doi:10.1182/blood-2007-06-096529 Prepublished online December 3, 2007; Luca Tamagnone and Alfonso Catalano Simona Moretti, Antonio Procopio, Raffaella Lazzarini, Maria Rita Rippo, Roberto Testa, Maurizio Marra, promoting Fas translocation into lipid rafts Semaphorin3A signaling controls Fas (CD95)-mediated apoptosis by (4217 articles) Neoplasia Articles on similar topics can be found in the following Blood collections http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requests Information about reproducing this article in parts or in its entirety may be found online at: http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprints Information about ordering reprints may be found online at: http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtml Information about subscriptions and ASH membership may be found online at: digital object identifier (DOIs) and date of initial publication. the indexed by PubMed from initial publication. Citations to Advance online articles must include final publication). Advance online articles are citable and establish publication priority; they are appeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet Copyright 2011 by The American Society of Hematology; all rights reserved. 20036. the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.org From

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

doi:10.1182/blood-2007-06-096529Prepublished online December 3, 2007;
Luca Tamagnone and Alfonso CatalanoSimona Moretti, Antonio Procopio, Raffaella Lazzarini, Maria Rita Rippo, Roberto Testa, Maurizio Marra, promoting Fas translocation into lipid raftsSemaphorin3A signaling controls Fas (CD95)-mediated apoptosis by
(4217 articles)Neoplasia �Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
digital object identifier (DOIs) and date of initial publication. theindexed by PubMed from initial publication. Citations to Advance online articles must include
final publication). Advance online articles are citable and establish publication priority; they areappeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet
Copyright 2011 by The American Society of Hematology; all rights reserved.20036.the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

1
Word Count: Abstract 156 Text 4409
Semaphorin3A signaling controls Fas (CD95)-mediated apoptosis by promoting Fas
translocation into lipid rafts
Simona Moretti1,2, Antonio Procopio1,2, Raffaella Lazzarini1,2, Maria Rita Rippo1,2, Roberto Testa3,
Maurizio Marra3, Luca Tamagnone4 and Alfonso Catalano1,2
1Department of Molecular Pathology and Innovative Therapies, Polytechnic University of Marche,
Ancona, Italy
2Center of Cytology and 3Diabetology Unit, Research Department, Italian National Research Centers
on Aging (INRCA-IRCCS), Ancona, Italy
4Institute for Cancer Research and Treatment, University of Turin, Candiolo, Italy
Short Title: Sema3A enhances Fas-mediated apoptosis
Corresponding author: Alfonso Catalano, Dipartimento di Patologia Molecolare, Politecnica delle
Marche, Via Tronto 10/A, 60100, Ancona, Italy. Phone: (39) 0712206245, Fax: (39) 0712206240, E-
mail: [email protected]; [email protected]
Blood First Edition Paper, prepublished online December 3, 2007; DOI 10.1182/blood-2007-06-096529
Copyright © 2007 American Society of Hematology
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

2
Abstract
Semaphorins and their receptors (plexins) have pleiotropic biological functions, including regulation of
immune responses. However, the role of these molecules inside the immune system and the signal
transduction mechanism(s) they utilize are largely unknown. Here, we show that Semaphorin3A
(Sema3A) triggers a proapoptotic programme that sensitizes leukemic T cells to Fas (CD95)-mediated
apoptosis. We found that Sema3A stimulation provoked Fas translocation into lipid raft microdomains
before binding with agonistic antibody or FasL (CD95L). Disruption of lipid rafts reduced sensitivity
to Fas-mediated apoptosis in the presence of Sema3A. Furthermore, we show that plexin-A1, together
with Sema3A-binding neuropilin-1, was rapidly incorporated into membrane rafts after ligand
stimulation, resulting in the transport of actin-linking proteins into Fas-enriched rafts. Cells expressing
a dominant-negative mutant of plexin-A1 did not show Fas clustering and apoptosis upon Sema3A/Fas
co-stimulation. This work identifies a novel biological function of semaphorins and presents an
unexpected signaling mechanism linking semaphorin to the tumor necrosis factor family receptors.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

3
Introduction
The semaphorin family comprises soluble and membrane-bound proteins that function during neuronal
development, organogenesis, angiogenesis and cancer progression1,2. This family has also attracted the
attention of immunologists as novel regulators of immune cell responses3. Various members of
semaphorins act as amplifiers of the immune response4, while others, the secreted semaphorins of class
3 in particular, may negatively control immune functions5. Previously, we have found that Semaphorin-
3A (Sema3A), a prototype member of secreted semaphorins of class 3, has the potential to inhibit T
cell functions by promoting growth arrest and/or blocking proinflammatory cytokine secretion6. The
downstream pathways by which Sema3A exerts its action on T cells are still unclear, but they are likely
to involve a receptor complex formed from one of two neuropilin (NP) proteins and one of four Plexin-
A proteins. NPs serve as the primary ligand binding sites and Plexin-As as the signal transducing
components7,8.
Recent evidence indicated that Sema3A/NP/PlexA signaling may regulate cell apoptosis9,10. In fact,
upon treatment with recombinant human Sema3A, neurons were found to undergo apoptosis.
Moreover, marked and prolonged protection from dopamine-induced apoptosis was achieved by co-
treatment with function-blocking anti-Sema3A antibodies. Therefore, Sema3A has been proposed to
act as an autocrine and paracrine “amplifier” signal for neuronal cell death. Apoptosis is a pivotal
process in immunological development that is frequently curbed in lymphoid leukemias. Interestingly,
the potential role of Sema3A in the regulation of apoptosis in immune cells has not been investigated
so far. To address this question, we assayed the potential regulatory function of Sema3A in Fas-
induced apoptosis of human leukemic cells.
Fas (CD95/APO-1) is the prototype of “death receptors” in the tumor necrosis factor (TNF) receptor
superfamily. It has been implicated in a wide range of apoptosis-based physiological processes, e.g. T-
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

4
cell-dependent cytotoxicity, deletion of autoreactive T and B cells, activation-induced cell death, tumor
surveillance, immune privilege, angiogenesis, as well as pathological degenerative diseases such as
autoimmunity, fulminant hepatitis, and neurodegeneration11,12. Upon ligation with its cognate ligand
FasL, or with agonistic antibodies, Fas rapidly recruits the Fas-associated death domain protein
(FADD) and procaspase 8, forming the so-called “death inducing signaling complex” (DISC). The
clustering of procaspase-8 molecules in the DISC complex results in their activation by self-cleavage,
triggering downstream effector caspases and eventually leading to apoptosis13. In certain cells
(indicated as “type I”), Fas is localized in detergent-resistant plasma membrane microdomains called
lipid rafts, and Fas-induced apoptosis depends on signaling pathways restricted to these
microdomains14. In contrast, in other cell types (i.e. “type II”), Fas is excluded from lipid rafts and its
signaling depends on the clustering with selected co-receptors15-17. Lipid rafts are cholesterol-rich
structures where adapters and kinases required for signal transduction are localized, including
glycosylphosphatidyl-inositol (GPI)-linked proteins and molecules connecting the actin cytoskeleton to
the plasma membrane (the so-called ERM proteins)18. The formation of membrane platforms where
Fas and its signal-transducers are brought in close proximity is thought to increase DISC formation and
therefore potentiate Fas signaling15-17. Here, we show a novel function of Sema3A, mediated by
plexins, whereby it increases Fas translocation into membrane rafts, sensitizing leukemic cells to Fas-
mediated apoptosis.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

5
Materials and methods
Cell culture and apoptosis
The human leukemic cell lines HUT78 and HUT78.B119 was kindly provided by R. Testi (University
of Rome, Tor Vergata, Italy). The other cell lines were obtained from American Type Culture
Collection and grown as described previously in RPMI-1640 culture medium supplemented with 10%
heat-inactivated fetal calf serum (FCS).6 Bone marrow aspirates, obtained from patients at the initial
diagnosis and after signing informed consent, were kindly provided by the Clinic of Hematology of the
Polytechnic University of the Marche (Ancona, Italy). Mononuclear cells isolated by Ficoll-Hypaque
density gradient centrifugation, consisting mostly of leukemia blasts (> 90%), were washed in
phosphate-buffered saline (PBS), resuspended in cell culture medium, and used immediately for
experimentation. Death receptor-mediated apoptosis was induced with either rhFasL
(rhsSuperFasLigandTM), TRAIL, anti-Fas mAb APO1-1 (Alexis Biochemicals, Milan, Italy), or
cytotoxic anti-Fas CH11 antibody (Upstate Biotechnology, Milan, Italy). Human recombinant Sema-
3A fused to human Fc fragment (Sema3A-Fc) were purchased from R&D Systems (Minneapolis, MN)
and added simultaneously to anti-Fas stimuli. For the NP1 functional blocking test, the blocking anti-
NP1 antibody, a generous gift of Dr. M. Tessier-Lavigne, was previously described.6 The control
antibody was a rabbit polyclonal anti-NP1 antibody (clone H286, Santa Cruz Biotechnology, Santa
Cruz, CA) with a nonblocking function.6 The caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp(Ome)-
fluoromethylketone (zVAD-fmk), the de novo RNA inhibitor actinomycin D or the protein synthesis
inhibitor cycloheximide were from Sigma (Milan, Italy). To disrupt lipid rafts, cells (5 x 105) were
pretreated with 15 µg/ml methyl-β-cyclodextrin (MBCD) (Sigma) for 1 hr at 37°C in serum-free
medium before Sema3A-Fc and Fas stimuli.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

6
For quantitative determination of apoptosis, cells (5 x 105) were fixed overnight in 70% ethanol at
4°C. Cells were then incubated for 1 hr with 1 mg/mL RNase A and 20 µg/mL propidium iodide at
room temperature, and analyzed with a Becton Dickinson (Milan, Italy) FACScan flow cytometer as
described previously20. Apoptotic cells were calculated as the percentage of cells in the sub-G1 region
in cell cycle analysis. Apoptotic cells were also identified using an annexin-V apoptosis detection kit
(BD-Pharmingen).
Immunofluorescence Flow Cytometry
Cell surface expression of Fas and NP1 was analyzed by immunofluorescence flow cytometry in 4 x
105 cells as described previously20 in a Becton Dickinson FACSCalibur™ flow cytometer using anti-
Fas DX2 mAb (a gift of Dr. R. Testi) and specific antibody against NP1 (Clone H286, Santa Cruz
Biotechnology, Santa Cruz, CA). FAM-VAD-fmk was obtained from Cell Technology, Inc. Isotype-
matched control antibodies were included in each staining.
Isolation of lipid rafts
Lipid rafts were isolated from 107 cells by nonionic detergent lysis and centrifugation on discontinuous
sucrose gradients exactly as described previously15. One ml fractions were collected from the top of the
gradient and 20 µl of each fraction was subjected to SDS-PAGE, immunoblotting, and enhanced
chemiluminescence detection. Location of lipid rafts was determined using cholera toxin (CTx) B
subunit conjugated to horseradish peroxidase (anti-GM1; Sigma) or by blotting fractions for the
tyrosine kinase Lck (obtained from Santa Cruz Biotechnology). Proteins were identified using the
following specific antibodies: anti-48-kDa Fas (C-20), anti-120-kDa-NP-1 (H286), anti-195-kDa-
plexin-A1 (H-60), anti-30-kDa RhoGDI (K-21) rabbit polyclonal antibodies and anti-21-kDa RhoA
mAb (Santa Cruz Biotechnology, Santa Cruz, CA.); anti-29-kDa FADD (clone-1) and anti-80-kDa
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

7
ezrin (clone-18) mAb (BD Transduction Laboratories, Lexington, KY); anti-55-kDa procaspase-8 (Ab-
3) mAb (Oncogene Research Products). Prestained protein molecular mass standards (Bio-Rad; Milan,
Italy) were run in parallel.
Confocal and immunofluorescence microscopy
To confocal microscopy, cells were settled onto poly-L-lysine-coated slides and analyzed with a Zeiss
LSM 510 laser scan confocal microscope (Oberkochen, Germany) for membrane raft and protein
visualization as described15,16. Colocalization assays were analyzed by excitation of the corresponding
fluorochromes in the same section. Negative controls, lacking the primary antibody or using an
irrelevant antibody, showed no staining. To immunofluorescence, cells (3 × 106) were washed and spun
onto glass slides at 900 rpm for 2 min. The cells were then fixed and exposed to purified anti-Fas
(DX2) and anti-plexin-A1 or anti-NP1 antibodies for 1 hr. Then, they were incubated for 30 min with
the secondary antibodies Cy3-conjugated goat anti-rabbit IgG and FITC-conjugated goat anti-mouse
IgG (Jackson ImmunoResearch Laboratories, Inc). F-actin filaments were stained with phalloidin-
TRITC and nuclei were stained with DAPI (Sigma, Milan, Italy). Each slide was mounted with
Fluoromount G (EMS, PA). Images were acquired and processed on a fluorescence microscope with
X40 objective. Image analysis and merging of images was done with Adobe PhotoShop 7.0 software
(Adobe System).
Real-Time RT-PCR
Real-Time RT-PCR analysis was done in a Chromo4 sequence detector (Bio-Rad, Milan, Italy) as
previously described6. The primers and probes of Plexin-A1, -A2, -A3 and -A4 genes were determined
by Laboratory Tools software analysis of Stratagene (Milan, Italy). As positive control for human
plexin-A1 and -A4, we used cDNAs derived from PBMC of healthy donors. Details of sequences and
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

8
thermal cycle conditions are available upon request. Data were acquired and analyzed with the
sequence detector Chromo4 software.
Immunoprecipitation and immunoblot analysis
Immunoprecipitation and immunoblots were performed from whole cell lysates as previously
described20-22. Briefly, cells were lysed with EB buffer (20 mM Tris-HCl at pH 7.4, 5 mM EDTA, 150
mM sodium chloride, 10% glycerol and 1% Triton X-100) in the presence of 1 µg/ml leupeptin, 3
µg/ml aprotinin, 1 µg/ml pepstatin, 2 mM phenylmethylsulphonyl fluoride and 1 mM sodium
orthovanadate. After immunoprecipitation with antibodies against NP1 and plexin-A1 (all by Santa
Cruz Biotechnology, Santa Cruz, CA), high-stringency washes were performed (EB buffer containing 1
M lithium chloride). Western blots were then performed and appropriate antibodies were detected
using enhanced chemiluminescence (Amersham Corp., Milan, Italy). In some experiments, blots were
reprobed with an anti-actin monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) as
loading control.
Dominant-negative plexin-A1 transfectants and RNA interference
Dominant-negative plexin-A1 expression lentiviral vector has been described previously8,21 and was
produced in 293T packaging cells, transiently cotransfected with a mixture of transfer, envelope, and
core-packaging plasmids. Conditioned media containing the vector was harvested 48 h after
transfection, and incubated with a fresh culture of sparse Jurkat cells, in the presence of 8 µg/µl
Polybrene (Sigma, Milan, Italy) for 16 h. The infected cells were previously exposed to 32 µM
genistein for 3 hrs to enhance lentiviral vector entry23. Genistein was purchased from Calbiochem
(Milan, Italy) and diluted in dimethyl sulfoxide (DMSO) prior to use. The final concentration of
DMSO in the cultures never exceeded 0.2% (vol/vol). The plexin construct included a vesicular
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

9
stomatitis virus (VSV) tag, detectable with an anti-VSV-G monoclonal antibody (V-5507; Sigma, St
Louis, MO) by immunoblotting.
Four siRNA sequences specific for human plexin-A1 were selected, synthesized and annealed by the
manufacturer (Dharmacon, Milan, Italy). Transfection was performed using RNAiFect (Qiagen)
according to the manufacturer’s protocol. After 48 h of incubation, the resulting cells were harvested,
washed and used for subsequent experiments. Transfection efficiencies were determined using
fluorescein-labelled non-silencing RNA (35-45%).
Statistical analysis
All values were expressed as mean ± s.e.m of no less than triplicate measurements of three independent
experiments. Comparison of results between different groups was performed by one-way analysis of
variance, paired t-test and ANOVA using StatView 5.0 (NET Engineering, Pavia, Italy). A P value ≤
0.05 was considered statistically significant.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

10
Results
Pro-apoptotic activity of Sema3A in association with Fas-stimulation
The pro-apoptotic activity of Sema-3A was assessed in a variety of human tumor cells. We first used
Fas-sensitive T leukemia cells Jurkat (as type II cell line) and HUT78 (as type I cell line), as well as the
Fas-resistant HUT78.B1 cells, previously derived from HUT7814. As reported for type I and type II
cells15, Jurkat cells expressed less Fas on their cell surface than HUT78 or HUT78.B1 cells (Figure
1A). rhFasL or the agonistic cytotoxic CH11 anti-Fas antibody induced apoptosis in Jurkat and HUT78
cells, but not in HUT78.B1 cells (Figure 1, B and C). In response to APO1-1, an IgG1 isotype switch
variant of anti-Fas antibody that induces apoptosis in type I but not in type II cells15, HUT78 cells
underwent apoptosis, whereas Jurkat and HUT78.B1 cells were almost completely resistant (Figure 1,
B and C). In contrast, all of the cells were sensitive to TNF-related apoptosis-inducing ligand (TRAIL)
(Figure 1, B and C). When Jurkat (type II) cells were treated with either rhFasL, CH11 or APO1-1, the
co-stimulation with Sema3A strongly increase the sensitivity to cell death (Figure 1B). Sema3A was
less effective in sensitizing HUT78 (type I) cells, whereas it had no effect on HUT78.B1 cells (Figure
1C). Moreover, Sema3A did not increase the sensitivity to cell death induced by TRAIL (Figure 1, B
and C). These data show that Sema3A sensitizes some cell-types to die via anti-Fas stimuli.
We next investigated the involvement of Sema3A-receptor NP1 in the pro-apoptotic activity of
Sema3A. Jurkat, HUT78 and HUT78.B1 expressed similar protein levels of NP1 on their cell surface
(Figure 2A). However, the addition of blocking antibodies directed against NP1 (anti-NP1) abrogated
the sensitizing effect mediated by Sema3A, but had no effect on Fas-induced cell death (Figure 2B).
These data suggest that Sema3A, through NP1 receptor, enhances the death signal induced by Fas
activation, especially in Jurkat (type II) cells.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

11
Sema3A increases Fas-mediated caspase activation in type II, but not in type I cells
We then analyzed the effect of Sema3A co-treatment on the cleavage of procaspase-8 and -3, which is
the hallmark of Fas-induced apoptosis11-13. In a time-course experiment, we observed full cleavage of
caspase-8 and -3 after only 2 hours of Sema3A/anti-Fas co-stimulation in Jurkat cells (Figure 3A),
while APO1-1 alone was only partly effective at this stage. In contrast, we did not find a substantial
shift in the activation of caspase-8 and -3 in HUT78 cells. In H9 T lymphoma cells, another type I cell
line, there was no change in caspases activation after Sema-3A co-treatment; whereas in the type II
CEM T leukemia cells we found complete cleavage of caspase-8 and -3, similar to Jurkat cells (data
not shown). Consistently, the enhancement of Fas-mediated apoptosis by Sema3A was blocked by pre-
incubating Jurkat or CEM cells with the broad-range caspase inhibitor zVAD-fmk (Figure 3B).
Therefore, the observed differences in Fas-mediated cell death in presence of Sema3A in type II cells
seem to correlate with the intracellular activation of caspases.
Increased Fas-mediated apoptosis by Sema3A does not require new protein synthesis or Rap1
activity
We next examined whether Sema3A could induce Fas up-regulation on the cell surface, resulting in
higher receptor density and signaling potential. However, Sema3A did not increase Fas expression on
the cell surface in Jurkat cells, after up to 6 hours of treatment (at the stage in which the increased
sensitivity to apoptosis was observed; Supplementary Figure 1A). To assess whether the pro-apoptotic
activity of Sema3A was dependent on new protein synthesis, we pretreated Jurkat cells with
actinomycin D (Act. D) or cycloheximide (CHX). Neither Act. D nor CHX were able to inhibit the
increased sensitivity to cell death mediated by Sema3A (Supplementary Figure 1B).
Moreover, since we have previously shown that the small G protein Rap1 is involved in NP1-
mediated Sema3A inhibitory signaling in T-cells6, we asked whether the same signal transducer could
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

12
be implicated in the regulation of Fas-induced apoptosis. We expressed a dominant-negative Rap1
(Rap1N17) in Jurkat cells; however Sema3A co-treatment promoted apoptosis in cells expressing
Rap1N17 in a comparable manner as in cells transfected with an empty vector (Supplementary Figure
1C). Therefore, the pro-apoptotic signaling of Sema3A occurred independently of Rap1 activation.
Sema3A stimulation recruits Fas into the lipid rafts, where semaphorin receptor NP1 is localized
In the absence of the cognate ligand, redistribution of Fas on lipid rafts is a potential mechanism to up-
regulate its signaling in type II cells15-17. Therefore, we asked whether Sema3A may enhance Fas-
mediated apoptosis in Jurkat (type II) cells by Fas redistribution in lipid rafts. The membrane proteins
from Jurkat cell lysates were separated by sucrose gradient ultra-centrifugation and analyzed by
western blotting as previously described15. Fractions containing the lipid rafts (4-5) were identified by
the presence of the tyrosine kinase Lck (Figure 4A). In un-stimulated cells, Fas was not found in lipid
rafts, whereas after 60 min treatment with Sema3A, a marked proportion of it translocated into lipid
rafts (Figure 4A). Neither FADD nor procaspase-8 were recruited into lipid rafts upon Sema3A
stimulation (Figure 4A), consistent with the notion that Fas re-localization into lipid rafts in type II
cells occurs before any stimulation with death receptor ligands15,16. The Sema3A receptor NP1 was
markedly present in lipid rafts before Sema3A treatment and its localization did not change upon
stimulation (Figure 4A). Also in this case, Fas translocation from soluble (S, fraction 11) to insoluble
lipid raft (R, fraction 4) was NP1 dependent, because it was entirely abrogated in the presence of anti-
NP1 blocking antibody (Figure 4B). The raft-associated glycosphingolipid GM1 showed localization of
the raft fraction in these experiments (Figure 4B). Therefore, a portion of NP1 is distributed into lipid
rafts, and its stimulation with Sema3A elicits the recruitment of Fas into lipid rafts.
As previously described16, the disruption of lipid rafts by cholesterol depletion using methyl-β-
cyclodextrin (MBCD) did not affect Fas-mediated apoptosis in Jurkat cells (Figure 4C). However, by
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

13
pre-treating with MBCD we completely abrogated Sema3A-mediated amplification of Fas apoptosis
(Figure 4C). These results indicated that Fas translocation into lipid rafts induced by a NP1-dependent
Sema3A signaling is required to increase Fas-mediated apoptosis.
Sema3A co-stimulation enhances Fas-induced apoptosis in primary leukemic cells
Our data indicated that Sema3A can sensitize some leukemic cell lines to Fas-mediated apoptosis. To
extend our findings to primary leukemic cells, we studied bone marrow cells derived from patients with
acute myeloblastic leukemia or acute promyelocytic leukemia (M2 or M3, respectively, following the
FAB classification). These M2 and M3 leukemia cells were positive for Fas and NP1 (>60% and >50%
positive cells, respectively; data not shown), and underwent apoptosis after Fas stimulation
(Supplementary Figure 2A). Consistent with that seen in Jurkat cells, primary leukemia cells were
sensitized to Fas-induced cell death by treatment with Sema3A, and this effect was inhibited by MBCD
(Supplementary Figure 2A). Staining of leukemia cells with the fluorescent caspase substrate FAM-
VAD-fmk showed that a higher percentage of cells receiving anti-Fas and Sema3A co-treatment
contained more activated caspases than cells receiving anti-Fas alone. Consistently, this effect was
blocked by MBCD pretreatment (Supplementary Figure 2B). Therefore, Sema3A and Fas stimuli may
act in synergy to enhance caspase activation and apoptosis in primary leukemic cells.
The Sema3A co-receptor plexin-A is involved in enhancing Fas-mediated cell death
In addition to NP1, members of the plexin-A family (plexin-A1, -A2, -A3, and -A4) serve as
components of receptor complexes for Sema3A.8 We found that plexin-A1 and -A3 mRNAs are
expressed in cell lines HUT78 and H9 (type I), Jurkat and CEM (type II), as well as in primary M2 and
M3 leukemic cells. In contrast, plexin-A2 and -A4 were virtually absent in these cells (Figure 5A).
Plexin-A1 was expressed more abundantly than plexin-A3, and it was confirmed at the protein level by
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

14
immunoblotting (Figure 5B). Moreover, by immunofluorescence microscopy, plexin-A1 and NP1 were
found to co-localize with Fas in Sema3A-treated Jurkat cells, but not in untreated cells (Figure 5, C and
D). A substantial proportion of plexin-A1 translocated into lipid rafts in cells stimulated with Sema3A
(Figure 6A) and the association of plexin-A1 with NP1 was increased in response to Sema3A (Figure
6B). Since we found that NP1 is constitutively present in these membrane microdomains (see Fig. 4),
our data indicate that Sema3A enhances NP1/plexin-A1 complex formation into lipid rafts.
To determine the role of plexin-A1 in Sema3A-mediated signaling, we generated a clone from the
leukemic Jurkat cell line that express a previously characterized plexin-A1 dominant-negative form8,21
(DN-plexin-A1). The control and DN-plexin-A1-transfected cells displayed similar sensitivities to Fas
stimulation alone. However, the DN-plexin-A1 cells was less sensitive to Fas/Sema3A co-triggering
than control cells (Figure 6C). We also selectively knocked down plexin-A1 gene expression. RNAi
against plexin-A1 reduced the pro-apoptotic activity of Sema3A on Jurkat cells (Figure 6D). These
results suggest that NP1 and plexin-A1 are functional receptor components for Sema3A.
Fas re-localization induced by Sema3A requires actin cytoskeleton
We next asked how Fas is concentrated in lipid rafts by Sema3A/NP/PlexA signaling. Clustered rafts
are often bound to the cytoskeleton, and Sema3A signaling is thought to alter actin reorganization in
immune cells24,25. To investigate whether Sema3A could modify the actin cytoskeleton in human
leukemic cells, Jurkat cells treated with Sema3A were stained with phalloidin-TRITC for F-actin and
with Hoechst 33258 to assess nuclear morphology and visualized by fluorescence microscopy. Similar
to FasL, Sema3A caused both cell shape change and a redistribution of the actin cytoskeleton (Figure
7A). In addition, by pre-treating Jurkat cells with the microfilament-disrupting agent cytochalasin-B,
we prevented the sensitization to Fas-induced apoptosis mediated by Sema3A (Figure 7B).
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

15
Ezrin, a major component of the ERM protein family (ezrin, radixin, moesin), can interact with Fas
and mediate Fas cell membrane localization during Fas-induced apoptosis18. We found that ezrin as
well as actin dynamics regulatory proteins, such as RhoA and RhoGDI, were accumulated in lipid rafts
following Sema3A stimulation (Figure 7C). In comparison, the amount of either ezrin, RhoA or
RhoGDI redistributed in the lipid rafts was very faint when the DN-plexin-A1 cell line was stimulated
with Sema3A alone (Figure 7D). In addition, Fas re-localization into lipid rafts was not observed in
these cells upon Sema3A treatment (Figure 7E). All together, these findings indicate that PlexinA-
driven Fas translocation into lipid rafts sensitizes leukemic cells to apoptosis and that this effect seems
to require actin-linking proteins accumulation in membrane rafts and cytoskeleton reorganization.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

16
Discussion
The recruitment of Fas to lipid rafts, elicited by independent non-apoptotic receptor signaling,
represents a non-completely-understood pathway that modulates Fas-induced cell death14-16.
Semaphorins play a central role in axonal guidance, and emerging evidence points to diverse functions
of several semaphorins (including Sema3A) in the immune system. For instance, class IV semaphorins
(e.g. Sema4D and Sema4A) play crucial roles in the reciprocal stimulation between T cells and APCs,
both in vitro and in vivo26. In addition, Sema3A inhibits the migration of human monocytes in response
to cytokine stimulation27 as well as T cell proliferation and cytokines production under stimulating
conditions6. In this study, we identify a novel biological function for semaphorins in immune cells and
an unexpected signaling mechanism, namely, the coupling of Sema3A to a death receptor.
A pivotal role of secreted semaphorins in the regulation of neuronal cell death has been previously
established28, although the implicated pathways have not been characterized as yet. The present study
demonstrates that secreted Sema3A and its receptor NP1 are important determinants of leukemic cells
sensitivity to Fas-mediated death signals (Figs. 1 and 2). De novo protein synthesis is not essential for
the pro-apoptotic activity of Sema3A, but the redistribution and clustering of membrane-bound Fas into
lipid raft microdomains is a pivotal step for cell death signaling (Fig. 4).
Membrane rafts could serve to generate high local concentration of Fas, as platforms for coupling
adaptor and effector proteins required for Fas downstream signaling, facilitating and amplifying
signaling processes by transient local assembly of various cross-interacting molecules15-17. This is of
particular importance in Fas-mediated signal transduction because death receptors lack enzymatic
activity and the pathway is triggered by protein/protein interactions. The presence of Fas in lipid rafts is
restricted to cells previously described as type I, which show more efficient formation of death
signaling complexes and greater sensitivity to Fas stimuli. In peripheral T cells, it has been shown that
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

17
the membrane distribution of Fas is dynamically regulated by antigen receptor signaling and possibly
by other signals 15-17. This can increase Fas-mediated apoptosis in type II cells and may be important to
promote the clonotypic elimination of chronically stimulated T cells. As some leukemic primary cells
are described as type II cells, in which Fas signaling is dependent on receptor-mediated clustering in
membrane microdomains18, our observations could be also relevant for apoptotic pathways in leukemia
cells. Notably, Sema3A expression is reduced in primary leukemic cells, whereas they express the
receptor NP1 at high levels.29,30 Therefore, the Sema3A-dependent regulatory mechanism described
here may be relevant to curb uncontrolled T-cell proliferation.
The intracellular domain of NP1 is short, and apparently unable to mediate functional responses to
Sema-3A.1 Plexin-A1 is a co-receptor for Sema-3A in the nervous system2-4 and it has a signaling role
in immune responses31,32. We detected plexin-A1 expression in our cells and a dominant negative
mutant of plexin-A1 or siRNA against plexin-A1 blocked the pro-apoptotic activity of Sema3A.
Therefore, Sema-3A may act through plexin-A1 in leukemic cells.
Interestingly, our data show the translocation of plexin-A1 into membrane rafts upon Sema3A
stimulation, while a remarkable fraction of NP1 appears to be constitutively localized in these
membrane microdomains. Since lipid rafts are specialized structures involved in several biological
processes, such as apoptosis, synaptic transmission, adhesion and migration15-18, the clustering of NP1
and Plexin-A1 into these microdomains on the cell surface may lead to a more effective Sema3A
signaling. The clustering of NP1 and plexin-A1 on the cell surface, upon Sema3A stimulation, had
been shown previously in neuronal cells33, however the identity of the implicated membrane
microdomains was not known. Notably, we found that Plexin-A1 is rapidly incorporated into lipid
rafts, after a few minutes of incubation with Sema3A (Fig. 6), and this process precedes Fas clustering
(our unpublished data). Thus, we propose that Sema3A/NP1/Plexin signaling rearranges membrane
rafts, promoting receptor clustering and Fas redistribution.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

18
How Plexin can affect Fas translocation into lipid rafts is presently unknown. This signal is distinct
from the Rap-1-dependent pathway leading to inhibition of T cell proliferation and cytokine
production6. It has been demonstrated that the cytoplasmic domain of plexins carries an intrinsic R-Ras
GAP activity34, and several groups have reported that plexin-associated effector molecules control
cytoskeletal dynamics and integrin function through monomeric G proteins35. Cytoskeletal
rearrangements or changes in the interaction between Fas and components of the actin cytoskeleton
may contribute to Fas translocation into lipid rafts36. Our findings indicate the involvement of actin-
network remodeling triggered by plexins in the translocation of Fas into lipid rafts (Fig. 7).
Alternatively, post-translational modification of Fas may also mediate translocation, and this point is
currently under investigation.
In conclusion, the data presented here indicate that Sema3A plays a relevant role by bolstering Fas-
mediated apoptosis in human leukemic cells. Moreover, since NP1 and Sema3A are constitutively
expressed in human thymus in both thymic epithelial cells and CD4/CD8-defined thymocytes37, our
data suggest a possible involvement of Sema3A in T cell homeostasis, a process in which the role of
tumor necrosis factor family receptors is well documented11,12.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

19
Acknowledgments
We acknowledge Dr. M. Tessier-Lavigne (Howard Hughes Medical Institute, Stanford University, CA)
for providing the blocking anti-NP1 antibody. Dr. L. Capparuccia (IRCC, Candiolo, Torino), Dr. G.
Fulgenzi (P.U. of Marche, Ancona, Italy) and Dr. M. Fanelli (Centre of Biotechnology, University of
Urbino, Fano, Italy) for technical assistance. This work has been supported by grants from the Italian
Association for Cancer Research (AIRC) to A.P. and to A.C.; the Ministry of University and the
Ministry of Health to A.P. S.M. and R.L. were supported by a fellowship from AIRC and FIRC,
respectively.
Author contributions
S.M. performed cell-culture experiments, generated figures, and helped to write the manuscript. A.P.
analyzed the data, and helped to design the study and write the manuscript. R.L. assisted with
experiments and provided expertise in molecular biology. R.T., M.M. and M.R.R. provided
methodological expertise and helped to design some experiments. L.T. analyzed the data, and helped to
design the study and write the manuscript. A.C. (principal investigator) designed the experiments,
interpreted and analyzed data, and drafted and edited the manuscript.
The authors declare no competing financial interests.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

20
References
1. Tamagnone L, Comoglio PM. Signalling by semaphorin receptors: cell guidance and beyond.
Trends Cell Biol. 2000;10:377-383.
2. Comoglio PM, Tamagnone L, Giordano S. Invasive growth: a two-way street for semaphorin
signalling. Nat Cell Biol. 2004;6:1155-1157.
3. Kikutani H, Kumanogoh A. Semaphorins in interactions between T cells and antigen-presenting
cells. Nat Rev Immunol. 2003;3:159-167.
4. Kumanogoh A, Marukawa S, Suzuki K, et al. Class IV semaphorin Sema4A enhances T-cell
activation and interacts with Tim-2. Nature. 2002;419:629-633.
5. Delaire S, Billard C, Tordjman R, et al. Biological activity of soluble CD100. II. Soluble
CD100, similarly to H-SemaIII, inhibits immune cell migration. J Immunol. 2001;166:4348-
5416.
6. Catalano A, Caprari P, Moretti S, Faronato M, Tamagnone L, Procopio A. Semaphorin-3A is
expressed by tumor cells and alters T-cell signal transduction and function. Blood.
2006;107:3321-3329.
7. He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin
III. Cell. 1997;90:739-751.
8. Tamagnone L, Artigiani S, Chen H, et al. Plexins are a large family of receptors for
transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 1999;99:71-80.
9. Bagnard D, Vaillant C, Khuth ST, et al. Semaphorin 3A-vascular endothelial growth factor-165
balance mediates migration and apoptosis of neural progenitor cells by the recruitment of
shared receptor. J Neurosci. 2001;21:3332-3341.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

21
10. Shirvan A, Kimron M, Holdengreber V, et al. Anti-semaphorin 3A antibodies rescue retinal
ganglion cells from cell death following optic nerve axotomy. J. Biol. Chem. 2002;277:49799-
49807.
11. Lambert CA, Landau M, Desbarats J. Fas-beyond death: a regenerative role for Fas in the
nervous system. Apoptosis. 2003;8:551-562.
12. Li-Weber M, Krammer PH. Function and regulation of the CD95 (APO-1/Fas) ligand in the
immune system. Semin. Immunol. 2003;15:145-157.
13. Siegel RM, Frederiksen JK, Zacharias DA, et al. Fas preassociation required for apoptosis
signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354-2357.
14. Scaffidi C, Fulda S, Srinivasan A, et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J.
1998;17:1675-1687.
15. Muppidi JR, and Siegel RM. Ligand-independent redistribution of Fas (CD95) into lipid rafts
mediates clonotypic T cell death. Nat. Immunol. 2004;5:182-189.
16. Legembre P, Daburon S, Moreau P, et al. Amplification of Fas-mediated apoptosis in type II
cells via microdomain recruitment. Mol. Cell. Biol. 2005;25:6811-6820.
17. Legembre P, Daburon S, Moreau P, Moreau JF, Taupin JL. Modulation of Fas-mediated
apoptosis by lipid rafts in T lymphocytes. J Immunol. 2006;176:716-720.
18. Gajate C, Mollinedo F. Cytoskeleton-mediated death receptor and ligand concentration in lipid
rafts forms apoptosis-promoting clusters in cancer chemotherapy. J Biol Chem.
2005;280:11641-11647.
19. Cascino I, Papoff G, De Maria R, Testi R, Ruberti G. Fas/Apo-1 (CD95) receptor lacking the
intracytoplasmic signaling domain protects tumor cells from Fas-mediated apoptosis. J
Immunol. 1996;156:13-17.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

22
20. Catalano A, Caprari P, Soddu S, Procopio A, Romano M. 5-lipoxygenase antagonizes
genotoxic stress-induced apoptosis by altering p53 nuclear trafficking. FASEB J.
2004;18:1740-1742.
21. Catalano A, Caprari P, Rodilossi S, et al. Cross-talk between vascular endothelial growth factor
and semaphorin-3A pathway in the regulation of normal and malignant mesothelial cell
proliferation. FASEB J. 2004;18:358-360.
22. Catalano A, Rodilossi S, Rippo MR, Caprari P, Procopio A. Induction of stem cell factor/c-
Kit/slug signal transduction in multidrug-resistant malignant mesothelioma cells. J Biol Chem.
2004;279:46706-46714.
23. Bergom C, Goel R, Paddock C, Gao C, Newman DK, Matsuyama S, Newman PJ. The cell-
adhesion and signaling molecule PECAM-1 is a molecular mediator of resistance to genotoxic
chemotherapy. Cancer Biol Ther. 2006;5:1699-1707.
24. Moran M, Miceli MC. Engagement of GPI-linked CD48 contributes to TCR signals and
cytoskeletal reorganization: a role for lipid rafts in T cell activation. Immunity. 1998;9:787-796.
25. Lepelletier Y, Moura IC, Hadj-Slimane R, et al. Immunosuppressive role of semaphorin-3A on
T cell proliferation is mediated by inhibition of actin cytoskeleton reorganization. Eur J
Immunol. 2006;36:1782-1793.
26. Kikutani H, Kumanogoh A. Semaphorins in interactions between T cells and antigen-presenting
cells. Nat Rev Immunol. 2003;3:159-167.
27. Moretti S, Procopio A, Boemi M, Catalano A. Neuronal semaphorins regulate a primary
immune response. Curr Neurovasc Res. 2006;3:295-305.
28. Shirvan A, Ziv I, Fleminger G, Shina R, He Z, Brudo I, Melamed E, Barzilai A. Semaphorins
as mediators of neuronal apoptosis. J Neurochem. 1999;73:961-971.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

23
29. Wang Z, Li Y, Wu X, Cheng S, Yang L, Wu Y. KDR and Sema3 genes expression in bone
marrow stromal cells and hematopoietic cells from leukemia patients and normal individuals.
Hematology. 2005;10:307-312.
30. Kreuter M, Woelke K, Bieker R, Schliemann C, Steins M, Buechner T, Berdel WE, Mesters
RM. Correlation of neuropilin-1 overexpression to survival in acute myeloid leukemia.
Leukemia. 2006;20:1950-1954.
31. Wong AW, Brickey WJ, Taxman DJ, et al. CIITA-regulated plexin-A1 affects T-cell-dendritic
cell interactions. Nat Immunol. 2003;4:891-898.
32. Takegahara N, Takamatsu H, Toyofuku T, et al. Plexin-A1 and its interaction with DAP12 in
immune responses and bone homeostasis. Nat Cell Biol. 2006;8:615-622.
33. Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H,
Strittmatter SM. Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell.
1999;99:59-69
34. Oinuma I, Ishikawa Y, Katoh H, Negishi M. The Semaphorin 4D receptor Plexin-B1 is a
GTPase activating protein for R-Ras. Science. 2004;305:862-865.
35. Tamagnone L, Giordano S. Semaphorin pathways orchestrate osteogenesis. Nat Cell Biol.
2006;8:545-547.
36. Parlato S, Giammarioli AM, Logozzi M, et al. CD95 (APO-1/Fas) linkage to the actin
cytoskeleton through ezrin in human T lymphocytes: a novel regulatory mechanism of the
CD95 apoptotic pathway. Embo. J. 2000;19:5123-5134.
37. Lepelletier Y, Smaniotto S, Hadj-Slimane R, Villa-Verde DM, Nogueira AC, Dardenne M,
Hermine O, Savino W. Control of human thymocyte migration by Neuropilin-1/Semaphorin-
3A-mediated interactions. Proc Natl Acad Sci U S A. 2007;104: 5545-5550.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

24
Figure legends
Figure 1. Fas-induced cell death varies with cell type in the presence of Sema3A. (A) Jurkat, HUT78
and HUT78.B1 cells were assayed by flow cytometry for their respective cell surface content of Fas. (B
and C) Jurkat, HUT78 or HUT78.B1 cells were stimulated with 50 ng/ml rhFasL, 200 ng/ml CH11,
150 ng/ml APO1-1, and 50 ng/ml TRAIL, in the presence of soluble control IgG or Sema3A-Fc (150
ng/ml) for 7 hrs. Cell death was assessed by staining using FITC-annexin V. Data are the mean ± s.e.m.
of three independent experiments. * indicates P < 0.05 vs. none and ** indicates P < 0.05 vs its
respective death receptor ligand.
Figure 2. The pro-apoptotic activity of Sema3A is dependent by NP1. (A) Surface expression of NP1
for Jurkat, HUT78 or HUT78.B1 cells. (B) Jurkat cells were incubated with CH11 plus Sema3A-Fc or
control IgG for 7 hrs, and cell death was determined as described in figure 1. Alternatively, cells were
first incubated with the blocking anti-NP1 antibody or a control Ab. Statistical analysis is shown.
Figure 3. Sema3A co-signal differentially increases the Fas-mediated caspase activation in cell lines.
(A) Jurkat (left) or HUT78 (right) cells were stimulated with 150 ng/ml APO1-1 in the presence of
control IgG or Sema3A-Fc (150 ng/ml) for indicated times. Activation of caspase 8 and caspase 3 was
analyzed by immunoblotting. The 18 kDa and 17 kDa bands correspond to active cleavage forms of
caspase-8 and -3, respectively. The results shown are representative of three independent experiments.
(B) Jurkat or CEM (type II) cells were incubated for 7 hrs as described in panel A with or without
zVAD-fmk (20 µM). Cell death was assessed by flow cytometry with propidium iodide.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

25
Figure 4. Sema3A stimulation induces redistribution of Fas into lipid rafts. (A) Jurkat cells (1 x 107)
were treated with control IgG or Sema3A-Fc for 60 min and subjected to density gradient fractionation.
Fractions were immunoblotted with antibodies for Lck, Fas, FADD, caspase-8 and NP1. Fractions
corresponding to lipid raft (R) and soluble (S) proteins are indicated. (B) Jurkat cells were pre-
incubated with the blocking anti-NP1 antibody or a control Ab. Then, cells were treated as in panel A.
Fractions 4 (R) and 11 (S) were immunoblotted with antibodies specific for GM1 and Fas. Data in A
and B are representative of at least three independent experiments. (C) Jurkat cells were stimulated
with Sema3A-Fc for 60 min, depleted of cholesterol with 15 µg/ml MBCD for 10 min at 37 °C and
then stimulated with CH11 for 6 hrs. Cell death was determined by flow cytometry with annexin V
staining. P value is shown.
Figure 5. Plexins expression in leukemic cells. (A) Total RNA (50 ng/µl) was isolated from the
indicated cells and real-time PCR was done with primers and probes specific for plexin-A1, -A2, -A3,
and -A4 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. Plexins mRNA expression
was normalized to GAPDH for each sample. (B) Total lysates (50 µg) from indicated cell lines were
immunoblotted (Blot Ab) with anti-plexin-A1 antibody. Expression of actin was used as loading
control. (C and D) Colocalization of Fas (green) with either plexin-A1 (C) or NP1 (D) (red) in
Sema3A-treated Jurkat cells in contrast to cells treated with control IgG. Images are magnified × 1600.
Figure 6. Involvement of Plexin in Sema3A signaling. (A) Jurkat cells were treated with control IgG or
Sema3A-Fc for 60 min, and subjected to density gradient fractionation. For anti-Lck immunoblots, 10
µg of protein was loaded per lane and for anti-plexin-A1 immunoblots, 40 µg of protein was loaded per
lane. (B) Jurkat cells (107 cells per test) were treated with control IgG or Sema3A-Fc (150 ng/ml) for 5
min and lysates were prepared. Equivalent amounts of whole-cell lysates were either
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

26
immunoprecipitated with anti-plexin-A1 antibody (IP: anti-plexin-A1) or anti-NP1 antibody (IP: anti-
NP1). Immune complexes were then immunoblotted (Blot Ab) as indicated. Data in A and B are
representative of three experiments. (C) The Jurkat cell line was engineered by lentiviral-mediated
gene-transfer of a truncated form of plexin-A1, lacking its cytoplasmic domain (DN-plexin-A1). These
cells or control Jurkat cells were incubated with CH11 or CH11 plus Sema3A-Fc for 6 hrs. (D)
Additionally, Jurkat cells transfected with siRNA specific for plexin-A1 or non-silencing siRNA were
cultured with control IgG or Sema3A-Fc for 6-8 hrs. plexin-A1-expression in RNAi-treated Jurkat cells
was determined by RT-PCR analysis. Cell death was quantified by flow cytometry with annexin V
staining. P values were also shown.
Figure 7. Plexin-mediated Sema3A stimulation induces cytoskeleton reorganization and redistribution
of actin-linking proteins into lipid rafts. (A) Jurkat cells were incubated with control IgG or Sema3A-
Fc (150 ng/ml) for 30 min. As positive control, Jurkat cells were also incubated with 50 ng/ml rhFasL.
Actin morphology was visualized by staining with phalloidin-TRITC. Nuclei were stained with
Hoechst 33258. Images are magnified × 1600. (B) Jurkat cells were pretreated with 5 µg/ml
cytochalasin B (CB) for 30 min and then incubated with anti-Fas or anti-Fas plus Sema3A-Fc for 6 hrs.
Cell death was quantified by flow cytometry with annexin V staining. P values were also shown. (C-E)
Control or dominant negative plexin-A1 expressing cells (DN-plexin-A1) were treated with control
IgG or Sema3A-Fc for 60 min and subjected to density gradient fractionation. (C and D) Fractions
were immunoblotted with antibodies for GM1, Ezrin, RhoA, RhoGDI. Western blots were also probed
with anti-VSV-G antibody to detect the VSV-G epitope tag of plexin construct. (E) Fractions derived
from DN-plexin-A1 were immunoblotted with antibodies for Lck and Fas. Lipid raft (R) and soluble
(S) fractions are indicated.
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
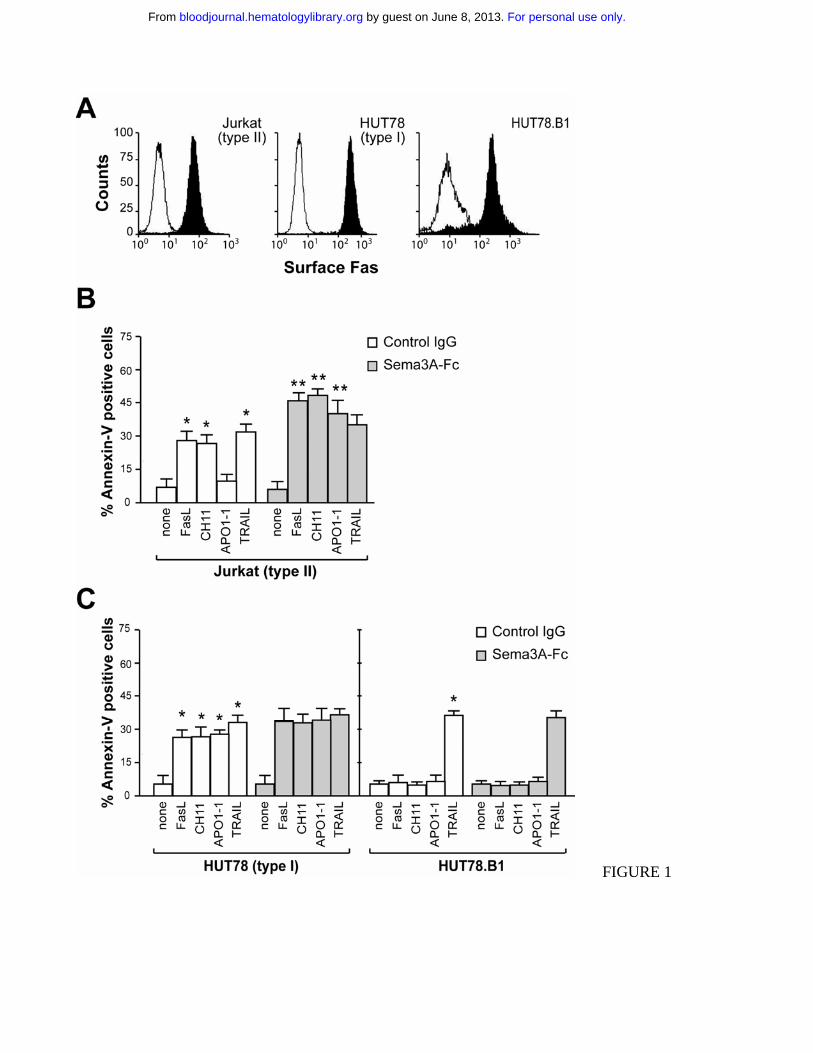
FIGURE 1
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

FIGURE 2
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
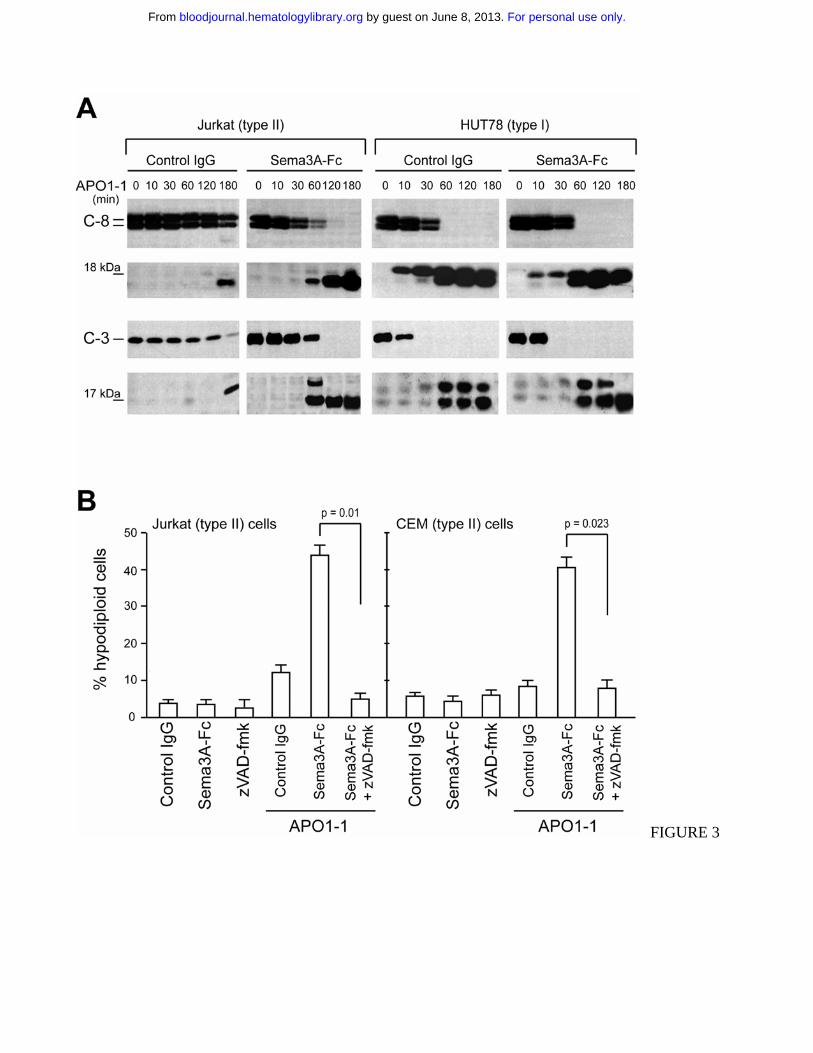
FIGURE 3
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

FIGURE 4-AB
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
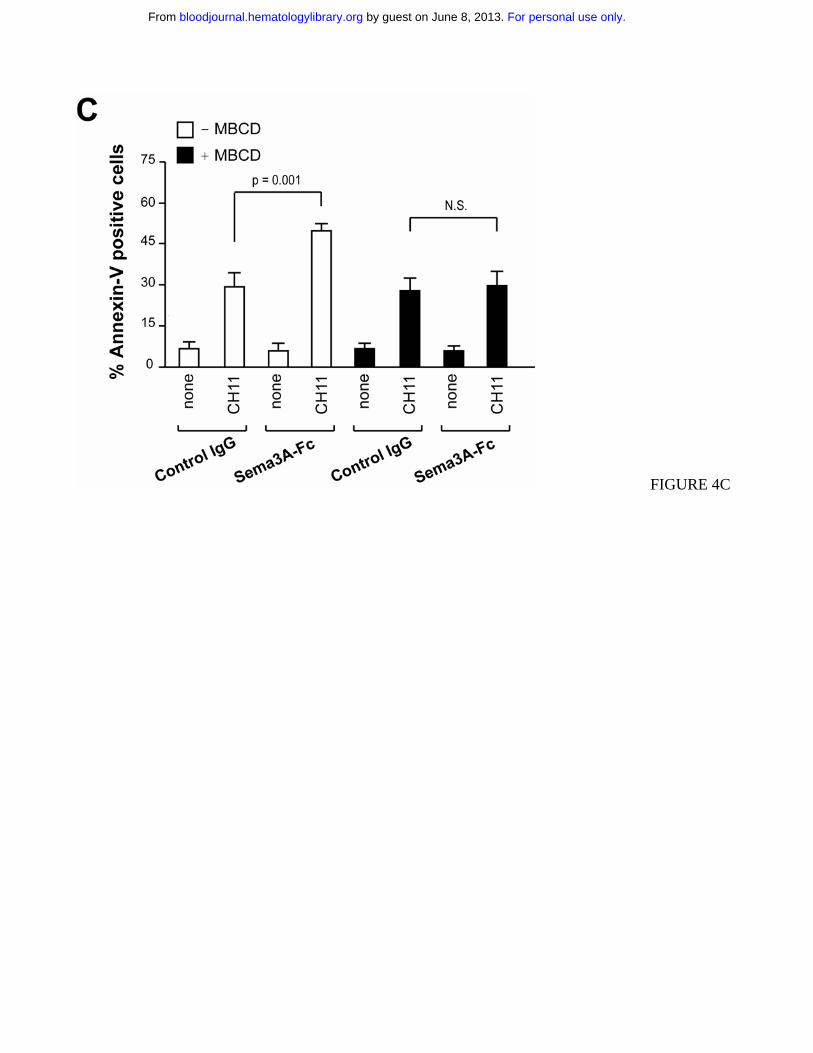
FIGURE 4C
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

FIGURE 5-AB
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

FIGURE 5-CD
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom

FIGURE 6
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
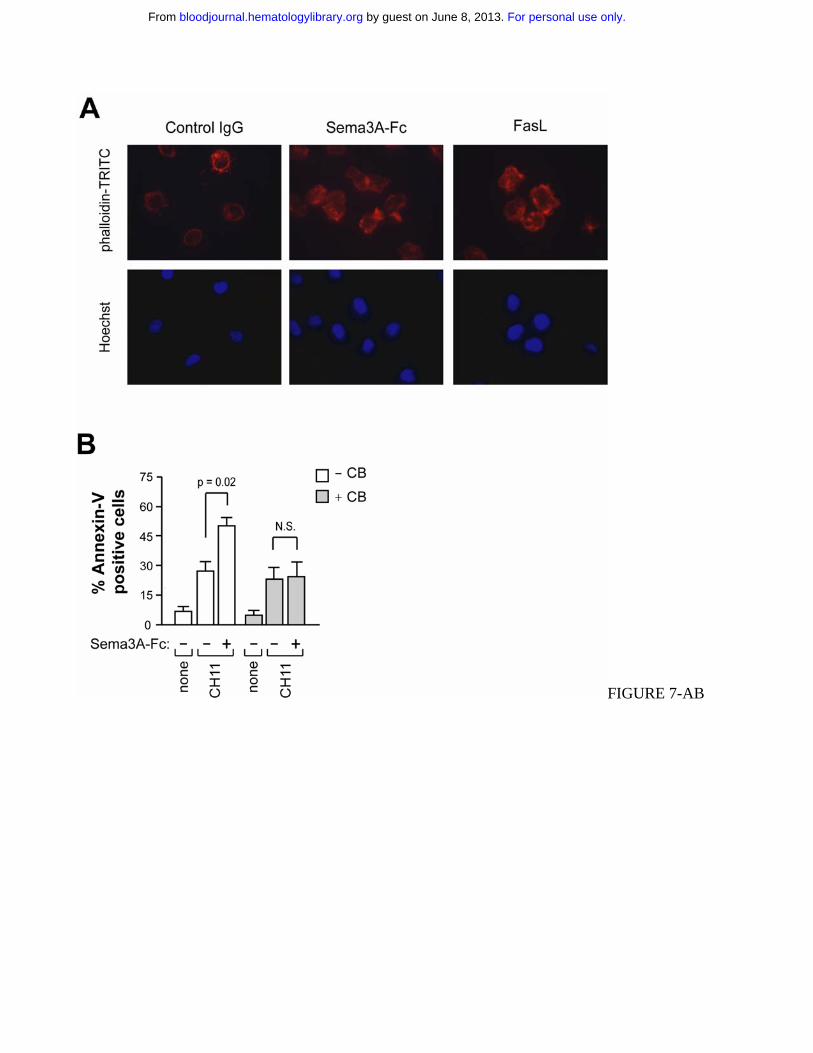
FIGURE 7-AB
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
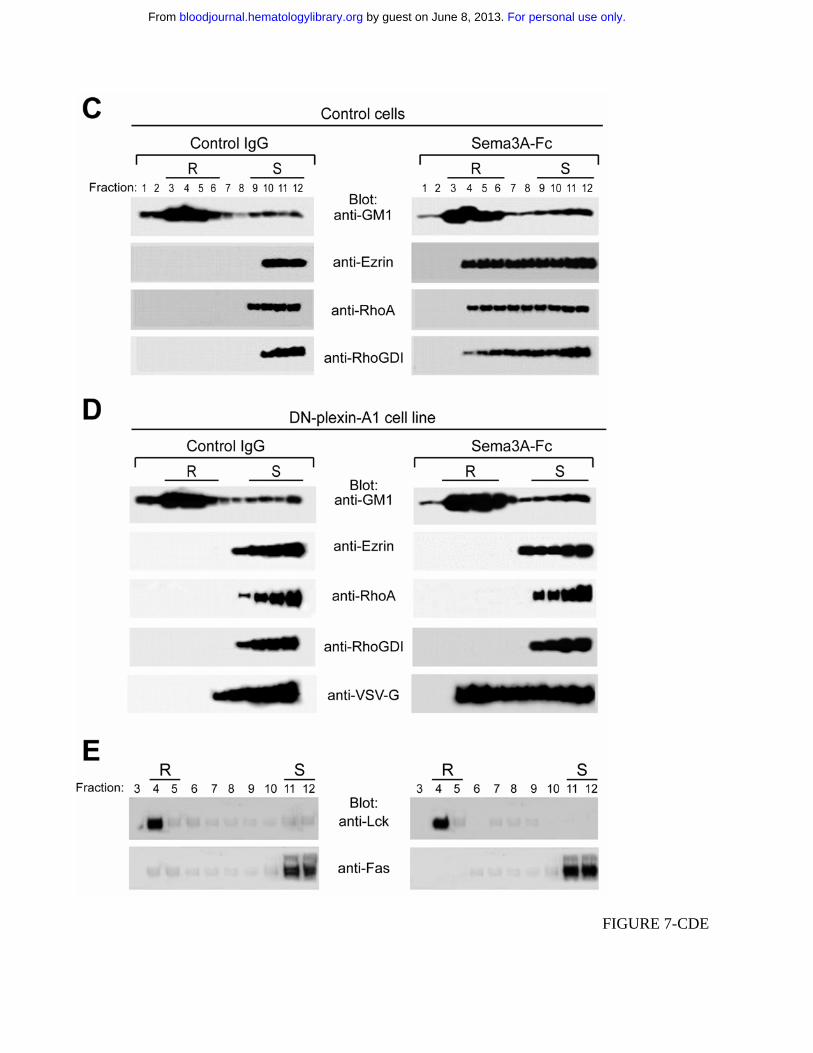
FIGURE 7-CDE
For personal use only. by guest on June 8, 2013. bloodjournal.hematologylibrary.orgFrom
Related Documents











