EUKARYOTIC CELL, June 2010, p. 906–914 Vol. 9, No. 6 1535-9778/10/$12.00 doi:10.1128/EC.00003-10 Copyright © 2010, American Society for Microbiology. All Rights Reserved. Rotenone Enhances the Antifungal Properties of Staurosporine Ana Castro, 1,2 Catarina Lemos, 1,2 Artur Falca ˜o, 1 Andreia S. Fernandes, 1 N. Louise Glass, 3 and Arnaldo Videira 1,4 * IBMC, Instituto de Biologia Molecular e Celular, Universidade do Porto, Rua do Campo Alegre 823, 4150-180 Porto, Portugal 1 ; UFP, Faculdade de Cie ˆncias da Sau ´de, Universidade Fernando Pessoa, Rua Carlos da Maia 296, 4200-150 Porto, Portugal 2 ; Department of Plant and Microbial Biology, University of California, Berkeley, California 94720-3102 3 ; and ICBAS, Instituto de Cie ˆncias Biome ´dicas de Abel Salazar, Universidade do Porto, Largo Prof. Abel Salazar 2, 4099-003 Porto, Portugal 4 Received 5 January 2010/Accepted 24 April 2010 We studied staurosporine-induced cell death in the filamentous fungus Neurospora crassa. The generation of reactive oxygen species during the process appears to be an important signaling event, since addition of the antioxidant glutathione prevents the effects of staurosporine on fungal growth. Selected mutants with muta- tions in respiratory chain complex I are extremely sensitive to the drug, stressing the involvement of complex I in programmed cell death. Following this finding, we determined that the complex I-specific inhibitor rotenone used in combination with staurosporine results in a synergistic and specific antifungal activity, likely through a concerted action on intracellular glutathione depletion. Paradoxically, the synergistic antifungal activity of rotenone and staurosporine is observed in N. crassa complex I mutants and in Saccharomyces cerevisiae, which lacks complex I. In addition, it is not observed when other complex I inhibitors are used instead of rotenone. These results indicate that the rotenone effect is independent of complex I inhibition. The combination of rotenone and staurosporine is effective against N. crassa as well as against the common pathogens Aspergillus fumigatus and Candida albicans, pointing to its usefulness as an antifungal agent. Programmed cell death (PCD) refers to a genetically con- trolled process of cellular suicide initiated by endogenous or extrinsic signals. Many of the genes involved are widely con- served from unicellular to multicellular organisms (46). Apop- tosis and autophagy, with its particular characteristics, have been recognized as the main categories of PCD (27). The process of PCD is crucial for the development and homeostasis of metazoan organisms and has been implicated in a number of human disorders, including cancer and neurodegenerative and infectious diseases (3, 10, 25, 55). The participation in PCD of mitochondria, the cellular or- ganelles responsible for the production of most cellular ATP in eukaryotes (30), has been well established. Particularly, these organelles have a central role in the intrinsic (mitochondrion- dependent) pathway of apoptosis, which includes production of reactive oxygen species (ROS), membrane depolarization, ultrastructural changes, and the release of cytochrome c and other proteins (18, 50, 55). Drugs like staurosporine (STS), an inhibitor of protein kinases, have been used to induce the mitochondrion-dependent pathway of apoptosis (24, 35). Stau- rosporine (48) and derivatives have been used in clinical trials for cancer therapy (63). The complex I inhibitor rotenone too has been widely used to induce PCD and also extensively applied as a pesticide (11, 39, 56). Thus, these types of drugs can be employed for the acquisition of fundamental knowledge and for more practical applications, like modulation of the progression of PCD. Modulation of PCD by targeting metabolic pathways in- volved in the process can be exploited to the benefit of human health in several very significant situations, from cancer ther- apy (4, 57) to the treatment of fungal infections (3, 52). How- ever, the molecular basis of PCD involves complex metabolic networks (32, 38, 59), and further work is required for their identification. Neurospora crassa has many advantages for bio- chemical and genetic experiments (14, 17, 23) and is thus a good model organism for the study of mechanisms of PCD. We are interested in identifying the cell molecular pathways associ- ated with PCD and using this knowledge to devise strategies to modulate the process. In this work, we analyze the effects of STS on the N. crassa wild type and mitochondrial complex I mutants and describe the synergistic effect of combining STS and rotenone on this organism and other human-pathogenic fungi. MATERIALS AND METHODS Strains and growth techniques. The wild-type N. crassa sequenced strain (FGSC 2489) and several deletion strains generated by the Neurospora Genome Project (17) were obtained from the Fungal Genetics Stock Center (43). Mutants with mutations in the respiratory chain complex I genes nuo14, nuo51, and nuo78 have been described previously (41). Standard procedures were employed for growth and handling of the Neurospora strains (13). Aspergillus fumigatus ATCC 46645 was a kind gift of Euge ´nia Pinto. Spore suspensions were prepared from 5-day-old cultures grown on Sabouraud’s glucose agar at 37°C. The cells were harvested by gentle agitation with 0.01% (vol/vol) Tween 80 and filtered through cheesecloth. Candida albicans SC5314 was a kind gift of Alexandra Correia. The cells were incubated in yeast Winge liquid medium at 30°C, agitated at 200 rpm overnight, harvested by centrifugation, and resuspended in saline buffer (51). Saccharomyces cerevisiae was a kind gift of Vitor Costa. The cells were incubated in yeast extract-peptone-dextrose (YPD) liquid medium and agitated at 140 rpm overnight at 26°C. Cell viability. Neurospora wild-type conidial cells were harvested and sus- pended in minimal medium at a concentration of 5 10 6 cells/ml and incubated for 30 min at 26°C. Then, cells were washed in water, and staurosporine (STS) from LC Laboratories was added from a stock solution of 5 mg/ml in dimethyl sulfoxide (DMSO) (D5879; Sigma-Aldrich). Cells incubated with DMSO were * Corresponding author. Mailing address: IBMC, Instituto de Bio- logia Molecular e Celular, Universidade do Porto, Rua do Campo Alegre 823, 4150-180 Porto, Portugal. Phone: 351 226074900. Fax: 351 226099157. E-mail: [email protected]. Published ahead of print on 30 April 2010. 906 Downloaded from https://journals.asm.org/journal/ec on 18 November 2021 by 178.140.172.18.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

EUKARYOTIC CELL, June 2010, p. 906–914 Vol. 9, No. 61535-9778/10/$12.00 doi:10.1128/EC.00003-10Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Rotenone Enhances the Antifungal Properties of Staurosporine�
Ana Castro,1,2 Catarina Lemos,1,2 Artur Falcao,1 Andreia S. Fernandes,1N. Louise Glass,3 and Arnaldo Videira1,4*
IBMC, Instituto de Biologia Molecular e Celular, Universidade do Porto, Rua do Campo Alegre 823, 4150-180 Porto, Portugal1;UFP, Faculdade de Ciencias da Saude, Universidade Fernando Pessoa, Rua Carlos da Maia 296, 4200-150 Porto, Portugal2;
Department of Plant and Microbial Biology, University of California, Berkeley, California 94720-31023; and ICBAS,Instituto de Ciencias Biomedicas de Abel Salazar, Universidade do Porto, Largo Prof. Abel Salazar 2,
4099-003 Porto, Portugal4
Received 5 January 2010/Accepted 24 April 2010
We studied staurosporine-induced cell death in the filamentous fungus Neurospora crassa. The generation ofreactive oxygen species during the process appears to be an important signaling event, since addition of theantioxidant glutathione prevents the effects of staurosporine on fungal growth. Selected mutants with muta-tions in respiratory chain complex I are extremely sensitive to the drug, stressing the involvement of complexI in programmed cell death. Following this finding, we determined that the complex I-specific inhibitorrotenone used in combination with staurosporine results in a synergistic and specific antifungal activity, likelythrough a concerted action on intracellular glutathione depletion. Paradoxically, the synergistic antifungalactivity of rotenone and staurosporine is observed in N. crassa complex I mutants and in Saccharomycescerevisiae, which lacks complex I. In addition, it is not observed when other complex I inhibitors are usedinstead of rotenone. These results indicate that the rotenone effect is independent of complex I inhibition. Thecombination of rotenone and staurosporine is effective against N. crassa as well as against the commonpathogens Aspergillus fumigatus and Candida albicans, pointing to its usefulness as an antifungal agent.
Programmed cell death (PCD) refers to a genetically con-trolled process of cellular suicide initiated by endogenous orextrinsic signals. Many of the genes involved are widely con-served from unicellular to multicellular organisms (46). Apop-tosis and autophagy, with its particular characteristics, havebeen recognized as the main categories of PCD (27). Theprocess of PCD is crucial for the development and homeostasisof metazoan organisms and has been implicated in a number ofhuman disorders, including cancer and neurodegenerative andinfectious diseases (3, 10, 25, 55).
The participation in PCD of mitochondria, the cellular or-ganelles responsible for the production of most cellular ATP ineukaryotes (30), has been well established. Particularly, theseorganelles have a central role in the intrinsic (mitochondrion-dependent) pathway of apoptosis, which includes productionof reactive oxygen species (ROS), membrane depolarization,ultrastructural changes, and the release of cytochrome c andother proteins (18, 50, 55). Drugs like staurosporine (STS), aninhibitor of protein kinases, have been used to induce themitochondrion-dependent pathway of apoptosis (24, 35). Stau-rosporine (48) and derivatives have been used in clinical trialsfor cancer therapy (63). The complex I inhibitor rotenone toohas been widely used to induce PCD and also extensivelyapplied as a pesticide (11, 39, 56). Thus, these types of drugscan be employed for the acquisition of fundamental knowledgeand for more practical applications, like modulation of theprogression of PCD.
Modulation of PCD by targeting metabolic pathways in-volved in the process can be exploited to the benefit of humanhealth in several very significant situations, from cancer ther-apy (4, 57) to the treatment of fungal infections (3, 52). How-ever, the molecular basis of PCD involves complex metabolicnetworks (32, 38, 59), and further work is required for theiridentification. Neurospora crassa has many advantages for bio-chemical and genetic experiments (14, 17, 23) and is thus agood model organism for the study of mechanisms of PCD. Weare interested in identifying the cell molecular pathways associ-ated with PCD and using this knowledge to devise strategies tomodulate the process. In this work, we analyze the effects of STSon the N. crassa wild type and mitochondrial complex I mutantsand describe the synergistic effect of combining STS and rotenoneon this organism and other human-pathogenic fungi.
MATERIALS AND METHODS
Strains and growth techniques. The wild-type N. crassa sequenced strain (FGSC2489) and several deletion strains generated by the Neurospora Genome Project(17) were obtained from the Fungal Genetics Stock Center (43). Mutants withmutations in the respiratory chain complex I genes nuo14, nuo51, and nuo78 havebeen described previously (41). Standard procedures were employed for growth andhandling of the Neurospora strains (13). Aspergillus fumigatus ATCC 46645 was akind gift of Eugenia Pinto. Spore suspensions were prepared from 5-day-old culturesgrown on Sabouraud’s glucose agar at 37°C. The cells were harvested by gentleagitation with 0.01% (vol/vol) Tween 80 and filtered through cheesecloth. Candidaalbicans SC5314 was a kind gift of Alexandra Correia. The cells were incubated inyeast Winge liquid medium at 30°C, agitated at 200 rpm overnight, harvested bycentrifugation, and resuspended in saline buffer (51). Saccharomyces cerevisiae was akind gift of Vitor Costa. The cells were incubated in yeast extract-peptone-dextrose(YPD) liquid medium and agitated at 140 rpm overnight at 26°C.
Cell viability. Neurospora wild-type conidial cells were harvested and sus-pended in minimal medium at a concentration of 5 � 106 cells/ml and incubatedfor 30 min at 26°C. Then, cells were washed in water, and staurosporine (STS)from LC Laboratories was added from a stock solution of 5 mg/ml in dimethylsulfoxide (DMSO) (D5879; Sigma-Aldrich). Cells incubated with DMSO were
* Corresponding author. Mailing address: IBMC, Instituto de Bio-logia Molecular e Celular, Universidade do Porto, Rua do CampoAlegre 823, 4150-180 Porto, Portugal. Phone: 351 226074900. Fax: 351226099157. E-mail: [email protected].
� Published ahead of print on 30 April 2010.
906
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.

used as a control. The viabilities of conidia at different times were determined byplating aliquots of the cells on GFS agar medium (13), which induces colonialgrowth, and counting CFU after incubation of the plates at 26°C for about 72 h.
Detection of ROS. Mitochondrial ROS production was monitored by usingdihydrorhodamine 123 from Sigma (St. Louis, MO). The reduced form of dihydro-rhodamine 123 does not fluoresce until entering an actively respiring cell, whereit is oxidized by ROS (mainly H2O2, but also HOCl and peroxinitrite) to a greenfluorescent compound that is sequestered in the mitochondria (36). N. crassaconidial cells were harvested and suspended (5 � 106 cells/ml) in minimalmedium containing 50 �g/ml of dihydrorhodamine 123. The cell suspensionswere incubated at 26°C for 30 min and washed with water before the addition ofSTS. Rhodamine 123 fluorescence was monitored at the single-cell level by useof a FACSCalibur cytometer (BD Biosciences), and data were analyzed withCELLQuest version 3.3 (BD Biosciences), with excitation at 480 nm and emis-sion at 530 nm. When employed, antioxidants were added in preincubation withdihydrorhodamine 123 and (after washes) added again in conjunction with STS.
Glutathione determination. N. crassa (107 conidia/ml) was grown in minimalmedium for 5 h at 30°C, followed by 60 min of incubation in the presence of 12.5�M staurosporine, 100 �g/ml rotenone, both, or DMSO as a control. The cellswere collected by filtration and disrupted by maceration. Then, EDTA was addedto a final concentration of 5 mM and the material was suspended in 1 M HClO4,50 mM potassium phosphate buffer, pH 7.2, and centrifuged for 5 min at 5,000� g at 4°C. The supernatant was neutralized to pH 6 to 7 with 4 M KOH and 0.6M MOPS (morpholinepropanesulfonic acid) and centrifuged for 1 min in max-imum rotation at 4°C. The sum of glutathione and glutathione disulfide (totalglutathione) was determined using a kinetic assay with glutathione reductase,described before (1). The reaction was followed spectrophotometrically using aShimadzu UV-160A instrument.
TUNEL assay. N. crassa conidia were preincubated in minimal medium for 30min, washed in water, and then treated with staurosporine for 60 min and washedagain. Spheroplasts were prepared, and fixation was performed 5 h after treat-ment with the drug. DNA strand breaks were analyzed by terminal deoxynucle-otidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) using an insitu cell death detection fluorescein kit (Roche Applied Science), essentially asdescribed previously (8). Flow cytometry, using 20,000 cells per sample, wascarried out as described above.
Spot assays. Cellular suspensions (conidia) from all fungi were prepared andadjusted to a concentration of 6.56 � 107 cells/ml. Five microliters from serialdilutions of each fungal cell preparation was spotted on agar plates containingvarious drugs. Images were taken by scanning the plates after 2 to 3 days of incu-bation at 26°C. Rotenone, the reduced form of L-glutathione, �-carotene, and L-ascorbic acid were obtained from Sigma-Aldrich. The agar media used were GFS forN. crassa, Sabouraud for A. fumigatus and C. albicans, and YPD for S. cerevisiae.
RESULTS
Selected complex I mutants are highly sensitive to stauro-sporine. Staurosporine is a cell death inducer for a variety ofcells. To assess whether it has antifungal activity against Neu-rospora, we studied the effects of various concentrations of STS(5 to 50 �M) on the viability of asexual spores (conidia).Conidial cells were exposed to STS for different time periodsand plated in GFS agar medium (which induces colonialgrowth of N. crassa), and the resulting colonies were counted.As shown in Fig. 1A, the viability of cells is not significantlyaffected by DMSO (control) or a low STS concentration(5 �M). In contrast, cell viability decreases drastically withhigher concentrations of STS, in a concentration-dependentmanner, showing that STS has potent antifungal activityagainst N. crassa conidia. As detected by the TUNEL assay(Fig. 1B), STS treatment also provokes chromatin fragmenta-tion, a hallmark of apoptosis, indicating that it induces anapoptosis-like cell death in N. crassa.
We found previously that mutants with mutations in respi-ratory chain complex I are more resistant to another cell deathinducer (phytosphingosine) than the wild-type strain (8). Wetherefore asked whether complex I is also involved in STS-induced death. We analyzed the STS sensitivities of conidial
cells from complex I mutant strains in spot assays. These mu-tants display different phenotypes in terms of complex I assem-bly (41, 58). Serial dilutions of conidial suspensions from eachstrain were spotted on media containing STS and incubated at26°C, and the growth levels of the strains were qualitativelyanalyzed (Fig. 2). All strains grew similarly on control GFSagar medium but showed differences when STS was included inthe medium. The wild-type and nuo78 mutant strains wereaffected by STS to similar extents. In contrast, nuo9.8, nuo14,nuo30.4, and nuo51 complex I mutants were extremely sensi-tive to the drug. These results show that complex I (and mito-chondria) is involved in PCD induced by both phytosphingo-sine and staurosporine but that the two drugs act by quitedifferent mechanisms, since all of these mutants were moreresistant to phytosphingosine than the wild-type strain (8).
Rotenone and STS display a specific and synergistic activityagainst Neurospora and other pathogenic fungi. Since we ob-served that selected complex I mutants are highly sensitive to
FIG. 1. Effects of staurosporine on N. crassa. (A) Cell survival.Conidial suspensions were incubated in the absence of STS (DMSO)or in the presence of the indicated concentrations of STS. At theindicated time points, aliquots were removed and plated onto GFSagar medium. The numbers of CFU were determined relative to thatat time zero. The experiments were repeated independently at leastthree times. (B) TUNEL assay. N. crassa conidia were incubated in thepresence (gray) or absence (black) of 12.5 �M staurosporine. TUNEL-positive cells were analyzed by flow cytometry as detailed in Materialsand Methods. The percentage of TUNEL-positive cells is indicated.FITC, fluorescein isothiocyanate.
VOL. 9, 2010 STAUROSPORINE, ROTENONE, AND FUNGAL DEATH 907
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.

STS (Fig. 2), we reasoned that inhibiting the respiratory chainenzyme in wild-type cells would also render them much moresensitive to STS-induced death. Indeed, Fig. 2 shows that stau-rosporine has a much stronger antifungal effect against wild-type N. crassa cells when the complex I inhibitor rotenone isincorporated in the medium. While rotenone alone has nodetectable effect, its use in combination with STS leads to adramatic synergistic effect on fungal growth.
To determine whether the synergistic interaction of rote-none and STS was specific for Neurospora or also affectedgrowth of other fungi, we tested the effects of these drugs ontwo common human pathogens, A. fumigatus and C. albicans.Figure 3 depicts the results obtained from spot assay experi-ments. Treatment with STS alone reduced viability in all threespecies, although rotenone alone had little effect on viability ofthese fungal species. In contrast, rotenone and STS highly
reduced the viability of all three fungi. Taken together, theseresults point out that this type of drug combination may be veryuseful as a potential antifungal agent.
In similar experiments, other combinations of death induc-ers and complex I inhibitors were also tested. As death induc-ers, we used amphotericin B, a polyene antibiotic often usedfor treatment of systemic candidiasis or aspergillosis (52);phytosphingosine, with established antifungal activity againstN. crassa and Aspergillus nidulans (8, 9); and the oxidativestress agent hydrogen peroxide (44). As complex I inhibitors,we employed diphenyleneiodonium (DPI), a rather nonspecificinhibitor of NADH dehydrogenases (2, 37), and piericidin A,which interferes with the ubiquinone binding site of the en-zyme, as does rotenone (20, 47). We used concentrations ofdeath inhibitors that led to a visible effect on fungal growth.The results are summarized in Table 1. We did not observe any
TABLE 1. Antifungal effects of drug combinations, as determinedby spot assaysa
Organism andcomplex I inhibitor
Effect achieved with death inducer:
STS PHS AMB H2O2
NeurosporaRotenone � � � �DPI � NDb ND NDPiericidin A � ND ND ND
Aspergillus or CandidaRotenone � � � �
a Different combinations of complex I inhibitors and cell death inducers weretested. A plus sign indicates that a synergistic effect was observed, and a minussign means that the drug combination did not display an antifungal activityhigher than that achieved by each drug alone. Several concentrations of mostdrugs were tested, in the following ranges: amphotericin B (AMB), 0.1 to 32�g/ml; diphenyleneiodonium (DPI), 5 to 100 �M; piericidin A, 5 to 10 �M;H2O2, 0.5 to 1.5 mM; phytosphingosine (PHS), 5 to 12.5 �g/ml; rotenone, 100 to300 �g/ml. The STS concentration was 5 �M.
b ND, not determined.
FIG. 2. Drug sensitivity profiles of N. crassa strains, determinedusing spot assays. Serial dilutions (indicated by arrows) of conidialsuspensions of the designated strains were spotted in GFS agar me-dium containing 5 �M staurosporine (STS), 100 �g/ml rotenone, orboth, as indicated. The less concentrated spot contained �50 conidia.Growth differences were evaluated after incubation of the plates forabout 72 h at 26°C. wt, wild type.
FIG. 3. Synergistic effect of rotenone and STS on pathogenic fungi. Se-rial 3-fold dilutions of conidial suspensions of the indicated wild-type fungalspecies were spotted onto medium containing rotenone, staurosporine (STS),or both, as shown. Growth differences were evaluated after incubation of theplates for about 72 h at 26°C. GFS agar medium was used for N. crassa andSabouraud medium for both A. fumigatus and C. albicans. Rotenone wasused at 100 �g/ml. STS was used at 5 �M for N. crassa and 15 �M for bothA. fumigatus and C. albicans.
908 CASTRO ET AL. EUKARYOT. CELL
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.

VOL. 9, 2010 STAUROSPORINE, ROTENONE, AND FUNGAL DEATH 909
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.

synergistic antifungal effect with any of these combinations ofcomplex I inhibitors and death inducers. These data suggestedthat the specific synergistic antifungal effect of rotenone andstaurosporine is not dependent on the inhibition of complex I(also see below).
The antioxidant glutathione prevents the effects of STS androtenone. The generation of ROS is a typical event occurringduring programmed cell death (55). In addition, we recentlyidentified an association between decreased ROS productionin N. crassa complex I mutants and the increased resistance ofthese mutants to cell death induced by phytosphingosine (8).Therefore, we decided to examine by flow cytometry the pro-duction of ROS during STS-induced cell death. We alsowanted to determine whether the differential susceptibilities ofthe complex I mutants were correlated with the production ofvariable amounts of ROS among these strains. Figure 4A re-veals that N. crassa conidia produce ROS upon exposure toSTS. A time course analysis shows that the kinetics of ROSproduction is both concentration and time dependent (Fig.4B). ROS were already detectable at 15 min, and a maximumof �50% cells producing ROS was reached around 60 minafter the drug treatment.
Next, we analyzed ROS production of complex I mutantsfollowing treatment with STS, in a similar time course exper-iment. The results shown in Fig. 4C revealed that all complexI mutants produce less ROS than the wild-type strain followingSTS treatment, similarly to what was observed when thesestrains were treated with the death inducer phytosphingosine(8). These results are in agreement with findings that complexI represents a major cellular source of ROS within the cell(26). The mutants display different phenotypes in terms ofcomplex I assembly. The nuo51 mutant assembles a nonfunc-tional enzyme (19), the nuo78 mutant assembles only the mem-brane arm (29), and the nuo14 mutant assembles the periph-eral arm and intermediates of the membrane arm of complexI (41). Thus, their sensitivity toward STS (Fig. 2) cannot becorrelated with the levels of ROS production following drugexposure (Fig. 4C) or with their complex I phenotypes.
In order to verify the significance of STS-induced ROS for-mation, we performed spot assay experiments that includedseveral antioxidants in the culture medium. Figure 5A showsthat addition of the antioxidant glutathione (or its precursorN-acetylcysteine [not shown]) reverted not only the antifungalactivity of STS but also the much stronger antifungal activity ofSTS plus rotenone in N. crassa cells. Other antioxidants testedwere ineffective, but unlike glutathione and N-acetylcysteine,they were also unable to prevent STS-induced ROS formation(Fig. 5B). These experiments suggest that ROS formation is animportant event for STS-induced cell death.
The rotenone effect is independent of complex I inhibition.The idea of combining rotenone and STS came from the find-
ings that some complex I mutants were very sensitive to STS(Fig. 2). The fact that the nuo78 mutant, which lacks the78-kDa protein, is as sensitive to STS as the wild-type straincould be explained. Cleavage of the 78-kDa homologue wasclaimed to be required for apoptosis in mammalian cells (54).Thus, we assumed that inhibition of complex I by rotenone wasresponsible for the synergistic antifungal effect of STS plusrotenone. This reasoning implies that STS plus rotenone wouldhave no synergy when applied to complex I mutants, becausecomplex I is already nonfunctional in these strains. However,replication of the previous experiments confirmed that com-bining STS and rotenone leads to a synergistic antifungal effecton complex I mutants (Fig. 5A). Application of glutathionereverted this effect and also prevented the extreme sensitivityof the nuo51 mutant to STS. Taken together with the obser-vations that rotenone enhances STS-induced death but thatother complex I inhibitors do not (Table 1), these results in-dicate that another property of rotenone is actually responsiblefor the synergistic antifungal effect of STS plus rotenone andthat this effect is independent of complex I inhibition.
To test this hypothesis, we evaluated the sensitivities to STS,rotenone, and STS plus rotenone in a species that lacks com-plex I, the yeast S. cerevisiae (49). As shown in Fig. 6, S.cerevisiae shows little sensitivity to either STS or rotenonealone. However, when S. cerevisiae cells were treated with acombination of STS and rotenone, viability was greatly re-duced. As with N. crassa, the addition of glutathione preventedthis antifungal effect and cell viability was restored to wild-typelevels. These results support the findings that the synergisticantifungal effect of STS plus rotenone is independent of com-plex I inhibition.
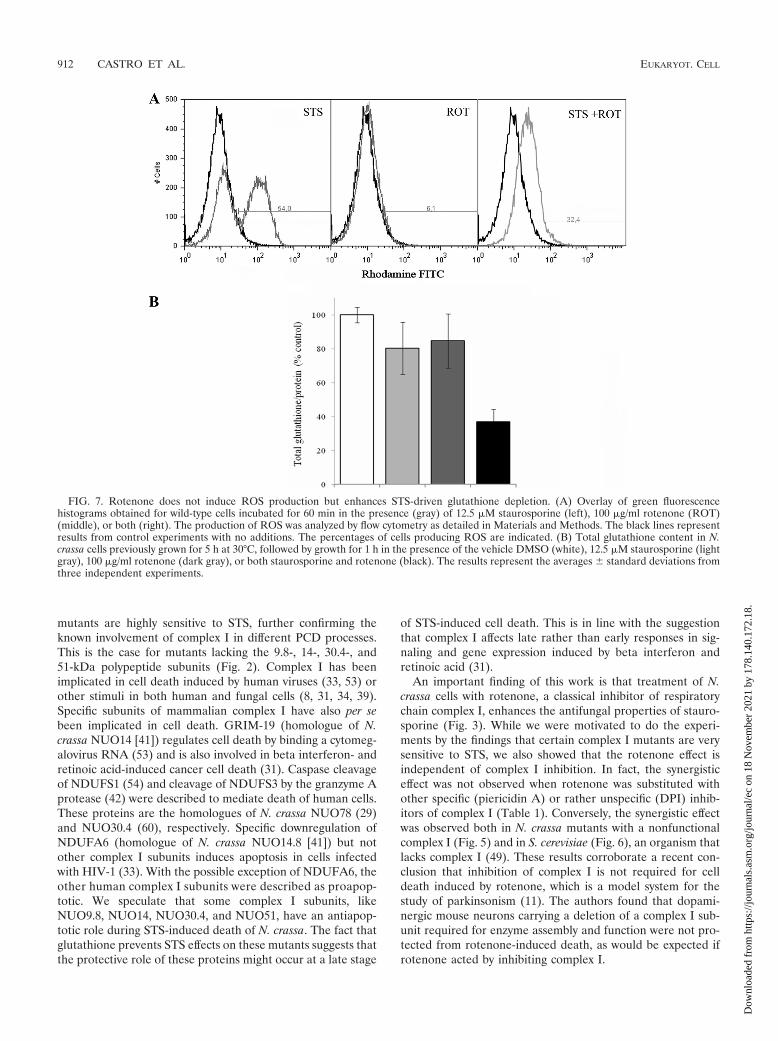
Unlike STS, rotenone does not induce ROS production in N.crassa (Fig. 7A). The specific protection by glutathione fromthe STS and rotenone effects led us to determine the intracel-lular levels of the antioxidant following drug treatment. Theresults shown in Fig. 7B reveal that treatment with either STSor rotenone caused a reduction in the intracellular levels ofglutathione of N. crassa conidia. Interestingly, treatment withboth STS and rotenone results in an increased depletion ofintracellular glutathione. Glutathione efflux and intracellulardepletion, besides the antioxidant properties of glutathione,appear to have an important role in cell death (21). Thus, theobserved synergistic effect on intracellular glutathione deple-tion following exposure of N. crassa to STS and rotenone likelyaccounts for the synergistic effect of the drug combination onfungal cell death.
DISCUSSION
The protein kinase inhibitor staurosporine has been shownto induce programmed cell death in a variety of organisms and
FIG. 4. STS-induced ROS production in N. crassa strains. (A) Overlay of green fluorescence histograms obtained for wild-type cells incubatedfor 60 min in the absence of STS (black) or in the presence of 12.5 �M or 25 �M STS (gray). The production of ROS was analyzed by flowcytometry as detailed in Materials and Methods. The percentages of cells producing ROS are indicated. (B) Time course production of ROS.Wild-type cells were monitored over a period of 90 min for changes in level of cellular ROS production, following no addition of STS (white) orthe addition of 12.5 �M STS (gray) or 25 �M STS (black). The error bars represent the standard deviations from three independent experiments.(C) ROS production in complex I mutants. Conidia from the indicated strains were incubated in the absence of STS (black) or in the presenceof 25 �M STS (gray) for 60 min. The production of ROS was analyzed by flow cytometry, and the percentages of cells producing ROS are shown.
910 CASTRO ET AL. EUKARYOT. CELL
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.

cell types (5, 12, 15, 24). It is able to activate a mitochondrion-dependent form of PCD, which includes ROS generation (24,35, 45), although the role of ROS in the process of PCDremains controversial. In this study, we show that STS induces
an apoptosis-like cell death in N. crassa, including a significantincrease in ROS production that occurs in a dose- and time-dependent manner (Fig. 1 and 4). The antioxidant glutathioneor its precursor N-acetylcysteine prevents ROS formation andalso completely reverts the effects of STS on N. crassa (Fig. 5).These data support the hypothesis that ROS are important forSTS-induced PCD. Furthermore, the inability of other testedantioxidants (�-carotene and ascorbic acid) to revert the ef-fects of STS on cell growth was correlated with their inability toprevent STS-induced ROS production. Thus, we believe thatan initial increase in ROS production is an important signalingevent in PCD, as was previously proposed for different systems(24, 34, 39, 55).
Recent work has indicated that glutathione efflux by multi-drug resistance-associated proteins (28) and the resulting cel-lular glutathione depletion, independent of ROS formation,are critical triggers of STS-induced PCD (12, 22). It was alsoobserved that glutathione depletion is required for ROS for-mation (22). Our results are in agreement with these findings,pointing to an important role of glutathione depletion in drug-induced fungal cell death (also see below). The addition ofglutathione could counteract glutathione loss (and also ham-per ROS generation) and thus prevent the induction of N.crassa cell death by STS. In addition, the nuo78 complex Imutant produces low levels of ROS upon STS exposure and yetis affected by the drug to an extent similar to that for thewild-type strain (Fig. 2 and 4).
A novel finding from this work is that selected complex I
FIG. 5. Glutathione prevents ROS formation and the synergistic effect of rotenone and STS. (A) Spot assays. Serial 3-fold dilutions (indicatedby arrows) of conidial suspensions of the designated N. crassa strains were spotted in GFS agar medium containing different drugs, as indicated,and incubated at 26°C. The upper panels depict control experiments. STS, rotenone, and glutathione were used at concentrations of 5 �M, 100�g/ml, and 10 mM, respectively. (B) The wild-type production of ROS induced by STS in the presence of the indicated antioxidants (gray) wasanalyzed by flow cytometry as detailed in Materials and Methods. The percentages of cells producing ROS are indicated. The black lines representresults from control experiments performed in the absence of STS. Glutathione (GSH), �-carotene, L-ascorbic acid, and N-acetylcysteine (NAC)were used at concentrations of 10 mM, 1 �M, 120 �M, and 10 mM, respectively.
FIG. 6. Effects of different drugs on S. cerevisiae. Serial 3-fold dilu-tions (indicated by arrows) of yeast cells were spotted in YPD mediumcontaining different drugs, as indicated, and incubated at 26°C. The upperpanels depict control experiments. STS, rotenone, and glutathione wereused at concentrations of 15 �M, 100 �g/ml, and 10 mM, respectively.
VOL. 9, 2010 STAUROSPORINE, ROTENONE, AND FUNGAL DEATH 911
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.
mutants are highly sensitive to STS, further confirming theknown involvement of complex I in different PCD processes.This is the case for mutants lacking the 9.8-, 14-, 30.4-, and51-kDa polypeptide subunits (Fig. 2). Complex I has beenimplicated in cell death induced by human viruses (33, 53) orother stimuli in both human and fungal cells (8, 31, 34, 39).Specific subunits of mammalian complex I have also per sebeen implicated in cell death. GRIM-19 (homologue of N.crassa NUO14 [41]) regulates cell death by binding a cytomeg-alovirus RNA (53) and is also involved in beta interferon- andretinoic acid-induced cancer cell death (31). Caspase cleavageof NDUFS1 (54) and cleavage of NDUFS3 by the granzyme Aprotease (42) were described to mediate death of human cells.These proteins are the homologues of N. crassa NUO78 (29)and NUO30.4 (60), respectively. Specific downregulation ofNDUFA6 (homologue of N. crassa NUO14.8 [41]) but notother complex I subunits induces apoptosis in cells infectedwith HIV-1 (33). With the possible exception of NDUFA6, theother human complex I subunits were described as proapop-totic. We speculate that some complex I subunits, likeNUO9.8, NUO14, NUO30.4, and NUO51, have an antiapop-totic role during STS-induced death of N. crassa. The fact thatglutathione prevents STS effects on these mutants suggests thatthe protective role of these proteins might occur at a late stage
of STS-induced cell death. This is in line with the suggestionthat complex I affects late rather than early responses in sig-naling and gene expression induced by beta interferon andretinoic acid (31).
An important finding of this work is that treatment of N.crassa cells with rotenone, a classical inhibitor of respiratorychain complex I, enhances the antifungal properties of stauro-sporine (Fig. 3). While we were motivated to do the experi-ments by the findings that certain complex I mutants are verysensitive to STS, we also showed that the rotenone effect isindependent of complex I inhibition. In fact, the synergisticeffect was not observed when rotenone was substituted withother specific (piericidin A) or rather unspecific (DPI) inhib-itors of complex I (Table 1). Conversely, the synergistic effectwas observed both in N. crassa mutants with a nonfunctionalcomplex I (Fig. 5) and in S. cerevisiae (Fig. 6), an organism thatlacks complex I (49). These results corroborate a recent con-clusion that inhibition of complex I is not required for celldeath induced by rotenone, which is a model system for thestudy of parkinsonism (11). The authors found that dopami-nergic mouse neurons carrying a deletion of a complex I sub-unit required for enzyme assembly and function were not pro-tected from rotenone-induced death, as would be expected ifrotenone acted by inhibiting complex I.
FIG. 7. Rotenone does not induce ROS production but enhances STS-driven glutathione depletion. (A) Overlay of green fluorescencehistograms obtained for wild-type cells incubated for 60 min in the presence (gray) of 12.5 �M staurosporine (left), 100 �g/ml rotenone (ROT)(middle), or both (right). The production of ROS was analyzed by flow cytometry as detailed in Materials and Methods. The black lines representresults from control experiments with no additions. The percentages of cells producing ROS are indicated. (B) Total glutathione content in N.crassa cells previously grown for 5 h at 30°C, followed by growth for 1 h in the presence of the vehicle DMSO (white), 12.5 �M staurosporine (lightgray), 100 �g/ml rotenone (dark gray), or both staurosporine and rotenone (black). The results represent the averages � standard deviations fromthree independent experiments.
912 CASTRO ET AL. EUKARYOT. CELL
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.

While rotenone has been described to induce cell deaththrough the production of ROS (34, 39), this appears not to bethe case with N. crassa. We could not detect ROS productionupon treatment of the fungal cells with rotenone (Fig. 7A),presumably because N. crassa contains alternative NADH de-hydrogenase enzymes (7, 16, 58) able to prevent ROS forma-tion. A similar situation was observed in rat models of Parkin-son’s disease, whereas transfection with a yeast NADHdehydrogenase prevented the overproduction of ROS inducedby rotenone (39, 40). Recently, rotenone was described tostimulate ROS, protein carbonylation (61), and protein nitra-tion, which was suggested to be a general signaling event un-derlying the formation of cytosolic protein aggregates, such asthe Lewy bodies characteristic of Parkinson’s disease (62).Rotenone also induces glutathione depletion (61; this work).These observations provide an attractive hypothesis to explainour results. In fact, we showed that the combination of rote-none and STS has a synergistic effect both on intracellularglutathione depletion (Fig. 7B) and on fungal cell death (Fig.2 and 3) and that glutathione or its precursor N-acetylcysteinecan revert the combined drug effects on N. crassa cells (Fig. 5).The fact that the two drugs act in synergy suggests that theyinduce different mechanisms of glutathione depletion.
Finally, we showed that rotenone also augments the activityof the death inducer staurosporine against other fungi, like S.cerevisiae, A. fumigatus, and C. albicans (Fig. 3 and 6). The lasttwo represent a threat, especially to immunocompromised in-dividuals (6). On the other hand, programmed cell death andtumor development are tightly linked phenomena. Modulationof cell death pathways is being explored to fight against cancer(57) and fungal infections (3), and the development of drugsthat target relevant metabolic pathways can increase the effectsof death inducers. For instance, staurosporine and its clinicallyrelevant analogue 7-hydroxystaurosporine (UCN-01) were re-cently found to increase the anticancer activity of valproic acid(63). Thus, the use of drug combinations, like staurosporineand rotenone, as antifungal and/or antitumor agents is likely tohave an impact in the medical field.
ACKNOWLEDGMENTS
This work was supported by research grants from FCT in Portugaland the European POCI program of QCAIII (coparticipant, FEDER)to A.V., by a sabbatical fellowship from Fundacao Luso-Americana toA.V., and by an NIH grant (GM60468) to N.L.G.
REFERENCES
1. Akerboom, T. P., and H. Sies. 1981. Assay of glutathione, glutathione disul-fide, and glutathione mixed disulfides in biological samples. Methods Enzy-mol. 77:373–382.
2. Aldieri, E., C. Riganti, M. Polimeni, E. Gazzano, C. Lussiana, I. Campia,and D. Ghigo. 2008. Classical inhibitors of NOX NAD(P)H oxidases are notspecific. Curr. Drug Metab. 9:686–696.
3. Almeida, B., A. Silva, A. Mesquita, B. Sampaio-Marques, F. Rodrigues, andP. Ludovico. 2008. Drug-induced apoptosis in yeast. Biochim. Biophys. Acta1783:1436–1448.
4. Armstrong, J. S. 2006. Mitochondria: a target for cancer therapy. Br. J.Pharmacol. 147:239–248.
5. Belmokhtar, C. A., J. Hillion, and E. Segal-Bendirdjian. 2001. Staurosporineinduces apoptosis through both caspase-dependent and caspase-independentmechanisms. Oncogene 20:3354–3362.
6. Cannon, R. D., E. Lamping, A. R. Holmes, K. Niimi, P. V. Baret, M. V.Keniya, K. Tanabe, M. Niimi, A. Goffeau, and B. C. Monk. 2009. Efflux-mediated antifungal drug resistance. Clin. Microbiol. Rev. 22:291–321.
7. Carneiro, P., M. Duarte, and A. Videira. 2007. The external alternativeNAD(P)H dehydrogenase NDE3 is localized both in the mitochondria andin the cytoplasm of Neurospora crassa. J. Mol. Biol. 368:1114–1121.
8. Castro, A., C. Lemos, A. Falcao, N. L. Glass, and A. Videira. 2008. Increasedresistance of complex I mutants to phytosphingosine-induced programmedcell death. J. Biol. Chem. 283:19314–19321.
9. Cheng, J., T.-S. Park, L.-C. Chio, A. S. Fischl, and X. S. Ye. 2003. Inductionof apoptosis by sphingoid long-chain bases in Aspergillus nidulans. Mol. Cell.Biol. 23:163–177.
10. Cheng, W. C., K. M. Leach, and J. M. Hardwick. 2008. Mitochondrial deathpathways in yeast and mammalian cells. Biochim. Biophys. Acta 1783:1272–1279.
11. Choi, W. S., S. E. Kruse, R. D. Palmiter, and Z. Xia. 2008. Mitochondrialcomplex I inhibition is not required for dopaminergic neuron death inducedby rotenone, MPP�, or paraquat. Proc. Natl. Acad. Sci. U. S. A. 39:15136–15141.
12. Circu, M. L., S. Stringer, C. A. Rhoads, M. P. Moyer, and T. Y. Aw. 2009. Therole of GSH efflux in staurosporine-induced apoptosis in colonic epithelialcells. Biochem. Pharmacol. 77:76–85.
13. Davis, R. H., and F. J. de Serres. 1970. Genetic and microbiological researchtechniques for Neurospora crassa. Methods Enzymol. 17A:79–143.
14. Davis, R. H., and D. D. Perkins. 2002. Timeline: Neurospora: a model ofmodel microbes. Nat. Rev. Genet. 3:397–403.
15. Deshmukh, M., and E. M. Johnson, Jr. 2000. Staurosporine-induced neuro-nal death: multiple mechanisms and methodological implications. CellDeath Differ. 7:250–261.
16. Duarte, M., and A. Videira. 2007. Mitochondrial NAD(P)H dehydrogenasesin filamentous fungi, p. 55–68. In M. I. Gonsalez Siso (ed.), Complex I andalternative dehydrogenases. Transworld Research Network, Trivandrum,Kerala, India.
17. Dunlap, J. C., K. A. Borkovich, M. R. Henn, G. E. Turner, M. S. Sachs, N. L.Glass, K. McCluskey, M. Plamann, J. E. Galagan, B. W. Birren, R. L. Weiss,J. P. Townsend, J. J. Loros, M. A. Nelson, R. Lambreghts, H. V. Colot, G.Park, P. Collopy, C. Ringelberg, C. Crew, L. Litvinkova, D. DeCaprio, H. M.Hood, S. Curilla, M. Shi, M. Crawford, M. Koerhsen, P. Montgomery, L.Larson, M. Pearson, T. Kasuga, C. Tian, M. Basturkmen, L. Altamirano,and J. Xu. 2007. Enabling a community to dissect an organism: overview ofthe Neurospora Functional Genomics Project. Adv. Genet. 57:49–96.
18. Eisenberg, T., S. Buttner, G. Kroemer, and F. Madeo. 2007. The mitochon-drial pathway in yeast apoptosis. Apoptosis 12:1011–1023.
19. Fecke, W., V. D. Sled, T. Ohnishi, and H. Weiss. 1994. Disruption of the geneencoding the NADH-binding subunit of NADH: ubiquinone oxidoreductasein Neurospora crassa. Formation of a partially assembled enzyme withoutFMN and the iron-sulphur cluster N-3. Eur. J. Biochem. 220:551–558.
20. Fendel, U., M. A. Tocilescu, S. Kerscher, and U. Brandt. 2008. Exploring theinhibitor binding pocket of respiratory complex I. Biochim. Biophys. Acta1777:660–665.
21. Franco, R., and J. A. Cidlowski. 2009. Apoptosis and glutathione: beyond anantioxidant. Cell Death Differ. 16:1303–1314.
22. Franco, R., M. I. Panayiotidis, and J. A. Cidlowski. 2007. Glutathione de-pletion is necessary for apoptosis in lymphoid cells independent of reactiveoxygen species formation. J. Biol. Chem. 282:30452–30465.
23. Galagan, J. E., S. E. Calvo, K. A. Borkovich, E. U. Selker, N. D. Read, D.Jaffe, W. FitzHugh, L.-J. Ma, S. Smirnov, S. Purcell, B. Rehman, T. Elkins,R. Engels, S. Wang, C. B. Nielsen, J. Butler, M. Endrizzi, D. Qui, P. Ianak-iev, D. Bell-Pedersen, M. A. Nelson, M. Werner-Washburne, C. P. Selitren-nikoff, J. A. Kinsey, E. L. Braun, A. Zelter, U. Schulte, G. O. Kothe, G. Jedd,W. Mewes, C. Staben, E. Marcotte, D. Greenberg, A. Roy, K. Foley, J. Naylor,N. Stange-Thomann, R. Barrett, S. Gnerre, M. Kamal, M. Kamvysselis, E.Mauceli, C. Bielke, S. Rudd, D. Frishman, S. Krystofova, C. Rasmussen,R. L. Metzenberg, D. D. Perkins, S. Kroken, C. Cogoni, G. Macino, D.Catcheside, W. Li, R. J. Pratt, S. A. Osmani, C. P. C. DeSouza, L. Glass,M. J. Orbach, J. A. Berglund, R. Voelker, O. Yarden, M. Plamann, S. Seiler,J. Dunlap, A. Radford, R. Aramayo, D. O. Natvig, L. A. Alex, G. Mannhaupt,D. J. Ebbole, M. Freitag, I. Paulsen, M. S. Sachs, E. S. Lander, C. Nusbaum,and B. Birren. 2003. The genome sequence of the filamentous fungus Neu-rospora crassa. Nature 422:859–868.
24. Gil, J., S. Almeida, C. R. Oliveira, and A. C. Rego. 2003. Cytosolic andmitochondrial ROS in staurosporine-induced retinal cell apoptosis. FreeRadic. Biol. Med. 35:1500–1514.
25. Green, D. R., and G. Kroemer. 2004. The pathophysiology of mitochondrialcell death. Science 305:626–629.
26. Grivennikova, V. G., and A. D. Vinogradov. 2006. Generation of superoxideby the mitochondrial complex I. Biochim. Biophys. Acta 1757:553–561.
27. Hamann, A., D. Brust, and H. D. Osiewacz. 2008. Apoptosis pathways infungal growth, development and ageing. Trends Microbiol. 16:276–283.
28. Hammond, C. L., R. Marchan, S. M. Krance, and N. Ballatori. 2007. Glu-tathione export during apoptosis requires functional multidrug resistance-associated proteins. J. Biol. Chem. 282:14337–14347.
29. Harkness, T. A., R. A. Rothery, J. H. Weiner, S. Werner, J. E. Azevedo, A.Videira, and F. E. Nargang. 1995. Disruption of the gene encoding the78-kilodalton subunit of the peripheral arm of complex I in Neurosporacrassa by repeat induced point mutation (RIP). Curr. Genet. 27:339–350.
30. Hatefi, Y. 1985. The mitochondrial electron transport and oxidative phos-phorylation system. Annu. Rev. Biochem. 54:1015–1069.
VOL. 9, 2010 STAUROSPORINE, ROTENONE, AND FUNGAL DEATH 913
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.

31. Huang, G., Y. Chen, H. Lu, and X. Cao. 2007. Coupling mitochondrialrespiratory chain to cell death: an essential role of mitochondrial complex Iin the interferon-beta and retinoic acid-induced cancer cell death. CellDeath Differ. 14:327–337.
32. Kimchi, A. 2007. Programmed cell death: from novel gene discovery tostudies on network connectivity and emerging biomedical implications.Cytokine Growth Factor Rev. 18:435–440.
33. Ladha, J. S., M. K. Tripathy, and D. Mitra. 2005. Mitochondrial complex Iactivity is impaired during HIV-1-induced T-cell apoptosis. Cell Death Dif-fer. 12:1417–1428.
34. Li, N., K. Ragheb, G. Lawler, J. Sturgis, B. Rajwa, J. A. Melendez, and J. P.Robinson. 2003. Mitochondrial complex I inhibitor rotenone induces apop-tosis through enhancing mitochondrial reactive oxygen species production.J. Biol. Chem. 278:8516–8525.
35. Li, Q., E. F. Sato, X. Zhu, and M. Inoue. 2009. A simultaneous release ofSOD1 with cytochrome c regulates mitochondria-dependent apoptosis. Mol.Cell. Biochem. 322:151–159.
36. Li, W., L. Sun, Q. Liang, J. Wang, W. Mo, and B. Zhou. 2006. Yeast AMIDhomologue Ndi1p displays respiration-restricted apoptotic activity and isinvolved in chronological aging. Mol. Biol. Cell 17:1802–1811.
37. Majander, A., M. Finel, and M. Wikstrom. 1994. Diphenyleneiodoniuminhibits reduction of iron-sulfur clusters in the mitochondrial NADH-ubiqui-none oxidoreductase (complex I). J. Biol. Chem. 269:21037–21042.
38. Majors, B. S., M. J. Betenbaugh, and G. G. Chiang. 2007. Links betweenmetabolism and apoptosis in mammalian cells: applications for anti-apopto-sis engineering. Metab. Eng. 9:317–326.
39. Marella, M., B. B. Seo, A. Matsuno-Yagi, and T. Yagi. 2007. Mechanism ofcell death caused by complex I defects in a rat dopaminergic cell line. J. Biol.Chem. 282:24146–24156.
40. Marella, M., B. B. Seo, E. Nakamaru-Ogiso, J. T. Greenamyre, A. Matsuno-Yagi, and T. Yagi. 2008. Protection by the NDI1 gene against neurodegen-eration in a rotenone rat model of Parkinson’s disease. PLoS One 3:e1433.
41. Marques, I., M. Duarte, J. Assuncao, A. V. Ushakova, and A. Videira. 2005.Composition of complex I from Neurospora crassa and disruption of two“accessory” subunits. Biochim. Biophys. Acta 1707:211–220.
42. Martinvalet, D., D. M. Dykxhoorn, R. Ferrini, and J. Lieberman. 2008.Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 133:681–692.
43. McCluskey, K. 2003. The Fungal Genetics Stock Center: from molds tomolecules. Adv. Appl. Microbiol. 52:245–262.
44. Miyoshi, N., H. Oubrahim, P. B. Chock, and E. R. Stadtman. 2006. Age-dependent cell death and the role of ATP in hydrogen peroxide-inducedapoptosis and necrosis. Proc. Natl. Acad. Sci. U. S. A. 103:1727–1731.
45. Muyderman, H., A. L. Wadey, M. Nilsson, and N. R. Sims. 2007. Mitochon-drial glutathione protects against cell death induced by oxidative and nitra-tive stress in astrocytes. J. Neurochem. 102:1369–1382.
46. Nedelcu, A. M. 2009. Comparative genomics of phylogenetically diverseunicellular eukaryotes provide new insights into the genetic basis for theevolution of the programmed cell death machinery. J. Mol. Evol. 68:256–268.
47. Okun, J. G., P. Lummen, and U. Brandt. 1999. Three classes of inhibitorsshare a common binding domain in mitochondrial complex I (NADH:ubiquinone oxidoreductase). J. Biol. Chem. 274:2625–2630.
48. Omura, S., Y. Iwai, A. Hirano, A. Nakagawa, J. Awaya, H. Tsuchya, Y.
Takahashi, and R. Masuma. 1977. A new alkaloid AM-2282 of Streptomycesorigin. Taxonomy, fermentation, isolation and preliminary characterization.J. Antibiot. (Tokyo) 30:275–282.
49. Overkamp, K. M., B. M. Bakker, P. Kotter, A. van Tuijl, S. de Vries, J. P. vanDijken, and J. T. Pronk. 2000. In vivo analysis of the mechanisms for oxi-dation of cytosolic NADH by Saccharomyces cerevisiae mitochondria. J.Bacteriol. 182:2823–2830.
50. Pereira, C., R. D. Silva, L. Saraiva, B. Johansson, M. J. Sousa, and M.Corte-Real. 2008. Mitochondria-dependent apoptosis in yeast. Biochim. Bio-phys. Acta 1783:1286–1302.
51. Phillips, A. J., I. Sudbery, and M. Ramsdale. 2003. Apoptosis induced byenvironmental stresses and amphotericin B in Candida albicans. Proc. Natl.Acad. Sci. U. S. A. 100:14327–14332.
52. Ramsdale, M. 2008. Programmed cell death in pathogenic fungi. Biochim.Biophys. Acta 1783:1369–1380.
53. Reeves, M. B., A. A. Davies, B. P. McSharry, G. W. Wilkinson, and J. H.Sinclair. 2007. Complex I binding by a virally encoded RNA regulates mi-tochondria-induced cell death. Science 316:1345–1348.
54. Ricci, J.-E., C. Munoz-Pinedo, P. Fitzgerald, B. Bailly-Maitre, G. A. Perkins,N. Yadava, I. E. Scheffler, M. H. Ellisman, and D. R. Green. 2004. Disruptionof mitochondrial function during apoptosis is mediated by caspase cleavageof the p75 subunit of complex I of the electron transport chain. Cell 117:773–786.
55. Skulachev, V. 2006. Bioenergetic aspects of apoptosis, necrosis and mitopto-sis. Apoptosis 11:473–485.
56. Tada-Oikawa, S., Y. Hiraku, M. Kawanishi, and S. Kawanishi. 2003. Mech-anisms of generation of hydrogen peroxide and change of mitochondrialmembrane potential during rotenone induced apoptosis. Life Sci. 73:3277–3288.
57. Tan, M. L., J. P. Ooi, N. Ismail, A. I. Moad, and T. S. Muhammad. 2009.Programmed cell death pathways and current antitumor targets. Pharm. Res.26:1547–1560.
58. Videira, A. 1998. Complex I from the fungus Neurospora crassa. Biochim.Biophys. Acta 1364:89–100.
59. Videira, A., T. Kasuga, C. Tian, C. Lemos, A. Castro, and N. L. Glass. 2009.Transcriptional analysis of the response of Neurospora crassa to phytosphin-gosine reveals links to mitochondrial function. Microbiology 155:3134–3141.
60. Videira, A., M. Tropschug, and S. Werner. 1990. Primary structure andexpression of a nuclear-coded subunit of complex I homologous to proteinsspecified by the chloroplast genome. Biochem. Biophys. Res. Commun.171:1168–1174.
61. Watabe, M., and T. Nakaki. 2007. Mitochondrial complex I inhibitor rote-none-elicited dopamine redistribution from vesicles to cytosol in humandopaminergic SH-SY5Y cells. J. Pharmacol. Exp. Ther. 323:499–507.
62. Watabe, M., and T. Nakaki. 2008. Mitochondrial complex I inhibitor rote-none inhibits and redistributes vesicular monoamine transporter 2 via nitra-tion in human dopaminergic SH-SY5Y cells. Mol. Pharmacol. 74:933–940.
63. Yeow, W. S., M. F. Ziauddin, J. B. Maxhimer, S. Shamimi-Noori, A. Baras,A. Chua, D. S. Schrump, and D. M. Nguyen. 2006. Potentiation of theanticancer effect of valproic acid, an antiepileptic agent with histone deacety-lase inhibitory activity, by the kinase inhibitor staurosporine or its clinicallyrelevant analogue UCN-01. Br. J. Cancer 94:1436–1445.
914 CASTRO ET AL. EUKARYOT. CELL
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/e
c on
18
Nov
embe
r 20
21 b
y 17
8.14
0.17
2.18
.
Related Documents











