Seminar 1: Seminar 1: Efficient Algorithms Efficient Algorithms for Molecular for Molecular Dynamics Simulation Dynamics Simulation Andrew Noske Andrew Noske

Seminar 1: Efficient Algorithms for Molecular Dynamics Simulation Andrew Noske.
Dec 16, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Seminar 1:Seminar 1:
Efficient Algorithms for Efficient Algorithms for Molecular Dynamics Molecular Dynamics
SimulationSimulation
Andrew NoskeAndrew Noske

About the ProjectAbout the Project
Part of Part of TOMSKTOMSK (Towards Molecular (Towards Molecular Structure Kinetics)Structure Kinetics)
Build an “Build an “engine layerengine layer” for particle ” for particle simulationssimulations

About Molecular SimulationsAbout Molecular Simulations
Real molecules obey Real molecules obey quantum lawsquantum laws… but … but can approximate with can approximate with classical lawsclassical laws..
e.g.: e.g.: Newton’s lawNewton’s law: force=mass*acceleration: force=mass*acceleration
Molecular Dynamics (MD)Molecular Dynamics (MD): : integrates equations of motion integrates equations of motion of atoms each of atoms each timesteptimestep.. Cannot predict precisely what will Cannot predict precisely what will
happen: generates statistical prediction.happen: generates statistical prediction.

Chosen Interaction ModelChosen Interaction Model
Lennard Jones Potential Pair EquationLennard Jones Potential Pair Equation::
I will use this to simulateI will use this to simulateatoms in a stable liquidatoms in a stable liquid..
NOTE: Will be easy to overwrite/change NOTE: Will be easy to overwrite/change interaction modelinteraction model & add statistical analysis functions (e.g.: calc & add statistical analysis functions (e.g.: calc Temperature).Temperature).
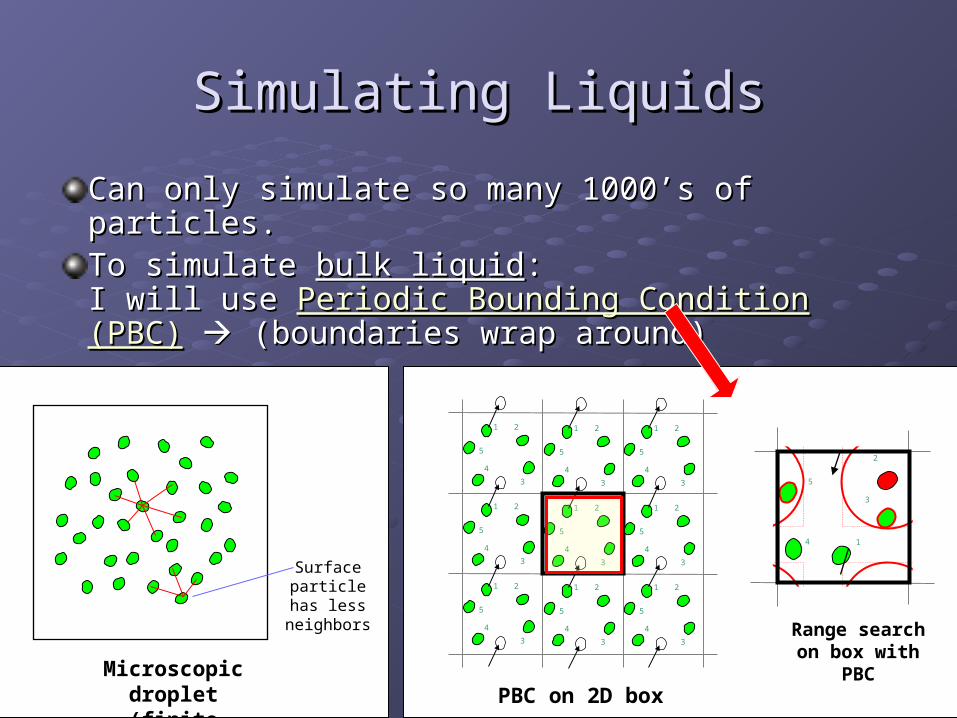
Simulating LiquidsSimulating Liquids
1
2
5
4
31
4
2
3
5
1
4
2
3
5
1
4
2
3
5
1
4
2
3
5
1
4
2
3
5
1
4
2
3
5
1
4
2
3
5
1
4
2
3
5
1
4
2
3
5
PBC on 2D box
Range search on box with PBC
Microscopic droplet(finite particles)
Can only simulate so many 1000’s of particles.Can only simulate so many 1000’s of particles.To simulate To simulate bulk liquidbulk liquid::I will use I will use Periodic Bounding Condition (PBC)Periodic Bounding Condition (PBC) (boundaries wrap around)(boundaries wrap around)
Surface particle has
less neighbors

N-body ProblemN-body Problem
N-body problemN-body problem: all (: all (NN) particles in a system ) particles in a system have pair-wise interaction.have pair-wise interaction.
Solutions:Solutions: Brute force approachBrute force approach
compare all pairs compare all pairs O(N O(N22)) Better approachBetter approach: approximate: approximate
distant forces distant forcesLead to many Lead to many specific solutions.specific solutions.
Consider ALL
Approximate
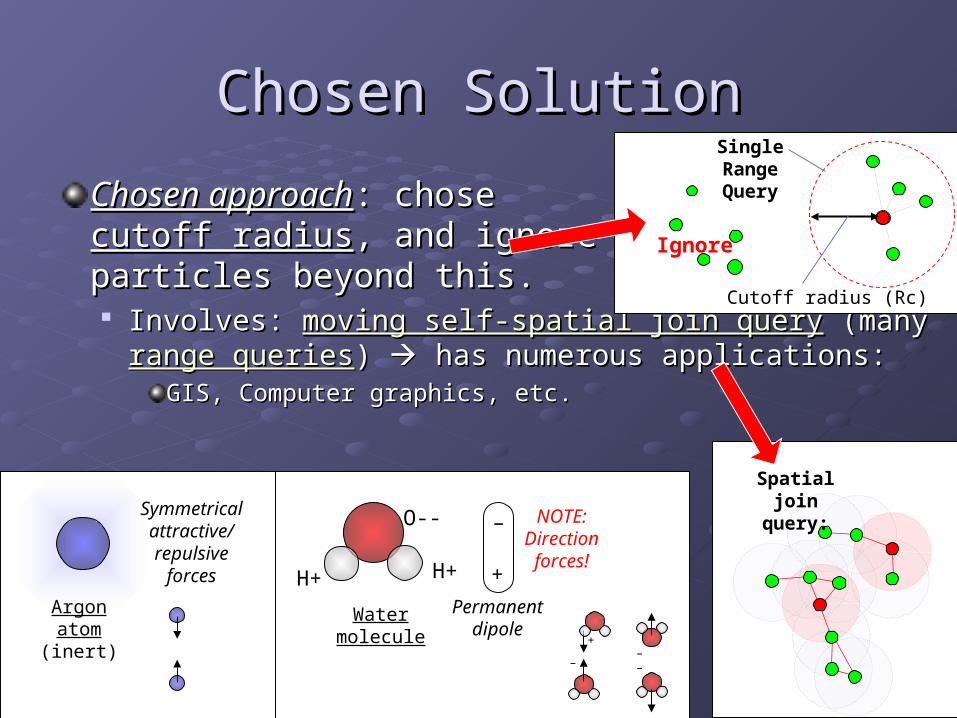
Chosen SolutionChosen Solution
Chosen approachChosen approach: chose : chose cutoff radiuscutoff radius, and ignore , and ignore particles beyond this.particles beyond this. Involves: Involves: moving self-spatial join querymoving self-spatial join query (many (many range range
queriesqueries) ) has numerous applications: has numerous applications:GIS, Computer graphics, etc.GIS, Computer graphics, etc.
Ignore
O--
H+ H+
–
+
Permanent dipole
Water molecule
Argonatom(inert)
Symmetrical attractive/ repulsive
forces
+
– ––
Cutoff radius (Rc)
Spatial join query:
NOTE:Direction forces!
Single Range Query

Spatial Data Structure: Fixed GridSpatial Data Structure: Fixed Grid
Reviewed many types of structures.Reviewed many types of structures.
Fixed gridFixed grid most effective for uniform particle most effective for uniform particle distribution.distribution. Time to Time to buildbuild (place (place
all pointsall pointsinto index) = O(N)into index) = O(N)
cell index = atom coordinate / cellLencell index = atom coordinate / cellLen
(along each dimension)(along each dimension)
Cell List technique
Cutoff radius(rc)
Fixed grid
cellLen
boxLen
rc
NOTE: cells per side (CPS)=5

Scientific ProcessScientific Process
Using Using Visual C++ console Visual C++ console applicationapplication..
Am using OO principles.Am using OO principles. Have read “Effective C++” & now Have read “Effective C++” & now
reading “More Effective C++”.reading “More Effective C++”.
Testing processTesting process:: Run a series of simulations in batch…Run a series of simulations in batch… Output results to Output results to CSVCSV… including:… including:
grid parameters,grid parameters,clock tics elapsed, clock tics elapsed, primary focus primary focusdistance calculates,distance calculates,& more& more
Analyze/graph CSV using Analyze/graph CSV using ExcelExcel.. Can be time consuming! Can be time consuming!

Simulation StepsSimulation Steps
Set-up:Set-up: #1) Setup grid structure#1) Setup grid structure #2) Setup atoms in offset lattice #2) Setup atoms in offset lattice #3) Assign random velocities. #3) Assign random velocities.
Iterate:Iterate: #1) #1) Build gridBuild grid (assign all atoms to cells). (assign all atoms to cells). #2) #2) Build neighbor listBuild neighbor list (for each atom in (for each atom in
each cell: find neighbors). each cell: find neighbors). can take 95% of time can take 95% of time #3) #3) Calculate force and move atomsCalculate force and move atoms implements implements
interaction model (can change).interaction model (can change). #4) Wrap atoms back into cell boundaries.#4) Wrap atoms back into cell boundaries. #5) Increment timestep.#5) Increment timestep.

Finding Good NeighboursFinding Good Neighbours
q
““Love thy neighbour” -- the bibleLove thy neighbour” -- the bible
q
q
Atom list approach:For each atom: check which cells are within
rc of atom
Cell list approach:Predetermine which cells are
within rc of each cell
NOTE: Volume sphere = NOTE: Volume sphere = 52%52% of it’s bounding cube of it’s bounding cube

Optimization Trick: Half-sphere QueryOptimization Trick: Half-sphere Query
i
j
i
j
Normal approach:
Search sphere
Faster approach:
Search upper hemi-sphere
NOTE: will capture each
neighbor twice (once from each end)

Smaller Optimization TricksSmaller Optimization Tricks
i
j
Early elimination if distanceEarly elimination if distancebetween atoms along anybetween atoms along anydimension > rc.dimension > rc.
Don’t calculate sqrt:Don’t calculate sqrt: Lennard-Jones can be doneLennard-Jones can be done
using distusing dist22
NOTE: if (NOTE: if (distdist2 2 <= cutoffRadius<= cutoffRadius22) ) is in range is in range
Determine optimal # of cells per size.Determine optimal # of cells per size. Is cell length = cutoff radius optimal?Is cell length = cutoff radius optimal?

Idea: Using MBRs in cellsIdea: Using MBRs in cells
For each cell, maintain a For each cell, maintain a Minimum Bounding Minimum Bounding Rectangle (MBR)Rectangle (MBR) around it’s atoms. around it’s atoms. For any cell JUST tipped by rc, check atom is inside For any cell JUST tipped by rc, check atom is inside
rc of MBR before considering atoms exhaustively.rc of MBR before considering atoms exhaustively.
This cell is just tipped”
MBR NOTE: can NOTE: can
also bealso beused inused inconjunction conjunction with “with “sub sub gridgrid””

Improving Cache Hits through Improving Cache Hits through Spatial LocalitySpatial Locality
Spatial locality principleSpatial locality principle: : objects objects close toclose to referred ones will referred ones will probablyprobably be requested again be requested again in the future.in the future. Unsorted atoms Unsorted atoms means many cache misses. means many cache misses.
13
2
4
5
6

Space Filling CurvesSpace Filling CurvesSpace-filling curveSpace-filling curve: : A line passing through A line passing through every point in a space, in some order (according every point in a space, in some order (according to some algorithm).to some algorithm).
Resort atom Resort atom periodicallyperiodically (group by cells & order using curve). (group by cells & order using curve). Improves CPU performance “>50% in 2D moving point query” Improves CPU performance “>50% in 2D moving point query”
worth trying.worth trying.
Row-wise Hilbert curveGray curve Z-ordering

Verlet Neighbour ListVerlet Neighbour List
Choose a “skin radius” greater than rc.Choose a “skin radius” greater than rc.
Build the “verlet neighbor list” using skin radiusBuild the “verlet neighbor list” using skin radius
Next few iterations: update list; check which neighbour pairs are Next few iterations: update list; check which neighbour pairs are inside rc.inside rc.
Refinement: only rebuild list when: sum of 2 max displacement (of Refinement: only rebuild list when: sum of 2 max displacement (of any 2 atoms) > skin “thickness”.any 2 atoms) > skin “thickness”.
1
4
2
5
6
Rl
7
7'
6'
3Rc
Cut-off sphere
Skin
Skin/verlet radius (Rv)
2 max displacements since rebuild

Some Verlet Specific IdeasSome Verlet Specific Ideas
Don’t check Don’t check displacementdisplacement each iteration: check at each iteration: check at decreasing periods.decreasing periods.
Don’t update Don’t update distances of atoms outside cut-off spheredistances of atoms outside cut-off sphere each iteration: check more frequently as it gets closer.each iteration: check more frequently as it gets closer.
Determine optimal skin radius.Determine optimal skin radius.
1
4
2
6
Rl
7
7'
6'
3Rc
Cut-off sphere
Skin
Skin/verlet radius (Rv)
2 max displacements since rebuild5

Performance vs. AccuracyPerformance vs. Accuracy
Some techniques that will Some techniques that will improve performance, but improve performance, but decrease “accuracy”:decrease “accuracy”: Don’t always rebuild/update Don’t always rebuild/update
neighbor list when necessary.neighbor list when necessary.
Increasing timestepIncreasing timestep
Decreasing cutoff radiusDecreasing cutoff radius
I can graph these by testing I can graph these by testing against a control.against a control.
larger timestep
small timestep
smaller rc
large rc

ProgressProgress
Basic grid is implementedBasic grid is implemented
Thesis started.Thesis started.
Several graphs obtained.Several graphs obtained.
Added front end:Added front end: Uses Uses MFCMFC (Microsoft Foundation (Microsoft Foundation
Class) using Class) using OpenGLOpenGL.. Demonstrates building on engine layer.Demonstrates building on engine layer. Lets me see particles animate (& work out problems)Lets me see particles animate (& work out problems) Code messyCode messy
Is still Is still MUCHMUCH to implement & test. to implement & test.

ConclusionConclusionMolecular dynamicsMolecular dynamics : an (approximated) : an (approximated) simulation of real worldsimulation of real worldparticle behaviour.particle behaviour.
Periodic Bounding ConditionPeriodic Bounding Condition used for “bulk” liquidused for “bulk” liquid
Best existing approach:Best existing approach: Verlet listVerlet list, ,
using using cell listcell list & & fixed gridfixed grid to build. to build.
Ideas for improvement:Ideas for improvement: Minimal half cell list templates.Minimal half cell list templates. MBRs inside cells.MBRs inside cells. Space filling curves.Space filling curves.
Thesis may resemble a guide to implementing/optimizing Thesis may resemble a guide to implementing/optimizing moving spatial join queriesmoving spatial join queries..
Still much to be done.Still much to be done.
1
4
2
5
6
Rl
7'
6'
3
Rc rc

Thank You…Thank You…
Any Questions?Any Questions?
cubic atom list vs cell list
0
500
1000
1500
2000
2500
0 2 4 6 8 10 12 14
cellSidesDivDistC
av
gT
ics
Pe
rIt
atoms=5000 - min atom lisboundary mbrsatoms=5000 - min atom list mbrs
atoms=5000 - min atom list ef f
atoms=5000 - cubic atom list
atoms=5000 - min cell list unloaded
atoms=5000 - min cell list
numAtoms: 1000-5000rc: 0.01-0.25boxLen: 1CPS: 10timeStep: 0.01timeStepsToExe: 20useVerlet: 0
Adjacency List Size Comparison
0
100
200
300
400
500
600
700
800
0 0.5 1 1.5 2 2.5 3 3.5 4 4.5
Cell sides per cutoff radius
# c
ell
s
cubic full
min full
cubic half
cubic half


Revised GoalRevised Goal
Implement engine as efficient as possible.Implement engine as efficient as possible.How it should work:How it should work: Pass in minimum parameters:Pass in minimum parameters:
# atoms, offset, max velocity & box size# atoms, offset, max velocity & box size(or load atom data from file).(or load atom data from file).cutoff radiuscutoff radius
Engine should set remaining details to Engine should set remaining details to optimal. Including:optimal. Including:
cells per side,cells per side,verlet radius,verlet radius,subgrid? mbr? algorithm? etc.subgrid? mbr? algorithm? etc.
rc
EXTRA SLIDE

Idea: Sub GridIdea: Sub Grid
““Sub grid adjacency list template guideSub grid adjacency list template guide”: break each cell ”: break each cell into (imaginary) sub-grid.into (imaginary) sub-grid.
When considering an atom, check with sub-cell it belongs to, When considering an atom, check with sub-cell it belongs to, that sub-cell will refine which adjacent cells to search.that sub-cell will refine which adjacent cells to search.
NOTENOTE:: My primary focus is: My primary focus is: reducing time per iterationreducing time per iteration……
but but main memory requirementsmain memory requirements (& (& setup timesetup time) also important.) also important. Choosing cell length equal Choosing cell length equal
to rc is typical. to rc is typical. But is it optimal?But is it optimal?
No one method/metric is No one method/metric is likely to be optimal in ALL likely to be optimal in ALL cases cases too many too many parameters.parameters.
EXTRA SLIDE
Related Documents











