1 THÈSE / UNIVERSITÉ DE RENNES 1 sous le sceau de l’Université Bretagne Loire pour le grade de DOCTEUR DE L’UNIVERSITÉ DE RENNES 1 Mention : CHIMIE Ecole doctorale SDLM Saravanakumar ELANGOVAN Préparée à l’unité de recherche UMR 6226 ISCR (Institut des Sciences Chimiques de Rennes) - UFR Sciences et Propriété de la Matière Well-defined Iron and Manganese Catalysts for Reduction and Dehydrogenation Reactions Thèse rapportée par : Noël LUGAN Directeur de recherche CNRS LCC Toulouse / rapporteur Jean-Cyrille HIERSO Professeur Université de Bourgogne / rapporteur et soutenue à RENNES le 19 Janvier 2017 devant le jury composé de : Matthias BELLER Professeur LIKAT Rostock Christian BRUNEAU IR CNRS ISCR Rennes Noël LUGAN DR CNRS LCC Toulouse Jean-Cyrille HIERSO Professeur Université de Bourgogne Armelle OUALI CR CNRS Institut Charles Gerhardt Montpellier Jean-Baptiste SORTAIS MCF-HDR, Université de Rennes 1 Christophe DARCEL Professeur, Université de Rennes 1

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
THÈSE / UNIVERSITÉ DE RENNES 1 sous le sceau de l’Université Bretagne Loire
pour le grade de
DOCTEUR DE L’UNIVERSITÉ DE RENNES 1 Mention : CHIMIE
Ecole doctorale SDLM
Saravanakumar ELANGOVAN Préparée à l’unité de recherche UMR 6226 ISCR (Institut des Sciences
Chimiques de Rennes) - UFR Sciences et Propriété de la Matière
Well-defined Iron and Manganese Catalysts for Reduction and Dehydrogenation Reactions
Thèse rapportée par : Noël LUGAN Directeur de recherche CNRS LCC Toulouse / rapporteur Jean-Cyrille HIERSO Professeur Université de Bourgogne / rapporteur et soutenue à RENNES le 19 Janvier 2017 devant le jury composé de :
Matthias BELLER Professeur LIKAT Rostock Christian BRUNEAU IR CNRS ISCR Rennes Noël LUGAN DR CNRS LCC Toulouse Jean-Cyrille HIERSO Professeur Université de Bourgogne Armelle OUALI CR CNRS Institut Charles Gerhardt Montpellier Jean-Baptiste SORTAIS MCF-HDR, Université de Rennes 1 Christophe DARCEL Professeur, Université de Rennes 1


Acknowledgements The following work was realized in the team of “Organometallics: Materials and
Catalysis (OMC)”, UMR 6226 CNRS-Université de Rennes 1, Institut des Sciences Chimiques
de Rennes, and Leibniz-Institut für Katalyse e.V. an der Universität Rostock, between Oct. 2013-
Sep. 2016. I have spent half of my PhD in Université de Rennes 1 and other half in LIKAT. I had
then an outsanding opportunity to work in different lab to learn new things.
I would like to express my deepest gratitude to my supervisors, Prof. Christophe Darcel,
Prof. Dr. Matthias Beller, Dr. Jean-Baptiste Sortais and Dr. Kathrin Junge for their excellent
guidance, caring, patience, and providing me with an excellent atmosphere for doing research. It
was my great pleasure to work with four supervisors. It helped me to improve my research career
in many ways and also to get different ideas. I could not have imagined having a better advisors
and mentors for my Ph.D study.
Prof. Christophe Darcel was my teacher, supervisor and motivator. He helped me a lot
from my master studies to present and taught me organometallic and catalysis. This work would
not have been possible without his guidance, support and encouragement. Under his guidance I
successfully overcame many difficulties and he offers many new things to learn. I had the most
difficult time while writing this thesis; he gave me the moral support and freedom I needed to
move on.
I want to express my deepest thank to Prof. Dr. Matthias Beller, a person with kind and
positive disposition. My cordial thanks for accepting me as a Ph.D student, your warm
encouragement, valuable guidance and extensive discussions around my work. I am taking him as
a role model to lead my career in life. Even with his busy schedule, he helped me for the fast
publication of the obtained results in a highly competitive area of research. He gave me an
opportunity to work an industrial project and to communicate with scientist in the company.
I would like to thank Dr. Jean-Baptiste Sortais who build me in complex synthesis. He
has been supportive both in the study and personal life since the days I began started to work in
the lab. Thanks for the insightful discussion, valuable advice and critical comments helped me
through the road of my thesis.

4
I thank Dr. Kathrin Junge, for her advices and friendly assistance with various problems
all the time, especially for her help with the paperwork and fruitful discussion. She supported me
to overcome many difficulties. I never forgot the moments that we had in the cake break.
My sincere thanks also go to Prof. Pierre H. Dixneuf and Dr. Christian Bruneau for their
encouragement, teaching and motivation in research at all the time.
Meanwhile, I would like to extend my appreciation to the jury members: Dr. Noël Lugan,
Prof. Jean-Cyrille Hierso, Dr. Armelle Ouali and Dr. Christian Bruneau, and thank them for
spending their valuable time in reading my thesis.
Then, I would like to extend my thanks to Dr. Cédric Fischmeister, Dr. Henri Doucet, Dr.
Mathieu Achard, Dr. Henrik Junge, Dr. Helfried Neumann Dr. Ralf Jackstell, and Dr. Lucie
Norel for their useful help in research.
It is also my pleasure to thank Dr. Thierry Roisnel and Dr. Vincent Dorcet from X-Ray
diffraction center, Université de Rennes 1, and Dr. Anke Spannenberg, LIKAT for their help in
X-Ray diffraction analysis, and to thank Dr. Haijun Jiao, LIKAT for DFT calculations and
valuable suggestions for my research. Also I thank Dr. Wolfgang Baumann for NMR studies.
I take this opportunity to sincerely acknowledge Région Bretagne, France and LIKAT
Ph.D fellowship for providing financial assistance.
Thank to our industrial collaborators Dr. Stephan Bachmann and Dr. Michelangelo
Scalone, Hoffmann la Roche, Switzerland.
In the meantime, I would like to take this opportunity to thank Prof. Jean-François
Carpentier, Prof. Domnique Lorcy, Prof. Muriel Hissler, Prof. Jean-Pierre Bazureau, Dr.
Florence Geneste, Dr. Sophie Guillaume and Dr. Evgueni Kirillov for their innovative and
informative teaching during my master study.
Bianca Wendt, thanks for your excellent technical assistance in the lab, particularly for
autoclave technique, and your kindly answers to my general questions.
It is also my pleasure to thank Mrs. Béatrice Mahi and Mrs. Françoise Toupet, Mrs. Anne
Tonn and Nicole Aulerich, for their kind help during my thesis.
I thank my lab mates both in Rennes and Rostock, Samuel Quintero-Duque, Thomas
Dombray, Jianxia Zheng, Duo Wei, Haoquan Li, Yanan Miao, Christoph Topf, Marcel Garbe,
Jacob Neumann, Yuehui Li Basudev Shaoo, Marc Perez, Feng Cheng, Kishore, Jagadeesh and

Xinjiang Cui for the stimulating discussions and for all the fun we have had in the last three
years.
Also I thank my Indian friends Aswin, Raghavendran, Surya, Manikandan, Murali,
Kathiravan, Swinton Darious, Apurba Shaoo, Linus Paulin. In particular, I am grateful to
Dr. Charles Beromeo Bheeter who suggested I should come for master in Rennes and
enlightening me the first glance of research. His unconditional support and encouragement
allowed me to work hard since my master studies.
Last but not the least; I would like to thank my family: my parents, sisters and brothers for
their unconditional love and supporting me spiritually throughout this thesis.

i

i
Table of Contents General Introduction ........................................................................................................................ 2
Part 1 ................................................................................................................................................ 6
Iron and manganese catalyzed reduction of C=O bonds: literature survey ...................................... 6
1. Introduction .................................................................................................................................. 8
2. Reduction of C=O bond in carbonyl derivatives .......................................................................... 9
2.1 Hydrogenation ........................................................................................................................ 9
2.1.1 Iron catalyzed hydrogenation of C=O bond .................................................................... 9
2.1.2 Manganese catalyzed hydrogenation of C=O bonds ..................................................... 15
2.2 Hydrosilylation ..................................................................................................................... 16
2.2.1 Iron-catalyzed hydrosilylation of aldehydes and ketones ............................................. 16
2.2.2 Iron-catalyzed hydrosilylation of carboxylic derivatives .............................................. 25
2.2.3 Manganese-catalyzed hydrosilylation of C=O bonds ................................................... 29
2.3 Hydrogen transfer ................................................................................................................. 32
3. Conclusion .................................................................................................................................. 38
4. References .................................................................................................................................. 38
Part 2 .............................................................................................................................................. 44
Synthesis and catalytic applicationsof Knölker’s NHC complexes ............................................... 44
1. Introduction ................................................................................................................................ 46
2. Discovery of iron cyclopentadienone complexes and their applications in catalysis ................ 46
3. Results and discussions .............................................................................................................. 53
3.1 Preparation of the complexes ............................................................................................... 53
3.2 Characterization of the complexes ....................................................................................... 54
3.2.1 NMR and IR studies ...................................................................................................... 55
3.2.2 Electrochemical studies. ................................................................................................ 56

ii
3.2.3 X-Ray diffraction studies .............................................................................................. 57
3.3 Catalytic applications ........................................................................................................... 60
4. Conclusion .................................................................................................................................. 65
5. Experimental Part ....................................................................................................................... 65
5.1 Synthesis of 1-mesityl-3-((R)-1-phenylethyl)imidazolium chloride salt ............................. 66
5.2 Synthesis of complexes ........................................................................................................ 67
5.3 Characterization data of the nitrile products. ....................................................................... 74
6. References .................................................................................................................................. 76
Part 3 .............................................................................................................................................. 80
Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid
derivatives………………………………………………………………………………………...80
1. Introduction ................................................................................................................................ 82
2. Iron pincer complexes ................................................................................................................ 83
2.1 Hydrogenation of aldehydes and ketones ............................................................................. 83
2.2 Hydrogenation of carboxylic acid derivatives...................................................................... 83
2.3 Dehydrogenation of alcohols ............................................................................................... 86
2.4 Dehydrogenation for the production of hydrogen and carbon dioxide ................................ 87
3. Cobalt pincer complexes ............................................................................................................ 88
3.1 Hydrogenation ...................................................................................................................... 88
3.2 Dehydrogenation of alcohols ............................................................................................... 90
4. Nickel PNP pincer complexes .................................................................................................... 92
5. Manganese pincer complexes ..................................................................................................... 93
6. References .................................................................................................................................. 97
Part 3 - Chapter 1 ......................................................................................................................... 100
Iron and manganese catalyzed nitrile hydrogenation ................................................................... 100

iii
1. Introduction .............................................................................................................................. 102
2. Ruthenium catalysts for hydrogenation of nitriles ................................................................... 103
3. Hydrogenation of nitriles with other metal complexes ............................................................ 108
4. Iron and cobalt catalyzed hydrogenation of nitriles ................................................................. 108
5. Results and Discussions ........................................................................................................... 109
5.1 Iron catalyzed nitrile hydrogenation .................................................................................. 109
5.1.1 Synthesis of iron pincer complexes ............................................................................. 109
5.1.2 Optimisation of the reaction parameters for catalysed nitrile hydrogenation ............. 114
5.1.3 Scope of the hydrogenation of aromatic nitriles ......................................................... 115
5.1.4 Hydrogenation of aliphatic and dinitriles .................................................................... 117
5.2. Manganese catalyzed nitrile hydrogenation ...................................................................... 118
5.2.1 Manganese pincer complexes preparation .................................................................. 118
5.2.2 Optimization of reaction parameters ........................................................................... 125
5.2.3 Hydrogenation of aromatic nitriles ............................................................................. 128
5.2.5 Mechanistic investigations .......................................................................................... 130
6. Conclusion ................................................................................................................................ 136
7. Experimental section ................................................................................................................ 137
7.1 General experimental details .............................................................................................. 137
7.2. Synthesis of iron pincer complexes ................................................................................... 137
7.3 X-ray Structural Analysis ................................................................................................... 140
7.4 Synthesis of manganese pincer complexes ........................................................................ 143
7.5. Computational details ........................................................................................................ 148
7.6. NMR for isolated products ................................................................................................ 150
8. References ................................................................................................................................ 152
Part 3 - Chapter 2 ......................................................................................................................... 154

iv
Iron and manganese catalyzed hydrogenation of esters to alcohols ............................................. 154
1. Introduction .............................................................................................................................. 156
1.1. Ruthenium catalyzed ester hydrogenation ........................................................................ 157
1.2 Iridium catalyzed ester hydrogenation ............................................................................... 164
1.3 Earth abundant metals for ester hydrogenation .................................................................. 165
2. Results and discussions ............................................................................................................ 167
2.1 Hydrogenation of esters to alcohols catalyzed by iron pincer complexes ......................... 167
2.1.1 Optimisation of the reaction parameters ..................................................................... 167
2.1.2 Hydrogenation of various aromatic and aliphatic esters ............................................. 169
2.1.3 Hydrogenation of diesters and lactones ...................................................................... 170
2.1.4 Computation studies .................................................................................................... 174
2.2. Manganese catalyzed ester hydrogenation ........................................................................ 176
2.2.1 New PNP manganese complex synthesis .................................................................... 176
2.2.3 Hydrogenation of aromatic esters ............................................................................... 181
2.2.4 Hydrogenation of benzylic and aliphatic esters .......................................................... 183
2.2.5 Hydrogenation of diesters and lactones ...................................................................... 184
2.3 Manganese catalyzed hydrogenation of aldehydes and ketones ........................................ 185
2.4 Mechanistic investigation ................................................................................................... 187
3. Conclusion ................................................................................................................................ 188
4. Experimental section ................................................................................................................ 189
4.1 General experimental details .............................................................................................. 189
4.2 General procedure for the iron catalyzed hydrogenation of esters ..................................... 189
4.3 Analytical data of the isolated products ............................................................................. 191
4.4. Synthesis of manganese pincer complexes. ...................................................................... 194
4.5 General procedure for the manganese catalyzed hydrogenation of esters ......................... 198

v
4.6 Computational studies ........................................................................................................ 199
4.7 Data for isolated products .................................................................................................. 203
Part 4 Chapter 1 .......................................................................................................................... 214
Iron catalyzed alkylation of ketones with alcohols ................................................................. 214
1. Introduction .............................................................................................................................. 216
1.1 C-C bond formation via alkylation of ketones with primary alcohols .......................... 217
1.2 Alkylation of ketones with propargylic alcohol ................................................................. 222
1.3 Alkylation of ketones with methanol ............................................................................. 223
2. Knölker type complex catalyzed C-C bond formation reactions ............................................. 225
2.1 Optimisation of the reaction conditions ......................................................................... 226
2.2 Scope of the -alkylation of ketones with alcohols ........................................................... 229
3. Friedländer annulation reaction ................................................................................................ 230
3.1 Introduction ................................................................................................................... 230
3.2. Iron catalyzed synthesis of quinolone derivatives ......................................................... 234
4. Conclusion ................................................................................................................................ 235
5. Experimental part ..................................................................................................................... 235
Part 4 Chapter 2 ............................................................................................................................ 248
Manganese catalyzed N-alkylation of amines with alcohols........................................................ 248
1. Introduction .............................................................................................................................. 250
1.1 Iron catalyzed N-alkylation of amines with alcohols ......................................................... 252
1.2 Cobalt based catalytic system ............................................................................................ 254
2. N-alkylation of aniline using PNP pincer manganese complexes ............................................ 255
2.1 Optimization of reaction parameters .................................................................................. 255
2.2. Selective N-alkylation of various anilines with benzyl alcohols ....................................... 260
2.3 N-Alkylation of amines with various (hetero)aromatic and aliphatic alcohols .................. 261

vi
2.4. Manganese catalyzed N-methylation of aniline with methanol ........................................ 265
3. Conclusion ................................................................................................................................ 268
4. Experimental part ..................................................................................................................... 268
4.1 General experimental details .............................................................................................. 268
4.2. NMR data for isolated products ........................................................................................ 270
6. References ................................................................................................................................ 288
General conclusion ....................................................................................................................... 292
Annexes ........................................................................................................................................ 296
1. List of synthesized complexes .............................................................................................. 296
2. List of publications ............................................................................................................... 296

1
General Introduction

2

2
General Introduction Transition-metal catalysis is a key technology for the advancement of green chemistry,
specifically for waste prevention, to reduce energy consumption, to achieve high atom efficiency,
and from an economical point of view. In industry, although the fundamental processes for
refining petroleum and its conversion to basic building blocks are based on heterogeneous
catalysts, many important value-added products are manufactured by homogeneous catalytic
processes. Transition metal homogeneous catalysts can be tunable both electronically and
sterically by varying the metal and/or ligands which is an important factor to achieve notable
selectivity in organic transformations. Even though this area of research is now well established,
the development of new catalytic systems is still important for both academic and industrial
applications.
In the reduction area, catalytic reactions using molecular hydrogen are highly attractive
since it is the cleanest reducing agent and hydrogenation is arguably one of the most important
catalytic method in synthetic organic chemistry both on the laboratory and the production scale.[1-
2] So far, mainly heterogeneous catalysts are known to promote such hydrogenations under harsh
conditions.[3-6] In contrast, homogeneous, molecular-defined organometallic complexes are often
considered to be more active at lower reaction temperatures and hydrogen pressures, which might
lead to higher selectivity.[7] Hence, the development of well-defined homogeneous complexes
which give mild and selective protocols for effective hydrogenation reactions is an actual and
highly desired research goal in the scientific community.[8-10] Without doubt the majority of the
contributions in organometallic catalysis have been performed applying noble metals mainly
based on platinium, palladium, rhodium and ruthenium complexes. Due to economic constraints,
limited availability, sometimes sensitivity and toxicity of precious metal complexes, there is an
increasing interest to substitute such catalysts by more easily available bio-relevant metals.[11]
In this respect, iron and manganese are the most attractive candidates due to their
abundance, price and eco-compatibility. Iron is the most abundant transition metal in the earth’s
crust and manganese is the third most after iron and titanium. In the recent years, more and more
research groups entered the iron age of homogeneous catalysis especially in the hydrogenation
and dehydrogenation area, iron pincer complexes being studied by many groups.[12-18]

3
During the past decade, manganese complexes were well studied in C-H activation,[19-22]
electrochemical CO2 reduction,[23-27] hydrosilylation,[28-31] oxidation[32] and cross coupling
reactions.[33] In 2016, manganese pincer complexes have been prepared and used for the
hydrogenation and dehydrogenation reactions since bifunctional mechanism involving the ligand
can operate for efficient H2 activation.
Like hydrogenation, hydrogen borrowing (hydrogen auto transfer) reactions are
considered to be a greener and atom economic reaction since only water is produced as a side
product. Based on these aspects, the borrowing hydrogen methodology received huge attention in
the last years.[34-38] The hydrogen auto transfer process involves an initial oxidative hydrogen
elimination, followed by different types of condensation reactions, and is completed with a final
reductive hydrogen addition to give the targeted product. Advantageously, no additional external
hydrogen source is needed in this domino process because the parent alcohol acts as the hydrogen
donor. In addition, it should be noted that a variety of alcohols is easily available from renewable
feedstock making this methodology especially suitable for the valorization of biomass or
biomass-derived building blocks.[39]
Thus, this field of research is still a growing area and main part of the present work will
be dedicated to the use of iron and manganese based complexes in hydrogenation and hydrogen
borrowing reactions.
The main goal of the thesis was to develop new catalytic systems based on earth abundant
metals for greener reactions such as hydrogenation and hydrogen borrowing using either reported
or new well-defined iron and manganese complexes. The manuscript is structured into four parts.
Part 1: bibliographic survey on reduction reactions based on earth abundant metals mainly iron
and manganese developed during the last decade. This chapter will focus on the catalytic
reductions where the hydride sources are molecular hydrogen (hydrogenation), silanes and
siloxanes (hydrosilylation), alcohols and formic acid (transfer hydrogenation).
Part 2: a new family of NHC-substituted iron Knölker type complexes have been prepared by
substitution of one CO-ligand by the corresponding NHC ligand using UV radiation. All the
complexes have been obtained in good yields and fully characterized. Their potential in catalysis
has been demonstrated in the case of the dehydration of primary benzamides into benzonitriles
derivatives using the inexpensive PMHS (polymethylhydrosiloxane) as the dehydrating reagent.

4
Part 3 deals with iron and manganese catalyzed hydrogenation of carboxylic acid derivatives and
will be divided in two chapters.
Chapter 1: the hydrogenation of nitriles is described using earth abundant iron and
manganese pincer complexes. Novel iron and manganese complexes were synthesized and these
catalytic systems allowed the hydrogenation of aromatic, aliphatic and dinitriles to the
corresponding amines.
Chapter 2: an effective and selective hydrogenation of esters is reported with iron and
manganese complexes. A second generation iron PNP iron pincer complexes were prepared and
used for the hydrogenation of esters. Similarly, manganese pincer complexes have been
synthesized and reported in the hydrogenation of various esters to the corresponding alcohols. In
addition, it was shown that these complexes were able to hydrogenate carbonyl compounds.
Part 4 concerns the use of iron and manganese complexes in hydrogen borrowing reactions.
Chapter 1: the iron-catalyzed -alkylation of ketones with primary alcohols in the
presence of a catalytic amount of base is described using an air stable Knölker type complex. The
optimized catalytic system permitted the development of the first iron-catalyzed Friedländer
annulation reaction starting from 2-aminobenzyl alcohols.
Chapter 2: N-monoalkylation of primary amines with alcohols, including methanol,
catalyzed by manganese pincer complexes is presented.
References
[1] P. N. Rylander, in Catalytic Hydrogenation in Organic Synthesis, Academic Press, New York, 1979.
[2] P. G. Andersson, I. J. Munslo, in Modern Reduction Methods, Wiley, New York, 2008. [3] S. Nishimura, in Handbook of Heterogeneous Hydrogenation for Ogranic Synthesis,
Wiley, New York 2001. [4] Y. Pouilloux, F. Autin, J. Barrault, Catal. Today 2000, 63, 87-100. [5] J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensenab, E. A. Pidko, Chem. Soc.
Rev. 2015, 44, 3808-3833. [6] H. G. Manyar, C. Paun, R. Pilus, D. W. Rooney, J. M. Thompsona, C. Hardacre, Chem.
Commun. 2010, 46, 6279-6281. [7] J. G. de Vries, C. J. Elsevier, in Handbook of Homogeneous Hydrogenation (Ed.: Wiley-
VCH), Weinheim 2007. [8] M. L. Clarke, Catal. Sci. Technol. 2012, 2, 2418-2423. [9] P. A. Dub, T. Ikariya, ACS Catal. 2012, 2, 1718-1741.

5
[10] S. Werkmeister, K. Junge, M. Beller, Org. Process Res. Dev. 2014, 18, 289-302. [11] R. M. Bullock, Science 2013, 342, 1054-1055. [12] I. Bauer, H.-J. Knolker, Chem. Rev. 2015, 115, 3170-3387. [13] A. Quintard, J. Rodriguez, Angew. Chem. Int. Ed. 2014, 53, 4044-4055. [14] S. Werkmeister, J. Neumann, K. Junge, M. Beller, Chem. Eur. J. 2015, 21, 12226-12250. [15] E. A. Bielinski, M. Förster, Y. Zhang, W. H. Bernskoetter, N. Hazari, M. C. Holthausen,
ACS Catal. 2015, 5, 2404-2415. [16] R. H. Morris, Acc. Chem. Res. 2015, 48, 1494-1502. [17] T. Zell, D. Milstein, Acc. Chem. Res. 2015, 48, 1979-1994. [18] G. Bauer, K. A. Kirchner, Angew. Chem. Int. Ed. 2011, 50, 5798-5800. [19] W. Liu, L. Ackermann, ACS Catal. 2016, 6, 3743-3752. [20] W. Liu, S. C. Richter, Y. Zhang, L. Ackermann, Angew. Chem. Int. Ed. 2016, 55, 7747-
7750. [21] B. Zhou, P. Ma, H. Chen, C. Wang, Chem. Commun. 2014, 50, 14558-14561. [22] R. He, Z.-T. Huang, Q.-Y. Zheng, C. Wang, Angew. Chem. Int. Ed. 2014, 53, 4950-4953. [23] H. Takeda, H. Koizumi, K. Okamoto, O. Ishitani, Chem. Commun. 2014, 50, 1491-1493. [24] F. Franco, C. Cometto, F. Ferrero Vallana, F. Sordello, E. Priola, C. Minero, C.Nervi, R.
Gobetto, Chem. Commun. 2014, 50, 14670-14673. [25] M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold, C. P. Kubiak,
J. Am. Chem. Soc. 2014, 136, 5460-5471. [26] J. Agarwal, T. W. Shaw, C. J. Stanton, G. F. Majetich, A. B. Bocarsly, H. F. Schaefer,
Angew. Chem. Int. Ed. 2014, 53, 5152-5155. [27] J. M. Smieja, M. D. Sampson, K. A. Grice, E. E. Benson, J .D. Froehlich, C. P. Kubiak,
Inorg. Chem. 2013, 52, 2484-2491. [28] T. K. Mukhopadhyay, M. Flores, T. L. Groy, R. J. Trovitch, J. Am. Chem. Soc. 2014, 136,
882-885. [29] J. Zheng, S. Elangovan, D. A. Valyaev, R. Brousses, V. César, J.-B. Sortais, C. Darcel, N.
Lugan, G. Lavigne, Adv. Synth. Catal. 2014, 356, 1093-1097. [30] V. K. Chidara, G. Du, Organometallics 2013, 32, 5034-5037. [31] J. Zheng, S. Chevance, C. Darcel, J.-B. Sortais, Chem. Commun. 2013, 49, 10010-10012. [32] P. Saisaha, J. W. de Boer, W. R. Browne, Chem. Soc. Rev. 2013, 42, 2059-2074. [33] D. A. Valyaev, G. Lavigne, N. Lugan, Coord. Chem. Rev. 2016, 308, 191-235. [34] Q. Yang, Q. Wanga, Z. Yu, Chem. Soc. Rev. 2015, 44, 2305-2329. [35] G. E. Dobereiner, R. H. Crabtree, Chem. Rev. 2010, 110, 681-703. [36] M. H. S. A. Hamid, P. A. Slatford, J. M. J. Williams, Adv. Synth. Catal. 2007, 349, 1555-
1575. [37] S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem 2011, 3,
1853-1864. [38] J. Leonard, A. J. Blacker, S. P. Marsden, M. F. Jones, K. R. Mulholland, R. Newton, Org.
Process Res. Dev. 2015, 19, 1400-1410. [39] K. Barta, P. C. Ford, Acc. Chem. Res. 2014, 47, 1503-1512.

6
Part 1
Iron and manganese catalyzed reduction of C=O
bonds: literature survey

7

8
1. Introduction Reduction reactions are of the utmost important transformations in molecular syntheses,
and are extensively used in both academic and industrial processes.[1] Traditionally such reactions
are performed using at least stoichiometric amount of metallic hydride reagents, such as lithium
aluminum hydride (LiAlH4) or sodium borohydride (NaBH4).[2] Although this method is always
efficient, major disadvantages of these reagents are the chemoselectivity for the reduction of
poly-functional derivatives in multi-step sequences and the coproduction of stoichiometric
amounts of waste metal salts, which may require difficult separation and post-treatment. Thus an
alternative method is still needed to overcome those problems especially the selectivity issue
(functional group tolerance, protection/deprotection sequences, etc.). Transition-metal
homogeneous catalyzed reduction reactions have then shown to be an encouraging approach to
reach such target.
The selective catalytic reduction of carbon-carbon and carbon-heteroatom multiple bonds
under mild conditions constitutes an important reaction in molecular synthesis, more particularly
in pharmaceutical as well as agrochemical industries.[3] Clearly, high regio-, chemo- or stereo-
selectivity and broad functional group tolerance are the key and crucial factors for the acceptance
and application of novel methodologies in large scale applications. Using transition metal
catalysts, molecular H2, hydrogen transfer reagents and hydrosilanes or siloxane are nowadays
commonly used as reducing agents in the reduction reactions.
On another hand, since the beginning of the 21st century, a huge increase in the use of
earth abundant transition metals such as iron, or manganese as powerful alternative catalysts to
classical precious ones such as rhodium, palladium, or platinum, in transformations for applied
chemistry.[4] Indeed, considering the current important concerns about climate change and
associated green chemistry principles, the substitution of these expensive noble transition metals
by more benign ones, such as the first row transition metals is highly desirable and is without any
doubt one of the important challenges of the 21st century.
This chapter will then focus mainly the developments made on catalytic reduction
reactions such as hydrogenation, transfer hydrogenation and hydrosilylation using iron and
manganese complexes as catalysts and their applications in organic synthesis.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
9
2. Reduction of C=O bond in carbonyl derivatives Reduction of polar functional bonds such as C=O, C=N or C=S with molecular hydrogen
is one of the most important reactions and plays a key role in the production of numerous bulk
products and intermediates in the chemical industry.[5-7] In this area, heterogeneous catalysts are
usually well established to perform the hydrogenation of non-demanding polar functional groups,
which often takes place at high temperatures and/or pressures and suffers in the selectivity
issues.[8] Hence, the development of well-defined homogeneous transition metal complexes
which can be able to work under milder temperature and pressure conditions constitutes a topical
cutting-edge target in modern reduction area.[9] Transition metals such as ruthenium, iridium and
rhodium have dominated this area of research for decades.[10] However, in terms of sustainability
such precious metals ought to be substituted by inexpensive, low toxic and widely abundant first
row base metals such as iron and manganese.
2.1 Hydrogenation Molecular hydrogen is one of the most efficient and cleanest reducing agent in chemical
processes and is also now considered to be the one of the fuel of the future. Hydrogen is already
widely used in the synthesis of ammonia and in petroleum refining process. For example, in the
Haber process hundreds of million tons of ammonia fertilizer are produced annually from H2 and
N2. Hydrogenation of substrates having a polar multiple C-heteroatom bonds such as carbonyl
derivatives (e.g. ketones or aldehydes) and carboxylic compounds (e.g. esters, amides, nitriles)
has attracted significant attention because the obtained products such as alcohols and amines are
important building blocks in industry. This reaction is well documented by using many transition-
metal complexes containing noble metals.[11-12] In recent years, iron complexes have emerged as
catalysts for the important hydrogenation of C=O bonds.[13-15] In this part we mainly focus on the
hydrogenation of carbonyl compounds and hydrogenation of carboxylic acid derivatives will be
discussed in part 3.
2.1.1 Iron catalyzed hydrogenation of C=O bond The first catalytic effort on the hydrogenation of carbonyl derivatives was reported by
Marko et al. in 1983. Reduction of acetophenone was carried out in the presence of Fe(CO)5 (10
mol%) under drastic conditions [150 °C - 100 bar of H2/CO (98.5:1.5) in triethylamine].[16-17]

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
10
In the 1990s, Knölker developed the first iron hydride hydroxycyclopentadienyl
complex[18] which is the analogue of Ru Shvo’s complex.[19] Later, in 2007 Casey et al. studied
their catalytic properties in hydrogenation and transfer hydrogenation reactions.[20-21] Complex 1
catalyzed the hydrogenation of ketones under mild conditions (3 atm, 25 oC, 1-68 h) with good
chemoselectivities in the presence of other reducible functional groups such as esters, isolated
alkenes and alkynes group (Scheme 1).
Scheme 1. First hydrogenation of ketones and imine with iron complex.
In 2011 Berkessel et al. synthesized a series of chiral iron complexes using a modified
Casey’s type catalyst Iron(II)-cyclopentadienone-tricarbonyl 2 substituting one carbonyl by a
chiral phosphoramidite ligand under UV irradiation and studied their catalytic activity in the
asymmetric hydrogenation of acetophenone.[22] Nevertheless, even if moderate to very good
conversions of acetophenone to 1-phenylethanol was obtained (up to 90%), the highest
enantiomeric excess obtained was 31% ee (S) (Scheme 2). Recently, Gennari has developed new
chiral cyclopentadienone iron complexes derived from (R)-Binol 3 which can be used as catalyst
(2 mol%) when treated with Me3NO (4 mol%) in the asymmetric hydrogenation of ketones
leading to the corresponding alcohols with ee upto 77% (22-99% conversion, 30 bar H2,
i-PrOH/H2O (5:2), 70 °C, 18 h).[23]
Scheme 2. Chiral Knölker type complexes in asymmetric hydrogenation of ketones.
In 2013, Renaud reported the use of Knölker type complexes with cationic fragments such
as ammonium salts and evaluated their catalytic activity in hydrogenation reactions in water

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
11
(Scheme 3).[24] The insertion of an ammonium salt allowed for a better solubility of the iron pre-
catalyst 4 in water, leading to improved reactivity under these conditions as compared to the
complex 1. A low reactivity was observed if the ammonium salt is placed too close to the reactive
site. The use of a catalytic amount of Me3NO was necessary for the formation of the active
catalyst. Using this protocol, a series of ketones, aldehydes, or imines could be efficiently
hydrogenated at 85 °C in H2O under 10 atm of H2. Cyano functionality can be tolerated, but the
hydrogenation of α,β-unsaturated ketones led primarily to a mixture of fully reduced compounds
resulting in the concomitant reduction of the C=C and C=O bonds.
Scheme 3. Hydrogenation of ketones and imines catalyzed by 4.
Simultaneously, Beller et al. reported that the air stable iron complex 5 was able to
catalyse efficiently and selectively the hydrogenation of carbonyl compounds under 30 atm of H2
at 100 °C in a mixture of i-PrOH/H2O in the presence of a catalytic amount of K2CO3. Notably,
low loading of the complex 5 (0.05 mol%) allowed the reduction of a variety of aldehydes and
ketones and good functional group tolerance was observed as esters, amides and heterocycles
were not reduced (Scheme 4). Interestingly, using this procedure the selective unsaturated
aldehydes were selectively reduced into the corresponding unsaturated alcohols.[25]
Scheme 4. In situ generated Knölker’s catalyst for the hydrogenation of aldehydes and ketones.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
12
In 2008, Morris and co-workers[26-27] prepared the P2N2 ligands containing iron complexes
for the asymmetric hydrogenation and transfer hydrogenation of ketones. This was the first
efficient iron catalyzed asymmetric hydrogenation reactions (Scheme 5). The use of the complex
[Fe(NCMe)2{(R,R)-cyP2N2}](BF4)2 6 as a catalyst (0.44 mol%), in the presence of 6.7 mol% of
tBuOK, in the hydrogenation of acetophenone led to 1-phenylethanol with a moderate conversion
(40%) and ee (27%) at rt after 18 h under 25 atm of H2 in iPrOH.[28] This catalytic system (TOF 5
h-1 at 50 °C) was found to be somewhat less active system than Casey’s iron catalyst (TOF 2 h-1
at 25 °C).
Scheme 5. Iron catalyzed asymmetric hydrogenation.
In 2013, Beller developed a highly selective iron-catalyzed hydrogenation of
unsaturated aldehydes to give the corresponding synthetically valuable allylic alcohols
(Scheme 6).[29] The cationic iron-tetraphos fluoride complex 7 is able to catalyze the reduction of
a broad range of aromatic and aliphatic aldehydes, including unsaturated aldehydes in 95-
99% yields at low catalyst loadings (0.2-0.4 mol%) in the presence of 1-5 mol% of TFA at 120 oC for 2-5 h under 20 bar of H2. Noteworthy, this reaction tolerated reducible groups such as
esters, sulfides, C=C bonds or ketones.
Scheme 6. Selective hydrogenation of unsaturated aldehydes using 7.
In 2011, following their previous contributions on ruthenium-pincer complexes involving
an original mode of cooperation between the pincer ligand and the metal center via
aromatization/de-aromatization of the pyridine moiety of the ligand,[30] Milstein synthesized well-

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
13
defined iron pincer complexes [(iPrPNP)Fe(CO)Br2] 8 and [(iPrPNP)FeH(CO)Br] 9 and
demonstrated that the iron hydride complex 9 (0.05 mol%) was the most efficient catalyst for
hydrogenation of a broad scope of aliphatic and aromatic ketones under mild conditions (4.1 bar
H2, RT) in the presence of catalytic amount of KOtBu (0.1 mol%) with turnover numbers of up to
1880 and TOF up to 425 h-1 at 40 °C (Scheme 7).[31] Interestingly, cyano or amino groups seemed
to inhibit the reaction whereas no chemoselectivity was observed for the reduction of α,β-
unsaturated ketones. Notably, the iron(II) pincer complex [Fe(η1-BH4)(H)(CO)(iPrPNP)] 10 (0.05
mol%) bearing both hydride and borohydride ligands, exhibited a similar catalytic activity, with
no additional base.[32] In 2015, the same group reported that this iron pincer complex 9 was also
an efficient precatalyst for the hydrogenation of secondary and tertiary aliphatic aldehydes and
aryl aldehydes (Scheme 7). These reactions proceed smoothly under mild conditions (30 bar H2,
40 °C) and notably with very low catalyst loadings (0.025 mol%) to give the corresponding
alcohols in good to quantitative yields. This protocol is not suitable for primary aldehydes R-
CH2-CHO, as aldol condensation proceeds faster than the hydrogenation of the primary
aldehydes.[33]
Scheme 7. Pincer iron complexes for catalyzed hydrogenation of ketones and aldehydes.
In 2015, Hu reported the synthesis and the characterizations of several diphosphinite
analogous PONOP complexes such as 11 which catalyzed the hydrogenation of aldehydes under

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
14
mild conditions (8 bar H2, MeOH, rt, 24 h) and with high functional group tolerance, notably the
chemoselectivity towards aldehyde versus alkenes, esters and ketones. Nevertheless, the complex
11 was less active as 10 mol% of catalyst were necessary to perform the reduction.[34] (Scheme 7)
In 2014, Kirchner reported Fe(II) PNP pincer hydride complexes [(iPr-PNP)FeH(CO)Br]
12 based on the 2,6-diaminiopyridine (Scheme 7). Complexes with labile ligands such as 12 are
efficient catalysts for the hydrogenation of ketones and aldehydes leading to alcohols under mild
conditions (30 bar H2, EtOH, 40 °C, 16 h).[35] The reduction reaction took place at room
temperature in the presence of 0.5 mol% of 12 with turnover frequencies up to 770 h−1 using 5
bar hydrogen pressure. Recently, they have shown that the N-Me spacer analogous complex
[(iPr-PNMeP)FeH(CO)Br] 13 (cis/trans mixture) exhibited outstanding efficiency under 30 bar of
hydrogen at 40 °C in the presence of 1 mol% of DBU (TON up 80,000 to and TOF up to 20,000
h-1 were reached). Notably, esters, alkenyl (included conjugated C=C ones), and alkynyl moieties
were tolerated.[36]
In asymmetric area, in 2014, Morris developed novel chiral unsymmetrical P-N-P’ iron
pincer complexes, mer-trans-[Fe(Br)(CO)2(P-CH=N-P’)][BF4] 14 (0.1 mol%) which were used
in the asymmetric hydrogenation of ketones in THF at 50 °C under 5 atm H2 in the presence of a
catalytic amount of tBuOK (1 mol%)[37-38] (Scheme 8). mer-[Fe(OR)(H)(CO)(Cy2P-CH2-NH-
PPh2)] iron hydride complexes were also used as pre-catalysts and were obtained by reacting the
bromo iron complex 14 with LiAlH4 followed by an alcohol. Various (hetero)aryl alkylketones
were hydrogenated with ees up to 85% (20-90% conv.). Noteworthy, high activities were
observed (TONs up to 990 and TOFs up to 1980 h–1).
Scheme 8. Asymmetric iron catalyzed hydrogenation of ketones.
Using an in situ generated catalyst (0.5 mol%) from chiral N4P2 22-membered
macrocyclic ligand L1 and Fe3(CO)12, Xiao and Gao reported the asymmetric hydrogenation of a

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
15
large variety of (hetero)arylalkyl ketones with ees up to 99%, and good functional group
tolerance (cyano, iodo, bromo and α,β-C=C, Scheme 9). It must be noted that the chemoselective
reduction of β-ketoesters affording the corresponding chiral hydroxyesters was successfully
achieved (50 bar of H2 at 65 °C for 30 h in methanol).[39]
Scheme 9. Fe3(CO)12/L1 based catalyst for asymmetric hydrogenation of ketones.
Dual iron-catalyzed process has been developed by Beller for the hydrogenation of α-keto
and α-imino esters to the corresponding α-hydroxyesters and α-aminoesters under 50 bar H2 at 65 oC using a combination of two catalysts, Fe3(CO)12 (6.7 mol%) and Fe(OTf)2 (7.2 mol%) in the
presence of phenanthridine L2 (20 mol%) (Scheme 10).[40] This method utilized the NAD(P)H
model dihydrophenanthridine (DHPD) as hydrogen transferring agent.
Scheme 10. Hydrogenation of keto esters using iron catalyst.
2.1.2 Manganese catalyzed hydrogenation of C=O bonds At the beginning of this Ph.D research project, there was no report dealing with the
hydrogenation of carbonyl or carboxylic derivatives using manganese based catalyst. Following
the works developed in both groups in Rennes and Rostock on manganese catalyzed
hydrogenation of nitriles, ketones and aldehydes using PNP manganese pincer complexes[41]
(detail see Part 3 chapter 1), very recently Kempe et. al reported another manganese catalyzed
hydrogenation of ketones in the presence of KOtBu (10 mol%) as a base to activate the PN5P
pincer manganese complex 15 (Scheme 11). The reaction occurs under mild conditions (20 bar
H2 and 80 oC) and allows broad substrate scope at low catalyst loading (0.1-1 mol%).[42]

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
16
Scheme 11. PN5P pincer manganese catalyzed hydrogenation of ketones.
2.2 Hydrosilylation Hydrosilylation is a promising alternative method for the catalytic reduction of organic
molecules in comparison with other reduction reactions such as hydrogenation and transfer
hydrogenation as well as reduction with stoichiometric aluminum and boron hydrides owing to its
operational simplicity and mild conditions, and good selectivity. While hydrosilanes are inert
towards non activated carbonyl compounds, a considerable number of transition metal complexes
are known to act as catalysts for hydrosilylation reactions. In this area, iron-catalyzed
hydrosilylation is one of the most popular reduction reactions owing to the high natural
abundance, low cost and low toxicity of metal.[43]
2.2.1 Iron-catalyzed hydrosilylation of aldehydes and ketones The very first example of the hydrosilylation of ketones using iron based complexes as
the catalysts was reported by Brunner and Fish in 1990. Using [Fe(Cp)(CO)(X)(L)] complexes
(16, 0.5-1 mol%), acetophenone gave quantitatively the silylated ether by reaction with Ph2SiH2
(1 equiv.) at 50-80 °C for 24 h. (Scheme 12).[44]
Scheme 12. Pioneering iron-catalysed hydrosilylation of acetophenone.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
17
Using in situ generated iron catalytic systems
It was only two decades later, in 2007, that Nishiyama and Furuta described the first
general and efficient hydrosilylation of ketones in the presence of Fe(OAc)2 (5 mol%) and
nitrogen based ligand TMEDA (N,N,N’,N’-tetramethylethylene-diamine, 10 mol%) using
(EtO)2MeSiH (2 equiv.) at 65 °C for 20-24 h yielding the corresponding alcohols in good yields
upon hydrolytic workup (50-94% yields).[45-46] A combination of iron(II) acetate and sodium
thiophene-2-carboxylate promotes the hydrosilylation of aromatic and aliphatic ketones even
more efficiently and leads to excellent yields for a variety of substrates. The asymmetric
hydrosilylation of ketones was also carried out using the chiral ligands such as N,N,N-
bis(oxazolinylphenyl)-(bopa) ligands (e.g. Bopa-dpm L3) (3 mol%) in combination with 2 mol%
of Fe(OAc)2 (Scheme 13). In most cases, the enantiomeric excesses of the obtained alcohols were
good (37-79% ee).[47]
Scheme 13. In situ generated iron catalyst in hydrosilylation reactions.
Later Beller described an iron/phosphine based catalytic system used in the
hydrosilylation of carbonyl compounds: an in situ generated catalyst from Fe(OAc)2 (5 mol%)
and PCy3 (10 mol%) as the ligand in the presence of PMHS (3 equiv.) as the hydride source in
THF at 65 °C for 16 h; the reduction of functionalized aromatic, heteroaromatic and alkyl
aldehydes were efficiently reduced into the corresponding alcohols with moderate to excellent
yields (35 examples, 60–99% yields).[48] The same catalytic system was also able to reduce a
wide range of ketones (17 examples, 58–96% yields) after 20 h at 65 °C.[49] Further, the

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
18
asymmetric hydrosilylation of ketones was also studied under similar conditions using [Fe(OAc)2
and chiral diphosphine (S,S)-Me-Duphos L4 with either (EtO)2MeSiH or PMHS as the hydrogen
source at room temperature or 65 °C, with yields up to 99% and an ees up to 99% (Scheme
13).[50] Using an in situ generated catalyst from PCy3 (1.1 mol% ) and the air- and moisture-stable
complex [Bu4N][Fe(CO)3(NO)] 17 (1-2.5 mol%), Plietker reported a highly active system for the
mild hydrosilylation of aldehydes and ketones using PMHS leading to the corresponding alcohols
in 65-99% yields at 30-50 °C for 14 h. The [FeH(CO)(NO)(dppp)] complex 18 [dppp:
bis(diphenylphosphino)propane] can also be used as a catalyst (1 mol%) for the reduction of
aldehydes and ketones, even if the conditions were more drastic (0.5 equiv. of NEt3, 1 equiv. of
PhSiH3, THF, 80 °C, 18 h, 66-98% yields).[51]
During the last decade, the use of well-defined NHC iron complexes and of in situ
generated catalysts from iron salts and NHC ligands has become popular.[52-53] These NHC iron
complexes have also been employed as catalysts for a reduction of carbonyl compounds. Thus,
selective hydrosilylation of aldehydes and ketones catalyzed by in situ generated iron(II)-NHC
complexes were reported by Adolfsson and co-workers.[54-55] An iron NHC complex, generated in
situ from iron(II) acetate (2.5 mol%), IPr.HCl L5 (3 mol%), and n-butyllithium (3 mol%), has
been successfully applied as catalyst for the hydrosilylation of ketones with
polymethylhydrosiloxane (PMHS, 3 equiv.) as reducing agent (Scheme 14). Importantly, the
exact stoichiometry between the base and the imidazolium salts is crucial: indeed simple alkoxide
salts can also promote such catalytic hydrosilylations in the presence of trisubstituted silanes.
This protocol demonstrates good functional group tolerance, giving chemoselective reductions on
substrates with more than one reducible group. Later, the same group studied the hydrosilylation
reactions using imidazolium salts L6 (1.1 mol%) with 1 mol% Fe(OAc)2 as the iron source, 2.2
mol% n-BuLi for carbene generation and 3 equiv. PMHS as the hydride source in THF at 65 °C
(Scheme 14). This catalytic system showed good chemoselectivity in presence of double
carbon-carbon bonds, ester and cyano groups.
Scheme 14. In situ generated iron NHC complexes in hydrosilylation reactions.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
19
In 2011 Thiel et. al reported iron(II) acetate and iron(II) octanoate together with various
pyrazol-3-yl-pyridines, 4,4’-bipyrimidines, bipyrazoles, and a pyridylpyrimidine as catalytic
system for the hydrosilylation of acetophenone.[56] The reaction could be performed in alkane
solution at 80 oC using polymethylhydrosiloxane (PMHS) as reductant.
Using well-defined iron complexes
In 2010, Tilley developed an efficient hydrosilylation of carbonyl derivatives using the
highly air-sensitive iron silylamide catalyst [Fe(N(SiMe3)2)2], in the presence of 1.6 equiv. of
diphenylsilane. The reaction was performed at 23 °C for 0.3-20 h using 0.01-2.7 mol% catalyst
loading giving TOFs up to 2400 h–1 for the reduction of 3-pentanone. Furthermore, the reduction
can tolerate nitrile, cyclopropyl and alkenyl units.[57] The related Fe(II) bis-(trimethylsilyl)amido
complexes 19 bearing a N-phosphinoamidate ligand (Figure 1) were also an efficient catalysts
(0.015-1 mol%) for the hydrosilylation of a large range of aldehydes and ketones in the presence
of 1 equiv. of PhSiH3. Noticeably, TOFs up to 23600 h–1 were obtained at rt for the reduction of
acetophenone, demonstrating the beneficial influence of the ligand (vs 1266 h–1 with
[Fe(N(SiMe3)2)2]).[58]
Figure 1. N-phosphinoamidinate iron complex in hydrosilylation.
Furthermore, cyclometallated iron complexes (Figure 1) can be applied in hydrosilylation
which were conducted with 0.3-0.6 mol% of the hydrido iron complex 20 in the presence of 1.2
equiv. of (EtO)3SiH in THF at 55 °C for 1-4 h for aldehydes and 4-12 h for ketones (65-92%
yields).[59-60] The use of hydrido iron complex 21 bearing P,S chelating ligand was reported with
similar activities under similar conditions (TOF up to 25 h-1).[61] In 2011, in Rennes, the iron
dihydride complex (dppe)2Fe(H)2 (1 mol%) was reported as a catalyst when used in combination
with sodium tetraethoxyborate (1 mol%) as cocatalyst for the hydrosilylation of several ketones

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
20
and aldehydes with inexpensive PMHS (2 equiv.) at 100 oC in toluene was carried out under
visible light irradiation to give the corresponding alcohols in good to excellent yields.[62]
Another important series of efficient iron complexes, cyclopentadienyl piano-stool
iron(II) complexes, were extensively developed for the hydrosilylation of carbonyl derivatives.
Following the pioneering contribution of Brunner,[63-64] Nikonov reported the reactivity of the
cationic CpFe-phosphine complex 22 (5 mol%) for the hydrosilylation of benzaldehyde using
H2SiMePh at 22 °C for 3 h (Figure 2).
Figure 2. Piano-stool iron complexes
In Rennes, a similar series of carbonyl complexes such as [CpFe(CO)2PPh3]PF6,
[CpFe(CO)(PPh3)I] were also described in hydrosilylation[65] (Figure 2). With 23 (5 mol%) and
1.1 equiv. of PhSiH3 at 30 °C in 16 h under visible light irradiation, aromatic aldehydes were
reduced in very good conversions (92-98% in THF, 91-97% under neat conditions). Notably,
PMHS (4 equiv.) could also promoted the reduction under similar conditions (conversions up to
95%). The neutral iron complex 24 was found to be the most efficient of the series for the
reduction of ketones when performed under neat conditions and visible light activation (1.2
equiv. PhSiH3, 70 °C, 30 h).
Piano-stool iron complexes bearing N-heterocyclic carbene ligands (NHC) are also useful
catalysts for hydrosilylation (Figure 3). In 2010, Royo showed that tethered Cp-NHC iron
complexes such as 25-26 (1 mol%) were suitable catalysts for the hydrosilylation of activated
aldehydes (EtO)2MeSiH (1.2 equiv.), 80 °C, 2-24 h).[66] This system showed a good
chemoselectivity in the presence of reducible functional groups such as nitro, cyano and ester.
The group in Rennes made a significant contribution in hydrosilylation using well defined
iron NHC complexes. In the same year mild conditions was developed by the group in Rennes
when using neutral and cationic cyclopentadienyl(NHC)iron complexes 27-28 as catalysts.[67]
Thus, aldehydes could be completely converted within 3 h at 30 oC while ketones required 16 h at
70 oC for a good conversion. The cationic complex activated by visible light irradiation. Further
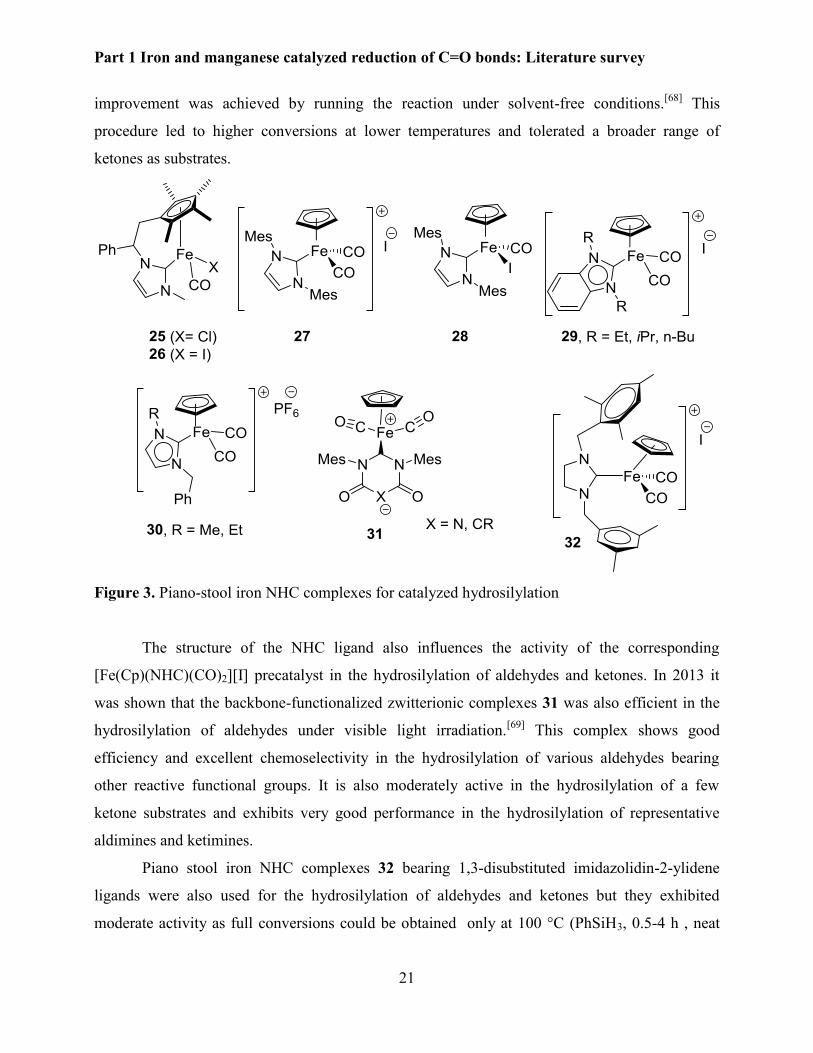
Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
21
improvement was achieved by running the reaction under solvent-free conditions.[68] This
procedure led to higher conversions at lower temperatures and tolerated a broader range of
ketones as substrates.
Figure 3. Piano-stool iron NHC complexes for catalyzed hydrosilylation
The structure of the NHC ligand also influences the activity of the corresponding
[Fe(Cp)(NHC)(CO)2][I] precatalyst in the hydrosilylation of aldehydes and ketones. In 2013 it
was shown that the backbone-functionalized zwitterionic complexes 31 was also efficient in the
hydrosilylation of aldehydes under visible light irradiation.[69] This complex shows good
efficiency and excellent chemoselectivity in the hydrosilylation of various aldehydes bearing
other reactive functional groups. It is also moderately active in the hydrosilylation of a few
ketone substrates and exhibits very good performance in the hydrosilylation of representative
aldimines and ketimines.
Piano stool iron NHC complexes 32 bearing 1,3-disubstituted imidazolidin-2-ylidene
ligands were also used for the hydrosilylation of aldehydes and ketones but they exhibited
moderate activity as full conversions could be obtained only at 100 °C (PhSiH3, 0.5-4 h , neat

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
22
conditions, without light activation).[70] Benzimidazole 29 and imidazole 30 based NHC iron
cationic complexes were also evaluated and were found to be active catalyst precursors for the
hydrosilylation of aldehydes (THF, 30 oC, 3 h) and ketones (Neat, 70 oC, 17 h).[71]
Figure 4. NHC iron complexes in hydrosilylation reactions.
In 2012 Glorius synthesized low valent bis(NHC) iron complexes 33-34 (Figure 4) and
studied their reactivity in hydrosilylation. They exhibited high efficiency for the reduction of 2’-
acetonaphthone in the presence of (EtO)3SiH (1.1 equiv., 25 °C, 5 h) or of Ph2SiH2 (1.1 equiv.,
40 °C, 5 h).[72]
In 2013, Royo described the use of iron(0) complexes Fe(NHC)(CO)4 35, prepared by the
direct reaction of Fe3(CO)12 with equimolecular amounts of NHC imidazolium halide precursors,
in catalyzed hydrosilylation of benzaldehyde derivatives with phenylsilane (1.2 equiv.) as
reducing agent using 1 mol% of catalyst at rt for 4 h with a broad functional group tolerance
including reducible ketones, nitriles or nitro moieties.[73] In 2015, bis(imino)acenaphthene iron
arene complex, BIAN-Fe(C7H8) 36, was found to be an efficient precatalyst for the
hydrosilylation of aldehydes and ketones using Ph2SiH2. This method exhibits broad functional
group tolerance and high activities at 70 °C for 0.5-18 h under solvent-free conditions.[74]
Iron(III) based complexes were recently reported as good candidates for the hydrosilylation.
Indeed, 1 mol% of amine-bis(phenolate) iron complex 37 in the presence of 3 equiv. of
triethoxysilane promoted the reduction of aldehydes and ketones at 80 °C for 3-24 h.[75]

23
Iron pincer complexes in hydrosilylation reactions
In 2008, Chirik reported bis(imino)pyridine iron precatalysts 38-39 for the hydrosilylation
of aldehydes and ketones with diphenylsilane and phenylsilane. Efficient carbonyl
hydrosilylation is observed at low (0.1-1.0 mol %) catalyst loadings and with 2 equiv of either
PhSiH3 or Ph2SiH2 at room temperature for 1 h.[76]
Figure 5. Bis(imino)pyridine iron complexes for hydrosilylation of aldehydes and ketones.
In 2011, Guan et al. reported the synthesis of new iron hydride complexes bearing
phosphinite-based pincer ligands (POCOP) 40-43 and their application in the catalytic
hydrosilylation of aldehydes and ketones (Fig. 6).[77] For the reduction of benzaldehyde, even if
full conversions were obtained for all the complexes at 50-65 °C using 1 mol% of catalyst in the
presence of 1.1 equiv. of (EtO)3SiH, the complex 40 was identified as the more active one as the
reaction could be performed in 1 h, versus 4 h for complex 41 and 68 and 96 h for 42 and 43,
respectively. With ketones such as acetophenone, a higher temperature (80 °C) was required for
4.5 h to reach full conversion and isolate 87% of the corresponding alcohol using complex 40.
Figure 6. Iron PCP and PSiP pincer complexes in hydrosilylation reactions.
Most recently, H. Sun et al. described a silyl iron pincer complexes 44 bearing a tridentate
bis(phosphine)silyl,[78] and a PCP ligands.[79] Their catalytic activities were evaluated in the

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
24
hydrosilylation of aldehydes using 1 mol% of 44a in the presence of (EtO)3SiH in THF at 60 °C
for 1 h; benzyl alcohol was obtained quantitatively whereas the reduction of ketones such as
acetophenone and cyclohexanone required a longer reaction time (6 h) to lead to the
corresponding alcohols. Slightly higher activities were obtained with the complex 44b at 50 °C
[aldehydes: 0.3-1 mol% 44b, 1-13 h; ketones: 1 mol% 44b, 16 h].
Interestingly, well-defined chiral pincer iron complexes can be efficiently used for asymmetric
hydrosilylation of ketones. In 2009, Chirik has shown that chiral tridentate
(S,S)-(iPrpybox)-Fe(CH2SiMe3)2 complex 45 (0.3 mol%) can catalyse the asymmetric
hydrosilylation of ketones in the presence of 2 equiv. of PhSiH3 and 0.95 equiv. of B(C6F5)3 at 23
°C in Et2O for 1 h leading to the corresponding alcohols with ees up to 54%.[80] Using (S,S)-
phebox-ip-Fe(CO)2Br complex 46 (2 mol%) in association with 2 mol% of Na(acac), Nishiyama
performed similar performances in the hydrosilylation of p-phenylacetophenone in the presence
of 1.5 equiv. of (EtO)2MeSiH at 50 °C in hexane for 24 h (66% ee).[81] Notably, when using a
chiral iminopyridine-oxazoline iron complex 47 (1 mol%) in combination with 2 mol% of
NaBEt3H as the catalyst, higher ees were obtained in the hydrosilylation of arylketones (ees up to
93%) in the presence of 1 equiv. of diphenylsilane at 25 °C.[82]
Using (S,S)-BOPA/FeCl2 complex 48 (5 mol%) in the presence of 2 equiv. of (EtO)3SiH,
the reduction of aromatic ketones led quantitatively to the corresponding alcohols with 32-95%
ees after 48 h at 65 °C, but low ees were observed with alkyl ketones.[83]
Figure 7. Well-defined chiral pincer iron complexes for catalyzed asymmetric hydrosilylation.
Up to now, Gade reported among the most efficient catalysts using isoindoles based iron
catalysts, tetraphenyl-carbpi-Fe(OAc) complex 49 (5 mol%) which can perform the

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
25
hydrosilylation of ketones with moderate to good enantioselectivity (56-93%) by reaction with 2
equiv. of (EtO)2MeSiH at 40 °C for 40 h.[84] The best results were obtained using the chiral iron
alkoxide boxmi pincer complex 50 as the catalyst (5 mol%) in the presence of 2 equiv. of
(EtO)2MeSiH in toluene for 6 h in a temperature range from - 78 °C to rt.[85]
2.2.2 Iron-catalyzed hydrosilylation of carboxylic derivatives Hydrosilylation of amides
Among the carboxylic acid derivatives, the most difficult ones to reduce are carboxamides
mainly due to chemoselection issues (C-N vs C=O cleavage) and their catalytic transition metal
reductions are well-exemplified.
In 2009, concomitantly, Beller and Nagashima reported the first iron-catalysed
hydrosilylation of secondary and tertiary amides leading specifically to the corresponding
amines. Beller described iron-catalyzed reduction of secondary and tertiary amides to the
corresponding amines with inexpensive PMHS (4-8 equiv.) as a reducing agent in the presence of
2-10 mol% of Fe3(CO)12 at 100 °C for 24 h (Scheme 15). This catalytic system allows a broad
substrate scope with high selectivity and good functional group tolerance giving a variety of
amines in good yields.[86]
Scheme 15. Hydrosilylation of amides using iron catalyst.
Nagashima and co-workers also demonstrated that pentacarbonyliron (10 mol%) or
dodecacarbonyltriiron were efficient catalysts for the reduction of tertiary carboxamides to
amines with TMDS (2.2 equiv.) under thermal or photolytic conditions (Scheme 16).[87] In 2011
the same authors employed a heptanuclear ironcarbonyl cluster [Fe3(CO)11(μ-H)]2Fe(DMF)4 (0.5
mol% for Fe) as catalyst for the hydrosilylation of tertiary amides with 1,2-bis-
(dimethylsilyl)benzene (2.2 equiv.).[88] This procedure found to be active system which is shorter
reaction times (30-180 mins), lower catalyst loadings (0.5 mol%), and higher yields (up to 96%)
for amide substrates.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
26
Scheme 16. Iron catalyzed hydrosilylation of tertiary amides.
The same year, the group in Rennes reported the hydrosilylation of carboxamides with
phenylsilane in the presence of 5 mol% of the complex [CpFe(CO)2(IMes)]I 51.[89] The reaction
was performed at 100 °C for 24 h under solvent-free conditions and visible light irradiation.
Tertiary and secondary amides were readily reduced to the corresponding amines in 77-98%
yields (Scheme 17). Using an in situ Fe/NHC complex, an efficient protocol for the reduction of
tertiary amides to the corresponding tertiary amines by iron-catalyzed hydrosilylation has been
reported by Adolfsson and Buitrago in 2013.[90] The method relied on an iron NHC catalyst
generated in situ from iron(II) acetate, [Ph-HEMIM][OTf] L7, and n-butyllithium. A variety of
aryl and heteroaryl amides could be converted to afford the amines in high yields. It must be
noted the important role of LiCl to increase both chemoselectivities and activity.
Scheme 17. NHC-iron catalysts for the hydrosilylation of amides to amines.
Furthermore, NHC-Fe(0) complex 52 (1 mol%) was shown to be able to catalyze the
hydrosilylation of tertiary amides by reaction with 3 equiv. of Ph2SiH2 at 70 °C for 24 h. (Scheme
17) By contrast, the hydrosilylation of primary amides to primary amines are more difficult to
perform as the only obtained products were the corresponding nitriles obtained by dehydration.[91]

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
27
In order to perform such reduction, Beller et al. used two different iron catalyst in a consecutive
manner with diethoxymethylsilane.[92] Initially, primary amides were dehydrated into
corresponding nitrile intermediates with 3 equiv. of silane in the presence of 2-5 mol% of iron
[Et3NH][HFe3(CO)11] 53. This nitrile intermediate was reduced into amines in the presence of 3.5
equiv. (EtO)2MeSiH and an in situ generated catalyst from 20 mol% of Fe(OAc)2 and 20 mol%
of the phenanthroline ligand L8 at 100 oC for 28 additional hours (Scheme 18).
Scheme 18. Two iron catalysts for hydrosilylation of amides.
Hydrosilylation of carboxylic acids and esters
The group in Rennes developed the first iron-catalyzed hydrosilylation of esters such as
alkanoates and 2-substituted acetates using a well-defined iron complex,
[CpFe(CO)2(PCy3)][BF4], as the catalyst, at 100 oC, in the presence of 4 equiv. of phenylsilane
under visible light irradiation and neat conditions. The corresponding alcohols were then obtained
in 51-88% isolated yields.[93] (Scheme 19)
Scheme 19. Hydrosilylation of esters using iron catalyst.
Shortly thereafter, Beller showed that the in situ generated Fe(stearate)2/ NH2CH2CH2NH2
[stereate = CH3(CH2)16COO] catalytic system was also efficient for the selective hydrosilylation
of carboxylic esters to alcohols using inexpensive polymethylhydrosiloxane (PMHS) as the
reducing agent.[94] The catalytic method exhibits broad substrate scope, including aliphatic,
aromatic, heterocyclic esters and lactones. (Scheme 20)

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
28
Scheme 20. Hydrosilylation of esters using an in situ iron and amine based catalyst.
The same transformation was also accomplished by Turculet, Stradiotto, and Sydora using
the (N-phosphinoamidinate)iron precatalyst 19 (0.01 1.0 mol%) and 1 equiv. of phenylsilane as
the reductant at rt for 4 h.[58]
The challenging transformation of esters to ethers was developed by Beller in 2013.[95]
Thus, triiron dodecacarbonyl (10 mol%) in combination with TMDS (3 equiv.) allows for the
selective reduction of variety of esters into the corresponding ethers in 50-85% yields at 100 oC
for 2 h. (Scheme 21)
Scheme 21. Esters to ethers catalyzed by triiron dodecacarbonyl.
The group in Rennes developed an efficient methodology for chemoselective reduction of
esters to aldehydes with N-heterocyclic-carbene iron complex [(IMes)Fe(CO)4], as the catalyst (1
mol%) in the presence of a secondary silane (diethylsilane or diphenylsilane) as the reducing
agent (Scheme 22). This reaction proceeded at room temperature under UV irradiation and
allowed the reduction of substituted aromatic, aliphatic esters and lactones.[96]
Scheme 22. Catalyzed selective reduction of esters to aldehydes.
The group in Rennes reported efficient examples of the selective reduction of carboxylic
acids to aldehydes or alcohols by switching the catalyst in combination with silane. Thus, the

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
29
combination of (COD)Fe(CO)3 (5 mol%) and phenylsilane (4 equiv.) under UV irradiation at rt
for 24 h yielded selectively alcohols after hydrolysis in good yields while aldehydes were
selectively obtained using TMDS (2 equiv.) as the reducing agent in association with 5 mol% of
(t-BPO)(Fe(CO)3 as the catalyst at 50 °C for 24 h.[97] (Scheme 23)
Scheme 23. Selective reduction of carboxylic acids using iron complexes.
2.2.3 Manganese-catalyzed hydrosilylation of C=O bonds The first example of manganese-catalyzed hydrosilylation of ketones was described by
Yates in 1982 using Mn2(CO)10 as the catalyst (2.4 mol%) in the presence of 1 equiv. of Et3SiH
in neat conditions under UV at 29 °C for 20 h: the corresponding isopropyl triethylsilyl ether was
then obtained in only 5% yield.[98] A breakthrough in this area was made by Curtler when he
performed the reaction between the acyl manganese complexes Mn(CO)5(COMe) 55 and 1-2
equiv. of HSiMePh2 at rt leading to 81% of the siloxyethyl complex (CO)5MnCH(OSiMePh2)CH3
56 and 2% of the siloxyvinyl byproduct (CO)5Mn-C(OSiMePh2)=CH2. It is interesting to note
that this reaction occured without adding a catalyst, in an autocatalytic fashion. Similarly, 56 can
catalyze the hydrosilylation of (η5-C5H5)Fe(CO)2(COR) complexes leading to the corresponding
(η5-C5H5)Fe(CO)2(COSiHPh2)(R) in up to 97% yield.[99]
Half-sandwich 1-hydronaphthene type manganese complexes 57 and cationic naphthalene
manganese complex 58 also catalyze the hydrosilylation of alkyl and aryl ketones at rt for 0.5-3 h
in the presence of diphenylsilane, 57 being the most active with a TOF of 100 h-1 for the
hydrosilylation of acetophenone.[100-101] (Figure 8)
Figure 8. Efficient half-sandwich manganese complexes for catalytic hydrosilylation of ketones.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
30
Half sandwich manganese cyclopentadienyl N-heterocyclic carbene complexes can be
also suitable catalysts for hydrosilylation of aryl and aliphatic aldehydes and ketones. The well
defined manganese N-heterocyclic carbene (NHC) complexes such as 59 were used under UV
irradiation (350 nm) in the presence of 1.5 equiv. of Ph2SiH2 at rt for 2 h, (> 99% conv. for the
hydrosilylation of acetophenone). Notably, cymanthrene CpMn(CO)3 showed very low activity
(4% conv.), showing the crucial role of the NHC ligand on the reactivity (Scheme 24). [102]
Interestingly, a huge functional group tolerance (heterocyclic moieties, halides, conjugated and
non-conjugated alkenyl, alkynyl nitrile, ester, etc.) was obtained.
Scheme 24. Hydrosilylation of aldehydes and ketones with manganese NHC complex.
Manganese complexes bearing multidentate ligands can also be active catalysts in
hydrosilylation. Indeed, salen-Mn complex 60 (0.5 mol%) can promote the hydrosilylation of
carbonyl derivatives in the presence of 0.5 equiv. of PhSiH3 at 80 °C for 1 min. - 3 h with yields
up to 95% and TOFs up to 8800 h-1 for the reduction of p-nitrobenzaldehyde.[103] (Figure 9)
Using non innocent redox bis(imino)pyridine manganese complexes, Trovitch developed
hydrosilylation of ketones. Using 0.01-1 mol% of 61 in the presence of 1 equiv. of phenylsilane,
aryl and alkyl ketones can be hydrosilylated at 25 °C for 4 min. - 24 h leading to a mixture of
tertiary and quaternary silanes in 80-99% conversions, with TOF up 76,800 h-1 (for the
hydrosilylation of cyclohexanone). The paramagnetic bis(enamide)tris pyridine manganese
complex 62 is also an efficient catalyst with TONs and TOFs up to 14170 and 2475 h-1,
respectively (PhCHO, 25 °C, 1 h, 1 equiv. PhSiH3).[104-105]
Figure 9. Multidentate ligand Mn complexes for catalytic hydrosilylation of ketones.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
31
Magnus has shown that aldehydes and cyclic five-and six-membered ketones can be
reduced in the presence of 0.4 equiv. of phenylsilane at rt in isopropanol under an atmosphere of
oxygen using 3 mol% of tris(dipivaloylmethanoto)manganese(III) (Mn(dpm)3 63) as the pre-
catalyst. By contrast, the reaction is less efficient for acyclic and macrocyclic ketones.[106]
Manganese catalysts can be also used for the hydrosilylation of carboxylic derivatives. In
1995, Cutler showed that (PPh3)(CO)5Mn(COMe) complex 64 can reduce alkanoic esters to
dialkyl ethers via a silyl acetal intermediate in the presence of phenylsilane in benzene at 24 °C
for 61-83% yields. Lactones can also be reduced leading to the corresponding cyclic ethers in 35-
65% yields.[107] In 2014, Trovitch investigated the pentadentate bis(imino)pyridine bis
diphosphine Mn(0) complex 61 as a catalyst (1 mol%) in hydrosilylation of alkyl and phenyl
acetates in the presence of 1 equiv. of phenylsilane. (Scheme 25) A mixture of tertiary and
quaternary silicon derivatives was obtained due to a dihydrosilylation process.[104]
Scheme 25. Manganese catalyzed reduction of esters.
The first selective reduction of carboxylic acids to aldehydes catalyzed by a commercially
available and inexpensive manganese carbonyl complex Mn2(CO)10 (5 mol%) was performed in
the presence of 3.3 equiv. of triethylsilane under UV irradiation (350 nm) at rt for 3 h. Alkenyl
and alkyl carboxylic acids were efficiently hydrosilylated leading to the corresponding
disilylacetals RCH(OSiEt3)2 (60-98% isolated yields) which were then quenched in acidic
conditions to produce aldehydes with a good functional group tolerance (hydroxyl, amino,
halides, internal C=C, heteroaromatic rings). Benzoic acid derivatives can be also reduced with
moderate yields (29-39%). [108]
Scheme 26. Hydrosilylation of acids under UV radiation.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
32
2.3 Hydrogen transfer In addition to hydrogenation with molecular hydrogen, transfer hydrogenation is another
alternative reduction method of C=O bonds. Mostly isopropanol and formic acid are used as a
good hydrogen donors, these reagents being easy to handle and cheap. In addition, the
experimental operation is easier and more attractive with regard to safety issues and equipment in
comparison to hydrogenation with pressurized H2 gas.
In the 80’s, iron carbonyl complexes were used in catalytic transfer hydrogenations of
ketones. Using 4 mol% of Fe3(CO)12 in the presence of 1-phenylethanol or i-PrOH as the hydride
source and benzyl-triethylammonium chloride and 18-crown-6 as phase transfer catalysts, the
reduction proceeded at 28 °C for 2.5 h leading to the corresponding alcohols with moderate
conversions (20-60%) and TOFs up to 13 h–1.[109-110] In 2006, Beller and co-workers reported a
general transfer hydrogenation of aliphatic and aromatic ketones in the presence of 1 mol%
[Fe3(CO)12]/terpy/PPh3 or FeCl2/terpy/PPh3 and of i-PrONa (2 mol%) and i-PrOH as the
hydrogen source (Scheme 27).[111] The same year, he reported the use of in situ generated iron
porphyrins catalysts from either Fe3(CO)12 or FeCl2 for the transfer hydrogenation of ketones.[112]
Using 2-propanol as the hydrogen source, various ketones were reduced to the corresponding
alcohols in good to excellent yield and selectivity (22-99% conv.; TOFs up to 642 h–1).
Scheme 27. Iron catalyzed transfer hydrogenation reactions.
The association of an iron precursor with diaminodiphosphine ligands (P2N2) led to
catalysts able to perform the reduction of carbonyl derivatives under milder conditions, and even
at rt. The first report by Gao in 2004 reported the asymmetric transfer hydrogenation of ketones

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
33
catalyzed by an in situ species (1 mol%) from [NHEt3][Fe3H(CO)11] and (S,S)-Ph-ethP2(NH)2
L10 or (S,S)-CyP2(NH)2 L11 (Figure 10). The reduction of Ph2CHCOCH3 with 80 mol% of KOH
in i-PrOH at 65 °C for 21 h furnished the best results with ees up to 98% but with low yields
(18%). Acetophenone led to 1-phenylethanol with 56% ee and 92% yield.[113]
Figure 10. Gao’s P-NH-NH-P ligands for asymmetric transfer hydrogenation of ketones.
Using similar ligands, in 2008 Morris reported the asymmetric transfer hydrogenation of
ketones by well-defined iron(II) complexes trans-(R,R)-[Fe(CyP2N2)(NCMe)(CO)](BF4)2 66 and
trans-(R,R)-[Fe(CyP2N2)(NCMe)(t-BuCN)](BF4)2 67, with 18-61% ees (Figure 11). 66 was
efficient at rt for the reduction of benzaldehyde (94% conv., 2.4 h) and N-benzylideneaniline
(>99%, 17 h). It also permitted the reduction of aromatic ketones with good conversions, but
moderate ees (e.g. acetophenone: TOF = 2600 h–1 and 35% ee at 24 °C). By contrast, 67
exhibited better enantioselectivity but lower activity. Using analogues of 66 such as 68 (0.17-0.5
mol%), ees up to 96% were observed, in particular with more hindered ketones, and notably with
similar catalytic activity (TOF up to 2000 h–1 for acetophenone).[114]
Figure 11. Morris’s catalysts for transfer hydrogenation from 2-propanol.
In an intense work, Morris reported a systematic study involving a new series of
diiminophosphine iron(II) complexes (17 examples, Figure 12).[115] As a representative example,
higher activity and stereoselectivity was obtained with 70: the reduction of acetophenone led to
90% conv. and ees up to 82%, (22 °C, 0.5 h, 0.05 mol% catalyst; TOF: 3600 h–1). For other
ketones, 35-90% conv. and 14-99% ees were obtained, hindered aromatic ketones such as t-
BuCOPh leading the best ee.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
34
Figure 12. Diiminophosphine iron(II) complexes for asymmetric transfer hydrogenation of ketones.
The nature of the phosphorus moiety had also a significant influence on the activity. Thus
replacing the diphenylphosphino groups by diethylphosphino ones, the corresponding complex
71 showed a moderate catalytic activity at 25 °C (TOF: 563 h-1). It can be underlined that the
homologous complexes bearing (c-C6H11)2P or (i-Pr)2P moiety were inactive. The p-
tolylphosphine based complex 72 showed the highest activity observed in this series (TOF: 30000
± 1500 h-1) with similar enantioselectivity (acetophenone: 82% ee, at 28 °C and 0.1 mol%
loading). The m-xylyl phosphine complex 73 also exhibited good activity (TOF: 26000 h-1) and
slightly better enantioselectivity (90% ee).
As to generate the active catalyst, 8 equiv. of KOtBu (with respect to [Fe] is required, a
new series of neutral ene-amido pentacoordinate iron(II)-N2P2 complexes 76-77 were prepared by
deprotonation with KOtBu at the α carbon of the phosphorus center in 74-75 (Scheme 28) and
tested under base-free conditions: they exhibited similar activity compared to the parent
complexes in transfer hydrogenation of acetophenone which indicated the base-activation of
iron(II)-N2P2 complexes in catalytic transfer hydrogenation.[116]
Scheme 28. Synthesis of neutral pentacoordinate Fe(II)-N2P2 complexes.
Morris also described a family of amine(imine)diphosphine chloro iron complexes for the
efficient asymmetric transfer hydrogenation of ketones. The use of 0.016-0.05 mol% of 78 and
0.033-0.4 mol% of KOtBu in i-PrOH permitted the reduction of acetophenone with ees up to

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
35
90% and TOFs up to 119 s–1 (Figure 13). Using the bromo analogue 79, ee up to 99% can be
reached with a catalyst/base/substrate ratio of 1:8:6121 in i-PrOH at 28 °C for 2-120 min. [117-118]
Figure 13. Amine(imine)diphosphine chloro and bromo iron complexes.
In transfer hydrogenation of ketones, Morris has shown that Fe(0) nanoparticles
functionalized with chiral and achiral PNNP ligands prepared from 68 and 69, respectively, can
be active catalysts in transfer hydrogenation of ketones; ee up to 64% were obtained with the
chiral Fe nanoparticles. Noticeably, the preparation of nanoparticles has a significant influence on
their activity.[119] It is important to underline that two structurally similar complexes can lead to
different catalytic species: the trans-[Fe(NCMe)CO(PPh2C6H4CH=NCHPh)2][BF4]2 precatalyst
68 generated Fe nanoparticles during the reduction, whereas the trans-
[Fe(CO)Br(PR2CH2CH=NCHPh)2][BF4] 80 remained homogeneous,[120] which underlined the
importance of a careful study of every catalytic systems.
Le Floch designed related iron complexes with tetradentate ligands bearing two
iminophosphorane moieties and two phosphines, thiophosphino, and phosphine oxide groups
(Figure 14).[121] Complexes [(FeCl2(P2N)] 81, [(FeCl2(O2N2)] 82 and [(FeCl2(N2)] 83, can be used
as precatalysts (0.1 mol%) with 4 mol% NaOiPr for 6-8 h in iPrOH: acetophenone can be reduced
at 82°C in 80-91% conversion.
Figure 14. Iminophosphorane diorganophosphorus iron complexes.
In the same manner as hydrogenation, using 0.5 mol% of an in situ generated catalyst
from the chiral macrocycle ligand L1 and Fe3(CO)12, the asymmetric transfer hydrogenation of
ketones was performed at 65 °C in i-PrOH in the presence of 12 mol% of KOH and 6 mol% of
NH4Cl with TOFs up to 1940 h–1, high conversions and excellent ees (both up to 99%) (Scheme
29). Recently, Mezzetti described iron complexes 84 bearing original C2-symmetric macrocyclic

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
36
P-chirogenic N2P2 ligands which were efficient in transfer hydrogenation of arylalkylketones at
75 °C in i-PrOH in the presence of 4 mol% of NaOt-Bu with ee up to 99%.[122-123]
Scheme 29. Macrocyclic ligand iron catalys for enantioselective TH of ketones.
In 2010 Royo et. al reported the use of tethered Cp-NHC iron complexes [(Cp*-
NHC)Fe(CO)]I complexes such as 27 in catalytic transfer hydrogenation of ketones such as
acetophenone, benzophenone and cyclohexanone using 2-propanol as hydrogen source in the
presence of 1 equiv. of KOH at 80 °C for 2-18 h.[66] Similarly, cationic [Fe(Cp)(NHC)(CO)2][I]
complexes 85-87 bearing 1,3-dialkylated N-heterocyclic carbene ligands were used as catalysts
for transfer hydrogenation of cyclohexanone (KOH, iPrOH, 82 °C, 9-11 h). The in-situ generated
active species (0.5 mol%) obtained from imidazolium salts and CpFe(CO)2I permitted the
reduction of several ketones in 21-99% conversions under similar conditions[124-125] (Scheme 30).
Scheme 30. NHC-Fe piano-stool for transfer hydrogenation of ketones.
In 2012, Funk synthesized air-stable, nitrile-ligated (cyclopentadienone) iron dicarbonyl
compounds and studied their activities as catalysts in the transfer hydrogenation of aldehydes and
ketones.
Scheme 31. Knölker type catalyst for for transfer hydrogenation.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
37
The acetonitrile complex 65 showed the best activity in the transfer hydrogenation of aldehydes
(2 mol% 65) and ketones (5 mol% 65) at 80 °C for 18 h.[126] (Scheme 31)
A series of chiral (cyclopentadienone)iron complexes such as 88 in combination with
trimethylamine N-oxide has been investigated by Wills and co-workers for the asymmetric
transfer hydrogenation of acetophenone. Using 10 mol% of 88 and 10 mol% of Me3NO in the
presence of formic acid and NEt3 (ratio 5:2) at 28 °C for 48 h, acetophenone can be reduced with
enantiomeric excesses up to 25%.[127] (Scheme 32)
Scheme 32. Chiral Knölker type complexes for catalyzed transfer hydrogenation reactions.
In 2013, Beller developed an efficient iron-based catalyst system (0.4 mol%) generated in
situ from Fe(BF4)2∙6H2O and P(CH2CH2PPh2)3 [tetraphos, (PP3)] in the presence of 1.1 equiv. of
HCO2H as the hydrogen source in THF at 60 °C for 2 h, for the highly selective transfer
hydrogenation of aromatic, aliphatic, and -unsaturated aldehydes with 96-99% yields.
Reducible functional groups such as chloro, bromo, ketone, ester and styryl were tolerated.[128]
(Scheme 33)
Scheme 33. Transfer hydrogenation of -unsaturated aldehydes.
In addition to hydrogenation, Hu et al reported the use of pincer PONOP iron complexes
such as 11 (5 mol%) in catalyzed chemoselective transfer hydrogenation of aldehydes in the
presence of 5 equiv. of HCOONa as hydrogen donor in methanol at 40 °C for 6 h. The
corresponding alcohols were obtained in 68-98% yields, and functional groups such as akenyl,
ester, nitro were tolerated. [34]

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
38
3. Conclusion In recent years, the substitution of expensive noble metals by earth abundant transition
metals such as iron or manganese is a hot topic in homogeneous catalysis. Despite a tremendous
progress has been developed with iron complexes in the field of reduction reactions, there is still
a continuing interest to advance a catalytic method in which the reaction works under milder
conditions with low catalyst loading to achieve high TON and TOF. Furthermore, the reduction
of carboxylic derivatives via hydrogenation is still a challenging task.
Unlike iron, the use of manganese based complexes is really less developed in the reduction area
except for the hydrosilylation of carbonyl derivatives. The development of efficient manganese
catalytic systems for reduction reactions including the hydrogenation of carbonyl and carboxylic
derivatives is a virgin territory.
4. References [1] S. D. Burke, R. L. Danheiser, in Handbook of Reagents for Organic Synthesis, Oxidising
and Reducing Agents, Wiley, New York, 1999. [2] J. Seyden-Penne, in Reductions by the Alumino- and Boro-hydrides in Organic Synthesis,
Vol. 105, 106, VCH, New York, 1991, p. 86−94. [3] H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner, M. Studer, Adv. Synth. Catal.
2003, 345, 103-151. [4] For representative recent reviews and books on iron catalysis, see: (a) C. Bolm, J. Legros,
J. L. Paih, L. Zani, Chem. Rev. 2004, 104, 6217-6254. b) B. Plietker (ed.), Iron Catalysis in Organic Chemistry, Wiley-VCH Verlag,Weinheim, 2008, pp 125-143; (c) E. B. Bauer (ed.), iron Catalysis II Topics in Organomet. Chem. 2015; (d) C. L. Sun, B. J. Li, Z. J. Shi, Chem. Rev. 2011, 111, 1293-1314; (e) K. Gopalaiah, Chem. Rev. 2013, 113, 3248-3296; (f) K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann, F. E. Kühn, Chem. Rev. 2014, 114, 5215-5272; (g) I. Bauer, H.-J. Knölker, Chem. Rev. 2015, 115, 3170-3387. For representative recent reviews on manganese catalysis, see: (h) D. A. Valyaev, G. Lavigne, N. Lugan, Coord. Chem. Rev. 2016, 308, 191-235; (i) R. I. Khusnutdinov, A. R. Bayguzina, U. M. Dzhemilev, Russ. J. Org. Chem. 2012, 48, 309-348; (j) J. R. Carney, B. R. Dillon, S. P. Thomas, Eur. J. Org. Chem. 2016, 3912-3929.
[5] P. N. Rylander, in Catalytic Hydrogenation in Organic Synthesis, Acadamic Press, New York, 1979.
[6] A. M. Smith, R. Whyman, Chem. Rev. 2014, 114, 5477-5510. [7] P. A. Dub, T. Ikariya, ACS Catal. 2012, 2, 1718−1741.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
39
[8] J. Pritchard, G. A. Filonenko, R. V. Putten, E. J. Hensen, E. A. Pidko, Chem. Soc. Rev. 2015, 44, 3808-3833.
[9] J. G. de Vries, C. J. Elsevier, Eds., in Handbook of Homogeneous Hydrogenation, Wiley, New York, 2006.
[10] E. N. Jacobsen, A. Pfaltz, H. Y. (eds), in Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999.
[11] H. U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner, M. Studer, Adv. Synth. Catal. 2003, 345, 103-151.
[12] F. Naud, F. Spindler, C. J. Rueggeberg, A. T. Schmidt, H. U. Blaser, Org. Process Res. Dev. 2007, 11, 519-523.
[13] K. Junge, K. Schroder, M. Beller, Chem. Commun. 2011, 47, 4849-4859. [14] I. Bauer, H.-J. Knölker, Chem. Rev. 2015, 115, 3170-3387. [15] S. Gaillard, J.-L. Renaud, ChemSusChem 2008, 1, 505-509. [16] L. Markó, M. A. Radhi, I. Ötvös, J. Organomet. Chem. 1981, 218, 369-376. [17] L. Markó, J. Palágyi, Trans. Met. Chem. 1983, 8, 207. [18] a) H.-J. Knölker, J. Heber, C. H. Mahler, Synlett, 1992, 1002-1004; (b) H.-J. Knölker, J.
Heber, Synlett, 1993, 924-926; (c) H.-J. Knölker, E. Baum, R. Klauss, Tetrahedron Lett. 1995, 36, 7647; (d) H.-J. Knölker, H. Goesmann, R. Klauss, Angew. Chem. Int. Ed. 1999, 38, 702-705; (e) H.-J. Knölker, E. Baum, H. Goesmann, R. Klauss, Angew. Chem. Int. Ed. 1999, 38, 2064-2066.
[19] B. L. Conley, M. K. Pennington-Boggio, E. Boz, T. Williams, Chem. Rev. 2010, 110, 2294-2312.
[20] C. P. Casey, H. Guan, J. Am. Chem. Soc. 2007, 129, 5816-5817. [21] C. P. Casey, H. Guan, J. Am. Chem. Soc. 2009, 131, 2499-2507. [22] A. Berkessel, S. Reichau, A. von der Höh, N. Leconte, J. r.-M. Neudörfl, Organometallics
2011, 30, 3880-3887. [23] P. Gajewski, M. Renom-Carrassco, S. Vailati Facchini, L. Pignatoro, L. Lefort, J. G. de
Vries, R. Ferraccioli, A. Forni, U. Piarulli, C. Gennari, Eur. J. Org. Chem. 2015, 1887-1893.
[24] D. S. Mérel, M. Elie, J.-F. Lohier, S. Gaillard, J.-L. Renaud, ChemCatChem 2013, 5, 2939-2945.
[25] S. Fleischer, S. Zhou, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2013, 52, 5120-5124. [26] C. Sui-Seng, F. N. Haque, A. Hadzovic, A.-M. Pütz, V. Reuss, N. Meyer, A. J. Lough, M.
Zimmer-De Iuliis, R. H. Morris, Inorg. Chem. 2009, 48, 735-743. [27] R. H. Morris, Acc. Chem. Res. 2015, 48, 1494-1502. [28] R. H. Morris, Chem. Soc. Rev. 2009, 38, 2282-2291. [29] G. Wienhçfer, F. A. Westerhaus, K. Junge, R. Ludwig, M. Beller, Chem. Eur. J. 2013, 19,
7701-7707. [30] (a) J. Zhang, G. Leitus, Y. Ben-David, D. Milstein, J. Am. Chem. Soc. 2005, 127, 10840-
10841; (b) J. Zhang, G. Leitus, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2006, 45, 1113-1115; (c) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon, D. Milstein, J.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
40
Am. Chem. Soc. 2010, 132, 16756-16758; (d) C. Gunanathan, D. Milstein, Acc. Chem. Res. 2011, 44, 588-602.
[31] R. Langer, G. Leitus, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2011, 50, 2120 -2124.
[32] R. Langer, M. A. Iron, L. Konstantinovski, Y. Diskin-Posner, G. Leitus, Y. Ben-David, D. Milstein, Chem. Eur. J. 2012, 18, 7196-7209.
[33] T. Zell, Y. Ben-David, D. Milstein, Catal. Sci. Technol. 2015, 5, 822-826. [34] S. Mazza, R. Scopelliti, X. Hu, Organometallics 2015, 34, 1538-1545. [35] N. Gorgas, B. Stöger, L. F. Veiros, E. Pittenauer, G. Allmaier, K. Kirchner,
Organometallics 2014, 33, 6905-6914. [36] N. Gorgas, B. Stöger, L. F. Veiros, K. Kirchner, ACS Catal. 2016, 6, 2664-2672. [37] P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough, R. H. Morris, J.
Am. Chem. Soc. 2014, 136, 1367-1380. [38] J. F. Sonnenberg, A. J. Lough, R. H. Morris, Organometallics 2014, 33, 6452-6465. [39] Y. Li, S. Yu, X. Wu, J. Xiao, W. Shen, Z. Dong, J. Gao, J. Am. Chem. Soc. 2014, 136,
4031-4039. [40] L.-Q. Lu, Y. Li, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2013, 52, 8382-8386. [41] S.Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K.
Junge, M. Beller, J. Am. Chem. Soc. 2016, 138, 8809-8814. [42] F. Kallmeier, T. Irrgang, T. Dietel, R. Kempe, Angew.Chem. Int. Ed. 2016, 55, 11806-
11809. [43] M. Zhanga, A. Zhangb, Appl. Organometal. Chem. 2010, 24, 751-757. [44] H. Brunner, K. Fisch, Angew. Chem. Int. Ed. Engl. 1990, 29, 1131-1132. [45] H. Nishiyama, A. Furuta, Chem. Commun. 2007, 760-762. [46] A. Furuta, H. Nishiyama, Tetrahedron Lett. 2008, 49, 110-113. [47] T. Inagaki, L. T. Phong, A. Furuta, J. Ito, H. Nishiyama, Chem. Eur. J. 2010, 16, 3090-
3096. [48] N. S. Shaikh, K. Junge, M. Beller, Org. Lett. 2007, 9, 5429-5432. [49] D. Addis, N. Shaikh, S. Zhou, S. Das, K. Junge, M. Beller, Chem. Asian J. 2010, 5, 1687-
1691. [50] N. S. Shaikh, S. Enthaler, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2008, 47, 2497-
2501. [51] A. P. Dieskau, J.-M. Begoui, B. Plietker, Eur. J. Org. Chem. 2011, 5291-5296. [52] D. Bèzier, J.-B. Sortais, C. Darcel, Adv. Synth. Catal. 2013, 355, 19-33. [53] K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann, F. E. Kühn,
Chem. Rev. 2014, 114, 5215-5272. [54] E. Buitrago, L. Zani, H. Adolfsson, Appl. Organomet. Chem. 2011, 25, 748-752. [55] E. Buitrago, F. Tinnis, H. Adolfsson, Adv. Synth. Catal. 2012, 354, 217-222. [56] K. Muller, A. Schubert, T. Jozak, A. Ahrens-Botzong, V. Schunemann, W. R. Thiel,
ChemCatChem 2011, 3, 887-892. [57] J. Yang, T. D. Tilley, Angew. Chem. Int. Ed. 2010, 49, 10186-10188.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
41
[58] A. J. Ruddy, C. M. Kelly, S. M. Crawford, C. A. Wheaton, O. L. Sydora, B. L. Small, M. Stradiotto, L. Turculet, Organometallics 2013, 32, 5581-5588.
[59] Z. Zuo, H. Sun, L. Wang, X. Li, Dalton Trans. 2014, 43, 11716-11722. [60] L. Wang, H. Sun, X. Li, Eur. J. Inorg. Chem. 2015, 2732-2743. [61] B. Xue, H. Sun, X. Li, RSC Adv. 2015, 5, 52000-52006. [62] L. C. Misal Castro, D. Bèzier, J.-B. Sortais, C. Darcel, Adv. Synth. Catal. 2011, 353,
1279-1284. [63] H. Brunner, K. Fisch, J. Organomet. Chem. 1991, 412, C11-C13. [64] H. Brunner, M. Rötzer, J. Organomet. Chem. 1992, 425, 119-124. [65] J. Zheng, L. C. Misal Castro, T. Roisnel, C. Darcel, J.-B. Sortais, Inorg. Chim. Acta 2012,
380, 301-307. [66] (a) V. V K. M. Kandepi, J. M. S. Cardoso, E. Peris, B. Royo, Organometallics 2010, 29,
2777-2782; (b) J. M. S. Cardoso, A. Fernandes, B. de P. Cardoso, M. D. Carvalho, L. P. Ferreira, M. J. Calhorda, B. Royo, Organometallics 2014, 33, 5670-5677.
[67] F. Jiang, D. Bézier, J.-B. Sortais, C. Darcel, Adv. Synth. Catal. 2011, 353, 239-244. [68] D. Bézier, F. Jiang, T. Roisnel, J.-B. Sortais, C. Darcel, Eur. J. Inorg. Chem. 2012, 2012,
1333-1337. [69] V. Cesar, L. C. Misal Castro, T. Dombray, J.-B. Sortais, C. Darcel, S. Labat, K. Miqueu,
J.-M. Sotiropoulos, R. Brousses, N. Lugan, G. Lavigne, Organometallics 2013, 32, 4643-4655.
[70] S. Demir, Y. Gökçe, N. Kaloğlu, J.-B. Sortais, C. Darcel, İ. Özdemira, Appl. Organometal. Chem. 2013, 27, 459-464.
[71] D. Kumar, A.P. Prakasham, L. P. Bheeter, J.-B. Sortais, M. Gangwar, T. Roisnel, A. C. Kalita, C. Darcel, P. Ghosh, J. Oganomet. Chem. 2014, 762, 81-87.
[72] T. Hashimoto, S. Urban, R. Hoshino, Y. Ohki, K. Tatsumi, F. Glorius, Organometallics 2012, 31, 4474−4479.
[73] S. Warratz, L. Postigo, B. Royo, Organometallics 2013, 32, 893−897. [74] F. S. Wekesa, R. Arias-Ugarte, L. Kong, Z. Sumner, G. P. McGovern, M. Findlater,
Organometallics 2015, 34, 5051-5056. [75] K. Zhu, M. P. Shaver, S. P. Thomas, Eur. J. Org. Chem. 2015, 2119-2123. [76] A. M. Tondreau, E. Lobkovsky, P. J. Chirik, Org. Lett. 2008, 10, 2789-2792. [77] P. Bhattacharya, J. A. Krause, H. Guan, Organometallics 2011, 30, 4720-4729. [78] S. Wu, X. Li, Z. Xiong, W. Xu, Y. Lu, H. Sun, Organometallics 2013, 32, 3227-3237. [79] H. Zhao, H. Sun, X. Li, Organometallics 2014, 33, 3535-3539. [80] A. M. Tondreau, J. M. Darmon, B. M. Wile, S. K. Floyd, E. Lobkovsky, P. J. Chirik,
Organometallics 2009, 28. 3928-3940. [81] S. Hosokawa, J.-I Ito, H. Nishiyama, Organometallics 2010, 29, 5773-5775. [82] Z. Zuo, L. Zhang, X. Leng, Z. Huang, Chem. Commun. 2015, 51, 5073-5076. [83] T. Inagaki, A. Ito, J.- i Ito, H. Nishiyama, Angew. Chem. Int. Ed. 2010, 49, 9384-9387. [84] B. K. Langlotz, H. Wadepohl, L. H. Gade, Angew. Chem. Int. Ed. 2008, 47, 4670-4674. [85] T. Bleith, H. Wadepohl, L. H. Gade, J. Am. Chem. Soc. 2015, 137, 2456-2459.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
42
[86] S. Zhou, K. Junge, D, Addis, S. Das, M. Beller, Angew. Chem. 2009, 121, 9671-9674. [87] Y. Sunada, H. Kawakami, T. Imaoka, Y. Motoyama, H. Nagashima, Angew. Chem. Int.
Ed. 2009, 48, 9511-9514. [88] H. Tsutsumi, Y. Sunada, H. Nagashima, Chem. Commun. 2011, 47, 6581-6583. [89] D. Bézier, G. T. Venkanna, J.-B. Sortais, C. Darcel, ChemCatChem 2011, 3, 1747-1750. [90] A. Volkov, E. Buitrago, H. Adolfsson, Eur. J. Org. Chem. 2013, 2066-2070. [91] S. Zhou, D. Addis, S. Das, K. Junge, M. Beller, Chem. Commun. 2009, 4883-4885. [92] S. Das, B. Wendt, K. Moller, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2012, 51, 1662-
1666. [93] D. Bézier, G. T. Venkanna, L. C. Misal Castro, J. Zheng, T. Roisnel, J.-B. Sortais, C.
Darcel, Adv. Synth. Catal. 2012, 354, 1879-1884. [94] K. Junge, B. Wendt, S. Zhou, M. Beller, Eur. J. Org. Chem. 2013, 2061-2065. [95] S. Das, Y. Li, K. Junge, M. Beller, Chem. Commun. 2012, 48, 10742-10744. [96] H. Li, L. C. Misal Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais, C. Darcel,
Angew. Chem. Int. Ed. 2013, 52, 8045-8049. [97] L. C. Misal Castro, H. Li, J.-B. Sortais, C. Darcel, Chem. Commun. 2012, 48, 10514-
10516. [98] R. L. Yates, J. Catal., 1982, 78, 111-115. [99] P. K. Hanna, B. T. Gregg, A. R. Cutler, Organometallics 1991, 10, 31-33. [100] S. U. Son, S.-J. Paik, I. S. Lee, Y.-A. Lee, Y. K. Chung, W. K. Seok, H. N. Lee,
Organometallics 1999, 18, 4114-4118. [101] S. U. Son, S.-J. Paik, Y. K. Chung, J. Mol. Catal. A, 2000, 151, 87-90. [102] J. Zheng, S. Elangovan, D. A. Valyaev, R. Brousses, V. César, J.-B. Sortais, C. Darcel, N.
Luganb, G. Lavigne, Adv. Synth. Catal. 2014, 356, 1093-1097. [103] V. K. Chidara, G. Du, Organometallics 2013, 32, 5034-5037. [104] T. K. Mukhopadhyay, M. Flores, T. L. Groy, R. J. Trovitch, J. Am. Chem. Soc. 2014, 136,
882−885. [105] C. Ghosh, T. K. Mukhopadhyay, M. Flores, T. L. Groy, R. J. Trovitch, Inorg. Chem.
2015, 54, 10398-10406. [106] P. Magnus, M. R. Fielding, Tetrahedron Lett. 2001, 42, 6633-6636. [107] Z. Mao, B. T. Gregg, A. R. Cutler, J. Am. Chem. Soc. 1995, 117, 10139-10140. [108] J. Zheng, S. Chevance, C. Darcel, J.-B. Sortais, Chem. Commun. 2013, 49, 10010-10012. [109] K. Jothimony, S. Vancheesen, J. Mol. Catal. 1989, 52, 301-304. [110] K. Jothimony, S. Vancheesen, J. C. Kuriacose, J. Mol. Catal. 1985, 32, 11-16. [111] S. Enthaler, B. Hagemann, G. Erre, K. Junge, M. Beller, Chem. Asian J. 2006, 1, 598-604. [112] S. Enthaler, G. Erre, M. K. Tse, K. Junge, M. Beller, Tetrahedron Lett. 2006, 47, 8095-
8099. [113] J. S. Chen, L. L. Chen, Y. Xing, G. Chen, W. Y. Shen, Z. R. Dong, Y. Y. Li, J. X. Gao,
Huaxue Xuebao 2004, 62, 1745-1750. [114] N. Meyer, A. J. Lough, R. H. Morris, Chem. Eur. J. 2009, 15, 5605-5610.

Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
43
[115] (a) A. A. Mikhailine, A. J. Lough, R. H. Morris, J. Am. Chem. Soc. 2009, 131, 1394-1395; (b) P. O. Lagaditis, A. J. Lough, R. H. Morris, Inorg. Chem. 2010, 49, 10057-10066; (c) A. A. Mikhailine, R. H. Morris, Inorg. Chem. 2010, 49, 11039-11044; (d) P. E. Sues, A. J. Lough, R. H. Morris, Organometallics 2011, 30, 4418-4431; (e) A. A. Mikhailine, E. Kim, C. Dingels, A. J. Lough, R. H. Morris, Inorg. Chem. 2008, 47, 6587-6589.
[116] P. O. Lagaditis, A. J. Lough, R. H. Morris, J. Am. Chem. Soc. 2011, 133, 9662-9665. [117] W. Zuo, A. J. Lough, Y. F. Li, R. H. Morris, Science 2013, 342, 1080-1083. [118] W. Zuo, S. Tauer, D. E. Demyan, E. Prokopchuk, R. H. Morris, Organometallics 2014,
33, 5791-5801. [119] J. F. Sonnenberg, N. Coombs, P. A. Dube, R. H. Morris, J. Am. Chem. Soc. 2012, 134,
5893-5899. [120] J. F. Sonnenberg, R. H. Morris, Catal. Sci. Technol. 2014, 4, 3426-3438. [121] A. Buchard, H. Heuclin, A. Auffrant, X. F. Le Goff, P. L. Floch, Dalton Trans. 2009,
1659-1667. [122] R. Biegler, R. Huber, A. Mezzetti, Angew. Chem. Int. Ed. 2015, 54, 5171-5174. [123] R. Biegler, R. Huber, A. Mezzetti, Synlett 2016, 831-847. [124] M. D. Bala, M. I. Ikhile, J. Mol. Catal. A, Chem. 2014, 385, 98-105. [125] P. Das, T. Elder, W.W. Brennessel, S. C. Chmely, RSC Adv. 2016, 6, 88050-88056. [126] T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal. 2012, 354, 597-601. [127] J. P. Hopewell, J. E. D. Martins, T. C. Johnson, J. Godfrey, M. Wills, Org. Biomol. Chem.
2012, 10, 134-145. [128] G. Wienhöfer, F.A. Westerhaus, K. Junge, M. Beller, J. Organomet. Chem. 2013, 744,
156-159.

44
Part 2
Synthesis and catalytic applications
of Knölker’s NHC complexes

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
45

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
46
1. Introduction The catalytic properties of metal complexes can be finely tuned by the modulation of the
electronic and steric properties of the ligands.[1] During the last decades, bifunctional catalysts,
which combine a metal center and a non-innocent ligand working together in close partnership,
have been proven as a powerful tool in molecular synthesis. These non-innocent ligands linked to
the metal center are then crucial to finely adjust the reactivity and the catalytic activity of metal
complexes.[2] As a representative example, the ruthenium cyclopentadienone complex, so-called
Shvo complex 1, was widely applied in redox reaction, hydrogen borrowing, dynamic kinetic
resolution and cascade reactions.[3-4] Nevertheless, even if ruthenium based catalysts were useful
in such transformations, the limited availability of precious transition metals, their rather high
price and their toxicity diminish their attractiveness for future use, and thus more earth abundant,
inexpensive and environmentally friendly metal based alternatives have to be found.[5] In this
perspective, iron fits well due to its huge abundance, low toxicity and attractive low cost.
Therefore the preparation of well-defined iron-based catalysts with comparable activity became
desirable for the development of more sustainable reduction reactions.
Figure 1. Analogy between Ru Shvo and Fe Knölker complexes.
2. Discovery of iron cyclopentadienone complexes and their applications in
catalysis The first tricarbonyl (4-cyclopentadienone)iron complexes 3 have been synthesized in
the 50’s by the reaction of Fe(CO)5 with a diyne[6-7] (Scheme 1) and studied in depth by Knölker
in the 90’s [8-11] Interestingly, Knölker and co-workers were able to isolate the first iron hydride

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
47
hydroxycyclopentadienyl complex resulting of a Hieber-type reaction[12] (Scheme 2) which is
structural analogue to the Shvo complex 1 (Figure 1). Advantageously, complex 3 is stable in air
for months and can even be purified by column chromatography, by contrast with the hydrido
complex 2 which slowly decomposes under aerobic conditions.
Scheme 1. Synthesis of tricarbonyl (4-cyclopentadienone)iron complexes 3.
Scheme 2. Synthesis of Knölker's iron hydride complex 2.
In 2007, a significant breakthrough was made by Casey and Guan who discovered the
potential application of the Knölker complex in reduction area.[13,14] Knolker’s iron hydrido
complex 2 has been demonstrated to be an efficient catalyst for the chemoselective hydrogenation
of aldehydes, ketones, and imines in toluene at room temperature under moderate 3 atm hydrogen
pressure (Scheme 3).
Scheme 3. First hydrogenation of ketones and imine with Knolker’s iron complex 2.
Interestingly, non-conjugated alkenyl, alkynyl, halides, nitro, epoxides, and ester
functional groups were tolerated under these conditions.
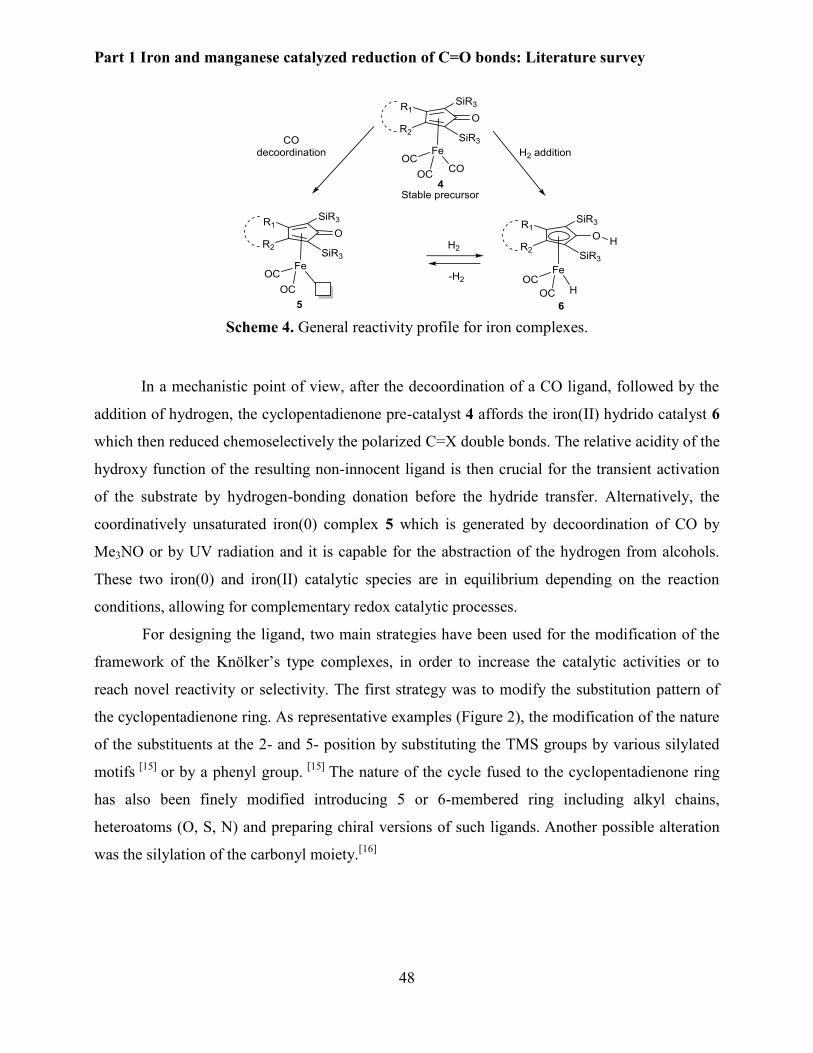
Part 1 Iron and manganese catalyzed reduction of C=O bonds: Literature survey
48
Scheme 4. General reactivity profile for iron complexes.
In a mechanistic point of view, after the decoordination of a CO ligand, followed by the
addition of hydrogen, the cyclopentadienone pre-catalyst 4 affords the iron(II) hydrido catalyst 6
which then reduced chemoselectively the polarized C=X double bonds. The relative acidity of the
hydroxy function of the resulting non-innocent ligand is then crucial for the transient activation
of the substrate by hydrogen-bonding donation before the hydride transfer. Alternatively, the
coordinatively unsaturated iron(0) complex 5 which is generated by decoordination of CO by
Me3NO or by UV radiation and it is capable for the abstraction of the hydrogen from alcohols.
These two iron(0) and iron(II) catalytic species are in equilibrium depending on the reaction
conditions, allowing for complementary redox catalytic processes.
For designing the ligand, two main strategies have been used for the modification of the
framework of the Knölker’s type complexes, in order to increase the catalytic activities or to
reach novel reactivity or selectivity. The first strategy was to modify the substitution pattern of
the cyclopentadienone ring. As representative examples (Figure 2), the modification of the nature
of the substituents at the 2- and 5- position by substituting the TMS groups by various silylated
motifs [15] or by a phenyl group. [15] The nature of the cycle fused to the cyclopentadienone ring
has also been finely modified introducing 5 or 6-membered ring including alkyl chains,
heteroatoms (O, S, N) and preparing chiral versions of such ligands. Another possible alteration
was the silylation of the carbonyl moiety.[16]
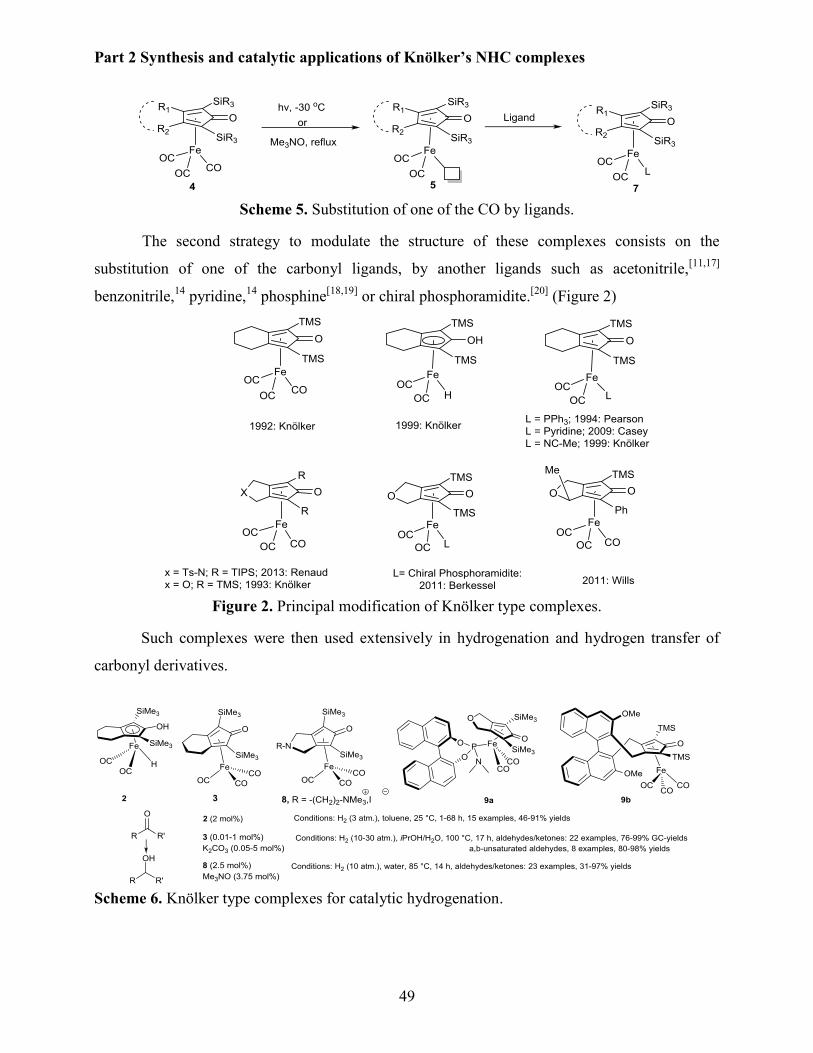
Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
49
Scheme 5. Substitution of one of the CO by ligands.
The second strategy to modulate the structure of these complexes consists on the
substitution of one of the carbonyl ligands, by another ligands such as acetonitrile,[11,17]
benzonitrile,14 pyridine,14 phosphine[18,19] or chiral phosphoramidite.[20] (Figure 2)
Figure 2. Principal modification of Knölker type complexes.
Such complexes were then used extensively in hydrogenation and hydrogen transfer of
carbonyl derivatives.
Scheme 6. Knölker type complexes for catalytic hydrogenation.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
50
Beller developed a series of air-stable Knölker type complexes such as 3 which catalyzed
the hydrogenation of aldehydes and ketones under 30 atm of H2 at 100 °C in a mixture of
i-PrOH/H2O, with a good functional group tolerance (esters, amides and heterocycles). More
impressively, α,β-unsaturated aldehydes were selectively reduced to the corresponding allylic
alcohols in high yields.[21] Under water-gas shift conditions (10 atm of CO in DMSO/H2O, 100
°C), 3 can also catalyze the reduction of aldehydes.[22]
Renaud simultaneously described the use of Knölker type complexes with cationic fragments
such as ammonium salts 8 for the catalytic hydrogenation of aldehydes and ketones at 85 °C in
H2O under 10 atm of H2.[23]
In 2011, using the modified Casey’s catalyst 9a in which one carbonyl ligand was substituted by
a chiral phosphoramidite ligand under UV-light irradiation,[24] Berkessel performed an
asymmetric hydrogenation of acetophenone, albeit only moderate ee (up to 31%) were obtained.
Recently, Gennari has developed new chiral cyclopentadienone iron complexes derived from (R)-
Binol 9b which can be used as catalyst (2 mol%) when treated with Me3NO (4 mol%) in the
asymmetric hydrogenation of ketones leading to the corresponding alcohols with ee up 77% (22-
99% conversion, 30 bar H2, i-PrOH/H2O (5:2), 70 °C, 18 h).[25]
Funk developed a family of air-stable, nitrile-ligated Knölker type complexes for transfer
hydrogenation. The acetonitrile complex 10 showed the best activity in the TH of aldehydes (2
mol% 10) and ketones (5 mol% 10) at 80 °C for 18 h. Noticeably, 10 exhibited similar activities
to the air-sensitive iron hydride complex 2 (1 mol%, 75 °C, 16 h, Scheme 7).[26]
Scheme 7. Knölker type catalyst for transfer hydrogenation.
These Knölker type complexes can be also useful in reduction of imines. Indeed, Beller
described the first iron-catalyzed hydrogenation of imines to amines,[27] using the resulting
catalyst from the in situ association of the Knölker complex 2 and the chiral phosphoric Brönsted

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
51
acid 11. Thus, the hydrogenation of numerous N-arylketimines led to the corresponding amines in
60-94% yields and 67-98% ees (Scheme 8).
Scheme 8. Knölker complex 2 / chiral phosphoric Brönsted acid 11-12 for the catalytic
asymmetric hydrogenation of C=N bonds.
Using the same in situ generated catalytic system (3-5 mol% of 2 and 1-2 mol% of 11-12), the
enantioselective hydrogenation of quinoxalines and 2H-1,4-benzoxamines led to the
tetrahydroquinoxalines and 3,4-dihydro-2H-1,4-benzoxamines with enantiomeric ratios up to
97:3 and 87:13, respectively (Scheme 8).[28]
Noticeably, Renaud has shown that the water soluble Knölker type complex 8 (Scheme 6, 2.5
mol%) in the presence of Me3NO (3.75 mol%) can perform the reduction of imines under 10 bar
of H2 in water at 100 °C for 24 h (yields 91-98%).[29]
The reduction of imines can also be performed by transfer hydrogenation (TH) reaction.
Indeed, Zhao succeeded to reduce N-aryl and N-alkyl imines by TH using a catalytic combination
of two iron species, Funk type complex 13 (5 mol%) and Fe(acac)3 (10 mol%). Thus, at 110 °C
for 48 h in iPrOH in the absence of a base, both aryl and alkyl ketimines were reduced in 38-99%
yields (Scheme 9). Notably the crucial role of Fe(acac)3 as a Lewis acid was underlined: in its
absence, at 100 °C, only 9% conversion was observed.[30]
Scheme 9. Knölker type catalysts for TH of imines.

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
52
Knölker type complexes were also efficient for direct reductive amination (DRA) of carbonyl
derivatives: using 5 mol% of 3 and 5 mol% of Me3NO, under 5 bar of H2 at 85 °C in EtOH,
aldehydes reacted with alkylamines leading to the corresponding alkylated amines in 38-94%
yields. A slight adaptation of the conditions permitted the reaction with ketones, as it was
conducted in MeOH with a catalytic amount of NH4PF6 (Scheme 10).[31]
Scheme 10. Knölker type complexes for catalysed DRA.
DRA reactions can also be conducted starting from primary alcohols using a hydrogen
borrowing catalysis methodology at rather high temperatures (Scheme 11). In a first report by
Feringa and Barta in 2014, the Knölker complex 3 (5 mol%) in the presence of 10 mol% of
Me3NO catalysed the autotransfer hydrogenation of numerous primary alkyl alcohols or diols
with aniline and benzylamine derivatives in cyclopentyl methyl ether at 120-140 °C leading to
the corresponding amines with yields up to 95%.[32] This reaction can be extended to allylic
alcohols leading to allylic amines without isomerization of the C=C bond.[33] Afterwards, Wills
used the modified Knölker complex 15 (10 mol%) associated to 10 mol% of Me3NO which
exhibited similar activity in toluene or xylene at 110-140 °C.[34] Concomitantly, Zhao reported
that Funk type complex 13 (10 mol%) in the presence of 40 mol% of AgF was particularly
efficient for the DRA of anilines with both primary and secondary alcohols.[35]
Scheme 11. Iron-catalyzed DRA reaction via hydrogen borrowing catalysis.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
53
Surprisingly, even if N-heterocyclic carbene (NHC) ligands are widely applied and well-
studied in organometallic chemistry, and in particular with iron (See part 1),[36-39] up to now,
Knölker’s complexes bearing a N-heterocyclic carbene (NHC) as a ligand have not been
described. Notably, these neutral two-electron donor ligands show pronounced σ-donor ability
with only little to no π-back-bonding, which permits to form strong bonds with the metal center
then making the corresponding complexes highly resistant toward decomposition.
The huge development of both Knölker type iron derivatives and NHC iron complexes
prompt us to develop new NHC substituted Knölker type complexes and test them in
hydrosilylation reactions.
3. Results and discussions
3.1 Preparation of the complexes Previously, two routes have been developed to introduce a phosphine or a nitrile by
exchange with a CO ligand of the Knölker complex 3, either (i) under thermal conditions in
refluxing Bu2O or acetone in the presence of Me3NO or (ii) under photochemical conditions
using U.V. irradiation. To introduce the N-heterocyclic carbene ligand in the coordination sphere
of the iron complex, we have chosen the photochemical pathway based on our experience in the
photochemistry of iron carbonyl complexes.[40,41] As a model reaction, a solution of 3 with the
free 1,3-dimesitylimidazol-2-ylidene carbene (IMes) (obtained in situ by deprotonation of the
corresponding chloride salt, IMes.HCl with potassium hexamethyldisilazide (KHMDS), was
irradiated under UV (350 nm) in toluene for 20 h at room temperature leading to the expected
ligand exchange of one CO by the N-heterocylic carbene ligand (Scheme 12).
Scheme 12. General synthesis of the complexes.
During the course of the reaction, the color of solution turned from brown to dark-deep
yellow. The 1H-NMR of the crude mixture indicated a conversion of 90% of 3 to the expected

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
54
complex C1 (10% of unreacted 3 was also detected). Interestingly, the complex was purified by
column chromatography on silica gel, under air, leading to the isolation of C1, as a golden yellow
solid, in 78% yield. It can also be pointed out that the reaction was performed on gram scale.
Figure 3. NHC-substituted Knölker complexes (isolated yield indicated under the structure).
The same procedure was applied for the preparation of complexes bearing various symmetrical
unsaturated NHC-ligand bearing respectively a methyl (IMe), an isopropyl (IPr), or a cyclohexyl
(ICy) substituent on each of the nitrogen atoms, leading to C2-C4 in good isolated yields (64-
84%) (Figure 3). It is noteworthy that the coordination of the saturated 1,3-dimesityl-4,5-
dihydroimidazol-2-ylidene (SIMes) N-heterocylic carbene on 3 also took place, as monitored by 1H NMR, but unfortunately this complex was not stable, and decomposed during the steps of
purification.
To synthesize unsaturated dissymmetric NHC ligands, we have followed the recent
procedure described by Baslé and Maudit[42] to prepare the N-mesityl-N-cyclohexylimidazolium
salt. In a similar fashion, starting from (R)-phenylethylamine and mesitylamine, a chiral
dissymmetric imidazolium chloride was prepared in moderate isolated yield (28%). Both ligands
have also been successfully coordinated to the iron center following the route described above
(C5 and C6 with 65 and 76% yield, respectively, Figure 3).
3.2 Characterization of the complexes All the prepared complexes have been fully characterized by classical techniques such 1H, 13C{1H} NMR, IR, X-Ray diffraction studies, cyclic voltammetry, HR-MS and elemental
analysis.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
55
3.2.1 NMR and IR studies The coordination of the N-heterocyclic carbene ligand is ascertained by 13C{1H} NMR,
where the carbene carbon atoms appear between 181.5 to 187.8 ppm. These chemical shifts are in
agreement with those of the carbene carbon of the analogue (NHC)Fe(CO)4 complexes.[43-45] In a
similar manner, the carbon of the carbonyl groups appear between 216.7 ppm and 219.3 ppm, in
accordance with the (NHC)Fe(CO)4 complexes. These signals are low-field shifted compared to
the CpFe(CO)2(NHC)+ cationic complexes, which display a chemical shift typically around 209-
210 ppm.[40,41,46] This is an agreement with the lower donating ability of the neutral
cyclopentadienone ligand compared to the anionic cyclopentadienide ligand. In the case of the
chiral complex C6, the two resonances appear for the diastereotopic CO ligands at 216.7 and
217.1 ppm. The 1H-NMR spectra of the complexes at room temperature are straightforward: all
of them show the presence of the cyclopentadienone ring and the NHC ligand.
The stretching frequencies of the carbonyl ligands in IR are reported in Table 1. Compared to the
non-substituted Knölker complex 3 (CO = 2061, 2053, 1987 cm-1, COav = 2033 cm-1),[24] or
substituted Knölker’s complexes with a benzonitrile (1971 cm-1),[14] a triphenylphosphine (1965
cm-1)[18] or an acetonitrile (1953.5 cm-1)[17] motif, the COav for all the complexes appear at lower
frequencies, which demonstrate the strong electron donating character of the NHC ligand. In
addition, the stretching frequencies were approximate to the reported TEP values for the
NHCs.[42],[47]
Table 1. Summary of relevant spectroscopic and electrochemical data
Complex CO
(cm-1)
COav
(cm-1)a
(V)
[Ep (mV)]b TEP value (cm-1)c
C1 1978, 1913 1945 0.11d [-] 2050.5 C2 1975, 1919 1947 0.16 [75] 2054.1 C3 1975, 1924 1949 0.12 [78] 2051.5 C4 1969,1907 1938 0.10 [93] 2049.7 C5 1980, 1919 1949 0.04 [108] 2050.8 C6 1973, 1915 1944 0.14d [91] -
a COav is the average value of the frequencies of the two CO-stretching bands (recorded in the solid state using an
ATR equipment). b Sample concentration: 1 mM, Bu4NPF6 (0.2 M) in CH2Cl2, v = 100 mV·s-1, potentials are
reported in V vs. [FeCp2]/[FeCp2]+, the internal decamethylferrocene standard showed a wave with Ep= 60-70 mV. c Tolman Electronic Parameters according to references 42 and 47. d measured at 800 mV.s-1.
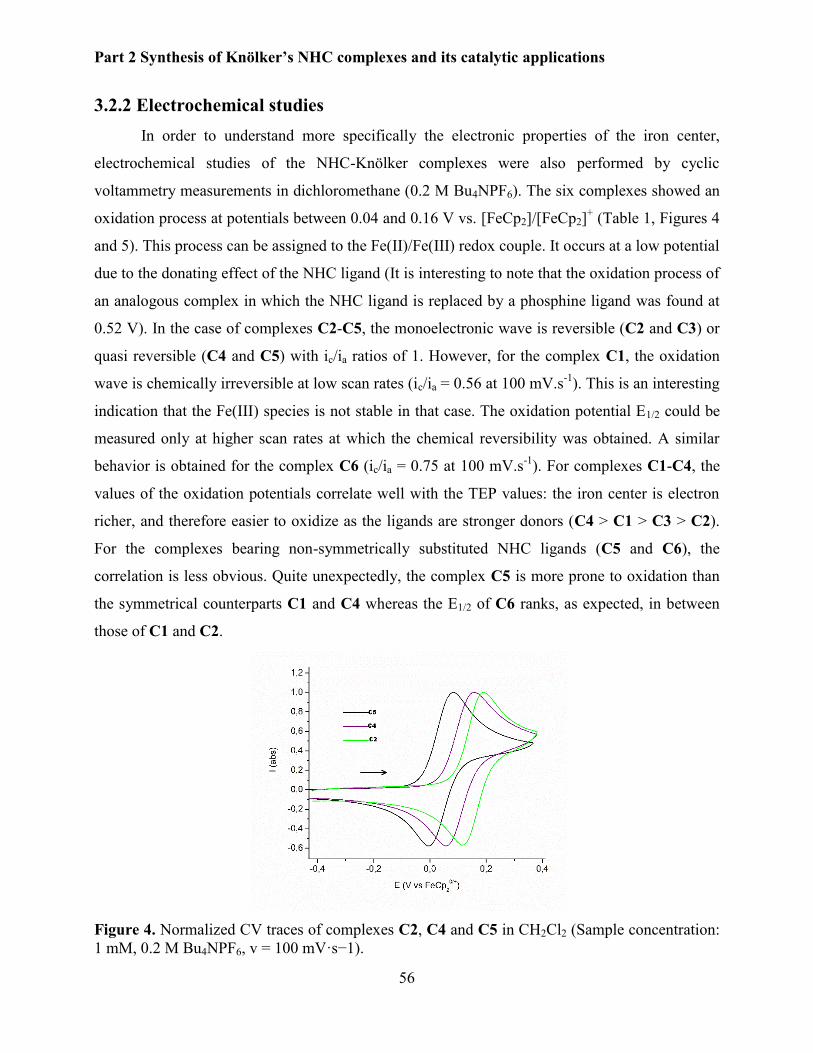
Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
56
3.2.2 Electrochemical studies In order to understand more specifically the electronic properties of the iron center,
electrochemical studies of the NHC-Knölker complexes were also performed by cyclic
voltammetry measurements in dichloromethane (0.2 M Bu4NPF6). The six complexes showed an
oxidation process at potentials between 0.04 and 0.16 V vs. [FeCp2]/[FeCp2]+ (Table 1, Figures 4
and 5). This process can be assigned to the Fe(II)/Fe(III) redox couple. It occurs at a low potential
due to the donating effect of the NHC ligand (It is interesting to note that the oxidation process of
an analogous complex in which the NHC ligand is replaced by a phosphine ligand was found at
0.52 V). In the case of complexes C2-C5, the monoelectronic wave is reversible (C2 and C3) or
quasi reversible (C4 and C5) with ic/ia ratios of 1. However, for the complex C1, the oxidation
wave is chemically irreversible at low scan rates (ic/ia = 0.56 at 100 mV.s-1). This is an interesting
indication that the Fe(III) species is not stable in that case. The oxidation potential E1/2 could be
measured only at higher scan rates at which the chemical reversibility was obtained. A similar
behavior is obtained for the complex C6 (ic/ia = 0.75 at 100 mV.s-1). For complexes C1-C4, the
values of the oxidation potentials correlate well with the TEP values: the iron center is electron
richer, and therefore easier to oxidize as the ligands are stronger donors (C4 > C1 > C3 > C2).
For the complexes bearing non-symmetrically substituted NHC ligands (C5 and C6), the
correlation is less obvious. Quite unexpectedly, the complex C5 is more prone to oxidation than
the symmetrical counterparts C1 and C4 whereas the E1/2 of C6 ranks, as expected, in between
those of C1 and C2.
Figure 4. Normalized CV traces of complexes C2, C4 and C5 in CH2Cl2 (Sample concentration: 1 mM, 0.2 M Bu4NPF6, v = 100 mV·s−1).
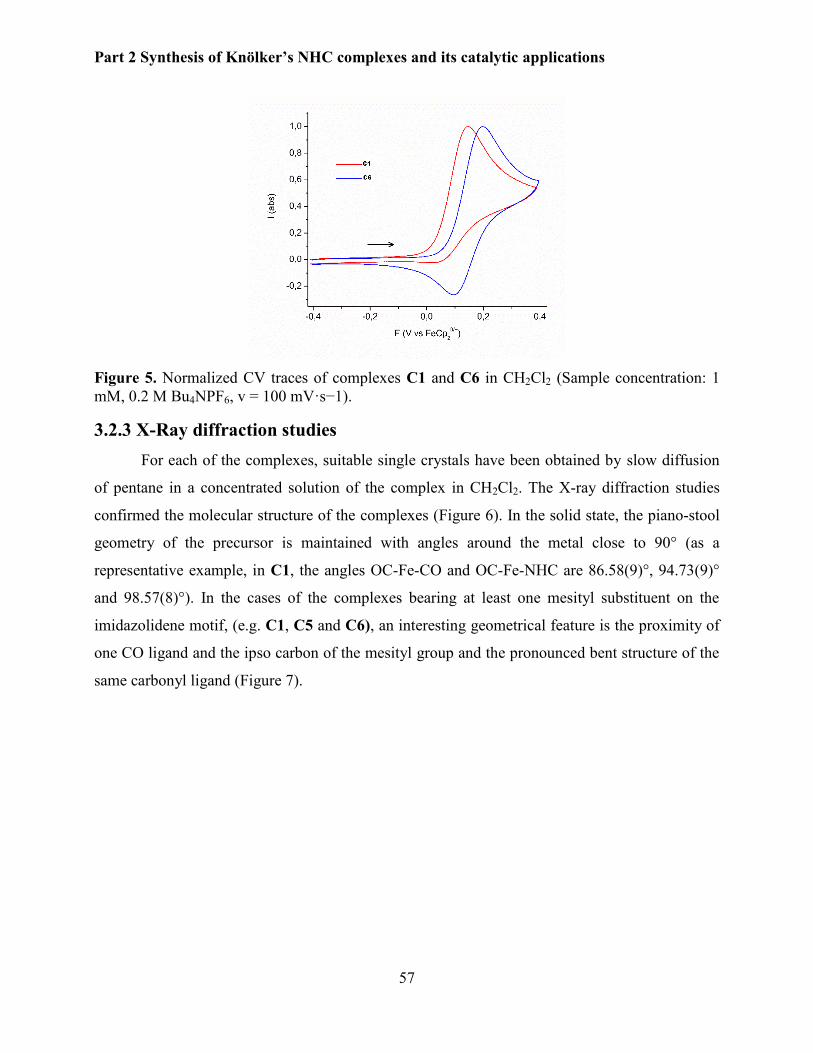
Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
57
Figure 5. Normalized CV traces of complexes C1 and C6 in CH2Cl2 (Sample concentration: 1 mM, 0.2 M Bu4NPF6, v = 100 mV·s−1).
3.2.3 X-Ray diffraction studies For each of the complexes, suitable single crystals have been obtained by slow diffusion
of pentane in a concentrated solution of the complex in CH2Cl2. The X-ray diffraction studies
confirmed the molecular structure of the complexes (Figure 6). In the solid state, the piano-stool
geometry of the precursor is maintained with angles around the metal close to 90° (as a
representative example, in C1, the angles OC-Fe-CO and OC-Fe-NHC are 86.58(9)°, 94.73(9)°
and 98.57(8)°). In the cases of the complexes bearing at least one mesityl substituent on the
imidazolidene motif, (e.g. C1, C5 and C6), an interesting geometrical feature is the proximity of
one CO ligand and the ipso carbon of the mesityl group and the pronounced bent structure of the
same carbonyl ligand (Figure 7).
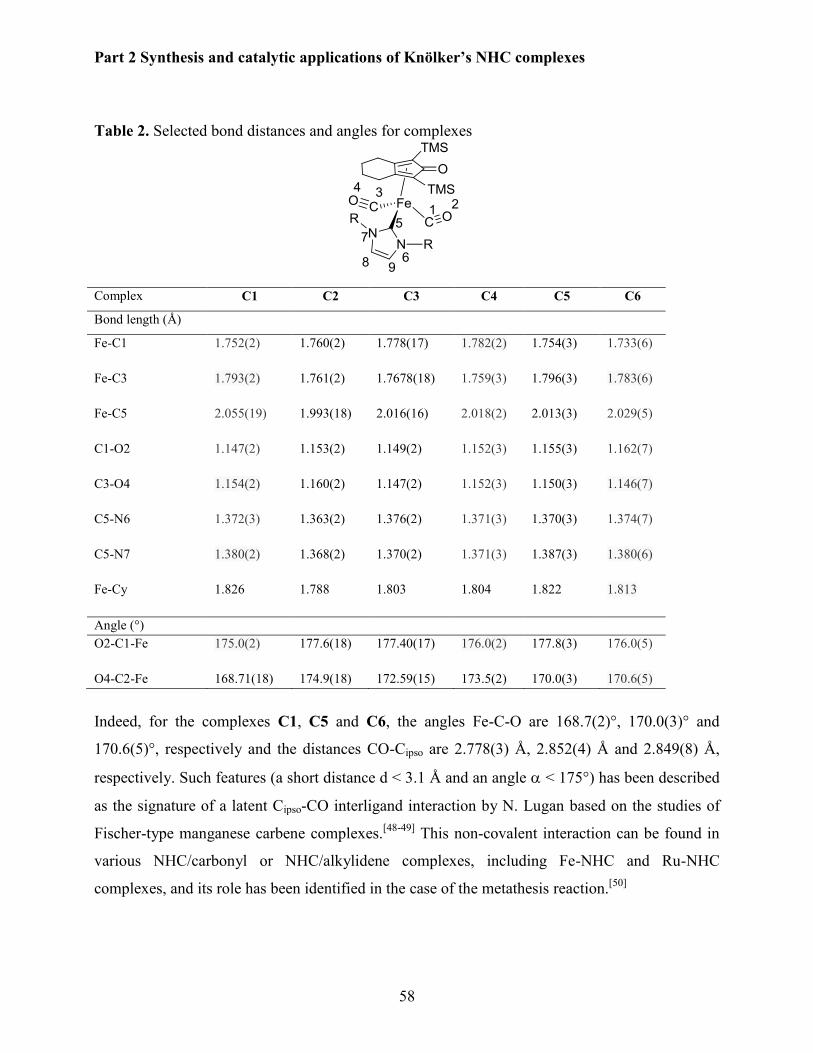
Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
58
Table 2. Selected bond distances and angles for complexes
Complex C1 C2 C3 C4 C5 C6
Bond length (Å) Fe-C1 1.752(2)
1.760(2) 1.778(17) 1.782(2)
1.754(3) 1.733(6)
Fe-C3 1.793(2)
1.761(2) 1.7678(18) 1.759(3)
1.796(3) 1.783(6)
Fe-C5 2.055(19)
1.993(18) 2.016(16) 2.018(2)
2.013(3) 2.029(5)
C1-O2 1.147(2)
1.153(2) 1.149(2) 1.152(3)
1.155(3) 1.162(7)
C3-O4 1.154(2)
1.160(2) 1.147(2) 1.152(3)
1.150(3) 1.146(7)
C5-N6 1.372(3)
1.363(2) 1.376(2) 1.371(3)
1.370(3) 1.374(7)
C5-N7 1.380(2)
1.368(2) 1.370(2) 1.371(3)
1.387(3) 1.380(6)
Fe-Cy
1.826 1.788 1.803 1.804 1.822 1.813
Angle (°) O2-C1-Fe 175.0(2)
177.6(18) 177.40(17) 176.0(2)
177.8(3) 176.0(5)
O4-C2-Fe 168.71(18) 174.9(18) 172.59(15) 173.5(2) 170.0(3) 170.6(5)
Indeed, for the complexes C1, C5 and C6, the angles Fe-C-O are 168.7(2)°, 170.0(3)° and
170.6(5)°, respectively and the distances CO-Cipso are 2.778(3) Å, 2.852(4) Å and 2.849(8) Å,
respectively. Such features (a short distance d < 3.1 Å and an angle < 175°) has been described
as the signature of a latent Cipso-CO interligand interaction by N. Lugan based on the studies of
Fischer-type manganese carbene complexes.[48-49] This non-covalent interaction can be found in
various NHC/carbonyl or NHC/alkylidene complexes, including Fe-NHC and Ru-NHC
complexes, and its role has been identified in the case of the metathesis reaction.[50]
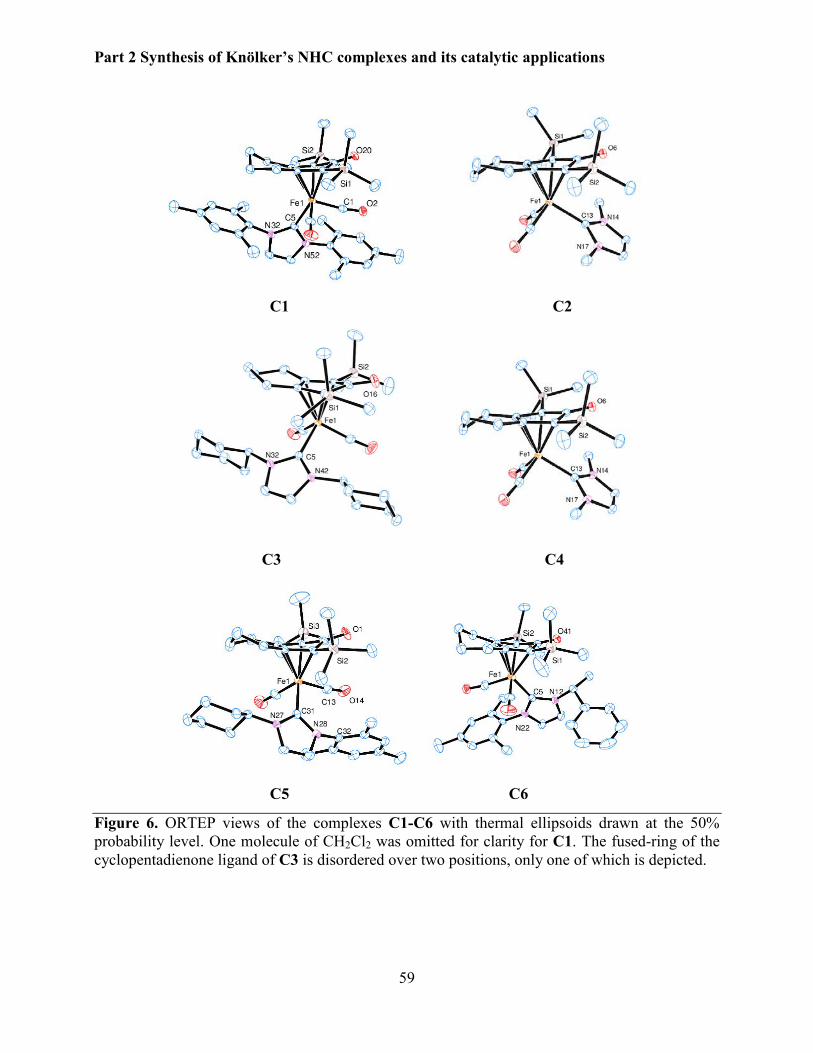
Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
59
C1 C2
C3 C4
C5 C6
Figure 6. ORTEP views of the complexes C1-C6 with thermal ellipsoids drawn at the 50% probability level. One molecule of CH2Cl2 was omitted for clarity for C1. The fused-ring of the cyclopentadienone ligand of C3 is disordered over two positions, only one of which is depicted.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
60
Figure 7. Non-covalent CO-Cipso interaction in the solid state.
To gain active hydride Fe-NHC complexes, we performed Hieber base reaction, on the
basis of previous work of Knölker,[12] by reaction of the complex C1 with sodium hydroxide then
hydrolysis. Unfortunately, we didn’t observe the formation of the Fe-H species even under
thermal conditions (up to 70 °C). (Scheme 13)
Scheme 13. Attempt to prepare Fe-H complex.
The substitution of one CO ligand by a nitrile ligand was also tested with the complex C1.
Conducting the reaction in toluene under experiment in UV activation at 350 nm, or in thermal
condition (110 oC), no coordination of nitrile was observed.
3.3 Catalytic applications With these new NHC-Knölker complexes in hand, we have then explored their potential
in catalytic reactions to enlarge the scope of applications of Knölker type derivatives in the
reduction area. Inspired from the work on selective hydrogenation of aldehydes and ketones, [21]
we evaluated the activity of these novel Knölker NHC complexes in hydrogenation of
acetophenone under similar conditions than the ones described by Beller (1 mol% 3, 5 mol%
K2CO3) in order to study the influence of the NHC ligand on the activity. To our delight, when
using 3 mol% of C1 in the presence of 15 mol% of K2CO3 in a mixture i-PrOH/water under 30
bar of H2 at 100 °C for 24 h, 97% conversion was observed, even if C1 was found to be less
active than 3 in hydrogenation of acetophenone (Scheme 14). Noteworthy, under similar

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
61
conditions, only the complex C3 gave 19% conversion, as with the other ones, no reaction was
observed.
Scheme 14. Hydrogenation of acetophenone with C1
Later, based on our research interest in hydrosilylation reactions with iron carbonyl
catalysts, we have evaluated the activity of these new iron NHC complexes in hydrosilylation
reactions. As an initial test reaction, the complexes were evaluated in the catalytic reduction of
acetophenone through hydrosilylation under UV irradiation using 1.5 equiv. phenylsilane as the
hydrosilane. Interestingly, using 1 mol% of C1 in toluene at rt, almost a full conversion (95%) of
acetophenone leading to 1-phenylethanol was observed by GC analysis. Notably, it was the first
efficient Knölker type complex in hydrosilylation reaction. Indeed, the hydrosilylation of
acetophenone with Knölker precursor 3 under the same condition gave only 26% conversion.
This last result clearly showed the beneficial influence of the NHC ligand on the efficiency of the
hydrosilylation reaction.
During the screening of the reactivity of the prepared NHC iron Knölker complexes in
hydrosilylation reactions, we discovered an interesting dehydration of primary benzamide to
benzonitrile when using 5 mol% of C1 and 3 equiv. of phenylsilane in toluene at 100 °C for 40 h.
Such dehydration under hydrosilylation conditions was already described with several transition
metals such as ruthenium,[51] or zinc.[52] To the best of our knowledge, only 3 examples of the
latter reaction have been reported catalyzed with iron catalysts. First, Beller[53] described the use
of 2-5 mol% of [Et3NH][HFe(CO)11] in the presence of 3 equiv. of diethoxymethylsilane as the
dehydrating reagent in toluene at 100 °C. Then, we reported similar reaction using 5 mol% of
[CpFe(CO)2(IMes)][I] in the presence of 4 equiv. of phenylsilane at 100 °C under visible light
activation.[54] Very recently, X. Li reported the used of hydrido thiophenolato iron(II) complexes
[cis-Fe(H)(SPh)(PMe3)4] (2 mol%) for the dehydration of primary amides in the presence of 3
equiv. of (EtO)3SiH at 60 °C for 24 h.[55]
Thus, we used new NHC complexes C1-C6 in the dehydration of primary amides into nitrile
derivatives and have selected the inexpensive PMHS (polymethylhydrosiloxane) as the

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
62
dehydration reagent. To optimize the reaction conditions, the complex C1 was selected as the
model precatalyst, and benzamide as the model substrate (Scheme 15, Table 3).
Scheme 15. Dehydration of benzamide into benzonitrile catalyzed by iron.
To our delight, benzamide can be converted to benzonitrile in high GC-yields (up to 97%)
using 5 mol% of the catalyst C1 at 100 °C for 24 h, in the presence of 5 equiv. of PMHS, in
different solvents, such as toluene, CPME (cyclopentyl methyl ether) and 1,4-dioxane (Table 3,
entries 1-3). The reaction did not proceed in dimethyl carbonate or ethanol and gave only 13%
yield when conducting in THF (Entries 11-13). It must be pointed out that decreasing the
temperature at 80 °C (Entry 4), the catalyst loading to 3 mol% (Entry 5) or the quantity of PMHS
(Entries 6-7) has a deleterious effect on the reaction efficiency. The effect of UV irradiation in
order to promote the reaction at lower temperature (35 °C) was also checked, but the conversion
was very low (Entry 8).
Table 3. Optimization of the parameters of the iron-catalyzed dehydration of benzamides a
Entry PMHS (equiv.) Solvent Temp
(°C)
Yield (%)b
1 5 Toluene 100 97 2 5 1,4 dioxane 100 94 3 5 CPME 100 97 4 5 CPME 80 18 5c 5 CPME 100 51 6 3 CPME 100 87 7 2 CPME 100 35 8d 5 CPME UV 6 9e 5 Toluene 100 2 10f 5 Toluene 100 57 11 5 EtOH 100 0 12 5 DMC 100 0 13 5 THF 70 13
[a] Typical conditions: catalyst C1 (5 mol%), benzamide (0.25 mmol), toluene (2 mL) and PMHS (5 equiv.) were added in this order under an argon atmosphere and the solution was heated at 100 °C for 24 h. [b] Determined by GC using dodecane as the internal standard. [c] 3 mol% of catalyst C1 was used. [d]. 350 nm, 35 °C. [e] 3 as the catalyst. [f] (IMes)Fe(CO)4 as the catalyst. The efficiency of the different catalysts was then evaluated: at 100 °C in toluene, under the
optimized conditions described above, the complexes C1-C5 displayed a similar activity, as full
conversions were obtained in each case (Table 4). Interestingly, under such conditions, the
original Knölker complex 3 was completely inactive (2% GC-yield, entry 6) and Fe(IMes)(CO)4
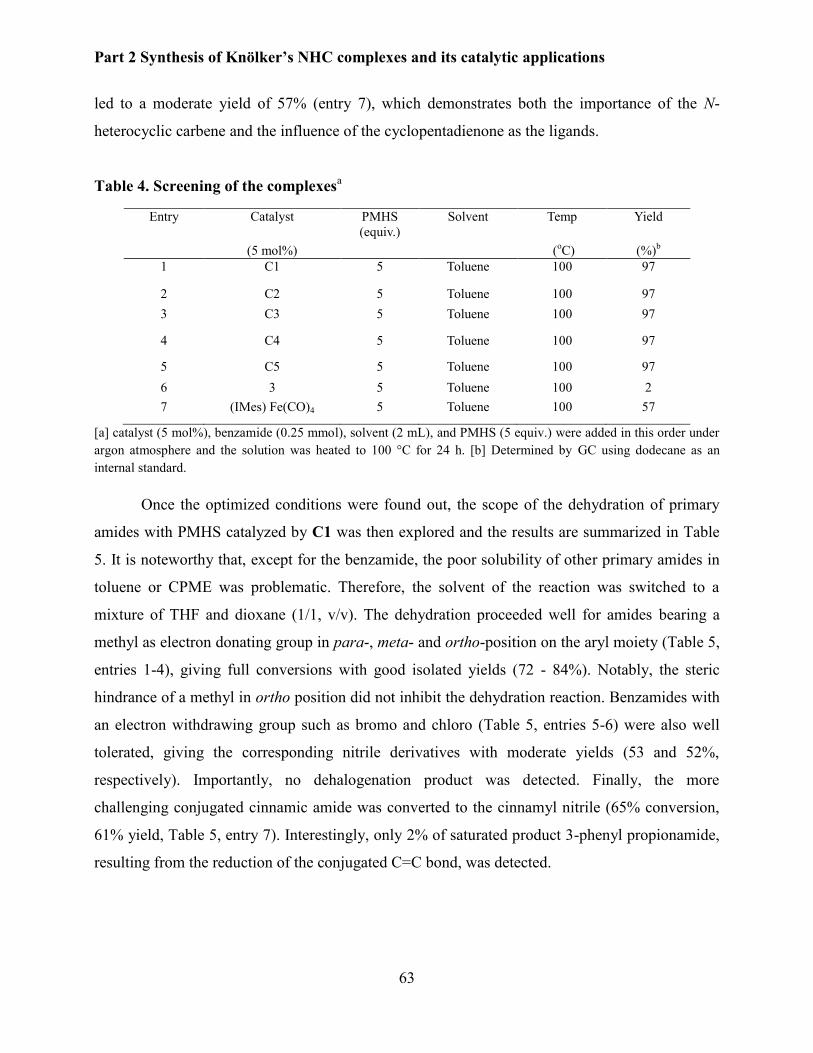
Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
63
led to a moderate yield of 57% (entry 7), which demonstrates both the importance of the N-
heterocyclic carbene and the influence of the cyclopentadienone as the ligands.
Table 4. Screening of the complexesa
Entry Catalyst
(5 mol%)
PMHS (equiv.)
Solvent Temp
(oC)
Yield
(%)b 1 C1 5 Toluene 100 97
2 C2 5 Toluene 100 97 3 C3 5 Toluene 100 97
4 C4 5 Toluene 100 97
5 C5 5 Toluene 100 97 6 3 5 Toluene 100 2 7 (IMes) Fe(CO)4 5 Toluene 100 57
[a] catalyst (5 mol%), benzamide (0.25 mmol), solvent (2 mL), and PMHS (5 equiv.) were added in this order under argon atmosphere and the solution was heated to 100 °C for 24 h. [b] Determined by GC using dodecane as an internal standard.
Once the optimized conditions were found out, the scope of the dehydration of primary
amides with PMHS catalyzed by C1 was then explored and the results are summarized in Table
5. It is noteworthy that, except for the benzamide, the poor solubility of other primary amides in
toluene or CPME was problematic. Therefore, the solvent of the reaction was switched to a
mixture of THF and dioxane (1/1, v/v). The dehydration proceeded well for amides bearing a
methyl as electron donating group in para-, meta- and ortho-position on the aryl moiety (Table 5,
entries 1-4), giving full conversions with good isolated yields (72 - 84%). Notably, the steric
hindrance of a methyl in ortho position did not inhibit the dehydration reaction. Benzamides with
an electron withdrawing group such as bromo and chloro (Table 5, entries 5-6) were also well
tolerated, giving the corresponding nitrile derivatives with moderate yields (53 and 52%,
respectively). Importantly, no dehalogenation product was detected. Finally, the more
challenging conjugated cinnamic amide was converted to the cinnamyl nitrile (65% conversion,
61% yield, Table 5, entry 7). Interestingly, only 2% of saturated product 3-phenyl propionamide,
resulting from the reduction of the conjugated C=C bond, was detected.

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
64
Table 5. Scope of the dehydration of primary amides to nitriles a Entry Amide Cat
(mol%) Time (h)
Conv.b (%)
Yield c (%)
1
5 24 >97 72
2
5 24 >97 84
3
5 48 >97 77
4
8 48 82 72
5
8 48 75 53
6
8 48 63 52
7 d
8 48 65
61
[a] Reaction conditions: catalyst C1 (5-8 mol%), amide (1 mmol), THF/1,4-dioxane (2 : 2 mL), and PMHS (5 equiv.), 100 °C, under an argon atmosphere. [b] The conversion was determined by 1H NMR. [c] Isolated yield. [d] 2% of 3-phenyl propionitrile was detected.
Scheme 16. Proposed reaction mechansim for the dehydration of amides using iron catalyst.
With respect to the reaction mechanism previously reported by Nagashima[51] and
Beller[53] using Ru and Fe respectively, the iron-catalyzed dehydration protocol may proceed via
a similar pathway. The reaction of the silane with the iron catalyst should generate an activated
species, which hydrosilylates the amide. Subsequent exchange of the N–H proton by another silyl
group under release of hydrogen leads to a mixture of the bis(silyl)amide and the N,O-
bis(silyl)imidate. Finally, elimination of the disiloxane produces the corresponding nitrile.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
65
4. Conclusion In conclusion, we have synthesized and fully characterized a new family of NHC-
substituted iron Knölker type complexes by UV promoted substitution of one CO-ligand by the
corresponding NHC ligand. Notably, these complexes are air-stable and are purified by column
chromatography. All the complexes have been obtained in good yields and fully characterized,
notably by electrochemical techniques and X-ray diffraction. Their potential in catalysis has been
evaluated in hydrogenation and hydrosilylation of acetophenone, and demonstrated in the case of
the dehydration of primary benzamides into benzonitrile derivatives using the inexpensive PMHS
as the dehydrating reagent. These results open the way to apply Knölker type complexes in
hydrosilylation reactions.
5. Experimental Part General Methods. All reactions were carried out with oven-dried glassware using standard
Schlenk techniques under an inert atmosphere of dry argon or in an argon-filled glove-box.
Toluene, THF, diethyl ether (Et2O), and CH2Cl2 were dried over Braun MB-SPS-800 solvent
purification system and degassed by thaw-freeze cycles. Technical grade pentane and diethyl
ether were used for column chromatography. Analytical TLC was performed on Merck 60F254
silica gel plates (0.25 mm thickness). Column chromatography was performed on Acros Organics
Ultrapure silica gel (mesh size 40-60 μm, 60Å). All reagents were obtained from commercial
sources and liquid reagents were dried on molecular sieves and degassed prior to use. 1H, and 13C{1H} NMR spectra were recorded in CDCl3, C6D6 at ambient temperature unless otherwise
stated, on Bruker AVANCE 500, AVANCE 400 and AVANCE 300 spectrometers at 500, 400
and 300 MHz, respectively. 1H and 13C spectra were calibrated using the residual solvent signal
as internal standard (1H: CDCl3 7.26 ppm, C6D6 7.16 ppm, 13C: CDCl3, central peak is 77.00
ppm, C6D6 central peak is 128.06 ppm). IR spectra were measured by Shimadzu IR-Affinity 1
with ATR equipment. HR-MS spectra and microanalysis were carried out by the corresponding
facilities at the CRMPO (Centre Regional de Mesures Physiques de l’Ouest), University of
Rennes 1.
X-ray diffraction data were collected on an APEXII, Bruker-AXS diffractometer equipped with a
CCD detector, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at T = 150(2) K.
The structure was solved by direct methods using the SIR97 pro-gram23, and then refined with

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
66
full-matrix least-square methods based on F2 (SHELXL-97)24 with the aid of the WINGX25
program.
Electrochemical studies were carried out under argon using an Eco Chemie Autolab PGSTAT 30
potentiostat, the working electrode was a Pt disk, and SCE electrode was used as reference
electrode. The sample concentration was ~1 mM (CH2Cl2, 0.2 M Bu4NPF6) and the solution was
carefully deoxygenated by argon bubbling. The applied potential was measured with
decamethylferrocene as the internal reference and calculated versus ferrocene with E° ([FeCp2]/
[FeCp2]+) = +0.542 V vs [FeCp*2]/ [FeCp*2]+. UV irradiations were performed in a Rayonet
RPR-100 apparatus at 350 nm.
The imidazolium salts NHC.HX,[78] and Knölker’s complex[32] were prepared according to the
published procedures.
5.1 Synthesis of 1-mesityl-3-((R)-1-phenylethyl)imidazolium chloride salt The imidazolium salt was prepared following the procedure described by Baslé and Maudit[64] In
a round-bottomed flask were placed 2,4,6-trimethylaniline (1.41 mL, 10 mmol), R-(+)--
methylbenzylamine (1.27 mL, 10 mmol) and acetic acid (45 mmol, 4.5 equiv.) then the mixture
was heated at 40 °C for 5 min (mixture A). In another round-bottomed flask were placed glyoxal
(10 mmol, 1.0 equiv., 40 % wt in aqueous solution), formaldehyde (10 mmol, 1.0 equiv., 37 % wt
in aqueous solution) and acetic acid (45 mmol, 4.5 equiv.) then the mixture was heated at 40 °C
for 5 min (mixture B). At 40 °C, mixture B was added to mixture A and the resulting mixture was
stirred at 40 °C for 10 min then cooled down to room temperature. Dichloromethane (100 mL)
was added and the organic layer was successively washed with water (200 mL) then brine (2 x
100 mL). The combined aqueous layers were extracted with dichloromethane (100 mL). The
combined organic layers were dried over magnesium sulfate, filtered and the volatiles were
removed under reduced pressure. Pure compound was obtained as a white solid after purification
by column chromatography on silica gel (CH2Cl2/MeOH, 9/1 as the eluant). 900 mg, yield 28%. 1H NMR (400 MHz, CDCl3): δ 10.86 (s, 1H), 7.85 (s, 1H), 7.61 (d, J = 6.5 Hz, 2H), 7.36-7.29 (m, 3H), 7.19 (s, 1H), 6.92 (s, 1H, CHIm), 6.91 (s, 1H, CHIm), 6.67 (q, J = 7.0 Hz, 1H), 2.27 (s, 3H, CH3Mes), 2.03 (s, 3H, CH3Mes), 2.02 (d, J = 7.0 Hz, 3H), 1.95 (s, 3H, CH3Mes). 13C{1H} NMR (101 MHz, CDCl3): δ 140.9, 138.1, 137.8, 134.0, 133.9, 130.7, 129.6, 129.2, 129.0, 127.1, 123.4, 120.9, 59.1 (CH), 20.9 (CH3Mes), 20.6 (CH3), 17.5 (CH3Mes), 17.4 (CH3Mes). Anal. calc for C20H23N2Cl: C, 73.49; H, 7.09; N, 8.57. Found: C, 73.31; H, 7.02; N, 8.32.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
67
5.2 Synthesis of complexes Synthesis of the complex C1
To a suspension of 1,3-dimesitylimidazolium chloride (1.20 g, 3.6 mmol, 1.5 equiv.) in toluene
(10 mL), KHMDS (0.5 M in toluene, 7.9 mL, 3.9 mmol, 1.6 equiv.) was added drop-wise at r.t.
and the solution was vigorously stirred for 30 min. The resulting white suspension was directly
filtered through celite into the Schlenk tube containing Knölker’s precursor 3 (1.00 g, 2.3 mmol,
1 equiv.) in toluene (10 mL). Then, the reaction mixture was irradiated under UV (350 nm) at
room temperature for 20 h. After completion of the reaction, the initial brown solution turned to
the dark yellow. The solvent was evaporated under vacuum. The resulting residue was purified
by column chromatograph on silica gel. A yellow colored fraction of the expected complex was
collected (petroleum ether/ethyl acetate, 80:20), concentrated under vacuum to give C1 (1.25 g,
78%) as a golden yellow powder. Single crystals suitable for X-ray diffraction studies were
grown by layering a concentrated solution of the complex in CH2Cl2 with pentane. 1H NMR (400 MHz, CDCl3): δ 6.99 (s, 4 H, CHMes), 6.85 (s, 2 H, CHNHC), 2.35 (s, 6 H, p-CH3), 2.19-2.17 (m, 4 H, CH2), 2.15 (s, 12 H, o-CH3), 1.34-1.31 (m, 2 H, CH2), 0.98-0.95 (m, 2 H, CH2), 0.05 (s, 18 H, SiMe3). 13C{1H} NMR (100 MHz, CDCl3): δ 217.4 (2 CO), 187.8 (NCN), 179.5 (C=O), 139.6 (Co), 138.2 (Ci), 136.4 (Cp), 129.4 (Cm), 126.2 (CHNHC), 104.9 (C=CSi), 67.5 (CSi), 24.6 (CH2), 21.9 (CH2), 21.0 (p- CH3), 19.1 (o -CH3), 0.5 (SiMe3). IR (ATR, cm-1): 1978, 1913, 1577. Anal. Calc. for C38H50N2O3FeSi2: C, 65.69; H, 7.25; N, 4.03. Found: C, 65.24; H, 7.18; N, 3.92. HR-MS [ESI]: m/z [M + H] + calcd for C38H51N2O3
56FeSi2 695.2787 found 695.2782 (0 ppm).

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
68
Synthesis of the complex C2
To a suspension of 1,3-dimethylimidazolium hexafluorophosphate (87 mg, 0.35 mmol) in toluene
(2 mL), KHMDS (0.5 M in toluene, 0.78 mL, 0.39 mmol) was added dropwise at RT and the
resulting suspension was vigorously stirred for 30 min. The resulting white suspension was
transferred through celite into the Schlenk tube containing a Knölker precursor 3 (100 mg, 0.24
mmol) and washed the celite bed with toluene (2 mL) three times. Then, the reaction mixture was
irradiated under UV (350 nm) light at room temperature for 20 h. After completion of the
reaction, the initial brown solution was changed into the dark yellow. Solvent was evaporated
under vacuum. The resulting residue was purified by column chromatography on silica gel. A
yellow color of the target NHC complex was collected by petroleum ether/ethyl acetate, 50:50
and it was concentrated under vacuum to give C2 (75 mg, 64%) as a yellow powder. X-ray-
quality crystals were grown by layering a solution of complex in CH2Cl2 with pentane. 1H NMR (500 MHz, CDCl3): δ 6.98 (s, 2 H, CHNHC), 3.91 (s, 6 H, MeNHC), 2.45 (m, 4 H, CH2), 1.85-1.78 (m, 4 H, CH2), 0.17 (s, 18 H, SiMe3). 13C{1H} NMR (125 MHz, CDCl3): δ 217.2 (2 CO), 185.0 (NCN), 176.8 (C=O), 123.6 (CHNHC), 103.7 (C=CSi), 70.9 (CSi), 39.9 (MeNHC), 24.6 (CH2), 22.6 (CH2), 0.3 (SiMe3). IR (ATR, cm-1): 1975, 1919, 1548. Anal. Calc. for C22H34N2O3FeSi2: C, 54.31; H, 7.04; N, 5.76. Found: C, 54.29; H, 7.03; N, 5.58. HR-MS [ESI]: m/z [M + H] + calcd for C22H35N2O3
56FeSi2 487.1530 found 487.1533 (1 ppm). Synthesis of the complex C3
To a suspension of 1,3-diisopropylimidazolium hexafluorophosphate (107 mg, 0.35 mmol) in
toluene (2 mL), KHMDS (0.5 M in toluene, 0.78 mL, 0.39 mmol) was added dropwise at RT and

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
69
the resulting suspension was vigorously stirred for 30 min. The resulting white suspension was
transferred through celite into the Schlenk tube containing a Knölker precursor 3 (100 mg, 0.24
mmol) and washed the celite bed with toluene (2 mL) three times. Then, the reaction mixture was
irradiated under UV (350 nm) light at room temperature for 20 h. After completion of the
reaction, the initial brown solution was changed into the orange red solution. Solvent was
evaporated under vacuum. The resulting residue was purified by column chromatography on
silica gel. A yellow color of the target NHC complex was collected by Petroleum ether/ethyl
acetate, 80:20 and it was concentrated under vacuum to give C3 (110 mg, 84%) as a yellow
powder. X-ray-quality crystals were grown by layering a solution of complex in CH2Cl2 with
pentane. 1H NMR (500 MHz, C6D6): δ 6.53 (s, 2 H, CHNHC), 5.13 (sept, 3J = 6.4 Hz, 2 H, CHIpr), 2.59-2.54 (m, 2H, CH2), 2.45-2.37 (m, 2 H, CH2), 1.83-1.79 (m, 2 H, CH2), 1.57-1.53 (m, 2 H, CH2), 1.24 (d, 12 H, 3J = 6.4 Hz, CH3Ipr), 0.32 (s, 18 H, SiMe3). 13C{1H} NMR (125 MHz, C6D6): δ 219.3 (2 CO), 183.1 (NCN), 177.7 (C=O), 119.0 (CHNHC), 103.4 (C=CSi), 72.6 (CSi), 51.8 (CHIpr), 25.5 (CH2), 24.7 (CH3Ipr), 22.8 (CH2), 0.5 (SiMe3). IR (ATR,cm-1): 1975, 1924, 1583. Anal. calc for C26H42N2O3FeSi2: C, 57.55; H, 7.80; N, 5.16. Found: C, 57.48; H, 7.85; N, 4.96. HR-MS [ESI]: m/z [M + H] + calcd for C26H43N2O3
56FeSi2 543.2156 found 543.2158 (0 ppm). Synthesis of the complex C4
To a suspension of 1,3-dicyclohexylimidazolium tetrafluoroborate (229 mg, 0.72 mmol) in
toluene (4 mL), KHMDS (0.5 M in toluene, 1.6 mL, 0.79 mmol) was added and the resulting
suspension was vigorously stirred for 30 min. The resulting white suspension was transferred
through celite to the Schlenk tube containing a Knölker precursor (200 mg, 0.48 mmol) and
washed the celite bed with toluene (2 mL) three times. Then, the reaction mixture was irradiated
under UV (350 nm) light at room temperature for 20 h. After completion of the reaction, the
initial brown solution was changed into the dark yellow. Solvent was evaporated under vacuum.
The resulting residue was purified by column chromatography on silica gel. A yellow color of the
target NHC complex was collected by Petroleum ether/ethyl acetate, 90:10 and it was

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
70
concentrated under vacuum to give C4 (210 mg, 70%) as a yellow powder. X-ray-quality
crystals were grown by layering a solution of complex in CH2Cl2 with pentane. 1H NMR (500 MHz, CDCl3): δ 7.08 (s, 2 H, CHNHC), 4.81-4.75 (m, 2 H, CHCy), 2.48-2.35 (m, 4 H, CH2), 2.06-1.20 (m, 24 H), 0.11 (s, 18 H, SiMe3). 13C{1H} NMR (125 MHz, CDCl3): δ 217.4 (2 CO), 181.5 (NCN), 178.3 (C=O), 119.6 (CHNHC), 104.1 (C=CSi), 68.5 (CSi), 58.9 (CHCy), 35.5 (CH2), 25.3 (CH2Cy), 25.2 (CH2Cy), 24.0 (CH2), 22.6 (CH2Cy), 0.01 (SiMe3). IR (ATR, cm-1): 1969, 1907, 1585. Anal. Calc. for C32H50N2O3FeSi2: C, 61.72; H, 8.09; N, 4.50. Found: C, 61.98; H, 8.32; N, 4.45. HR-MS [ESI]: m/z [M + H] + calcd for C32H51N2O3
56FeSi2 623.2782 found 623.2781 (0 ppm). Synthesis of the complex C5
. To a suspension of 3-cycloheptyl-1-mesitylimidazolium chloride (220 mg, 0.72 mmol) in toluene
(4 mL), KHMDS (0.5 M in toluene, 1.6 mL, 0.79 mmol) was added and the resulting suspension
was vigorously stirred for 30 min. The resulting white suspension was transferred through celite
to the Schlenk tube containing a Knölker precursor (200 mg, 0.48 mmol) and washed the celite
bed with toluene (2 mL) three times. Then, the reaction mixture was irradiated under UV (350
nm) light at room temperature for 20 h. After completion of the reaction, the initial brown
solution was changed into the dark yellow. Solvent was evaporated under vacuum. The resulting
residue was purified by column chromatography on silica gel. A yellow color of the target NHC
complex was collected by petroleum ether/ethyl acetate, 80:20 and it was concentrated under
vacuum to give C5 (203 mg, 65%) as a yellow powder. X-ray-quality crystals were grown by
layering a solution of complex in CH2Cl2 with pentane. 1H NMR (400 MHz, CDCl3): δ 7.30 (s, 1 H, CHNHC), 6.95 (s, 2 H, CHMes), 6.86 (s, 1 H, CHNHC), 4.79-4.73 (m, 1 H, CHCy), 2.33-2.25 (m, 8 H), 1.97 (s, 6H, o-CH3), 1.85-1.56 (m, 9 H), 1.28-1.21 (m, 4 H); 0.14 (s, 18 H, SiMe3). 13C{1H} NMR (125 MHz, CDCl3): δ 217.6 (2 CO), 186.9 (NCN), 177.5 (C=O), 139.3 (CqMes), 137.9 (CqMes), 136.3 (CqMes), 129.2 (CHMes), 125.1 (CHNHC), 120.5 (CHNHC), 102.9 (C=CSi), 70.4 (CSi), 58.3 (CHCy), 35.2 (CH2), 29.7 (CH2), 25.4 (CH2), 24.8 (CH2), 24.4 (CH2), 22.3 (CH2), 21.0 (p-CH3), 18.9 (o-CH3), 0.71 (SiMe3). IR (ATR, cm-1): 1980, 1919, 1575, 1548.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
71
IR (CH2Cl2, cm-1): 1979, 1919, 1570. In the solid state, two stretching bands for the C=O of the ketone are present, probably due to the presence of two rotamers. In solution, only one band was observed. Anal. Calc. for C35H50N2O3FeSi2: C, 63.61; H, 7.65; N, 4.25. Found: C, 64.09; H, 7.80; N, 3.89. HR-MS [ESI]: m/z [M + H] + calcd for C35H51N2O3
56FeSi2 659.2782 found 659.2779 (0 ppm).

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
72
Synthesis of the complex C6
To a suspension of 1-mesityl-3-(1-phenylethyl)imidazolium chloride (235 mg, 0.32 mmol) in
toluene (4 mL), KHMDS (0.5 M in toluene, 1.6 mL, 0.79 mmol) was added and the resulting
suspension was vigorously stirred for 30 min. The resulting white suspension was transferred
through celite to the Schlenk tube containing a Knölker precursor (200 mg, 0.48 mmol) and
washed the celite bed with toluene (2 mL) three times. Then, the reaction mixture was irradiated
under UV (350 nm) light at room temperature for 20 h. After completion of the reaction, the
initial brown solution was changed into the dark yellow. Solvent was evaporated under vacuum.
The resulting residue was purified by column chromatography on silica gel. A yellow color of the
target NHC complex was collected by petroleum ether/ethyl acetate, 90:10 and it was
concentrated under vacuum to give C6 (250 mg, 76%) as a yellow powder. X-ray-quality crystals
were grown by layering a solution of complex in CH2Cl2 with pentane.
1H NMR (400 MHz, CDCl3): δ 7.47 (d, 3J = 7.8 Hz, 2H), 7.36 (d, 3J = 7.5 Hz, 2H), 7.28 (m, 1H), 6.98 (s, 1H, CHMes), 6.97 (s, 1H, CHMes), 6.91 (d, 3J = 1.8 Hz, 1H, CHNHC), 6.84 (d, 3J = 1.8 Hz, 1H, CHNHC), 6.06 (q, 3J = 6.5 Hz, 1 H, CH), 2.34 (s, 3H, CH3Mes), 2.40-2.21 (m, 4H, CH2), 2.08 (s, 3H, CH3Mes), 2.05 (s, 3H, CH3Mes), 1.94 (d, 3J = 6.5 Hz, 3H, CH3), 1.70-1.40 (m, 4H, CH2), 0.11 (s, 9H, SiMe3), 0.06 (s, 9H, SiMe3). 13C{1H} NMR (125 MHz, CDCl3): δ 217.1 (CO), 216.7 (CO), 187.4 (NCN), 180.0 (C=O), 142.2 (CqAr), 139.5 (CqMes), 138.2 (CqMes), 136.5(CqMes), 136.2(CqMes), 129.3 (CHMes), 129.2 (CHMes), 128.3 (m-CHAr), 127.3 (p-CHAr), 127.2 (o-CHAr), 125.0 (CHNHC), 121.8 (CHNHC), 104.6 (C=CSi), 99.7 (C=CSi), 70.6 (CSi), 69.5 (CSi), 57.7 (CH), 25.0 (CH2), 23.8 (CH2), 22.2 (CH2), 22.0 (CH2), 21.9 (CH3), 21.0(CH3Mes), 19.1(CH3Mes), 18.4 (CH3Mes), 0.5 (SiMe3), 0.4 (SiMe3). IR (ATR, cm-1): 1973, 1915, 1585, 1552. IR (CH2Cl2, cm-1): 1980, 1921, 1574. In the solid state, two stretching bands for the C=O of the ketone are present, probably due to the presence of two rotamers. In solution, only one band was observed Anal. Calc. for C37H48N2O3FeSi2: C, 65.28; H, 7.11; N, 4.11. Found: C, 65.39; H, 7.04; N, 4.07. HR-MS [ESI]: m/z [M + Na] + calcd for C37H48N2O3NaSi256Fe 703.2450 found 703.2467 (2 ppm).
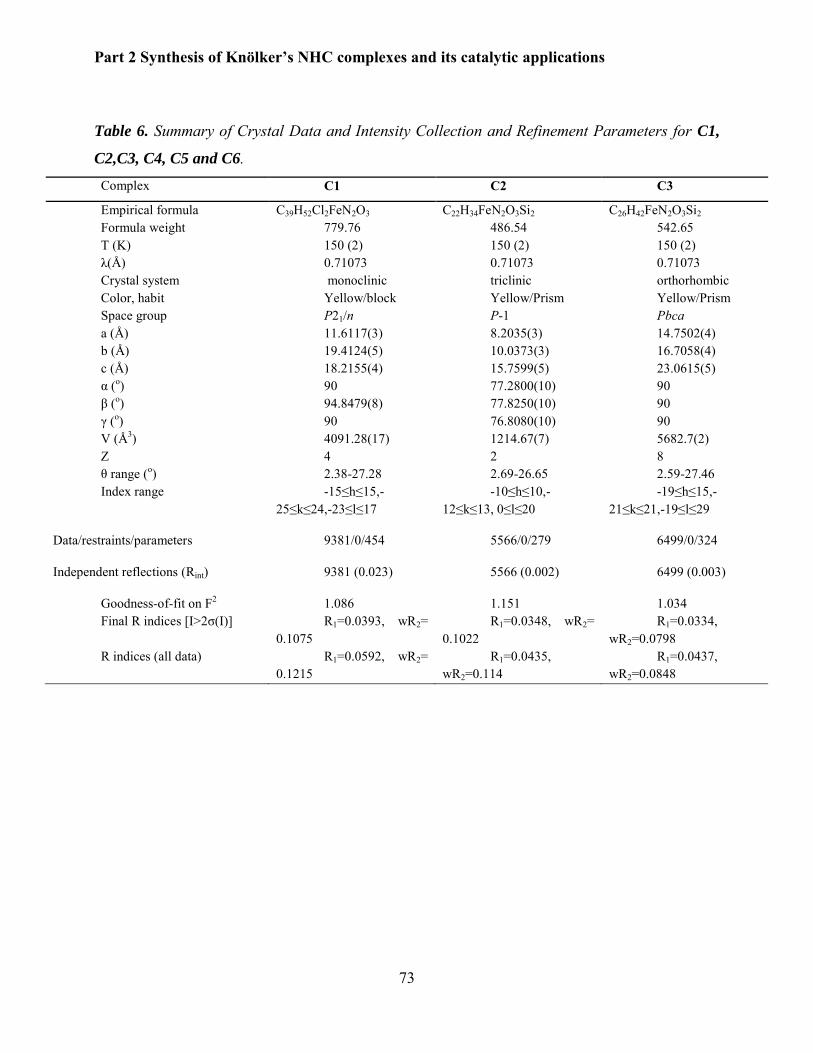
Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
73
Table 6. Summary of Crystal Data and Intensity Collection and Refinement Parameters for C1,
C2,C3, C4, C5 and C6. Complex C1 C2 C3
Empirical formula C39H52Cl2FeN2O3 C22H34FeN2O3Si2 C26H42FeN2O3Si2 Formula weight 779.76 486.54 542.65 T (K) 150 (2) 150 (2) 150 (2) λ(Å) 0.71073 0.71073 0.71073 Crystal system monoclinic triclinic orthorhombic Color, habit Yellow/block Yellow/Prism Yellow/Prism Space group P21/n P-1 Pbca a (Å) 11.6117(3) 8.2035(3) 14.7502(4) b (Å) 19.4124(5) 10.0373(3) 16.7058(4) c (Å) 18.2155(4) 15.7599(5) 23.0615(5) α (o) 90 77.2800(10) 90 β (o) 94.8479(8) 77.8250(10) 90 γ (o) 90 76.8080(10) 90 V (Å3) 4091.28(17) 1214.67(7) 5682.7(2) Z 4 2 8 θ range (o) 2.38-27.28 2.69-26.65 2.59-27.46 Index range -15≤h≤15,-
25≤k≤24,-23≤l≤17 -10≤h≤10,-
12≤k≤13, 0≤l≤20 -19≤h≤15,-
21≤k≤21,-19≤l≤29
Data/restraints/parameters 9381/0/454 5566/0/279 6499/0/324
Independent reflections (Rint) 9381 (0.023) 5566 (0.002) 6499 (0.003)
Goodness-of-fit on F2 1.086 1.151 1.034 Final R indices [I>2σ(I)] R1=0.0393, wR2=
0.1075 R1=0.0348, wR2=
0.1022 R1=0.0334,
wR2=0.0798 R indices (all data) R1=0.0592, wR2=
0.1215 R1=0.0435,
wR2=0.114 R1=0.0437,
wR2=0.0848
Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
74
Complex C4 C5 C6
Empirical formula C32H50FeN2O3Si2 C35H50FeN2O3Si2 C37H48FeN2O3Si2 Formula weight 622.77 658.8 680.8 T (K) 150 (2) 150 (2) 150 (2) λ(Å) 0.71073 0.71073 0.71073 Crystal system monoclinic monoclinic monoclinic Color, habit Yellow/Plate Yellow/Prism Yellow/Prism Space group P21/n P21/n P21 a (Å) 10.4166(4) 11.2175(4) 10.356(3) b (Å) 21.1251(7) 20.1571(6) 15.454(4) c (Å) 16.6337(7) 15.8102(5) 11.602(3) α (o) 90 90 90 β (o) 93.9530(10) 93.404(2) 101.012(9) γ (o) 90 90 90 V (Å3) 3651.6(2) 3568.6(2) 1822.6(9) Z 4 4 2
θ range (o) 2.44-26.02 2.5-25.67 2.42-21.71 Index range -13≤h≤13,-
27≤k≤19,-21≤l≤21 -14≤h≤14,-
20≤k≤26,-20≤l≤20 -13≤h≤12,-
13≤k≤19,-14≤l≤14
Data/restraints/parameters 8342/0/367 8110/0/397 6839/0/416
Independent reflections (Rint) 8342 (0.002) 8110 (0.01) 6839 (0.01)
Goodness-of-fit on F2 1.086 1.025 1.009 Final R indices [I>2σ(I)] R1=0.0472,
wR2=0.116 R1=, 0.054 wR2= 0.1022
R1=0.0656, wR2=0.1438
R indices (all data) R1=0.0708, wR2=0.1243
R1=0.0896, wR2=0.1308
R1=0.0939, wR2=0.1611
5.3 Characterization data of the nitrile products. Benzonitrile
The compound was prepared as described in the general procedure. Purification by flash chromatography (Petroleum ether/ethyl acetate, 80:20), colorless oil, 75 mg, 72% isolated yield. 1H NMR (400 MHz, CDCl3): δ 7.47 (t, J = 7.7 Hz, 2H), 7.60 (t, J = 7.6 Hz, 1H), 7.65 (d, J = 7.7 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 112.4, 118.8, 129.0, 132.1, 132.7.

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
75
4-Methylbenzonitrile
The compound was prepared as described in the general procedure. Purification by flash chromatography (Petroleum ether/ethyl acetate, 80:20), colorless oil, 98 mg, 84% isolated yield. 1H NMR (400 MHz, CDCl3): δ 2.40 (s, 3H), 7.25 (d, J = 8.1 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 21.7, 109.1, 119.0, 129.7, 131.9, 143.6. 3-Methylbenzonitrile
The compound was prepared as described in the general procedure. Purification by flash chromatography (Petroleum ether/ethyl acetate, 80:20), colorless oil, 90 mg, 77% isolated yield. 1H NMR (400 MHz, CDCl3): δ 2.39 (s, 3H), 7.32-7.46 (m, 4H). 13C{1H} NMR (100 MHz, CDCl3): δ 21.1, 112.2, 119.0, 128.9, 129.2, 132.5, 133.6, 139.2. 2-Methylbenzonitrile
The compound was prepared as described in the general procedure. Purification by flash chromatography (Petroleum ether/ethyl acetate, 80:20), colorless oil, 85 mg, 72% isolated yield. 1H NMR (400 MHz, CDCl3): δ 2.55 (s, 3 H), 7.26 (t, J = 7.6 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.48 (t, J = 7.7 Hz, 1H), 7.59 (d, J = 7.7 Hz, 1H). 13C{1H} NMR (75 MHz, CDCl3): δ 20.4, 112.8, 118.0, 126.2, 130.2, 132.5, 132.6, 141.9. 4-Bromobenzonitrile
The compound was prepared as described in the general procedure. Purification by flash chromatography (Petroleum ether/ethyl acetate, 80:20), white powder, 96 mg, 53% isolated yield. 1H NMR (400 MHz, CDCl3): δ 7.53 (d, J = 8.5 Hz, 2H), 7.64 (d, J = 8.5 Hz, 2H). 13C{1H} NMR (75 MHz, CDCl3): δ 111.2, 118.0, 127.9, 132.6, 133.4.

Part 2 Synthesis and catalytic applications of Knölker’s NHC complexes
76
p-Chlorobenzonitrile
The compound was prepared as described in the general procedure. Purification by flash chromatography (Petroleum ether/ethyl acetate, 80:20), white powder, 71 mg, 52% isolated yield. 1H NMR (400 MHz, CDCl3): δ 7.47 (d, J = 8.6 Hz, 2 H), 7.60 (d, J = 8.6 Hz, 2 H). 13C{1H} NMR (100 MHz, CDCl3): δ 110.8, 118.0, 129.7, 133.3, 139.5. Cinnamonitrile
The compound was prepared as described in the general procedure. Purification by flash chromatography (Petroleum ether/ethyl acetate, 80:20), white powder, 80 mg, 61% isolated yield. 1H NMR (400 MHz, CDCl3): δ 5.89 (d, J = 16.7 Hz, 1H), 7.39-7.46 (m, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 96.3, 118.1, 127.3, 129.1, 131.2, 133.5, 150.5.
6. References [1] H. Grut macher, Angew. Chem. Int. Ed. 2008, 47, 1814-1818 [2] T. Ikariya, S. Masakatsu, Topics in Organometallic Chemistry, Bifunctional Molecular
Catalysis; Springer: Berlin, Heidelberg, 2011; Vol. 37. [3] A. Quintard, J. Rodriguez, Angew. Chem. Int. Ed. 2014, 53, 4044-4055. [4] B. L. Conley, M. K. Pennington-Boggio, E. Boz, T. J. Williams, Chem. Rev. 2010, 110,
2294-2312. [5] G. Bauer, K. A. Kirchner, Angew. Chem. Int. Ed. 2011, 50, 5798-5800. [6] W. Reppe, H. Vetter, Liebigs Ann. Chem. 1953, 582, 133-161. [7] G. N. Schrauzern, J. Am. Chem. Soc. 1959, 81, 5307-5310. [8] H.-J. Knölker, J. Heber, C. H. Mahler, Synlett 1992, 1992, 1002-1004. [9] H.-J. Knölker, J. Heber, Synlett 1993, 1993, 924-926. [10] H.-J. Knölker, E. Baum, R. Klauss, Tetrahedron Lett. 1995, 36, 7647-7650. [11] H.-J. Knölker, H. Goesmann, R. Klauss, Angew. Chem. Int. Ed. 1999, 38, 702-705. [12] H.-J. Knölker, E. Baum, H. Goesmann, R. Klauss, Angew. Chem. Int. Ed. 1999, 38, 2064-
2066. [13] C. P. Casey, H. Guan, J. Am. Chem. Soc. 2007, 129, 5816-5817. [14] C. P. Casey, H. Guan, J. Am. Chem. Soc. 2009, 131, 2499-2507. [15] A. Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard, J.-L. Renaud, Angew.
Chem. Int. Ed. 2012, 51, 4976-4980. [16] M. Kamitani, Y. Nishiguchi, R. Tada, M. Itazaki, H. Nakazawa, Organometallics 2014, 33, 1532-1535. [17] T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal. 2012, 354, 597-601.
[18] A. J. Pearson, R. J. Shively, R. A. Dubbert, Organometallics 1992, 11, 4096-4104.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
77
[19] A. J. Pearson, R. J. Shively, Organometallics 1994, 13, 578-584. [20] A. Berkessel, S. Reichau, A. von der Höh, N. Leconte, J. M. Neudörfl, Organometallics
2011, 30, 3880-3887. [21] S. Fleischer, S. Zhou, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2013, 52, 5120-5124. [22] A. Tlili, J. Schranck, H. Neumann, M. Beller, Chem. Eur. J. 2012, 18, 15935-15939. [23] D. S. Mérel, M. Elie, J. F. Lohier, S. Gaillard, J.-L. Renaud, ChemCatChem 2013, 5, 2939-
2945. [24] A. Berkessel, S. Reichau, A. von der Höh, N. Leconte, J. M. Neudörfl, Organometallics
2011, 30, 3880-3887. [25] P. Gajewski, M. Renom-Carrassco, S. Vailati Facchini, L. Pignatoro, L. Lefort, J. G. de
Vries, R. Ferraccioli, A. Forni, U. Piarulli, C. Gennari, Eur. J. Org. Chem. 2015, 1887-1893.
[26] T. N. Plank, J. L. Drake, D. K. Kim, T. W. Funk, Adv. Synth. Catal. 2012, 354, 597-601. [27] S. Zhou, S. Fleischer, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2011, 50, 5120-5124. [28] S. Fleischer, S. Zhou, S. Werkmeister, K. Junge, M. Beller, Chem. Eur. J. 2013, 19, 4997-
5003. [29] D. S. Mérel, M. Elie, J. F. Lohier, S. Gaillard, J. L. Renaud, ChemCatChem 2013, 5, 2939-
2945. [30] H.-J. Pan, T. W. Ng, Y. Zhao, Org. Biomol. Chem. 2016, 14, 5490-5493. [31] (a) A. Pagnoux-Ozherelyeva, N. Pannetier, M. Diagne Mbaye, S. Gaillard, J. L. Renaud,
Angew. Chem. Int. Ed. 2012, 51, 4976-4980. (b) S. Moulin, H. Dentel, A. Pagnoux-Ozherelyeva, S. Gaillard, A. Poater, L. Cavallo, J. F. Lohier, J. L. Renaud, Chem. Eur. J. 2013, 19, 17881-17890I.
[32] Y. Tao, B. L. Feringa, K. Barta, Nat. Commun 2014, 5, 5602. [33] B. Emayavaramban, M. Roy, B. Sundararaju, Chem. Eur. J. 2016, 22, 3952-3955. [34] A. J. Rawlings, L. J. Diorazio, M. Wills, Org. Lett. 2015, 17, 1086-1089. [35]. H. J. Pan, T. Wei Ng, Y. Zhao, Chem. Commun. 2015, 51, 11907-11910. [36] I. Bauer, H.-J. Knölker, Chem. Rev. 2015, 115, 3170-3387. [37] K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann, F. E. Kühn,
Chem. Rev. 2014, 114, 5215-5272. [38] D. Bézier, J.-B. Sortais, C. Darcel, Adv. Synth. Catal. 2013, 355, 19-33. [39] M. J. Ingleson, R. A. Layfield, Chem. Commun. 2012, 48, 3579-3589. [40] D. Bézier, F. Jiang, J.-B. Sortais, C. Darcel, Eur. J. Inorg. Chem. 2012, 1333-1337. [41] H. Jaafar, H. Li, L. C. Misal Castro, J. Zheng,T. Roisnel, J.-B. Sortais, C. Darcel. Eur. J.
Inorg. Chem. 2012, 3546-3550. [42] P. Queval, C. Jahier, M. Rouen, I. Artur, J.-C. Legeay, L. Falivene, L. Toupet, C. Crévisy, O.
Baslé, M. Maudit, Angew. Chem. Int. Ed. 2013, 52, 14103-14107. [43] H. Li, L. C. Misal Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais, C. Darcel, Angew.
Chem. Int. Ed. 2013, 52, 8045-8049. [44] T. Hashimoto, R. Hoshino, T. Hatanaka, Y. Ohki, K. Tatsumi, Organometallics 2014, 33,
921-929. [45] S. Warratz, L. Postigo, B. Royo, Organometallics 2013, 32, 893-897. [46] V.V K.M. Kandepi, J. M. S. Cardoso, E. Peris, B. Royo, Organometallics 2010, 29, 2777-
2782. [47] D. G. Gusev, Organometallics 2009, 28, 6458-6461.

Part 2 Synthesis of Knölker’s NHC complexes and its catalytic applications
78
[48] (a) I. Fernández, N. Lugan, G. Lavigne, Organometallics 2012, 31, 1155-1160; (b) D. A. Valyaev, R. Brousses, N. Lugan, I. Fernández, M.A. Sierra, Chem. Eur. J. 2011, 17, 6602-6605.
[49] V. César, L. C. Misal Castro, T. Dombray, J.-B. Sortais, C. Darcel, S. Labat, K. Miqueu, J.-M. Sotiropoulos, R. Brousses, N. Lugan, G. Lavigne, Organometallics 2013, 32, 4643-4655.
[50] R. Credendino, L. Falivene, L Cavallo, J. Am. Chem. Soc. 2012, 134, 8127-8135. [51] S. Hanada, Y. Motoyama, H. Nagashima, Eur. J. Org. Chem. 2008, 24, 4097-4100. [52] S. Enthaler, S. Inoue, Chem. Asian J. 2012, 7, 169-175. [53] (a) S. Zhou, D. Addis, S. Das, K. Junge, M. Beller, Chem. Commun. 2009, 4883-4885; (b) S.
Enthaler, Eur. J. Org. Chem., 2011,4760-4763. [54] D. Bezier, G. T. Venkanna, J.-B. Sortais, C. Darcel, ChemCatChem 2011, 11, 1747-1750. [55] B. Xue, H. Sun, Y. Wang, T. Zheng, X. Li, O. Fuhr, D. Fenske, Catal. Commun., 2016, 86,
148–150. [56] L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz, V. César, Chem. Rev. 2011,
111, 2705-2733.

79

80
Part 3
Non noble metal pincer complexes in hydrogenation of
carbonyl and carboxylic acid derivatives

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
81

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
82
1. Introduction The catalytic performances of transition-metal complexes are strongly dependent on the
electronic and steric properties of the associated ligands.[1] As such, ligand design is nowadays a
cornerstone in organometallic chemistry for the conception of efficient transition metal
complexes. During the last decade, the field of bifunctional catalysis, which deals with the
cooperation of a metal center with the linked ligands during the catalytic cycle, became one
important area of research. Indeed, bifunctional catalysts became a powerful tool in molecular
synthetic chemistry,[2] and more particularly, transition metal complexes associated with active
ligand sites, have shown enhancement of catalytic activities and new catalytic reactions have
been also developed. On another hand, in the area of homogeneous catalysis, the reactivity of a
catalyst increases at the expense of its stability. Interestingly, pincer-based metal complex
catalysts had become popular as they are able to exhibit an exceptional balance between stability
and reactivity.[3] This balance can be well tuned by a judicious choice of the metal center and/or
systematic ligand modifications, in order to reach better reactivity, stability, and reaction
selectivity, these aspects being crucial in catalyst design. Thus to develop original “pincer
ligands” which are tridentate ligands bind in a meridional fashion to the metal center, the
selection of the right backbone, side arms A and axial E groups are crucial as each parameter has
an influence on the steric restrictions imposed on the remaining coordination sites. The electronic
properties of the complex can be easily modified by the modification of the electron-donating or
–withdrawing character of the substituents on the E moieties, as well as of the nature of the
ligand backbone. Classically, E moieties are two electron donor ligands such as NR2, PR2, OR,
SR, etc. linked to the central backbone by ether, amino or methylene side arm A able also to
modify the stereo-electronic properties of the pincer ligand. (Figure 1).
Figure 1. General structure of a pincer complex.
Since, the pioneering works of Shaw and co-workers [4-5] in the 1970s, pincer-type complexes
have played a very important role in organometallic chemistry and more particularly in
homogeneous catalysis.

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
83
On the basis of the easy pincer ligand modification, the remarkable metal complex stability and
reactivity, numerous pincer complexes of transition metals such as Ru, Fe, Co, Rh, Ir, Ni, Pd, Pt,
and Re, were developed during the last decades. Furthermore, since the discovery of synergic
remarkable effects of bifunctional metal-ligand catalysis by Noyori in hydrogenation[6-8], the NH
effect[9] and the metal ligand cooperation via dearomatization/aromatization of the ligand
backbone,[10-11] PNP pincer complexes have been shown to be versatile catalysts, in particular,
with applications in hydrogenation, dehydrogenation, and related reactions, notably in the
ruthenium series.[12-14] A large number of novel and valuable reactions has been developed using
both stoichiometric and catalytic amounts of ruthenium pincer complexes. The aim of this
introduction part is not to give a complete survey of the pincer complexes but to give a summary
of the developments achieved in this field with first row non-noble metals used in hydrogenation
reactions.
2. Iron pincer complexes After an intensive studies on ruthenium-based pincer complexes, due to the important
concerns about the sustainability and the substitution of noble transition metals by inexpensive
eco-friendly ones, more and more interest was focused towards non-noble metal catalysts in the
last decade. Indeed, iron represents an abundant, inexpensive and low-toxic alternative to
precious metal catalysts.[15-16]
2.1 Hydrogenation of aldehydes and ketones A detailed description of iron catalyzed hydrogenation of aldehydes and ketones are shown in the
part 1.
2.2 Hydrogenation of carboxylic acid derivatives Even if less studied area, iron catalyzed hydrogenation of esters succeeded due to the use
of iron pincer complexes as the catalysts. Indeed, in 2014, Milstein reported the selective
hydrogenation of trifluoroacetic esters to the corresponding alcohols using [(t-Bu-
PNP)FeH2(CO)] 1 as the catalyst (1 mol%) (Scheme 1). These reactions proceeded under mild
conditions (5-25 bar H2 and 40 oC) in the presence of 5 mol% of NaOMe to reduce activated
trifluoroacetic esters to 2,2,2-trifluoroethanol and alcohols corresponding to the ester alkoxy

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
84
groups (52-99% NMR-yields).[17] Noteworthy, no reduction was observed with difluoroacetic
esters.
Scheme 1. Iron catalysed hydrogenation of esters.
Later, in concomitant contributions in 2014, Beller[18] and Guan[19] described the use of
the bifunctional PNP iron pincer complex 2 as an efficient catalyst for the hydrogenation of both
aliphatic and aromatic esters into alcohols under base-free conditions (Scheme 1). Beller has
shown that using 1 mol% of 2 under 30 bar of H2 at 100-120 °C, the efficient, base-free
hydrogenation of different non-activated esters can be performed with a good functional group
tolerance including halides, heteroaromatic motifs (such as furans, pyridines or benzothiazoles),
and non-conjugated alkenyl moieties. Impressively, a pharmaceutical intermediate (used for the
synthesis of Alisporivir, Novartis) which contains 12 amide moieties, a double bond and an
acetyl group has been selectively hydrogenated leading to the corresponding alcohol. Lactones
were also efficiently hydrogenated to the corresponding diols. Based on B3PW91 DFT
calculations a suitable mechanism was proposed which indicated a trans-dihydride species as
catalytically active intermediate and involved a concerted hydrogen transfer from the iron center
and the PNP ligand.
In the meantime, Guan[19] developed a similar hydrogenation procedure using the same
complex 2 although with higher catalyst loading (3 mol%) but at lower pressure (10-16 bar) of H2
at 115 °C. Under neat conditions, an industrial important substrate (CE-1270) derived from

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
85
coconut oils and containing mixtures of C12-C16 esters [methyl laurate, methyl myristate and
C10 and C16 methyl esters (73:26:1)] was successfully hydrogenated.
In 2014, the first iron PNP pincer 2 catalyzed nitrile hydrogenation was presented by
Beller.[20] Notably, without any additives such as a base, aryl, alkyl, heterocyclic nitriles and
dinitriles were efficiently hydrogenated to the corresponding amines (Scheme 2). Excellent
functional group tolerance was achieved for ,-unsaturated compounds as well as for substrates
containing halogens, ester, ether, amino and acetamido substituents (40-99% yields).
Scheme 2. Hydrogenation of nitriles catalyzed by iron pincer complex.
Very recently, Milstein and coworkers synthesized PNP iron complex 3 and studied its
catalytic applications in hydrogenation of nitriles (Scheme 3).[21] This complex, in the presence of
KHMDS and NaHBEt3 effectively catalyzed the hydrogenation of various (hetero) aromatic,
benzylic, and aliphatic nitriles yielding the corresponding primary amines selectively in good to
excellent yields at 140 oC and 60 bar H2.
Scheme 3. Hydrogenation of nitriles catalyzed by 3.
A series of iron pincer complexes with the general formula [(R-PNHP)Fe(H)(CO)(BH4)]
(where R = tBu, Cy, iPr, Ph, Et) were synthesized by Langer in 2016 and were used as catalysts
for hydrogenation of esters and amides (Scheme 4).[22] A small variation on the phosphorous
ligand structure has a surprising influence on the catalytic properties and stability of the pincer
complexes. The complex bearing the less sterically hindered and the stronger σ-donating

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
86
diethylphosphino moities lead to an improved activity in the hydrogenation of esters to alcohols,
compared to that of the previously reported iPr-substituted complexes.[18] The highly active EtPNP complex was then used in the direct hydrogenation of amides to alcohols and amines under
mild conditions.
Scheme 4. Hydrogenolysis of amides using iron pincer complexes.
Very recently, Milstein reported the catalytic hydrogenation of activated amides to
alcohols and amines using the iron pincer complex 4 in the presence of 3 equiv. of KHMDS at
140 oC under 60 bar H2.[23] Thus, the iron pincer complex [(iPr-PNP)Fe(H)(BH4)(CO)] and [(iPr-
PNP)Fe(H)(Br)(CO)] are effective pre-catalysts for the selective hydrogenation of a wide range
of N-substituted 2,2,2, trifluoroacetamides to trifluoroalcohol and the corresponding amines.
Scheme 5. Hydrogenolysis of activated amides by Milstein.
2.3 Dehydrogenation of alcohols The reverse dehydrogenation reaction can be also promoted by the same PNP iron
complexes. Indeed, in 2014, Schneider and Jones reported the acceptorless dehydrogenation of
alcohols using the well-defined PNP iron-based catalyst 2 (Scheme 6). Using 0.1-1 mol% of 2, in
refluxing THF or toluene for 8-48 h, 1-arylethanol compounds were dehydrogenated to the
corresponding acetophenone derivatives in good isolated yields upon release of dihydrogen.[24]
Starting from primary alcohols, esters were obtained in 60-85% yields. The same year, they

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
87
reported an acceptorless dehydrogenation and a hydrogenation of N-heterocycles with a
molecular-defined iron complex 2.[25] Under optimized conditions (3 mol% 2, 140 °C, reflux, 30
h) several heterocycles such as 1,2,3,4-tetrahydroqinaldine, 2-methylindoline and 2,6-
dimethylpiperidine were dehydrogenated giving the corresponding compounds in 58-91% yields
(Scheme 6). Notably the reverse hydrogenation reaction can be performed using 3 mol% of 2, 10
mol% of KOtBu in THF at 80 °C under 5-10 bar of H2.
Scheme 6. Iron catalyzed dehydrogenation reactions.
2.4 Dehydrogenation for the production of hydrogen and carbon dioxide From a sustainable point of view, carbon dioxide is a nontoxic, abundant, and economical
C1 building block in chemical commodities area (urea, formic acid, formaldehyde, methanol,
etc.).[26-27] The direct hydrogenations of carbon dioxide to formaldehyde, methanol and methane
or the reverse reaction are an important area of research, and more particularly, the carbon
dioxide hydrogenation to formic acid as a reservoir of hydrogen. In iron catalysis, following the
pioneering contributions of Jessop[28]and Beller & Laurenczy[29] Milstein reported the use of the
well-defined iron complex [(t-BuPNP)FeH2(CO)] as a catalyst for the selective decomposition of
formic acid to dihydrogen and carbon dioxide in the presence of Et3N (50 mol%).[30]
In 2013, Beller reported the use of the PNP pincer complex 2 for the dehydrogenation of
methanol in the presence of water (MeOH/H2O 9:1) to carbon dioxide and hydrogen at 91 °C in
the presence of KOH as a base with TON up to 10,000 (Scheme 7).[31] Later in 2015, Hazari
reported a base free methanol dehydrogenation using a PNP iron ccomplex [Fe(PNP-

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
88
iPr)(OCHO)(H)(CO)] 5 (0.1 mol%) and Lewis acid co-catalyst (LiBF4, 10 mol%) and achieved
TON up to 50,000.[32] (Scheme 7)
Scheme 7. Iron-catalyzed dehydrogenation of methanol.
Hazari and Schneider reported the use of the same complex 5 as an active catalyst (0.1
mol%) for the highly efficient dehydrogenation of formic acid to dihydrogen and carbon dioxide
in the presence of LiBF4 in refluxing dioxane (TON up to 106, TOF up to 196000 h-1). [33]
3. Cobalt pincer complexes
3.1 Hydrogenation In 2012 and 2013, the group of Hanson reported several contributions on PNP pincer
cobalt-based complexes for the hydrogenation of alkenes, imines, aldehydes and ketones.[34-35]
The cobalt(II) hydride PNHP 6 and PNMeP 7 pincer complexes exhibited high activities,
especially in C=C double bond hydrogenation (Scheme 8). At room temperature, using 2 mol% 6
or 7, associated to 2 mol% of H[BArF4](Et2O)2 in the case of 6, 1 bar H2 within 24-40 hours,
more than 20 styrenes and aliphatic alkenes have been hydrogenated leading to the corresponding
alkanes in 66-99% yields (Scheme 8). Notably, carboxylic acid and ester were tolerated. The
association of the complex 6 with H[BArF4](Et2O)2 (2 mol%) also catalysed the hydrogenation of
aldehydes, ketones and aldimines under 1-4 bar of H2 at 25-60 °C (70-99% yields). Unsaturated
ketones and aldehydes were fully reduced under such conditions.
Scheme 8. Cobalt catalyzed hydrogenation of alkenes and carbonyl derivatives.

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
89
In 2015, Kempe reported a highly active PNNNP pincer Co catalyst 8 for the
homogeneous hydrogenation of C=O bonds.[36] Using 0.25-3 mol% of 8 associated to 0.5-6 mol%
of NaOtBu (2 equiv. with respect to the precatalyst) in 2-methylbutanol under 20 bar of hydrogen
at RT, various dialkyl, diaryl, and aryl-alkyl ketones were hydrogenated in 64-99% GC-yields.
Conjugated and non-conjugated aldehydes were chemoselectively reduced leading to the
corresponding unsaturated primary alcohols (Scheme 9).
Scheme 9. Cobalt catalyzed hydrogenation of ketones.
In 2015, Milstein described PNNEt2 and PNNH supported cobalt (II) pincer complexes for
application in the hydrogenation of esters to alcohols (Scheme 10). The PNNH-based cobalt
pincer complex 9 was found to be the best catalyst. Notably, phenyl and alkyl alkanoate can be
reduced in 50-87%. -valerolactone was also reduced leading to 1,4-pentanediol in 50% yield. By
contrast, no reaction occurred with methyl benzoate and 2,2,2-trifluoroethyl trifloroacetate.[37]
Scheme 10. Cobalt catalyzed hydrogenation of nitriles and esters.
The same group reported the catalyzed hydrogenation of nitriles to primary amines promoted by
the same complex 9 (Scheme 10). Indeed, using 2 mol% of 9 in the presence of NaEt3BH (2
mol%) and NaOEt (4.4 mol%), the hydrogenation of various (hetero)-aromatic, benzylic, and

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
90
aliphatic nitriles to the corresponding primary amines was performed at 135 oC in benzene under
30 bar H2.[38] Interesting functional group tolerance (halide, -NH2, nitro) was observed.
3.2 Dehydrogenation of alcohols In 2013, Hanson developed the acceptorless dehydrogenation of alcohols to ketones using
the cationic pincer cobalt complex 10. Using 5 mol% of 10 generated from the association of the
complex 6 with H[BArF4](Et2O)2 in toluene at 120 °C for 24 h, secondary alcohols led to the
corresponding ketones in 56-95% GC-yields. Notably the dehydrogenation of 4-methoxybenzyl
alcohol led to 4-meyhoxybenzaldehyde in only 24% yield. When performing the reaction in the
presence of 1.1 equiv. of alkyl or benzyl amines for 27-52 h at 120 °C, the corresponding imines
was obtained as the major products (56-98% GC-yield) with trace amount of the amine resulting
from the reduction of the imine.[39] (Scheme 11)
Scheme 11. Cobalt catalyzed dehydrogenation of secondary alcohols and reductive amination.
Similarly, based on this work, and using hydrogen borrowing technology, several
contributions described the synthesis of secondary amines starting from alcohols and primary
amines. Zhang then reported the direct N-alkylation of both aromatic and aliphatic amines by
alkyl and benzyl alcohols catalyzed by the pincer complex 10 (2 mol%) under base-free
conditions but in the presence of 4 Å molecular sieves in refluxing toluene for 48 h (Scheme 12).
A range of alcohols and amines including both aromatic and aliphatic substrates were efficiently
converted to secondary amines in 74-98% yields with only small amount of imine intermediates
in specific examples.[40a] Kempe also developed the N-alkylation of primary (hetero)anilines with
alcohols catalyzed by PN5P pincer cobalt complexes such as 11. Using 2 mol% of 11 in the
presence of 1.2 equivalents of KOtBu in toluene at 80 °C, the corresponding secondary alkylated
anilines were obtained in 51-96% yields.[40b] Only halides were reported as tolerated functional
groups. Interestingly, using a sequential procedure, unsymmetrical secondary diamines can be
prepared from 1,3-diaminobenzene in good yields (Scheme 12).

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
91
Using PNCNP pincer cobalt complexes 12, in 2016, Kirchner described a general method
for the N-alkylation of primary (hetero)anilines with alcohols. With 2 mol% of 12a as the
catalyst, in the presence of 1.3 equiv. of KOtBu in toluene at 80 °C, primary anilines were
efficiently transformed into the corresponding secondary anilines. The reaction is more difficult
with secondary aniline as only low yields (10%) were observed. Halides, heteroaromatic moieties
(pyridyl, furanyl), conjugated C=C bond and remote C=C bond can be tolerated. The reaction can
be conducted under base free conditions when performing the reaction using 2 mol% of 12b in
the presence of 3 Å molecular sieves in toluene at 130 °C for 16 h.[41]
Scheme 12. N-alkylation of amines with alcohols catalysed by cobalt PNP complexes.
Very recently, Kempe described the alkylation of unactivated acetates and acetamides
catalyzed by cobalt pincer complexes 11 and 13 in the presence of t-BuOK (1.2 -1.5 equiv.)
under mild conditions (80-100 oC) (Scheme 13). Starting from acetamide derivatives, 2.5 mol%
of 11 promoted the catalytic alkylation using primary alcohols in the presence of 1.2 equiv. of
t-BuOK in THF at 100 °C for 24 h. The corresponding alkylated esters were obtained in 55-93%
yields. The alkylation of acetate compounds is catalyzed by the complex 13 (5 mol%) at 80 °C,
1.2 equiv. of t-BuOK in toluene (48-82% yields).[42]
Scheme 13. Alkylation of acetates and acetamides catalyzed by cobalt complexes.

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
92
In 2015, Jones reported the reversible dehydrogenation/hydrogenation of N-heterocycles
catalysed by cobalt pincer complexes in the absence of an acceptor.[43] To perform efficiently the
dehydrogenation reaction, 10 mol% of the complex 10 was used at 150 oC for 3-4 days in xylene
(5 examples, 65-98% GC-yields). The reverse hydrogenation reaction took place at 120 oC for 48
h in THF with 5 mol% of 10 under 10 bar of hydrogen (19-99% GC-yields) (Scheme 14).
Scheme 14. Cobalt catalyzed hydrogenation and dehydrogenation reactions.
4. Nickel PNP pincer complexes The area of pincer nickel complexes is well developed,[44] more particularly, nickel PNP
pincer complexes were also efficiently used in reduction area.
In 2009, Guan reported that the hydrosilylation of aldehydes can be catalyzed using well-
defined PONOP pincer hydrido nickel complex 14 (0.2 mol%) in the presence of 1.3 equiv. of
PhSiH3 as a reducing agent at RT or 70 °C for 1-24 h (Scheme 15).[45] This catalytic system was
less efficient for the hydrosilylation of ketones as 1 mol% of catalyst has to be used at 70 °C for
24 h to obtained the corresponding alcohols with low to moderate conversions.
Scheme 15. Nickel catalyzed hydrosilylaton of aldehydes.
Alkene hydrogenations with cationic and neutral nickel PNP pincer complexes as the
catalysts have been developed by Hanson et al. (Scheme 16). More specifically, the cationic
nickel hydride complex 15 was able to hydrogenate a short scope of non-functionalized alkenes
in 48-99% NMR-yields using 10 mol% of 15 under relatively mild conditions (80 °C, 4 bar H2,
24-48 h, THF-d8).[46] Noticeably, aldehydes were reduced in low yields under similar conditions.

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
93
Scheme 16. Nickel catalyzed hydrogenation of styrene.
Reduction of carbon dioxide to formaldehyde via to bis(silyl)acetal derivatives can be
also performed using a catalytic system based on a bis(phosphino)boryl nickel hydride complex
16 combined with B(C6F5)3, in the presence of triethylsilane at 60 °C. Interesting activity was
obtained with TON up to 1200 and TOF up to 56 h-1.[47] (Scheme 17)
Scheme 17. Nickel catalyzed hydrosilylation of CO2.
Enthaler and Junge have reported the use of a pincer hydrido complex 17 for the
hydrogenation of sodium bicarbonate to formate. Using 0.025 mol% of 17 in methanol under 55
bar of H2 at 150 °C for 20 h, sodium bicarbonate was reduced to sodium formate with TON up to
3038. 17 was also able to perform the reverse reaction, namely the decomposition of formic acid
to CO2 and H2. Running the reaction with 17 as the catalyst in propylene carbonate in the
presence of n-octylamine (HCHO/ amine = 11:10) at 80 °C, TON up to 626 was reached.[48]
Figure 2. Nickel PNP pincer complex.
5. Manganese pincer complexes Manganese complexes are frequently employed in oxidations, cross coupling reactions
and C-H activations.[49-50] By contrast, in comparison with the other first row transition metals,
manganese is less employed for catalytic reductions with the main applications in hydrosilylation
and electrocatalytic reactions. Scarce examples of manganese were also known in hydrogenation

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
94
area since several decades. Hydrogenation of alkenes can be performed using Mn2(CO)10 as the
catalyst at elevated temperature and pressure (160 °C, 200 atm H2).[51] cis-(CO)4(PPh3)MnH was
reported as a catalyst for the hydrogenation of 1-octene under milder conditions (1 atm H2, neat
conditions, RT) under UV light activation with TOF up to 10 h−1.[52]
On another hand, the great majority of reports on the use of manganese in homogeneous
catalysis have been carried out with simple commercially available inorganic salts or
organometallic manganese precursors. However, during the last decade, it was clearly shown that
a fine and rational design of the ligand bound to manganese center led to active and selective
catalysts. This paragraph will describe the use of PNP pincer manganese complexes in
homogeneous catalytic reduction.
In 2009, Ozerov and Nocera reported the synthesis of pincer meridionally ligated
complexes (PNP)Mn(CO)3 18 and (PNP)Mn(CO)2 19. These complexes exhibited reversible
electrochemical oxidation events at low potentials (Scheme 18).[53]
Scheme 18. PNP Mn(CO)x complexes.
In 2014, Kuwata and Ikariya reported the synthesis of iron, cobalt, and manganese
complexes [MCl2(NNN)] (M = Co, Mn) bearing a 2,6-bis(5-tert-butyl-1H-pyrazol-3-yl)pyridine
NNN pincer-type ligand by the treatment of the ligand with divalent cobalt and manganese
chloride salts[54] (Scheme 19).
Scheme 19. Synthesis of NNN-pincer manganese and cobalt complexes.

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
95
In 2013, Trovitch reported the synthesis of a redox-active PNP Mn complex 22 which was
an active catalyst in the hydrosilylation of ketones and esters under mild conditions (Scheme
20).[55] Using 0.01-1 mol% of 22 in the presence of 1 equiv. of phenylsilane, aryl and alkyl
ketones can be hydrosilylated at 25 °C for 4 min.-24 h leading to a mixture of tertiary and
quaternary silanes in 80-99% conversions. It must be pointed out the exceptional activity of this
catalyst as TOF up to 76,800 h-1 can be reached for the hydrosilylation of cyclohexanone.
Hydrosilylation of esters was also performed using 1 mol% of 22 in the presence of 1 equiv. of
phenylsilane leading to a mixture of tertiary and quaternary silicon derivatives with relatively
modest TOFs.
Scheme 20. Hydrosilylation of ketones by a manganese bisiminopyridine diphosphine complex.
In 2016, Kirchner reported the synthesis of a series of vanadium, chromium, and
manganese PNP complexes of the types including [Mn(PNP)Cl2] 23 for application in oxidative
homo-coupling of aryl Grignard reagents leading to symmetrical biaryls in the presence of MeI as
oxidizing agents (Scheme 21).[56]
Scheme 21. Synthesis of PNNNP pincer complexes.
Recently, Milstein[57] and Kirchner[58] independently reported the efficient coupling of
alcohols and amines leading selectively to the corresponding imines catalyzed by well-defined
pincer Mn(I) complexes 24 and 25 (3 mol%) at 135-140 °C (Scheme 22).

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
96
Scheme 22. Manganese catalysed dehydrogenative coupling of alcohols and amines.
Recently, the (PNHP)Mn(CO)2 (I) carboxylate complex 27 was prepared via 1,2-addition
of either formic or oxalic acid to (PNP)Mn(CO)2 26. The catalytic activity of the carboxylate
complex 26 was studied in the decomposition of formic acid (Scheme 23).[59] 26 was active for
both dehydration and dehydrogenation of formic acid.
Scheme 23. Manganese complex for catalyzed decomposition of formic acid.
In summary, pyridine and aliphatic backbone supported pincer complexes have been well
studied with Ru, Fe and Co (Figure 3). Interestingly, such pincer complexes are active in
hydrogenation and dehydrogenation reactions. Especially, aliphatic PNP pincer complexes
showed excellent activity in hydrogenation of nitriles and esters with Fe[18, 20] and Ru[60]. Inspired
from these reported works, the aim of this part is to describe the synthesis of iron- and
manganese- PNP pincer complexes and their applications in catalytic hydrogenation of carbonyl
and carboxylic acid derivatives.

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
97
Figure 3. Selected examples of pyridine and aliphatic supported pincer complexes in
hydrogenation and dehydrogenation reactions.
6. References [1] K. Hindson, B. de Bruin, Eur. J. Inorg. Chem 2012, 340-342. [2] T. Ikariya, S. Masakatsu, in Topics in Organometallic Chemistry, Bifunctional Molecular
Catalysis;, Vol. 37, Springer: Berlin, Heidelberg, 2011. [3] D. W. Lee, C. M. Jensen, D. M. Morales, Organometallics 2003, 22, 4744-4749. [4] C. J. Moulton, B. L. Shaw, J. Chem. Soc. Dalton Trans. 1976, 1020–1024. [5] G. V. Koten, K. Timmer, J. G. Noltes, A. L. Spek, J. Chem. Soc. Chem. Commun. 1978,
250–252. [6] H. Doucet, T. Ohkuma, K. Murata, T. Yokozawa, M. Kozawa, E. Katayama, A. F.
England, T. Ikariya, R. Noyori, Angew. Chem. Int. Ed. 1998, 37, 1703-1707. [7] R. Noyori, T. Ohkuma, Angew. Chem. Int. Ed. 2001, 40, 40-73. [8] R. Noyori, M. Yamakawa, S. Hashiguchi, J. Org. Chem. 2001, 66, 7931-7944. [9] B. Zhao, Z. Han, K. Ding, Angew. Chem. Int. Ed. 2013, 52, 4744-4788. [10] T. Zell, D. Milstein, Acc. Chem. Res. 2015, 48, 1979-1994. [11] C. Gunanathan, D. Milstein, Chem. Rev. 2014, 114, 12024-12087. [12] H. A. Younus, W. Su, N. Ahmad, S. Chen, F. Verpoort, Adv. Synth. Catal. 2015, 357,
283–330. [13] E. Balaraman, D. Milstein, Top. Organomet. Chem. 2014, 48, 19-43. [14] C. Gunanathan, D. Milstein, Chem. Rev. 2014, 114, 12024-12087. [15] C. Bolm, J. Legros, J. L. Paih, L. Zani, Chem. Rev. 2004, 104, 6217-6254. [16] S. Enthaler, K. Junge, M. Beller, Angew. Chem. Int. Ed. 2008, 47, 3317-3321. [17] T. Zell, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2014, 53, 4685-4689. [18] S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F.
Gallou, M. Beller, Angew. Chem. Int. Ed. 2014, 53, 8722-8726. [19] S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause,
H. Guan, J. Am. Chem. Soc. 2014, 136, 7869-7872.

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
98
[20] C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge, K. Junge, M. Beller, Nat. Commun. 2014, 5, 4111.
[21] S. Chakraborty, G. Leitus, D. Milstein, Chem. Commun. 2016, 52, 1812-1815. [22] F. Schneck, M. Assmann, M. Balmer, K. Harms, R. Langer, Organometallics 2016, 35,
1931-1943. [23] J. A. Garg, S. Chakraborty, Y. Ben-David, D. Milstein, Chem. Commun. 2016, 52, 5285-
5288. [24] S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen,
W. D. Jones, S. Schneider, ACS Catal. 2014, 4, 3994−4003. [25] S. Chakraborty, W. W. Brennessel, W. D. Jones, J. Am. Chem. Soc. 2014, 136, 8564-
8567. [26] E. A. Quadrelli, G. Centi, J.-L. Duplan, S. Perathoner, ChemSusChem 2011, 4, 1194-
1215. [27] W. Wang, S. Wang, X. Ma, J. Gong, Chem. Soc. Rev. 2011, 40, 3703-3727. [28] C.-C. Tai, T. Chang, B. Roller, P. G. Jessop, Inorg. Chem. 2003, 42, 7340-7341. [29] C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G.
Laurenczy, M. Beller, Angew. Chem. Int. Ed. 2010, 49, 9777-9780. [30] T. Zell, B. Butschke, Y. Ben-David, D. Milstein, Chem. Eur. J. 2013, 19, 8068-8072. [31] E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge,
M. Beller, Angew. Chem. Int. Ed. 2013, 52, 14162-14166. [32] E. A. Bielinski, M. Förster, Y. Zhang, W. H. Bernskoetter, N. Hazaria, M. C.
Holthausenb, ACS Catal. 2015, 5, 2404-2415. [33] E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Wurt le, W. H.
Bernskoetter, N. Hazari, S. Schneider, J. Am. Chem. Soc. 2014, 136, 10234-10237. [34] G. Zhang, B. L. Scott, S. K. Hanson, Angew. Chem. Int. Ed. 2012, 51, 12102-12106. [35] G. Zhang, K. V. Vasudevan, B. L. Scott, S. K. Hanson, J. Am. Chem. Soc. 2013, 135,
8668-8681. [36] S. Rösler, J. Obenauf, R. Kempe, J. Am. Chem. Soc. 2015, 137, 7998-8001. [37] D. Srimani, A. Mukherjee, A. F. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. Shimon, Y.
Ben David, D. Milstein, Angew. Chem. Int. Ed. 2015, 54, 12357-12360. [38] A. Mukherjee, D. Srimani, S. Chakraborty, Y. Ben-David, D. Milstein, J. Am. Chem. Soc.
2015, 137, 8888-8891. [39] G. Zhang, S. K. Hanson, Org. Lett. 2013, 15, 650-653. [40] a) G. Zhang, Zhiwei Yin, S. Zheng, Org. Lett. 2016, 18, 300-303. b) S. Rosler, M. Ertl, T.
Irrgang, R. Kempe, Angew. Chem. Int. Ed. 2015, 54, 15046-15050. [41] M. Mastalir, G. Tomsu, E. Pittenauer, G. Allmaier, K. Kirchner, Org. Lett. 2016, 18,
3462. [42] N. Deibl, R. Kempe, J. Am. Chem. Soc. 2016, 138, 10786-10789. [43] R. Xu, S. Chakraborty, H. Yuan, W. D. Jones, ACS Catal. 2015, 5, 6350-6354. [44] D. Zargarian, A. Castonguay, D. M. Spasyukand, Top. Organomet. Chem. 2013, 40, 131-
174. [45] S. Chakraborty, J. A. Krause, H. Guan, Organometallics 2009, 28, 582-586. [46] K. V. Vasudevan, B. L. Scott, S. K. Hanson, Eur. J. Inorg. Chem. 2012, 4898-4906. [47] P. Ríos, N. Curado, J. López-Serrano, A. Rodríguez, Chem. Commun. 2016, 52, 2114-
2117.

Part 3 Non noble metal pincer complexes in hydrogenation of carbonyl and carboxylic acid derivatives
99
[48] S. Enthaler, A. Brueck, A. Kammer, K. Junge, E. Irran, S. Guelak, ChemCatChem 2015, 7, 65-69.
[49] D. A. Valyaev, G. Lavigne, N. Lugan, Coord. Chem. Rev. 2016, 308, 191-235. [50] J. R. Carney, Barry. R. Dillon, S. P. Thomas, Eur. J. Org. Chem.2016, 23, 3912-3929. [51] T.A. Weil, S. Metlin, I. Wender, J. Organomet. Chem. 1973, 49, 227-232. [52] P. L. Bogdan, P. J. Sullivan, T. A. Donovan Jr., J. D. Atwood, J. Organomet. Chem.
1984, 269, 51-54. [53] A. T. Radosevich, J. G. Melnick, S. A. Stoian, D. Bacciu, C.-H. Chen, B. M. Foxman, O.
V. Ozerov, D. G. Nocera, Inorg. Chem. 2009, 48, 9214-9221. [54] K. Umehara, S. Kuwata, T. Ikariya, Inorg. Chim. Acta 2014, 413, 136-142. [55] T. K. Mukhopadhyay, M. Flores, T. L. Groy, R. J. Trovitch, J. Am. Chem. Soc. 2014, 136,
882-885. [56] M. Mastalir, M. Glatz, B. Stöger, M. Weil, E. Pittenauer, G. Allmaier, K. Kirchner, Inorg.
Chim. Acta 2016, 455, 707–714. [57] A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben-David, N.-A. Espinosa-
Jalapa, D. Milstein, J. Am. Chem. Soc. 2016, 138, 4298-4301. [58] M. Mastalir, M. Glatz, N. Gorgas, B. Stöger, E. Pittenauer, G. Allmaier, L. F. Veiros, K.
Kirchner, Chem. Eur. J. 2016, 22, 12316-12320. [59] A. M. Tondreau, J. M. Boncella, Organometallics 2016, 35, 2049-2052. [60] S. Werkmeister, K. Junge, M. Beller, Org. Process Res. Dev. 2014, 18, 289-302.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
100
Part 3 Chapter 1
Iron and manganese catalyzed nitrile hydrogenation

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
101

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
102
1. Introduction Catalytic reduction of nitriles with molecular hydrogen is a potential method and
straightforward approach to obtain primary amines that are most important in industry for the
synthesis of both fine and bulk chemicals and for pharmaceuticals.[1-2] For example, the key
constituent of Nylon-6,6, hexamethylenediamine, is derived from reduction of adiponitrile. In
addition, nitrile functionalities occur in several natural and synthetic organic compounds, which
include pharmaceuticals, and can be synthesized by the nucleophilic substitution reactions of
alkyl halides with metal cyanides,[3] the dehydration of primary amides,[4-6] and the cross-
metathesis reaction with acrylonitriles.[7] More importantly, the catalytic hydrogenation of nitriles
represents an atom-economic and valuable route for the synthesis of amines. However, compared
to reductions of C=C, C=O, and C=N bonds, the selective hydrogenation of nitriles to primary
amines is difficult due to crucial selectivity problem arises concomitant with the formation of
other side products such as primary, secondary and tertiary amines, as well as intermediate
imines (Scheme 1). The reaction of the primary amine 3 with the imine intermediate 2 gives the
secondary imine 4 with elimination of ammonia. Subsequent reduction of the secondary imine 4
gives the diamine 5. The formation of significant amounts of side products such as secondary
imines/amines lowers the selectivity and overall atom efficiency of the reaction. Thus, the control
of the selectivity is the challenging factor for this reaction.
Conventional stoichiometric reduction methods involve strong reducing reagents, such as LiAlH4
and borane,[8] and hydrosilylation in the presence of tetramethyldisiloxane and titanium
isopropoxide.[9] These methods are not environmentally benign due to the coproduction of
stoichiometric amounts of waste metal salts. On the other hand, in industry, heterogeneous
catalysts, usually Raney-Ni and -Co, are used for performing such transformation.[10-11] However,
limitations of these catalysts are not tolerating towards functional groups and need additives, such
as ammonia, to avoid the formation of side products. Thus, the development of well-defined
homogeneous transition metal catalysts is more interesting in order to reach better chemo-
selectivity. In this line, several ruthenium, iridium and rhenium based catalyst are known for this
transformation.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
103
Scheme 1. Hydrogenation of nitriles and possible side reactions.
2. Ruthenium catalysts for hydrogenation of nitriles Numerous ruthenium complexes have been reported for the catalytic hydrogenation of
nitriles whether as an in situ generated catalytic system or well defined complexes. The group in
Rostock made a significant contribution using a combination of [Ru(cod)(methylallyl)2] 6 and
phosphine/carbene based catalysts for the selective hydrogenation of nitriles to primary amines.
In 2008, the group in Rostock developed a Ru/PPh3 based catalytic system for nitrile
hydrogenation in the presence of 10 mol% of KOtBu as a base.[12] This protocol permits the
selective hydrogenation of various nitriles into primary amines at 80-140 oC and 50 bar H2 with
good to excellent yields (Scheme 2).
Scheme 2. Hydrogenation of nitriles by using Ru/PPh3 catalytic system.
The same year an in situ catalyst obtained from [Ru(cod)(methylallyl)2] and DPPF[1,2-
bis-(diphenylphosphino)ferrocene] was developed for the hydrogenation of various nitriles to
give primary amines.[13] Under optimized conditions, aromatic, heteroaromatic and aliphatic
nitriles were hydrogenated chemoselectively into corresponding primary amines (Scheme 3).

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
104
Scheme 3. Hydrogenation of nitriles using an in situ generated ruthenium/DPPF catalytic system.
Later, an efficient ruthenium catalytic system generated by combination of the
[Ru(cod)(methylallyl)2] with imidazolyl phosphines 8 or 9 was developed in Rostock for the
hydrogenation of aromatic and aliphatic nitriles leading the corresponding primary amines in
good to excellent yields (Scheme 4).[14]
Scheme 4. Reduction of nitriles by an in situ catalyst obtained from ruthenium and imidazolyl phosphine.
Recently, the same group described a novel catalytic system based on the combination of
[Ru(cod)(methylallyl)2] and the ligand 10 for the selective hydrogenation of nitriles to primary
amines (Scheme 5).[15] The main advantage of this protocol is that no basic additive was
necessary to achieve good chemoselectivities to the primary amine. Furthermore, a variety of
aromatic and aliphatic nitriles can be hydrogenated at mild conditions (50 oC, 15 bar H2).
Scheme 5. Reduction of nitriles by ruthenium and 10.
In 2009, Beller et al. reported a ruthenium / carbene catalytic system for the selective
hydrogenation of nitriles (Scheme 6). Using this catalyst, different aromatic nitriles were reduced
with excellent chemoselectivity at ambient temperature (40 oC) and pressure (35 bar H2).

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
105
Noticeably, one aliphatic nitrile could be also reduced with >99% selectivity and 12% yield.
However, the substrate scope of the Ru / carbene catalyst is lower compared to that of the Ru /
phosphine systems but achieves good selectivity.[15a]
Scheme 6. Hydrogenation of nitriles using ruthenium/NHC 11 catalytic system.
In 2007, Morris and co-workers reported the use of ruthenium hydride complexes for the
hydrogenation of benzonitrile.[16] After activation with t-BuOK, the complex 12 shows excellent
activity for the hydrogenation of benzonitrile to benzylamine in toluene (Scheme 7).
Scheme 7. Hydrogenation of benzonitrile using PNNP ruthenium hydride complex 12.
In 2011, Leitner and co-workers studied PNP pincer supported ruthenium hydride
complex for nitrile reduction.[17] This catalytic system allows for the hydrogenation of various
aliphatic and aromatic nitriles to the corresponding primary amines under optimized reaction
conditions. To achieve full conversion, high pressure (75 bar of H2) and high temperature (135 oC) were necessary. Addition of a small amount of water (5 equiv. relative to the catalyst)
provided increased conversions and selectivity towards primary amines (Scheme 8).
Scheme 8. Hydrogenation of nitriles by using the PNP ruthenium hydride complex 13.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
106
The selective hydrogenation of aromatic and aliphatic nitriles into amines and imines was
described by Prechtl in 2015.[17a] Using a ruthenium pincer complex, the selectivity towards
amines or imines could be controlled by simple parameter changes. The reactions were conducted
under very mild conditions between 50–100 oC at 0.4 MPa of H2 pressure without any additives
at low catalytic loadings of 0.5–1 mol%, which results in quantitative conversions and high
selectivity.
Scheme 9. Hydrogenation of nitriles catalyzed by 14 and 15.
In 2015, Beller and coll. developed an efficient ruthenium-catalyzed protocol for the
reduction of various nitriles (Scheme 10). By applying the commercially available Ru-Macho-BH
complex 16, a variety of aliphatic, aromatic and (hetero)cyclic nitriles including the industrially
important substrate adipodinitrile are selectively transformed to the corresponding primary
amines at 100 oC under 30 bar H2 without addition of base. Modelling studies suggest an outer-
sphere mechanism.[17b]
Scheme 10. Hydrogenation of nitriles using Ru MACHOBH complex 16.
In 2012, Bruneau et al. reported the use of ruthenium-benzylidene and ruthenium-
indenylidene catalysts for the selective hydrogenation of nitriles into the corresponding primary
amines in the presence of 3 mol% of KOtBu at 80 oC under 20 bar H2 (Scheme 11). The catalytic
amount of base favours the reduction of the nitrile to the amine and strongly inhibits the
formation of secondary amines.[18]
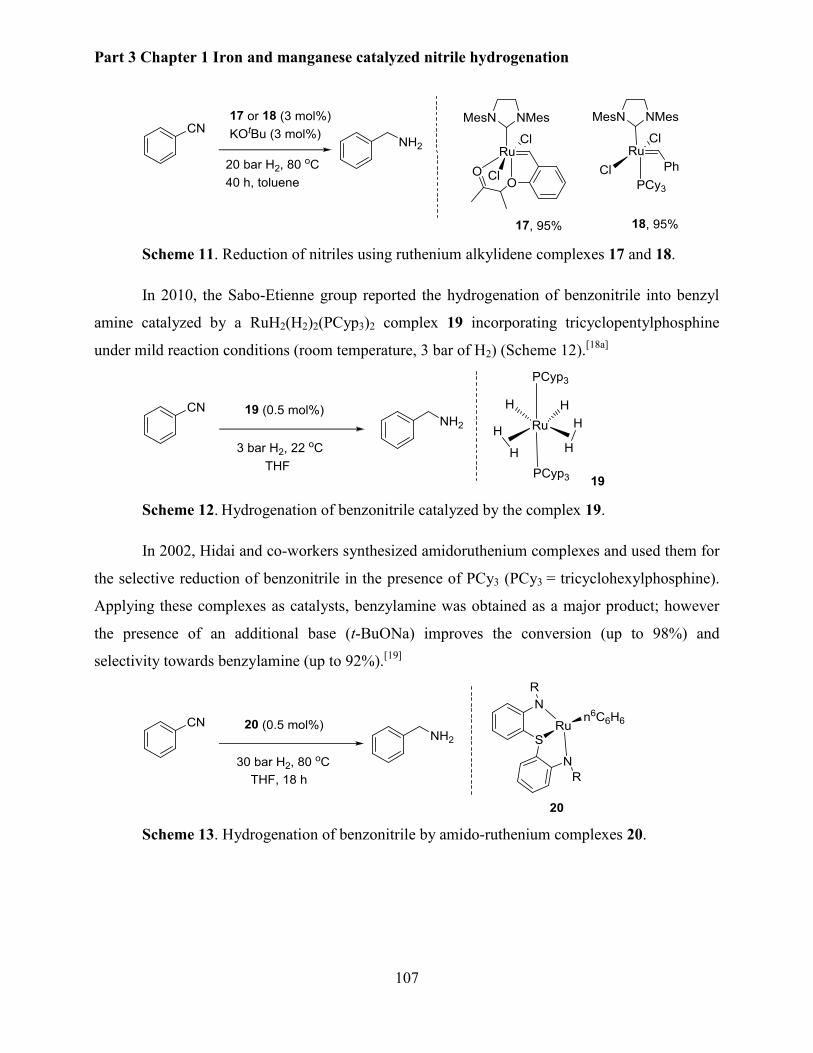
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
107
Scheme 11. Reduction of nitriles using ruthenium alkylidene complexes 17 and 18.
In 2010, the Sabo-Etienne group reported the hydrogenation of benzonitrile into benzyl
amine catalyzed by a RuH2(H2)2(PCyp3)2 complex 19 incorporating tricyclopentylphosphine
under mild reaction conditions (room temperature, 3 bar of H2) (Scheme 12).[18a]
Scheme 12. Hydrogenation of benzonitrile catalyzed by the complex 19.
In 2002, Hidai and co-workers synthesized amidoruthenium complexes and used them for
the selective reduction of benzonitrile in the presence of PCy3 (PCy3 = tricyclohexylphosphine).
Applying these complexes as catalysts, benzylamine was obtained as a major product; however
the presence of an additional base (t-BuONa) improves the conversion (up to 98%) and
selectivity towards benzylamine (up to 92%).[19]
Scheme 13. Hydrogenation of benzonitrile by amido-ruthenium complexes 20.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
108
3. Hydrogenation of nitriles with other metal complexes Low-valent molybdenum and tungsten amides M(NO)(CO)(PNP) {M = Mo, 21; W, 22)
were found to be active catalysts for the hydrogenation of various nitriles to the corresponding
imines, primary amines and N-substituted imines with high selectivity for the latter type of
product.[20] A wide range of nitriles could be hydrogenated to corresponding imines along with
primary amines when performing the reaction at 140 °C under 60 bar of H2 in THF.
Scheme 14. Hydrogenation of nitriles using Mo and W pincer complexes 21 and 22.
Berke et al. developed an efficient homogeneous rhenium-catalyzed hydrogenation of
nitriles with good selectivity for symmetrical secondary amines or tertiary amines relatively at
high pressure (75 bar H2) and temperature (140 oC). Addition of triethylsilane could increase the
TOFs and suppress over-alkylation of the amines.[21]
Scheme 15. Hydrogenation of nitriles catalyzed by Rhenium complexes.
4. Iron and cobalt catalyzed hydrogenation of nitriles The replacement of expensive noble metals by earth abundant transition metals is a hot topic in
catalytic community. In this respect Iron, cobalt and manganese received more attention more
recently. Non noble metal catalyzed hydrogenations of nitriles are very scarcely reported in the

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
109
literature. Recently, Beller[22] and Milstein[23] made a significant advance in this field. A recent
report on nitrile hydrogenation is described in the introduction part. (Non-noble metal pincer
complexes in hydrogenation of carbonyl and carboxylic acid derivatives)
Figure 1. Non noble metal complexes in nitrile catalyzed hydrogenations.
5. Results and Discussions
5.1 Iron catalyzed nitrile hydrogenation Encouraged by the recent contributions reported on hydrogenation of nitriles to primary
amines conducted with the iron PNP pincer complex C7, we planned to evaluate the influence of
the alkyl substituents at the phosphorous binding site on the catalytic performance of the
complexes in the hydrogenation of nitriles. Therefore, we synthesized the new iron pincer
complexes C8 and C9, bearing different substituents on the phosphine moiety.
Figure 2. Pincer iron complexes used for this study.
5.1.1 Synthesis of iron pincer complexes Synthesis of of Cy2 NH PNP Fe complex (C8)
First, the dibromo complex [(PNPCy)Fe(CO)Br2] 29 is prepared by the treatment of
FeBr2·2THF with 1 equivalent of the PNP ligand, bis(2-dicyclohexyl phosphinoethyl)amine 28 in
THF/EtOH at room temperature overnight then exposure to an atmosphere of CO for 1 h. The
complex 29 was then isolated as dark blue solid compound in 96% yield. The 31P NMR spectra

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
110
exhibits a singlet at = 61.2 ppm. In the IR spectra of 29, a strong CO stretching band at 1943
cm-1 and the N-H vibrations at 3117 cm-1 are observed. The hydridoborato complex C8 was
prepared in 54% yield by the reaction of the complex 29 with an excess of NaBH4 (10 equiv.) in
dry ethanol at room temperature for 2 h. The bright yellow complex C8 was then characterized
by different spectroscopic methods (NMR, IR, MS). In the 31P NMR spectra of the dihydride
complex C8, a mixture of two isomers was detected at = 91.5 ppm (major isomer) and = 92.7
ppm (minor isomer). Additionally, the 1H-NMR spectrum shows a sharp triplet at = -19.6 ppm
(major isomer) for the hydride ligand, whereas the BH4 ligand resonates as a broad signal at = -
2.9 ppm. In the IR spectrum bands at 1905 cm-1 indicate the coordination of CO to the metal
centre.
Scheme 16. Synthesis of the complex C8.
Synthesis of Et2 NH PNP Fe complex (C9)
The ligand was synthesized by a three step procedure. First, the deprotonation by phenyllithium
(1.5 equiv.) of the secondary phosphine Et2PH led the lithium diethylphosphide after a reaction
overnight at room temperature and 18 h at 60 °C. This phosphide then reacted with 0.5 equiv. of
N,N-bis(2-chloroethyl)-1,1,1-trimethylsilylamine at -40 °C and the N-trimethylsilylated PNP
ligand was obtained after 8 h at reflux. The hydrolysis using aqueous TBAF (1.1 equiv.) at reflux
gave the PNP ligand in 93% yield. The crude bis(2-diethylphosphinoethyl)amine 30 was then
used directly to prepare the complex 31 by reaction with 1.05 equiv. of FeBr2·2THF in ethanol
under an atmosphere of CO at room temperature. The dark blue iron dibromo complex 31 was
isolated in 68% yield after washing with ethanol. Crystals suitable for X-ray analysis were
obtained by slow diffusion of CH2Cl2 into a solution of 31 in pentane. Representation of this
molecule is shown in Figure 3 and selected bond lengths and bond angles are listed in Table 1.
The bright blue complex was fully characterized by NMR, IR and X-ray analysis. Notably, IR
spectrum showed the CO frequency at 1936 cm-1 and N-H band appeared at 3182 cm-1.
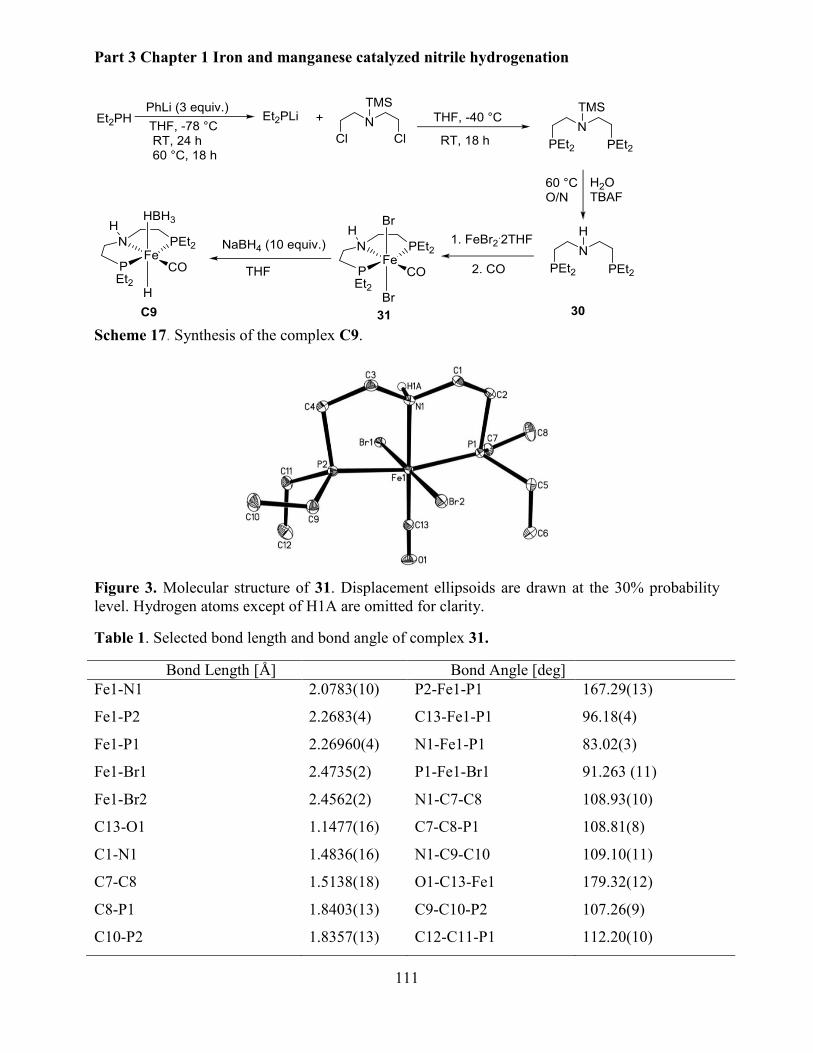
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
111
Scheme 17. Synthesis of the complex C9.
Figure 3. Molecular structure of 31. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms except of H1A are omitted for clarity.
Table 1. Selected bond length and bond angle of complex 31.
Bond Length [Å] Bond Angle [deg] Fe1-N1 2.0783(10) P2-Fe1-P1 167.29(13)
Fe1-P2 2.2683(4) C13-Fe1-P1 96.18(4)
Fe1-P1 2.26960(4) N1-Fe1-P1 83.02(3)
Fe1-Br1 2.4735(2) P1-Fe1-Br1 91.263 (11)
Fe1-Br2 2.4562(2) N1-C7-C8 108.93(10)
C13-O1 1.1477(16) C7-C8-P1 108.81(8)
C1-N1 1.4836(16) N1-C9-C10 109.10(11)
C7-C8 1.5138(18) O1-C13-Fe1 179.32(12)
C8-P1 1.8403(13) C9-C10-P2 107.26(9)
C10-P2 1.8357(13) C12-C11-P1 112.20(10)

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
112
The hydridoborato complex C9 was then prepared in 56% yield by treating the complex 31 with
10 equiv. of NaBH4 in THF/EtOH at room temperature. The initial blue homogeneous Fe(II)
complex 31 was turned into a yellow suspension. Crystals suitable for X-ray analysis were
obtained by slow diffusion of heptane into a solution of C9 in THF. The bright yellow complex
C9 has been fully characterized by multinuclear NMR spectroscopy, high resolution mass
spectrometry, IR spectroscopy (ATR), X-ray and elemental analysis. In the 1H NMR spectrum,
the hydride ligand resonated at = -19.6 ppm (JHP = 50.0 Hz) as a sharp triplet, whereas the BH4
ligand appears as a broad signal at = -3.0 ppm. The IR spectrum exhibited a band at 1898 cm-1
indicating the presence of a CO ligand and a band at 3201 cm-1 characteristic of N-H band. X-ray
analysis of C9 reveals a distorted octahedral coordination geometry around the Fe(II) center, with
the CO ligand located trans to the nitrogen atom and the hydride ligand located trans to the 1-
coordinated hydroborate ligand. Besides, the hydrogen atoms at Fe and N are arranged anti to
each other.
Figure 4. Molecular structure of the complex C9. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms except those on Fe, N and B are omitted for clarity.
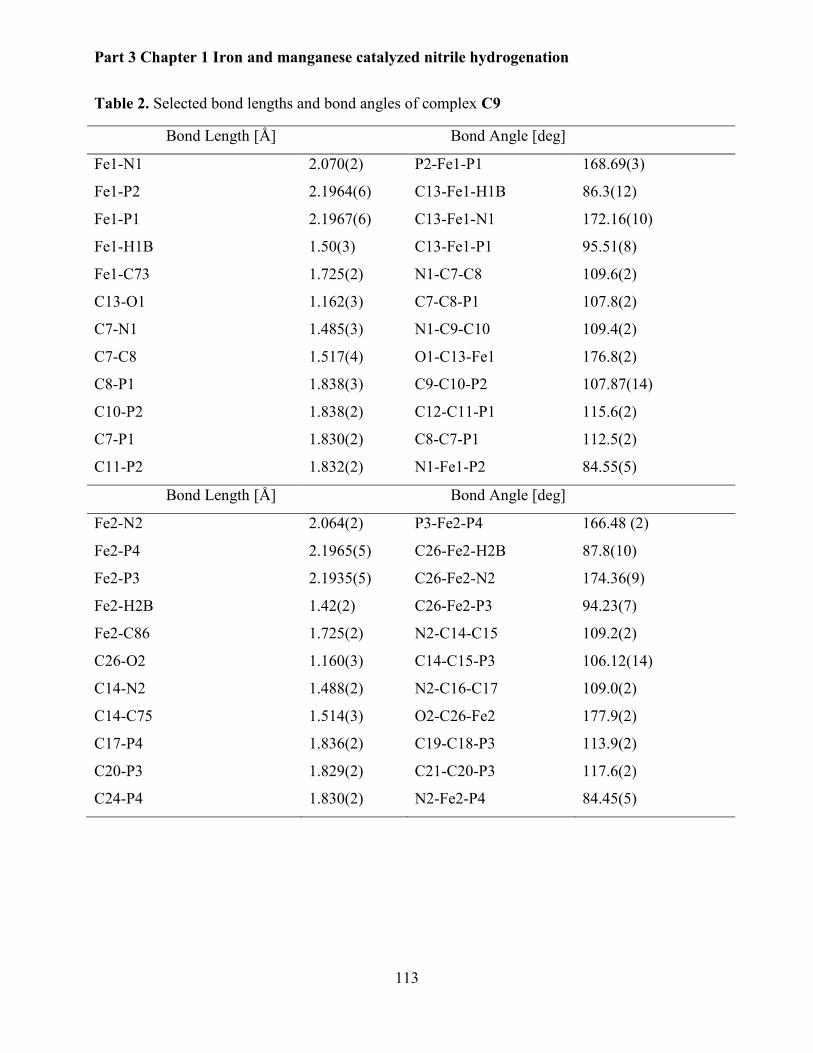
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
113
Table 2. Selected bond lengths and bond angles of complex C9
Bond Length [Å] Bond Angle [deg]
Fe1-N1 2.070(2) P2-Fe1-P1 168.69(3)
Fe1-P2 2.1964(6) C13-Fe1-H1B 86.3(12)
Fe1-P1 2.1967(6) C13-Fe1-N1 172.16(10)
Fe1-H1B 1.50(3) C13-Fe1-P1 95.51(8)
Fe1-C73 1.725(2) N1-C7-C8 109.6(2)
C13-O1 1.162(3) C7-C8-P1 107.8(2)
C7-N1 1.485(3) N1-C9-C10 109.4(2)
C7-C8 1.517(4) O1-C13-Fe1 176.8(2)
C8-P1 1.838(3) C9-C10-P2 107.87(14)
C10-P2 1.838(2) C12-C11-P1 115.6(2)
C7-P1 1.830(2) C8-C7-P1 112.5(2)
C11-P2 1.832(2) N1-Fe1-P2 84.55(5)
Bond Length [Å] Bond Angle [deg]
Fe2-N2 2.064(2) P3-Fe2-P4 166.48 (2)
Fe2-P4 2.1965(5) C26-Fe2-H2B 87.8(10)
Fe2-P3 2.1935(5) C26-Fe2-N2 174.36(9)
Fe2-H2B 1.42(2) C26-Fe2-P3 94.23(7)
Fe2-C86 1.725(2) N2-C14-C15 109.2(2)
C26-O2 1.160(3) C14-C15-P3 106.12(14)
C14-N2 1.488(2) N2-C16-C17 109.0(2)
C14-C75 1.514(3) O2-C26-Fe2 177.9(2)
C17-P4 1.836(2) C19-C18-P3 113.9(2)
C20-P3 1.829(2) C21-C20-P3 117.6(2)
C24-P4 1.830(2) N2-Fe2-P4 84.45(5)

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
114
5.1.2 Optimisation of the reaction parameters for catalysed nitrile
hydrogenation
After preparing two novel complexes C8 and C9, the influence of structural variation of
the phosphine moiety in the hydrogenation of nitriles was studied. Indeed, the reaction of
benzonitrile to benzylamine was chosen as benchmark system to study on the activity of this type
of catalysts. In preliminary experiments, different amounts of catalyst loadings (0.25-1 mol%) of
C7-C9 were tested under 30 bar H2 at 70 °C for 3 h (Scheme 18). At 1 mol% catalyst loading, the
three complexes produced benzylamine in high yields, while the catalytic performance of
Fe(PNPEt) complex C9 completely dropped down with half the amount of catalyst. To our
delight, using 0.5 mol% of the complex C8 afforded 90% yield of benzylamine at 70 °C under 30
bar of hydrogen.
Scheme 18. Yields of benzylamine using different catalyst loadings of C7-C9. Reaction conditions: 1.0 mmol benzonitrile, 0.25-1 mol% C7-C9, 2 mL iPrOH, 30 bar H2, 3 h, 70 °C.
Using the active complex C8, the optimization of the reaction parameters (time, solvent,
temperature) was carried out (Table 3). Notably, the temperature and the solvent played a
significant role for the catalyst efficiency. Good to excellent yields of benzylamine were
produced even using half of the catalyst loading (0.5-1 mol%) at 70 °C in 3 h (Table 3, entries 1
and 2). The reaction runs fast yielding complete conversion and 86% of amine after 2 h.
Interestingly, when decreasing the temperature to 55 °C, even if the conversion of the starting
material is still high (92%), the major detected product is the corresponding secondary imine.
(Entry 4) When toluene was used as solvent, traces of benzaldehyde was observed (Table 3, entry
6).
0
20
40
60
80
100
1 0,5 0,25
Yie
ld [%
]
catalyst loading [mol%]
cat. 1 cat. 2 cat. 3
cat 1 = C7 cat 2 = C8 cat 3 = C9

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
115
Table 3. Optimization of the reaction conditions for the hydrogenation of benzonitrile
Entry Catalyst[mol%]
Temp [°C]
Time [h]
Conv.[b]
[%] Yield[b]
[%]
1 0.5 70 3 99 90 2 1 70 3 99 89 3 1 70 2 99 86 4 1 55 3 92 --- 5 1 40 3 44 ---
6[c] 1 70 3 17 ---
[a] Conditions: 1.0 mmol benzonitrile, 1 mol% C8, 2 mL iPrOH, 30 bar H2, 2-3 h, 40-70 °C. [b] Determined by GC analysis. [c] 2 mL toluene.
5.1.3 Scope of the hydrogenation of aromatic nitriles
Next, we investigated the scope of the nitrile reduction with 1 mol% of the complex C8 to
illustrate the generality of this iron-catalyzed procedure. Therefore, various aromatic, aliphatic
and heterocyclic nitriles (Table 4 & Scheme 19) were hydrogenated to their corresponding
amines in high yields under 30 bar H2 at 70-100 °C for 3 h. Nitriles with electron withdrawing
substituents such as fluoro, trifluoromethyl and esters were hydrogenated in good yields (84-
95%, Table 4, entries 2, 3 and 6). Notably, the hydrogenation of nitriles selectively took place
even in the presence of ester group (32f) producing the amino-ester in excellent yield (95%). 4-
phenylbenzonitrile 32d provided 93% of the corresponding amine. Unfortunately, easily
reducible ketone substituent was not tolerated and the 4-acylbenzonitrile led to the corresponding
amino alcohol 33e in 63% yield (Table 4, entry 5). Para- and ortho-aminobenzonitriles 32g and
32h were efficiently reduced leading to the corresponding diamines in 72 and 94% yields,
respectively (Table 4, entries 7, 8). In addition, nitrogen- and sulfur-containing heteroaromatic
nitriles (Table 4, entries 9, 10) were hydrogenated to their corresponding amines with moderate
to good yields up to 90%.
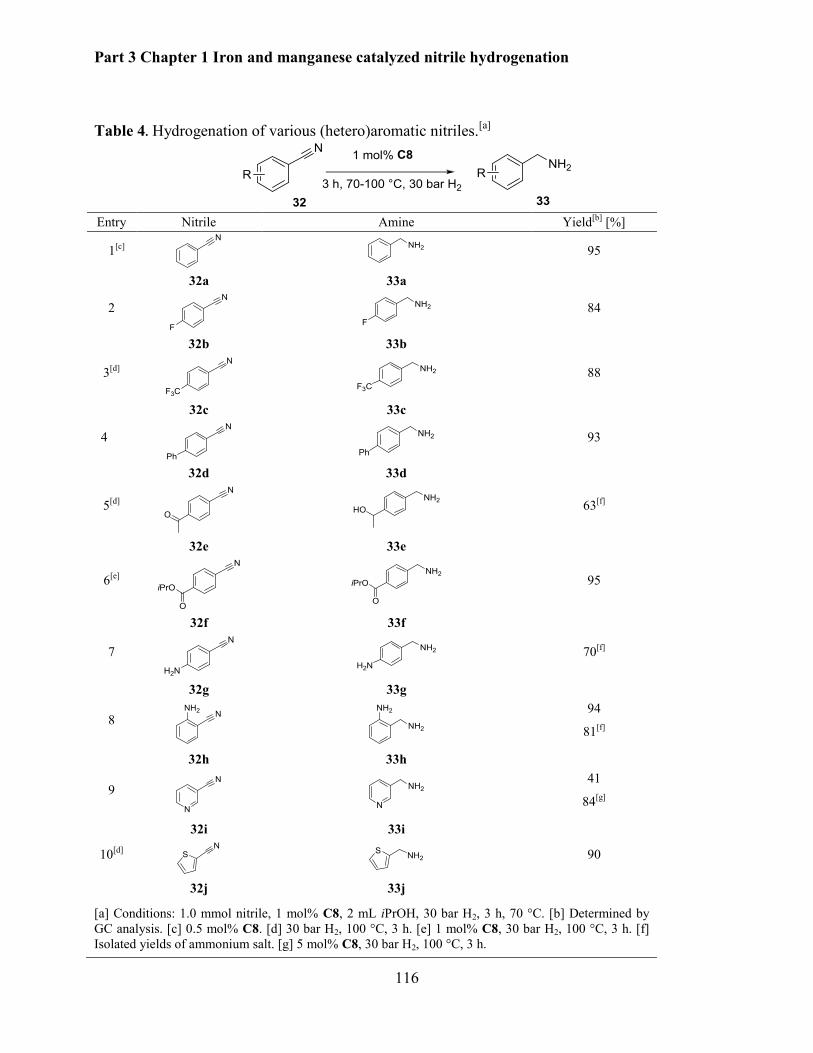
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
116
Table 4. Hydrogenation of various (hetero)aromatic nitriles.[a]
Entry Nitrile Amine Yield[b] [%]
1[c]
95
32a 33a
2
84
32b 33b
3[d]
88
32c 33c
4
93
32d 33d
5[d]
63[f]
32e 33e
6[e]
95
32f 33f
7
70[f]
32g 33g
8
94
81[f]
32h 33h
9
41
84[g]
32i 33i
10[d]
90
32j 33j
[a] Conditions: 1.0 mmol nitrile, 1 mol% C8, 2 mL iPrOH, 30 bar H2, 3 h, 70 °C. [b] Determined by GC analysis. [c] 0.5 mol% C8. [d] 30 bar H2, 100 °C, 3 h. [e] 1 mol% C8, 30 bar H2, 100 °C, 3 h. [f] Isolated yields of ammonium salt. [g] 5 mol% C8, 30 bar H2, 100 °C, 3 h.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
117
5.1.4 Hydrogenation of aliphatic and dinitriles
Furthermore, the applicability of the complex C8 as suitable and selective homogeneous
hydrogenation catalyst was demonstrated for the transformation of aliphatic nitriles and
adipodinitrile to the corresponding amines (Scheme 19). Cyclic as well as linear alkylnitriles
were smoothly reduced in good yields under mild conditions (70 °C, 30 bar, 3 h) generating
exclusively the primary amines. In the case of the hydrogenation of cinnamyl nitrile, higher
temperature (100°C) was needed in order to obtain full conversion. However, under these
conditions, small amounts (8%) of the C=C double bond were reduced. It should be pointed out
that the synthesis of the industrially important hexane-1,6-diamine was performed with excellent
selectivity and quantitative yield. This is one of the rare examples for a homogeneously catalyzed
hydrogenation of adipodinitrile to the corresponding diamine which proceeds without further side
reactions.
Scheme 19. Hydrogenation of alkylamine using C8. Reaction conditions: 1.0 mmol nitrile, 1 mol% C8, 2 mL iPrOH, 30 bar H2, 3 h, 70 °C; * 100 °C, ca. 8% of C=C hydrogenation.
In summary, the synthesis of the new iron PNP pincer complexes C8 and C9 and its efficient
application for the nitrile reduction was presented in this chapter. We compare the activity of the
complexes C7-C9 for nitrile hydrogenation. C7 and C8 are showing almost the same activity
whereas C9 was found to be less active catalytic system for the nitrile hydrogenation. This C8
catalytic system results in the selective hydrogenation of various aromatic, aliphatic and
heterocyclic nitriles including adipodinitrile to primary amines under mild conditions in short
time.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
118
5.2. Manganese catalyzed nitrile hydrogenation Inspired from the recent work on non-noble metal catalyzed nitrile hydrogenation the
group in Rostock became interested to develop the manganese catalysts for such transformation.
Based on the expertise of the Rostock team on the preparation and the use of PNP pincer Fe and
Ru complexes for hydrogenation and dehydrogenation reactions,[24] four manganese pincer
complexes bearing two different PNP ligands were prepared (Figure 5).
Figure 5. Manganese complexes used for this study.
5.2.1 Manganese pincer complexes preparation PNPMnCl2 complex C13
An initial attempt, i.e. the reaction of MnCl2 with 1 equiv. of the isopropyl-tagged PNP ligand in
THF at room temperature overnight afforded the five-coordinate 15e complex [Mn(PNP)Cl2]
C13 in 83% isolated yield. (Scheme 20)
Scheme 20. Synthesis of the (PNP)MnCl2 complex C13.
As the complex C13 was a paramagnetic compound, in order to unequivocally establish the
ligand arrangement around the manganese center, the molecular structure of this complex was
determined by X-ray diffraction analysis (Figure 6). Crystals suitable for analysis was grown by
slow diffusion of heptane into CH2Cl2. The X-ray structure reveals a trigonal bipyramidal
structure and the ligands are arranged facially.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
119
Figure 6. Molecular structure of the complex C13 in the crystal. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms except of that attached to nitrogen and the solvent molecule are omitted for clarity.
Table 5. Selected bond lengths and bond angles of complex C13.
Bond Length [Å] Bond Angle [deg]
Mn1 N1 2.3556(13) N1 Mn1 P1 75.38(3)
Mn1 P2 2.6171(5) P1 Mn1 P2 145.265(15)
Mn1 P1 2.6530(5) Cl1 Mn1 P1 96.249(16)
Cl1 Mn1 2.3649(4) Cl2 Mn1 P1 102.108(16)
Cl2 Mn1 2.3638(4) N1 Mn1 Cl2 100.04(3)
C14 P2 1.8459(15) N1 Mn1 Cl1 143.47(3)
C11 P2 1.8443(16) Cl2 Mn1 Cl1 116.485(17)
C8 P1 1.8469(17) Cl2 Mn1 P2 101.552(16)
C5 P1 1.8462(16) Cl1 Mn1 P2 95.546(15)
In comparison to the reported bisaminophosphine pyridine complex (iPrPNNNP)Mn(Cl)2
34, the phosphorus-metal bond lengths of 2.6171(5) and 2.6530(5) Å) are significantly longer
than in the ones observed in 34 (2.5931(5) and 2.5794(5) Å). The Mn(1)−Cl(1) and Mn(1)−Cl(2)
bond distances of 2.3556(13) and 2.3638(4) Å), respectively, are on the same range than the ones
already described in the complex 34 (2.3940(5) and 2.3658(5) Å).[25]

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
120
PNPMn(CO)2Br complexes C10 and C11
In order to prepare the potentially active carbonyl-ligated manganese hydride complex,
C13 was reacted in THF under CO atmosphere, but unfortunately no reaction occurred. Hence an
alternative strategy was envisioned: upon reaction of commercially available [MnBr(CO)5] (1.5
mmol) with the corresponding PNP ligands (1.6 mmol) in toluene at 100 °C for 20 h, air-stable
dicarbonyl manganese complexes C10 and C11 were conveniently prepared in a straightforward
manner in 93 and 80%, respectively, without the necessity of additional CO treatment (Scheme
21).
Scheme 21. Synthesis of (PNP)Mn(CO)2Br complexes C10 and C11.
The resulting bright yellow complexes were fully characterized by NMR and IR spectroscopy,
high resolution mass spectrometry, X-ray and elemental analysis. The IR (ATR) spectra of C10
and C11 contain strong CO stretching vibrations at 1903 cm-1, 1815 cm-1 and 1913 cm-1, 1826
cm-1, respectively, which clearly indicate the coordination of two CO ligands to the metal center. 31P spectrum shows a singlet at =81.8 and =73.6 ppm for C10 and C11 complexes
respectively, indicating that the two phosphorus atoms of the ligand are equivalent. The X-ray
analysis of C10 and C11 show a distorted-octahedral manganese center surrounded by three
meridionally placed donor atoms of the PNP ligand. It reveals that one of the CO ligand is
located trans to the nitrogen atom (e.g. C-Mn-N angle is 78.21(9)° in C11) and the other CO is
located trans to the bromide ligand (C-Mn-Br angles are 178.87(6)° in C10 and 179.39(8)° in
C11. The N-H ligand and the bromide ligand present in cis position (N-Mn-Br angles are
84.81(4)° in C10 and 85.89(6)° in C11). Representations of these molecules are shown in Figure
7.
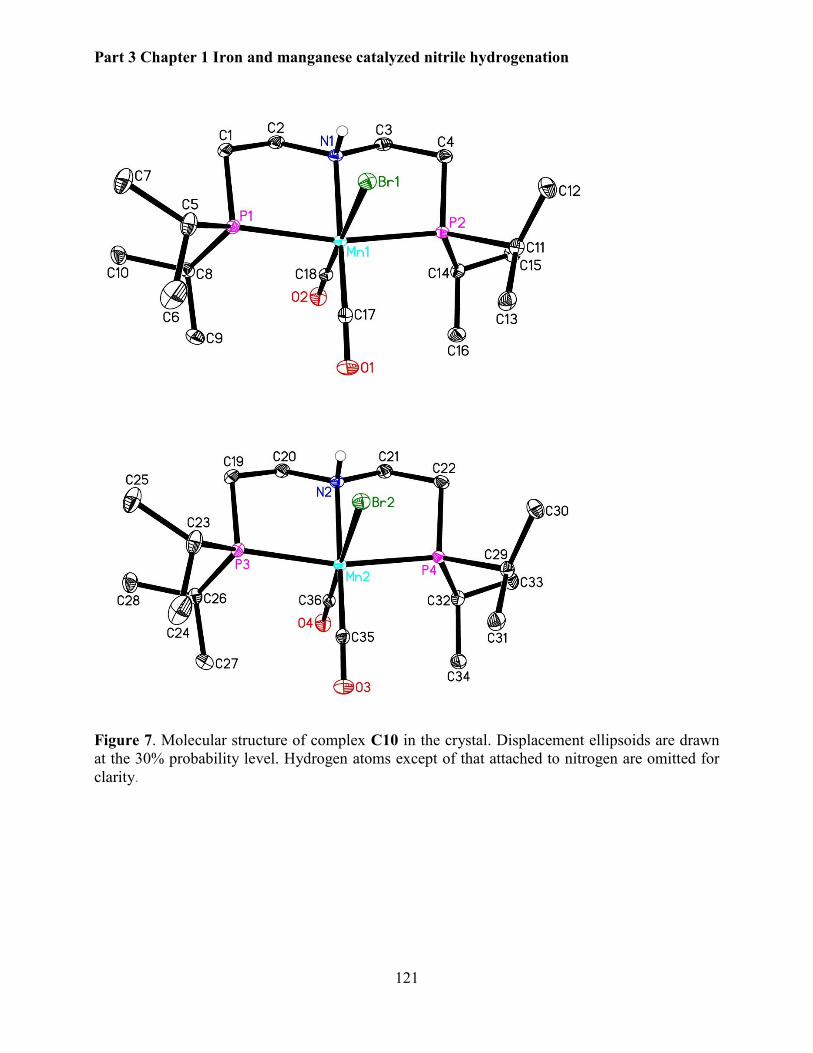
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
121
Figure 7. Molecular structure of complex C10 in the crystal. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms except of that attached to nitrogen are omitted for clarity.
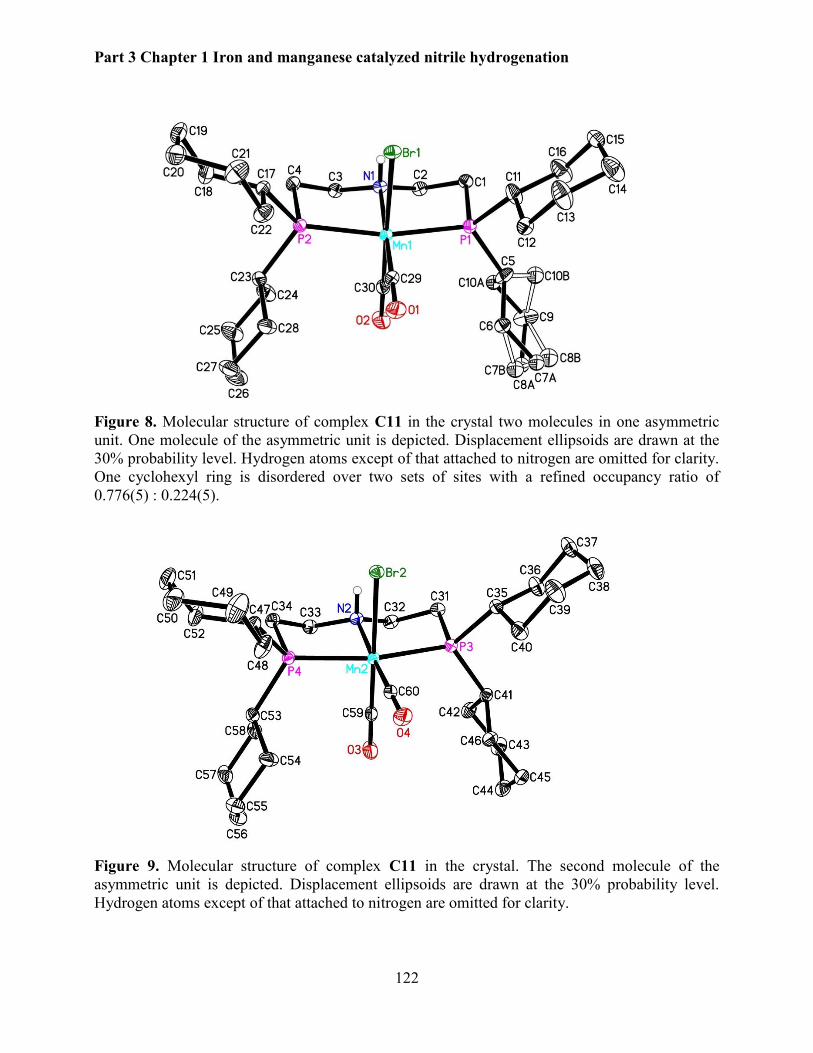
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
122
Figure 8. Molecular structure of complex C11 in the crystal two molecules in one asymmetric unit. One molecule of the asymmetric unit is depicted. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms except of that attached to nitrogen are omitted for clarity. One cyclohexyl ring is disordered over two sets of sites with a refined occupancy ratio of 0.776(5) : 0.224(5).
Figure 9. Molecular structure of complex C11 in the crystal. The second molecule of the asymmetric unit is depicted. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms except of that attached to nitrogen are omitted for clarity.
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
123
Table 6. Selected bond lengths and bond angles of complex C10.
Bond Length [Å] Bond Angle [deg]
Mn1 N1 2.1217(13) N1 Mn1 P1 83.08(4)
Mn1 P2 2.2988(5) P2 Mn1 P1 165.831(19)
Mn1 P1 2.3008(5) C18 Mn1 Br1 178.87(6)
C18 Mn1 1.7507(16) C17 Mn1 Br1 91.98(5)
C17 Mn1 1.7810(17) N1 Mn1 Br1 84.81(4)
C17 O1 1.162(2) P2 Mn1 Br1 89.327(12)
C18 O2 1.1649(18) P1 Mn1 Br1 90.517(13)
Br1 Mn1 2.5774(3) C17 Mn1 C18
C17 Mn1 N1
88.82(7)
176.77(6)
Table 7. Selected bond lengths and bond angles of complex C11.
Bond Length [Å] Bond Angle [deg]
Mn1 N1 2.1255(19) N1 Mn1 P1 82.64(6)
Mn1 P2 2.3048(7) P1 Mn1 P2 164.73(3)
Mn1 P1 2.3046(7) C30 Mn1 Br1 179.39(8)
C29 Mn1 1.787(2) C29 Mn1 Br1 92.34(8)
C30 Mn1 1.754(3) N1 Mn1 Br1 85.89(6)
C29 O1 1.154(3) P2 Mn1 Br1 87.41(2)
C30 O2 1.157(3) P1 Mn1 Br1 87.91(2)
Br1 Mn1 2.5630(5) C30 Mn1 C29 87.62(11)
C1 P1 1.846(2) C30 Mn1 N1 94.15(10)
C4 P2 1.850(2) C29 Mn1 N1 178.21(9)
In comparison with other PNP manganese carbonyl complexes described by Kempe
(35),[26] and Nocera (36),[27] in the complexes C10-C11, the Mn-phosphorus bond distances
[C10, 2.2988(5) Å, 2.3008(5) Å, C11, 2.3048(7)Å, 2.3046(7)Å] are not significantly different
from the ones in 35 and 36 [2.265(2)-2.286(1) Å]. The Mn–N distance of 2.1217(13) Å in C10
and 2.1255(19) Å in C11 are slightly longer than the one reported for an amido-Mn bond

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
124
[2.076(2) Å, 36] but shorter than the corresponding amino-Mn bond in the cationic [HN-36][OTf]
(2.286(1) Å). Finally, the Mn-CO distances 1.7507(16) and 1.7810(17) Å in C10 and 1.787(2)
and 1.754(3) Å in C11 are not important significantly different with the ones described for 35 and
36 [1.759(6)-1.844(3) Å]. (Figure 10)
Figure 10. Kempe and Nocera PNP Mn complexes.
PNPMn(H)(CO)2 complex C12
The manganese hydride complex C12 was then synthesized by treatment of C10 with one
equivalent of sodium triethylborohydride in THF at room temperature for 2 h. The yellow color
suspension was slowly changed into red solution. The orange red solid complex was then isolated
in 36% yield and it was characterized by NMR and IR studies. (Scheme 22)
Scheme 22. Preparation of the PNP pincer hydrido manganese complex C12.
31P{1H} NMR spectrum shows a singlet at =109.6 ppm. Interestingly, the presence of the
hydride was clearly observable in 1H NMR as a triplet was present at δ = -5.53 (t, 2JPH 50.7 Hz).
The IR spectrum of C12 contains strong CO stretching vibrations at 1889 cm-1, 1815 cm-1 and the
N-H vibration showed broad stretching vibration at 3271 cm-1. Noticeably, the complex C12
slowly decomposed at room temperature and should be stored at -30 oC.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
125
Advantageously, complexes C10-C11 were highly stable and do not decompose easily in the
presence of air. In fact, exposing the complex C10 as a solid for 25 days to air did not result in
any decomposition which is confirmed by NMR and IR.
5.2.2 Optimization of reaction parameters Having some potential manganese catalysts in hand, we started to investigate their
behavior in the hydrogenation of nitriles. Based on the recent interest in the selective reduction of
carboxylic acid derivatives and more particularly of nitriles leading primary amines,[28-30] our
initial attempts focused on the hydrogenation of benzonitrile as benchmark substrate (Table 8).
Catalytic experiments were performed with complexes C10-C13 and commercially available
Mn(CO)5Br and CpMn(CO)3 (3 mol%) in i-PrOH at 120 °C for 24 h under 50 bar of H2. Using
the complex C10 in the absence of base, only 10% conversion was detected, but no desired
product was obtained and only 5% of benzyl alcohol was formed (Table 9, entry 1). In contrast,
combining 3 mol% of C10 with 10 mol% of base (such as t-BuONa), under similar conditions,
the benzonitrile was selectively transformed into benzylamine (98% GC-yield) (Table 8, entry 1).
To the best of our knowledge, this represents the first example of catalytic hydrogenation of
nitriles using a molecularly-defined manganese complex. The dicyclohexylphosphino pincer
complex C11 was also efficient for this transformation, but led to the benzylamime with a
slightly lower yield (87%) (Table 8, entry 2).
Interestingly, the complex C12, which is prepared from C10 by treatment with sodium
triethylborohydride or which can be generated in situ by addition of a base under H2 pressure
(vide infra), permitted the full conversion of benzonitrile producing benzylamine in 81% GC-
yield. It must be underlined that using 3 mol% of C12 in the absence of a catalytic amount of
base, only 42% of benzylamine was obtained. These lower yields in the benchmark reaction
using the hydrido complex C12 can be easily explained by its lower stability.
It must also be pointed out that the dichloro Mn complex C13 and the commercially available
complexes Mn(CO)5Br and CpMn(CO)3 under similar conditions did not lead to any conversion,
which shows the crucial importance of the PNP ligand on the activity of the manganese catalysts.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
126
Table 8. Manganese-catalyzed hydrogenation of benzonitrile[a]
Entry Complex Conv (%)[b] Yield (%)[b]
1 C10 > 99 98 2 C11 > 99 87
3[c] C12 > 99 81 4 C13 > 99 0 6 [MnBr(CO)5] 0 0 7 CpMn(CO)3 0 0
[a] Reaction conditions: Substrate (0.5 mmol), C10-C13 (0.015 mmol, 3 mol%), t-BuONa (0.05 mmol, 10 mol%), i-PrOH (1 mL), 24 h, 120 °C, 50 bar H2. [b] Conversion and yield were determined by GC analysis using hexadecane as an internal standard. [c] Under similar conditions in the absence of a base, benzylamine was obtained in only 42% yield.
Next, the nature of the solvent was examined. In toluene, when 3 mol% of C10 was used
for the hydrogenation of benzonitrile in the presence of 10 mol% of t-BuOK at 120 oC for 24 h,
>99% conversion and 98% of benzylamine was detected (Table 9, entry 2). With polar solvents
such as THF and EtOH, even if the conversion was full, only moderate GC-yields of benzylamine
was obtained (60 and 59%, respectively, Table 9, entries 4-5). When CPME and benzene was
used, 75 and 79% GC yields of benzylamine were observed, respectively (Table 9, entries 6-7).
When decreasing the catalyst loading from 3 mol% to 2 mol%, toluene was found to be a better
solvent than isopropanol for the hydrogenation of benzonitrile to benzylamine (89 vs 75% CG-
yields, Table 9, entries 8-9).
The nature of the catalytic amount of base was then evaluated. With 3 mol% of C10, the
use of 10 mol% t-BuOK as the base permitted to obtain benzylamine in 68% GC-yield (Table 9,
entry 10). With sodium alkoxide bases such as NaOMe and NaOEt, full conversions were
observed and the benzylamine was produced in 53 and 32% GC yield, respectively (Table 9,
entries 11-12). Sodium hydroxide can also be used as a base for this hydrogenation of
benzonitrile yielding 63% of benzylamine (Table 9, entry 14). When 10 mol% of NaEt3BH was
used in association of 3 mol% catalyst C10, 59% of benzylamine was obtained with 5% of benzyl
alcohol as by-product (Table 9, entry 13). When decreasing the amount of t-BuONa to 5 mol%, a
significant decrease of the yield was then observed (69%, Table 9, entry 15).
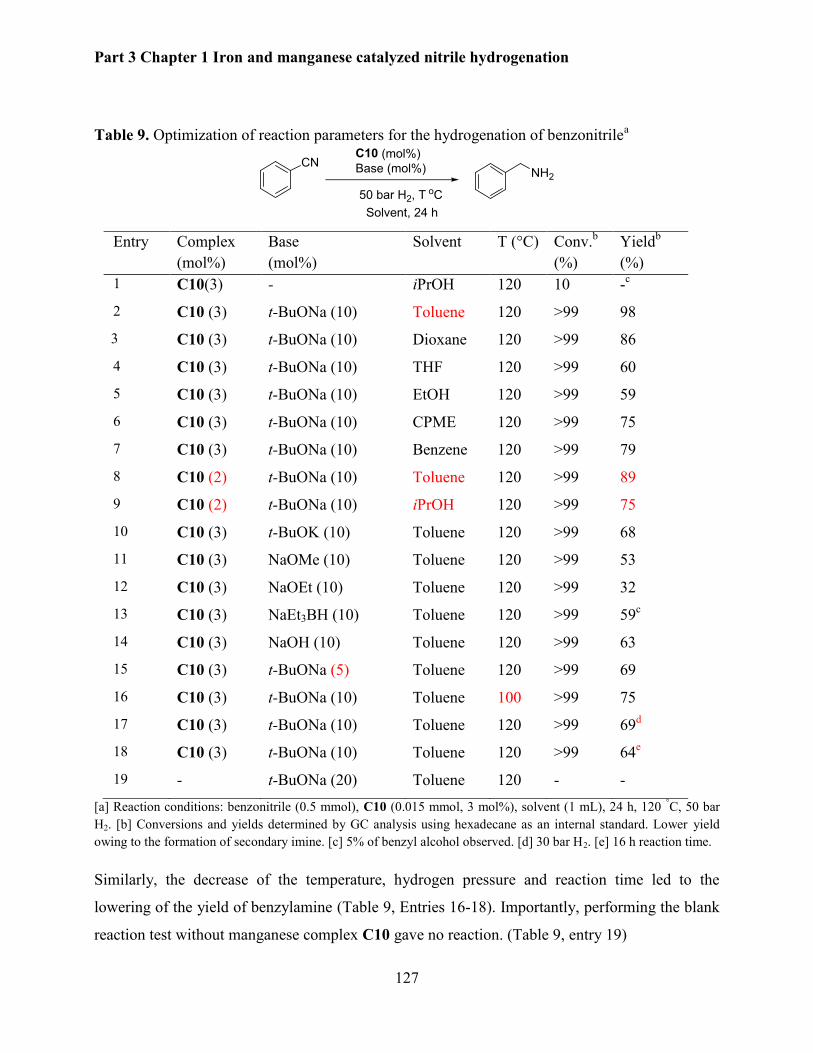
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
127
Table 9. Optimization of reaction parameters for the hydrogenation of benzonitrilea
Entry Complex
(mol%) Base (mol%)
Solvent T (°C) (oC)
Conv.b
(%) Yieldb (%)
1 C10(3) - iPrOH 120 10 -c
2 C10 (3) t-BuONa (10) Toluene 120 >99 98 3 C10 (3) t-BuONa (10) Dioxane 120 >99 86
4 C10 (3) t-BuONa (10) THF 120 >99 60 5 C10 (3) t-BuONa (10) EtOH 120 >99 59
6 C10 (3) t-BuONa (10) CPME 120 >99 75 7 C10 (3) t-BuONa (10) Benzene 120 >99 79
8 C10 (2) t-BuONa (10) Toluene 120 >99 89 9 C10 (2) t-BuONa (10) iPrOH 120 >99 75
10 C10 (3) t-BuOK (10) Toluene 120 >99 68 11 C10 (3) NaOMe (10) Toluene 120 >99 53
12 C10 (3) NaOEt (10) Toluene 120 >99 32 13 C10 (3) NaEt3BH (10) Toluene 120 >99 59c
14 C10 (3) NaOH (10) Toluene 120 >99 63 15 C10 (3) t-BuONa (5) Toluene 120 >99 69
16 C10 (3) t-BuONa (10) Toluene 100 >99 75 17 C10 (3) t-BuONa (10) Toluene 120 >99 69d 18 C10 (3) t-BuONa (10) Toluene 120 >99 64e
19 - t-BuONa (20) Toluene 120 - - [a] Reaction conditions: benzonitrile (0.5 mmol), C10 (0.015 mmol, 3 mol%), solvent (1 mL), 24 h, 120 °C, 50 bar H2. [b] Conversions and yields determined by GC analysis using hexadecane as an internal standard. Lower yield owing to the formation of secondary imine. [c] 5% of benzyl alcohol observed. [d] 30 bar H2. [e] 16 h reaction time.
Similarly, the decrease of the temperature, hydrogen pressure and reaction time led to the
lowering of the yield of benzylamine (Table 9, Entries 16-18). Importantly, performing the blank
reaction test without manganese complex C10 gave no reaction. (Table 9, entry 19)

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
128
The evaluation of the critical reaction parameters of the hydrogenation showed that the optimal
condition was the use of 3 mol% C10 in toluene with 10 mol% of t-BuONa as the base under 50
bar of H2 at 120 oC for 24 h.
Scheme 23. Catalytic hydrogenation of various (hetero)aromatic nitriles
Reaction conditions: Substrate (0.5 mmol), C10 (0.015 mmol, 3 mol%), t-BuONa (0.05 mmol, 10 mol%), toluene (1 mL), 24 h, 120 °C, 50 bar H2. Conversion (>99%) and yield were determined by GC analysis using hexadecane as an internal standard. [a] Isolated yield.
5.2.3 Hydrogenation of aromatic nitriles The applicability of the catalyst C10 was demonstrated in the selective hydrogenation of
various nitriles including substituted aromatic, benzylic, aliphatic and di-nitriles (Schemes 23 and
24). In general, electron-donating (Me, OMe, 37a-b) and electron-withdrawing (Cl, Br and CF3,
37c-i) substituted benzonitriles were hydrogenated into the corresponding amines with moderate
to good yields (62-96%). Noticeably, the ortho substitution slightly inhibited the reactivity as
o-chlorobenzonitrile led to the corresponding o-chlorobenzylamonium salt in 62% yield. (Scheme
23, 38e). 1-napthonitrile was also reduced into the naphthalen-1-ylmethanamine and isolated as a
hydrochloride salt in 62% yield (Scheme 23, 38i). Heterocyclic aromatic nitriles such as
nicotinonitrile and furan-2-carbonitrile were successfully hydrogenated to the corresponding

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
129
amines with moderate yields (69 and 65%, respectively, Scheme 23, 38m-n). The reaction can
tolerate an amino functional group as p-anminobenzonitrile led to p-aminobenzylamine in
moderate 42% GC-yield. Notably, the reduction of 4-(trifluoromethyl)benzonitrile was easily up
scaled to 5g and 38i.HCl was isolated in 95% yield (Scheme 23, 38i)
Scheme 24. Catalytic hydrogenation of aliphatic and di-nitriles.
Reaction conditions: substrate (0.5 mmol), C10 (0.015 mmol, 3 mol%), t-BuONa (0.05 mmol, 10 mol%), toluene (1 mL), 24-60 h, 100-120 °C, 50 bar H2. Isolated as a HCl salt. Conversion (>99%) was determined by GC analysis using hexadecane as an internal standard. [a] GC yield. [b] 25% of the corresponding saturated amine was also detected as a by-product.
5.2.4 Hydrogenation of aliphatic nitriles
Next, the activity of the manganese catalyst C10 towards a collection of more demanding
aliphatic nitriles was explored (Scheme 24). As an example, 2-(4-methoxyphenyl)acetonitrile was
successfully hydrogenated to the corresponding ammonium salt 40a in 96% isolated yield.
Cyclohexyl methanenitrile was hydrogenated yielding 87% of the desired product 40c after 36 h
of reaction. Interestingly, a long chain C6-C19 (Scheme 24, 39d-i) containing nitriles were
reduced into the corresponding aliphatic amines with good yields. Noteworthy, the hydrogenation
of the conjugated nitrile led to a mixture of the corresponding unsaturated (53 %) and saturated
(25 %) amines. Remarkably, the isolated C=C double bond-containing 5-hexenenitrile was
selectively hydrogenated to the unsaturated amine 40j without affecting the double bond (78%
isolated yield of the ammonium salt). Finally, terephthalonitrile was reduced to the corresponding

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
130
diamine isolated as its hydrochloride salt 40k in 80% yield under the standard reaction
conditions. Even though this catalytic system was efficient for hydrogenation of aromatic and
aliphatic nitriles, it had some limitation in the selectivity (Scheme 25). When adiponitrile was
used as a substrate, 22% of the desired diamine product was obtained. In addition, 35% of the
mono-reduction product 6-aminocapronitrile and 5% of caprolactam detected as side products.
Notably, this catalytic system did not tolerate a reducible functional group such as nitro and
ketone. When these substrates are examined for the hydrogenation, the corresponding imines and
other side products are obtained along with amine products.
Scheme 25. Limitations in the Mn-catalyzed hydrogenation of nitriles.
5.2.5 Mechanistic investigations In order to obtain information on the nature of the catalytic active manganese species,
NMR investigations were conducted using the complex C10 in the presence 3 equiv. of t-BuONa
as the base in toluene-d8 under 5 bar H2 at room temperature. After 1 h, a red-orange solution was
formed which clearly exhibited in the 1H NMR spectrum the signal of the manganese hydride as
a triplet at -5.67 ppm with a coupling constant 2JP,H of 50.7 Hz, indicating that the manganese
center is coordinated to two equivalent phosphorus moieties. Additionally, the in situ IR
measurements in toluene of the reaction mixture showed full conversion of the complex
PNPMn(CO)2Br C10 into the corresponding hydrido complex PNPMnH(CO)2 C12 and an
unchanged coordination of the pincer ligand and the two CO ligands (Figure 11). As compared to
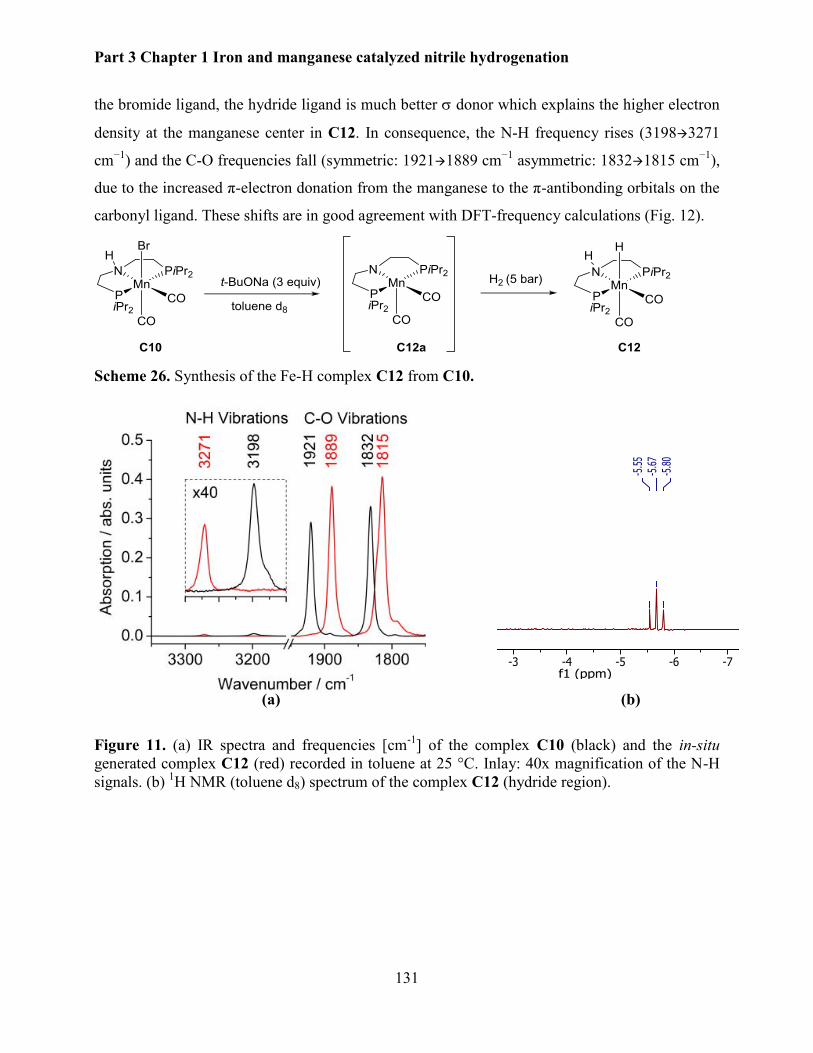
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
131
the bromide ligand, the hydride ligand is much better donor which explains the higher electron
density at the manganese center in C12. In consequence, the N-H frequency rises (31983271
cm−1) and the C-O frequencies fall (symmetric: 19211889 cm−1 asymmetric: 18321815 cm−1),
due to the increased π-electron donation from the manganese to the π-antibonding orbitals on the
carbonyl ligand. These shifts are in good agreement with DFT-frequency calculations (Fig. 12).
Scheme 26. Synthesis of the Fe-H complex C12 from C10.
(a) (b)
Figure 11. (a) IR spectra and frequencies [cm-1] of the complex C10 (black) and the in-situ generated complex C12 (red) recorded in toluene at 25 °C. Inlay: 40x magnification of the N-H signals. (b) 1H NMR (toluene d8) spectrum of the complex C12 (hydride region).
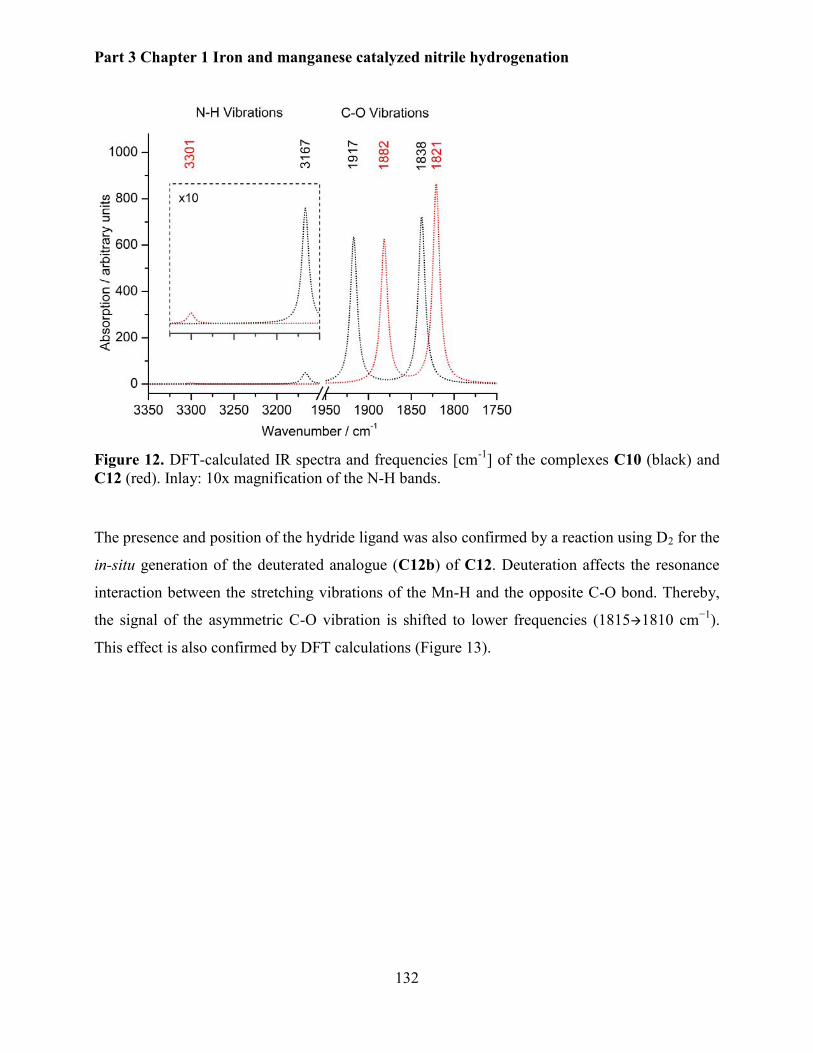
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
132
Figure 12. DFT-calculated IR spectra and frequencies [cm-1] of the complexes C10 (black) and C12 (red). Inlay: 10x magnification of the N-H bands.
The presence and position of the hydride ligand was also confirmed by a reaction using D2 for the
in-situ generation of the deuterated analogue (C12b) of C12. Deuteration affects the resonance
interaction between the stretching vibrations of the Mn-H and the opposite C-O bond. Thereby,
the signal of the asymmetric C-O vibration is shifted to lower frequencies (18151810 cm−1).
This effect is also confirmed by DFT calculations (Figure 13).
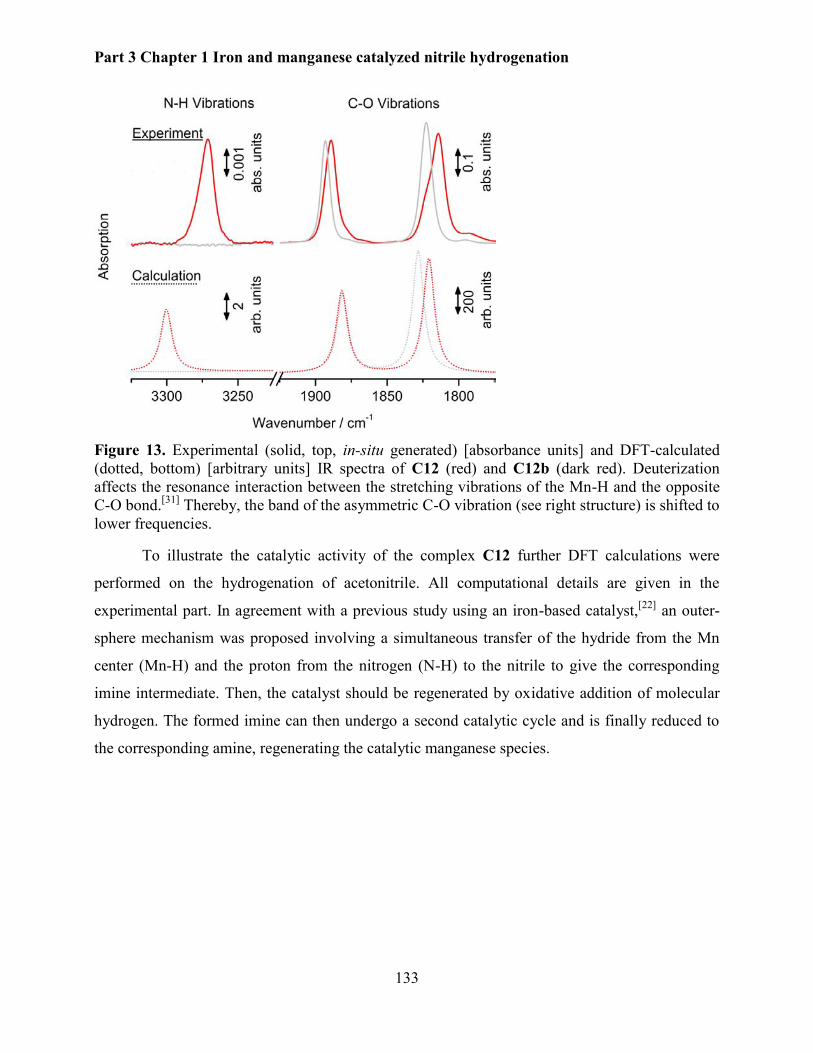
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
133
Figure 13. Experimental (solid, top, in-situ generated) [absorbance units] and DFT-calculated (dotted, bottom) [arbitrary units] IR spectra of C12 (red) and C12b (dark red). Deuterization affects the resonance interaction between the stretching vibrations of the Mn-H and the opposite C-O bond.[31] Thereby, the band of the asymmetric C-O vibration (see right structure) is shifted to lower frequencies.
To illustrate the catalytic activity of the complex C12 further DFT calculations were
performed on the hydrogenation of acetonitrile. All computational details are given in the
experimental part. In agreement with a previous study using an iron-based catalyst,[22] an outer-
sphere mechanism was proposed involving a simultaneous transfer of the hydride from the Mn
center (Mn-H) and the proton from the nitrogen (N-H) to the nitrile to give the corresponding
imine intermediate. Then, the catalyst should be regenerated by oxidative addition of molecular
hydrogen. The formed imine can then undergo a second catalytic cycle and is finally reduced to
the corresponding amine, regenerating the catalytic manganese species.
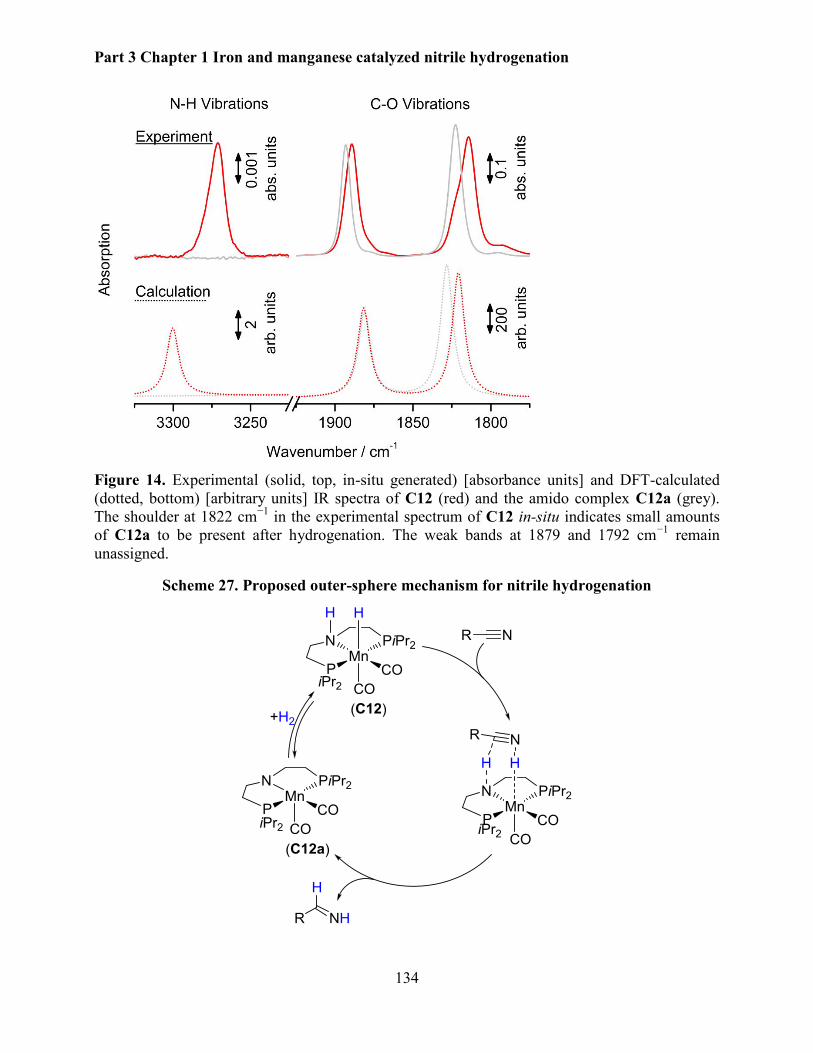
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
134
Figure 14. Experimental (solid, top, in-situ generated) [absorbance units] and DFT-calculated (dotted, bottom) [arbitrary units] IR spectra of C12 (red) and the amido complex C12a (grey). The shoulder at 1822 cm−1 in the experimental spectrum of C12 in-situ indicates small amounts of C12a to be present after hydrogenation. The weak bands at 1879 and 1792 cm−1 remain unassigned.
Scheme 27. Proposed outer-sphere mechanism for nitrile hydrogenation

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
135
To show the reversibility of the step between the hydrido manganese C12 and amido
manganese C12a complexes, the energetic parameters including the barrier and reaction energy
of the concerted H2 elimination were calculated. For the hydrido Mn complex C12, the computed
Gibbs free energy barrier for H2 elimination is 20.2 kcal/mol. In general, the reaction is slightly
exergonic by 0.2 kcal/mol. Hence, a well-balanced equilibrium can be established under H2
atmosphere. For the regeneration of the catalyst C12 via the addition of H2, the barrier is 20.4
kcal/mol. Interestingly, the dissociation of the equatorial CO ligand was found to be endergonic
by 41.0 kcal/mol, which clearly indicates the stability of this CO coordination. This agrees well
with the experimental results: in situ spectroscopic studies of the reaction mixture vide supra
proved that the CO ligands were still intact. Comparing the hydrogenation of acetonitrile
(CH3CN) and benzonitrile (PhCN) using the Mn complex C12 as the catalyst gave similar results,
but the computed Gibbs free energy barrier is slightly higher (20.9 vs 17.8 kcal/mol,
respectively).
Figure 15. Catalytic intermediates: (a) transition state of the transfer of the protic hydrogen to the
nitrile-nitrogen and hydride coordination to the corresponding carbon; (b) amido complex
(C12a).
Finally, to evaluate the homogeneity of the catalytic system, poisoning experiments were
performed in the presence of varying amounts of PMe3, PPh3 and Hg.[32-33] In all the cases, no
significant effects in the hydrogenation of benzonitrile were observed (see table 10). When we
used 10 mol% PMe3 as an additive, full conversion was observed with 86% GC yield of
benzylamine which is the similar yield when the reaction performed without any additive. Next,
we examined the reaction with bulky phosphine PPh3 and Hg also didn’t show any difference in
the resulted yields of the benzylamine obtained. The constant activity in the presence of PMe3 or
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
136
Hg can be explained by an outer sphere hydrogenation mechanism of a molecular-defined
catalyst species. The observed similar reduction rates in the presence of Hg33a indicate the
homogeneous nature of the catalyst.
Table 10. Homogeneity test
Entry Additives Quantity
(equiv.) Conv. (%)
Yield (%)
1 - - >99 86
2 PMe3 0.1 >99 86
3 PMe3 0.5 >99 88
4 PMe3 1 >99 87
5 PPh3 0.1 >99 87
6 PPh3 0.5 >99 84
7 PPh3 1 >99 85
8 Hg 0.1 >99 88
9 Hg 0.5 >99 83
10 Hg 1 >99 77
6. Conclusion In summary, novel manganese PNP supported pincer complexes C10-C13 were
synthesized and fully characterized by spectroscopic and X-ray studies. The complexes C10-C11
were easy to prepare in an one step procedure without the use of toxic CO and were activated
with a catalytic amount of t-BuONa as the base or under H2 pressure. Importantly, the developed
precatalyst C10 is air-stable and easy to handle. These pincer manganese complexes were shown
to be efficient and versatile catalysts in hydrogenation of nitriles with a nice functional group
tolerance. Interestingly, molecular-defined manganese-amide centers permitted the practical
activation of molecular di-hydrogen. A mechanistic and DFT study suggested that an outer-
sphere mechanism by a simultaneous transfer of the proton from the amino part of the ligand and

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
137
from the hydrido metal centre. Furthermore, poisoning experiments seem to prove the
homogeneous nature of the catalyst.
In general conclusion, iron and manganese catalyzed nitrile hydrogenations are effective
transformations. Iron (30 bar H2, 70 oC) catalytic system is more efficient than the manganese
one (50 bar H2 and 120 oC). Although manganese is less active, the pre-catalyst is easy to prepare
in one step procedure and is air-stable, which is a more value for scale up applications. In a
mechanistic point of view, outer sphere mechanisms were described for both iron and manganese
catalyzed nitrile hydrogenation.
7. Experimental section
7.1 General experimental details Unless otherwise stated, all reactions were performed under an argon atmosphere with exclusion
of moisture from reagents and glassware using standard techniques for manipulating air-sensitive
compounds. All isolated products were characterized by 1H NMR and 13C NMR spectroscopy as
well as high resolution mass spectrometry (HRMS). NMR spectra were recorded on a Bruker AV
300 or 400. All chemical shifts (δ) are reported in ppm and coupling constants (J) in Hz. All
chemical shifts are related to residual solvent peaks [CDCl3: 7.26 (1H), 77.16 (13C); C6D6: 7.16
(1H), 128.06 (13C)], respectively. All measurements were carried out at room temperature unless
otherwise stated. Mass spectra were in general recorded on a Finnigan MAT 95-XP (Thermo
Electron) or on a 6210 Time-of-Flight LC/MS (Agilent). Gas chromatography was performed on
a HP 6890 with a HP5 column (Agilent).
Reagents: Unless otherwise stated, commercial reagents were used without purification.
7.2. Synthesis of iron pincer complexes Synthesis of [FeBr2(CO)(HN(CH2CH2PCy2)2)]
The ligand HN(CH2CH2PCy2)2 (1.21 g, 2.61 mmol) was dissolved in 25 mL THF (abs.) at room
temperature in a 100 mL Schlenk tube under argon. 932.7 mg FeBr2·2THF (2.61 mmol) in 20 mL
of ethanol was added dropwise over a period of 5-10 minutes. A white fluffy precipitate was
formed and the mixture was stirred over night at room temperature. CO was bubbled through the
suspension for 1 hour until the precipitate was dissolved completely and the solution turned to

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
138
dark blue. After removing the CO by bubbling argon though the solution, the solvents were
removed under reduced pressure to obtain a blue crystalline solid. The product was washed 3
times with 10 mL of ethanol and dried under vacuum. 1.78 g (2.5 mmol, Yield: 96 %). 1H NMR (400 MHz, CD2Cl2): δ 5.39 (t, J = 12.8 Hz, 1H, NH), 3.76 – 3.60 (m, 2H, CH2), 3.51 – 3.26 (m, 2H, CH2), 2.59 – 2.32 (m, 6H), 2.20 (dd, J = 22.5, 12.6 Hz, 4H), 2.08 – 1.57 (m, 26H), 1.38 – 1.04 (m, 13H). 31P NMR (162 MHz, CD2Cl2): δ 59.92. 13C NMR (101 MHz, CD2Cl2): δ 227.50 (s, CO), 50.66 (s, CH2), 36.34 (t, JCP = 10.4 Hz, CH), 34.66 (t, JCP = 8.9 Hz, CH), 30.72 (s), 29.94 (s), 29.27 (s), 28.72 – 27.49 (m), 26.94 (d, J CP = 12.2 Hz), 24.84 (t, J CP = 7.3 Hz). IR (ATR): 3229,9 cm-1 (NH), 3177.4 cm-1 (NH), 2918.1 cm-1, 2845,3 cm-1, 1942.7 cm-1 (CO). HRMS: calc. for C28 H53 Br Fe N P2 602.21648, found 602.21635 (Fe –HBr -CO). Synthesis of [Fe(CO)(H)(HBH3)(HN(CH2CH2PCy2)2)] C8
254.1 mg (0.36 mmol) of [FeBr2(CO)(HN(C2H4PCy2)2)] were placed in 50 mL Schlenk-tube
under argon and 10 mL of dry ethanol were added. NaBH4 (136.2 mg, 3.6 mmol, 10 equiv.) was
gradually added to the suspension. During 2 hours of stirring at room temperature, the color of
the solution changed from blue to green and finally to yellow. The solvent was evaporated under
vacuum. The yellow solid was dissolved in toluene to give a bright yellow solution with white
precipitate. After the solution was filtered, the toluene was evaporated under vacuum. The yellow
solid was recrystallized from a mixture of THF and n-heptane and washed 5 times with 10 mL of
n-heptane. The product was obtained as a yellow powder. 154.7 mg (0.27 mmol, Yield: 76 %) 1H NMR (300 MHz, Benzene-d6) δ 3.97 (br, 1H, NH), 2.88 (d, JHP = 11.7 Hz, 2H), 2.54 (m, 4H), 2.22 – 2.02 (m, 2H), 2.00 – 1.44 (m, 27H), 1.44 – 1.02 (m, 14H), 0.95 – 0.84 (m, 2H), -2.79 (br, 4H, BH4), major isomer δ -19.57 (t, JHP = 52 Hz, 1H, FeH, 72 %), minor isomer δ -20.44 (t, J = 52.1 Hz, 1H, FeH, 28%). 31P NMR (122 MHz, C6D6, 298 K) minor isomer δ = 92.70 (d, J = 5 Hz, 31%), major isomer δ 91.54 (d, J = 11.2 Hz, 69 %), impurities 47.47. 13C NMR (101 MHz, Benzene-d6): δ 54.17 (t, JCP = 5.6 Hz, CH2), 40.10 (t, JCP = 9.3 Hz, CH), 36.60 (t, JCP = 12.6 Hz, CH), 32.11 (s), 31.32 (s), 30.31 (s), 29.12 (t, JCP = 52.0 Hz), 28.26 (s), 28.20 (s), 28.05 (s), 27.96 (s), 27.85 (t, JCP = 4.3 Hz), 27.39 (t, JCP = 4.6 Hz), 27.17 (t, JCP = 6.3 Hz), 26.83 (s), 26.69 (s), 23.12 (s), 14.38 (s), CO not observed. IR (ATR): 3198.0 cm-1 (NH), 2917.9 cm-1, 2847.3 cm-1, 2360.3 cm-1 (BH3), 1905.0 cm-1 (CO). HRMS: calc. for C29H57BFeNOP2 564.33581, found 564.33648 (Fe – H).

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
139
Synthesis of bis(2-diethylphosphinoethyl)amine
To a cooled solution of THF (20 mL, -78 oC) containing Et2PH (1.0 g, 11.1 mmol) was added 9.2
mL (16.6 mmol) of 1.8 M PhLi in dibutylether. The resulting black solution was stirred at
ambient temperature overnight and hereafter the reaction mixture was heated to 60 oC and stirred
for additional 18 h. The reaction was monitered by NMR to identify the formation of Et2PLi.
(31P{1H}NMR (THF-d8): -54.6 ppm). To the well stirred Et2PLi solution, a mixture of N,N-bis(2-
chloroethyl)-1,1,1-trimethylsilylamine (1.2 g, 5.6 mmol) in THF (5 mL) was added at -40 °C
within 10 min. After completion of the addition, the mixture was allowed to reach room
temperature and after that was then refluxed for 8 h. The solution was then cooled to room
temperature, treated with a TBAF (6.1 mL, 6.11 mmol) / water (10 mL) mixture and again
refluxed for 8 h. After cooling to room temperature, the phases were seperated and the aqueous
layer was washed with diethyl ether (3 x 10 mL). The combined organic phases were dried over
MgSO4 and after filtration, the volatiles were removed in vacuo. The title compound was isolated
as a pale brown viscous liquid. The pincer ligand was used without further purification. 1.300 g
(Yield: 93% with respect to silylamine). 1H NMR (300 MHz, C6D6) δ: 1.02-0.89 (m, 12H), 1.34-1.25 (m, 8H), 1.55-1.42 (m, 4H), 2.75-2.54 (m, 4H). 31P{1H} NMR (122 MHz, C6D6) δ: -26.41. 13C{1H} NMR (75 MHz, C6D6) δ: 47.6, 47.3, 28.1, 28.0, 19.7, 19.6, 10.0, 9.8. Synthesis of {FeBr2(CO)[HN(CH2CH2P(CH2CH3)2)2]}
A solution of HN(CH2CH2PEt2)2 (250 mg, 1 mmol) in THF (2.5 mL) was added dropwise to a
suspension of FeBr2·2THF (0.375 g, 1.05 mmol) in EtOH (6 mL). The resulting mixture was
stirred at room temperature overnight and then the argon atmosphere was replaced by carbon
monoxide. A clear blue solution was formed within a period of 15 min. Upon removal of the
solvent, the residue was washed with EtOH (3 x 10 mL) to remove unreacted FeBr2·2 THF. The
title compound was isolated as a blue solid which was dried in vacuo. 330 mg (Yield: 68%).
Crystals suitable for X-ray analysis were obtained by slow diffusion of CH2Cl2 into a solution of
31 in pentane. 1H NMR (400 MHz, THF-d8, 297 K) δ: 1.27-1.33 (m, 12H, P(CH2CH3)4), 2.08-2.28 (overlapping m, 12H, CH2CH3, PCH2), 3.15-3.24 (m, 2H, NCH2), 3.43-3.50 (m, 2H, NCH2), 4.98 (bt, 1H, NH);

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
140
13C{1H} NMR (101 MHz, THF-d8, 297 K) δ 7.5 (s, P(CH2CH3)2), 7.7 (s, P(CH2CH3)2), 15.8-16.2 (m, CH2CH3), 27.8 (t, JC-P = 8.7 Hz, PCH2), 49.5 (t, JC-P = 4.8 Hz, NCH2), 225.4 (CO) ; 31P{1H} NMR (162 MHz, THF-d8, 297 K) δ: 65.5 (s). ESI-HRMS (m/z pos): Calculated for [C13H29NO2P2Fe]: 350.10958; found: 350.11005[M-Br2+OH]+ IR ATR (solid): ῡ [cm-1] 1936 (s, ῡ CO), 3182 (bs, ῡ N-H)
Synthesis of {Fe(H)(HBH3)(CO)[HN(CH2CH2P(CH2CH3)2)2]} C9
{{FeBr2(CO)[HN((CH2CH2)P(CH2CH3))2]} (330 mg, 0.67 mmol) was suspended in THF (10
mL) and NaBH4 (256 mg, 6.7 mmol) in EtOH (5 mL) was added at room temperature. The
reaction mixture was stirred for 2 h at room temperature to produce a bright yellow solution. The
solvent was removed in vacuo and toluene (10 mL) was added. The resulting suspension was
filtered and the liquid portion was concentrated in vacuo. The resulting solid was repeatedly
washed with n-heptane to afford the title compound as a bright yellow solid. 131 mg (Yield:
56%).
Crystals suitable for X-ray analysis were obtained by slow diffusion of heptane into a solution of
C9 in benzene. 1H{31P} NMR (400 MHz, C6D6, 297 K) major isomer δ: -19.67 (s, 1H, FeH), minor isomer δ: -20.30 (s, 1H, FeH ), -3.23 (br, FeBH4, 4H), 0.98 (t, J = 7.6 Hz, 6H, P(CH2CH3)2), 1.19 (t, J = 7.6 Hz, 6H, P(CH2CH3)2), 1.34-1.59 (overlapping m, 8H, P(CH2CH3)2, PCH2, NCH2), 1.67 (m, 2H, PCH2), 1.93 (m, 2H, P(CH2CH3), 2.20 (m, 2H, P(CH2CH3), 2.46 (m, 2H, NCH2), 3.78 (bt, 1H, NH); The additional minor hydride signals in the regions -15 to -26 ppm are attributed to related iron (di)hydride species. 13C{1H} NMR (101 MHz, C6D6, 297 K) δ: 8.5 (s, P(CH2CH3)2), 9.0 (s, P(CH2CH3)2), 20.2 (t, JC-P
= 11.2 Hz, P(CH2CH3)2), 23.2 (t, JC-P = 13.7 Hz, P(CH2CH3)2), 28.0 (t, JC-P = 8.3 Hz, PCH2), 53.3 (t, JC-P = 6.2 Hz, NCH2), 221.1 (CO); 31P{1H} NMR (162 MHz, C6D6, 298 K) major isomer δ: 81.8; minor isomer δ 78.8. IR ATR (solid): ῡ [cm-1] 2339 (b, ῡ BH), major isomer1898 (s, ῡ CO). minor isomer 1809 (s, ῡ CO). ESI-HRMS (m/z pos): Calculated for [C13H33BNOP2Fe]: 348.14771; found: 348.14804. Elemental Analysis: Anal. Calculated for [C13H34BNOP2Fe]: C, 44.74; H, 9.82; N, 4.01. found: C, 44.61; H, 9.05; N, 3.75.
7.3 X-ray Structural Analysis: Data were collected on a Bruker Kappa APEX II Duo
diffractometer. The structure was solved by direct methods (SHELXS-97)[34] and refined by full-
matrix least-squares procedures on F2 (SHELXL-2014)[35]. XP (Bruker AXS) was used for
graphical representation.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
141
Crystal data for complex 31. C13H29Br2FeNOP2, M = 492.98, triclinic, space group P1, a =
9.0871(4), b = 10.6078(5), c = 11.4672(5) Å, α = 102.8749(10), , = 109.6577(10)°, V = Å3, T
= 150(2) K, Z = 2, 32196 reflections measured, 5091 independent reflections (Rint =), final R
values (I > 2σ(I)): R1 = 0.0158, wR2 = 0.0374, final R values (all data): R1 = 0.0193, wR2 =
0.0387, GOF on F2: 1.037, 189 parameters.
Table 6. Crystallographic data for 31
Complex 31
Empirical formula C13H29Br2FeNOP2 Formula weight 492.98 T (K) 150 (2) λ(Å) 0.71073 Crystal system Triclinic Color, habit Blue, prism Space group P1 a (Å) 9.0871(4) b (Å) 10.6078(5) c (Å) 11.4672(5) α (o) 102.8749(10) β (o) 101.3372(10) γ (o) 109.6577(10) V (Å3) 969.37(8) Z 2, 32196 θ range (o) 2.38-28.82 Index range -12≤h≤12, -14≤k≤14,
-15≤l≤15
Data/restraints/parameters 5091/0/189
Independent reflections (Rint) 5091 (0.0223)
Goodness-of-fit on F2 1.037 Final R indices [I>2σ(I)] R1=0.0158,
wR2=0.0374 R indices (all data) R1=0.0193,
wR2=0.0387

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
142
Crystal Data for Complex C9: a = b = 13.1712(3), c = 17.7424(5) Å, β = 98.2275(6)°, V =
1887.74(8) Å3, T = 150(2) K, Z = 4, 36485 reflections measured, 9117 independent reflections
(Rint = 0.0196), final R values (I > 2σ(I)): R1 = 0.0206, wR2 = 0.0507, final R values (all data):
R1 = 0.0217, wR2 = 0.0514, GOF on F2: 1.038, 400 parameters. The crystal studied was found to
be a racemic twin.
Table 7. Crystallographic data for C9
Complex C9
Empirical formula C13H34BFeNOP2 Formula weight 349.01 T (K) 150 (2) λ(Å) 0.71073 Crystal system Monoclinic Color, habit Yellow, prism Space group P21 a (Å) 8.1620(2) b (Å) 13.1712(3) c (Å) 17.7424(5) α (o) 90 β (o) 98.2275(6) γ (o) 90 V (Å3) 1887.74(8) Z 4, 36485 θ range (o) 2.79-28.87 Index range -10≤h≤10, -17≤k≤17,
-23≤l≤21
Data/restraints/parameters 9117/1/400
Independent reflections (Rint) 9117 (0.0196)
Goodness-of-fit on F2 1.038 Final R indices [I>2σ(I)] R1=0.0206,
wR2=0.0507 R indices (all data) R1=0.0217,
wR2=0.0514

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
143
7.4 Synthesis of manganese pincer complexes Synthesis and characterization of {MnBr(CO)2[NH(CH2CH2P(iPr)2)2 C10
To the orange-yellow suspension of [MnBr(CO)5] (427 mg, 1.5 mmol) in toluene (25 mL) was
added [HN(CH2CH2P(iPr)2)2] (500 mg, 1.6 mmol, 10%wt in THF) dropwise. The suspension
turned into a clear yellow solution within 10 min. After the gas evolution had ceased the reaction
mixture was heated to 100 oC and further stirred for 20 h under argon. During the heating yellow
precipitate was formed. The reaction mixture was cooled to room temperature and concentrated
in vacuo. The crude was thoroughly washed with heptane and dried at the pump affording the
title compound as yellow powder (740 mg, 93%). Crystals suitable for X-ray diffraction analysis
were obtained by slow diffusion of pentane into a solution of C10 in CH2Cl2. 1H NMR (400 MHz, C6D6, 297 K): δ 3.27 (m, 2H, PCH(CH3)2), 2.83 (bt, J = 12.5 Hz, 1H, NH), 2.47 (m, 2H, NCH2CH2), 2.22 (m, 2H, PCH(CH3)2), 1.95 (m, 2H, NCH2CH2), 1.66 (m, 2H, NCH2CH2), 1.52 (m, 6H, PCH(CH3)2), 1.32 (m, 6H, PCH(CH3)2), 1.25 (m, 2H, NCH2CH2), 1.22 (m, 6H, PCH(CH3)2), 1.07 (m, 6H, PCH(CH3)2). 31P{1H} NMR (162 MHz, C6D6, 297 K): δ = 81.8 (s). 13C{1H} NMR (101 MHz, C6D6, 297 K): δ 232.5 (br, CO), 229.6 (br, CO), 52.7 (vt, 5.2 Hz, NCH2CH2), 27.1 (vt, 5.9 Hz, NCH2CH2,), 26.1 (vt, 9.3 Hz, PCH(CH3)2), 24.4 (vt, 9.5 Hz, PCH(CH3)2), 20.3 (vt, 1 Hz, PCH(CH3)2), 20.1 (vt, 1 Hz, PCH(CH3)2), 18.9 (s, PCH(CH3)2), 18.4 (vt, 2 Hz, PCH(CH3)2 ). IR-ATR (solid): ῡ [cm-1] 1903 (s, ῡ CO), 1815 (s, ῡ CO). ESI-HRMS (m/z pos): Calculated for [C18H36MnNO2P2]: 416.16745; found: 416.16752[M+H-Br]+. Elemental analysis: Calculated for [C18H37BrMnNO2P2]: C, 43.56; H, 7.51; N, 2.82. Found: C, 43.38; H, 7.45; N, 2.62. Table 8. Selected bond lengths and bond angles of the complex C10. Bond Length [Å] Bond Angle [deg]
Mn1 N1 2.1217(13) N1 Mn1 P1 83.08(4)
Mn1 P2 2.2988(5) P2 Mn1 P1 165.831(19)
Mn1 P1 2.3008(5) C18 Mn1 Br1 178.87(6)
C18 Mn1 1.7507(16) C17 Mn1 Br1 91.98(5)
C17 Mn1 1.7810(17) N1 Mn1 Br1 84.81(4)
C17 O1 1.162(2) P2 Mn1 Br1 89.327(12)
C18 O2 1.1649(18) P1 Mn1 Br1 90.517(13)
Br1 Mn1 2.5774(3) C17 Mn1 C18 88.82(7)
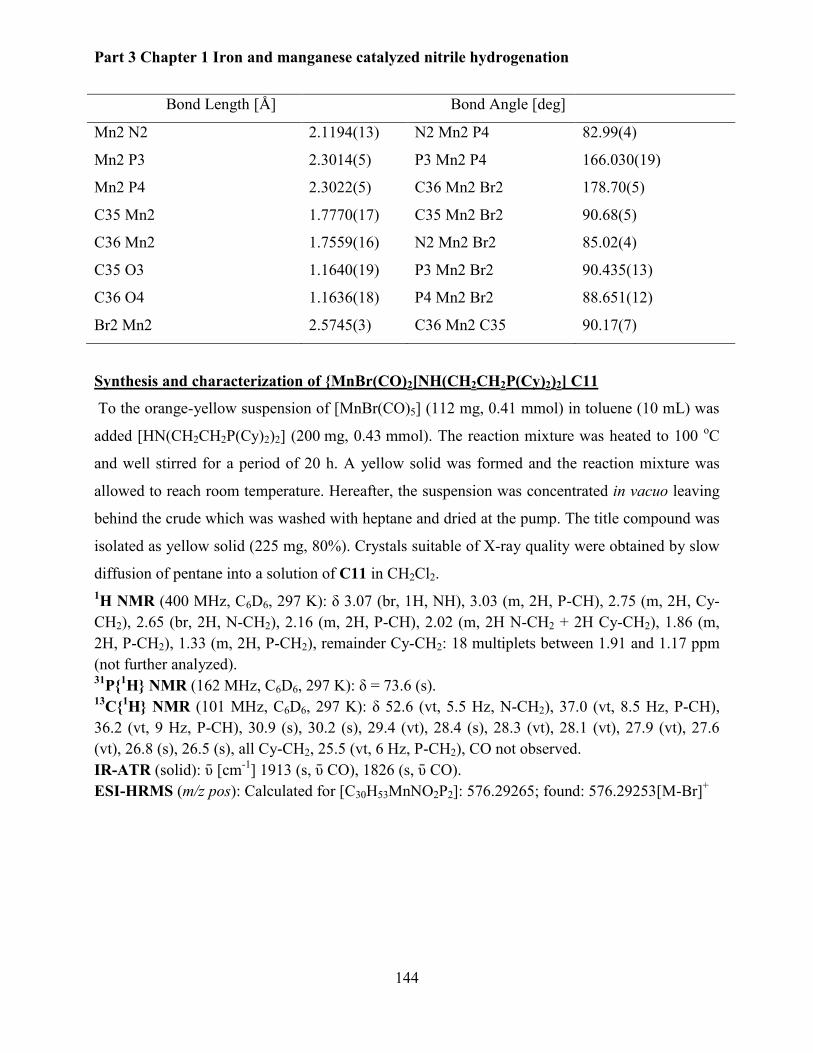
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
144
Bond Length [Å] Bond Angle [deg]
Mn2 N2 2.1194(13) N2 Mn2 P4 82.99(4)
Mn2 P3 2.3014(5) P3 Mn2 P4 166.030(19)
Mn2 P4 2.3022(5) C36 Mn2 Br2 178.70(5)
C35 Mn2 1.7770(17) C35 Mn2 Br2 90.68(5)
C36 Mn2 1.7559(16) N2 Mn2 Br2 85.02(4)
C35 O3 1.1640(19) P3 Mn2 Br2 90.435(13)
C36 O4 1.1636(18) P4 Mn2 Br2 88.651(12)
Br2 Mn2 2.5745(3) C36 Mn2 C35 90.17(7)
Synthesis and characterization of {MnBr(CO)2[NH(CH2CH2P(Cy)2)2] C11
To the orange-yellow suspension of [MnBr(CO)5] (112 mg, 0.41 mmol) in toluene (10 mL) was
added [HN(CH2CH2P(Cy)2)2] (200 mg, 0.43 mmol). The reaction mixture was heated to 100 oC
and well stirred for a period of 20 h. A yellow solid was formed and the reaction mixture was
allowed to reach room temperature. Hereafter, the suspension was concentrated in vacuo leaving
behind the crude which was washed with heptane and dried at the pump. The title compound was
isolated as yellow solid (225 mg, 80%). Crystals suitable of X-ray quality were obtained by slow
diffusion of pentane into a solution of C11 in CH2Cl2. 1H NMR (400 MHz, C6D6, 297 K): δ 3.07 (br, 1H, NH), 3.03 (m, 2H, P-CH), 2.75 (m, 2H, Cy-CH2), 2.65 (br, 2H, N-CH2), 2.16 (m, 2H, P-CH), 2.02 (m, 2H N-CH2 + 2H Cy-CH2), 1.86 (m, 2H, P-CH2), 1.33 (m, 2H, P-CH2), remainder Cy-CH2: 18 multiplets between 1.91 and 1.17 ppm (not further analyzed). 31P{1H} NMR (162 MHz, C6D6, 297 K): δ = 73.6 (s). 13C{1H} NMR (101 MHz, C6D6, 297 K): δ 52.6 (vt, 5.5 Hz, N-CH2), 37.0 (vt, 8.5 Hz, P-CH), 36.2 (vt, 9 Hz, P-CH), 30.9 (s), 30.2 (s), 29.4 (vt), 28.4 (s), 28.3 (vt), 28.1 (vt), 27.9 (vt), 27.6 (vt), 26.8 (s), 26.5 (s), all Cy-CH2, 25.5 (vt, 6 Hz, P-CH2), CO not observed. IR-ATR (solid): ῡ [cm-1] 1913 (s, ῡ CO), 1826 (s, ῡ CO). ESI-HRMS (m/z pos): Calculated for [C30H53MnNO2P2]: 576.29265; found: 576.29253[M-Br]+
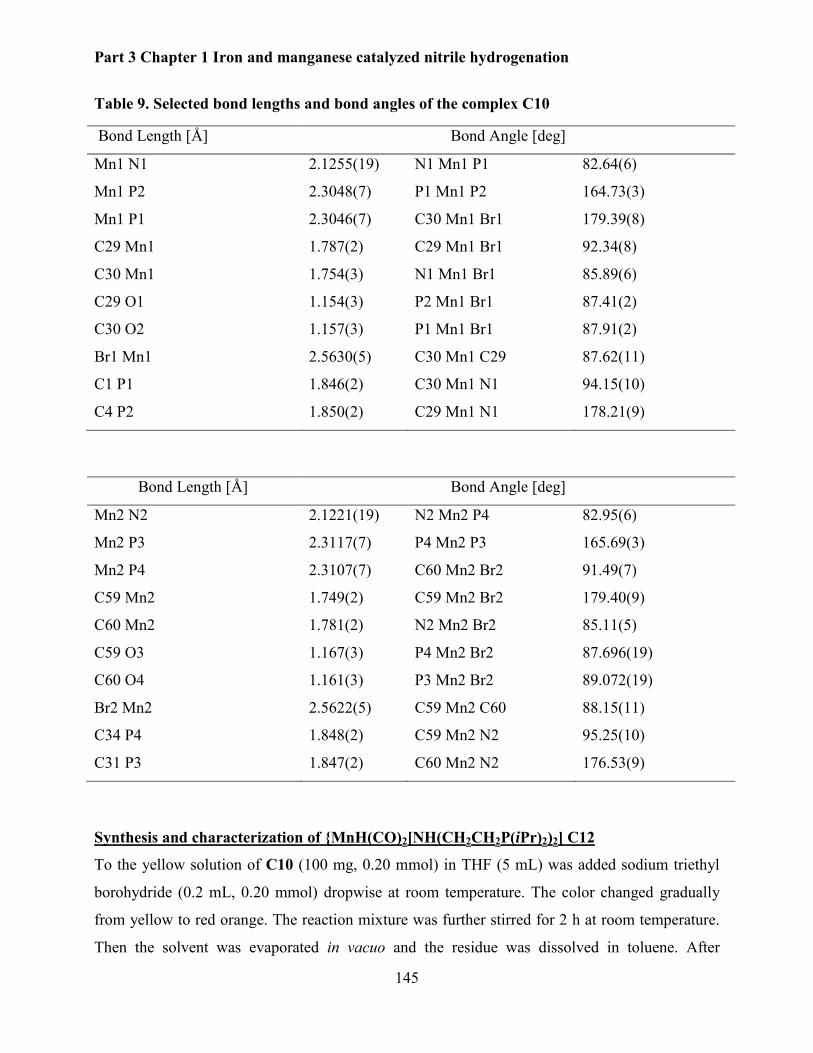
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
145
Table 9. Selected bond lengths and bond angles of the complex C10
Bond Length [Å] Bond Angle [deg]
Mn1 N1 2.1255(19) N1 Mn1 P1 82.64(6)
Mn1 P2 2.3048(7) P1 Mn1 P2 164.73(3)
Mn1 P1 2.3046(7) C30 Mn1 Br1 179.39(8)
C29 Mn1 1.787(2) C29 Mn1 Br1 92.34(8)
C30 Mn1 1.754(3) N1 Mn1 Br1 85.89(6)
C29 O1 1.154(3) P2 Mn1 Br1 87.41(2)
C30 O2 1.157(3) P1 Mn1 Br1 87.91(2)
Br1 Mn1 2.5630(5) C30 Mn1 C29 87.62(11)
C1 P1 1.846(2) C30 Mn1 N1 94.15(10)
C4 P2 1.850(2) C29 Mn1 N1 178.21(9)
Bond Length [Å] Bond Angle [deg]
Mn2 N2 2.1221(19) N2 Mn2 P4 82.95(6)
Mn2 P3 2.3117(7) P4 Mn2 P3 165.69(3)
Mn2 P4 2.3107(7) C60 Mn2 Br2 91.49(7)
C59 Mn2 1.749(2) C59 Mn2 Br2 179.40(9)
C60 Mn2 1.781(2) N2 Mn2 Br2 85.11(5)
C59 O3 1.167(3) P4 Mn2 Br2 87.696(19)
C60 O4 1.161(3) P3 Mn2 Br2 89.072(19)
Br2 Mn2 2.5622(5) C59 Mn2 C60 88.15(11)
C34 P4 1.848(2) C59 Mn2 N2 95.25(10)
C31 P3 1.847(2) C60 Mn2 N2 176.53(9)
Synthesis and characterization of {MnH(CO)2[NH(CH2CH2P(iPr)2)2] C12
To the yellow solution of C10 (100 mg, 0.20 mmol) in THF (5 mL) was added sodium triethyl
borohydride (0.2 mL, 0.20 mmol) dropwise at room temperature. The color changed gradually
from yellow to red orange. The reaction mixture was further stirred for 2 h at room temperature.
Then the solvent was evaporated in vacuo and the residue was dissolved in toluene. After

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
146
filtration through a frit the orange-red solution was evaporated to dryness leaving behind the
crude. Washing with pentane afforded orange yellow solid (36% yield) which was stored at -30 oC. The title compound slowly decomposed upon standing which indicate that this complex is
only moderately stable. 1H NMR (400 MHz, C6D6, 297 K): δ 2.20 (m, 2H, N-CH2), 2.16 (m, 2H, P-CH), 1.91 (m, 2H, P-CH), 1.69 (m, 2H, N-CH2), 1.50 (m, 2H, P-CH2), 1.45 (m, 6H, CH3), 1.35 (m, 6H, CH3), 1.28 (m, 6H, CH3), 1.26 (m, 6H, CH3), 0.78 (m, 2H, P-CH2), -5.53 (t, 2JP,H 50.7 Hz, Mn-H); N-H not found but presence of N-H confirmed by IR spectroscopy. 31P{1H} NMR (162 MHz, C6D6, 297 K): δ = 109.6 (s). 13C{1H} NMR (101 MHz, C6D6, 297 K): δ 53.1 (vt, 6.8 Hz, N-CH2), 30.2 (vt, 11.8 Hz, P-CH), 29.0 (vt, 7.7 Hz, P-CH), 27.0 (vt, P-CH2), 19.9 (s, CH3), 19.6 (s, CH3), 19.4 (s, CH3), 19.3 (s, CH3), CO not observed. In situ generation of {MnH(CO)2[NH(CH2CH2P(iPr)2)2]
Upon treatment of complex C10 (1 equiv.) with t-BuONa (3 equiv.) in toluene-d8 (1 mL) an
orange red solution was formed immediately. This sample was stirred under H2 atmosphere (5
bar) for 1 h. 1H-NMR and IR analysis were performed immediately. 1H NMR (400 MHz, C7D8, 297 K): δ = -5.67 (t, 2JP,H 50.7 Hz, Mn-H). 31P{1H} NMR (162 MHz, C7D8, 297 K): δ = 109.7 (s).
Synthesis and characterization of {MnCl2[NH(CH2CH2P(iPr)2)2] C13
A dried Schlenk tube was charged with MnCl2 (41 mg, 0.33 mmol), THF (5 mL) and
[HN(CH2CH2P(iPr2)2] (100 mg, 0.33 mmol, 10%wt in THF) in this order and the reaction
mixture was stirred at RT overnight. A white precipitate was formed and the volatiles were
removed in vacuo. The white solid was characterized by X-ray analysis whereas crystals suitable
for analysis were grown by slow diffusion of CH2Cl2 into heptane. Yield: 122 mg, 83%.
ESI-HRMS (m/z pos): Calculated for [C16H37Cl2MnNP2]: 431.12315; found: 431.12334. Elemental analysis: Calculated for [C16H37Cl2MnNP2]: C, 44.56; H, 8.65; N, 3.25. Found: C, 44.67; H, 8.68; N, 3.40.
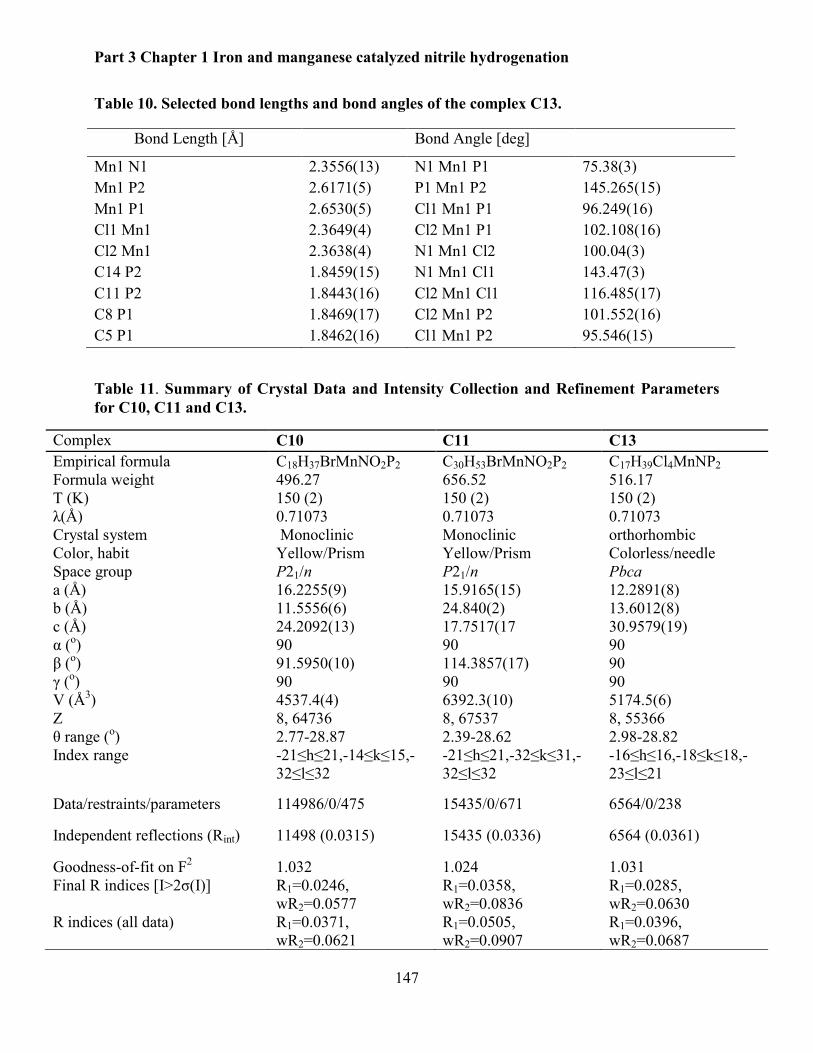
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
147
Table 10. Selected bond lengths and bond angles of the complex C13.
Bond Length [Å] Bond Angle [deg]
Mn1 N1 2.3556(13) N1 Mn1 P1 75.38(3) Mn1 P2 2.6171(5) P1 Mn1 P2 145.265(15) Mn1 P1 2.6530(5) Cl1 Mn1 P1 96.249(16) Cl1 Mn1 2.3649(4) Cl2 Mn1 P1 102.108(16) Cl2 Mn1 2.3638(4) N1 Mn1 Cl2 100.04(3) C14 P2 1.8459(15) N1 Mn1 Cl1 143.47(3) C11 P2 1.8443(16) Cl2 Mn1 Cl1 116.485(17) C8 P1 1.8469(17) Cl2 Mn1 P2 101.552(16) C5 P1 1.8462(16) Cl1 Mn1 P2 95.546(15)
Table 11. Summary of Crystal Data and Intensity Collection and Refinement Parameters for C10, C11 and C13.
Complex C10 C11 C13 Empirical formula C18H37BrMnNO2P2 C30H53BrMnNO2P2 C17H39Cl4MnNP2 Formula weight 496.27 656.52 516.17 T (K) 150 (2) 150 (2) 150 (2) λ(Å) 0.71073 0.71073 0.71073 Crystal system Monoclinic Monoclinic orthorhombic Color, habit Yellow/Prism Yellow/Prism Colorless/needle Space group P21/n P21/n Pbca a (Å) 16.2255(9) 15.9165(15) 12.2891(8) b (Å) 11.5556(6) 24.840(2) 13.6012(8) c (Å) 24.2092(13) 17.7517(17 30.9579(19) α (o) 90 90 90 β (o) 91.5950(10) 114.3857(17) 90 γ (o) 90 90 90 V (Å3) 4537.4(4) 6392.3(10) 5174.5(6) Z 8, 64736 8, 67537 8, 55366 θ range (o) 2.77-28.87 2.39-28.62 2.98-28.82 Index range -21≤h≤21,-14≤k≤15,-
32≤l≤32 -21≤h≤21,-32≤k≤31,-32≤l≤32
-16≤h≤16,-18≤k≤18,-23≤l≤21
Data/restraints/parameters 114986/0/475 15435/0/671 6564/0/238
Independent reflections (Rint) 11498 (0.0315) 15435 (0.0336) 6564 (0.0361)
Goodness-of-fit on F2 1.032 1.024 1.031 Final R indices [I>2σ(I)] R1=0.0246,
wR2=0.0577 R1=0.0358, wR2=0.0836
R1=0.0285, wR2=0.0630
R indices (all data) R1=0.0371, wR2=0.0621
R1=0.0505, wR2=0.0907
R1=0.0396, wR2=0.0687
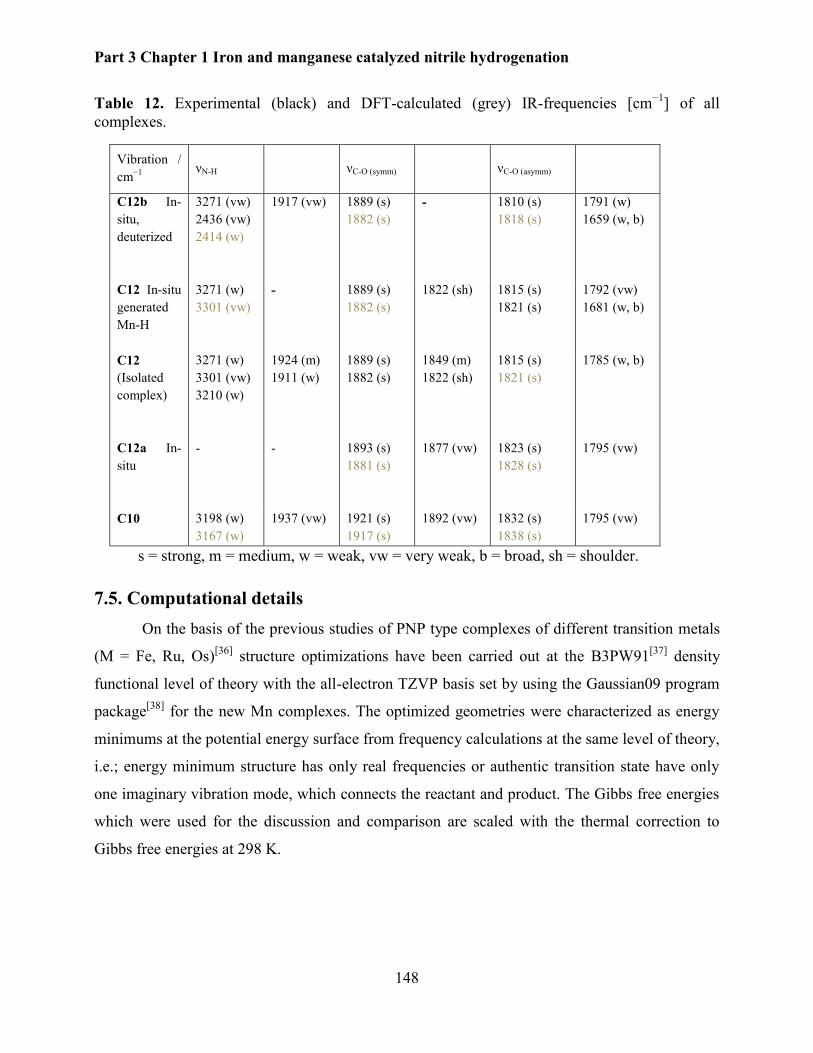
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
148
Table 12. Experimental (black) and DFT-calculated (grey) IR-frequencies [cm−1] of all complexes.
Vibration / cm−1
νN-H νC-O (symm) νC-O (asymm)
C12b In-situ, deuterized
3271 (vw) 2436 (vw) 2414 (w)
1917 (vw)
1889 (s) 1882 (s)
- 1810 (s) 1818 (s)
1791 (w) 1659 (w, b)
C12 In-situ generated Mn-H
3271 (w) 3301 (vw)
- 1889 (s) 1882 (s)
1822 (sh)
1815 (s) 1821 (s)
1792 (vw) 1681 (w, b)
C12 (Isolated complex)
3271 (w) 3301 (vw) 3210 (w)
1924 (m) 1911 (w)
1889 (s) 1882 (s)
1849 (m) 1822 (sh)
1815 (s) 1821 (s)
1785 (w, b)
C12a In-situ
- - 1893 (s) 1881 (s)
1877 (vw)
1823 (s) 1828 (s)
1795 (vw)
C10 3198 (w) 3167 (w)
1937 (vw)
1921 (s) 1917 (s)
1892 (vw) 1832 (s) 1838 (s)
1795 (vw)
s = strong, m = medium, w = weak, vw = very weak, b = broad, sh = shoulder.
7.5. Computational details On the basis of the previous studies of PNP type complexes of different transition metals
(M = Fe, Ru, Os)[36] structure optimizations have been carried out at the B3PW91[37] density
functional level of theory with the all-electron TZVP basis set by using the Gaussian09 program
package[38] for the new Mn complexes. The optimized geometries were characterized as energy
minimums at the potential energy surface from frequency calculations at the same level of theory,
i.e.; energy minimum structure has only real frequencies or authentic transition state have only
one imaginary vibration mode, which connects the reactant and product. The Gibbs free energies
which were used for the discussion and comparison are scaled with the thermal correction to
Gibbs free energies at 298 K.
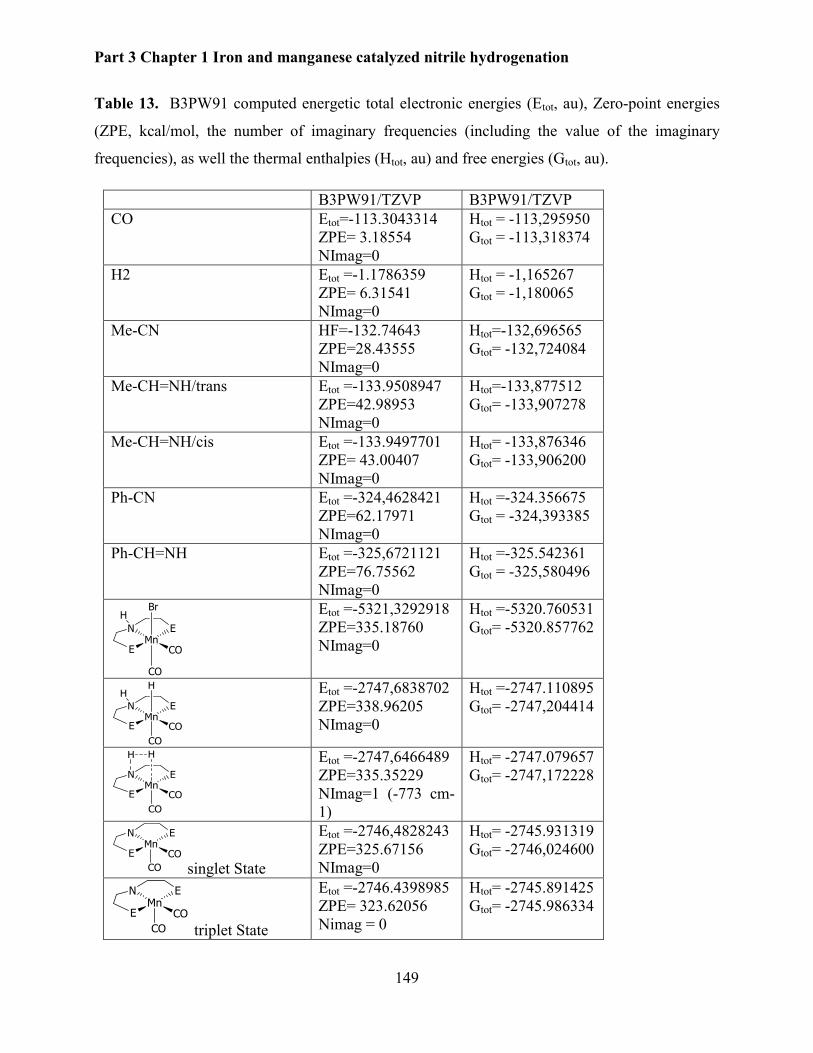
Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
149
Table 13. B3PW91 computed energetic total electronic energies (Etot, au), Zero-point energies
(ZPE, kcal/mol, the number of imaginary frequencies (including the value of the imaginary
frequencies), as well the thermal enthalpies (Htot, au) and free energies (Gtot, au).
B3PW91/TZVP B3PW91/TZVP CO Etot=-113.3043314
ZPE= 3.18554 NImag=0
Htot = -113,295950 Gtot = -113,318374
H2 Etot =-1.1786359 ZPE= 6.31541 NImag=0
Htot = -1,165267 Gtot = -1,180065
Me-CN
HF=-132.74643 ZPE=28.43555 NImag=0
Htot=-132,696565 Gtot= -132,724084
Me-CH=NH/trans
Etot =-133.9508947 ZPE=42.98953 NImag=0
Htot=-133,877512 Gtot= -133,907278
Me-CH=NH/cis
Etot =-133.9497701 ZPE= 43.00407 NImag=0
Htot= -133,876346 Gtot= -133,906200
Ph-CN Etot =-324,4628421 ZPE=62.17971 NImag=0
Htot =-324.356675 Gtot = -324,393385
Ph-CH=NH Etot =-325,6721121 ZPE=76.75562 NImag=0
Htot =-325.542361 Gtot = -325,580496
Etot =-5321,3292918 ZPE=335.18760 NImag=0
Htot =-5320.760531 Gtot= -5320.857762
Etot =-2747,6838702 ZPE=338.96205 NImag=0
Htot =-2747.110895 Gtot= -2747,204414
Etot =-2747,6466489 ZPE=335.35229 NImag=1 (-773 cm-1)
Htot= -2747.079657 Gtot= -2747,172228
singlet State
Etot =-2746,4828243 ZPE=325.67156 NImag=0
Htot= -2745.931319 Gtot= -2746,024600
triplet State
Etot =-2746.4398985 ZPE= 323.62056 Nimag = 0
Htot= -2745.891425 Gtot= -2745.986334

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
150
7.6. NMR for isolated products
(3-Chlorophenyl)methylammonium chloride
1H-NMR (300 MHz, DMSO-d6): δ 8.33 (s, 3H), 7.62 (s, 1H), 7.45-7.42 (m, 3H), 4.01 (s, 2H). 13C-NMR (75 MHz, DMSO- d6): δ 136.7, 132.9, 130.3, 128.7, 128.2, 127.7, 41.5. (2-Chlorophenyl)methylammonium chloride
1H-NMR (300 MHz, MeOD): δ 7.54-7.51 (m, 2H), 7.45-7.41 (m, 2H), 4.28 (s, 2H). 13C-NMR (75 MHz, MeOD): δ 135.2, 132.1, 132.1, 131.0, 128.8, 41.6. (3,4-Dichlorophenyl)methylammonium chloride
1H-NMR (300 MHz, MeOD): δ 7.68 (s, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.42-7.39 (m, 1H), 4.12 (s, 2H). 13C-NMR (75 MHz, MeOD): δ 134.9, 134.1, 133.8, 132.2, 132.1, 129.9, 43.0. (4-Bromophenyl)methylammonium chloride
1H-NMR (300 MHz, DMSO-d6): δ 8.47 (s, 3H), 7.62 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 3.98 (s, 2H). 13C-NMR (75 MHz, DMSO-d6): δ 133.5, 131.4, 131.3, 121.5, 41.4. (4-(Trifluoromethyl)phenyl)methanaminium chloride
1H NMR (300 MHz, DMSO-d6) δ 8.70 (s, 3H), 8.01 – 7.45 (m, 4H), 4.26 – 3.89 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 138.8, 129.7, 125.4 (q, J = 31.5 Hz), 125.3 (q, J = 4.6 Hz), 125.2 (q, J = 272.6 Hz), 41.5. 19F NMR (282 MHz, DMSO-d6) δ -56.20.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
151
Naphthalen-1-ylmethylammonium chloride
1H-NMR (300 MHz, MeOD): δ 8.11 (d, J = 8.3 Hz, 1H), 7.98-7.95 (m, 2H), 7.69-7.52 (m, 4H), 4.63 (s, 2H). 13C-NMR (75 MHz, MeOD): δ 135.3, 132.2, 131.0, 130.1, 130.0, 128.7, 128.2, 127.4, 126.4, 123.6, 41.3. 2-(4-Methoxyphenyl)ethylammonium chloride
1H-NMR (300 MHz, D2O): δ 7.26 (d, J = 8.6 Hz, 2H), 6.98 (d, J = 8.6 Hz, 2H), 3.81 (s, 3H), 3.22 (t, J = 7.3 Hz, 2H), 2.92 (t, J = 7.2 Hz, 2H). 13C-NMR (75 MHz, D2O): δ 157.9, 130.0, 129.0, 114.4, 55.3, 40.6, 31.7. (E)-3-Phenylprop-2-en-1-ylammonium chloride
1H-NMR (300 MHz, D2O): δ 7.53-7.31 (m, 8H), 6.84-6.78 (m, 1H), 6.32-6.27 (m, 1H), 3.77 (d, J = 6.8 Hz, 2H), 2.99 (t, J = 7.8 Hz, 1H), 2.74-2.69 (m, 1H), 2.02-1.92 (m, 1H). 13C-NMR (75 MHz, D2O): δ 136.0, 128.9, 128.7, 126.6, 126.7, 120.1, 41.2, 1-Decanylammonium chloride
1H NMR (300 MHz, D2O): δ 2.93 (t, J = 7.6 Hz, 2H), 1.65-1.55 (m, 2H), 1.38-1.10 (m, 14H), 0.82-0.78 (m, 3H). 13C NMR (75 MHz, D2O): δ 39.5, 31.2, 28.6, 28.5, 28.4, 28.1, 26.6, 25.5, 22.0, 13.4. 1-Heptadecanylammonium chloride
1H-NMR (300 MHz, MeOD): δ 2.90 (t, J = 7.6 Hz, 2H), 1.69-1.60 (m, 2H), 1.40-1.28 (m, 28H), 0.91-0.87 (m, 3H). 13C-NMR (75 MHz, MeOD): δ 40.7, 33.0, 30.7 (large peak), 30.6, 30.5, 30.4, 30.2, 28.5, 27.4, 23.7, 14.4.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
152
1-Nonadecanylammonium chloride
1H-NMR (300 MHz, MeOD): δ 2.90 (t, J = 7.5 Hz, 2H), 1.67-1.59 (m, 2H), 1.35-1.27 (m, 31H), 0.91-0.68 (m, 3H). 13C-NMR (75 MHz, MeOD): δ 40.7, 32.9, 30.7 (large peak), 30.6, 30.4, 30.4, 30.2, 28.5, 27.4, 23.6, 14.4. Hex-5-en-1-ylammonium chloride
1H-NMR (400 MHz, MeOD): δ 5.88-5.81 (m, 1H), 5.09-4.98 (m, 2H), 2.95 (t, J = 7.5 Hz, 2H), 2.17-2.12 (m, 2H), 1.74-1.67 (m, 2H), 1.56-1.48 (m, 2H). 13C-NMR (101 MHz, MeOD): δ 138.9, 115.5, 40.6, 33.9, 27.8, 26.5. p-Xylyl bisammonium chloride
1H-NMR (300 MHz, D2O): δ 7.48 (s, 4H), 4.17 (s, 4H). 13C-NMR (75 MHz, D2O): δ 133.4, 129.5, 42.6.
8. References [1] K. S. Hayes, Appl. Catal. A 2001, 221, 187-195. [2] S. A. Lawrence, in Amines: Synthesis, Properties, and Application, Cambridge University
Press; Cambridge, 2004. [3] D. W. Kim, C. E. Song, D. Y. Chi, J. Org. Chem. 2003, 68, 4281-4285. [4] C.-W. Kuo, J.-L. Zhu, J.-D. Wu, C.-M. Chu, C.-F. Yao, K.-S. Shia, Chem. Commun.
2007, 301-303. [5] S. Hanada, Y. Motoyama, H. Nagashima, Eur. J. Org. Chem. 2008, 24, 4097-4100. [6] S. Zhou, D. Addis, S. Das, K. Junge, M. Beller, Chem. Commun. 2009, 4883-4885. [7] X. Miao, P. H. Dixneuf, C. Fischmeister, C. Bruneau, Green Chem. 2011, 13, 2258-2271. [8] J. Seyden-Penne, in 2nd ed. Reduction by the Alumino- and Borohydrides in Organic
Synthesis, Wiley-VCH, 1997, pp. 149-154. [9] S. Laval, W. Dayoub, A. Favre-Reguillon, M. Berthod, P. Demonchaux, G. Mignani, M.
Lemaire, Tetrahedron Lett. 2009, 50, 7005-7007. [10] P. Kukula, M. Studer, H.-U. Blaser, Adv. Synth. Catal. 2004, 346, 1487-1493. [11] J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensenab, E. A. Pidko, Chem. Soc.
Rev. 2015, 44, 3808-3833. [12] S. Enthaler, K. Junge, D. Addis, G. Erre, M. Beller, ChemSusChem 2008, 1, 1006-1010. [13] S. Enthaler, D. Addis, K. Junge, G. Erre, M. Beller, Chem. Eur. J 2008, 14, 9491-9494. [14] S. Werkmeister, K. Junge, B. Wendt, A. Spannenberg, H. Jiao, C. Bornschein, M. Beller,
Chem. Eur. J. 2014, 20, 4227-4231. [15] R. Adam, C. B. Bheeter, R. Jackstell, M. Beller, ChemCatChem 2016, 8, 1329-1334.

Part 3 Chapter 1 Iron and manganese catalyzed nitrile hydrogenation
153
[15a] D. Addis, S. Enthaler, K. Junge, B. Wendt, M. Beller, Tetrahedron Lett. 2009, 50, 3654-3656.
[16] I. B. T. Li, F. N. Haque, M. Z. Iuliis, D. Song, R. H. Morris, Organometallics 2007, 26, 5940-5949.
[17] C. Gunanathan, M. Hçlscher, W. Leitner, Eur. J. Inorg. Chem. 2011, 3381-3386. [17a] J.-H. Choi, M. H. G. Prechtl, ChemCatChem 2015, 7, 1023-1028. [17b] J. Neumann, C. Bornschein, H. Jiao, K. Junge, M. Beller, Eur. J. Org. Chem. 2015, 5944-
5948. [18] J. B. X. Miao, P. H. Dixneuf, C. Fischmeister, C., J.-L. D. Bruneau, J.-L. Couturier,
ChemCatChem 2012, 4, 1911-1916. [18a] R. Reguillo, M. Grellier, N. Vautravers, L. Vendier, S. Sabo-Etienne, J. Am. Chem. Soc.
2010, 132, 7854–7855. [19] S. Takemoto, H. Kawamura, Y. Yamada, T. Okada, A. Ono, E. Yoshikawa, Y. Mizobe,
M. Hidai, Organometallics 2002, 21, 3897-3904. [20] S. Chakraborty, H. Berke, ACS Catal. 2014, 4, 2191-2194. [21] K. Rajesh, B. Dudle, O. Blacque, H. Berke, Adv. Synth. Catal. 2011, 353, 1479-1484. [22] C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge,
K. Junge, M. Beller, Nat. Commun. 2014, 5, 4111. [23] S. Chakraborty, G. Leitus, D. Milstein, Chem. Commun. 2016, 52, 1812-1815. [24] S. Werkmeister, J. Neumann, K. Junge, M. Beller, Chem. Eur. J. 2015, 21, 12226-12250. [25] A. M. Tondreau, J. M. Boncella, Polyhedron 2016, 116, 96-104. [26] F. Kallmeier, T. Irrgang, T. Dietel, R. Kempe, Angew.Chem. Int. Ed. 2016, 55, 11806-
11809. [27] A. T. Radosevich, J. G. Melnick, S. A. Stoian, D. Bacciu, C.-H. Chen, B. M. Foxman, O.
V. Ozerov, D. G. Nocera, Inorg. Chem. 2009, 48, 9214-9221. [28] M. L. Clarke, Catal. Sci. Technol. 2012, 2, 2418-2423. [29] P. A. Dub, T. Ikariya, ACS Catal. 2012, 2, 1718-1741. [30] S. Werkmeister, K. Junge, M. Beller, Org. Process Res. Dev. 2014, 18, 289-302. [31] H. W. Walker, P. C. Ford, Inorg. Chem. 1982, 2509-2510. [32] J. F. Sonnenberg, N. Coombs, P. A. Dube, R. H. Morris, J. Am. Chem. Soc. 2012, 134,
5893-5899. [33] Y. Li, S. Yu, X. Wu, J. Xiao, W. Shen, Z. Dong, J. Gao, J. Am. Chem. Soc. 2014, 136,
4031-4039. a)G. T. Britton, J. W. McBain, J. Am. Chem. Soc. 1926, 48, 593–598. [34] G. M. Sheldrick, Acta Cryst 2008, A64, 112. [35] G. M. Sheldrick, Acta Cryst 2015, C71, 3. [36] H. Jiao, K. Junge, E. A. Alberico, M. Beller, J. Comput. Chem. 2016, 37, 168-176. [37] J. P. Perdew, Phys. Rev. B 1986, 33, 8822-8824. [38] A. Schaefer, C. Huber, R. Ahlrichs, J. Chem. Phys. 1994, 100, 5829-5835.

154
Part 3 Chapter 2
Iron and manganese catalyzed hydrogenation of esters
to alcohols

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
155

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
156
1. Introduction The reduction of carboxylic acid derivatives constitutes one of the most important
transformations both on laboratory and industry applications.[1] Obviously, reduction with
molecular hydrogen is an atom efficient and powerful tool for the development of successful
process in the field of fine chemicals and pharmaceuticals.[2] Although the catalytic
hydrogenation of aldehydes and ketones are well reported, reduction of carboxylic acids and their
ester derivatives using molecular H2 are still a challenging task due to less electrophilicity of the
carbonyl carbon (Scheme 1). In this respect, the catalytic hydrogenation of esters to alcohols is
receiving considerable interest as they represent a key industrial process for the production of
agrochemicals, flavors and fragrance. Mostly, this transformation relied on the use of
stoichiometric amounts of inorganic metal hydrides such as LiAlH4 and related reagents resulting
in the formation of stoichiometric amount of waste by-products especially on the large scale; the
handling and disposal of the generated waste is usually problematic in industry.[3] So the
development of efficient and general method for the reduction of esters is more interesting in a
greener chemistry point of view. Particularly, catalytic hydrogenation of esters and lactones
employing H2 constitutes a completely atom economic, waste-free and environmentally more
benign transformation.[4-6] In this area, heterogeneous catalysts were applied for more than a
century in the hydrogenation of fatty acids and esters. Indeed, the field of hydrogenation of fats
and oils was awarded in 1912 by the Nobel prize to Paul Sabatier and one of the first commercial
hydrogenation processes for fats and oils was patented by W. Normann in 1902 (then developed
by Procter and Gamble since 1909) and in 1903 by J. Crosfield & sons Ldt. However, these
catalysts usually require high temperature and pressure (usually in the range 200-300 °C and 140-
300 bar of hydrogen) and these methods are limited by poor functional group tolerance.[7-9]
Hence, the development of milder and more selective catalytic protocols for ester hydrogenation
was facilitated by the use of well-defined homogeneous complexes and still constitutes an actual
and highly desired research goal. More particularly, noble transition metals such as ruthenium,
iridium, and osmium complexes were used advantageously for the reduction of esters.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
157
Figure 1. Challenge level in catalytic hydrogenation: carboxylic acids and their derivatives.
1.1. Ruthenium catalyzed ester hydrogenation Ruthenium monophosphine based catalysts
Indeed, in the 1980’s, the first homogenous hydrogenation of esters to alcohols was
described using anionic ruthenium dihydride complexes
[(Ph3P)Ph2P(C6H4)RuH2−K+(Et2O)·C10H18]2 and [(Ph3P)(Ph2P)RuH2
−K+·diglyme]2 under mild
conditions (90 °C, 6.2 bar).[10-11] Notably, the hydrogenation works better with activated esters
[C2H5CO2CH3 (5%) < CH3CO2CH3 (22%) < CF3CO2CH3 (88%) < CF3CO2CH2CF3
(quantitative)]. (Scheme 1)
Scheme 1. First homogeneous catalyzed hydrogenation of activated esters.
Simultaneously, Matteoli and Bianchi reported that the ruthenium cluster
[H4Ru4(CO)8(PBu3)4] catalyzed the hydrogenation of dicarboxylic acid esters to the
corresponding hydroxyesters under very harsh conditions (180 °C, 130 bar H2, 144 h) in
moderate activities for a selection of dicarboxylic acid esters such as dimethyl oxalate and diethyl
malonate. Ru(CO)2(μ-OAc)2(PBu3)2 was shown to catalyze efficiently the hydrogenation of
dialkyl oxalates under similar conditions yielding quantitatively the corresponding alkyl
glycolate.[12-14]

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
158
Ruthenium bidentate ligand based catalysts
In 2007, Saudan from Firmenich company made a significant contribution for the ester
hydrogenation using highly efficient Ru complexes with bidentate N,P ligands (Scheme 2). The
reaction proceeded in milder conditions (50 bar of H2, 100 °C). Using this protocol, aromatic
esters, aliphatic esters and lactones were hydrogenated successfully. Interestingly, esters with a
di- or tri-substituted C=C bond were reduced to the corresponding unsaturated alcohols with high
chemoselectivity. By contrast, esters with terminal alkenyl moiety or methyl cinnamate led to the
corresponding saturated alcohols.[15] Interestingly, the tetradentate PNNP ligand/Ru complex 2
exhibited similar reactivity.
Scheme 2. P,N ligand Ru based complexes for ester hydrogenation.
In 2008, Ikariya found that the bifunctional catalyst Cp*RuCl(P-N) 3 (1 mol%) in the
presence of a catalytic amount of the base (25 equiv. / Ru) promoted the hydrogenation of
lactones and simple esters.[16] As a representative example, phthalide was hydrogenated to the
corresponding alcohol in quantitative yield under 50 bar H2 at 100 °C. (Scheme 3)
Scheme 3. P-N and N-N ligand based ruthenium catalysts for hydrogenation of lactones.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
159
Similarly, Beller reported the use of an in situ generated catalyst from [Ru(benzene)Cl2]2
(0.25 mol%) and a phosphine-imidazoyl ligand 5 (1 mol%) in the presence of 10 mol% of
t-BuOK in THF at 100 °C under 50 bar of hydrogen to promote the hydrogenation of benzoate
derivatives in 17-99% yields. Phthalide was then hydrogenated in 84% yield.[17]
In 2011, Ikariya modified the complex structure from P-N to N-N type ligand and they
showed a better activity for the hydrogenation of lactones. [18] A large variety of lactones were
then reduced into the corresponding diols in the presence of 1 mol% of the ruthenium complex 4
and 25 mol% of t-BuOK as the base under 50 bar H2 at 100 °C for 6-72 h. Notably, using a chiral
optically active 1,2-diamine ligands, the corresponding Cp*RuCl(N-N) complex can be applied
for an enantioselective hydrogenation of racemic lactone via a dynamic kinetic resolution (full
conversion, ee up to 32% under 50 bar of H2 at 80 °C).
In 2010, Morris demonstrated that [RuCp*(C-NH2)(py)]PF6 6 bearing a chelating N-
heterocyclic carbene ligand with a pendant NH2 group was an active catalyst for the
hydrogenation of methyl benzoate to benzyl alcohol and methanol at 25 °C under 8 bar of H2
with a TOF of 209 h-1, or at 50 °C under 25 bar of H2 with a TOF of 838 h-1 (Scheme 4).
Interestingly, this catalyst is active for the hydrogenation of other polar bonds.[19]
Scheme 4. NHC-Ru catalyzed hydrogenation of methyl benzoate.
Bis-NHC ruthenium catalytic species can be also prepared from [Ru(p-cymene)Cl2]2 (0.5
mol%) and bis-carbene 7 (2 mol%) and has also shown a nice activity for the reduction of
benzoate and alkanoate derivatives and lactones using 30 mol% of t-BuOK in 1.4-dioxane at 100
°C under 50 bar of hydrogen (32-92% yields).[20]
Ruthenium-triphos based catalysts
The use of triphos ligands associated to ruthenium permitted to make a breakthrough in
the hydrogenation of carboxylic acid derivatives, in particular of esters. In 1997, Elsevier and co-
workers reported a more reactive ruthenium-phosphine catalyst generated in situ from a triphos

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
160
ligand, MeC(CH2PPh2)3, and Ru(acac)3 in the presence of zinc in MeOH which was found to be
one of the most efficient homogeneous catalytic system for the hydrogenation of dimethyl oxalate
to ethylene glycol (full conversion, 84% yield under 70 bar of hydrogen at 100 °C for 16 h)
(Scheme 5).[21] Using the same catalytic system, Frediani succeeded to hydrogenate of fumaric
acid, succinic acid and γ-butyrolactone to the 1,4-butanediol (best result: 81% of 1,4-butanediol
from fumaric acid under 80 bar of hydrogen at 180 °C for 48 h).[22]
Scheme 5. Elsevier’s Ru/triphos catalyst for hydrogenation of dimethyl oxalate.
Hara and Wada demonstrated that the catalytic system based on Ru (acac)3 / tris-n-
octylphosphine / p-toluenesulfonic acid was able to hydrogenate cyclic esters to α,ω-diol under
less drastic pressure conditions (50 bar H2, 200 °C, 3 h).[23] In 2001, the in situ generated catalyst
from [Ru(acac)3 (2 mol%)/P(n-C8H17)3 (20 mol%)] permitted to perform the hydrogenation of
methyl phenylacetate to phenylethanol (21%) in the presence of a catalytic amount of Zn (5
mol%) in tetraglyme under low hydrogen pressure (10 bar H2), but still at 200 °C.[24] Notably the
transesterification product, Ph-CH2-COO-CH2-CH2-Ph, was also formed in 27% yield.
Notably, the hydrogenation of unactivated esters were successfully developed using Ru-
triphos system and HBF4 or Et3N as an additive (in replacement of zinc) under 85 bar H2 at 120
°C (Scheme 6). This catalytic system allowed to hydrogenate benzyl benzoate (86%), an aliphatic
ester (methyl palmitate, quantitative) and an aliphatic di-ester (dimethyl maleate, 94%).[25]
Scheme 6. Reduction of benzyl benzoate.
Similarly, in 2011, Hanton has developed a ruthenium triphosphine system using a
tripodal phosphine ligand, N(CH2PPh2)3, N-TriPhosPh in association with Ru(acac)3 which was
an active catalyst (1 mol%) for the hydrogenation of esters such as dimethyl oxalate (to ethylene
glycol, quantitative conversion) under 80 bar of H2 at 100 °C in 5.7 h. By contrast, this system
showed very low activity towards simple aliphatic esters.[26]

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
161
Pincer ruthenium based catalysts
After the significant progress made by Ru-triphos system for the ester hydrogenation, in
2006, Milstein reported that the well-defined Ru NNP pincer complex 8 was able to hydrogenate
non-activated esters to the corresponding alcohols under relatively mild and neutral conditions,
without any additive (5.3 bar of hydrogen, 115 °C, 7 h) (Scheme 7).[27] Interestingly, in a
mechanism point of view, the pincer PNN ligand in the complex 8 has a crucial role for the
hydrogen cleavage via an aromatization/dearomatization sequence.
Scheme 7. Ru NNP pincer complex catalyzed ester hydrogenation.
In 2012, Takasago company has shown that the commercially available Ru-MACHO PNP
pincer complex 9 (0.1 mol%) could effectively catalyzed the hydrogenation of various kinds of
esters with good yields and selectivities in the presence of NaOMe as a base (10 mol%) under 50
bar of H2 at 100 for 16 h.[28] Interestingly, this system can hydrogenate optically pure (R)-methyl
lactate using 0.05 mol% of the complex 9 at 28 °C for 12 h under 42 bar of H2 giving 1,2
propandiol in 92% yield and 99% ee. To prove the viability of ester hydrogenation in practical
large-scale, the reduction of methyl lactate was scaled up to 2200 Kg (Scheme 8). This complex 9
(0.05 mol%) could also be used for the hydrogenation of L-menthyloxyacetic acid methyl ester to
the cooling flavor 2-(L-menthoxy)ethanol by reaction at 80 °C under 45 bar of H2 for 5 h.
Interestingly, using an in situ generated catalyst from hydrazine-MACHO like ligand 10 and
[RuCl2(p-cymene)]2 (0.01-0.1 mol%) in the presence of 5 mol% of NaOEt as the base,
hydrogenation of esters can be conducted at 80 °C under 50 bar of H2 for 16 h (TON up to 17200
for the hydrogenation of methyl benzoate).[29]
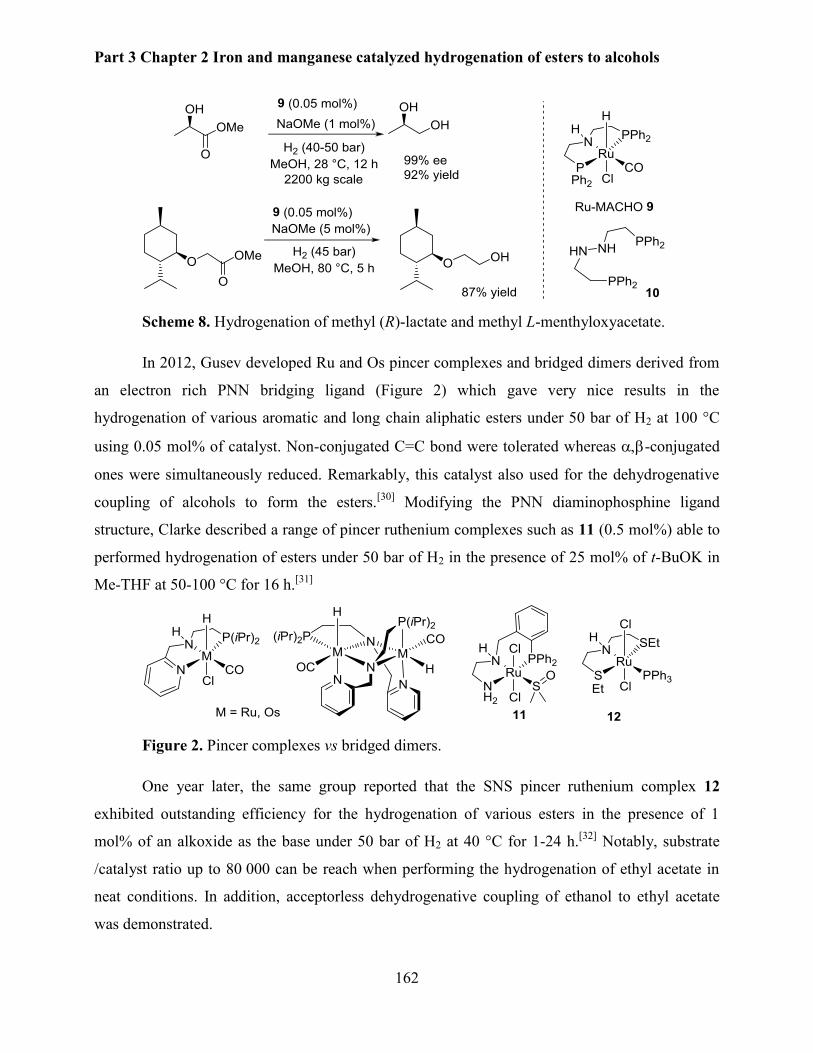
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
162
Scheme 8. Hydrogenation of methyl (R)-lactate and methyl L-menthyloxyacetate.
In 2012, Gusev developed Ru and Os pincer complexes and bridged dimers derived from
an electron rich PNN bridging ligand (Figure 2) which gave very nice results in the
hydrogenation of various aromatic and long chain aliphatic esters under 50 bar of H2 at 100 °C
using 0.05 mol% of catalyst. Non-conjugated C=C bond were tolerated whereas -conjugated
ones were simultaneously reduced. Remarkably, this catalyst also used for the dehydrogenative
coupling of alcohols to form the esters.[30] Modifying the PNN diaminophosphine ligand
structure, Clarke described a range of pincer ruthenium complexes such as 11 (0.5 mol%) able to
performed hydrogenation of esters under 50 bar of H2 in the presence of 25 mol% of t-BuOK in
Me-THF at 50-100 °C for 16 h.[31]
Figure 2. Pincer complexes vs bridged dimers.
One year later, the same group reported that the SNS pincer ruthenium complex 12
exhibited outstanding efficiency for the hydrogenation of various esters in the presence of 1
mol% of an alkoxide as the base under 50 bar of H2 at 40 °C for 1-24 h.[32] Notably, substrate
/catalyst ratio up to 80 000 can be reach when performing the hydrogenation of ethyl acetate in
neat conditions. In addition, acceptorless dehydrogenative coupling of ethanol to ethyl acetate
was demonstrated.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
163
In 2015, Pidko described a nice example of ester hydrogenation involving a bis-N-
heterocyclic carbene ligand (NHC) Ru CNC pincer complex 15 (0.025-0.067 mol%) in the
presence of 2 mol% of t-BuOK at 70 °C for 16 h under 50 bar of H2 (Figure 3). Both aromatic
and aliphatic esters are efficiently reduced with TOF up to 78 600 h-1 for ethyl hexanoate at
substrate/catalyst ratio of 10 000, which makes this complex one of the most active in
hydrogenation of esters.[33] Notably, the hydrogenation of methyl cinnamate yielded a mixture of
the saturated (10%) and the unsaturated alcohol (72%), whereas the reduction of dimethyl
itaconate led chemospecifically to the saturated diester resulting from the hydrogenation of only
the C=C bond. The pyridine bis-NHC ruthenium complex 16 was also active in ester
hydrogenation under similar conditions (0.5 mol% 16, 70 °C for 4 h under 50 bar of H2).[34]
Recently, following the works of Song,[35] Chianese[36] reported the use of NHC-based
CNN pincer ruthenium complexes 13 and 14 bearing amino pyridine arm (0.1-0.8 mol%) for the
hydrogenation of both alkyloate and benzoate esters at 105 °C under low hydrogen pressure (6
bar) in the presence of 0.6-5 mol% of NaOt-Bu. The reduction of lactones to diol need higher
hydrogen pressure (30 bar).
Figure 3. Ru-NHC based pincer complexes.
Ruthenium polydentate based catalysts
In 2014, Zhou described ruthenium complexes bearing tetradentate bipyridine ligands 17
which were found to be active catalysts for hydrogenation of various aromatic, aliphatic esters
and lactones under mild conditions (50 bar H2, 25 °C) at low catalyst loading (0.01-0.1 mol%)
(Figure 4). Particularly, biomass derived valerolactone (GVL) was hydrogenated to useful 1,4-
pentanediol with excellent TON (91 000) and TOF (1896 h-1).[37]

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
164
In 2015, a new tetradentate ruthenium complex 18 was reported as an efficient catalyst for
the hydrogenation of esters, including fatty acid esters to fatty alcohols.[38] Using 0.001-0.02
mol% of 18 in the presence of 2.5 mol% of KOtBu under 50 atm of H2 at 80 °C for 5 h, a large
variety of esters including ε-caprolactone and diesters can be reduced in 80-99% yields with TON
up to 80 000 for the hydrogenation of ethyl acetate. Noticeably, the hydrogenation of methyl
cinnamate yielded exclusively to the saturated alcohol (72%), whereas the reduction of
unconjugated unsaturated esters such as methyl 3-cyclohexenecarboxylate gave a mixture of the
saturated (3%) and the unsaturated (94%) alcohols. Using similar tetradentate ruthenium complex
19 bearing a bipyridine moiety,[39] they obtained a less active catalyst (100 °C, 50 bar H2, 16 h at
0.01-0.02 mol%).
Figure 4. Recently developed Ru complexes for effective ester hydrogenation.
1.2 Iridium catalyzed ester hydrogenation Beller reported iridium-catalyzed hydrogenation of esters in the presence of base
(NaOMe, 10 mol%) at 50 bar H2 and 130 oC. By using a commercially available iridium pincer
complex, different aromatic, aliphatic, and (hetero)cyclic esters were reduced to the
corresponding alcohols in moderate to very good yields.[39a]
Scheme 9. Iridum catalyzed ester hydrogenation.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
165
1.3 Earth abundant metals for ester hydrogenation Albeit the ester hydrogenation was successful with precious ruthenium complexes, the
substitution of expensive and potentially toxic noble metal catalysts by inexpensive, abundant,
and environmentally benign metals is a prime goal in chemistry. In particular, iron and
manganese, are attractive alternatives because of their high abundance, low cost, and low
toxicity.
Iron catalyzed ester hydrogenation
In 2014, Milstein developed the first ester hydrogenation catalyst using PNP Fe catalytic
system (Scheme 10). The dihydride iron pincer complex 21 (1 mol%) was effective for the
hydrogenation of activated esters such as trifluoroacetates.[40] The selective hydrogenation of
trifluoroacetic esters to the corresponding alcohols proceeded smoothly under relatively mild
reaction conditions (5-25 bar and 40 °C) to give the products in 52-99% NMR yields when
NaOAc (5 mol%) was used. Unfortunately, no reduction was observed with difluoroacetic ester
derivatives, and the report did not describe the reduction of unactivated esters.
Scheme 10. First iron catalyzed hydrogenation of esters.
In the same year, a significant breakthrough was made by Beller[41] and Guan[42-43] in this
field using iron MACHO type PNP complex C7. Both groups reported independently the
effective and selective hydrogenation of aromatic, aliphatic, diesters and lactones in base free
conditions (1-3 mol% of C7, 10-30 bar H2, 100-120 °C) (Scheme 11). Carboxylic amides,
heteroaromatic motifs such as furans, pyridines, benzothiazoles, and non-conjugated alkenyl
moieties were tolerated whereas nitriles were reduced to the corresponding primary amines.
Under neat conditions, crude industrial mixtures of C12-C16 esters were also reduced.
Interestingly Beller showed the selective hydrogenation of ester moiety in a dodecapeptide which
is the intermediate for the synthesis of Alisporivir. In this case only the terminal methyl ester was
hydrogenated even in the presence of sensitive acetate and double bond. DFT calculation showed

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
166
that Fe dihydride is the active species and involves the outer-sphere mechanism which is
simultaneously transfer the hydride from the metal center as well as from the PNP ligand.
Scheme 11. Fe PNP system for effective ester hydrogenation.
Cobalt catalyzed ester hydrogenation
In 2015, Milstein reported the first cobalt pincer complex able to catalyze the
hydrogenation of esters to the corresponding alcohols in the presence of NaEt3BH (8 mol%) and
t-BuOK (25 mol%) using 4 mol% of the PNN pincer cobalt complex 22 under 50 bar of H2 at 130
°C for 38-72 h.[44] (Scheme 12) Importantly, only alkanoate derivatives and lactones were
reduced to the corresponding alcohols (50-87% GC-yields) and both methyl benzoate and 2,2,2-
trifluoroethyl trifluoroacetate were not reduced. The mechanism thus suggested that the reaction
proceeded through an enolate intermediate, explaining the limitation of the scope.
Scheme 12. Co PNN catalyst for ester hydrogenation.
Recently, Bruin and Elsevier reported a Co/triphos system for the highly effective
hydrogenation of esters and acids.[45] Using an in situ generated catalyst obtained from

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
167
Co(BF4)26H2O and MeC(CH2Ph2)3 in methanol under 80 bar of hydrogen at 100 °C for 22 h,
lactones, alkanoate and benzoate derivatives were reduced quite efficiently.
In the light of these results, the development of catalytic system which works under
milder conditions is continuing interest in research area. In this chapter, we described the well-
defined iron and manganese pincer complexes catalyzed ester hydrogenation.
2. Results and discussions
2.1 Hydrogenation of esters to alcohols catalyzed by iron pincer complexes Encouraged by the recent contributions on the hydrogenation of esters to alcohols[41-42]
conducted with the iron PNP pincer complex C7, we focused on the use of two novel well-
defined complexes C8 and C9 bearing phosphine motifs with different alkyl substituents in order
to evaluate their influence on the catalytic performance of the iron complexes in the
hydrogenation of esters and lactones. (Figure 5)
Figure 5. PNP Pincer iron complexes used for this study.
2.1.1 Optimisation of the reaction parameters The efficiency of these well-defined iron complexes C7-C9 were investigated in ester
hydrogenation. For the catalytic benchmark reaction, methyl benzoate was chosen as the tested
substrate using 1 mol% of the complexes C7-C9 under 30 bar of H2 at 60 °C. Interestingly, with
the complex C9, benzyl alcohol was obtained in 99% GC-yield (Table 1, entry 3) whereas under
the same reaction conditions, the complexes C7 and C8 only gave benzyl alcohol in 50 and 30%
GC-yields, respectively (Table 1, entries 1-2). There was no other side product was observed.
Noteworthy, the complex C9 also permitted the hydrogenation of methyl benzoate even at 40 °C
but with a significant lower yield of 44% (Table 1, entry 4).
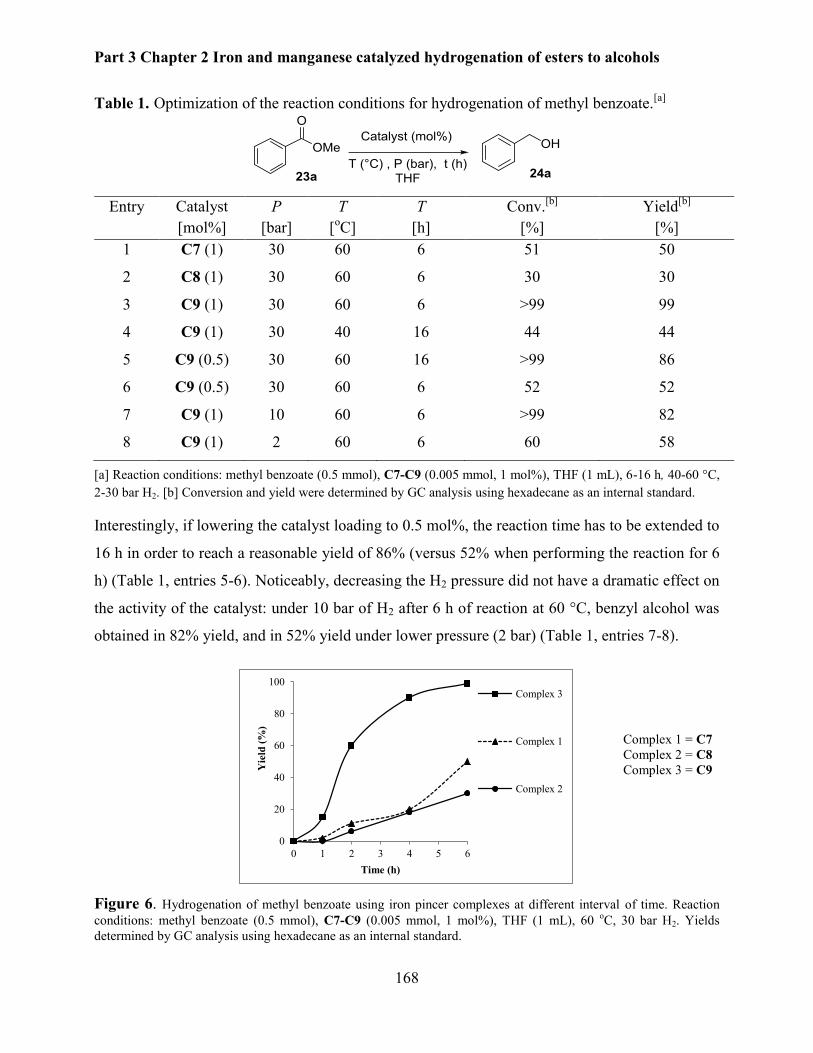
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
168
Table 1. Optimization of the reaction conditions for hydrogenation of methyl benzoate.[a]
[a] Reaction conditions: methyl benzoate (0.5 mmol), C7-C9 (0.005 mmol, 1 mol%), THF (1 mL), 6-16 h, 40-60 °C, 2-30 bar H2. [b] Conversion and yield were determined by GC analysis using hexadecane as an internal standard.
Interestingly, if lowering the catalyst loading to 0.5 mol%, the reaction time has to be extended to
16 h in order to reach a reasonable yield of 86% (versus 52% when performing the reaction for 6
h) (Table 1, entries 5-6). Noticeably, decreasing the H2 pressure did not have a dramatic effect on
the activity of the catalyst: under 10 bar of H2 after 6 h of reaction at 60 °C, benzyl alcohol was
obtained in 82% yield, and in 52% yield under lower pressure (2 bar) (Table 1, entries 7-8).
Figure 6. Hydrogenation of methyl benzoate using iron pincer complexes at different interval of time. Reaction conditions: methyl benzoate (0.5 mmol), C7-C9 (0.005 mmol, 1 mol%), THF (1 mL), 60 oC, 30 bar H2. Yields determined by GC analysis using hexadecane as an internal standard.
0
20
40
60
80
100
0 1 2 3 4 5 6
Yie
ld (%
)
Time (h)
Complex 3
Complex 1
Complex 2
Entry Catalyst [mol%]
P [bar]
T [oC]
T [h]
Conv.[b] [%]
Yield[b] [%]
1 C7 (1) 30 60 6 51 50
2 C8 (1) 30 60 6 30 30
3 C9 (1) 30 60 6 >99 99
4 C9 (1) 30 40 16 44 44
5 C9 (0.5) 30 60 16 >99 86
6 C9 (0.5) 30 60 6 52 52
7 C9 (1) 10 60 6 >99 82
8 C9 (1) 2 60 6 60 58
Complex 1 = C7 Complex 2 = C8 Complex 3 = C9

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
169
To evaluate the most active catalyst, we studied the performance of the complexes C7-C9
in the hydrogenation of methyl benzoate at 60 oC under 30 bar of H2 applying at different interval
times (Figure 6). The hydrogenation of methyl benzoate performed with complex C9 afforded
benzyl alcohol in 90% yield within 4 h. However, the use of the complexes C7 and C8 under the
same reaction conditions resulted in significantly lower yields of 20% and 18% respectively.
These results clearly showed that the complex C9 bearing diethylphosphino moiety is the most
efficient iron catalyst of the series for ester hydrogenation. The best optimised conditions found
for the hydrogenation of methyl benzoate is 1 mol% of C9 under 30 bar of H2 at 60 oC for 6 h.
2.1.2 Hydrogenation of various aromatic and aliphatic esters
Next, the scope of the hydrogenation reaction was examined with the diethylphosphine
tagged complex C9. Various aromatic and aliphatic esters, diesters (Tables 2 and 3) as well as
lactones (Table 3) were hydrogenated to their corresponding alcohols/diols with excellent yields
at 30 bar H2 and 60-100 °C using 1-2 mol% of C9.
Benzoate derivatives with both electron-donating 23b and electron-withdrawing (23c,
23d) substituents were converted into the corresponding alcohols with excellent yields under 30
bar H2 at 60 °C for 18 h (Table 2, entries 2-4). Noticeably, with strong electron-withdrawing
substituent such as CF3, 2 mol% of C9 was necessary to obtain a good yield. Additionally, the
hydrogenation of aliphatic esters was also possible: as a representative example, the long chain
aliphatic ester methyl octanoate 23e produced 1-octanol in quantitative yield by reaction in the
presence of 1 mol% of C9 under 30 bar H2 at 60 °C for 6 h (Table 2, entry 5). Then, the
chemoselectivity of this hydrogenation was studied utilizing ester derivatives bearing alkenyl
moieties (Table 2, entries 6-9). Noticeably, esters having conjugated C=C bonds such as methyl
1-cyclohexene-1-carboxylate 23f or methyl cinnamate 23g were fully reduced and the
corresponding saturated alcohols were obtained in 95% and 98% yields, respectively.
Importantly, the isolated C=C bond both in methyl 3-cyclohexene-1-carboxylate 23h and the
aliphatic unsaturated ester 23i was not hydrogenated during this catalytic reduction and the
corresponding unsaturated alcohols were specifically obtained. Heteroaromatic esters such as
furfuryl ester 23j and methyl nicotinate 23k were smoothly reduced to the desired alcohols in 92-
95% yields (Table 2, entries 10-11).

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
170
Hydrogenation of menthyl acetate 23l led to menthol in 63% yield, showing that this iron
hydrogenation may be a potential deprotection method of acetate protecting group. The
hydrogenation of methyl levulinate afforded 1,4-pentane diol 24m in 90% isolated yield, showing
that a ketone moiety was not tolerated under these conditions. Moreover, the reduction of an
alkanoate bearing a N,N-dimethylamino substituent such as ethyl 3-(N,N-dimethylamino)-
propanoate led to the corresponding 3-(N,N-dimethylamino)propan-1-ol 24n in 86% GC yield
(Table 2, entry 14). Note that the homogeneous hydrogenation of this substrate 23n to 24n was
not possible with the well described Ru-MACHO-BH catalyst.[10]
2.1.3 Hydrogenation of diesters and lactones
The efficiency of the complex C9 as selective homogeneous hydrogenation catalyst was
further demonstrated in the preparation of classical diols from diesters and lactones, respectively.
Indeed, aromatic diesters such as dimethyl terephthalate 23o and naphthalene 2,6-
dimethylcarboxylate 23p were cleanly reduced to form the diols in 96 and 89% yields,
respectively, using 1 mol% of C9 under 30 bar H2 at 100 °C for 18 h (Table 3, entries 1-2).
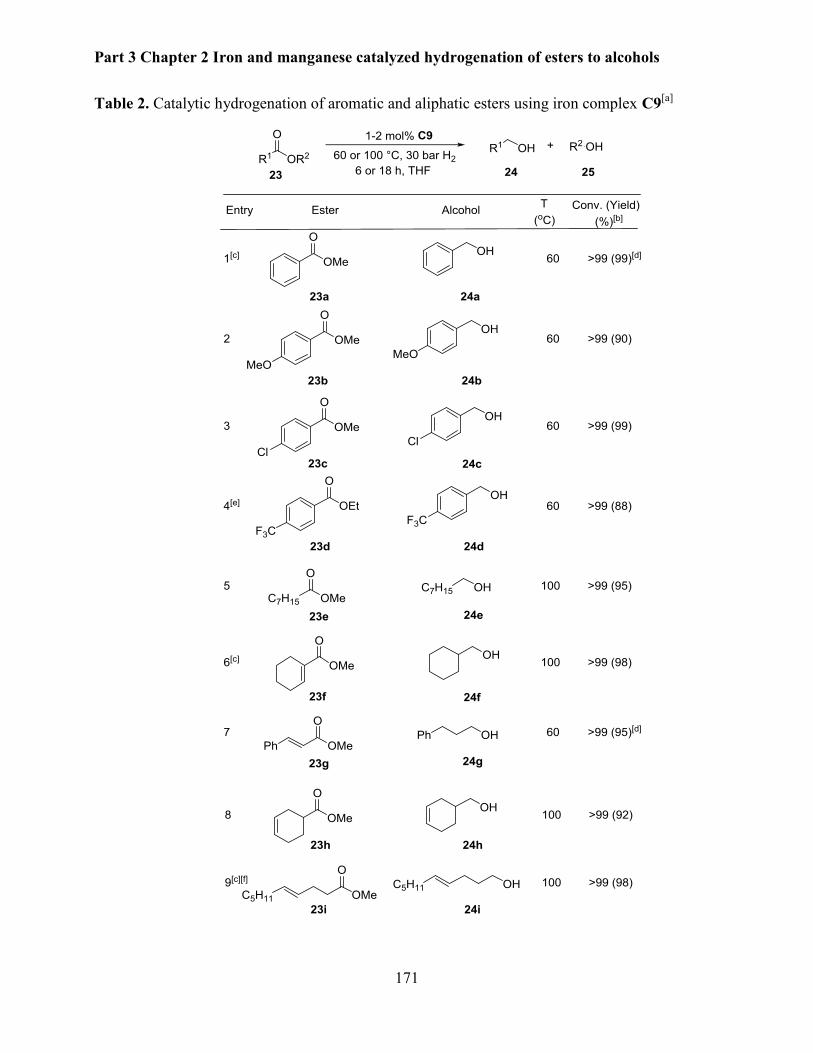
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
171
Table 2. Catalytic hydrogenation of aromatic and aliphatic esters using iron complex C9[a]

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
172
[a] Substrate (1 mmol), C9 (0.01 mmol, 1 mol%), THF (1 mL), 60 or 100 °C, 30 bar H2, 18 h. [b] Conversion was determined by GC analysis using hexadecane as an internal standard (Isolated yield in parentheses). [c] Substrate (0.5 mmol), C9 (0.005 mmol, 1 mol%), yield was determined GC analysis using hexadecane as an internal standard. [d] Reaction time: 6 h. [e] 2 mol% C9. [f] cis-4-Decen-1-ol was used for the product calibration.
The catalytic system was tolerant towards pyridine ring as the 3,5-dicarboxylatopyridine
derivative 23q was reduced selectively to the corresponding diol 24q in 91% isolated yield when
using 2 mol% of the complex C9 (Table 3, entry 3). Aliphatic dimethylsuccinate was also
reduced to 1,4-butanediol in 97% yield under such conditions (Table 3, entry 4).
Lactones can be fully reduced leading to the corresponding diols (Table 3, entries 5-7)
butyrolactone was easily reduced quantitatively to 1,4-butanediol using 1 mol% of C9 under
30 bar H2 at 60 °C for 18 h. The valerolactone (GVL), an important bio-mass derivative, was
successfully reduced in more drastic conditions, as 2 mol% of the complex C9 were necessary at
100 °C under 30 bar H2 for 18 h to lead to the 1-methylbutane-1,4-diol 24t in 98% isolated yield.
The mixture of cis and trans isomers of the whiskey lactone 23u (one of the important ingredient
in the aroma of whiskey) was hydrogenated to the corresponding diol 24u with 97% isolated
yield (mixture of stereoisomers).
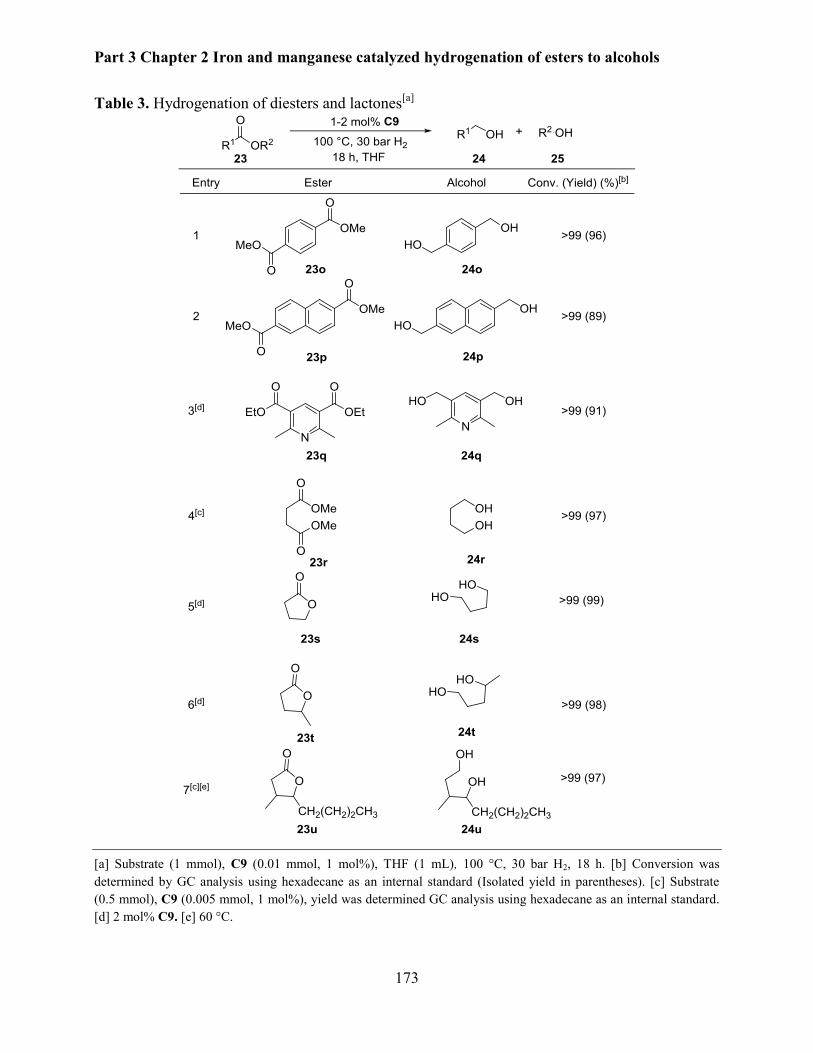
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
173
Table 3. Hydrogenation of diesters and lactones[a]
[a] Substrate (1 mmol), C9 (0.01 mmol, 1 mol%), THF (1 mL), 100 °C, 30 bar H2, 18 h. [b] Conversion was determined by GC analysis using hexadecane as an internal standard (Isolated yield in parentheses). [c] Substrate (0.5 mmol), C9 (0.005 mmol, 1 mol%), yield was determined GC analysis using hexadecane as an internal standard. [d] 2 mol% C9. [e] 60 °C.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
174
Limitations were also observed: indeed, under the optimised conditions (1 mol% C9, 30
bar H2, 100 oC, 18 h) difficult substrates such as esters bearing benzothiazoline or phenol,
dimethyl oxalate, and enol ester were not reduced.
Figure 7. Non working substrates.
2.1.4 Computation studies In the previous studies on methyl benzoate hydrogenation [23] using catalyst C7A at the
B3PW91/TZVP DFT level, it was found that the reaction follows an outer-sphere mechanism in
two successive cycles. The first one is the formation of the corresponding hemiacetal followed by
the dissociation into benzaldehyde and methanol; and the second one is benzaldehyde
hydrogenation to benzyl alcohol. After each step, the active catalyst C7A is regenerated by H2
addition from the amido intermediate C7B (Scheme 13).
Scheme 13. First step of methyl benzoate hydrogenation.
On the basis of the computed Gibbs free energy barriers, hemiacetal formation has the
highest barrier and therefore represents the rate-determining step. To compare the hydrogenation
activity of catalysts C7A-C9A, we computed only the first step reaction by using the same
method (B3PW91/TZVP) and procedure. As reported previously, the Gibbs free energy barrier
[ΔGa(1)] for hemiacetal formation by using catalyst C7A is 21.51 kcal/mol. Using catalysts C8A
and C9A, the free energy barrier is 24.12 and 20.15 kcal mol-1, respectively. This shows that the

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
175
catalyst C8A has the highest free energy barrier, while the barrier of catalyst C9A is the lower.
Our computed order of free energy barriers is in line with the observed activity shown in scheme
13. This can be ascribed to the steric effect of the substituents at the P centers. In addition to the
hydrogenation barriers, it is also interesting to compare the Gibbs free energy barrier [ΔGa(2)] of
the catalyst regeneration. Moving from C7B to C7A, the reaction has a Gibbs free energy barrier
of 17.14 kcal mol-1 and is slightly exergonic by 0.33 kcal mol-1. Moving from C8B to C8A, the
reaction has a Gibbs free energy barrier of 16.46 kcal mol-1 and is exergonic by 3.86 kcal mol-1.
Moving from C9B to C9A, the Gibbs free energy barrier is 18.12 kcal mol-1 and the reaction is
slightly exergonic by 0.37 kcal mol-1.
Based on these results, the following mechanism was proposed for the hydrogenation of methyl
benzoate 23a (Scheme 14). The ester is hydrogenated in a concerted way by the simultaneous
transfer of the hydride from the iron center and the proton from the nitrogen ligand in complex to
give the corresponding hemiacetal and amido complex C9B. Next, the dissociation of hemiacetal
to benzaldehyde and methanol takes place, while C9A is regenerated from C9B by addition of
H2. In the final step, benzaldehyde is hydrogenated to give the benzyl alcohol.
Scheme 14. Proposed mechanism for the Fe-catalyzed ester hydrogenation: methyl benzoate was used as the representative example.
In summary, hydrogenation of esters to alcohols with a well-defined iron Et2PNP pincer
complex has been developed. This sterically less hindered Et2PNP complex provides superior

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
176
catalytic activity in the hydrogenation of various carboxylic acid esters and lactones compared to
the known iPr2PNPFe complex. Successful hydrogenation proceeds under relatively mild
conditions (60 °C) with lower catalyst loadings.
2.2. Manganese catalyzed ester hydrogenation Inspired from the iron catalysed ester hydrogenation conducted with iPr2PNP, Et2PNP
complexes and recent developments with manganese pincer complexes (Part 3 Chapter1), PNP
manganese catalyzed nitrile hydrogenation was then studied. To the best of our knowledge,
manganese based catalyst were not used for the hydrogenation of esters to alcohols. In this
paragraph, the four molecular defined manganese complexes C10-11 and C14-15 were evaluated
in the hydrogenation of various esters leading to the corresponding alcohols (Figure 8).
Figure 8. Manganese pincer complexes used for this study.
2.2.1 New PNP manganese complex synthesis The initial attempts were focused on the catalytic activity of the manganese pincer
complexes in hydrogenation of methyl benzoate in the presence of 2 mol% of catalyst under 30
bar of H2 at 100 °C (Table 5). Unfortunately, the complex C10, which showed high activity in the
hydrogenation of ketones, afforded only very low yield (6%) of the desired alcohol (Table 5,
entry 1). Under harsher conditions (under 80 bar H2 at 120 °C with 3 mol% of catalyst), the
complex C10 gave satisfactory yield (38%, Table 5, entry 3). Similarly, the cyclohexyl-
substituted manganese pincer complex C11 proved to be not suitable (2% yield, Table 5, entry 2).
To improve the catalyst activity, a focus was made on the synthesis of less hindered manganese
complexes such as C14 and C15.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
177
Scheme 15. Synthesis of manganese complexes C14 and C15.
A reaction of Mn(CO)5Br with the Et2PNP ligand in toluene at 100 °C for 20 h gave a
mixture of di- and tri-carbonyl manganese complexes which were isolated separately leading to
the complexes C14 and C15 with 64% and 22% yields, respectively and characterized by
spectroscopic methods. The IR (ATR) spectrum of C14 contains strong CO stretching vibrations
at 2007 cm-1, 1933 cm-1 and 1891 cm-1 which clearly indicate the coordination of three CO
ligands to the metal center. The IR spectrum of the complex C15 exhibited two strong CO
stretching vibrations at 1908 cm-1 and 1823 cm-1 showing the coordination of two CO ligands to
the metal center. 31P NMR spectra show a singlet at = 63.1 and = 68.73 ppm for C14 and C15
complexes respectively, indicating that the two phosphorus atoms of the ligand are equivalent. X-
ray analysis of the tricarbonyl complex C14 reveals that the Mn center has a distorted octahedral
coordination sphere, where both P atoms and the N atom as well as the three CO ligands are
located cis to each other (Figure 9). This configuration with both P atoms in cis-position is
surprising, since PNP pincer complexes, such as C10, C11 and C15 as well as related Fe, Ru and
Os or Ir pincer complexes usually show trans-orientation for the two P atoms.
Table 4. Summary of relevant spectroscopic datas
Complexes Molecular formula νCO (cm-1) 31P (ppm) Mass
C10 C18H36MnNO2P2 1903, 1815 81.8 416.16745
C11 C30H53MnNO2P2 1913, 1826 73.6 576.29265
C14 C15H29NO3P2Mn 2007, 1933,
1891
63.1 388.09977
C15 C14H29MnNO2P2 1908, 1823 68.73 360.10517
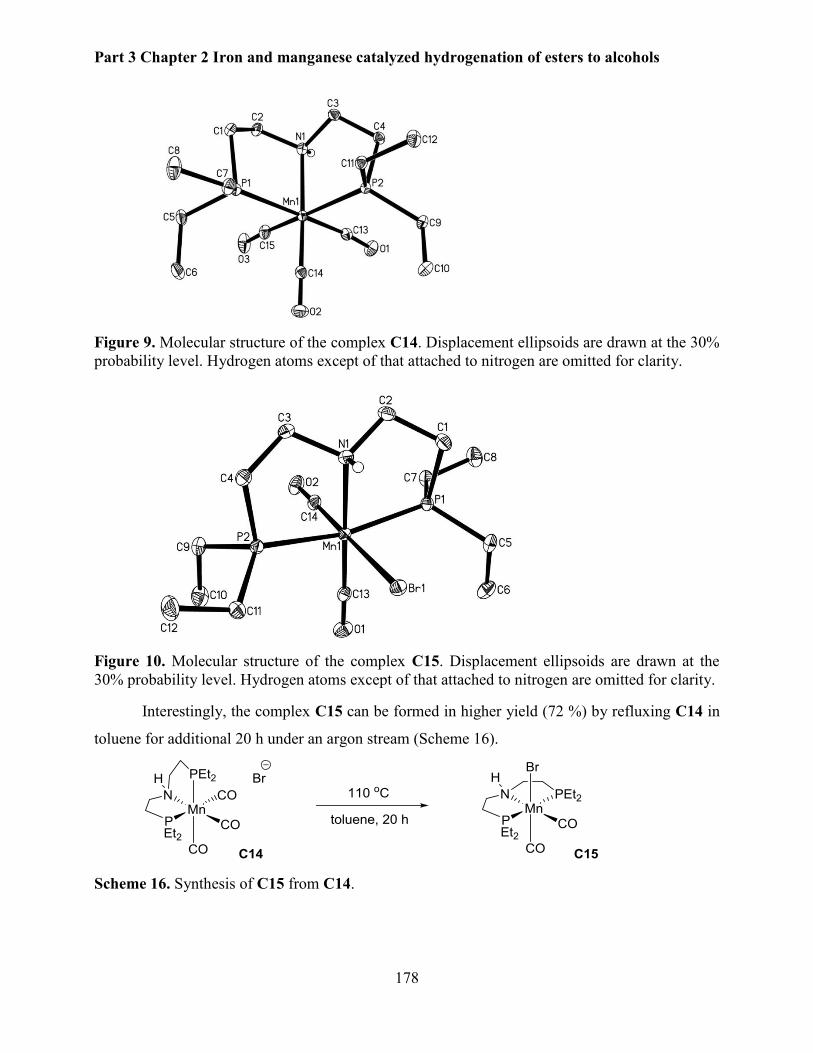
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
178
Figure 9. Molecular structure of the complex C14. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms except of that attached to nitrogen are omitted for clarity.
Figure 10. Molecular structure of the complex C15. Displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms except of that attached to nitrogen are omitted for clarity.
Interestingly, the complex C15 can be formed in higher yield (72 %) by refluxing C14 in
toluene for additional 20 h under an argon stream (Scheme 16).
Scheme 16. Synthesis of C15 from C14.
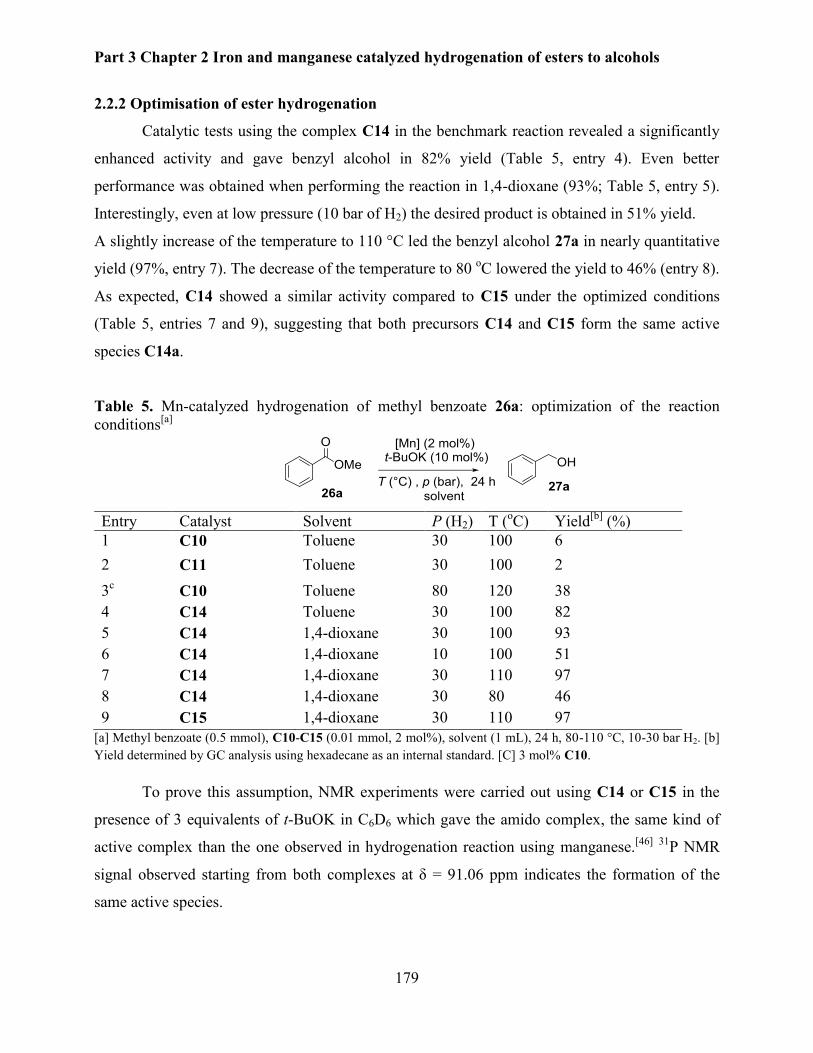
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
179
2.2.2 Optimisation of ester hydrogenation
Catalytic tests using the complex C14 in the benchmark reaction revealed a significantly
enhanced activity and gave benzyl alcohol in 82% yield (Table 5, entry 4). Even better
performance was obtained when performing the reaction in 1,4-dioxane (93%; Table 5, entry 5).
Interestingly, even at low pressure (10 bar of H2) the desired product is obtained in 51% yield.
A slightly increase of the temperature to 110 °C led the benzyl alcohol 27a in nearly quantitative
yield (97%, entry 7). The decrease of the temperature to 80 oC lowered the yield to 46% (entry 8).
As expected, C14 showed a similar activity compared to C15 under the optimized conditions
(Table 5, entries 7 and 9), suggesting that both precursors C14 and C15 form the same active
species C14a.
Table 5. Mn-catalyzed hydrogenation of methyl benzoate 26a: optimization of the reaction conditions[a]
Entry Catalyst Solvent P (H2) T (oC) Yield[b] (%) 1 C10 Toluene 30 100 6 2 C11 Toluene 30 100 2 3c C10 Toluene 80 120 38 4 C14 Toluene 30 100 82 5 C14 1,4-dioxane 30 100 93 6 C14 1,4-dioxane 10 100 51 7 C14 1,4-dioxane 30 110 97 8 C14 1,4-dioxane 30 80 46 9 C15 1,4-dioxane 30 110 97
[a] Methyl benzoate (0.5 mmol), C10-C15 (0.01 mmol, 2 mol%), solvent (1 mL), 24 h, 80-110 °C, 10-30 bar H2. [b] Yield determined by GC analysis using hexadecane as an internal standard. [C] 3 mol% C10.
To prove this assumption, NMR experiments were carried out using C14 or C15 in the
presence of 3 equivalents of t-BuOK in C6D6 which gave the amido complex, the same kind of
active complex than the one observed in hydrogenation reaction using manganese.[46] 31P NMR
signal observed starting from both complexes at δ = 91.06 ppm indicates the formation of the
same active species.
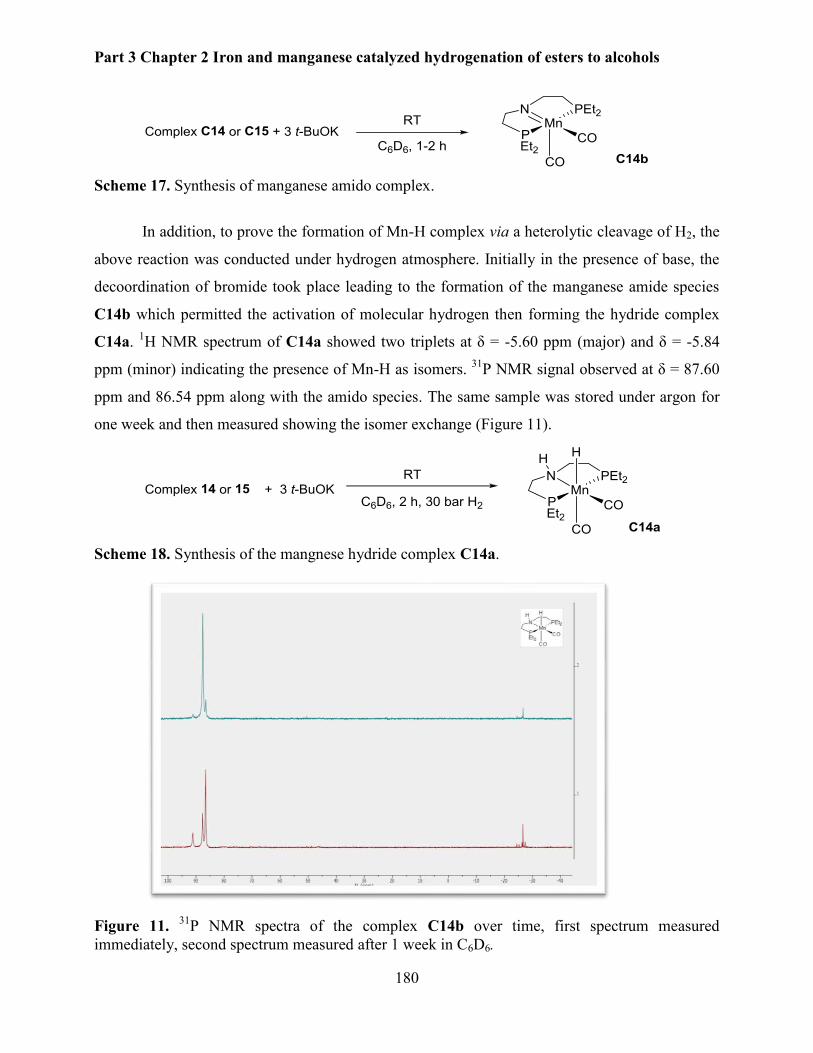
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
180
Scheme 17. Synthesis of manganese amido complex.
In addition, to prove the formation of Mn-H complex via a heterolytic cleavage of H2, the
above reaction was conducted under hydrogen atmosphere. Initially in the presence of base, the
decoordination of bromide took place leading to the formation of the manganese amide species
C14b which permitted the activation of molecular hydrogen then forming the hydride complex
C14a. 1H NMR spectrum of C14a showed two triplets at δ = -5.60 ppm (major) and δ = -5.84
ppm (minor) indicating the presence of Mn-H as isomers. 31P NMR signal observed at δ = 87.60
ppm and 86.54 ppm along with the amido species. The same sample was stored under argon for
one week and then measured showing the isomer exchange (Figure 11).
Scheme 18. Synthesis of the mangnese hydride complex C14a.
Figure 11. 31P NMR spectra of the complex C14b over time, first spectrum measured immediately, second spectrum measured after 1 week in C6D6.
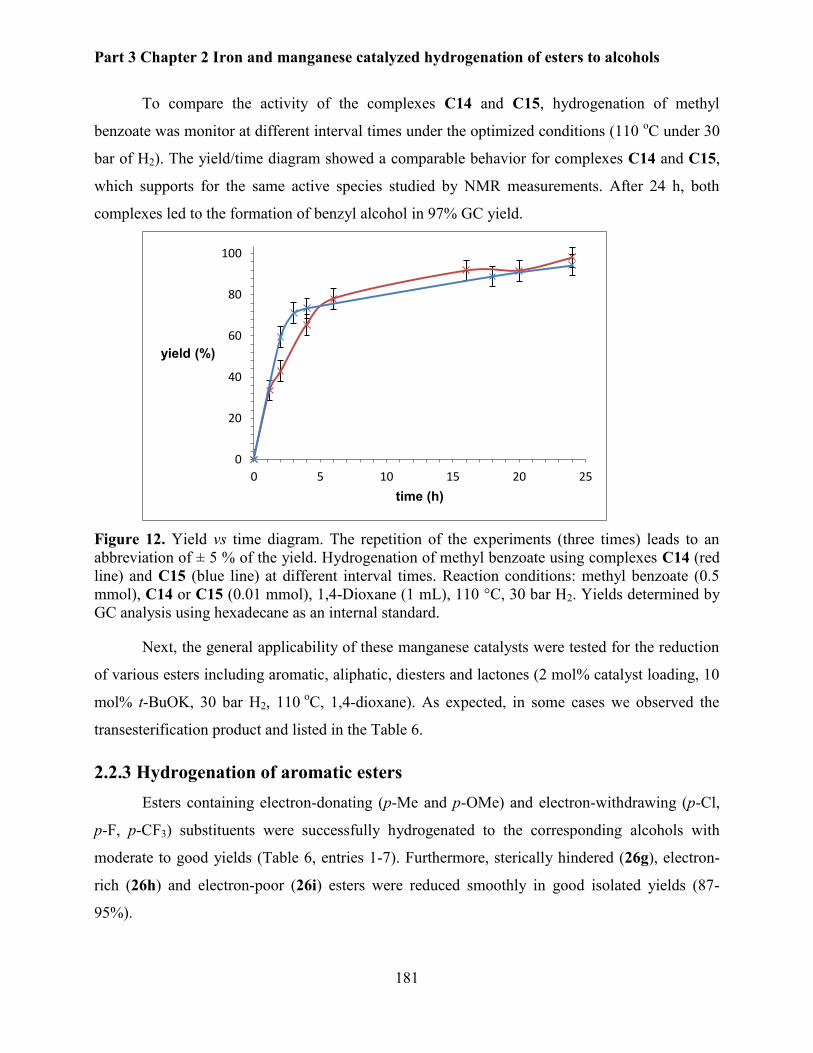
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
181
To compare the activity of the complexes C14 and C15, hydrogenation of methyl
benzoate was monitor at different interval times under the optimized conditions (110 oC under 30
bar of H2). The yield/time diagram showed a comparable behavior for complexes C14 and C15,
which supports for the same active species studied by NMR measurements. After 24 h, both
complexes led to the formation of benzyl alcohol in 97% GC yield.
Figure 12. Yield vs time diagram. The repetition of the experiments (three times) leads to an abbreviation of ± 5 % of the yield. Hydrogenation of methyl benzoate using complexes C14 (red line) and C15 (blue line) at different interval times. Reaction conditions: methyl benzoate (0.5 mmol), C14 or C15 (0.01 mmol), 1,4-Dioxane (1 mL), 110 °C, 30 bar H2. Yields determined by GC analysis using hexadecane as an internal standard.
Next, the general applicability of these manganese catalysts were tested for the reduction
of various esters including aromatic, aliphatic, diesters and lactones (2 mol% catalyst loading, 10
mol% t-BuOK, 30 bar H2, 110 oC, 1,4-dioxane). As expected, in some cases we observed the
transesterification product and listed in the Table 6.
2.2.3 Hydrogenation of aromatic esters Esters containing electron-donating (p-Me and p-OMe) and electron-withdrawing (p-Cl,
p-F, p-CF3) substituents were successfully hydrogenated to the corresponding alcohols with
moderate to good yields (Table 6, entries 1-7). Furthermore, sterically hindered (26g), electron-
rich (26h) and electron-poor (26i) esters were reduced smoothly in good isolated yields (87-
95%).
0
20
40
60
80
100
0 5 10 15 20 25
yield (%)
time (h)

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
182
Table 6. Manganese-catalyzed hydrogenation of aromatic esters.a,b
Entry Ester Alcohol Conv. (Yield)
(%)[b] Trans-
esterification product (%)
traces
1 2 3 4 5 6
R = H, 26a R = Me, 26b R = OMe, 26c R = CF3, 26d R = Cl, 26e R = F, 26f
>99(97) >99(86) >99(95) >99(75) >99(91) >99(89)
7 26g
27g
>99(95)
-
8
26h
27h
>99(89) -
9
26i
27i
>99(87) -
10 26j
27j
>99(86) -
11 26k
27k
>99(87)[c] -
12
26l
27l
>99(78)[c 4
[a] Substrate (1 mmol), C14 (2 mol%), 1,4-dioxane (2 mL), 24 h, 110 °C, 30 bar H2. [b] Conversion was determined by GC (Isolated yield in parenthesis). [c] GC yield was determined by hexadecane as an internal standard.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
183
Methyl 2-naphthoate (26j) was hydrogenated into naphthalen-2-ylmethanol with 86%
isolated yield. Interestingly, heteroaromatic esters such as 3-pyridine (26l) and 2-furan methyl
(26k) esters were fully reduced into the corresponding alcohols with good yields.
2.2.4 Hydrogenation of benzylic and aliphatic esters Next, the hydrogenation of benzylic and aliphatic esters under the optimised conditions
was examined (Table 7, entries 1-9). Indeed, 2-phenylethanol and its derivatives are important
chemicals, which are widely used in food materials, fine chemical industries and it’s also a
potentially valuable alcohol for next-generation biofuel. Based on this interest of 2-phenylethanol
derivatives, different benzylic esters were tested. In all the cases, esters were fully hydrogenated
and led to the corresponding alcohols with good yields (87-95%, Table 7, 26m-26p).
Interestingly, the isolated double bond remained intact, when 3-cyclohexene-1-carboxylate 26q
as well as the bio-based methyl oleate 26u were used as substrates. By contrast, the conjugated
ester methyl cinnamate 26r was fully reduced leading to the saturated alcohol in 93% yield.
Menthyl acetate 26s was also hydrogenated to menthol 27s in 88% yield. In addition, long chain
aliphatic ester methyl octanoate 26t gave 1-octanol with 71% yield.
Table 7. Manganese-catalyzed hydrogenation of benzylic, aliphatic, diesters and lactones.a
Entry Ester Alcohol Conv. (Yield) [%]d
Trans-esterifi-cation product (%)
1 2 3 4
26m R = H 26n R = Cl
26o R = F 26p R = t-Bu
>99(79) >99(87) >99(93) >99(95)
6 3 3 -
5
26q
27q
98(83)b,e 6
6
26r
27r
>99(93) -
7
97(88)c -

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
184
26s 27s 8
26t
27t
92(71)e 11
9
26u
27u
95(89)c -
10
26v
27v
>99(82) -
11
26x
27x
>99(57) -
12
26y
27y
>99(88)e -
13
26z
27z
>99(98)e -
14
26aa
27aa
>93(66)c,e 20 Methyl 4-
hydroxybutanoate
15
26ab
27ab
>99(95) Methyl(4-hydroxymethyl)-benzoate (1%) and 4-Methylbenzyl alcohol (1%)
16
26ac
27ac
>99(58)c Hydroxymethyl-2-furoic acid methyl ester (37%)
[a] Substrate (1 mmol), C14 (2 mol%), 1,4-dioxane (2 mL), 24 h, 110 °C, 30 bar H2. [b] 48 h [c] C14 (3 mol%), 48 h, 120 °C. [d] Conversion was determined by GC (Isolated yield in parenthesis). [e] GC yield was determined by hexadecane as an internal standard.
2.2.5 Hydrogenation of diesters and lactones The versatility of the complex C14 was further demonstrated for the reduction of lactones
and diesters to diols (Table 7, entries 10-16). For example, the flavour and fragrance agent -
octalactone was converted to the corresponding diol in good yield (82%, Table 7, entry 10).
Notably, the clean reduction of bio-mass derived -valerolactone 26z (GVL), which is of actual

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
185
interest for the concept of a biorefinery, was successfully achieved to give 1,5-pentandiol in
excellent yield (98%, entry 13). 5-Bromophthalide 26x was converted into the diol 27x in 57%
yield. Interestingly, butyrolactone 26y was quantitatively transformed into 1,4-butanediol 27y
with good yield (88%). Finally, aromatic and aliphatic diesters were reduced to the corresponding
diols with moderate to good yields (Table 7, entries 14-16). Notably, when dimethyl furan-2,5-
dicarboxylate used as a substrate, 37% of hydroxymethyl-2-furoic acid methyl ester and 58% of
the fully reduced furan-2,5-diyldimethanol were obtained (Table 7, entry 16).
2.3 Manganese catalyzed hydrogenation of aldehydes and ketones In addition to the carboxylic acid derivatives such as nitriles and esters, it was shown that
the selective hydrogenation of ketones (Scheme 19) and aldehydes (Scheme 20) to the
corresponding alcohols was possible in the presence of 1 mol% of the complex C10 and 3 mol%
t-BuONa under 10-30 bar of H2 at 60-100 °C for 24 h. Under these conditions, heteroaromatic
ketone (3-pyridyl methyl ketone) and cyclic ketone (1-indanone) were reduced into the
corresponding alcohols 29a and 29b in 91 and 95% yields, respectively. Notably, the cyclopropyl
phenyl ketone furnished 95% yield of the corresponding cyclopropyl containing alcohol 29b
indicating that the reaction may not proceed via stable radical intermediates. Importantly, the
manganese-based catalyst C10 can tolerate other reducible functional groups such as lactams
(29e), esters (29f) and isolated C=C bonds (29i, 29j). Additionally, the heterocyclic substrate 1-
benzylpiperidin-4-one and 4-chromanone were converted into the corresponding alcohols 29g
and 29h, in 77 and 90% yields, respectively.
Aldehydes can be reduced under similar conditions and chemoselectivity issues were
more particularly studied. Thus, unsaturated aldehydes reacted chemoselectively under mild
conditions in the presence of 1 mol% of the catalyst C10 and 3 mo% of t-BuONa under 10 bar H2
at 60 °C for 24 h; (Scheme 25). In the 3 selected unsaturated aldehydes, only the carbonyl
group was reduced selectively leading to the corresponding allylic alcohols 31a-c in 81-93%
yields. Accordingly, 2-octynal gave oct-2-yn-1-ol 31d in 96% isolated yield. In addition, several
natural products, e.g. citronellal (3,7-dimethyloct-6-enal), perillaldehyde and 5-
hydroxymethylfurfural (HMF) gave the corresponding primary alcohols 31e-g in moderate to
good yields (64-89%). These substrates are also applied on larger scale as monomers for
polymers (HMF) or intermediates in the flavor and fragrance industry.
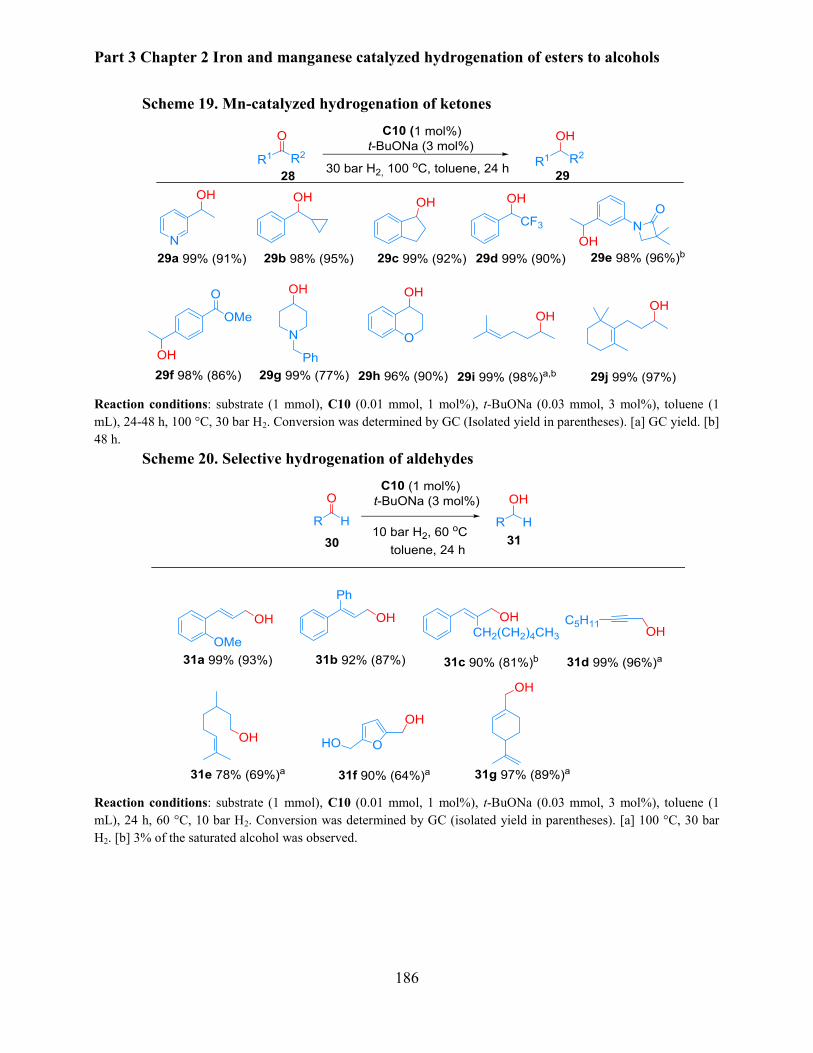
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
186
Scheme 19. Mn-catalyzed hydrogenation of ketones
Reaction conditions: substrate (1 mmol), C10 (0.01 mmol, 1 mol%), t-BuONa (0.03 mmol, 3 mol%), toluene (1 mL), 24-48 h, 100 °C, 30 bar H2. Conversion was determined by GC (Isolated yield in parentheses). [a] GC yield. [b] 48 h.
Scheme 20. Selective hydrogenation of aldehydes
Reaction conditions: substrate (1 mmol), C10 (0.01 mmol, 1 mol%), t-BuONa (0.03 mmol, 3 mol%), toluene (1 mL), 24 h, 60 °C, 10 bar H2. Conversion was determined by GC (isolated yield in parentheses). [a] 100 °C, 30 bar H2. [b] 3% of the saturated alcohol was observed.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
187
2.4 Mechanistic investigation In order to elucidate the reaction mechanism and to understand the different performance
of catalysts C10a, C11a and C14a, which can be deduced from the corresponding pre-catalysts,
C10, C11 and C14, B3PW91 DFT computations were carried out. The rational proposal for the
formation C14a is based on the fact that the cationic ethyl complex C14 and the neutral ethyl
complex C15 exhibited the same catalytic performance. This assumption is supported by NMR
experiments where the active amido species C14b is observed when pre-catalysts C14 and C15
are treated with a base.
Modelling studies show that hydride complexes C10a, C11a and C14a were very stable
towards CO dissociation. In addition, a well-balanced equilibrium was established for the
concerted H2 elimination from C10a, C11a and C14a to the respective amido complexes C10b,
C11b and C14b. In all complexes, the barrier of H2 elimination is around 20 kcal/mol and the
reactions are slightly exergonic by 0.2-1.5 kcal/mol.
Scheme 21. Proposed mechanism for the Mn-catalysed ester hydrogenation.
On the base of the computations, the following outer sphere mechanism was postulated
(Scheme 21). Methyl benzoate 26a is reduced in two cycles via the formation of the
corresponding hemiacetal 27c. A stepwise process was identified for the first cycle (26a to 27c).
The hydrogen atoms are transferred stepwise as hydride from the manganese center and as proton
from the nitrogen ligand in C10a (C11a or C14a) resulting in the amido complex C10b (C11b or

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
188
C14b) formation. In the first step, Mn-HC transfer results in an intermediate and the second
step N-HO transfer results in 27c. Additionally, it was found that for all catalysts the first step
has a higher barrier than the second step (32.7 vs. 30.1, 34.1 vs. 27.9 and 31.8 vs. 25.8 kcal/mol;
respectively, for C10a, C11a and C14a).
Next, the hemiacetal 27c is dissociated to benzaldehyde 27b and methanol, while the
amido complex is regenerated by addition of H2. In the second cycle, benzaldehyde 27b is
hydrogenated stepwise to benzyl alcohol 27a. This cycle also undergoes a similar stepwise
process. Here, the first step has a higher barrier than the second step (15.6/11.9, 18.5/13.5 and
15.9/12.8 kcal/mol, respectively, for C10a, C11a and C14a). Based on the energy barriers of
both cycles, the initial reduction of the ester was proposed to be rate-determining. Comparing the
different catalysts for this first hydrogenation cycle, C14a/C14b has the lowest barrier followed
by that of C10a/C10b and C11a/C11b, which is in agreement with the observed catalytic activity
(C14 > C10 > C11).
In conclusion, the first molecularly defined manganese complex catalysed hydrogenation of
esters to alcohols was described. This catalytic system permitted effective and selective
hydrogenation of various aromatic, aliphatic, diesters and lactones. Experimental evidence and
DFT calculations showed that the reaction proceeded via an outer sphere mechanism.
3. Conclusion The sterically less demanding Et2 substituted iron and manganese PNP pincer complexes
were prepared and the activity of these complexes was compared with the ones of iron and
manganese PNP complexes with two different substitutions on phosphorous moiety (isopropyl or
cyclohexyl). The Et2PNP iron and manganese complexes were found to be efficient catalyst for
ester hydrogenation. Interestingly, combination of Mn(CO)5Br with [HN(CH2CH2P(Et)2)2] leads
to a mixture of cationic and neutral Mn PNP pincer complexes C14 and C15 and both of them
led to the same catalytic active species under the reaction conditions. This catalytic system gave
a broad substrate scope for the selective hydrogenation of various aromatic and aliphatic esters
including diester motifs and lactones.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
189
4. Experimental section
4.1 General experimental details Unless otherwise stated, all reactions were performed under an argon atmosphere with
exclusion of moisture from reagents and glassware using standard techniques for manipulating
air-sensitive compounds. All isolated products were characterized by 1H NMR and 13C NMR
spectroscopy as well as high resolution mass spectrometry (HRMS). NMR spectra were recorded
on a Bruker AV 300 or 400. All chemical shifts (δ) are reported in ppm and coupling constants
(J) in Hz. All chemical shifts are related to residual solvent peaks [CDCl3: 7.26 (1H), 77.16 (13C);
C6D6: 7.16 (1H), 128.06 (13C)], respectively. All measurements were carried out at room
temperature unless otherwise stated. Mass spectra were in general recorded on a Finnigan MAT
95-XP (Thermo Electron) or on a 6210 Time-of-Flight LC/MS (Agilent). Gas chromatography
was performed on a HP 6890 with a HP5 column (Agilent).
Reagents: Unless otherwise stated, commercial reagents were used without purification.
4.2 General procedure for the iron catalyzed hydrogenation of esters All catalytic hydrogenation experiments using molecular hydrogen were carried out in a Parr
Instruments autoclave (300 mL) advanced with an internal alloy plate to include seven uniform
reaction vials (4 mL) equipped with a cap and needle penetrating the septum.
Representative Experiment: Under an argon atmosphere, a vial was charged with C9 (0.01
mmol), which was dissolved in dry THF (1 mL). The yellow solution was stirred briefly before
the ester or lactone (1 mmol) was added. The vial was placed in the alloy plate which was then
placed into the autoclave. Once sealed, the autoclave was purged 3 times with hydrogen, then
pressurized to 30 bar and heated to 60 or 100 °C for 6 or 18 h. Afterwards, the autoclave was
cooled to RT, depressurized, and the reaction mixture was analyzed by GC-FID and GC-MS.
Product isolation was performed via column chromatography using silica gel as stationary phase
and an n-pentane / ethyl acetate mixture (2:1) as eluent.
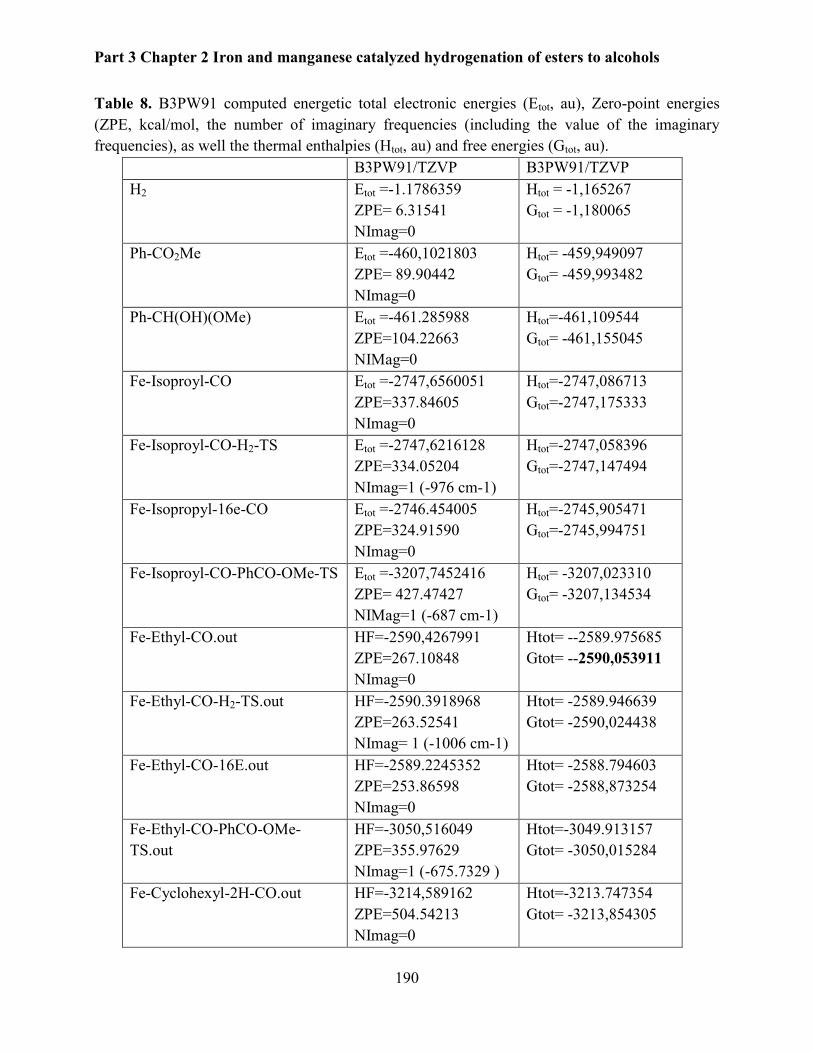
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
190
Table 8. B3PW91 computed energetic total electronic energies (Etot, au), Zero-point energies (ZPE, kcal/mol, the number of imaginary frequencies (including the value of the imaginary frequencies), as well the thermal enthalpies (Htot, au) and free energies (Gtot, au).
B3PW91/TZVP B3PW91/TZVP H2 Etot =-1.1786359
ZPE= 6.31541 NImag=0
Htot = -1,165267 Gtot = -1,180065
Ph-CO2Me Etot =-460,1021803 ZPE= 89.90442 NImag=0
Htot= -459,949097 Gtot= -459,993482
Ph-CH(OH)(OMe)
Etot =-461.285988 ZPE=104.22663 NIMag=0
Htot=-461,109544 Gtot= -461,155045
Fe-Isoproyl-CO Etot =-2747,6560051 ZPE=337.84605 NImag=0
Htot=-2747,086713 Gtot=-2747,175333
Fe-Isoproyl-CO-H2-TS Etot =-2747,6216128 ZPE=334.05204 NImag=1 (-976 cm-1)
Htot=-2747,058396 Gtot=-2747,147494
Fe-Isopropyl-16e-CO Etot =-2746.454005 ZPE=324.91590 NImag=0
Htot=-2745,905471 Gtot=-2745,994751
Fe-Isoproyl-CO-PhCO-OMe-TS Etot =-3207,7452416 ZPE= 427.47427 NIMag=1 (-687 cm-1)
Htot= -3207,023310 Gtot= -3207,134534
Fe-Ethyl-CO.out HF=-2590,4267991 ZPE=267.10848 NImag=0
Htot= --2589.975685 Gtot= --2590,053911
Fe-Ethyl-CO-H2-TS.out HF=-2590.3918968 ZPE=263.52541 NImag= 1 (-1006 cm-1)
Htot= -2589.946639 Gtot= -2590,024438
Fe-Ethyl-CO-16E.out HF=-2589.2245352 ZPE=253.86598 NImag=0
Htot= -2588.794603 Gtot= -2588,873254
Fe-Ethyl-CO-PhCO-OMe-TS.out
HF=-3050,516049 ZPE=355.97629 NImag=1 (-675.7329 )
Htot=-3049.913157 Gtot= -3050,015284
Fe-Cyclohexyl-2H-CO.out HF=-3214,589162 ZPE=504.54213 NImag=0
Htot=-3213.747354 Gtot= -3213,854305

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
191
Fe-Cyclohexyl-2H-CO-TS.out HF=-3214.5528196 ZPE=501.39010 NImag=0
Htot=-3213.716410 Gtot= -3213,821918
Fe-Cyclohexyl-H-CO-16E HF=-3213.3836256 ZPE=491.90821 NImag=0
Htot=-3212.562392 Gtot= -3212,668090
Fe-Cy-CO-PhCO-OMe-TS.out HF=-3674,6745119 ZPE=593.94280 NImag=1 (-711.4911 )
Htot=-3673.680289 Gtot=-3673,809337
4.3 Analytical data of the isolated products (4-Methoxyphenyl)methanol
The compound was prepared as described above (125 mg, 90% isolated yield). Colorless liquid. 1H NMR (300 MHz, CDCl3) δ: 7.27-7.22 (m, 2H), 6.88-6.83 (m, 2H), 4.55 (s, 2H), 3.77 (s, 3H), 2.11 (s, 1H). 13C NMR (75 MHz, CDCl3) δ: 159.1, 133.1, 128.5, 113.8, 64.8, 55.2. (4-Chlorophenyl)methanol
The compound was prepared as described above (142 mg, 99% isolated yield). Off-white solid. 1H NMR (300 MHz, CDCl3) δ: 7.28-7.20 (m, 4H), 4.59 (s, 2H), 1.78 (s, 1H). 13C NMR (75 MHz, CDCl3) δ: 139.2, 133.3, 128.6, 128.3, 64.5. (4-(Trifluoromethyl)phenyl)methanol
The compound was prepared as described above (155 mg, 88% isolated yield). Colorless liquid. 1H NMR (300 MHz, CDCl3) δ: 7.60 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 4.72 (s, 2H), 2.38 (s, 1H). 13C NMR (75 MHz, CDCl3) δ: 144.7, 129.9 (J = 33.0 Hz), 126.8, 125.4 (q, J = 3.8 Hz), 122.3 (q, J = 274.2 Hz), 64.3. Octan-1-ol
The compound was prepared as described above (123 mg, 95% isolated yield). Colorless oil.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
192
1H NMR (300 MHz, CDCl3) δ: 3.63 (t, J = 6.6 Hz, 2H), 1.58-1.28 (m, 3H), 1.27-1.25 (m, 10H), 0.90-0.85 (m, 3H). 13C NMR (75 MHz, CDCl3) δ: 63.1, 32.7, 31.8, 29.4, 29.3, 25.7, 22.6, 14.1. 3-Phenylpropan-1-ol
The compound was prepared as described above (129 mg, 95% isolated yield). Colorless oil. 1H NMR (300 MHz, CDCl3) δ: 7.36-7.23 (m, 5H), 3.71 (t, J = 6.4 Hz, 2H), 2.75 (dd, J = 8.7, 6.8 Hz, 2H), 1.99 - 1.83 (m, 2H), 1.83 (s, 1H). 13C NMR (75 MHz, CDCl3) δ: 141.7, 128.4, 128.3, 125.8, 62.2, 34.1, 32.0. Cyclohex-3-en-1-ylmethanol
The compound was prepared as described above (103 mg, 92% isolated yield). Colorless liquid. 1H NMR (300 MHz, CDCl3) δ: 5.68-5.66 (m, 2H), 3.60 (s, 2H), 2.12-2.03 (m, 4H), 1.84 -1.75 (m, 3H), 1.25 (s, 1H). 13C NMR (75 MHz, CDCl3) δ: 127.0, 125.8, 67.9, 36.4, 28.1, 25.1, 24.5. Furan-2-ylmethanol
The compound was prepared as described above (93 mg, 95% isolated yield). Colorless oil. 1H NMR (300 MHz, CDCl3) δ: 7.14 (s, 1H), 6.20-6.15 (m, 2H), 4.45 (s, 2H), 2.20 (s, 2H). 13C NMR (75 MHz, CDCl3) δ: 153.9, 142.4, 110.2, 107.6, 57.2. Pyridin-3-ylmethanol
The compound was prepared as described above (100 mg, 92% isolated yield). Colorless oil. 1H NMR (300 MHz, CDCl3) δ: 8.48-8.41 (m, 2H), 7.73-7.70 (m, 1H), 7.29-7.25 (m, 1H), 4.69 (s, 2H), 4.00 (s, 1H). 13C NMR (75 MHz, CDCl3) δ: 148.2, 148.0, 136.9, 135.1, 123.6, 62.2. 1,4-Pentanediol
The compound was prepared as described above (94 mg, 90% isolated yield). Colorless liquid. 1H NMR (300 MHz, CDCl3) δ: 4.24 (br s, 2H), 3.84-3.76 (m, 1H), 3.66 -3.50 (m, 2H), 1.66-1.49 (m, 4H), 1.18 (d, J = 6.2 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 67.7, 62.6, 36.1, 29.0, 23.4.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
193
1,4-Phenylenedimethanol
The compound was prepared as described above (133 mg, 92% isolated yield). White crystals. 1H NMR (300 MHz, MeOD) δ: 7.33 (s, 4H), 4.87 (s, 2H), 4.59 (s, 4H). 13C NMR (75 MHz, MeOD) δ:141.7, 128.0, 65.0. Naphthalene-2,6-diyldimethanol
The compound was prepared as described above (169 mg, 92% isolated yield). Off-white solid. 1H NMR (300 MHz, DMSO-d6) δ: 7.86-7.80 (m, 4H), 7.45 (d, J = 8.4 Hz, 2H), 5.31 (t, J = 5.8 Hz, 2H), 4.66 (d, J = 5.6 Hz, 4H). 13C NMR (75 MHz, DMSO-d6) δ: 139.7, 132.1, 127.4, 125.4, 124.2, 63.0. (2,6-Dimethylpyridine-3,5-diyl)dimethanol
The compound was prepared as described above (153 mg, 91% isolated yield). Off-white solid. 1H NMR (400 MHz, DMSO-d6) δ: 7.63 (s, 1H), 5.16 (s, 2H), 4.47 (d, J = 5.1 Hz, 4H), 2.35 (s, 6H). 13C NMR (101 MHz, DMSO- d6) δ: 152.3, 133.2, 132.1, 60.1, 20.8. 3-Methyloctane-1,4-diol
The compound was prepared as described above (153 mg, 97% isolated yield). Colorless liquid. Obtained as a mixture of diastereoisomers. 1H NMR (300 MHz, CDCl3) δ: 3.74 – 3.46 (m, 3H), 1.67 (m, 2H), 1.56 – 1.17 (m, 7H), 0.96 – 0.78 (m, 6H). 13C NMR (75 MHz, CDCl3) δ: 75.6, 74.7, 60.3, 60.0, 36.3, 35.9, 35.9, 35.1, 33.9, 33.1, 29.6, 28.6, 27.9, 22.7, 22.7, 16.4, 14.1, 13.8.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
194
4.4. Synthesis of manganese pincer complexes Synthesis of the manganese complexes C14 and C15
To the orange-yellow suspension of Mn(CO)5Br (461 mg, 1.7 mmol) in toluene (20 mL)
[HN(CH2CH2P(Et)2)2] (440 mg, 1.8 mmol, dissolved in 2 mL toluene) was added. The solid was
formed during this time and it was heated to 100 °C and further stirred for 20 h under argon flow.
The reaction mixture was cooled to room temperature and concentrated in vacuum. The crude
was thoroughly washed with toluene several times and the solvent was transferred to another
Schlenk tube. The pale yellow solid was dried in vacuum and yielding C14 (530 mg, 64%). Then,
the dark yellow solvent was evaporated and dried at the pump gave dark orange yellow sticky
solid. It was washed with heptane thrice and afterwards twice with a small amount of methanol (3
mL) affording C15 as an orange yellow solid (22%).
For complex C14:
Crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of pentane into a
concentrated solution of C14 in methanol. 1H NMR (300 MHz, DMSO-d6) δ 1.11-1.25 (m, 12H, P-(CH2CH3)4), 1.90-2.30 (overlapping m,
12H, CH2CH3, PCH2), 2.84-2.61 (m, 4H, NCH2), 6.62 (1H, NH). 31P{1H} NMR (122 MHz, DMSO-d6) δ 63.1. 13C {1H} NMR (101 MHz, DMSO-d6) δ 8.3 (s, P(CH2CH3)2), 8.6 (s, P(CH2CH3)2), 18.1 (t, JC-P =
10.6 Hz, P(CH2CH3)2), 19.7 (t, JC-P = 16.1 Hz, P(CH2CH3)2), 26.2 (t, JC-P = 10.6 Hz, PCH2), 51.5
(s, NCH2) 216.1 (CO).
IR-ATR (solid) ῡ [cm-1]: 2007 (s, ῡ CO), 1933 (s, ῡ CO), 1891 (s, ῡ CO).
ESI-HRMS (m/z pos) Calculated for [C15H29NO3P2Mn]: 388.09977; found: 388.09985.
Elemental analysis calc for C15H29BrMnNO3P2, M = 468.19 g/mol: C 38.48 (38.44); H 6.24
(6.22) N 2.99 (3.19).
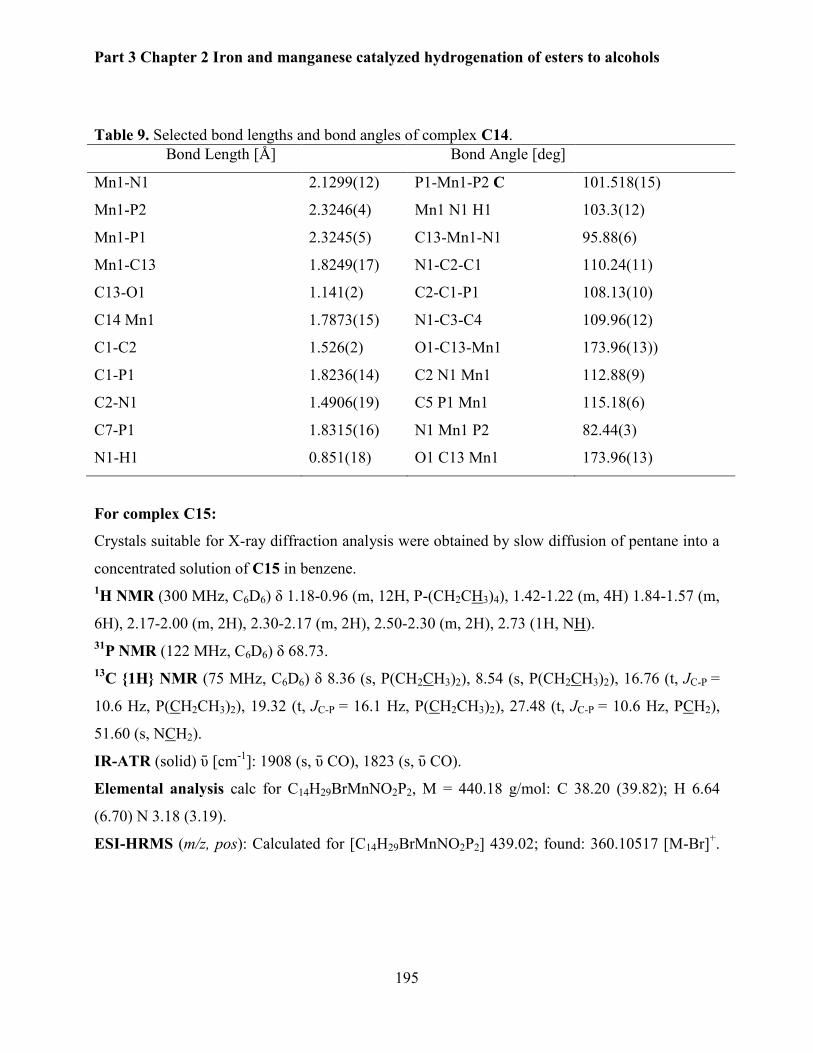
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
195
Table 9. Selected bond lengths and bond angles of complex C14. Bond Length [Å] Bond Angle [deg]
Mn1-N1 2.1299(12) P1-Mn1-P2 C 101.518(15)
Mn1-P2 2.3246(4) Mn1 N1 H1 103.3(12)
Mn1-P1 2.3245(5) C13-Mn1-N1 95.88(6)
Mn1-C13 1.8249(17) N1-C2-C1 110.24(11)
C13-O1 1.141(2) C2-C1-P1 108.13(10)
C14 Mn1 1.7873(15) N1-C3-C4 109.96(12)
C1-C2 1.526(2) O1-C13-Mn1 173.96(13))
C1-P1 1.8236(14) C2 N1 Mn1 112.88(9)
C2-N1 1.4906(19) C5 P1 Mn1 115.18(6)
C7-P1 1.8315(16) N1 Mn1 P2 82.44(3)
N1-H1 0.851(18) O1 C13 Mn1 173.96(13)
For complex C15:
Crystals suitable for X-ray diffraction analysis were obtained by slow diffusion of pentane into a
concentrated solution of C15 in benzene. 1H NMR (300 MHz, C6D6) δ 1.18-0.96 (m, 12H, P-(CH2CH3)4), 1.42-1.22 (m, 4H) 1.84-1.57 (m,
6H), 2.17-2.00 (m, 2H), 2.30-2.17 (m, 2H), 2.50-2.30 (m, 2H), 2.73 (1H, NH). 31P NMR (122 MHz, C6D6) δ 68.73. 13C {1H} NMR (75 MHz, C6D6) δ 8.36 (s, P(CH2CH3)2), 8.54 (s, P(CH2CH3)2), 16.76 (t, JC-P =
10.6 Hz, P(CH2CH3)2), 19.32 (t, JC-P = 16.1 Hz, P(CH2CH3)2), 27.48 (t, JC-P = 10.6 Hz, PCH2),
51.60 (s, NCH2).
IR-ATR (solid) ῡ [cm-1]: 1908 (s, ῡ CO), 1823 (s, ῡ CO).
Elemental analysis calc for C14H29BrMnNO2P2, M = 440.18 g/mol: C 38.20 (39.82); H 6.64
(6.70) N 3.18 (3.19).
ESI-HRMS (m/z, pos): Calculated for [C14H29BrMnNO2P2] 439.02; found: 360.10517 [M-Br]+.
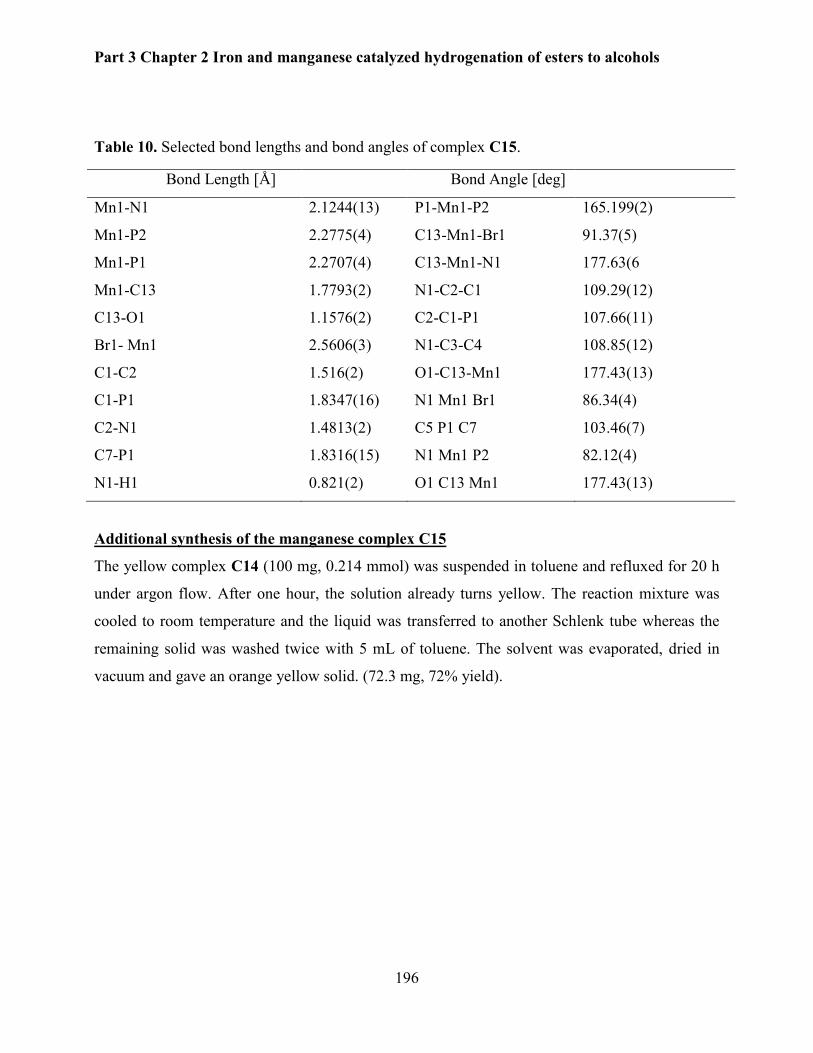
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
196
Table 10. Selected bond lengths and bond angles of complex C15.
Bond Length [Å] Bond Angle [deg]
Mn1-N1 2.1244(13) P1-Mn1-P2 165.199(2)
Mn1-P2 2.2775(4) C13-Mn1-Br1 91.37(5)
Mn1-P1 2.2707(4) C13-Mn1-N1 177.63(6
Mn1-C13 1.7793(2) N1-C2-C1 109.29(12)
C13-O1 1.1576(2) C2-C1-P1 107.66(11)
Br1- Mn1 2.5606(3) N1-C3-C4 108.85(12)
C1-C2 1.516(2) O1-C13-Mn1 177.43(13)
C1-P1 1.8347(16) N1 Mn1 Br1 86.34(4)
C2-N1 1.4813(2) C5 P1 C7 103.46(7)
C7-P1 1.8316(15) N1 Mn1 P2 82.12(4)
N1-H1 0.821(2) O1 C13 Mn1 177.43(13)
Additional synthesis of the manganese complex C15
The yellow complex C14 (100 mg, 0.214 mmol) was suspended in toluene and refluxed for 20 h
under argon flow. After one hour, the solution already turns yellow. The reaction mixture was
cooled to room temperature and the liquid was transferred to another Schlenk tube whereas the
remaining solid was washed twice with 5 mL of toluene. The solvent was evaporated, dried in
vacuum and gave an orange yellow solid. (72.3 mg, 72% yield).
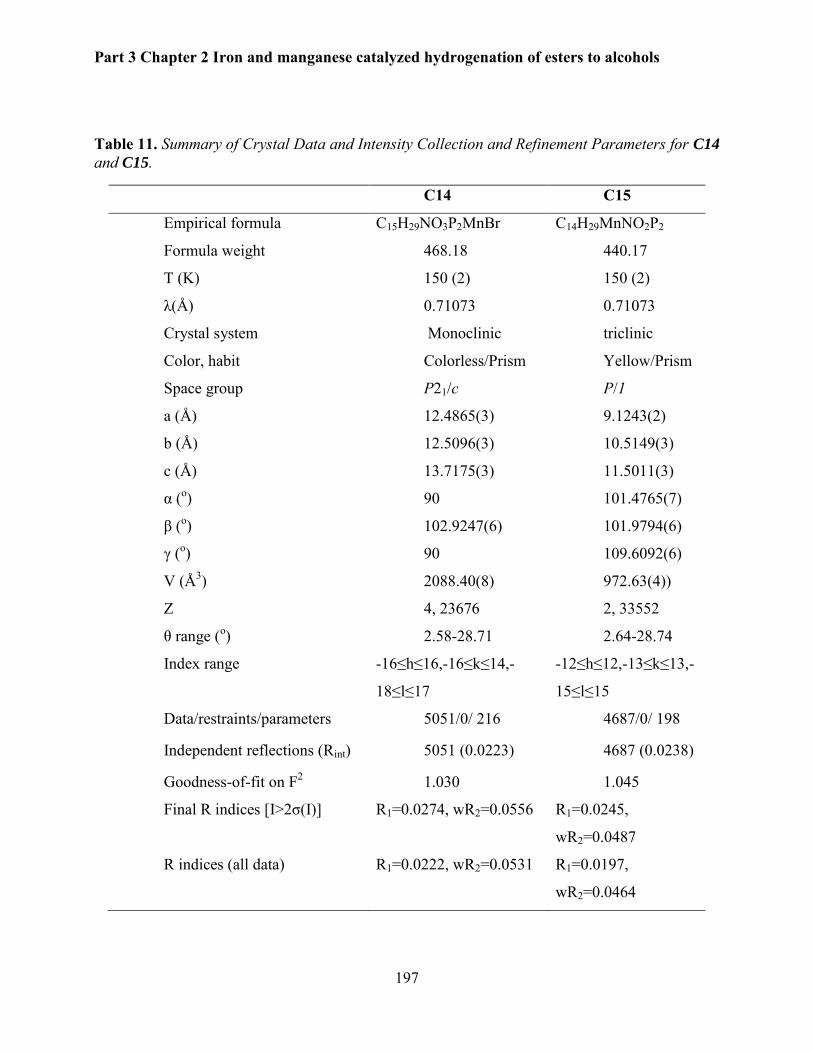
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
197
Table 11. Summary of Crystal Data and Intensity Collection and Refinement Parameters for C14
and C15.
C14 C15
Empirical formula C15H29NO3P2MnBr C14H29MnNO2P2
Formula weight 468.18 440.17
T (K) 150 (2) 150 (2)
λ(Å) 0.71073 0.71073
Crystal system Monoclinic triclinic
Color, habit Colorless/Prism Yellow/Prism
Space group P21/c P/1
a (Å) 12.4865(3) 9.1243(2)
b (Å) 12.5096(3) 10.5149(3)
c (Å) 13.7175(3) 11.5011(3)
α (o) 90 101.4765(7)
β (o) 102.9247(6) 101.9794(6)
γ (o) 90 109.6092(6)
V (Å3) 2088.40(8) 972.63(4))
Z 4, 23676 2, 33552
θ range (o) 2.58-28.71 2.64-28.74
Index range -16≤h≤16,-16≤k≤14,-
18≤l≤17
-12≤h≤12,-13≤k≤13,-
15≤l≤15
Data/restraints/parameters 5051/0/ 216 4687/0/ 198
Independent reflections (Rint) 5051 (0.0223) 4687 (0.0238)
Goodness-of-fit on F2 1.030 1.045
Final R indices [I>2σ(I)] R1=0.0274, wR2=0.0556 R1=0.0245,
wR2=0.0487
R indices (all data) R1=0.0222, wR2=0.0531 R1=0.0197,
wR2=0.0464

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
198
Investigation of the active species
Amido complex (C14b)
Complex C14 or C15 (15 mg) was placed with 3 equivalents of t-BuOK as the base (3.6 mg) in a
10 mL Schlenk tube and 1 mL of C6D6 was added. The complex C15 was immediately solved
and a red solution was obtained. The solution of the complex C14 was at first yellow and got red
after about one hour. 31P NMR (122 MHz, C6D6) δ 91.06.
Hydride complex (C14a)
Under an argon atmosphere, the complex C14 or C15 (15 mg) was placed with 3 equivalents of
t-BuOK as the base (3.6 mg) in a 3 mL vial and 1 mL of C6D6 was added. The vial was placed in
the alloy plate which was then placed into the autoclave. Once sealed, the autoclave was purged 5
times with hydrogen, then pressurized to 30 bar and stirred for two hours. The pale red solution
was transferred to a Young tube and analysed by NMR. 1H NMR (122 MHz, C6D6) δ -5.60 (t, J = 49.15, 1 H), -5.84 (t, J = 50.35, 1H). 31P NMR (122 MHz, C6D6) δ 91.06 (amido complex), 87.60, 86.54, -26.65 (free ligand).
4.5 General procedure for the manganese catalyzed hydrogenation of esters All catalytic hydrogenation experiments using molecular hydrogen were carried out in a Parr
Instruments autoclave (300 mL) advanced with an internal alloy plate include up to 7 uniform
reaction vials (8 mL) equipped with a cap and needle penetrating the septum.
Representative experiment: Under an argon atmosphere, a vial was charged with C14 or C15
(0.03 mmol) and t-BuOK (0.1 mmol) which was dissolved in dry 1,4-dioxane (2 mL). The yellow
solution of complex C14 was stirred briefly before the ester or lactone (1 mmol) was added
whereas the solution of complex C14 was immediately red by adding the solvent which indicates
a fast formation of the active species. The vial was placed in the alloy plate which was then
placed into the autoclave. Once sealed, the autoclave was purged 5 times with hydrogen, then
pressurized to 30 bar and heated to 110 °C or 120 °C for 24 h or 48 h. Afterwards, the autoclave
was cooled to RT, depressurized, and the reaction mixture was analyzed by GC-FID and GC-MS.
Product isolation was performed via column chromatography using silica gel as stationary phase
and an n-pentane / ethyl acetate or n-pentane / isopropanol mixture as eluent.
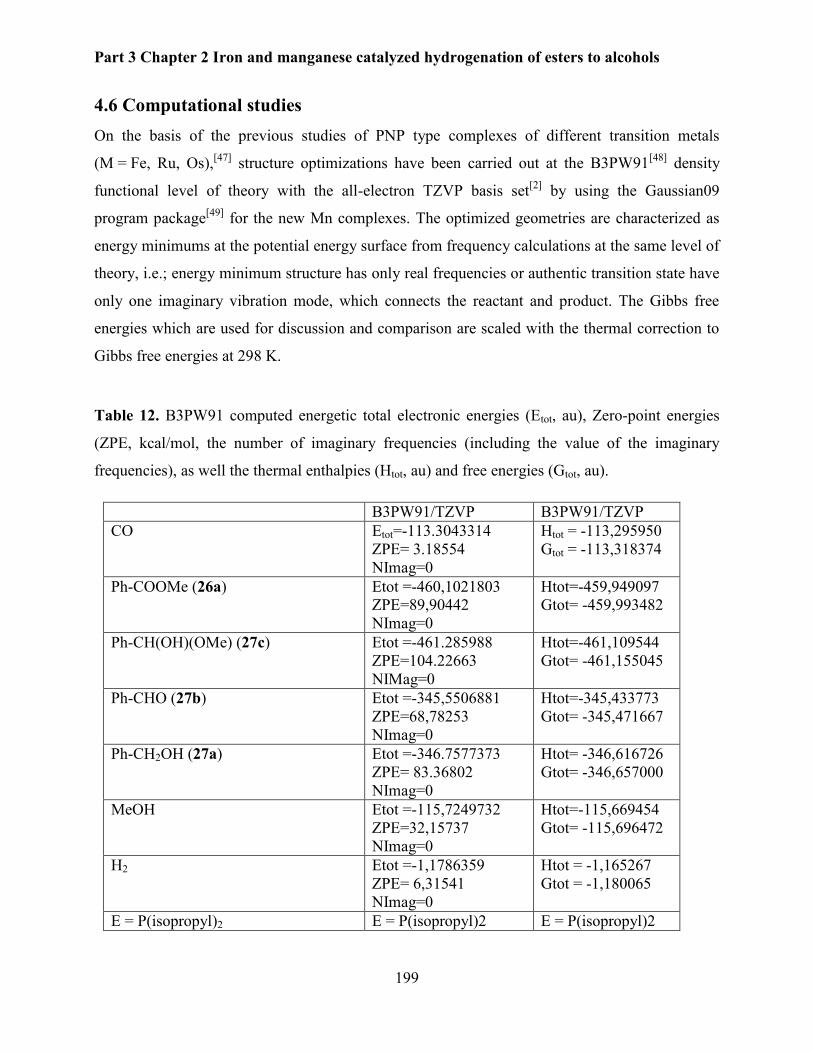
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
199
4.6 Computational studies On the basis of the previous studies of PNP type complexes of different transition metals
(M = Fe, Ru, Os),[47] structure optimizations have been carried out at the B3PW91[48] density
functional level of theory with the all-electron TZVP basis set[2] by using the Gaussian09
program package[49] for the new Mn complexes. The optimized geometries are characterized as
energy minimums at the potential energy surface from frequency calculations at the same level of
theory, i.e.; energy minimum structure has only real frequencies or authentic transition state have
only one imaginary vibration mode, which connects the reactant and product. The Gibbs free
energies which are used for discussion and comparison are scaled with the thermal correction to
Gibbs free energies at 298 K.
Table 12. B3PW91 computed energetic total electronic energies (Etot, au), Zero-point energies
(ZPE, kcal/mol, the number of imaginary frequencies (including the value of the imaginary
frequencies), as well the thermal enthalpies (Htot, au) and free energies (Gtot, au).
B3PW91/TZVP B3PW91/TZVP CO Etot=-113.3043314
ZPE= 3.18554 NImag=0
Htot = -113,295950 Gtot = -113,318374
Ph-COOMe (26a) Etot =-460,1021803 ZPE=89,90442 NImag=0
Htot=-459,949097 Gtot= -459,993482
Ph-CH(OH)(OMe) (27c) Etot =-461.285988 ZPE=104.22663 NIMag=0
Htot=-461,109544 Gtot= -461,155045
Ph-CHO (27b) Etot =-345,5506881 ZPE=68,78253 NImag=0
Htot=-345,433773 Gtot= -345,471667
Ph-CH2OH (27a) Etot =-346.7577373 ZPE= 83.36802 NImag=0
Htot= -346,616726 Gtot= -346,657000
MeOH Etot =-115,7249732 ZPE=32,15737 NImag=0
Htot=-115,669454 Gtot= -115,696472
H2 Etot =-1,1786359 ZPE= 6,31541 NImag=0
Htot = -1,165267 Gtot = -1,180065
E = P(isopropyl)2 E = P(isopropyl)2 E = P(isopropyl)2
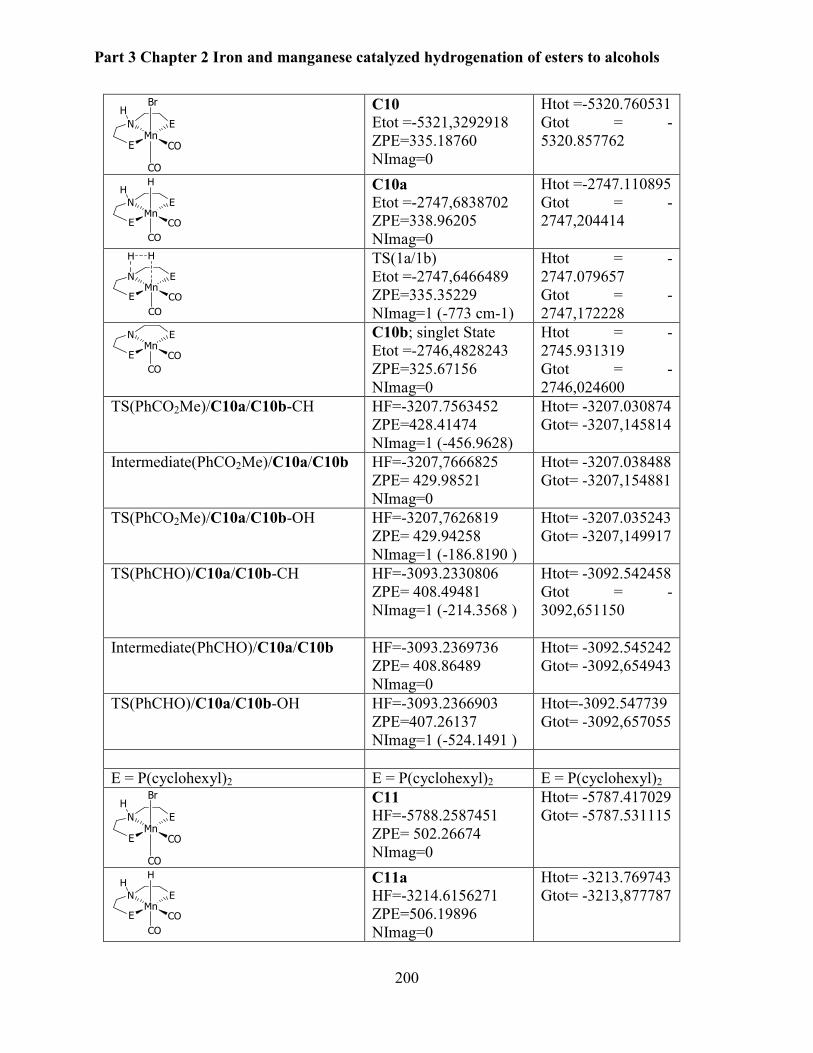
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
200
C10 Etot =-5321,3292918 ZPE=335.18760 NImag=0
Htot =-5320.760531 Gtot = -5320.857762
C10a Etot =-2747,6838702 ZPE=338.96205 NImag=0
Htot =-2747.110895 Gtot = -2747,204414
TS(1a/1b) Etot =-2747,6466489 ZPE=335.35229 NImag=1 (-773 cm-1)
Htot = -2747.079657 Gtot = -2747,172228
C10b; singlet State Etot =-2746,4828243 ZPE=325.67156 NImag=0
Htot = -2745.931319 Gtot = -2746,024600
TS(PhCO2Me)/C10a/C10b-CH HF=-3207.7563452 ZPE=428.41474 NImag=1 (-456.9628)
Htot= -3207.030874 Gtot= -3207,145814
Intermediate(PhCO2Me)/C10a/C10b HF=-3207,7666825 ZPE= 429.98521 NImag=0
Htot= -3207.038488 Gtot= -3207,154881
TS(PhCO2Me)/C10a/C10b-OH HF=-3207,7626819 ZPE= 429.94258 NImag=1 (-186.8190 )
Htot= -3207.035243 Gtot= -3207,149917
TS(PhCHO)/C10a/C10b-CH HF=-3093.2330806 ZPE= 408.49481 NImag=1 (-214.3568 )
Htot= -3092.542458 Gtot = -3092,651150
Intermediate(PhCHO)/C10a/C10b HF=-3093.2369736 ZPE= 408.86489 NImag=0
Htot= -3092.545242 Gtot= -3092,654943
TS(PhCHO)/C10a/C10b-OH HF=-3093.2366903 ZPE=407.26137 NImag=1 (-524.1491 )
Htot=-3092.547739 Gtot= -3092,657055
E = P(cyclohexyl)2 E = P(cyclohexyl)2 E = P(cyclohexyl)2
C11 HF=-5788.2587451 ZPE= 502.26674 NImag=0
Htot= -5787.417029 Gtot= -5787.531115
C11a HF=-3214.6156271 ZPE=506.19896 NImag=0
Htot= -3213.769743 Gtot= -3213,877787
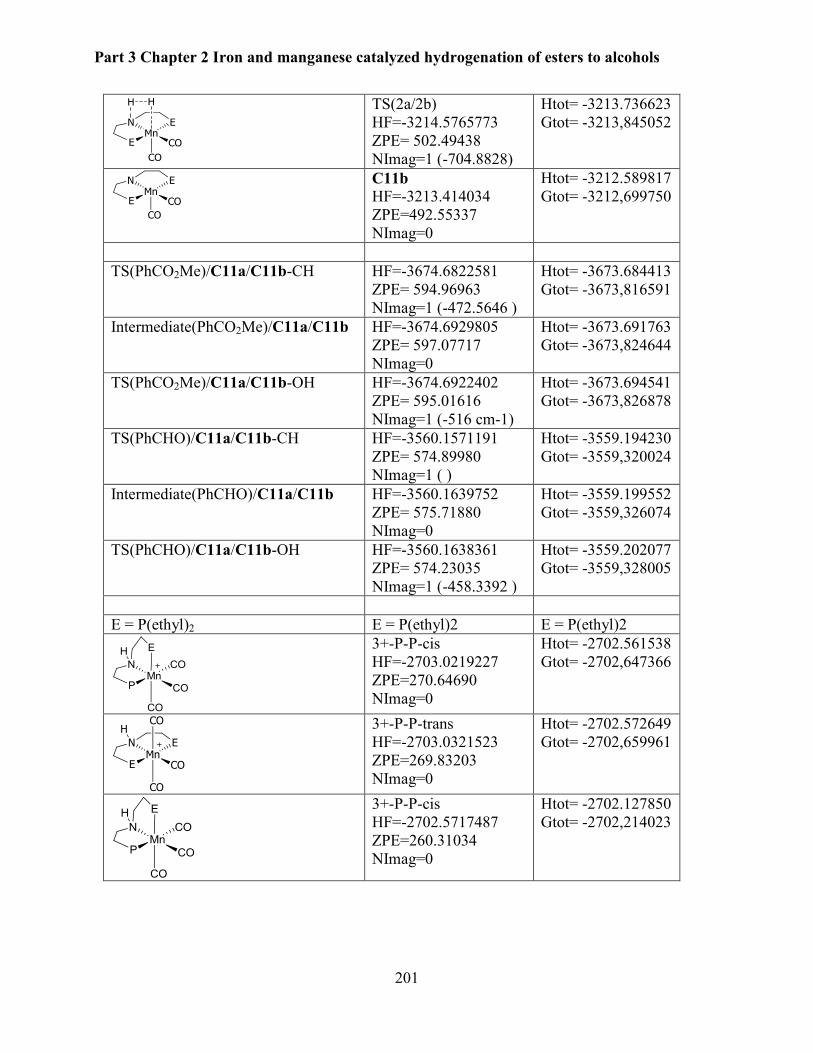
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
201
TS(2a/2b) HF=-3214.5765773 ZPE= 502.49438 NImag=1 (-704.8828)
Htot= -3213.736623 Gtot= -3213,845052
C11b HF=-3213.414034 ZPE=492.55337 NImag=0
Htot= -3212.589817 Gtot= -3212,699750
TS(PhCO2Me)/C11a/C11b-CH HF=-3674.6822581
ZPE= 594.96963 NImag=1 (-472.5646 )
Htot= -3673.684413 Gtot= -3673,816591
Intermediate(PhCO2Me)/C11a/C11b HF=-3674.6929805 ZPE= 597.07717 NImag=0
Htot= -3673.691763 Gtot= -3673,824644
TS(PhCO2Me)/C11a/C11b-OH HF=-3674.6922402 ZPE= 595.01616 NImag=1 (-516 cm-1)
Htot= -3673.694541 Gtot= -3673,826878
TS(PhCHO)/C11a/C11b-CH HF=-3560.1571191 ZPE= 574.89980 NImag=1 ( )
Htot= -3559.194230 Gtot= -3559,320024
Intermediate(PhCHO)/C11a/C11b HF=-3560.1639752 ZPE= 575.71880 NImag=0
Htot= -3559.199552 Gtot= -3559,326074
TS(PhCHO)/C11a/C11b-OH HF=-3560.1638361 ZPE= 574.23035 NImag=1 (-458.3392 )
Htot= -3559.202077 Gtot= -3559,328005
E = P(ethyl)2 E = P(ethyl)2 E = P(ethyl)2
3+-P-P-cis HF=-2703.0219227 ZPE=270.64690 NImag=0
Htot= -2702.561538 Gtot= -2702,647366
3+-P-P-trans HF=-2703.0321523 ZPE=269.83203 NImag=0
Htot= -2702.572649 Gtot= -2702,659961
3+-P-P-cis HF=-2702.5717487 ZPE=260.31034 NImag=0
Htot= -2702.127850 Gtot= -2702,214023
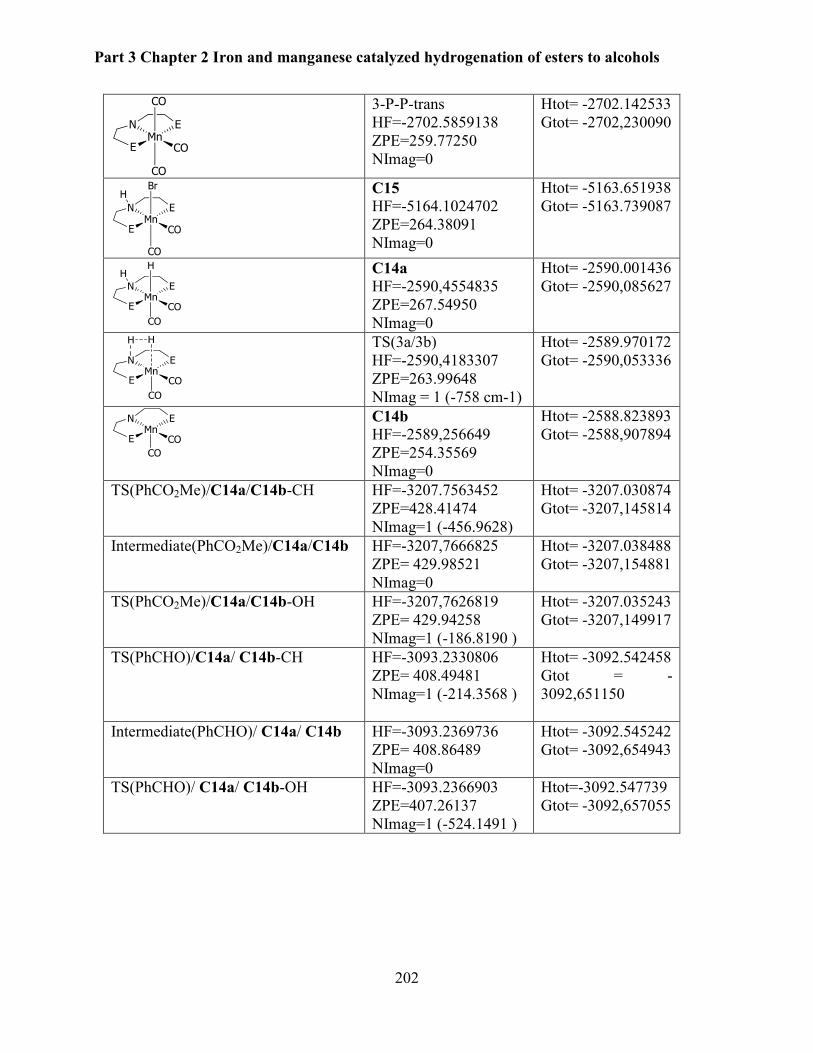
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
202
3-P-P-trans HF=-2702.5859138 ZPE=259.77250 NImag=0
Htot= -2702.142533 Gtot= -2702,230090
C15 HF=-5164.1024702 ZPE=264.38091 NImag=0
Htot= -5163.651938 Gtot= -5163.739087
C14a HF=-2590,4554835 ZPE=267.54950 NImag=0
Htot= -2590.001436 Gtot= -2590,085627
TS(3a/3b) HF=-2590,4183307 ZPE=263.99648 NImag = 1 (-758 cm-1)
Htot= -2589.970172 Gtot= -2590,053336
C14b HF=-2589,256649 ZPE=254.35569 NImag=0
Htot= -2588.823893 Gtot= -2588,907894
TS(PhCO2Me)/C14a/C14b-CH HF=-3207.7563452 ZPE=428.41474 NImag=1 (-456.9628)
Htot= -3207.030874 Gtot= -3207,145814
Intermediate(PhCO2Me)/C14a/C14b HF=-3207,7666825 ZPE= 429.98521 NImag=0
Htot= -3207.038488 Gtot= -3207,154881
TS(PhCO2Me)/C14a/C14b-OH HF=-3207,7626819 ZPE= 429.94258 NImag=1 (-186.8190 )
Htot= -3207.035243 Gtot= -3207,149917
TS(PhCHO)/C14a/ C14b-CH HF=-3093.2330806 ZPE= 408.49481 NImag=1 (-214.3568 )
Htot= -3092.542458 Gtot = -3092,651150
Intermediate(PhCHO)/ C14a/ C14b HF=-3093.2369736 ZPE= 408.86489 NImag=0
Htot= -3092.545242 Gtot= -3092,654943
TS(PhCHO)/ C14a/ C14b-OH HF=-3093.2366903 ZPE=407.26137 NImag=1 (-524.1491 )
Htot=-3092.547739 Gtot= -3092,657055

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
203
4.7 Data for isolated products Benzyl alcohol
1H NMR (300 MHz, CDCl3): δ 7.40 – 7.27 (m, 5H), 4.70 (s, 2H), 1.75 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 140.8, 128.6, 127.7, 127.0, 77.0, 65.4.
4-Methylbenzyl alcohol
1H NMR (300 MHz, CDCl3): δ 7.39 – 7.04 (m, 4H), 4.60 (s, 2H), 2.33 (s, 3H), 1.69 (bs, 1H). 13C NMR (75 MHz, CDCl3): δ 138.0, 137.4, 129.3, 127.2, 65.2, 21.2. 4-Methoxybenzyl alcohol
1H NMR (300 MHz, CD2Cl2): δ 7.27 (d, J = 8.3 Hz, 2H), 6.89 (d, J = 8.1 Hz, 2H), 4.57 (s, 2H), 3.79 (s, 3H), 2.38 (s, 1H). 13C NMR (75 MHz, CD2Cl2): δ 159.6, 134.0, 129.0, 114.2, 65.2, 55.7. 4-(Trifluoromethyl)benzyl alcohol
1H NMR (300 MHz, CDCl3): δ 7.61 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 7.9 Hz, 2H), 4.75 (s, 2H), 2.02 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 144.8, 129.8 (q, J = 32.5 Hz), 126.9, 125.5 (q, J = 3.8 Hz), 122.4
(q, J = 272.8 Hz), 64.5.
4-Chlorobenzyl alcohol
1H NMR (300 MHz, CD2Cl2): δ 7.52 – 7.18 (m, 4H), 4.64 (d, J = 6.8 Hz, 2H), 2.03 (s, 1H). 13C NMR (75 MHz, CD2Cl2): δ 140.3, 133.5, 129.0, 128.8, 64.7.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
204
4-Fluorobenzyl alcohol
1H NMR (300 MHz, CDCl3): δ 7.49 – 7.16 (m, 2H), 7.20 – 6.94 (m, 2H), 4.62 (s, 2H), 2.85 – 2.56 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 160.7 (d, J = 245.5 Hz), 136.6 (d, J = 2.8 Hz), 128.8 (d, J = 7.8
Hz), 115.2 (d, J = 7.8 Hz), 64.4.
2-Methoxybenzyl alcohol
1H NMR (300 MHz, CD2Cl2): δ 7.60 – 7.24 (m, 2H), 7.30 – 7.00 (m, 2H), 4.82 (s, 1H), 4.02 (s, 2H), 2.75 (s, 1H). 13C NMR (75 MHz, CD2Cl2): δ 157.9, 130.0, 129.2, 128.9, 120.9, 110.7, 62.0, 55.8. 3,4,5-Trimethoxybenzyl alcohol
1H NMR (300 MHz, CDCl3): δ 6.60 (s, 2H), 4.63 (s, 2H), 3.86 (s, 6H), 3.83 (s, 3H).
13C NMR (75 MHz, CDCl3): δ 153.4, 137.4, 136.7, 103.9, 65.7, 60.9, 56.2. 2,5-Dichlorobenzyl alcohol
1H NMR (300 MHz, CDCl3): δ 7.50 (s, 1H), 7.27 (d, J = 8.5 Hz, 1H), 7.20 (d, J = 8.5 Hz, 1H), 4.74 (s, 2H), 2.13 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 140.0, 133.1, 130.5, 130.4, 128.7, 128.4, 62.3. 2-Naphthalenemethanol
1H NMR (300 MHz, CDCl3): δ 7.86 – 7.80 (m, 4H), 7.51 – 7.46 (m, 3H), 4.85 (s, 2H), 1.89 13C NMR (101 MHz, CDCl3): δ 133.3, 132.8, 128.3, 127.8, 127.6, 126.1, 125.8, 125.4, 125.1, 124.9, 65.4.
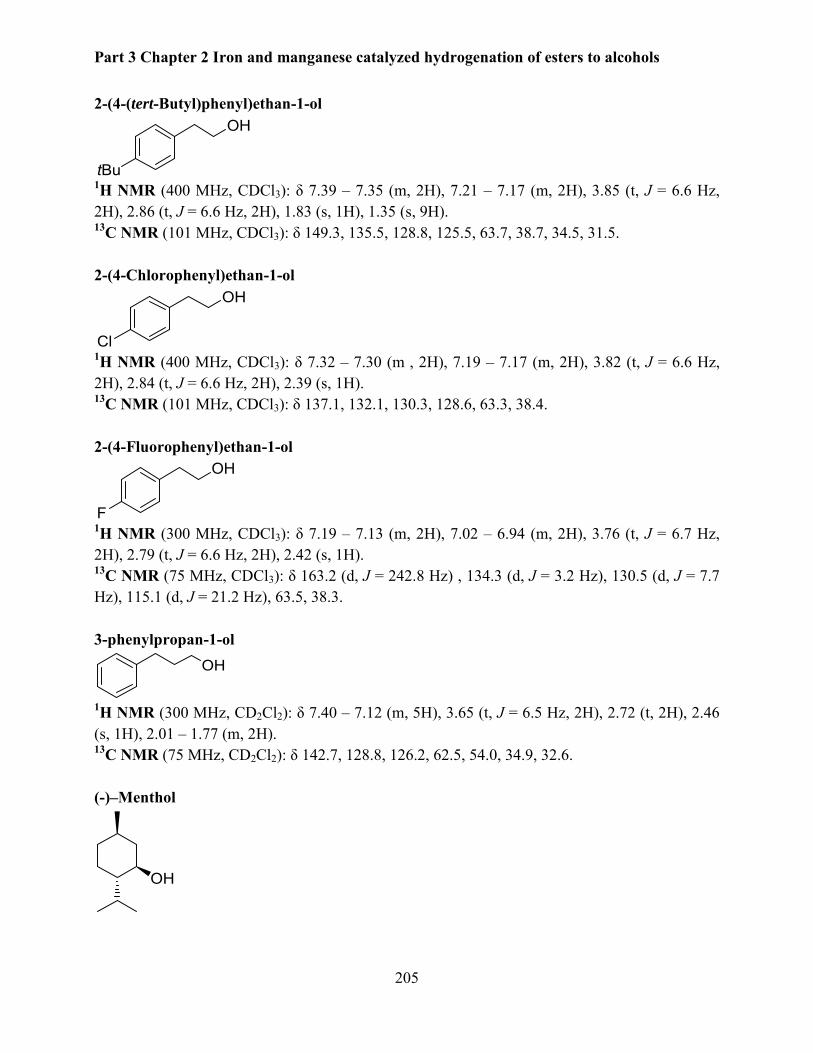
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
205
2-(4-(tert-Butyl)phenyl)ethan-1-ol
1H NMR (400 MHz, CDCl3): δ 7.39 – 7.35 (m, 2H), 7.21 – 7.17 (m, 2H), 3.85 (t, J = 6.6 Hz, 2H), 2.86 (t, J = 6.6 Hz, 2H), 1.83 (s, 1H), 1.35 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 149.3, 135.5, 128.8, 125.5, 63.7, 38.7, 34.5, 31.5. 2-(4-Chlorophenyl)ethan-1-ol
1H NMR (400 MHz, CDCl3): δ 7.32 – 7.30 (m , 2H), 7.19 – 7.17 (m, 2H), 3.82 (t, J = 6.6 Hz, 2H), 2.84 (t, J = 6.6 Hz, 2H), 2.39 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 137.1, 132.1, 130.3, 128.6, 63.3, 38.4. 2-(4-Fluorophenyl)ethan-1-ol
1H NMR (300 MHz, CDCl3): δ 7.19 – 7.13 (m, 2H), 7.02 – 6.94 (m, 2H), 3.76 (t, J = 6.7 Hz, 2H), 2.79 (t, J = 6.6 Hz, 2H), 2.42 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 163.2 (d, J = 242.8 Hz) , 134.3 (d, J = 3.2 Hz), 130.5 (d, J = 7.7 Hz), 115.1 (d, J = 21.2 Hz), 63.5, 38.3. 3-phenylpropan-1-ol
1H NMR (300 MHz, CD2Cl2): δ 7.40 – 7.12 (m, 5H), 3.65 (t, J = 6.5 Hz, 2H), 2.72 (t, 2H), 2.46 (s, 1H), 2.01 – 1.77 (m, 2H). 13C NMR (75 MHz, CD2Cl2): δ 142.7, 128.8, 126.2, 62.5, 54.0, 34.9, 32.6. (-)–Menthol

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
206
1H NMR (300 MHz, CDCl3): δ 3.46 – 3.35 (m, 1H), 2.25 – 2.08 (m, 1H), 2.02 – 1.89 (m, 1H), 1.74 – 1.54 (m, 2H), 1.50 – 1.32 (m, 2H), 1.16 – 0.96 (m, 2H), 0.93 (d, J = 4.7 Hz, 1H), 0.90 (d, J = 4.4 Hz, 2H), 0.81 (d, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 77.1, 50.3, 45.2, 34.7, 31.8, 25.9, 23.3, 22.3, 21.1, 16.2. Oleyl alcohol
1H NMR (300 MHz, CDCl3): δ 5.43 – 5.25 (m, 2H), 3.64 (t, J = 6.6 Hz, 2H), 2.01 (q, J = 6.6 Hz, 4H), 1.63 – 1.50 (m, 2H), 1.42 – 1.17 (m, 23H), 0.94 – 0.79 (m, 3H). 13C NMR (75 MHz, CDCl3): δ 129.9, 129.8, 63.1, 32.8, 31.9, 29.8, 29.7, 29.5, 29.5, 29.4, 29.3, 29.2, 27.2, 27.2, 25.7, 22.7, 14.1. 1,4-Octanediol
1H NMR (300 MHz, CDCl3): δ 3.75 – 3.50 (m, 3H), 2.92 (s, 2H), 1.70 – 1.16 (m, 10H), 0.89-0.84 (m, 3H). 13C NMR (75 MHz, CDCl3): δ 71.9, 63.0, 37.4, 34.5, 29.2, 28.0, 22.8, 14.2. (3,4,5-Trimethoxyphenyl)methanol
1H NMR (300 MHz, DMSO-d6): δ 7.56 (s, 1H), 7.42 (d, J = 8.1 Hz, 1H), 7.33 (d, J = 8.1 Hz, 1H), 5.25 (t, J = 5.5 Hz, 1H), 5.17 (t, J = 5.4 Hz, 1H), 4.52 (d, J = 5.4 Hz, 2H), 4.47 (d, J = 5.4 Hz, 2H). 13C NMR (75 MHz, DMSO-d6): δ 142.2, 138.5, 132.1, 129.0, 128.7, 119.7, 59.6, 59.4. 1,4-Octanediol
1H NMR (300 MHz, DMSO-d6): δ 7.25 (s, 4H), 5.12 (t, J = 5.7, 2H), 4.47 (d, J = 5.7, 4H). 13C NMR (75 MHz, DMSO-d6): δ 140.8, 126.2, 62.7.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
207
2,5-Bis(hydroxymethyl)furan
1H NMR (300 MHz, DMSO-d6): δ 6.19 (s, 2H), 5.16 (t, J = 5.7 Hz, 2H), 4.35 (d, J = 5.7 Hz, 4H). 13C NMR (75 MHz, DMSO-d6): δ 154.6, 107.3, 55.6. Methyl 5-(Hydroxymethyl)furan-2-carboxylate
1H NMR (400 MHz, CDCl3): δ 7.11 (d, J = 3.4 Hz, 1H), 6.39 (d, J = 3.4 Hz, 1H), 4.65 (s, 2H), 3.87 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 159.3, 158.6, 144.0, 119.0, 109.5, 57.5, 52.1. 1-(Pyridin-3-yl)ethan-1-ol
1H-NMR (400 MHz, CDCl3): δ 8.44-8.36 (m, 2H), 7.69 (d, J = 7.5 Hz, 1H), 7.24- 7.22 (m, 1H), 4.86 (q, J = 6.4 Hz, 1H), 4.55 (s, 1H), 1.45 (d, J = 6.2 Hz, 3H). 13C-NMR (101 MHz, CDCl3): δ 148.0, 147.0, 141.8, 133.5, 123.6, 67.4, 25.1. Cyclopropyl(phenyl)methanol
1H-NMR (300 MHz, CDCl3): δ 7.25-7.11 (m, 5H), 3.79 (d, J = 8.1 Hz, 1H), 2.34 (s, 1H), 1.06-0.98 (m, 1H), 0.46-0.17 (m, 4H). 13C-NMR (75 MHz, CDCl3): δ 143.7, 128.1, 127.3, 125.9, 78.9, 19.0, 3.5, 2.6. 2,3-Dihydro-1H-inden-1-ol
1H-NMR (300 MHz, CDCl3): δ 7.42-7.39 (m, 1H), 7.27-7.23 (m, 3H), 5.23-5.19 (m, 1H), 3.08- 3.00 (m, 1H), 2.86-2.76 (m, 1H), 2.47-2.45 (m, 1H), 2.36 (s, 1H), 1.98-1.87 (m, 1H). 13C-NMR (75 MHz, CDCl3): δ 144.9, 143.2, 128.2, 126.5, 124.7, 124.1, 76.2, 35.7, 29.7.
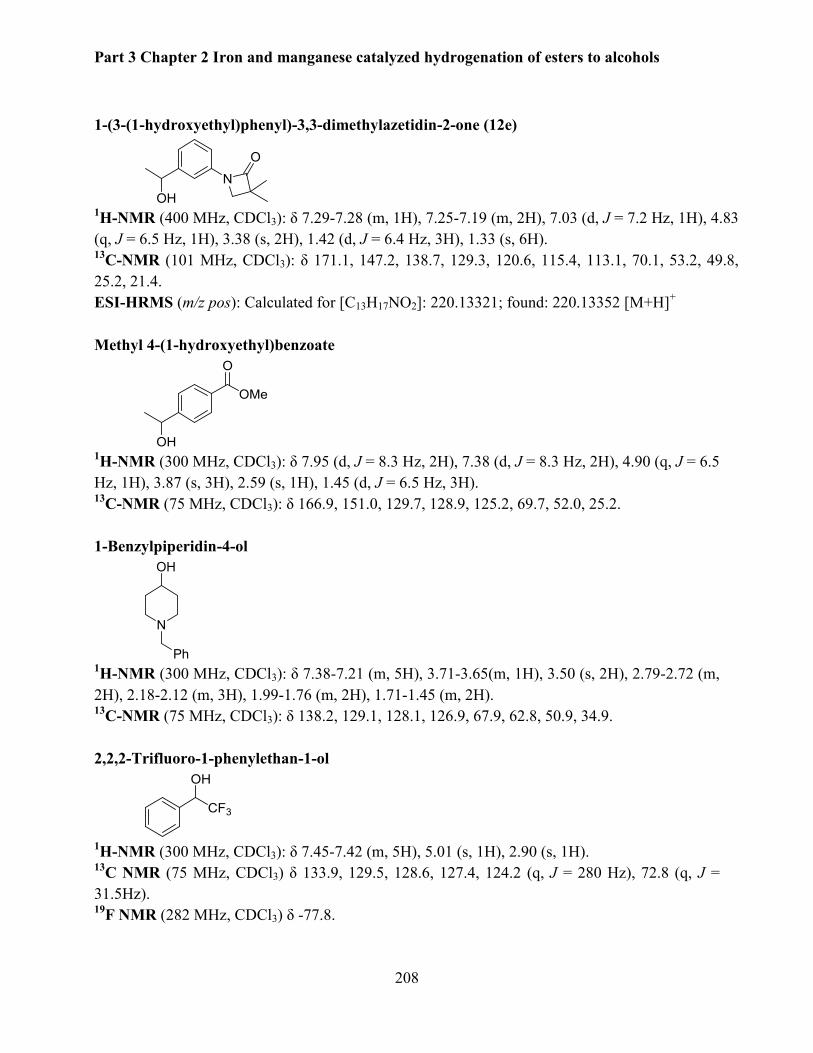
Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
208
1-(3-(1-hydroxyethyl)phenyl)-3,3-dimethylazetidin-2-one (12e)
1H-NMR (400 MHz, CDCl3): δ 7.29-7.28 (m, 1H), 7.25-7.19 (m, 2H), 7.03 (d, J = 7.2 Hz, 1H), 4.83 (q, J = 6.5 Hz, 1H), 3.38 (s, 2H), 1.42 (d, J = 6.4 Hz, 3H), 1.33 (s, 6H). 13C-NMR (101 MHz, CDCl3): δ 171.1, 147.2, 138.7, 129.3, 120.6, 115.4, 113.1, 70.1, 53.2, 49.8, 25.2, 21.4. ESI-HRMS (m/z pos): Calculated for [C13H17NO2]: 220.13321; found: 220.13352 [M+H]+
Methyl 4-(1-hydroxyethyl)benzoate
1H-NMR (300 MHz, CDCl3): δ 7.95 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 8.3 Hz, 2H), 4.90 (q, J = 6.5 Hz, 1H), 3.87 (s, 3H), 2.59 (s, 1H), 1.45 (d, J = 6.5 Hz, 3H). 13C-NMR (75 MHz, CDCl3): δ 166.9, 151.0, 129.7, 128.9, 125.2, 69.7, 52.0, 25.2. 1-Benzylpiperidin-4-ol
1H-NMR (300 MHz, CDCl3): δ 7.38-7.21 (m, 5H), 3.71-3.65(m, 1H), 3.50 (s, 2H), 2.79-2.72 (m, 2H), 2.18-2.12 (m, 3H), 1.99-1.76 (m, 2H), 1.71-1.45 (m, 2H). 13C-NMR (75 MHz, CDCl3): δ 138.2, 129.1, 128.1, 126.9, 67.9, 62.8, 50.9, 34.9. 2,2,2-Trifluoro-1-phenylethan-1-ol
1H-NMR (300 MHz, CDCl3): δ 7.45-7.42 (m, 5H), 5.01 (s, 1H), 2.90 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 133.9, 129.5, 128.6, 127.4, 124.2 (q, J = 280 Hz), 72.8 (q, J = 31.5Hz). 19F NMR (282 MHz, CDCl3) δ -77.8.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
209
Chroman-4-ol
1H-NMR (300 MHz, CDCl3): δ 7.31-7.18 (m, 2H), 6.94-6.82 (m, 2H), 4.67 (t, J = 4.0 Hz, 1H), 4.27-4.23 (m, 2H), 2.11-2.02 (m, 2H). 13C-NMR (75 MHz, CDCl3): δ 154.5, 129.6, 129.6, 124.2, 120.5, 116.9, 63.2, 61.8, 30.7. 4-(2,6,6-Trimethylcyclohex-1-en-1-yl)butan-2-ol
1H-NMR (300 MHz, CDCl3): δ 3.84-3.74 (m, 1H), 1.91-1.87 (m, 4H), 1.59 -1.51 (m, 8H), 1.42-1.39 (m, 2H), 1.21 (d, J = 6.2 Hz, 3H), 0.98 (s, 6H). 13C-NMR (75 MHz, CDCl3): δ 136.8, 126.9, 77.2, 68.8, 39.9, 39.8, 34.9, 32.7, 28.6, 24.7, 23.3, 19.8, 19.5. (E)-3-(2-Methoxyphenyl)prop-2-en-1-ol
1H-NMR (300 MHz, CDCl3): δ 7.44 (dd, J = 7.6, 1.7 Hz, 1H), 7.30-7.18 (m, 1H), 7.01-6.83 (m, 3H), 6.39 (dt, J = 16.0, 5.9 Hz, 1H), 4.33 (dd, J = 5.9, 1.5 Hz, 2H), 3.85 (s, 3H), 1.65 (s, 1H). 13C-NMR (75 MHz, CDCl3): δ 156.7 129.3, 128.7, 126.9, 126.2, 125.7, 120.6, 110.8, 64.3, 55.4. 3,3-Diphenylprop-2-en-1-ol
1H-NMR (300 MHz, CDCl3): δ 7.49-7.40 (m, 3H), 7.40-7.31 (m, 5H), 7.28-7.22 (m, 2H), 6.33 (t, J = 6.7 Hz, 1H), 4.29 (d, J = 6.7 Hz, 2H), 2.14 (s, 1H). 13C-NMR (75 MHz, CDCl3): δ 143.9, 141.7, 138.9, 129.6, 128.3, 128.2, 128.1, 127.6, 127.6, 127.5, 60.5. (E)-2-Benzylideneoctan-1-ol
1H-NMR (400 MHz, CDCl3): δ 7.41-7.34 (m, 2H), 7.31-7.24 (m, 3H), 6.57 (s, 1H), 4.26 (s, 2H), 2.42-2.25 (m, 2H), 2.03 (s, 1H), 1.57-1.50 (m, 2H), 1.39-1.24 (m, 6H), 0.99-0.77 (m, 3H).

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
210
13C-NMR (101 MHz, CDCl3): δ 142.3, 137.5, 128.5, 128.1, 126.3, 125.1, 66.9, 31.5, 29.5, 28.7, 28.3, 22.5, 14.0. Oct-2-yn-1-ol)
1H-NMR (300 MHz, CDCl3): δ 4.25 (s, 2H), 2.20 (t, J = 7.1 Hz, 2H), 1.51-1.31 (m, 6H), 0.89 (t, J = 6.9 Hz, 3H). 13C-NMR (75 MHz, CDCl3): δ 86.5, 78.2, 51.3, 30.9, 28.2, 22.2, 18.6, 13.9. 3,7-Dimethyloct-6-en-1-ol
1H-NMR (300 MHz, CDCl3): δ 5.13-5.06 (m, 1H), 3.78-3.53 (m, 2H), 2.15-1.84 (m, 2H), 1.75-1.49 (m, 9H), 1.44-1.15 (m, 3H), 0.97-0.84 (m, 3H). 13C-NMR (75 MHz, CDCl3): δ 131.2, 124.6, 61.1, 39.8, 37.2, 29.1, 25.7, 25.4, 19.5, 17.6. Furan-2,5-diyldimethanol
1H NMR (300 MHz, MeOD): δ 6.22 (s, 2H), 4.48 (s, 4H). 13C NMR (75 MHz, MeOD): δ 155.9, 109.3, 57.6, 49.3. (4-(Prop-1-en-2-yl)cyclohex-1-en-1-yl)methanol (Perillyl alcohol)
1H NMR (300 MHz, CDCl3): δ 5.69-5.66 (m, 1H), 4.71-4.69 (m, 2H), 3.97-3.96 (m, 2H), 2.17-1.81 (m, 7H), 1.72 (s, 3H), 1.50-1.45 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 149.7, 137.1, 122.2, 108.5, 67.0, 41.1, 30.3, 27.4, 26.0, 20.7.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
211
5. References [1] P. G. Andersson, I. J. Munslo, in in Modern Reduction Methods, Wiley, New York, 2008. [2] P. N. Rylander, in in Catalytic Hydrogenation in Organic Syntheses, Academic Press,
New York, 1979. [3] S. N. Ege, D. C. Health Company, 1989, p. 596. [4] M. L. Clarke, Catal. Sci. Technol. 2012, 2, 2418-2423. [5] P. A. Dub, T. Ikariya, ACS Catal. 2012, 2, 1718-1741. [6] S. Werkmeister, K. Junge, M. Beller, Org. Process Res. Dev. 2014, 18, 289-302. [7] J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen, E. A. Pidko, Chem. Soc.
Rev. 2015, 44, 3808-3833. [8] H. G. Manyar, C. Paun, R. Pilus, D. W. Rooney, J. M. Thompson, C. Hardacre, Chem.
Commun. 2010, 46, 6279-6281. [9] Y. Pouilloux, F. Autin, J. Barrault, Catal. Today 2000, 63, 87-100. [10] R. A. Grey, G. P. Pez, A. Wallo, J. Corsi, J. Chem. Soc, Chem. Commun. 1980, 783-784. [11] R. A. Grey, G. P. Pez, A. Wallo, J. Am. Chem. Soc. 1981, 103, 7536-7542. [12] U. Matteoli, M. Blanchi, G. Menchi, P. Prediani, F. Piacenti, J. Mol. Catal. 1984, 22, 353-
362. [13] U. Matteoli, G. Menchi, M. Bianchi, F. Piacenti, J. Organomet. Chem. 1986, 299, 233-
238. [14] U. Matteoli, G. Menchi, M. Bianchi, F. Piacenti, S. Ianelli, M. Nardelli, J. Organomet.
Chem. 1995, 498, 177-186. [15] L. A. Saudan, C. M. Saudan, C. Debieux, P. Wyss, Angew. Chem. Int. Ed. 2007, 119,
7617-7620. [16] M. Ito, T. Ikariya, J. Synth. Org. Chem. Jpn. 2008, 66, 1042-1048. [17] K. Junge, B. Wendt, F. A. Westerhaus, A. Spannenberg, H. Jiao, M. Beller, Chem. Eur. J.
2012, 18, 9011 – 9018. [18] M. Ito, T. Ootsuka, R. Watari, A. Shiibashi, A. Himizu, T. Ikariya, J. Am. Chem. Soc.
2011, 133, 4240-4242. [19] W. W. N. O, A. J. Lough, R. H. Morris, Chem. Commun. 2010, 46, 8240-8242. [20] F. A. Westerhaus, B. Wendt, A. Dumrath, G. Wienhöfer, K. Junge, M. Beller,
Chemsuschem 2013, 1001-1005. [21] H. T. Teunissen, C. J. Elsevier, Chem. Commun. 1997, 667-668. [22] L. Rosi, M. Frediani, P. Frediani, J. Organomet. Chem. 2010, 695, 1314-1322 [23] Y. Hara, H. Inagaki, S. Nishimura, K. Wada, Chem. Lett. 1992, 21, 1983-1986. [24] K. Nomura, H. Ogura, Y. Imanishi, J. Mol. Catal. 2001, 166, 345-349. [25] H. T. Teunissen, C. J. Elsevier, Chem. Commun. 1998, 1367-1368. [26] M. J. Hanton, S. Tin, B. J. Boardman, P. Miller, J. Mol. Catal. 2011, 346, 70-78. [27] J. Zhang, G. Leitus, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2006, 45, 1113-
1115. [28] W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T.
Kobayashi, N. Sayo, T. Saito, Org. Process Res. Dev. 2012, 16, 166-171. [29] X. Tan, Q. Wang, Y. Liu, F. Wang, H. Lv, X. Zhang, Chem. Commun. 2015, 51, 12193-
12196. [30] D. Spasyuk, S. Smith, D. G. Gusev, Angew. Chem. Int. Ed. 2012, 51, 2772-2775.

Part 3 Chapter 2 Iron and manganese catalyzed hydrogenation of esters to alcohols
212
[31] J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, David B. Cordes, A. M. Z. Slawin, M. L. Clarke, Chem. Eur. J. 2015, 27, 10851-10860.
[32] D. Spasyuk, S. Smith, D. G. Gusev, Angew. Chem. Int. Ed. 2013, 52, 2538-2542. [33] G. A. Filonenko, M. J. B. Aguila, E. N. Schulpen, R. van Putten, J. Wiecko, C. Müller, L.
Lefort, E. J. M. Hensen, E. A. Pidko, J. Am. Chem. Soc. 2015, 137, 7620-7623. [34] G. A. Filonenko, E. Cosimi, L. Lefort, M. P. Conley, C. Coperet, M. Lutz, E. J. M.
Hensen, E. A. Pidko, ACS Catal. 2014, 4, 2667-2671. [35] Y. Sun, C. Koehler, R. Tan, V. T. Annibale, D. Song, Chem. Commun. 2011, 47, 8349-
8351. [36] D. Kim, L. Le, M. J. Drance, K. H. Jensen, K. Bogdanovski, Tia N. Cervarich, M. G.
Barnard, N. J. Pudalov, S. M. M. Knapp, A. R. Chianese, Organometallics 2016, 35, 982-989.
[37] W. Li, J.-H. Xie, M.-L. Yuan, Q.-L. Zhou, Green Chem. 2014, 16, 4081-4085. [38] X. Tan, Y. Wang, Y. Liu, F. Wang, L. Shi, K. H. Lee, Z. Lin, H. Lv, X. Zhang, Org. Lett.
2015, 17, 454-457. [39] F. Wang, X. Tan, H. Lv, X. Zhang, Chem. Asian J. 2016, 11,2103-2106. [39a] K. Junge, B. Wendt, H. Jiao, M. Beller, ChemCatChem 2014, 6, 2810-2814. [40] T. Zell, Y. Ben-David, D. Milstein, Angew. Chem. Int. Ed. 2014, 53, 4685-4689. [41] S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F.
Gallou, M. Beller, Angew. Chem. Int. Ed. 2014, 53, 8722-8726. [42] S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause,
H. Guan, J. Am. Chem. Soc. 2014, 136, 7869-7872. [43] S. Qu. H. Dai, Y. Dang, C. Song, Z.-X. Wang, H. Guan, ACS Catal. 2014, 4, 4377-4388. [44] D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W.
Shimon, Y. Ben David, D. Milstein, Angew. Chem. Int. Ed. 2015, 54, 12357-12360. [45] T. J. Korstanje, J. Ivar van der Vlugt, C. J. Elsevier, B. de Bruin, Science 2015, 350, 298-
302. [46] S. Elangovan, B. Wendt, C. Topf, S. Bachmann, M. Scalone, A. Spannenberg, H. Jiao, W.
Baumann, K. Junge, M. Beller, Adv. Synth. Catal. 2016, 358, 820-825. [47] H. Jiao, K. Junge, E. Alberico, M. Beller, J. Comput. Chem. 2016, 37, 168-176. [48] J. P. Perdew, Phys. Rev. B 1986, 33, 8822-8824. [49] A. Schaefer, C. Huber, R. Ahlrichs, J. Chem. Phys. 1994, 100, 5829-5835.

213

214
Part 4
Hydrogen borrowing reactions
Chapter 1
Iron catalyzed alkylation of ketones with
alcohols

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
215

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
216
1. Introduction The development of efficient and selective methodologies, the rise of green chemistry has
led organic chemists and chemical industry to take into account the impact of the processes on
the environment. Among the different ecofriendly approaches, borrowing hydrogen
methodologies are of increasing importance in organic synthesis and catalysis as they represent
prime examples of green chemistry.[1-4] In this process, the alcohols play an important role as H2
donor, no external reductant is needed and water is generated as the only side product. With the
gradual depletion of fossil resources, the utilization of abundant and sustainable alcohols as the
feedstocks for chemical production is highly desirable in sustainable chemistry. On the one hand,
alcohols could serve as alternative reductants for reducing a number of functional groups.[5-7]
Comparing to the traditional hydrogenation processes using high-pressure hydrogen gas, the
transfer hydrogenation with alcohols no need for special equipment such as autoclaves, offering
more convenient and safer operations. On the other hand, alcohols have been employed as
coupling agents for various synthetic purposes via an acceptorless dehydrogenative process.[8-10]
Moreover, in line with the principles of green chemistry, the application of alcohols as both
hydrogen suppliers and coupling components for the benign construction of carbon-carbon and
carbon-heteroatom bonds in synthetic chemistry is of significant importance.
Since the groups of Grigg and Watanabe reported transition-metal catalyzed N-alkylations of
amines by means of alcohols as the alkylating agents,[11-12] continuous efforts have been made
towards both N-alkylation of amines and C-alkylation of ketones and related compounds with
alcohols as the alkylating agents, through a borrowing-hydrogen (BH) or hydrogen-autotransfer
(HA) strategy. A typical BH or HA process is demonstrated in Scheme 1. In such a process, an
alcohol transfers hydrogens to a transition-metal catalyst and forms the corresponding aldehyde
or ketone intermediate. This intermediate is then transformed into either an imine by
condensation with an amine or an olefin by condensation with the -C-H unit of a ketone.
Subsequent addition of hydrogen to either the imine or olefin intermediate, from the transition-
metal hydride species, gives the product with a newly formed C-N or C-C bond, respectively,
thus forming water as the only by-product.
In this chapter, we are mainly focused on the formation of C-C bond with ketones and alcohols.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
217
Scheme 1. Transition metal catalyzed -alkylation of ketones with alcohols.
1.1 C-C bond formation via alkylation of ketones with primary alcohols Carbon-carbon bond formation by means of efficient, selective, and environmentally
benign processes has been a challenging task in synthetic chemistry.[13] Although the alkylation
of ketones with alcohols in the presence of mixed metal oxide catalysts at very high temperatures
is well known,[14-15] its synthetic applicability was decreased as a result of the very low chemical
yields obtained and the presence of many by-products. However, in 1969, a French patent
revealed the possible usefulness of this approach.[16]
To build a C-C bond by the selective -functionalization of ketones with organohalides in the
presence of bases is one of the most fundamental reactions.[17] This methodology usually suffers
from the use of stoichiometric amount of bases, and the use of halides which leads to the
formation of (over)stoichiometric amounts of waste.
Scheme 2. -functionalization of ketones with organohalides.
By contrast, due to their wide availability, even from the biomass and often lower prices,
alcohols have emerged as interesting greener alternative alkylating reagents in the presence of
suitable catalysts,[18-20] an alcohol can be dehydrogenated to the corresponding aldehyde or
ketone, which in situ reacts with an enolate to form after dehydration the -unsaturated ketone.
Finally, this latter product is reduced to the desired alkylated ketone. Notably, in the overall
process, the catalyst plays the role of a hydrogen shuttle. In the process of -alkylation of

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
218
ketones 1 with alcohols 2 through a borrowing hydrogen, various products can be also obtained
such as alkylated ketones 3, -unsaturated ketones 4, or corresponding alcohols 5-7 and
polyalkylated product 8, when choosing the appropriate starting ketones or functionalized
alcohols.[21] Although, this method suffers in the selectivity problems because it produces more
side products (Scheme 3). Pioneering results using alcohols as alkylation reagents have been
reported by Guerbet already more than hundred years ago. Here, the “self” β-alkylation of
primary alcohols proceeds in the presence of copper salts and base.[22]
Scheme 3. Possible side products of alkylation of ketones with alcohols.
Later, in 2002 Cho, Sim and co-workers reported that ketones could be regioselectively
alkylated with various primary alcohols in the presence of a ruthenium catalyst RuCl2(PPh3)3.[23]
Advantageously, with this ruthenium complex, not only alkyl aryl ketones but also dialkyl
ketones could be successfully alkylated (Table 1, entry 1). In the case of substituted aryl alkyl
ketones, yields were not affected by the electronic properties of the substituents at the aromatic
ring. Although with dialkyl ketones, yields were lower than those for the related aryl compounds.
Interestingly, for the dialkyl ketones, alkylation took place specifically at the less-hindered
position. Notably, hydrogen acceptor (1-dodecene) was compulsory to prevent the over-reduction
of the final alkylated ketone to the corresponding alcohol.
Interestingly, in 2005, Yus et al. demonstrated that the use of [RuCl2(dmso)4] catalyzed
the C-C formation in the absence of any type of additive (Table 1, entry 2).[24] This catalytic
system allowed the use of heteroaromatic ketones and benzylic alcohol derivatives. Surprisingly,
the alkylation of benzo-fused cyclic ketones gave only -unsaturated ketones. Later, the same
group adjust the reaction parameters and used additives with the same ruthenium catalyst, for the
synthesis of the corresponding alcohols, quinolines, and -unsaturated ketones.[25]

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
219
Other metals have also been used such alkylation process. For example, palladium on
charcoal has been used as a heterogeneous catalyst in the presence of a large excess of 1-decene
and base (Table 1, entry 3). Notably, the ratio of the starting ketone and alcohol was critical to
prevent the over reduction of product 3.[26]
More interesting results were obtained using palladium nanoparticles entrapped in
aluminum hydroxide (Table 1, entry 4).[27] For this system, to maintain the reaction rate, the
addition of one equivalent of base was required in each cycle. Furthermore, inert-gas conditions
were necessary to avoid the oxidation of the palladium hydride intermediate; otherwise the
reaction stopped at the formation of the -unsaturated ketone. (Table 1, entry 5).[28]
Table 1. Comparision of alkylation of ketones with alcohols by different catalysts.
NDMP: 2-(2-naphthyl)-4,6- dimethyl-pyrimidine
In 2004, Ishii made significant advances in this field. The dimeric complex [IrCl(cod)]2 has been
used as a catalyst for the -alkylation of ketones using alcohols as electrophiles.[29] The reactions
could be performed in the absence of organic solvents using PPh3 as the ligand and only
substoichiometric amounts of base to afford the expected products at 100 oC for 24 h (Table 1,
entry 6). Under these conditions, several alkyl aryl ketones and dialkyl ketones could be alkylated
Entry Catalyst (mol%) Conditions Yields(%) Ref
1 [RuCl2(PPh3)3] (2) 1 (1 equiv.), 2 (1 equiv.), KOH (1 equiv.), 1-dodecene (1 equiv),
dioxane, 80 oC, 20 h
48-86 23
2 [RuCl2(dmso)4] (2) 1 (1 equiv.), 2 (1 equiv.), KOH (1 equiv.), dioxane, 80 oC, 24 h 41-93 24
3 Pd/C (5) 1 (1 equiv.), 2 (2 equiv.), KOH (3 equiv.), 1-decene (4 equiv.),
dioxane, 100 oC, 20 h
40-88 26
4 Pd/AlO(OH) (0.2) 1 (1 equiv.), 2 (1.2 equiv ), K3PO4 (3 equiv.), toluene, 110 oC, 8 h 80-98 27
5 Pd/viologen polymers(5) 1 (1 equiv.), 2 (2 equiv.), Ba(OH)2·H2O (1 equiv.), H2O (7
equiv.), neat, 100 oC, 24 h
82-95 28
6 [IrCl(cod)]2 (0.1) 1 (1 equiv.), 2 (2 equiv.), KOH (0.1 equiv.), PPh3 (0.04), neat,
100 oC, 4 h
47-96 29
7 RuHCl(CO)(PPh3)3 (1) 1 (1.3 equiv.), 2 (1 equiv.), Cs2CO3 (1.5 equiv.), toluene, 140 oC,
20 h
22-92 30
8 Ir(NDMP)(PPh3)2Cl2 1 (1 equiv.), 2 (2 equiv.), NaOH (1 equiv.), dioxane, 110 oC, 12 h 45-85 31

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
220
selectively with good yields. The catalytic system was also active to peform the reaction between
acetone and two molar equivalents of alcohol leading the ’-dialkylated ketone in high yield.
In 2012, Ryu’s group showed that alkylation reactions can be effectively catalyzed by
RuHCl(CO)(PPh3)3 in the presence of 1.5 equiv. of Cs2CO3 as the base.[30] They also reported
that a catalytic amount of 1,10-phenanthroline dramatically promoted -alkylation using
nonbenzylic aliphatic alcohols (Table 1, entry 7). The disadvantage of this system was the
necessity of a stoichiometric amount of base (1.5 equiv.) to achieve good yields of the expected
product.
In the meantime, Ir(NDMP)(PPh3)2Cl2 (NDMP = 2-(2-naphthyl)-4,6-dimethyl-
pyrimidine) catalyst was used for the alkylation of ketones with alcohols (2 equiv.) in the
presence of 1 equiv. of NaOH as a base at 110 oC for 3 h (Table 1, entry 8).[31]
In 2011, Pullarkat reported a ruthenacycle catlayzed one-pot -alkylation of secondary
alcohols with primary alcohols in the presence of 1 equiv. of NaOt-Bu at 110 °C for 17 h
(Scheme 4). This studies revealed the importance of the amino hydrogen in controlling the
catalytic activity of the ruthenacycle in such hydrogen transfer processes.[32]
Scheme 4. Ruthenium catalyzed alkylation of ketones with secondary alcohols.
In 2012, Xu and co-workers successfully developed a ligand-free Cu-catalyzed aerobic C-
alkylation of secondary alcohols or methyl ketones by primary alcohols at 120 °C for 24-30 h in
the presence of 30 mol% of KOH to achieve selectively the alkylated alcohols with only trace
amounts of alkylated ketones in main of the exemples (Scheme 5).[33]
Scheme 5. Copper catalyzed alkylation of ketones with alcohols.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
221
In 2014, Adolfsson and co-workers demonstrated that the in situ generated catalyst based
on [Ru(p-cymene)Cl2]2 and amino acid hydroxyamide ligand mediated the tandem
α-alkylation/asymmetric transfer hydrogenation process (Scheme 6). A series of substituted
acetophenones were successfully alkylated with simple primary alcohols with low to moderate
yields and ee up to 89%.[34]
Scheme 6. Alkylation of ketones with alcohols catalyzed by ruthenium.
In 2014, iridium CNP complexes were reported as catalysts (1 mol%) for the effective
alkylation of ketones with primary alcohols in the presence of 1 equiv. of Cs2CO3 in tert-amyl
alcohol at 120 oC for 24 h (Scheme 7).[35]
Scheme 7. Alkylation of ketones with alcohols catalyzed by irdium.
Recently, a rhodium complex bearing a ortho-phosphinoanilido ligand was used as a
catalyst (2 mol%) to develop the alkylation of acetophenone derivatives or secondary alcohols by
primary alcohols in the presence of a stoichiometric amount of Cs2CO3 as the base in refluxing
xylene for 24 h leading to the corresponding alkylated alcohols in 42-91% yields (Scheme 8).[36]
Scheme 8. Alkylation of ketones with alcohols catalyzed by rhodium.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
222
Very recently, Glorius reported the ruthenium-NHC catayzed alkylation of methylene
ketones with variety of primary alcohols as alkylating agent in the presence of 2 equiv. of
LiOtBu. In addition one spot double alkylation of acetophenone derivatives were reported by
sequential addition of primary alcohols.[37]
Scheme 9. Ru NHC catalyzed alkylation of methylene ketones with alcohols.
An osmium complex was also developed for the alkylation of arylacetonitriles and methyl
ketones with primary alcohols in the presence of 20 mol% of KOH under toluene refluxing
conditions for 0.5-6 h (Scheme 10).[38] Notably, as the catalyst was also efficient for the hydration
of nitrile to primary amide, the generated water has to be removed by Dean-Stark technique.
Turnover frequencies between 675 and 176 h−1 for nitriles and between 194 and 28 h−1 for
ketones were obtained.
Scheme 10. Osmium catalyzed alkylation of nitriles and ketones with alcohols.
1.2 Alkylation of ketones with propargylic alcohols In 2001, propargylic alkylated products were synthesised in high yields with complete
regioselectivities catalyzed by Cp*RuCl(µ2-SMe)2RuCp*Cl ruthenium complex (5 mol%) and
NH4BF4 (10 mol%) from propargylic alcohols and various ketones (used in excess) under mild
and neutral reaction conditions.[39]
Scheme 11. Alkylation of ketones with propargylic alcohols catalyzed by ruthenium.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
223
A recent report showed that silver hexafluoroantimonate(V) (AgSbF6) can be used as a
catalyst (10 mol%) for the -alkylation of unactivated ketones by propargylic alcohols. The silver
catalyst promoted the convertion of the ketone into an enol ether via acetal and the generation of
a carbocationic center at the benzylic position of the benzylic alcohol. The C-C coupling then
proceeded by reaction of the enol ether with the carbocation. The used alcohols included benzylic
alcohols, propargyl alcohols, cinnamyl alcohols, and diarylmethanols. The in situ formed acetals
are the key for the success of the reaction.[40]
Scheme 12. Silver hexafluoroantimonate catalyzed propargylation of ketones.
1.3 Alkylation of ketones with methanol All the methods described above failed to use methanol as a C1 alkylating agent. Indeed,
methanol oxidation is relatively difficult; the reaction energy for methanol dehydrogenation (H
= 84 kJ. mol-1) is higher than those for the dehydrogenation of higher alcohols such as ethanol
(H = 68 kJ. mol-1).[41] In very recent years, alkylation of ketones with methanol was studied. In
2014, iridium complex [Cp*IrCl2]2 (5 mol%) in combination with KOH (50 mol%) as a base was
successfully developed for an efficient -methylation of ketones or phenylacetonitriles using
methanol at 120 °C for 15 h (Scheme 13). Additionally, this catalytic system was allowed three-
component cross -methylalkylations of methyl ketones using methanol and primary alcohols
(Scheme 14).[42]
Scheme 13. Ir catalyzed alkylation of ketones with methanol.
Scheme 14. Three component cross -methylalkylations.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
224
In the same year, Li described the α-methylation of ketones with methanol using
Cp*Ir complex bearing a functional bipyridonate ligand as the catalyst (1 mol%) (Scheme
15). Moreover high catalytic activity was found for the α-alkylation of ketones with primary
alcohols, including methanol under mild conditions (0.3 equiv. of Cs2CO3, refluxing
methanol under air for 12-18 h).[43] In 2015, the same group reported that this bifunctional
iridium complexes can be effectively used for the alkylation of ketones by tandem aceptorless
dehydrogenation /-alkylation from secondary and primary alcohols.[44]
Scheme 15. Ir catalyzed alkylation of ketones with alcohols.
Donohoe reported the C-alkylation of ketones with methanol as an alkylating agent
using rhodium complex as the catalyst. The reaction proceeded smoothly in the presence of 5
mol% Cp*RhCl2 and 5 equiv. of Cs2CO3 as a base under oxygen atmosphere at 65 °C for 48
h. (Scheme 16). Double alkylation was also successfully developed with methyl ketones
using methanol under similar conditions (Scheme 16).[45]
Scheme 16. Rhodium catalyzed methylation of ketones with methanol.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
225
2. Knölker type complex catalyzed C-C bond formation reactions Eventhough effective alkylation reactions were already reported with noble transition
metals, obviously, in terms of sustainability, such precious transition metals should be replaced
by more eco-friendly, inexpensive and widely abundant first row based metals. Among these
metals, iron attracts significant attention and is considered as a valuable alternative.[46-49] In fact,
in the last decade iron catalysts are increasingly used in C-C cross coupling reactions[50-55] and
especially reductions.[56-58] With respect to the catalyst, Knölker-type complexes 16 constitute
convenient and stable precursors for hydrogenation and hydrogen transfer reactions but also for
selective oxidations of alcohols. (for details, see chapter 1). So far, such complexes were scarcely
used in redox neutral processes. Meanwhile, acceptorless alcohol dehydrogenation reactions were
recently described with iron pincer complexes using the so-called MACHO (PNP) ligand.[59-60]
In addition, the synthesis of substituted amines by alkylation of the corresponding primary
or secondary amines by alcohols was also just reported. (See chapter 2 for N-alkylation reactions)
In this chapter, we will report for the first time an iron-catalyzed -alkylation of ketones with
alcohols in the presence of the complex 16 using a hydrogen borrowing strategy (Scheme 17).
Based on the precedent results using Knölker type catalysts for both reduction and oxidation
reactions[61] and for alkylation of amines,[61] we envisioned that such complex can promote
hydrogen transfer reactions using alcohols as alkylating reagent in -functionalization of ketones.
Scheme 17. Iron-catalyzed -alkylation of ketones with alcohols.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
226
2.1 Optimisation of the reaction conditions In a preliminary experiment with acetophenone (1 equiv.) and benzylalcohol (1.5 equiv.)
in the presence of 5 mol% of the complex 16 as the pre-catalyst and 30 mol% of K2CO3 as a base
at 140 °C, 1,3-diphenylpropan-1-one 14 was obtained in 55% GC-yield together with 1-
phenylethanol 15 (32% GC-yield), resulting from the reduction of acetophenone which clearly
indicates that hydrogen transfer did occurred during the reaction (Table 2, entry 1).
Table 2. Iron-catalyzed α-alkylation of acetophenone with benzylalcohol: Variation of the
reaction parameters[a]
Entry Complex
(mol%) Base (mol%)
Time (h)
Yield (%) 14[b]
Yield (%) 15[b]
1 16 (5) K2CO3 (30) 36 55 32
2 16 (5) Cs2CO3 (30) 24 62 38
3 17 (2) Cs2CO3 (10) 24 46 24
4 18 (2) Cs2CO3 (10) 24 33 18
5 19 (2) Cs2CO3 (10) 24 45 30
6 Fe2(CO)9 (5) Cs2CO3 (10) 48 0 0
7 16 (2) Cs2CO3 (10) 24 61 29
8[c] 16 (2) Cs2CO3 (10) 24 74 20
9[c] 16 (2) Cs2CO3 (10) 24 44 10
10[c,d] 16 (2) Cs2CO3 (10) 24 80 6
11[c,e] 16 (2) Cs2CO3 (10) 24 0 0
12 16 (10) - 24 0 5
13 - Cs2CO3 (15) 24 0 0
[a] acetophenone (0.5 mmol), benzylalcohol (0.75 mmol, 1.5 equiv.), [Fe] (2-10 mol%), base (10-30 mol%), toluene (1 mL), 140 °C. [b] Yields determined by GC analysis. [c] 1.3 equiv. of benzylalcohol were used. [d] 2 mol% of PPh3 was used as an additive. [e] Reaction under neat conditions at 140 °C.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
227
When using Cs2CO3 (30 mol%) as the base, the reactivity was increased and 62% of the desired
-alkylated product 14 was obtained after 24 h at 140 °C with 38% of 15 (Table 1, entry 2).
Variation of the nature of the iron complex by substituting one CO ligand by PPh3 (17), and
acetonitrile (18) or by modifying the cyclopentadienone ligand (19) led to active catalysts, but
with lower chemoselectivities (yields 14/15 from 33/18 to 45/30) (Table 2, entries 3-5). Notably,
the cyclopentadienone motif is crucial for the activity of the catalyst as with Fe2(CO)9 as the
precatalyst, no activity was observed, even at prolonged reaction time (48 h) (Table 2, entry 6).
Interestingly, when decreasing the catalytic amount of the iron complex 16 to 2 mol% and of the
base Cs2CO3 10 mol% in the presence of 1.5 equiv. of benzylalcohol, 61% of 14 and 29% of 1-
phenylethanol 15 was obtained (Table 2, entry 7). When lowering the amount of benzylalcohol to
1.3 equiv., the chemoselectivity was improved notably with a decrease of the amount of 1-
phenylethanol 15: with the catalyst 16 (14: 74% and 15: 20%) and with the catalyst 17 (14: 44%
and 15: 10%) (Table 2, entries 8 and 9 vs 7 and 3, respectively). Noticeably, under similar
experimental conditions, the temperature of the reaction had a significant influence. Indeed at 100
°C, only 11% conversion was observed whereas at 120 °C, 85% conversion can be reached with
72% of 14 and 6% of 1-phenylethanol 15 were obtained.
The influence of the nature of the solvent was also examinated. In dioxane, in the presence of 2
mol% of 16 and 2 mol% of PPh3 with 10 mol% Cs2CO3 only 9% conversion was detected in THF
and no reaction took place at 70 oC. Noteworthy, in neat conditions at 140 oC for 24 h, no
reactivity was observed. Toluene was then selected as the solvent of the reaction (Table 3).
Table 3. Optimization with different solvents.[a]
Entry 16 (mol%) Temp. (oC) Solvent Yield 14 (%)[b] Yield 15 (%)[b]
1 2 120 Toluene 72 6
2 2 100 Toluene 8 3
2 2 140 Dioxane 9 54
3[c] 2 70 THF - -
4[c] 2 140 - 0 0
[a] PhCOMe (0.5 mmol), BnOH (0.75 mmol), [Fe] (2 mol%), Cs2CO3 (10 mol%), solvent (1 mL), 70-140 °C, 24 h. [b] Yield determined by GC. [c] PPh3 (2 mol%) used as an additive and 1.3 equiv. of alcohol used.
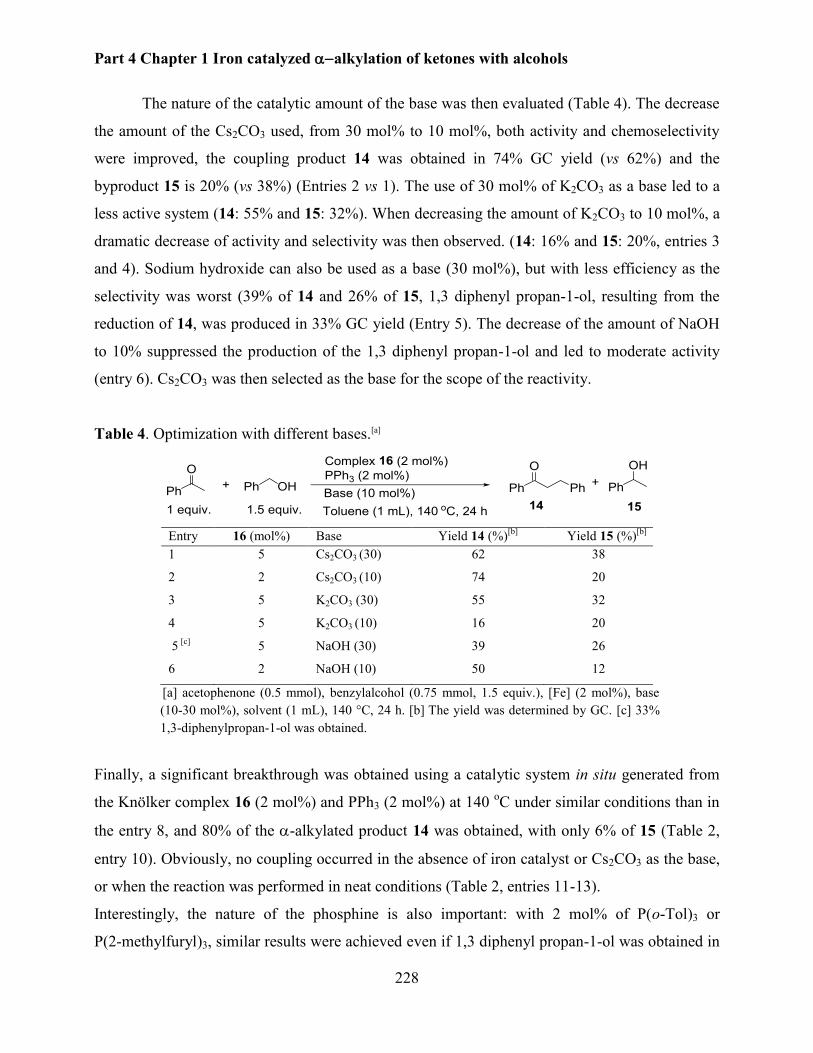
Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
228
The nature of the catalytic amount of the base was then evaluated (Table 4). The decrease
the amount of the Cs2CO3 used, from 30 mol% to 10 mol%, both activity and chemoselectivity
were improved, the coupling product 14 was obtained in 74% GC yield (vs 62%) and the
byproduct 15 is 20% (vs 38%) (Entries 2 vs 1). The use of 30 mol% of K2CO3 as a base led to a
less active system (14: 55% and 15: 32%). When decreasing the amount of K2CO3 to 10 mol%, a
dramatic decrease of activity and selectivity was then observed. (14: 16% and 15: 20%, entries 3
and 4). Sodium hydroxide can also be used as a base (30 mol%), but with less efficiency as the
selectivity was worst (39% of 14 and 26% of 15, 1,3 diphenyl propan-1-ol, resulting from the
reduction of 14, was produced in 33% GC yield (Entry 5). The decrease of the amount of NaOH
to 10% suppressed the production of the 1,3 diphenyl propan-1-ol and led to moderate activity
(entry 6). Cs2CO3 was then selected as the base for the scope of the reactivity.
Table 4. Optimization with different bases.[a]
Entry 16 (mol%) Base Yield 14 (%)[b] Yield 15 (%)[b] 1 5 Cs2CO3 (30) 62 38
2 2 Cs2CO3 (10) 74 20
3 5 K2CO3 (30) 55 32
4 5 K2CO3 (10) 16 20
5 [c] 5 NaOH (30) 39 26
6 2 NaOH (10) 50 12 [a] acetophenone (0.5 mmol), benzylalcohol (0.75 mmol, 1.5 equiv.), [Fe] (2 mol%), base (10-30 mol%), solvent (1 mL), 140 °C, 24 h. [b] The yield was determined by GC. [c] 33% 1,3-diphenylpropan-1-ol was obtained.
Finally, a significant breakthrough was obtained using a catalytic system in situ generated from
the Knölker complex 16 (2 mol%) and PPh3 (2 mol%) at 140 oC under similar conditions than in
the entry 8, and 80% of the -alkylated product 14 was obtained, with only 6% of 15 (Table 2,
entry 10). Obviously, no coupling occurred in the absence of iron catalyst or Cs2CO3 as the base,
or when the reaction was performed in neat conditions (Table 2, entries 11-13).
Interestingly, the nature of the phosphine is also important: with 2 mol% of P(o-Tol)3 or
P(2-methylfuryl)3, similar results were achieved even if 1,3 diphenyl propan-1-ol was obtained in
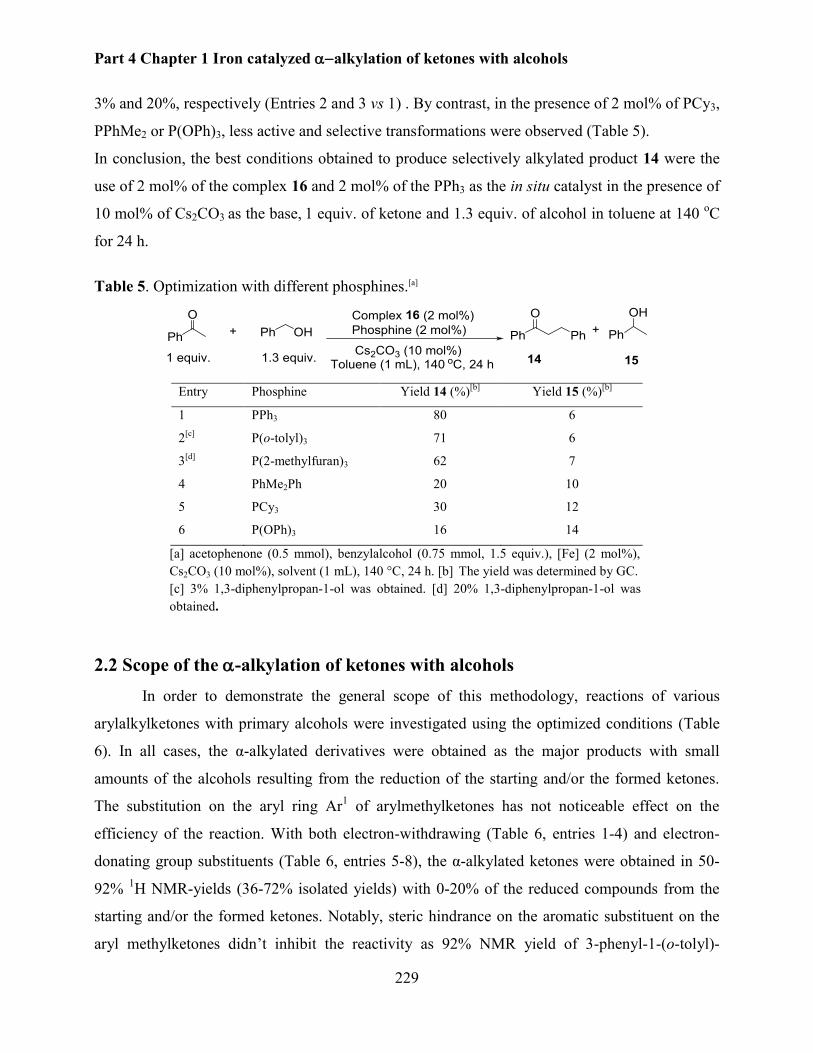
Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
229
3% and 20%, respectively (Entries 2 and 3 vs 1) . By contrast, in the presence of 2 mol% of PCy3,
PPhMe2 or P(OPh)3, less active and selective transformations were observed (Table 5).
In conclusion, the best conditions obtained to produce selectively alkylated product 14 were the
use of 2 mol% of the complex 16 and 2 mol% of the PPh3 as the in situ catalyst in the presence of
10 mol% of Cs2CO3 as the base, 1 equiv. of ketone and 1.3 equiv. of alcohol in toluene at 140 oC
for 24 h.
Table 5. Optimization with different phosphines.[a]
Entry Phosphine Yield 14 (%)[b] Yield 15 (%)[b]
1 PPh3 80 6
2[c] P(o-tolyl)3 71 6
3[d] P(2-methylfuran)3 62 7
4 PhMe2Ph 20 10
5 PCy3 30 12
6 P(OPh)3 16 14
[a] acetophenone (0.5 mmol), benzylalcohol (0.75 mmol, 1.5 equiv.), [Fe] (2 mol%), Cs2CO3 (10 mol%), solvent (1 mL), 140 °C, 24 h. [b] The yield was determined by GC. [c] 3% 1,3-diphenylpropan-1-ol was obtained. [d] 20% 1,3-diphenylpropan-1-ol was obtained.
2.2 Scope of the -alkylation of ketones with alcohols In order to demonstrate the general scope of this methodology, reactions of various
arylalkylketones with primary alcohols were investigated using the optimized conditions (Table
6). In all cases, the α-alkylated derivatives were obtained as the major products with small
amounts of the alcohols resulting from the reduction of the starting and/or the formed ketones.
The substitution on the aryl ring Ar1 of arylmethylketones has not noticeable effect on the
efficiency of the reaction. With both electron-withdrawing (Table 6, entries 1-4) and electron-
donating group substituents (Table 6, entries 5-8), the α-alkylated ketones were obtained in 50-
92% 1H NMR-yields (36-72% isolated yields) with 0-20% of the reduced compounds from the
starting and/or the formed ketones. Notably, steric hindrance on the aromatic substituent on the
aryl methylketones didn’t inhibit the reactivity as 92% NMR yield of 3-phenyl-1-(o-tolyl)-

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
230
propan-1-one was obtained starting from 2’-methyl acetophenone. Nevertheless, starting with the
most hindered 2’,4’,6’-trimethylacetophenone, only 59% of 1-mesityl-3-phenylpropan-1-one 20i
was obtained as the sole product. Additionally, no dehalogenation was observed when starting
from p-fluoro-, p-chloro- and p-bromo-acetophenone.
α-Tetralone in the presence of benzyl alcohol led specifically to the α-alkylated α-tetralone 20q in
50 % isolated yield after 48 h in the presence of t-BuOK used as the base. When Cs2CO3 was
used as a base, no coupling product was formed. Similarly, there was no significant effect of the
substitution of the aryl ring R2 of the benzyl alcohol derivatives. Consequently, alcohols with
both electron-donating (Table 6, entries 11 and 12) and electron-withdrawing substituents (Table
6, entries 13 and 14) gave the corresponding α-alkylated ketones in 51-62% isolated yields.
Furthermore, bio-based and synthetically more interesting alcohols such as butan-1-ol and 3-
phenylpropan-1-ol can be used and led to the corresponding ketones in 55 and 42% isolated
yields (Table 6, entries 19 and 20). In contrast, methanol was, unfortunately, not suitable for this
transformation, probably due to the more difficult dehydrogenation reaction of this alcohol.
Gratifyingly with respect to organic synthesis, heteroaromatic methylketones such as 3-
pyridylmethylketone or heteroaromatic alcohols such as 2-thienylethanol and 2-furylethanol were
performed well in this reaction (Table 6, entries 22 and 23).
3. Friedländer annulation reaction
3.1 Introduction Using this general hydrogen borrowing concept, iron-catalyzed methodology can be used for the
synthesis of quinoline derivatives. Understandably, the quinoline ring system is present in a
number of natural and synthetic products often endowed with interesting pharmacological or
physical properties.[62] Consequently, a number of methods for the synthesis of quinolines have
been reported.
Scheme 18. Friedländer annulation reaction.
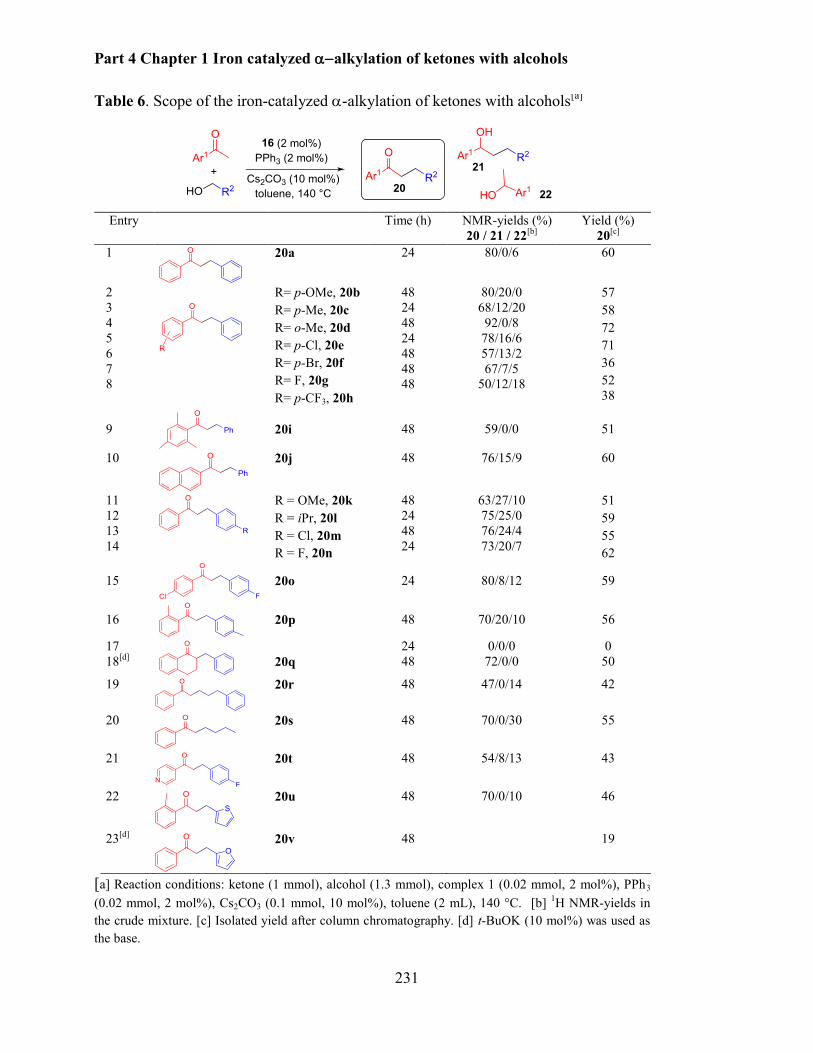
Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
231
Table 6. Scope of the iron-catalyzed -alkylation of ketones with alcohols[a]
Entry Time (h) NMR-yields (%)
20 / 21 / 22[b] Yield (%)
20[c] 1
20a 24 80/0/6 60
2 3 4 5 6 7 8
R= p-OMe, 20b R= p-Me, 20c R= o-Me, 20d R= p-Cl, 20e R= p-Br, 20f R= F, 20g R= p-CF3, 20h
48 24 48 24 48 48 48
80/20/0 68/12/20 92/0/8
78/16/6 57/13/2 67/7/5
50/12/18
57 58 72 71 36 52 38
9
20i 48 59/0/0 51
10
20j 48 76/15/9 60
11 12 13 14
R = OMe, 20k R = iPr, 20l R = Cl, 20m R = F, 20n
48 24 48 24
63/27/10 75/25/0 76/24/4 73/20/7
51 59 55 62
15
20o 24 80/8/12 59
16
20p 48 70/20/10 56
17 18[d]
20q
24 48
0/0/0 72/0/0
0 50
19
20r 48 47/0/14 42
20
20s 48 70/0/30 55
21
20t 48 54/8/13 43
22
20u 48 70/0/10 46
23[d]
20v 48 19
[a] Reaction conditions: ketone (1 mmol), alcohol (1.3 mmol), complex 1 (0.02 mmol, 2 mol%), PPh3 (0.02 mmol, 2 mol%), Cs2CO3 (0.1 mmol, 10 mol%), toluene (2 mL), 140 °C. [b] 1H NMR-yields in the crude mixture. [c] Isolated yield after column chromatography. [d] t-BuOK (10 mol%) was used as the base.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
232
Among them, the Friedländer reaction is still one of the simplest methods. Based on the classic
Friedländer annulation reaction, quinolines can be prepared in a straightforward way from 2-
aminobenzaldehyde and various ketones.[63] Generally, self-aldol condensation by-products and
the low stability of 2-aminobenzaldehyde are some of the challenges of this reaction. Indeed
using the more stable 2-aminobenzylalcohol in the presence of a catalytic amount of base under
hydrogen borrowing conditions is obviously an advantage to perform this reaction compared to
the reactions in the presence of stoichiometric amounts of base which can promote self aldol
condensation reactions.
In 2001, Cho and Sim reported that 2-aminobenzyl alcohol can be oxidatively cyclised in
the presence of ketones in high yields (Scheme 19). In the presence of 1 mol% of
RuCl2(=CHPh)(PCy3)2 as the catalyst and 1 equiv. of potassium hydroxide at 80 oC, the reaction
showed a nice scope with various ketones.[64]
Scheme 19. Ru catalyzed synthesis of quinolines.
In 2005, Ishii showed that the different quinolone derivatives were synthesized in good
yields starting from 2-aminobenzyl alcohol and ketones in the presence of catalytic amounts of an
iridium complex such as [IrCl(cod)]2 (1 mol%), PPh3 (4 mol%) and KOH (20 mol%) under
solvent free conditions (8 examples, 4-75% isolated yields). [65]
In 2006, Cho and Sim reported again a Friedländer annulation reaction from 2-
aminobenzyl alcohol with an array of ketones in dioxane at 100 oC in the presence of a catalytic
amount of CuCl2 (10 mol%), 3 equiv. of KOH under 1 atm of O2 at 100 oC to afford the
corresponding quinolines in good yields.[66] In 2009, Cho and co-workers reported that reusuable
CuCl2/molecular sieves 4Å or PEG-2000 catalytic system could able to promote Friedländer
synthesis to give quinolines in good yields. The copper catalytic system could be easily recovered
from the reaction mixture and reused 10 times without any loss of catalytic activity.[67]
In 2007, Ramon and Yus reported the synthesis of polysubstituted quinoline derivatives
from aromatic or aliphatic alcohols in the presence of RuCl2(dmso)4 as the catalyst under solvent-
free conditions.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
233
Scheme 20. Synthesis of quinoline derivatives from amino ketones and alcohols.
A few years later, in 2008, the second-generation Grubbs catalyst was found to be even an
active catalyst for a modification of the Friedländer reaction (Scheme 21). This catalytic system
(1 mol%) in combination with KOtBu as a base gave good to excellent yields for the preparation
of a variety of quinoline derivatives at 80 °C for 1 h. Notably, the presence of a hydrogen
acceptor is required to regenerate the catalyst species.
Scheme 21. Synthesis of quinoline derivatives catalyzed by a 2nd generation Grubbs’ catalyst.
Ying described that Ag-Pd alloy nanoparticles supported on carbon catalyzed the effective
synthesis of polysubstitited quinolines through a one-pot, two-step tandem reaction. In the first
step, the nanoparticles catalyzed the alkylation of ketones with primary alcohols through a
hydrogen borrowing process. Interestingly, this -alkylated ketones again reacted with 2-
aminobenzyl alcohols in a modified Friedländer synthesis to give polysubstituted quinolines.[68]
Scheme 22. One pot synthesis of polysubstituted quinoline.
Recently, using the combination of Ru3(CO)12 (1 mol%) and dppf (3 mol%) a convenient
synthesis of quinolines has been established (Scheme 23). Different 2-nitroaryl benzyl alcohols
reacted with a variety of alcohols via a hydrogen-transfer strategy leading to various substituted
quinoline products in moderate to good yields (40-81%) at 150 oC for 18 h.[69]

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
234
Scheme 23. Synthesis of quinoline from nitro aryl alcohols.
In 2013, Milstein reported that bipyridyl-based ruthenium pincer complex catalyzed one-
step synthesis of substituted pyridine- and quinolone derivatives by acceptorless dehydrogenative
coupling of -aminoalcohols with secondary alcohols in the presence of a base (Scheme 24). The
reaction involves consecutive C–N and C–C bond formation.
Scheme 24. Ru catalyzed synthesis of pyridines and quinoline from amino alcohols.
3.2. Iron catalyzed synthesis of quinolone derivatives Based on the background of the reported Friedländer synthesis, using the optimal
conditions developed for α-alkylation of ketones (2 mol% of the complex 16, 2 mol% of PPh3, 10
mol% of Cs2CO3, in toluene at 140 °C), 2-aminobenzylalcohol 23 (1.3 equiv.) reacted with 1
equiv. of acetophenone to give the corresponding quinoline derivative 24 in 63% isolated yield.
The only other product detected in this sequence being 2-phenylethanol resulting for the
reduction of acetophnone (Scheme 25).
Scheme 25. Iron-catalyzed modified Friedländer annulation reaction. [a] Isolated yields. [b] 30
mol% of t-BuOK was used.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
235
Using t-BuOK (10 mol%) as the base permitted to isolate 65% of 24a. In addition,
2-aminobenzylalcohol 23 reacted with p-OMe- and p-Cl-substituted acetophenones to afford the
corresponding quinolines 24b and 24c in 55 and 67% yields, respectively. Even if we obtained
comparable results in the case of acetophenone with Cs2CO3 and KOtBu (10 mol%) [Cs2CO3
:quinoline 24a (68%), 1-phenylethanol 15 (32%); KOtBu: 24a (66%), 15 (34%)], with substituted
acetophenone derivatives, KOtBu was more efficient [for example, 4-Cl-C6H4-COMe: Cs2CO3,
24a (35%), 15 (20%); KOtBu, 24a (72%), 15 (28%)]. Similarly, propiophenone yielded the
corresponding quinoline 24d in 56% yield. Finally, 5,6-dihydrobenzo[c]acridine 24e was
obtained in 65% starting from α-tetralone.
4. Conclusion The hydrogen autotransfer process is a more efficient and cleaner strategy than some
classical alkylation protocols, substituting hazardous and expensive alkyl halides, sulfonates, or
sulfates, strong bases, and extreme conditions by simple alcohols, metal hydroxides, and milder
conditions. Furthermore, the only waste generated through the overall process is water. Using this
methodology, we have developed the first iron-catalyzedalkylation of ketones with primary
alcohols using 2 mol% of the Knölker complex associate with 2 mol% of the PPh3 as the catalyst
in the presence of a catalytic amount of base (10 mol%). A variety of ketones were alkylated with
different alcohols. The optimized catalytic system permitted the development of the first iron-
catalyzed Friedländer annulation reaction starting from 2-aminobenzyl alcohols. Notably, this
method is not only of interest for organic synthesis, but also permits the green valorization of bio-
based alcohols.
5. Experimental part All reagents were obtained from commercial sources and used as received, except
acetophenone which was distilled prior to use. All reactions were carried out with flame-dried
glassware using standard Schlenk techniques under an inert atmosphere of dry argon. Toluene
and THF were dried over Braun MB-SPS-800 solvent purification system. Technical grade
pentane, diethylether, dichloromethane and methanol were used for column chromatography.
Analytical TLC was performed on Merck 60F254 silica gel plates (0.25 mm thickness). Column
chromatography was performed on Acros Organics Ultrapure silica gel (mesh size 40-60 μm,

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
236
60Å). 1H-NMR spectra were recorded in CDCl3 or CD3OD at ambient temperature on Bruker
AVANCE 300 and 400 spectrometers at 300.1, and 400.1 MHz respectively, using the solvent as
internal standard (CDCl3 7.26 ppm, CD3OD 3.31 ppm). 13C-NMR spectra were obtained at 75.5,
100.6 or 125.7 MHz and referenced to the internal solvent signals (CDCl3, central peak is 77.00
ppm, CD3OD central peak is 49.00 ppm). Chemical shifts (δ) and coupling constants (J) are given
in ppm and in Hz, respectively. The peak patterns are indicated as follows: (s, singlet; d, doublet;
t, triplet; q, quartet; o, octet; m, multiplet, and br. for broad). GC analyses were performed with
GC-2014 (Shimadzu) 2010 equipped with a 30-m capillary column (Supelco, SPBTM-20, fused
silica capillary column, 30 m × 0.25 mm × 0.25 mm film thickness), which was used with N2/air
as vector gas. The following GC conditions were used: initial temperature 80 °C, for 2 minutes,
then rate 10 °C/min. until 220 °C and 220 °C for 15 minutes.
GCMS were measured by GCMS-QP2010S (Shimadzu) with GC-2010 equipped with a 30 m
capillary column (Supelco, SLBTM-5ms, fused silica capillary column, 30 m × 0.25 mm × 0.25
mm film thickness), which was used with helium as vector gas. The following GC-MS conditions
were used: Initial temperature 100 °C, for 2 minutes, then rate 10 °C/min. until 250 °C and 250
°C for 10 minutes.
Typical procedure (A) for the iron catalyzed α -alkylation of ketones with primary alcohols
An oven-dried 10-mL Schlenk tube, equipped with a stirring bar, was charged with acetophenone
(119 mg, 1 mmol), alcohol (140 mg, 1.3 mmol), iron complex (8.4 mg, 0.02 mmol), PPh3 (5.2 mg
0.02 mmol,), Cs2CO3 (32.4 mg, 0.1 mmol) and toluene (2 mL). Solid materials were weighed into
the Schlenk tube under air, and the Schlenk tube was subsequently connected to an argon line and
vacuum-argon exchange was performed three times. Liquid starting materials and solvent were
charged under an argon stream. The Schlenk tube was capped and the mixture was rapidly stirred
at room temperature for 2 min, then was placed into a pre-heated oil bath at 140 oC and stirred for
a given time. After the reaction, the reaction mixture was cooled to room temperature then diluted
with ethyl acetate and washed with brine solution and concentrated under reduced pressure. The
residue was purified by flash chromatography on silica gel (n-pentane/diethylether) to afford 60%
yield of the desired product.
Typical procedure (B) for the iron catalyzed α-alkylation of ketones with primary alcohols
An oven-dried 10 mL Schlenk tube, equipped with a stirring bar, was charged with acetophenone
(119 mg, 1 mmol), alcohol (160 mg, 1.3 mmol), iron complex (8.4 mg, 0.02 mmol), PPh3 (5.2 mg

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
237
0.02 mmol,), t-BuOK, (11.2 mg, 0.1 mmol) and toluene (2 mL). Solid materials were weighed
into the Schlenk tube under air, and the Schlenk tube was subsequently connected to an argon
line and vacuum-argon exchange was performed three times. Liquid starting materials and
solvent were charged under an argon stream. The Schlenk tube was capped and the mixture was
rapidly stirred at room temperature for 2 min, then was placed into a pre-heated oil bath at 140 oC
and stirred for a given time. After completion of the reaction, the reaction mixture was cooled to
room temperature then diluted with ethyl acetate and washed with brine solution and
concentrated under reduced pressure. The residue was purified by flash chromatography on silica
gel (n-pentane/diethylether) to afford 65% yield of the desired product.
5.1 NMR characterization data for the alkylation products
1,3-Diphenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 126 mg, 60% isolated yield). White solid. 1H NMR (400 MHz, CDCl3) δ: 8.05 – 7.93 (m, 2H), 7.58 (t, J = 7.4, 1H), 7.48 (t, J = 7.6, 2H), 7.29 (m, 5H), 3.38 – 3.28 (m, 2H), 3.10 (t, J = 7.7, 2H). 13C NMR (100 MHz, CDCl3) δ: 199.1, 141.2, 136.8, 133.0, 128.5, 128.5, 128.4, 128.0, 126.1, 40.4, 30.1. 1-(4-Methoxyphenyl)-3-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 137 mg, 57% isolated yield). Pale yellow solid. 1H NMR (400 MHz, CDCl3) δ: 7.96 (d, J = 8.9, 2H), 7.31 – 7.22 (m, 5H), 6.94 (d, J = 8.9, 2H), 3.88 (s, 3H), 3.31 – 3.23 (m, 2H), 3.11 – 3.04 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 197.8, 163.4, 141.4, 130.2, 129.9, 128.4, 128.4, 126.0, 113.7, 55.4, 40.1, 30.3. 3-Phenyl-1-(p-tolyl)propan-1-one
The compound was prepared as described in the general procedure A (m = 130 mg, 58% isolated yield). White solid.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
238
1H NMR (400 MHz, CDCl3) δ: 7.82 (d, J=8.1, 2H), 7.30 – 7.14 (m, 7H), 3.28 – 3.19 (m, 2H), 3.02 (t, J=7.7, 2H), 2.36 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 198.8, 143.7, 141.6, 134.3, 129.2, 128.4, 128.4, 128.1, 126.0, 40.3, 30.2, 21.5. 3-Phenyl-1-(o-tolyl)propan-1-one
The compound was prepared as described in the general procedure A (m = 162 mg, 72% isolated yield). Colorless liquid. 1H NMR (400 MHz, CDCl3) δ: 7.58 – 7.46 (m, 1H), 7.32 – 7.09 (m, 8H), 3.15 (t, J = 7.6, 2H), 2.98 (t, J = 7.6, 2H), 2.41 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 203.2, 141.1, 137.9, 137.8, 131.8, 131.1, 128.4, 128.3, 128.2, 126.0, 125.5, 43.0, 30.2, 21.1. 1-Mesityl-3-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 129 mg, 51% isolated yield). Colorless liquid. 1H NMR (400 MHz, CDCl3) δ: 7.41 – 7.26 (m, 5H), 6.89 (s, 2H), 3.14-3.07 (m , 4H), 2.34 (s, 3H), 2.20 (s, 6H). 13C NMR (100 MHz, CDCl3) δ: 209.5, 140.8, 139.4, 138.2, 132.4, 128.4, 128.3, 126.0, 46.2, 29.4, 20.9, 18.9. 1-(4-Chlorophenyl)-3-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 173 mg, 71% isolated yield). Yellow solid. 1H NMR (400 MHz, CDCl3) δ: 7.82 (d, J = 8.6, 2H), 7.35 (d, J = 8.6, 2H), 7.25 – 7.14 (m, 5H), 3.20 (t, J = 7.6, 2H), 3.00 (t, J = 7.6, 2H). 13C NMR (75 MHz, CDCl3) δ: 197.8, 141.0, 139.4, 135.1, 129.4, 128.8, 128.5, 128.3, 126.1, 40.3, 30.0. 1-(4-Bromophenyl)-3-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 105 mg, 36% isolated yield). Yellow solid.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
239
1H NMR (400 MHz, CDCl3) δ: 7.73 (d, J = 8.4, 1H), 7.51 (d, J = 8.4, 1H), 7.19 (m, 3H), 3.19 (t, J = 7.6, 1H), 2.98 (t, J = 7.6, 1H). 13C NMR (100 MHz, CDCl3) δ: 198.0, 141.0, 135.5, 131.8, 129.5, 128.5, 128.3, 126.1, 40.3, 30.0. 1-(4-Fluorophenyl)-3-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 120 mg, 52% isolated yield). Colorless liquid. 1H NMR (400 MHz, CDCl3) δ: 8.00-7.96 (m, 2H), 7.37 – 7.17 (m, 5H), 7.13-7.09 (t, m, 2H), 3.27 (t, J= 7.7, 2H), 3.08 (d, J = 7.9, 2H). 13C NMR (100 MHz, CDCl3) δ: 197.4, 166.9 (d, J = 254.7 Hz), 141.0, 133.2 (d, J = 3.0 Hz), 130.6 (d, J = 9.3 Hz), 129.0, 128.3, 126.1, 115.7 (d, J = 21.4 Hz), 40.2, 30.0. 3-Phenyl-1-(4-(trifluoromethyl)phenyl)propan-1-one
The compound was prepared as described in the general procedure A (m = 105 mg, 38% isolated yield). Orange solid. 1H NMR (400 MHz, CDCl3) δ: 8.05 (d, J=8.2, 2H), 7.72 (d, J = 8.3, 2H), 7.34 – 7.23 (m, 5H), 3.33 (t, J = 7.6, 2H), 3.09 (t, J = 7.6, 2H). 13C NMR (100 MHz, CDCl3) δ: 198.1, 140.8, 139.4, 134 (q, J=32), 128.6, 128.4, 128.3, 126.2, 125.7, 123.6 (q, J = 271), 40.7, 29.9. 2-Benzyl-3,4-dihydronaphthalen-1(2H)-one
The compound was prepared as described in the general procedure A (m = 117 mg, 50% isolated yield). Gummy solid. 1H NMR (400 MHz, CDCl3) δ: 8.12 (d, J = 7.7, 1H), 7.48 (dd, J = 7.4, 0.9, 1H), 7.34 (t, J = 7.5, 3H), 7.30 – 7.22 (m, 4H), 3.56-3.52 (m, 1H), 2.98-2.95 (m, 2H), 2.82 – 2.73 (m, 1H), 2.72-2.69 (m, 1H), 2.21 – 2.09 (m, 1H), 1.90 – 1.74 (m, 1H). 13C NMR (100 MHz, CDCl3) δ: 199.2, 143.9, 139.9, 133.1, 132.4, 129.1, 128.6, 128.3, 127.4, 126.5, 126.0, 49.3, 35.6, 28.5, 27.6. 1-(Naphthalen-2-yl)-3-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 155 mg, 60% isolated yield). Yellow solid; 1H NMR (400 MHz, CDCl3) δ: 8.40 (s, 1H), 7.99 (dd, J = 8.6, 1.6, 1H), 7.91

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
240
– 7.76 (m, 3H), 7.51 (m, 2H), 7.30 – 7.17 (m, 5H), 3.45 – 3.32 (m, 2H), 3.13 – 3.03 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 199.0, 141.3, 135.5, 134.1, 132.4, 129.6, 129.4, 128.5, 128.4, 128.4, 128.3, 127.7, 126.7, 126.1, 123.7, 40.4, 30.2. 3-(4-Fluorophenyl)-1-(pyridin-4-yl)propan-1-one
The compound was prepared as described in the general procedure A (m = 98 mg, 43% isolated yield). White crystal. 1H NMR (400 MHz, CDCl3) δ: 8.79 (d, J = 5.5, 1H), 7.80 – 7.61 (m, 1H), 7.19 (dd, J = 8.4, 5.5, 1H), 6.97 (t, J = 8.7, 1H), 3.27 (t, J=7.4, 1H), 3.04 (t, J = 7.4, 1H). 13C NMR (100 MHz, CDCl3) δ: 198.3, 161.5 (d, J = 242.0), 151.0, 142.5, 136.2 (d, J = 3.0), 129.8 (d, J = 8.0), 120.9, 115.3 (d, J = 21.0), 40.7, 28.8. 3-(4-Methoxyphenyl)-1-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 122 mg, 51% isolated yield). White solid. 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J = 7.3, 2H), 7.61 – 7.39 (m, 3H), 7.21 (t, J = 16.7, 2H), 6.86 (d, J = 8.6, 2H), 3.79 (s, 3H), 3.28 (t, J = 7.6, 2H), 3.03 (t, J = 7.6, 2H). 13C NMR (100 MHz, CDCl3) δ: 199.2, 157.8, 136.8, 133.2, 132.9, 129.2, 128.4, 127.9, 113.8, 55.1, 40.5, 29.1. 1-(o-Tolyl)-3-(p-tolyl)propan-1-one
The compound was prepared as described in the general procedure A (m = 133 mg, 56% isolated yield). Colorless liquid. 1H NMR (400 MHz, CDCl3) δ: 7.68 – 7.60 (m, 1H), 7.40 (dd, J = 10.8, 4.1, 1H), 7.28 (t, J = 6.8, 2H), 7.22 – 7.12 (m, 4H), 3.24 (d, J = 8.0, 2H), 3.07 (d, J = 7.9, 2H), 2.53 (s, 3H), 2.37 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 203.3, 138.0, 137.9, 137.8, 135.4, 131.8, 131.1, 129.1, 128.3, 128.2, 125.5, 43.3, 29.8, 21.1, 20.9. 3-(4-Isopropylphenyl)-1-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 148 mg, 59% isolated yield). Yellow liquid.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
241
1H NMR (400 MHz, CDCl3) δ: 8.09 – 7.90 (m, 2H), 7.57 (t, J = 7.4, 1H), 7.47 (t, J = 7.6, 2H), 7.20-7.17 (m, 4H), 3.41 – 3.21 (m, 2H), 3.14 – 3.00 (m, 2H), 2.94-2.87 (m, 1H), 1.27 (d, J = 6.9, 6H). 13C NMR (75 MHz, CDCl3) δ: 199.2, 146.6, 138.5, 136.9, 132.9, 128.5, 128.3, 128.0, 126.5, 40.4, 33.6, 29.7, 24.0. 3-(4-Chlorophenyl)-1-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 134 mg, 55% isolated yield). Yellow solid. 1H NMR (400 MHz, CDCl3) δ: 8.13 – 7.89 (m, 2H), 7.63 (t, J = 7.4, 1H), 7.52 (t, J = 7.6, 2H), 7.35 – 7.30 (m, 2H), 7.25 (d, J = 8.4, 2H), 3.35 (t, J = 7.5, 2H), 3.11 (t, J=7.5, 2H). 13C NMR (100 MHz, CDCl3) δ: 198.7, 139.6, 136.6, 133.0, 131.7, 129.7, 128.5, 128.5, 127.9, 40.0, 29.2. 3-(4-Fluorophenyl)-1-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 142 mg, 62% isolated yield). Yellow solid. 1H NMR (400 MHz, CDCl3) δ: 7.97 – 7.94 (m, 2H), 7.56 (t, J = 7.4, 1H), 7.46 (t, J = 7.6, 2H), 7.21 (dd, J = 8.4, 5.5, 2H), 6.98 (t, J = 8.7, 2H), 3.28 (t, J = 7.6, 2H), 3.05 (t, J = 7.5, 2H). 13C NMR (75 MHz, CDCl3) δ: 199.0, 161.4 (d, J = 242), 136.8, 133.1, 129.8, (d, J = 7.5), 128.6, 128.0, 115.2 (d, J = 21), 40.4, 29.2 1-(4-Chlorophenyl)-3-(4-fluorophenyl)propan-1-one
The compound was prepared as described in the general procedure A (m = 156 mg, 59% isolated yield). Yellow solid. 1H NMR (400 MHz, CDCl3) δ: 7.88 (d, J=8.6, 1H), 7.42 (d, J=8.6, 1H), 7.19 (dd, J=8.5, 5.5, 1H), 6.97 (t, J=8.7, 1H), 3.24 (t, J=7.5, 1H), 3.04 (t, J=7.5, 1H). 13C NMR (101 MHz, CDCl3) δ: 197.7, 161.4 (d, J=243), 139.6, 136.6 (d, J=3), 135.1, 129.7 (d, J=8.0), 129.4, 128.9, 115.2 (d, J=21), 115.1, 40.4, 29.2. 1-Phenylhexan-1-one

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
242
The compound was prepared as described in the general procedure A (m = 99 mg, 55% isolated yield). Colorless liquid. 1H NMR (400 MHz, CDCl3) δ: 7.96 (d, J=7.4, 2H), 7.55 (t, J=7.3, 1H), 7.45 (t, J=7.6, 2H), 2.96 (t, J=7.4, 2H), 1.75 (dq, J=14.7, 7.3, 2H), 1.37 (dq, J=7.1, 3.6, 4H), 0.91 (t, J=7.0, 3H). 13C NMR (100 MHz, CDCl3) δ: 200.5, 137.1, 132.8, 128.5, 128.0, 38.5, 31.5, 24.0, 22.5, 13.9. 1,5-Diphenylpentan-1-one
The compound was prepared as described in the general procedure A (m = 98 mg, 42% isolated yield). Colorless liquid. 1H NMR (400 MHz, CDCl3) δ = 7.95 (d, J=7.3, 2H), 7.56 (t, J=7.3, 1H), 7.46 (t, J=7.6, 2H), 7.28 (dd, J=12.8, 5.6, 2H), 7.24 – 7.15 (m, 3H), 3.00 (t, J=7.1, 2H), 2.68 (t, J=7.4, 2H), 1.88 – 1.67 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 200.2, 142.2, 137.0, 132.8, 128.5, 128.3, 128.2, 127.9, 125.7, 38.3, 35.7, 31.0, 23.9. 3-(Furan-2-yl)-1-phenylpropan-1-one
The compound was prepared as described in the general procedure A (m = 38 mg, 19% isolated yield). Brown liquid. 1H NMR (400 MHz, CDCl3) δ : 7.98 (d, J=7.5, 2H), 7.57 (t, J=7.3, 1H), 7.47 (t, J=7.6, 2H), 7.31 (s, 1H), 6.29 (d, J=2.4, 1H), 6.06 (d, J=2.6, 1H), 3.34 (t, J=7.5, 2H), 3.10 (t, J=7.5, 2H). 13C NMR (100 MHz, CDCl3) δ: 198.6, 154.7, 141.0, 136.7, 133.1, 128.6, 128.0, 110.2, 105.3, 36.9, 22.5. 3-(Thiophen-2-yl)-1-(o-tolyl)propan-1-one
The compound was prepared as described in the general procedure A (m = 105 mg, 46% isolated yield). Colorless liquid. 1H NMR (400 MHz, CDCl3) δ: 7.65 (d, J=7.4, 1H), 7.40 (t, J=7.1, 1H), 7.28 (t, J=6.7, 2H), 7.15 (dd, J=5.1, 0.6, 1H), 6.94 (dd, J=5.0, 3.5, 1H), 6.87 (d, J=3.2, 1H), 3.31 (s, 4H), 2.52 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 202.5, 143.8, 138.2, 137.7, 132.0, 131.3, 128.4, 126.8, 125.7, 124.7, 123.4, 43.2, 24.4, 21.3.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
243
2-Phenylquinoline
The compound was prepared as described in the general procedure B (m = 134 mg, 65% isolated yield). Yellow solid. 1H NMR (400 MHz, CDCl3) δ: 8.21 (dd, J=8.3, 7.3, 4H), 7.85 (dd, J=19.3, 8.4, 2H), 7.74 (t, J=7.4, 1H), 7.64 – 7.41 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 157.2, 148.2, 139.6, 136.6, 129.6, 129.5, 129.2, 128.7, 127.5, 127.3, 127.1, 126.1, 118.9. 2-(4-Methoxyphenyl)quinoline
The compound was prepared as described in the general procedure B (m = 130 mg, 55% isolated yield). Pale brown solid. 1H NMR (400 MHz, CDCl3) δ: 8.15 (dd, J=13.5, 5.7, 4H), 7.80 (t, J=8.8, 2H), 7.72 (d, J=7.4, 1H), 7.49 (t, J=7.4, 1H), 7.05 (d, J=8.5, 2H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.7, 156.7, 148.2, 136.5, 132.1, 129.4, 129.4, 128.8, 127.3, 126.8, 125.8, 118.4, 114.1, 55.2. 2-(4-Chlorophenyl)quinoline
The compound was prepared as described in the general procedure B (m = 161 mg, 67% isolated yield). White solid. 1H NMR (400 MHz, CDCl3) δ: 8.26 – 8.07 (m, 4H), 7.83 (d, J=8.5, 2H), 7.74 (t, J=7.6, 1H), 7.60 – 7.44 (m, 3H). 13C NMR (100 MHz, CDCl3) δ: 155.9, 148.2, 138.0, 136.9, 135.5, 129.8, 129.6, 128.9, 128.7, 127.4, 127.1, 126.4, 118.5. 3-Methyl-2-phenylquinoline
The compound was prepared as described in the general procedure B (m = 122 mg, 56% isolated yield). Yellow liquid. 1H NMR (400 MHz, CDCl3) δ: 8.15 (d, J=8.5, 1H), 8.01 (s, 1H), 7.78 (d, J=8.1, 1H), 7.74 – 7.63 (m, 1H), 7.64 – 7.57 (m, 2H), 7.56 – 7.39 (m, 4H), 2.47 (s, 3H). 13C NMR (100 MHz, CDCl3) δ: 160.4, 146.5, 140.8, 136.6, 129.2, 129.1, 128.8, 128.6, 128.2, 128.1, 127.5, 126.6, 126.3, 20.5.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
244
5,6-Dihydrobenzo[c]acridine
The compound was prepared as described in the general procedure B (m = 149 mg, 65% isolated yield). Yellow solid. 1H NMR (400 MHz, CDCl3) δ: 8.64 (d, J=7.3, 1H), 8.19 (d, J=8.4, 1H), 7.93 (s, 1H), 7.77 (d, J=8.1, 1H), 7.73 – 7.63 (m, 1H), 7.56 – 7.37 (m, 3H), 7.35 – 7.26 (m, 1H), 3.19 – 3.11 (m, 2H), 3.09 – 3.00 (m, 2H). 13C NMR (100 MHz, CDCl3) δ: 153.3, 147.5, 139.3, 134.6, 133.6, 130.5, 129.6, 129.3, 128.5, 127.8, 127.8, 127.2, 126.8, 126.1, 125.9, 28.7, 28.3. 6. References
[1] P. N. Rylander, in in Catalytic Hydrogenation in Organic Syntheses, Academic Press, New York, 1979.
[2] A. J. A. Watson, J. M. J. Williams, Science 2010, 329, 635-636. [3] J. Leonard, A. J. Blacker, S. P. Marsden, M. F. Jones, K. R. Mulholland, R. Newton, Org.
Process Res. Dev. 2015, 19, 1400-1410. [4] Q. Yang, Q. Wanga, Z. Yu, Chem. Soc. Rev. 2015, 44, 2305-2329. [5] W. Zuo, A. J. Lough, Y. F. Li, R. H. Morris, Science 2013, 342, 1080-1083. [6] S. Werkmeister, C. Bornschein, K. Junge, M. Beller, Chem. Eur. J. 2013, 19, 4437-4440. [7] R. V. Jagadeesh, G. Wienhofer, F. A. Westerhaus, A. E. Surkus, H. Junge, K. Junge, M.
Beller, Chem. Eur. J. 2011, 17, 14375-14379. [8] T. Hille, T. Irrgang, R. Kempe, Chem. Eur. J. 2014, 20, 5569-5572. [9] E. Balaraman, E. Khaskin, G. Leitus, D. Milstein, Nat. Chem. 2013, 5, 122-125. [10] X. Wang, M. Chen, F. Xiea, M. Zhang, RSC Adv. 2014, 4, 14744-14751. [11] R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit, N. Tongpenyai, J. Chem. Soc., Chem.
Commun. 1981, 611. [12] Y. Tsuji, R. Takeuchi, H. Ogawa, Y. Watanabe, Chem. Lett 1986, 293. [13] G. Guillena, D. J. Ramon, M. Yus, Angew. Chem. Int. Ed. 2007, 46, 2358-2364. [14] V. IpatiFe'v, N. Kliukvin, Ber. Dtsch. Chem. Ges. 1925, 58, 4-12. [15] V. N. Ipatieff, V. Haensel, J. Org. Chem. 1942, 7, 189-198. [16] P. Chabardes, F. Y. Querou (Rhone-Poulenc SA), [Chem. Abstr. 1970, 73, 61760]. 1969. [17] M. T. Reetz, Angew. Chem. Int. Ed. Engl. 1982, 21, 96-108. [18] G. E. Dobereiner, R. H. Crabtree, Chem. Rev. 2010, 110, 681-703. [19] S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem 2011, 3,
1853-1864. [20] Y. Obora, ACS Catal. 2014, 4, 3972-3981. [21] F. Huang, Z. Liu, Z. Yu, Angew. Chem. Int. Ed. 2016, 55, 865-872. [22] A. Haller, M. Guerbet, C. R. Hebd. Seances Acad. Sci. 1909, 129. [23] C. S. Cho, B. T. Kim, T.-J. Kim, S. C. Shim, Tetrahedron Lett. 2002, 43, 7987-7989. [24] R. Martínez, G. J. Brand, D. J. Ramón, M. Yus, Tetrahedron Lett. 2005, 46, 3683-3686. [25] R. Martínez, D. J. Ramón, M. Yus, Tetrahedron 2006, 62, 8988-9001. [26] C. S. Cho, J. Mol. Catal. A. 2005, 240, 55-60.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
245
[27] M. S. Kwon, N. Kim, S. H. Seo, I. S. Park, R. K. Cheedrala, J. Park, Angew. Chem. Int. Ed. 2005, 44, 6913-6915.
[28] Y. M. A. Yamada, Y. Uozumi, Org. Lett. 2006, 8 1375-1378. [29] K. Taguchi, H. Nakagawa, T. Hirabayashi, S. Sakaguchi, Y. Ishii, J. Am. Chem. Soc.
2004, 126, 72-73. [30] T. Kuwahara, T. Fukuyama, I. Ryu, Org. Lett. 2012, 14, 4703-4705. [31] C. Xu, X.-M. Dong, Z.-Q. Wang, X.-Q. Hao, Z. Li, L.-M. Duan, B.-M. Ji, M.-P. Song, J.
Organomet. Chem. 2012, 700, 214-218. [32] X. Chang, L. W. Chuan, L. Yongxin, S. A. Pullarkat, Tetrahedron Lett. 2012, 53, 1450–
1455. [33] S. Liao, K. Yu, Q. Li, H. Tian, Z. Zhang, X. Yu, Q. Xu, Org. Biomol. Chem. 2012, 2973–
2978. [34] O. O. Kovalenko, H. Lundberg, D. Hübner, H. Adolfsson, Eur. J. Org. Chem. 2014,
6639–6642. [35] D. Wang, K. Zhao, P. Ma, C. Xu, Y. Ding, Tetrahedron Lett. 2014, 55, 7233–7235. [36] X. Yu, Q. Y. Wang, Q. J. Wu, D. W. Wang, Rus. J. Gen. Chem. 2016, 86, 178-183. [37] C. Schlepphorst, B. Maji, F. Glorius, ACS Catal. 2016, 6, 4184−4188. [38] M. L. Buil, M. A. Esteruelas, J. Herrero, S. Izquierdo, I. M. Pastor, M. Yus, ACS Catal.
2013, 3, 2072−2075. [39] Y. Nishibayashi, I. Wakiji, Y. Ishii, S. Uemura, M. Hidai, J. Am. Chem. Soc. 2001, 123,
3393-3394. [40] N. Naveen, S. R. Koppolu, R. Balamurugan, Adv. Synth. Catal. 2015, 357, 1463–1473. [41] M. Qian, M. A. Liauw, G. Emig, Appl. Catal. A 2003, 238, 211-222. [42] S. Ogawa, Y. Obora, Chem. Commun. 2014, 50, 2491-2493. [43] F. Li, J. Ma, N. Wang, J. Org. Chem. 2014, 79, 10447-10455. [44] R. Wang, J. Ma, F. Li, J. Org. Chem. 2015, 80, 10769−10776.
[45] L. K. Chan, D. L. Poole, D. Shen, M. P. Healy, T. J. Donohoe, Angew. Chem. Int. Ed. 2014, 53, 761-765.
[46] I. Bauer, H.-J. Knölker, Chem. Rev. 2015, 115, 3170-3387. [47] K. Gopalaiah, Chem. Rev. 2013, 113, 3248-3296. [48] D. Bézier, J.-B. Sortais, C. Darcel, Adv. Synth. Catal. 2013, 355, 19-33. [49] B. Plietker, Iron Catalysis in Organic Chemistry., Wiley-VCH Verlag,Weinheim, 2008. [50] C. Darcel, J.-B. Sortais, S. Q. Duque in From C-H to C-C bonds: Cross Dehydrogenative
Coupling Vol 26 (Ed.: C.-J. Li), RSC Green Chemistry Series, London 2015, pp.67-92. [51] F. Jia, Z. Li, Org. Chem. Front. 2014, 1, 194-214. [52] R. Jana, T. P. Pathak, M. S. Sigman, Chem. Rev. 2011, 111, 1417-1492. [53] P. Knochel, T. Thaler, C. Diene, Isr. J. Chem. 2010, 50, 547-557. [54] W. M. Czaplik, M. Mayer, J. Cvengros, A. J. V. Wangelin, ChemSusChem 2009, 2, 396-
417. [55] B. D. Sherry, A. Fürstner, Acc. Chem. Res. 2008, 41, 1500-1511. [56] K. Junge, K. Schröder, M. Beller, Chem. Commun. 2011, 47, 4849-4859. [57] B. A. F. L. Bailly, S. P. Thomas, RSC Adv. 2011, 1435-1445. [58] R. H. Morris, Chem. Soc. Rev. 2009, 38, 2282-2291. [59] M. Peña-López, H. Neumann, M. Beller, ChemCatChem 2015, 7, 865-871. [60] S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C.
Holthausen, W. D. Jones, S. Schneider, ACS Catal. 2014, 4, 3994-4003. [61] A. Quintard, J. Rodriguez, Angew. Chem. Int. Ed. 2014, 53, 4044-4055.

Part 4 Chapter 1 Iron catalyzed alkylation of ketones with alcohols
246
[62] J. P. Michael, Nat. Prod. Rep. 1997, 14, 605-618. [63] J. Marco-Contelles, E. Pe´rez-Mayoral, A. Samadi, M. A. D. C. Carreiras, E. Soriano,
Chem. Rev. 2009, 109, 2652–2671. [64] C. S. Cho, B. T. Kim, T.-J. Kim, S. C. Shim, Chem. Commun. 2001, 2576-2577. [65] K. Taguchi, S. Sakaguchi, Y. Ishii, Tetrahedron Lett. 2005, 46, 4539-4542. [66] C. S. Cho, W. X. Renb, S. C. Shimb, Tetrahedron Lett. 2006, 47, 6781–6785. [67] C. S. Choa, W. X. Rena, N. S. Yoonb, J. Mol. Catal. A: Chem. 2009, 299, 177-120. [68] B. W. J. Chen, L. L. Chng, J. Yang, Y. Wei, J. Yang, J. Y. Ying, ChemCatChem 2013, 5,
277-283. [69] F. Xie, M. Zhang, M. Chen, W. Lv, H. Jiang, ChemCatChem 2015, 7, 349-353.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
247

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
248
Part 4 Chapter 2
Manganese catalyzed N-alkylation of amines with
alcohols

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
249

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
250
1. Introduction Amines and their derivatives are of significant importance for bulk and fine chemical
industry, agrochemicals, dyes and natural products.[1-2] Indeed, the role of amines in
pharmaceuticals is immense and the relative science of using amines as pharmaceutical active
drugs goes back at least a century. (Selected representative examples of pharmaceutically
important molecules are shown in Figure 1) Hence, the development of new and improved
methods for the selective preparation of amines is of continuing interest. There are a huge
number of powerful methods describing the synthesis of amines such as the classical nucleophilic
substitution, the catalytic Buchwald-Hartwig[3-5] and Ullmann[6-7] cross-coupling reactions,
hydroamination,[8-9] hydroaminomethylation,[10] reduction of nitriles,[11] and nitro compounds,[12-
13] or reductive amination of carbonyl derivatives.[14] Most of these well-known and routinely
used procedures usually require transition metal based catalysts, most of them being derived from
noble metals. Although these methods proved their high efficiency and versatility in numerous
examples, they often suffer from the co-production of considerable amounts of side products or
metallic waste, which is not compatible with sustainability issues.
Among the different methodologies used for the synthesis of amines, borrowing hydrogen
(auto transfer) methodologies are of increasing prominence in molecular synthesis and catalysis;
indeed, they represent prime examples of green chemistry and are nice examples of a highly atom
economical reactions.[15-21] In this area, heterogeneous catalysts are well-known to perform this
transformation by reaction at high temperature and pressure.[22-24] As a representative example,
alkylation of aliphatic amines can be catalyzed by Raney Ni,[25] alumina, silica, or
montmorillonite at temperatures greater than 200 oC.[26] Classically, these catalysts are applied in
industry for the preparation of methylamines from methanol on bulk scale. Unfortunately, most
of these materials needing drastic reaction conditions (250-500 °C) make them not suitable for
the synthesis of more advanced amines bearing sensible functional moieties, especially in life
science area.
Thus hydrogen borrowing process is based on first the oxidation of alcohols to carbonyl
derivatives and then reductive amination without external hydrogen source because alcohols play
Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
251
an important role as H2 donor. The catalyst will also play a crucial role as a shuttle of dihydrogen
during the redox process (Scheme 1)
Scheme 1. “Hydrogen autotransfer” technology in the alkylation of amines with alcohols.
Figure 1. Selected examples of amines containing pharmaceutical molecules.
The pioneering reports dealing with the N-alkylation of amines by alcohols in the
presence of homogeneous catalysts were reported at the beginning of the 1980´s by Watanabe
using 0.5-2 mol% of RuCl2(PPh3)3 as a catalyst at 180 °C for 5 h[27] and by Grigg with
RhH(PPh3)4 (5 mol%) in refluxing methanol.[28] Thus, a tremendous progress has been made in
this field using precious metal complexes. During the last decade, numerous applications were
reported,[16-17, 19, 29] in particular using iridium[30-34] and ruthenium[35-41] based catalysts for the

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
252
synthesis of amines using hydrogen auto transfer technology. In this line, asymmetric versions
were also reported for the synthesis of chiral amines.[42-46]
Similarly to hydrogenation or hydrosilylation areas, since less than one decade, earth
abundant transition metal based complexes started to be described in auto-transfer reaction, more
particularly in N-alkylation of amines which will be reported hereafter.
1.1 Iron catalyzed N-alkylation of amines with alcohols In 2010, Deng reported the use of the catalytic system based on FeCl2 (5 mol%) in the
presence of K2CO3 (20 mol%) for the successful N-alkylation of sulfonamides with an excess of
benzylic alcohols via borrowing hydrogen methodology at 135 oC for 20 h (Scheme 2).[47]
Scheme 2. Iron catalyzed N-alkylation of sulfonamides.
In 2014, Feringa and Barta reported an efficient and general iron catalyzed N-alkylation
of amines with alcohols by hydrogen auto transfer technology (Scheme 3).[48] The air stable
Knölker complex 2 was used as a pre-catalyst without addition of any base but in the presence of
trimethylamine N-oxide (Me3NO) which is required to activate the complex by decoordination of
one CO ligand. A variety of amines were alkylated using different type alcohols at 120-130 oC
using 5 mol% of catalyst. Noticeably, this catalytic system was also used efficiently for the
synthesis of Piribedil 3 which is used for the treatment of Parkinson’s disease.
Scheme 3. Iron catalyzed N-alkylation of amines with alcohols.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
253
Notably, when starting from diols and benzylamines, N-benzyl 5-, 6- or 7-membered
nitrogen-containing heterocycles were obtained with 60-85% yields. Later, the same group
developed the synthesis of various benzylamines through amination of benzyl alcohols using
borrowing hydrogen pathway.[49] Interestingly, pharmaceutically relevant compounds were
prepared by using renewable building block 2,5-furan-dimethanol.
Scheme 4. Iron catalyzed alkylation of amines with benzylalcohols.
In 2015, Wills reported the use of the iron-tetraphenylcyclopentadienone tricarbonyl
complex 4 as a pre-catalyst (10 mol%) in the association with 10 mol% of Me3NO for the N-
alkylation of aniline derivatives with various benzyl alcohols and alkanols at 110 oC in toluene
for 48 h or at 140 °C in xylene for 24 h (Scheme 5).[50]
Scheme 5. N-alkylation of amine with iron tetraphenylcyclopentadienone tricarbonyl complex 4.
In 2015, further improved method was developed by Zhao and coworkers using Lewis acid
assisted iron catalyzed amination with secondary alcohols (Scheme 6).[51] In particular, silver
fluoride (40 mol%) was used as an additive to increase the reaction rate in the amination of
secondary alkanols catalyzed by the Knӧlker’s complex 5, even if it is used in a high catalyst
loading (10 mol%).
Scheme 6. N-alkylation of amines with secondary alkanols.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
254
Using the same catalytic system (10 mol% of the complex 2, 20 mol% Me3NO ), Sundararaju and
coworkers reported an iron catalyzed N-allylation of amines using various allylic alcohols
(Scheme 7).[52] Using 10 mol% catalyst, Me3NO (20 mol%) at 130 oC and 24 h various
heterocyclic amines, benzylic, aliphatic and amines were alkylated by different allylic alcohols.
This methodolody allows for the synthesis of drugs such as Cinnarizine (antihistaminic) and
Nafetifine (antifungal).
Scheme 7. N-allylation of amines with allyllic alcohols.
1.2 Cobalt based catalytic system Zhang reported direct N-alkylation of both aromatic and aliphatic amines by primary
alcohols catalyzed by a PNP pincer cobalt complex 7 (2 mol%) under base-free conditions in the
presence of molecular sieves in refluxing toluene (Scheme 8). A range of primary alcohols and
primary amines including both aromatic and aliphatic substrates were efficiently converted to
secondary amines in good to excellent yields.[53] Notably, the reaction was less selective with
secondary alcohols and did work with tert-butanol. Interestingly, the simple addition of
molecular sieves permitted to selectively obtain the N-alkylated amines whereas in its absence,
the imine intermediates were obtained as the major products.
Scheme 8. Cobalt catalyzed N-alkylation of amines with alcohols.
Kempe also reported that the N-alkylation of (hetero)aniline derivatives with alcohols
catalyzed by Cobalt complexes stabilized by a pincer PN5P ligand under mild conditions (80 oC,

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
255
24 h) in the presence of 1.2 equivalent of KOtBu with 2 mol% of catalyst (Scheme 9). [54]
Notably, benzenediamine can be efficiently alkylated in a sequential manner with two different
alcohols.
Scheme 9. N-alkylation of anilines with alcohols catalyzed by 8.
2. N-alkylation of aniline using PNP pincer manganese complexes The PNP pincer manganese complexes C10 and C11 being able to efficiently perform
hydrogenation of carbonyl and carboxylic derivatives (see part 3),[55] the potential of PNP pincer
complexes were then explored in hydrogen borrowing reactions, more specifically the N-
alkylation of amines by alcohols.
2.1 Optimization of reaction parameters At the start of this study, four different manganese pincer complexes C10-C16 (Table 1)
were tested with aniline (1 equiv.) and benzyl alcohol (1.2 equiv.) as benchmark substrates.
Gratifyingly, in the presence of 2 mol% of C10 or C11 and 1 equiv. of t-BuOK in toluene at
80 °C for 24 h, N-benzylaniline 9a was obtained in 78% and 56% yields, respectively (Table 1,
entries 1 and 2). In contrast, the Mn(II) complex C13 exhibited less activity (11% yield, Table 1,
entry 3). With 2 mol% of the NNN bispyridine amino cationic pincer complex C16,[56] only 7%
of 9a was obtained. Noticeably, the commercially available manganese precursor Mn(CO)5Br
gave only 2% conversion, showing the importance of the PNP ligand on the efficiency of the
catalyst. It must be pointed out that the corresponding imine derivatives were the only observed
“by-products” formed with our described catalytic system.
To show the effect of the base in this alkylation reactions, different type of bases were
tested (Table 2). Potassium and sodium tert-butoxide led to 91 and 65% conversion of aniline and
the corresponding N-benzylaniline 9a was obtained in 78 and 45% GC yields, respectively
(Entries 1 and 2). While using Cs2CO3, KOH and NaOEt as the bases, only poor conversions of
aniline were observed (14-36%, entries 3-5). When NaOMe was used as a base, moderate
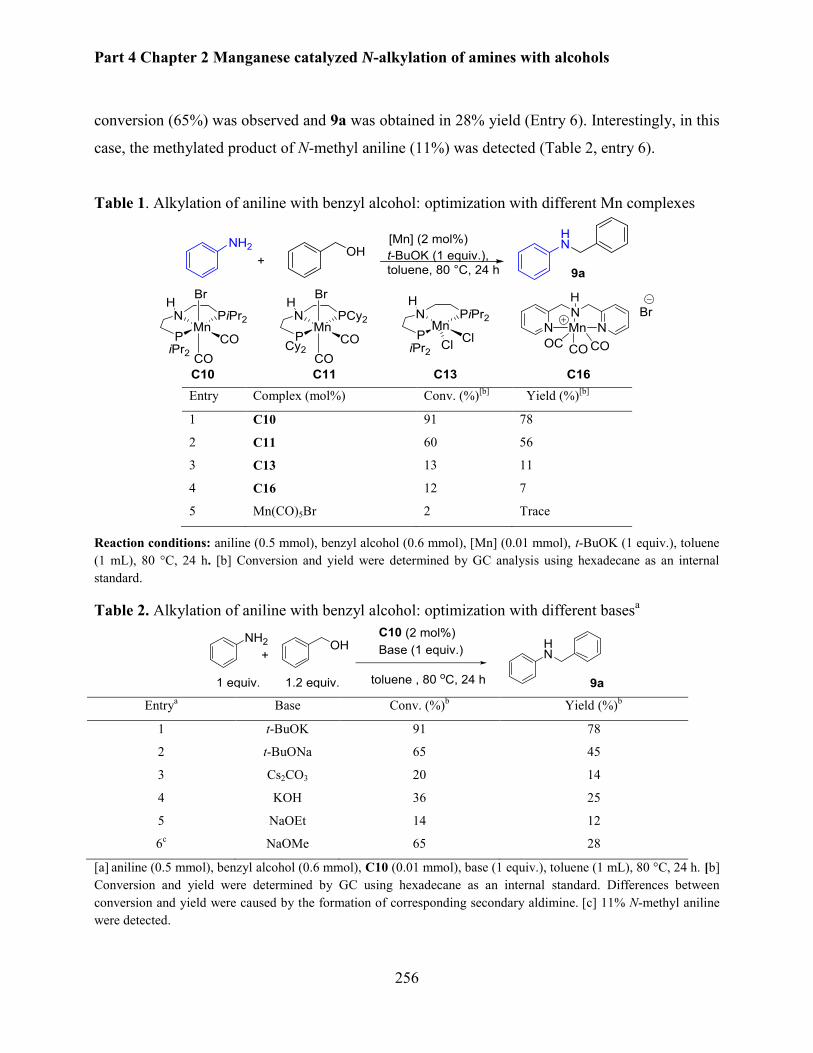
Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
256
conversion (65%) was observed and 9a was obtained in 28% yield (Entry 6). Interestingly, in this
case, the methylated product of N-methyl aniline (11%) was detected (Table 2, entry 6).
Table 1. Alkylation of aniline with benzyl alcohol: optimization with different Mn complexes
Entry Complex (mol%) Conv. (%)[b] Yield (%)[b]
1 C10 91 78
2 C11 60 56
3 C13 13 11
4 C16 12 7
5 Mn(CO)5Br 2 Trace
Reaction conditions: aniline (0.5 mmol), benzyl alcohol (0.6 mmol), [Mn] (0.01 mmol), t-BuOK (1 equiv.), toluene (1 mL), 80 °C, 24 h. [b] Conversion and yield were determined by GC analysis using hexadecane as an internal standard.
Table 2. Alkylation of aniline with benzyl alcohol: optimization with different basesa
Entrya Base Conv. (%)b Yield (%)b
1 t-BuOK 91 78
2 t-BuONa 65 45
3 Cs2CO3 20 14
4 KOH 36 25
5 NaOEt 14 12
6c NaOMe 65 28
[a] aniline (0.5 mmol), benzyl alcohol (0.6 mmol), C10 (0.01 mmol), base (1 equiv.), toluene (1 mL), 80 °C, 24 h. [b]
Conversion and yield were determined by GC using hexadecane as an internal standard. Differences between conversion and yield were caused by the formation of corresponding secondary aldimine. [c] 11% N-methyl aniline were detected.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
257
Next the influence of the solvent and various organic solvents was investigated in this
reaction. As can be seen from Table 3, toluene appears to be the most suitable solvent, giving the
highest conversion (91%) and yield (78%). Using THF, dioxane and CPME, good conversions
(72-87%) and moderate yields (55-74%) were observed (Table 3, entries 2-4). Notably, polar
solvents such as t-amyl alcohol and DMF led to moderate conversions (43-44%) and N-
benzylaniline 9a was obtained in low yields (15-18%, Table 3, entries 1 and 5).
Table 3. Alkylation of aniline with benzyl alcohol: solvent screeninga
Entrya Solvent Conv.(%)b Yield (%)b
1 t-amyl alcohol 44 15
2 THF 72 55
3 1,4 Dioxane 82 65
4 CPME 87 74
5 DMF 43 18
6 toluene 91 78
[a] aniline (0.5 mmol), benzyl alcohol (0.6 mmol), C10 (0.01 mmol), t-BuOK (1 equiv.), solvent (1 mL), 80 °C, 24
h. [b] Conversion and yield were determined by GC using hexadecane as an internal standard. Differences between
conversion and yield were caused by the formation of corresponding secondary aldimine. CPME = cyclopentyl
methyl ether.
After these optimizations, we were interested to see if the addition of stoichiometric amounts of
base was necessary to allow complete conversion or whether a catalytic amount of base can be
used. Therefore, the quantity of base was screened. The result shown in Table 4, entry 4,
suggested that a complete conversion can be obtained using 0.75 equivalent of t-BuOK leading to
the N-benzylaniline 9a with 97% yield after 24 h at 80 °C. However, the decrease of the base
loading to 50, 20 or 10 mol% has a deleterious effect on the activity as only 46, 20 and 12%,
respectively, of the N-benzylaniline 9a were obtained (Table 1, entries 1-3). Then, the reaction
time using 10 mol% of t-BuOK was investigated to evaluate if it is possible to increase the
activity. To this end, the reaction time was increased to 48 h (Entry 6). The conversion (52%)

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
258
and the yield of 9a (30%) (versus 14% and 12%, respectively at 24 h, entry 1) showed that a
prolonged reaction time permitted to increase the activity, without reaching the results obtained
with 0.75 equivalent of t-BuOK after 24 h.
Table 4. Alkylation of aniline with benzyl alcohol: screening of the quantity of basea
Entrya t-BuOK (equiv.) Conv. (%)b Yield (%)b
1 0.1 14 12
2 0.2 27 20
3 0.5 73 46
4 0.75 99 97
5
6c
1
0.1
99
52
96
30
[a] aniline (0.5 mmol), benzyl alcohol (0.6 mmol), C10 (0.01 mmol), t-BuOK (0.1-1.0 equiv.), toluene (1 mL), 80 °C, 24 h. [b] Conversion and yield were determined by GC using hexadecane as an internal standard. Differences between conversion and yield were caused by the formation of corresponding secondary aldimine. [c] 48 h.
Further investigation was performed by the variation of reaction conditions with the
complex C10. (Table 5) When the reaction was carried out without addition of the base (Table 5,
entry 1) using 3 mol% of C10 at high temperature (140 oC), no reaction occurred. Furthermore, a
blank reaction without catalyst C10 in the presence of 1.0 equiv. of t-BuOK at 80 oC for 24 h
gave no conversion (Table 5, entry 2). The minimum loading of catalyst C10 was then evaluated.
(Table 5, entries 3, 8 and 9 vs 4). When the catalyst loading was reduced to 2 mol%, 78% yield of
9a was obtained (versus 96% with 3 mol% of C10, entry 4). Noticeably, the catalyst loading can
be further reduced to 1 mol%, but the yield of the obtained N-benzylaniline 9a decreased to 54%
after 24 h at 80 °C and to 88% after 48 h. There was no significant influence when modifying the
amount of benzyl alcohol (1 equiv.) and benzylamine (1.2 equiv.) (Table 5, entry 5). As already
shown in Table 4, decreasing the quantity of base to 0.5 mol% and reducing the temperature to 60 oC, significantly lowered the yield of 9a (46%, 52% respectively, Table 5, entries 6 and 10). The
reaction was also carried out under neat conditions at 100 oC for 24 h using 2 mol% of C10 (9a:
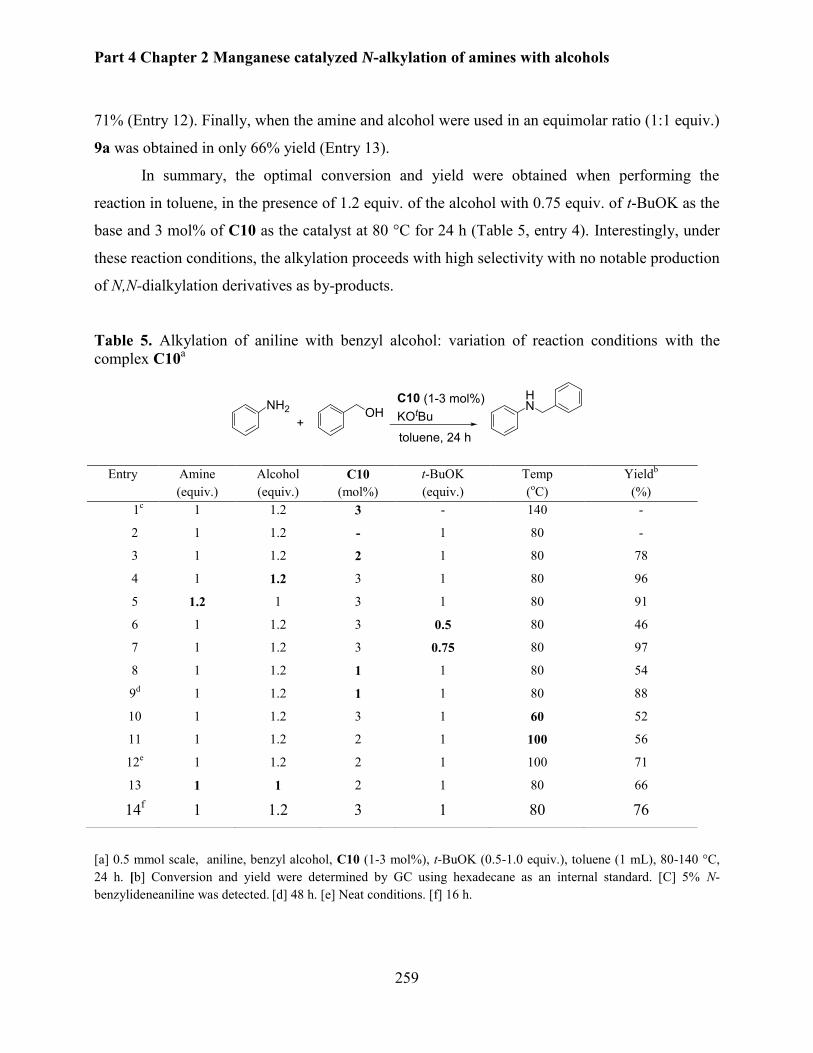
Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
259
71% (Entry 12). Finally, when the amine and alcohol were used in an equimolar ratio (1:1 equiv.)
9a was obtained in only 66% yield (Entry 13).
In summary, the optimal conversion and yield were obtained when performing the
reaction in toluene, in the presence of 1.2 equiv. of the alcohol with 0.75 equiv. of t-BuOK as the
base and 3 mol% of C10 as the catalyst at 80 °C for 24 h (Table 5, entry 4). Interestingly, under
these reaction conditions, the alkylation proceeds with high selectivity with no notable production
of N,N-dialkylation derivatives as by-products.
Table 5. Alkylation of aniline with benzyl alcohol: variation of reaction conditions with the complex C10a
[a] 0.5 mmol scale, aniline, benzyl alcohol, C10 (1-3 mol%), t-BuOK (0.5-1.0 equiv.), toluene (1 mL), 80-140 °C, 24 h. [b] Conversion and yield were determined by GC using hexadecane as an internal standard. [C] 5% N-benzylideneaniline was detected. [d] 48 h. [e] Neat conditions. [f] 16 h.
Entry Amine (equiv.)
Alcohol (equiv.)
C10 (mol%)
t-BuOK (equiv.)
Temp (oC)
Yieldb (%)
1c 1 1.2 3 - 140 -
2 1 1.2 - 1 80 -
3 1 1.2 2 1 80 78
4 1 1.2 3 1 80 96
5 1.2 1 3 1 80 91
6 1 1.2 3 0.5 80 46
7 1 1.2 3 0.75 80 97
8 1 1.2 1 1 80 54
9d 1 1.2 1 1 80 88
10 1 1.2 3 1 60 52
11 1 1.2 2 1 100 56
12e 1 1.2 2 1 100 71
13 1 1 2 1 80 66
14f 1 1.2 3 1 80 76

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
260
2.2. Selective N-alkylation of various anilines with benzyl alcohols To demonstrate the applicability of this catalytic system, various aromatic anilines and
alcohols were evaluated under optimized conditions. First, the alkylation of different substituted
anilines with benzyl alcohol was explored.
Scheme 10. N-alkylation of different amine with alcoholsa
General reaction conditions: [a] aniline derivatives (1 mmol), benzyl alcohol (1.2 mmol), C10 (3 mol%), t-BuOK (0.75 equiv.), toluene (2 mL), 80 °C, 24 h. Conversion was determined by GC (isolated yield in parentheses). [b] Traces of reduction (< 2%) of double bond were observed. [c] 11% of N,9-dibenzyl-9H-fluoren-2-amine 9q’ was detected.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
261
Substrates bearing both electron-donating (Scheme 10, 9b-d, 9g) and electron-
withdrawing substituents on the aryl ring of the aniline were selectively alkylated to afford the N-
monoalkylated anilines in good yields (typically 80-90%). Even the hindered o-chloro-aniline
and di-m-tert-butyl-aniline provided the corresponding monoalkylated amines 9i and 9g with
excellent selectivity which indicated that the steric factor didn’t inhibit the reaction.
Advantageously to the reported cobalt[54] and iron complexes,[48] aniline compounds containing
C=C bonds (4-amino-styrene and 3-amino-styrene) were alkylated leading to 9k-9l with high
isolated yields (89-90%) and chemoselectivity as the the C=C double bond was not affected.
Notably, heteroaromatic amines such as 2-amino-pyridine and 3-picolylamine led to the N-
alkylkated compounds 9m and 9n in good isolated yields (92 and 83%, respectively).
Furthermore, important building blocks for pharmaceuticals, e.g. aminobenzodioxane derivatives,
are also effectively transformed (9o-9p). 2-Aminofluorene was alkylated with benzylic alcohol
leading to N-benzyl-9H-fluoren-2-amine 9q with 74% yield and the C- and N-alkylated product,
N,9-dibenzyl-9H-fluoren-2-amine 9q’ in 11% yield. (Figure 2)
Figure 2 - N,9-dibenzyl-9H-fluoren-2-amine
Under similar conditions, phenanthren-9-amine and biphenylamine were converted into
the corresponding alkylated products 9r and 9s in 91 and 75% yields, respectively. To
demonstrate the synthetic utility of this method, the alkylation of 2-aminopyridine with benzyl
alcohol was also performed on gram scale under optimized conditions leading to 9m in 89%
isolated yield as a white solid.
2.3 N-Alkylation of amines with various (hetero)aromatic and aliphatic
alcohols Next, the possibility to apply different primary alcohols as the coupling partner was explored.
(Scheme 11) By reaction with aniline, p-Me, p-OMe, m,m’-(Me)2, p-Cl and p-SMe substituted
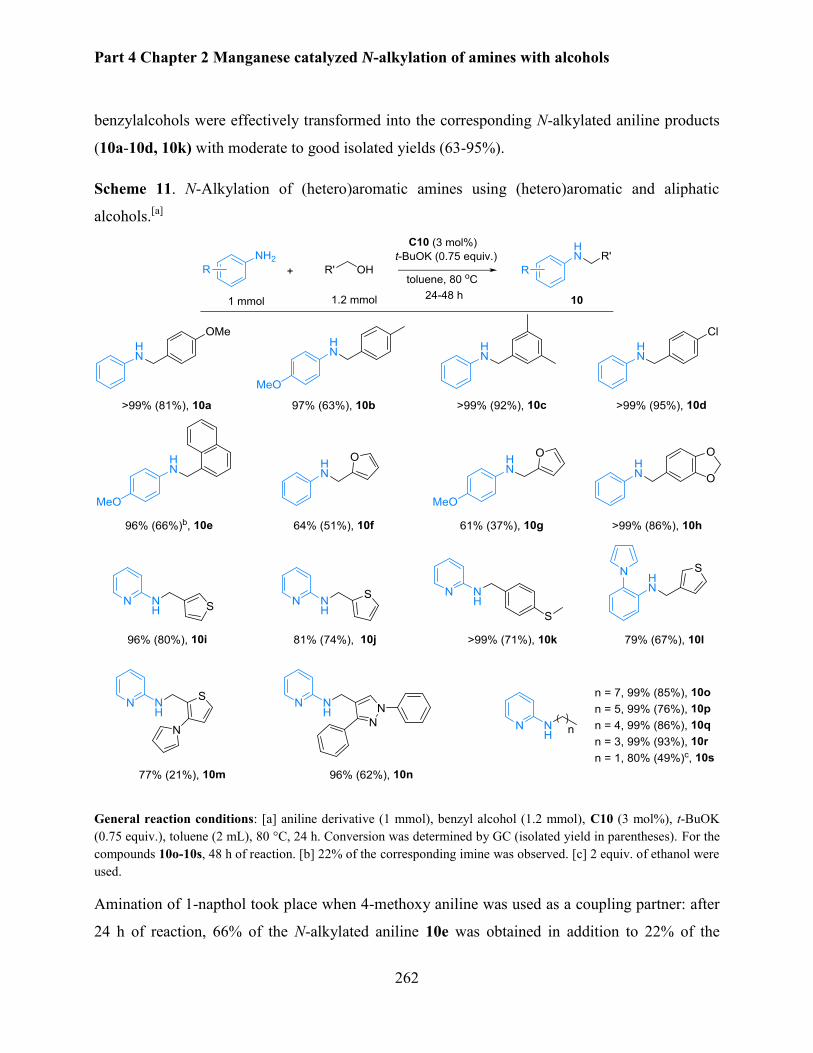
Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
262
benzylalcohols were effectively transformed into the corresponding N-alkylated aniline products
(10a-10d, 10k) with moderate to good isolated yields (63-95%).
Scheme 11. N-Alkylation of (hetero)aromatic amines using (hetero)aromatic and aliphatic
alcohols.[a]
General reaction conditions: [a] aniline derivative (1 mmol), benzyl alcohol (1.2 mmol), C10 (3 mol%), t-BuOK (0.75 equiv.), toluene (2 mL), 80 °C, 24 h. Conversion was determined by GC (isolated yield in parentheses). For the compounds 10o-10s, 48 h of reaction. [b] 22% of the corresponding imine was observed. [c] 2 equiv. of ethanol were used.
Amination of 1-napthol took place when 4-methoxy aniline was used as a coupling partner: after
24 h of reaction, 66% of the N-alkylated aniline 10e was obtained in addition to 22% of the

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
263
corresponding imine. The reaction of heteroaromatic alcohols, including biomass derived furfuryl
alcohols, also proceeded smoothly and furnished the desired N-alkylated products with moderate
to good yields (37-96%, Scheme 11, 10f-g, 10i-j, 10l-n). Long (C8) as well as short chain (C2)
aliphatic alcohols were successfully applied to alkylate 2-aminopyridine yielding the
corresponding N-alkylated derivatives in 49-93% yields after 48 h of reaction at 80 °C (10o-10s).
Resveratrol derived amines 11 (see Figure 3) are known to be active for the treatment of
Alzheimer’s disease.[57] In light of the high chemo-selectivity observed in the presence of vinyl
groups (see products 9k and 9l, Scheme 10), the catalytic system based on C10 seems to be
appropriate to perform N-alkylation of 4-amino-trans-stilbene. To proof this statement, 4-amino-
trans-stilbene was alkylated with different various alcohols in the presence of 3 mol% of C10
under the standard conditions (0.75 equiv. KOtBu, 80 oC for 24 h). Noticeably, in all cases,
alkylated derivatives 11a-11e were isolated in good to excellent yields (88-97%, Figure 3) with
high chemoselectivity.
Figure 3. Synthesis of resveratrol derivatives. Reaction conditions: 4-aminostilbene (1 mmol), alcohol (1.2 mmol), C10 (0.03 mmol), t-BuOK (0.75 equiv.), toluene (2 mL), 80 °C, 24 h.
In some cases, functional group tolerance issues were observed. When 4-cyano-aniline
was used under standard conditions, even if a full conversion was observed, only 27% of the N-
alkylated 4-cyanoaniline product 12 was obtained in association with 14% of the product 13
resulting of the hydration of the cyano moiety of 12, due to the in-situ formation of water during
alkylation reaction. Furthermore, the reaction was not selective as many unidentified by-products
were detected in GC in small quantities. The methyl p-aminobenzoate can be also alkylated using

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
264
benzyl alcohol: a mixture of the N-alkylated amine 14 (23%) and of the p-(N-benzylamino)-
benzoic acid 15 (15%) resulting from the hydrolysis of 14 was obtained.
Scheme 12. Selectivity issues in alkylation of amine.
Mainly due to solubility issues in toluene, the N-alkylation of the aniline derivatives 16, 17 and
18 did not lead to the corresponding N-alkylated compounds, even if low conversions were
observed (10-25%). (Figure 4)
Figure 4. Non-working substrates
Noticeably, when the hydrogen borrowing reaction was performed with benzylamine and
aliphatic amines, the dehydrogenative coupling occurred with full conversion but mainly led to
the formation of corresponding imines such as 19 under the optimized conditions. Only traces of
alkylated amine were observed. (Scheme 13)
Scheme 13. Dehydrogenative coupling of benzyl alcohol and benzylamine.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
265
2.4. Manganese catalyzed N-methylation of aniline with methanol Among the different aliphatic alcohols, the selective monomethylation of amines with
methanol is most challenging. Apart from the formation of side products such as N,N-dimethyl
amines, methanol is more difficult to dehydrogenate.[58] However, N-methylated amines are very
important drug molecules and natural products.[1] Therefore, this transformation is still performed
using toxic and/or hazardous methyl halides or sulfates in the agent.[59] Despite this importance,
to date only limited studies on transition metal-catalyzed N-methylation of amines using
methanol have been reported employing precious metals such as Ru[60-61] and Ir.[62-63]
In 2015, Seayad and coworkers reported an effective N-methylation of aniline derivatives
using methanol (Scheme 14). By employing an in situ-generated complex from [RuCp*Cl2]2 and
dpePhos ligand in the presence of 5 mol % of LiOtBu, various aromatic amines were selectively
alkylated in the presence of reducible functional group such as ester, ketone, amide, cyano, or
nitro at 100 oC. When aliphatic amines are used as the substrate, N,N-dimethylation was
selectively observed.[60]
Scheme 14. Ruthenium catalyzed N-methylation of anilines.
In 2004, the ruthenium(II) half-sandwich complex [RuCl(η5-C5H5)(PPh3)2] (1 mol%) was
used to catalyze the N-methylation reaction with methanol at 100 oC. Secondary amines were
converted into the corresponding monomethylated products and primary amines were alkylated
leading to the dimethylated products (Scheme 14).[52]
In 2012 Li and coworkers reported that the iridium complex, [Cp*IrCl2]2, can be used as
an efficient catalyst (0.1 mol%) for the N-methylation of anilines in the presence of one
equivalent of NaOH as a base at high temperature 150 oC (Scheme 15). This method showed

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
266
broad substrate scope including methylation of arylamines, arylsulfonamides and amino-
azoles.[53]
Scheme 15. Iridium catalyzed N-methylation of anilines.
Recently, iridium catalyst 21 was reported for the monomethylation of aniline derivatives.
The bis(N-heterocyclic carbene) iridium complex (5 mol%) catalyzed the reaction which
proceeded at 120 oC for 5 h (Scheme 15). [54]
To the best of our knowledge, there is no report dealing with on manganese catalyzed N-
methylation of amines by methanol. The methylation reaction using the active PNP manganese
complex C10 was then studied. As shown in Scheme 16, aniline derivatives can be efficiently
N-methylated using methanol as the C1 source. Using 3 mol% of C10 in the presence of 1 mol%
of t-BuOK in methanol at 100 °C for 24 h, the corresponding N-methylated products were
obtained in good to excellent isolated yields (22a-22k: 52-94%). Notably, electron donating and
electron withdrawing substituted anilines were converted into the desired products 22a-22f with
good yields (85-92%, Scheme 16). Importantly, the catalyst C10 showed very good
chemoselectivity as Br- and I- substituents were well tolerated (22f, 22i), albeit in the case of
sterically hindered 2-iodo N-methyl aniline 22j, 15% of the dehalogenated product, namely
N-methylaniline, was observed. Interestingly, 3-aminopyridine was methylated yielding 89% of
22g. Interestingly, 3-amino-styrene was chemoselectively transformed into the corresponding
methylated product 22h without affecting the double bond. Under the standard conditions 2-(1H-
pyrrol-1-yl)aniline and 2-aminofluroene were methylated with moderate to good isolated yields
(52 and 71%, respectively).

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
267
Scheme 16. N-Methylation of primary anilines using methanol.a
General reaction conditions: [a] aniline derivative (1 mmol), C10 (3 mol%), t-BuOK (1 equiv.), methanol (2 mL), 100 °C, 24 h. Conversion was determined by GC ( isolated yield in parentheses). [b] Traces of reduction of double bond. [c] 15% dehalogenation was observed.
As already mentioned before, any trace of dialkylation products was observed during the
transformation. In agreement with these observations, the reaction of N-methylaniline with
benzyl alcohol did not lead to the tertiary aniline N-benzyl-N-methylaniline.
Apart from intermolecular reactions, an intramolecular cyclisation was also performed. Indeed,
under the optimized conditions (3 mol% of C10, 1 equiv. of t-BuOK, toluene, 100 °C, 48 h),
2-(2-aminophenyl)ethanol led to the corresponding indole in 98% yield (Scheme 17).
Scheme 17. Synthesis of indole via hydrogen auto transfer methodology.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
268
3. Conclusion In summary, the direct C–N bond formation with manganese complex as a catalyst is
possible through a hydrogen borrowing pathway using a PNP pincer complex C10 as the catalyst.
Such catalyst is able to exhibit suitable activity for both steps of the transformation, the alcohol
dehydrogenation leading to carbonyl derivative and imine hydrogenation in an overall hydrogen
borrowing process. In this chapter, it is clearly demonstrated for the first time that molecular-
defined manganese pincer complexes (Mn-PNP) C10-C11 are efficient catalysts for inter- and
intramolecular amination reactions. Advantageously, these complexes are stable and can be
conveniently handled in the presence of air. This protocol for the N-alkylation of aniline
derivatives with primary benzyl alcohols and alkanols proceeds under mild conditions (80-100
°C) with good chemoselectivity. Importantly, this catalytic system is also effective for the
selective monoalkylation of anilines and tolerates functional groups, including olefins, halides,
thioether, benzodioxane and heteroaromatic groups. By contrast, aliphatic amines led to the
formation of imines with alcohols. Of special importance are the N-methylations of various
amines using methanol, which constitute the first example of this transformation using non-noble
metal complexes under mild conditions in the presence of base.
4. Experimental part
4.1 General experimental details Unless otherwise stated, all reactions were performed under an argon atmosphere with
exclusion of moisture from reagents and glassware using standard techniques for manipulating
air-sensitive compounds. All isolated products were characterized by 1H NMR and 13C NMR
spectroscopy as well as high resolution mass spectrometry (HRMS). NMR spectra were recorded
on a Bruker AV 300 or 400. All chemical shifts (δ) are reported in ppm and coupling constants
(J) in Hz. All chemical shifts are related to residual solvent peaks [CDCl3: 7.26 (1H), 77.16 (13C);
C6D6: 7.16 (1H), 128.06 (13C)], respectively. All measurements were carried out at room
temperature unless otherwise stated. Mass spectra were in general recorded on a Finnigan MAT
95-XP (Thermo Electron) or on a 6210 Time-of-Flight LC/MS (Agilent). Gas chromatography
was performed on a HP 6890 with a HP5 column (Agilent).
Reagents: Unless otherwise stated, commercial reagents were used without purification

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
269
Synthesis and characterization of complex C16
A commercially available 2-(picolyl)amine (213 mg, 1.00 mmol) was added in a 50 mL Schlenk
flask and dissolved in a dry toluene (30 mL) under argon. Mn(CO)5Br (275 mg, 1.00 mmol) was
added to the solution, the mixture was refluxed for 24 h under a steam of argon. A yellow solid
precipitate which was formed after cooling down the reaction mixture to room temperature, was
filtered under argon and washed with 3 portions of Et2O (10 mL each). The removal of solvent
afforded [Mn(dpa)(CO)3]Br (250 mg, 58% yield) as pale yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 8.94 (d, J = 5.0 Hz, 2H), 7.90 (t, J = 7.7 Hz, 2H), 7.56 (t, J = 7.0 Hz, 1H), 7.50 (d, J = 7.8 Hz, 2H), 7.42 (t, J = 6.6 Hz, 2H), 4.80-4.72 (m, 2H), 4.43 (br d, J = 17.5 Hz, 2H). 13C NMR (75 MHz, DMSO-d6): δ 219.4, 160.9, 151.9, 139.1, 125.0, 122.4, 61.0. IR-ATR (solid): ῡ [cm-1] 2026 (s, ῡ CO), 1913 (s, ῡ CO), 1888 (s, ῡ CO).
Typical procedure (A) for the manganese catalyzed N-alkylation of amines with alcohols
An oven-dried 25-mL Schlenk tube, prepared with a stirring bar, was charged with the
yellow complex C10 (14.9 mg, 0.03 mmol), t-BuOK (84 mg, 0.75 mmol) and dry toluene (2 mL).
Then, the corresponding alcohol (1.2 mmol) and amine (1 mmol) were added to the red colored
suspension. Solid materials were weighed into the Schlenk tube under air, and the Schlenk tube
was subsequently connected to a Schlenk line and vacuum-argon exchange was done for three
times. Liquid compounds and solvent were charged under an argon flow. The Schlenk tube was
placed into an aluminum block and heated to 80 °C and stirred for a given time. The reaction
mixture was cooled to RT, quenched with water and extracted with ethyl acetate. The organic
layers were then dried over MgSO4 and concentrated under reduced pressure. The residue was
purified by flash chromatography on silica gel (n-pentane/diethylether) to afford the desired
product. The product was analyzed by 1H- and 13C-NMR spectroscopy.
General procedure (B) for the N-methylation of amines using methanol
An oven dried pressure tube was charged with Mn complex C10 (14.9 mg, 0.03 mmol),
t-BuOK (112 mg, 1 mmol). and amine (1 mmol) were weighed into the pressure tube under air,
and the pressure tube was connected to a Schlenk line and vacuum-argon exchange was
performed three times. Liquid amines and dry, degassed methanol (1 mL) were charged under an
argon stream after the three vacuum-argon exchanges. The pressure tube was closed with a
Teflon® stopper and was heated to 100 °C. After 24 h, the reaction mixture was cooled to room

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
270
temperature and extracted with ethyl acetate / water. The organic layer was dried over MgSO4
and transferred into a round bottom flask. SiO2 (350 mg) was added to the mixture. The solvent
was removed in vacuo and the product was purified by column chromatography using heptane
and ethyl acetate. The product was analyzed by 1H- and 13C-NMR spectroscopy.
4.2. NMR data for isolated products N-Benzyl-4-methoxyaniline
The compound was prepared as described in the general procedure A (m = 191 mg, 90% isolated yield).Yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.42 – 7.30 (m, 5H), 6.84 – 6.81 (m, 2H), 6.80 – 6.61 (m, 2H), 4.31 (s, 2H), 3.77 (s, 4H). 13C NMR (75 MHz, CDCl3): δ 152.3, 142.4, 139.6, 128.7, 127.7, 127.3, 114.9, 114.9, 55.8, 49.4. N-Benzyl-4-methylaniline
The compound was prepared as described in the general procedure A (m = 171 mg, 87% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.58 – 7.33 (m, 5H), 7.21 – 7.01 (m, 2H), 6.79 – 6.58 (m, 2H), 4.41 (s, 2H), 2.47 – 2.27 (m, 3H). 13C NMR (75 MHz, CDCl3): δ 145.9, 138.6, 129.8, 128.6, 127.5, 127.2, 126.7, 113.1, 48.6, 20.5. N-Benzyl-4-ethoxyaniline
The compound was prepared as described in the general procedure A (m = 180 mg, 79% isolated yield). White solid; 1H NMR (300 MHz, CDCl3): δ 7.44 – 7.32 (m, 5H), 6.87 – 6.84 (m, 2H), 6.83 – 6.62 (m, 2H), 4.32 (s, 2H), 4.00 (q, J = 7.0 Hz, 2H), 3.77 (s, 1H), 1.43 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 151.5, 142.4, 139.7, 128.6, 127.6, 127.2, 115.8, 114.2, 64.1, 49.3, 15.1.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
271
N-Benzyl-4-(methylthio)aniline
The compound was prepared as described in the general procedure A (m = 161 mg, 71% isolated yield). Yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.28 – 7.16 (m, 5H), 7.15 – 7.08 (m, 2H), 6.50 – 6.45 (m, 2H), 4.20 (s, 2H), 3.97 (s, 1H), 2.30 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 146.9, 139.2, 131.4, 128.7, 127.4, 127.3, 124.4, 113.5, 48.2, 19.1. N-Benzyl-4-bromoaniline
The compound was prepared as described in the general procedure A (m = 231 mg, 88% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.29-7.12 (m, 7H), 6.42-6.37 (m, 2H), 4.19 (s, 2H), 3.96 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 147.1, 138.9, 131.9, 128.7, 127.4, 127.4, 114.5, 109.2, 48.2. N-Benzyl-2-chloroaniline
The compound was prepared as described in the general procedure A (m = 152 mg, 70% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.45 – 7.34 (m, 6H), 7.19 – 7.14 (m, 1H), 6.72 (td, J = 7.6, 1.1 Hz, 2H), 4.84 (s, 1H), 4.46 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 143.9, 138.8, 129.2, 128.8, 128.7, 127.9, 127.4, 127.3, 119.2, 111.6, 47.9. N-Benzyl-3-(trifluoromethyl)aniline

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
272
The compound was prepared as described in the general procedure A (m = 212 mg, 84% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.47 – 7.28 (m, 6H), 7.05-7.02 (m, 1H), 6.93-6.91 (m, 1H), 6.82 – 6.81 (m, 1H), 4.39 (s, 2H), 4.33 – 4.19 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 148.4, 138.7, 131.7 (q, J = 31.6 Hz), 129.8, 128.9, 127.6, 127.6, 124.5 (q, J = 272.5 Hz), 115.8 (q, J = 1.4 Hz), 114.0 (q, J = 3.9 Hz), 109.2 (q, J = 4.0 Hz), 48.2. N-Benzyl-3,5-dimethoxyaniline
The compound was prepared as described in the general procedure A (m = 221 mg, 91% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.27 – 7.13 (m, 5H), 5.81-5.73 (m, 3H), 4.18 (s, 2H), 4.00 (br.s, 1H), 3.62 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 161.8, 150.1, 139.3, 128.7, 127.6, 127.3, 91.8, 90.0, 55.2, 48.4. N-Benzyl-3,5-di-tert-butylaniline
The compound was prepared as described in the general procedure A (m = 181 mg, 61% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.59– 7.44 (m, 5H), 7.04 (t, J = 1.6 Hz, 1H), 6.72 (d, J = 1.6 Hz, 2H), 4.50 (s, 2H), 4.09 (br. s, 1H), 1.50 (s, 18H). 13C NMR (75 MHz, CDCl3): δ 151.7, 147.7, 139.8, 128.6, 127.9, 127.3, 112.4, 107.7, 48.9, 34.9, 31.6. GCMS-EI (70 eV): m/z (%) = 295 (M+, 100), 280 (11), 207 (3), 91 (36). HRMS (EI, m/z) calcd. for C21H29N: 295.22945; found: 295.22920.
N-Benzyl-4-vinylaniline
The compound was prepared as described in the general procedure A (m = 185 mg, 89% isolated yield). Yellow sticky solid;

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
273
1H NMR (300 MHz, CDCl3): δ 7.58 – 7.24 (m, 7H), 6.79 – 6.60 (m, 3H), 5.64 (dd, J = 17.6, 1.1 Hz, 1H), 5.13 (dd, J = 10.9, 1.1 Hz, 1H), 4.40 (s, 2H), 4.17 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 147.9, 139.3, 136.7, 128.7, 127.5, 127.4, 127.4, 127.3, 112.7, 109.5, 48.2. GCMS-EI (70 eV): m/z (%) = 209 (M+, 82), 132 (11), 91 (100), 77 (12), 65 (17). HRMS (EI, m/z) calcd. for C15H15N: 209.11990; found: 209.11981. N-Benzyl-3-vinylaniline
The compound was prepared as described in the general procedure A (m = 189 mg, 90% isolated yield). Colorless liquid; 1H NMR (300 MHz, CDCl3): δ 7.49 – 7.22 (m, 6H), 6.94 – 6.72 (m, 4H), 5.81 (dd, J = 17.6, 1.1 Hz, 1H), 5.32 (dd, J = 10.9, 1.1 Hz, 1H), 4.42 (s, 2H), 4.09 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 148.3, 139.4, 138.6, 137.3, 129.4, 128.8, 127.6, 127.3, 115.9, 113.5, 112.6, 110.7, 48.3. GCMS-EI (70 eV): m/z (%) = 209 (M+, 92), 208 (43), 132 (18), 91 (100), 77 (14), 65 (18). HRMS (EI, m/z) calcd. for C15H15N: 209.11990; found: 209.11960. N-Benzylpyridin-2-amine
The compound was prepared as described in the general procedure A (m = 170 mg, 92% isolated yield). White solid; 1H NMR (300 MHz, CDCl3): δ 7.96-7.94 (m, 1H), 7.29 – 7.23 (m, 6H), 6.47-6.43 (m, 1H), 6.24 (dt, J = 8.4, 0.9 Hz, 1H), 5.18 (s, 1H), 4.38 (d, J = 5.7 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 158.7, 148.1, 139.2, 137.5, 128.6, 127.4, 127.2, 113.1, 106.7, 46.3. N-Benzyl-2,3-dihydrobenzo[b][1,4]dioxin-6-amine
The compound was prepared as described in the general procedure A (m = 203 mg, 84% isolated yield). White crystal;

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
274
1H NMR (300 MHz, CDCl3): δ 7.45 – 7.28 (m, 5H), 6.76 (d, J = 8.5 Hz, 1H), 6.26 – 6.20 (m, 2H), 4.28 (s, 2H), 4.25-4.17 (m, 4H), 3.79 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 144.0, 143.2, 139.6, 135.7, 128.6, 127.6, 127.2, 117.6, 106.7, 101.6, 64.7, 64.2, 49.0. GCMS-EI (70 eV): m/z (%) = 241 (M+, 100), 184 (11), 150 (52), 122 (37), 91 (46). HRMS (EI, m/z) calcd. for C15H15O2N: 241.10973; found: 241.10968. N-Benzylbenzo[d][1,3]dioxol-5-amine
The compound was prepared as described in the general procedure A (m = 204 mg, 89% isolated yield). White solid; 1H NMR (400 MHz, CDCl3): δ 7.41 – 7.27 (m, 5H), 6.65 (d, J = 8.2 Hz, 1H), 6.30 (d, J = 2.1 Hz, 1H), 6.11 (dd, J = 8.2, 2.1 Hz, 1H), 5.85 (s, 2H), 4.27 (s, 2H). 13C NMR (101 MHz, CDCl3): δ 148.3, 143.2, 140.1, 138.9, 128.6, 127.7, 127.4, 108.6, 105.1, 100.6, 96.5, 49.6. GCMS-EI (70 eV): m/z (%) = 227 (M+, 100), 136 (72), 91 (64), 65 (13). N-Benzyl-9H-fluoren-2-amine
The compound was prepared as described in the general procedure A (m = 200 mg, 74% isolated yield). Yellow solid; 1H NMR (300 MHz, CDCl3): δ 7.66-7.63 (m, 1H), 7.57 (d, J = 8.1 Hz, 1H), 7.38 – 7.29 (m, 6H), 7.22 – 7.12 (m, 2H), 6.70 (dd, J = 8.1, 2.1 Hz, 1H), 6.51 (d, J = 2.1 Hz, 1H), 4.18 (t, J = 7.6 Hz, 1H), 3.63 (s, 2H), 3.12 (d, J = 7.6 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 148.8, 146.0, 145.7, 141.3, 140.1, 131.9, 129.6, 128.3, 127.1, 126.4, 125.1, 124.6, 120.6, 118.6, 114.3, 111.8, 48.5, 40.3. GCMS-EI (70 eV): m/z (%) = 271 (M+, 48), 180 (100), 152 (21), 91 (8). HRMS (EI, m/z) calcd. for C20H17N: 271.13555; found: 271.13545. N-Benzylphenanthren-9-amine
The compound was prepared as described in the general procedure A (m = 257 mg, 91% isolated yield). White solid;

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
275
1H NMR (300 MHz, CDCl3): δ 8.75 (dd, J = 8.2, 1.4 Hz, 1H), 8.59 (dd, J = 8.3, 1.3 Hz, 1H), 7.94 – 7.89 (m, 1H), 7.74 – 7.60 (m, 3H), 7.56 – 7.51 (m, 3H), 7.48 – 7.38 (m, 4H), 6.88 (s, 1H), 4.71 (s, 1H), 4.59 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 140.9, 139.1, 133.7, 131.2, 128.9, 128.0, 127.6, 127.0, 126.8, 126.6, 126.4, 125.5, 125.4, 123.6, 123.1, 122.5, 120.5, 102.8, 48.8. GCMS-EI (70 eV): m/z (%) = 283 (M+, 100), 192 (12), 165 (86), 91 (27). HRMS (EI, m/z) calcd. for C21H17N (M)+ 283.13555; found 283.13516. N-Benzyl-[1,1’-biphenyl]-2-amine
The compound was prepared as described in the general procedure A (m = 194 mg, 75% isolated yield).Colorless crystals; 1H NMR (400 MHz, CDCl3): δ 7.54 – 7.44 (m, 4H), 7.42 – 7.31 (m, 5H), 7.31 – 7.26 (m, 1H), 7.25 – 7.19 (m, 1H), 7.15 (dd, J = 7.4, 1.7 Hz, 1H), 6.82 (td, J = 7.4, 1.1 Hz, 1H), 6.70 (dd, J = 8.2, 1.1 Hz, 1H), 4.44 (s, 1H), 4.37 (s, 2H). 13C NMR (101 MHz,CDCl3): δ 145.0, 139.6, 130.4, 129.5, 129.1, 128.8, 128.7, 127.8, 127.4, 127.2, 124.9, 117.3, 114.3, 110.9, 48.3. GCMS-EI (70 eV): m/z (%) = 260 (20), 259 (M+, 100), 258 (37), 182 (12), 168 (28), 167 (38), 166 (10), 152 (11), 91 (50), 65 (10). N-(4-Methoxybenzyl)aniline
The compound was prepared as described in the general procedure A (m = 173 mg, 81% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.42 – 7.38 (m, 2H), 7.33 – 7.26 (m, 2H), 7.02 – 6.97 (m, 2H), 6.84 (tt, J = 7.3, 1.1 Hz, 1H), 6.79 – 6.70 (m, 2H), 4.35 (s, 2H), 3.90 (s, 3H, NH). 13C NMR (75 MHz, CDCl3): δ 158.9, 148.2, 131.4, 129.3, 128.8, 117.6, 114.1, 112.9, 55.3, 47.8. N-(4-Chlorobenzyl)aniline

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
276
The compound was prepared as described in the general procedure A (m = 207 mg, 95% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.26-7.78 (m, 4H), 7.17 – 7.12 (m, 2H), 6.73 – 6.71 (m, 1H), 6.69 – 6.56 (m, 2H), 4.25 (s, 2H), 4.02 (m, 1H). 13C NMR (75 MHz, CDCl3): δ 147.9, 138.1, 132.9, 129.4, 128.8, 128.8, 117.9, 113.0, 47.7. 4-Methoxy-N-(4-methylbenzyl)aniline
The compound was prepared as described in the general procedure A (m = 142 mg, 63% isolated yield). White solid; 1H NMR (300 MHz, CDCl3): δ 7.35 – 7.25 (m, 2H), 7.24 – 7.21 (m, 2H), 6.88 – 6.83 (m, 2H), 6.69 – 6.64 (m, 2H), 4.30 (s, 2H), 3.80 (s, 4H), 2.43 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 152.2, 142.6, 136.7, 136.7,129.3, 127.6, 114.9, 114.1, 55.8, 48.9, 21.2. N-(3,5-Dimethylbenzyl)aniline
The compound was prepared as described in the general procedure A (m = 194 mg, 92% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.37-7.32 (m, 2H), 7.16 – 7.09 (m, 3H), 6.92-6.89 (s, 1H), 6.87-6.86 (dt, J = 7.7, 1.1 Hz, 2H), 4.37 (s, 2H), 4.20 – 3.96 (m, 1H), 2.48 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 148.3, 139.4, 138.2, 129.3, 128.9, 125.4, 117.5, 112.8, 48.4, 21.3. GCMS-EI (70 eV): m/z (%) = 211 (M+, 63), 196 (8), 119 (100), 104 (9), 91 (15), 77 (19). HRMS (EI, m/z) calcd. for C25H17N (M)+ 211.13555; found 211.13547. 4-Methoxy-N-(naphthalen-1-ylmethyl)aniline

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
277
The compound was prepared as described in the general procedure A (m = 174 mg, 66% isolated yield). Yellow solid; 1H NMR (300 MHz, CDCl3): δ 8.20 – 8.08 (m, 1H), 8.03 – 7.90 (m, 1H), 7.91 – 7.82 (m, 1H), 7.65 – 7.52 (m, 3H), 7.48 (dd, J = 8.1, 7.0 Hz, 1H), 7.01 – 6.79 (m, 2H), 6.81 – 6.52 (m, 2H), 4.72 (s, 2H), 3.82 (s, 3H (OMe)+1H (NH)). 13C NMR (75 MHz, CDCl3): δ 152.2, 142.6, 134.7, 133.9, 131.6, 128.8, 128.1, 126.3, 126.0, 125.8, 125.6, 123.6, 115.0, 114.0, 55.8, 47.3. N-(Furan-2-ylmethyl)aniline
The compound was prepared as described in the general procedure A (m = 88mg, 51% isolated yield). Colorless oil; 1H NMR (300 MHz, CDCl3): δ 7.27-7.26 (m, 1H), 7.12 – 7.06 (m, 2H), 6.65-6.58 (m, 1H), 6.56 – 6.55 (m, 2H), 6.23-6.21(m, 1H), 6.14-6.12 (m, 1H), 4.20 (s, 2H), 3.83 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 152.8, 147.6, 141.9, 129.3, 118.1, 113.2, 110.4, 107.1, 41.5. N-(Furan-2-ylmethyl)-4-methoxyaniline
The compound was prepared as described in the general procedure A (m = 75 mg, 37% isolated yield). Yellow oil; 1H NMR (300 MHz, CDCl3): δ 7.39-7.38 (m, 1H), 6.84 – 6.79 (m, 2H), 6.69 – 6.64 (m, 2H), 6.35-6.33 (s, 1H), 6.25-6.23 (m, 1H), 4.28 (s, 2H), 3.76 (s, 4H). 13C NMR (75 MHz, CDCl3): δ 153.1, 152.6, 141.9, 141.8, 114.8, 114.7, 110.4, 106.9, 55.7, 42.5. N-(Benzo[d][1,3]dioxol-5-ylmethyl)aniline
The compound was prepared as described in the general procedure A (m = 195 mg, 86% isolated yield). White solid; 1H NMR (300 MHz, CDCl3): δ 7.29 – 7.18 (m, 2H), 6.95 – 6.74 (m, 4H), 6.72 – 6.63 (m, 2H), 5.97 (s, 2H), 4.27 (s, 2H), 4.04 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 148.1, 147.9, 146.8, 133.4, 129.3, 120.6, 117.6, 112.9, 108.4, 108.1, 101.0, 48.2.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
278
N-(Thiophen-3-ylmethyl)pyridin-2-amine
The compound was prepared as described in the general procedure A (m = 151 mg, 80% isolated yield). White solid; 1H NMR (300 MHz, CDCl3): δ 8.21 – 7.90 (m, 1H), 7.39 (ddd, J = 8.5, 7.1, 1.5 Hz, 1H), 7.31 – 7.23 (m, 1H), 7.19 – 7.13 (m, 1H), 7.06 (dd, J = 4.9, 1.3 Hz, 1H), 6.60-6.56 (m, J = 7.2, 5.0, 1.0 Hz, 1H), 6.38 (d, J = 7.9 Hz, 1H), 5.12 (s, 1H), 4.49 (d, J = 5.3 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 158.6, 148.2, 140.3, 137.5, 127.2, 126.2, 121.6, 113.2, 106.9, 41.8. GCMS-EI (70 eV): m/z (%) = 190 (M+, 100), 189 (23), 157 (11), 112 (57), 97 (79), 78 (38). N-(Thiophen-2-ylmethyl)pyridin-2-amine
The compound was prepared as described in the general procedure A (m = 140 mg, 74% isolated yield). White solid; 1H NMR (300 MHz, CDCl3): δ 8.13-8.10 (m, 1H), 7.44-7.38 (m, 1H), 7.20 (dd, J = 5.0, 1.3 Hz, 1H), 7.02-7.00 (m, 1H), 6.97-6.94 (m, 1H), 6.63-6.58 (m, 1H), 6.42 (d, J = 8.4 Hz, 1H), 5.08 (s, 1H), 4.69 (dd, J = 5.9, 1.0 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 158.2, 148.2, 142.7, 137.5, 126.8, 125.2, 124.7, 113.5, 107.4, 41.3. GCMS-EI (70eV): m/z (%) = 190 (M+, 95), 157 (20), 112 (41), 97 (100), 78 (30). N-(4-(Methylthio)benzyl)pyridin-2-amine
The compound was prepared as described in the general procedure A (m = 162 mg, 71% isolated yield). White solid; 1H NMR (300 MHz, CDCl3): δ 8.18 (ddd, J = 5.2, 1.9, 0.9 Hz, 1H), 7.49 (ddd, J = 8.4, 7.1, 1.9 Hz, 1H), 7.44 – 7.29 (m, 4H), 6.68 (ddd, J = 7.2, 5.0, 0.9 Hz, 1H), 6.46 (dt, J = 8.5, 0.9 Hz, 1H), 5.19 (s, 1H), 4.56 (d, J = 5.7 Hz, 2H), 2.57 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 158.6, 148.1, 137.5, 137.2, 136.2, 128.0, 127.0, 113.2, 106.8, 45.8, 16.1. GCMS-EI (70 eV): m/z (%) = 230 (M+, 82), 215 (7), 152 (28), 137 (100), 122 (18), 78 (26). HRMS (EI, m/z) calcd. for C13H14N2S (M)+ 230.08722; found 230.08720.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
279
2-(1H-Pyrrol-1-yl)-N-(thiophen-3-ylmethyl)aniline
The compound was prepared as described in the general procedure A (m = 170 mg, 67% isolated yield). Colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.39 – 7.33 (m, 2H), 7.28 – 7.23 (m, 1H), 7.19 – 7.17 (m, 1H), 7.08 (dd, J = 5.0, 1.3 Hz, 1H), 6.96 – 6.90 (m, 2H), 6.90 – 6.80 (m, 2H), 6.46-6.44 (m, 2H), 4.41 (s, 2H), 4.26 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 143.5, 140.2, 128.9, 127.4, 127.2, 126.7, 126.3, 121.9, 121.3, 116.9, 111.6, 109.6, 43.4. GCMS-EI (70 eV): m/z (%) = 254 (M+, 100), 253 (30), 169 (18), 157 (59), 135 (11), 97 (65), 77 (10). N-((3-(1H-Pyrrol-1-yl)thiophen-2-yl)methyl)pyridin-2-amine
The compound was prepared as described in the general procedure A (m = 52 mg, 21% isolated yield). Colorless oil. 1H NMR (300 MHz, CDCl3): δ 8.13-8.11 (m , 1H), 7.43 – 7.37 (m, 1H), 7.20 (dd, J = 5.3, 0.7 Hz, 1H), 7.00 (d, J = 5.3 Hz, 1H), 6.90-6.89 (m, 2H), 6.64 – 6.60 (m, 1H), 6.36 – 6.31 (m, 3H), 4.83 (s, 1H), 4.64 (d, J = 5.8 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 157.8, 148.2, 137.5, 136.9, 132.2, 125.1, 123.7, 121.9, 113.8, 109.5, 107.6, 38.7. HRMS (EI, m/z) calcd. for C14H13N3S (M+1)+ 256.09029; found 256.09076. N-((1,3-Diphenyl-1H-pyrazol-4-yl)methyl)pyridin-2-amine
The compound was prepared as described in the general procedure A. 1H NMR (300 MHz, CDCl3): δ 8.15 – 8.13 (m, 1H), 7.98 (s, 1H), 7.80 – 7.71 (m, 4H), 7.47 – 7.37 (m, 6H), 7.30 – 7.25 (m, 1H), 6.65-6.60 (m, 1H), 6.41 (d, J = 8.4 Hz, 1H), 4.82 (s, 1H), 4.59 (d, J = 5.2 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 158.2, 151.2, 148.1, 139.9, 137.5, 133.0, 129.3, 129.2, 128.6, 128.0, 127.7, 127.5, 126.3, 118.8, 113.4, 107.2, 37.3. HRMS (EI, m/z) calcd. for C21H18N4 (M+1)+ 327.16042; found 327.16058.

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
280
N-Octylpyridin-2-amine
The compound was prepared as described in the general procedure A (m = 175 mg, 85% isolated yield). White solid. 1H NMR (300 MHz, CDCl3): δ 8.05-8.03 (m, 1H), 7.39-7.33 (m, 1H), 6.52 (dd, J = 6.7, 5.4 Hz1H), 6.33 (d, J = 8.4 Hz, 1H), 4.69 (s, 1H), 3.23-3.17 (m, 2H), 1.63 – 1.53 (m, 2H), 1.29 – 1.23 (m, 10H), 0.99 – 0.54 (m, 3H). 13C NMR (75 MHz, CDCl3): δ 159.0, 148.2, 137.3, 112.5, 106.3, 42.3, 31.8, 29.6, 29.4, 29.3, 27.1, 22.7, 14.1. N-Hexylpyridin-2-amine
The compound was prepared as described in the general procedure A (m = 135 mg, 76% isolated yield). Colorless oil. 1H NMR (300 MHz, CDCl3): δ 8.04-8.02 (m, 1H), 7.38-7.33 (m, 1H), 6.52-6.47 (m, 1H), 6.32 (d, J = 8.5 Hz, 1H), 4.70 (s, 1H), 3.23-3.16 (m, 2H), 1.62 – 1.52 (m, 2H), 1.40 – 1.24 (m, 6H), 0.88 – 0.83(m, 3H). 13C NMR (75 MHz, CDCl3): δ 159.0, 148.2, 137.4, 112.5, 106.3, 42.3, 31.6, 29.5, 26.7, 22.6, 14.1. N-Pentylpyridin-2-amine
The compound was prepared as described in the general procedure A (m = 141 mg, 86% isolated yield White solid; 1H NMR (300 MHz, CDCl3): δ 8.03 – 78.01 (m, 1H), 7.34 (ddd, J = 8.7, 7.1, 1.8 Hz, 1H), 6.50-6.45 (m, 1H), 6.31 (d, J = 8.5 Hz, 1H), 4.77 (s, 1H), 3.21-3.15 (m, 2H), 1.61-1.51 (m, 2H), 1.34 – 1.28 (m, 4H), 0.88 – 0.83 (m, 3H). 13C NMR (75 MHz, CDCl3): δ 159.0, 148.1, 137.3, 112.4, 106.2, 42.2, 31.4, 29.2, 22.4, 13.9. GCMS-EI (70 eV): m/z (%) = 164 (M+, 15), 121 (28), 107 (100), 94 (32), 78 (33). N-Butylpyridin-2-amine

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
281
The compound was prepared as described in the general procedure A (m = 140 mg, 93% isolated yield). White solid. 1H NMR (300 MHz, CDCl3): δ 8.03-8.00 (m, 1H), 7.33 (ddd, J = 8.8, 7.1, 1.9 Hz, 1H), 6.50-6.44 (m, 1H), 6.30 (d, J = 8.5 Hz, 1H), 4.78 (s, 1H), 3.34 – 3.00 (m, 2H), 1.62 – 1.46 (m, 2H), 1.44 – 1.29 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 159.0, 148.0, 137.3, 112.4, 106.3, 41.9, 31.6, 20.2, 13.8. GCMS-EI (70 eV): m/z (%) = 150 (M+, 18), 121 (30), 107 (100), 94 (30), 78 (39). N-Ethylpyridin-2-amine
The compound was prepared as described in the general procedure A (m = 59 mg, 49% isolated yield). Yellow oil. 1H NMR (300 MHz, CDCl3): δ 8.07 – 7.80 (m, 1H), 7.40 (ddd, J = 8.6, 7.0, 1.8 Hz, 1H), 6.57 – 6.50 (m, 1H), 6.39 (d, J = 8.5, 1H), 4.59 (br. s, 1H), 3.32-3.24 (m, 2H), 1.24 (t, J = 7.2 Hz, 3H).13C NMR (75 MHz, CDCl3): δ 158.8, 148.0, 137.6, 112.7, 106.5, 37.0, 14.9. GCMS-EI (70 eV): m/z (%) = 122 (M+, 39), 107 (100), 94 (31), 78 (52), 67 (18). (E)-N-Benzyl-4-styrylaniline
The compound was prepared as described in the general procedure A (m = 251 mg, 88% isolated yield). Yellow solid. 1H NMR (300 MHz, CDCl3): δ 7.49 – 7.46 (m, 2H), 7.38 – 7.21 (m, 10H), 7.10 – 6.88 (m, 2H), 6.68 – 6.66 (m, 2H), 4.88 (s, 1H), 4.37 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 147.2, 138.8, 138.1, 128.8, 128.7, 127.8, 127.7, 127.5, 126.9, 126.2, 125.3, 125.1, 120.7, 113.6, 48.7. GCMS-EI (70 eV): m/z (%) = 285 (M+, 100), 194 (32), 167 (21), 165 (19), 91 (28). HRMS (EI, m/z) calcd. for C21H19N (M)+ 285.15120; found 285.15112. (E)-N-(4-Chlorobenzyl)-4-styrylaniline
The compound was prepared as described in the general procedure A (m = 307 mg , 96% isolated yield). Yellow solid. 1H NMR (300 MHz,CDCl3): δ 7.51 – 7.44 (m, 2H), 7.39 – 7.33 (m, 3H), 7.32 – 7.29 (m, 4H), 7.25 – 7.18 (m, 1H), 7.03 (d, J = 16.3 Hz, 1H), 6.91 (d, J = 16.3 Hz, 1H), 6.70 – 6.55 (m, 2H), 4.34 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 147.1, 138.1, 137.5, 133.2, 129.6, 129.3, 128.9,

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
282
128.7, 127.8, 127.0, 126.2, 125.1, 120.9, 113.5, 47.9. GCMS- EI (70 eV): m/z (%) = 319 (M+, 100), 194 (51), 178 (12), 167 (27), 165 (24), 152 (17), 127 (12), 125 (30), 89 (13). (E)-N-(3,5-Dimethylbenzyl)-4-styrylaniline
The compound was prepared as described in the general procedure A (m = 306 mg, 97% isolated yield). Yellow solid; 1H NMR (300 MHz, CDCl3): δ 7.53 – 7.43 (m, 2H), 7.41 – 7.29 (m, 4H), 7.25 – 7.17 (m, 1H), 7.09 – 6.86 (m, 5H), 6.66 (dd, J = 9.0, 2.1 Hz, 2H), 4.28 (s, 2H), 2.32 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 148.5, 138.9, 138.4, 138.2, 129.2, 128.9, 128.7, 127.9, 127.4, 126.9, 126.2, 125.6, 124.8, 113.4, 48.7, 21.4. GCMS-EI (70 eV): m/z (%) = 313 (M+, 100), 194 (18), 167 (11), 165 (11), 119 (49). (E)-N-(Benzo[d][1,3]dioxo-5-ylmethyl)-4-styrylaniline
The compound was prepared as described in the general procedure A (m = 306 mg, 93% isolated yield). Yellow solid. 1H NMR (300 MHz,CDCl3): δ 7.54 – 7.43 (m, 2H), 7.41 – 7.28 (m, 4H), 7.25 – 7.17 (m, 1H), 7.04 (d, J = 16.3 Hz, 1H), 6.98 – 6.74 (m, 4H), 6.67 – 6.55 (m, 2H), 5.96 (s, 2H), 4.27 (s, 2H), 4.15 (s, 1H). 13C NMR (75 MHz, CDCl3): δ 148.1, 147.8, 146.9, 138.2, 133.1, 128.9, 128.7, 127.9, 127.2, 126.9, 126.2, 124.8, 120.7, 113.1, 108.5, 108.1, 101.2, 48.2. GCMS-EI (70 eV): m/z (%) = 329 (M+, 62), 165 (9), 135 (100), 77(13). (E)-4-Styryl-N-(thiophen-2-ylmethyl)aniline
The compound was prepared as described in the general procedure A (m = 263 mg, 90% isolated yield). Yellow solid. 1H NMR (300 MHz,CDCl3): δ 7.53 – 7.44 (m, 2H), 7.43 – 7.29 (m, 5H), 7.25 – 7.17 (m, 2H), 7.12 – 6.85 (m, 3H), 6.69 – 6.61 (m, 2H), 4.38 (s, 2H), 4.11 (s, 1H). 13C NMR (75 MHz,CDCl3): δ 147.8, 140.3, 138.2, 128.9, 128.7, 127.9, 127.3, 127.2, 126.9, 126.4, 126.2, 124.8, 121.9, 113.1, 43.8. GCMS-EI (70 eV): m/z (%) = 291 (M+, 100), 194 (35), 178 (10), 165 (17), 152 (12), 97 (49).

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
283
Indole
The compound was prepared as described in the general procedure (m = 115 mg, 98% isolated yield).White crystals. 1H NMR (400 MHz, CDCl3): δ 7.81 – 7.74 (s, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.34 – 7.28 (m, 1H), 7.28 – 7.22 (m, 1H), 7.21 – 7.16 (m, 1H), 6.69 – 6.62 (m, 1H). 13C NMR (101 MHz, CDCl3): δ 135.8, 127.9, 124.3, 122.0, 120.8, 119.9, 111.2, 102.6. GCMS-EI (70 eV): m/z (%) = 117 (M+, 100), 116 (9.66), 90 (41), 89 (34), 63 (14). N-Methyl-p-toluidine
The compound was prepared as described in the general procedure B (m = 104 mg, 86% isolated yield). Yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 6.89 (d, J = 8.1 Hz, 2H), 6.44 (d, J = 8.4 Hz, 2H), 5.36 – 5.19 (m, 1H), 2.62 (d, J = 5.1 Hz, 3H), 2.14 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 147.9, 129.5, 124.1, 112.0, 30.2, 20.3. GCMS-EI (70 eV): m/z (%) = 121 (M+, 70), 120 (100), 106 (8), 91 (18), 77 (10), 65 (7). 4-Ethyl-N-methylaniline
The compound was prepared as described in the general procedure B (m = 115 mg, 85% isolated yield). Brownish oil. 1H NMR (300 MHz, DMSO-d6): δ = 7.03 – 6.80 (m, 2H), 6.60 – 6.35 (m, 2H), 5.53 – 5.05 (m, 1H), 2.63 (d, J = 4.1 Hz, 3H), 2.44 (q, J = 7.6 Hz, 2H), 1.10 (t, J = 7.6 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ = 148.1, 130.9, 128.3, 112.0, 30.2, 27.6, 16.4. GCMS-EI (70 eV): (EI, 70eV): m/z (%) = 135 (M+, 37), 120 (100), 119 (9), 91 (8), 77 (9). 4-tert-Butyl-N-methylaniline

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
284
The compound was prepared as described in the general procedure B (m = 139 mg, 85% isolated yield). Yellow oil. 1H NMR (300 MHz, CDCl3): δ 7.35 – 7.27 (m, 2H), 6.70 – 6.59 (m, 2H), 3.62 (s, 1H), 2.88 (s, 3H), 1.37 (s, 9H). 13C NMR (75 MHz, CDCl3): δ 147.1, 140.1, 126.0, 112.3, 33.9, 31.7, 31.0. GCMS-EI (70 eV): m/z (%) = 163 (M+, 21), 149 (11), 148 (100), 133 (15), 132 (11), 120 (25).
4-Methoxy-N-methylaniline
The compound was prepared as described in the general procedure B (m = 126 mg, 92% isolated yield). Yellow solid. 1H NMR (300 MHz, CDCl3): δ = 6.91 – 6.75 (m, 2H), 6.69 – 6.53 (m, 2H), 3.77 (s, 3H), 3.46 (bs, 1H), 2.82 (s, 3H). 13C NMR (75 MHz, CDCl3): δ = 152.1, 143.7, 114.9, 113.7, 55.9, 31.7. GCMS-EI (70 eV): m/z (%) = 137 (M+, 51), 122 (100), 94 (48), 65 (17). 4-Ethoxy-N-methylaniline
The compound was prepared as described in the general procedure B (m = 133 mg, 88% isolated yield). Yellow oil. 1H NMR (300 MHz, DMSO-d6): δ 6.75 – 6.64 (m, 2H), 6.56 – 6.37 (m, 2H), 5.08 (s, 1H), 3.86 (q, J = 7.0 Hz, 2H), 2.61 (s, 3H), 1.25 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 150.0, 144.5, 115.6, 112.8, 63.6, 30.7, 15.1. GCMS-EI (70 eV): m/z (%) = 151 (M+, 35), 123 (16), 122 (100), 94 (20), 65 (8). 3-Iodo-N-methylaniline
The compound was prepared as described in the general procedure B (m = 168 mg, 94% isolated yield). Yellow oil. 1H NMR (300 MHz, DMSO-d6): δ 6.94 – 6.74 (m, 3H), 6.60 – 6.43 (m, 1H), 5.80 (q, J = 4.6 Hz, 1H), 2.62 (d, J = 5.1 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 151.5, 131.0, 124.0, 119.8, 111.3, 95.7, 29.6. GCMS-EI (70 eV): m/z (%) = 233 (M+, 100), 232 (22), 106 (17), 105 (8), 104 (9), 79 (12), 77 (18).

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
285
N-Methyl-3-vinylaniline
The compound was prepared as described in the general procedure B (m = 89 mg, 67% isolated yield). Yellow oil. 1H NMR (300 MHz,CDCl3): δ 7.19 (t, J = 7.7 Hz, 1H), 6.94-6.79 (m, 1H), 6.79-6.63 (m, 2H), 6.63-6.53 (m, 1H), 5.76 (dd, J = 17.6, 1.0 Hz, 1H), 5.25 (dd, J = 10.9, 1.0 Hz, 1H), 3.68 (s, 1H), 2.87 (s, 3H). 13C NMR (75 MHz,CDCl3): δ 149.5, 138.6, 137.4, 129.4, 115.7, 113.5, 112.3, 110.1, 30.9. GCMS-EI (70 eV): m/z (%) = 133 (M+, 100), 132 (99), 117 (11), 104 (12), 103 (12), 77 (17). 4-Bromo-N-methylaniline
The compound was prepared as described in the general procedure B (m = 162 mg, 87% isolated yield). Brownish oil. 1H NMR (300 MHz,CDCl3): δ 7.39 – 7.14 (m, 2H), 6.59 – 6.38 (m, 2H), 3.70 (s, 1H), 2.81 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 148.3, 131.9, 114.1, 108.9, 30.8. GCMS-EI (70 eV): m/z (%) = 186 (M+, 85), 185 (100), 106 (11), 105 (37), 104 (26), 91 (19), 79 (15), 78 (17), 77 (28), 76 (15), 75 (16), 74 (17), 65 (11), 64 (12), 63 (22), 51 (13), 50 (20). N-Methylpyridin-3-amine
The compound was prepared as described in the general procedure B (m = 96 mg, 89% isolated yield). Yellow oil. 1H NMR (300 MHz,CDCl3): δ 8.00 (d, J = 2.6 Hz, 1H), 7.92 (d, J = 4.2 Hz, 1H), 7.06 (dd, J = 8.3, 4.6 Hz, 1H), 6.94 – 6.73 (m, 1H), 3.87 (s, 1H), 2.81 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 145.3, 138.4, 135.7, 123.8, 118.1, 30.3. GCMS-EI (70 eV): m/z (%) = 108 (M+, 100), 107 (97), 80 (16), 78 (22), 66 (12), 52 (14), 51 (20), 50 (14), 39 (24), 38 (11). 2-Iodo-N-methylaniline

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
286
The compound was prepared as described in the general procedure B (m = 133 mg, 57% isolated yield). Yellow oil. 1H NMR (300 MHz, CDCl3): δ 7.68 (dd, J = 7.8, 1.5 Hz, 1H), 7.33 – 7.17 (m, 1H), 6.58 (dd, J = 8.2, 1.4 Hz, 1H), 6.52 – 6.42 (m, 1H), 4.27 (s, 1H), 2.90 (s, 3H). 13C NMR (75 MHz,CDCl3): δ 148.2, 138.9, 129.5, 118.6, 110.1, 85.2, 31.1. GCMS-EI (70 eV): m/z (%) = 233 (M+, 100), 232 (34), 127 (28), 106 (16), 105 (12), 104 (15), 79 (15), 78 (12), 77 (32), 51 (10). 2-Ethyl-N-methylaniline
The compound was prepared as described in the general procedure B (m = 99 mg, 73% isolated yield). Yellow oil. 1H NMR (300 MHz, CDCl3): δ 7.18 (td, J = 7.8, 1.6 Hz, 1H), 7.09 (dd, J = 7.4, 1.5 Hz, 1H), 6.74 (td, J = 7.4, 1.0 Hz, 1H), 6.69 – 6.61 (m, 1H), 3.88 (s, 1H), 2.91 (s, 3H), 2.50 (q, J = 7.5 Hz, 2H), 1.26 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ 147.2, 127.8, 127.2, 117.3, 109.8, 31.1, 23.9, 13.0. GCMS-EI (70 eV): m/z (%)= 135 (M+, 46), 120 (100), 119 (12), 118 (13), 91 (25), 77 (13).
N-Methyl-2,3-dihydrobenzo[b][1,4]dioxin-6-amine
The compound was prepared as described in the general procedure B (m = 143 mg, 86% isolated yield). Brown oil. 1H NMR (400 MHz, CDCl3): δ = 6.73 (d, J = 7.8 Hz, 1H), 6.27 – 6.03 (m, 2H), 4.44 – 3.97 (m, 4H), 3.48 (s, 1H), 2.80 (s, 3H). 13C NMR (101 MHz, CDCl3): δ = 144.7, 144.1, 135.6, 117.6, 106.5, 101.1, 64.8, 64.2, 31.5. GCMS-EI (70 eV): m/z (%) = 165 (M+, 100) 110 (13), 109 (91), 108 (99), 81 (12), 80 (26), 55 (12), 52 (12), 51 (11).
N-Methyl-9H-fluoren-2-amine
The compound was prepared as described in the general procedure B (m = 137 mg, 70% isolated yield).Yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.64 (dt, J = 7.7, 0.9 Hz, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.48 (dt, J = 7.5, 1.0 Hz, 1H), 7.37 – 7.29 (m, 1H), 7.19 (td, J = 7.4, 1.1 Hz, 1H), 6.82 (q, J = 1.1 Hz, 1H),

Part 4 Chapter 2 Manganese catalyzed N-alkylation of amines with alcohols
287
6.65 (dd, J = 8.2, 2.2 Hz, 1H), 3.84 (s, 3H; NH + Ar-CH2-Ar), 2.91 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 149.0, 145.3, 142.5, 142.3, 131.9, 126.7, 124.9, 124.8, 120.7, 118.5, 111.8, 108.9, 37.1, 31.2 . GCMS-EI (70 eV): m/z (%) = 196 (14), 195 (M+, 100), 194 (59), 180 (15), 165 (30), 152 (21). N-Methyl-2-(1-H-pyrrol-1-yl)aniline
The compound was prepared as described in the general procedure B (m = 90 mg, 52% isolated yield). White solid. 1H NMR (400 MHz,CDCl3): δ 7.32 (td, J = 7.8, 1.6 Hz, 1H), 7.18 (dd, J = 7.7, 1.4 Hz, 1H), 6.84 (t, J = 2.1 Hz, 2H), 6.81 – 6.72 (m, 2H), 6.39 (t, J = 2.1 Hz, 2H), 3.88 (s, 1H), 2.83 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 144.9, 129.1, 127.3, 127.0, 122.0, 116.4, 110.6, 109.4, 30.4. GCMS-EI (70 eV): m/z (%) = 172 (M+, 65), 171 (100), 156 (14), 145 (14), 77 (12).

288
6. References [1] S. A. Lawerence, in in Amines: Synthesis properties, and applications,, Cambridge,
Cambridge University, 2004. [2] A. S. Travis, E. Z. Rappoport), in in the Chemistry of Anilines, Vol. Vol. 1 Wiley-
Interscience, New York, 2007, p. 717. [3] J. Magano, J. R. Dunetz, Chem. Rev. 2011, 111, 2177-2250. [4] Y. Aubin, C. Fischmeister, C. M. Thomas, J.-L. Renaud, Chem. Soc. Rev. 2010, 39, 4130-
4145. [5] S. L. Buchwald, C. Mauger, G. Mignani, U. Scholz, Adv. Synth. Catal. 2006, 348, 23-39. [6] E. Sperotto, G. P. M. van Klink, G. van Koten, J. G. de. Vries, Dalton Trans. 2010, 39,
10338-10351. [7] F. Monnier, M. Taillefer, Angew. Chem. Int. Ed. 2008, 47, 3096-3099. [8] L. Huang, M. Arndt, K. Gooßen, H. Heydt, L. J. Gooßen, Chem. Rev. 2015, 115, 2596-
2697. [9] K. Alex, A. Tillack, N. Schwarz, M. Beller, ChemSusChem 2008, 1, 333-338. [10] D. Crozet, M. Urrutigoïty, P. Kalck, ChemCatChem 2011, 3, 1102-1118. [11] D. B. Bagal, B. M. Bhanage, Adv. Synth. Catal. 2015, 357, 883-900. [12] R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V.
Schünemann, A. Brückner, M. Beller, Science 2013, 342, 1073-1076. [13] K. Junge, B. Wendt, N. Shaikh, M. Beller, Chem. Commun. 2010, 46, 1769-1771. [14] T. C. Nugent, M. El-Shazly, Adv. Synth. Catal. 2010, 352, 753-819. [15] Q. Yang, Q. Wanga, Z. Yu, Chem. Soc. Rev. 2015, 44, 2305-2329. [16] G. E. Dobereiner, R. H. Crabtree, Chem. Rev. 2010, 110, 681-703. [17] M. H. S. A. Hamid, P. A. Slatford, J. M. J. Williams, Adv. Synth. Catal. 2007, 349, 1555-
1575. [18] S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann, M. Beller, ChemCatChem 2011, 3,
1853-1864. [19] G. Guillena, D. J. Ramón, M. Yus, Chem. Rev. 2010, 110, 1611-1641. [20] C. Gunanathan, D. Milstein, Science 2013, 341, 249-260. [21] A. Nandakumar, S. P. Midya, V. G. Landge, E. Balaraman, Angew. Chem. Int. Ed. 2015,
54, 11022-11034. [22] K.-i. Shimizu, N. Imaiida, K. Kon, S. M. A. Hakim Siddiki, A. Satsuma, ACS Catal.
2013, 3, 998-1005. [23] H. Yang, X. Cui, X. Dai, Y. Deng, F. Shi, Nat. Commun. 2015, 6, 6478. [24] X. Liu, P. Hermange, J. Ruiz, D. Astruc, ChemCatChem 2016, 8, 1043-1045. [25] N. Botta, D. de Angelis, R. Nicoletti, Synthesis 1977, 722-723. [26] R. E. Vultier, A. Baiker, A. Wokaun, Appl. Catal. 1987, 30, 167-176. [27] Y. Watanabe, Y. Tsuji, Y. Ohsugi, Tetrahedron Lett. 1981, 22, 2667-2670. [28] R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit, N. Tongpenyai, J. Chem. Soc., Chem.
Commun. 1981, 611. [29] R. H. Crabtree, Organometallics 2011, 30, 17-19. [30] S. Wöckel, P. Plessow, M. Schelwies, M. K. Brinks, F. Rominger, P. Hofmann, M.
Limbach, ACS Catal. 2014, 4, 152-161.

289
[31] Y. Zhang, C.-S. Lim, D. S. B. Sim, H.-J. Pan, Y. Zhao, Angew. Chem., Int. Ed. 2014, 53, 1399-1403.
[32] S. Ruch, T. Irrgang, R. Kempe, Chem. Eur. J. 2014, 20, 13279-13285. [33] P. Qu, C. Sun, J. Ma, F. Li, Adv. Synth. Catal. 2014, 356, 447-459. [34] Y.-H. Chang, Y. Nakajima, F. Ozawa, Organometallics 2013, 32, 2210-2215. [35] V. R. Jumde, L. Gonsalvi, A. Guerriero, M. Peruzzini, M. Taddei, Eur. J. Org. Chem.
2015, 1829-1833. [36] V. R. Jumde, E. Cini, A. Porcheddu, M. Taddei, Eur. J. Org. Chem. 2015, 1068−1074. [37] E. Balaraman, D. S. Y. Diskin-Posner, D. Milstein, Catal. Lett. 2015, 145, 139-144. [38] S. P. Shan, X. Xie, G. Boopathy, T. D. Tuan, R. Balamurugan, V. H. Han, M. S. Abdul,
RSC Adv. 2015, 5, 4434-4442. [39] N. J. Oldenhuis, V. M. Dong, Z. Guan, J. Am. Chem. Soc. 2014, 136, 12548-12551. [40] A. B. Enyong, B. Moasser, J. Org. Chem. 2014, 79, 7553-7563. [41] M. Chen, M. Zhang, F. Xie, X. Wang, H. Jiang, ChemCatChem 2014, 6, 2993−2997. [42] Y. Zhang, C.-Si Lim, D. S. B. Sim, H.-J. Pan, Y. Zhao, Angew. Chem. Int. Ed. 2014, 53,
1399-1403. [43] D. Hollmann, ChemSusChem 2014, 7, 2411-2413. [44] Y. Z. Z.-Q. Rong, R. H. B. Chua, H.-J. Pan, Y. Zhao, J. Am. Chem. Soc. 2015, 137, 4944-
4947. [45] C.-S. L. Y. Zhang, ; D. S. B. Sim, H.-J. Pan, Y. Zhao, Angew. Chem. Int. Ed. 2014, 53,
1399-1403. [46] H. N. M. Pena-Lopez, M. Beller, Angew. Chem. Int. Ed. 2016, 55, 7826-7830. [47] X. Cui, F. Shi, Yan Zhang, Y. Deng, Tetrahedron Lett. 2010, 51, 2048-2051. [48] Y. Tao, B. L. Feringa, K. Barta, Nat. Commun. 2014, 5, 5602. [49] T. Yan, B. L. Feringa, K. Barta, ACS Catal. 2016, 6, 381-388 [50] A. J. Rawlings, L. J. Diorazio, M. Wills, Org. Lett. 2015, 17, 1086-1089. [51] H.-J. Pan, T. W. Ng, Y. Zhao, Chem. Commun. 2015, 51, 11907-11910. [52] B. Emayavaramban, M. Roy, B. Sundararaju, Chem. Eur. J. 2016, 22, 3952-3955. [53] G. Zhang, Z. Yin, S. Zheng, Org. Lett. 2016, 18, 300-303. [54] S. Rosler, M. Ertl, T. Irrgang, R. Kempe, Angew. Chem. Int. Ed. 2015, 54, 15046-15050. [55] S.Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K.
Junge, M. Beller, J. Am. Chem. Soc. 2016, 138, 8809-8814. [56] M. A. Gonzalez, M. A. Yim, S. Cheng, A. Moyes, A. J. Hobbs, P. K. Mascharak, Inorg.
Chem. 2012, 51, 601-608. [57] C. Lu, Y. Guo, J. Yan, Z. Luo, H.-B. Luo, M. Yan, L. Huang, X. Li, J. Med. Chem. 2013,
56, 5843-5859. [58] W. H. Lin, H. F. Chang, Catal. Today 2004, 97, 181-188. [59] G. Lamoureux, C. Aguero, ARKIVOC 2009, 251-264. [60] T. T. Dang, B. Ramalingam, A. M. Seayad, ACS Catal. 2015, 5, 4082-4088. [61] A. Del Zotto, W. Baratta, M. Sandri, G. Verardo, P. Rigo, Eur. J. Inorg. Chem. 2004,
2004, 524-529. [62] F. Li, J. Xie, H. Shan, C. Sun, L. Chen, RSC Adv. 2012, 2, 8645-8652. [63] J. Campos, L. S. Sharninghausen, M. G. Manas, R. H. Crabtree, Inorg. Chem. 2015, 54,
5079-5084.

290
General conclusion

291

292
General conclusion The main objective of this thesis was to develop new catalytic systems based on earth
abundant metals for hydrogenation and hydrogen borrowing reactions using either reported or
new well-defined iron and manganese complexes.
A new family of NHC-substituted iron Knölker type complexes has been synthesized by
UV promoted substitution of one CO-ligand by the corresponding NHC ligand. All the
complexes have been obtained in good yields and fully characterized. Their potential in catalysis
has been demonstrated in the case of the dehydration of primary benzamides into benzonitrile
derivatives using the inexpensive PMHS (polymethylhydrosiloxane) as the dehydrating reagent.
Novel Fe pincer complexes have been synthesized and used the effective ester
hydrogenation by means of the second generation iron PNP pincer complexes. These catalytic
systems gave improved results for the selective hydrogenation of various aromatic and aliphatic
esters including diester motifs and lactones. The same catalytic systems were used for the nitrile
hydrogenation.

293
A new family of manganese pincer complexes has been developed and their catalytic
applications have been studied in nitrile (C10 and C11) and ester hydrogenation (C10-C15).
These manganese catalytic systems allowed for the selective reduction of various nitriles and
esters in the presence of catalytic amount of base.
The first iron-catalyzed -alkylation of ketones with primary alcohols has been developed
in the presence of a catalytic amount of base. The key to success for this novel transformation is
the use of a Knölker-type iron complex as catalyst. The optimized catalytic system permitted the
development of the first iron-catalyzed Friedländer annulation reaction starting from 2-
aminobenzyl alcohols.

294
N-alkylation of amines with alcohols catalyzed by manganese pincer complexes is
presented. This catalytic system is effective for the monoalkylation of anilines and tolerates a
variety of functional groups. N-methylations of amines using methanol was also performed and it
is the first examples of this transformation using non-noble metal complexes under mild
conditions in the presence of base.
Let’s compare activity of the Fe, Mn pincer complexes with well-known Ru pincer
complex for hydrogenation reactions such as esters and nitriles reduction reactions. Iron
hydroborato and manganese cationic complexes were found to be less active in ester
hydrogenation compared to the ruthenium complex (TON = 4000) and also reaction proceeded
under mild conditions with ruthenium complex. However earth abundant metal complexes
reported for ester hydrogenation just few months ago. So now the door is opened to use non
noble metal complexes for such ester hydrogenation reactions. In future, these metals will reach
the same level success like noble metal complexes.
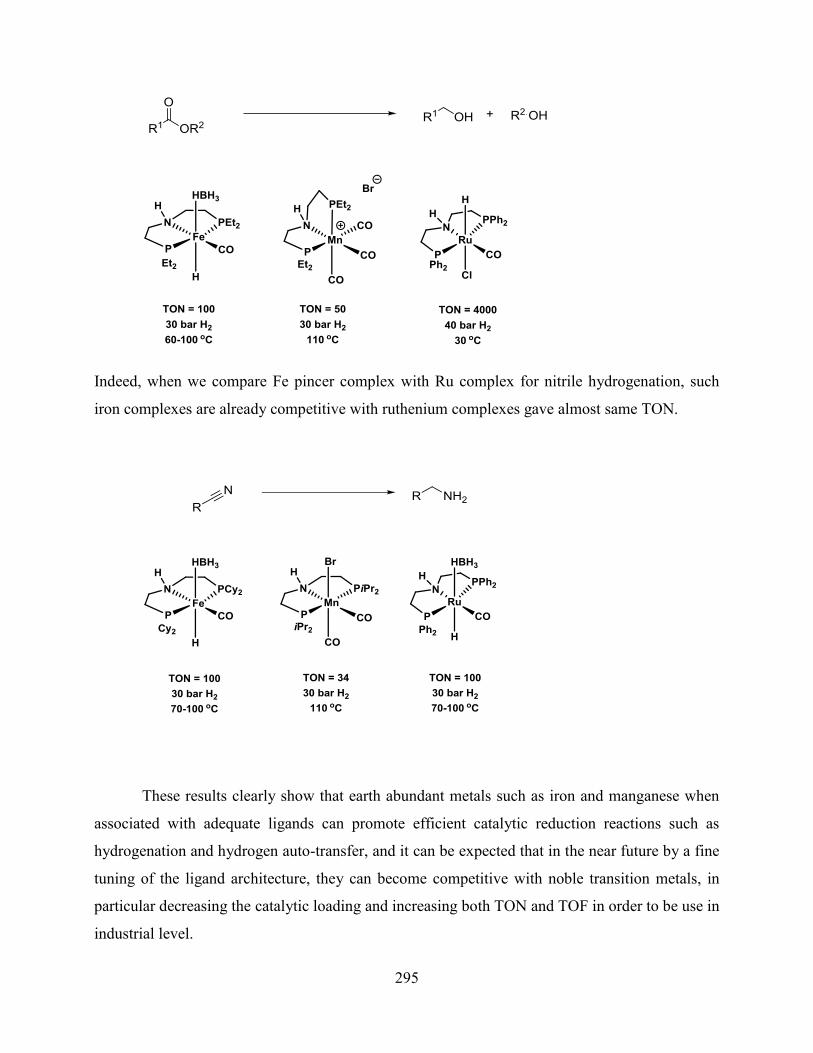
295
Indeed, when we compare Fe pincer complex with Ru complex for nitrile hydrogenation, such
iron complexes are already competitive with ruthenium complexes gave almost same TON.
These results clearly show that earth abundant metals such as iron and manganese when
associated with adequate ligands can promote efficient catalytic reduction reactions such as
hydrogenation and hydrogen auto-transfer, and it can be expected that in the near future by a fine
tuning of the ligand architecture, they can become competitive with noble transition metals, in
particular decreasing the catalytic loading and increasing both TON and TOF in order to be use in
industrial level.

296
Annexes
1. List of synthesized complexes
2. List of publications

Annexe1. List of synthesized complexes
297
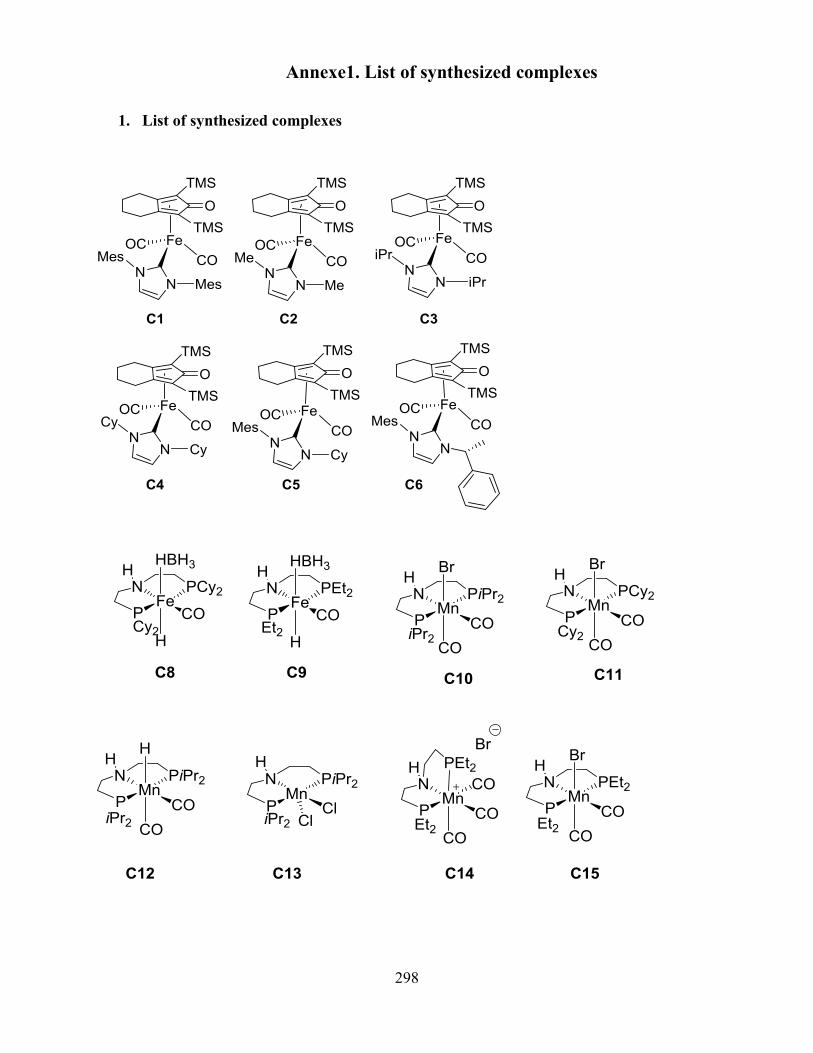
Annexe1. List of synthesized complexes
298
1. List of synthesized complexes

299

300
2. List of publications
1. Knölker’s iron complexes bearing a N-heterocyclic carbene ligand: synthesis,
characterization, and catalytic dehydration of primary amides.
S. Elangovan, S. Quintero-Duque, V. Dorcet, T. Roisnel, L. Norel, C. Darcel, J.-B.
Sortais, Organometallics 2015, 34, 4521-4528.
2. Iron-catalyzed -alkylation of ketones with alcohols.
S. Elangovan, J.-B. Sortais, M. Beller, C. Darcel, Angew. Chem. Int. Ed. 2015, 54, 14483-
14486.
3. Improved second generation iron pincer complexes for effective ester hydrogenation.
S. Elangovan, B. Wendt, C. Topf, S. Bachmann, M. Scalone, A. Spannenberg, H. Jiao,
W. Baumann, K. Junge, M. Beller, Adv. Synth. Catal. 2016, 358, 820-825.
4. Selective catalytic reductions of nitriles, ketones and aldehydes by well-defined
manganese pincer complexes.
S.Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K.
Junge, M. Beller, J. Am. Chem. Soc. 2016, 138, 8809-8814.
5. Efficient and selective N-alkylation of amines with alcohols catalyzed by manganese
pincer complexes.
S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel, M. Beller, Nat. Commun.
2016, 7, 12641.
6. Selective catalytic hydrogenation of nitriles to primary amines using iron pincer
complexes.
S. Lange, S. Elangovan, C. Cordes, A. Spannenberg, H. Junge, S. Bachmann, M. Scalone,
C. Topf, K. Junge, M. Beller, Cat. Sci. Technol. 2016, 6, 4768-4772.
7. Hydrogenation of esters to alcohols catalyzed by defined molecular manganese
pincer complexes.
S. Elangovan, M. Garbe, K. Junge, H. Jiao, A. Spannenberg, M. Beller, Angew. Chem.
Int. Ed. 2016, 55, 15364–15368.

301
8. Molecularly Defined Manganese Pincer Complexes for Selective Transfer
Hydrogenation of Ketones.
M. Perez, S. Elangovan, A. Spannenberg, K. Junge, M. Beller, ChemSusChem 2017, 10,
83-86.
9. Manganese catalyzed hydrogen autotransfer C-C bond formation: α-Alkylation of
ketones with primary alcohols.
M. Pena-Lopez, P. Piehl, S. Elangovan, H. Neumann, M. Beller, Angew. Chem. Int. Ed.
2016, 55, 14967-14971.
10. Selective catalytic two-step process for ethylene glycol from carbon monoxide.
K. Dong, S. Elangovan, R. Sang, A. Spannenberg, R. Jackstell, K. Junge, Y. Li, M.
Beller, Nat. Commun. 2016, 7, 12075.
11. Half-Sandwich Manganese Complexes Bearing Cp Tethered
N-Heterocyclic Carbene Ligands: Synthesis and Mechanistic Insights into the
Catalytic Ketone Hydrosilylation
D. A. Valyaev, D. Wei, S. Elangovan, M. Cavailles, V. Dorcet, J.-B. Sortais, C. Darcel,
N. Lugan, Organometallics 2016, 35, 4090–4098.
12. Improved and general manganese catalyzed N-methylation of aromatic amines using
methanol
J. Neumann, S. Elangovan, A. Spannenberg, K. Junge, Matthias Beller, Chem. Eur. J.
2017, 23, 5410-5413.
13. A stable manganese pincer catalyst for the selective dehydrogenation of methanol
M. A-Fernandez, L. K. Vogt, S. Fischer, W. Zhou, H. Jiao, M. Garbe, S. Elangovan, K.
Junge, H. Junge, R. Ludwig, M. Beller, Angew. Chem. Int. Ed. 2017, 56, 559–562.

302
Résumé de la thèse en français
Ces travaux de thèse ont ete realises dans l’UMR 6226 CNRS – Université de Rennes 1
« Institut des Sciences Chimiques de Rennes » dans l’equipe « Organométalliques : Matériaux et
Catalyse » sous les directions de Christophe Darcel et Jean-Baptiste Sortais ainsi qu’au LIKAT
Rostock sous les directions de Matthias Beller et Kathrin Junge dans le cadre du LIA
ChemSusCat Rennes-Rostock entre Septembre 2013 et Aout 2016.
L’objectif principal de ce travail de thèse a ete de développer de nouveaux systèmes
catalytiques dérivés de métaux de transition abondants et bon marché pour effectuer des réactions
d’hydrogenations et de prêt d’hydrogène (hydrogen borrowing) de derives carbonyles ou
carboxyliques. Les deux métaux de transition ciblés dans ce travail ont été le fer et le manganèse
(respectivement premier et troisième métal de transition par son abondance dans l’ecorce
terrestre).
En effet, dans le contexte actuel du développement durable et de la chimie verte, la
catalyse homogène par les complexes de métaux est un des axes de recherche les plus innovants
tant au niveau academique qu’industriel. Depuis l'avènement de la catalyse homogène dans les
années 1970, les métaux nobles tels le palladium, le platine, le rhodium ou le ruthénium ont été
principalement utilisés. Par contre, le cours erratique des métaux précieux dû principalement à
leur raréfaction ou à des conjonctures géopolitiques tendues a des conséquences sur le prix de
revient d’un procede catalytique et peut causer des problèmes de perennites de procedes
industriels. Ainsi, la mise au point de catalyseurs à base de métaux de transition peu onéreux, peu
toxiques, sans perte d’activite par rapport aux catalyseurs correspondants derives de met ux
precieux est toujours un domaine de recherche d’actualite dans lequel la competi ion
internationale est intense.
Le document de thèse est divisé en 4 parties, une introduction générale resituant les
principales realisations dans le domaine des reductions (hydrogenation, transfert d’hydrogène et
hydrosilylation) de liaisons C=O catalysés par des complexes de fer et de manganèse, 3 parties de
résultats et une conclusion générale.

303
Dans la seconde partie, une nouvelle famille de complexes de fer de type Knölker
associés à des ligands carbènes N-hétérocycliques (NHC) a ete syntheti ee par substitution d’un
ligand CO par un ligand NHC promue par une activation UV. Les complexes obtenus ont été
caractérisés par spectroscopie RMN et IR, électrochimie et diffraction RX. Leurs potentiels en
tant que catalyseurs ont été démontrés dans la réaction de déshydratation de benzamides
primaires Ar-CONH2 en dérivés benzonitriles Ar-CN en utilisant le polyméthylhydrosiloxane
(PMHS), silane bon marché, comme agent de déshydratation. (Schéma 1)
Schéma 1 Déshydratation des amides primaires en nitriles catalysée par des complexes de fer.
La troisième partie de ces travaux de thèse a été consacrée aux reactions d’hydrogenation
de dérivés carbonylés et carboxyliques catalysées par des complexes bien définis de métaux non
nobles tels que le fer et le manganèse.
Après une introduction décrivant les principales utilisations de complexes pinces de
met ux de transition de la première periode, le premier chapitre est consacre à l’hydrogenation de
dérivés nitriles catalysée par des complexes pinces de fer et de manganèse.

304
Dans un premier temps, une deuxième génération de complexes pinces de fer C7-C9 possédant
une pince de type aminodiphosphine a été développée. Ces complexes ont prouvé leur efficacité
dans la réaction d’hydrogenation selective de nitriles aromatiques ou hétéroaromatiques en
amines primaires correspondantes dans des conditions modérées (30 bar d’hydrogène à 70-100
°C pendant 3 h, 1 mol% du complexe C8). Dans ces conditions, les groupements type ester sont
tolérés alors que les groupements cétone sont simultanément réduits en alcool. L’hydrogenation
de nitriles aliphatiques en alkylamines primaires est également effectuée avec de bons
rendements. (Schéma 3)
Schéma 3 Hydrogénation des nitriles en amines catalysée par des complexes pinces de fer
Une nouvelle famille de complexes pinces de manganèse a également été développée et leurs
propriet s catalytiques evalue s dans la reaction d’hydrogenation des nitriles. Les complexes C10
et C11 (3 mol%) catalysent la réduction sélective des dérivés nitriles (hétéro)aromatiques et
aliphatiques en amines primaires en présence de 10 mol% de t-BuONa comme base dans le
toluène à 120 °C pendant 24 h sous 50 bar de H2. (Schéma 4)
Schéma 4 Hydrogénation des nitriles en amines catalysée par des complexes pinces de
manganèse
Des études théoriques par calculs DFT suggère un mécanisme par sphère externe, la partie azotée
du ligand pince ayant un rôle actif dans pour le transfert simultane d’un proton alors que le centre
manganèse délivre un hydrure. (Schéma 5)

305
Schéma 5 Mecanisme propose pour l’hydrogenation des nitriles catalysee par des complexes
pinces de manganèse
Le second chapitre decrit l’hydrogenation d’esters aromatiques et aliphatiques en alcools
catalysée par les complexes pinces de fer et de manganèse préparés précédemment. Le complexe
de fer C9 s’est revele le plus actif dans la réaction d’hydrogenation selective d’esters aromatiques
et aliphatiques en alcools dans des conditions moderees (30 bar d’hydrogène à 60-100 °C pendant
6-18 h). (Schéma 6) Dans ces conditions réactionnelles, les cétones et les doubles liaisons C=C
conjuguées sont simultanément réduites, alors que les doubles liaisons non conjuguées sont
tolérées. Les diesters et les lactones sont également réduits en diols dans les mêmes conditions.
Schéma 6 Hydrogénation des esters en alcools catalysée par des complexes pinces de fer
Les complexes pinces de manganèse C14 et C15 ont également montré de bonnes réactivités
pour l’hydrogenation des esters en alcools. (2 mol% de complexe C14 en présence de 10 mol%
de t-BuONa comme base dans le 1,4-dioxane à 110 °C pendant 24 h sous 30 bar de H2). (Schéma
7)
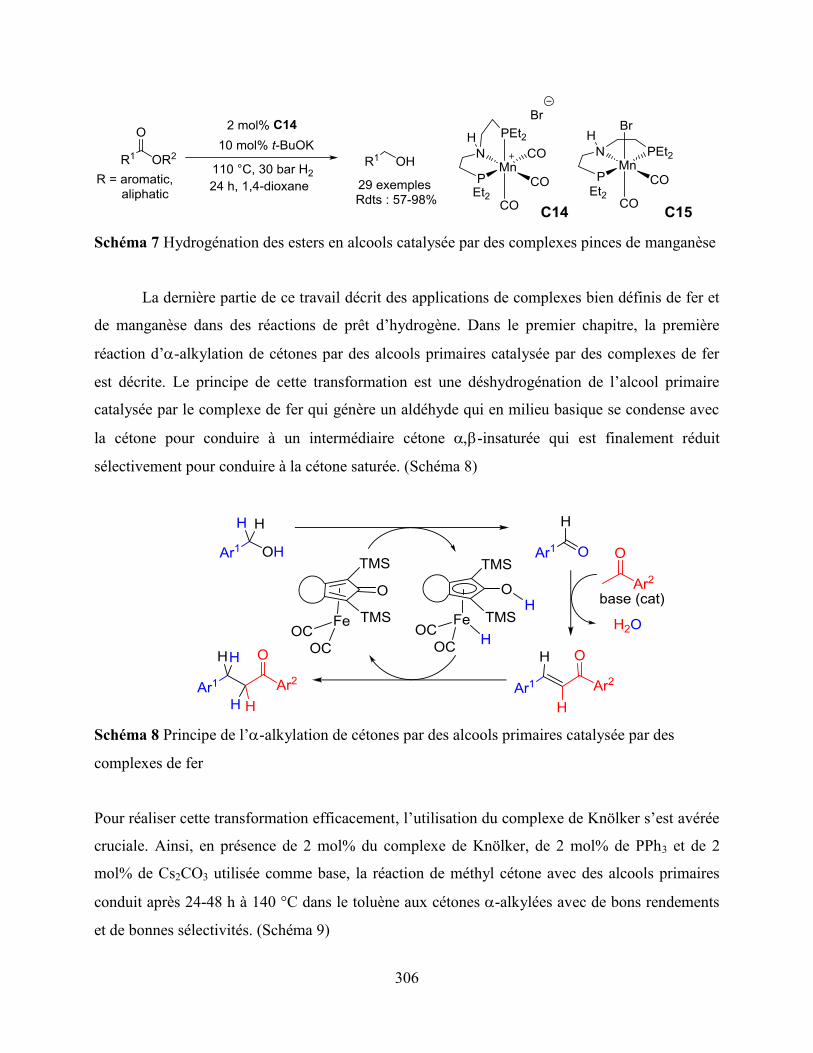
306
Schéma 7 Hydrogénation des esters en alcools catalysée par des complexes pinces de manganèse
La dernière partie de ce travail décrit des applications de complexes bien définis de fer et
de manganèse dans des réactions de prêt d’hydrogène. Dans le premier chapitre, la première
reaction d’-alkylation de cétones par des alcools primaires catalysée par des complexes de fer
est décrite. Le principe de cette transformation est une deshydrogenation de l’alcool primaire
catalysée par le complexe de fer qui génère un aldéhyde qui en milieu basique se condense avec
la cétone pour conduire à un intermédiaire cétone -insaturée qui est finalement réduit
sélectivement pour conduire à la cétone saturée. (Schéma 8)
Schéma 8 Principe de l’-alkylation de cétones par des alcools primaires catalysée par des
complexes de fer
Pour réaliser cette transformation efficacement, l’utilisation du complexe de Knölker s’est averee
cruciale. Ainsi, en présence de 2 mol% du complexe de Knölker, de 2 mol% de PPh3 et de 2
mol% de Cs2CO3 utilisée comme base, la réaction de méthyl cétone avec des alcools primaires
conduit après 24-48 h à 140 °C dans le toluène aux cétones -alkylées avec de bons rendements
et de bonnes sélectivités. (Schéma 9)

307
Schéma 9 -Alkylation de cétones par des alcools primaires catalysée par des complexes de fer
de type Knölker.
Dans les conditions opératoires optimisées précédentes, la première annulation de type
Friedländer catalysée au fer a été décrite à partir de 2-aminobenzylalcool et de cétones. La
meilleure base pour effectuer cette transformation est t-BuOK (10 mol%). (Schéma 10)
Schéma 10 Annulation de Friedländer catalysée par des complexes de fer de type Knölker
Le second chapitre est consacré à la réaction de N-alkylation des amines par des alcools
catalysée par des complexes pinces de manganèse. Le principe de cette transformation est une
deshydrogenation de l’alcool primaire catalysee par le complexe de manganèse qui genère un
alde yde qui se condense avec l’amine pour conduire à un intermediaire aldimine qui est
finalement réduit selectivement pour conduire à l’amine N-alkylée. (Schéma 11)
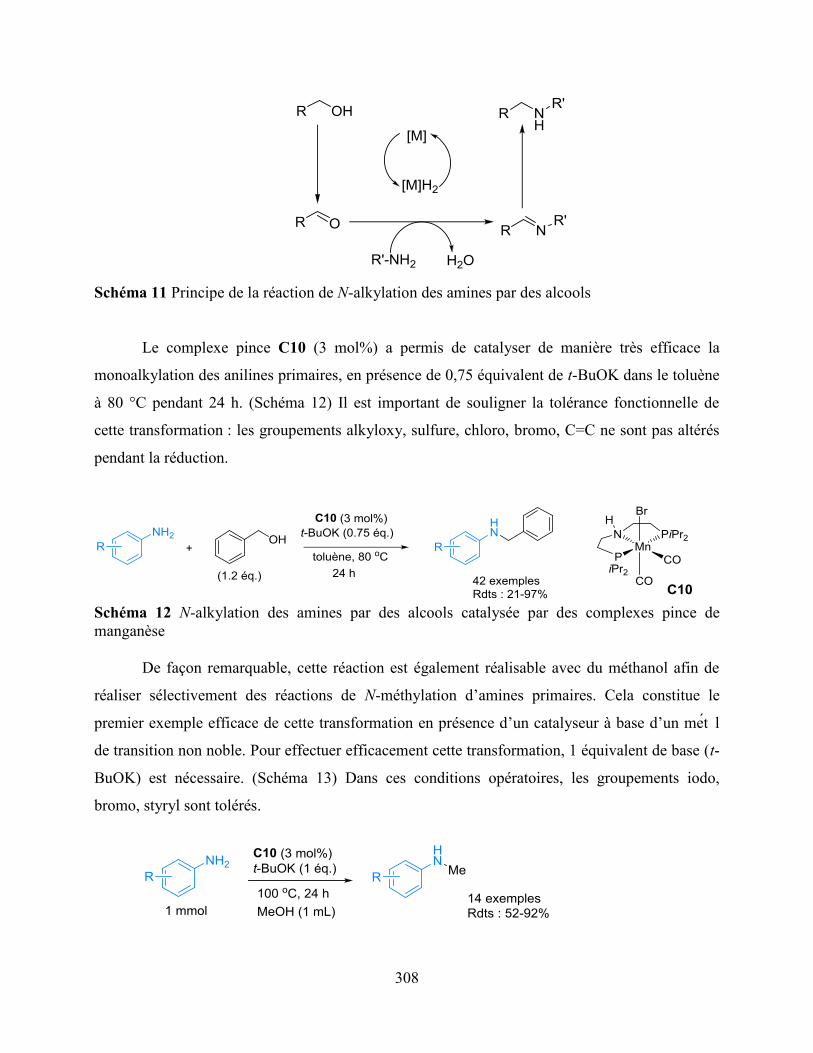
308
Schéma 11 Principe de la réaction de N-alkylation des amines par des alcools
Le complexe pince C10 (3 mol%) a permis de catalyser de manière très efficace la
monoalkylation des anilines primaires, en présence de 0,75 équivalent de t-BuOK dans le toluène
à 80 °C pendant 24 h. (Schéma 12) Il est important de souligner la tolérance fonctionnelle de
cette transformation : les groupements alkyloxy, sulfure, chloro, bromo, C=C ne sont pas altérés
pendant la réduction.
Schéma 12 N-alkylation des amines par des alcools catalysée par des complexes pince de manganèse
De façon remarquable, cette réaction est également réalisable avec du méthanol afin de
réaliser sélectivement des réactions de N-méthylation d’amines primaires. Cela constitue le
premier exemple efficace de cette transformation en présence d’un catalyseur à base d’un met l
de transition non noble. Pour effectuer efficacement cette transformation, 1 équivalent de base (t-
BuOK) est nécessaire. (Schéma 13) Dans ces conditions opératoires, les groupements iodo,
bromo, styryl sont tolérés.

309
Schéma 13 N-méthylation des amines par le méthanol catalysée par des complexes pince de manganèse
Par reaction intramole ulaire, l’indole peut être obtenu à partir du
2-(2-aminophényl)éthanol en présence de 3 mol% du complexe C10, 1 équivalent de t-BuOK,
dans le toluène à 100 °C pendant 48 h. (Schéma 14)
Schéma 14 Formation d’indole à partir de 2-(2-aminophényl)éthanol
En conclusion, ces résultats montrent très clairement que les métaux de transition
abondants, peu toxiques et peu onéreux tels que le fer et le manganèse lorsqu’ils sont associes à
un ligand adéquat peuvent promouvoir efficacement des réactions de réductions catalytiques
telles que les hydrogénations et des réductions par prêt d’hydrogène ou des réactions de
déshydratations.
Les perspectives de ce travail sont nombreuses. En particulier, une fine modulation de
l’architecture des ligands autour du fer ou du manganèse devrait permettre, dans un futur proche,
d’obtenir des catalyseurs plus efficaces (diminution des quantités de catalyseurs utilisés, des
pressions et des températures de réactions, etc.) alors capables de devenir compétitifs avec ceux
issus de métaux nobles, encore classiquement utilisés au niveau industriel.

310
Related Documents











