Send Orders for Reprints to [email protected] Combinatorial Chemistry & High Throughput Screening, 2015, 18, 151-187 151 Discovery of Anti-Malarial Agents Through Application of In Silico Studies Mahesh A. Barmade, Prashant R. Murumkar, Mayank Kumar Sharma, Kaushik P. Shingala, Rajani R. Giridhar and Mange Ram Yadav * Pharmacy Department, Faculty of Technology and Engineering, Kalabhavan, The M. S. University of Baroda, Vadodara- 390 001, Gujarat, India Abstract: Among the various parasitic diseases, malaria is the deadliest one. Due to the emergence of high drug resistance to the existing drug candidates there is a global need for development of new drug candidates which will be effective against resistant strains of malaria parasite. In silico molecular modeling approaches have been playing an important role in the discovery of novel lead molecules having antimalarial activity. Present review is an effort to cover all the developments related to the application of computational techniques for the design and discovery of novel antimalarial compounds since the year 2011 onwards. Keywords: Antimalarial agents, drug resistance, docking, homology model, malaria, pharmacophore, QSAR, virtual screening. 1. INTRODUCTION Despite its complete preventability and treatability, malaria is one of the leading causes of mortality and morbidity among the parasitic diseases. As per the latest report of WHO, malaria was responsible for a burden of 207 million cases in 2012 with an estimated 6,27,000 deaths amongst which 4,82,000 were children under the age of five. Due to undeveloped protective immunity, young children are the main target. It is speculated to cause death of 1300 children every day. The African region contributed the highest (90%) number of all malaria deaths and it is referred as the disease of the poor [1-3]. The main causative agent of malaria is Plasmodium, a genus belonging to the phylum protozoa. Plasmodium has four main species i.e. Plasmodium falciparum, P. vivax, P. malarie and P. ovale which have the potential of inducing malaria disease in humans. Among these species, P. falciparum contributed for 90% of malaria cases worldwide and was responsible for the highest number of malaria deaths annually [1, 4, 5]. The fifth species of Plasmodium i.e. P. Knowlesi is responsible for malarial infection in both humans as well as monkeys in certain areas of Southeast Asia [6]. The transmission of malaria occurs mainly through the bites of infected female anopheles mosquitoes and depends on a number of climatic factors (rain fall patterns, temperature and humidity), and tropical regions are more favorable than temperate ones for its spread [5, 7]. In 1955 WHO launched Global Malaria Eradication Programme using two basic tools i.e. chloroquine (CQ, 1; Fig. 1) for prophylaxis and treatment purposes and dichlorodiphenyltrichloroethane (DDT) for the control of the vector. Suddenly, there was a tremendous increase in malaria burden in many parts of the world *Address correspondence to this author at the Pharmacy Department, Faculty of Technology and Engineering, Kalabhavan, The M. S. University of Baroda, Vadodara- 390 001, Gujarat, India; Tel: +91 265 2434187; Fax: +91 265 2418927; E-mail: [email protected] (especially in Thailand) as soon as Global Malaria Eradication Programme was terminated [8]. More recently in August 2014, with the aim to provide complete information regarding prevention of reintroduction of malaria, WHO regional office for Europe has launched regional framework for prevention of malaria reintroduction and certification of malaria elimination 2014-2020 [9]. The present recommended regimen for the treatment of malaria is about two decades old and almost all of the agents suffer from the drawback of parasitic drug resistance [10]. According to the available literature [4] it has been concluded that for better treatment of malaria infection, drug candidates must have some essential criteria that mainly include potent activity against the asexual form in the blood stage infection, fast acting response, low toxic profile and easy availability. Many drugs are available for the treatment of malaria which are active against different strains of the parasite as depicted in Fig. (1) [11]. Among the various therapeutic agents, quinine (2) obtained from cinchona tree has been known from ancient times as an effective drug against number of Plasmodium species. As mechanism of action it is reported to have hemozoin biocrystallization inhibitory activity. As a blood schizonticidal agent it proved to be less effective and more toxic than chloroquine (1) [12]. Chloroquine belongs to aminoquinoline class of drugs and it was one of the most successful agent responsible for malaria eradication program in the 1950s [11, 13]. It was one of the safest drugs that could be used during pregnancy. However, after ten years of its introduction, emergence of drug resistance has been reported in Southeast Asia, but it is still used as the first line agent in sub-Saharan African regions [14]. Other aminoquinolines such as amodiaquine (3), mefloquine (4) and halofantrine (5) are effective against CQ-resistant strains that proved to be effective antimalarial agents [15]. Pyrimethamine (6) in combination with sulfadoxine (7) was 1875-5402/15 $58.00+.00 © 2015 Bentham Science Publishers

Send Orders for Reprints to [email protected] Discovery of Anti-Malarial Agents Through Application of In Silico Studies
May 06, 2023
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Send Orders for Reprints to [email protected]
Combinatorial Chemistry & High Throughput Screening, 2015, 18, 151-187 151
Discovery of Anti-Malarial Agents Through Application of In Silico Studies
Mahesh A. Barmade, Prashant R. Murumkar, Mayank Kumar Sharma, Kaushik P. Shingala, Rajani R. Giridhar and Mange Ram Yadav*
Pharmacy Department, Faculty of Technology and Engineering, Kalabhavan, The M. S. University of Baroda, Vadodara- 390 001, Gujarat, India
Abstract: Among the various parasitic diseases, malaria is the deadliest one. Due to the emergence of high drug resistance to the existing drug candidates there is a global need for development of new drug candidates which will be effective against resistant strains of malaria parasite. In silico molecular modeling approaches have been playing an important role in the discovery of novel lead molecules having antimalarial activity. Present review is an effort to cover all the developments related to the application of computational techniques for the design and discovery of novel antimalarial compounds since the year 2011 onwards.
Keywords: Antimalarial agents, drug resistance, docking, homology model, malaria, pharmacophore, QSAR, virtual screening.
1. INTRODUCTION
Despite its complete preventability and treatability, malaria is one of the leading causes of mortality and morbidity among the parasitic diseases. As per the latest report of WHO, malaria was responsible for a burden of 207 million cases in 2012 with an estimated 6,27,000 deaths amongst which 4,82,000 were children under the age of five. Due to undeveloped protective immunity, young children are the main target. It is speculated to cause death of 1300 children every day. The African region contributed the highest (90%) number of all malaria deaths and it is referred as the disease of the poor [1-3]. The main causative agent of malaria is Plasmodium, a genus belonging to the phylum protozoa. Plasmodium has four main species i.e. Plasmodium falciparum, P. vivax, P. malarie and P. ovale which have the potential of inducing malaria disease in humans. Among these species, P. falciparum contributed for 90% of malaria cases worldwide and was responsible for the highest number of malaria deaths annually [1, 4, 5]. The fifth species of Plasmodium i.e. P. Knowlesi is responsible for malarial infection in both humans as well as monkeys in certain areas of Southeast Asia [6]. The transmission of malaria occurs mainly through the bites of infected female anopheles mosquitoes and depends on a number of climatic factors (rain fall patterns, temperature and humidity), and tropical regions are more favorable than temperate ones for its spread [5, 7]. In 1955 WHO launched Global Malaria Eradication Programme using two basic tools i.e. chloroquine (CQ, 1; Fig. 1) for prophylaxis and treatment purposes and dichlorodiphenyltrichloroethane (DDT) for the control of the vector. Suddenly, there was a tremendous increase in malaria burden in many parts of the world
*Address correspondence to this author at the Pharmacy Department, Faculty of Technology and Engineering, Kalabhavan, The M. S. University of Baroda, Vadodara- 390 001, Gujarat, India; Tel: +91 265 2434187; Fax: +91 265 2418927; E-mail: [email protected]
(especially in Thailand) as soon as Global Malaria Eradication Programme was terminated [8]. More recently in August 2014, with the aim to provide complete information regarding prevention of reintroduction of malaria, WHO regional office for Europe has launched regional framework for prevention of malaria reintroduction and certification of malaria elimination 2014-2020 [9]. The present recommended regimen for the treatment of malaria is about two decades old and almost all of the agents suffer from the drawback of parasitic drug resistance [10]. According to the available literature [4] it has been concluded that for better treatment of malaria infection, drug candidates must have some essential criteria that mainly include potent activity against the asexual form in the blood stage infection, fast acting response, low toxic profile and easy availability. Many drugs are available for the treatment of malaria which are active against different strains of the parasite as depicted in Fig. (1) [11]. Among the various therapeutic agents, quinine (2) obtained from cinchona tree has been known from ancient times as an effective drug against number of Plasmodium species. As mechanism of action it is reported to have hemozoin biocrystallization inhibitory activity. As a blood schizonticidal agent it proved to be less effective and more toxic than chloroquine (1) [12]. Chloroquine belongs to aminoquinoline class of drugs and it was one of the most successful agent responsible for malaria eradication program in the 1950s [11, 13]. It was one of the safest drugs that could be used during pregnancy. However, after ten years of its introduction, emergence of drug resistance has been reported in Southeast Asia, but it is still used as the first line agent in sub-Saharan African regions [14]. Other aminoquinolines such as amodiaquine (3), mefloquine (4) and halofantrine (5) are effective against CQ-resistant strains that proved to be effective antimalarial agents [15]. Pyrimethamine (6) in combination with sulfadoxine (7) was
1875-5402/15 $58.00+.00 © 2015 Bentham Science Publishers

152 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
successfully used in CQ-resistant P. falciparum strains as an alternative. Pyrimethamine (6) acts by inhibiting dihydro-folate reductase enzyme which plays a major role in biosynthesis of purines and pyrimidines and ultimately stops the DNA replication, cell division and reproduction cascade.
Sulfadoxine (7) is reported to act through inhibition of dihydropteroate synthetase, essential in tetrahydrofolate synthesis pathway of the parasite [16-18]. Atovaquone (8) a therapeutic agent of benzoquinone class is mainly used as prophylactic agent for travellers. A combination of
OOH
OO
Me
Me
H
H
OMe
N
ClN
N
Me
H2N
NH2
CF3
N CF3
OHNH
CF3
Cl
Cl
OH
N(C4H9)2 OMe
NN
OMe
NHSO
OH2N
OH O
O
ClCl
NHNNMe
Me NH2 NH2
(1) (2) (3) (4)
(5) (6) (7)
(8) (9) (10)
(14) (15)
O
OHMe
H
Me
OO
O
O
O
OH
Me
H O
OHMe
H
Me
OO
OMeMe
H
(11) (12) (13)
Chloroquine Quinine Amodiaquine Mefloquine
Halofantrine Pyrimethamine Sulfadoxine
Atovaquone Proguanil Artemisinin
Artesunate Artemether Lumefantrine
Tetracycline Doxycycline
HN
MeN(Et)2
Cl N
HON
N
HN
Cl
N(Et)2
OH
MeO
OH
NH2
O
N
OH O
MeMe
OH
HO Me
OH O O O
OH
O
NH2
OH OH
Me NMe
OH
OH
Me
HON(C4H9)2
Cl
Cl Cl
CH2
H
Fig. (1). Marketed antimalarial agents.

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 153
atovaquone (8) with proguanil (9) minimized the resistance occurring in case of atovaquone monotherapy [19]. Recently, parasitic resistance to artemisinin (10) has also been reported in Cambodia, Myanmar, Thailand and Vietnam [20]. As per WHO conclusion artemisinin resistance is a result of oral artemisinin-monotherapy which was discontinued as soon as resistance was observed. WHO recommended alternative approaches for uncomplicated malaria caused by P. falciparum that are artemisinin-based combination therapies (ACTs) e.g. artesunate (11)+amodiaquine, artesunate+meflo-quine, artemether (12)+lumefantrine (13) and artesunate and sulfadoxine/pyrimethamine. The non-artemisinin based com-binations mainly include sulfadoxine-pyrimethamine (SP), SP+chloroquine; SP+amodiaquine; SP+ mefloquine and quinine+tetracycline (14)/doxycycline (15) [1, 21, 22]. Malaria chemotherapy lacks vaccine approach and Malaria Vaccine Technology Roadmap (MVTR) is playing an important role in development of malaria vaccines. A mosquirix (RTS, S) vaccine against P. falciparum currently is under phase III clinical trials [23]. A brief discussion on these therapeutic agents has concluded that complete eradication of malaria is not a simple task and we needs to look forward for the development of new antimalarial agents. In light of the emerging resistance to the existing antimalarial drugs there is an urgent need to develop new antimalarial agents and to necessitate validation of newer chemotherapeutic targets for attacking the parasite. The potential chemotherapeutic targets have been classified in three main categories: 1) The Plasmodium digestive vacuole which includes mainly hemoglobin digestion and heme detoxification along with some antioxidant defence mechanism, and drug accumulation and extrusion 2) Macromolecular and metabolite synthesis cascade e.g. glycolysis and phospholipid metabolism, and tubulin assembly 3) Trafficking, drug transporter and signaling cascades for membrane processes. Different targets along with their location in the cell, enzymes involved and targeting drug molecules have been precisely discussed in literature [24-27]. In our laboratory, we have also been making efforts to design and develop some novel antimalarial agents [28, 29].
1.1. Computer Aided Drug Design (CADD)
CADD is always a point of discussion in the discovery process of novel leads having potential bioactivity. It is aimed to predict sort of binding involved between a drug candidate and the target and uses computational chemistry to enhance, discover and study lead molecules of biological interest [30-32]. The discovery of anhydrase inhibitor, dorzolamide in 1995 was the first successful attempt of CADD in the drug discovery process [33]. In the past few years HIV/AIDS has killed huge number of people infected with human immunodeficiency virus. Quite often many people infected with this deadly virus have to depend on initially developed drugs like Relenza (Zanamivir) and Viracept (Nelfinavir) which were developed using application of CADD [34, 35]. Other examples of therapeutic agents developed using CADD approach are cimetidine, imatinib, enfluritide, zolpidem, zopiclone, probenecid, raltitrexed, amprenavir, raltegravir etc. [32]. Hence, due to early success of CADD in development of lead molecules of
biological interest, it is considered as an integral part of drug discovery process. Now a days almost every area of drug development including nanotechnology, molecular biology and biochemistry is dependent on computational chemistry [36]. On the basis of availability of structure and other information of the target, CADD is broadly classified in two main categories i.e. structure based drug design and ligand based drug design [37]. The structure based drug design depends on the identification of three dimensional structure of the target site based on X-ray crystallography, NMR data or molecular and quantum mechanics calculations, for the designing of lead molecules. Docking is carried out for a molecule which satisfies the geometric constrains with good chemical match. Its binding strength is calculated using scoring functions [38]. Homology modeling plays an important role in identification of unknown structures based on alignment of unknown receptor sequence with template structures through database search [39, 40]. Virtual high-throughput screening approach is one of the most widely chemometric tool of interest to pharmaceutical companies. This allows screening of available library of compounds against a target and identifies the strongly bonded molecules which ultimately are extracted from the available dataset for further evaluation and optimization [41-43]. Ligand based drug design techniques can be used in the absence of crystal structure of the biological target. In this technique knowledge of the structures of the molecules which bind to the biological target is exploited. QSAR is a key chemometric tool used for identification of structural features which tend to affect the biological activity. QSAR model can be developed for compounds of therapeutic use which are responsible for establishing a relationship between biological effect i.e. the activity and the chemistry i.e. the structure [44, 45]. A simple mathematical relationship equation for QSAR can be represented as: Biological activity (BA) = f [Physiochemical and/or structural parameters] Where: o BA is a measure of both binding affinities (Ki, Km)
and inhibitory activities (IC50, ED50) o Physicochemical parameters account for hydrophobicity,
electronic properties, topology and steric effects The availability of trustworthy biological data, choice of suitable descriptors like physico-chemical parameters and application of statistical methods forms the basis for the development of a robust QSAR model. Stepwise multiple linear regression (MLR), Genetic Function Approximation (GFA), Principal Component Analysis (PCA) or genetic PLS are used for development of QSAR models [46]. Development of the best QSAR model requires the use of appropriate set of compounds. There are reports [47-49] indicating that Simplified Molecular Input Line Energy System (SMILES) can be used as similarity criteria for selecting right molecular set for the QSAR studies. SMILES was designed to have good human readability as molecular file format which is most commonly used for storage and retrieval of compounds across multiple computer platforms.

154 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
Pharmacophore mapping, another ligand based drug design approach is used mainly for determination of binding orientation of structurally diverse ligands to a common receptor site. It is also considered as one of the starting points for the development of 3D-QSAR models. It is also used to define the structural features (e.g. hydrophobic center, aromatic rings, hydrogen bond acceptor/donor and cations/anions) of the ligands with similar activity profile. The step wise process for pharmacophore model development starts with selection of a set of molecules, conformational analysis, molecular superimposition and finally validation of the developed pharmacophore model [50, 51]. As far as application of structure and ligand based drug designing techniques are concerned our group has been actively engaged in designing and development of some biologically active analogs i.e. COX-2 inhibitors [52, 53], TACE inhibitors [54-57], Telomerase inhibitors [58, 59] etc. The present review is a humble attempt to cover all chemometric studies related to antimalarial agents from the year 2011 till date, through the application of computer assisted drug design techniques. Following sections review the literature reported CADD studies of antimalarials based upon structure and ligand based drug design techniques.
2. APPLICATION OF STRUCTURE BASED DRUG DESIGN TECHNIQUES FOR THE DISCOVERY OF ANTIMALARIAL AGENTS
2.1. Studies Based on Homology Modeling
Rawat et al. [60] reported the sequence homology and structural analysis of P. vivax plasmepsin 4 (PvPM4). The sequence of PvPM4 showed 100% homology with the isolated P. vivax, collected from seven different geographical regions of India. The theoretical 3D-strucutral model of PvPM4 was constructed having a typical aspartic protease structure which was bi-lobed, compact and had a distinct peptide binding cleft as shown in Fig. (2). It has been found Fig. (2). 3D-model of Indian P. vivax plasmepsin 4 (PvPM4) protein showing the complete active site pocket. Used with permission from Rawat et al., Infect. Genet. Evol., 2011, 11, 924-933 [Copyright© 2011, Elsevier B.V.] (color images are available online).
from Ramachandran plot by Rawat et al. that 99.2% of the amino acid residues of the main chain conformations were observed in the favored or allowed region with an overall G factor of -0.14 indicating that the constructed 3D model was having a good quality molecular geometry. The developed structure also showed more beta sheets as compared to alpha helices indicating a rigid and stable structure under various conditions having the C score of 1.98, expected TM score and RMSD value of 0.48 ± 0.15 and 11.8 ± 4.5, respectively. Inter Pro Scan was applied for conserved domain search analysis of PvPM4 protein which revealed typical aspartic protease like domain structure with a predicted molecular function of aspartic-type endopeptidase activity. As per the authors’ claim this kind of structural studies might open new avenues for designing of new inhibitors and substrate interaction simulations. The 3D-strucutre of ribosomal protein P1 (RPP1) of P. falciparum is still unknown. So, Kumari et al. [61] reported an ab initio model of RPP1 of P. falciparum by using the amino acid sequence retrieved from NCBI database (Accession number AAN35632). In the template selection, BLASTP was carried out against PDB database but no appropriate template was found during this study. The top hit of BLASTP showed 53 % query coverage and 37 % identity. The obtained hits were not aligned with sequence of C-terminal conserved domain of RPP1. So, tertiary structure prediction was carried out by ab initio method. As per the authors’ claim the best model was chosen on the basis of Ramachandran plots generated using PROCHECK and packing score which was calculated using WHATIF consisting of 5 helices as shown in Fig. (3). Further, molecular dynamics simulation was also carried out to examine the conformational variation of the model protein within solvent system. The RMSD of backbone with respect to the modelled structure was calculated, and it was found out that the protein was stable during the simulation period of 30 ns.
Fig. (3). Model of 60S ribosomal protein P1 (RPP1) [organism: P. falciparum] generated by ab initio modeling using I-TASSER server. Used with permission from Kumari et al., J. Theor. Biol., 2014, 343, 113-119 [Copyright© 2013, Elsevier Ltd.] (color images are available online).

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 155
The 3D-homology model with ATP bound and unbound form of PfD0975w (RIO-like kinase in the P. falciparum kinome) was developed using template model of Archaeoglobus fulgidus right open reading frame (AfRIO-2) crystal structure (PDB code: 1TQI, 1ZAO) with MODELLER9v7 program. The quality of the developed model was evaluated through Ramachandran plots that gave residues value below 95% and modelled structure quality factor of 91.667. The structural features for ATP binding and kinase activity is shown in the Fig. (4) depicting presence of N-terminal winged helix domain (1-84) and C-terminal kinase domain (148-275) along with some other residues. The overall structural features agreed highly with those of AfRIO-2. The developed 3D model was claimed to superimpose on ATP-bound structure of AfRIO-2 to highlight the interactions of ATP and nucleotide binding within PfD0975w. These results showed the presence of hydrophobic residues A171, V177 and F236, supporting the ribose ring, and A157, I161 and M186 interacted with adenine ring. The negatively charged P04 group remained on positively-charged tunnel and interacted with I148, S150, R152, P237 and other nearby residues [62].
Fig. (4). 3D-model of PFD0975w with structural domains. Used with permission from Trivedi et al., Chem. Biol. Drug Des., 2012, 79, 600-609 [Copyright© 2012, John Wiley & Sons Als.] (color images are available online).
2.2. Studies Based on Docking
The bioactive interactions between aspartic protease enzyme (plasmepsin-II) with ditert.amine inhibitors have been reported by Ojha and Roy [63]. This ligand-receptor interaction study explored the presence of non-polar and polar amino acid residues into the active site of aspartic protease enzyme. With the help of docking studies the authors have concluded that the most active compound (16) shows three hydrogen bonding, one with Val170 and remaining two with Ser218 which made the molecule the most active one. Although the most active compound forms three bumps but these were nullified due to the presence of three strong hydrogen bonds. On the other hand the poorly active analog (17) forms only one hydrogen bond with Gly216 and has 10 bumps which clearly indicated the dominance of hydrogen bonding over bumps as was the case with the most active compound. In this study, the authors have concluded that presence of proper substituent on biphenyl ring was essential for designing of novel antimalarial analogs of di-tertiary amine derivatives. Comparative docking analysis of a series of 95 nucleoside analogs as inhibitors of P. falciparum deoxy-uridine-5’-triphosphate nucleotidohydrolase (PfdUTPase), using three different chemometric softwares were carried out by Ojha and Roy [64]. The docking studies of the most active compound (18) using Discovery Studio 2.1 (Fig. 5a), Maestro 9.3 (Fig. 5b) and MOE (Fig. 5c) showed that -NH and keto groups of uracil moiety formed hydrogen bonding with Ile117 and Asn103 amino acid residues and π -π stacking with Phe46. The docking results of the three different softwares revealed that the least active compound (19) formed hydrogen bonds with Ile117 while the uracil ring functionalities (-NH and keto) did not form hydrogen bonds with Ile117 and Asn103 [Discovery studio 2.1; (Fig. 5d)], Phe464 [Maestro 9.3; (Fig. 5e)] and Ile74 [MOE; (Fig. 5f)] along with the absence of π -π interactions due to changed orientation. The docking studies also pointed out the selective interaction of compounds for Phe46 which was absent in humans and showed presence in the parasite. On the basis of structural similarity indices between cytochrome bc1 inhibitors (atovaquone and stigmatellin) with quinolone derivatives, Villalobos et al. [65] carried out a study in such a way that quinolone derivatives would interact with the same binding site as that of cytochrome bc1 inhibitors. Reliability of the docking approach was validated by i) extracting co-crystallized stigmatellin from the complex and redocking within cytochrome bc1 enzyme
O
N
O OMe
Me
N
O
N
Me
N(C4H9)2
N
N
N44
(16) (17)

156 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
N
NH
O
ONH(Ph)3C
HN
N
O
OO
O
O
H2NO2SHN
HO (18) (19)
ii) optimization of 3D structure of stigmatellin through PM6 and docking within cytochrome bc1 enzyme. The docked (Fig. 6a-c) three most active compounds (20-22) showed similar orientation as that of crystalline ligand (Fig. 6d) and formed hydrophobic interactions through long linear alkyl chain with hydrophobic tunnel of amino acids. The -NH group of quinolone ring formed hydrogen bond with the side chain of His181 that played a key role in electron transport. The two least active compounds (23, 24) among the series, showed lack of interaction with hydrophobic tunnel due to the absence of long linear alkyl chain.
Fig. (5). Docked conformations of the most active compound (18; a-c) and the least active compound (19; d-f) using Discovery studio 2.1, Maestro 9.3 and MOE, respectively. Used with permission from Ojha and Roy Comb. Chem. High Throughput Screen., 2013, 16, 739-757 [Copyright© 2014, Bentham Science Publisher] (color images are available online).
(a) (b)
(c) (d)
(e) (f)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 157
Fig. (6). Docked poses of the three most active compounds (20) (a, green), (21) (b, pink) and (22) (c, blue), superimposed compounds (20-22) and stigmatellin (d, purple). Used with permission from Villalobos et al., J. Mol. Graph. Model., 2013, 46, 105-124 [Copyright© 2013, Elsevier Inc.] (color images are available online).
NH
O
R3
R4 Me
R1
R2
Compounds R1 R2 R3 R4
(20) Heptyl H H H
(21) Nonyl H H H
(22) Hept-2-yne H Cl OCH3
(23) Benzyl H H H
(24) Phenyl OCH3 H H(20-24)
(a) (b)
(c) (d)

158 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
Adane et al. [66] designed a class of novel S-benzylated guanylthiourea derivatives as potential PfDHFR inhibitors using in silico methods such as molecular electrostatic potentials and molecular docking. The docking studies were carried out using X-ray crystal structures of wild type (PDB: 1J3I and 3DGA) and quadruple mutant type (PDB: 1J3K and 3DG8) PfDHFR enzymes. Similar docking results were obtained for guanylthiourea moiety as those previously obtained from pyrimethamine, WR99210 and biguanide derivatives. Hydrogen bonds were formed by the guanylthiourea moiety with Asp54, Leu164 and Ile14, and hydrophobic interactions were observed with Phe58 and Phe116 in the active site of PfDHFR enzyme. To prevent potential steric clashes with the amino acid Asn108 in the active site of the mutant PfDHFR enzyme, it was observed that flexibility was maintained by introducing linker unit with 1 to 3 carbon atoms between the hydrophobic aromatic tail and the guanylthiourea moiety. The highest G-score was obtained for compound (25) having additional hydrophobic interactions with Phe116. Bi-substituted guanylthiourea (26) showed a G-score comparable to that of standard compound WR99210. Additional interactions of compound (26) observed with Ser111, Lys49 and Trp48 might be due to six bonds present between the two guanylthiourea moieties, facilitating strong interactions as shown in Fig. (7b). Likewise, compound (27) formed hydrogen bonding with Asp54 and exhibited hydrophobic interactions with Phe58 as shown in Fig. (7b). The hydrophobic pocket of the enzyme
was occupied by the bulkier iodine group attached to the meta-position of the benzyl ring in compound (27). Gahtori et al. [67] reported docking studies for chlorophenylthiazolyl-1,3,5-triazines into three different sites viz. α, β and γ of PfDHFR-TS. The α and β sites were responsible for the interdomain interaction between effective DHFR inhibitors such as pyrimethamine and WR-99210, and the last γ site was the largest cavity present inside the receptor. The main aim of this study was to identify different binding patterns of the chlorophenylthiazolylamino-1,3,5-triazine hybrid analogs in different catalytic sites and also to find out the structural requirements around the basic skeleton. In case of site α, compound (28) showed the best H-bond score (-12.3326) with the minimum interatomic H-bond distance (2.1678 Å). Nitrogen of thiazole ring formed H-bond with Arg122 indicating that Arg122 is responsible for the only interaction in site α. Orientation of compound (28) in binding site α is shown in Fig. (8a). In case of site β, due to the least interatomic distance for H-bond formed by oxygen of Ile164 residue with compound (29) it was found to have four different poses with dissimilar H-bond scoring viz. -11.1078 to -9.6169. This interaction helped the compound to perfectly fit within the receptor through different conformations. Compound (29) was reported to bind with minimum distance of 1.8790 Å to carbonyl oxygen of Ile164 and it showed comparatively weak H-bonding with Cys15. It has also been observed by the authors that the
S N
NH
NH2
NH2MeO
OF3C
S N
NH
NH2
NH2
SN
NH
H2N
NH2
S N
NH
NH2
NH2
I
(25) (26) (27)
Fig. (7). Molecular docking in the active site of quadruple mutant PfDHFR enzyme (a) Guanylthiourea derivative (25) showing H-bond interaction with Trp48, Lys49, Asp54 and Ser111; (b) Compound (27) showing interactions with Asp54 and Phe58. Used with permission from Adane et al., Bioorg. Med. Chem. Lett., 2014, 24, 613-617 [Copyright© 2013, Elsevier Ltd] (color images are available online).
(a) (b)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 159
diethyl fragment was an indispensable trait for the compounds to bind effectively to the binding site β. Amine as the connecting bridge between 1,3,5-triazine and the terminal substituent showed more affinity towards the binding site than the mercapto analogs. Butyl group as the terminal substituent attached to the triazine ring was found to be more efficient than the allyl group. 4-Chloro substituent on phenyl ring was found to reveal higher affinity than the 3,4-dichloro substituent although the 3,4-dichloro substituent formed good contacts with Asp54 and Arg122. In case of γ binding site, minimum interatomic distance (1.8919Å) for H-bond was formed by compound (30) with carbonyl oxygen of His323 in chain D. For hydrophobic interactions, pi-pi interaction was also considered for ligand binding to residues such as Ile331 at the right distance of 2.00 Å to chain D. The heterocyclic ring of the ligand was also compatible with the aromatic moieties like Phe, Tyr etc. to have pi-pi interactions in compound (31) with a minimum distance of 3.90Å to chain C of Tyr322 (Fig. 8b). From this study the authors have concluded that residues such as Asp54, Arg 122, Ile164 (chain A), His 323 (Chain C), His 323 and Lys 359 (chain D)
were involved in making contacts between PfDHFR-TS and the ligands. Kalani et al. [68] isolated and characterized 18β-glycyrrhetinic acid (GA, 32) as a major constituent from the roots of G. glabra as an anti-malarial agent. Molecular docking studies were performed by the authors using the structure of P. falciparum lactate dehydrogenase (PfLDH) (PDB: 1CEQ). GA (32) showed moderate docking (LibDock) score of 71.18 whereas the standard anti-malarial drug chloroquine (CQ, 1) showed a docking score of 131.15 (Fig. 9a). GA formed a single hydrogen bond with Thr101 (Fig. 9b), whereas CQ formed three H-bonds with Phe100, Asn108 and Asn140. The residues of NADH binding site on PfLDH interacted with both GA and CQ. Superimposition of co-crystallized NADH and docked GA (32) and CQ (1) on PfLDH showed similar binding affinities. Docking and superimposition results revealed that GA was bound well within the NADH binding pocket of PfLDH, but exhibited slightly less binding affinity than the standard CQ.
N N
N
N(C2H5)2
SHN
S
N
Cl
ClC4H9
N N
N
N(C2H5)2
HN
HN
S
N ClC4H9
(28) (29)
N N
N
NH2
SHN
S
N
Cl
ClO
N N
NHN
HN
S
N ClO
N(C2H5)2 (30) (31)
Fig. (8). (a) Predicted binding mode of compound (28) in the active site α of PfDHFR-TS, (b) pi-pi interactions for compound (31). Used with permission from Gahtori et al., Exp. Parasitol., 2012, 130, 292-299 [Copyright© 2012, Elsevier Inc.] (color images are available online).
(a) (b)

160 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
Me
OH
O
O
HO
Me Me H
Me
Me
Me
MeH
(32)
Chadha et al. [69] investigated inclusion of artesunate in the cavities of β -cyclodextrin (β-CD) and its methyl and hydroxypropyl derivatives by molecular modeling studies as shown in Fig. (10a). The 3D structures of β -CD and artesunate were obtained from the Protein Data Bank for docking purpose. The obtained β -CD-artesunate complex was further taken up for MD simulations. During the trajectory analysis, it was observed that the complex of β -CD-artesunate retained its structure and was stable during the whole simulation time. The RMSD of the complex over the whole trajectory of 5 ns was computed as 1.33 while final frame was 1.56 indicating that the β -CD-artesunate complex was not separated out and remained steady throughout the simulation time. In the β -CD-artesunate complex, two H-bond interactions were observed between β-CD and artesunate. The first H-bond was formed between “o” position proton (Fig. 10a) and the primary OH group of β-CD, and the second one was seen between the carboxylate group and secondary OH group of the β-CD. The artesunate core moiety was snugly fitted into the β-CD cavity showing an excellent interaction with the pyran rings of the sugar residue as shown in Fig. (10b). The comparative analysis of human and plasmodial translationally controlled tumor protein (PfTCTP) interaction with artemisinin has been reported by Eichhorn et al. [70].
This study also investigated crystal structure of PfTCTP (Fig. 11a) and its superimposed interaction study with human TCTP (Fig. 11b). Although there was similarity between secondary structures and folding of the human and PfTCTP crystal structures; the primary structures showed different sequence of amino acids. Alignment of human and PfTCTP on comparison showed eleven identical matches. The blind docking study of artemisinin as ligand on PfTCTP crystal structure gave birth to thirteen active binding sites (Fig. 11c) and the best two binding positions 1 (Fig. 11d) and 2 (Fig. 11e) showed binding energies of -6.5 and -5.68 kcal/mol, respectively. Finally it was concluded that single cysteine residue of PfTCTP did not have active role in interaction with artemisinin. It was reported that trioxane ring of artemisinin (10) exerted its bioactivity against malaria. The peroxide bond interaction between artemisinin and heme took place through highly negative charge on artemisinin and highly positive charge of heme [71]. Keeping this in mind Cristino et al. [72] have carried out computational modeling of nineteen 10-substituted deoxoartemisinin analogs. In this study the authors have calculated molecular electrostatic potentials (MEP) which played an essential role in obtaining structural insights for potent antimalarial activity. The use of molecular electrostatic potentials showed that the artemisinins have a reactive site for electrophilic attack by the Fe2+ ion of heme against an electronegative region of the studied artemisinins. Reduction of dimensionality and the basis of selection of descriptors used to classify the most and the least active artemisinin analogs were applied by using various chemometric methods such as PCA, Hierarchical Cluster Analysis (HCA), K-Nearest Neighbor (KNN), Soft Independent Modeling Class of Analogy (SIMCA) and Stepwise Discriminant Analysis (SDA). The results obtained through these chemometric methods showed that electronic (LUMO energy) and steric features i.e. distance between O1 atom in the ligand and iron from heme (DFeO1) and Molecular Representation of Structure Based on Electron
Fig. (9). 2D-diagrams of protein-ligand interactions: (a) CQ (1); (b) GA (32) (color images are available online).
(a) (b)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 161
Diffraction code of signal 15 (Mor15u) were responsible for differentiation into the most (33) and the least (34) active compounds. In addition to these, hydrophobic features such as Average Connectivity Index Chi-1A were also found to be important for the activity. The developed chemometric models were used to predict the activity of eight artemisinins. The prediction results showed that five of them were most active against D-6 stains of P. falciparum. Docking studies concluded that O1 atom rather than O2 of artemisinin interacted with heme more preferentially due to higher negative charge on O1 and also due to steric hindrance on O2 atom.
O
O
CH2COPh
Me
Me
H
O
O
HMe
H
O
OMe
Me
H
O
O
HMe
H
MeMeMe
OH
12
12
(33) (34)
Chouhan et al. [73] reported a plausible putative binding site for the ligands into the P. falciparum RIO2 (right open reading frame) kinase to clarify the involvement of highly conserved N-terminal winged (wHTH) domain and the flexible loop. Molecular recognition study revealed that Ser105 was one of the major contributing residues of PfRIO2 for auto-phosphorylation. The DNA sitemap analysis resulted into four possible ligand binding sites with major binding in wHTH and flexible loop as indicated by blue and green regions respectively in Fig. (12a). The reported plausible binding site was considered as combination of blue and green regions as the other two regions were smaller in size and fragmented ones. The prediction of residues available on the protein which were responsible for DNA binding was carried out by the authors through online server of Meta-DBSite which supported the
current study. The closeness of DNA binding site and wHTH domain was supported by domain studies analysis (Fig. 12b). On the basis of previously disclosed [62] 3D-structural aspects of PfRIO2 kinase, Parveen et al. [74] reported the identification of various binding sites within PfRIO2 kinase (Fig. 13a-d) with the help of sitemap module of Schrodinger chemometric software. This study gave rise to four new binding sites named as S1, S2, S3 and S4 (Fig. 13a). S1 site is situated near to the DNA binding region within the flexible loop. The S2 site comprises of mainly a major groove and has capability for binding to ATP. The S3 and S4 sites are small in size and nearer to the surface which clearly indicates their non-suitability for binding to inhibitors. On this basis Parveen et al. have carried out molecular interaction studies of α-pyrone analogs to S1 and S2 sites of PfRIO2. The molecular interaction studies of the α -pyrone analogs were claimed to be carried out with two complex models of the protein. The first complex model comprised of both DNA and ATP whereas the second model contained only the DNA. The obtained results were not significant because of the rigidity of the enzyme and it required enzyme flexibility. Hence, to acquire enzyme flexibility, induced fit docking (IFD) protocol was used for docking and the side chain refinement was done with Prime module. The docking results were significant with respect to the first complex model and all docking experiments were carried out further within the flexible loop of the first complex model. The surface diagram for the most active compound (35)
O O
HN
Br
O
N
(35)
Fig. (10). (a) Inclusion mode of artesunate into cyclodextrin cavity, (b) The space-fill model of artesunate (guest) and β-cyclodextrin (host) showing the intermolecular interactions. Used with permission from Chadha et al., Results in Pharma Sciences, 2011, 1, 38-48 [Copyright© 2011, Elsevier B.V.] (color images are available online).
(a) (b)

162 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
Fig. (11). (a) Crystal structure of PfTCTP, (b) superimposition of human TCTP with PfTCTP, (c) artemisinin binding sites of PfTCTP, best two binding positions 1 (d) and 2 (e) with artemisinin as the binding ligand. Used with permission from Eichhorn et al., Biochem. Pharm., 2013, 85, 38-45 [Copyright© 2012, Elsevier Inc.] (color images are available online).
(a) (b)
(c) (d)
(e)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 163
(Fig. 13d) and its docked pose within S1 site is depicted in Fig. (13b, c), respectively. The ligand-receptor interactions with the major hot spot residues of the flexible loop are showed in (Fig. 13d). Further, to evaluate energetic preference and binding interaction of compounds within S1
active site IFD dockings were scored by extra precision scoring function. The results obtained from Multi-ligand Biomolecular Association with Energetic (eMBrAcE) supported the S1 site as the plausible active site.
Fig. (12). (a) The sitemap analysis of PfRIO2 showing four possible sites for ligand binding. The blue and green colors represent wHTH and flexible loop regions respectively, (b) electrostatic surface shows major positive region near wHTH and flexible loop region and supporting site-map analysis to identify DNA binding region. Used with permission from Chouhan et al., J. Mol. Model., 2013, 19, 485-496 [Copyright© 2012, Springer-Verlag] (color images are available online).
Fig. (13). (a) PfRIO2 kinase sitemap analysis with four plausible binding sites, (b) the surface diagram of the most active compound showing binding sites and (c) its docked conformation within S1 site, (d) ligand-receptor interaction with the most active compound (35) showing major hot spot residues within the flexible loop. Used with permission from Parveen et al., Eur. J. Med. Chem., 2013, 70, 607-612 [Copyright© 2014, Elsevier Masson SAS] (color images are available online).
(a) (b)
(a) (b)
(c) (d)

164 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
Shah et al. [75] carried out structure-based virtual screening of a database library of focused cysteine protease inhibitors consisting of ~ 65000 compounds. A four-featured (ADHH) pharmacophore was applied as the first filter through which about 30000 compounds were filtered out. Further, docking was applied as the second filter and top 3000 compounds (top 10 %) ranked on the basis of Emodel score were selected for eMBrAce minimization calculation (Macromodel) as the third filter and out of that top 500 compounds were selected. On the basis of visual inspection, 200 molecules were chosen and subjected to diversity analysis. Finally, fifty diverse compounds were selected which were evaluated for inhibition of FP-2, FP-3 and against chloroquine resistant (W2 strain) P. falciparum parasites. Twenty one compounds were found to be active against FP-2 and four against FP-3. The most active compound (36) showed IC50 value of 1.39 µM. The 4-oxophthalazine core of compound (36) appeared to protrude into the S1’ pocket with the carboxylic acid group interacting with the residues of an active site-forming oxyanion hole like interaction with NH of Cys42 and the amide side chain of Gln36 as shown in Fig. (14). Trp206 and Val152 of the S1’ pocket were also involved in the van der Waals interaction with the phthalazine moiety. NH of indole in Trp206 formed a hydrogen bond with carboxylate moiety of compound (36). The α -ketoamide, a soft-electrophile showed H-bonding with -NH of Gly82 and the backbone carbonyl of Asn173. The phenyl ring at the terminus of the amide linker and terminal 2,4-dimethylphenyl moiety were involved in hydrophobic interactions with Ala175 and Leu84/Ile85 respectively. A tetrazole containing compound (37) showed IC50 value of 2.18 µM. Compound (38) having triazole ring was found to be selective FP-2 inhibitor having IC50 value of 4.59 µM. Compounds (39 and 40) showed inhibition of the cultured parasites (IC50 value = 7.51 and 7.85 µM respectively), indicating a need for further structural optimization of these compounds.
Fig. (14). Predicted binding mode of the virtual screening hit (36) in the FP-2 binding pocket. Used with permission from Shah et al., J. Chem. Inf. Model., 2011, 51, 852-864 [Copyright© 2011, American Chemical Society] (color images are available online).
Fugel et al. [76] identified and developed a novel class of selective inhibitors of PfGSK-3 as antimalarial agents through high-throughput screening (HTS) campaign from a commercial library. A total of 10,480 compounds were evaluated at a concentration of 10 µM against recombinant PfGSK-3. Only 18 molecular entities were found to be active with more than 50 % inhibition at 10 µM. Interestingly, five of these inhibitors were having a common heterocyclic ring system as the parent scaffold i.e. 3-amino-4-arylthieno[2,3-b]pyridine motif (41). Further, a small library consisting of 427 compounds related to compound (41) were tested for PfGSK-3 inhibitors, amongst which congener (42) was obtained as a selective and active PfGSK-3 inhibitor with IC50 value in micromolar concentration range. Thus, the initial hit was identified as 3,6-diamino-4-(2-halophenyl)-2-benzoylthieno[2,3-b]pyridine-5-carbonitrile (42). Molecular
N
N
OHO
OON
H
S
Me
Me
HN
OF
N N
NN
HN
O
ON
Me
(36) (37)
N
NN
S
N
O
OCH3
NH
O O CH3
O
O
O
O
NNS
NH
O
NO
N
Me
(38) (39) (40)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 165
modifications of compound (42) were carried out with the aim to increase the PfGSK-3 inhibitory activity and selectivity. The highest activity was found in compound (43) in which the benzoyl and 2-iodophenyl groups of compound (41) were replaced with cyano and 2-chlorophenyl groups respectively, having an IC50 value of 0.13 µM. Compound (43) was co-crystallized with the human enzyme. Hexagonal crystals of the complex (pTyr216)-GSK3β-43 exhibited poor diffraction quality. To obtain a crystal form with better diffraction properties, (pTyr216)-GSK3β and compound (43) were co-crystallized with an axin-derived peptide [axin(383-401)]. Binding mode of the compound (43) is depicted in Fig. (15). H-bonds were formed between the 6-amino group and the pyridine nitrogen of compound (43) with the carbonyl oxygens of Val135 and Asp133 respectively. Likewise, 3-amino-2-cyano groups of compound (43) also formed H-bonds with Lys85 and Asp200 as shown in Fig. (15). Fig. (15). Docked pose of compound (43) (carbon scaffold shown in green) in the active site of HsGSK-3. The red spheres represent crystallographically observed water molecules, one of which mediates the contact between the ligand and Asp133. Used with permission from Fugel et al., J. Med. Chem., 2013, 56, 264-275 [Copyright© 2013, American Chemical Society] (color images are available online).
Marom et al. [77] studied β-hematin crystal structure and its heme dimer as a repeat unit on the basis of van der Waals (vdW) corrected density functional theory (DFT) (Fig. 16a, b). This study was carried out on the basis of previously
reported pseudopolymorphic behavior of β -hematin which was analyzed using X-ray diffraction (XRD). The significant binding of the heme dimer and the β -hematin crystal highlighted the significant role of vdW through different behavior of DFT vs DFT+vdW along the three crystal axes. In case of the hydrogen bonding along the c-axis, the DFT and DFT+vdW gave the minimum lattice with experimental support. On the other hand, dispersive bonding as in case of α- and β -axes showed little or no bonding in DFT, and DFT+vdW gave good experimental background. The obtained difference between geometry of dimers in crystal and with that of the isolated dimers further signified the essential role of dispersion. Finally the authors concluded that the first stage of nucleation is formation of cyclic dimers with formation of cd12 as the most stable dimer in two phases of β-hematin. The mechanism of falcipain-2 (FP2) inhibition by epoxysuccinate E64 has been reported by Arafet et al. [78] with the help of quantum and molecular mechanics studies. The complete free energy reaction cascade was obtained with the help of molecular dynamics simulations using hybrid AM1d/MM and M06-2x/MM potentials. This study reveled that Cys42 irreversibly attacked both the carbon atoms i.e. C2 and C3 of the epoxy ring of the E64 inhibitor. Attack on the C2 took place in single step whereas on C3 it took place in two steps. There was a slightly smaller barrier on C2 carbon than on the much more stabilized C3 as derived from ligand-receptor complex. The amino acids, Gln171, Asp170, Gln36, Trp43, Asn81 and His174 played essential role in proper orientation of the inhibitor. This study concluded that QM and MM simulations were coincidental to give two different conformations which differed in their orientation in active site of the protein. One of the conformer (corresponding to initial X-ray structure) has guanidine group of E64 which interacted with Tyr78 (Fig. 17a) and in another conformation, guanidine of E64 interacted with Asp170 (Fig. 17b) and it was energetically favored one.
2.3. Studies Based on Virtual Screening Approach
Sullivan et al. [79] reported virtual screening of a database consisting of 5.8 million compounds. A quantum anti-malarial model was constructed first with the 26 known anti-malarials and 1730 FDA drugs with antimalarial activity at 10 µM concentration. The developed model produced 12 quantum components that were positively correlated to the activity and more than 20 quantum components were produced with negative correlation to antimalarial action.
N S
NH2Aryl
EWG
N S
NH2NC
H2N
O
Cl
I
N S
NH2
CN
NC
H2N
Cl
(41) (42) (43)

166 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
Each quantum component can be expressed by various chemical substructures. Another filter was generated to ensure that the selected compounds were non-toxic. A number of molecules were identified from the quantum database that ranked them according to their quantum anti-malarial attributes. Top twelve compounds were selected on the basis of commercial availability, and in vitro validation was performed in both anti-P. falciparum and mammalian cytotoxicity assays. Compound (44) showed the highest activity with an IC50 value of 0.027 µM and was found to be non-toxic.
N
Me
N
Me
6
(44)
Kumari et al. [61] carried out virtual screening of zinc database by using different filters. Docking studies were used as the first filter in which homology model of ribosomal phosphoprotein P1 (RPP1) was utilized for the study. The obtained top hits were further filtered by “Lipinski’s rule of five”. A small library of 45 compounds was obtained which was further analyzed by ligand-RPP1 binding interactions. Top three compounds were selected on the basis of the least binding energy, good cavity coverage and satisfactory number of hydrogen bonds. Ethyl loflazepate (45) showed the best interactions with the RPP1. Two hydrogen bonds were formed by ethyl loflazepate with Ser74 and electrostatic bond with Ile4 along with van der Waals interactions with residues at Ala2, Val3, Ile4, Glu16, Thr20, Phe57, Leu70, Leu73, Val85 and Leu112 positions as shown in Fig. (18).
Fig. (16). Structures predicted by DFT+vdW calculation (a) isolated heme dimer, (b) heme dimer in β-hematin crystal. Used with permission from Marom et al., Cryst. Growth Des., 2011, 11, 3332-3341 [Copyright© 2011, American Chemical Society] (color images are available online).
Fig. (17). Relative orientation of two possible conformations of E64 in the active site. (a) interacting with Tyr78, (b) interacting with Asp170. Used with permission from Arafet et al., Biochemistry, 2014, 53, 336-3346 [Copyright© 2014, American Chemical Society] (color images are available online).
(a) (b)
(a) (b)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 167
F
N
HN
Cl
O
OEt
O
(45)
Fig. (18). 3D-view of ethyl loflazepate (45) interactions with RPP1. Ethyl loflazepate is showed in solid green and interacting residues of RPP1 in gray wire frame. The ligand forms two hydrogen bonds with Ser74, one electrostatic bond with Ile4 and van der Walls interaction with Ala2, Val3, Ile4, Glu16, Thr20, Phe57, Leu70, Leu73, Val85 and Leu112. Used with permission from Kumari et al., J. Theor. Biol., 2014, 343, 113-119 [Copyright© 2013, Elsevier Ltd.] (color images are available online).
A structure comparison analysis approach has been reported by Franco et al. [80] for the designing of novel antimalarial agents. The authors have successfully screened a data set of 3,20,000 compounds against crystal structure of PfUCHL3 using Autodock vina module. Top ten hits were screened against UCHL3 human ortholog (PDB 1UCH) to identify the compounds that could define the target (PfUCHL3) over its human counterpart. The virtual screening approach gave two potential lead compounds i.e. 46 and 47 with respective IC50 values of 603.1 and 378.5 ng/ml against PfUCHL3.
3. APPLICATION OF LIGAND BASED DRUG DESIGN TECHNIQUES FOR THE DISCOVERY OF ANTIMALARIAL AGENTS
3.1. QSAR Studies on Antimalarial Compounds
QSAR studies on the antimalarial compounds carried out by various research groups worldwide have been discussed on the basis of chemical classes of the compounds.
3.1.1. QSAR Studies of Artemisinin Derivatives
Abbasitabar et al. [81] in 2012 reported a QSAR model developed with a set of 179 artemisinin analogs having potent antimalarial activity. Various descriptor selection methods were employed to build predictive QSAR models. The selection of molecular descriptors was carried out stepwise, successive projection of algorithm and memorized_ACS methods. Two matrices of independent variables (D1 and D2) were used where D1 matrix contained the descriptors calculated using Dragon and MOE softwares, and the D2 matrix contained the 1st to 3rd order of the calculated descriptors and the logarithm of their absolute values. The memorized_ACS algorithm resulted into the best QSAR model. The model obtained from D2 matrix showed better quality. This study concluded that a better QSAR model may be obtained by using higher orders of the molecular descriptors coupled with a powerful algorithm to select the right descriptors. Calculation of molecular descriptors for the development of novel artemisinin analogs (48) with the help of Hartree-Fock method was reported by Santos et al. [82] in 2013. In this study the QSAR model showed 89.55 % of variance with q2 of 0.83 and r2 value of 0.92. The authors have proposed 10 novel compounds and revealed that lipophilicity has major role to play in antimalarial activity of artemisinin analogs. This study focused on 1,2,13-trioxane endoperoxide ring of artemisinin and on geometrical parameters.
O
O
OR
Me
H
O
O
HMe
HH
Me
(48)
A concurrent application of three approaches i.e. pharmacophore, CoMFA/CoMSIA and HQSAR studies, for the development of QSAR model of substituted 1,2,4-
NH
NH
NH
NH2 NH2
OHOH
OHOO
O
O
HO
OH OHMe
MeMe
Me
(46) (47)

168 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
trioxanes, the reactive part of artemisinin (10) has been reported by Gupta et al. in 2013 [83]. The quantitative pharmacophore based alignment was reported to have good statistical data for both CoMFA and CoMSIA analysis with q2 of 0.72 for both the analyses with r2 values of 0.83 and 0.87, respectively. Further, to get structural insights into antimalarial activity, the authors also applied HQSAR analysis which helped in finding the substructural fingerprints for the substituted 1,2,4-trioxanes.
3.1.2. QSAR Studies of 4-Aminoquinoline Derivatives
To get structural information for potent antimalarial activity against both chloroquine sensitive (HB3) and resistant (Dd2) P. falciparum strains Sahu et al. [84] have reported a QSAR model for a series of 4-amino-7-chloroquinolines with varying side chain substituents (49-52). The best statistical model for compounds active against HB3 was found to be statistically acceptable having r2=0.9024, cross validated squared correlation coefficient (q2) and r2
pred equal to 0.8089 and 0.7463 respectively. In case of Dd2 strain, the best model was obtained with statistical values of r2= 0.9188, q2= 0.8349 and predictive r2= 0.7258. The authors used MLR methodology with the use of various methods for features’ selection such as Genetic Algorithm (GA), Simulated Annealing (SA) and Stepwise (SW) forward-backward methods. Deshpande et al. [85] have reported a QSAR model by using a series of 4-anilinoquinolines (53) with the help of CP-MLR and GA approaches. Different models were developed by both CP-MLR and GA approaches with the use of five common descriptors, namely H-052, MATS4m, MATS7e, Mor30p, and R7m. The PLS model of molecular descriptors from combined CP-MLR and GA approaches explored 73.1% of variance with r2, q2 values of 0.731 and 0.688 respectively. Najafi et al. [86] in 2013 studied inhibition of cytochrome P450s (CYPs) by chloroquine and 4-aminoquinoline analogs (54) by developing a QSAR model
using genetic algorithms-multiple linear regression methods. The developed model was found to be robust with predictive r2 value of 0.74. This study concluded that size, shape, atomic weight and flexibility of the molecules played key roles in interaction of the ligands with P450 isoenzymes. Gupta M. K. [87] evaluated antimalarial and cytotoxic activity of 4-aminoquinolines (55) with the help of QSAR model. Among the various descriptors, N-067 a descriptor representing sec.aliphatic amines was found to have positive contribution for antimalarial activity with minimal cytotoxicity. The obtained model proved that the presence of primary aromatic amines and sec.aliphatic amines contributed positively while secondary aliphatic amides contributed negatively for potent antimalarial activity. This study also revealed that aromatic ethers, CH2R2 and CH3X were lead fragments for cytotoxicity of the resulting compounds. Masand et al. [88] reported a QSAR model in 2014 developed by using a set of 4-aminoquinoline analogs (56) using CORrelation and Logic (CORAL) as a chemometric tool. In this study statistically robust and highly predictive QSAR models for six random various splits into training and test sets of 112 compounds were developed by using GA-MLR and optimal SMILES-based descriptors. In three cases i.e. split 1, 3 and 5 preferable models were developed by GA-MLR and in other three cases i.e. split 2,4 and 6 preferable models were developed by optimal descriptors. All of the six splits gave significant statistical data with r2 values of >0.85 and >0.78 for sub-training and validation sets, respectively. This study revealed that the topological distance of six bonds between N and O has significant correlation with antimalarial activity as shown in compound (57).
3.1.3. QSAR Studies of Cyclic Peroxy Analogs
Topological and physico-chemical parameters were used for the prediction of antimalarial activity of cyclic peroxy
N
HN
Cl
N N(C2H5)2
R
n
N
NH
Cl
RHN NHRn n
N
HN
Cl
NRR'
N
HN
Cl
NRR'
(49) (50) (51) (52)
N
HN
R1
R2
R3
Cl1
4
3'
5'
N
HN
R2
R1
N
HN
Cl
R/Ar
(53) (54) (55)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 169
analogs (58) by Shaik et al. [89]. Among the various models, the best model was developed with five-parameters i.e. molecular weight (MW), surface tension (ST), equalized electronegativity (χeq), Kier and Hall valence connectivity index (oχv
) and indicator parameter (IP2) with r2 value of 0.9582 for the prediction of log IC50 activity. The results obtained from this study concluded that low molecular weight, high equalized electronegativity, less branching and low ST of the molecules were essential features for designing of new antimalarial agents.
3.1.4. QSAR Studies of 4(1H)-Quinolone (Endochin) Analogs
The theoretical justification for the importance of “descriptors’ thinning” in the development of QSAR models were reported by Ojha and Roy in 2011 [90]. In this study QSAR model was developed according to the organization for economic co-operation and development (OECD) principles using 53 endochin (59) analogs [4(1-H)-quinolone derivatives] which were evaluated for antimalarial activity against TM-90-C2B strain. Five different chemometric tools namely, stepwise regression, factor analysis followed by MLR (FA-MLR), factor analysis followed by PLS (FA-PLS), GFA and G/PLS were carried out for the development of robust QSAR models. Out of these five models, a statistically acceptable model was developed with the help of SW-GFA and G/PLS techniques having q2 values of 0.749 and 0.735, and r2 values of 0.797 and 0.776, respectively. These QSAR models concluded that freely rotatable bonds at R3 and R5 positions have positive effect on antimalarial activity.
O OR
R
O
ArMe
(58)
N
R1 O
R5
Me
R6R4
R3
R2
(59)
Li et al. [91] have reported QSAR models of a set of 3-carboxy-4(1H)-quinolone analogs (60) with the aim to gain structural insights for potent antimalarial activity. The explored QSAR models gave significant statistical data with r2 values of 0.977, 0.973 and q2
Loo values of 0.650, 0.777 for CoMFA and CoMSIA analysis, respectively. The obtained results for CoMFA analysis concluded that for steric factors, substitution at R5 with higher ethers e.g. Ph-O-Ph (Fig. 19a; Green color) and lower ethers at R1 position (Fig. 19a; Yellow color) offered good antimalarial activity. On the other hand for electrostatic factors, electropositive and electron withdrawing groups diminish activity (Fig. 19b; Red color) whereas electronegative and electron donating groups at R2 markedly increase activity (Fig. 19b; Blue color). The results obtained from CoMSIA analysis were similar to CoMFA results particularly at R5 and additionally inclusion of hydrophobic field at this position improved the model (Fig. 19C; Yellow color).
NH
R4
OR2
R3
R1R5
(60)
Ojha and Roy [92] successfully applied group based QSAR (G-QSAR) approach to correlate antimalarial activity contribution of various chemical groups at different sites of endochin (61) scaffold. The developed G-QSAR model was more robust as compared to the previously reported [90] QSAR model using the same set of analogs. The reliability of G-QSAR model was proved with cross-validated correlation coefficient (q2) of 0.806 and predictivity (r2
pred) of 0.793. This study concluded that differing substitution of R1 and R2 positions have the key role to play in antimalarial activity. The substituent next to carbonyl group, R6 with increasing molecular weight and polarizability afforded far better antimalarial activity that diminished with high molecular weight alkyne functionality.
N
HN
R1
NH
R2
1
9 13
N
HN
HN
HN
F
MeO
(56) (57)

170 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
N
OR4R1
R2R3
R6
MeR5
(61)
Villalobos et al. [65] in 2013 have successfully developed 2D and 3D-QSAR models of 4(1H)-quinolone analogs having potential antimalarial activity. The 2D-QSAR model showed 78.6% variance in bioactivity with r2 and q2 values of 0.784 and 0.674 respectively. The results obtained for 3D-QSAR model were more promising with r2 and q2 values of 0.970 and 0.635 for CoMFA analysis and 0.921 and 0.537 for CoMSIA analysis, respectively. The CoMFA model showed 68.3% variance for steric factors (Fig. 20a) and 31.7% variance with respect to the electrostatic (Fig. 20b) fields. CoMSIA model proved to have the highest 39.4% variance for electrostatic field, 34% variance for steric and 26.65% variance for hydrophobic fields, respectively. This study gave rise to a new potent antimalarial molecule TJ5 (62) with the highest pEC50 values calculated by both CoMFA (10.4458) and CoMSIA (10.8553) analyses. In (Fig. 20) green color represents increase in activity with bulkier groups (steric field; Fig. 20a); yellow color represents decrease in activity with bulkier groups (steric field; Fig. 20a); blue color represents increase in activity with electropositive groups (electrostatic field; Fig. 20b).
NH
ONH
MeCl
O
NNH
Me
TJ5 (62)
3.1.5. QSAR Studies of Aurone and Azaaurone Derivatives
Gupta et al. [93] in 2014 have successfully explored a series of aurones (63) for quantification of antimalarial activity and their binding interactions with macromolecules. The developed QSAR model showed the best statistical data with r2 value of 0.941 and represented 86.9% variance in the activity with more than 99.9% internal statistical significance level. The assessment of QSAR model with various molecular descriptors revealed that H6m descriptor contributed positively whereas R2v and Mor30v contributed negatively to the inhibitory activity. H6m and R2v belong to GETAWAY descriptors’ class which represents geometry, topology and atom-weight assembly. Mor30v is a representa-tive of Morse-Code descriptors which describes 3D-structures with the help of electron diffraction.
XR1
R2O
R
(63)
The chemoinformatic QSAR approach for a series of aurone and azaaurone analogs (64) to gain structural insights for potential antimalarial activity has been reported by Sharma et al. [94] in 2014. Structural requirements for designing of novel antimalarial agents were explored through 2D-QSAR, group-based and k-Nearest Neighbor QSAR studies. The developed QSAR model was robust with r2 and predictive r2 values of 0.9061 and 0.8719, respectively. The reported QSAR study revealed that halogens with higher electronegativity (Cl, F) at R3 and R5 positions, and bulky substituents at R1 and R2 positions tended to increase bioactivity of the designed compounds.
Fig. (19). Contour plots for CoMFA (a) Steric, (b) Electrostatic; (c) CoMSIA Hydrophobic fields. Used with permission from Li et al., J. Mol. Graph. Model., 2013, 44, 266-277 [Copyright© 2013, Elsevier Inc.] (color images are available online).
(a) (b)
(c)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 171
XR3
R4
R5R6
R7
OR2
R1
(64)
3.1.6. QSAR Studies of Tetrahydroacridinone (THA) Analogs
Ojha and Roy [95] reported QSAR model of 1,2,3,4-tetrahydroacridin-9(10H)-one (65) analogs with the application of four different chemoinformatic techniques i.e. GFA, CoMFA and CoMSIA analysis (Fig. 21a-c), HQSAR technique of fragment based QSAR analysis and fragment contribution of group based QSAR techniques. Various models were developed by using different techniques to get
Fig. (20). Contour plots for (a) CoMFA steric fields, (b) CoMFA electrostatic fields. Used with permission from Villalobos et al., J. Mol. Graph. Model., 2013, 46, 105-124 [Copyright© 2013, Elsevier Inc.] (color images are available online).
Fig. (21). (a) Steric, (b) Electrostatic and (c) Hydrophobic contour maps for CoMSIA analysis. (For steric, electrostatic and hydrophobic contours green, blue and yellow colors represent favored regions whereas yellow, red and white present disfavored regions, respectively). Used with permission from Ojha and Roy, Comb. Chem. High Throughput Screen., 2013, 16, 7-21 [Copyright© 2014, Bentham Science Publishers] (color images are available online).
(a) (b)
(a) (b)
(c)

172 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
similarity indices of cross validated and non-cross validated correlations. The authors have suggested some key positions for the development of potential antimalarial activity. The substitution of R2 position with bulky groups, R3 with hydrophobic and sterically disfavored groups, and R4 with positive charge without carbon chain were reported to have positive contributions toward antimalarial activity whereas C-6 position showed decreased bioactivity with bulky groups.
NH
O
R2
R3
R4
R1
R5
R6
(65)
Sharma et al. [96] in 2013 reported contributions of different substituents on 1,2,3,4-tetrahydroacridin-9(10H)-one scaffold (66) for the antimalarial activity (W2 and TM90 strains) through Free-Wilson matrix approach. PLS analysis was carried out for development of a joint model and the developed model was best suited for W2 strain as compared to TM90 strain with r2 and q2 values of 0.72 and 0.67, respectively. The derived model suggested that 5th and 8th positions have key roles in governing the bioactivity and should be free from substituents.
NH
OR4
R3
R2
R1In
A B C
In = Variable ring size (66)
3.1.7. QSAR Studies of Oligo-Pyrrole Analogs
Singh et al. [97] in 2012 have successfully correlated, predicted and observed antimalarial activity (P. falciparum,
D6 strain) of naturally occuring prodiginines (67-70) along with their synthetic derivatives (71). A QSAR model was developed signifying the role of different chemometric descriptors. In this study the authors have concluded that increasing the value of the descriptors’ index with 5-order of neighborhood symmetry (IC5) and mean topological charge indices of order 5 (JGI5), and lowering the value of the Moran autocorrelation -lag 6/weighted by atomic masses (MATS6m) and Geary autocorrelation-lag 5/weighted by atomic Sanderson electronegativities (GATS5e) could be considered as key factors for further increasing the biological activity. In 2013 Singh et al. [98] reported QSAR model of a series of natural and synthetic prodiginine derivatives (71) having antimalarial activity against both chloroquine sensitive D6 and multi-drug resistant Dd2 strains of P. falciparum. The developed model was robust and was validated using both internal and external validation methods showing significant statistical data with r2>0.7, higher F value (F >20) and predictivity value (Q2) of >0.6. As per the authors’ claim the developed model could prove significant for development of new antimalarial agents through virtual screening approach.
3.1.8. QSAR Studies of Miscellaneous Antimalarial Scaffolds
Application of multivariate image analysis (MIA) descriptors for the development of validated QSAR model for a series of 2-aziridinyl and 2,3-bisaziridinyl-1,4-naphthoquinonyl sulfate and acylate derivatives (72) was reported by Goodarz and Freitas in 2011 [99]. The predictivity of the developed QSAR model was analyzed through PLS, N-PLS, and LS-SVM regression methods. Among the analyzed regression methods, non-linear LS-SVM proved to be the best with r2 and q2
loo values of 0.947 and 0.914, respectively.
NH N
HN
Me
Me
OMe
14
NH N
HN
OMe
HN
N
NH
OMe Me
Me10
(67) (68) (69)
N
HN
R3
R2
OCH3
R1HN
N
NH
OMeMe
(70) (71)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 173
R2
O
O
R1
R3 (72)
Mahajan et al. [100] deduced a predictive QSAR of a set of fourteen benzophenone derivatives (73). The developed QSAR model revealed that compounds with low potential energy and attached electron donating groups were responsible for diminished antimalarial activity. The positive correlation in the sense of antimalarial activity was observed with increased polarity of the compounds. The pharmacokinetic and drug-likeliness studies were also carried out with the help of “Quikprop”.
C
O
RR'1
23
4
5
1'2' 3'
4'
5'
(73)
Analysis of a series of thiourea/urea analogs (74-76) for antimalarial activity with statistically significant QSAR model was reported by Beheshti et al. [101] in 2012. The molecular descriptors were selected through Genetic algorithm (GA) method. The explored model was validated with leave one out method having r2 value of 0.801 and q2 value of 0.722. In this study authors have concluded that eight variables namely volume and hydrophobicity of molecule, distance between electronegative atoms (oxygen), reactivity of atom, HOMO-1 energy, H atom with minimum net atomic charge and XY shadow descriptors play key roles in antimalarial activity. Liang et al. [102] reported 3D-QSAR CoMFA and CoMSIA models for a series of 1,2,4,5-tetraoxanes (77) for their antimalarial activity. Both the models, CoMFA and CoMSIA showed good statistical results with significant values of 0.719 and 0.739 for cross-validated coefficient (q2), 0.855 and 0.847 for non-cross-validated coefficient (r2) and 0.335 and 0.344 standard error of estimate (SEE), respectively. CoMFA model showed 92% contribution in the favor of steric field and the model could help in differentiating molecules on the basis of their steric interactions having potent biological activity. On the other hand, CoMSIA model showed similar results as CoMFA model for steric field but gave more importance to hydrophobicity.
O O
OO
R1 R2 (77)
Worachartcheewan et al. [103] in 2013 have successfully reported QSAR model of a set of amidino-bis-benzimidazole derivatives (78) having potent antimalarial activity. In this study the researchers have successfully used MLR and ANN methods for the development of predictive QSAR models. Both the models of MLR and ANN approaches showed significant statistical data by leave-one-out method with correlation coefficient values of 0.976 and 0.982 and root mean square error of 0.130 and 0.110, respectively.
N
HN
H2N
NH
N
HN R
(78)
Ibezim et al. [104] reported a QSAR model of a set of substituted arylpiperazine derivatives (79) with the aim of establishing relationship between the structural characteristics and their exhibited biological activity. The linear modeling approaches were used for development of predictive QSAR model. Multivariable linear regressions were used for searching molecular descriptors and CORAL program for calculating flexible descriptors. The developed QSAR model was validated with the help of leave-one-out procedure. This study also reported that r2
loo should not necessarily be >0.7 for a good model rather r2
loo <0.7 can also provide a satisfactory model.
N N ArR
(79)
Saghaie et al. [105] in 2013 reported a QSAR model of a set of 3-hydroxypyridinones (80) and successfully compared experimental biological activity with the predicted ones through application of B3LYP/6-311++G** optimized geometries for various descriptors by using MLR and PCR analysis. The results obtained from both, MLR and PCR analyses were promising but PCR analysis resulted into good statistics in the form of regression and predictivity. On the basis of the obtained results, six new compounds were designed and their pIC50 values calculated, of which the two compounds (81, 82) showed higher pIC50 values compared to the compounds existing in the already studied set of compounds.
N
N
HNHN
HN
S
R2
R1
N
HNHN
HN
O
R2
O
Me N
N
HNHN
HN
OR2
R1
R1
(74) (75) (76)

174 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
Wang et al. [106] have reported 3D-QSAR model using a set of 247 compounds having 2-pyrimidinecarbonitrile analogs with the application of three different alignment rules i.e. ligand-based, receptor-based and scaffold based. Ligand based alignment gave the best QSAR model with a more significant CoMSIA model over the CoMFA model. The explored CoMSIA model (Fig. 22a-c) of the most active compound (83) showed the best statistical values with q2 value of 0.501with non-cross validated correlation coefficient of 0.821. It showed greater contribution for the hydrophobic field i.e. 42.4% over steric (20.8%) and electrostatic (36.9%) fields. As per the report, reliability of the model developed using PCA analysis was carried out by comparing results obtained through random division method and it was found out that both were significantly reliable. The authors revealed that substitution of bulky group at 3-position and on rings B and D was contributing for potent antimalarial activity, as well as electronegative/positive groups at B, C and D rings and hydrophobic groups at B and D rings were also favored for bioactivity. In (Fig. 22a) green
and yellow contours indicate respectively increase and decrease in activity with respect to bulky groups. Red and blue contours indicate increase in activity respectively with negative and positive charges (Fig. 22b). Yellow and white contours indicate, increase and decrease in activity respectively, with respect to the hydrophobic groups (Fig. 22c).
N
N
N
HN
N
N
Me
ON
Br
Me
(83)
N
O
R6 R2
R1
OH
N
O
CH3
H
OH
N
NH2
N
O
CH3
H
OH
N
NH2
(80) (81) (82)
Fig. (22). Contour maps for CoMSIA (a) Steric, (b) Electrostatic and (c) Hydrophobic fields. Used with permission from Wang et al., Mol. Biosyst. 2013, 9, 2296-2310 [Copyright© 2014, Royal Society of Chemistry].
(a) (b)
(c)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 175
Mishra et al. [107] in 2014 reported a QSAR model for a set of 51 compounds having 4-substituted quinazoline derivatives (84) with potential antimalarial activity against P. falciparum. The 2D-QSAR model was developed using PLS method, and kNNMF approach was used for development of 3D-QSAR model. The statistical data obtained from 3D-QSAR model showed results with q2 value of 0.760 and predicted r2 value of 0.710. The explored 3D-QSAR model concluded that the molecular weight descriptor had positive effect on antimalarial activity.
N
N
R1
R2 (84)
Sharma et al. [108] in 2014 have successfully carried out chemoinformatic study on quinoline-based analogs having potential antimalarial activity. The results obtained from this study suggested that CoMFA analysis was superior over CoMSIA analysis. The CoMFA model offered the best statistical data with internal cross validation of 0.70 and non-cross validation of 0.80. HQSAR analysis showed strong positive correlation of molecular fragments towards the quinoline ring and NH functionality. The contour maps of the most active compound (85) showed positive contribution in the bioactivity by the electron withdrawing substituents on C-2 carbon of the quinoline ring.
N
N
Me
O
MeMe
HN
Me
CNCl
NH2
(85)
Aher and Roy [109] have reported QSAR model of set of aminothiazoles (86) and aminopyridines (87) using different chemometric tools like stepwise regression, genetic function algorithm (GFA) spline and GFA linear+spline. Among these methods, the best model was developed with GFA linear+spline analysis that showed internal cross validated squared correlation (q2) value of 0.675 and external prediction (r2
pred) value of 0.720. The molecular descriptor SC-2, highlighted the positive significance of increasing the size of the molecules and presence of trifluoromethyl group
for antimalarial activity. Increasing the number of tertiary carbon atom on benzene ring of the molecules had positive effects on bioactivity. The diaryl and arylalkyloxy groups were responsible for diminished activity. The effect of two tautomers (keto and enol) of a set of phosphoramidate and phosphorothioamidate analogs (88) on QSAR models was reported by Masand et al. [110]. The best model was developed with GA-MLR analysis having r2, r2
ex values of 0.79 and 0.786 for keto tautomers and 0.785 and 0.770 for enol tautomers, respectively. The effects of molecular fields responsible to cause differences in bioactivity were analyzed using fields similarity analysis. The field similarity analysis study concluded that lipophilicity has a key role to play in enhancing the bioactivity as indicated by a positive field in the tautomers of the most active compounds. It also showed its effect in the least active enol tautomers contrary to the keto tautomers. The overall study concluded that tautomerism was devoid of any effect on QSAR model but tautomerism showed its importance in identification of structural and pharmacophoric features responsible for bioactivity.
3.2. QSAR Studies of Antimalarial Compounds with Specific Targets
3.2.1. Deoxyuridine-5’-Triphosphate Nucleotidohydrolase (PfdUTPase) Inhibitors
To gain structural insights of molecular descriptors, the first QSAR report on nucleoside analogs (89) with potential bioactivity was published by Ojha and Roy in 2013 [64]. The developed model was validated internally (q2 value of 0.73) and externally (r2
predicted value of 0.75). The obtained results clearly defined the positive correlation of aromatic carbon fragment, presence of linear chain rather than cyclic oxolane ring and of hydroxyl functionality in the molecules toward deoxyuridine-5’-triphosphate nucleotidohydrolase (PfdUTPase) inhibition.
N
NH
O
O
R (89)
O
HO
NNH
O
O
Me
RO
HO
NNH
O
O
MeR
(90) (91)
N
SNH
R'
OR
NR
R'
H2N
O
PNHR2
X
OR3R1
(86) (87) (88)

176 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
3.2.2. P. falciparum Thymidylate Kinase (PfTMPK) Inhibitors
In continuation of their work on chemoinformatic approaches for development of novel antimalarial agents Ojha and Roy [111] in 2013 reported a QSAR model of a series of alpha (90) and beta (91) thymidine analogs with potential activity against P. falciparum thymidylate kinase
(PfTMPK) enzyme. The reliability and robustness of the model was validated internally (q2 of 0.806) as well as externally (r2
pred of 0.818). The results obtained from this study concluded that compounds having NH group tend to exhibit higher bioactivity. More precisely, compounds with more electrophilic character and increased oxygen atoms have prominent effect on PfTMPK enzyme inhibition.
SN
R2
O
SNH
N SN R
O
S
HO
HO
(92)
(93)
(94)
1-Chloro-2-(2-methylprop-1-enyl)benzene
3,4,5-Trimethylcyclohex-1-ene
2,3-Dihydro-4-vinyl-1H-indene
R
R1
(92-94) (95)
Fig. (23). (a) CoMFA and (b-f) CoMSIA contour maps. (a). Steric fields: Green and yellow colors respectively indicate favored and disfavored regions for bulky groups; (b). Electrostatic field: Red and blue colors respectively indicate, favored and disfavored regions for electronegative groups; (c). Hydrophobic field: Favorable and unfavorable regions for hydrophobicity indicated with yellow and gray colors, respectively; (d). Hydrogen bond donor: cyan and purple colors indicate, favorable and unfavorable regions for hydrogen bond donor groups; (e). Hydrogen bond acceptor: Orange and magenta represent favorable and unfavorable regions for hydrogen bond acceptors, respectively. Used with permission from Neves et al., Bioorg. Med. Chem. Lett., 2013, 23, 2436-2441 [Copyright© 2013, Elsevier Ltd.] (images are available online).
(a) (b)
(c) (d)
(e) (f)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 177
3.2.3. P. falciparum Enoyl-ACP Reductase (PfENR) Inhibitors
Neves et al. [112] in 2013 discovered three novel hits (92-94) against P. falciparum enoyl-ACP reductase (PfENR) through the application of chemoinformatic approach for a series of rhodanine analogs (95). The authors have successfully carried out HQSAR, CoMFA and CoMSIA analysis for development of a robust and validated QSAR model. Among the 3D-QSAR techniques, ligand based CoMFA model proved to have a key role in designing of novel antimalarial agents having q2
LOO of 0.806 and r2 value of 0.955. This study disclosed structural requirements for the development of potent antimalarial agents and concluded that hydrophobic, electrostatic and hydrogen bond donors play important roles in governing the biological activity. The contour maps (Fig. 23a-f) indicated that electronegative and hydrogen bond donor substituents at R1-phenyl ring were contributing positively for higher bioactivity.
3.2.4. Plasmepsin-II Inhibitors
Qidwai et al. [113] reported a QSAR model for artemisinin derivatives (96) as plasmepsin II inhibitors for antimalarial activity. The developed model proved to be robust with a non-cross validated r2
ncv 0.90 and predicted accuracy r2pred of 0.82.
Besides this, the model also proved beneficial in determining correlations of all atoms, ring counts, shape indices and connectivity indices with antimalarial activity.
O
OH
OO
CH3
H3C
HH
CH3R
H
(96)
Ojha and Roy [63] explored a series of di-tertiary amines (97) as plasmepsin-II inhibitors. Various topological, physicochemical and three-dimensional descriptors were used for the development of robust QSAR models which showed q2 values in the range of 0.487 to 0.664 and r2
pred values ranging in between 0.534 to 0.579. The AlogP98, log of partition coefficient was strongly contributing towards plasmepsin-II inhibition. Increasing the Alogp98 value increased biological activity significantly. The results obtained from this study also concluded that more number of “=O” was essential for higher activity, whereas increasing the number of rotatable bonds markedly diminished activity.
Me
O
N
R
R'
3 (97)
3.2.5. P. falciparum Cysteine Protease (Falcipain-2) Inhibitors
Teixeira et al. [114] reported 3D-QSAR analysis of a set of 39 peptidyl vinyl sulfone analogs (98) with potential
antimalarial activity. Since the predictivity and quality of the models depend mainly on alignment methods, alignments were defined in this study using ligand based and receptor-docked structure based approaches. Receptor-docked structure alignment method yielded the best statistical data in favor of CoMFA (q2 value of 0.696 and r2 of 0.980) and CoMSIA models along with electrostatic and hydrophobic fields (q2 value of 0.711 and r2 of 0.992).
HNR3
O
NH
S
O
R2
R1
O O
R1'
(98)
HNN
O
NH
S
O
O
O
Ph
Me
MeHO
(99)
CoMSIA (EH) model showed statistically superior results than CoMFA. The developed model gave insight into the structural requirements for designing of potent antimalarial agents. Substitution of bulky groups at R1’ and R3 positions, electropositive groups at R1 and R2 side chains and electronegative groups near to the SO2-R1’ moiety were essential for FP2 inhibitory activity. Substitution of bulkier groups at R2 position tended to diminish FP2 inhibitory activity. The best QSAR models developed by various research groups for antimalarial agents have been summarized in the Table 1.
3.3. Application of Pharmacophore Modeling Techniques for Discovery of Antimalarial Agents
Singh et al. [98] have successfully carried out pharmacophore modeling studies on natural and synthetic prodiginines for obtaining structural insights for their antimalarial activity against D6 and Dd2 Plasmodium strains. The common pharmacophore hypothesis (CPH) was generated using PLS. The five-featured best pharmacophore model (ADHRR, Fig. 24a) for D6 strain and six-featured (AADDRR, Fig. 24b) model for Dd2 strain has been reported. Validation of the developed pharmacophore models were carried out with QSAR analysis having good predictive ability. A ligand based approach for pharmacophore mapping has been reported with the aim to highlight the spatial bioactive arrangement of various chemical features of thymidine analogs as potent antimalarials by Ojha and Roy [111]. On the basis of the number of conformers generated, the best 10 hypotheses were developed out of which hypothesis 2 (Fig. 25a) with HBA, HBD and HBD features proved to be a significant one. External validation through mapping of test set compounds to individual hypotheses showed r2
pred of

178 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
Table 1. Summary of QSAR models for antimalarial agents.
Sr. No.
No. of Comp. Used Type of Model Descriptors Used
Validation Parameters Ref.
r r2 q2 r2pred.
1 56 • GFA (Linear+Spline)
• MSA descriptors • Spatial (Jurs-RNCG, Jurs-TASA) • Shape (NCOSV) • Electronic • Thermodynamic
- 0.70 0.66 0.57 [63]
2 95 • GFA (Spline) • DRAGON descriptors (S_aaSC, nOxolanes, S_sOH and SC-3_P)
- 0.76 0.73 0.75 [64]
3 48
• SW-MLR (2D-QSAR)
• Quantum mechanical (EHOMO, ELUMO, DM, polarizability, atomic charge on selected atoms) • Electronic (η, χ, ω, S) • Reactivity (electrophilicity index) • Physicochemical (Log P, GGUT_SLOGP_0 and SLogP_VSA_5) • Topological (KierFlex)
- 0.78 0.67 - [65]
• PLS
CoMFA • Steric • Electrostatic - 0.97 0.63
(Loo) 0.81
CoMSIA • Steric • Electrostatic • Hydrophobic - 0.92 0.53
(Loo) 0.77
4 179 • MLR
• Structural • Electronic • TOPO (D/Dr1) • RDF (RDF020u, RDF02030u) • ACF (C- 011) • BCUT (BCUT_SLOGP_3) • PEOE
- 0.81 - 0.88 [81]
5 21 • PLS
• CONST • Quantity and trajectory • Information indexes • Adjacency indexes • Molecular radic profiles • RDF • FUNCT • Charge • D2 • Connectivity indexes • 2D-AUTO • BCUT • 3D-Morse • ACF • WHIM • GETAWAY (RTe+) • HE • QO11
- 0.92 0.83 - [82]
6 88 • PLS
HQSAR • Molecular hologram - 0.87 0.57 -
[83] CoMFA • Steric • Electrostatic - 0.83 0.72 0.63
CoMSIA • Steric • Electrostatic • Hydrophobic - 0.87 0.72 0.71
7 18
• SW- MLR
HB3 strain • Quantum (HOMO) • kappa shape index (kappa3) • T_N_N_5 • EA • T_2_C_4
- 0.90 0.80 0.74
[84]
• GA- MLR
Dd2 strain • Quantum (XY polarizability) - 0.91 0.83 0.72
8 90 • PLS
• 2D-AUTO (MATS4m, MATS7e) • ACF(H-052) • 3D-Morse (Mor30p) • GETAWAY (R7m)
- 0.73 0.68
(Loo) 0.67 (test)
[85]
9 21 • GA-MLR • 2D-AUTO (MATS2P) • 3D-Morse (Mor11v) • WHIM (G2v) • GETAWAY (R4m)
- 0.98 - 0.74 [86]

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 179
(Table 1) contd…..
Sr. No.
No. of Comp. Used Type of Model Descriptors Used
Validation Parameters Ref.
r r2 q2 r2pred.
10 31 • PLS
3D7 strain
• 2D-AUTO (MATS1p, MATS5m, MATS1e, MATS1P ) • FUNCT (nCoNHR, nNH2Ph) • ACF (N-069) • Hydrophilic factor • Third order chi (X3A, X3Av)
0.92 - 0.82 -
[87] KI strain
• 2D-AUTO (MATS8e, MATS1p, MATS8p, MATS8v) • TOPO (IVDE) • FUNCT (nCoNHR, nNH2Ph, nNHR) • ACF (N-069, N-067)
0.90 - 0.75 -
KB cell lines
• TOPO (TI2, CIC0, CIC1) • MWC (MWC10) • FUNCT (nRORPh) • ACF (C-002, C-005, C- 008, H-046, H-052, N-067)
0.85 - 0.61 -
11 112 • GA-MLR • SMILES-based descriptors (RDF040v, Mor13e, FO6[N-o])
- 0.84 0.83 - [88]
12 20 • MLR • TOPO (χeq, oχv) • Physico-chemical parameters (MW, ST) • IP2
- 0.95 0.95
(Loo) - [89]
13 53 • SW-GFA
• Balaban index (Jx_2.4723) • STRU (Rotlbonds) • Thermodynamic (MR_24.7006) • Atom type descriptor (Atype_C_22)
- 0.79 0.74
(Loo) 0.80 [90]
14 49
• GA-MLR (2D-QSAR)
• Edge adjacency indices(EEig12d) • GETEWAY (HATS5p) • Information indices (CIC3) • TOPO (BLI) • 2D-FREQFING (F10[O-O] • MOE i3D (E_strain] • 2D-BIFING (B08[O-F] • Burden eigenvalue (BEHv7)
- 0.86 0.76
(Loo) -
[91]
• PLS
CoMFA • Steric • Electrostatic - 0.97 0.65
(Loo) -
CoMSIA • Steric • Electrostatic • Hydrophobic - 0.97 0.77
(Loo) -
15 53 • G/PLS • Physicochemical [Individual (MW, Volume), E-state contributions (SsOHE index, StsCE-index)]
- 0.86 0.80 0.79 [92]
16 35 • SEQ-MLR • GETAWAY (H6M, R2v) • 3D-Morse (Mor30v) • FUNCT (nNHR, NROR) • EMP (ARR)
- 0.94 0.81
(Loo) 0.75 [93]
17 35
• SW-PLS (2D-QSAR)
• Physicochemical (Individual, chi, path count, chi chain, chiV chain, chain path count, cluster, path cluster, kappa, element count, E-state numbers, E-state contributions, contributions, PSA)
- 0.90 0.81
(Loo) 0.87
[94]
• SA-PLS (GQSAR)
• Descriptors used in 2D QSAR along with Electro- TOPO and Bauman alignment TOPO
- 0.76 0.73 0.74
• SA-kNNMFA • Steric • Electrostatic • Hydrophobic - 0.78 0.69 0.74

180 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
(Table 1) contd…..
Sr. No.
No. of Comp. Used Type of Model Descriptors Used
Validation Parameters Ref.
r r2 q2 r2pred.
18 73
• GFA (Descriptor based QSAR)
• Radofgyration • Jurs-FPSA-2 • Jurs-PNSA-3 • TOPO (3χv
p) - 0.77
0.67 (Loo)
0.75
[95]
• GQSAR • Fragment based descriptors ( R1- MW, R2-oχv
, R4-carbons count, R3- slogp, R6-slogp)
- 0.73 0.67 0.70
• HQSAR • Fragment based descriptors (fragment size, Bin length)
- 0.91 0.53 0.62
• PLS
CoMFA • Steric • Electrostatic - 0.68 0.53
(Loo) 0.52
CoMSIA • Steric • Electrostatic • Hydrophobic • H-Don • H-Acc
- 0.77 0.50
(Loo) 0.63
19 109 • PLS
W2 strain • 2D-AUTO (MATS3e, MATS2v, MATS6e, GATS5v, GATS8e) • FUNCT (nRORPh, nPhX)
- 0.72 0.67
(Loo) 0.65
[96]
TM90 strain • CONST (Me, nO, nX) • TOPO [T(S.F.), LP1, IDDE] • EMP (ARR)
- 0.66 0.60
(Loo) 0.51
20 60 • CP-MLR • TOPO (IC5, MSD) • GVZ (JGI5, JGI6) • 2D-AUTO (MATs6m, GATS5e)
0.86 - 0.69
(Loo) - [97]
21 60
• MRA (Descriptor based)
D6 strain
• Inertia moment 2 length • Kier Chi6 (path) index • kappa 3 index • Wiener topological Index
0.88 0.77 0.61 0.60
[98] Dd2 strain
• Inertia moment 2 length • ADME H-bond donors • VAMP polarization XX component • VAMP quadpole XZ component
0.85 0.73 0.63 0.63
• PLS (Pharmaco-phore based)
D6 strain • H-Acc • H-Don • Aromatic • Hydrophobic
- 0.99 0.65 -
Dd2 strain
• H-Acc • H-Don • Aromatic - 0.99 0.85 -
22 63 • LS-SVM • Multivariate image analysis descriptors
- 0.94 0.91
(Loo) -
[99]
23 14 • MLR • Physicochemical (MW, MV, DM, EA, IP, PE, LogPo/w)
0.94 0.89 0.73
(Loo) - [100]
24 68 • GA-MLR
• TOPO [T(O--O)] • Geometrical (XY shadow) • Electrostatic (ESP-MNACH) • Quantum-chemical (MERIC, HOMO 1) • GETEWAY (HATS5e) • 2D-AUTO (MATS2v) • M logP
- 0.80 0.72
(Loo) 0.80 [101]
25 39 • PLS
CoMFA • Steric • Electrostatic - 0.85 0.71 0.84
[102] CoMSIA
• Steric • Electrostatic • Hydrophobic • H-Don • H-Acc
- 0.84 0.73 0.83
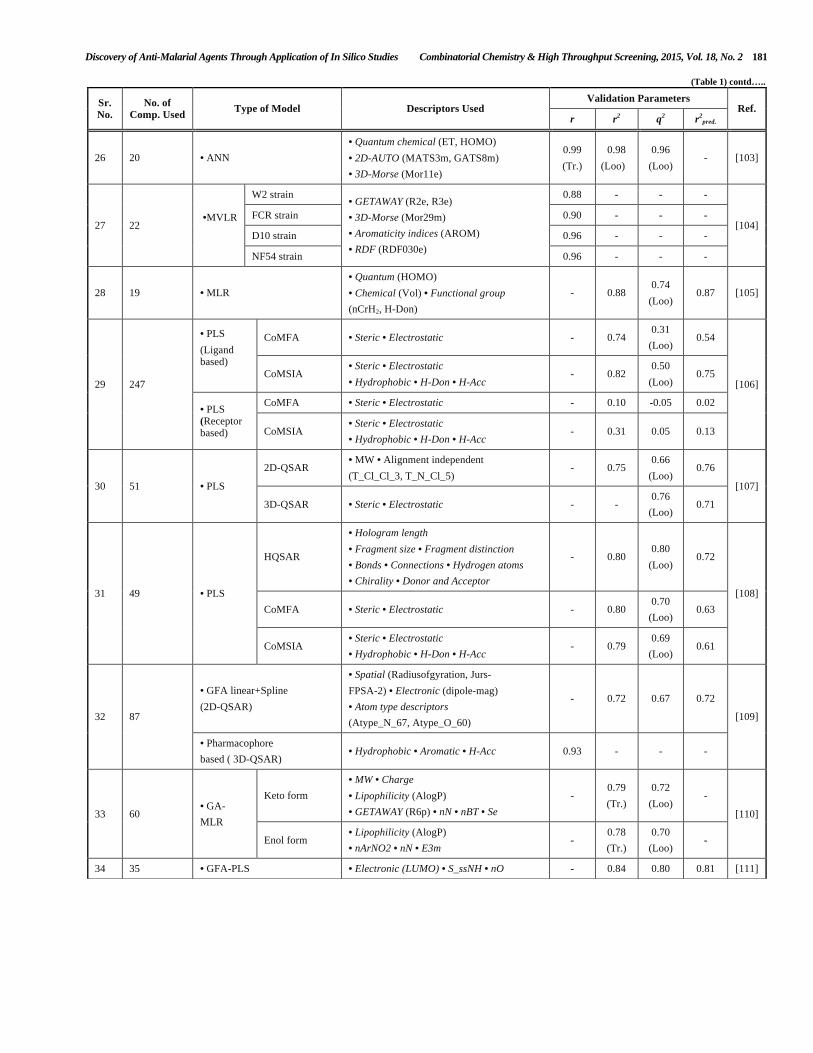
Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 181
(Table 1) contd…..
Sr. No.
No. of Comp. Used Type of Model Descriptors Used
Validation Parameters Ref.
r r2 q2 r2pred.
26 20 • ANN • Quantum chemical (ET, HOMO) • 2D-AUTO (MATS3m, GATS8m) • 3D-Morse (Mor11e)
0.99 (Tr.)
0.98 (Loo)
0.96 (Loo)
- [103]
27 22 •MVLR
W2 strain • GETAWAY (R2e, R3e) • 3D-Morse (Mor29m) • Aromaticity indices (AROM) • RDF (RDF030e)
0.88 - - -
[104] FCR strain 0.90 - - -
D10 strain 0.96 - - -
NF54 strain 0.96 - - -
28 19 • MLR • Quantum (HOMO) • Chemical (Vol) • Functional group (nCrH2, H-Don)
- 0.88 0.74
(Loo) 0.87 [105]
29 247
• PLS (Ligand based)
CoMFA • Steric • Electrostatic - 0.74 0.31
(Loo) 0.54
[106] CoMSIA
• Steric • Electrostatic • Hydrophobic • H-Don • H-Acc
- 0.82 0.50
(Loo) 0.75
• PLS (Receptor based)
CoMFA • Steric • Electrostatic - 0.10 -0.05 0.02
CoMSIA • Steric • Electrostatic • Hydrophobic • H-Don • H-Acc
- 0.31 0.05 0.13
30 51 • PLS
2D-QSAR • MW • Alignment independent (T_Cl_Cl_3, T_N_Cl_5)
- 0.75 0.66
(Loo) 0.76
[107]
3D-QSAR • Steric • Electrostatic - - 0.76
(Loo) 0.71
31 49 • PLS
HQSAR
• Hologram length • Fragment size • Fragment distinction • Bonds • Connections • Hydrogen atoms • Chirality • Donor and Acceptor
- 0.80 0.80
(Loo) 0.72
[108] CoMFA • Steric • Electrostatic - 0.80
0.70 (Loo)
0.63
CoMSIA • Steric • Electrostatic • Hydrophobic • H-Don • H-Acc
- 0.79 0.69
(Loo) 0.61
32 87
• GFA linear+Spline (2D-QSAR)
• Spatial (Radiusofgyration, Jurs- FPSA-2) • Electronic (dipole-mag) • Atom type descriptors (Atype_N_67, Atype_O_60)
- 0.72 0.67 0.72
[109]
• Pharmacophore based ( 3D-QSAR)
• Hydrophobic • Aromatic • H-Acc 0.93 - - -
33 60 • GA- MLR
Keto form • MW • Charge • Lipophilicity (AlogP) • GETAWAY (R6p) • nN • nBT • Se
- 0.79 (Tr.)
0.72 (Loo)
-
[110]
Enol form • Lipophilicity (AlogP) • nArNO2 • nN • E3m
- 0.78 (Tr.)
0.70 (Loo)
-
34 35 • GFA-PLS • Electronic (LUMO) • S_ssNH • nO - 0.84 0.80 0.81 [111]

182 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
0.638. The mapping of HBA, HBD and HBD features matched with the respective 4-positioned ketonic group, 3’- positioned hydroxyl group and the side chain -NH group of aryl substituent of thymidines. The mapping of hypothesis 2 on the most active compound was carried out as shown in (Fig. 25b). A pharmacophore hypothesis was reported by Sharma et al. [94] with the aim to get information about chemical features of azaaurone derivatives required for antimalarial activity. The best model was developed with four pharmacophore features i.e. one hydrogen bond acceptor (HBA), one hydrogen bond donor (HBD) and two hydrophobic rings. Among the various pharmacophoric features, hydrogen bond donor groups contributed mainly for the highest antimalarial activity. In the pharmacophore
model inter-group distances have been measured in order to clarify their role in bioactivity. As per the suggestion of the authors potential antimalarial agents could be designed with increasing the distance between aromatic and polar groups. Another four-featured pharmacophore model was reported by Aher and Roy [109] having significant correlation coefficient (r) of 0.932 for a diverse set of aminothiazoles and aminopyridines against multi-drug resistant strain (k1) of P. falciparum. The best defined hypothesis was reported to have one hydrogen bond acceptor, one hydrophobic (HYAI) and two aromatic rings. The developed model was validated externally with mapping of the most active (100) and the least active (101) compounds using the defined hypotheses. Statistics for the developed model showed close correlation value between the
(Table 1) contd…..
Sr. No.
No. of Comp. Used Type of Model Descriptors Used
Validation Parameters Ref.
r r2 q2 r2pred.
35 36 • PLS
HQSAR
• Hologram length • Fragment size • Fragment distinction • Bonds • Connections • Hydrogen atoms • Chirality • Donor and Acceptor
- 0.96 0.75
(Loo) 0.87
[112]
CoMFA • Steric • Electrostatic - 0.95 0.80
(Loo) 0.82
CoMSIA • Steric • Electrostatic • Hydrophobic • H-Don • H-Acc
- 0.96 0.65
(Loo) 0.74
36 36 • MLR
• Atom count (all atoms) • SASA • Connectivity index (order 1, standard) • Ring count (all rings) • Shape index (basic kappa, order 2)
- 0.90 0.82 0.82 [113]
37 39 • PLS
CoMFA • Steric • Electrostatic - 0.98 0.69 0.76
[114] CoMSIA
• Steric • Electrostatic • Hydrophobic • H-Don • H-Acc
- 0.99 0.71 0.74
ACF: Atom-centered fragments; ALogP: Ghose/Crippen octanol/water partition coefficient; ANN: Artificial neural network; AROM: Measures the degree of Aromaticity; Mor29m: Signal 29/weighted by atomic masses; BCUT: Eigen value of modified adjacency matrix; CoRAL: CoRrelation And Logic; CONST: Constitutional; CP-MLR: Combinatorial protocol in multiple linear regression; DM: Dipole moment; D2: torsion angle of O1-O2_Fe-N2; EA: Electron affinity; ET: Total energy; ESp-MNACH: Electrostatic potential-minimum net atomic charge for an H atom; EHOMO: Energy for HOMO; ELUMO: Energy of LUMO; FA: Factor analysis; FUNCT: Functional; Fo6[N-O]: 3D cum indicator variable; GVZ: Galvez topological charge; GETAWAY: Geometry, topology and atom-weights assembly; GA: Genetic algorithm; GATS5e: Geary aurocorrelation-lag5/weighted by atomic Sanderson electronegativities; GFA: Genetic function approximation; G/PLS: Genetic partial least squares; Η: Hardness; H-Don: Hydrogen donor; H-Acc: Hydrogen acceptor; HE: Hydration energy; HQSAR: Hologram QSAR; HOMO: Highest occupied molecular orbital energy; HOMO-1: Energy of the molecular orbital below HOMO; HATS5e: Leverage-weighted autocorrelation of lag 5 / weighted by atomic Sanderson electronegativities; IP: Ionization potential ; IP2: Indicator parameter for presence of halogen on aromatic ring; IC5: Information content index; JGI5: Mean topological charge index of order 5; JGI6: Mean topological charge index of order 6; Jurs-FPSA-2: Fractional charged partial positive surface area; Jurs-TASA: Total hydrophobic surface area; Jurs-RNCG: Relative negative charge; Jurs-FNSA-3: Fractional charged partial negative surface area; kappa3: Third kappa shape index; KNNMF: K-nearest neighbor molecular field analysis; LS-SVM: Least square support vector machine; Loo- Leave one out; LogP o/w: Partition coefficient in octanol/water; MATS2v: Moran autocorrelation -lag 2/weighted by atomic van der Waals volumes; MATS2p: Moran autocorrelation - lag 2/weighted by atomic polarizabilities; MATS6m: Moran autocorrelation-lag6/weighted by atomic masses; MERIC: Maximum electrophilic reactivity index for a C atom; MR: Molar refractivity; MLR: Multiple linear regression; MSA: Molecular shape analysis; MFA: Molecular field analysis; MRA: Multiple regression analysis; Mor2gm: Signal2g/weighted by atomic masses; MSD: Mean surface distance index; MOE i3D: Molecular operating environment descriptors based on internal coordinates; MV: Molecular volume; MVLR: Multivariable linear regressions; Mor11v: 3D-MoRSE - signal 11 / weighted by avv; MSD: Mean square distance index (Balaban); MlogP: Moriguchi octanol-water partition coefficient; MWC: Molecular walks counts; MW: Molecular weight; NBT: Number of bonds; NCOSV: Non-common overlap steric volume; N-PLS: Multilinear partial least squares; nCrH2: Number of ring secondary carbon, C (sp3), atoms; nBT: Number of bonds; nO: Number of oxygen atoms present in the molecules; nOxolanes: Number of oxolanes functional groups present in the molecules; PCA: Principle component analysis; PCR: Principle component regression; PE: Potential energy; PEOE: Partial equalization of orbital electronegativity; PLS: Partial least squares; PSA: Polar surface area; Q_O11: Charge on Oxygen atom of O11; Qm: Mean absolute atomic charge; R1u+ : R maximal autocorrelation of lag 1/unweighted; R4m: R autocorrelation of lag 4/weighted by atomic masses; R5m+ R: Maximal autocorrelation of lag 5/weighted by atomic masses; Radofgyration: Measure of the size of an object; RDF: Radial distribution function; RDF030e: RDF 3.0/weighted by atomic Sanderson electronegativities; RDF030v: RDF-4.0/weighted by atomic van der Walls volume; R2e: The R autocorrelation of lag 2; R3e: R autocorrelation of lag 3; R6p: R autocorrelation of lag 6/weighted by atomic polarizabilities; RTe+: maximum rate of R/Sanderson electronegativity; S: Softness; SA: Simulated annealing; SASA: Solvent accessibility surface area; SMILES: Simplified molecular input line energy system; Se: Sum of atomic Sanderson electronegativities; ST: Surface tension; STRU: Structural; SPA: Successive projection algorithm; SW: Stepwise regression; S_sOH: Summation of E-state index of fragment -OH; S_aaSC: Summation of E-state values for the aromatic carbon fragments; S_ssNH: Summation of E-state values for fragments; SC-3_P: Number of third-order subgraphs in the molecular graph; Tr: Training set; TOPO: Topological; T(O- -O): Summation of topological distance between Oxygen atoms within the molecule; T_C_N_4: Count of number of Carbon atoms separated from any nitrogen atom by four bond distance; T_N_N_5: Count of number of Nitrogen atoms separated from any other Nitrogen atom by five bonds; WHIM: Weighted holistic invariant molecular design; XY Shadow: Characterizing shape and extent of the molecule in terms of its 3D coordinates; 2D-AUTO: 2D-Autocorrelations; 2D-BIFING: 2D-Binary fingerprints; 2D-FREQFING: 2D-Frequency fingerprints; 3D-Morse: 3D-Molecule representation of structure based on electron diffraction signals; ω: Electrophilicity; ωi: Electrophilicity index; χ eq: Equalized electronegativity; oχv: Kier & hall valence connectivity indices; 3χp
v: Third order valence modified path connectivity index.

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 183
actual and the fixed costs and large difference between the actual and the null costs. Results obtained with the reported model concluded that for designing of novel antimalarial agents, the two aromatic rings were essential out of which one should be either a pyrazole/pyridine/thiazole and the other a phenyl or some heterocycle.
N
ON
HN
NH2NF3C
HNO
O
N
SNH2
(100) (101)
3.4. Studies Based on Virtual Screening
Zhang et al. [115] have discovered novel anti-malarial compounds by using QSAR-based virtual screening. Firstly, QSAR models were developed and validated by using a dataset of 3133 compounds. The dataset was screened for its anti-malarial activity by using in vitro P. falciparum growth inhibition assay. The assay results showed that 158 compounds out of 3133 exhibited reproducible potency (EC50) better than 2 µM and were considered as active compounds, while the remaining 2975 compounds were considered inactives. For virtual screening, ChemBridge databaset of 4,54,638 organic compounds were used. By using different filters like Applicability Domain (AD), Drug-likeness, QSAR model and Consensus Prediction Thresholds (CPT), 176 organic compounds were predicted as putative hits possessing antimalarial activity, along with a set of 42 inactive compounds as negative controls. Total 25 computational hits were tested experimentally and out of this 18 compounds showed moderate anti-malarial activity having EC50 range from 2 to 8 µM and 7 compounds with
Fig. (24). The best pharmacophore models a) ADHRR (D6 strain) and b) AADDRR (Dd2 strain). Used with permission from Singh et al., Curr. Comput. Aided Drug Des., 2013, 9, 350-359 [Copyright© 2014, Bentham Science Publishers] (images are available online).
Fig. (25). (a) The best featured hypothesis 2 pharmacophore, (b) mapping on the most active compound. Used with permission from Ojha and Roy, Biosystems, 2013, 13, 177-195 [Copyright© 2013, Elsevier Inc.] (images are available online).
(a) (b)
(a) (b)

184 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
EC50 values less than 2 µM. The most potent compound (102) showed EC50 value of 95.6 nM. The 42 inactive compounds which were used as a negative control were also tested for growth inhibition of P. falciparum and for mammalian cell drug susceptibility assays. All these 42 inactives were found to possess EC50 values of more than 10 µM, which ultimately confirmed the high specificity of the model.
N
O
O
N
H2C
(102)
3.5. Studies Based on Combined Ligand and Structure Based Virtual Screening
Shah et al. [116] applied a combined ligand and structure-based virtual screening approach wherein mining of the compounds bearing the substructures of compounds (37, 38 and 40) was carried out from Asinex and Chembridge databases having 2009 compounds. After the docking, approximately 1500 compounds were matching the subgraphs of compounds (37, 38 and 40). Further, approximately 300 compounds (top 20 %) were selected on the basis of top Emodel score. Finally 69 compounds were evaluated and a total of 28 compounds were identified as low micromolar inhibitors of FP-2 and FP-3. The best analogs obtained through virtual screening from tetrazole, triazole and quinazoline series were compounds (103-105) exhibiting IC50 values of 6.67, 18.61 and 4.64 µM, respectively. This study emphasized three important factors that contributed to potent inhibition of FP-2 and FP-3: • Strong H-bond network with the key residues of the
active site of FP-2 such as Gly83, Cys42, Gln36 and Asn173.
• Displacement of unfavorable hydration sites by complementary groups of the ligands.
• Chemical reactivity of the electrophiles toward the catalytic cysteine of the active site.
CONCLUSION
Malaria is one of the deadliest diseases among the parasite infections that contributed for infection of more than 40% of the world population. It affected mainly the people living in economically backward countries. The countries of
Asia and Africa have suffered from lack of controlled trials due to availability of only small number of patients. Current antimalarial therapy is based on the use of old drugs. Development of drug resistance in the parasite puts a question mark on the treatment of malaria in the future. In the last few years number of lead molecules has been reported as antimalarial agents but to establish their potential as drug candidates remains a major task. Issues related to drug resistance, rapid onset of action, safety particularly in children and pregnant women and curing malaria in a single dose needed to be addressed for these new molecules. Due to lack of above mentioned features these chemotypes have failed to become successful drug candidates [117]. To address this global issue, there is a need to develop lead molecules with diverse scaffolds to which the parasite has not been exposed before. The molecular hybridization approach which combines scaffolds of two active drugs together into one significant molecule for the same or different target sites has proved to be one of the best strategies to overcome drug resistance. The basis of this strategy depends on the hypothesis that during treatment, one of the drugs might be prone to resistance but the other drug has the ability to kill the parasite [118, 119]. Currently world’s eyes are focused on the development of malaria vaccine which at the moment suffers from the drawbacks of route of administration and the need for its cryopreservation. CADD has already established its role as a live platform for the development of novel leads of biological interest. Hence, a concerted approach by medicinal chemists, computational chemists and pharmacologists may prove helpful for the development of novel antimalarial agents. The present review focuses mainly on the efforts made by researchers around the world reinvigorating drug discovery programme with the application of in silico techniques. In this review we have discussed about chemometric methods used for the designing of antimalarials under the heading of structure based and ligand based drug design techniques. Docking studies of different compounds with parasite enzymes like PfDHFR, PfLDH, PfTCTP, PfRIO2 kinase, PfGSK-3 and FP2 have been performed. In ligand based drug design, QSAR studies of various scaffolds along with pharmacophore modeling have been discussed with concluding remarks. Among the QSAR studies of different analogs it has been observed that 4-aminoquinoline is still a live scaffold for antimalarial drug discovery studies. Some of the researchers have also been engaged in designing of novel leads based on natural and synthetic prodiginine scaffold. The research is also aimed at designing of heterocyclic rings (i.e. pyrazole, tetrazole, thiazole and pyridine) containing lead molecules for antimalarial activity.
HN
OF
N N
NNHN
O
ON
Me
NN
MeMe Me
S
HNN
S
O
N
NN
SNH
Me
O
HNMe
(103) (104) (105)

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 185
CONFLICT OF INTEREST
The authors confirm that contents of this article have no conflict of interests.
ACKNOWLEDGEMENTS
The authors thank the University Grant commission (UGC), New Delhi for the sanction of Major Research Project entitled, “Design and development of novel antimalarial agents” and for the award of Junior Research Fellowship to Mahesh A. Barmade.
REFERENCES
[1] Malaria fact sheet N°94, Updated March 2014. http://www.who.int/ mediacentre/factsheets/fs094/en/. (Accessed on August 30, 2014)
[2] Worrall, E.; Basu, S.; Hanson, K. Is malaria a disease of poverty? A review of the literature. Trop. Med. Int. Health, 2005, 10, 1047-1059.
[3] Renslo, A. R.; Mckerrow, J.H. Drug discovery and development for neglected parasite diseases. Nat. Chem. Biol., 2006, 2, 701-710.
[4] Greenwood, B.B.; Fidock, D.A.; Kyle, D.E.; Kappe, S.H.I.; Alonso, P.L.; Collins, F.H.; Duffy, P.E. Malaria: progress, peris, and prospects for eradication. J. Clin. Invest., 2008, 118, 1266-1276.
[5] Greenwood, B.M.; Bojang, K.; Whitty, C.J.M.; Targett, G.A.T. Malaria. Lancet, 2005, 365, 1487-1498.
[6] White, N.J. Plasmodium knowlesi: the fifth human malaria parasite. Clin. Infect. Dis., 2008, 46, 172-173.
[7] Paul, E.P.; Céline, C.; Diane, P.; Edwin, M. Understanding and Modelling the Impact of Climate Change on Infectious Diseases - Progress and Future Challenges, Climate Change - Socioeconomic Effects, Kheradmand, H., Ed.; In tech: China, 2011; pp.43-66
[8] Chareonviriyaphap, T.; Bangs, M.J.; Ratanatham, S. Status of malaria in Thailand. Southeast Asian J. Trop. Med. Public Health, 2000, 31, 225-237.
[9] Immunization, Vaccines and Biologicals. http://www.who.int/ immunization/diseases/malaria/en/. (Accessed on August 30, 2014)
[10] Baird, J.K. Effectiveness of antimalarial drugs. N. Engl. J. Med., 2005, 352, 1565-1577.
[11] Wiesner, J.; Ortmann, R.; Jomaa, H.; Schlitzer, M. New antimalarial drugs. Angew. Chem. Int. Ed., 2003, 42, 5274-5293.
[12] Reyburn, H.; Mtove, G.; Hendriksen, I.; Von Seidlein, L. Oral quinine for the treatment of uncomplicated malaria. Brit. J. Med., 2009, 339, b2066.
[13] Gregson, A.; Plowe, C.V. Mechanisms of resistance of malaria parasites to antifolates. Pharmacol. Rev., 2005, 57, 117-145.
[14] Uhlemann, A.C.; Krishna, S. Antimalarial multi-drug resistance in Asia: mechanisms and assessment. Curr. Top. Microbiol. Immunol., 2005, 295, 39-53.
[15] Fitch, C.D. Ferriprotoporphyrin IX, phospholipids, and the antimalarial actions of quinoline drugs. Life Sci., 2004, 74, 1957-1972.
[16] Abdul-Ghania, R.; Faraga, H.F.; Allama, A.F. Sulfadoxine-pyrimethamine resistance in Plasmodium falciparum: A zoomed image at the molecular level within a geographic context. Acta Tropica., 2013, 125, 163-190.
[17] Basco, L.K.; Tahar, R.; Ringwald, P. Molecular basis of in vivo resistance to sulfadoxine-pyrimethamine in African adult patients infected with Plasmodium falciparum malaria parasites. J. Antimicrob. Chemother., 1998, 42, 1811-1814.
[18] White, N.J. Antimalarial drug resistance. J. Clin. Invest., 2004, 113, 1084-1092.
[19] Srivastava, I.K.; Rottenberg, H.; Vaidya, A.B. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J. Biol. Chem., 1997, 272, 3961-3966.
[20] Ashley, E.A.; Dhorda, M.; Spread of Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med., 2014,371, 411-423.
[21] Q&A on artemisinin resistance. http://www.who.int/malaria/media/ artemisinin_resistance_qa/en/. (Accessed on August 30, 2014)
[22] Angus, B. Novel anti-malarial combinations and their toxicity. Expert Rev. Clin. Pharmacol., 2014, 7, 299-316.
[23] Salvador, A.; Hernadez, R.M.; Pedraz, J.L.; Igartua, M. Plasmodium falciparum malaria vaccines: current status, pitfalls and future directions. Expert Rev. Vaccines, 2012, 11, 1071-1086.
[24] Sahu, N.K.; Sahu, S.; Kohli, D.V. Novel molecular targets for antimalarial drug development. Chem. Biol. Drug. Des., 2008, 71, 287-297.
[25] Olliaro, P.L.;Yuthavong, Y. An overview of chemotherapeutic targets for antimalarial drug discovery. Pharmacol. Ther., 1999, 81, 91-110.
[26] Roy, K.; Ojha, P.K. Advances in quantitative structure-activity relationship models of antimalarials. Expert Opin. Drug Disco., 2010, 5, 751-778.
[27] Mane, U.R.; Gupta, R.C.; Nadkarni, S.S.; Giridhar, R.R.; Naik, P.P.; Yadav, M.R. Falcipain inhibitors as potential therapeutics for resistant strains of malaria: a patent review. Expert Opin. Ther. Patents, 2013, 23, 165-187.
[28] Mane, U.R.; Li, H.; Huang, J.; Gupta, R.C.; Nadkarni, S.S.; Giridhar, R.; Naik, P.P.; Yadav, M.R. Pyrido[1,2-α]pyrimidin-4-ones as antiplasmodial falcipain-2 inhibitors. Bioorg. Med. Chem., 2012, 20, 6296-6304.
[29] Mane, U.R.; Mohanakrishnan, D.; Sahal, D.; Murumkar, P.R.; Giridhar, R.; Yadav, M.R. Synthesis and biological evaluation of some novel pyrido[1,2-α] pyrimidin-4-ones as antimalarial agents. Eur. J. Med. Chem., 2014, 79, 422-435.
[30] Satyanarayanajois, S.D. Drug design and discovery: Methods and protocols. Humana Press, New York, 2011.
[31] Veselovsky, A.V.; Ivanov, A.S. Strategy of computer-aided drug design. Curr. Drug Targets Infect. Disord., 2003, 3, 33-40.
[32] Fong, P.; Lei, H. A Systematic review on computer-aided drug design, docking and scoring. Journal of Macao Polytechnic Institute, 2010, 47-51.
[33] Kubinyi, H. success Stories of Computer-Aided Design. In: Computer Applications in Pharmaceutical Research and Development; Sean Ekins, Ed.; John Wiley & Sons, Inc: New York, 2006; pp. 377-424.
[34] Cobb, K. Dock this: in silico drug design feeds drug development. Biomed. Comput. Rev., 2007, 20-30.
[35] Murumkar, P.R.; Le, L.; Truong, T.N.; Yadav, M.R. Determination of structural requirements of influenza neuraminidase type A inhibitors and binding interaction analysis with the active site of A/H1N1 by 3D-QSAR CoMFA and CoMSIA modeling. Med. Chem. Commun., 2011, 2, 710-719.
[36] Nag, A.; Dey, B. Computer-aided Drug Design and Delivery Systems. The McGraw-Hill Companies. New York; 2011.
[37] Martin, Y.C. Computer assisted rational drug design. Methods Enzymol., 1991, 203, 587-613.
[38] Kroemer, R.T. Structure-based drug design: docking and scoring. Curr. Protein Pept. Sci., 2007, 8, 312-328.
[39] Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J., 1986, 5, 823-826.
[40] Ginalski, K. Comparative modeling for protein structure prediction. Curr. Opin. Struct. Biol., 2006, 16, 172-177.
[41] Murumkar, P.R.; Sharma, M.K.; Giridhar, R.; Yadav, M.R. Virtual screening-based identification of lead molecules as selective TACE inhibitors. Med. Chem. Res., 2014, DOI 10.1007/s00044-014-1097-7.
[42] Schneider, G. Virtual screening: an endless staircase? Nat. Rev. Drug Discov., 2010, 9, 273-276.
[43] Rester, U. From virtuality to reality-Virtual screening in lead discovery and lead optimization: a medicinal chemistry perspective. Curr. Opin. Drug Disco. Devel., 2008, 11, 559-568.
[44] Cronin, M.T.D. In: Challenges and Advances in Computational Chemistry and Physics; Jerzy leszczynski, Ed.; Springer Science B.V: New York, 2010; Vol.8, pp. 3-11.
[45] Selassie, C.D. Burger’s Medicinal Chemistry and Drug Discovery; Donald Abraham, Ed.; John Wiley & Sons, Inc. New York, 2003; Vol. 1, pp. 1-42.
[46] Azevedo, W.F.; Dias, R. Computational methods for calculation of ligand-binding affinity. Curr. Drug Targets, 2009, 9, 1031-1039.
[47] Putz, M.V.; Dudas, N.A. Variational principles for mechanistic quantitative structure activity relationship (QSAR) studies: application on uracil derivatives’ anti-HIV action. Struct. Chem., 2013, 24, 1873-1893.

186 Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 Barmade et al.
[48] Putz, M.V.; Dudas, N.A. Determining chemical reactivity driving biological activity from SMILES transformations: The bonding mechanism of anti-HIV pyrimidines. Molecules, 2013, 18, 9061-9116.
[49] Putz, M.V.; Tudoran, M.; Putz, A. Structure properties and chemical-Bio/ecological of PAH interactions: From synthesis to cosmic spectral lines, nanochemistry, and lipophilicity-drives reactivity. Curr. Org. Chem., 2013, 17, 2845-2871.
[50] Tollenaere, J.P. The role of structure-based ligand design and molecular modeling in drug discovery. Pharm. World Sci., 1996, 18, 56-62.
[51] Yang, S. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today, 2010, 15, 444-450.
[52] Puntambekar, D.S.; Giridhar, R.; Yadav, M.R. 3D-QSAR CoMFA/CoMSIA studies on 5-aryl-2,2-dialkyl-4-phenyl-3(2H)-furanone derivatives as selective COX-2 inhibitors. Acta. Pharm., 2006, 56, 157-174.
[53] Yadav, M.R.; Shirude, S.T.; Puntambekar, D.S.; Patel, P.J.; Prajapati, H.B.; Parmar, A.; Balaraman, R.; Giridhar, R. Studies in 3,4-diaryl-1,2,5-oxadiazoles and their N-oxides: Search for better COX-2 inhibitors. Acta Pharm., 2007, 57, 13-30.
[54] Murumkar, P.R.; Giridhar, R.; Yadav, M.R. 3D-quantitative structure-activity relationship studies on benzothiadiazepine hydroxamates as inhibitors of tumor necrosis factor-α converting enzyme. Chem. Biol. Drug Des., 2008, 71, 363-373.
[55] Murumkar, P.R.; Gupta, S.D.; Zambre, V.P.; Giridhar, R.; Yadav, M.R. Development of predictive 3D-QSAR CoMFA and CoMSIA models for β-aminohydroxamic acid-derived tumor necrosis factor-α converting enzyme inhibitors. Chem. Biol. Drug Des., 2009, 73, 97-107.
[56] Murumkar, P.R.; Zambre, V.P.; Yadav, M.R. Development of predictive pharmacophore model for in silico screening, and 3D QSAR CoMFA and CoMSIA studies for lead optimization, for designing of potent tumor necrosis factor alpha converting enzyme inhibitors. J. Comput. Aided Mol. Des., 2010, 24, 143-156.
[57] Gupta, S.D.; Murumkar, P.R.; Giridhar, R.; Yadav, M.R. Studies on novel 2-imidazolidinones and tetrahydropyrimidin-2(1H)-ones as potential TACE inhibitors: Design, synthesis, molecular modeling, and preliminary biological evaluation. Bioorg. Med. Chem., 2009, 17, 3604-3617.
[58] Zambre, V.P.; Murumkar, P.R.; Giridhar, R.; Yadav, M.R. Structural investigations of acridine derivatives by CoMFA and CoMSIA reveal novel insight into their structures toward DNA G-quadruplex mediated telomerase inhibition and offer a highly predictive 3D-model for substituted acridines. J. Chem. Inf. Model., 2009, 49, 1298-1311.
[59] Zambre, V.P.; Murumkar, P.R.; Giridhar, R.; Yadav, M.R. Development of highly predictive 3D-QSAR CoMSIA models for anthraquinone and acridone derivatives as telomerase inhibitors targeting G-quadruplex DNA telomere. J. Mol. Graph. Model., 2010, 29, 229-239.
[60] Rawat, M.; Vijay, S.; Gupta, Y.; Dixit R.; Tiwari, P.K.; Sharma, A. Sequence homology and structural analysis of plasmepsin4 isolated from Indian Plasmodium vivax isolates. Infect. Genet. Evol., 2011, 11, 924-933.
[61] Kumari, S.; Priya A.M.; SajithaLulu, Tauqueer, M. Molecular modeling, simulation and virtual screening of ribosomal phosphoprotein P1 from Plasmodium falciparum. J. Theor. Biol., 2014, 343, 113-119.
[62] Trivedi, V.; Nag, S. In Silico characterization of atypical kinase PFD0975w from Plasmodium kinome: A suitable target for drug discovery. Chem. Biol. Drug Des., 2012, 79, 600-609.
[63] Ojha, P.K.; Roy, K. Exploring molecular docking and QSAR studies of plasmepsin-II inhibitor di-tertiary amines as potential antimalarial compounds. Mol. Simulat., 2011, 37, 779-803.
[64] Ojha, P.K.; Roy, K. Exploring structural requirements for a class of nucleoside inhibitors (PfdUTPase) as antimalarials: First report on QSAR, pharmacophore mapping and multiple docking studies. Comb. Chem. High Throughput Screen., 2013, 16, 739-757.
[65] Villalobos, T.P.; Ricardo, G.; Joel, J.A.; 2D, 3D-QSAR and molecular docking of 4(1H)-quinolones analogues with antimalarial activities. J. Mol. Graph. Model., 2013, 46, 105-124.
[66] Adane, L.; Bhagat, S.; Arfeen, M.; Bhatia, S.; Sirawaraporn, R.;Sirawaraporn, W.; Chakraborti, A.; Bharatam, P. Design and synthesis of guanylthiourea derivatives as potential inhibitors of
Plasmodium falciparum dihydrofolate reductase enzyme. Bioorg. Med. Chem. Lett., 2014, 24, 613-617.
[67] Gahtori, P.; Ghosh, S.K.; Parida, P.; Prakash, A.; Gogoi, K.; Bhat, H.; Singh, U. Antimalarial evaluation and docking studies of hybrid phenylthiazolyl-1,3,5-triazine derivatives: A novel and potential antifolate lead for Pf-DHFR-TS inhibition. Exp. Parasitol., 2012, 130, 292-299.
[68] Kalani, K.; Agarwal, J.; Alam, S.; Khan, S.; Pal, A.; Srivastava, S.K. In silico and in vivo anti-malarial studies of 18β glycyrrhetinic acid from Glycyrrhiza glabra. Plos One., 2013, 8, e74761.
[69] Chadha, R.; Gupta, S.; Shukla, G.; Jain, D.V.S.; Pissurlenkar, R.S.; Coutinho, E.C. Interaction of artesunate with β -cyclodextrin: Characterization, thermodynamic parameters, molecularmodeling, effect of PEG on complexation and antimalarial activity. Results in Pharma Sciences, 2011, 1, 38-48.
[70] Eichhorn, T.; Dominic, W.; Berthold, B.; Natalie, D.; Martin, F.; Wolf-Dieter, L.; Rolf, M.; Luise, K.; Thomas, S.; Joachim, G.; Thomas, E. Molecular interaction of artemisinin with translationally controlled tumor protein (TCTP) of Plasmodium falciparum. Biochem. Pharm., 2013, 85, 38-45.
[71] Bernardinelli, G.; Jefford, C.W.; Maric, D.; Thomson, C.; Weber, J. Computational studies of the structures and properties of potential antimalarial compounds based on the 1,2,4-trioxane ring structure. I. Artemisinin-like molecules. Int. J. Quant. Chem., 1994, 52, 117-131.
[72] Cristino, M.G.; Meneses, C.C.; Soeiro, M.M.; Ferreira, J.E.; Figueiredo, A.F.; Barbosa, J.P.; Almeida, R.C.O.; Pinheiro, J.C.; Pinheiro, A.L. Computational modeling of antimalarial 10-substituted deoxoartemisinins. J. Theor. Comut. Chem., 2012, 11, 241-263.
[73] Chouhan, D.K.; Sharon, A.; Bal, C. Molecular and structural insight into Plasmodium falciparum RIO2 kinase. J. Mol. Model., 2013, 19, 485-496.
[74] Parveen, A.; Chakraborty, A.; Konreddy, A.; Chakravarty, H.; Sharon, A.; Trivedi, V.; Bal, C. Skeletal hybridization and PfRIO-2 kinase modeling for synthesis of α-pyrone analogs as anti-malarial agent. Eur. J. Med. Chem., 2013, 70, 607-612.
[75] Shah, F.; Mukherjee, P.; Gut, J.; Legac, J.; Rosenthal, P. J.; Tekwani, B.L.; Avery M.A. Identification of novel malarial cysteine protease inhibitors using structure-based virtual screening of a focused cysteine protease inhibitor library. J. Chem. Inf. Model., 2011, 51, 852-864.
[76] Fugel, W.; Anselm, E.; Bernhard, G.; Ron, D.; Narkiss, P.; Lutz, P.; Laurence, H.P.; Blandine, B.; Morgane, R.; Ilya, O.; Christian, D.; Sebastian, K.; Thomas, L.; Laurent, M.; Conrad, K. 3,6-Dia-mino-4-(2-halophenyl)-2-benzoylthieno[2,3‑b]pyridine-5-carbonitriles are selective inhibitors of Plasmodium falciparum glycogen synthase kinase3. J. Med. Chem., 2013, 56, 264-275.
[77] Marom, N.; Tkatchenko, A.; Kapishnikov, S.; Kronik, L.; Leisterowitz, L. Structure and formation of synthetic hemozoin: insights from first-principles calculations. Cryst. Growth Des., 2011, 11, 3332-3341.
[78] Arafet, K.; Silvia, F.; Sergio, M.; Vicent, M. Quantum Mechanics/Molecular Mechanics Studies of the Mechanismof Falcipain 2 inhibition by the Epoxysuccinate E64. Biochemistry, 2014, 53, 3336-3346.
[79] Sullivan, D.J.; Kaludov, N.; Martinov, M.N. Discovery of potent, novel, non-toxic anti-malarial compounds via quantum modelling, virtual screening and in vitro experimental validation. Malar. J., 2011, 10, 274.
[80] Franco, J.; Blackie, M.A.L.; Toth, D.; Smith, P.J.; Capuano, J.; Fastnacht, K.; Berkes, C. A structural comparative approach to identifying novel antimalarial inhibitors. Comput. Bio. Chem., 2013, 45, 42-47.
[81] Abbasitabar, F.; Zare-Shahabadi, V. Development predictive QSAR models for artemisinin analogues by various feature selection methods: A comparative study. SAR QSAR Environ. Res., 2012, 23, 1-15.
[82] Santos, C.R.; Josinete, B.V.; Cleison, C. L.; Lorane, I. S.; Raimundo, N. P.; Clarissa, S. L.; Elizabeth, V. M.; Davi, S. B.; Williams, C. M.; José Carlos, T. C. A SAR and QSAR study of new artemisinin compounds with antimalarial activity. Molecules, 2014, 19, 367-399.
[83] Gupta, A.K.; Saxena A.K. Triple-layered QSAR studies on substituted 1,2,4-trioxanes as potential antimalarial agents: superiority of the quantitative pharmacophore-based alignment

Discovery of Anti-Malarial Agents Through Application of In Silico Studies Combinatorial Chemistry & High Throughput Screening, 2015, Vol. 18, No. 2 187
over common substructure based alignment. SAR QSAR Environ Res., 2013, 24, 119-134.
[84] Sahu, N.K.; Sharma, M.C.; Mourya, V.; Kohli, D.V. QSAR studies of some side chain modified 7-chloro-4-aminoquinolines as antimalarial agents. Arabian J. Chem., 2011, DOI:10.1016/j. arabjc.2010.12.005.
[85] Deshpande, S.; Goodarzi, M.; Katti, S.; Prabhakar, Y.S. Topological features in profiling the antimalarial activity landscape of anilinoquinolines: A multipronged QSAR study. J. Chem., 2013, Article ID 154629.
[86] Najafi, A.; Sobhanardakani, S. Multivariate modeling of cytochrome P450 enzymes for 4-aminoquinoline antimalarial analogues using genetic-algorithms multiple linear regression. Trop. J. Pharm. Res., 2013, 12, 905-912.
[87] Gupta, M.K. CP-MLR/PLS-directed QSAR studies on the antimalarial activity and cytotoxicity of substituted 4-aminoquinolines. Med. Chem. Res., 2013, 22, 3497-3509.
[88] Masand, V.H.; Toropov A.A.; Toropova A.P.; Mahajan D.T. QSAR models for anti-malarial activity of 4-aminoquinolines. Curr. Comput. Aided Drug Des., 2014, 10, 75-82.
[89] Shaik, B.; Singh, J.; Agrawal V.K. Prediction of antimalarial activity of some cyclic peroxy ketals using physico-chemical and topological indices. Med. Chem. Res., 2012, 21, 2097-2104.
[90] Ojha, P.K.; Roy, K. Comparative QSARs for antimalarial endochins: Importance of descriptor-thinning and noise reduction prior to feature selection. Chemometr. Intell. lab. Syst., 2011, 109, 146-161.
[91] Li, J.; Li, S.; Bai, C.; Liu, H.; Gramatica, P. Structural requirements of 3-carboxyl-4(1H)-quinolones as potential antimalarials from 2D and 3D QSAR analysis. J. Mol. Graph. Model., 2013, 44, 266-277.
[92] Ojha, P.K.; Roy, K. Exploration of important sites of antimalarial endochins for optimum structural modification using group-based QSAR (G-QSAR) modeling. Curr. Comput. Aided Drug Des., 2013, 9, 336-349.
[93] Gupta, A.K.; Singh, P.; Sabarwal, N. Rationalization of Molecular descriptor of aurone analogs toward antimalarial activity. Asian J. Pharm. Clin. Res., 2014, 7, 186-192.
[94] Sharma, M.C.; Sharma, S.; Sharma, P.; Kumar, A. Pharmacophore and QSAR modeling of some structurally diverse azaaurones derivatives as anti-malarial activity. Med. Chem. Res., 2014, 23,181-198.
[95] Ojha, P.K.; Roy, K. First report on exploring structural requirements of 1,2,3,4-tetrahydroacridin-9(10H)-one analogs as antimalarials using multiple QSAR approaches: descriptor-based QSAR, CoMFA-CoMSIA, 3D-QSAR, HQSAR and G-QSAR approaches. Comb. Chem. High Throughput Screen., 2013, 16, 7-21.
[96] Sharma, B.K.; Verma, S.; Prabhakar, Y.S. Topological and physicochemical characteristics of 1,2,3,4-tetrahydroacridin-9(10H)-ones and their antimalarial profiles: a composite insight to the structure-activity relation. Curr. Comput. Aided Drug Des., 2013, 9, 317-335.
[97] Singh, P.; Shekhawat N. Chemometric descriptors in the rationale of antimalarial activity of natural and synthetic prodiginines. J. Curr. Chem. Pharm. Sci., 2012, 40, 244-260.
[98] Singh, B.; Vishwakarma, R.A.; Bharate, S.B. QSAR and pharmacophore modeling of natural and synthetic antimalarial prodiginines. Curr. Comput. Aided Drug Des., 2013, 9, 350-359.
[99] Goodarz, M.; Freitas, M.P. MIA-QSAR coupled to different regression methods for the modeling of antimalarial activities of 2-aziridinyl and 2,3-bis-(aziridinyl)-1,4-naphtoquinonyl sulfate and acylate derivatives. Medicinal Chemistry, 2011, 7, 645-654.
[100] Mahajan, S.; Kamath, V.; Nayak, S.; Vaidya, S. QSAR analysis of benzophenone derivatives as antimalarial agents. Indian J. Pharm. Sci., 2012, 74, 41-47.
[101] Beheshti, A.; Eslam, P.; Mehdi, N.; Saadat, V. QSAR modeling of antimalarial activity of urea derivatives using genetic algorithm-multiple linear regressions. J. Saudi. Chem. Soc., 2012, DOI.org/10.1016/j.jscs.2012.07.019.
[102] Liang, T.; Luhui, R.; Qingshan, L. 3D-QSAR studies of tetraoxanes derivatives as antimalarial agents using CoMFA and CoMSIA approaches. Bull. Korean Chem. Soc., 2013, 34, 1823-1828.
[103] Worachartcheewan, A.; Chanin, N.; Chartchalerm, I.; Virapong, P. QSAR study of amidino bis-benzimidazole derivatives as potent anti-malarial agents against. Chem. Pap., 2013, 67, 1462-1473.
[104] Ibezim, E.; Duchowicz, P.R.; Ortiz, E.V.; Castro, E.A. QSAR on aryl-piperazine derivatives with activity on malaria, Chemometr. Intell. Lab. Syst., 2012,110, 81-88.
[105] Saghaie, L.; Hamidreza, S.; Hassan, S.; Mohsen, S.; Danial, S. Stepwise MLR and PCR QSAR study of the pharmaceutical activities of antimalarial 3-hydroxypyridinone agents using B3LYP/6-311++G** descriptors. Med. Chem. Res., 2013, 22, 1679-1688.
[106] Wang, L.; Yan, L.; Yinfeng, Y.; Shuwei, Z.; Ling, Y. Structural features of falcipain-3 inhibitors: an in silico study. Mol. BioSyst., 2013, 9, 2296-2310.
[107] Mishra, M.; Mishra, V.; Senger, P.; Pathak, A.; Kashaw, S.K. Exploring QSAR studies on 4-substituted quinazoline derivatives as antimalarial compounds for the development of predictive models. Med. Chem. Res., 2014, 23, 1397-1405.
[108] Sharma, R.; Patil, S.; Maurya, P. Drug discovery studies on quinolinebased derivatives as potential antimalarial agents. SAR QSAR Environ. Res., 2014, 25, 189-203.
[109] Aher, B.; Roy, K. QSAR and pharmacophore modeling of diverse aminothiazoles and aminopyridines for antimalarial potency against multidrug-resistant Plasmodium falciparum. Med. Chem. Res., 2014. Article in press (DOI 10.1007/s00044-014-0997-x)
[110] Masand, V.H.; Mahajan, D.T.; Gramatica, P.; Barlow, J. Tautomerism and multiple modeling enhance the efficacy of QSAR: antimalarial activity of phosphoramidate and phosphorothioamidate analogues of amiprophos methyl. Med. Chem. Res., 2014, DOI 10.1007/s00044-014-1043-8.
[111] Ojha, P.K.; Roy, K. First report on exploring structural requirements of alpha and beta thymidine analogs for PfTMPK inhibitory activity using in silico studies. Biosystems., 2013, 13, 177-195.
[112] Neves, B.J.; Bueno, R.V.; Braga, R.C.; Andrade, C.H. Discovery of new potential hits of Plasmodium falciparum enoyl-ACP reductase through ligand- and structure-based drug design approaches. Bioorg. Med. Chem. Lett., 2013, 23, 2436-2441.
[113] Qidwai, T.; Yadav, D.K.; Khan, F.; Dhawan, S.; Bhakuni, R.S. QSAR, docking and ADMET Studies of artemisinin derivatives for antimalarial activity targeting plasmepsin II, a Hemoglobin-Degrading Enzyme from P. falciparum. Curr. Pharm. Des., 2012, 18, 6133-6154.
[114] Teixeira, C.; Gomes, R.B.; Couesnon, T.; Gomes P. Molecular docking and 3D-quantitative structure activity relationship analyses of peptidyl vinyl sulfones: Plasmodium Falciparum cysteine proteases inhibitors. J. Comput. Aided Mol. Des., 2011, 25,763-775.
[115] Zhang, L.; Denis, F.; Alexander, S.; Hao, Z.; Alexander, G.; Sean, E.; Julie, C.; Michele, C. C.; Martina, S.; Dena, H.; Armand, G.; Kiplin, G.; Alexander, T. Discovery of novel antimalarial compounds enabled by QSAR-based virtual screening, J. Chem. Inf. Model., 2013, 53, 475-492.
[116] Shah, F.; Gut, J.; Legac, J.; Shivakumar, D.; Sherman, W.; Rosenthal, P.J.; Avery, M.A. Computer-aided drug design of falcipain inhibitors: virtual screening, structure-activity relationships, hydration site thermodynamics, and reactivity analysis. J. Chem. Inf. Model., 2012, 52, 696-710.
[117] Biamonte, M.A.; Wanner, J.; Le Roch, K.G. Recent advances in malaria drug discovery. Bioorg. Med. Chem. Lett., 2013, 23, 2829-2843.
[118] Sunny, M.; Rajesh, C. U.; Shabana, I. K.; Babu, L. T.; Diwan, S. R. Novel 4-aminoquinoline-pyrimidine based hybrids with improved in vitro and in vivo antimalarial activity. ACS Med. Chem. Lett., 2012, 3, 555-559.
[119] Shashi, P.; Pooja, A.; Kumkum, S.; RajaKumar, S.; Sunil, K. P.; Pravesh, V.; Saxena, J. K.; Abhisheak, S.; Jawahar, L.; Prem, M. C. Synthesis and bioevaluation of novel 4-aminoquinoline-tetrazole derivatives as potent antimalarial agents. Eur. J. Med. Chem., 2013, 66, 69-81.
Received: September 23, 2014 Revised: November 26, 2014 Accepted: November 27, 2014
Related Documents











