Epilepsia, 46(Suppl. 9):34–47, 2005 Blackwell Publishing, Inc. C International League Against Epilepsy Seizures of Idiopathic Generalized Epilepsies ∗ †‡Reyna M. Dur´ on, †‡Marco T. Medina, ∗ ‡Iris E. Mart´ ınez-Ju´ arez, ∗ ‡§Julia N. Bailey, ∗ ‡Katerina Tanya Perez-Gosiengfiao, ¶‡Ricardo Ramos-Ram´ ırez, ¶‡Minerva L´ opez-Ruiz, ‡Mar´ ıa Elisa Alonso, #‡Ram´ on H. Castro Ortega, ∗∗ ‡Ignacio Pascual-Castroviejo, ∗ ‡Jes´ us Machado-Salas, ††‡Lizardo Mija, and ∗ ‡Antonio V. Delgado-Escueta ∗ California Comprehensive Epilepsy Program, David Geffen School of Medicine at UCLA, Los Angeles, CA, U.S.A.; †National Autonomous University, Tegucigalpa, Honduras; ‡Genetic Epilepsy Studies (GENESS) International Consortium; §Neuropsychiatric Institute, David Geffen School of Medicine at UCLA, Los Angeles, CA, U.S.A.; National Institute of Neurology and Neurosurgery, Mexico City, Mexico; ¶Neurology and Neurosurgery Unit, Mexico General Hospital, Mexico City, Mexico; #Autonomous University of Sonora, Hermosillo, Mexico; ∗∗ Pediatric Neurology, University Hospital La Paz, Madrid, Spain; and ††Institute of Neurological Sciences, Lima, Peru Summary: Idiopathic generalized epilepsies (IGEs) comprise at least 40% of epilepsies in the United States, 20% in Mexico, and 8% in Central America. Here, we review seizure pheno- types across IGE syndromes, their response to treatment and advances in molecular genetics that influence nosology. Our review included the Medline database from 1945 to 2005 and our prospectively collected Genetic Epilepsy Studies (GENESS) Consortium database. Generalized seizures occur with differ- ent and similar semiologies, frequencies, and patterns, ages at onset, and outcomes in different IGEs, suggesting common neuroanatomical pathways for seizure phenotypes. However, the same seizure phenotypes respond differently to the same treatments in different IGEs, suggesting different molecular defects across syndromes. De novo mutations in SCN1A in sporadic Dravet syndrome and germline mutations in SCN1A, SCN1B, and SCN2A in generalized epilepsies with febrile seizures plus have unraveled the heterogenous myoclonic epilep- sies of infancy and early childhood. Mutations in GABRA1, GABRG2, and GABRB3 are associated with absence seizures, while mutations in CLCN2 and myoclonin/EFHC1 substanti- ate juvenile myoclonic epilepsy as a clinical entity. Refined understanding of seizure phenotypes, their semiology, frequen- cies, and patterns together with the identification of molecu- lar lesions in IGEs continue to accelerate the development of molecular epileptology. Key Words: Seizures—Epilepsy— Generalized—Classification—Treatment. In this millennium, the epilepsies remain the most com- mon serious neurological problem worldwide, afflicting approximately 40–100 million persons (1). The lifetime prevalence of epilepsy in the United States is 2–5%, affect- ing an estimated 3 million Americans. The public health importance of the epilepsies is unquestionable; epilepsy costs in the United States total approximately $11.1–12.5 billion a year (2). Of all epilepsies, the idiopathic gen- eralized epilepsies (IGEs) comprise at least 40% in the United States, 20% in Mexico, and 8% in Central Amer- ica (3,4). The IGEs comprise a larger percentage of the epilepsies in the United States because some of the causes of symptomatic epilepsies have decreased. Address correspondence and reprint requests to Antonio V. Delgado- Escueta at Comprehensive Epilepsy Program, David Geffen School of Medicine at UCLA, Epilepsy Center of Excellence, VA GLAHS Medical Center, 11301 Wilshire Blvd., Bldg. 500, Suite 3405, Los Angeles, CA 90073, U.S.A. E-mail: [email protected] This supplement has been supported through an unrestricted grant from UCB S.A., manufacturers of levetiracetam (Keppra ). Among the IGEs, various authors consider juvenile myoclonic epilepsy (JME), childhood absence epilepsy (CAE), and pure grand mal on awakening as common varieties of epilepsies (5). JME is probably the most com- mon form of IGE and the most common cause of pri- mary grand mal seizures. Most hospital and clinical-based reports calculate that JME accounts for at least 12% of all epilepsies (6), while a house-to-house population sur- vey estimated that 3% were myoclonic epilepsies (7). Be- cause pure grand mal epilepsy is almost always associated with myoclonias or absences on closed-circuit television- electroencephalogram (CCTV-EEG) studies, data from our program and others suggest that JME syndromes and absence epilepsy syndromes account for the majority of IGEs. Livingston (1975) and Lennox (1945) estimate the prevalence of absence seizures to be 2.3–37.7% of all epilepsies (8–10). Epilepsies with typical absence seizures are certainly common forms of IGEs, as exemplified by CAE, juve- nile absence epilepsy (JAE), and eyelid myoclonia with 34

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Epilepsia, 46(Suppl. 9):34–47, 2005Blackwell Publishing, Inc.C© International League Against Epilepsy
Seizures of Idiopathic Generalized Epilepsies
∗†‡Reyna M. Duron, †‡Marco T. Medina, ∗‡‖Iris E. Martınez-Juarez, ∗‡§Julia N. Bailey,∗‡Katerina Tanya Perez-Gosiengfiao, ¶‡Ricardo Ramos-Ramırez, ¶‡Minerva Lopez-Ruiz,‖‡Marıa Elisa Alonso, #‡Ramon H. Castro Ortega, ∗∗‡Ignacio Pascual-Castroviejo,∗‡Jesus Machado-Salas, ††‡Lizardo Mija, and ∗‡Antonio V. Delgado-Escueta
∗California Comprehensive Epilepsy Program, David Geffen School of Medicine at UCLA, Los Angeles, CA, U.S.A.; †NationalAutonomous University, Tegucigalpa, Honduras; ‡Genetic Epilepsy Studies (GENESS) International Consortium; §NeuropsychiatricInstitute, David Geffen School of Medicine at UCLA, Los Angeles, CA, U.S.A.; ‖National Institute of Neurology and Neurosurgery,
Mexico City, Mexico; ¶Neurology and Neurosurgery Unit, Mexico General Hospital, Mexico City, Mexico; #Autonomous Universityof Sonora, Hermosillo, Mexico; ∗∗Pediatric Neurology, University Hospital La Paz, Madrid, Spain; and ††Institute of Neurological
Sciences, Lima, Peru
Summary: Idiopathic generalized epilepsies (IGEs) compriseat least 40% of epilepsies in the United States, 20% in Mexico,and 8% in Central America. Here, we review seizure pheno-types across IGE syndromes, their response to treatment andadvances in molecular genetics that influence nosology. Ourreview included the Medline database from 1945 to 2005 andour prospectively collected Genetic Epilepsy Studies (GENESS)Consortium database. Generalized seizures occur with differ-ent and similar semiologies, frequencies, and patterns, agesat onset, and outcomes in different IGEs, suggesting commonneuroanatomical pathways for seizure phenotypes. However,the same seizure phenotypes respond differently to the sametreatments in different IGEs, suggesting different molecular
defects across syndromes. De novo mutations in SCN1A insporadic Dravet syndrome and germline mutations in SCN1A,SCN1B, and SCN2A in generalized epilepsies with febrileseizures plus have unraveled the heterogenous myoclonic epilep-sies of infancy and early childhood. Mutations in GABRA1,GABRG2, and GABRB3 are associated with absence seizures,while mutations in CLCN2 and myoclonin/EFHC1 substanti-ate juvenile myoclonic epilepsy as a clinical entity. Refinedunderstanding of seizure phenotypes, their semiology, frequen-cies, and patterns together with the identification of molecu-lar lesions in IGEs continue to accelerate the development ofmolecular epileptology. Key Words: Seizures—Epilepsy—Generalized—Classification—Treatment.
In this millennium, the epilepsies remain the most com-mon serious neurological problem worldwide, afflictingapproximately 40–100 million persons (1). The lifetimeprevalence of epilepsy in the United States is 2–5%, affect-ing an estimated 3 million Americans. The public healthimportance of the epilepsies is unquestionable; epilepsycosts in the United States total approximately $11.1–12.5billion a year (2). Of all epilepsies, the idiopathic gen-eralized epilepsies (IGEs) comprise at least 40% in theUnited States, 20% in Mexico, and 8% in Central Amer-ica (3,4). The IGEs comprise a larger percentage of theepilepsies in the United States because some of the causesof symptomatic epilepsies have decreased.
Address correspondence and reprint requests to Antonio V. Delgado-Escueta at Comprehensive Epilepsy Program, David Geffen School ofMedicine at UCLA, Epilepsy Center of Excellence, VA GLAHS MedicalCenter, 11301 Wilshire Blvd., Bldg. 500, Suite 3405, Los Angeles, CA90073, U.S.A. E-mail: [email protected]
This supplement has been supported through an unrestricted grantfrom UCB S.A., manufacturers of levetiracetam (Keppra�).
Among the IGEs, various authors consider juvenilemyoclonic epilepsy (JME), childhood absence epilepsy(CAE), and pure grand mal on awakening as commonvarieties of epilepsies (5). JME is probably the most com-mon form of IGE and the most common cause of pri-mary grand mal seizures. Most hospital and clinical-basedreports calculate that JME accounts for at least 12% ofall epilepsies (6), while a house-to-house population sur-vey estimated that 3% were myoclonic epilepsies (7). Be-cause pure grand mal epilepsy is almost always associatedwith myoclonias or absences on closed-circuit television-electroencephalogram (CCTV-EEG) studies, data fromour program and others suggest that JME syndromes andabsence epilepsy syndromes account for the majority ofIGEs. Livingston (1975) and Lennox (1945) estimate theprevalence of absence seizures to be 2.3–37.7% of allepilepsies (8–10).
Epilepsies with typical absence seizures are certainlycommon forms of IGEs, as exemplified by CAE, juve-nile absence epilepsy (JAE), and eyelid myoclonia with
34

SEIZURES IN IGE 35
absence. CAE accounts for 2–10% of children with epilep-sies, while JAE constitutes 10% of all IGEs (5,11,12). Eye-lid myoclonia with absence contributes 2% to all epilep-sies and 11% to all IGEs (13). It is difficult to estimatethe contributions of rare to infrequent epilepsy syndromesto all epilepsies in general. Severe myoclonic epilepsyof Dravet, benign myoclonic epilepsy, myoclonic ab-sence, photogenic epilepsies, idiopathic myoclonic astaticepilepsy (MAE) or the Doose syndrome, and early child-hood myoclonic epilepsies could conservatively add an-other 2–3% to the IGEs (8).
It is the purpose of this article to review seizure phe-notypes across the IGE syndromes, discuss how the sameseizure types in various IGEs respond differently to thesame treatment, and review how advances in molecularepileptology have influenced nosology.
FREQUENCY OF SEIZURES IN IDIOPATHICGENERALIZED EPILEPSY SYNDROMES
ACCORDING TO THE LITERATURE
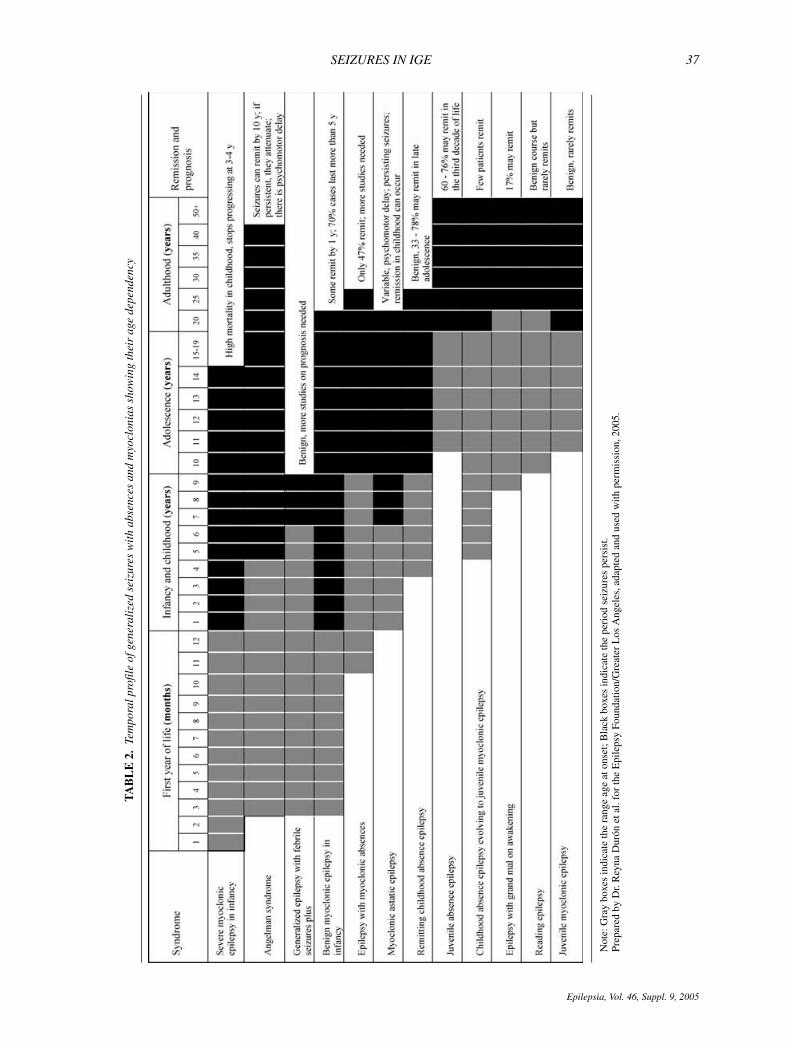
Table 1 shows the frequency of different seizure typesin generalized epilepsy syndromes, and Table 2 showsthe temporal profile of seizures in most of these syn-dromes (6,13,14–42). Tonic–clonic, absence, and my-oclonic seizures occur in variable frequencies and patternsin the different IGEs, suggesting common neurochemicalor anatomical pathways. In the last two decades, separatesubtypes of several IGE syndromes have been identifiedand several phenotypes (syndromes and subsyndromes)have been characterized with specific molecular defects(5,8,11). For example, after long-term video-EEG record-ings of MAE, Oguni et al. reported that GABA mutationswere associated with a higher percentage of absences, andthat unfavorable cases of MAE showed more absencesthan those with favorable or intermediate outcome (17).
Grand mal seizuresGrand mal clonic–tonic–clonic or tonic–clonic seizures
are common to all epilepsy syndromes. Clonic–tonic–clonic seizures are almost always genetic or biochemicalin origin, while tonic–clonic seizures can have a geneticor lesional etiology. The percentage of pure grand malis low if seizures are studied with videotape and EEGtelemetry polygraphy (11). Most of the time, grand malseizures are preceded or accompanied by absences or my-oclonic seizures that are not reported or noted by patientsand clinicians (43). Myoclonic seizures are often mis-taken as tics and nervousness (44). Additionally, grandmal seizures can be the first seizure type in up to 30% ofpatients with JME, with myoclonias reported later (35).Epidemiological studies commonly undercount absenceswith grand mal and JME, listing them instead under gen-eralized tonic–clonic seizures.
Although many epidemiological studies from industri-alized and nonindustrialized countries reported a higherproportion of primary generalized epilepsy with grand
mal seizures in the distant past (1,4), recent studies in thedeveloping world using the seizure and syndromatic clas-sification of the International League Against Epilepsy(ILAE) (45,46) show a higher frequency of partial seizureswith secondary generalization, which seems to indicate ahigh incidence of symptomatic epilepsy in the developingworld (4,47).
AbsencesThe International Classification of Epilepsies and
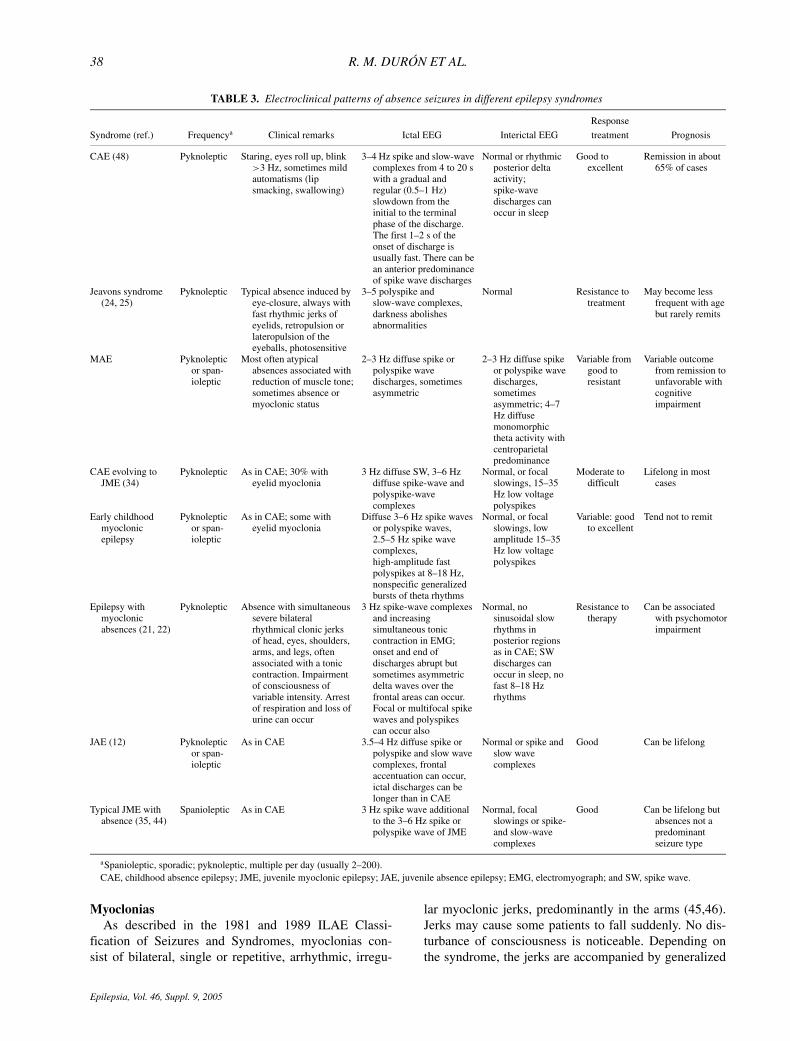
Epileptic Syndromes includes four types of IGEs with typ-ical absences: CAE, JAE, JME, and epilepsy with general-ized grand mal on awakening (EGMA) (45,46). Several at-tempts have been made to better understand the syndromeswith typical absence seizures, and several series indicatethe existence of even more absence syndromes in additionto the four listed above. Table 3 and Figs. 1A–D show thatabsence seizures may exhibit different patterns across syn-dromes reported in literature (5,12,21,22,24,25,34,44,48).
Several varieties of absence syndromes are differenti-ated from the typical CAE that occurs between 4 and 10years of age and that remits in adolescence. These ab-sence syndromes are absence epilepsy of early childhoodstarting before 5 years of age (49,50); MAE, in whichmyoclonias occur simultaneously with absence seizures(21,22); eyelid myoclonia with absences (23–25); CAEthat persists into adulthood (29–31,51–54); and CAEthat evolves into JME (34,35). Pyknoleptic CAEs per-sisting into adolescence and adulthood with grand malseizures are reported to occur in 12–32% of all absencecases (29,51,52,55,56). Some authors have reported thatCAE can precede JME: Panayiotopoulos reported 7 of65 cases (11%) (25), Asconape and Penry reported 3of 11 cases (25%) (57), and Wirrel et al. reported 17%(31,52,58), while our studies with the Genetic EpilepsyStudies (GENESS) Consortium reported 45 of 258 cases(18%) (35). However, until recently, no family studieshave been published that determine the seizure pheno-types of family members, sex preponderance, mode oftransmission, and long-term outcomes in the syndrome ofCAE evolving to JME. So far, some authors have consid-ered absences starting in childhood as an incidental com-ponent of JME (59). As discussed later, this subgroup thatwe call CAE evolving to JME shows different features andseems to constitute a different subsyndrome.
Many have emphasized how absence seizures are fre-quently unrecognized or misdiagnosed as complex par-tial seizures. Electroencephalography, particularly video-electroencephalography, is invaluable in separating thetwo forms of seizures. Sometimes focal EEG abnormal-ities are seen in some absence epilepsies and are erro-neously interpreted as evidence of partial epilepsy (34,60).Additionally, symptomatic typical absences can occur ingenetically predisposed adults with brain damage such asencephalopathies produced by the Lafora disease and theUnverricht–Lundborg epilepsy (61,62).
Epilepsia, Vol. 46, Suppl. 9, 2005

36 R. M. DURON ET AL.
TA
BL
E1.
Fre
quen
cyof
the
diffe
rent
seiz
ure
type
sin
pati
ents
wit
hIG
Esy
ndro
mes
Toni
c–cl
onic
orFe
brile
Eye
lidPe
rior
alcl
onic
–A
bsen
ceM
yocl
onic
seiz
ures
Myo
clon
icM
yocl
onic
myo
clon
iam
yocl
onia
sA
toni
cTo
nic
Foca
la
Epi
leps
ysy
ndro
mes
Ref
.to
nic–
clon
ic(%
)(%
)(%
)(%
)ab
senc
e(%
)as
tatic
(%)
(%)
(%)
(%)
(%)
(%)
Ben
ign
myo
clon
icep
ileps
yof
infa
ncy
15R
are
—10
0R
are
——
Rar
e—
——
—
Seve
rem
yocl
onic
epile
psy
ofin
fanc
y(D
rave
tsyn
drom
e)14
,16
1440
–95b
100
28–4
8—
—C
omm
on—
—14
53
Fam
ilial
myo
clon
icep
ileps
yw
ithon
seti
nin
fanc
y18
100
—10
063
——
——
——
—
MA
E(D
oose
synd
rom
e)19
,20
75–9
562
–79
100
11–2
8—
100
——
—30
–95
—M
yocl
onic
abse
nces
21,2
245
4—
Rar
e10
0—
4—
——
—E
yelid
myo
clon
iaw
ithab
senc
e(J
eavo
nssy
ndro
me)
13,2
3–25
5010
0%34
–55
Rar
e10
0—
——
——
—
GE
FS+
with
GA
BR
2m
utat
ion
2634
297
86—
4—
—4
—4
GE
FS+
with
SCN
1Am
utat
ion
2664
219
83—
9—
—12
—9
Typi
calC
AE
27–3
330
–60
100
—20
—22
——
——
——
—C
AE
evol
ving
toJM
E34
9810
010
03
——
30—
——
—Ty
pica
lJM
E6,
35–3
880
–95
7–38
100
13—
4—
23—
——
JAE
12,3
9,40
Com
mon
100
15–2
0—
37–8
0—
——
——
—G
rand
mal
onaw
aken
ing
6,41
,42
100
13–6
36–
29—
——
——
——
4–22
GE
FS+,
gene
raliz
edep
ileps
yw
ithfe
brile
seiz
ures
plus
;GA
BR
2,G
AB
Are
cept
orsu
buni
t2;S
CN
1,so
dium
chan
nela
lfa
1su
buni
t;C
AE
,chi
ldho
odab
senc
eep
ileps
y;JM
E,j
uven
ilem
yocl
onic
epile
psy;
JAE
,juv
enile
abse
nce
epile
psy;
dash
esm
ean
the
seiz
ure
type
has
notb
een
repo
rted
inth
esy
ndro
me.
a Con
side
rth
atfo
cals
eizu
res
and
segm
enta
lmyo
clon
ias
are
ofte
nco
nfus
edw
ithea
chot
her.
bR
efer
ring
tofa
mily
mem
bers
,not
prob
ands
.
Epilepsia, Vol. 46, Suppl. 9, 2005
SEIZURES IN IGE 37
TA
BL
E2.
Tem
pora
lpro
file
ofge
nera
lize
dse
izur
esw
ith
abse
nces
and
myo
clon
ias
show
ing
thei
rag
ede
pend
ency
Not
e:G
ray
boxe
sin
dica
teth
era
nge
age
aton
set;
Bla
ckbo
xes
indi
cate
the
peri
odse
izur
espe
rsis
t.Pr
epar
edby
Dr.
Rey
naD
uron
etal
.for
the
Epi
leps
yFo
unda
tion/
Gre
ater
Los
Ang
eles
,ada
pted
and
used
with
perm
issi
on,2
005.
Epilepsia, Vol. 46, Suppl. 9, 2005
38 R. M. DURON ET AL.
TABLE 3. Electroclinical patterns of absence seizures in different epilepsy syndromes
Response
Syndrome (ref.) Frequencya Clinical remarks Ictal EEG Interictal EEG treatment Prognosis
CAE (48) Pyknoleptic Staring, eyes roll up, blink>3 Hz, sometimes mildautomatisms (lipsmacking, swallowing)
3–4 Hz spike and slow-wavecomplexes from 4 to 20 swith a gradual andregular (0.5–1 Hz)slowdown from theinitial to the terminalphase of the discharge.The first 1–2 s of theonset of discharge isusually fast. There can bean anterior predominanceof spike wave discharges
Normal or rhythmicposterior deltaactivity;spike-wavedischarges canoccur in sleep
Good toexcellent
Remission in about65% of cases
Jeavons syndrome(24, 25)
Pyknoleptic Typical absence induced byeye-closure, always withfast rhythmic jerks ofeyelids, retropulsion orlateropulsion of theeyeballs, photosensitive
3–5 polyspike andslow-wave complexes,darkness abolishesabnormalities
Normal Resistance totreatment
May become lessfrequent with agebut rarely remits
MAE Pyknolepticor span-ioleptic
Most often atypicalabsences associated withreduction of muscle tone;sometimes absence ormyoclonic status
2–3 Hz diffuse spike orpolyspike wavedischarges, sometimesasymmetric
2–3 Hz diffuse spikeor polyspike wavedischarges,sometimesasymmetric; 4–7Hz diffusemonomorphictheta activity withcentroparietalpredominance
Variable fromgood toresistant
Variable outcomefrom remission tounfavorable withcognitiveimpairment
CAE evolving toJME (34)
Pyknoleptic As in CAE; 30% witheyelid myoclonia
3 Hz diffuse SW, 3–6 Hzdiffuse spike-wave andpolyspike-wavecomplexes
Normal, or focalslowings, 15–35Hz low voltagepolyspikes
Moderate todifficult
Lifelong in mostcases
Early childhoodmyoclonicepilepsy
Pyknolepticor span-ioleptic
As in CAE; some witheyelid myoclonia
Diffuse 3–6 Hz spike wavesor polyspike waves,2.5–5 Hz spike wavecomplexes,high-amplitude fastpolyspikes at 8–18 Hz,nonspecific generalizedbursts of theta rhythms
Normal, or focalslowings, lowamplitude 15–35Hz low voltagepolyspikes
Variable: goodto excellent
Tend not to remit
Epilepsy withmyoclonicabsences (21, 22)
Pyknoleptic Absence with simultaneoussevere bilateralrhythmical clonic jerksof head, eyes, shoulders,arms, and legs, oftenassociated with a toniccontraction. Impairmentof consciousness ofvariable intensity. Arrestof respiration and loss ofurine can occur
3 Hz spike-wave complexesand increasingsimultaneous toniccontraction in EMG;onset and end ofdischarges abrupt butsometimes asymmetricdelta waves over thefrontal areas can occur.Focal or multifocal spikewaves and polyspikescan occur also
Normal, nosinusoidal slowrhythms inposterior regionsas in CAE; SWdischarges canoccur in sleep, nofast 8–18 Hzrhythms
Resistance totherapy
Can be associatedwith psychomotorimpairment
JAE (12) Pyknolepticor span-ioleptic
As in CAE 3.5–4 Hz diffuse spike orpolyspike and slow wavecomplexes, frontalaccentuation can occur,ictal discharges can belonger than in CAE
Normal or spike andslow wavecomplexes
Good Can be lifelong
Typical JME withabsence (35, 44)
Spanioleptic As in CAE 3 Hz spike wave additionalto the 3–6 Hz spike orpolyspike wave of JME
Normal, focalslowings or spike-and slow-wavecomplexes
Good Can be lifelong butabsences not apredominantseizure type
aSpanioleptic, sporadic; pyknoleptic, multiple per day (usually 2–200).CAE, childhood absence epilepsy; JME, juvenile myoclonic epilepsy; JAE, juvenile absence epilepsy; EMG, electromyograph; and SW, spike wave.
MyocloniasAs described in the 1981 and 1989 ILAE Classi-
fication of Seizures and Syndromes, myoclonias con-sist of bilateral, single or repetitive, arrhythmic, irregu-
lar myoclonic jerks, predominantly in the arms (45,46).Jerks may cause some patients to fall suddenly. No dis-turbance of consciousness is noticeable. Depending onthe syndrome, the jerks are accompanied by generalized
Epilepsia, Vol. 46, Suppl. 9, 2005

SEIZURES IN IGE 39
FIG. 1. Ictal electroencephalograms (EEGs) of absence seizures in several absence syndromes. A: Typical CAE: 9- and 10-year-oldpatient with staring episodes and 3 Hz spike-wave complexes. B: CAE evolving to JME: myoclonia followed by absence while reading and4 Hz spike-wave and polyspike-wave complexes. C: Myoclonic absences. Nine-year-old patient with absences simultaneous to jerks inboth arms and head: 4 Hz spike-wave and polyspike-wave complexes followed by diffuse slowing. D: Left . Absence with eyelid myoclonia(Jeavons syndrome): 17-year-old patient who started having absences with eyelid myoclonias at age 4 persisting together with grandmal seizures in adolescence. Ictal EEG during photic stimulation shows 2.5–3 Hz spike and polyspike followed by slow wave. Right :Ten-year-old boy with staring episodes with fast blinking had ictal 5 Hz spike-wave complexes.
tonic–clonic seizures or absences. The interictal EEG has4–6 Hz spike or polyspike and slow-wave complexes,while high voltage 12–30 polyspikes accompanied my-oclonic seizures (Fig. 2) (44,63).
Myoclonias are the sine qua non of JME, a commonepilepsy syndrome (6,37,38,43,64,65). Myoclonias mayalso mark the various syndromes of childhood myoclonic
FIG. 2. EEGs in patients with juvenile epilepsy (JME). A: Diffuse polyspike followed by slow-wave during myoclonias early in the morningin two adolescent patients with classic JME. B: Interictal diffuse 3 Hz polyspike and slow-wave discharge in a female adolescent patientwith JME, spanioleptic absences and astatic seizures.
epilepsies, which represent a broad group of heteroge-neous clinical and electroencephalographic entities. Be-tween infancy and 5 years of age, myoclonias can appeartogether with generalized clonic, tonic–clonic, atonic-drop or astatic, and absence seizures. Although seizuresappear in various combinations, the predominant type ofepileptic attacks has often determined the name given to
Epilepsia, Vol. 46, Suppl. 9, 2005

40 R. M. DURON ET AL.
the syndromes. For instance, several authors recognizedcryptogenic myoclonic epilepsies based on the predomi-nance of myoclonic seizures (66–68). Doose et al. (69),Doose (19), and Doose and Baier (70) separated primar-ily generalized seizures with generalized irregular spikesand waves from symptomatic West’s infantile spasms andhypsarrhythmia, and from symptomatic Lennox–Gastautsyndrome, initially designated as myoclonic petit mal ofearly childhood. These authors have recently used theterm primarily generalized myoclonic astatic petit mal ofearly childhood to encompass an epilepsy syndrome char-acterized by myoclonic, astatic, myoclonic astatic, andmostly tonic–clonic seizures with EEG generalized spikesand waves, photosensitivity, and 4–7 Hz biparietal thetarhythms (70). With a high incidence of seizures and ge-netic EEG patterns in relatives, these children have mostlynormal development and no neurological deficits beforethe onset of seizures. Reporting in 117 cases, Doose notedin 1992 their variable course ranging from spontaneousremission to persistence with neurological deterioration(71).
Because primarily generalized myoclonic seizures inchildhood are underrecognized and underdiagnosed, theirtrue prevalence is unknown. Pazzaglia et al. (1979) cal-culated that 1.4% of all epilepsies have myoclonic astaticseizures (MAS) (72), while Doose and Sitepu in an epi-demiologic study in Germany reported that 1–2% of allchildhood epilepsies up to 9 years of age have MAS (73).In the population-based epilepsy study performed by Med-ina et al. in Salama, Honduras, only 1% of epilepsies be-longed to this syndrome (3).
SEIZURES IN SPECIFIC IDIOPATHICGENERALIZED EPILEPSY SYNDROMES
ACCORDING TO THE GENESS DATABASE
Early childhood myoclonic epilepsy
Seizures in probands and relativesWe prospectively studied 22 probands with early child-
hood myoclonic epilepsy (ECME) or the idiopathic varietyof MAS epilepsy that started during late infancy or earlychildhood. All probands had myoclonias (100%), whilea majority had astatic drops (91%), generalized tonic–clonic seizures (72%), and absences (68%). Less fre-quently clonic–tonic–clonic and generalized clonic (27%)seizures were present. Bilateral 2–6 Hz spike or polyspikewaves appeared in all probands, and classic 3 Hz spikewave was present in three (14%), and 2–3 Hz spike wavecomplexes were also present in five probands (23%).There was a slight male preponderance (9 female; 12male) among probands, but affected nonproband familymembers were often female (31 female, 9 male), with aratio of 3.4:1. Familial history of epilepsy was present in50% of probands. Maternal transmission was more com-mon than paternal transmission (67% vs. 33%), but in
both cases, clinically or EEG-affected nonproband mem-bers were more often female (75% and 82%, respectively).In nonproband family members, grand mal (72.5%), ab-sences (42.5%), and myoclonic seizures (22.5%) werecommon, and astatic seizures were infrequently reported(10%). We prefer the term ECME, because MAS decreasein frequency and eventually remit in childhood, but in 47%of patients, myoclonic or clonic–tonic–clonic seizures per-sist into adolescence. Valproate was effective in seizurecontrol in most cases.
JME subsyndromesJME starts at adolescence with myoclonic seizures that
usually occur after awakening and are triggered by sleepdeprivation and fatigue. In addition to myoclonic jerks,95% of patients with JME have generalized tonic–clonicgrand mal seizures, and about one-third also have absenceor petit mal seizures. We have subdivided 258 JME fam-ilies into four groups according to the combination ofseizure types afflicting the proband; the age of probands atonset of seizures; the results of physical, neurological, andbrain imaging examinations; and the long-term outcomeafter treatment with valproate and/or the new generationof antiepileptic drugs (AEDs) (34,35).
Classic JME
Seizures in probandsThis subsyndrome accounts for 73% (n = 186) of all
our cases, starting in adolescence as isolated awaken-ing myoclonic seizures or generalized clonic–tonic–clonicseizures. Myoclonias were most often the first seizure typeto appear (117 of 170 cases; 68%). In 30% of probands (52of 170) grand mal seizures preceded myoclonias. Almostall (95%) had tonic–clonic or clonic–tonic–clonic grandmal seizures. Sometimes rare isolated absence seizures(spanioleptic or sporadic absences) or rare episodes ofabsences appeared in clusters. In nine cases, the typeof seizure at onset was not ascertained. The interictalEEG had 4–6 Hz spike or polyspike and slow-wave com-plexes, while high voltage 12–30 polyspikes accompaniedmyoclonic seizures. Although females were numericallymore common than males among probands (103 females,83 males; 1.25:1 ratio), the difference was not statisticallysignificant (χ2 = 1.078, p = 0.29).
Myoclonias started on average at age 15.1 years (range7–28 years) in 170 patients. Grand mal seizures appearedfor the first time at an average age of 15.9 years (range 8–33 years). Absences started at the mean age of 16.8 years(range 11–30 years) and were present in 33% of the pa-tients (57 of 170). Absences were spanioleptic in all casesand were the first seizure type reported only in one case(onset at age 16 years). The most common trigger factorsfor seizures were sleep deprivation, stress, alcohol, non-compliance, and menses. Other less common causes ofseizures were fatigue, hyperventilation, photosensitivity,
Epilepsia, Vol. 46, Suppl. 9, 2005

SEIZURES IN IGE 41
and physical activity. Sixty-one percent of probandsshowed 4–6 Hz spike-wave or polyspike-wave complexesin a 30- to 40-minute interictal EEG. Fourteen percenthad 3 Hz spike-wave complexes, either alone or associ-ated with 4–6 Hz polyspike-wave complexes. Focal slow,sharp, or spike waves were found in 10% of patients. Gen-eralized bursts of 4–6 Hz slow waves without spikes werefound in 2%, and 13% of the interictal EEGs were normal.
Seizures in relativesSeizures were present in family members in 92 of
186 cases of classic JME (49%). Epileptic seizures af-fected 188 of 1764 (11%) of nonproband family mem-bers. Specific seizure types could be determined in only136 of the affected relatives; 11.5% of first-degree rela-tives had seizures. The most common (46%) were my-oclonias with or without grand mal. Rarely were absenceseizures present in relatives. Only 4% of second-degreerelatives had seizures. Most common were grand malseizures (41.4%), followed by myoclonic seizures (28%).Abnormal EEGs were found in 24 asymptomatic relatives.Of these 24 relatives, 15 (63%) had 4–6 Hz polyspikewaves, four (17%) had bursts of focal or diffuse slowing,three (12%) had 3–5 Hz spike and wave complexes, andtwo (8%) had bursts of focal or diffuse sharp waves. Mostasymptomatic family members with EEG polyspike wavewere female (10 females, 5 males).
Childhood absence epilepsy evolving to JMEThis group accounts for 17% of all the idiopathic my-
oclonic epilepsies in our series and consisted of 45 pa-tients who had absence epilepsy that started in childhoodand persisted with myoclonic and tonic–clonic grand malseizures into adulthood (34). We studied the clinical andEEG features of 985 persons belonging to these fami-lies. Of the families, 29% (13 of 45) families were mul-tiplex/multigenerational, 27% (12 of 45) were multigen-erational only, and 16% (7 of 45) were multiplex. Of thefamilies, 28% (13 of 45) were simplex. We collected dataup to late adolescence or adulthood in 38 cases (84%).Thirty-eight families (62%) were of Caucasian ethnic ori-gin, while 17 (38%) were mixtures of European and Amer-ican Indians.
Seizures in probandsFemales were numerically preponderant among
probands (29 females, 16 males; 1.8:1 ratio), but this wasnot statistically significant (χ2 = 1.91, p = 0.166) becauseof the small number of cases. Childhood absence seizureswere documented as the first seizure in all probands. Ab-sences started at an average age of 6.9 years (1–11 years) in41 probands. In four probands, parents could only describeabsences as starting during or before elementary school.Among those with persisting seizures, absences continuedto be the chief complaint in 44.4%, and absences mixedwith tonic–clonic or myoclonic seizures were the chief
complaint in 16%. Absences occurred at least once a day(range 1–200 per day), namely pyknoleptic.
Myoclonias started at a mean age of 14 years (range8–47 years). Myoclonic seizures preceded grand mal con-vulsions in 18%, but more often they started in adoles-cence simultaneously with tonic–clonic seizures (40% ofpatients). Tonic–clonic seizures appeared for the first timeon average at age 12 years (range 2–37 years).
All probands had the classic 3 Hz single-spike and slow-wave complexes. Seventy-eight percent (29 probands) alsohad 2–5 Hz single-spike and slow-wave complexes. In ad-dition, 20 individuals, or 54%, also had 4–6 Hz polyspike-wave complexes. Thirty-four percent of all cases had bothsingle-spike wave and polyspike waves. We also founddiffuse, fast low-amplitude 15–25 Hz rhythms in 22% ofcases. Bursts of 3–5 Hz single-spike wave and/or 4–6 Hzpolyspike wave were induced during hyperventilation in29% of probands and during photostimulation in 22%.Background activity was normal in all cases.
Seizures in relativesAbsence seizures most often affected nonproband fam-
ily members. Myoclonias only or myoclonias plus grandmal affected only five persons (6%). CAE evolving toJME was documented in 13 (16%) of nonproband affectedmembers (eight females, five males). When checking forparental affectedness in multiplex/multigenerational pedi-grees, 9 of 32 mothers had seizures more often (28%)than fathers (2 of 32, or 6%). Sixteen families (306 non-proband members) had EEG studies. Ten percent (30members) had single-spike wave or polyspike-wave com-plexes; 22 of these persons were asymptomatic. Overall,almost 20% of relatives reported seizures. Twenty-one per-cent of first-degree relatives had seizures. Most commonwere absences (56%). Second-degree relatives commonlyreported absences and tonic–clonic seizures.
JME with adolescent-onset pyknoleptic absenceThis group accounted for 7% of all JME cases (18
families). Pyknoleptic absences started commonly at age16 years, usually between the ages of 11 and 32 years,and were combined with myoclonia and clonic–tonic–clonic grand mal seizures. Myoclonic seizures were thefirst seizure type in 61%. There was a female prepon-derance in probands (2.6:1 female:male ratio, χ2 = 1.87,p = 0.171). Myoclonic seizures were the main seizuretype in 72% of the probands, but in eight cases with persis-tent seizures, absences with or without myoclonic seizureswere predominant (five of eight cases). Interictal EEGsof 14 probands were available for analysis. Seven of 14had normal 4–5 Hz spike-wave or polyspike waves. Twoshowed 4–6 Hz polyspike-wave complexes mixed with3 Hz spike-wave complexes. One had generalized bursts ofslow waves, and one had 3 Hz spike-wave complexes. Pho-toparoxysmal response was found in two of 14 probands.Three probands had normal EEGs. Neurological exams
Epilepsia, Vol. 46, Suppl. 9, 2005

42 R. M. DURON ET AL.
were normal in all patients. Neuroimaging studies alsowere normal.
Of 18 probands, 61% (11 of 18) had a family history ofepilepsy. Five families were multiplex/multigenerationalfamilies, five families were multigenerational, and onewas multiplex. Maternal transmission was as common (4of 11) as paternal transmission (5 of 11), and two familieswere bilineal. Mode of inheritance could not be deter-mined in one family with monozygotic twins. Epilepticseizures affected 28 nonproband family members. Thirtypercent had JME as well, while 30% had grand malseizures only. Three nonprobands were asymptomatic butwere affected by EEG 4–6 Hz polyspike-wave traits. Threepersons had absences only. Febrile seizures were presentin three probands, and partial seizures were present in onenonproband.
JME with astatic seizuresThis group accounted for 3% of all JME cases (nine
families). Aside from the adolescent-onset myoclonia andtonic–clonic or clonic–tonic–clonic grand mal seizures,nine patients exhibited astatic seizures between the agesof 8 and 17 years, with a mean age of onset of 14.3years. No gender predominance was found (five females,four males). Only one of our patients has spaniolepticabsences with onset also in adolescence. EEGs of four pa-tients showed 4–6 Hz spike-wave or polyspike and slow-wave complexes. One patient had the same pattern, butpolyspike-waves were combined with 3 Hz spike-wavecomplexes. One patient’s EEG consisted of generalizedbursts of slow waves. General and neurological exami-nations were normal in all patients. Four probands hadcomputed tomography (CT) scans, and one had positionemission tomography with fluorodeoxyglucose. One pa-tient had bilateral hippocampal atrophy on magnetic res-onance imaging (MRI).
Four probands (44%) had family histories ofepilepsy. Five families were simplex, and four weremultiplex/multigenerational. Both maternal and pater-nal transmission were equally common in multiplex/multigenerational families, and no bilineality was evidentin this group. Among nonprobands family members, fivewere affected. Two had JME, but one also presented withMAS during adolescence. One had grand mal only. Onewho was asymptomatic was affected by EEG polyspikeand slow waves, and one had absence seizures only.
THE SAME SEIZURE TYPES IN VARIOUSIDIOPATHIC GENERALIZED EPILEPSIESRESPOND DIFFERENTLY TO THE SAME
TREATMENT
Primarily generalized seizures share a particular phar-macological sensitivity not found in seizure types pro-duced by lesions. They respond well to valproate andthe new generation AEDs levetiracetam, topiramate, zon-
isamide, and often lamotrigine. They can be exacerbatedby phenytoin, carbamazepine, oxcarbazepine, gabapentin,and primidone (74). Yet, the same absences and my-oclonic, tonic–clonic, and astatic seizures that respondwell to valproate in one syndrome, for example, CAE,classic JME, or ECME, do not respond in another syn-drome, for example, Dravet syndrome, CAE evolving toJME, or MAE of Doose (34,35). This is why there is gen-eral agreement that selection of the best treatment op-tions should be based not only on the correct determi-nation of seizure types but also on the correct diagno-sis of epilepsy syndrome (75). According to the experi-ence of most epileptologists, the more combinations ofseizure types a patient has, the harder it is to controlseizures with AEDs, especially if absences persist fromchildhood (52,58,76).
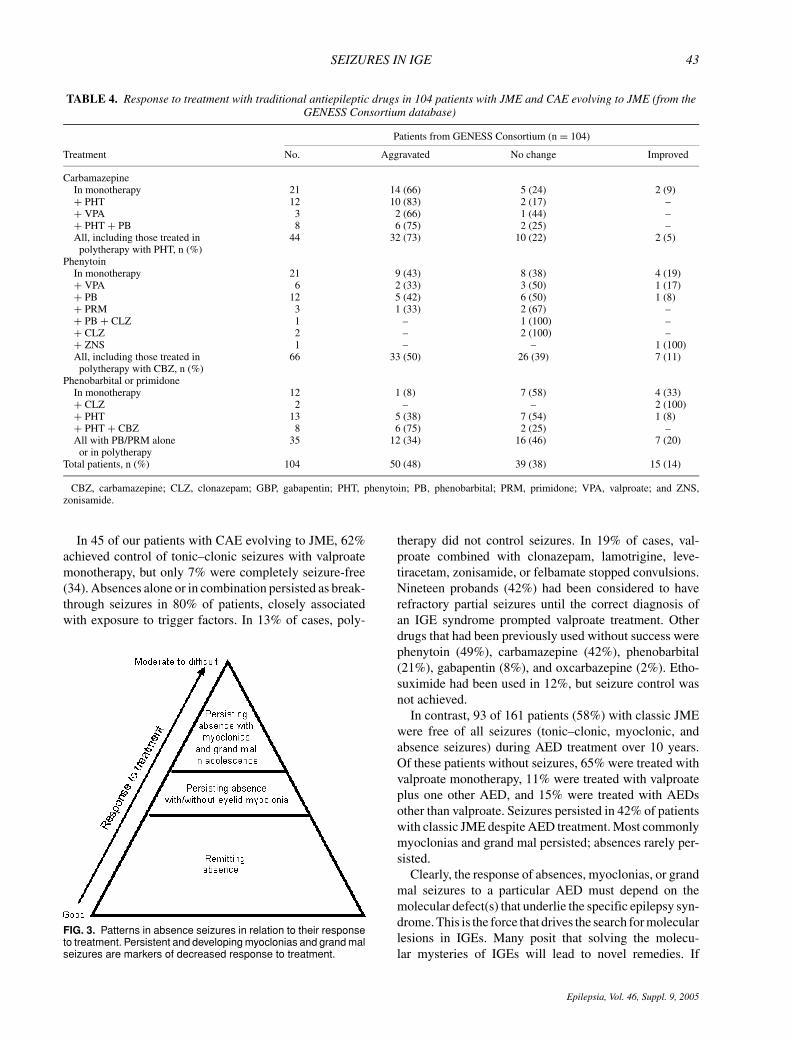
Another important observation, only recently empha-sized, has been the aggravation of IGEs by the older-generation AEDs. Table 4 shows the response to treat-ment with traditional AEDs in our series of JME and CAEcases evolving to JME. Among 161 JME patients, 104(64%) received carbamazepine, phenytoin, phenobarbi-tal, or primidone prior to referral to our clinics. Fourteenpercent reported some improvement of seizures, mostlytonic–clonic seizures. In contrast, 48% blamed carba-mazepine and phenytoin for increasing frequencies oftonic–clonic grand mal, absence, and myoclonic seizures.Phenytoin as monotherapy or in polytherapy showed theworst profile, aggravating seizures in two-thirds of cases.In two patients, the combination of carbamazepine andphenytoin triggered the first appearance of absence status.Elimination of both AEDs eliminated absence seizures.Other series have reported similar findings in myoclonicand absences epilepsies (74). Vigabatrin, tiagabine, andgabapentin can also aggravate IGEs and have been re-ported to induce myoclonic and absence status (77,78).In infrequent cases, lamotrigine has aggravated seizurefrequency in myoclonic epilepsies (79).
Fig. 3 depicts the variable response to treatment of ab-sence seizures according to their different patterns. Usu-ally, satisfactory control of absences is achieved withsodium valproate or ethosuximide, but some subtypesof absence epilepsy such as CAE evolving to JME andJAE show higher risk of chronicity and relapses (34). TheSwedish epidemiological study performed by Olsson andHagberg (51) from 1978 to 1982 first helped to discerntwo distinct groups among 134 children with absences.The first group is composed of patients with CAE thatstarts before the age of 12, quickly responds to therapy,and has little or no risk of grand mal and a high remissionrate. The second group comprises patients with JAE thatstarts at age 12 years or later who have a very high risk ofgrand mal and usually a good response to therapy, but ahigh risk of relapse on withdrawal of therapy. Classifyingabsence epilepsy into subgroups is useful for prognosis.
Epilepsia, Vol. 46, Suppl. 9, 2005
SEIZURES IN IGE 43
TABLE 4. Response to treatment with traditional antiepileptic drugs in 104 patients with JME and CAE evolving to JME (from theGENESS Consortium database)
Patients from GENESS Consortium (n = 104)
Treatment No. Aggravated No change Improved
CarbamazepineIn monotherapy 21 14 (66) 5 (24) 2 (9)+ PHT 12 10 (83) 2 (17) –+ VPA 3 2 (66) 1 (44) –+ PHT + PB 8 6 (75) 2 (25) –All, including those treated in 44 32 (73) 10 (22) 2 (5)
polytherapy with PHT, n (%)Phenytoin
In monotherapy 21 9 (43) 8 (38) 4 (19)+ VPA 6 2 (33) 3 (50) 1 (17)+ PB 12 5 (42) 6 (50) 1 (8)+ PRM 3 1 (33) 2 (67) –+ PB + CLZ 1 – 1 (100) –+ CLZ 2 – 2 (100) –+ ZNS 1 – – 1 (100)All, including those treated in 66 33 (50) 26 (39) 7 (11)
polytherapy with CBZ, n (%)Phenobarbital or primidone
In monotherapy 12 1 (8) 7 (58) 4 (33)+ CLZ 2 – – 2 (100)+ PHT 13 5 (38) 7 (54) 1 (8)+ PHT + CBZ 8 6 (75) 2 (25) –All with PB/PRM alone 35 12 (34) 16 (46) 7 (20)
or in polytherapyTotal patients, n (%) 104 50 (48) 39 (38) 15 (14)
CBZ, carbamazepine; CLZ, clonazepam; GBP, gabapentin; PHT, phenytoin; PB, phenobarbital; PRM, primidone; VPA, valproate; and ZNS,zonisamide.
In 45 of our patients with CAE evolving to JME, 62%achieved control of tonic–clonic seizures with valproatemonotherapy, but only 7% were completely seizure-free(34). Absences alone or in combination persisted as break-through seizures in 80% of patients, closely associatedwith exposure to trigger factors. In 13% of cases, poly-
FIG. 3. Patterns in absence seizures in relation to their responseto treatment. Persistent and developing myoclonias and grand malseizures are markers of decreased response to treatment.
therapy did not control seizures. In 19% of cases, val-proate combined with clonazepam, lamotrigine, leve-tiracetam, zonisamide, or felbamate stopped convulsions.Nineteen probands (42%) had been considered to haverefractory partial seizures until the correct diagnosis ofan IGE syndrome prompted valproate treatment. Otherdrugs that had been previously used without success werephenytoin (49%), carbamazepine (42%), phenobarbital(21%), gabapentin (8%), and oxcarbazepine (2%). Etho-suximide had been used in 12%, but seizure control wasnot achieved.
In contrast, 93 of 161 patients (58%) with classic JMEwere free of all seizures (tonic–clonic, myoclonic, andabsence seizures) during AED treatment over 10 years.Of these patients without seizures, 65% were treated withvalproate monotherapy, 11% were treated with valproateplus one other AED, and 15% were treated with AEDsother than valproate. Seizures persisted in 42% of patientswith classic JME despite AED treatment. Most commonlymyoclonias and grand mal persisted; absences rarely per-sisted.
Clearly, the response of absences, myoclonias, or grandmal seizures to a particular AED must depend on themolecular defect(s) that underlie the specific epilepsy syn-drome. This is the force that drives the search for molecularlesions in IGEs. Many posit that solving the molecu-lar mysteries of IGEs will lead to novel remedies. If
Epilepsia, Vol. 46, Suppl. 9, 2005
44 R. M. DURON ET AL.
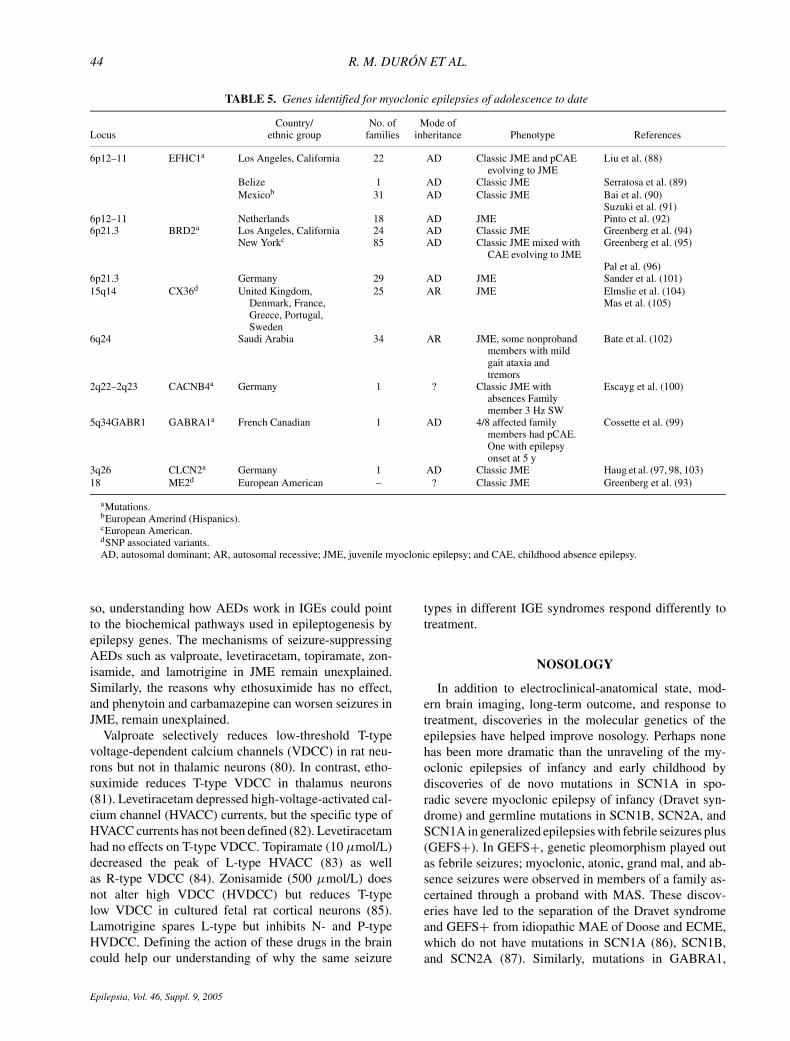
TABLE 5. Genes identified for myoclonic epilepsies of adolescence to date
Country/ No. of Mode ofLocus ethnic group families inheritance Phenotype References
6p12–11 EFHC1a Los Angeles, California 22 AD Classic JME and pCAEevolving to JME
Liu et al. (88)
Belize 1 AD Classic JME Serratosa et al. (89)Mexicob 31 AD Classic JME Bai et al. (90)
Suzuki et al. (91)6p12–11 Netherlands 18 AD JME Pinto et al. (92)6p21.3 BRD2a Los Angeles, California 24 AD Classic JME Greenberg et al. (94)
New Yorkc 85 AD Classic JME mixed withCAE evolving to JME
Greenberg et al. (95)
Pal et al. (96)6p21.3 Germany 29 AD JME Sander et al. (101)15q14 CX36d United Kingdom,
Denmark, France,Greece, Portugal,Sweden
25 AR JME Elmslie et al. (104)Mas et al. (105)
6q24 Saudi Arabia 34 AR JME, some nonprobandmembers with mildgait ataxia andtremors
Bate et al. (102)
2q22–2q23 CACNB4a Germany 1 ? Classic JME withabsences Familymember 3 Hz SW
Escayg et al. (100)
5q34GABR1 GABRA1a French Canadian 1 AD 4/8 affected familymembers had pCAE.One with epilepsyonset at 5 y
Cossette et al. (99)
3q26 CLCN2a Germany 1 AD Classic JME Haug et al. (97, 98, 103)18 ME2d European American – ? Classic JME Greenberg et al. (93)
aMutations.bEuropean Amerind (Hispanics).cEuropean American.dSNP associated variants.AD, autosomal dominant; AR, autosomal recessive; JME, juvenile myoclonic epilepsy; and CAE, childhood absence epilepsy.
so, understanding how AEDs work in IGEs could pointto the biochemical pathways used in epileptogenesis byepilepsy genes. The mechanisms of seizure-suppressingAEDs such as valproate, levetiracetam, topiramate, zon-isamide, and lamotrigine in JME remain unexplained.Similarly, the reasons why ethosuximide has no effect,and phenytoin and carbamazepine can worsen seizures inJME, remain unexplained.
Valproate selectively reduces low-threshold T-typevoltage-dependent calcium channels (VDCC) in rat neu-rons but not in thalamic neurons (80). In contrast, etho-suximide reduces T-type VDCC in thalamus neurons(81). Levetiracetam depressed high-voltage-activated cal-cium channel (HVACC) currents, but the specific type ofHVACC currents has not been defined (82). Levetiracetamhad no effects on T-type VDCC. Topiramate (10 µmol/L)decreased the peak of L-type HVACC (83) as wellas R-type VDCC (84). Zonisamide (500 µmol/L) doesnot alter high VDCC (HVDCC) but reduces T-typelow VDCC in cultured fetal rat cortical neurons (85).Lamotrigine spares L-type but inhibits N- and P-typeHVDCC. Defining the action of these drugs in the braincould help our understanding of why the same seizure
types in different IGE syndromes respond differently totreatment.
NOSOLOGY
In addition to electroclinical-anatomical state, mod-ern brain imaging, long-term outcome, and response totreatment, discoveries in the molecular genetics of theepilepsies have helped improve nosology. Perhaps nonehas been more dramatic than the unraveling of the my-oclonic epilepsies of infancy and early childhood bydiscoveries of de novo mutations in SCN1A in spo-radic severe myoclonic epilepsy of infancy (Dravet syn-drome) and germline mutations in SCN1B, SCN2A, andSCN1A in generalized epilepsies with febrile seizures plus(GEFS+). In GEFS+, genetic pleomorphism played outas febrile seizures; myoclonic, atonic, grand mal, and ab-sence seizures were observed in members of a family as-certained through a proband with MAS. These discov-eries have led to the separation of the Dravet syndromeand GEFS+ from idiopathic MAE of Doose and ECME,which do not have mutations in SCN1A (86), SCN1B,and SCN2A (87). Similarly, mutations in GABRA1,
Epilepsia, Vol. 46, Suppl. 9, 2005

SEIZURES IN IGE 45
GABRG2, and GABRB3 are associated with syndromesthat have in common the presence of absence seizures.Mutations in CLCN2 and myoclonin/EFHC1 substantiatethe entity of JME, the latter no longer recognized as asingle entity by the 2001 ILAE classification of epilepsysyndromes.
Finding separate chromosomal loci in specific IGEscan also help nosology. Three subvarieties of absencesyndromes previously suggested by clinical epileptolo-gists have different chromosome loci indicating differ-ent molecular lesions, namely, remitting CAE in chro-mosome 15q11–12 (5); CAE that persists into adulthoodwith tonic–clonic convulsions in chromosome 8q24 (54);and CAE evolving to JME, nonallelic to all presentlyknown epilepsy loci (34). Future molecular genetic studiesshould tell us if the other subsyndromes of absence epilep-sies such as absence epilepsy of early childhood startingbefore 5 years of age (49), myoclonic absence epilepsyin which myoclonias occur simultaneously with absenceseizures (21,22), eyelid myoclonia with absences (24,25),and juvenile absence epilepsy (12) truly exist as separateentities.
Similar advances in JME have occurred, providing usa glimpse of its complex genetics. Table 5 shows theloci and genes currently associated with JME (88–105).Three mutation-harboring genes for JME have been iden-tified to date. One is the GABA-receptor gene, which isunique to two large French–Canadian families with JMEand one index case with absence epilepsy (99). The sec-ond gene is CLNC2, which was found in a large Ger-man family (97,98,103). These two genes appear to beunique to these families and have not been found inother samples. The third gene found for JME was re-cently discovered in our laboratories (91). Missense mu-tations in Myoclonin/EFHC1 in 6p12 cosegregated with21 affected members from 6 of 24 unrelated families ofEuropean-Amerind or Hispanic ethnicity. Most recently,novel missense, nonsense, and deletion mutations in my-oclonin/EFHC1 have been found in Mexican, Honduran,and Japanese families. However, mutations and polymor-phisms in Myoclonin/EFHC1 do not account for all ourJME families in America. There are obviously additionalloci to be found that work alone or in conjunction withother loci to produce the JME phenotype and related syn-dromes. To date, there is no locus identified for the syn-drome of CAE evolving to JME which is nonallelic to allknown epilepsy loci (35,106,107).
REFERENCES
1. Hauser WA, Hesdorffer DC. Epilepsy—frequency, causes andconsequences. New York: Demos Medical Publishing, 1990:1–51.
2. Begley CE, Famulari M, Annegers JF, et al. The cost of epilepsy inthe United States: an estimate from population-based clinical andsurvey data. Epilepsia 2000;41:342–51.
3. Medina MT, Duron RM, Martınez L, et al. Prevalence, incidenceand causes of epilepsy in rural Honduras: the Salama study. Epilep-sia 2005;46:1–8.
4. Jallon P. Epilepsy in developing countries. Epilepsia1997;38:1143–51.
5. Delgado-Escueta AV, Alonso M, Medina MT, et al. The search forepilepsy genes in juvenile myoclonic epilepsy: discoveries alongthe way. In: Schmitz B, Sander T, eds. Juvenile myoclonic epilepsy:the Janz syndrome. Petersfield: Wrightson Biomedical Publishing,2000:145–70.
6. Janz D. Die epilepsien. Stuttgart: George Thieme Verlag, 1969.7. Nicoletti A, Reggio A, Bartoloni A, et al. Prevalence of epilepsy
in rural Bolivia: a door-to-door survey. Neurology 1999;53:2064–9.
8. Genton P, Roger J, Guerrini R, et al. History and classification of“myoclonic” epilepsies: from seizures to syndromes to diseases.Adv Neurol 2005;95:1–14.
9. Livingston S, Torres I, Pauli LL, et al. Petit mal epilepsy. Results ofa prolonged follow-up study of 117 patients. JAMA 1965;194:227–32.
10. Lennox W. The petit mal epilepsies. JAMA 1945;129:1069–73.11. Delgado-Escueta AV, Greenberg D, Weissbecker K, et al. Gene
mapping in the idiopathic generalized epilepsies. Juvenile my-oclonic epilepsy, childhood absence epilepsy, epilepsy with grandmal seizures and early childhood myoclonic epilepsy. Epilepsia1990;31(suppl 3):S19–29.
12. Wolf P, Inoue Y. Juvenile absence epilepsy. In: Roger J, BureauM, Dravet C, et al, eds. Epileptic syndromes in infancy, childhoodand adolescence. 3rd ed. London: John Libbey, 2002:331–4.
13. Giannakodimos S, Panayiotopoulos CP. Eyelid myoclonia withabsences in adults: a clinical and video-EEG study. Epilepsia1996;37:36–44.
14. Dravet C, Bureau M, Oguni H, et al. Severe myoclonic epilepsy ininfancy: Dravet syndrome. Adv Neurol 2005;95:71–102.
15. Dravet C, Bureau M. Benign myoclonic epilepsy in infancy. AdvNeurol 2005;95:127–37.
16. Ohki T, Watanabe K, Negoro K, et al. Severe myoclonic epilepsyin infancy: evolution of seizures. Seizure 1997;6:219–24.
17. Oguni H, Tanaka T, Hayashi K, et al. Treatment and long-termprognosis of myoclonic–astatic epilepsy of early childhood. Neu-ropediatrics 2002;33:122–32.
18. Zara F, De Falco F. Autosomal recessive benign myoclonicepilepsy of infancy. Adv Neurol 2005;95:139–45.
19. Doose H. Myoclonic-astatic epilepsy. Epilepsy Res 1992;6(suppl):163–8.
20. Kaminska A, Ickowicz A, Plouin P, et al. Delineation of cryp-togenic Lennox-Gastaut syndrome and myoclonic astatic epilepsyusing multiple correspondence analysis. Epilepsy Res 1999;36:15–29.
21. Bureau M, Tassinari C. Myoclonic absences: the seizure and thesyndrome. In: Roger J, Bureau M, Dravet C, et al, eds. Epilepticsyndromes in infancy, childhood and adolescence. 2nd ed. London:John Libbey, 2002:175–83.
22. Tassinari CA. Bureau M. Thomas P. Epilepsy with myoclonic ab-sences. In: Roger J, Bureau M, Dravet C, et al, eds. Epileptic syn-dromes in infancy, childhood and adolescence. 2nd ed. London:John Libbey, 1992:151–60.
23. Appleton RE, Panayiotopoulos CP, Acomb BA, et al Eyelid my-oclonia with typical absences: an epilepsy syndrome. J NeurolNeurosurg Psychiatry 1993;56:1312–6.
24. Covanis A. Eyelid myoclonia and absence. Adv Neurol2005;95:185–96.
25. Panayiotopoulus CP, Agathonikou A, Koutroumanidis M, et al.Eyelid myoclonia with absences: the symptoms. In: Duncan J,Panayiotopoulos CP, eds. Eyelid myoclonia with absences. Lon-don: John Libbey, 1996:17–26.
26. Baulac M G-AI, Baulac S, Leguern E. Myoclonic seizures in thecontext of generalized epilepsy with febrile seizures plus (GEFS+).Adv Neurol 2005;95:119–25.
27. Oller-Daurella L. 5000 epilepticos-clınica y evolucion. Barcelona:ESPAXS, 1994:169–70.
28. Loiseau P, Pestre M, Dartigues JF, et al. Long-term prognosisin two forms of childhood epilepsy: typical absence seizures and
Epilepsia, Vol. 46, Suppl. 9, 2005

46 R. M. DURON ET AL.
epilepsy with rolandic (centrotemporal) EEG foci. Ann Neurol1983;13:642–8.
29. Charlton M, Yahr M. Long-term follow-up of patients with petitmal. Arch Neurol 1967;16:595–8.
30. Currier RD, Kooi K, Saidman LJ. Prognosis of pure petit mal.Neurology 1963;13:959–67.
31. Wirrel EC, Camfield CS, Camfield PR, et al. Long-term prognosisof typical childhood absence epilepsy: remission of progression tojuvenile myoclonic epilepsy. Neurology 1996;47:912–8.
32. Shian WF, Chi C. Childhood absence epilepsy. Zhonghua Yi XueZa Zhi (Taipei) 1994;53:298–301.
33. Loiseau P, Duche B, Pedespan JM. Absence epilepsies. Epilepsia1995;36:1182–6.
34. Medina MT, Duron RM, Alonso ME, et al. Childhood absenceevolving to juvenile myoclonic epilepsy: electroclinical and geneticfeatures. Adv Neurol 2005;95:197–216.
35. Alonso ME, Medina MT, Martınez Juarez IE, et al. Familial juve-nile myoclonic epilepsy. Adv Neurol 2004;95:227–44.
36. Genton P, Gelisse P, Thomas P. Juvenile myoclonic epilepsy today:current definitions and limits. In: Schmitz B, Sander T, eds. Juve-nile myoclonic epilepsy: the Janz syndrome. Petersfield: WrightsonBiomedical Publishing, 2000:11–32.
37. Tsuboi T. Primary generalized epilepsy with sporadic myocloniasof myoclonic petit mal type. Stuttgart: Thieme, 1977.
38. Tsuboi T, Christian W. Epilepsy: a clinical electroencephalo-graphic and statistical study of 466 patients. Berlin: Springer,1976.
39. Obeid T. Clinical and genetic aspects of juvenile absence epilepsy.J Neurol 1994;241:487–91.
40. Covanis A, Skiadas K, Loli N, et al. Absence epilepsy: early prog-nostic signs. Seizure 1992;1:281–9.
41. Wolf P. Epilepsy with grand mal on awakening. In: Schmitz B,Sander T, eds. Juvenile myoclonic epilepsy: the Janz syndrome.Petersfield: Wrightson Biomedical Publishing, 2000:357–67.
42. Wolf P, Meyer T. Juvenile myoclonic epilepsy: a syndrome chal-lenging syndromic concepts? In: Schmitz B, Sander T, eds. Juve-nile myoclonic epilepsy: the Janz syndrome. Petersfield: WrightsonBiomedical Publishing, 2000:33–9.
43. Jain S, Padma M, Maheshwari MC. Occurrence of only my-oclonic jerks in juvenile myoclonic epilepsy. Acta Neurol Scand1997;95:263–7.
44. Delgado-Escueta AV, Serratosa JM, Medina MT. Juvenile my-oclonic epilepsy. In: Elaine Wyllie, ed The treatment of epilepsy:principles and practice. 2nd ed. Baltimore: Williams and Wilkins,1996;33:484–501.
45. Commission on Classification and Terminology of the Interna-tional League Against Epilepsy. Proposal for revised classificationof epilepsies and epileptic syndromes. Epilepsia 1989;30:389–99.
46. Commission on Classification and Terminology of the Interna-tional League Against Epilepsy. Proposal for revised clinicaland electrographic classification of epileptic seizures. Epilepsia1981;22:489–501.
47. Medina MT, Duron RM, Martınez, et al. Prevalence, incidence andcauses of epilepsy in rural Honduras: the Salama study. Epilepsia2005;46(5):1–8.
48. Loiseau P, Pnatiotopoulos CP, Hirsch E. Childhood absenceepilepsy and related syndromes. In: Roger J, Bureau M, DravetC, et al, eds. Epileptic syndromes in infancy, childhood and ado-lescence. 2nd ed. London: John Libbey, 2002:285–304.
49. Chaix Y, Daquin G, Monteiro F, et al. Absence epilepsy with onsetbefore age three years: a heterogeneous and often severe condition.Epilepsia 2003;44(7):944–9.
50. Cavazzuti GB, Ferrari F, Galli B, et al. Epilepsy with typical ab-sence seizures with onset during the first year of life. Epilepsia1989;30:802–6.
51. Olsson I, Hagberg G. Epidemiology of absence epilepsy. III. Clin-ical aspects. Acta Paediatr Scand 1991;80:1066–2.
52. Wirrell E. Natural history of absence epilepsy in children. Can JNeurol Sci 2003;30:184–8.
53. Gordon N, Aird R. Idiopathic childhood absences, a system dis-order: its diagnosis and differentiation. Dev Med Child Neurol1991;33:744–8.
54. Fong C, Shan P, Huang Y, et al. Childhood absence epilepsy in
and Indian (Bombay) family maps to chromosome 8q24. Neurology1998;50:A357.
55. Bouma PA, Westendorp RG, van Dijk JG, et al. The outcome ofabsence epilepsy: a meta-analysis. Neurology 1996;47:802–8.
56. Sato S, Dreifuss FE, Penry JK, et al. Long-term follow-up of ab-sence seizures. Neurology 1983;33:1590–5.
57. Asconape J, Penry J. Some clinical and EEG aspects of benignjuvenile myoclonic epilepsy. Epilepsia 1984;25:108–14.
58. Wirrel E, Camfield C, Camfield D, et al. Prognostic significanceof failure of the initial antiepileptic drugs in children with absenceepilepsy. Epilepsia 2001;42:760–3.
59. Thomas P, Genton P, Gelisse P, et al. In: Roger J, Bureau M,Dravet C, et al, eds. Epileptic syndromes in infancy, childhood andadolescence. London: John Libbey, 2002:335–55.
60. Panayiotopoulos CP, Chroni E, Daskalopoulos C, et al. Typicalabsence seizures in adults: clinical, EEG, video-EEG findings anddiagnostic/syndromic considerations. J Neurol Neurosurg Psychi-atry 1992;55:1002–8.
61. Ferrie CD, Giannakodimos S, Robinson RO, et al Symptomatictypical absence seizures. In: Duncan JS, Panayiotopoulos CP,eds. Typical absences and related epileptic syndromes. Edinburgh:Churchill Livingstone, 1995:241–52.
62. Ganesh S, Delgado-Escueta AV, Suzuki T, et al. Genotype-phenotype correlations for EPM2A mutations in Lafora’s pro-gressive myoclonus epilepsy: exon 1 mutations associate withan early-onset cognitive deficit subphenotype. Hum Mol Genet2002;11:1263–71.
63. Delgado-Escueta AV, Enrile BF. Juvenile myoclonic epilepsy ofJanz. Neurology 1984;34:285–95.
64. Jha S, Mathur VN, Mishra VN. Pitfalls in diagnosis of epilepsy ofJanz and its complications. Neurol India 2002;50:467–9.
65. Murthy JM, Yangala R, Srinivas M. The syndromic classification ofthe International League Against Epilepsy: a hospital-based studyfrom South India. Epilepsia 1998;39:48–54.
66. Aicardi J. Myoclonic epilepsies of infancy and childhood. AdvNeurol 1986;43:11–31.
67. Aicardi J, Chevrie J. Myoclonic epilepsies of childhood. Neu-ropadiatrie 1971;3:177–90.
68. Aicardi J. Course and prognosis of certain childhood epilepsieswith predominantly myoclonic seizures. In: Wada JA, Penry JK,eds. Advances in epileptology: XIIth Epilepsy International Sym-posium. New York: Raven Press, 1980:159–63.
69. Doose H, Gerken H, Leonhardt R, et al. Centrencephalicmyoclonic-astatic petit mal. Clinical and genetic investigations.Neuropadiatrie 1970;2:59–78.
70. Doose H, Baier WK. Epilepsy with primarily generalizedmyoclonic-astatic seizures: a genetically determined disease. EurJ Pediatr 1987;146:550–4.
71. Doose H, Volzke E. Petit mal status in early childhood and demen-tia. Neuropadiatrie 1979;10:10–14.
72. Pazzaglia P, Giovanardi R, Cirignotta F, et al. Nosografia delleepilessie miocloniche. Rivista Italiana di EEG e NeurofisiologıaClinica 1978;2:245–52.
73. Doose H, Sitepu B. Childhood epilepsy in a German city. Neuro-pediatrics 1983;14:220–4.
74. Genton P, McMenamin J. Aggravation of seizures by antiepilepticdrugs: what to do in clinical practice. Epilepsia 1998;38:26–9.
75. Camfield P, Camfield C. Childhood epilepsy: what is the evidencefor what we think and what we do? J Child Neurol 2003;18:272–87.
76. Medina MT, Martınez-Juarez IE, Duron RM, et al. The treatmentof myoclonic epilepsies of childhood, adolescence and adulthood.Adv Neurol 2005;95:307–24.
77. Knake S, Hamer HM, Schomburg U, et al. Tiagabine-inducedabsence status in idiopathic generalized epilepsy. Seizure1999;8:314–7.
78. Pedersen SA, Klosterskow P, Gram L, et al. Long-term study ofgamma-vinyl GABA in the treatment of epilepsy. Acta NeurolScand 1985;72:295–8.
79. Biraben A, Allain H, Scarbin JM, et al. Exacerbation of juvenilemyoclonic epilepsy with lamotrigine. Neurology 2000;55:1758.
80. Kelly KM, Gross RA, MacDonald RL. Valproic acid selectively re-duces the low-threshold (T) calcium current in rat nodose neurons.Neurosci Lett 1990;116:233–8.
Epilepsia, Vol. 46, Suppl. 9, 2005

SEIZURES IN IGE 47
81. Coulter DA, Huguenard JR, Prince DA. Characterization of etho-suximide reduction of low-threshold calcium current in thalamicneurons. Ann Neurol 1989;25:582–93.
82. Niespodziany I, Klitgaard H, Margineanu DG. Levetiracetam:modulation of high-voltage-activated Ca+ currents in CA1 pyra-midal neurons of rat hyppocampal slices. Epilepsia 2000;31:347.
83. Zhang X, Velumian AA, Jones OT, et al. Modulation of highvoltage-activated calcium channels in dentate granule cells by top-iramate. Epilepsia 2000;41(suppl):S52–60.
84. Kuzmiski JB, Barr W, Zamponi GW, et al. Topiramate inhibitsthe initiation of plateau potentials in CA1 neurons by depressingR-type calcium channels. Epilepsia 2005;46:481–9.
85. Suzuki S, Kawahami K, Nishimura S, et al. Zonisamide blocksT-type calcium channel in cultured neurons of rat cerebral cortex.Epilepsy Res 1992;12:21–7.
86. Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in thesodium channels gene SCN1A cause severe myoclonic epilepsy ofinfancy. Am J Hum Genet 2001;68:1327–32.
87. Wallace RH, Wang D, Singh R, et al. Febrile seizures and general-ized epilepsy associated with a mutation in the Na+ -channel beta1 subunit gene SCN1B. Nat Genet 1998;19:366–70.
88. Liu AW, Delgado-Escueta AV, Gee MN, et al. Juvenile myoclonicepilepsy in chromosome 6p12-p11: locus heterogeneity and re-combinations. Am J Hum Genet 1996;63:438–46.
89. Serratosa JM, Delgado-Escueta AV, Medina MT, et al. Clinicaland genetic analysis of a large pedigree with juvenile myoclonicepilepsy. Ann Neurol 1996;39:187–95.
90. Bai D, Alonso M, Medina MT, et al. Juvenile myoclonic epilepsy:linkage to chromosome 6p12 in Mexico families. Am J Med Genet2002;113:268–74.
91. Suzuki T, Delgado-Escueta A, Aguan K, et al. Mutations in EFHC1cause juvenile myoclonic epilepsy. Nat Genet 2004;36:842–9.
92. Pinto D, Westland B, de Haan GJ, et al. Genome-wide linkage scanof epilepsy-related photoparoxysmal electroencephalographic re-sponse: evidence for linkage on chromosomes 7q32 and 16p13.Hum Mol Genet 2005;14:171–8.
93. Greenberg DA, Cayanis E, Strug L, et al. Malic enzyme 2 mayunderlie susceptibility to adolescent-onset idiopathic generalizedepilepsy. Am J Hum Genet 2005;76:139–46.
94. Greenberg DA, Delgado-Escueta AV, Wideliz H, et al. Juvenile
myoclonic epilepsy may be linked to the BF and HLA loci onhuman chromosome 6. Am J Med Genet 1988;31:185–92.
95. Greenberg DA, Durner M, Keddache M, et al. Reproducibility andcomplications in gene searches: linkage on chromosome 6, het-erogeneity, association, and maternal inheritance in juvenile my-oclonic epilepsy. Am J Hum Genet 2000;66:508–16.
96. Pal DK, Evgrafov OV, Tabares P, et al. BRD2(RING3) is a probablemajor susceptibility gene for common juvenile myoclonic epilepsy.Am J Hum Genet 2003;73(2):261–70.
97. Haug K, Warnstedt M, Alekov AK, et al. Mutations in CLCN2encoding a voltage-gated chloride channel are associated with id-iopathic generalized epilepsies. Nat Genet 2003;33(4):527–32.
98. Heils A. CLCN2 and idiopathic generalized epilepsy. Adv Neurol2005;95:265–71.
99. Cossette P, Loi L, Brisebois K, et al. Mutation of GABRA1 inan autosomal dominant form of juvenile myoclonic epilepsy. NatGenet 2002;31:184–9.
100. Escayg A, DeWaard M, Lee DD, et al. Coding and noncodingvariation of the human calcium-channel B4-subunit gene CACNB4in patients with idiopathic generalized epilepsy and episodic ataxia.Am J Hum Genet 2000;66:1531–9.
101. Sander T, Bockenkamp B, Hildmann T, et al. Refined mapping ofthe epilepsy susceptibility locus EJM1 on chromosome 6. Neurol-ogy 1997;49:842–7.
102. Bate L, Mitchell W, Williamson M, et al. Molecular genetic analy-sis of juvenile myoclonic epilepsy in the Saudi Arabian population[Abstract]. Epilepsia 2000;41(7):72.
103. Haug K, Warnstedt M, Alekov AK, et al. Mutations in CLCN2encoding a voltage-gated chloride channel are associated with id-iopathic generalized epilepsies. Nat Genet 2003;33:527–32.
104. Elmslie FV, Rees M, Williamson MP, et al. Genetic mapping ofa major susceptibility locus for juvenile myoclonic epilepsy onchromosome 15q. Hum Mol Genet 1997;8:1329–34.
105. Mas C, Taske N, Deutsch S, et al. Association of the connexin36gene with juvenile myoclonic epilepsy. J Med Genet 2004;41:e93.
106. Crunelli V, Leresche N. Childhood absence epilepsy: genes, chan-nels, neurons and networks. Nature 2002;3:371–82.
107. Delgado-Escueta AV, Medina MT, Bai DS, et al. Geneticsof idiopathic myoclonic epilepsies: an overview. Adv Neurol2002;89:161–84.
Epilepsia, Vol. 46, Suppl. 9, 2005
Related Documents











