Biol. Rev.(), , pp. – Printed in Great Britain ROLE OF FETAL AND INFANT GROWTH IN PROGRAMMING METABOLISM IN LATER LIFE B M. DESAI C. N. HALES Department of Clinical Biochemistry, University of Cambridge, Cambridge CBQR (Received June ; revised September ; accepted October ) ABSTRACT Fetal growth and development is dependent upon the nutritional, hormonal and metabolic environment provided by the mother. Any disturbance in this environment can modify early fetal development with possible long-term outcomes as demonstrated by extensive work on ‘ programming ’. Growth restriction resulting from a deficit in tissue}organ cell number (as measured by tissue DNA content) is irrecoverable. However, when the cell size (or cell protein content) is reduced, the effects on growth may not be permanent. Recent epidemiological studies using archival records of anthropometric measurements related to early growth in humans have shown strong statistical associations between these indices of early development and diseases in later life. It has been hypothesised that the processes explaining these associations involve adaptive changes in fetal organ development in response to maternal and fetal malnutrition. These adaptations may permanently alter adult metabolism in a way which is beneficial to survival under continued conditions of malnutrition but detrimental when nutrition is abundant. This hypothesis is being tested in a rat model which involves studying the growth and metabolism in the offspring of rat dams fed a low-protein diet during pregnancy and}or lactation. Using this rat model, it has been demonstrated that there is : (i) Permanent growth retardation in offspring nursed by dams fed a low-protein diet. (ii) Permanent and selective changes in organ growth. Essential organs like the brain and lungs are relatively protected from reduction in growth at the expense of visceral organs such as the liver, pancreas, muscle and spleen. (iii) Programming of liver metabolism as reflected by permanent changes in activities of key hepatic enzymes of glycolysis and gluconeogenesis (glucokinase and phosphoenolpyruvate carboxykinase) in a direction which would potentially bias the liver towards a ‘ starved ’ setting. We have speculated that these changes could be a result of altered periportal and perivenous regions of the liver which may also affect other aspects of hepatic function. (iv) Deterioration in glucose tolerance with age. (v) An increase in the life span of offspring exposed to maternal protein restriction only during the lactation period, and a decrease in life span when exposed to maternal protein restriction only during gestation. These studies show that hepatic metabolism and even longevity can be programmed by events during early life. Key words : maternal nutrition, birth weight, nutritional programming, non-insulin-dependent diabetes.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Biol. Rev. (), , pp. –
Printed in Great Britain
ROLE OF FETAL AND INFANT GROWTH IN PROGRAMMING
METABOLISM IN LATER LIFE
B M. DESAI C. N. HALES
Department of Clinical Biochemistry, University of Cambridge, Cambridge CB QR
(Received June ; revised September ; accepted October )
ABSTRACT
Fetal growth and development is dependent upon the nutritional, hormonal and metabolic
environment provided by the mother. Any disturbance in this environment can modify early
fetal development with possible long-term outcomes as demonstrated by extensive work on
‘programming’. Growth restriction resulting from a deficit in tissue}organ cell number (as
measured by tissue DNA content) is irrecoverable. However, when the cell size (or cell protein
content) is reduced, the effects on growth may not be permanent.
Recent epidemiological studies using archival records of anthropometric measurements
related to early growth in humans have shown strong statistical associations between these
indices of early development and diseases in later life. It has been hypothesised that the
processes explaining these associations involve adaptive changes in fetal organ development in
response to maternal and fetal malnutrition. These adaptations may permanently alter adult
metabolism in a way which is beneficial to survival under continued conditions of malnutrition
but detrimental when nutrition is abundant.
This hypothesis is being tested in a rat model which involves studying the growth and
metabolism in the offspring of rat dams fed a low-protein diet during pregnancy and}or
lactation. Using this rat model, it has been demonstrated that there is :
(i) Permanent growth retardation in offspring nursed by dams fed a low-protein diet.
(ii) Permanent and selective changes in organ growth. Essential organs like the brain and
lungs are relatively protected from reduction in growth at the expense of visceral organs such
as the liver, pancreas, muscle and spleen.
(iii) Programming of liver metabolism as reflected by permanent changes in activities of key
hepatic enzymes of glycolysis and gluconeogenesis (glucokinase and phosphoenolpyruvate
carboxykinase) in a direction which would potentially bias the liver towards a ‘starved’
setting. We have speculated that these changes could be a result of altered periportal and
perivenous regions of the liver which may also affect other aspects of hepatic function.
(iv) Deterioration in glucose tolerance with age.
(v) An increase in the life span of offspring exposed to maternal protein restriction only
during the lactation period, and a decrease in life span when exposed to maternal protein
restriction only during gestation.
These studies show that hepatic metabolism and even longevity can be programmed by
events during early life.
Key words : maternal nutrition, birth weight, nutritional programming, non-insulin-dependent
diabetes.

M. D C. N. H
CONTENTS
I. Introduction . . . . . . . . . . . . . . .
II. Nutrition, growth and development . . . . . . . . . .
() Maternal nutrition and fetal growth . . . . . . . . .
() Fetal adaptation to malnutrition . . . . . . . . . .
III. Programming. . . . . . . . . . . . . . .
() Nutritional programming in animals . . . . . . . . .
() Nutritional programming in humans . . . . . . . . .
() The mechanisms underlying programming . . . . . . . .
IV. Early growth and adult onset diseases . . . . . . . . . .
() Recent epidemiological studies . . . . . . . . . .
() Thrifty phenotype hypothesis . . . . . . . . . . .
() Recent animal studies . . . . . . . . . . . .
V. Conclusions . . . . . . . . . . . . . . .
VI. Acknowledgements . . . . . . . . . . . . .
VII. References . . . . . . . . . . . . . . .
I. INTRODUCTION
Recent epidemiological studies in people whose birth weights were recorded many
years ago suggest links between impaired growth during early life and the development
much later of diseases such as ischaemic heart disease (Barker et al., ), hypertension
(Barker et al., ), diabetes (Hales et al., ) and features of insulin resistance
syndrome (Barker et al., a). The long-term effects of retarded early growth are
proposed to result from maternal and fetal malnutrition at critical periods of fetal or
infant development leading to reduction in the growth of organs and permanent
changes in their metabolism or structure, or both. Furthermore, transition from early
nutrition deprivation to nutrition abundance in later life might reduce the ability to
produce and respond to insulin. The concept may explain the high prevalence of non-
insulin-dependent diabetes (NIDDM) in human populations that have experienced a
transition from an undernourished environment to one of overnutrition, for example,
in the Pima Indian (Bennett et al., ; Jarret, ), Nauruans of the Pacific Islands
(Zimmet et al., ) and migrant populations (Cheah & Tan, ; Zimmet et al.,
; Cohen et al., ). Evidence that maternal and infant undernutrition itself may
be diabetogenic comes mainly from studies in both animals and humans suffering from
protein–energy malnutrition in which functional abnormalities in insulin secretion and
glucose tolerance have been demonstrated (reviewed in Section IV).
The view that early malnutrition can impair β-cell growth and function is consistent
with early studies on malnourished children from the developing countries, in whom
impaired glucose tolerance was found even after recovery from the nutritional insult
(James & Coore, ; Milner, ). Studies in experimental animals show clearly
that these changes can be reproduced by subjecting either fetal or early postnatal
animals to general protein–calorie malnutrition (Weinkove, Weinkove & Pimstone,
) or, interestingly, to protein deficiency alone (Pimstone, ; Swenne, Crace &
Milner, ). Moreover, the degree of the loss of insulin secretion in protein–calorie
malnutrition is much more severe than would be expected from the degree of reduction
of islet volume (Weinkove et al., ). This, of course, is reminiscent of the situation
in human NIDDM. Poor insulin secretion may occur not only from less β-cells, but
also as a result of abnormal islet structure and vascularization (Snoeck et al., ).

Programming fetal and infant growth
The paradox of the failure of natural selection to eliminate a genetically determined
lethal condition was addressed several years ago by Neel’s ‘ thrifty genotype’ hypothesis
(Neel, ). It suggests that the high incidence of diabetes in Western or recently
affluent societies results from the existence of diabetogenic genes which confer a
survival advantage in the condition of subsistence living, although are detrimental to
survival under conditions of overnutrition.
Hales & Barker () have proposed a hypothesis concerning the aetiology of
NIDDM, the ‘thrifty phenotype’ which provides an alternative to the ‘thrifty
genotype’ hypothesis. The essence of this hypothesis is that poor fetal and early
postnatal nutrition imposes mechanisms of nutritional thrift upon the growing
individual. One of the major long-term consequences of inadequate early nutrition is
suggested to be impaired development of the endocrine pancreas and increased
susceptibility to the development of NIDDM.
II. NUTRITION, GROWTH AND DEVELOPMENT
Growth and development of the mammalian embryo and fetus is a highly complicated
sequence of events whereby a single-cell zygote develops into a complex yet organized,
multicellular, multisystem, fully developed animal. Tissue or organ development
involves three different processes, namely, ‘hyperplasia’ also known as replication or
proliferation; differentiation, which involves the orderly and programmed migration of
cells into definable tissues and organ rudiments; and, ‘hypertrophy’, in which the cells
increase in size and acquire the capacity to synthesize macromolecules for specialized
functions. In mammals, as in many other species, the major part of the developmental
process pertaining to cell division occurs during intrauterine life. In some organs and
tissues, it continues after birth. For example, at birth, a virtually full complement of
brain neurones and of renal glomeruli are present (Hinchliffe et al., a) and
available data suggest that at the age of yr at least half the adult complement of β-cells
is present (Stefan et al., ).
Fetal growth and development is controlled by two major factors, namely, genetic
factors as determined by the fetal genome, and environmental factors, such as maternal
nutrition, that alter the expression of the fetal genome. Recent studies into the control
of fetal growth appear to suggest that the genome regulates the growth and development
of the fetus in a predetermined pathway in which specific genes are turned on and off
at specific stages of fetal development, and the environmental factors influence growth
by their effect on this normal pattern of genetic expression (Han, ).
() Maternal nutrition and fetal growth
The relationship between the diet of the mother and the well-being of the fetus and
infant continues to be a matter of great importance, uncertainty and controversy. It has
been known for many years that birth weight is strongly maternally determined and
that genetic factors play a relatively weak role (Carr-Hill et al., ). In Walton &
Hammond’s () well-known experiments, in which Shetland and Shire horses were
crossed, the foals were smaller at birth when a Shetland pony was the mother than when
a Shire horse was the mother. As the genetic composition of the two crosses was similar,
this implied that the Shetland mother had constrained the growth of the fetus. These

M. D C. N. H
findings from cross-breeding experiments are supported by recent embryo-transfer
experiments. Once again, the size at birth of animal embryos removed from their
mothers’ uteri is related to the size of the uterus into which they are transferred (Snow,
).
In human studies, comparisons of birth weights of half-siblings show that those
related through the mother tend to have similar birth weights whereas those related
through the father do not (Morton, ). Ounsted & Ounsted () showed that the
mothers of growth-retarded babies had themselves had low mean birth weight. Further
studies have shown that the birth weight of mothers is not only related to that of their
children but also to that of the subsequent generation (Hackman et al., ; Klebanoff,
Meirik & Berendes, ; Emmanuel et al., ). This has led to the conclusion that
mothers constrain fetal growth and that the degree of constraint they exert is set when
they themselves are in utero (Ounsted, Scott & Ounsted, ).
‘Maternal constraint ’ is thought to reflect the limited capacity of the mother to
deliver nutrients to her fetus, which in turn limits the growth of the fetus (McCance &
Widdowson, ; Gluckman et al., ). From human (Eastman & Jackson, ;
Hackman et al., ) and animal (Owens, Owens & Robinson, ; Zeman, ;
Chow & Lee, ) studies, it is known that both the plane of nutrition at the onset of
pregnancy (previous nutritional exposure) and the nutrition during the pregnancy itself
can exert an influence on fetal growth. Not only this, but possibly the nutritional status
of the mother’s own mother in the past may influence fetal growth as demonstrated by
a study of survivors of the Dutch ‘hunger winter’. A famine occurred in the western
part of the Netherlands during the Second World War (Stein & Susser, ). It began
in October after the German occupation forces imposed an embargo on all
transport and food supplies – a reprisal for a general strike. It ended suddenly, in May
, when the Allied armies liberated the Netherlands. An account of the famine,
written immediately after liberation, stated that ‘the average food intake from all
sources, including extra-legal, in October was approximately calories. This
was reduced to calories or less in April ’. Dutch women who were born in
Amsterdam during and after the famine, and who experienced famine during the first
and second trimesters of their intrauterine lives, have been followed up. Although they
themselves had normal birth weight the mean birth weight of their own babies was
reduced (Lumey, ). In this study, clear effects on the reproductive outcomes are
seen in the generation following an environmental exposure in utero. Furthermore, a
study on Swedish women has shown that women who were themselves small for
gestational age (growth retarded in utero) had an increased risk of giving birth to either
intrauterine growth retarded or preterm infants, again emphasizing the importance of
maternal factors and the intrauterine milieu. Moreover, women who were preterm at
birth did not demonstrate a similar risk (Klebanoff, Meirik & Berendes ). Studies
on pregnant women from developing countries indicate that women with low pre-
pregnancy weight and short stature had infants with low birth weight (Kramer, ).
Therefore, adverse maternal nutrition can be viewed as a long-term environmental
adversity affecting the rapidly developing fetus, and the process of adaptation to this
milieu could be considered as a response to this adversity.

Programming fetal and infant growth
() Fetal adaptation to malnutrition
The plasticity of the fetus enables it to adapt to conditions of adverse maternal
nutrition by reducing its growth and development (Widdowson & McCance, ;
Owens et al., ). Whilst adaptation allows a highly effective means of survival under
the situation of nutritional deprivation, it raises a number of issues which need to be
addressed. First, what are the limits of this adaptation, and at what point does it break
down? For instance, the simplest way of adapting to undernutrition is to reduce body
weight, thereby reducing the use of substrates which leads to a fall in basal metabolic
rate. However, there is obviously a limit below which weight loss becomes excessive,
and morbidity and mortality increases. Secondly, what are the costs of the adaptation?
Almost by definition, adaptation confers benefts compared with the unadapted state,
but it also usually involves costs. A number of studies have shown that undernutrition
causes a reduction in cell numbers in certain organs which is irrecoverable (Widdowson,
; Winick, ; Hinchliffe et al., ). Thirdly, what kind of relationship does the
adapting organism’s response have to the stress imposed? Does any degree of growth
deficit carry some risk? And finally, what are the mechanisms involved?
III. PROGRAMMING
The possibility that nutrition in early life could influence a propensity to adult
diseases invokes the concept of ‘programming’. The principle that the nutritional,
hormonal, and metabolic environment provided by the mother may permanently change
the structure and physiology of her offspring was established long ago. Recently, Lucas
() defined programming as a process whereby a stimulus or insult, at a critical or
sensitive period of development, has lasting or lifelong significance.
There are four essential principles which underlie the concept of programming. (i)
Nutritional (or non-nutritional) manipulations cause different effects at different times
in early life. (ii) Rapidly growing fetuses and neonates are more vulnerable to these
manipulations. (iii) Manipulation in early life has permanent effects. (iv) The permanent
effects include reduced cell numbers, altered organ structure and resetting of hormonal
axes.
() Nutritional programming in animals
Animal evidence for programming during critical periods of development dates back
over years (Spalding, ). Non-nutritional influences, for example, hormonal
signals operating during critical periods, also have numerous programming effects. The
classic example of such a phenomenon is the exposure of female rats at a critical period
of fetal life to a single exogenous dose of testosterone, which permanently reorientated
sexual behaviour (Angelbeck & Du Brul, ). A similar dose of testosterone in -
day-old females had no effect. Thus, there is a critical time at which the animal’s sexual
physiology is sensitive and can be permanently changed. Exposure of animals to an
excess of thyroxine during the neonatal period changes the pituitary-hypothalamic
responses linked to the secretion of thyroid-stimulating hormone in later life (Besa &
Pascula-Leone, ).
Some of the earliest work relating developmental physiology to nutrition and to its
outcome were the studies of McCance and Widdowson. Employing the ‘ large and small

M. D C. N. H
litter ’ technique, they produced cohorts of well-nourished and undernourished
suckling animals which subsequently paved the way for future studies. The intriguing
features of their studies were that the rats raised in large litters were small at weaning,
continued to eat less and remained smaller as adults. Also, these studies provided some
of the early information linking chronological, somatic and behavioural development in
retarded growth. More precisely, they identified critical stages of development at which
interference with an animal’s growth could have permanent and far-reaching
consequences (McCance & Widdowson, , ; Widdowson & McCance, ,
).
In concert with this work were the studies of Winick and colleagues which put
forward a hypothesis to explain growth retardation. They suggested that essentially
growth encompasses two stages, namely, cell division and cell enlargement. In a series
of studies on malnourished rats from birth until days of age, they demonstrated that
the cellular effects of malnutrition depend upon the phase of growth of the animals at
the time of onset of malnutrition. Very early in life, malnutrition would impede cell
division, organ growth and differentiation which may be irrecoverable. Later in life, it
would lead to changes in cell composition and cell size (Winick & Noble, ; Winick,
Fish & Rosso, ; Winick ). Conversely, overfeeding rats by raising the animals
in litters of – pups compared to – pups during the proliferative phase of growth
caused acceleration of the rate of cell division, and produced a higher DNA content per
organ (Winick & Noble, ). Thus, these studies collectively indicate that a reduced
supply of nutrients during early life (prenatal and postnatal) interferes with the rate of
cell multiplication in the various organs and that the effect is proportionally more
deleterious in tissues with a faster rate of cell multiplication.
Nutritional programming has been demonstrated convincingly in a range of
mammals, such as rats, mice, sheep, pigs and primates. It has further been shown that
poor nutrition may permanently programme the structure and physiology of a range of
organs and tissues, although such effects may remain latent until the animal is mature.
For instance, nutrition at a vulnerable period of brain development has permanent
effects on brain size, with the cerebellum being more affected than the cerebrum, brain
cell number (Winick, ) and performance (Katz, ; Smart, ).
Early studies have unequivocally demonstrated that alteration of cholesterol
metabolism during development, either by changing the amount or composition of the
diet or by giving cholestyramine which sequesters bile salts and increases cholesterol
excretion, has permanent effects. Thus, treatment of rat dams during gestation up-
regulated cholesterol synthesis in the adult offspring, whereas treatment of the dams
after birth permanently changed the body’s capacity to excrete cholesterol in bile
(Innis, , ; Little & Hahn, ).
The age at which an animal is weaned also appears to have a longlasting influence on
metabolism. Rats that are weaned prematurely have a raised plasma cholesterol
concentration in later life, which becomes apparent only after months (Hahn & Kirby,
).
Mott, Lewis & McGill () have shown that early overfeeding in primates may
programme later obesity and that breast-fed and bottle-fed baboons have long-term
differences in their lipid metabolism and in the degree of atherosclerosis. Similarly,
Duff & Snell () have shown that overnutrition in rats caused by litter manipulation

Programming fetal and infant growth
resulted in alteration in hepatic enzyme activities that reflected an increased capacity
for lipid synthesis by the liver.
These results raise the question of where the ‘memory’ of the early event had been
stored in the intervening period and further demonstrate that early diet could play a role
in the later development of disease states that have considerable significance in humans.
In addition, recent studies on rats have shown that the quality of nutrition in early
life could have permanent consequences on carbohydrate metabolism. For instance,
maternal protein-undernourished fetuses show long-term morphological changes in β-
cells (Snoeck et al., ), insulin secretion (Dahri et al., ), blood pressure
(Langley & Jackson ) and hepatic metabolism (Desai et al., ; Ozanne et al., ).
() Nutritional programming in humans
Given the evidence for programming in general and the evidence for nutritional
programming in mammals, the same phenomena in humans might be predicted.
Childhood obesity may predispose to adult obesity (Charney et al., ). This is
supported by animal data cited above (Mott, Lewis & McGill, ). Paradoxically,
however, early starvation of mothers may also be followed by adult obesity in offspring.
Among men born during the Dutch ‘hunger winter’ of –, those whose mothers
were exposed to famine in the first half of gestation became obese as adults (Stein &
Susser, ; Ravelli, Stein & Susser, ). It is thought that, during early gestation,
hypothalamic control of appetite becomes set in relation to body size (Widdowson &
McCance, ). An inappropriate setting of the hypothalamus could have led to
obesity in men whose mothers were exposed to famine in utero. Alternatively, early
nutrition could influence the adipocyte number (Faust et al., ) or size (Lewis et al.,
).
Recently, Hinchcliffe et al. () have shown that asymmetrical retardation
(disproportionate growth) is associated with a substantial reduction in the number of
nephrons, and that there is no postnatal compensation for this.
Iron deficiency in infancy, common both in the West and in developing countries, is
related to poor developmental performance (Ankett et al., ), which could have
long-lasting consequences (Dallman, Siimes & Stekel, ; Walter et al., ).
A prospective dietary study on preterm infants undertaken by Lucas & Morley
() has shown that a brief period of early dietary management has a major impact
about years later on brain growth, neurodevelopment and bone mineralization.
Compared to formula feed, human milk appeared to promote brain growth, brain
development and bone mineralisation in these infants.
() The mechanisms underlying programming
Collectively, the animal and human studies provide convincing evidence that
nutrition at a critical or sensitive period in early life may influence a wide variety of
metabolic, developmental and pathological processes in adulthood. If this is so, defining
the mechanisms involved could be crucial to understanding the pathogenesis of adult
disease. Some programming events may have immediate effects, for example, neuronal
growth at a critical stage with subsequent failure of catch-up. Other programming
effects, as cited previously, are deferred. In this instance, the question is how the

M. D C. N. H
memory of early events is stored and later expressed, despite continuous cellular
replication and replacement. Lucas () has postulated three cellular mechanisms. It
could occur at the level of an individual gene, with induction or repression of gene
expression. Another possibility is a permanent reduction in cell numbers. Alternatively,
specific individual cells might be deleted or clonally expanded.
IV. EARLY GROWTH AND ADULT ONSET DISEASES
() Recent epidemiological studies
Epidemiological and animal studies have suggested that long-term health can be
influenced by events in early life. Early evidence came from geographical studies which
revealed differences in disease rates in different places or at different times. Forsdahl
() first reported from Norway that a close correlation existed between current death
rates from ischaemic heart disease and past infant mortality rates in different areas of
the country. However, the current infant mortality rate was less strongly related to
current ischaemic heart disease deaths. This was attributed to adverse influences in
childhood and adolescence, associated with poor living standards, which increased
susceptibility to other influences associated with affluence and encountered in later life.
These observations were confirmed in the UK by Williams, Roberts & Davies
(). Moreover, in their study, there was an equally good correlation between past
and present infant mortality and ischaemic heart disease deaths, thus they questioned
the role of living conditions in early life. It is possible in the UK (unlike Norway) that
geographical variations in infant mortality persist (Markowe, ).
In , studies from different areas of England and Wales reported that infant
mortality in – predicted the incidence of death from ischaemic heart disease in
– (Barker & Osmond, ). It was suggested that the mechanisms which linked
infant mortality and later ischaemic heart disease could be operating during fetal life.
Moreover, since infant mortality is known to be strongly and inversely related to birth
weight (McCormick, ; Dollfus et al., ), it became important to determine
whether birth weight itself was related to ischaemic heart disease.
Therefore, Barker and his colleagues undertook a search for old records of birth
weight and discovered a few populations in the United Kingdom where such records
existed, stretching back for over years. In certain places, such as Hertfordshire,
Preston and Sheffield, records existed of early anthropometry such as birth weight,
weight at yr, head circumference and abdominal circumference for people who are
currently in the age range – yr. This was followed by a more detailed series of
studies which established statistical links between indices of infant and fetal growth and
the risk of death from ischaemic heart disease and hypertension. For example, in a
group of Hertfordshire men, death from ischaemic heart disease increased as birth
weight or weight at yr decreased (Barker et al., ). Subsequent studies showed
linkages of indices of early growth with hypertension (Barker et al., ), plasma
fibrinogen concentration, factor VII (Barker et al., ), cholesterol and apolipoprotein
B (Fall et al., ). These associations parallel those with death rates from ischaemic
heart disease in that high body weight in early life is associated with a low incidence of
each risk factor. The associations are strong and graded, and are independent of social
class, either at birth or currently, and of influences in adult lifestyle such as smoking and
alcohol consumption.

Programming fetal and infant growth
A striking feature of these findings is that different risk factors are related to different
patterns of early growth. It was observed that adult blood pressure increased in relation
to decreasing birth weight but not independently to weight at yr. The strongest
predictor of adult blood pressure was seen in adults who at birth had a large placenta
in relation to birth weight (Barker et al., ). Plasma fibrinogen concentrations were
related in men to weight at yr but not to birth weight. In fact, those babies who were
short in relation to head size tended to develop hypertension and high plasma
fibrinogen concentrations (Barker et al., ). Increased placental size has been
implicated as a sign of maternal, and possibly fetal, malnutrition. This is supported by
a study from Oxford which showed that maternal anaemia is associated with increased
placental size and with an increased ratio of placental weight to birth weight (Godfrey
et al., ).
In view of the known association between ischaemic heart disease, hypertension and
NIDDM, and the fact that very rapid growth of β-cells of the islets of Langerhans
occurs during fetal life (at least in rats), the possibility that loss of glucose tolerance
could itself be a consequence of poor early growth and development was explored.
In the same population of Hertfordshire men of mean age years, glucose tolerance
(either judged by the h plasma glucose concentration at the end of the test or the
percentage of men with impaired glucose tolerance or newly discovered NIDDM) was
strongly related to birth weight. The smallest infants had the worst glucose tolerance.
Furthermore, these men had an eightfold increased risk of having impaired glucose
tolerance or NIDDM if their weight at yr was equal to or less than ± kg compared
with those whose weight at yr was equal to or greater than ± kg (Hales et al., ).
This was true in each social class and at each level of body weight. A similar relationship
with birth weight was seen in men and women of mean age from Preston (Phipps et
al., ). These trends were barely changed by adjustments for duration of gestation,
and therefore reflected differences in fetal growth rates.
Recent advances in assay methodology make it possible to measure specifically
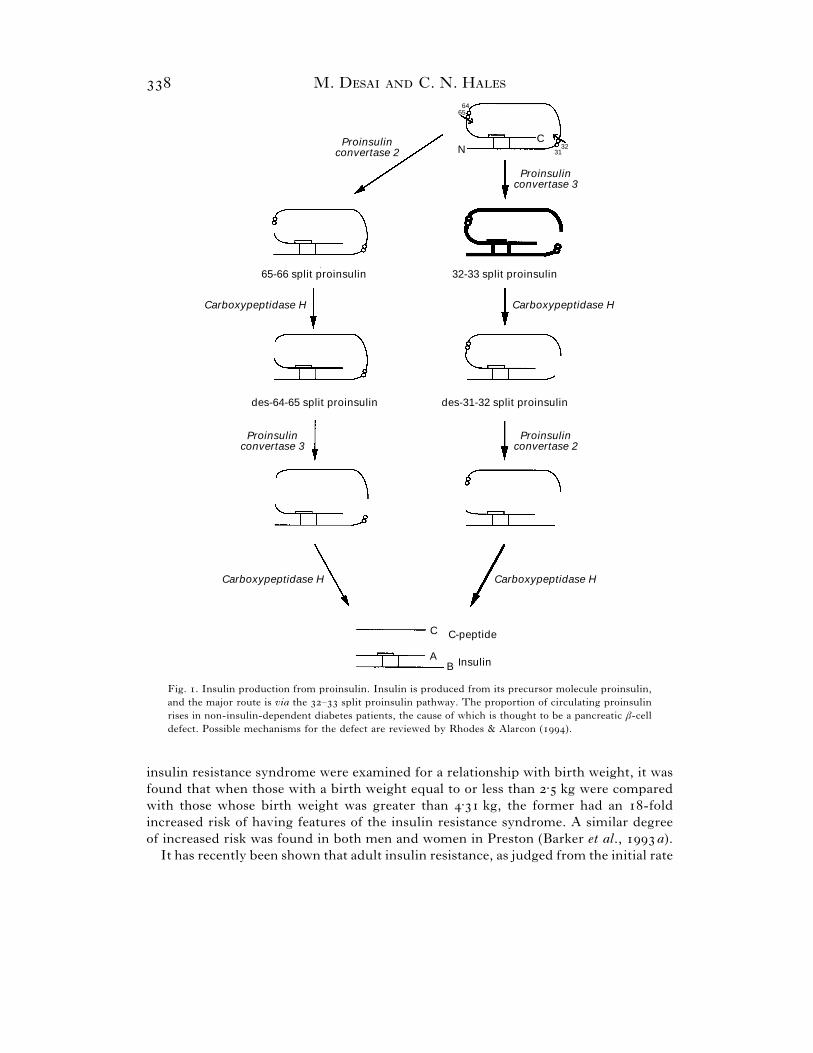
plasma concentrations of the precursor of insulin, – split proinsulin (Sobey et al.,
; Temple et al., ). A raised plasma – split proinsulin concentration is
thought to indicate β-cell dysfunction (see Fig. ). Concentrations of plasma – split
proinsulin were higher in Hertfordshire men with lower birth weight and weight at yr
of age. Therefore, this suggests that the relationship between low birth weight and
NIDDM depends partly on impaired β-cell function. This interpretation is consistent
with the occurrence of impaired development of the endocrine pancreas in babies with
intrauterine growth retardation (Van Assche et al., ). However, in studies of
middle-aged adults, no strong relationships between indices of β-cell function and early
anthropometry have been observed (Phillips et al., b ; Alvarsson, Efendic & Grill,
). The lack of such correlations may reflect the adaptation of β-cell responses to
insulin resistance in later adult life. There is a relationship between min plasma
glucose levels and birth weight in young men (Robinson et al., ) which is consistent
with this conclusion.
The association between early growth and NIDDM raised the possibility of whether
the combination of reduced glucose tolerance, hypertension and hypertriglyceridaemia,
components of what is now referred to as the ‘ insulin resistance syndrome’ or
‘syndrome X’, could be linked to birth weight. When Hertfordshire men with the
M. D C. N. H
32-33 split proinsulin
CN 32
31
6564
Proinsulinconvertase 3
Carboxypeptidase H
des-31-32 split proinsulin
Proinsulinconvertase 2
Carboxypeptidase H
C
AB
C-peptide
Insulin
Carboxypeptidase H
des-64-65 split proinsulin
Proinsulinconvertase 3
65-66 split proinsulin
Carboxypeptidase H
Proinsulinconvertase 2
Fig. . Insulin production from proinsulin. Insulin is produced from its precursor molecule proinsulin,
and the major route is via the – split proinsulin pathway. The proportion of circulating proinsulin
rises in non-insulin-dependent diabetes patients, the cause of which is thought to be a pancreatic β-cell
defect. Possible mechanisms for the defect are reviewed by Rhodes & Alarcon ().
insulin resistance syndrome were examined for a relationship with birth weight, it was
found that when those with a birth weight equal to or less than ± kg were compared
with those whose birth weight was greater than ± kg, the former had an -fold
increased risk of having features of the insulin resistance syndrome. A similar degree
of increased risk was found in both men and women in Preston (Barker et al., a).
It has recently been shown that adult insulin resistance, as judged from the initial rate

Programming fetal and infant growth
of fall of plasma glucose levels after a bolus injection of insulin, was related to an index
of thinness at birth. Thus, infants of low ponderal index (weight divided by length
cubed) were most insulin resistant as adults. Adult obesity was an added risk such that
infants who were thin at birth and obese (as judged by body mass index) as adults were
approximately twice as insulin resistant as those who were heavier at birth but thin as
adults (Phillips et al., a). The processes which link thinness at birth with insulin
resistance in adult life are not known. Studies of patients with NIDDM, using
euglycaemic clamp, have shown that peripheral tissues, particularly skeletal muscle, are
an important site of insulin resistance (DeFronzo, ). Muscle biopsies have shown
that insulin resistance is associated with a lower density of capillaries in muscle, a lesser
proportion of Type muscle fibres and a greater proportion of Type B fibres (Lillioja
et al., ). Babies born at term with a low ponderal index have a reduced mid-arm
circumference which implies that they have a low muscle bulk as well as less
subcutaneous fat (Robinson et al., ). The authors therefore suggested that thinness
in fetal life may be associated with abnormalities in muscle structure and function that
persist into adult life and interfere with the ability of insulin to promote glucose uptake.
These observations have been independently confirmed in divergent populations.
For example, the relationship of low birth weight to increased risk of adult NIDDM
has been confirmed in Pima Indians (McCance et al., ) and the relationship of low
birth weight to features of the insulin resistance syndrome has been confirmed in
Mexican Americans (Valdez et al., ) and in Swedish men (Lithell et al., ).
Interestingly, in the Pima Indians, there is also an increased risk associated with higher
birth weight (McCance et al., ). Presumably, this is because a large proportion of
pregnancies are associated with gestational diabetes which is also a factor known to
predispose to subsequent NIDDM in the offspring. Earlier work on rats which had
diabetes expeirmentally induced during pregnancy has also shown that there are long-
term consequences to the offspring – mild diabetes during pregnancy was associated
with decreased insulin secretion in the offspring whereas severe diabetes caused insulin
resistance (Van Assche, Aerts & Holemans, ; Holemans, Aerts & Van Assche,
).
Additional evidence that processes leading to NIDDM operate early in life is the
finding that individuals with impaired glucose tolerance are significantly shorter than
those with normal glucose tolerance in East Anglia in England (Williams et al., ).
Confirmation of this association has also been observed in a very different population
in Tanzania in which impaired glucose tolerance was found to be more common in
shorter individuals, even though the population was underweight by Western standards
and NIDDM itself very uncommon (Swai et al., ).
Studies on younger populations, including children aged and years from
Salisbury (Law et al., ), and -year-old men in Southampton (Robinson et al.,
) show a similar association between fetal growth, blood pressure and insulin
response to that observed previously in older people. Therefore, these associations
provide further evidence that the pathogenesis of NIDDM is set in sequence in early
life, and that metabolism becomes impaired within a few years of birth.
The variations in birth weight in the Hertfordshire and Preston studies may be
markers of more subtle but more important changes in organ structure and function
which determine later disease. For example, recent studies have shown that raised

M. D C. N. H
serum cholesterol concentrations in adult life are associated with impaired growth
during late gestation. It is suggested that impaired fetal growth leads to under-
development of the liver and a resultant permanent change in low-density lipoprotein
cholesterol metabolism (Barker et al., b).
Hence, the findings are reproducible and applicable to widely different ethnic groups
and may provide important insights into the underlying pathogenic processes. In order
to incorporate these findings into a pathogenic mechanism leading to impaired glucose
tolerance, NIDDM and insulin resistance syndrome, it is necessary to understand the
major processes determining fetal growth and development. These associations between
early growth and long-term health are thought to reflect ‘programming’, whereby
influences which impair fetal growth have permanent effects on the structure and
function of particular organs and tissues.
() Thrifty phenotype hypothesis
The proposed role of the fetal environment in the pathogenesis of non-insulin-
dependent diabetes and its associated features has been incorporated into the ‘thrifty
phenotype’ hypothesis (Hales & Barker, ). The essence of this hypothesis is that
poor fetal nutrition imposes growth and developmental constraints and changes upon
the fetus which may be considered as achieving metabolic thrift. It is envisaged that the
changes adopted serve at least two functions. First, they operate by selective nutritional
distribution between organs, such that the overall brain growth and development is
preserved relative to certain other organs such as the liver and pancreas. Secondly, the
changes which occur adapt metabolism in postnatal life, such that the chances of
survival under conditions of poor nutrition postnatally are enhanced. There is a
growing body of work on experimental animals which strongly supports the existence
of such mechanisms.
A further development of the ‘thrifty phenotype’ hypothesis is the proposal that
whilst these adaptive changes are beneficial to survival under conditions of poor
nutrition, they may be detrimental under conditions of normal or overnutrition. Under
these conditions, there would be a reduced ability to produce and respond to insulin
with subsequent increased risk of impaired glucose tolerance or NIDDM. Other
changes may also be entrained, leading to hypertension and disordered lipid metabolism
depending on the timing and type of nutrient deficiency. It is envisaged that poor
nutrition during early life need not inevitably lead to NIDDM in adult life. It is rather
the transition from nutritional deprivation to nutritional abundance which leads to the
metabolic conflicts resulting in NIDDM.
This hypothesis also draws particular attention to the potential role of fetal amino
acid deficiency in the aetiology of NIDDM. This is because of the key role of amino
acids as substrates for fetal energy production and their being essential for fetal growth
(reviewed by Jones, ). Furthermore, insulin, a major fetal growth hormone
(Fowden, ), is predominantly regulated by amino acids rather than by glucose
during fetal life. Hence, both the growth of β-cells and the triggering of insulin
secretion are affected.

Programming fetal and infant growth
() Recent animal studies
In recent years, the key role of nutritional protein in ensuring proper development
of the islets of Langerhans has been demonstrated by Hoet, Remacle and their
colleagues in Louvain. It has been shown that pregnant rats fed a diet containing
slightly less than half the normal protein content produced pups with reduced neonatal
β-cell proliferation as measured by tritiated thymidine levels, islet size and
vascularization (Snoeck et al., ). The reduced vascularization may relate to
observations in an earlier study in which impairment of insulin secretion following
protein–calorie deficiency was found to be more severe than would have been expected
from the reduction in numbers of islet cells alone (Weinkove et al., ). In other
studies, rats weaned on a low-protein diet for only weeks produced an insulin
response to glucose that was permanently impaired, leading to the suggestion that ‘early
malnutrition may predispose to diabetes’ (Swenne et al., ). It was subsequently
shown that offspring from protein-restricted mothers, maintained on low protein to the
adult age of d had reduced glucose tolerance, associated with reduced insulin
secretion. A group of animals fed normally after birth had an intermediate response
(Dahri et al., ).
Other studies have shown that prenatal exposure to a maternal low-protein diet
induced hypertension in the young adult offspring (Langley & Jackson, ). The
finding that the activity of placental enzyme β-hydroxy-steroid dehydrogenase,
which protects the fetus from deleterious effects of maternal corticosteroids, was lower
in these maternal-protein-restricted offspring suggested a role for glucocorticoid
hormones (Phillips et al., ).
We have studied the offspring of protein-restricted rats during pregnancy and}or
lactation. Permanent growth retardation was seen in offspring nursed by mothers on a
low-protein diet despite the fact that they were weaned on a normal % protein diet.
Conversely, a complete catch-up in growth was demonstrated by offspring born to
protein-restricted mothers but subsequently nursed by mothers fed an adequate
protein diet (Fig. ). Sex-dependent, selective and permanent organ weight changes
were also observed. For instance, the brain and lungs were relatively protected from
reduction in growth whilst the liver and pancreas were more disadvantaged in these
offspring at days of age (i.e. before weaning) (Table ). In -week-old male rats
which had been weaned on a normal diet, the relative weight of muscle was significantly
lower whereas in the female rats, the relative weight of the pancreas was increased
(Desai et al., ).
At days postpartum, changes in liver enzyme activity in the offspring from low-
protein-fed mothers (approximately % decrease in glucokinase and about %
increase in phosphoenolpyruvate carboxykinase) shifted the enzyme setting of the liver
by a factor of % in favour of glucose production rather than utilization. These
changes were still apparent in adult offspring, demonstrating permanency (Desai et al.,
). It is known that glucokinase and phosphoenolpyruvate carboxykinase are
predominantly located in different metabolic zones of the liver. The former is expressed
around the perivenous zone and the latter around the periportal zone (Jungermann &
Katz, ). We have therefore hypothesized that, during development, different
hepatic cells may have multiplied differentially according to the nutritional status of the
M. D C. N. H
0
100
200
300
400
500
600
Bo
dy w
eig
ht
(g)
0 8 24 32 40 48
Weeks
Female rats
0
100
200
300
400
500
600
Bo
dy w
eig
ht
(g)
0 8 24 32 40 48
Weeks
Male rats
700
800
900
16
16
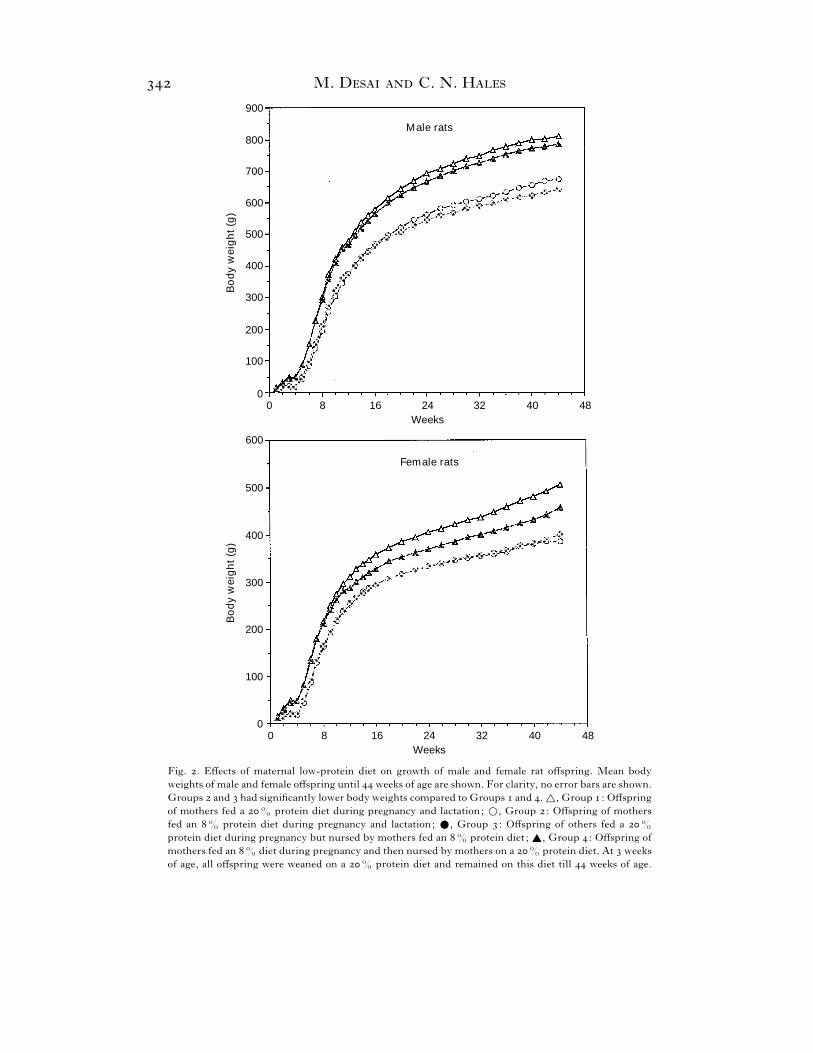
Fig. . Effects of maternal low-protein diet on growth of male and female rat offspring. Mean body
weights of male and female offspring until weeks of age are shown. For clarity, no error bars are shown.
Groups and had significantly lower body weights compared to Groups and . ^, Group : Offspring
of mothers fed a % protein diet during pregnancy and lactation; D, Group : Offspring of mothers
fed an % protein diet during pregnancy and lactation; E, Group : Offspring of others fed a %
protein diet during pregnancy but nursed by mothers fed an % protein diet ; _, Group : Offspring of
mothers fed an % diet during pregnancy and then nursed by mothers on a % protein diet. At weeks
of age, all offspring were weaned on a % protein diet and remained on this diet till weeks of age.

Programming fetal and infant growth
Table . Selective changes in organ growth in offspring of rats fed a low-protein diet
(The body and organ weights of -day-old offspring from groups , and (see legend to Fig. for details) are
expressed as percentage Group weights. Groups and show similar reduction in body weight whereas Group
exhibits partial recovery. In all three groups, the brain and lung are relatively preserved, the kidney and thymus
show similar weight reduction to the body whereas the pancreas, liver, muscle and spleen are more disadvantaged.
The heart is the only organ which shows variation in reduction in weight depending upon when the exposure to
the maternal low-protein diet occurred.)
Percentage of control weight
Organs Group Group Group
Body
Brain
Lung
Heart
Kidney
Thymus
Pancreas
Liver
Spleen
Muscle
fetus, resulting in the population of perivenous cells being contracted and that of
periportal cells being expanded. Consistent with this, we found that the activity of
another perivenous enzyme, glutamine synthetase, decreased and another periportal
enzyme, carbomyl phosphate synthetase, increased in the offspring from low-protein-
fed mothers (Desai et al., ). These livers when isolated and perfused with glucagon
and insulin showed a marked resistance to the action of insulin in inhibiting glucagon-
stimulated glucose output (Ozanne et al., ). The offspring of rat dams protein-
restricted during pregnancy and lactation were not glucose intolerant at months of
age. However, at months of age, the worsening of glucose tolerance in these offspring
was more extensive than that of control offspring. In the former group of offspring, this
was mainly attributable to insulin resistance in the males and insulin deficiency in the
females (Hales et al., ). This reflects the human situation in which males are more
insulin resistant than females (Phillips et al., a).
We also investigated the influence of a maternal low-protein diet on longevity in the
offspring (Hales et al., ). Our findings show that a brief period of exposure to
lactating dams which have been protein deprived has a beneficial effect on longevity.
This brief period is also capable of permanently reducing the growth trajectory of
offspring. Conversely, a highly detrimental effect on longevity was observed when the
pups growth was retarded in utero, but was then accelerated as a result of good postnatal
nutrition by being suckled by unrestricted mothers. The rate of growth of such pups
when expressed relative to their initial body weight was faster than in the controls
during early life. On the other hand, longevity was unchanged when the pups growth
was retarded in utero and the same rate of growth was maintained during the postnatal
period by being nursed by the same low-protein-fed mothers (Fig. ). It has been
known for many years that undernutrition increases longevity in a variety of animals
(Berg and Simms, ). Our findings demonstrate that nutrition during the first three
weeks of postnatal life affects not only growth but also longevity in rats.
M. D C. N. H
0
200
600
400
Bo
dy w
eig
ht/
weig
ht
at
day 3
(%
)
0 7 21 28
Days
14
800
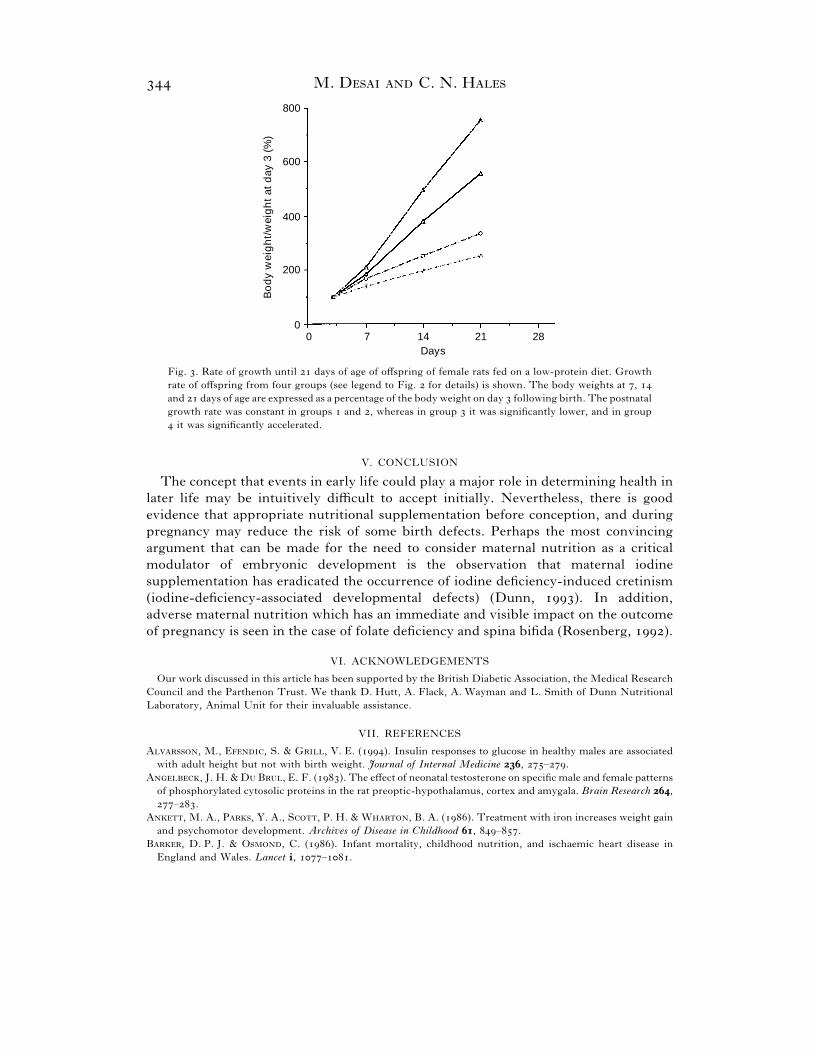
Fig. . Rate of growth until days of age of offspring of female rats fed on a low-protein diet. Growth
rate of offspring from four groups (see legend to Fig. for details) is shown. The body weights at ,
and days of age are expressed as a percentage of the body weight on day following birth. The postnatal
growth rate was constant in groups and , whereas in group it was significantly lower, and in group
it was significantly accelerated.
V. CONCLUSION
The concept that events in early life could play a major role in determining health in
later life may be intuitively difficult to accept initially. Nevertheless, there is good
evidence that appropriate nutritional supplementation before conception, and during
pregnancy may reduce the risk of some birth defects. Perhaps the most convincing
argument that can be made for the need to consider maternal nutrition as a critical
modulator of embryonic development is the observation that maternal iodine
supplementation has eradicated the occurrence of iodine deficiency-induced cretinism
(iodine-deficiency-associated developmental defects) (Dunn, ). In addition,
adverse maternal nutrition which has an immediate and visible impact on the outcome
of pregnancy is seen in the case of folate deficiency and spina bifida (Rosenberg, ).
VI. ACKNOWLEDGEMENTS
Our work discussed in this article has been supported by the British Diabetic Association, the Medical Research
Council and the Parthenon Trust. We thank D. Hutt, A. Flack, A. Wayman and L. Smith of Dunn Nutritional
Laboratory, Animal Unit for their invaluable assistance.
VII. REFERENCES
A, M., E, S. & G, V. E. (). Insulin responses to glucose in healthy males are associated
with adult height but not with birth weight. Journal of Internal Medicine , –.
A, J. H. & D B, E. F. (). The effect of neonatal testosterone on specific male and female patterns
of phosphorylated cytosolic proteins in the rat preoptic-hypothalamus, cortex and amygala. Brain Research ,
–.
A, M. A., P, Y. A., S, P. H. & W, B. A. (). Treatment with iron increases weight gain
and psychomotor development. Archives of Disease in Childhood , –.
B, D. P. J. & O, C. (). Infant mortality, childhood nutrition, and ischaemic heart disease in
England and Wales. Lancet i, –.

Programming fetal and infant growth
B, D. P. J., O, C., W, P. D. & M, B. (). Weight in infancy and death from ischaemic
heart disease. Lancet ii, –.
B, D. J. P., B, A. R., O, C. & S, S. J. (). Fetal and placental size and risk of
hypertension in adult life. British Medical Journal , –.
B, D. J. P., M, T. W., F, C. H. D., L, A., O, C., P, K. & S, Y. ().
Relation of fetal and infant growth to plasma fibrinogen and factor VII concentrations in adult life. British
Medical Journal , –.
B, D. J. P., H, C. N., F, C. H. D., O, C., P, K. & C, P. M. S. (a). Type
(non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced
fetal growth. Diabetologia , –.
B, D. J. P., M, C. N., O, C., H, C. N. & F, C. H. D. (b). Growth in utero and
serum cholesterol concentrations in adult life. British Medical Journal , –.
B, P. H., R, N. B., M, M. & L C, P. M. (). Epidemiologic studies of diabetes
in the Pima Indians. Recent Progress in Hormone Research , –.
B, B. N. & S, H. S. (). Nutrition and longevity in the rat. II. Longevity and onset of disease with
different levels of food intake. Journal of Nutrition , –.
B, M. E. & P-L, A. M. (). Effect of neonatal hyperthyroidism upon the regulation of TSH
secretion in rats. Acta Endocrinologica , –.
C-H, R., C, D. M., H, M. H. & M, A. (). Is birthweight determined genetically?
British Medical Journal , –.
C, E., G, H. C., MB, M., L, B. & P, R. (). Childhood antecedents of adult
obesity. The New England Journal of Medicine , –.
C, J. S. & T, B. Y. (). Diabetes and different races in a similar environment. In Diabetes (ed.
W. Waldhaust), pp. –. Excerpta Medica, Elsevier, Amsterdam.
C, B. F. & L, C. F. (). Effect of dietary restrition of pregnant rats on body weight gain of the offspring.
Journal of Nutrition , –.
C, M. P., S, E., R, Y. & Z, A. (). High prevalence of diabetes in young adult Ethiopian
immigrants to Israel. Diabetes , –.
D, S., S, A., R-B, B., R, C. & H, J. J. (). Islet function in offspring of
mothers on low-protein diet during gestation. Diabetes , –.
D, P. R., S, M. A. & S, A. (). Iron deficiency in infancy and childhood. American Journal
of Clinical Nutrition , –.
DF, R. A. (). The triumvirate: beta cell, muscle, liver. A collusion responsible for NIDDM. Diabetes
, –.
D, M., C, N. J., O, S. E., L, A. & H, C. N. (). Adult glucose and lipid metabolism
may be programmed during fetal life. Biochemical Society Transactions , –.
D, M., C, N. J., L, A. & H, C. N. (). Organ-selective growth in the offspring of protein
restricted mothers. British Journal of Nutrition , –.
D, C., P, M., S, E. & C, A. (). Infant mortality: A practical approach to the analysis
of the leading causes of death and risk factors. Pediatrics , –.
D, D. A. & S, K. (). Effect of altered neonatal nutrition on the development of enzymes of lipid and
carbohydrate metabolism in the rat. Journal of Nutrition , –.
D, J. T. (). Iodine supplementation and the prevention of cretinism. In Maternal Nutrition and Pregnancy
Outcome (ed. C. L. Keen, A. Bendich and C. C. Willhite), pp. –. Annals of The New York Academy of
Sciences, volume , USA.
E, N. J. & J, E. (). Weight relationship in pregnancy. I. The bearing of maternal weight gain
and pre-pregnancy weight on birth weight in full term pregnancies. Obstetrical and Gynecological Survey ,
–.
E, I., F, H., A, E. & E, S. J. W. (). Intergenerational studies of human
birthweight from birth cohort. I. Evidence for a multigenerational effect. British Journal of Obstetrics and
Gynaecology , –.
F, C. H. D., B, D. J. P., O, C., W, P. D., C, P. M. S. & H, C. N. (). Relation
of infant feeding to adult serum cholesterol concentration and death from ischaemic heart disease. British
Medical Journal , –.
F, I. M., J, P. R., S, J. S. & H, J. (). Diet-induced adipocyte number increase in adult
rats : a new model of obesity. American Journal of Physiology , E–.
F, A. (). Are poor living conditions in childhood and adolescence an important risk factor for
ateriosclerotic heart disease? British Journal of Preventive and Social Medicine , –.

M. D C. N. H
F, A. L. (). The role of insulin in prenatal growth. Journal of Developmental Physiology , –.
G, P. D., B, B. H., O, M., H, J. E. & B, N. S. (). Fetal growth in late
gestation – A constrained pattern of growth. Acta Paediatrica Scandinavia Suppl. , –.
G, K. M., R, C. W. G., B, D. J. P. & O, C. (). The effect of maternal anaemia and
iron deficiency on ratio of fetal weight to placental weight. British Journal of Obstetrics and Gynaecology ,
–.
H, E., E, I., V B, G. & D, J. (). Maternal birth weight and subsequent
pregnancy outcome. Journal of the American Medical Association , –.
H, P. & K, L. (). Immediate and late effects of premature weaning and of feeding a high fat or high
carbohydrate diet to weaning rats. Journal of Nutrition , –.
H, C. N., B, D. J. P., C, P. M. S., C, L. J., F, C., O, C. & W, P. D. (). Fetal
and infant growth and impaired glucose tolerance at age years. British Medical Journal , –.
H, C. N. & B, D. J. P. (). Type (non-insulin-dependent) diabetes mellitus : the thrifty phenotype
hypothesis. Diabetologia , –.
H, C. N., D, M., O, S. E. & C, N. J. (). Fishing in the steam of Diabetes : from
measuring Insulin to the control of fetal organogenesis. Biochemical Society Transactions , –.
H, V. K. M. (). Genetic mechanisms of regulation of fetal growth. In Fetal Growth (ed. F. Sharp, R. B.
Fraser and R. D. G. Milner), pp. –. Proceedings of th study group of the Royal College of Obstetricians
and Gynaecologists, Royal College of Obstetricians and Gynaecologists, London.
H, S. A., S, P. H., H, C. V., C, Y. F. & V, D. V (a). Human
intrauterine renal growth expressed in absolute number of glomeruli assessed by the ‘Disector’ method and
Cavalieri principle. Laboratory Investigation , –.
H, S. A., L, M. R. J., S, P. H., H, C. V. & V, D. V (). The effect of
intrauterine growth retardation on the development of renal nephrons. British Journal of Obstetrics and
Gynaecology , –.
H, K., A, L. & V A, F. A. (). Evidence for an insulin resistance in the adult offspring
of pregnant streptozotocin-diabetic rats. Diabetologia , –.
I, S. M. (). Influence of maternal cholestyramine treatment on cholesterol and bile metabolism in adult
offspring. Journal of Nutrition , –.
I, S. M. (). The role of diet during development on the regulation of adult cholesterol homeostasis.
Canadian Journal of Physiology Pharmacology , –.
J, W. P. T. & C, H. G. (). Persistent impairment of insulin secretion and glucose tolerance after
malnutrition. American Journal of Clinical Nutrition , –.
J, R. J. (). The epidemiology of diabetes mellitus. In Textbook of diabetes. (ed. J. C. Pickup and
G. Williams), pp. –. Blackwell Scientific Publication.
J, C. T. (). Fetal metabolism and fetal growth. Journal of Reproduction and Fertility , –.
J, K. & K, N. (). Functional specialization of different hepatocyte populations. Physiological
Review , –.
K, H. B. (). The influence of undernutrition on learning performance in rodents. Nutrition Abstract Review
, –.
K, M. A., M, O. & B, H. W. (). Second-generation consequences of small-for-dates
birth. Pediatrics , –.
K, M. S. (). Determinants of low birth weight: methodological assessment and meta-analysis. Bulletin
of the World Health Organizaiton , –.
L, S. C. & J, A. A. (). Increased systolic pressure in adult rats induced by fetal exposure to
maternal low protein diets. Clinical Science , –.
L, C. M., B, D. J. P., B, A. R. & O, C. (). Maternal and fetal influences on blood pressure.
Archives of Disease in Childhood , –.
L, D. S., B, H. A., MM, C. A., MG, H. C., C, K. D. & M, E. J. ().
Preweaning food intake influences the adiposity of young adult baboons. Journal of Clinical Investigation ,
–.
L, S., Y, A. A., C, C. L., I, J. L., A, W. G. H., Z, J. K., Y-J$ , H.,
C, L., S, T. W. & B, C. (). Skeletal muscle capillary density and fibre type are
possible determinants of in vivo insulin resistance in man. Journal of Clinical Investigation , –.
L, H. O., MK, P. M., B, L., M, R., L, U. B. & L, D. A. (). Relation
of size at birth to non-insulin dependent diabetes and insulin concentrations in men aged – years. British
Medical Journal , –.

Programming fetal and infant growth
L, M. T. & H, P. (). Diet and metabolic development. The FASEB Journal , –.
L, A. & M, R. (). Influence of early diet on outcome in preterm infants. Acta Paediatrica
Supplement , –.
L, A. (). Programming by early nutrition in man. In The Childhood Environment and Adult Disease (ed.
G. R. Bock and J. Whelan), pp. –. John Wiley, Ciba Foundation symposium, number , Chichester.
L, L. H. (). Decreased birthweights in infants after maternal in utero exposure to the Dutch famine of
–. Paediatric and Perinatal Epidemiology , –.
M, H. (). Health trends in the past years. Health Trends , –.
MC, D. R., P, D. J., H, R. L., J, L. T. H., K, W. C. & B, P. H. ().
Birthweight and non-insulin-dependent diabetes : ‘‘ thrifty genotype’’, ‘‘ thrifty phenotype’’, or ‘‘surviving
small baby genotype’’. British Medical Journal , –.
MC, R. A. & W, E. M. (). Protein deficiencies and calorie deficiencies. Lancet ii, –.
MC, R. A. & W, E. M. (). The determinants of growth and form. Proceedings of The Royal
Society of London , –.
MC, M. C. (). The contribution of low birth weight to infant mortality and childhood morbidity. The
New England Journal of Medicine , –.
M, R. D. G. (). Metabolic and hormonal responses to glucose and glucagon in patients with infantile
malnutrition. Pediatric Research , –.
M, N. E. (). The inheritance of human birthweight. Annals of Human Genetics , –.
M, G. E., L, D. S. & MG, H. C. (). Programming of cholesterol metabolism by breast or formula
feeding. In The Childhood Environment and Adult Disease (ed. G. R. Bock and J. Whelan), pp. –. John
Wiley, Ciba Foundation symposium, number , Chichester.
N, J. V. (). Diabetes mellitus : a ‘ thrifty’ genotype rendered detrimental by ‘progress ’? American Journal
of Human Genetics , –.
O, M. & O, C. (). Maternal regulation of intra-uterine growth. Nature , –.
O, M., S, A. & O, C. (). Transmission through the female line of a mechanism constraining
human fetal growth. Annals of Human Biology , –.
O, J. A., O, P. C. & R, J. S. (). Experimental fetal growth retardation: metabolic and
endocrine aspects. In Advances in Fetal Physiology (ed. P. D. Gluckman, B. M. Johnston and P. W. Nathanielsz),
pp. –. Perinatology Press, Ithaca.
O, S. E., S, G. D., T, J. & H, C. N. (). Altered regulation of hepatic glucose output
in the male offspring of protein-malnourished rat dams. American Journal of Physiology , E–E.
P, D. I. W., B, D. J. P., H, C. N., H, S. & O, C. (a). Thinness at birth and
insulin resistance in adult life. Diabetologia , –.
P, D. I. W., H, S., C, P. M. S., H, C. N. & O, C. (b). Fetal growth and insulin
secretion in adult life. Diabetologia , –.
P, G. J., L-E, S. C., B, R., S, J. R., E, C. R. W. & J, A. A.
(). The role of dietary protein restriction during pregnancy on the activity of placental β-hydroxysteroid
dehydrogenase. Proceedings of The Nutrition Soceity A, .
P, K., B, D. J. P., H, C. N., F, C. H. D., O, C. & C, P. M. S. (). Fetal growth
and impaired glucose tolerance in men and women. Diabetologia , –.
P, B. L. (). Endocrine function in protein-calorie malnutrition. Clinical Endocrinology , –.
R, G. P., S, Z. A. & S, M. W. (). Obesity in young men after famine exposure in utero and
early infancy. The New England Journal of Medicine , –.
R, C. J. & A, C. (). What β-cell defect could lead to hyperproinsulinemia in NIDDM? Some
clues from recent advances made in understanding the proinsulin-processing mechanism. Diabetes , –.
R, S. M., W, T., H, M. C., B, D. J. P. & O, C. (). Fetal heart rate and
intrauterine growth. British Journal of Obstetrics and Gynaecology , –.
R, S., W, R. J., C, P. M., B, D. J. P., H, C. N. & O, C. (). The relation
of fetal growth to plasma glucose in young men. Diabetologia , –.
R, I. H. (). Folic acid and neural tube-defects – Time for action? The New England Journal of
Medicine , –.
S (). Undernutrition, learning and memory: a review of experimental studies. In Proceedings of XIII
International Congress of Nutrition (ed. T. G. Taylor and N. K. Jenkins), pp. –. John Libbey, London.
S, A., R, C., R, B. & H, J. J. (). Effect of a low protein diet during pregnancy on the
fetal rat endocrine pancreas. Biology of the Neonate , –.
S, M. H. L. (). Effects of genome on size at birth. In Fetal Growth (ed. F. Sharp, R. B. Fraser and
R. D. G. Milner), pp. –. Royal College of Obstetricians and Gynaecologists, London.

M. D C. N. H
S, W. J., B, S. F., C, C. A., C, P. M., F. B. H., G, I. P., L, S. D., O,
D. R., S, A. E., S, K., T, R. C. & H, C. N. (). Sensitive and specific two-site
immunoradiometric assays for human insulin, proinsulin, - split and - split proinsulins. Biochemical
Journal , –.
S, D. A. (). Instinct with original observations on young animals. Macmillan’s Magazine , –.
(Reprinted in British Journal of Animal Behaviour , –).
S, Y., G, S., P, A. & O, L. (). Quantitative immunofluorescent study of the endocrine
cell population in the developing human pancreas. Diabetes , –.
S, Z. A. & S, M. W. (). The Dutch Famine, –, and the reproductive process. I. Effects on
six indices at birth. Pediatric Research , –.
S, A. B., K, H. M., M, G., K, P. M., A, K. G. M. M. & ML, D. G. (). Is
diabetes mellitus related to undernutrition in rural Tanzania? British Medical Journal , –.
S, I., C, C. J. & M, R. D. G. (). Persistent impairment of insulin secretory response to glucose
in adult rats after limited periods of protein-calorie malnutrition early life. Diabetes , –.
T, R. C., C, C. A., L, S. D., O, D. R., S, A. E., S, W. J. & H, C. N.
(). Insulin deficiency in non-insulin-dependent diabetes. Lancet i, –.
V, R., A, M. A., T, G. H., B, B. S. & S, M. P. (). Birthweight and adult
health outcomes in a biethnic population in the USA. Diabetologia , –.
V A, F. A., D, F., A, L. & V, M. (). The endocrine pancreas in the small-for-dates
infants. British Journal of Obstetrics and Gynaecology , –.
V A, F. A., A, L. & H, K. (). Metabolic alterations in adulthood after intrauterine
development in mothers with mild diabetes. Diabetes , –.
W, A. & H, J. (). The maternal effects on growth and conformation in Shire horse–Shetland
pony crosses. Proceedings of The Royal Society of London , –.
W, T., A, I., C, P. & P, C. G. (). Iron deficinecy anemia: adverse effects on
infant psychomotor development. Pediatrics , –.
W, C., W, E. A. & P, B. L. (). Insulin release and pancreatic islet volume in
malnourished rats. South African Medical Journal , .
W, E. M. (). Intra-uterine growth retardation in the pig. . Organ size and cellular development at
birth and after growth to maturity. Biology of the Neonate , –.
W, E. M. & MC, R. A. (). Some effects of accelerating growth. . General somatic
development. Proceedings of The Royal Society of London , –.
W, E. M. & MC, R. A. (). A Review: New thoughts on growth. Pediatric Research ,
–.
W, D. R. R., B, C., C, P. M. S., C, L., H, C. N., D, N. E. & W, T. (). Impaired
glucose tolerance and height. British Medical Journal , .
W, D. R. R., R, S. J. & D, T. W. (). Deaths from ischaemic heart disease and infant
mortality in England and Wales. Journal of Epidemiology and Community Health , –.
W, M. & N, A. (). Cellular response in rats during malnutrition at various ages. Journal of Nutrition
, –.
W, M. & N (). Cellular response with increased feeding in neonatal rats. Journal of Nutrition ,
–.
W, M., F, I. & R, P. (). Cellular recovery in rat tissues after a brief period of neonatal
malnutrition. Journal of Nutrition , –.
W, M. (). Cellular changes during placental and fetal growth. American Journal of Obstetrics Gynecology
, –.
Z, F. J. (). Effect on the young rat of maternal protein restriction. Journal of Nutrition , –.
Z, P., T, R., R, P., K, H., S, G., R R, L. & H, D. (). Prevalence of
diabetes and impaired glucose tolerance in the biracial (Melanesian and Indian) population of Fiji : A rural-
Urban comparison. American Journal of Epidemiology , –.
Z, P., K, H., T, R., R, L. R., B, B., B, J., H, W. & T, K. (). The
high prevalence of diabetes mellitus, impaired glucose tolerance and diabetic retinopathy in Nauru: the
survey. Diabetes Research , –.
Related Documents











