)
c = ASA 4-5h (mg/l)
c = SA 4h (mg/l)
c = ASA 8h (mg/l)
c = SA 8h (mg/l)
5 100,75 64,30 7,95 61,77 8,68 10 200,10 132,82 11,78 138,33 16,22
12,5 250,05 171,09 14,93 179,61 20,58 15 314,28 206,79 14,05 213,88 20,31
17,5 350,81 221,10 14,69 228,69 21,41 20 400,28 243,46 13,19 251,87 20,59 30 601,02 357,95 19,84 379,99 32,11 40 800,48 454,11 23,68 483,56 38,28
Kuvassa 29 on eri sekoituskäytännöillä tehtyjen spektrofotometrimittausten
pyyhkäisyspektrit tutkitulta aallonpituusalueelta. Pyyhkäisyspektrit osoittavat, että 24
tunnin sekoituksen aikana matalia pitoisuuksia asetyylisalisyylihappoa sisältävät
näytteet hajoavat salisyylihapoksi ja peittävät asetyylisalisyylihapon aallonpituuden
alleen. Ainoastaan vahvimmalla 1000,2 mg/l pitoisuudella sekoitettujen ASA-
näytteiden spektrit vaikuttavat luotettavilta. 100,2 mg/l pitoisuuden 24 tunnin
sekoituksen kohdalla täytyy jo miettiä, kuinka paljon SA:n piikki vaikuttaa ASA:n
tulokseen.

71
Kuva 29. Liuotettavan ASA:n määrän ja hydrolysoitumisen vaikutukset spektrofotometrin spektriin eri sekoitusajoilla, kun liuotustilavuus pysyy samana. Kunkin näytteen laimennettu ASA pitoisuus oli spektrofotometrimittauksissa noin 100 mg/l.
7.2 Eri esikäsittelyiden vaikutukset malliin ja
ennustettavuuteen
7.2.1 Asetyylisalisyylihappo-dikalsiumfosfaattiseos
Taulukossa 6 on koottuna eri esikäsittelyiden vaikutukset asetyylisalisyylihapon ja
dikalsiumfosfaatin seoksien Raman-spektrometrimittauksista saataviin malleihin ja
niiden ennustavuuteen. Puhdas asetyylisalisyylihappo ei pysynyt suuren
pitoisuuseron vuoksi luotettavuusrajojen sisäpuolella, mistä johtuen se on poistettu
mittauspisteistä. Tiiviimmällä näytejoukolla saavutettiin myös parempi ennustusteho.
Asetyylisalisyylihapon ja dikalsiumfosfaatin seokset käyttäytyivät hyvin loogisesti,
mikä voidaan todeta hyvistä tuloksista ilman minkäänlaista esikäsittelyäkin. Paras
ennustettavuus numeerisesti tuli Row centering -käsittelyllä, jolla saatiin RMSEP-
arvoksi 0,77 kahdella merkitsevällä komponentilla. Ennustettavuudeltaan samalle

72
tasolle voidaan tulkita myös esikäsittelemättömän datan kahden komponentin
RMSEP-arvo 0,83 ja toisen asteen derivaatalla saatu yhden komponentin RMSEP-
arvo 0,91 niiden mallien yksinkertaisuuden vuoksi. Lyhyempi mittausaika paransi
sekä esikäsittelemättömän mallin että derivoitujen mallien ennustavuutta, kun taas
sirontaa korjaavilla käsittelyillä lyhyempi mittausaika antoi huonommat tulokset. Erot
mallien välillä eivät kuitenkaan olleet suuria.
7.2.2 Ibuprofeeni-laktoosiseos
Ibuprofeenin ja laktoosin seossarjasta poistettiin mittauspiste 55 % mittausvirheenä.
Tälle seokselle tehokkaimmiksi käsittelyiksi osoittautui toisen asteen derivointi
RMSEP-arvolla 3,71 yhdellä vaikuttavalla komponentilla, käsittelemätön data
kahdella vaikuttavalla komponentilla (RMSEP 3,79) ja Row centering yhdellä
komponentilla (RMSEP 3,78) (taulukko 7). Mittauksen kestolla ei näytä olevan tässä
seoksessa suurta merkitystä mallien ennustavuuteen.
7.2.3 Ibuprofeeni-mannitoliseos
Ibuprofeenin ja mannitolin seosksissa kolmella komponentilla parhaimmat tulokset
antoivat tasoituspohjainen Savitzky-Golay -käsittely (RMSEP 6,24) ja ilman
esikäsittelyä suoritettu ennustus (RMSEP 7,14) (taulukko 8). Sirontaa korjaava MSC-
käsittely saavutti kahdella vaikuttavalla komponentilla RMSEP-arvon 7,76.
Mittausajoilla ei ollut vaikutusta ennustustehoon. Mittauspiste 40 % poistettiin kaikista
käsittelyistä mittausvirheenä. Myös 85 %:n näyte erottui SIMCA:ssa muista näytteistä
selvästi, mutta sirontaa korjaavat käsittelyt pystyivät korjaamaan tämän eron.
7.2.4 Parasetamoli-mannitoliseos
Parasetamolin ja mannitolin seoksissa 90 % näyte poistettiin malleista
mittausvirheenä (taulukko 9). Myös 60 %:n näyte erottui muista näytteistä

73
SIMCA:ssa. Sirontaa korjaavat esikäsittelyt pystyivät kuitenkin korjaamaan tämän
eron näytteissä, toisin kuin derivointipohjaiset esikäsittelyt. SNV- ja MSC-käsittelyt
toimivat parhaiten tälle seossarjalle antaen RMSEP-tulokset SNV 3,24 ja MSC 3,14
kolmella vaikuttavalla komponentilla. Vertailukelpoisena tuloksena voidaan pitää
ensimmäisen asteen derivaatan kahden komponentin RMSEP-arvoa 4,72.
Mittausajan pituudella ei ollut vaikutusta seossarjan ennustettavuuteen.
7.2.5 Parasetamoli-laktoosiseos
Parasetamolin ja laktoosin seoksilla hajontaa korjaavat MSC ja SNV nousevat
soveltuvimmiksi esikäsittelyiksi (taulukko 10). MSC antaa RMSEP-arvon 2,14 ja
SNV-käsittely 2,28. Mittausajan vaikutus jäi pieneksi tällä seoksella. Ainoastaan
esikäsittely-yhdistelmät MSC-dydx ja SNV-dydx aiheuttavat eroa yhden ja kuuden
sekunnin mittauksien välille. Yhden sekunnin mittauksessa MSC-dydx -esikäsittelyllä
saatiin RMSEP-tulos 5,17. Vastaava tulos kuuden sekunnin mittauksessa oli 2,55.
SNV-dydx -esikäsittelyllä vastaavat RMSEP-arvot olivat 4,82 ja 2,77.

74
Taulukko 6. Esikäsittelyiden vaikutukset acetyylisalisyylihapon ja dikalsiumfosfaatin seoksiin. dydx: ensimmäisen asteen derivaatta esikäsittely, dydx2: toisen asteen derivaatta esikäsittely, SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely, Row: rivien keskitys esikäsittely.
Asetyylisalisyylihappo ja dikalsiumfosfaatti 5–40%
Malli Ennuste Ennuste
(q2≥0,93)
Yhden sekunnin sarjat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustus- pisteet
q2- summa
Ilman käsittelyä 1,29 1,91 0,83 425;550;750;784;985;1044;1191;1293;1606 2 100 5:12,5;17,5;30 0,99
dydx 1,46 1,84 1,02 750;784;1044;1191;1293;1606 1 100 5:12,5;17,5;30 0,983
SNV-dydx 1,57 - 4,83 550;750;784;985;1044;1191;1293;1606 4 100 5:12,5;17,5;30 0,962
MSC-dydx 1,12 - 3,48 550;750;784;1044;1191;1293;1606 4 100 5:12,5;17,5;30 0,974
Savitzky-Golay 1,29 1,91 0,83 425;550;750;784;1044;1191;1293;1606 2 100 5:12,5;17,5;30 0,984
dydx2 1,29 1,66 0,91 750;784;1044;1191;1293;1606 1 100 5:12,5;17,5;30 0,995
MSC 3,47 4,93 3,78 425,550;750;784;1191;1293; 3 100 5:12,5;17,5;30 0,94
SNV 2,20 - 4,81 425;550;750;784;1044;1293;1606 4 100 5:12,5;17,5;30 0,973
Row 1,20 1,78 0,77 425;550;750;784;985;1044;1191;1293;1606 2 100 5:12,5;17,5;30 0,979
Kuuden sekunnin sarjat
Ilman käsittelyä 1,26 1,79 0,90 425;550;750;784,5;1044;1293;1606 2 100 5:12,5;17,5;30 0,986
dydx 1,45 1,70 1,07 425;550;750;784;1044;1191;1293;1606 1 100 5:12,5;17,5;30 0,993
dydx2 1,29 1,61 0,98 287,425,550,639,702,750,784,1044,1150,1191,1257,1043,1606 1 100 5:12,5;17,5;30 0,988
SNV-dydx 1,72 - 3,97 550;750;784;1044;1191;1293;1606 4 100 5:12,5;17,5;30 0,973
MSC-dydx (4,34) - 2,86 750;784;1044;1191;1606 (3)4 100 5:12,5;17,5;30 0,977
SNV (4,41) - 3,26 425;550;750;784;1044;1293;1606 (3)5 100 5:12,5;17,5;30 1,0
MSC (3,29) - 1,91 425;550;750;784;1044;1191;1293;1606 (3)4 100 5:12,5;17,5;30 0,945
Row 1,21 1,73 0,84 425;550;750;784;1044;1293;1606 2 100 5:12,5;17,5;30 0,981
Row-dydx 1,45 1,80 1,07 425;550;750;784;1044;1191;1293;1606 1 100 5:12,5;17,5;30 0,983

75
Taulukko 7. Esikäsittelyiden vaikutukset ibuprofeenin ja laktoosin seoksiin. dydx: ensimmäisen asteen derivaatta esikäsittely, dydx2: toisen asteen derivaatta esikäsittely, SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely, Row: rivien keskitys esikäsittely.
Ibuprofeeni ja laktoosi 5–95 %
Malli Ennuste Ennuste (q2≥0,93)
Yhden sekunnin sarjat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustus- pisteet
q2- summa
Ilman käsittelyä 7,48 9,14 3,79 636,744,820,833,1115,1181,957,1608 2 55 10,20,40,70,90 0,978
dydx 7,82 9,36 3,78 636;744;783;820;833;1007;1115;1181;1207;1608 1 55 10,20,40,70,90 0,974
dydx2 7,77 9,33 3,71 636;744;783;820;833;1007;1115;1181;1207;1608 1 55 10,20,40,70,90 0,974
SNV 11,94 13,27 9,52 636;744;783;820;833;1007;1115;1181;1207;1608 2 55 10,20,40,70,90 0,953
SNV-dydx 11,90 16,25 13,61 636;744;783;820;833;1007;1115;1181;1207;1608 1 55 10,20,40,70,90 0,93
dydx-SNV 6,95 7,48 5,44 636;744;783;820;833;1007;1115;1181;1207;1608 5 55 10,20,10,70,90 0,96
MSC 10,27 11,43 8,36 636;744;820;833;1007;1115;1181;1207;1608 2 55 10,20,40,70,90 0,966
MSC-dydx 14,51 14,61 13,65 636;744;783;820;833;1007;1115;1181;1207;1608 1 55 10,20,40,70,90 0,949
dydx-Row 7,82 9,36 3,78 636;744;783;820;833;1007;1115;1181;1207;1608 1 55 10,20,40,70,90 0,974
Kuuden sekunnin sarjat
Ilman käsittelyä 7,50 9,16 3,78 636;744;783;820;833;1007;1115;1181;1207;1608 2 55 10,20,40,70,90 0,977
dydx 7,80 9,37 3,65 636;744;783;820;833;1007;1115;1181;1207;1608 1 55 10,20,40,70,90 0,974
dydx2 7,76 9,34 3,61 636;744;783;820;833;1007;1115;1181;1207;1608 1 55 10,20,40,70,90 0,974
SNV-dydx 15,51 16,12 13,95 636;744;783;820;833;1007;1115;1181;1207;1608 1 55 10,20,40,70,90 0,931
SNV 11,95 13,19 9,71 636;744;783;820;833;1007;1115;1181;1207;1608 2 55 10,20,40,70,90 0,954
dydx-SNV (0,92) 6,12 11,22 636;744;783;820;833;1007;1115;1181;1207;1608 (6)5 55 10,20,40,70,90 0,94
MSC 10,32 11,39 8,66 636;744;783;820;833;1007;1115; 1181;1207;1452;1608 2 55 10,20,40,70,90 0,964

76
Taulukko 8. Esikäsittelyiden vaikutukset ibuprofeenin ja mannitolin seoksiin. dydx: ensimmäisen asteen derivaatta esikäsittely, SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely.
Ibuprofeeni ja mannitoli 10–90 % malli ennuste ennuste (q2≥0,93)
Yhden sekunnin sarjat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustus- pisteet
q2-summa
Ilman käsittelyä 7,29 11,08 7,14 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 3 40 10,20,50,70,80 0,976
dydx (8,34) 9,95 8,47 636;745;783;820;833;876;1007;1115;1181;1206;1608 (1)2 40 10,20,50,70,80 0,965
MSC-dydx (7,52) 17,24 15,23 636;745;783;820;833;876;1007;1115;1181;1206;1608 (2)1 40 10,20,50,70,80 0,934
Savitzky-Golay 7,29 9,38 6,24 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 3 40 10,20,50,70,80 0,976
SNV-dydx 10,93 12,52 10,98 636;745;783;820;833;876;1007;1115;1181;1206;1608 2 40 10,20,50,70,80 0,931
SNV 10,95 12,49 11,08 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 2 40 10,20,50,70,80 0,932
MSC 7,53 8,53 7,76 636;745;783;820;833;876;1007;1115;1181;1206;1452:1608 2 40 10,20,50,70,80 0,978
Kuuden sekunnin sarjat
Ilman käsittelyä 6,72 8,10 6,35 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 3 40 10,20,50,70,80 0,978
dydx 8,04 9,57 8,28 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 3 40 10,20,50,70,80 0,971
MSC-dydx (7,47) 17,11 15,14 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 (2)1 40 10,02,50,70,80 0,935
Savitzky-Golay 6,72 8,10 6,35 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 3 40 10,20,50,70,80 0,978
SNV-dydx 10,93 12,52 10,97 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 2 40 10,20,50,70,80 0,931
MSC 7,46 8,45 7,70 636;745;783;820;833;876;1007;1115;1181;1206;1452;1608 2 40 10,20,50,70,80 0,979

77
Taulukko 9. Esikäsittelyiden vaikutukset parasetamolin ja mannitolin seoksiin. dydx: ensimmäisen asteen derivaatta esikäsittely, SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely, SG: Savitzky-Golay esikäsittely
Parasetamoli ja mannitoli 10–90 %
malli ennuste ennuste (q2≥0,93)
Yhden sekunnin sarjat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustus- pisteet
q2- summa
Ilman käsittelyä (12,18) 8,84 9,04 391;651;797;858;1168;1237;1324;1610 (2)3 90 10,20,40,70,85 0,967
dydx (6,97) 3,95 4,72 391;651;797;608;1168;1237;1324;1371;1313;1610;1398 (1)2 90 10,20,40,70,85 0,975
SNV 2,69 2,42 3,24 391;651;797;608;1168;1237;1324;1371;1313;1610;1398 3 90 10,20,40,70,85 0,995
SNV-dydx 4,55 4,37 5,61 391;651;797;858;1168;1237;1277;1324;1371;1610;1649 3 90 10,20,40,70,85 0,988
MSC 2,44 1,94 3,14 391;651;797;858;1168;1237;1277;1324;1371;1610;1649 3 90 10,20,40,70,85 0,996
MSC-dydx 5,42 3,94 5,72 327;391;651;797;858;1168;1237;1277;1324;1371;1561;1610;1649 3 90 10,20,40,70,85 0,991
Savitzky-Golay 12,18 8,84 9,04 327;391;651;797;858;1168;1237;1277;1324;1371;1561;1610;1649 1 90 10,20,40,70,85 0,967
SG-dydx (5,57) 8,38 12,50 327;391;651;797;858;1168;1237;1277;1324;1371;1561;1610;1649 (1)3 90 10,20,40,70,85 0,994
Kuuden sekunnin sarjat
Ilman käsittelyä (12,01) 10,45 10,75 391,651,797,858,1168,1237,1324,1371,1610,1649 (2)3 90 10,20,40,70,85 0,961
dydx 12,59 9,06 12,79 391,651,797,858,1168,1237,1277,1324,1610,1649 1 90 10,20,40,70,85 0,952
SNV 2,90 2,24 3,46 391,651,797,858,1168,1237,1324,1371,1610 3 90 10,20,40,70,85 0,997
SNV-dydx 4,16 4,12 4,85 391,651,797,858,1168,1237,1324,1371,1610,1649 3 90 10,20,40,70,85 0,991
SNV-SG 2,90 2,24 3,46 391,651,797,858,1168,1237,1324,1371,1610 3 90 10,20,40,70,85 0,997
MSC 2,55 2,13 3,07 391,651,797,858,1168,1237,1324,1610 3 90 10,20,40,70,85 0,997
MSC-dydx 3,56 2,93 4,61 391,651,797,858,1168,1237,1324,1610 3 90 10,20,40,70,85 0,996
Savitzky-Golay (12,01) 10,45 10,75 327;391;651;797;858;1168;1237;1277;1324;1371;1561;1610;1649 (2)3 90 10,20,40,70,85 0,961
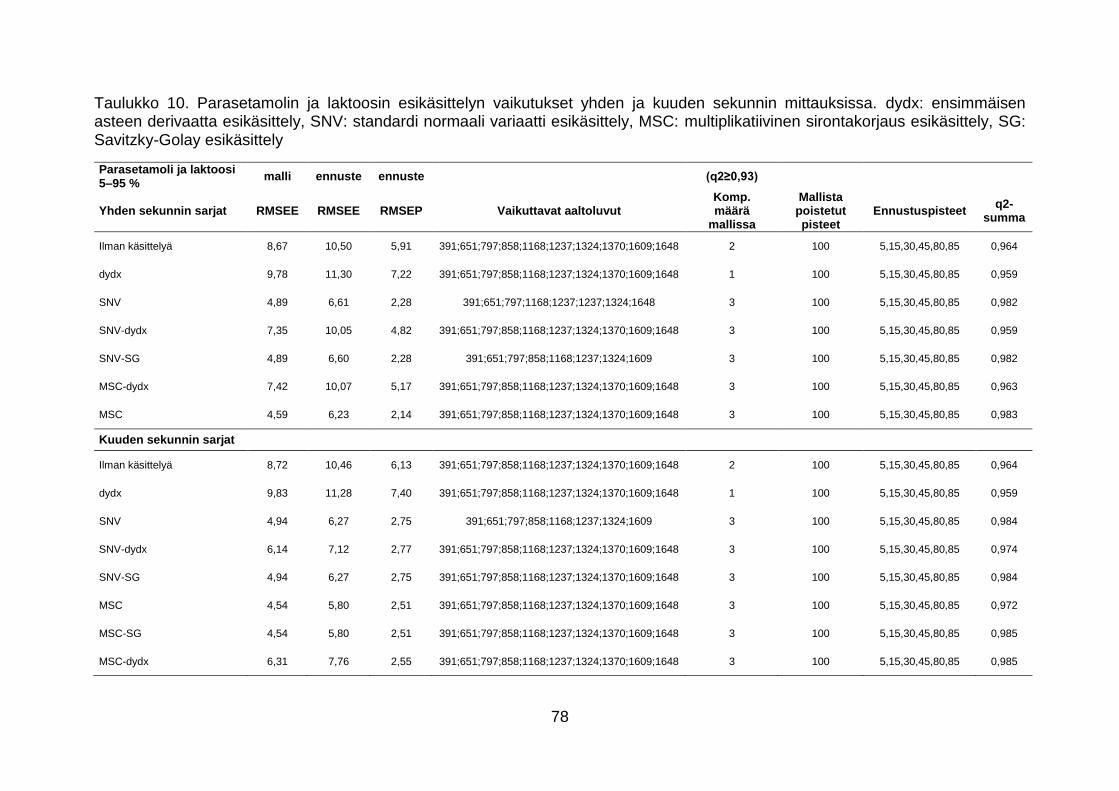
78
Taulukko 10. Parasetamolin ja laktoosin esikäsittelyn vaikutukset yhden ja kuuden sekunnin mittauksissa. dydx: ensimmäisen asteen derivaatta esikäsittely, SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely, SG: Savitzky-Golay esikäsittely
Parasetamoli ja laktoosi 5–95 %
malli ennuste ennuste (q2≥0,93)
Yhden sekunnin sarjat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustuspisteet q2-
summa
Ilman käsittelyä 8,67 10,50 5,91 391;651;797;858;1168;1237;1324;1370;1609;1648 2 100 5,15,30,45,80,85 0,964
dydx 9,78 11,30 7,22 391;651;797;858;1168;1237;1324;1370;1609;1648 1 100 5,15,30,45,80,85 0,959
SNV 4,89 6,61 2,28 391;651;797;1168;1237;1237;1324;1648 3 100 5,15,30,45,80,85 0,982
SNV-dydx 7,35 10,05 4,82 391;651;797;858;1168;1237;1324;1370;1609;1648 3 100 5,15,30,45,80,85 0,959
SNV-SG 4,89 6,60 2,28 391;651;797;858;1168;1237;1324;1609 3 100 5,15,30,45,80,85 0,982
MSC-dydx 7,42 10,07 5,17 391;651;797;858;1168;1237;1324;1370;1609;1648 3 100 5,15,30,45,80,85 0,963
MSC 4,59 6,23 2,14 391;651;797;858;1168;1237;1324;1370;1609;1648 3 100 5,15,30,45,80,85 0,983
Kuuden sekunnin sarjat
Ilman käsittelyä 8,72 10,46 6,13 391;651;797;858;1168;1237;1324;1370;1609;1648 2 100 5,15,30,45,80,85 0,964
dydx 9,83 11,28 7,40 391;651;797;858;1168;1237;1324;1370;1609;1648 1 100 5,15,30,45,80,85 0,959
SNV 4,94 6,27 2,75 391;651;797;858;1168;1237;1324;1609 3 100 5,15,30,45,80,85 0,984
SNV-dydx 6,14 7,12 2,77 391;651;797;858;1168;1237;1324;1370;1609;1648 3 100 5,15,30,45,80,85 0,974
SNV-SG 4,94 6,27 2,75 391;651;797;858;1168;1237;1324;1370;1609;1648 3 100 5,15,30,45,80,85 0,984
MSC 4,54 5,80 2,51 391;651;797;858;1168;1237;1324;1370;1609;1648 3 100 5,15,30,45,80,85 0,972
MSC-SG 4,54 5,80 2,51 391;651;797;858;1168;1237;1324;1370;1609;1648 3 100 5,15,30,45,80,85 0,985
MSC-dydx 6,31 7,76 2,55 391;651;797;858;1168;1237;1324;1370;1609;1648 3 100 5,15,30,45,80,85 0,985

79
7.3 Liikkeessä tehdyt mittaukset
7.3.1 Asetyylisalisyylihappo-dikalsiumfosfaattiseokset
ASA:n ja DCP:n seoksilla tehtiin mittaukset kahdelle erilaiselle mittausjoukolle.
Mittausjoukossa 1 käytettiin ASA:n pitoisuuksia 10 %, 15 % ja 20 % (taulukko 11).
Mittausjoukossa 2 ASA:n pitoisuudet olivat lähempänä toisiaan – 12,5 %, 15 % ja 17,5 %
(taulukko 12). Näin saatiin tarkasteltua laitteiston erotuskykyä paremmin. Tutkittaessa
mittausjoukon 1 näytteitä omana ryhmänään, jossa ennustusmalli muodostettiin osasta
mittauspisteistä, saavutettiin hyviä ennustustuloksia mallilla. Eri esikäsittelyillä saavutettiin
0,96–1,53 RMSEP-arvoja yhdellä tai kahdella komponentilla. Mittausjoukolla 2 saavutettiin
vastaavalla järjestelyllä RMSEP-arvoiksi 1,2–1,96. Mittausjoukon 1 mittaukset pysyivät
esikäsittelemättömässä datassa omina ryhminään, eikä ohjelma arvioinut niitä observed
vs. predicted kuvaajassa sekaisin (kuva 30). 10 % näytteet (n=3) asettuivat välille 11,7–12
%, 15 % näytteet (n=11) taas välille 13,2–17,1 % ja 20 % näytteet (n=2) välille 18,2–18,3
%. Mittausjoukossa 2 ryhmät eivät pysyneet samoilla asetuksilla omina ryhminään. 12,5 %
näytteet (n=3) asettuivat ohjelman arvioinnissa 14,3–15 % välille. 15 % näytteet (n=11)
arvioitiin 12,7–16,4 % välille ja 17,5 % näytteet (n=2) 16,7–17,4 % välille (kuva 31). SNV-
ja MSC-esikäsittelyiden mallit täyttivät tutkimuksen määritysrajan q2≥0,93 jo yhdellä
komponentilla. Muodostetut mallit eivät osanneet kuitenkaan määrittää eroa seoksille
vasta kuin kahdella komponentilla, joten määritysrajasta poiketaan näissä kohdissa.
Ongelma koski mittauksia, joissa liikkuvat näytteet mitattiin keskenään ja liikkuvat näytteet
oli yhdistetty paikallaan mitattujen 10 %, 12,5 %, 15 %, 17,5 % ja 20 % näytteiden kanssa.

80
Kuva 30. Mittausjoukon yksi 10, 15 ja 20 % ASA näytteet erottuivat omina ryhminään x-akselilla
Kuva 31. Mittausjoukon kaksi 12,5 %, 15 % ja 17,5 % ASA näytteet eivät erottuneet enää täysin omina ryhminään mittausjoukossa
Kun liikkuvien mittausten spektrit yhdistettiin paikallaan mitattujen kanssa, erottuivat
paikallaan mitatut spektrit selvästi korkeamman intensiteetin johdosta niin piikkien kuin
taustankin osalta. Sirontaa korjaavat esikäsittelyt kykenivät poistamaan pääosin liikkeestä

81
johtuvan eron mittausten välillä. Tämä oli havaittavissa sarjoista muodostettujen mallien
antamista ennustustuloksista. Muut kuin sirontaa korjaavat esikäsittelyt antoivat
huonompia RMSEP-arvoja, kun niitä verrattiin pelkästään liikkeessä mitattuihin ryhmiin.
Sama ilmiö havaittiin molemmilla mittausjoukoilla.
Liikkuvat näytteet sovitettiin myös laajempiin, koko pitoisuusvaihtelun omaaviin, paikallaan
mitattuihin malleihin. Ainoastaan sirontaa korjaavat esikäsittelyt ja niiden yhdistelmät
antoivat järkeviä tuloksia, joten muut esikäsittelyt jätettiin tässä huomioimatta.
Tehokkaimmiksi esikäsittelyiksi sopivat SNV (RMSEP=2,96) ja MSC (RMSEP=3,10)
kahdella vaikuttavalla komponentilla. SNV-käsittelyllä RMSEP-kuvaajassa 10 % näytteet
(n=3) olivat 11,8–12,4 %, 15 % näytteet (n=11) sijoittuivat mallin avulla 15,1–19,4 % ja
20 % näytteet (n=2) vastaavasti 23,6–23,8 %.
7.3.2 Ibuprofeeni-laktoosiseokset
Ibuprofeenin ja laktoosin seoksilla tehtiin sama mittaus ja vastaavat esikäsittelyt
ibuprofeenin pitoisuuksilla 10 %, 15 % ja 20 % (taulukko 13). Tällä seoksella paras
RMSEP-tulos saavutettiin esikäsittely-yhdistelmällä SNV ja toisen asteen derivaatta, joka
antoi tuloksen 1,84 liikkuvien näytteiden keskinäisessä vertailussa ja 1,82 yhdistettynä
samojen pitoisuuksien paikallaan oleviin näytteisiin. MSC- ja SNV-käsittelyt antoivat
pienimmät RMSEP-arvot (0,45 ja 0,50). Arvot eivät kuitenkaan ole vertailukelpoisia muihin
esikäsittelyihin, koska näissä jouduttiin poikkeamaan mittauskäytännöistä. q2-summa
täyttyi mallilla jo ensimmäisellä komponentilla, mutta malli tunnisti vasta kahdella
komponentilla näytteiden eri pitoisuudet. Ohjelmisto arvioi esikäsittelemättömässä datassa
näytevahvuudet omiksi ryhmiksensä. 10 % näytteet (n=3) sijoittuivat 10,7–10,9 % arvoille,
15 % näytteet (n=11) taas 11,7–17,5 % arvoille ja 20 % näytteet (n=2) välille 20–20,4 %.
SNV-dydx2 -esikäsittelyillä vastaavat arvot olivat 12,8–13,3 (10 % näytteet), 13,8–15,5 (15
% näytteet) ja 16,9–17 (20 % näytteet).
Käytettäessä laajempaa paikallaan olevien näytteiden mallia pohjana paras sovitus saatiin
MSC-esikäsittelyllä. Se antaa RMSEP-arvoksi 2,83 kahdella komponentilla ja arvioi
näytteiden paikat selvästi omiksi ryhmikseen. 10 % näytteet (n=3) sijoittuivat 4–5 %
arvoille, 15 % näytteet (n=11) taas 12–17,5 % arvoille ja 20 % näytteet (n=2) välille 20–

82
20,4 %. Vaikka MSC-esikäsittelyn avulla ryhmät saatiin eriteltyä omiksi ryhmikseen, ei
mallilla saavutettu kaikille pitoisuuksille hyvää tarkkuutta.
7.3.3 Parasetamoli-laktoosiseokset
Pelkästään liikkuvia näytteitä arvioitaessa paras RMSEP-tulos saatiin esikäsittely-
yhdistelmällä MSC–dydx2 (2,55) ja SNV–dydx2 (2,35) yhdellä vaikuttavalla komponentilla
(taulukko 14). SNV-dydx2 -esikäsittelyllä ohjelma arvioi 10 % näytteiden (n=3) sijoittuvan
13,35–13,5 % väliin, 15 % näytteet (n=11) 14,1–15,2 % väliin ja 20 % näytteet (n=2) 15,4–
15,8 %. MSC–dydx2 -käsittelyllä ohjelma ei pystynyt arvioimaan näytteitä omiksi
ryhmikseen. 10 % näytteet sijoittuivat 13,8–14 % välille, 15 % näytteet 14–15 % välille ja
20 % näytteet sijoittuivat 15–15,4 % väliin.
Paikallaan mitattujen näytteiden käyttäminen mallina saa liikkuvien näytteiden arvioinnin
leviämään. SNV–dydx2 -käsittelyllä 10 % näytteet ennustetaan 5,8–6,6 % pitoisuuksille,
15 % näytteet 19,3–21,7 % välille ja 20 % näytteet pitoisuuksille 31,1–31,2 %

83
Taulukko 11. ASA- ja DCP-seokset liikkuvalla alustalla 1 sekunnin mittausajoilla ja 10 sekunnin väleillä, ASA-pitoisuudet näytteissä 10 %, 15 %, 20 %. dydx: ensimmäisen asteen derivaatta esikäsittely, dydx2: toisen asteen derivaatta esikäsittely, SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely.
ASA, DCP 1s, 10s välein 1RPM, 2x15_10_2x15_20
Malli Ennuste Ennuste
(q2≥0,93)
Pelkät liikkuvat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustuspisteet q2-
summa
ilman käsittelyä 1,48 1,48 1,49 424,550,751,989,1046,1192,1544,1607 1 10p,15p,20p 15,2;15,5;20,6;15,14,10,17;15,18 0,991
dydx 1,44 1,39 1,53 424,550,751,786,989,1046,1155,1192,1260,1544,1607 1 “ “ 0,991
MSC* 1,15 1,28 0,97 424,550,751,786,989,1046,1192,1544,1607 2* “ “ 0,991
SNV* 1,15 1,28 0,98 424,550,751,989,1046,1192,1544,1607 2* “ “ 0,991
Savitzky-Golay 1,48 1,48 1,49 424,550,751,989,1046,1192,1544,1607 1 “ “ 0,991
MSC-dydx2 1,55 1,61 1,49 751,786,1046,1192,1544,1607 1 “ “ 0,99
Liikkuvat ja 10, 15, 20 %
ilman käsittelyä 3,05 3,38 2,61 424,550,751,989,1046,1192,1544,1607 1 – 15,2;15,5;20,6;15,14,10,17;15,18 0,947
dydx 1,00 1,07 2,75 424,550,751,786,989,1046,1155,1192,1260,1544,1607 4 – “ 0,982
MSC* 1,08 1,13 0,98 424,550,751,786,989,1046,1192,1544,1607 2* – “ 0,994
SNV* 1,10 1,16 0,99 424,550,751,989,1046,1192,1544,1607 2* – “ 0,994
Savitzky-Golay 3,08 3,42 2,62 424,550,751,989,1046,1192,1544,1607 3 – “ 0,946
MSC-dydx2 1,58 1,63 1,47 751,786,1046,1192,1544,1607 1 – “ 0,99
Liikkuvat + 5–40 %
MSC 3,01 5,04 3,10 424,550,751,786,989,1046,1192,1544,1607 2 0,100 koko liikkuva näytejoukko 0,96
MSC-dydx2 2,77 4,84 3,28 751,786,1046,1192,1544,1607 2 “ “ 0,949
SNV 3,17 5,44 2,96 424,550,751,786,989,1046,1192,1544,1607 2 “ “ 0,949
SNV-dydx2 2,94 5,19 3,19 751,786,1046,1192,1544,1607 2 “ “ 0,944
* Käsittelyssä enemmän komponentteja kuin q2≥0,93 määritys ohjaisi

84
Taulukko 12. ASA- ja DCP-seokset liikkuvalla alustalla 1 sekunnin mittausajoilla ja 10 sekunnin väleillä, ASA-pitoisuudet näytteissä 12,5 %, 15 %, 17,5 %. SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely, dydx2: toisen asteen derivaatta esikäsittely.
ASA, DCP 1s, 10s välein 1RPM,2x15_12,5_2x15_17,5
Malli Ennuste Ennuste
(q2≥0,93)
Pelkät liikkuvat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustuspisteet q2-
summa
ilman käsittelyä 1,55 1,69 1,37 423,550,750,784,985,1045,1191,1292,1606 1 12,5p,15p,17,5p 15b,15e,17,5f,15j,15n,12,5o 0,987
dydx2 1,87 1,81 1,96 750,784,1045,1191,1292,1606 1 “ ” 0,986
MSC* 1,15 1,18 1,20 423,550,750,784,985,1045,1191,1292,1606 2 “ ” 0,991
SNV* 1,16 1,18 1,21 423,550,750,784,985,1045,1191,1292,1606 2 “ ” 0,991
MSC-dydx2 1,15 1,11 1,26 750,784,1045,1191,1292,1606 1 “ ” 0,994
Liikkuvat ja 12,5; 15; 17,5 %
ilman käsittelyä (1,49) 0,23 1,22 423,550,750,784,985,1045,1191,1292,1606 (4)6 - 15b,15e,17,5f,15j,15n,12,5o 0,961
dydx2 1,91 2,26 1,44 750,784,1045,1191,1292,1606 3 - ” 0,935
MSC* 1,08 1,02 1,29 423,550,750,784,985,1045,1191,1292,1606 2 - ” 0,987
SNV 1,09 1,02 1,31 423,550,750,784,985,1045,1191,1292,1606 2 - ” 0,995
MSC-dydx2 1,19 1,17 1,25 750,784,1045,1191,1292,1606 1 - ” 0,994
Liikkuvat + 5–40 %
MSC 2,71 4,40 2,81 423,550,750,784,985,1045,1191,1292,1606 2 0,100 koko liikkuva näytejoukko 0,97
MSC-dydx2 (4,04) 4,13 3,10 750,784,1045,1191,1292,1606 (1)2 ” ” 0,963
SNV 3,21 5,44 3,11 423,550,750,784,985,1045,1191,1292,1606 2 ” ” 0,949
SNV-dydx2 2,99 5,19 3,38 750,784,1045,1191,1292,1606 2 ” ” 0,944
dydx2 (1,16) 1,41 10,58 750,784,1045,1191,1292,1606 (4)1 ” ” 0,995
* Käsittelyssä enemmän komponentteja kuin q2≥0,93 määritys ohjaisi

85
Taulukko 13. Ibuprofeenin ja laktoosin seokset liikkuvalla alustalla 1 sekunnin mittausajoilla ja 10 sekunnin väleillä, Ibuprofeenin pitoisuudet 10 %, 15 % ja 20 %. SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely, dydx2: toisen asteen derivaatta esikäsittely.
IBU LAKTO 1s,10s välein, 1RPM, 2x15_10_2x15_20
Malli Ennuste Ennuste
(q2≥0,93)
Pelkät liikkuvat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustuspisteet q2-
summa
ilman käsittelyä 2,25 2,42 2,23 356,377,745,833,851,876,1086,1118,
1141,1180,1207,1263,1338,1608 (2)1 – 15b,15e,20f,15j,15n,10o 0,973
dydx2 3,10 3,47 2,69 356,377,476,636,745,782,819,833,851,876,915,
1086,1118,1141,1180,1207,1608 1 “ 0,946
MSC* 0,56 0,60 0,54 356,377,476,636,745,833,851,876,915,1086,1118,
1141,1180,1207,1263,1338,1608 2* “ 0,999
SNV* 0,55 0,61 0,51 “ 2* “ 0,998
SNV-dydx2 1,89 1,96 1,84 “ 1 “ 0,981
MSC-dydx2 2,52 2,61 2,45 “ 1 “ 0,968
Liikkuvat + 10, 15 ja 20 %
ilman käsittelyä 2,38 2,57 2,85 356,377,476,636,745 ,833,851,876,915,1086,1118,
1141,1180,1207,1263,1338,1608 3 – 15b,15e,20f,15j,15n,10o 0,962
dydx2 2,99 3,22 2,72 356,377,476,636,745,782,819,833,851,876,915,
1086,1118,1141,1180,1207,1608 3 “ 0,931
MSC* 0,74 0,86 0,50 356,377,476,636,745,833,851,876,915,1086,1118,
1141,1180,1207,1263,1338,1608 2* “ 0,996
SNV* 0,73 0,85 0,45 “ 2* “ 0,997
SNV-dydx2 2,10 2,26 1,82 “ 1 “ 0,996
MSC-dydx2 2,76 2,95 2,43 “ 1 “
Liikkuvat + 5–95 %
Ilman käsittely’ 7,46 2,41 10,52 636,746,783,820,833,1008,1115,1207,1453,1608 2 0,55,100 kaikki liikkeessä olevat 0,998
MSC* 5,35 7,77 2,83 387,494,532,567,583,755,863,931,958,1357 2 “ 0,98
MSC-dydx2 (5,21) 11,77 11,84 637,746,783,819,833,1007,1115,1181,12071608 (2)1 “ 0,963
SNV 7,51 10,94 5,48 “ 2 “ 0,964
SNV-dydx2 (7,38) 14,49 10,40 “ (2)1 “ 0,943
dydx2 7,90 2,47 10,35 636,746,783,820,833,1008,1115,1181,1207,1453,1608 1 “ 0,998
* Käsittelyssä enemmän komponentteja kuin q2≥0,93 määritys ohjaisi

86
Taulukko 14. Parasetamolin ja laktoosin seokset liikkuvalla alustalla 1 sekunnin mittausajoilla ja 10 sekunnin väleillä, parasetamolin pitoisuudet 10 %, 15 % ja 20 %. SNV: standardi normaali variaatti esikäsittely, MSC: multiplikatiivinen sirontakorjaus esikäsittely, dydx2: toisen asteen derivaatta esikäsittely, SG: Savitzky-Golay esikäsittely
ParaLakto 1s_10s välein 1 RPM, 2x15_10_2x15_20
Malli Ennuste Ennuste
(q2≥0,93)
Pelkät liikkuvat RMSEE RMSEE RMSEP Vaikuttavat aaltoluvut Komp. määrä
mallissa
Mallista poistetut pisteet
Ennustuspisteet q2-
summa
ilman käsittelyä 2,26 2,18 2,41 107,228,403,548,608,837,919,987,1075,1361 2 – 15v,15e,20f,15j,15n,10o 0,992
dydx2 2,15 1,98 2,45 107,124,402,546,610,624,665,769,837,919,987,1076,1362 2 “ “ 0,97
MSC* 0,34 0,38 0,31 107,124,142,225,403,548,609,836,914,982,1072,1362 2 “ “ 0,97
MSC–dydx2 2,57 2,64 2,55 107,124,402,546,610,624,665,769,837,919,987,1076,1362 1 “ “ 0,97
SNV-dydx2 2,28 2,35 2,35 “ 1 “ “ 0,984
Liikkuvat + 10,15 ja 20 %
ilman käsittelyä 1,10 1,09 0,98 107,228,403,548,608,837,919,987,1075,1361 3 – 15v,15e,20f,15j,15n,10o 0,964
dydx2 2,22 2,25 4,18 107,124,402,546,610,624,665,769,837,919,987,1076,1362 3 “ “ 0,951
MSC* 0,54 0,62 0,36 107,124,142,225,403,548,609,836,914,982,1072,1362 2 “ “ 0,998
MSC–dydx2 2,77 2,94 2,50 107,124,402,546,610,624,665,769,837,919,987,1076,1362 1 “ “ 0,963
SNV-dydx2 2,47 2,62 2,21 “ 1 “ “ 0,991
Liikkuvat + 5–95 %
Ilman käsittelyä 9,24 8,61 9,04 107,228,403,548,608,837,919,987,1075,1361 2 0,100 Kaikki liikkeessä olevat
näytteet 0,961
MSC 9,19 12,59 6,70 141,402,548,608,919,988,1029,1075,1121,1361,1399 2 “ “ 0,924
MSC-dydx2 9,13 12,62 6,08 “ 2 “ “ 0,938
SNV 9,24 12,87 5,71 “ 2 “ “ 0,952
SNV-dydx2 9,22 12,90 5,23 “ 2 “ “ 0,95
dydx2 9,46 9,99 9,04 “ 1 “ “ 0,957
* Käsittelyssä enemmän komponentteja kuin q2≥0,93 määritys ohjaisi

87
7.4 Spektrimuutokset siirryttäessä puhdasaineesta
seokseen
Suurimmat erot yksittäisten spektrien välillä syntyvät, kun siirrytään puhdasaineesta
seoksiin. Erityisesti spektrin pohjataso saattaa laskea tai nousta huomattavasti tässä
siirtymässä (kuva 32). Ibuprofeenin ja laktoosin tapauksessa seoksissa korostuu
laktoosin aiheuttama pohjatason nouseminen. Siirryttäessä puhtaasta laktoosista
seokseen, jossa on 95 % laktoosia ja 5 % ibuprofeenia, laskee pohjan intensiteetti
1000–1500 yksikköä käsittelemättömällä datalla. Puhtaan ibuprofeenin pohjataso on
matalalla ja siirryttäessä seokseen, jossa on 95 % ibuprofeenia ja 5 % laktoosia, se
ei nouse yhtä paljon, kuin se laski siirryttäessä puhtaasta laktoosista seokseen. 95 %
ibuprofeeniseoksen piikit ovat kuitenkin vastaavia puhdasaineen piikkejä
korkeammat (kuva 32).
Esikäsittelyillä, kuten MSC (kuva 33 ja 35), saadaan pohjatasoja yhtenäistettyä, ja
erot on helpompi havaita. Silmämääräisesti spektrejä tutkittaessa siirtymä puhtaasta
laktoosista seokseen on helpompi havaita kuin siirtyminen puhtaasta ibuprofeenista
seokseen. Tämä johtuu laktoosin pienien pitoisuuksien vähäisestä piikkien
muodostuksesta. Selkeimmin ibuprofeenin osuuden aiheuttamat muutokset piikkeihin
ovat havaittavissa aallonpituuksilla 1185 ja 1210 MSC-käsitellyssä datassa (kuva
33). Muissa kohdissa spektriä puhdasaineet ja niitä lähinnä olevien seosten spektrit
muistuttavat liian paljon toisiaan eron havaitsemiseksi pelkällä spektrin visuaalisella
tulkinnalla. MSC-käsittely saa ibuprofeenin puhdasainespektrin piikit 1185 ja 1210
aaltoluvuilla seoksia korkeammiksi. Laktoosin aiheuttama fluoresenssi haittaa myös
pienten laktoosimäärien havaitsemista. Pitoisuushavainnointeja ei voi kuitenkaan
tehdä luotettavasti yksittäisen piikin perusteella. Tietokonepohjaisesti tutkittaessa,
kaikki pitoisuudet huomioon otettaessa ja vertailtaessa koko spektrin alaa
käsittelemätön data antoikin selkeästi paremman ennustettavuuden ibuprofeenin ja
laktoosin seoksille (taulukko 7). Ibuprofeenin ja mannitolin seoksissa, joissa
kumpikaan ei aiheuta puhdasaineina korkeaa pohjatasoa, piikit tulevat selvemmin
esiin spektristä (kuva 34 ja 35). Tälläkin seoksella mannitolin pienet määrät on
vaikeampi havaita kuin ibuprofeenin. Suurimman osalla aaltolukualueesta 100 % ja
90 % ibuprofeenin spektrit kulkivat päällekkäin ja seokset aiheuttavat suuremmat
piikit kuin puhdasaineet (kuva 34). Tietokonepohjaisissa pitoisuusmäärityksissä

88
tehdyissä tutkimuksissa MSC-käsittely antoi vähemmillä komponenteilla saman
ennustettavuuden kuin käsittelemätön data (taulukko 8).
Kuva 32. Ibuprofeenin ja laktoosin puhdasaineiden sekä 5 %, 50 % ja 95 % ibuprofeenia sisältävien seoksien käsittelemättömät spektrit yhden sekunnin kuvausajalla. Spektristä on otettu tarkasteluun 850–1250 aaltolukualueella cm-1.
Kuva 33. Ibuprofeenin ja laktoosin seokset 0 %, 5 %, 50 %, 95 % ja 100 % ibuprofeenin osuudella. Spektrit MSC-käsitelty aaltolukualueella 860–1240.
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
13000
14000
15000
850 900 950 1000 1050 1100 1150 1200 1250
Inte
nsiteett
i
Aaltoluku (cm-1)
550951000

89
Kuva 34. Ibuprofeenin ja mannitolin puhdasaineiden ja seoksien spektrit 10 %, 50 % ja 90 % ibuprofeenin osuudella. Spektrit aaltolukualueella 800–1050.
Kuva 12. Ibuprofeenin ja mannitolin puhdasaineiden ja 10 %, 50 % ja 90 % ibuprofeenia sisältävät MSC-käsitellyt spektrit.
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
800 850 900 950 1000 1050
Inte
nsiteett
i
Aaltoluku (cm-1)
10
50
90
100
0

90
7.5 Raman-spektrikirjasto
Spektrikirjastoon kerättiin 41 eri ainetta (liitetaulukko 1). Mittaukset tehtiin pääasiassa
yhden ja kuuden sekunnin mittausajoilla. Mitatuista aineista 21:llä oli matala
pohjataso, ja 20 aineista fluoresoi, joista 12 voimakkaasti. Spektreistä 23 oli
silmämääräisesti helposti tunnistettavia ja ne saivat arvon hyvä, kymmenen oli
tyydyttävästi tunnistettavissa ja kahdeksan huonosti. Eniten referenssispektrejä löytyi
parasetamolille (6), asetyylisalisyylihapolle (4), salisyylihapolle (4) ja propranololille
(4). Yhteensä 26 aineelle löytyi jonkinlainen referenssispektri aikaisemmin
julkaistuista tutkimuksista. Suurin osa tässä työssä tutkituista spektreistä oli
muodoltaan samanlaisia tai melkein samanlaisia kuin muissa tutkimuksissa
esiintyneet spektrit. Joissain tapauksissa referenssispektrien tulkintoja hankaloittivat
artikkelien erilaiset spektrien asettelut. Artikkelien säteilylähteiden tehot vaihtelivat
0,6 ja 1000 mW:n välillä ja käytetyt aallonpituudet 514 ja 1064 nm:n välillä.
Artikkelien Raman-järjestelmät perustuivat myös eri Raman-tekniikoihin, mikä saattoi
vaikuttaa spektrien muotoon ja tulkittavuuteen.
Artikkeleissa esiintyneet parasetamolin spektrit olivat hyvin lähellä tutkimuksen
mittaustuloksia. Piikkien intensiteetti vaihteli jonkin verran, mutta ne olivat hyvin
tunnistettavissa. Samoin asetyylisalisyylihapon spektrit olivat hyvin tai melko hyvin
tunnistettavissa samanlaisiksi tämän tutkimuksen mittauksen kanssa. Nikotiiniamidin
kohdalla löytyi sekä tämän tutkimuksen spektriä tukeva artikkeli, että sen spektristä
osittain eroavia artikkeleita (Kumar ym. 2010, Ramanlingman ym. 2010, Rajesh ym.
2012). Kaikki sulfaniiliamidit olivat joko hyvin hankalia tai mahdottomia tunnistaa
referenssiartikkelin perusteella (Sutherland ym. 1990). Spektrien tulkintaa
hankaloittivat artikkelin erilaiset spektrin mittakaava-asettelut ja omassa
tutkimuksessa esiintynyt voimakas sulfametasiinin fluoresointi. Mikrokiteisen
selluloosan tulkintaa haittasi voimakas fluoresenssi ja erilainen asettelu kuin
artikkeleissa (Widjaja ja Seah 2008, Maltas ym. 2013). Propranololin osalta
hankaloittava tekijä oli sen eri isomeerit. Artikkeleissa oli käytetty
peilikuvaisomeereistä potentimpaa propranolin S-muotoa, jonka spektri eroaa
raseemisen seoksen spektristä piikkien intensiteetin osalta sekä ylimääräisellä piikillä
aaltoluvulla 200 (kuva 36). Kuvasta on hyvä huomata kuitenkin, että isomeerit ja
raseeminen seos on mitattu eri Raman-tekniikoilla. Verrattaessa tutkimuksen spektriä

91
raseemiseen referenssispektriin oli tunnistettavuus parempi. Tutkimuksessa oli
mukana kahden eri valmistajan parasetamolia. Mittaukset antoivat niille hyvin
samanlaiset spektrit, mutta pohjataso mittauksissa oli jostain syystä erilainen (kuva
26).
Kuva 13. Propranololin isomeerien ja raseemisen seoksen Raman-spektrit (Bodoki ym. 2012)
Kuva 14. Kahden eri valmistajan syntetisoiman parasetamolin Raman-spektri.
0
5000
10000
15000
20000
25000
30000
35000
250 350 450 550 650 750 850 950 1050 1150 1250 1350 1450 1550 1650 1750 1850
Inte
nsiteett
i
Aaltoluku (cm-1)
Parasetamoli
Acetaminophen

92
8 POHDINTA
Paikallaan tehtyjen mittausten tulokset antoivat viitteitä derivointipohjaisten
esikäsittelyjen paremmasta soveltuvuudesta pitoisuuden määrittämiseen. Ne ovat
kuitenkin herkkiä taustan vaihtelulle, jonka takia tulos voi olla täysin vääränlainen.
SNV- ja MSC-esikäsittelyt sietävät paremmin taustan muutoksia, mutta antavat
huonompia ennustustuloksia mittauksilla, joissa spektreissä ei esiinny normaalien
muutoksien lisäksi poikkeamia. Paikallaan tehdyissä mittauksissa esikäsittely-
yhdistelmien käyttäminen ei osoittautunut kannattavaksi yhdelläkään seoksella, vaan
ennustustulokset pysyivät joko lähes samoina tai huonontuivat yksittäisiin
esikäsittelyihin verrattuna. Käsittelemättömässä datassa osa havaintopisteistä ei
asettunut datajoukossa oletetulle kohdalle. Mahdollisia syitä tähän saattavat olla
näytteen huono sekoitus, mittausvirhe tai mittaamista varten tehty näytteen tiheyteen
vaikuttava pinnan tasoittaminen, joka vaikuttaa Raman-spektrin muodostumiseen.
Tätä vaihtelua pystyttäisiin vähentämään standardoimalla näytteen valmistus ja
mittaus. Ongelmaa voisi vähentää myös useampien mittausten ottaminen näytteestä,
jolloin tulos saattaisi tarkentua. Paikallaan mitatuista seossarjoista tarkimmat tulokset
saavutettiin ASA–DCP -sarjalla, joka koostui 0–40 % asetyylisalisyylihappoa
sisältävistä seoksista. Tälle seossarjalle muodostetulla ennustusmallilla onnistuttiin
saavuttamaan mittaustarkkuus, jossa ennustettujen tulosten ero näytteiden
teoreettisiin pitoisuuksiin oli alle kaksi prosenttia. Kaikki seosyhdistelmät eivät
saavuttaneet kuitenkaan näin hyviä tuloksia. Paikallaankin tehdyissä mittauksissa
sekä mittausjärjestelmä että onnistuneiden ennustusmallien luominen vaativat vielä
lisää tutkimusta, jotta pystytään luomaan nopeita, tarkkoja, luotettavia ja toistettavia
malleja erilaisille lääkeaineseoksille erilaisissa tuotantoympäristöissä.
Tutkimuksen aikana mallien luotettavuuden arviointia hankaloittivat näytejoukkojen
pienuudet sekä ennustusmallin komponenttien määrän rajaavan q2-summan
arvottaminen siten, ettei se yli- tai alikäsittelisi dataa. Myös virheellisten mittausten
tunnistaminen normaalista spektrin vaihtelusta tarvitsee lisää arviointia.
Ennustemallien luotettavuutta pystyttäisiin parantamaan suurentamalla niiden
tekemiseen ja arviointiin käytettäviä näytejoukkoja. Tämä voisi auttaa myös
tunnistamaan helpommin virheelliset mittaukset. Sen lisäksi kullekin esikäsittelylle

93
soveltuvan q2-raja-arvon määrittelyperusteen luominen voisi lisätä ennustusmallin
tarkkuutta säilyttäen kuitenkin mallin toistettavuuden.
Liikkeessä tehtyjen mittausten tuloksia voidaan tutkia kahdelta suunnalta, joko
saavutetun pitoisuustarkkuuden perusteella tai järjestelmän kyvyllä erottaa eri
pitoisuusryhmät toisistaan. Tässä tutkimuksessa esikäsittely-yhdistelmät soveltuivat
paremmin liikkeessä tehtyjen mittausten esikäsittelyyn kuin aikaisempiin paikallaan
tehtyjen. Niillä saavutettiin usein parempi tai yhtä hyvä ennustettavuus vähemmillä
mallin komponenteilla kuin yksittäisillä esikäsittelyillä. Sirontaa korjaavat esikäsittelyt
sopivat paremmin näytteiden ennustamiseen, ja niiden avulla pystyttiin yhdistämään
paikallaan mitattujen näytteiden ennustusmalleja liikkeessä mitattujen näytteiden
tunnistamiseen. Paikallaan mitattujen näytteiden ennustusmallien käyttö paransi eri
pitoisuusryhmien erottelua toisistaan, mutta huononsi mallien kykyä tunnistaa
näytteiden pitoisuudet. Tämä saattoi johtua paikallisten mallien suurempi-
intensiteettisistä piikeistä, jotka painottivat siten ennustusta eri tavalla kuin liikkeessä
mitattujen ennustusmallit. Seokset, joissa laktoosi oli toisena aineena, antoivat
huonompia RMSEP-arvoja ennustuksissa, kun ne yhdistettiin paikallaan tehtyjen
mittausten kanssa. Tämä voi johtua liikkeen aiheuttamasta piikkien intensiteetin
laskusta sekä laktoosin aiheuttaman fluorisenssin yhdistelmästä, molemmat näistä
vaikuttavat spektrin muotoon merkittävästi. Liikkeessä tehtyjen mittausten
näytejoukot osoittautuivat liian pieniksi ja painottuivat mittausryhmien sisällä liikaa,
mikä esti tarkempien arviointien ja johtopäätösten tekemisen.
Mittauksissa haluttiin jäljitellä jatkuvatoimisen linjaston toimintaympäristöä.
Puutteelliset koejärjestelyt mittauksien paikannuksessa sekä näytteiden asettelussa
kuitenkin aiheuttivat, ettei kaikkea tutkimusdataa voitu käyttää Raman-spektrometrin
soveltuvuuden arviointiin reaaliaikaisessa analytiikassa. Järjestelmä vaatisikin uusia
tutkimuksia, joissa keskitytään tarkemmin jäljittelemään oikean jatkuvatoimisen
prosessin toimintaympäristöä.
Spektrikirjasto näyttää olevan pääosin vertailukelpoinen muihin tutkimuksiin nähden,
joten se antaa hyvää pohjaa myös spektreille, joille ei vertailukohtaa löytynyt.
Spektrikirjasto toimii hyvänä pohjana aineiden tunnistamiseen pienessä
mittakaavassa sekä valmistuneen jatkuvatoimisen laitteiston tutkimuksiin.
Spektrikirjaston tarkempi arviointi kaipaisi spektrien normalisointia, jotta niiden

94
tarkempaa rakennetta voitaisiin vertailla. Tarkempaan arviointia varten spektrien
käsittelyn pitäisi olla järjestelmällistä ja niiden tunnistaminen tulisi suorittaa
tietokonepohjaisesti, kuten nykyaikaiset kannettavat Raman-spektrometrit tekevät.
Kemiallinen pitoisuuden määrittäminen ei onnistunut käytetyllä menetelmällä, vaan
24 tuntia lämmitettyjen seosten pitoisuudet olivat yli laskennallisten arvojen.
Määritykset tulisi vielä toistaa, koska laimennosten tekeminen lisää helposti
epätarkkuutta tuloksissa. Eri sekoitusmenetelmillä saadut spektrofotometri spektrit
kuitenkin antavat osviittaa, että pienillä ASA-pitoisuuksilla näytteet hajoavat suurelta
osin salisyylihapoksi jo sekoituksessa, ja ASA:n aiheuttama absorbanssipiikki (276
nm) jää salisyylihapon piikin (300 nm) alle. Neljän ja kahdeksan tunnin liuotusajat
eivät kuitenkaan näyttäneet riittävän saavuttamaan täysin liuennutta tilaa liuoksissa.
Asetyylisalisyylihapon ongelmana on huono vesiliukoisuus sekä lämmön että
kosteuden nopeuttama hajoaminen salisyylihapoksi, mikä tekee koejärjestelyistä
monimutkaisempia (Lin-Li ym. 2008). Tulokset osoittavat, että kemiallista
vertailuanalyysiä varten pitää kehittää uusi paremmin soveltuva koejärjestely.
Pitoisuusmääritykset voisi suorittaa esimerkiksi vesi-etanoliliuoksella, johon ASA
liukenee paremmin, tai jollakin eri seosyhdistelmällä (Iwunze 2008). Näiden lisäksi
voisi tutkia soveltuvimpia sekoitusolosuhteita ja sekoituksen kestoa, erityisesti
pienempipitoisuuksisille ASA-seoksille.
Raman-spektrometrilaitteistojen on todettu kykenevän hyvinkin tarkkoihin
lääkeainepitoisuusmäärityksiin (Eliasson ym. 2008, Mazurek ja Szostak 2008,
Townshend ym. 2012), ja paikoitellen tässäkin tutkimuksessa laitteisto osoitti hyvää
tarkkuutta ottaen huomioon vähäisen tuntemuksen PLS-mallien muodostuksesta ja
Raman-kuvantamisesta – tutkimuksessa ilmenneistä suurista puutteista huolimatta.
Koejärjestelyiden suurimmat puutteet Raman-spektrometrian jatkuvatoimiseen
analytiikkaan soveltuvuuden arvioinnissa olivat pienet ja epätasapainossa olevat
näytemäärät liikkuvissa mittauksissa sekä nopeammin liikkuvien näytteiden puute.
Raman-spektroskopia on periaatteeltaan erittäin hyvin jatkuvatoimiseen analytiikkaan
soveltuva järjestelmä nopeutensa ja spesifisyytensä takia. Suoraan tällaista
järjestelmää ja näitä ennustusmalleja ei voisi tehtyjen testien perusteella harkita
jatkuvatoimiseen analytiikkaan. Kokeet antoivat kuitenkin viitteitä Raman-
spektrometrian potentiaalista, ja tarkemmalla perehtymisellä mallien muodostukseen

95
sekä paremmilla koejärjestelyillä voitaisiin ennustustehoa saada huomattavasti
paremmaksi.

96
9 KIRJALLISUUS:
Aaltonen J, Strachan C.J, Pöllänen K, Yliruusi J, Rantanen J: Hyphenated
spectroscopy as a polymorph screening tool. Journal of Pharmaceutical and
Biomedical Analysis 44: 477–483, 2007
Abdi H. Partial Least Square (PLS) Regression. Kirjassa: Encyclopedia of Social
Sciences Research Methods. Toimittajat: Lewis-Beck M., Bryman A., Futing T.
Thousand Oaks (CA): Sage. 2003
Agalloco J., ja Akers J.: Sterile Product Manufacturing, kirjassa: Pharmaceutical
Manufacturing Handbook: Production and Processes. sivut:102-120, Toimittaja: Cox
G.S, Wiley, NJ USA: 2008.
Agalloco J.: Steam sterilization-in-place technology and validation. Sivut: 201,
Kirjassa: Validation of Pharmaceutical Processes, kolmas versio. Toimittajat:
Agalloco J ja Carleton J.F. Informa Healtcare: NY USA: 2007
Agalloco J.: Thinking Inside the Box: The Application of Isolation Technology for
Aseptic Processin, Pharmaceutical Technology 8-11, 2006
Al-Achi A, Gupta M.R, Stagner W.C: Integrated Pharmaceutics: Applied
Preformulation, Product Design, and Regulatory Science sivut: 561-577. Wiley,
Somerset, NJ, USA: 2013.
Al-Ghananeem A. ja Crooks P.A: Phase I and Phase II Ocular Metabolic Activities
and the Role of Metabolism in Ophthalmic Prodrug and Codrug Design and Delivery.
Molecules 12, 373–388 2007
Ali Y. ja Lehmussaari K: Industrial perspective in ocular drug delivery. Advanced
Drug Delivery Reviews 58: 1258–1268, 2006
Alvarez-Ros M.C, Sánchez-Cortés S. García-Ramos J.V, Vibrational study of the
salicylate interaction with metallic ions and surfaces. Spectrochimica Acta Part A 56:
2471–2477, 2000
Amo E.M. ja Urtti A: Current and future ophthalmic drug delivery systems A shift to
the posterior segment. Drug Discovery Today, 13(3) 135–143, 2008

97
Boczar M, Wójcik M.J, Szczeponek K, Jamróz D, Zieba A, Kawalek B, Theoretical
modeling of infrared spectra of aspirin and its deuterated derivative. Chemical
Physics 286: 63–79, 2003
Bodoki E, Oltean M, Bodoki A, Stiufiuc R, Chiral recognition and quantification of
propranolol enantiomers by surface enhanced Raman scattering through
supramolecular interaction with b-cyclodextrin. Talanta 101: 53–58, 2012
Bompart M, Gheber L.A, De Wilde Y, Haupt K. Direct detection of analyte binding to
single molecularly imprinted polymer particles by confocal Raman spectroscopy.
Biosensors and Bioelectronics 25: 568–571, 2009
Braun D.E, Maas S.G, Zencirci N, Langes C, Urbanetz N.A, Griesser U.J,
Simultaneous quantitative analysis of ternary mixtures ofd-mannitol polymorphs by
FT-Raman spectroscopy and multivariate calibration models. International Journal of
Pharmaceutics 385: 29–36, 2010
Bregni C, Chiappetta D, Faiden N, Carlucci A, García R, Pasquali R: Release study
of diclofenac from new carbomer gels. Pakistan journal of pharmaceutical sciences
21(1), 2–16, 2008
Brizuela A.B, Bichara L.C, Romano E, Yurquina A, Locatelli S, Brandán S.A, A
complete characterization of the vibrational spectra of sucrose. Carbohydrate
Research 361: 212–218, 2012
Bugay D.E. ja Brush R.C. Chemical identity testing by remote-based dispersive
raman spectroscopy. Journal of applied spectroscopy 64(5): 468-475, 2010
Burstein N.L.: Preservative Alteration of Corneal Permeability in Humans and
Rabbits. Investigative ophthalmology & visual science, 25(12) 1453-1457, 1984
Campbell S.N, Williams A.C, Grimsey I.M, Booth S.W. Quantitative analysis of
mannitol polymorphs. FT-Raman spectroscopy. Journal of Pharmaceutical and
Biomedical Analysis 28: 1135–1147, 2002

98
Castro J.L, Lopez-Ramirez M.R, Arenas J.F, Soto J, Otero J,C. Evidence of
Deprotonation of Aromatic Acids and Amides Adsorbed on Silver Colloids by
Surface-Enhanced Raman Scattering. Langmuir 28: 8926−8932, 2012
Chiarello K: Pharma industry drives innovation in barrier/isolation design.
Pharmaceutical Technology, 28(3) 44–54, 2004
Chrai S.S, Patton T.F, Mehta A, Robinson J.R: Lacrimal and instilled fluid dynamics
in rabbit eye. Journal of Pharmaceutical Sciences 62, 1112–1121, 1973
Danion A., Aesenault I. ja Vermette P.: Antibacterial Activity of Contact Lenses
Bearing Surface-Immobilized Layers of Intact Liposomes Loaded with Levofloxacin.
Journal of pharmaceutical sciences, 96(9), 2350- 2363, 2007
De Beer T.R.M, Alleso M, Goethals F, ym: Implementation of a Process Analytical
Technology System in a Freeze-Drying Process Using Raman Spectroscopy for In-
Line Process Monitoring. Analytical Chemistry, 79(21): 7992-8003, 2007
De Beer T.R.M, Baeyens W.R.G, Vander Hayden Y, Remon J.P, Vervaet C, Verpoort
F, Influence of particle size on the quantitative determination of salicylic acid in a
pharmaceutical ointment using FT-Raman spectroscopy. European journal of
pharmaceutical sciences 30: 229–235, 2007
De Beer T.R.M, Burggraeve A., Fonteyne M., Saerens L., Remon J.P., Vervaet C.:
Near infrared and Raman spectroscopy for the in-process monitoring of
pharmaceutical production processes, International Journal of Pharmaceutics,
417: 32–47, 2011
Drinkwater J.L. Restricted Access Barriers - RABS definitions and performance.
http://www.pharmaceutical-int.com/article/restricted-access-barriers-rabs-definitions-
and-performance.html, haettu internetistä: 8.12.2013
Eliasson C., Macleod N.A., Jayes L.C., Clarke F.C., Hammond S.V., Smith M.R.,
Matousek P.: Non-invasive quantitative assessment of the content of pharmaceutical
capsules using transmission Raman spectroscopy. Journal of Pharmaceutical and
Biomedical Analysis 47: 221–229, 2008

99
Eljarrat-Binstock E, Raiskup F., Frucht-Pery J. ja Domb A.J.: Transcorneal and
transscleral iontophoresis of dexamethasone phosphate using drug loaded hydrogel.
Journal of Controlled Release, 106, 386 – 390, 2005
EMEA: Decision trees for the selection of sterilisation methods (cpmp/qwp/054/98)
Lontoo 2000
Etälukio, Pitkä matematiikka Derivaatta. Haettu internetistä: 26.11.2013.
http://www.edu.fi/etalukioetusivu
Euroopan komissio: EU Guidelines to Good Manufacturing Practice Medicinal
Products for Human and Veterinary Use: Annex 1 Manufacture of Sterile Medicinal
Products, Brysseli 2008
Ferraro J.R, Nakamoto K, Brown C.W: Introductory Raman Spectroscopy sivut: 1-
16,18-27,95-97,105,106,112-115,267-274 , 2. painos. Academic Press, Burlington,
MA, USA, 2002
FDA Food and Drug Administration: Guidance for industry, sterile drug products
produced by aseptic processing – current good manufacturing practice.2004
Frucht-Pery, J. , Mechoulam, H. , Siganos, C. S. , Ever-Hadani, P. , Shapiro, M. , ja
Domb, A. Iontophoresis-gentamicin delivery into the rabbit cornea, using a hydrogel
delivery probe, Experimental Eye Research.78(3), 745–749, 2004.
Furrer P. ja Mayer J.M.: Ocular Tolerance of Absorption Enhancers in Ophthalmic
Preparations. American association of pharmaceutical scientists, 4(1)1-5, 2002
Furrer P., Delie F. ja Plazonnet B.Opthalmic Drug Delivery Kirjassa: Drugs and the
Pharmaceutical Sciences, Volume 184: Modified Release Drug Delivery Technology,
painos 2 (2 versio). sivut 68-75 Toim. Hadgraft J., Lane M.E., Rathbone M. J.,
Roberts M.S. CRC Press, New York 2008
Garbwal R., Sbady S.F., Ellis E.J., ym. Sustained Ocular Delivery of Ciprofloxacin
Using Nanospheres and Conventional Contact Lens Material. Investigative
Ophthalmology & Visual Science, 53(3), 1341-1352, 2012

100
Gemperline P.J, Principal Component Analysis Kirjassa: Practical guide to
chemometrics, sivut: 2-3, 77-86, 2. painos. Toim. Gemberline P.J, Taylor&Francis,
Boca Raton, 2006
Genta I, Conti B, Perugini P, Pavanetto F, Spadaro A,Puglisi G: Bioadhesive
Microspheres for Ophthalmic Administration of Acyclovir. Journal of Pharmacy and
Pharmacology, 49(8), 737-742, 1997
Gibson M: Opthalmic Dosage Forms. Kirjassa: Pharmaceutical Preformulation and
Formulation, 2. painos. sivut: 431-453, Toim. Gibson M, informa healthcare, New
York 2009
Gulsen D. ja Chauhan A. Dispersion of microemulsion drops in HEMA hydrogel: a
potential ophthalmic drug delivery vehicle. International Journal of Pharmaceutics,
292, 95–117, 2005
Gupta H, Jain S, Rashi M, Mishra P, Mishra AK: Sustained ocular drug delivery from
a temperature and pH triggered novel in situ gel system. Drug Delivery, 14: 507–515,
2007
Gurtu, J.N. Khera H.C: Physical Chemistry, Volume III, sivut: 53,54,58,59 1. painos.
Global Media, Meerut, IND, 2008
Haaser M, Windbergs M, Mcfoverin C.M, ym. Analysis of Matrix Dosage Forms
During Dissolution Testing Using Raman Microscopy, Journal of pharmaceutical
sciences, 100: 4452–4459, 2011
Halhal M., Renard G., Courtois Y., BenEzra D., Behar-Cohen F.: Iontophoresis: from
the lab to the bed side. Experimental Eye Research, 78, 751–757, 2004
Heckert R.A., Best M., Jordan L.T., Dulac G.,C., Eddinton D.L. ja Sterritt W.G.:
Efficacy of Vaporized Hydrogen Peroxide against Exotic Animal Viruses. Applied
andenvironmentalmicrobiology,63(10), 3916–3918, 1997
Holly F.J. ja Lemp M.A.: Tear physiology and dry eyes. Survey of ophthalmology,
22(2) 69-87, 1977

101
Hornof M.D. ja Bernkop-Schnürch A.: In VitroEvaluation of the Permeation
Enhancing Effect of Polycarbophil–Cysteine Conjugates on the Cornea of Rabbits
Journal of pharmaceutical sciences,91(12) 2588-2592, 2002
Hämäläinen K.M., Määttä E., Piirainen H., Sarkola M., Väisänen A., Ranta V.P, Urtti
A.: Roles of acid/base nature and molecular weight in drug release from matrices of
gelfoam and monoisopropyl ester of poly vinyl methyl ether–maleic anhydride.
Journal of Controlled Release, 56, 273–283, 1998
International Organization for Standardization Eurooppalainen standardisointi
komitea: Aseptic processing of health care products - Part 5: Sterilization in place
(ISO 13408-5:2006)( EN ISO 13408-5:2011), Brysseli 2011
Jabeen S, Dines T.J, Leharne S.A, Chowdhry B.Z, Raman and IR spectroscopic
studies of fenamates – Conformational differences in polymorphs of flufenamic acid,
mefenamic acid and tolfenamic acid. Spectrochimica Acta Part A: Molecular and
Biomolecular Spectroscopy 96: 972–985, 2012
Johansson j, Pettersson S, Taylor L.S, Infrared imaging of laser-induced heating
during Raman spectroscopy of pharmaceutical solids, Journal of Pharmaceutical and
Biomedical Analysis 30: 1223–1231, 2002
Järvinen K, Järvinen T, Urtti A: Ocular absorption following topical delivery.
Advanced Drug Delivery Reviews 16: 3–19, 1995
Järvinen T., Rautio J., Masson M. ja Loftsson T.: Design and Pharmaceutical
Applications of Prodrugs. Kirjassa: Drug discovery handbook. sivut: 738-739,
Toimittaja Gad S.C., Wiley-Interscience, NJ, USA, 2005
Kačer P., Švrček J., Syslová K., ym.: Vapor Phase Hydrogen Peroxide – Method for
Decontamination of Surfaces and Working Areas from Organic Pollutants, Kirjassa:
Organic Pollutants Ten Years After the Stockholm Convention - Environmental and
Analytical Update, sivut 404-406, Toimittaja Dr. Tomasz Puzyn, Intech, Rijeka
Croatia, 2012

102
Kachrimanis K, Braun D.E, Griesser U.J, Quantitative analysis of paracetamol
polymorphs in powder mixtures by FT-Raman spectroscopy and PLS regression.
Journal of Pharmaceutical and Biomedical Analysis 43: 407–412, 2007
Kaiser optical systemssin internetsivut Haettu internetistä 12.2.2013:
http://www.kosi.com/Raman_Spectroscopy/rtr-ramantutorial.php?ss=800
Kaiser optical systemssin internetsivut Haettu internetistä 21.3.2013:
http://www.kosi.com/Raman_Spectroscopy/phat-probehead.php
Kantorovich K, Tsarfati I, Gheber L.A, Haupt K, Bar I, Writing Droplets of Molecularly
Imprinted Polymers by Nano Fountain Pen and Detecting Their Molecular
Interactions by Surface-Enhanced Raman Scattering. Analytical Chemistry 81: 5686–
5690, 2009
Kantorovich K, Tsarfati I, Gheber L.A, Haupt K, Bar I. Reading microdots of a
molecularly imprinted polymer by surface-enhanced Raman spectroscopy.
Biosensors and Bioelectronics 26: 809–814, 2010
Kaur I.P. ja Kanwar M.: Ocular Preparations: The Formulation Approach. Drug
Development and Industrial Pharmacy, 28(5), 473–493 2002
Keith P. Mastering VHP Sterilization. Global BioPharmaceutical Resources, Inc.
Newsletter January 2012
Kim M., Chung H., Woo Y. ja Kemper M.: New reliable Raman collection system
using the wide area illumination (WAI) scheme combined with the synchronous
intensity correction standard for the analysis of pharmaceutical tablets. Analytica
Chimica Acta 579: 209-216, 2006
Kogerman K, Aaltonen J, Strachan C. J, ym: Establishing Quantitative In-Line
Analysis of Multiple Solid-State Transformations During Dehydration. Journal Of
Pharmaceutical Sciences 97, (11): 4983-4999, 2008
Kramer, R.: Chemometric Techniques for Quantitative Analysis. sivu. 190 CRC
Press, NY, USA: 1998

103
Kumar M Jaiswal S, Singh R, ym. Ab initiostudies of molecular structures, conformers
and vibrational spectra of heterocyclic organics: I. Nicotinamide and its N-oxide.
Spectrochimica Acta Part A75: 281–292 2010
Lang J.C.: Ocular drug delivery conventional ocular formulations. Advanced Drug
Delivery Reviews 16: 39–43, 1995
Larkin P.J, Santangelo M, Šašic S. Internal Multiple-Scattering Hole-Enhanced
Raman Spectroscopy: Improved Backscattering Fourier Transform Raman Sampling
in Pharmaceutical Tablets Utilizing Cylindrical–Conical Holes. Applied Spectroscopy
66: 892–902, 2012
Lauderback J., Fraser J., Gustin E., McDonnell G., ja Williams K.: Point-of-
Manufacture Sterilization: A look at the material compatibility of Eastar 6763
copolyester with vaporized hydrogen peroxide sterilization. Haettu internetistä:
http://www.devicelink.com/pmpn/archive/02/10/005.html
Lin-Li L., Xian-Cheng Z. ja Jian-Lin T.: Evaluation of the Stability of Aspirin in Solid
State by the Programmed Humidifying and Non-isothermal Experiments. Archives of
Pharmacal Research 31 (3), 381-389, 2008
Listgarten J. ja Emili A.: Statistical and Computational Methods for Comparative
Proteomic Profiling Using Liquid Chromatography-Tandem Mass Spectrometry.
Molecular & Cellular Proteomics 4 (4): 419-434, 2005
Liu Y, Liu J, Zhang X, Zhang R, Huang Y, Wu C: In Situ Gelling Gelrite/Alginate
Formulations as Vehicles for Ophthalmic Drug Delivery. American Association of
Pharmaceutical Scientists 11: 610–620, 2010
Lopez-Heredia M.A, Sariibrahimoglu K, Yang W, ym. Influence of the pore generator
on the evolution of the mechanical properties and the porosity and interconnectivity
of a calcium phosphate cement. Acta Biomaterialia 8: 404–414, 2012
López-Sánchez M, Ruedas-Rama M.J, Ruiz-Medina A, Molina-Díaz A, Ayora-
Cañada M.J. Pharmaceutical powders analysis using FT-Raman spectrometry:
Simultaneous determination of sulfathiazole and sulfanilamide. Talanta 74: 1603–
1607, 2008

104
Lysfjord J. ja Porter M.: Esitys: Barrier Isolation History and Trends 2008 Final Data.
Haettu internetistä 9.12.2013: http://getfinefocus.com/sites/default/files/Isolator
%20ISPE%20study%202008%20copy.pdf
Löfgren A, Brodin A, Sjönkvist-Saers E: Blow-Fill-Seal Pharmaceutical Packaging –
Towards Safe and Convenient Medical Container. Business briefing: pharmatech
may: 214–216, 2003
Maltas D.C, Kwok K, Wang P, Taylor L.S, Ben-Amotz D. Rapid classification of
pharmaceutical ingredients with Raman spectroscopy using compressive detection
strategy with PLS-DA multivariate filters. Journal of Pharmaceutical and Biomedical
Analysis 80: 63–68, 2013
Mazuel C. ja Friteyre M.-C: United States Patent Nro. 4 861 760: Ophthalmological
composition of the type which undergoes liquid-gel phase transition. 1989
Mazurek S. ja Szostak R.: Quantitative determination of diclofenac sodium in solid
dosage forms by FT-Raman spectroscopy. Journal of Pharmaceutical and
Biomedical Analysis 48: 814–821, 2008
Meko D.M: Applied Time Series Analysis kurssimateriaali: Notes for week 4. Haettu
internetistä 26.11.2013: http://www.ltrr.arizona.edu/~dmeko/geos585a.html
Merkli , Cyrus T, Robert G: Use of insoluble biodegradable polymers in ophthalmic
systems for the sustained release of drugs. 41(5), 271–283, 1995
Microsoft excel help. Haettu internetistä 6.11.2013: http://office.microsoft.com/fi-
fi/excel-help/trendin-tai-keskiarvoviivan-lisaaminen-kaavioon-HA102809798.aspx.
Mundapa A.S.: Ocular inserts Kirjassa: Update on Polymers for Ocular Drug
Delivery, sivut: Toimittaja: Mundada A.S., iSmithers, Shrewsbury, GBR: 2011
Naes T, Isaksson T, Fearn T, Davies T: A user-friendly guide to Multivariate
Calibration and Classification, sivut:107,108,110,111,114,115,124,125,1. painos. NIR
Publicationas Chischester UK 2002

105
Nayak S.N, Sogali B.S, Thakur R.S: Formulation and evaluation of ph triggered in
situ ophthalmic gel of moxifloxacin hydrochloride. International Journal of Pharmacy
and Pharmaceutical Sciences. 4: 452-459, 2012
Negvesky G.J., Butrus S.I., Abifarah H.A, Yung C.L ja Yalkowsky S.H.: Ocular
Gelfoam Disc-Applicator for Pupillary Dilation in Human. Journal of ocular
pharmacology and therapeutics, 16(4) 311-316, 2000
Noonan K.Y, Beshire M, Darnell J, Frederick K.A. Qualitative and Quantitative
Analysis of Illicit Drug Mixtures on Paper Currency Using Raman Microspectroscopy.
Applied Spectroscopy 59: 1493–1497, 2005
Noonan K.Y, Tonge L.A, Fenton O.S, Damiano D.B, Frederick K.A. Rapid
Classification of Simulated Street Drug Mixtures Using Raman Spectroscopy and
Principal Component Analysis. Applied Spectroscopy 63: 742–747, 2009
Olds W.J, Sundarajoo S, Shelby M, Cletus B, Fredericks P.M, Izake E.M.
Noninvasive, Quantitative Analysis of Drug Mixtures in Containers Using Spatially
Offset Raman Spectroscopy (SORS) and Multivariate Statistical Analysis. Applied
Spectroscopy 66: 530–537, 2012
Pal I. ja Kanwar M: Ocular Preparations: The Formulation Approach. Drug
Development and industrial Pharmacy 28(5), 473–493, 2002
Patane M.A., Schubert W., Sanford T., Gee R., Burgos M., Isom W.P. ja Ruiz-Perez
B. Evaluation of Ocular and General Safety Following Repeated Dosing of
Dexamethasone Phosphate Delivered by Transscleral Iontophoresis in Rabbits.
Journal of ocular pharmacology and therapeutics, 29(8), 760-770, 2013
Peng C.-C. ja Chauhan A. Extended cyclosporine delivery by silicone–hydrogel
contact lenses. Journal of Controlled Release, 154, 267–274, 2011
Rajesh Goud N, Suresh K, Sanphui P, Nangia A. Fast dissolving eutectic
compositions of curcumin. International Journal of Pharmaceutics 439: 63–72, 2012.
Raman C.V. ja Krishnan K.S.: A New type of secondary radiation. Nature 121: 501-
502, 1928

106
Reed C. Recent technical advancements in blow-fill-seal technology. Bussines
briefing: Pharmagenerics 2002
Reed C. PharmaManufacturing.com 2008 Haettu internetistä
http://www.pharmamanufacturing.com/articles/2009/093.html
Rinnan Å, Berg, F.V.D, Engelsen S.B: Review of the most common pre-processing
techniques for near-infrared spectra. Trends in Analytical Chemistry, 28(10): 1201-
1222, 2009
Robinson J.R. ja Mlynek G.M: Bioadhesive and phase-change polymers for ocular
drug delivery. Advanced Drug Delivery Reviews 16: 45–50, 1995
Rudolph W.W ja Irmer G, Raman and Infrared Spectroscopic Investigations on
Aqueous Alkali Metal Phosphate Solutions and Density Functional Theory
Calculations of Phosphate–Water Clusters. Applied Spectroscopy 61: 1312–1327,
2007
Rupenthal, I.A. ja Alany R.G Ocular Drug Delivery Kirjassa: Pharmaceutical
Manufacturing Handbook: Production and Processes. Sivut: 750 Toimittaja Cox.
S.C., Wiley,Hoboken, NJ, USA: 2008.
Saerens L, Dierickx L, Lenain B, Vervaet C, Remon J.P, De Beer T: Raman
spectroscopy for the in-line polymer–drug quantification and solid state
characterization during a pharmaceutical hot-melt extrusion process. European
Journal of Pharmaceutics and Biopharmaceutics 77: 158–163, 2011
Saettone M.F. Salminen L: Ocular inserts for topical delivery. Advanced Drug
Delivery Reviews. 16 95–106, 1995
Schell D: What You Should Know About Blow-Fill-Seal Technology. Pharmaceutical
online Haettu internetistä 17.12.2013: http://www.pharmaceuticalonline.com/doc/
what-you-should-know-about-blow-fill-seal-0001: 2010
Schwartz D.T: Raman Spectroscopy: Introductory Tutorial. haettu internetistä
23.11.2013: https://depts.washington.edu/ntuf/facility/docs/NTUF-Raman-Tutorial.pdf
2005

107
Sieg J.W. ja Robinson J.R.: Vehicle effects on ocular drug bioavailability I: Evalution
of fluorometholone. Journal of pharmaceutical sciences 64(6), 631-936, 1975
Simamora P., Nadkarni S.R., Lee Y.C. ja Yalkosky S.H.: Controlled delivery of
pilocarpine. 2. In-vivo evaluation of Gelfoam® device, International Journal of
Pharmaceutics, 170, 209–214, 1998
Singh ja Shah: Surface chemical aspects of ocular drug delivery systems. Kirjassa:
Ocular Therapeutics and Drug Delivery. Toimittaja Reddy I.K. sivut 40-41, Technomic
publication, Pensylvania, USA: 1996
Souza J.G., Dias K., Pereira A.T., Bernardi D.S. ja Lopez R.F.V: Topical delivery of
ocular therapeutics: carrier systems and physical methods. Journal of pharmacy and
pharmacology 1-24, 2013
Sutherland W.S, Laserna J.J, Angebranndt M.J, Winefordner J.D, Surface-Enhanced
Raman Analysis of Sulfa Drugs on Colloidal Silver Dispersion. Analytical Chemistry
62: 689–693, 1990
Townshend N., Nordon A., Littlejohn D., Myrick M., Andrews J. ja Dallin P.:
Comparison of the Determination of a Low-Concentration Active Ingredient in
Pharmaceutical Tablets by Backscatter and Transmission Raman Spectrometry.
Analytical Chemistry 82: 4671–4676, 2012
Vadnere M., Amoidon G., Lindenbaum S.,ja Haslam J.L. Thermodynamic studies on
the gel-sol transition of some pluronic polyols. International journal of Pharmaceutics,
22, 207-218, 1984
Van Santvilet L. ja Ludwig A: Determinants of eye drop size, Survey of
ophthalmology, 49(2), 198-213, 2004
Wang C, Vickers T.J, Mann C.K, Direct assay and shelf-life monitoring of aspirin
tablets using Raman spectroscopy. Journal of Pharmaceutical and Biomedical
Analysis 16: 87–94, 1997
Wang Y, Li YS, Zhang Z, An D, Surface-enhanced Raman scattering of some water
insoluble drugs in silver hydrosols. Spectrochimica Acta Part A 59: 589–594, 2003

108
Varghese H.T, Panicker C.Y, Philip D. Vibrational spectroscopic studies and ab initio
calculations of sulfanilamide. Spectrochimica Acta Part A 65: 155–158, 2006
Vergote G.J, De Beer T.R.M, Vervaet C. ym: In-line monitoring of a pharmaceutical
blending process using FT-Raman spectroscopy. European Journal of
Pharmaceutical Sciences 21: 479–485, 2004
Whyte W: Cleanroom Technology: Fundamentals of design, testing and operation
John Wiley & Sons Ltd, Baffins Lane 2001
Widjaja E ja Seah R.K.H. Application of Raman microscopy and band-target entropy
minimization to identify minor components in model pharmaceutical tablets. Journal
of Pharmaceutical and Biomedical Analysis 46: 274–281, 2008
Wikström H, Lewis I.R, Taylor Lynne: Comparison of Sampling Techniques for In-
Line Monitoring Using Raman Spectroscopy. Applied spectroscopy, 59(7): 934-941
2005
Wilson CG, Zhu YP, Kurmala P, Rao LS, Dhillon B. Ophthalmic drug delivery.
Kirjassa: Drug Delivery and Targeting for Pharmacists and Pharmaceutical Scientists
sivut: 298–318. 300,301: Toimittajat Hillery AM, Lloyd AW, Swarbrick J. CRC Press,
Boca Raton, FL, USA: 2001.
Windbergs M, Haaser M, Mcgoverin C.M, Gordon K.C, Kleinbudde P, Strachan C.J.
Investigating the Relationship between Drug Distribution in Solid Lipid Matrices and
Dissolution Behaviour Using Raman Spectroscopy and Mapping. Journal of
Pharmaceutical Sciences 99: 1464–1475, 2010
Wirges M, Funke A, Serno P, Knop K, Kleinebudde P: Development and in-line
validation of a Process Analytical Technology to facilitate the scale up of
coating processes. Journal of Pharmaceutical and Biomedical Analysis 78–79: 57–
64, 2013
Vueba M.L, Pina M.E, Batista de Carvalho L.A.E. Conformational Stability of
Ibuprofen: Assessed by DFT Calculations and Optical Vibrational Spectroscopy.
Journal of Pharmaceutical Sciences 97, 845–859, 2008

109
Zimmer A. ja Kreuter J.: Microspheres and nanoparticles used in ocular delivery
systems. Advanced Drug Delivery Reviews, 16, 61-73, 1995

110
LIITETAULUKOT Liitetaulukko 1. Raman-spektrikirjasto
Aine Erä Mittausajat Fluoresoi Spektri Valmistaja R* Tunnusomaiset piikit
Acetaminophen (Parasetamoli) 020M0203V 1,6 ei Hyvä Sigma-
Aldrich
X 390,651,797,857,1168,1237,1324,1370,1609,1647
Asetyylisalisyyliahappo 048K0015 1,6,12 ei Hyvä Sigma X 424,549,750,784,1044,1190,1292,1606
Bentsokaiini 116K0017 1,3,6 ei Hyvä Sigma X 862,1172,1280,1604,1681
Carbopoli 971P NF CC69LAJ260 1,6 osittain Tyydyttävä Noveon 499,837,1080,1320,1452,1663 (pyöreät piikit)
Carbopoli 974P NF CC76NAB668 1,6 osittain Tyydyttävä Noveon 492,828,10821318,1452,1659
(pyöreät matalat piikit korkea pohja)
Hydroksietyyliselluloosa 282553988 1,6 on Huono Fluka Ei tunnuspiikkejä
Ibuprofeeni IB1M485 1,6 ei Hyvä BASF corp X 636,745,833,1180,1206,1607
Kalsiumfosfaatti-dihydraatti pHEur B10941A 1,6,12 ei Tyydyttävä Emcopress
premium
389,415,587,896,983,1087,1127
Kalsiumsilikaatti 1344-95-2 1,6,30 osittain Tyydyttävä Huber 966,1350,1411
Klooripropamidi 031H0722 1,6 ei Hyvä Sigma 259,625,754,911,1084,1159,1587
Klotrimatsoli BCBB5668 1,6 ei Hyvä Sigma 266,317,446,620,659,1001,1033,1079,1156,1585

111
Kofeiini (vedetön) 81612 1,6 ei Hyvä Cheminent X 482,554,642,740,1327
Laktoosi Pharmatose 90M pH.Eur 10407255 1,3,6 osittain Hyvä DFE Pharma X 356,361,382,475,850,875,1085,1119,1140,1260,
Magnesiumkloridi 8xH2O 12341983420
5143 1,6 osittain Huono Fluka
572,1000,1583(Ei selkeitä vahvoja piikkejä)
Magnesiumstearaatti pH Eur 15883 1,2,3,4,
6,1AdVp osittain Hyvä
X 1062,1129,1295,1436,1456
Maissitärkkelys 129K0076V 1,6 Osittain Tyydyttävä Sigma-
Aldrich
477,865,937,1049,1079,1125,1335,1457
Mannitoli D (-) K262 1,6 ei Hyvä Prolabo X 475,875,1036,1132
Mefenaamihappo 055K1555 1,6 on Huono Sigma X 621,772,807,1037,1159,1243,1332(ei selkeitä
tunnistepiikkejä)
Methocel® MC 2864831289 1,6 on Huono Fluka 937,1095,1358,1446(ei selkeitä tunnistepiikkejä)
Mikrokiteinen selluloosa 50M pH.Eur 5S8571 1,3,6 on Huono EMCOCEL X 1091 (ei kunnollisia piikkejä)
Mikrokiteinen selluloosa 90M pH.Eur 9S1191 1,3,6 on Huono EMCOCEL X 1092 (ei kunnollisia piikkejä) isoin piikki
Mikrokiteinen selluloosa PH101
pH.Eur 60935C 1,3,6Vp on Huono Avicel
X 1092 (ei kunnollisia piikkejä)
Nikotiiniamidi 1380397 1,6 ei Hyvä Fluka X 385,785,1041,1596
Nitrofurantoiini 067K0737 1,6 on Hyvä Sigma X 1017,1169,1247,1348,1378,1492,1609

112
Parasetamoli 1005402-
2002 1,3,6 ei Hyvä Oriola
X 390,651,797,857,1168,1237,1324,1370,1609,1647
Polyetyleeniglykoli 044K0092 1,6 ei Hyvä Sigma X 277,361,533,843,(858),1061,(1124),1140,1231,1279,147
9
Polyvinyylipyrrolidoni BCBb7859 1,6 ei Hyvä Fluka 752,850,933,1018,1226,1424,1670
Propranololi HCL 95.334 1,6 ei Hyvä S.I.M.S X 488,736,760,1385,1443,1578
Pyratsiinikarboksamidi 068K1129 1,6 ei Hyvä Sigma 413,806,1024,1052,1453,1524,1577
Salisyyliamidi S51885-358 1,6 ei Hyvä Sigma-
Aldrich
X 562,746,1039,1125,1143,1166,1246,1432,1622
Salisyylihappo 097K3401 1,6 ei Hyvä Sigma-
Aldrich
X 451,566,772,1030,1153,1247,1323,1634
Sukroosi 40K0201 1,6 Osittain Hyvä Sigma X 401,524,548,639,849,920,1037,1124,1237,1461
Sulfadiatsiini 027k2064 1,3,6 ei Hyvä Sigma X 285,544,633,824,847,993,1097,1149,1598
Sulfameratsiini 068K1130 1,6 ei Hyvä Sigma X 546,633,819,838,998,1093,1151,1594
Sulfametasiini 118K0898 1,3,6 on Tyydyttävä Sigma X 577,635,823,840,996,1088,1142,1593
Sulfametoksatsoli 048K0124 1,6 on Tyydyttävä Sigma 291,544,687,830,924,1092,1142,1156,1507,1592
Sulfaniiliamidi 126K001 1,3,6 on Tyydyttävä Sigma X 296,561,633,819,840,897,1090,1134,1155,1593

113
Teofylliini vedetön 057K0691 1,6 ei Hyvä Sigma X 445,502,554,666,926,968,1248,1285,1314,1425,1663,17
04
Tiosalisyylihappo 106K3671 1,6 on Huono Sigma 801,1043,1121,1307,1562,1585(1043 piikki kunnollinen)
Tolbutamidi 058K1071 1,6 ei Hyvä Sigma 290,634,796,811,881,1093,1147,1159,1599
Trikalsiumfosfaatti 605044 1,6 ei Tyydyttävä Prolabo X 426,586,961 (ainoastaan 961 voimakas piikki.)
TriNatriumfosfaatti-dodekahydraatti A326478133 1,6 on Tyydyttävä Merck X 408,544,939,1000
*R: Aineelle löytyy kirjallisuudesta verrattava Ramanspektri
Liitetaulukko 3. Asetyylisalisyylihapon standardiliuosten valmistus kantaliuoksesta, suluissa laimennokseen käytetty pitoisuus ja lisätyn tislatun veden määrä.
Standardi ASA 0 ASA 1 ASA 2 ASA 3 ASA 4 ASA 5
Pitoisuus 50,2 mg/l 101 mg/l 201 mg/l 301 mg/l 402 mg/l 502 mg/l
Laimennusohje 1ml
(1004,8mg/l) + 19ml
1ml (1004,8mg/l)
+9 ml
2ml (1004,8mg/l)
+8 ml
3ml (1004,8mg/l)
+7 ml
4ml (1004,8mg/l)
+6 ml
5ml (1004,8mg/l)
+5 ml

114
Liitetaulukko 4. Salisyylihapon standardiliuosten valmistus kantaliuoksesta, suluissa laimennokseen käytetty pitoisuus ja lisätyn tislatun veden määrä.
Standardi SA 0 SA 1 SA 2 SA 3 SA 4 SA 5
Pitoisuus 5,038 mg/l l
37,785 mg/l
50,38 mg/l
75,57 mg/l
100,76 mg/l
151,14 mg/l
Laimennosohje 1ml
(50,38mg/l) + 9ml
5ml (151,14mg/l)
+ 15ml
1ml (1007,6mg/l)
+ 19ml
5ml (151,14mg/l)
+ 5ml
1ml (1007,6mg/l)
+ 9ml
3ml (1007,6mg/l)
+17 ml
Liitetaulukko 4. Kantaliuoksen valmistaminen asetyylisalisyylihapon ja dikalsiumfosfaatin seoksista ja laimentaminen edelleen spektrofotometrillä mitattaviksi näytteiksi
Näytteen ASA (%) 5 10 12,5 15 17,5 20 30 40
Näytteen paino (g) 1,0167 1,0214 1,000 1,0118 1,0030 1,0124 1,0150 1,0102
ASA:n teoreettinen pitoisuus (mg/l)
101,67 204,28 250 303,54 351,05 404,96 609 808,16
Mittausnäytteiden laimennosohje
– 5+5 4+6 3+7 3+7 5+15 2+8 2+8
Mittausnäytteiden ASA pitoisuus (mg/l)
(3 rinnakkaista näytettä) 101,67 102,14 100 91,062 105,315 134,987 121,8 161,632
Related Documents











