Selective Bypass of a Lagging Strand Roadblock by the Eukaryotic Replicative DNA Helicase Yu V. Fu, 1 Hasan Yardimci, 1 David T. Long, 1,7 The Vinh Ho 2,4,7 Angelo Guainazzi, 2,5,7 Vladimir P. Bermudez, 3 Jerard Hurwitz, 3 Antoine van Oijen, 1,6 Orlando D. Scha ¨ rer, 2 and Johannes C. Walter 1, * 1 Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA 2 Department of Pharmacological Sciences and Chemistry, Stony Brook University, Stony Brook, NY 11794, USA 3 Program of Molecular Biology, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, New York, NY 10021 4 Present address: Tecan Group Ltd., 8708 Ma ¨ nnedorf, Switzerland 5 Present address: Helsinn Therapeutics (U.S.) Inc., Bridgewater, NJ 08807, USA 6 Present address: The Zernike Institute for Advanced Materials, University of Groningen, 9747 AG Groningen, The Netherlands 7 These authors contributed equally to this work *Correspondence: [email protected] DOI 10.1016/j.cell.2011.07.045 SUMMARY The eukaryotic replicative DNA helicase, CMG, un- winds DNA by an unknown mechanism. In some models, CMG encircles and translocates along one strand of DNA while excluding the other strand. In others, CMG encircles and translocates along duplex DNA. To distinguish between these models, repli- somes were confronted with strand-specific DNA roadblocks in Xenopus egg extracts. An ssDNA tran- slocase should stall at an obstruction on the translo- cation strand but not the excluded strand, whereas a dsDNA translocase should stall at obstructions on either strand. We found that replisomes bypass large roadblocks on the lagging strand template much more readily than on the leading strand template. Our results indicate that CMG is a 3 0 to 5 0 ssDNA tran- slocase, consistent with unwinding via ‘‘steric exclu- sion.’’ Given that MCM2-7 encircles dsDNA in G1, the data imply that formation of CMG in S phase involves remodeling of MCM2-7 from a dsDNA to a ssDNA binding mode. INTRODUCTION In eukaryotic cells, DNA replication initiates at many chromo- somal locations called origins. At each origin, two ‘‘sister’’ repli- somes are assembled that move away from the origin in opposite directions. An essential component of the replisome is the repli- cative DNA helicase, which unwinds parental DNA, generating substrates for leading and lagging strand DNA polymerases. Current evidence indicates that the eukaryotic replicative DNA helicase contains at least three components, a heterohexameric ATPase called MCM2-7 and two cofactors, Cdc45 and GINS (Bochman and Schwacha, 2009; Ilves et al., 2010; Moyer et al., 2006; Pacek et al., 2006; Takahashi et al., 2005). The complex of Cdc45, MCM2-7, and GINS is called CMG (Moyer et al., 2006). A striking feature of eukaryotic DNA replication is the intricate, bi-phasic assembly of CMG, which underlies the cell cycle regu- lation of DNA replication (reviewed in Arias and Walter, 2007; La- bib, 2010). CMG assembly is best understood in yeast. In the G1 phase, when cyclin-dependent kinase (CDK) activity is low, ORC, Cdc6, and Cdt1 recruit MCM2-7 onto origins of DNA replication, forming a prereplication complex (pre-RC). Within the pre-RC, MCM2-7 complexes bind to double-stranded DNA (dsDNA) as inactive double hexamers (Evrin et al., 2009; Gambus et al., 2011; Remus et al., 2009) At the G1/S transition, when CDK activity rises, numerous additional factors cooperate to convert the MCM2-7 double hexamer into two CMG complexes. In particular, Cdc7-Dbf4 protein kinase (DDK) phosphorylates MCM2-7 (Randell et al., 2010 and references therein). CDK phos- phorylates Sld2 and Sld3 (Treslin/Ticrr in metazoans), promoting their interaction with Dpb11 (TopBP1 in metazoans). The Sld3- Sld2-Dpb11 complex enables the stable binding of Cdc45 and GINS to phosphorylated MCM2-7. Once formed, CMG unwinds the origin, allowing replisome assembly. Importantly, the high CDK activity present in the S and G2 phases inhibits the functions of ORC, Cdc6, and Cdt1, such that de novo MCM2-7 recruitment is blocked. As a result, CMG complexes that leave the origin during the first initiation event cannot be replaced, and each origin fires only once. Although the regulation of CMG assembly in metazoans is very similar, it appears to be even more compli- cated, depending on several additional factors, including Mcm9, DUE-B, GEMC1, Geminin, and CRL4 Cdt2 . A crucial question in the field is why so many proteins are required to convert bound MCM2-7 complexes into the active CMG helicase. This question is inextricably connected to an- other open question, which is how CMG unwinds DNA in S phase. Only by describing the final disposition of CMG on DNA will it be possible to understand how it is assembled and acti- vated. MCM2-7, the core molecular motor of CMG, is composed of six related AAA+ ATPase subunits, Mcm2-Mcm7 (Tye, 1999). Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. 931

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Selective Bypass of a Lagging StrandRoadblock by the EukaryoticReplicative DNA HelicaseYu V. Fu,1 Hasan Yardimci,1 David T. Long,1,7 The Vinh Ho2,4,7Angelo Guainazzi,2,5,7 Vladimir P. Bermudez,3
Jerard Hurwitz,3 Antoine van Oijen,1,6 Orlando D. Scharer,2 and Johannes C. Walter1,*1Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA2Department of Pharmacological Sciences and Chemistry, Stony Brook University, Stony Brook, NY 11794, USA3Program of Molecular Biology, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, New York, NY 100214Present address: Tecan Group Ltd., 8708 Mannedorf, Switzerland5Present address: Helsinn Therapeutics (U.S.) Inc., Bridgewater, NJ 08807, USA6Present address: The Zernike Institute for Advanced Materials, University of Groningen, 9747 AG Groningen, The Netherlands7These authors contributed equally to this work
*Correspondence: [email protected]
DOI 10.1016/j.cell.2011.07.045
SUMMARY
The eukaryotic replicative DNA helicase, CMG, un-winds DNA by an unknown mechanism. In somemodels, CMG encircles and translocates along onestrand of DNA while excluding the other strand. Inothers, CMG encircles and translocates along duplexDNA. To distinguish between these models, repli-somes were confronted with strand-specific DNAroadblocks in Xenopus egg extracts. An ssDNA tran-slocase should stall at an obstruction on the translo-cation strand but not the excluded strand, whereasa dsDNA translocase should stall at obstructions oneither strand. We found that replisomes bypass largeroadblocks on the lagging strand template muchmore readily than on the leading strand template.Our results indicate that CMG is a 30 to 50 ssDNA tran-slocase, consistent with unwinding via ‘‘steric exclu-sion.’’ Given that MCM2-7 encircles dsDNA in G1, thedata imply that formation of CMG in S phase involvesremodeling of MCM2-7 from a dsDNA to a ssDNAbinding mode.
INTRODUCTION
In eukaryotic cells, DNA replication initiates at many chromo-
somal locations called origins. At each origin, two ‘‘sister’’ repli-
somes are assembled thatmove away from the origin in opposite
directions. An essential component of the replisome is the repli-
cative DNA helicase, which unwinds parental DNA, generating
substrates for leading and lagging strand DNA polymerases.
Current evidence indicates that the eukaryotic replicative DNA
helicase contains at least three components, a heterohexameric
ATPase called MCM2-7 and two cofactors, Cdc45 and GINS
(Bochman and Schwacha, 2009; Ilves et al., 2010; Moyer et al.,
2006; Pacek et al., 2006; Takahashi et al., 2005). The complex
of Cdc45, MCM2-7, andGINS is called CMG (Moyer et al., 2006).
A striking feature of eukaryotic DNA replication is the intricate,
bi-phasic assembly of CMG, which underlies the cell cycle regu-
lation of DNA replication (reviewed in Arias andWalter, 2007; La-
bib, 2010). CMG assembly is best understood in yeast. In the G1
phase, when cyclin-dependent kinase (CDK) activity is low,ORC,
Cdc6, and Cdt1 recruit MCM2-7 onto origins of DNA replication,
forming a prereplication complex (pre-RC). Within the pre-RC,
MCM2-7 complexes bind to double-stranded DNA (dsDNA) as
inactive double hexamers (Evrin et al., 2009; Gambus et al.,
2011; Remus et al., 2009) At the G1/S transition, when CDK
activity rises, numerous additional factors cooperate to convert
the MCM2-7 double hexamer into two CMG complexes. In
particular, Cdc7-Dbf4 protein kinase (DDK) phosphorylates
MCM2-7 (Randell et al., 2010 and references therein). CDKphos-
phorylates Sld2 and Sld3 (Treslin/Ticrr in metazoans), promoting
their interaction with Dpb11 (TopBP1 in metazoans). The Sld3-
Sld2-Dpb11 complex enables the stable binding of Cdc45 and
GINS to phosphorylated MCM2-7. Once formed, CMG unwinds
the origin, allowing replisome assembly. Importantly, the high
CDKactivity present in the S andG2phases inhibits the functions
of ORC, Cdc6, andCdt1, such that de novoMCM2-7 recruitment
is blocked. As a result, CMG complexes that leave the origin
during the first initiation event cannot be replaced, and each
origin fires only once. Although the regulation of CMG assembly
in metazoans is very similar, it appears to be even more compli-
cated, depending on several additional factors, including
Mcm9, DUE-B, GEMC1, Geminin, and CRL4Cdt2.
A crucial question in the field is why so many proteins are
required to convert bound MCM2-7 complexes into the active
CMG helicase. This question is inextricably connected to an-
other open question, which is how CMG unwinds DNA in S
phase. Only by describing the final disposition of CMG on DNA
will it be possible to understand how it is assembled and acti-
vated. MCM2-7, the core molecular motor of CMG, is composed
of six related AAA+ ATPase subunits, Mcm2-Mcm7 (Tye, 1999).
Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. 931

Thus, GINS and Cdc45 have been proposed to bind to the
outside of the MCM2-7 complex and to regulate its ATPase
activity (Ilves et al., 2010). Crystallography and electron micros-
copy reveal that eukaryotic and archaeal MCM complexes form
ring-shaped hexamers whose central channel can accommo-
date single-stranded DNA (ssDNA) or dsDNA (Evrin et al.,
2009; Fletcher et al., 2003; Pape et al., 2003; Remus et al.,
2009). Thus, DNA unwinding likely involves one or both strands
of the DNA passing through the MCM central channel.
Most well-studied replicative DNA helicases, such as E. coli
DnaB and Bacteriophage T7 gene product 4 (T7 gp4), function
as single hexamers that encircle and translocate along one
strand of DNA while excluding the other strand (‘‘steric exclu-
sion’’). In biochemical experiments using purified proteins,
archaeal and eukaryotic MCMproteins including the CMG trans-
located along ssDNA in the 30 to 50 direction (Bochman and
Schwacha, 2008; Ilves et al., 2010; Kelman et al., 1999; Moyer
et al., 2006; Shechter et al., 2000). Using elegant manipulation
of DNA templates, steric exclusion by a purified Mcm4/6/7 sub-
complex was directly demonstrated (Kaplan et al., 2003). How-
ever, the physiological relevance of these studies is not clear
since the reactions bypassed the endogenous helicase loading
and activation pathway.
More recently, several considerations led to speculation that
CMG might unwind DNA by translocating along dsDNA (Laskey
and Madine, 2003; Mendez and Stillman, 2003; Takahashi et al.,
2005). First, Mcm4/6/7 can translocate along dsDNA with
considerable force (Kaplan et al., 2003). Second, MCM2-7 inter-
acts with dsDNA as part of the pre-RC (Evrin et al., 2009; Remus
et al., 2009). It is therefore attractive to propose that MCM2-7
does not fundamentally change its interaction with DNA upon
assembly into the CMG. Several models propose how CMG
might unwind DNA while translocating along dsDNA. One idea
is that CMG functions as an obligate dimer that pumps dsDNA
toward its dimer interface, and that single-stranded DNA is
extruded through lateral channels, analogous to a mechanism
proposed for SV40 large T-antigen (Li et al., 2003; Wessel
et al., 1992). However, there is no need for helicase dimerization
during eukaryotic DNA replication, arguing against this model
(Yardimci et al., 2010). Alternatively, a single CMG complex
motors along dsDNA. As DNA emerges from the rear of CMG’s
central channel, a proteinaceous pin or ‘‘plougshare’’ bisects
the duplex, leading to strand separation (Takahashi et al.,
2005). In a variation of this idea, ssDNA exits through side chan-
nels midway along the longitudinal axis of MCM (Brewster et al.,
2008). In all of these models, DNA enters the MCM central
channel as a duplex.
To understand how the replicative helicase interacts with
DNA in S phase, we examined the collision of replisomes with
various DNA roadblocks in Xenopus egg extracts. We previously
showed that when two replisomes converge on a DNA inter-
strand crosslink (ICL), the 30 ends of the leading strands in-
itially arrest 20-40 nucleotides (nt) from the ICL (Raschle et al.,
2008). Here, we provide evidence that the footprint of CMG on
DNA underlies these distal arrest points. We had also speculated
(Raschle et al., 2008) that the wide distribution of arrest points
might suggest that CMG translocates along dsDNA, since the
first replisome to arrive at the lesion could engulf the ICL and
932 Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc.
impose a more distal arrest on the second replisome. However,
we show here that blocking the arrival of one replisome using
biotin-streptavidin (biotin-SA) roadblocks has no effect on the
leading strand arrest points of the other converging replisome.
To directly examine the CMG translocation mode, we employed
strand-specific roadblocks. We reasoned that if CMG translo-
cates along dsDNA, it should be arrested by a bulky roadblock
on either the leading or lagging strand templates. In contrast, if
it moves along ssDNA in the 30 to 50 direction, it should be ar-
rested by a roadblock on the leading strand template but not
on the excluded, lagging strand template. We found that the
nascent leading strand stalls �30 nucleotides from a biotin-SA
complex on the leading strand template, consistent with CMG
stalling, whereas a biotin-SA complex on the lagging strand
template induced little arrest. Similar results were obtained in
a single-molecule assay using strand-specific quantum dot
(QDot) roadblocks. Together with previous biochemical anal-
yses, our data strongly suggest that CMG unwinds DNA by
translocating along ssDNA in the 30 to 50 direction, consistentwith DNA unwinding by steric exclusion. The data imply a series
of discrete molecular events that underlie the conversion of
MCM2-7 to an active CMG in S phase.
RESULTS
To understand how the eukaryotic replicative DNA helicase is
configured on DNA in S phase, we examined how the repli-
some interacts with specific DNA lesions. To this end, we em-
ployed nucleus-free Xenopus egg extracts (Walter et al., 1998),
which support efficient DNA replication of plasmids or l DNA.
In this system, DNA is first incubated with a high speed super-
natant of egg cytoplasm (HSS), which chromatinizes the
template and also promotes sequence nonspecific MCM2-7
recruitment to the DNA by ORC, Cdc6, and Cdt1 (Arias and
Walter, 2004). Subsequently, a concentrated nucleoplasmic
extract (NPE) is added, which supports CMG assembly depen-
dent on Cdc7-Drf1, Cdk2-Cyclin E, Mcm10, and Cdc45 (Taka-
hashi and Walter, 2005; Walter and Newport, 2000; Wohlschle-
gel et al., 2002). The Xenopus CMG travels with the replisome
and unwinds DNA throughout S phase (Pacek et al., 2006;
Pacek and Walter, 2004). These observations indicate that
nucleus-free Xenopus egg extracts promote activation of the
replicative DNA helicase by the same events that occur in
cells.
CMG Binding Correlates with Leading Strand Arrest 20Nucleotides from a DNA Interstrand CrosslinkWe first sought to detect the CMG footprint on DNA in S phase.
To this end, we examined the collision of the replisome with two
different lesions: a DNA inter-strand crosslink, which should
arrest CMG, and a DNA intra-strand crosslink, which should stall
the DNA polymerase but not CMG. Plasmids containing a site-
specific cisplatin inter-strand crosslink (pICLInter; Figure 1A, red
line) or 1,2 cisplatin intra-strand crosslink (pICLIntra; Figure 1B,
red bracket) were incubated sequentially in HSS and NPE con-
taining [a�32P]dATP. At different times after NPE addition,
DNA was extracted and digested with StuI, which cuts the
plasmid once 268 nt to the right of both lesions (Figure 1A and

D
Rel
ativ
e am
ount
Time (min)
ICL ICL Region
Control Region
C G A T C
pICLInter pICLIntra
0
10
20
30
40
50
-40
-20
Time (min): 0 10 15 20 30 60 90 120 0 10 15 20 30 60 90 120
Primer M
-1
2 1 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Stu
I D
ige
st - L
eftw
ard
F
ork
A
CAAGAAGAGCACGCGCCGGCGCTAGGCGACGTAA TCCGGA
pICLIntra
5’-CCCTCTCCTTGGTTCTTCTCGTGCGCGGCCGCGATCCGCTGCATT AGGCCT-3’
3’-GGGAGAGGAACCAAGAAGAGCACGCGCCGGCGCTAGGCGACGTAA TCCGGA-5’
pICLInter
5’-CCCTCTTCCGCTCTTCTTTCGTGCGCGGCCGCGATCCGCTGCATT AGGCCT-3’
3’-GGGAGAAGGCGAGAAGAAAGCACGCGCCGGCGCTAGGCGACGTAA TCCGGA-5’
GAGAAGAAAGCACGCGCCGGCGCTAGGCGACGTAA TCCGGA
0 10 30 20
B 0 10 20 30
Leftward Leading Strand
Stu I (-268)
Leftward Leading Strand
Stu I (-268)
0
20
40
60
80
100
120
0 20 40 60 80 100 120
Mcm7 ChIP – Control Region Mcm7 ChIP – ICL Region -20 Arrest -1 Arrest
Figure 1. CMG Causes Leading Strand
Stalling �20 Nucleotides from a DNA Inter-
strand Crosslink
(A) Cartoon depicting the DNA sequence sur-
rounding the ICL in pICLInter. Red line, inter-strand
crosslink. Blue arrow, StuI cleavage site, which is
used to map leftward leading strands in (C).
The sequence of the longest leading strand de-
tected at the 10min time point (see [C]) is shown in
red letters, and the product generated after the
leading strand advances toward the ICL after
15 min (see [C]) is shown in green letters. Blue
letters, sequence differences between pICLInter
and pICLIntra.
(B) Same as (A), except for pICLIntra. Red bracket,
1,2 intra-strand crosslink. The sequence of the
longest leading strand seen in (C) is shown in red
letters.
(C) Mapping leading strands near DNA inter- and
intrastrand crosslinks. pICLInter (lanes 1-8) or
pICLIntra (lanes 9–16) was incubated sequentially
in HSS and NPE containing [a�32P]dATP. At the
indicated times after NPE addition, replication
intermediates were digested with StuI, and sepa-
rated on a DNA sequencing gel alongside a
sequencing ladder generated with primer M (see
Figure S1A). The distance of sequencing products
from the bold G in panel (A) and T in panel (B) is
indicated on the right.
(D) Kinetics of Mcm7 binding to an ICL. pICLInter
was replicated as in (C) but lacking radioactivity,
and samples were withdrawn for Mcm7 ChIP
using ICL proximal (pink) and control (purple)
primer pairs (see plasmid cartoon). The relative
ChIP signal adjacent to the ICL (pink circles) and
distal to the ICL (purple triangles) was plotted. In
parallel reactions containing [a�32P]dATP, repli-
cation intermediates were digested with AflIII, and
separated on a DNA sequencing gel alongside
a sequencing ladder generated with primer S (see
Figure S1A). The leading strands stalled between
�20 and �40 and at the �1 position (see Fig-
ure S1B) were quantified and plotted (blue dia-
monds and gray squares). Error bars represent the
standard deviation of three experiments.
Figure S1A available online). Leftward leading strands were
visualized on a sequencing gel alongside an appropriate
sequencing ladder. On pICLInter, the 30 ends of the leading
Cell 146, 931–941, Se
strands initially stalled 20-40 nucleotides
from the ICL (Figure 1C, lane 2, and Fig-
ure 1A, red strand)(Raschle et al., 2008).
Subsequently, DNA synthesis resumed
and the leading strand stalled again 1 nt
from the ICL (Figure 1C, green arrow
and Figure 1A, green strand) (Raschle
et al., 2008). In contrast, during replica-
tion of pICLIntra, the 30 end of the leading
strand immediately advanced all the
way to the lesion, where it stalled (Fig-
ure 1C, lanes 10–16; Figure 1B, red
strand). These data imply that leading
strands arrest 20-40 nt from an inter-strand crosslink due to
the footprint of the replicative DNA helicase, which we denote
as CMG.
ptember 16, 2011 ª2011 Elsevier Inc. 933

To test this idea, we performed chromatin immunoprecipita-
tion (ChIP) for Mcm7, a CMG subunit. pICLInter was replicated,
and at different times, the reaction was crosslinked with formal-
dehyde, sonicated, immunoprecipitated with Mcm7 antibody,
and the recovered DNA amplified with ICL-proximal and control
primer pairs (Figure 1D). ChIP revealed that soon after replication
began, Mcm7 was depleted from the control region, as ex-
pected if CMG travels toward the ICL while displacing any latent
MCM2-7 complexes (Figure 1D, purple triangles). In contrast,
Mcm7 initially accumulated at the ICL, concurrent with the arrival
of leading strands at the �20 position (Figure 1D, compare pink
circles and blue diamonds). The subsequent disappearance of
Mcm7 from the ICL after 10 min correlated with the disappear-
ance of the �20 to �40 leading strand cluster (Figure 1D), and
the advance of the leading strand to the �1 position (Figure 1D,
gray squares). These data indicate that arrest of the leading
strand 20-40 nt from a cisplatin ICL is caused by the footprint
of CMG on DNA (and any dead volume of the DNA polymerase)
and that dissociation of CMG facilitates resumption of leading
strand synthesis toward the ICL.
Converging Replisomes Do Not Interfere with EachOther at an ICLWe postulated that the wide range of leading strand arrest points
near a DNA inter-strand crosslink (Figure 1C, red bracket) could
reflect heterogeneity in the CMG footprint and/or interference by
converging CMG complexes. In the latter view, the first CMG to
arrive at the ICL would impose a more distal stoppage point on
the second replisome, giving rise to the distribution of leading
strand products. Such interference might be particularly pro-
nounced if CMG travels along dsDNA, since an ICL might
enter deep into the central channel of CMG, allowing the first
replisome to prevent approach of the second replisome
(Figure S2A, top).
To test whether replisome interference occurs, we designed
a plasmid, pICLLead/Lag, in which the arrival of replication forks
on one side of an ICL can be blocked. pICLLead/Lag contains
a nitrogen mustard (NM)-like ICL [which causes a �24 position
arrest (Raschle et al., 2008)], as well as four biotinylated thymi-
dine nucleotides placed 34-40 nt to the right of the ICL, two on
the leading strand template and two on the lagging strand
template of the leftward replication fork (Figure 2A). In the pres-
ence of SA, the leftward DNA replication fork should not be able
to reach the ICL. To test whether this is the case, pICLLead/Lag
was preincubated with and without SA and then replicated in
the presence of [a�32P]dATP. At different times, DNA was di-
gested with StuI and nascent strands of the leftward replication
fork were visualized on sequencing gels (Figure 2B). In the
absence of SA, the leftward leading strand initially advanced to
within 24 nt of the ICL (Figure 2B, lane 1, red bracket), after which
it crept forward, nearly reaching the ICL (Figure 2B, lanes 5, 7, 9,
11, 13, green arrow), as expected based on our previous results
(Raschle et al., 2008). In the presence of SA, the�24 cluster was
largely absent, and we observed a new set of products starting
at the �70 position (Figure 2B, lanes 2, 4, 6, 8, 10, 12, orange
bracket), which is 30 nt from the outermost biotin-SA complex
at the �40 position (Figure 2B, bottom white arrow on DNA
sequencing ladder). The arrest of the leading strand 30 nt from
934 Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc.
the biotin-SA complex is similar to the arrest observed at
cisplatin (�20) and NM ICLs (�24). After the leading strand
paused for 10-15 min at �70, it was further extended to within
one nucleotide of the outermost biotin-SA complex located at
the �41 position (Figure 2B, lanes 4, 6, 8, 10, 12, 14, black
arrow). We infer that the leading strand arrest at �70 is due to
the footprint of CMG, which has stalled at the outermost
biotin-SA complex, and that the advance of the leading strand
to the �41 position reflects CMG dissociation and advance of
DNA polymerase ε to the lesion. This interpretation is supported
by the fact that purified DNA polymerase ε advanced to within
a single nucleotide of a biotin-SA complex in primer extension
reactions (Figure S2B). In summary, biotin-SA complexes placed
to the right of the ICL efficiently arrest the leftward CMG complex
for 10-30 min.
To look for interference of the rightward fork by the leftward
fork, pICLLead/Lag was replicated in the presence and absence
of SA and then digested with AflIII, which cuts 151 nt to the
left and 581 nt to the right of the ICL (Figure 2A). Strikingly,
the �24 arrest pattern of the rightward fork was unchanged
by the addition of SA (Figure 2C, blue bracket, compare ± SA
lanes), even though the leftward fork was efficiently arrested
by the roadblock (Figure S2C, even lanes, orange arrow). We
conclude that the presence of a replisome on one side of the
ICL does not affect the position of the second replisome on
the other side of the lesion. This result can be interpreted in
two ways. First, CMG is a dsDNA translocase, but the ICL
cannot enter its central channel, preventing the first replisome
from engulfing the ICL. However, this explanation is unlikely
since the NM-like ICL is nondistorting (A.G., Z. T., S.C., and
O.D.S., unpublished data) and does not expand the diameter
of the duplex (Angelov et al., 2009). The second interpretation
is that MCM2-7 translocates along ssDNA, such that both repli-
somes arrest just before the ICL, avoiding interference (Fig-
ure S2A, bottom).
Preferential Bypass of a Lagging Strand Roadblockby the Replicative DNA HelicaseTo interrogate directly the translocation mode of CMG on DNA,
we confronted the replisome with a roadblock on only the
leading or lagging strand templates. To this end, we prepared
two derivatives of pICLLead/Lag called pICLLead and pICLLag,
which contain biotins on the leading or lagging strand templates,
respectively, for the leftward replication fork (Figures 3B and 3C).
Using theseDNA templates, we could address how the biotin-SA
roadblocks placed on the leading or lagging strand templates
influence the leftward moving CMG without interference from
the rightward moving CMG, whose arrival is prevented by the
ICL (Figure 2C). If CMG translocates along ssDNA in the 30 to50 direction, it will stall at a biotin-SA complex located on the
leading strand template, yielding the same �70 leading strand
arrest as seen on pICLLead/Lag (Figures 3A and 3B). In contrast,
a biotin-SA complex located on the lagging strand template
might not affect CMG progression since this strand would be
excluded from the central channel of the helicase (Figure 3C).
In this case, the leftward CMG should only stall once it hits the
ICL, manifesting as a �24 arrest (Figure 3C). On the other
hand, if CMG translocates along dsDNA, it is predicted to stall

-35 -40
-34 -39
AflIII (151) AflIII (-581)
Primer S Primer M
Rightward Fork
LeftwardFork
ICL
Stu I (-309) A
0
Biotin-SA
B
C
10
20
30
40
50
ICL 0
5 : Time (min)
: SA G A T C
Primer S
+ + + + + + + 10 15 20 25 30 60
1 2 3 4 5 6 7 8 9 10 11 12 13 14
AflIII D
ige
st - R
igh
tw
ard
F
ork
-1
-24
-50
-70
G A T C
5 10 15 20 25 30 60
1 2 3 4 5 6 7 8 9 10 11 12 13 14
-90
-80
-70
-60
-50
-40
-30
-20
-10
ICL 0
Primer M
-1
-24
-50
-70
-80
-41
: Time (min) : SA
Stu
I D
ige
st - L
eftw
ard
F
ork
+ + + + + + +
Figure 2. Replisomes Converging on an ICL Do Not Interfere with Each Other
(A) Locations of restriction sites, the nitrogen-mustard-like ICL, biotins, and sequencing primers on pICLLead/Lag. Figure S2A presents two alternative scenarios for
how replication forks might interact at an ICL.
(B) pICLLead/Lag was preincubated with buffer or streptavidin, as indicated, and replicated in egg extracts in the presence of [a�32P]dATP. At the indicated times
after NPE addition, replication intermediates were digested with Stu I and separated on a DNA sequencing gel alongside a sequencing ladder generated with
primer M. The distance of products from the ICL is indicated on the left of the gel. White arrows on the DNA sequencing ladder indicate the location of biotins.
Red bracket, leading strand arrest 24–50 nt from the ICL in the absence of SA. Orange bracket, leading strand arrest 70-80 nt from the ICL in the presence of SA
(30–40 nt from the outermost biotin). Green arrow (�1 postion), leading strands that have advanced to the ICL. Black arrow (�41 position), leading strands that
have advanced to the outermost biotin-SA complex. Figure S2B shows that DNA polymerase ε can advance to within one nt of a biotin-SA complex on the leading
strand template.
(C) pICLLead/Lag was preincubated with buffer or streptavidin, as indicated, and replicated in egg extracts in the presence of [a�32P]dATP. At the indicated times
after NPE addition, replication intermediates were digested with AflIII (see Figure 2A) and separated on a DNA sequencing gel alongside a sequencing ladder
generated with primer S. Products of the rightward fork are shown. The leftward fork was efficiently arrested by the biotin-SA (see Figure S2C).
Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. 935
-24 0 -40 -70 -24
G A T C
10 15 30 20 : Time (min)
: SA
ICL 0
-10
-20
-30
-40
-50
-60
-70
-80
-90
Primer M : Plasmid
- 70
- 80
- 41
- 50
- 24
- 1
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
+ + + + + + + + + + + +
A
ICL
Bio-SA
pICLLead/Lag
B
pICLLead
C
pICLLag
3’ to 5’ ssDNA translocation
dsDNA translocation
pICLLead/Lag
pICLLead
pICLLag
ICL
ICL
ICL
ICL
ICL
Bio-SA D
E
F
Position (nt): 0 -40 -70
G
Stu
I D
ige
st - L
eftw
ard
F
ork
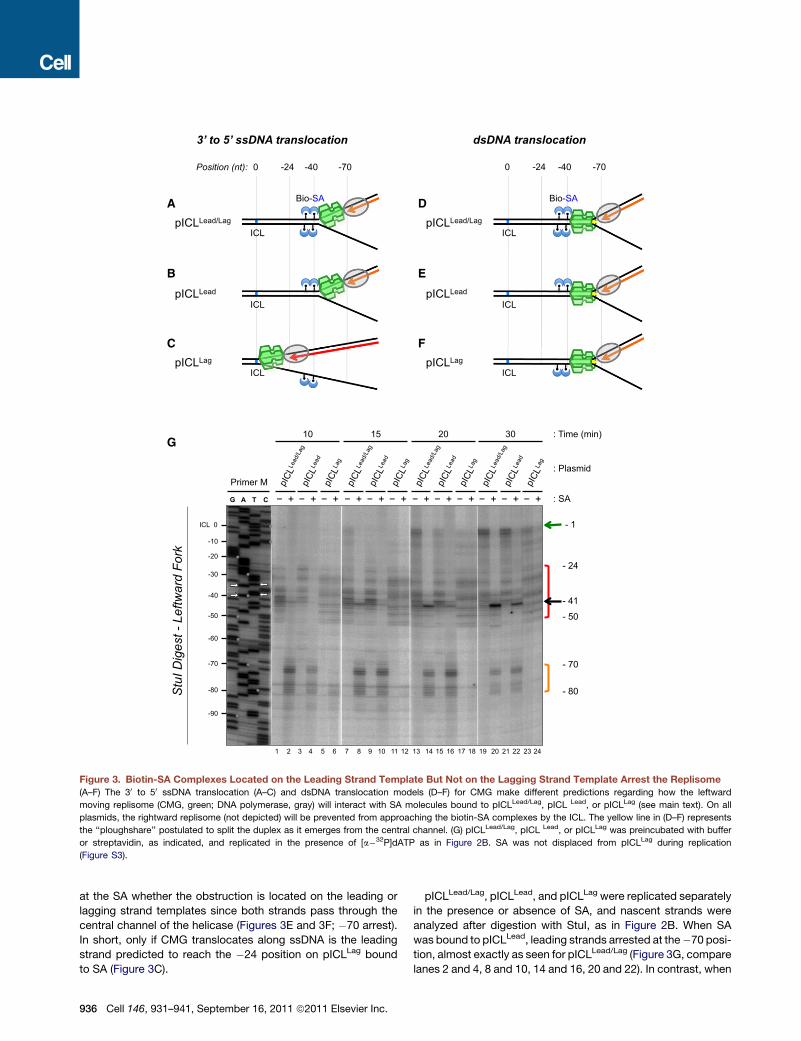
Figure 3. Biotin-SA Complexes Located on the Leading Strand Template But Not on the Lagging Strand Template Arrest the Replisome
(A–F) The 30 to 50 ssDNA translocation (A–C) and dsDNA translocation models (D–F) for CMG make different predictions regarding how the leftward
moving replisome (CMG, green; DNA polymerase, gray) will interact with SA molecules bound to pICLLead/Lag, pICL Lead, or pICLLag (see main text). On all
plasmids, the rightward replisome (not depicted) will be prevented from approaching the biotin-SA complexes by the ICL. The yellow line in (D–F) represents
the ‘‘ploughshare’’ postulated to split the duplex as it emerges from the central channel. (G) pICLLead/Lag, pICL Lead, or pICLLag was preincubated with buffer
or streptavidin, as indicated, and replicated in the presence of [a�32P]dATP as in Figure 2B. SA was not displaced from pICLLag during replication
(Figure S3).
at the SA whether the obstruction is located on the leading or
lagging strand templates since both strands pass through the
central channel of the helicase (Figures 3E and 3F; �70 arrest).
In short, only if CMG translocates along ssDNA is the leading
strand predicted to reach the �24 position on pICLLag bound
to SA (Figure 3C).
936 Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc.
pICLLead/Lag, pICLLead, and pICLLag were replicated separately
in the presence or absence of SA, and nascent strands were
analyzed after digestion with StuI, as in Figure 2B. When SA
was bound to pICLLead, leading strands arrested at the�70 posi-
tion, almost exactly as seen for pICLLead/Lag (Figure 3G, compare
lanes 2 and 4, 8 and 10, 14 and 16, 20 and 22). In contrast, when

A B
C
Figure 4. Single-Molecule Analysis of Repli-
some Collision with Leading and Lagging
Strand-Specific Roadblocks
(A) Reaction scheme for the replication of immo-
bilized l DNA in a microfluidic flow cell using a
single pair of diverging replisomes. SYTOXOrange
and dig-dUTP detection of replicated DNA are
indicated schematically. (B and C) l DNA con-
taining a QDot on the bottom strand (19 kb from
the end) or top strand (15 kb from the end), as
indicated, was immobilized within a microfluidic
flow cell and replicated as depicted in (A). After
protein removal, total DNA (SYTOX Orange), dig-
dUTP (fluorescein-labeled anti-dig Antibody), and
the QDot, were visualized and presented individ-
ually or as a merged image. Each dig tract was
classified as a rightward or leftward moving fork
depending on the location of the origin. If the tract
ended within 2 pixels (�0.3 mm) of the QDot, it was
considered arrested. Cartoons depicting each
type of collision, the expected outcome based on
the 30 to 50 ssDNA translocation model (check-
marks), representative examples of the raw data,
and the frequency of each outcome are included.
A hypothetical model in which a dsDNA translo-
case bypasses a lagging strand roadblock is
presented in Figure S4.
biotin-SA was located on the lagging strand template (pICLLag),
the result was very different. While a small fraction (maximally
28% of total signal) of leading strands transiently arrested near
the �80 position (Figure 3G, lane 6), the majority advanced
directly to the �24 position (Figure 3G, lanes 6, 12, 18, and 24,
red bracket). Therefore, CMG bypasses a lagging strand road-
block much more readily than a leading strand roadblock.
Using gel shift analysis, we verified that pICLLead and pICLLag
bound equally to SA (Figure S3A). To assess the retention of
biotin-SA complexes on DNA after replication in egg extracts,
we immunoprecipitated SA and quantified the associated
plasmid by real-time PCR. Importantly, pICLLag was recovered
at least as efficiently as pICLLead, indicating that the absence of
replisome arrest at the �70 position on pICLLag was not due to
reduced binding by SA to this DNA template in extracts (Fig-
ure S3B). To rule out that SA dissociated from biotin and
then reassociated, we added excess free biotin to the replica-
tion extract to trap dissociated SA. This manipulation had no
significant effect on the retention of SA on either template
(compare Figure S3B and Figure S3C; Figure S3D), arguing
there was no transient dissociation of the roadblock. The arrest
of the CMG by a leading strand but not a lagging strand-
specific roadblock clearly favors the 30 to 50 ssDNA transloca-
tion model.
Single-Molecule Analysis of Replisome Collisionswith Strand-Specific RoadblocksTo examine the interaction of replisomes with strand-specific
DNA roadblocks by an independent approach where replisomes
do not converge, we employed single-molecule analysis. l DNA
(48.5 kb) biotinylated at one or both 30 ends was attached to the
streptavidinated surface of a microfluidic flow cell. We recently
showed that these substrates are extensively replicated by
HSS/NPE in an MCM2-7 dependent manner (Yardimci et al.,
2010). DNA replication can be limited to a single pair of diverging
replisomes when replication initiation is restricted by adding
the Cdk2 inhibitor p27Kip shortly after NPE addition (Fig-
ure 4A)(Yardimci et al., 2010). Replicated DNA is detected using
SYTOX Orange (‘‘SYTOX’’), a nonspecific DNA dye, which gives
rise to a fluorescence signal that is twice as strong in regions
containing two daughter duplexes as in regions containing
a single parental duplex (Figure 4A; red molecule). We also
added dig-dUTP for the last 25 min of the reaction and then
stained the DNA with fluorescent anti-dig antibody (Figure 4A,
blue tracts). This allowed us to determine the approximate posi-
tion of the replication origin, which is located roughly halfway
between the stained dig-dUTP tracts, as well as the directionality
of each replication fork (Figure 4A) (Yardimci et al., 2010).
For the present study, we attached a quantum dot (QDot;
�20 nmdiameter) to the ‘‘bottom’’ strand of lDNA (seeMaterials
and Methods), 19 kb from the end (Figure 4Bi; green dot). Since
DNA replication initiates randomly on DNA in Xenopus egg
extracts, forks hit the QDot from both directions. The QDot
resides on the leading strand template for rightward moving
replication forks (Figure 4Bi and 4Bii) and on the lagging strand
template for leftward moving forks (Figures 4Biii and 4Biv). If
CMG translocates on ssDNA, as indicated by the biotin-SA
experiments presented above, leftward forks should bypass
the QDot (Figure 4Biv), whereas rightward forks should be ar-
rested (Figure 4Bi). In contrast, if CMG is a dsDNA translocase,
Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. 937

neither rightward nor leftward forks should be able to bypass the
QDot (Figures 4Bi and 4Biii).
In agreement with the 30 to 50 ssDNA translocation model, we
found that all rightward DNA replication forks stalled at the QDot
(Figure 4Bi), whereas 80% of leftward forks bypassed the QDot
(Figure 4Biv). We also attached the QDot to the ‘‘top’’ strand,
15 kb from the end of l DNA (Figure 4Ci, green dot). On this
DNA template, all polarities are reversed. Thus, if CMG is a 30
to 50 ssDNA translocase, leftward forks should stall at the QDot
(Figure 4Ciii) and rightward forks should bypass it (Figure 4Cii).
In agreement with this prediction, 93% of leftward forks stalled
at the QDot (Figure 4Ciii), whereas 74% of rightward forks by-
passed it (Figure 4Cii). The fact that 20%–25% of forks stalled
at a QDot on the lagging strand template suggests that while
CMG is a 30 to 50 ssDNA translocase, it can be transiently
arrested by a lesion on the excluded strand (see Discussion).
DISCUSSION
This study addresses whether the eukaryotic replicative DNA
helicase (CMG) travels along ssDNA or dsDNA in S phase,
a fundamental, unanswered question in cell biology. Our strategy
was based on the supposition that both 30 to 50 ssDNA and
dsDNA translocases should arrest at an obstruction on the
leading strand template (Figures 3B and 3E), whereas only a 30
to 50 ssDNA should be able to bypass a roadblock on the lagging
strand template (Figure 3C versus Figure 3F). Using the arrest
point of the leading strand as a marker for CMG stalling, we
observed that the helicase was arrested much more efficiently
by a leading strand biotin-SA roadblock than a lagging strand
roadblock. Analogous results were obtained using single-
molecule assays, in which replisomes were collided with QDots.
These data support the idea that CMG translocates along ssDNA
in the 30 to 50 direction. We cannot formally rule out an alternative
interpretation of the data in which MCM2-7 translocates along
dsDNA and transient opening of the helicase central channel
allows bypass of a lagging strand roadblock. However, this
model is improbable, principally because it does not explain
how the breached helicase could bypass the lesion (see Fig-
ure S4 and legend thereof).
Importantly, other aspects of our data are also consistent with
CMG being a ssDNA translocase. First, we observed no interfer-
ence between converging replisomes at an ICL, as might have
been expected in the dsDNA translocation model (see Results).
Second, the apparent size of the CMG footprint is consistent
with a ssDNA translocation model. Thus, upon initial collision
of the replisome with a cisplatin ICL, the 30 end of the leading
strand can advance to within 20 nucleotides of the lesion. Given
that the longitudinal axis of the eukaryotic MCM2-7 complex
comprises �115 A (Remus et al., 2009), MCM2-7 should protect
�34 basepairs of dsDNA (115 AO 3.4 A per basepair = 34 base-
pairs), which likely occurs in the context of prereplication com-
plexes. Importantly, ssDNA can be extended to 1.7 times the
length of B-formDNA (Smith et al., 1996; vanOijen, 2007). There-
fore, 20 nt (34 O 1.7) of fully extended ssDNA would suffice to
traverse the entire MCM2-7 channel. Thus, a ssDNA transloca-
tion model is compatible with the �20 nt arrest point seen for
the leading strand if the active site of DNA polymerase is located
938 Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc.
immediately behind the rear exit channel of CMG. Although
stretching ssDNA to this extent is energetically unfavorable,
this might occur as a result of interactions between DNA and
the CMG central channel and/or the pulling force exerted by
DNA polymerase. It is also worth noting that most of the leading
strands arrest further than 20 nt from the ICL, indicating that only
a small fraction of stalled CMG complexes encircle fully ex-
tended ssDNA. The reason why leading strands arrest 4 nucleo-
tides further away from a nitrogen-mustard like ICL versus
a cisplatin ICL (Raschle et al., 2008) is likely that the former lesion
stabilizes the duplex, preventing full approach to the lesion by
CMG. In the case of SA roadblocks, a steric clash between
CMG and SA might explain the more distal arrest point
(�30 nt) observed for this lesion.
Finally, our conclusion that the replicative helicase moves
along ssDNA is supported by extensive biochemical analysis
of purified MCM complexes. Archaeal MCMs (Kelman et al.,
1999; Shechter et al., 2000), Mcm4/6/7 (Ishimi, 1997), and
CMG (Moyer et al., 2006) exhibit 30 to 50 ssDNA translocation
activity on model DNA templates. In light of our findings, the
observation that MCM complexes can slide (Evrin et al., 2009;
Remus et al., 2009) or actively translocate along dsDNA (Kaplan
et al., 2003) could have other implications. As previously pro-
posed (Remus et al., 2009), the collision of a replisome with
latent MCM2-7 complexes could induce the latter to slide or
translocate along dsDNA ahead of the fork. These mobilized
MCMs might represent a cadre of reserve helicases that are
deployed to rescue stalled replication forks (Remus et al.,
2009), or they might serve to increase the local concentration
of pre-RCs in unreplicated DNA to potentiate an increased initi-
ation frequency late in S phase (Lucas et al., 2000).
In summary, the data aremost consistent with CMG transloca-
tion along ssDNA in the 30 to 50 direction. The evidence thus indi-
cates that CMG unwinds DNA via steric exclusion, and it strongly
disfavors models in which dsDNA passes through any part of the
CMG central channel. Future experiments will be required to
address the precise molecular mechanism by which CMG trans-
locates along ssDNA and how this leads to strand separation.
Implications for CMG and Replisome ArchitectureOur results have implications for the overall architecture of the
DNA replication fork. As discussed above, the �20 nt leading
strand arrest point matches precisely what is expected if ssDNA
is fully extended within the central channel of MCM2-7. Barring
rapid dissociation of replication proteins upon collision with the
ICL or amajor difference in the dimensions of yeast and Xenopus
MCM2-7, this observation implies that the C-terminal ATPase
domains of the MCM2-7 complex comprise the leading edge
of the replisome and that Cdc45, GINS, and any other helicase-
associated factors do not substantially alter the footprint of
MCM2-7 on DNA. Moreover, the data imply that the active site
of DNA polymerase epsilon resides immediately behind the rear
exit channel of CMG, which is consistent with our finding that
purified pol ε can advance to within 1 nucleotide of a biotin-SA
complex on the leading strand template. This configuration is
advantageous, as emerging ssDNA will be immediately repli-
cated, minimizing the possibility of strand reannealing or
cleavage.

Split Dimer
Open 2/5 gate
Extrude strand
Activate ATPase with GINS/Cdc45
Side View End-on view
(left hexamer)
Latent MCM2-7
double hexamer
DNA unwinding
G1 Phase
S Phase
(1)
(2)
(3)
(4)
Figure 5. Model for Replication Initiation
Model for helicase activation in which MCM2-7 encircles dsDNA in G1 and
ssDNA in S phase. See text for details.
We noted that when a biotin-SA complex was placed on the
lagging strand template, there was a low level of transient stalling
(Figure 3G, lane 6). Similarly, in single-molecule analysis, QDots
placed on the lagging strand template induced �20% stalling.
These results suggest that the excluded strand might interact
intimately with the outer face of the CMG, leading to steric
clashes with the bulky lesions that cause occasional or transient
arrest. Recent structural analysis of the Drosophila CMG com-
plex supports this conclusion since it reveals a potential
groove/channel on the outer surface of CMG that might interact
with the excluded strand (Costa et al., 2011).
Interactions of the Replisome with DNA DamageOur data have implications for the early steps of DNA interstrand
crosslink repair, which is initiated by the collision of two DNA
replication forkswith an ICL inXenopus egg extracts (Knipscheer
et al., 2009; Raschle et al., 2008). It was previously unknownwhy
leading strands pause at the �20 to �40 positions and then
undergo further elongation toward the ICL, followed by lesion
bypass. Our experiments indicate that the delay involves the
dissociation of CMG from the site of the lesion. Another implica-
tion is that when the replisome first arrives at an ICL, the DNA
immediately adjacent to the lesion is single stranded since one
strand is sequestered within the CMG central channel while
the other strand is excluded.
Implications for the Mechanism of CMG AssemblyIn G1, latent MCM2-7 double hexamers appear to interact with
dsDNA, as evidenced by the absence of ssDNA within pre-RCs
(our unpublished results; Bowers et al., 2004; Geraghty et al.,
2000) and the ability of latent MCM2-7 complexes to slide along
DNA (Evrin et al., 2009; Remus et al., 2009). Our data indicate
that in S phase, CMG encircles ssDNA with no duplex DNA re-
maining inside the central channel of the helicase. Furthermore,
CMG likely functions as a monomer (Gambus et al., 2006; Ilves
et al., 2010; Moyer et al., 2006; Yardimci et al., 2010). Assuming
these starting and end points, a number of discrete stages can be
envisioned that convert MCM2-7 to CMG, not necessarily in the
following order (Figure 5). (1) The MCM2-7 double hexamer is
split. (2) The latent MCM2-7 rings straddling dsDNA are opened,
perhaps via regulation of the Mcm2-Mcm5 gate (Bochman and
Schwacha, 2008). Both events might depend on MCM2-7 phos-
phorylation byDDK. (3) One strand of the DNAduplex is extruded
from the central channel, perhaps by the binding of Mcm10 or
Sld2 to ssDNA (Kanter and Kaplan, 2010; Warren et al., 2008).
(4) The gate is reclosed and the helicase motor is jump-started
by Cdc45 and GINS (Ilves et al., 2010). Whatever the precise
sequence of events, the reconfiguration of theMCM2-7 complex
from a dsDNA binding mode in G1 to a ssDNA binding mode
in S phase helps explain why so many initiation factors are
required to assemble CMG. This view of initiation establishes
a roadmap for future investigations into the functions of replica-
tion initiation factors. Notably, the DNA tumor virus initiator
proteins E1 and possibly Large T Antigen initially bind to dsDNA
within the viral origin of replication but unwind DNA via steric
exclusion (Enemark and Joshua-Tor, 2006). Thus, the transition
from dsDNA to ssDNA binding appears to be widely conserved
among eukaryotic replicative DNA helicases.
EXPERIMENTAL PROCEDURES
Preparation of Plasmids
To make pICLLead/Lag, pICLLead, and pICLLag, various ICL-biotin oligonucleo-
tides (see Supplementary Methods) were purified by polyacrylamide gel
electrophoresis, and ligated into pSVRLuc to form the three plasmids (see Fig-
ure 3) (Guainazzi et al., 2010; Raschle et al., 2008). pICLControl contains the
identical sequence as pICLLead/Lag, except for the four biotinylated thymidine
nucleotides. pICLInter and pICLIntra were constructed as previously described
(Raschle et al., 2008; Tremeau-Bravard et al., 2004). Compared to pICLInter
and pICLIntra, the plasmids pICLControl, pICLLead/Lag, pICLLead, and pICLLag
contain a 41 nt insert near the ICL.
Xenopus Egg Extracts and Replication
The preparation of Xenopus egg extracts (NPE and HSS) was as described
(Walter et al., 1998). For DNA replication, the plasmids (75ng/ml) were first
incubated with an equal volume of Streptavidin (5 mg/ml) (SouthernBiotech,
Birmingham, AL, USA) or buffer for 1 hr at room temperature (RT), after which
this mixture was added to HSS for 5 min at 22�C (12–15 ng/ml final plasmid
concentration), followed by addition of two volumes of NPE containing
[a�32P]dATP. Replication was stopped with Stop Solution (0.5% SDS,
25mM EDTA, 50 mM Tris, [pH 7.5]) at different time points. The purification
of DNA replication products was as described (Raschle et al., 2008).
Nascent Strand Analysis
The nascent strand analysis was carried out as described (Raschle et al.,
2008). Briefly, purified DNA replication products were isolated and digested
with the indicated enzymes. Restriction fragments were separated on 5% or
7% polyacrylamide sequencing gels. Gels were transferred to filter paper,
dried, and nascent strands visualized with a phosphorimager. Sequencing
ladders using primers S and M (see Figure 2A) were generated using the
Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. 939

Cycle Sequencing kit from USB (USB Corporation, Cleveland, OH, USA).
Quantification was performed by Image Gauge V4.22 (Fuji Photo Film Corpo-
ration, Tokyo, Japan).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) was modified from our existing proce-
dure (Pacek et al., 2006). After the addition of NPE, aliquots of the reaction
were crosslinked through the addition of 1% formaldehyde in ELB. After
10 min incubation at RT, glycine was added to stop the crosslinking reaction.
The crosslinked material was spun through Micro Bio-Spin 6 Chromatography
Columns (BIO-RAD, Hercules, CA, USA) to remove formaldehyde. The flow-
through was diluted with sonication buffer and subjected to sonication. Immo-
noprecipitation with the indicated antibodies and quantitative real-time PCR
were performed as described (Pacek et al., 2006). Anti-Mcm7 antibody was
previously described (Walter and Newport, 2000); anti-Streptavidin antibody
was purchased from Spring Bioscience (Spring Bioscience, Pleasanton,
CA, USA).
Single-Molecule Assays
A QDot was attached to the top strand 15 kb from the right end of l DNA
or to the bottom strand 19 kb away from the end of l DNA through modifi-
cation of a previous procedure (Kochaniak et al., 2009; Kuhn and Frank-
Kamenetskii, 2008). Anti-digoxigenin antibody (Roche Applied Sciences,
Indianapolis, IN, USA) was attached to QDot605 using Invitrogen QDot
Antibody Conjugation Kit. After immobilizing l DNA in the flow cell, 1 nM
of anti-digoxigenin conjugated QDot was injected. After 10-20 min incuba-
tion, the flow cell was washed with buffer and extracts were introduced.
Replication of surface immobilized l DNA from single initiations in Xenopus
egg extracts and visualization of replicated products was performed as
described (Yardimci et al., 2010). SYTOX, fluorescein labeled anti-dig, and
the QDot were imaged using 568 nm, 488 nm, and 405 nm laser light,
respectively. To assess replication fork arrest near QDots, replication forks
were labeled with dig-dUTP for 25 min. Since the average fork rate in
these experiments was 268 bp/minute (Yardimci et al., 2010), uninterrupted
replication would yield average dig-dUTP tracts of 6.7 kb (25 min 3
0.268 kb/minute). Therefore, only dig-dUTP tracts that were significantly
shorter than expected (%4.5 kb), which ended at a QDot, were considered
to represent stalled forks.
SUPPLEMENTAL INFORMATION
Supplemental Information contains Extended Experimental Procedures and
four figures and can be found with this article online at doi:10.1016/j.cell.
2011.07.045.
ACKNOWLEDGMENTS
We thankMilica Enoiu for the gift of pICLIntra, Anna Loveland, PuckKnipscheer,
and Tatsuro Takahashi for helpful discussions, and the members of our labo-
ratory for valuable feedback on the manuscript. J.C.W was supported by
National Institutes of Health (NIH) grant GM62267, by a Leukemia and
Lymphoma Scholar Award, and by the American Cancer Society grant RSG-
08-234-01-GMC. Y.V.F. was supported by a Human Frontier Science Program
Long-Term Fellowship (LT000307/2009). D.T.L was supported by a postdoc-
toral fellowship (PF-10-146-01-DMC) from the American Cancer Society.
O.D.S. was supported by grants from the New York State Office of Science
and Technology and Academic Research NYSTAR (C040069) and the NIH
(GM08454 and CA092584). A.M.v.O. acknowledges support from the Amer-
ican Cancer Society grant RSG-08-234-01 and Searle Scholarship 05-L-104.
J.H. was supported by NIH grant GM034559 and American Cancer Society
grant RSG-08-234-01-GMC.
Received: December 15, 2010
Revised: May 17, 2011
Accepted: July 29, 2011
Published: September 15, 2011
940 Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc.
REFERENCES
Angelov, T., Guainazzi, A., and Scharer, O.D. (2009). Generation of DNA
interstrand cross-links by post-synthetic reductive amination. Org. Lett. 11,
661–664.
Arias, E.E., andWalter, J.C. (2004). Initiation of DNA replication in Xenopus egg
extracts. Front. Biosci. 9, 3029–3045.
Arias, E.E., and Walter, J.C. (2007). Strength in numbers: preventing rereplica-
tion via multiple mechanisms in eukaryotic cells. Genes Dev. 21, 497–518.
Bochman, M.L., and Schwacha, A. (2008). The Mcm2-7 complex has in vitro
helicase activity. Mol. Cell 31, 287–293.
Bochman, M.L., and Schwacha, A. (2009). The Mcm complex: unwinding the
mechanism of a replicative helicase. Microbiol. Mol. Biol. Rev. 73, 652–683.
Bowers, J.L., Randell, J.C., Chen, S., and Bell, S.P. (2004). ATP hydrolysis by
ORC catalyzes reiterative Mcm2-7 assembly at a defined origin of replication.
Mol. Cell 16, 967–978.
Brewster, A.S., Wang, G., Yu, X., Greenleaf, W.B., Carazo, J.M., Tjajadia, M.,
Klein, M.G., and Chen, X.S. (2008). Crystal structure of a near-full-length
archaeal MCM: functional insights for an AAA+ hexameric helicase. Proc.
Natl. Acad. Sci. USA 105, 20191–20196.
Costa, A., Ilves, I., Tamberg, N., Petojevic, T., Nogales, E., Botchan, M.R., and
Berger, J.M. (2011). The structural basis for MCM2-7 helicase activation by
GINS and Cdc45. Nat. Struct. Mol. Biol. 18, 471–477.
Enemark, E.J., and Joshua-Tor, L. (2006). Mechanism of DNA translocation in
a replicative hexameric helicase. Nature 442, 270–275.
Evrin, C., Clarke, P., Zech, J., Lurz, R., Sun, J., Uhle, S., Li, H., Stillman, B., and
Speck, C. (2009). A double-hexameric MCM2-7 complex is loaded onto origin
DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA
106, 20240–20245.
Fletcher, R.J., Bishop, B.E., Leon, R.P., Sclafani, R.A., Ogata, C.M., and Chen,
X.S. (2003). The structure and function of MCM from archaeal M. Thermoauto-
trophicum. Nat. Struct. Biol. 10, 160–167.
Gambus, A., Jones, R.C., Sanchez-Diaz, A., Kanemaki, M., van Deursen, F.,
Edmondson, R.D., and Labib, K. (2006). GINS maintains association of
Cdc45 with MCM in replisome progression complexes at eukaryotic DNA
replication forks. Nat. Cell Biol. 8, 358–366.
Gambus, A., Khoudoli, G.A., Jones, R.C., and Blow, J.J. (2011). MCM2-7 form
double hexamers at licensed origins in Xenopus egg extract. J. Biol. Chem.
286, 11855–11864.
Geraghty, D.S., Ding, M., Heintz, N.H., and Pederson, D.S. (2000). Premature
structural changes at replication origins in a yeast minichromosome mainte-
nance (MCM) mutant. J. Biol. Chem. 275, 18011–18021.
Guainazzi, A., Campbell, A.J., Angelov, T., Simmerling, C., and Scharer, O.D.
(2010). Synthesis and molecular modeling of a nitrogen mustard DNA inter-
strand crosslink. Chemistry (Easton) 16, 12100–12103.
Ilves, I., Petojevic, T., Pesavento, J.J., and Botchan, M.R. (2010). Activation of
the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol. Cell
37, 247–258.
Ishimi, Y. (1997). A DNA helicase activity is associated with an MCM4, �6,
and �7 protein complex. J. Biol. Chem. 272, 24508–24513.
Kanter, D.M., and Kaplan, D.L. (2010). Sld2 binds to origin single-stranded
DNA and stimulates DNA annealing. Nucleic Acids Res. 39, 2580–2592.
Kaplan, D.L., Davey, M.J., and O’Donnell, M. (2003). Mcm4,6,7 uses a ‘‘pump
in ring’’ mechanism to unwind DNA by steric exclusion and actively translocate
along a duplex. J. Biol. Chem. 278, 49171–49182.
Kelman, Z., Lee, J.K., and Hurwitz, J. (1999). The single minichromosome
maintenance protein of Methanobacterium thermoautotrophicum DeltaH
contains DNA helicase activity. Proc. Natl. Acad. Sci. USA 96, 14783–14788.
Knipscheer, P., Raschle, M., Smogorzewska, A., Enoiu, M., Ho, T.V., Scharer,
O.D., Elledge, S.J., and Walter, J.C. (2009). The Fanconi anemia pathway
promotes replication-dependent DNA interstrand cross-link repair. Science
326, 1698–1701.

Kochaniak, A.B., Habuchi, S., Loparo, J.J., Chang, D.J., Cimprich, K.A.,
Walter, J.C., and van Oijen, A.M. (2009). Proliferating cell nuclear antigen
uses two distinct modes to move along DNA. J. Biol. Chem. 284, 17700–
17710.
Kuhn, H., and Frank-Kamenetskii, M.D. (2008). Labeling of unique sequences
in double-stranded DNA at sites of vicinal nicks generated by nicking endonu-
cleases. Nucleic Acids Res. 36, e40.
Labib, K. (2010). How do Cdc7 and cyclin-dependent kinases trigger the initi-
ation of chromosome replication in eukaryotic cells? Genes Dev. 24, 1208–
1219.
Laskey, R.A., and Madine, M.A. (2003). A rotary pumping model for helicase
function of MCM proteins at a distance from replication forks. EMBO Rep. 4,
26–30.
Li, D., Zhao, R., Lilyestrom, W., Gai, D., Zhang, R., DeCaprio, J.A., Fanning, E.,
Jochimiak, A., Szakonyi, G., and Chen, X.S. (2003). Structure of the replicative
helicase of the oncoprotein SV40 large tumour antigen. Nature 423, 512–518.
Lucas, I., Chevrier-Miller, M., Sogo, J.M., and Hyrien, O. (2000). Mechanisms
ensuring rapid and complete DNA replication despite random initiation in
Xenopus early embryos. J. Mol. Biol. 296, 769–786.
Mendez, J., and Stillman, B. (2003). Perpetuating the double helix: molecular
machines at eukaryotic DNA replication origins. Bioessays 25, 1158–1167.
Moyer, S.E., Lewis, P.W., and Botchan, M.R. (2006). Isolation of the Cdc45/
Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication
fork helicase. Proc. Natl. Acad. Sci. USA 103, 10236–10241.
Pacek, M., Tutter, A.V., Kubota, Y., Takisawa, H., and Walter, J.C. (2006).
Localization of MCM2-7, Cdc45, andGINS to the site of DNA unwinding during
eukaryotic DNA replication. Mol. Cell 21, 581–587.
Pacek, M., and Walter, J.C. (2004). A requirement for MCM7 and Cdc45 in
chromosome unwinding during eukaryotic DNA replication. EMBO J. 23,
3667–3676.
Pape, T., Meka, H., Chen, S., Vicentini, G., van Heel, M., and Onesti, S. (2003).
Hexameric ring structure of the full-length archaeal MCM protein complex.
EMBO Rep. 4, 1079–1083.
Randell, J.C., Fan, A., Chan, C., Francis, L.I., Heller, R.C., Galani, K., and Bell,
S.P. (2010). Mec1 Is One of Multiple Kinases that Prime the Mcm2-7 Helicase
for Phosphorylation by Cdc7. Mol. Cell 40, 353–363.
Raschle, M., Knipscheer, P., Enoiu, M., Angelov, T., Sun, J., Griffith, J.D.,
Ellenberger, T.E., Scharer, O.D., and Walter, J.C. (2008). Mechanism of repli-
cation-coupled DNA interstrand crosslink repair. Cell 134, 969–980.
Remus, D., Beuron, F., Tolun, G., Griffith, J.D., Morris, E.P., and Diffley, J.F.
(2009). Concerted loading of Mcm2-7 double hexamers around DNA during
DNA replication origin licensing. Cell 139, 719–730.
Shechter, D.F., Ying, C.Y., and Gautier, J. (2000). The intrinsic DNA helicase
activity of Methanobacterium thermoautotrophicum delta H minichromosome
maintenance protein. J. Biol. Chem. 275, 15049–15059.
Smith, S.B., Cui, Y., and Bustamante, C. (1996). Overstretching B-DNA: the
elastic response of individual double-stranded and single-stranded DNA
molecules. Science 271, 795–799.
Takahashi, T.S., andWalter, J.C. (2005). Cdc7-Drf1 is a developmentally regu-
lated protein kinase required for the initiation of vertebrate DNA replication.
Genes Dev. 19, 2295–2300.
Takahashi, T.S., Wigley, D.B., and Walter, J.C. (2005). Pumps, paradoxes and
ploughshares: mechanism of the MCM2-7 DNA helicase. Trends Biochem.
Sci. 30, 437–444.
Tremeau-Bravard, A., Riedl, T., Egly, J.M., and Dahmus, M.E. (2004). Fate of
RNA polymerase II stalled at a cisplatin lesion. J. Biol. Chem. 279, 7751–7759.
Tye, B.K. (1999). MCM proteins in DNA replication. Annu. Rev. Biochem. 68,
649–686.
van Oijen, A.M. (2007). Honey, I shrunk the DNA: DNA length as a probe for
nucleic-acid enzyme activity. Biopolymers 85, 144–153.
Walter, J., and Newport, J. (2000). Initiation of eukaryotic DNA replication:
origin unwinding and sequential chromatin association of Cdc45, RPA, and
DNA polymerase alpha. Mol. Cell 5, 617–627.
Walter, J., Sun, L., and Newport, J. (1998). Regulated chromosomal DNA
replication in the absence of a nucleus. Mol. Cell 1, 519–529.
Warren, E.M., Vaithiyalingam, S., Haworth, J., Greer, B., Bielinsky, A.K.,
Chazin, W.J., and Eichman, B.F. (2008). Structural basis for DNA binding by
replication initiator Mcm10. Structure 16, 1892–1901.
Wessel, R., Schweizer, J., and Stahl, H. (1992). Simian virus 40 T-antigen DNA
helicase is a hexamer which forms a binary complex during bidirectional
unwinding from the viral origin of DNA replication. J. Virol. 66, 804–815.
Wohlschlegel, J.A., Dhar, S.K., Prokhorova, T.A., Dutta, A., and Walter, J.C.
(2002). Xenopus Mcm10 binds to origins of DNA replication after Mcm2-7
and stimulates origin binding of Cdc45. Mol. Cell 9, 233–240.
Yardimci, H., Loveland, A.B., Habuchi, S., van Oijen, A.M., and Walter, J.C.
(2010). Uncoupling of sister replisomes during eukaryotic DNA replication.
Mol. Cell 40, 834–840.
Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. 941

Supplemental Information
EXTENDED EXPERIMENTAL PROCEDURES
Preparation of pICLLead/Lag, pICLLead, and pICLLag
To make pICLLead/Lag, pICLLead and pICLLag, two oligonucleotides (M1: 50-CCCTGTCACTGGTAG*ACAGCATTGGAATTCCC
CCCTTTTCTTTTAAACAT*GGAAT*GCAGACTGGC-30 and M2: 50-GCACGCCAGTCTGCAT*TCCAT*GTTTAAAAGAAAAGGGGGGA
ATTCCAATGCTG*TCTACCAGTGAC-30) containing a single modified guanosine base, 7-deaza-7-(2,3-dihydroxy-propyl)-guanine
(G*), were annealed and processed to generate the ICL (Angelov et al., 2009). M1 (for pICLLead), M2 (for pICLLag), or M1 and M2
(for pICLLead/Lag) were internally biotinylated at two positions (T*) by incorporation of a Biotin-dT phosphoramidite (Glen Research,
Sterling, VA, USA). The ICL-biotin duplexwas purified by polyacrylamide gel electrophoresis, extracted from the gel by electroelution,
and ligated into pSVRLuc to form the three plasmids. Compared to pICLInter and pICLIntra, the plasmids pICLControl, pICLLead/Lag,
pICLLead, and pICLLag contain a 41 nt insert near the ICL (underlined sequence in M1 and M2), such that the StuI site is located at
different positions on the two groups of plasmids (�268 versus �309).
Chromatin ImmunoprecipitationAfter the addition of NPE, 3 ml aliquots of the reaction were crosslinked through the addition of 47 ml 1% formaldehyde in ELB (10 mM
HEPES-KOH, [pH 7.7], 2.5 mM MgCl2, 50 mM KCl, 250 mM sucrose, and 1 mM DTT). After 10 min incubation at RT, 5 ml of 1.25M
glycine was added to stop the crosslinking reaction. The crosslinked material was spun through Micro Bio-Spin 6 Chromatography
Columns (BIO-RAD, Hercules, CA, USA). The flow-through was diluted to 500 ml with sonication buffer (20mMTris 7.5, 150mMNaCl,
2 mMEDTA, 0.5%NP-40, 5 mg/mL Aprotinin+Leupeptin, 2 mMPMSF) and subjected to sonication until the average final size of DNA
fragments is �300-500 bp. Immonoprecipitation with the indicated antibodies and quantitative real-time PCR were performed as
described in Pacek et al., 2006.
Primer Extension AssayThe primer extension assay was modified from Bermudez et al., 2010. A 50-32P-labeled primer (50-GCGTTGGCCGATTCAT
TAATGCA-30) was anneal to a 68-mer oligonucleotide (50-CTTTTAAACAT*GGAAT*GCAGACTGGCGTGCGCGGCCGCGATCC
GCTGCATTAATGAATCGGCCAACGC-30), which contains two biotinylated thymidines (T*), forming the biotin template. The control
template was identical to the biotin template except that it lacked biotinylated thymidine. The templates (0.5 mM)were incubated with
8 mM Streptavidin or buffer for 1 hr at room temperature. All primer extension reactions were performed in 10 ml containing 10 nM
templates and 100 mM dNTPs. The reaction for Taq polymerase contained 10mM Tris-HCl (pH 8.3), 50mM KCl, 1.5 mM MgCl2,
and 4 units Taq polymerase (New England Biolabs, Ipswich, MA, USA). Reactions were incubated at 72�C for the indicated time.
Reactions with recombinant human DNA polymerase epsilon (Bermudez et al., 2011) contained 20mM Tris (pH7.5), 1mM DTT,
100 ug/ml BSA, 10 mM Mg acetate, 30mM potassium glutamate, 5nM RFC, 50 nM PCNA, 5 nM DNA polymerase epsilon and
were performed at 37�C. The reactions were halted with 5 ml loading buffer (95% Formamide, 18mM EDTA, and 0.025% SDS)
and heated to 95�C for 3-4 min. The extension products were separated on a 7% polyacrylamide sequencing gel and visualized
by autoradiography.
Attaching a QDot to l DNATo attach a QDot to the top strand 15 kb from the right end of l DNA, l DNA was nicked at positions 33779 and 33791 on the top
strand with Nt.BstNBI (New England BioLabs, Ipswich, MA, USA). The oligonucleotide 50-TTCAGAGT*CTGAC-30 (Biosynthesis Inc.,Lewisville, TX, USA), which contains a digoxigenin at a single thymidine base (T*), was annealed and ligated to the nicked l DNA.
Alternatively, for QDot labeling of the bottom strand 19 kb away from the end, we used Nb.BsrDI which nicks l DNA at positions
29675 and 29707 on the bottom strand and the oligonucleotide 50-CATTGCTGATACCGTT*TAGCTGAAACGACATA-30 containinga digoxinenin at T*.
SUPPLEMENTAL REFERENCES
Bermudez, V.P., Farina, A., Tappin, I., and Hurwitz, J. (2010). Influence of the human cohesion establishment factor Ctf4/AND-1 on DNA replication. J. Biol. Chem.
285, 9493–9505.
Bermudez, V.P., Farina, A., Raghavan, V., Tappin, I., and Hurwitz, J. (2011). Studies on Human DNA Polymerase ε and GINS Complex and Their Role in DNA
Replication. J. Biol. Chem. 286, 28963–28977.
Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. S1
G A T C 0 10 15 20 30 60 90 120
Primer S pICLPt
ICL 0
10
20
30
40
-1
-20
-40
AflIII D
ige
st - R
igh
tw
ard
F
ork
AflIII (151) AflIII (-540)
Primer S Primer M
Rightward
Fork
Leftward
Fork
ICL
Stu I (-268) A 0
B
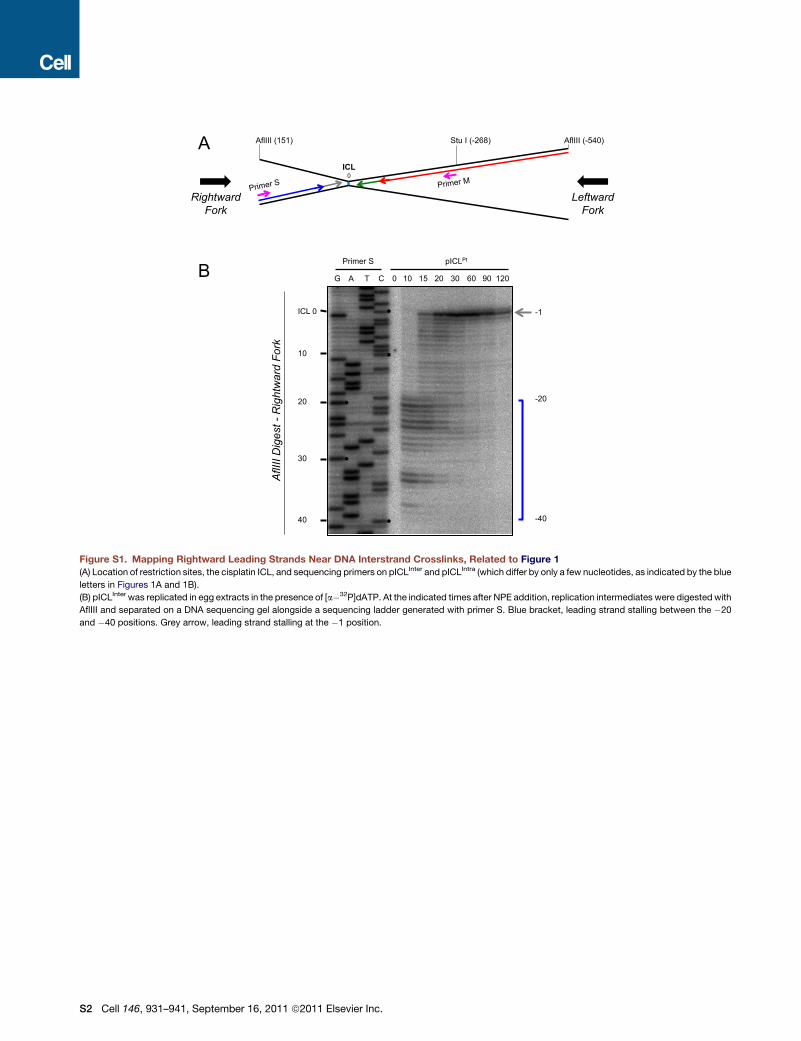
Figure S1. Mapping Rightward Leading Strands Near DNA Interstrand Crosslinks, Related to Figure 1
(A) Location of restriction sites, the cisplatin ICL, and sequencing primers on pICLInter and pICLIntra (which differ by only a few nucleotides, as indicated by the blue
letters in Figures 1A and 1B).
(B) pICLInter was replicated in egg extracts in the presence of [a�32P]dATP. At the indicated times after NPE addition, replication intermediates were digestedwith
AflIII and separated on a DNA sequencing gel alongside a sequencing ladder generated with primer S. Blue bracket, leading strand stalling between the �20
and �40 positions. Grey arrow, leading strand stalling at the �1 position.
S2 Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc.

Figure S2. CMG Complexes Converging on an ICL Do Not Interfere with Each Other, Related to Figure 2
(A) Model for the convergence of two replisomes on an ICL. Outcomes of replisome convergence assuming dsDNA translocation (top) or 30 to 50 ssDNAtranslocation (bottom) are depicted. The yellow line in the top panel represents the pin postulated to split the duplex as it emerges from the central channel.
(B) The �70 arrest cannot be explained by the footprint of DNA polymerase ε. (top) Cartoon depicting the biotinylated primer-template used for extension with
DNA pol ε. The extension product generated byDNA pol ε is shown in blue. Asterisk, location of radioactive label. (bottom) The primer template shown above or an
unbiotinylated control template was incubated with streptavidin or buffer and then incubated with Taq DNA polymerase at 72�C or DNA pol ε at 37�C, asindicated. At different times, extension products were analyzed on a 7% sequencing gel. The length of key extension products is indicated on the left of the gel.
Blue arrow, 52 nt extension products.
(C) The top half of the autoradiograph shown in primary Figure 2C is presented. Arrest of the leftward leading strand at the�70/-80 position and the�41 position
are indicated by orange and black arrows, respectively. Green arrow, approximate position of ICL. Light blue arrow, extension product. The data show that the
leftward fork is efficiently delayed at the �70 position by the biotin-SA complex.
Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. S3

Figure S3. SA Binds with Similar Efficiency to pICLLead/Lag, pICL Lead, and pICLLag Plasmids, Related to Figure 3
(A) To verify that pICLLead and pICLLag bound equally to SA, we performed gel shift analysis. pICLControl, pICLLead/Lag, pICLLead and pICLLag were preincubatedwith
streptavidin or buffer and digested with StuI and AflIII, which yields a 0.46 kb DNA fragment containing the biotin locus (green arrow). The digestion products were
separated on a 1%native agarose gel. SA caused efficient gel retardation of the 0.46 kbDNA fragments from pICLLead/Lag, pICLLead, and pICLLag (see lanes 4, 6, 8,
red and blue arrows). The SA-induced shift of the 0.46 kb fragment from pICLLead/Lag (red arrow) was greater than the shift for the fragments from pICLLead and
pICLLag (blue arrow), likely because the former contains 4 biotins instead of 2.
(B) Quantification of SA bound to plasmids during replication. pICLControl, pICLLead/Lag, pICLLead, and pICLLag were preincubated with streptavidin and replicated
in egg extracts. At the indicated times after NPE addition, reaction products were immunoprecipitated with anti-SA antibody. The quantity of plasmid associated
with SA was quantified by qPCR. The average of three independent experiments was graphed. Error bars indicate standard deviations.
(C) Same as (B), except that NPE contained 160 mM free biotin trap (the concentration of SA in the extract was 5 mM). The average of three independent
experiments was graphed. Error bars indicate standard deviations.
(D) Mapping of leading strands in the experiment shown in (C). pICLControl, pICLLead/Lag, pICLLead, and pICLLag were replicated as in (C) but in presence of [a�32P]
dATP. Replication products were digested with StuI, and analyzed on a 7% sequencing gel. The distance of products from the ICL is indicated on the left of the
gel. White arrows on the DNA sequencing ladder indicate the location of biotins. Orange bracket, leading strand arrest 70-80 nt from the ICL in the presence of SA.
S4 Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc.

Figure S4. Hypothetical Model for Bypass of a Lagging Strand Roadblock by a dsDNA Translocase, Related to Figure 4
In order to explain the bypass of a lagging strand roadblock by a dsDNA translocase, the following model would have to be invoked. When CMG collides with
a Biotin-SA complex on the lagging strand template, the central channel of the helicase opens along its entire length (A). Subsequently, the breached helicase
motors past the roadblock (B). Finally, the helicase channel recloses to enable continued unwinding by the enzyme (C). For the following reasons, this model is
improbable. If the pin or ‘‘ploughshare’’ (blue bar) remains lodged between the two strands of the duplex, the bypass will require strand separation. It is unlikely
that a helicase with an open channel could still carry out unwinding since interruption of the ring will disrupt coordinated cycles of ATP hydrolysis. In addition, the
rotation of the helicase around DNA that occurs during unwinding would be prevented by the presence of a large steric obstacle on one strand. This mechanism
also does not readily explain why only a lagging strand roadblock can be bypassed by CMG. A related model to the one presented in the figure is that after the
helicase opens upon encountering the roadblock, the pin disengages (not shown). Although this mechanism would facilitate movement past the roadblock
because unwinding by the breached helicase is not required, reactivation of the helicase downstream of the roadblock now requires reinsertion of the pin into the
duplex. How this could occur is unknown. Indeed, our single molecule analysis showed that that lesion bypass is independent of Cdk2 activity (Figure 4), which
promotes a late step in replication initiation (Walter, 2000). Therefore, the pin would have to be reinserted via a different mechanism from the one that normally
operates during initiation. In summary, neither mechanism discussed here involves a plausible scenario for how a lagging strand roadblock could be bypassed.
Cell 146, 931–941, September 16, 2011 ª2011 Elsevier Inc. S5
Related Documents











