ORIGINAL ARTICLE Sgk1 activates MDM2-dependent p53 degradation and affects cell proliferation, survival, and differentiation Rosario Amato & Lucia D’Antona & Giovanni Porciatti & Valter Agosti & Miranda Menniti & Cinzia Rinaldo & Nicola Costa & Emanuele Bellacchio & Stefano Mattarocci & Giorgio Fuiano & Silvia Soddu & Marco G. Paggi & Florian Lang & Nicola Perrotti Received: 26 February 2009 / Revised: 5 August 2009 / Accepted: 20 August 2009 / Published online: 11 September 2009 # Springer-Verlag 2009 Abstract Serum and glucocorticoid regulated kinase 1 (Sgk1) is a serine–threonine kinase that is activated by serum, steroids, insulin, vasopressin, and interleukin 2 at the transcriptional and post-translational levels. Sgk1 is also important in transduction of growth factors and steroid- dependent survival signals and may have a role in the development of resistance to cancer chemotherapy. In the present paper, we demonstrate that Sgk1 activates MDM2- dependent p53 ubiquitylation. The results were obtained in RKO cells and other cell lines by Sgk1-specific RNA silencing and were corroborated in an original mouse model as well as in transiently and in stably transfected HeLa cells expressing wild-type or dominant negative Sgk1 mutant. Sgk1 contributes to cell survival, cell-cycle progression, and epithelial de-differentiation. We also show that the effects of Sgk1 on the clonogenic potential of different cancer cells depend on the expression of wild-type p53. Since transcription of Sgk1 is activated by p53, we propose a finely tuned feedback model where Sgk1 down-regulates the expression of p53 by enhancing its mono- and polyubiquitylation. Keywords Sgk1 . p53 . Cell death . Cell signaling . MDM2 Introduction The serum and glucocorticoid regulated kinase 1 (Sgk1) is a serine–threonine kinase finely regulated by steroids, insulin, and vasopressin at the transcriptional and post- translational levels, through PDK1/2 [1, 2]. PDK1 binds phospho-S422 in the hydrophobic motif (H-motif) of SGK1 to phosphorylate T256. The H-motif kinase that phosphorylates SGK1 at S422 to prime it for phosphor- ylation by PDK1 has been recently identified as mTOR [3]. Activation of the kinase contributes to the Na + - retaining effects of insulin, vasopressin, and aldosterone [ 4] and is critical for the hypertensive effects of hyperinsulinism [5–7]. Sgk1 is also involved in mediating growth factor and steroid-dependent survival signals [8, 9]. We have recently demonstrated that Sgk1 is essential for transducing inter- Electronic supplementary material The online version of this article (doi:10.1007/s00109-009-0525-5) contains supplementary material, which is available to authorized users. R. Amato : L. D’Antona : G. Porciatti : V. Agosti : M. Menniti : G. Fuiano : N. Perrotti (*) Department of Experimental and clinical Medicine “G. Salvatore”, Faculty of Medicine, University Magna Graecia at Catanzaro, Catanzaro, Italy e-mail: [email protected] N. Costa Department of Pharmacobiology, University “Magna Graecia” Catanzaro, Catanzaro, Italy E. Bellacchio CSS-Mendel Institute, Rome, Italy C. Rinaldo : S. Soddu Department of Experimental Oncology, Molecular Oncogenesis Laboratory, Regina Elena Cancer Institute, Rome, Italy E. Bellacchio : S. Mattarocci : M. G. Paggi Department for the Development of Therapeutic Programs, Regina Elena Cancer Institute, Rome, Italy F. Lang Department of Physiology, University of Tübingen, Tübingen, Germany J Mol Med (2009) 87:1221–1239 DOI 10.1007/s00109-009-0525-5

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ORIGINAL ARTICLE
Sgk1 activates MDM2-dependent p53 degradationand affects cell proliferation, survival, and differentiation
Rosario Amato & Lucia D’Antona & Giovanni Porciatti & Valter Agosti &Miranda Menniti & Cinzia Rinaldo & Nicola Costa & Emanuele Bellacchio &
Stefano Mattarocci & Giorgio Fuiano & Silvia Soddu & Marco G. Paggi & Florian Lang &
Nicola Perrotti
Received: 26 February 2009 /Revised: 5 August 2009 /Accepted: 20 August 2009 /Published online: 11 September 2009# Springer-Verlag 2009
Abstract Serum and glucocorticoid regulated kinase 1(Sgk1) is a serine–threonine kinase that is activated byserum, steroids, insulin, vasopressin, and interleukin 2 atthe transcriptional and post-translational levels. Sgk1 is alsoimportant in transduction of growth factors and steroid-dependent survival signals and may have a role in thedevelopment of resistance to cancer chemotherapy. In the
present paper, we demonstrate that Sgk1 activates MDM2-dependent p53 ubiquitylation. The results were obtained inRKO cells and other cell lines by Sgk1-specific RNAsilencing and were corroborated in an original mouse modelas well as in transiently and in stably transfected HeLa cellsexpressing wild-type or dominant negative Sgk1 mutant.Sgk1 contributes to cell survival, cell-cycle progression,and epithelial de-differentiation. We also show that theeffects of Sgk1 on the clonogenic potential of differentcancer cells depend on the expression of wild-type p53.Since transcription of Sgk1 is activated by p53, we proposea finely tuned feedback model where Sgk1 down-regulatesthe expression of p53 by enhancing its mono- andpolyubiquitylation.
Keywords Sgk1 . p53 . Cell death . Cell signaling .MDM2
Introduction
The serum and glucocorticoid regulated kinase 1 (Sgk1)is a serine–threonine kinase finely regulated by steroids,insulin, and vasopressin at the transcriptional and post-translational levels, through PDK1/2 [1, 2]. PDK1 bindsphospho-S422 in the hydrophobic motif (H-motif) ofSGK1 to phosphorylate T256. The H-motif kinase thatphosphorylates SGK1 at S422 to prime it for phosphor-ylation by PDK1 has been recently identified as mTOR[3]. Activation of the kinase contributes to the Na+-retaining effects of insulin, vasopressin, and aldosterone[4] and is critical for the hypertensive effects ofhyperinsulinism [5–7].
Sgk1 is also involved in mediating growth factor andsteroid-dependent survival signals [8, 9]. We have recentlydemonstrated that Sgk1 is essential for transducing inter-
Electronic supplementary material The online version of this article(doi:10.1007/s00109-009-0525-5) contains supplementary material,which is available to authorized users.
R. Amato : L. D’Antona :G. Porciatti :V. Agosti :M. Menniti :G. Fuiano :N. Perrotti (*)Department of Experimental and clinical Medicine“G. Salvatore”, Faculty of Medicine,University Magna Graecia at Catanzaro,Catanzaro, Italye-mail: [email protected]
N. CostaDepartment of Pharmacobiology,University “Magna Graecia” Catanzaro,Catanzaro, Italy
E. BellacchioCSS-Mendel Institute,Rome, Italy
C. Rinaldo : S. SodduDepartment of Experimental Oncology, Molecular OncogenesisLaboratory, Regina Elena Cancer Institute,Rome, Italy
E. Bellacchio : S. Mattarocci :M. G. PaggiDepartment for the Development of Therapeutic Programs,Regina Elena Cancer Institute,Rome, Italy
F. LangDepartment of Physiology, University of Tübingen,Tübingen, Germany
J Mol Med (2009) 87:1221–1239DOI 10.1007/s00109-009-0525-5

leukin 2-dependent survival signals and that ectopicexpression of the IL-2 receptor in kidney cancer cellsprotects the cells from doxorubicin-induced apoptosis,thus demonstrating a possible mechanism underlyingdrug resistance [10]. A role for Sgk1 in tumor develop-ment is supported by its capability to promote G1 cell-cycle progression through p27 phosphorylation [3] andby its over-expression in several human tumor cells, evenin tissues where the cognate kinase Akt is not signifi-cantly activated [11, 12]. More recently, Sgk1 has beenfound to be essential and limiting in the inhibition ofGsk3, thus allowing the stabilization of c-myc in tumormodels lacking a functional RhoB [13]. Based on theserecent observations, Sgk1 antagonistic molecules areconsidered in the experimental therapy of prostatic cancer[14], a tumor that is also finally regulated by steroidhormones [15].
Some of these Sgk1-dependent functions might beexplained by the modulation of the activity of p53, acritical player in cell survival and cell-cycle progression.As a matter of fact, a link between Sgk1 and p53 hasbeen suggested [16]. Sgk1 is regulated by p53 at thetranscriptional level. The promoter region of the genecoding for Sgk1 contains p53 DNA binding elements, andSgk1, but not Akt, is necessary for p53-dependentsuppression of FKHRL1 in mouse embryo fibroblasts[17, 18].
p53 is mutated in at least 50% of human tumors [19].The protein can be inactivated by ubiquitylation [20, 21].The cellular p53 protein abundance is mainly modulated byMDM2, a negative regulator of p53 that binds p53 withinits N-terminal transactivation domain [22, 23]. Bindingtriggers the MDM2-dependent E3 ligase activity thatcatalyzes the ubiquitylation of p53, leading to itsproteasome-mediated degradation [24, 25].
p53 is able to regulate several caretaker functions,like cell-cycle exit at the G1-S checkpoint [26], orcyclin B1 and related cdks inhibition at the G2-Mcheckpoint [27]. Moreover, p53 is one of the principalregulators of apoptosis since it integrates both intrinsic/mitochondrial and extrinsic apoptosis pathways [28].More recently, p53 has been found capable of regulatinglimiting genes in differentiation and in cellular architec-tural control [29, 30].
The present paper aims to investigate the functionaland structural relationship between Sgk1 and p53 bymeans of Sgk1-specific RNA silencing and dominantnegative [4, 10, 31] technologies. We present evidencethat Sgk1, a gene regulated by p53 at the transcriptionallevel [32], affects cell proliferation and differentiation andthat the expression and the half-life of p53 is regulated bySgk1 through MDM2 that directs p53 to ubiquitylationand proteosomal degradation.
Experimental procedures
Commercially available reagents and antibodies wereobtained as indicated in the Electronic supplementarymaterials.
Recombinant DNAs
The sequences coding for myc-tagged wild-type Sgk1 anddominant negative D222A Sgk1 were cloned into thePcDNA4TO expression vector (Invitrogen, Milan, Italy)as previously described [10]. pGEX4T3-MDM2 (fulllength) was produced by one of us [33]. The CMV-drivenubiquitin expression construct pMT107 (His6-ubiquitin)has been described [34] and was kindly provided byD. Bohmann.
pCMV-Mdm2, GST-MDM2, GST-MDM2 S166A, andGST-MDM2 S188A were kindly provided by Dr. KarenVoudsen and pCMV-Mdm2ΔRing by Dr. Moshe Oren.pBABE empty vector was used together with PcDNA4-TO(empty vector) or PcDNA4-TO (myc) Sgk1 wild type (WT)or PcDNA4-TO (myc) Sgk1 D222A (DN) for colony assayexperiments. pcDNA3 UB-Ha, coding for the HA-taggedubiquitin protein, was obtained through the courtesy ofDr. Luca Ulianich.
Cell culture, transient transfection, and establishmentof continuous cell lines
The methods for cell culture, transient transfection, andestablishment of continuous cell lines are detailed inthe Electronic supplementary materials, according to apreviously published paper [10]. Continuous cell lineswere obtained from HeLa cells stably transfected withvectors PcDNA4-TO (empty vector), PcDNA4-TO (myc)Sgk1 WT, or PcDNA4-TO (myc) Sgk1 D222A (DN) andnamed empty vector cells, Sgk1WT, and Sgk1 D222ADN cells.
Virus generation and infection for RNA interferenceexperiments
The human sgk1 (NM_005627 GenBank) MISSIONshRNA set (five individual hairpins individually clonedinto pLKO.1-puro; Sigma) were used to generate lentiviralparticles in HEK293T packaging cells. SubconfluentHEK293T cells were cotransfected with 13 µg p27MISSION shRNA set, 18 µg pCMV-deltaR8.91, and12 µg pCMV-VSVG per 100-mm tissue culture plate byLipofectamine™ 2000. After 48 h from transfection,supernatants were collected at 8-h intervals, filtered, andused for three rounds of transduction of HeLa and RKO celllines in the presence of 8 µg/mL polybrene (Sigma).
1222 J Mol Med (2009) 87:1221–1239

Transduced cells were selected by puromycin (1.2 μg/mL),lysed 72 h after infection, and analyzed by immunoblottingwith anti-Sgk1, anti-p53 (DO-1), and anti-MDM2 anti-bodies. The Sgk1 knockdown in transduced HeLa andRKO cells was verified by comparison with HeLa andRKO cells, transduced with Mission non-target controltransduction virus (scrambled RNA; Sigma SHC002V).Both HeLa and RKO cells were transduced with each of thefive clones expressing different shRNAs. As indicated inthe “Results” section, clones 2294-Sgk1 and 458-Sgk1were effective in HeLa cells, whereas clone 458-Sgk1 waseffective in RKO cells.
Colony assays
Cervical carcinoma HeLa cells, human RKO cells,transformed human embryonic kidney Hek293A cells,and human non-small cell lung carcinoma H1299cells were resuspended at a density of 2×106 cells/mLand cotransfected with either PcDNA4-TO (emptyvector), PcDNA4-TO (myc) Sgk1 WT, or PcDNA4-TO(myc) Sgk1 D222A (DN) and with pBABE empty vectorcarrying the puromycin resistance gene by means ofLipofectamine 2000. After 3 weeks of selection in thepresence of puromycin (5 μg/mL), the colonies obtainedwere washed three times with PBS and stained withcrystal violet 0.5% W/V solution in methanol. Singlecolonies in every plate were counted and captured bydigital camera.
Cell stimulation and treatment with inhibitors
Cell cultures were serum-starved 24 h before stimulationwith any effector, unless otherwise specified in the figurelegends. Hormones, buffer, and inhibitors are detailed in theElectronic supplementary materials.
Immunofluorescence
Empty vector cells, Sgk1 WT cells, and Sgk1 D222A DNcells, grown on cover slips, were fixed with 3.7%formaldehyde for 20 min. Fixed cells were then permeabi-lized with 0.5% Triton X-100 for 1 min and washed withPBS (pH 7.4). The prepared samples were incubated withprimary antibodies to Sgk1, p53, MDM2, ZO-1, alphacatenin, vimentin, and myc-tag and stained as detailed inthe Electronic supplementary materials.
FACS analysis of synchronized and unsynchronized cells
Cell-cycle analysis was performed according to standardmethods as indicated in the Electronic supplementarymaterials.
Mice models and animal procedures
For cellular inoculations in mice, a cohort of six male,9-week-old BALB/c mice were inoculated subcutaneouslywith either 2×106 cells of empty vector or Sgk1 D222ADN cells on the back. The mice were killed after 17 days.The inoculated cells were recovered, lysed, and analyzed byWestern blot with anti-p53 DO-1 antibody, anti-α-catenin,and anti-c-myc tag antibodies.
Transgenic animals
Tet-CMV Sgk1 D222A DN single transgenic founderswere generated in collaboration with Murinus GmbH|Falkenried 88|D-20251 Hamburg as indicated in theElectronic supplementary materials. The single transgenicfounders were crossed with MUP-tTA mice obtained byDr. T. Jake Liang [35]. The experiments were performedon double transgenic, single transgenic, and wild-typeanimals as indicated in the Electronic supplementarymaterials.
p53 ubiquitylation experiments
In vitro and in vivo assays were carried out as previouslypublished [33, 36] with minor modifications, detailed in theElectronic supplementary materials.
p53 half-life experiments
Cells were pulse labeled with [35S]methionine and [35S]cysteine, as detailed in the Electronic supplementarymaterials.
Results
Sgk1-specific RNA silencing in RKO cells: effectson the level of expression, mono- and polyubiquitylationof p53
Sgk1 silencing was achieved by means of retrovirallyencoded small RNAs. In order to obtain an efficientsilencing of the endogenous Sgk1, RKO cells were infectedtwice.
A significant silencing of the endogenous Sgk1 wasachieved using the 458-Sgk1 siRNA, and a concomi-tant increase of p53 expression was observed (Fig. 1a,lane 2, top). In a separate experiment, depletion ofendogenous Sgk1 was almost complete after the firstinfection (Fig. 1b, lane 2, middle) and was associatedwith a significant increase of p53 expression (Fig. 1b,lane 2, top). After a second round of infection with the
J Mol Med (2009) 87:1221–1239 1223

458-Sgk1 silencer, endogenous Sgk1 was virtually unde-tectable (Fig. 1b, lane 3, middle), whereas a furtherincrease in the expression of p53 was achieved (Fig. 1b,lane 3, top). The expression of p53 can be modifiedthrough its proteosomal degradation driven by mono- andpolyubiquitylation. Thus, we compared the p53 ubiqui-tylation status in RKO cells in the presence or absence ofendogenous Sgk1. As shown in Fig. 1c (lane 2, upperpanel), Sgk1 depletion significantly decreased bothmono- and polyubiquitylation of p53. This decrease inp53 ubiquitylation was associated with the expectedincrease in expression levels of p53 (Fig. 1c, lane 2,lower panel).
Sgk1-specific RNA silencing in RKO cells: effects on cellproliferation
The inhibition of the endogenous kinase in RKO cells wasalso associated with a significant decrease of proliferationparalleled by an increase in the number of dead cells, asassessed by direct cell counting and Trypan blue exclusion,48 and 72 h after cell plating (Fig. 1d).
Sgk1 increases MDM2-dependent mono-and polyubiquitylation of p53 in vitro
In order to verify whether Sgk1 directly affected MDM2,thus causing mono- and polyubiquitylation of p53, an in
vitro assay was assembled where in vitro ubiquitylationreactions were performed in the presence of recombinant E1,E2, and ubiquitin (Fig. 2a, lanes 1–8) with addition of eitherGST (Fig. 2a, lanes 1 and 2) or GST-p53 (Fig. 2a, lanes4–7) as substrates, Sgk1 active kinase (Fig. 2a, lanes 3, 5,and 6), and the immunocomplexes obtained by dKO MEFstransduced with either MDM2 (Fig. 2a, lanes 1, 4, and 5),or its Δ-RING deletion mutant (Fig. 2a, lanes 6 and 7) assource of WT and defective E3 activity.
As shown in Fig. 2a, slowly migrating bands recognizedby anti-Ub Abs were detectable only in the presence of WTMDM2 and the active kinase Sgk1 (lane 5), while theΔ-RING deletion mutant was unable to modify GST-p53(lane 6), indicating that full-length MDM2, not its deletionmutant, catalyzes p53 ubiquitylation in vitro in the presenceof active Sgk1.
Sgk1 phosphorylates MDM2 at serine 166
The amount of MDM2 can be regulated by phosphorylation-dependent protein stability, and serine 166 is considered acrucial residue in this regulation. Thus, we asked whetherSgk1 might regulate p53 expression through direct phosphor-ylation of its negative regulator, MDM2.
Phosphorylation of MDM2 by Sgk1 was clearly detectedin an in vitro kinase assay using GST-MDM2 as a substratein the presence of the active kinase and γ32P-ATP (Fig. 2b,lane 5). In order to identify the residue(s) phosphorylated
Fig. 1 Effects of Sgk1-specific RNA silencing in RKO cells. RKOcells were infected with a retrovirus encoding Sgk1-specific RNAsilencer. a p53, Sgk1, and β-tubulin were detected by immunoblottingwith specific antibodies in RKO cells infected with retrovirus codingfor scrambled RNA (lane 1) or Sgk1 silencing RNA 458 (lane 2). bp53, Sgk1, and GAPDH were detected by immunoblotting withspecific antibodies in RKO cells infected twice with retrovirus codingfor scrambled (lane 1) or subjected to a single (lane 2) or double(lane 3) infection of the retrovirus encoding Sgk1 silencing RNA 458.
c RKO cells infected with retrovirus coding for scrambled RNA(lane 1) or Sgk1 silencing RNA 458 (lane 2) were transfected withHis–tagged ubiquitin. Cell extracts were subjected to nickel agarosepurification. Proteins were analyzed by immunoblotting with anantibody to p53. d Cells expressing either Sgk1-specific silencingRNA 458 or scrambled RNAwere plated at a density of 250,000 cellsper plate and incubated in the presence of FBS 10% for 48 and 72 hbefore counting. Dead cells were detected and counted manually byTrypan blue exclusion. Data are means ± SE obtained from triplicates
1224 J Mol Med (2009) 87:1221–1239
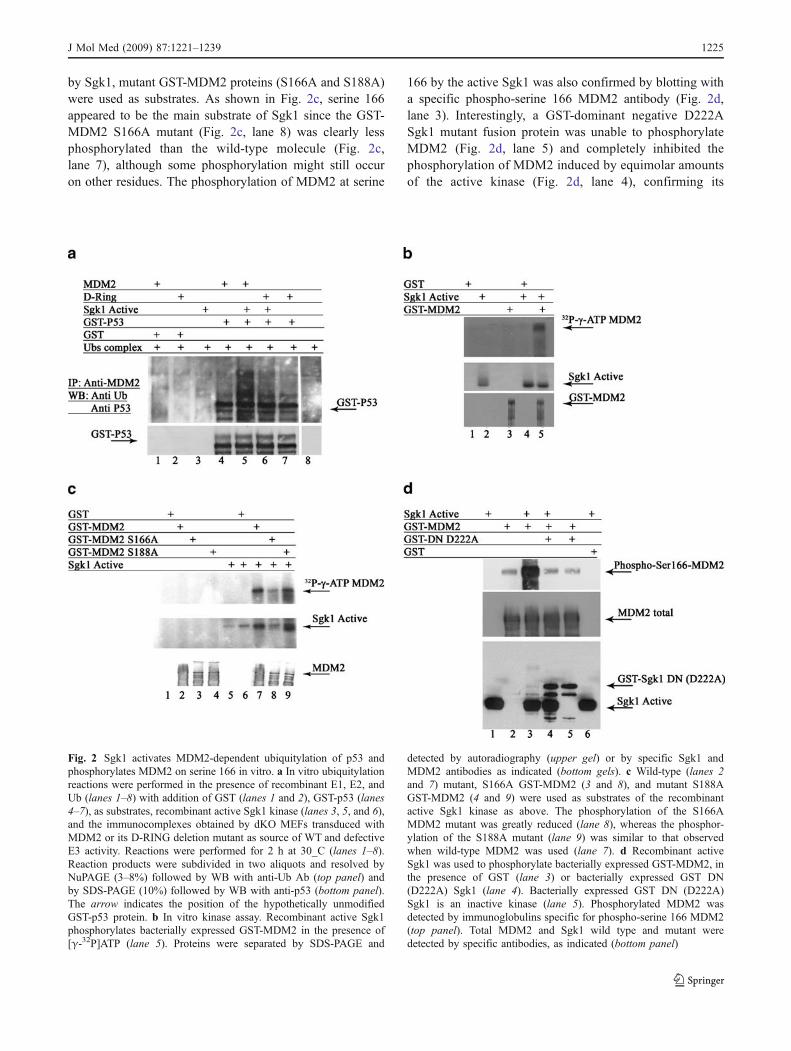
by Sgk1, mutant GST-MDM2 proteins (S166A and S188A)were used as substrates. As shown in Fig. 2c, serine 166appeared to be the main substrate of Sgk1 since the GST-MDM2 S166A mutant (Fig. 2c, lane 8) was clearly lessphosphorylated than the wild-type molecule (Fig. 2c,lane 7), although some phosphorylation might still occuron other residues. The phosphorylation of MDM2 at serine
166 by the active Sgk1 was also confirmed by blotting witha specific phospho-serine 166 MDM2 antibody (Fig. 2d,lane 3). Interestingly, a GST-dominant negative D222ASgk1 mutant fusion protein was unable to phosphorylateMDM2 (Fig. 2d, lane 5) and completely inhibited thephosphorylation of MDM2 induced by equimolar amountsof the active kinase (Fig. 2d, lane 4), confirming its
Fig. 2 Sgk1 activates MDM2-dependent ubiquitylation of p53 andphosphorylates MDM2 on serine 166 in vitro. a In vitro ubiquitylationreactions were performed in the presence of recombinant E1, E2, andUb (lanes 1–8) with addition of GST (lanes 1 and 2), GST-p53 (lanes4–7), as substrates, recombinant active Sgk1 kinase (lanes 3, 5, and 6),and the immunocomplexes obtained by dKO MEFs transduced withMDM2 or its D-RING deletion mutant as source of WT and defectiveE3 activity. Reactions were performed for 2 h at 30_C (lanes 1–8).Reaction products were subdivided in two aliquots and resolved byNuPAGE (3–8%) followed by WB with anti-Ub Ab (top panel) andby SDS-PAGE (10%) followed by WB with anti-p53 (bottom panel).The arrow indicates the position of the hypothetically unmodifiedGST-p53 protein. b In vitro kinase assay. Recombinant active Sgk1phosphorylates bacterially expressed GST-MDM2 in the presence of[γ-32P]ATP (lane 5). Proteins were separated by SDS-PAGE and
detected by autoradiography (upper gel) or by specific Sgk1 andMDM2 antibodies as indicated (bottom gels). c Wild-type (lanes 2and 7) mutant, S166A GST-MDM2 (3 and 8), and mutant S188AGST-MDM2 (4 and 9) were used as substrates of the recombinantactive Sgk1 kinase as above. The phosphorylation of the S166AMDM2 mutant was greatly reduced (lane 8), whereas the phosphor-ylation of the S188A mutant (lane 9) was similar to that observedwhen wild-type MDM2 was used (lane 7). d Recombinant activeSgk1 was used to phosphorylate bacterially expressed GST-MDM2, inthe presence of GST (lane 3) or bacterially expressed GST DN(D222A) Sgk1 (lane 4). Bacterially expressed GST DN (D222A)Sgk1 is an inactive kinase (lane 5). Phosphorylated MDM2 wasdetected by immunoglobulins specific for phospho-serine 166 MDM2(top panel). Total MDM2 and Sgk1 wild type and mutant weredetected by specific antibodies, as indicated (bottom panel)
J Mol Med (2009) 87:1221–1239 1225

function as a powerful inhibitor of Sgk1 in severalsystems.
Effects of the inhibition of the endogenous Sgk1 kinasein different tissues and cell lines
The results obtained in RKO cells were confirmed indifferent cell lines with different approaches. HeLa cellsrepresent a widely used tool to study cell proliferation anddifferentiation [37] with the caveat that they the express thehigh risk human papillomavirus E6 protein that binds p53and targets it for accelerated degradation and is expected toconfer resistance to the inhibition of the Sgk1 and MDM2-dependent degradation of p53 [38]. In our hands, RKO andHeLa cells express comparable amounts of p53 althoughboth cell lines show a significantly reduced expression ofp53 when compared with Hek293 cells, that express a wild-type p53, not inhibited by papillomavirus and characterizedby a completely different mechanism of cell-cycle regula-tion [39] (Fig. 3a).
Sgk1 silencing in HeLa cells
In HeLa cells, two RNA molecules resulted in complete(2294-Sgk1) or almost complete (458-Sgk1) Sgk1silencing (Fig. 3b, lanes 2 and 3). Even in HeLa cells,characterized by papillomavirus-induced down-regulationof p53, the inhibition of endogenous Sgk1 was associatedwith an impressive increase in the expression of p53. Theenhancement of p53 was correlated with the efficiency ofSgk1 inhibition since it was more evident when 2294-Sgk1 was used. Interestingly, the inhibition of Sgk1 wasparalleled by a marked decrease in the expression ofMDM2, the E3 ubiquitin ligase that is considered the mainregulator of p53.
Dominant negative technology is an alternative toolto Sgk1-specific RNA silencing
Expression of p53 protein was detected by Westernblotting in HeLa cells transiently transfected with anempty vector (PcDNA4-TO), or with vectors coding foreither Sgk1 wild-type (PcDNA4-TO (myc) Sgk1 wildtype) or the dominant negative mutant of Sgk1(PcDNA4-TO (myc) Sgk1 D222A). p53 was clearlydetectable in cells transfected with the empty controlvector (Fig. 3c, lane 2, top), virtually undetectable in cellsover-expressing wild-type Sgk1 (Fig. 3c lane 1 top), andclearly increased in cells over-expressing the dominantnegative mutant of Sgk1 (Fig. 3c, lane 3, top), demon-strating again that even in HeLa cells, with the dominantnegative technology, the inhibition of the endogenousSgk1 induces accumulation of p53.
Expression of the dominant negative mutant of Sgk1(Sgk1-DN) in livers of double transgenic mice (MUP1-TA/pTRE-D222A) enhances p53 levels
The effect of endogenous Sgk1 inhibition on p53 levelswas studied in a double transgenic mouse modelexpressing the dominant negative D222A Sgk1 mutantunder the control of a Tet-regulated promoter (Tet-off).A small cohort of five 25-week-old male mice (threedouble transgenics not treated with doxicycline, onesingle transgenic for the tTA, and one wild-type mouse)were selected (Fig. 3d, top panels). On the day of theexperiment, mice were killed, and the expression of thetransgene was detected in lysed livers by Western blottingusing an anti-Myc antibody (Fig. 3d, bottom panel). Theliver expression of the dominant negative D222A Sgk1was associated with a dramatic increase of p53 (Fig. 3d,lanes 3,4, and 5) when compared with the wild-type(Fig. 3d, lane 1) and the single transgenic mice (Fig. 3d,lane 2). The increased expression of p53 in the doubletransgenic mice paralleled the increased expression ofp21, indicating that the p53 protein expressed in liversunder these conditions is transcriptionally active andfunctional (Fig. 3d, lanes 3, 4, and 5 vs. 1 and 2).
Figure 3e shows a representative gel that demonstrates, byreverse PCR, that the expression of cDNA coding for themyc-Sgk1 D222A DN mutant transgene was detected onlyin double transgenic animals in the absence of doxicycline(lane 3), whereas it was not detected in double transgenicmice treated with doxicycline (lane 1) as well as in singletransgenic (lane 2) and in wild-type animals (lane 4).
Establishment of stably transfected HeLa cell linesexpressing wild-type and dominant negative mutantsof Sgk1
Expression of the transgenes in empty vector cells,Sgk1WT cells, and Sgk1 D222A DN cells was detectedby Western blot, reverse PCR, and immunofluorescence(Suppl. file Fig. 1). Anti-myc antibodies were able todetect a specific ∼50 kDa migrating band only in Sgk1WT cells (Suppl. file Fig. 1 A, lane 1) and in Sgk1 D222ADN cells (Suppl. file Fig. 1 A, lane 2), not in the emptyvector cells (Suppl. file Fig. 1 A, lane 3).
Similar results were obtained when the expression of thetransgene was assessed by reverse PCR. The expected450 bp PCR product, identified by direct sequencing, wasdetected only when primers specific for myc Sgk1 wereused to amplify cDNA from Sgk1WT and Sgk1 D222A DNcells (Suppl. file Fig. 1 B, lanes 3 and 4). No amplificationproduct was detected in the presence of cDNA obtainedfrom empty vector cells (Suppl. file Fig. 1 B, lane 2) or inthe absence of cDNA (Suppl. file Fig. 1 B, lane 1).
1226 J Mol Med (2009) 87:1221–1239
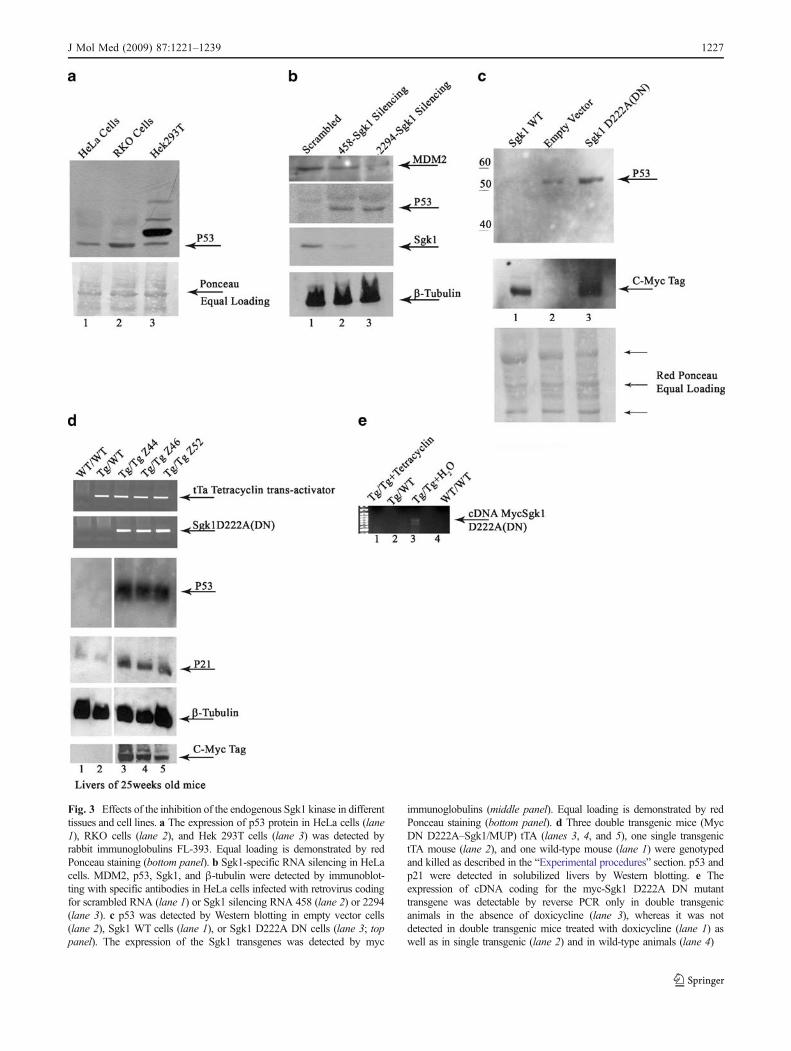
Fig. 3 Effects of the inhibition of the endogenous Sgk1 kinase in differenttissues and cell lines. a The expression of p53 protein in HeLa cells (lane1), RKO cells (lane 2), and Hek 293T cells (lane 3) was detected byrabbit immunoglobulins FL-393. Equal loading is demonstrated by redPonceau staining (bottom panel). b Sgk1-specific RNA silencing in HeLacells. MDM2, p53, Sgk1, and β-tubulin were detected by immunoblot-ting with specific antibodies in HeLa cells infected with retrovirus codingfor scrambled RNA (lane 1) or Sgk1 silencing RNA 458 (lane 2) or 2294(lane 3). c p53 was detected by Western blotting in empty vector cells(lane 2), Sgk1 WT cells (lane 1), or Sgk1 D222A DN cells (lane 3; toppanel). The expression of the Sgk1 transgenes was detected by myc
immunoglobulins (middle panel). Equal loading is demonstrated by redPonceau staining (bottom panel). d Three double transgenic mice (MycDN D222A–Sgk1/MUP) tTA (lanes 3, 4, and 5), one single transgenictTA mouse (lane 2), and one wild-type mouse (lane 1) were genotypedand killed as described in the “Experimental procedures” section. p53 andp21 were detected in solubilized livers by Western blotting. e Theexpression of cDNA coding for the myc-Sgk1 D222A DN mutanttransgene was detectable by reverse PCR only in double transgenicanimals in the absence of doxicycline (lane 3), whereas it was notdetected in double transgenic mice treated with doxicycline (lane 1) aswell as in single transgenic (lane 2) and in wild-type animals (lane 4)
J Mol Med (2009) 87:1221–1239 1227

Expression of wild-type and dominant negative(D222A) Sgk1 transgenes in the cells was confirmed byimmunofluorescence with anti-myc tag immunoglobulins(Suppl. file Fig. 1 C). The morphology of the Sgk1WTcells (Suppl. file Fig. 1 C, panel 1) was different from thatof Sgk1 D222A DN cells (Suppl. file Fig. 1 C, panel 2).The former cells appeared swollen with a Sgk1 preferen-tially localized at the cytoplasm, whereas the latter cellsappeared smaller for cytoplasmic regression. The findingswere confirmed when cells were observed by lighttransmission (Suppl. file Fig. 1 D). Parental and emptyvector cells showed the typical HeLa cell morphologywith a fusiform, fibroblast-like appearance, with poorcontact between cells (Suppl. file Fig. 1 D, panels 3 and4). The Sgk1 WT cells appeared swollen with abundantcytoplasm (Suppl. file Fig. 1D, panel 1), whereas the Sgk1D222A DN cells (Suppl. file Fig. 1D, panel 2) formed amore coherent epithelial monolayer with several contactsamong neighboring cells.
Sgk1 down-regulates p53 in stably transfected HeLa celllines
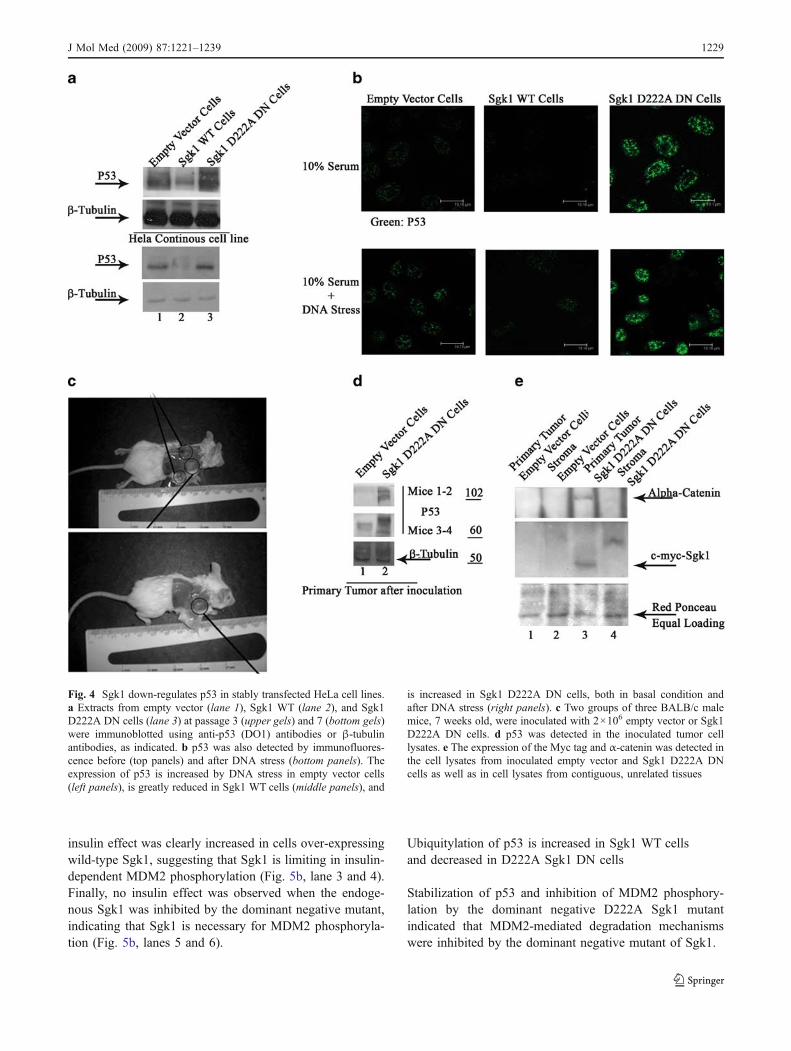
The p53 protein expression was evaluated by Westernblotting in empty vector, Sgk1 WT, and Sgk1 D222A DNcells. In Sgk1 WT cells, the expression of p53 was reducedcompared with the empty vector cells. On the contrary, inSgk1 D222A DN cells, the expression of p53 greatlyincreased. The difference in p53 expression was maintainedin culture and detected at different passages (Fig. 4a).Comparable results were obtained when p53 expressionwas assessed by immunofluorescence in empty vector,Sgk1 WT, and Sgk1 D222A DN cells (Fig. 4b). Expressionof p53 protein was detected in cells cultured in the presenceof 10% serum and clearly increased when cells wereexposed to genotoxic stress, as expected [20] (Fig. 4b, leftpanels). The increased expression of wild-type Sgk1 wasassociated with a decreased expression of p53, even in cellsexposed to genotoxic stress (Fig. 4 b, middle panels),whereas inhibition of the endogenous kinase was associatedwith a significant increase in the expression of p53, in cellsgrown in basal conditions as well as in cells exposed togenotoxic stress (Fig. 4b, right panels).
p53 expression in subcutaneous tumors obtainedby inoculation with HeLa cells
The expression of p53 was evaluated in tumors thatdeveloped 17 days after the subcutaneous inoculation ofempty vector and Sgk1 D222A DN cells in a smallcohort of BALB/c mice (Fig. 4c; three animals for eachgroup). In the tumors obtained by Sgk1 D222A DN cells,the expression of p53 was increased compared with the
tumors obtained by inoculating empty vector cells(Fig. 4d). Myc Sgk1 was also detectable in the epithelialcomponent of the Sgk1 D222A DN tumor cells (Fig. 5e).Interestingly, the expression of α-catenin was increased inthe epithelial component of Sgk1 D222A DN cellscompared with the empty vector cells (Fig. 4e), a resultconsistent with the epithelial appearance of the Sgk1D222A DN cells described above (Suppl. File Fig. 1 D,panel 2).
The over-expression of wild-type Sgk1 and the inhibitionof the endogenous kinase by the dominant negative mutant(Sgk1-DN) affect the abundance of MDM2 in stablytransfected HeLa cell lines
The expression of p53 can be increased by regulating eitherthe transcription of the gene or the degradation of theprotein through MDM2 or other E3 ubiquitin ligases.
An evident decrease in the expression of MDM2, themain regulator of p53, was observed upon inhibition ofSgk1 by specific RNA silencing (Fig. 3b). An effect ofSgk1 on the p53 gene transcription was ruled out since nosignificant difference in p53 RNA levels was detected byquantitative PCR on cDNA prepared from empty vector[1], Sgk1 WT (1.5±0.6) and Sgk1 DN D222A cells(1.5±0.1).
In the attempt to elucidate the mechanism by whichSgk1 affects the expression of p53 and MDM2, thecellular localization of MDM2 and p53 was studied byimmunofluorescence. p53 expression was slightly de-creased in Sgk1 WT cells (Fig. 5a, middle panel) andclearly increased in Sgk1 DN D222A cells (Fig. 5a,bottom panel), as expected. The expression of MDM2followed the opposite pattern. It was slightly increased inSgk1 WT cells (Fig. 5a, middle panel), whereas it wasclearly decreased or even abolished in D222A Sgk1cells(Fig. 5a, bottom panel), supporting the idea that afunctional Sgk1 is necessary to preserve the abundanceof MDM2, presumably by regulating the stability of theprotein. Nuclear colocalization of p53 and MDM2 wasevident in the control empty vector cells as well as inSgk1 WT cells (Fig. 5a, upper and middle panels),whereas no colocalization could be detected in D222ASgk1 cells since MDM2 was virtually undetectable(Fig 5a, bottom panel).
MDM2 phosphorylation in intact cells
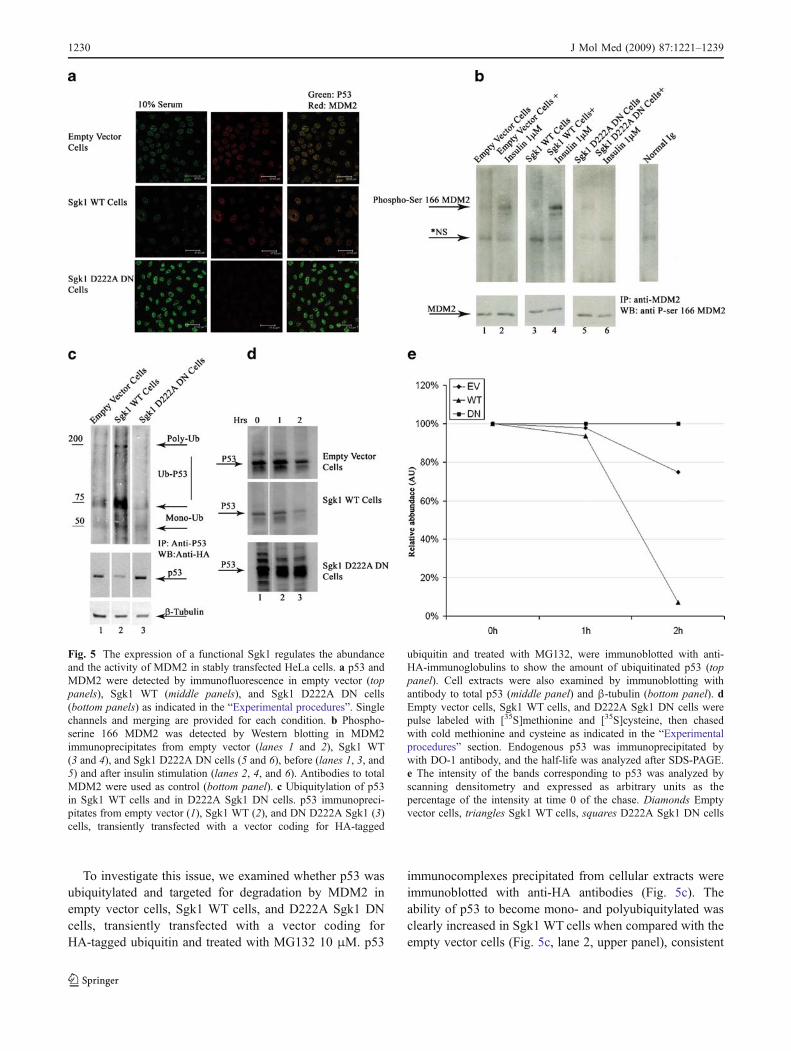
Sgk1-dependent phosphorylation of MDM2 was finallydemonstrated in intact empty vector, Sgk1 WT cells andSgk1 D222A DN cells, before and after insulin stimulation.
Insulin increased MDM2 phosphorylation in emptyvector cells, as expected (Fig. 5b, lanes 1 and 2). The
1228 J Mol Med (2009) 87:1221–1239
insulin effect was clearly increased in cells over-expressingwild-type Sgk1, suggesting that Sgk1 is limiting in insulin-dependent MDM2 phosphorylation (Fig. 5b, lane 3 and 4).Finally, no insulin effect was observed when the endoge-nous Sgk1 was inhibited by the dominant negative mutant,indicating that Sgk1 is necessary for MDM2 phosphoryla-tion (Fig. 5b, lanes 5 and 6).
Ubiquitylation of p53 is increased in Sgk1 WT cellsand decreased in D222A Sgk1 DN cells
Stabilization of p53 and inhibition of MDM2 phosphory-lation by the dominant negative D222A Sgk1 mutantindicated that MDM2-mediated degradation mechanismswere inhibited by the dominant negative mutant of Sgk1.
Fig. 4 Sgk1 down-regulates p53 in stably transfected HeLa cell lines.a Extracts from empty vector (lane 1), Sgk1 WT (lane 2), and Sgk1D222A DN cells (lane 3) at passage 3 (upper gels) and 7 (bottom gels)were immunoblotted using anti-p53 (DO1) antibodies or β-tubulinantibodies, as indicated. b p53 was also detected by immunofluores-cence before (top panels) and after DNA stress (bottom panels). Theexpression of p53 is increased by DNA stress in empty vector cells(left panels), is greatly reduced in Sgk1 WT cells (middle panels), and
is increased in Sgk1 D222A DN cells, both in basal condition andafter DNA stress (right panels). c Two groups of three BALB/c malemice, 7 weeks old, were inoculated with 2×106 empty vector or Sgk1D222A DN cells. d p53 was detected in the inoculated tumor celllysates. e The expression of the Myc tag and α-catenin was detected inthe cell lysates from inoculated empty vector and Sgk1 D222A DNcells as well as in cell lysates from contiguous, unrelated tissues
J Mol Med (2009) 87:1221–1239 1229
To investigate this issue, we examined whether p53 wasubiquitylated and targeted for degradation by MDM2 inempty vector cells, Sgk1 WT cells, and D222A Sgk1 DNcells, transiently transfected with a vector coding forHA-tagged ubiquitin and treated with MG132 10 μM. p53
immunocomplexes precipitated from cellular extracts wereimmunoblotted with anti-HA antibodies (Fig. 5c). Theability of p53 to become mono- and polyubiquitylated wasclearly increased in Sgk1 WT cells when compared with theempty vector cells (Fig. 5c, lane 2, upper panel), consistent
Fig. 5 The expression of a functional Sgk1 regulates the abundanceand the activity of MDM2 in stably transfected HeLa cells. a p53 andMDM2 were detected by immunofluorescence in empty vector (toppanels), Sgk1 WT (middle panels), and Sgk1 D222A DN cells(bottom panels) as indicated in the “Experimental procedures”. Singlechannels and merging are provided for each condition. b Phospho-serine 166 MDM2 was detected by Western blotting in MDM2immunoprecipitates from empty vector (lanes 1 and 2), Sgk1 WT(3 and 4), and Sgk1 D222A DN cells (5 and 6), before (lanes 1, 3, and5) and after insulin stimulation (lanes 2, 4, and 6). Antibodies to totalMDM2 were used as control (bottom panel). c Ubiquitylation of p53in Sgk1 WT cells and in D222A Sgk1 DN cells. p53 immunopreci-pitates from empty vector (1), Sgk1 WT (2), and DN D222A Sgk1 (3)cells, transiently transfected with a vector coding for HA-tagged
ubiquitin and treated with MG132, were immunoblotted with anti-HA-immunoglobulins to show the amount of ubiquitinated p53 (toppanel). Cell extracts were also examined by immunoblotting withantibody to total p53 (middle panel) and β-tubulin (bottom panel). dEmpty vector cells, Sgk1 WT cells, and D222A Sgk1 DN cells werepulse labeled with [35S]methionine and [35S]cysteine, then chasedwith cold methionine and cysteine as indicated in the “Experimentalprocedures” section. Endogenous p53 was immunoprecipitated bywith DO-1 antibody, and the half-life was analyzed after SDS-PAGE.e The intensity of the bands corresponding to p53 was analyzed byscanning densitometry and expressed as arbitrary units as thepercentage of the intensity at time 0 of the chase. Diamonds Emptyvector cells, triangles Sgk1 WT cells, squares D222A Sgk1 DN cells
1230 J Mol Med (2009) 87:1221–1239

with the decrease in total p53 (Fig. 5c, lane 2, bottompanel). Interestingly, in D222A Sgk1 DN cells, a decreasein p53 mono- and polyubiquitylation was observed (Fig. 5c,lane 3, upper panel), consistent with the increase in totalp53 (Fig. 5c, lane 3, bottom panel).
Sgk1 affects the half-life of p53
The final result of the activation of MDM2-dependent p53ubiquitylation is predicted to be a decrease in the half-lifeof p53.
Empty vector cells, Sgk1 WT cells, and Sgk1 D222ADN cells were pulse-labeled by [35S]methionine and [35S]cysteine and chased as indicated in the method section.
Figure 5d shows a representative result of two independentexperiments. In comparison with the empty vector cells(Fig. 5d, top gel), the half-life of p53 was shortened, andthe abundance of p53 was clearly reduced in cellsexpressing wild-type Sgk1 (Fig. 5d middle gel), whereasthe half-life was prolonged and the abundance of p53 wasincreased when the endogenous kinase was inhibited by theexpression of the dominant negative mutant Sgk1 (Fig. 5d,bottom panel).
Figure 5e shows a graphical representation of the resultwhere the intensity of the bands corresponding to p53 wasanalyzed by scanning densitometry and expressed inarbitrary units as the percentage of the intensity at time 0of the chase.
Fig. 6 Importance of p53 in the Sgk1-dependent clonogenic potentialof cancer cells. a–d A colony assay representative experiment in HeLacells (a), RKO cells (b), Hek293T cells (c), and H1299 cells (d). Eachcell line was treated as indicated in the “Experimental procedures”
section. The clones of several independent experiments were countedand graphically represented as histograms (bottom panel). Data are theaverage ± SEM of five independent experiments. The differences wereevaluated by t test. P values are presented at the bottom of each panel
J Mol Med (2009) 87:1221–1239 1231

Sgk1 and clonogenic capacity of cancer cells
Since p53 is a crucial regulator of cell proliferation andsurvival, we evaluated the consequences of the transient over-expression of wild-type and mutant Sgk1 on both cell cycleand survival by colony assay. The clonogenic capacity of p53expressing cells, i.e., RKO, HeLa, and Hek293 cells, wassignificantly enhanced by the expression of Sgk1 wild type,whereas it was inhibited by the expression of the dominantnegative D222A Sgk1 mutant (Fig. 6a–c), demonstrating thatthe effect of Sgk1 on the clonogenic capacity of cancer cellsis independent from the mechanism of transformation.
Interestingly, the dominant negative D222A mutant ofSgk1 had no effect in the p53-null H1299 cells, suggestingthat p53 expression is necessary for inhibition of theclonogenic capacity (Fig. 6d). Moreover, in these cells,Sgk1 wild type appeared to inhibit clonogenesis, suggestingthe possibility that Sgk1 also acts on a secondary pathwaywith growth inhibition potentiality, and this pathway isprevalent in the absence of p53.
These results suggest that the effects of Sgk1 on theclonogenic potential of cancer cells may be explained inlight of the ability of Sgk1 to inhibit p53.
Apoptosis and proliferation
The peculiar morphology of the Sgk1 WT cells and the Sgk1D222ADN cells, compared with the empty vector cells, mightreflect specific phenotypes related to apoptosis and prolifer-ation. Cell proliferation in stably transfected cell lines was firstassessed by cell counting and Trypan blue exclusion. Thenumber of living cells at 48 and 72 h was significantly reducedin Sgk1 D222A DN cells when compared with the emptyvector and the Sgk1 WT cells that showed, instead, asignificantly increased number of living cells. Conversely,an increased number of dead cells was detected in Sgk1D222A DN cells, at the same time points (Fig. 7a).
The profiles of cell-cycle distribution of unsynchronizedcells were further studied by fluorescence-activated cellsorting (FACS) analysis. The Sgk1 WT cells showed asignificant reduction in the percentage of apoptotic cellsand an increase in the percentage of cells in the G2-Mphase, when compared with the empty vector cells.Inhibition of endogenous Sgk1 by dominant negativemutant D222A resulted in a significant increase of basalapoptosis and in a clear-cut reduction in the percentage ofcells in the G2-M phase, suggesting that the expression of afunctional Sgk1 is essential for cell-cycle progression andsurvival (Fig. 7b, c). Accordingly, the expression of cyclinB1, an accepted hallmark of the G2-M phase, was inhibitedin the Sgk1 D222A DN cells, whereas it was not modifiedin Sgk1 WT cells (Fig. 7d).
The inhibition of Sgk1 by its dominant negative mutantis responsible for a consistent delay in the G2-M entryin a synchronized HeLa continuous cell line
In order to analyze the timing of cell-cycle progression forthe Sgk1 D222A DN cells, compared with the empty vectorcells and the Sgk1WT cells, cell cycle was studied in cellssynchronized by single hit thymidine and serum starvation.The analysis of cell cycle in synchronized cells revealedthat cell-cycle progression was similar in the empty vectorand in the Sgk1 WT cells, with a trend to a faster entrancein the G2-M for the Sgk1 WT cells already at 8 h. On theother hand, the Sgk1 D222A DN cells showed a strikingdelay in the G2-M entry with only 23% of cells in theG2-M phase after 12 h (Fig. 7e). The difference in thepercentage of cells in the G2-M phase between emptyvector cells and Sgk1 D222A DN, as well as betweenSgk1WT cells and Sgk1 D222A DN at 8 and 12 h, wasstatistically significant (Fig. 7f), thus confirming the crucialrole of a functional endogenous Sgk1 in cell-cycleprogression.
Sgk1 positively modulates epithelial dedifferentiation:the inhibition of the endogenous kinase by the dominantnegative mutant (Sgk1 DN) induces re-expressionof epithelial hallmarks
The different morphology described for the cell lines stablytransfected with vectors coding for wild-type and mutantSgk1 (Suppl. File Fig. 1 D) can be related to the differentialexpression of epithelial or mesenchymal markers, as wasalso suggested by the increased expression of α-catenin inD222A dominant negative tumors subcutaneously inocu-lated in mice (Fig. 4e). The transition from the expressionof epithelial to mesenchymal markers (EMT) has beenassociated with tumor formation and progression. Wefocused our attention on the expression of ZO-1,α-catenin, and vimentin, detected by immunofluorescencein empty vector, Sgk1WT, and in Sgk1 D222A DN cells(Fig. 8a–c). We observed that the epithelial markers ZO-1and α-catenin were completely down-regulated in Sgk1WT cells, when compared with the empty vector cells,whereas the inhibition of the endogenous Sgk1 in the Sgk1D222A DN cells resulted in the re-expression of these twomarkers, suggesting a role of Sgk1 in down-regulatingepithelial differentiation (Fig. 8a, b). Besides the epithelialmarkers, the expression of the mesenchymal markervimentin was studied in the same cells. We found thatvimentin was increased in the Sgk1WT cells, comparedwith the empty vector cells, whereas it was clearly down-regulated in the Sgk1 D222A DN cells, thus confirming therole of Sgk1 in inducing mesenchymal transition (Fig. 8c).
1232 J Mol Med (2009) 87:1221–1239
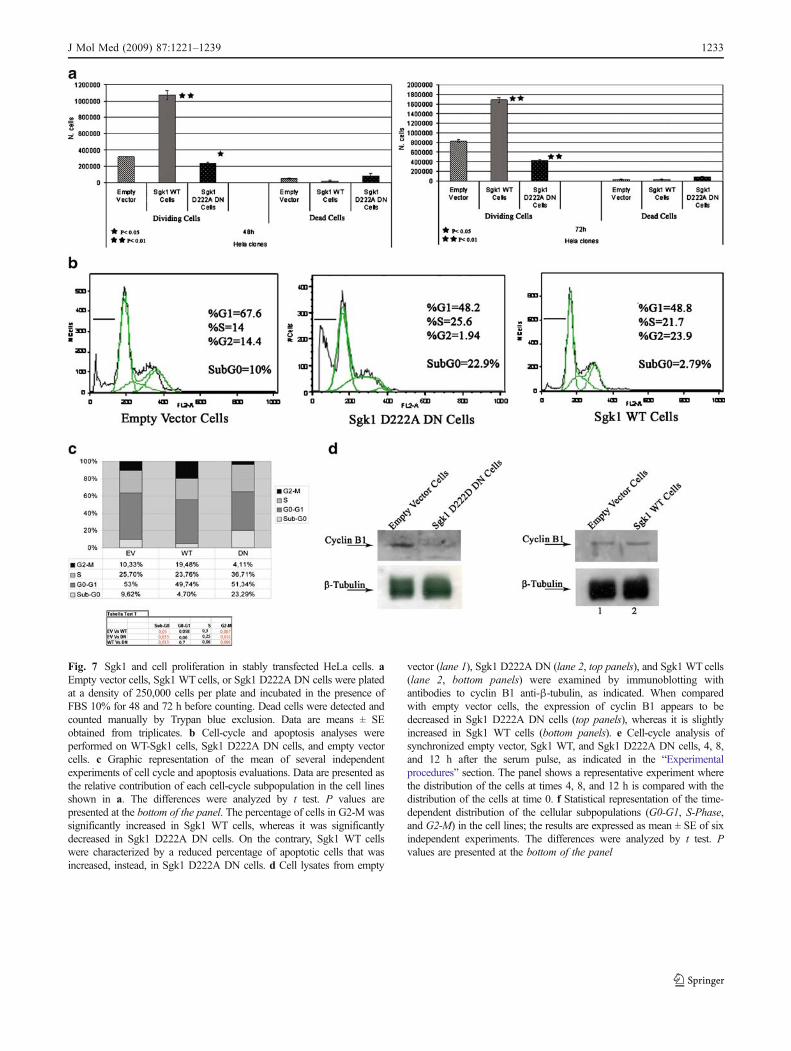
Fig. 7 Sgk1 and cell proliferation in stably transfected HeLa cells. aEmpty vector cells, Sgk1 WT cells, or Sgk1 D222A DN cells were platedat a density of 250,000 cells per plate and incubated in the presence ofFBS 10% for 48 and 72 h before counting. Dead cells were detected andcounted manually by Trypan blue exclusion. Data are means ± SEobtained from triplicates. b Cell-cycle and apoptosis analyses wereperformed on WT-Sgk1 cells, Sgk1 D222A DN cells, and empty vectorcells. c Graphic representation of the mean of several independentexperiments of cell cycle and apoptosis evaluations. Data are presented asthe relative contribution of each cell-cycle subpopulation in the cell linesshown in a. The differences were analyzed by t test. P values arepresented at the bottom of the panel. The percentage of cells in G2-M wassignificantly increased in Sgk1 WT cells, whereas it was significantlydecreased in Sgk1 D222A DN cells. On the contrary, Sgk1 WT cellswere characterized by a reduced percentage of apoptotic cells that wasincreased, instead, in Sgk1 D222A DN cells. d Cell lysates from empty
vector (lane 1), Sgk1 D222A DN (lane 2, top panels), and Sgk1 WT cells(lane 2, bottom panels) were examined by immunoblotting withantibodies to cyclin B1 anti-β-tubulin, as indicated. When comparedwith empty vector cells, the expression of cyclin B1 appears to bedecreased in Sgk1 D222A DN cells (top panels), whereas it is slightlyincreased in Sgk1 WT cells (bottom panels). e Cell-cycle analysis ofsynchronized empty vector, Sgk1 WT, and Sgk1 D222A DN cells, 4, 8,and 12 h after the serum pulse, as indicated in the “Experimentalprocedures” section. The panel shows a representative experiment wherethe distribution of the cells at times 4, 8, and 12 h is compared with thedistribution of the cells at time 0. f Statistical representation of the time-dependent distribution of the cellular subpopulations (G0-G1, S-Phase,and G2-M) in the cell lines; the results are expressed as mean ± SE of sixindependent experiments. The differences were analyzed by t test. Pvalues are presented at the bottom of the panel
J Mol Med (2009) 87:1221–1239 1233
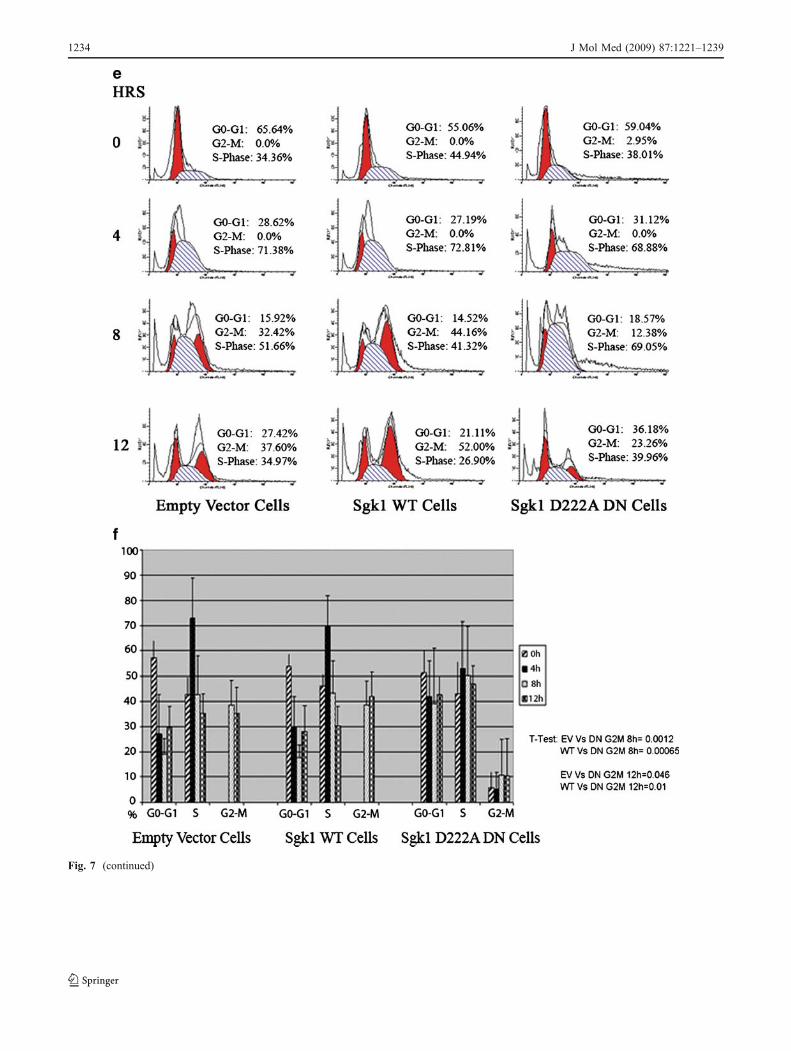
Fig. 7 (continued)
1234 J Mol Med (2009) 87:1221–1239
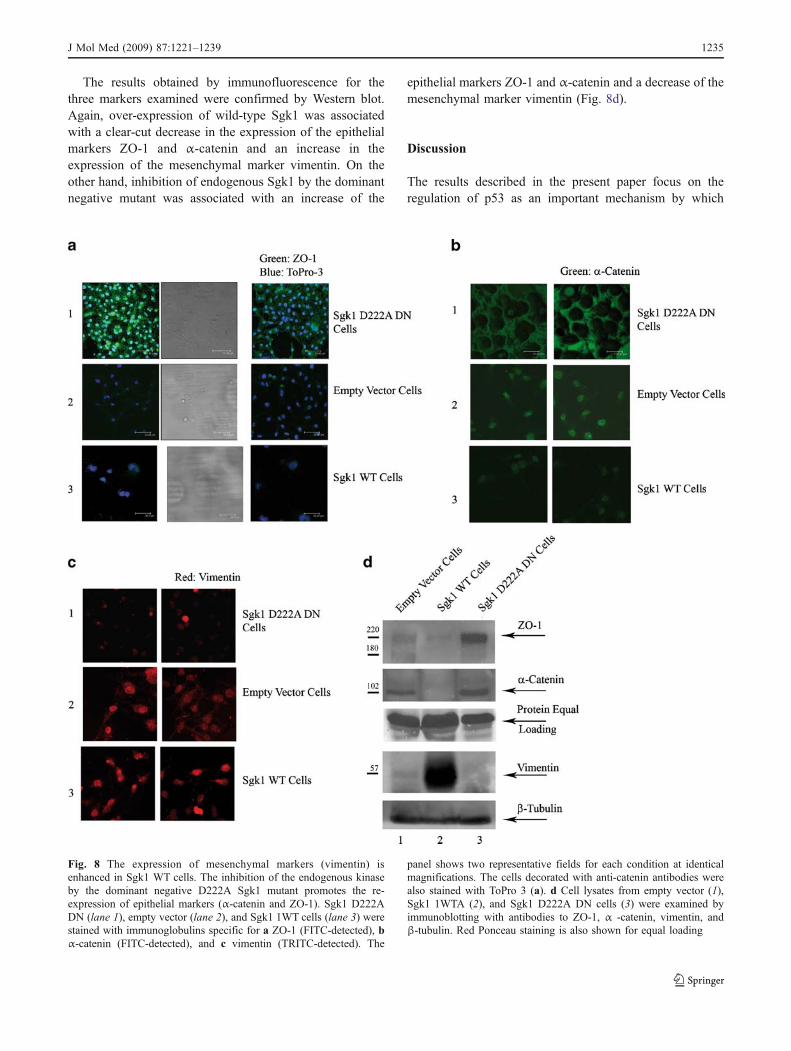
The results obtained by immunofluorescence for thethree markers examined were confirmed by Western blot.Again, over-expression of wild-type Sgk1 was associatedwith a clear-cut decrease in the expression of the epithelialmarkers ZO-1 and α-catenin and an increase in theexpression of the mesenchymal marker vimentin. On theother hand, inhibition of endogenous Sgk1 by the dominantnegative mutant was associated with an increase of the
epithelial markers ZO-1 and α-catenin and a decrease of themesenchymal marker vimentin (Fig. 8d).
Discussion
The results described in the present paper focus on theregulation of p53 as an important mechanism by which
Fig. 8 The expression of mesenchymal markers (vimentin) isenhanced in Sgk1 WT cells. The inhibition of the endogenous kinaseby the dominant negative D222A Sgk1 mutant promotes the re-expression of epithelial markers (α-catenin and ZO-1). Sgk1 D222ADN (lane 1), empty vector (lane 2), and Sgk1 1WT cells (lane 3) werestained with immunoglobulins specific for a ZO-1 (FITC-detected), bα-catenin (FITC-detected), and c vimentin (TRITC-detected). The
panel shows two representative fields for each condition at identicalmagnifications. The cells decorated with anti-catenin antibodies werealso stained with ToPro 3 (a). d Cell lysates from empty vector (1),Sgk1 1WTA (2), and Sgk1 D222A DN cells (3) were examined byimmunoblotting with antibodies to ZO-1, α -catenin, vimentin, andβ-tubulin. Red Ponceau staining is also shown for equal loading
J Mol Med (2009) 87:1221–1239 1235

Sgk1 controls cell proliferation, survival, and differentia-tion. This regulation can take place at the transcriptional orpost-translational levels. We were unable to demonstrate aneffect of wild-type and mutant Sgk1 on the transcription ofp53 by quantitative PCR. On the other hand, we presentedevidence demonstrating that Sgk1 regulated the expressionof p53 at the post-translational level.
The results were obtained in RKO cells, a not virallytransformed human colon carcinoma, by Sgk1-specificRNA silencing. The experiments in RKO cells, corroborat-ed by in vitro reconstituted kinase and ubiquitylationassays, led to the original conclusion that Sgk1 kinaseactivates MDM2-dependent mono- and polyubiquitylationof p53 protein and that the inhibition of the endogenouskinase increases the expression of p53 protein. The E3ubiquitin ligase MDM2 is considered the main regulator ofp53. Even subtle perturbations in MDM2 stoichiometryhave profound effects on p53 activity [40]. The interactionbetween p53 and MDM2 is probably the limiting factor inp53 degradation. For instance, upon genotoxic and oxida-tive stress, p53 can be stabilized by a series of interdepen-dent phosphorylations on serine and threonine residues atits N-terminal side that contribute to the disruption of theinteraction with MDM2 [41]. On the other hand, MDM2can be activated and stabilized by phosphorylation onseveral serine residues (serines 166, 186, and 473) by Akt[42, 43] or other kinases including mitogen-activatedprotein kinase-activated kinase 2 [44].
We believe that modifications in the activity of MDM2 canbe explained by the specific phosphorylation of MDM2 onserine 166, a reaction that was effectively inhibited by thedominant negative mutant D222A of Sgk1, in our cell-freeexperiments. This phosphorylation induces activation andstabilization of MDM2, increasing the E3 ligase functionrequired for p53 ubiquitylation [45]. In fact the inhibition ofthe endogenous kinase in RKO cells resulted in a significantdecrease in the mono- and polyubiquitylation of p53 thatparalleled the increase in the expression of the protein.
The results obtained in RKO cells were confirmed bydominant negative technology in different cell lines, even inHeLa cells, which are widely used cells [38], that areexpected to be more resistant to the inhibition of proteoso-mal degradation since they express the E6 protein ofpapillomavirus. In fact the induction of p53 by proteasomeinhibition is attenuated in E6 expressing RKO cells, whencompared with the parental RKO cells [38]. The resultswere confirmed in stably transfected HeLa cell lines and intransiently transfected cells, where the effect of the geneticbackground is generally considered negligible, given thestoichiometry of the high level of expression of thetransgene. These observations pointed to Sgk1 as amolecule that is limiting and necessary for the proteinexpression of p53. In fact the expression of p53 is increased
by the inhibition of the endogenous kinase by both Sgk1-specific RNA silencing and dominant negative technology.On the contrary the expression of p53 was decreased by theover-expression of exogenous kinase.
Ubiquitylation experiments in intact HeLa cell linesconfirmed the results obtained in vitro and in intact in RKOcells. Wild-type Sgk1 over-expression appeared to beassociated with mono- and polyubiquitylation of p53.Conversely, in cells expressing the dominant negativemutant of Sgk1, p53 ubiquitylation was greatly reduced.
The overall result of the Sgk1-dependent activation ofMDM2 is expected to be decreased in the half-life of p53.Accordingly, in our stably transfected HeLa cells, the half-life of p53 was clearly shortened in Sgk1 WT cells, whereasit was clearly prolonged in Sgk1 D222A DN cells, whencompared with the empty vector cells, thus confirming thefunctional consequences of the hyper-expression of Sgk1 orthe inhibition of the endogenous kinase.
Finally the findings were confirmed in vivo, in tumorsobtained in BALB/c mice 17 days after the subcutaneousinoculation of the cells expressing the dominant negativemutant of Sgk1 and in liver extracts from MUP1-TAD222A DN Sgk1 double transgenic mice characterized bytissue-specific and inducible expression of the transgene.We must here report that, despite the fundamental role ofSgk1 in cell-cycle progression, our MUP1-TA D222A DNSgk1 double transgenic mice have virtually no phenotype.This is not at all surprising if we consider that even inSGK1 knockout mice the phenotype is mild [46]. Appar-ently, Sgk1 plays a critical role in cancer cells with up-regulated Sgk1 but can be circumvented in normalproliferating tissues. Mice that either are made knockoutor harbor null mutations for cancer-related genes do notindeed always show a phenotype [47]. The reproducibilityof the observation in different settings proves that the effectof Sgk1 on the expression of p53 can have a generalmeaning, not limited to a specific cell line or a singlemethodology. The importance of p53 in Sgk1 signaling wasfurther emphasized by colony assays, a tool designed tostudy the overall result of both proliferation and survival.The increase in the expression of wild-type Sgk1 wasassociated with an enhanced clonogenic potential in eachcell line expressing wild-type p53, both virus-transformed(HeLa cells and Hek293A) and not virus-transformed(RKO cells). In these cells, the inhibition of the endogenousSgk1 by a dominant negative mutant greatly reduced theclonogenic potential, indicating that Sgk1 is limiting andnecessary for the full expression of the clonogenicphenotype in these cancer cells. On the other hand,inhibition of the endogenous Sgk1 had no effect on theclonogenic potential of cells that did not express p53(HI299 cells) showing that the clonogenic potentialregulated by Sgk1 may depend on the expression of p53.
1236 J Mol Med (2009) 87:1221–1239

Moreover, in these cells, the increased expression of Sgk1seemed to decrease the clonogenic potential, suggestingthat Sgk1 might activate different pathways leading togrowth inhibition, in the absence of p53. As a matter of factSgk1 has been demonstrated by others to inhibit Rafsignaling, thus down-regulating MAP and MAP kinase-dependent proliferation [48].
FACS analysis of our stably transfected cells wasparticularly informative. When compared with the emptyvector control cells, the number of hypo-diploid cells,commonly identified as apoptotic cells, was significantlydecreased in Sgk1WT cells, whereas it was significantlyincreased in Sgk1 DN D222A cells, a result that wasconsistent with what we have previously shown in differentcell lines [10]. Sgk1WT cells showed a significant increasein the relative percentage in the number of cells in G2/M,whereas in Sgk1 DN D222A cells, this percentage wassignificantly decreased together with the expression ofcyclin B1, an important marker for G2/M. Cell-cycleanalysis of synchronized cells demonstrated that theexpression of a functional Sgk1 was essential for cell-cycle progression. In fact, the inhibition of the endogenousSgk1 by the dominant negative mutant was associated witha delay in the entrance to G2/M and an increase in thenumber of apoptotic cells. This is, per se, a novelobservation since both Sgk1 and Akt have already beendescribed as proteins involved in the transduction ofsurvival signals, but neither of them had been associatedwith regulation of the cell-cycle progression.
Interestingly, over-expression of either wild-type Sgk1 ordominant negative mutant had significant consequences onthe cellular phenotype. Compared with the empty vectorcontrol cells, the Sgk1 WT cells appeared swollen, with alarge cytoplasm that we interpreted in the light of the abilityof Sgk1 to regulate a wide variety of Na+ coupledtransporters [9], which are expected to swell the cells[49]. The Sgk1 DN D222A cells were small, with a highnuclear cytoplasmic ratio and several contacts betweenneighboring cells, like an epithelial monolayer.
As a matter of fact, the expression of mesenchymalmarkers was found to be increased in Sgk1 WT cells,whereas the expression of epithelial markers was increased,instead, in the Sgk1 DN D222A cells. This observation wasconfirmed both by immunofluorescence and Westernblotting that support a crucial role of Sgk1 in EMT, atleast in HeLa cells. Sgk1 was limiting for EMT since theexpression of mesenchymal markers was increased in cellsover-expressing the kinase. Sgk1 was also essential forEMT since the expression of epithelial markers wasincreased when the endogenous kinase was inhibited bythe dominant negative mutant.
The loss of epithelial markers like α-catenin and ZO-1,necessary for monolayer adhesion and cellular polarization,
together with the increase in the expression of mesenchy-mal markers, like vimentin, can be interpreted in the light ofepithelial de-differentiation and transformation, a result thatwas consistent with the increased expression of Sgk1detected in several epithelial tumors [50, 51].
Taken together, our data allowed us to formulate a modelby which Sgk1 activates MDM2 by phosphorylation, thusdriving p53 to proteosomal degradation. According to thismodel, a novel auto-regulative feedback links Sgk1, p53,and MDM2, whereby p53 exerts a transcriptional regulationof Sgk1, whereas MDM2 and Sgk1 down-regulate theexpression of p53 by means of MDM2-mediated ubiquity-lation. Since Sgk1 is also regulated by mTOR [3] that isinvolved in a p53/MDM2 cross talk [52, 53], it is possiblethat this regulatory loop is part of a more complicatednetwork that includes mTOR, MDM2, and p53.
Acknowledgments We thank the American Journal Experts for theeditorial revision of the manuscript. This work was supported by theFondazione Carical, Cofin 2005/068017_004, Interlink II04C0G4EM,Cofin 2006/prot. 2006065339_005, and Firb 2001/RBNE01724C_007.Rosario Amato was partially supported by the Fondazione “Lilli Funaro”.
Conflict of interest statement The authors declare that they have noconflict of interests.
References
1. Perrotti N, He RA, Phillips SA, Haft CR, Taylor SI (2001)Activation of serum- and glucocorticoid-induced protein kinase(Sgk) by cyclic AMP and insulin. J Biol Chem 276:9406–9412
2. Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL (1993)Characterization of sgk, a novel member of the serine/threonineprotein kinase gene family which is transcriptionally induced byglucocorticoids and serum. Mol Cell Biol 13:2031–2040
3. Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R,Slingerland JM (2008) mTOR-Raptor binds and activates SGK1to regulate p27 phosphorylation. Mol Cell 30:701–711
4. Faletti CJ, Perrotti N, Taylor SI, Blazer-Yost BL (2002) Sgk: anessential convergence point for peptide and steroid hormoneregulation of ENaC-mediated Na+ transport. Am J Physiol CellPhysiol 282:C494–C500
5. Busjahn A, Aydin A, Uhlmann R, Krasko C, Bahring S, SzelesteiT, Feng Y, Dahm S, Sharma AM, Luft FC, Lang F (2002) Serum-and glucocorticoid-regulated kinase (SGK1) gene and bloodpressure. Hypertension 40:256–260
6. von Wowern F, Berglund G, Carlson J, Mansson H, Hedblad B,Melander O (2005) Genetic variance of SGK-1 is associated withblood pressure, blood pressure change over time and strength ofthe insulin-diastolic blood pressure relationship. Kidney Int68:2164–2172
7. Huang DY, Boini KM, Osswald H, Friedrich B, Artunc F, UllrichS, Rajamanickam J, Palmada M, Wulff P, Kuhl D, Vallon V, LangF (2006) Resistance of mice lacking the serum- andglucocorticoid-inducible kinase SGK1 against salt-sensitive hy-pertension induced by a high-fat diet. Am J Physiol Renal Physiol291:F1264–F1273
8. Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD(2001) Glucocorticoid receptor-mediated protection from apopto-
J Mol Med (2009) 87:1221–1239 1237

sis is associated with induction of the serine/threonine survivalkinase gene, sgk-1. J Biol Chem 276:16649–16654
9. Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N,Vallon V (2006) (Patho)physiological significance of the serum-and glucocorticoid-inducible kinase isoforms. Physiol Rev286:1151–1178. Review
10. Amato R, Menniti M, Agosti V, Boito R, Costa N, Bond HM,Barbieri V, Tagliaferri P, Venuta S, Perrotti N (2007) IL-2 signalsthrough Sgk1 and inhibits proliferation and apoptosis in kidneycancer cells. J Mol Med 85:707–721
11. Iwata S, Nomoto M, Morioka H, Miyata A (2004) Geneexpression profi l ing in the midbrain of str iatal 6-hydroxydopamine-injected mice. Synapse 51:279–286
12. Simon P, Schneck M, Hochstetter T, Koutsouki E, Mittelbronn M,Merseburger A, Weigert C, Niess A, Lang F (2007) Differentialregulation of serum- and glucocorticoid-inducible kinase 1(SGK1) splice variants based on alternative initiation of transcrip-tion. Cell Physiol Biochem 20:715–728
13. Huang M, Kamasani U, Prendergast GC (2006) RhoB facilitatesc-Myc turnover by supporting efficient nuclear accumulation ofGSK-3. Oncogene 25:1281–1289
14. Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ,Trizna W, Hammond M, Patterson JR, Thompson SK, Kazmin D,Norris JD, McDonnell DP (2008) Development of a small-molecule serum- and glucocorticoid- regulated kinase-1 antagonistand its evaluation as a prostate cancer therapeutic. Cancer Res68:7475–7483
15. Papadopoulou N, Papakonstanti EA, Kallergi G, AlevizopoulosK, Stournaras C (2009) Membrane androgen receptor activation inprostate and breast tumor cells: molecular signaling and clinicalimpact. IUBMB Life 61:56–61
16. Luft FC (2007) SGK1 survival through various lives may save usall. J Mol Med 85:657–659
17. You H, Jang Y, You-Ten AI, Okada H, Liepa J, Wakeham A,Zaugg K, Mak TW (2004) p53-dependent inhibition of FKHRL1in response to DNA damage through protein kinase SGK1. ProcNatl Acad Sci U S A 01:14057–14062
18. Cornez I, Creppe C, Gillard M, Hennuy B, Chapelle JP, DejardinE, Merville MP, Close P, Chariot A (2008) Deregulated expressionof pro-survival and pro-apoptotic p53-dependent genes uponElongator deficiency in colon cancer cells. Biochem Pharmacol75:2122–2134
19. Hollstein M, Sidransky D, Vogelstein B, Harris CC (1991) p53mutations in human cancers. Science 253:49–53
20. Jimenez GS, Khan SH, Stommel JM, Wahl GM (1999) p53regulation by post translational modification and nuclear retentionin response to diverse stresses. Oncogene 18:7656–7665
21. Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Wei Gu (2003)Mono- versus polyubiquitination: differential control of p53 fateby Mdm2. Science 302:1972–1975
22. Midgley CA, Lane DP (1997) p53 protein stability in tumour cellsis not determined by mutation but is dependent on Mdm2 binding.Oncogene 15:1179–1189
23. Moll UM, Petrenko O (2003) The MDM2–p53 interaction. MolCancer Res 1:1001–1008. Review
24. Roth J, Dobbelstein M, Freedman DA, Shenk T, Levine AJ (1998)Nucleo-cytoplasmic shuttling of the mdm2 oncoprotein regulatesthe levels of the p53 protein via a pathway used by the humanimmunodeficiency virus rev protein. EMBO J 17:554–564
25. Chen Z, Knutson E, Wang S, Martinez LA, Albrecht T (2007)Stabilization of p53 in human cytomegalovirus-initiated cells isassociated with sequestration of MDM2 and decreased p53ubiquitination. J Biol Chem 282:29284–29295
26. Li Y, Jenkins CW, Nichols MA, Xiong Y (1994) Cell cycleexpression and p53 regulation of the cyclin-dependent kinaseinhibitor p21. Oncogene 9:2261–2268
27. Taylor WR, Stark GR (2001) Regulation of the G2/M transitionby p53. Oncogene 20:1803–1815
28. Soengas MS, Alarcón RM, Yoshida H, Giaccia AJ, Hakem R,Mak TW, Lowe SW (1999) Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 284:156–159
29. Porrello A, Cerone MA, Coen S, Gurtner A, Fontemaggi G,Cimino L, Piaggio G, Sacchi A, Soddu S (2000) p53 regulatesmyogenesis by triggering the differentiation activity of pRb. J CellBiol 6:1295–1304
30. Cam H, Griesmann H, Beitzinger M, Hofmann L, Beinoraviciute-Kellner R, Sauer M, Hüttinger-Kirchhof N, Oswald C, Friedl P,Gattenlöhner S, Burek C, Rosenwald A, Stiewe T (2006) p53family members in myogenic differentiation and rhabdomyosar-coma development. Cancer Cell 10:281–293
31. Mies F, Spriet C, Heliot L, Sariban-Sohraby S (2007) EpithelialNa channel stimulation by n-3 fatty acids requires proximity to amembrane-bound A-kinase-anchoring protein complexed withprotein kinase A and phosphodiesterase. J Biol Chem282:18339–18347
32. Maiyar AC, Phu PT, Huang AJ, Firestone GL (1997) Repressionof glucocorticoid receptor transactivation and DNA binding of aglucocorticoid response element within the serum/glucocorticoid-inducible protein kinase (sgk) gene promoter by the p53 tumorsuppressor protein. Mol Endocrinol 11:312–329
33. Rinaldo C, Prodosmo A, Mancini F, Iacovelli S, Sacchi A, MorettiF, Soddu S (2007) MDM2-regulated degradation of HIPK2prevents p53Ser46 phosphorylation and DNA damage-inducedapoptosis. Mol Cell 25:739–750
34. Treier M, Staszewski LM, Bohmann D (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the deltadomain. Cell 78:787–798
35. Manickan E, Satoi J, Wang TC, Liang TJ (2001) Conditionalliver-specific expression of simian virus 40 T antigen leads toregulatable development of hepatic neoplasm in transgenic mice. JBiol Chem 276:13989–13994
36. Bloom J, Pagano M (2005) Experimental tests to definitivelydetermine ubiquitylation of a substrate. Methods Enzymol399:249–266
37. Bhushan S, Malik F, Kumar A, Kaur Isher H, Pal Kaur I, Taneja SC,Singh J (2009) Activation of p53/p21/PUMA alliance and disruptionof PI-3/Akt in multimodal targeting of apoptotic signaling cascadesin cervical cancer cells by a pentacyclic triterpenediol from Boswelliaserrata. Mol Carcinog. doi:10.1002/mc.20559
38. Chen F, Chang D, Goh M, Klibanov SA, Ljungman M (2000)Role of p53 in cell cycle regulation and apoptosis followingexposure to proteasome inhibitors. Cell Growth Differ 11:239–246
39. Lober C, Lenz-Stoppler C, Dobbelstein M (2002) Adenovirus E1-transformed cells grow despite the continuous presence oftranscriptionally active p53. J Gen Virol 83:2047–2057
40. Geyer RK, Yu ZK, Maki CG (2000) The MDM2 RING-fingerdomain is required to promote p53 nuclear export. Nat Cell Biol2:569–573
41. Lee JH, Kim HS, Lee SJ, Kim KT (2007) Stabilization andactivation of p53 induced by Cdk5 contributes to neuronal celldeath. J Cell Sci 120:2259–2271
42. Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D,Hemmings BA (2004) Stabilization of Mdm2 via decreasedubiquitination is mediated by protein kinase B/Akt-dependentphosphorylation. J Biol Chem 279:35510–35517
43. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC (2001)HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2phosphorylation. Nat Cell Biol 3:973–982
44. Weber HO, Ludwig RL, Morrison D, Kotlyarov A, Gaestel M,Vousden KH (2005) HDM2 phosphorylation by MAPKAP kinase2. Oncogene 24:1965–1972
1238 J Mol Med (2009) 87:1221–1239

45. Ashcroft M, Ludwig RL, Woods DB, Copeland TD, Weber HO,MacRae EJ, Vousden KH (2002) Phosphorylation of MDM2 byAkt. Oncogene 21:1955–1962
46. Wulff P, Vallon V, Huang DY, Völkl H, Yu F, Richter K, JansenM, Schlünz M, Klingel K, Loffing J, Kauselmann G, Bösl MR,Lang F, Kuhl D (2002) Impaired renal Na(+) retention in the sgk1-knockout mouse. J Clin Invest 110:1263–1268
47. Bera TK, Pastan I (2000) Mesothelin is not required fornormal mouse development or reproduction. Mol Cell Biol20:2902–2906
48. Zhang BH, Tang ED, Zhu T, Greenberg ME, Vojtek AB, Guan KL(2001) Serum- and glucocorticoid-inducible kinase SGK phos-phorylates and negatively regulates B-Raf. J Biol Chem276:31620–31626
49. Waldegger S, Barth P, Raber G, Lang F (1997) Cloning andcharacterization of a putative human serine/threonine proteinkinase transcriptionally modified during anisotonic and isotonicalterations of cell volume. Proc Natl Acad Sci 94:4440–4445
50. Chung EJ, Sung YK, Farooq M, Kim Y, Im S, Tak WY, HwangYJ, Kim YI, Han HS, Kim JC, Kim MK (2002) Gene expressionprofile analysis in human hepatocellular carcinoma by cDNAmicroarray. Mol Cell 14:382–387
51. Leong ML, Maiyar AC, Kim B, O'Keeffe BA, Firestone GL(2003) Expression of the serum- and glucocorticoid-inducibleprotein kinase, Sgk, is a cell survival response to multiple types ofenvironmental stress stimuli in mammary epithelial cells. J BiolChem 278:5871–5882
52. Kojima K, Shimanuki M, Shikami M, Samudio IJ, Ruvolo V,Corn P, Hanaoka N, Konopleva M, Andreeff M, Nakakuma H(2008) The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53induction by Mdm2 inhibition but enhances p53-mediatedmitochondrial apoptosis in p53 wild-type AML. Leukemia22:1728–1736
53. Feng Z, Zhang H, Levine AJ, Jin S (2005) The coordinateregulation of the p53 and mTOR pathways in cells. Proc NatlAcad Sci U S A 102:8204–8209
J Mol Med (2009) 87:1221–1239 1239
Related Documents











