University of Groningen Role of extracellular vesicles in hypoxia-induced hepatic injury in non-alcoholic fatty liver disease Hernandez Villanueva, Alejandra DOI: 10.33612/diss.180853744 IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2021 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Hernandez Villanueva, A. (2021). Role of extracellular vesicles in hypoxia-induced hepatic injury in non- alcoholic fatty liver disease. University of Groningen. https://doi.org/10.33612/diss.180853744 Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license. More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne- amendment. Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 26-07-2022

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

University of Groningen
Role of extracellular vesicles in hypoxia-induced hepatic injury in non-alcoholic fatty liverdiseaseHernandez Villanueva, Alejandra
DOI:10.33612/diss.180853744
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2021
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Hernandez Villanueva, A. (2021). Role of extracellular vesicles in hypoxia-induced hepatic injury in non-alcoholic fatty liver disease. University of Groningen. https://doi.org/10.33612/diss.180853744
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 26-07-2022

Roleofextracellularvesiclesinhypoxia-
inducedhepaticinjuryinnon-alcoholic
fattyliverdisease
PhDThesis
toobtainthedegreeofPhDatthe
UniversityofGroningen
ontheauthorityofthe
RectorMagnificusProf.C.Wijmenga
andinaccordancewith
thedecisionbytheCollegeofDeans.
Thisthesiswillbedefendedinpublicon
Wednesday6October2021at14.30hours
by
AlejandraAndreaHernándezVillanueva
bornon22March1990
inSantiago,Chile

2
ROLEOFEXTRACELLULARVESICLESIN
HYPOXIA-INDUCEDHEPATICINJURYINNON-
ALCOHOLICFATTYLIVERDISEASE
TESISPARAOPTARALGRADODEDOCTOR
ENCIENCIASMÉDICASENCOTUTELACONLA
UNIVERSIDADDEGRONINGEN
ALEJANDRAHERNÁNDEZVILLANUEVA
SUPERVISORES:
MARCOARRESEJIMENEZ
HANMOSHAGE
SANTIAGODECHILE
2020

3
Supervisors
Prof.A.J.Moshage
Prof.M.Arrese
Assessmentcommittee
Prof.J.W.Jonker
Prof.H.vanGoor
Prof.A.Feldstein

4
TomyparentsVictorandGloriaforpushingmetospreadmywingsandforalwaystrustingme,
Tomylovelyfamily,brothers,nephewsandauntsfortheirunconditionalsupport,
TomysoulfriendswhoaremyotherfamilythatIchose,
Tomytutor,speciallyHanandMarco,thatIoweeveryeducationandprofessionaltraining,
TomyboyfriendPedroforbeingthehappinessofmylife,
Toeachoneofyou...Ialwayscarryinmyheart.

5
Amispadresporimpulsarmeaabrirmisalasyporconfiarsiempreenmí,
Atodamifamiliaporsuamorincondicional,
Amisamigasdelalmaquesonmiotrafamiliaqueelegí,
Amisprofesoresquelesdebocadaenseñanzayformaciónprofesional,
Amipololoporserlaalegríademivida,
Acadaunodeustedes...losquieroyllevosiempreenmicorazón.

6
TableofContentsPag
Preface&Scopeofthisthesis 7
Chapter1:GeneralIntroductionandaimthethesis 9
Chapter2:ExtracellularvesiclesinNAFLD/ALD:frompathobiologytotherapy.
Cells9,817.2020 15
Chapter3:Chemicalhypoxiainducespro-inflammatorysignalsinfat-laden
hepatocytesandcontributestocellularcrosstalkwithKupffercellsthrough
extracellularvesicles.(BBA)MolecularBasisofDisease1866,165753.2020
65 30
Chapter4:Extracellularvesiclesderivedfromfat-ladenhepatocytesundergoing
chemicalhypoxiapromoteapro-fibroticphenotypeinhepaticstellatecells.
(BBA)MolecularBasisofDisease1866,165857.202061
Chapter5:HypoxiaincreasedCaspase-1inextracellularvesiclesderived
fromexperimentalnon-alcoholicsteatohepatitismodelsandpromotes
inflammasomeactivationinKupffercells.82
Chapter6:Discussion,ConclusionandFuturePerspectives99
Appendices:Summaries(EnglishandSpanish)andNederlandse 104
samenvatting,Acknowledgements;Abbreviations;
Supplemmentarymaterial,andListofpublications.

7
Preface&Scopeofthisdoctoralthesis
PREFACE
Non-alcoholicfattyliverdisease(NAFLD)isahighlyprevalentchronicliverdiseasethataffects30%of
thegeneralpopulation.TheincidenceofNAFLDisstillincreasing.NAFLDencompassesapathological
spectrumof liver injury, ranging from isolatedsteatosis toan inflammatorycondition termednon-
alcoholic steatohepatitis (NASH), that can progress to liver fibrosis and subsequent cirrhosis and
hepatocellular carcinoma (HCC). Clinical observations have indicated that obstructive sleep apnea
syndrome (OSAS) is a significant risk factor that predisposes patients to the progression of liver
steatosis,inflammationandfibrosis.OSASisasleepbreathingdisordercharacterizedbyintermittent
hypoxia(IH)duringsleep.IthasbeenshownthatIHinpatientsandinexperimentalrodentmodelsis
linkedtooxidativestress,steatosis,inflammationandliverfibrosis.
In recent years, extracellular vesicles (EV) have been implicated in intercellular communication in
variouspathophysiologicalconditions,includingNASH.However,themechanismsunderlyingtherole
ofEVinNASHinthecontextof(intermittent)hypoxiaremainsunexplored.
This doctoral thesis focuses on the role of extracellular vesicles in the pathogenesis of hypoxia-
induced hepatic injury in different models of NASH. To validate our hypothesis we used in vitro
modelstoinvestigatecellularcrosstalkbetweenfat-ladenhepatocytesexposedtochemicalhypoxia
andnon-parenchymalcells,suchashepaticstellatecellsandKupffercells.Also,weusedan invivo
model of NASH to evaluate whether IH promotes liver injury and increases circulating EV. Our
findings reveal new insights on the pathophysiological effects of hypoxia on lipotoxicity,
inflammation and fibrosis. Moreover, we provide novel insights with regard to the presence of
caspase-1inEV,suggestingcaspase-1asapotentialnovelbiomarkertomonitorNAFLDandOSA.
SCOPEOFTHETHESIS
Theresearchdescribedinthisthesisfocusesonthecombinationoflipotoxicityandhypoxiaininvitro
and in vivo models of NASH. We investigate whether hypoxia promotes the activation of
inflammatory and fibrotic pathways, with a special emphasis on the role of EV in inflammasome
activation.
InChapter1wepresentageneralintroductionofNAFLD,intermittenthypoxia-relatedOSA,therole
ofEVinNAFLD/NASHandthegeneralaimsofthisthesis.
In Chapter 2, the role of EV in liver pathophysiology is reviewed and discussed, including their
potentialapplicationsasbiomarkersandtherapeutics.

8
InChapter3we introduceourexperimental invitromodelofNASHusingprimary rathepatocytes
exposed to free fatty acids (FFA) and subjected to chemically-induced hypoxia (CH) using the
hypoxia-induciblefactor1alpha(HIF-1α)stabilizercobaltchloride(CoCl2).Weobservedthathypoxia
aggravateshepatocellular injuryviaamechanismthat involves inflammasome/caspase-1activation
infat-ladenhepatocytesandcellularcrosstalkwithKupffercellsthroughEV.Futhermore,we
InChapter4,westudythecellularcrosstalkbetweenhypoxichumanHepG2cellsexposedtoFFAand
humanhepaticLX-2stellatecells.Weobservedthathypoxiaexacerbateshepatocellulardamageand
pro-fibroticsignalingandthatthisiscorrelatedwithincreasedEVreleasefromfat-ladenHepG2cells.
Interestingly,EVfromhypoxicfat-ladenHepG2evokedapro-fibroticresponseinLX-2cells.
InChapter5weshowanimportantfindingaboutcaspase-1asoneofthecargocomponentsofEV
from both hypoxic fat-laden hepatocytes as well as from serum of mice fed the CDAA diet and
exposed to IH. Moreover, we observed that silencing of HIF-1α in hepatocytes abolished the
induction of inflammasome-related genes in Kupffer cells. These data suggest that IH could be an
aggravatingfactorintheprogressionofNASHviaHIF-1αinduction
Finally,inChapter6wesummarizeanddiscussourresultsandprovideanoutlookforfuturestudies.

9
Chapter1
1.-GENERALINTRODUCTIONANDAIMSOFTHEDOCTORALTHESIS
Non-alcoholicfattyliverdisease(NAFLD)isthemostcommonliverdiseaseworldwide,affectingupto
30% of the current population (1). NAFLD is used as an umbrella term that describes a clinico-
pathologicalentitydefinedbythepresenceofaspectrumofhepatichistologicalchanges.Observed
phenotypes vary in severity from non-inflammatory isolated steatosis (also termed non-alcoholic
fattyliver[NAFL])toamoreaggressiveformnamedsteatohepatitis(ornon-alcoholicsteatohepatitis,
NASH), which is characterized by inflammatory changes and hepatocellular ballooning associated
withvaryingdegreesofliverfibrosis(2,3).NASHisamultisystemdiseaseassociatedwithasignificant
risk for thedevelopmentof cirrhosisandhepatocellular carcinoma,aswell as liver transplantation
andliver-relateddeath(4,5).
The pathogenesis of NAFLD involves a complex interaction between nutritional factors, obesity,
insulin resistance, changes in the microbiota, genetic and epigenetic factors. These factors are
involved inthedevelopmentandprogressionofsteatosisresulting inanexcessiveaccumulationof
lipids, especially triglycerides, in the hepatocytes (6-8).Moreover, these factors contribute, either
simultaneouslyorsequentially,totheinflammatoryandfibroticresponseofthelivertosteatosis(9).
At the cellular level, the increased influx of free fatty acids (FFA) into the liver exceeds the
physiological capacity, leading to reactive oxygen species (ROS) overproduction, mitochondrial
dysfunctionandcellulardeathinaprocesscalledlipotoxicity,ultimatelyleadingtoinflammationand
fibrosis(10,11).
Several studies have shown that lipotoxicity appears to be the central driver of hepatocellular
damagethatcontributestotheinflammatoryresponseandthedevelopmentofliverfibrosis(12,13).
The crosstalk between hepatocytes and non-parenchymal cells plays a crucial role in this
inflammatory and fibrotic response (14-17). It is believed that fat-laden (steatotic) hepatocytes
release damage signals to the extracellular environment, resulting in paracrine effects on
neighboring cells such as resident macrophages of the liver (Kupffer cells) and stellate cells that
promote the activationof inflammatory and fibrogenicpathways, respectively (18,19). In addition,
fat-laden hepatocytes trigger various pathways leading to hepatocellular dysfunction by autocrine
effectsthatinitiateandperpetuatelipotoxicity(20-22).
Inrecentyears,obstructivesleepapneasyndrome(OSAS),acommonsleepdisordercharacterizedby
recurrentclosureoftheupperairwaysduringsleep,hasbeenassociatedwiththedevelopmentand
progressionofNAFLD(23)inbothobeseandnon-obesesubjects(24-25).Themostnotedhallmarkof
OSAS is intermittent hypoxia (IH), which leads to tissue hypoxia and can result in ROS

10
overproduction,mitochondrialdysfunctionandinflammation(26).Theseeventsarealsoinvolvedin
theinitiationofNASH(27).SeveralstudieshaveindicatedalinkbetweenOSASorIHwithincreased
liver TG accumulation in NAFLD through modulation of β-oxidation of fatty acids and de novo
lipogenesis inhepatocytes (28-31). Inaddition, inexperimentalmodelsofNASH ithasbeenshown
that mimicking OSAS with IH has pro-inflammatory effects as indicated by increased levels of
inflammatorycytokinessuchastumornecrosisfactoralpha(TNF-α)andinterleukin-6(IL-6)(30,31).
Likewise,invitrostudiesusinghepatocytesdemonstratedthathypoxia-induciblefactor1alpha(HIF-
1α)increaseshepatocyteapoptosisandgenerationofpro-inflammatorysignalsininflammatorycells
(32-34). Moreover, a link between HIF-1α and hepatic inflammation and fibrosis has also been
described inanimalstudiesof IH (35-37).Takentogether, theexperimentalevidencesuggests that
hypoxia,inaHIF-1αdependentmanner,contributestothetransitionfromisolatedsteatosistomore
advancedstagesofNAFLDandcanaggravatelipotoxicity,inflammationand/orfibrosise.
ThepathophysiologyofNASHisknowntoinvolvehepatocellulardamageassociatedwithlipotoxicity,
triggeringlocalinflammatoryresponsesandcontributingtoliverfibrosis.However,thereisalackof
knowledgeregardingthemechanismsofthedetrimentaleffectofhypoxiaonfat-ladenhepatocytes
andtheroleof intercellularcommunicationbetweenhepatocytesandnon-parenchymalcellstypes
suchasKupfferandstellatecells.
Recent studies have indicated that extracellular vesicles (EV) play a key role in intercellular
communicationinliverpathobiology(38-40).Inparticular,EVhavebeenimplicatedinhepatocellular
injury,inflammationandhepaticfibrosisinNAFLD,bothinhumansaswellasinexperimentalmodels
ofNASH(41-45).
EVarenanoparticlesdefinedbyalipidbilayer.ThecargoofEVcanhavemanyeffectsontargetcells,
bothphysiologicalaswellaspathophysiological.ClassificationofEVdependontheirsizeandsiteof
biogenesis:Exosomes(40-150nm)arereleasedfrommultivesicularbodies,microvesicles(MVs)(50-
1000nm)arereleasedfromthebuddingplasmamembraneandapoptoticbodies(50-5000 nm)are
releasedfromblebbingcells(47).ExosomesandMVsmaycontainavarietyofbioactivemolecules,
including cytoplasmic proteins, lipids, specific lipid raft-interacting proteins, messenger RNAs,
noncodingRNAsandmetabolites(39-46).Ingeneral,apoptoticbodiesareexcludedfromstudieson
EV (46). EVhavebeen implicated inmanypathophysiologicalprocessesand recently, the fieldhas
expandedintoEV-baseddiagnostics,prognosisandtherapeutics(48).
SeveralresearchgroupshavedemonstratedtheinvolvementofEVinintercellularcommunicationin
invivoaninvitromodelsofliverdiseases(42-44,48,49).Recentstudieshaveshownthathepatocyte-
derivedEV activatepro-inflammatory signals andpro-fibrotic signals in non-parenchymal cells (48,

11
49). Additionally, increased levels of circulating EV in in vivo models of NAFLD correlate with
histologicfeaturesofNASHthatindicateliverdamage(42-44,50).Therefore,itisimportanttostudy
thecontentofEVthatexertparacrineeffectsontargetcells.Interestingly,recentdatasuggestthat
EV released from fat-laden hepatocytes activate the inflammasome via caspase-1 activation in
hepatocytesandmacrophagesleadingtoaninflammatoryresponse(51).
TheNOD-likereceptorPyrinDomainContaining3 (NLRP3) inflammasome isspecificandcritical for
the activation of caspase-1 and the processing of pro-inflammatory cytokines. The NLRP3
inflammasomehasrecentlybeendemonstratedtocontributetothetransitionfromNAFLDtoNASH
(52,53). Studies have also shown that expression of inflammasome components is increased in
mousemodelsofNASHand inhumanswithNASH(54,55).Moreover,downregulationofNLRP3or
caspase-1inflammasomecomponentsalleviatehepaticsteatosis, inflammationandfibrosis(55-58),
suggestingthattheinflammasomeisapotentialtherapeutictargetinNASH.
Theresearchpresentedinthisdoctoralthesisinvestigateswhetherhypoxiaexacerbateslipotoxicity
in fat-laden hepatocytes and liver injury in a mice model of non-alcoholic steatohepatitis via the
release of extracellular vesicles and cellular crosstalk between hepatocytes and non-parenchymal
cells.Additionally,weexaminedinflammasomeactivationinconditionsofhypoxia,bothinvitroand
invivo.Takentogether, theresultsof this thesisestablisha linkbetweenextracellularvesiclesand
hepatocellular damage in hypoxia models that mimics OSA. This link involves EV-mediated
intercellular communication between hepatocytes and non-parenchymal cells in non-alcoholic
steatohepatitis.

12
2.-REFERENCES
1. EstesC,RazaviH,LoombaR,YounossiZ,SanyalAJ.Modelingtheepidemicofnonalcoholicfattyliverdiseasedemonstratesan
exponentialincreaseinburdenofdisease.Hepatology2018;67:123-133.
2. ArabJP,ArreseM,TraunerM.RecentInsightsintothePathogenesisofNonalcoholicFattyLiverDisease.AnnuRevPathol2018;
13:321-350.
3. KoyamaY,BrennerDA.Liverinflammationandfibrosis.J.Clin.Investig2017;127:55-64
4. SanyalAJ. Past, present and futureperspectives innonalcoholic fatty liverdisease.NatRevGastroenterolHepatol 2019; 16:
377-386.
5. YounossiZM.Non-alcoholicfattyliverdisease-Aglobalpublichealthperspective.JHepatol2019;70:531-544.
6. GohGB,McCulloughAJ.Naturalhistoryofnonalcoholicfattyliverdisease.Dig.Dis.Sci2016;61:1226–33
7. MussoG,CassaderM,GambinoR.Non-alcoholicsteatohepatitis:emergingmoleculartargetsandtherapeuticstrategies.Nat.
Rev.Drug.Discov.2016;15:249–74
8. HardyT,OakleyF,AnsteeQM,DayCP.Nonalcoholicfattyliverdisease:pathogenesisanddiseasespectrum.Annu.Rev.Pathol.
2016;11:451–96
9. Buzzetti E, PinzaniM, Tsochatzis EA. Themultiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD).Metabolism
2016;65:1038–48
10. Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, Mott JL, et al. Mechanisms of
lysophosphatidylcholine-inducedhepatocytelipoapoptosis.AmJPhysiolGastrointestLiverPhysiol2012;302:G77-84.
11. Hirsova P, Ibrahim SH, Gores GJ,Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH
pathogenesis.J.LipidRes.2016;57:1758–70
12. Geng Y, Hernandez A, Faber KN, de Meijier VE, Blokzijl H, Moshage H. Lipotoxicity in non-alcoholic fatty liver diseases:
mechanismsandclinicalimplications.BBA-MolCellBioll.October2019.Submitted
13. Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of
nontriglyceridefattyacidmetabolites.Hepatology2020;52:774–88
14. WobserH.,DornC.,WeissT.S.,etal.Lipidaccumulationinhepatocytesinducesfibrogenicactivationofhepaticstellatecells.
CellResearch.2009;19:996–1005.
15. Brenner C., Galluzzi L., KeppO., KroemerG. Decoding cell death signals in liver inflammation. Journal ofHepatology. 2013;
59:583–594.
16. LebensztejnD.M.,Flisiak-JackiewiczM.,Białokoz-Kalinowska I.,Bobrus-ChociejA.,Kowalska I.Hepatokinesandnon-alcoholic
fattyliverdisease.ActaBiochimicaPolonica.2016;63:459–467.
17. GanzM.,SzaboG.ImmuneandinflammatorypathwaysinNASH.HepatologyInternational.2013;7:771–781.
18. WobserH,DornC,WeissTS,AmannT,BollheimerC,BüttnerR,SchölmerichJ,HellerbrandC.Lipidaccumulationinhepatocytes
inducesfibrogenicactivationofhepaticstellatecells.CellRes.2009;19:996-1005.
19. WanJ,BenkdaneM,Teixeira-ClercF,BonnafousS,LouvetA,LafdilF,PeckerF,TranA,GualP,MallatA,LotersztajnS,PavoineC.
M2 Kupffer cells promoteM1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver
disease.Hepatology.2014;59:130-42.
20. BrennerC,GalluzziL,KeppO,KroemerG.Decodingcelldeathsignalsinliverinflammation.JHepatol.2013;59:583-94.
21. LiuJ,HanL,ZhuL,YuY.Freefattyacids,nottriglycerides,areassociatedwithnon-alcoholicliverinjuryprogressioninhighfat
dietinducedobeserats.LipidsHealthDis.201611;15:27.
22. Bellanti F, Villani R, FacciorussoA, VendemialeG, ServiddioG. Lipid oxidation products in the pathogenesis of non-alcoholic
steatohepatitis.FreeRadicBiolMed2017;111:173-185.
23. Mesarwi,O, Loomba,R,Malhotra,A.Obstructive sleepapnea,hypoxia,andnonalcoholic fatty liverdisease.AmJRespirCrit
CareMed.20191;199:830-841.
24. ChenLD,ZhangLJ,LinXJ,QiJC,LiH,WuZ,XuQZ,HuangYP,LinL.Associationbetweencontinuouspositiveairwaypressureand
serumaminotransferasesinpatientswithobstructivesleepapnea.EurArchOtorhinolaryngol2018;275:587-594.
25. Qi JC, Huang JC, Lin QC, Zhao JM, Lin X, Chen LD, Huang JF, Chen X. Relationship between obstructive sleep apnea and
nonalcoholicfattyliverdiseaseinnonobeseadults.SleepBreath2016;20:529-535.

13
26. May AM, Mehra R. Obstructive sleep apnea: role of intermittent hypoxia and inflammation. Semin Respir Crit Care Med.
2014;35:531-44.
27. NoureddinM,SanyalAJ.PathogenesisofNASH:TheImpactofMultiplePathways.CurrHepatolRep.2018;17:350-360.
28. Buttacavoli M, Gruttad'Auria CI, Olivo M, Virdone R, Castrogiovanni A, Mazzuca E, Marotta AM, Marrone O, Madonia S,
BonsignoreMR.LiverSteatosisandFibrosis inOSApatientsAfterLongtermCPAPTreatment:APreliminaryUltrasoundStudy.
UltrasoundMedBiol2016;42:104-109.
29. Aron-WisnewskyJ,MinvilleC,TordjmanJ,LevyP,BouillotJL,BasdevantA,BedossaP,ClementK,PepinJL.Chronicintermittent
hypoxiaisamajortriggerfornon-alcoholicfattyliverdiseaseinmorbidobese.JHepatol2012;56:225-233.
30. Briancon-MarjolletA,MonneretD,HenriM,etal.IntermittenthypoxiainobeseZuckerrats:cardiometabolicandinflammatory
effects.ExpPhysiol.2016;101:1432-1442.
31. Kang HH, Kim IK, Lee HI, et al. Chronic intermittent hypoxia induces liver fibrosis in mice with diet-induced obesity via
TLR4/MyD88/MAPK/NF-kBsignalingpathways.BiochemBiophysResCommun.2017;490:349-355.
32. ShiYF,FongCC,ZhangQ,CheungPY,TzangCH,WuRS,YangM.Hypoxiainducestheactivationofhumanhepaticstellatecells
LX-2throughTGF-betasignalingpathway.FEBSLett2007;581:203-210.
33. CoppleBL,BaiS,BurgoonLD,MoonJO.Hypoxia-inducible factor-1alpharegulatestheexpressionofgenes inhypoxichepatic
stellatecellsimportantforcollagendepositionandangiogenesis.LiverInt2011;31:230-244.
34. Roth KJ, Copple BL. Role of Hypoxia-Inducible Factors in the Development of Liver Fibrosis. CellMol Gastroenterol Hepatol
2015;1:589-597.
35. SavranskyV,BevansS,NanayakkaraA,LiJ,SmithPL,TorbensonMS,PolotskyVY.Chronicintermittenthypoxiacauseshepatitis
inamousemodelofdiet-inducedfattyliver.AmJPhysiolGastrointestLiverPhysiol2007;293:G871-877.
36. Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J
PhysiolGastrointestLiverPhysiol2009;296:G582-592.
37. MesarwiOA,ShinMK,Bevans-FontiS,SchlesingerC,ShawJ,PolotskyVY.HepatocyteHypoxiaInducibleFactor-1Mediatesthe
DevelopmentofLiverFibrosisinaMouseModelofNonalcoholicFattyLiverDisease.PLoSOne2016;11:e0168572.
38. Hirsova P, Ibrahim SH, Verma VK, Morton LA, Shah VH, LaRusso NF, Gores GJ, Malhi H. Extracellular vesicles in liver
pathobiology:Smallparticleswithbigimpact.Hepatology(Baltimore,Md)64:2219-2233,2016.
39. StahlPD,andRaposoG.ExtracellularVesicles:ExosomesandMicrovesicles, IntegratorsofHomeostasis.Physiology2019;34:
169-177.
40. ArreseM,EguchiA,FeldsteinAE.CirculatingmicroRNAs:emergingbiomarkersofliverdisease.SeminLiverDis2015;35:43-54
41. KakazuE,MauerAS,YinM,andMalhiH.Hepatocytesreleaseceramide-enrichedpro-inflammatoryextracellularvesiclesinan
IRE1alpha-dependentmanner.JLipidRes2016;57:233-245.
42. PoveroD,EguchiA,LiH,JohnsonCD,PapouchadoBG,WreeA,MesserK,andFeldsteinAE.Circulatingextracellularvesicleswith
specificproteomeand livermicroRNAsarepotentialbiomarkers for liver injury inexperimental fatty liverdisease.PLoSOne
2014;9:e11365.
43. PoveroD,EguchiA,NiesmanIR,AndronikouN,deMolleratduJeuX,MulyaA,BerkM,LazicM,ThapaliyaS,ParolaM,PatelHH,
and Feldstein AE. Lipid-induced toxicity stimulates hepatocytes to release angiogenicmicroparticles that require Vanin-1 for
uptakebyendothelialcells.SciSignal2013;6:ra88.
44. WelshJA,ScorlettiE,CloughGF,EnglystNA,andByrneCD.Leukocyteextracellularvesicleconcentrationisinverselyassociated
withliverfibrosisseverityinNAFLD.JLeukocBiol2018;104:631-639.
45. KornekM,LynchM,MehtaSH,LaiM,ExleyM,AfdhalNH,SchuppanD.Circulatingmicroparticlesasdisease-specificbiomarkers
ofseverityofinflammationinpatientswithhepatitisCornonalcoholicsteatohepatitis.Gastroenterology2012;143:448-458.
46. MalhiH.EmergingRoleofExtracellularVesiclesinLiverDiseases.AmJPhysiolGastrointestLiverPhysiol2019;317:G739–G749.
47. SzaboG,Momen-HeraviF.Extracellularvesicles in liverdiseaseandpotentialasbiomarkersandtherapeutic targets.NatRev
GastroenterolHepatol2017;14:455-466.
48. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF,WerneburgNW, CharltonMR, et al. Lipid-Induced Signaling Causes
ReleaseofInflammatoryExtracellularVesiclesFromHepatocytes.Gastroenterology2016;150:956-967.
49. LeeYS,KimSY,KoE,LeeJH,YiHS,YooYJ,JeJ,etal.Exosomesderivedfrompalmiticacid-treatedhepatocytesinducefibrotic
activationofhepaticstellatecells.SciRep2017;7:3710.

14
50. BaronM,LeroyerAS,MajdZ,LalloyerF,VallezE,BantubungiK,Chinetti-GbaguidiG,DeleriveP,BoulangerCM,StaelsB,and
TailleuxA.PPARalphaactivationdifferentlyaffectsmicroparticlecontentinatheroscleroticlesionsandliverofamousemodelof
atherosclerosisandNASH.Atherosclerosis2011;218:69-76.
51. CannitoS,MorelloE,BoccaC,FogliaB,BenettiE,NovoE,ChiazzaF,etal.Microvesiclesreleasedfromfat-ladencellspromote
activation of hepatocellular NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic
steatohepatitis.PLoSOne2017;12:e0172575.
52. Wan X, Xu C, Yu C, Li Y. Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can J Gastroenterol Hepatol.
2016;2016:6489012.
53. LuanJ,JuD.Inflammasome:ADouble-EdgedSwordinLiverDiseases.FrontImmunol.2018;9:2201.
54. WreeA,McGeoughMD,InzaugaratME,EguchiA,SchusterS,JohnsonCD,PeñaCA,GeislerLJ,PapouchadoBG,HoffmanHM,
FeldsteinAE.NLRP3inflammasomedrivenliverinjuryandfibrosis:RolesofIL-17andTNFinmice.Hepatology.2018;67:736-
749.
55. WreeA,McGeoughMD,PeñaCA,SchlattjanM,LiH, InzaugaratME,MesserK,CanbayA,HoffmanHM,FeldsteinAE.NLRP3
inflammasomeactivationisrequiredforfibrosisdevelopmentinNAFLD.JMolMed(Berl).2014;92:1069-82.
56. Dixon LJ, BerkM, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced
steatohepatitis.LabInvest.2012;92:713-23.
57. Yang G, Lee HE, Lee JY. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a
mousemodelinducedbyhighfatdiet.SciRep.2016;6:24399
58. CabreraD,WreeA,PoveroD,SolísN,HernandezA,PizarroM,MoshageH,TorresJ,FeldsteinAE,Cabello-VerrugioC,BrandanE,
Barrera F, Arab JP, Arrese M. Andrographolide Ameliorates Inflammation and Fibrogenesis and Attenuates Inflammasome
ActivationinExperimentalNon-AlcoholicSteatohepatitis.SciRep201714;7:3491.

15
Chapter2
EXTRACELLULARVESICLESINNAFLD/ALD:FROMPATHOBIOLOGYTOTHERAPY
ABSTRACT
In recent years, knowledge on the biology and pathobiology of extracellular vesicles (EV) has
exploded. EV are submicron membrane-bound structures secreted from different cell types
containingawidevarietyofbioactivemolecules[i.e.,proteins, lipids,andnucleicacids(codingand
non-codingRNA,andmitochondrialDNA].EVhaveimportantfunctionsincell-to-cellcommunication
and are found in awide variety of tissues and body fluids. Better delineation of EV structure and
advancesintheisolationandcharacterizationoftheircargohaveallowedtoexplorediagnosticand
therapeutic implications of these particles. In the field of liver diseases, EV are emerging as key
playersinpathogenesisofbothalcoholicliverdisease(ALD)andnonalcoholicliverdisease(NAFLD),
the most prevalent liver diseases worldwide and their complications, including development of
hepatocellularcarcinoma.Inthesediseases,stressed/damagedhepatocytesreleaselargequantities
ofEVthatcontributetotheoccurrenceofinflammation,fibrogenesisandangiogenesisthatarekey
pathobiologicalprocessesinliverdiseaseprogression.Moreover,thespecificmolecularsignaturesof
released EV in biofluids have allowed to consider EV as promising candidates to serve as disease
biomarkers. Also, different experimental studies have shown that EV may have potential for
therapeutic use as a liver-specific delivery method of different agents taking advantage of their
hepatocellularuptake through interactionswith specific receptors. In this review,wewill focuson
the most recent findings concerning the role of EV as new structures mediating autocrine and
paracrine intercellular communication in both ALD and NAFLD as well as their potential use as
biomarkers of disease severity and progression. Emerging therapeutic applications of EV in these
liverdiseasesarealsoexaminedandthepotentialforsuccessfultransitionfrombenchtoclinic.
1.-INTRODUCTION
Knowledgeofthepathobiologyofextracellularvesicles(EV)hasexpandedsignificantlyinthelastdecade
(1,2).Indeed,significantadvanceshavebeenmadeindelineatingthemechanismsofassemblyandrelease
ofEVaswellastheirsubsequentmembranefusionwithtargetcells(3,4).Moreover,powerfulanalytical
techniqueshavemadepossibletheextensivecharacterizationofthecargoofEVthatincludesamyriadof
molecules including growth factors, metabolic enzymes, microRNAs and transcription factors, certain
proteins,lipidsandmetabolites,amongothers,thatmodulateintercellularandinter-organcommunication
(3,5,6).Ofnote,high-throughputdatasetsofvesicularcomponentsarenowavailableinpublicdatabases,
whichstronglysupportsEVresearch(7,8).

16
NovelinsightsintothebiologyofEVshowthattheseparticlesregulatecriticalbiologicalfunctionsandmay
act as contributors to disease pathogenesis andmay also serve as disease biomarkers in virtue of the
relative simplicityofEV isolation fromdifferentbiofluids (9). Inaddition,EVaregaining interest froma
therapeuticpointofviewduetotheirpotentialasauniquedrugdeliverysystem(10).
InthefieldofhepatologyEVhaverecentlyemergedasnovelplayersinthepathogenesisandprogression
ofseveralconditions(11-13)includingthetwomostcommonliverdiseasesworld-wide:non-alcoholicliver
disease(NAFLD)andalcoholicliverdisease(ALD)(14).Specifically,recentstudiespointtoasignificantrole
ofEVinmodulating injury,amplifying inflammationandpromoting liverfibrosis inbothNAFLDandALD
(15-17).Sinceinformationonthistopicisdynamicandrapidlyevolving,weaimtoprovideanup-to-date
overviewofthecurrentknowledgeontheroleofEVinthecontextofbothNAFLDandALDwithemphasis
ontheirpotentialdiagnosticandtherapeuticimpactinthesediseases.Weexcludefromthisreviewdata
regardingEVinlivercancersincethishasbeenrecentlyreviewedelsewhere(18,19).
2.-GENERALCONCEPTSOFEVINTHELIVER:EVBIOGENESIS,SECRETIONANDCARGO
DetailsontheformationandsecretionpathwaysofEVhavebeenrecentlyreviewedelsewhere(15,20,21)
andcanalsobefoundinothercontributionsinthisspecialissueofCells(22).Onlybasicconceptswillbe
providedhereaswellasinformationonaspectsthatareofparticularimportanceforliverphysiologyand
pathophysiology.
Ingeneral,EVareclassifiedaccordingtosizeandbiogeneticpathway,suchasexosomes,microvesiclesand
apoptoticbodies(23).Exosomesarebilayerlipidvesicleswithadiameterof30-150 nmthatarederived
fromendosomalmultivesicularbodies(MVBs)(15,23).Itsformationresultsfromtheinvaginationofthe
plasmamembrane (early endosome) and the subsequent fusion of endocytic vesiclesmediated by the
endosomalsortingcomplexresponsiblefortransport(ESCRTs)andothercomponents(suchasceramides
andtetraspanins)(24).TheMVBscanreleasetheintraluminalvesiclesknownasexosomesbythefusionof
MVBstotheplasmamembrane,aprocessmediatedinpartbyRabGTPases(25).Microvesicles(MVs)have
adiameterof50-1000nmandoriginatefromtheplasmamembranebybuddingandfissionfollowedby
releaseintotheextracellularspace(15,23).MVscontainasubsetofcellsurfaceproteinsdependingonthe
compositionoftheparentalplasmamembrane(26,27).Apoptoticbodieshaveadiameterof100-5000 nm
and originate from the budding of cellmembranes andmay contain nuclearmaterial,which is quickly
phagocytosedduringprogrammedcelldeath (15,28).UnlikeexosomesandMVs, the roleof apoptotic
bodiesisnotrelatedtointercellularcommunication.Therefore,whenstudyingEVundercellulardamage
conditions,apoptoticbodiesareexcluded(29).
All EV transport a variety of bioactive molecules, including cytoplasmic proteins, lipids, specific lipid
raft-interactingproteins,messengerRNA(mRNA),microRNA(miRNA),ribosomalRNA(rRNA),transferRNA

17
(tRNA),noncodingRNAs(ncRNAs),DNA,mitochondrialDNA(mtDNA)andmetabolites(24,30).Lipidomic
analysis has shown that EV, independently of their biogenesis, contain a myriad of lipids such as
cholesterol, sphingomyelin, ceramide, saturated fatty acids, phosphatidylcholine,
phosphatidylethanolamine and phosphatidylserine (4, 23, 29, 30). In addition, proteomic analysis has
shown that EV contains different types of proteins, such as heat shock proteins (Hsp70 and Hsp90),
tetraspanins (CD9,CD63,CD81,CD82), endosomal sorting complexproteins required for transport (Alix
andTsg101),receptorsincludingepidermalgrowthfactorreceptor(EGFR),membranetraffickingproteins
(GTPases,FlotillinandAnnexins),cytoskeletalproteins(tubulinandactin)andcytosolicproteins(5,26).Of
note, thecargoofEVvariesdependingnotonlyontheircellularoriginbutalsoontheconditionunder
whichtheyarereleased(i.e.physiologicalvs.pathological).
In the liver, both parenchymal (hepatocytes) and non-parenchymal cells (i.e. hepatic stellate cells,
endothelial,cholangiocytes,Kupfercellsandliverendothelialcells)havebeenfoundtoreleaseEVinboth
physiologicalandpathologicalstates(20).However, informationontargetcell repertoire, receptorsor
other specific actions is still limited and incomplete. It has been shown that healthy hepatocytes
producelimitedamountsofexosomescontainingproteinspotentiallyrelevantforcellsurvival,growthand
proliferation(31),whereasstressedhepatocytesboostexosomerelease(32)andenrichtheircontent in
specific proteins, lipids and microRNAs that modulate the transcriptional program of neighboring
hepatocytesandnon-parenchymalcells,thusmodulatinginflammationandfibrosiswhicharecriticalfor
theprogressionofliverdiseases(Figure1).InterestinglyrecentevidencesuggeststhatEVfromfat-laden
hepatocytecanalsosignaltootherorganssuchasadiposetissueinfluencingadipogenesisandtissue
remodeling(33).
2.2EVandliverinflammation
Hepatocellular damage determines the release of a number of signals into the extracellular
environment that can contribute to tissue inflammation (34). Some of these signals (collectively
termed damage-associated molecular patterns [DAMPs]) are packaged in EV and signal between
hepatocytesandnon-parenchymalcells suchas liver-residentmacrophages (Kupffercells,KC) (35).
Indeed, EVmay evoke synthesis and release of proinflammatory cytokines such as pro-interleukin
(IL)-1b and IL-6 (36) by KCs, thus contributing to local inflammation (37). Also, EV released from
hepatocytes can promote therecruitmentof additional immune cells (i.e.
proinflammatorymonocyte-derived macrophages) into theliver maintaining and amplifying
inflammation. EV can also signal to endothelialcells and can contribute to vascular inflammation
(38). Furthermore, to add complexity, EV canalsobe secreted fromother cells and influence liver
inflammation. In this regard, some evidence suggests that platelet-derived EV may have

18
proinflammatoryeffects inthe liverbutthisneedsfurtherconfirmation(39,40).Finally, ithasalso
been shown that EV are released frommonocytic cells and induce polarization towards the anti-
inflammatory M2 phenotype of neighboring naive monocytes by delivering cargo miR-27a, thus
contributingtoresolutionofinflammation(41).Collectively,thesefindingssuggestthatEVreleased
from injuredhepatocyteshavean important role inmodulating the inflammatory responseduring
liver damage through intercellular communication between different cell types with potential
contributions fromother cell-derivedEV (35). Specificmechanisms involved inNAFLDandALDare
reviewedbelow.
2.3EVandliverfibrosis
Persistent liver fibrogenesis and the development of cirrhosis are responsible for the liver-related
morbidityandmortalityassociatedwithchronic liverdiseases (42).Activationor trans-differentiationof
HSCsresultingininsolublecollagendepositionanddistortionofthenormalmacro-andmicro-anatomical
structureoftheliveristhemajordriverofliverfibrogenesis(43).Theroleofparacrinesignalsoriginating
from injuredepithelial cells (hepatocytes) that candirectlyor indirectly induceHSCactivationhasbeen
recognized in recent years (44). In this regard, EV seem to play a role as shown by previous reported
studies.Ofnote,lipid-inducedhepatocyte-derivedEVseemtoregulateHSCactivationbyshuttlingspecific
microRNAs (i.e. miR-128-3p) that suppress PPAR-γ expression in HSC leading to a marked increase of
profibrogenicgeneexpression(45).Otherauthorshaveshownthatinternalizationofendothelial-derived
exosomesbyHSCsenhancescellmigrationinaprocessmediatedbysphingosine1-phosphate(S1P)(46).
Additionally, intercellular communicationbetweenquiescent andactivatedHSCs via exosomes canalso
modulatefibrosis.Inthisregard,aroleforshuttledmicroRNA214(miR-214)inregulatingtheexpressionof
alpha-smoothmuscleactinandcollageninactivatedHSCshasbeendemonstrated(47,48).Moreover,EV
derived from non-resident cells such as platelets or granulocytes have been shown to increase
angiogenesis and tohaveprocoagulantproperties, thuspromoting fibrogenesis (49, 50). These findings
suggest that EV appear to be key modulators in fibrosis as signals from both parenchymal and non-
parenchymallivercellscaneitherdriveorslowdownHSCactivation.Inaddition,circulatingEVmaybe
usedasabiomarkerofhepaticfibrosisandhavepotentialimplicationsforthedevelopmentofnovelanti-
fibrotictargets(51)asreviewedinthefollowingsections.
3.-EVASBIOMARKERSINLIVERDISEASES
DynamicchangesofEVgenerationinpathologicalconditionsandtheaccessibilitytomeasureandanalyze
theminbiologicalsamples(i.e.blood,urine,bileandotherbiofluids)renderEVgoodcandidatesasdisease
biomarkers.Therelativeaccessibilityfortestingandperformingrepeatedmeasurementsovertimecould
facilitate early diagnosis, disease monitoring and development of personalized medicine.

19
Furthermore,refinementoftechniquesallowingisolationandindepthcharacterizationofEVcargoes
can lead to identification of disease-specific molecular signatures or profiles and provide ample
opportunitiesforEVtobeusedassuitable,non-invasivebiomarkers(52).
EV have been studied as potential biomarkers of liver injury in the setting of ALD, NAFLD, drug-
inducedliverdiseaseandcholangiopathies(13,27,53,54)aswellasdiagnostictoolsforlivercancer
(i.e.hepatocellularcarcinomaandcholangiocarcinoma)(19,54).Inthisregard,determinationofthe
numberofcirculatingEVandtheuseofnucleicacidbased,lipid-basedandprotein-baseddiagnostics
have been performed tomeasure EV enriched in liver-derivedDNA,microRNAs, lipids or proteins
(13,27).However,beforeimplementingEV-basedbiomarkersintheclinic,standardizationofsample
processing(i.e.collection,transportation,storageandhandling)andassaysystemsisneededaswell
aslargereplicativestudiestoallowEVmolecularsignaturestobeconclusivelylinkedtospecificliver
diseases.RecentadvancesrelatedtoNAFLDandALDarereviewedbelow.
4.-THERAPEUTICPOTENCIALOFEV
From a therapeutic perspective, EV either unmodified or engineered, can be utilized for therapeutic
purposes(55).Withregardtoliverdiseases,effortshavebeenfocusedontwomajorareas:a)theuseof
EVasdeliveryvehiclesofdrugstotheliver(56)andb)theuseofEVthemselvesastherapeuticagentsto
stimulateliverregeneration,modulateinflammation,reduceliverfibrosisorhalthepatocarcinogenesis(57,
58). The formerapproach involves theuseofdifferent techniques to loadEVwithadesired cargo (i.e.
miRNA, siRNA, chemotherapeutic agents) to act as a “Trojan horse” on target cells. Theoretically,
membranepropertiesofEValloworganandcell-specificdelivery,immune-evasionandtargetingofdistinct
intracellulartraffickingpathways.However,anumberofchallengesrelatedtothemanufacturingofEV(i.e.
production,coating, loading,etc.)stillneedtobesolvedbeforecontrolledclinicalstudiescanbecarried
out(59,60).WithregardtotheuseofEVastherapeuticagents,mostoftheavailableevidencehasbeen
generatedusingmesenchymalstemcell(MSC)-derivedEVobtainedfromhumanumbilicalcordorhuman
embryos that have been tested in numerous preclinical liver diseasemodels (i.e. carbon tetrachloride,
thioacetamide,D-galactosamine/TNF-α-inducedlethalhepaticfailureandbileductligation)withpromising
results.DataforNAFLDandALDaremorelimitedandarereviewedinthecorrespondingsectionsbelow.
Athird,andstillnascent,approachrelatedtoEV-basedtherapyisbasedontheconceptthat interfering
withEVsecretionoruptakemayattenuateharmfuleffectsontargetcells(11,61).Inthisregard,several
pharmacological agents are being explored that havebeen shown to inhibit EV trafficking,modify lipid
metabolism or decrease EV secretion. However, the complexity of EV biogenesis poses significant
challengestothedevelopmentofspecificagentsabletoblockEVproductionselectively[seeref.(61)for
anin-depthreviewofthistopic].

20
5.-EVINNAFLD
AnumberofstudieshavedemonstratedaroleofEVinbothpathogenesisandprogressionofNAFLD
(14,15).Triggeringof inflammationandfibrosisdevelopmentarekeyforprogressionfromisolated
steatosis (also referred as NAFL or non-NASH fatty liver) to nonalcoholic steatohepatitis (NASH),
which ishallmarkedbythepresenceofhepatocyteballooningasreflectofongoing liver injuryand
death (36). Data from different diet-induced animal models of NASH have shown that EV
concentration increases with disease progression in a time-dependent manner.(17, 62, 63). This
seemstobearesponsetotheaccumulationof toxic lipidsandtheirdownstreammediators in the
liver,which increasethecapacityofhepatocytestoformandreleasedifferenttypesofEV(37,62-
64). In vitro treatment of hepatocytes with non-esterified fatty acids evokes the release of EV
containingnumerousmolecules includingC-X-Cmotif ligand10 (CXCL10), sphingosine-1-phosphate
(S1P), mitochondrial DNA (mtDNA), micro-RNAs, ceramides and tumor necrosis factor-related
apoptosis-inducingligand(TRAIL)(15).Thesemoleculesmayamplifyinflammationthroughmultiple
mechanisms such as macrophage activation and monocyte chemotaxis as well as inflammasome
activationandmodulationoftheNF-κBpathwayintargetcells(64,65).Asmentionedearlier,EVmay
bereleasedbydifferentmechanismsincludingacaspase-3-dependentmechanism(63)oractivation
ofdeath receptor5 (DR5) inhepatocytes.Hepatocyte-derivedEVare able to induceexpressionof
pro-inflammatorycytokinesandpromoteM1polarizationofhepaticmacrophages(37,66).Ofnote,
CXCL10-bearing EV can also serve as chemotactic stimuli formacrophages as shown recently (64).
Moreover,EVreleasedfromhepatocytescancontributetohepaticrecruitmentofmonocyte-derived
macrophages by promotingmonocyte adhesion via integrin β1(ITGβ1)-dependentmechanisms as
showninmurineNASH(67).AdditionalstimulicanalsostimulateEVreleasefromhepatocytes.Inthis
regard,ithasbeenobservedthathypoxiadeterminesthatfat-ladenhepatocytesreleaseEVableto
signal KC evoking proinflammatory phenotypes in this cells, a phenomena that may explain may
underlietheaggravatingeffectofobstructivesleepapneasyndromeonNAFLD(68).Thus, itseems
clear that lipotoxic injury of hepatocytes determines EV release, promoting inflammation through
activation and recruitment of macrophages (14) with clear implications for the triggering of
inflammationinNAFLD/NASH(36). Inaddition,hepatocyte-derivedEVmaypromoteHSCactivation
(45,69)inexperimentalmodelsofNAFLD/NASH.Interestingly,bothmouseandhumanHSCsrelease
EVthattargethepatocytesandHSCsthemselves(47,48,70).Collectively,thesedataimplicateEVas
partofthecellulareventstriggeringhepaticfibrogenesis,akeyprocessinNAFLDprogression(71).In
additiontosignalingtomacrophagesandHSCs,EVmayactonendothelialcells (38,63)promoting
vascularinflammationwithpotentialimplicationsforNAFLD-relatedatherosclerosis.Inthisregard,it
has been shown that EV carryingmiR-1 as cargomediates proinflammatory effects in endothelial

21
cells in mice via downregulation of KLF4 and activation of the NF-κB pathway (38). Finally, other
organ-derived EV (i.e. visceral adipose tissue-derived exosomes) can contribute to NAFLD
pathogenesisandprogressionandinfluencefibrogenicpathwaysinbothhepatocytesandHSCs(72)
underscoringtheroleofotherinsulin-sensitiveorgansinNAFLD.
Studiescarriedout inmousemodelsofNASHhaveshownthat totalcirculatingEVandparticularly
hepatocyte-derived EV are elevated early in the disease process while other cell-derived EV (i.e.
macrophage-andneutrophil-derivedEV)appearinthecirculationlater,likelyreflectingtheongoing
inflammatoryprocess(62,73).ProteomicprofilingofcirculatingEVinexperimentalNAFLDhasbeen
demonstrated to allow differentiation between NAFLD vs. control animals (17). These findings
underscorethepotentialofEVasminimallyinvasivebiomarkersforNAFLD(74),whichareurgently
forclinicaltrialsandintheclinic.SincecirculatingEV(mainlyexosomes)arealsoincreasedinhuman
NAFLDandhavebeenfoundbysomeauthorstocorrelatewithdiseasehistologicalfeatures(75),EV
analysis in serum involving quantitative and qualitative determinations (including cell surface
markersassessmentandmeasurementofdifferentcargoes [i.e.proteins, lipidsandmicroRNAs]) is
nowafocusofintenseresearch.Severalstudieshavebeenpublishedinthisregardshowingthatthe
number of CD14+ and CD16+ EV is inversely associated with the severity of NAFLD-related liver
fibrosis,while also increasing the diagnostic capability of the enhanced liver fibrosis score (LFS) in
patientswithNAFLD(AUC:0.948and0.967forCD14+andCD16+EV,respectively,vs0.915forLFS
alone) (76). Other efforts include detection of circulating EV containing C16:0 ceramide- and S1P-
enriched lipid species that progressively increase in the plasma of obese patients with simple
steatosis and inNASHpatientswithearly fibrosis (62).Unfortunately,diagnostic accuracyof these
determinationsremainsincompletelyexploredinthefieldofNAFLD/NASHandrigorousvalidationof
this approach is needed (13, 74). Moreover, significant challenges remain regarding isolation,
reproducibilityanddefinitionofnormalcontrols(13,77).
Therapeutic efforts involving EV in the field of NAFLD/NASH are nascent. Attempts to halt
inflammation and fibrosis in rodent models of NAFLD/NASH using EV as therapeutic agents have
been published recently. EV obtained from amnion-derivedMSC (AMSC) to treat rats with either
NASHor liver fibrosis inducedby the hepatic toxicant CCL4. AMSC-EVwere given intravenously in
one or two doses and amelioration of inflammation and fibrogenesis was observed (78). More
recently,humanliverstemcells(HLSCs)-derivedEVhavebeenusedtotreatmicewithdiet-induced
steatohepatitis(79).TheauthorsfoundthatEV-HLSCtreatmentsignificantlydownregulatedhepatic
pro-fibroticandpro-inflammatorygeneexpressionandamelioratedthehistologicalabnormalitiesin
mice with NASH. Proteomic analysis of EV-HLSCs showed that their cargo included various anti-

22
inflammatoryproteinssuchasInterleukin-10,thatmaycontributetotheobservedbeneficialeffects.
TheseresultsunderscoretheconceptthatEVcanbeexploitedfortherapyinNAFLD/NASH.
6.-EVANDALD
Recentstudieshave focusedon the roleofEV inALD (11,14,80,81).Bothhepatocyteandmonocyte-
derivedEVhavebeenpostulatedtoregulatemacrophagedifferentiation,therebypromotinginflammation
inalcoholichepatitis(AH)(24,41,53,69,82).Severalmoleculeshavebeenproposedtoberesponsiblefor
EV-mediatedcell-to-cellsignaling, includingmiRNAs(inparticular,miR-122andmiR-155)(41,81,83-88).
TheCD40ligandwasproposedasanEVcargothatcouldpromotemacrophageactivationinvitroandin
vivoinexperimentalmodelsofAH(81).Also,inamousemodelofALDinvolvinggastricinfusionofethanol,
amicroRNAbarcode (let7f,miR-29a, andmiR-340) that can be detected in blood,which is specific for
alcohol-relatedliverinjury(16).
Fromaclinicalpointofview,thereiscurrentlynobiomarkerabletoassessearlystagesofALD.EVhave
beenshowntocorrelatewiththediagnosisandprognosisofalcoholichepatitis(89).Additionally,EVhave
beenproposedaspotentialbiomarkerstodifferentiatemildandsevereformsofALDandtheircargo,such
assphingolipids,todiscriminatebetweendifferentliverdiseaseetiologies(89).Earlyidentificationofthese
subjectsmightleadtotimelyinterventioninthediseaseprocess.Atpresent,thediagnosisofAHreliesona
historyofalcoholconsumptioninacompatibleclinicalscenario.Therearenobiomarkerswhichpermitthe
earlydiagnosisofAHin“at-risk”populationsnorscreeningteststhatcananticipatethoseatriskformore
severe manifestations of AH. This at-risk group is particularly important to identify since there is no
effectivetherapyforsevereAHandeffortstoavoid itcouldbe implemented.Severalclinicalprognostic
markershavebeenproposedbutthevariablesusedreflecttheseverityofliverdisease.Theseindices(i.e.
theMaddreydiscriminantfunction,modelforend-stageliverdisease(MELD)andtheLilleScore)arebased
on the non-specific biochemical assessment of liver and renal function and rely heavily on serum total
bilirubin,prothrombintime,andcreatinine(90).Therelianceonbilirubinlimitstheirdiagnosticutility,as
specificity is confounded by hyperbilirubinemia in co-existent cholestatic liver diseases such as drug-
induced liver disease or cholestatic hepatitis such as primary sclerosing cholangitis and primary biliary
cholangitis. None of these prognostic scores utilizes a pathophysiologically validated biomarker that
reflectstheunderlyingmolecularandsignalingmechanismsofthedisease.Furthermore,theinflammatory
responseispredictiveofmortalitybutisnottakenintoaccountinmathematicalmodels.Theycanidentify
subjectsathighestmortalityriskwithanareaunderthereceiveroperatingcurve(AUROC)ofunder0.8;
idealsurvivalmodelsshouldhaveanAUROC>0.8(91).Thereasonthesescoresareimperfectmaybethat
they are not based on pathophysiologic mechanisms that mediate liver injury. Liver biopsy, the gold
standard for diagnosis ofAH, remainsunderutilizeddue to the concurrent coagulopathy,which greatly

23
increasestheriskofbiopsy-relatedcomplications(92).Liverbiopsy-basedhepatichistologyscoreperforms
asanAUROCof0.73inpredicting90-daymortality(91).InthisregardtheuseofEVasbiomarkerinAHis
promising.Thus,ithasbeenrecentlyreportedthatthetotalnumberofEVwassignificantlyincreasedin
patientswithAHandthattwomicroRNAs(i.e.miRNA-192andmiRNA-30a)weresignificantlyincreasedin
plasmaofsubjectswithAH(82).Mostrecently,EVhavebeenusedasasurrogatemarkerofimprovement
inclinicaltrialsofpatientswithAH(93).FurtherstudieswillberequiredtovalidateEVasabiomarkerfor
AHdiagnosisand/orprognosis.
7.-CONCLUDINGREMARKSANDFUTUREPERSPECTIVES
ThefieldofEVinfattyliverdiseasesisrapidlyevolvingastheimportantfunctionsoftheseparticlesincell-
to-cellcommunicationandinthepathogenesisofbothALDandNAFLD,themostprevalentliverdiseases
worldwide, are unveiled. The release of large quantities of EV by stressed/damaged hepatocytes
contributestoinflammation,fibrogenesisandangiogenesis,fuelingliverdiseaseprogressionandprovides
opportunities for intervention. The identification of specific molecular signatures of released EV is
promising in the search fordisease-specificbiomarkersalthoughmoredata isneeded tovalidate these
markersinlargercohortsandinarigorousmanner.EVmayhavepotentialfortherapeuticusebutthisfield
isstillnascent.MoreresearchisneededforasuccessfultransitionofcurrentEVknowledgefromthebench
totheclinic.

24
Figure1.Extracellularvesicles(EV)canbereleasedbyhepatocytesuponlipotoxicoralcohol-inducedinjury.EVcargoes
includeamyriadofmoleculesthatcanactontargetcellsevokinginflammatoryandfibrogeniceventsandpromoting
neoplastictransformationthuscontributingtotheprogressionofbothalcoholicliverdisease(ALD)andnonalcoholic
liverdisease (NAFLD) to their inflammatoryandmoreaggressive formsnonalcoholic steatohepatitis (NASH) and
alcoholicsteatohepatitis(ASH).
8.REFERENCES

25
1. Stahl PD, Raposo G. Extracellular Vesicles: Exosomes and Microvesicles, Integrators of Homeostasis. Physiology (Bethesda).
2019;34(3):169-77.
2.CocucciE,RacchettiG,MeldolesiJ.Sheddingmicrovesicles:artefactsnomore.TrendsCellBiol.2009;19(2):43-51.
3.AbelsER,BreakefieldXO.IntroductiontoExtracellularVesicles:Biogenesis,RNACargoSelection,Content,Release,andUptake.CellMol
Neurobiol.2016;36(3):301-12.
4.vanNielG,D'AngeloG,RaposoG.Sheddinglightonthecellbiologyofextracellularvesicles.NatRevMolCellBiol.2018;19(4):213-28.
5. Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to
characterizeheterogeneouspopulationsofextracellularvesiclesubtypes.ProcNatlAcadSciUSA.2016;113(8):E968-77.
6.ZivkoC,FuhrmannG,LucianiP.Liver-derivedextracellularvesicles:Acellbycelloverviewto isolationandcharacterizationpractices.
BiochimBiophysActaGenSubj.2020:129559.
7. Kim DK, Lee J, Kim SR, Choi DS, Yoon YJ, Kim JH, et al. EVpedia: a community web portal for extracellular vesicles research.
Bioinformatics.2015;31(6):933-9.
8.KimDK,KangB,KimOY,ChoiDS,LeeJ,KimSR,etal.EVpedia:anintegrateddatabaseofhigh-throughputdataforsystemicanalysesof
extracellularvesicles.JExtracellVesicles.2013;2.
9. Shao H, Im H, Castro CM, Breakefield X, Weissleder R, Lee H. New Technologies for Analysis of Extracellular Vesicles. Chem Rev.
2018;118(4):1917-50.
10.MaasSLN,BreakefieldXO,WeaverAM.ExtracellularVesicles:UniqueIntercellularDeliveryVehicles.TrendsCellBiol.2017;27(3):172-
88.
11. Urban SK, Mocan T, Sanger H, Lukacs-Kornek V, Kornek M. Extracellular Vesicles in Liver Diseases: Diagnostic, Prognostic, and
TherapeuticApplication.SeminLiverDis.2019;39(1):70-7.
12.LemoinneS,ThabutD,HoussetC,MoreauR,VallaD,BoulangerCM,etal.Theemergingrolesofmicrovesiclesinliverdiseases.NatRev
GastroenterolHepatol.2014;11(6):350-61.
13. Szabo G, Momen-Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat Rev
GastroenterolHepatol.2017;14(8):455-66.
14.EguchiA,FeldsteinAE.Extracellularvesiclesinnon-alcoholicandalcoholicfattyliverdiseases.LiverRes.2018;2(1):30-4.
15.MalhiH.Emergingroleofextracellularvesiclesinliverdiseases.AmJPhysiolGastrointestLiverPhysiol.2019;317(5):G739-G49.
16.EguchiA, LazaroRG,Wang J,Kim J,PoveroD,WillliamsB,etal.Extracellularvesicles releasedbyhepatocytes fromgastric infusion
modelofalcoholicliverdiseasecontainaMicroRNAbarcodethatcanbedetectedinblood.Hepatology.2017;65(2):475-90.
17.PoveroD,EguchiA,LiH,JohnsonCD,PapouchadoBG,WreeA,etal.Circulatingextracellularvesicleswithspecificproteomeandliver
microRNAsarepotentialbiomarkersforliverinjuryinexperimentalfattyliverdisease.PLoSOne.2014;9(12):e113651.
18.XieF,FengS,YangH,MaoY.Extracellularvesiclesinhepatocellularcancerandcholangiocarcinoma.AnnTranslMed.2019;7(5):86.
19.SasakiR,KandaT,YokosukaO,KatoN,MatsuokaS,MoriyamaM.ExosomesandHepatocellularCarcinoma:FromBenchtoBedside.Int
JMolSci.2019;20(6).
20.SungS,KimJ,JungY.Liver-DerivedExosomesandTheirImplicationsinLiverPathobiology.IntJMolSci.2018;19(12).
21. LatifkarA,HurYH, Sanchez JC,CerioneRA,AntonyakMA.New insights intoextracellular vesiclebiogenesis and function. J Cell Sci.
2019;132(13).

26
22.DoyleLM,WangMZ.OverviewofExtracellularVesicles,TheirOrigin,Composition,Purpose,andMethodsforExosomeIsolationand
Analysis.Cells.2019;8(7).
23.MoranL,CuberoFJ.Extracellularvesiclesinliverdiseaseandbeyond.WorldJGastroenterol.2018;24(40):4519-26.
24.Devhare PB, Ray RB. Extracellular vesicles:Novelmediator for cell to cell communications in liver pathogenesis.Mol AspectsMed.
2018;60:115-22.
25.HirsovaP,IbrahimSH,VermaVK,MortonLA,ShahVH,LaRussoNF,etal.Extracellularvesiclesinliverpathobiology:Smallparticleswith
bigimpact.Hepatology.2016;64(6):2219-33.
26.HarasztiRA,DidiotMC,SappE,LeszykJ,ShafferSA,RockwellHE,etal.High-resolutionproteomicandlipidomicanalysisofexosomes
andmicrovesiclesfromdifferentcellsources.JExtracellVesicles.2016;5:32570.
27.BanalesJM,FeldsteinAE,SangerH,Lukacs-KornekV,SzaboG,KornekM.ExtracellularVesiclesinLiverDiseases:MeetingReportfrom
theInternationalLiverCongress2018.HepatolCommun.2019;3(2):305-15.
28.AizawaS,BrarG,TsukamotoH.CellDeathandLiverDisease.GutLiver.2019.
29.TheryC,WitwerKW,AikawaE,AlcarazMJ,Anderson JD,AndriantsitohainaR,etal.Minimal information forstudiesofextracellular
vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014
guidelines.JExtracellVesicles.2018;7(1):1535750.
30. Royo F, Gil-CartonD,Gonzalez E,Mleczko J, Palomo L, Perez-CormenzanaM, et al. Differences in themetabolite composition and
mechanicalpropertiesofextracellularvesiclessecretedbyhepaticcellularmodels.JExtracellVesicles.2019;8(1):1575678.
31. Nojima H, Freeman CM, Schuster RM, Japtok L, Kleuser B, Edwards MJ, et al. Hepatocyte exosomes mediate liver repair and
regenerationviasphingosine-1-phosphate.JHepatol.2016;64(1):60-8.
32. Chen L, Chen R, Kemper S, Brigstock DR. Pathways of production and delivery of hepatocyte exosomes. J Cell Commun Signal.
2018;12(1):343-57.
33.ZhaoY,ZhaoMF,JiangS,WuJ,LiuJ,YuanXW,etal.Livergovernsadiposeremodellingviaextracellularvesicles inresponsetolipid
overload.NatCommun.2020;11(1):719.
34.ArreseM,CabreraD,KalergisAM,FeldsteinAE.InnateImmunityandInflammationinNAFLD/NASH.DigDisSci.2016;61(5):1294-303.
35.SatoK,KennedyL,LiangpunsakulS,KusumanchiP,YangZ,MengF,etal.IntercellularCommunicationbetweenHepaticCellsinLiver
Diseases.IntJMolSci.2019;20(9).
36. Schuster S, CabreraD, ArreseM, Feldstein AE. Triggering and resolution of inflammation inNASH.Nat RevGastroenterol Hepatol.
2018;15(6):349-64.
37.HirsovaP,IbrahimSH,KrishnanA,VermaVK,BronkSF,WerneburgNW,etal.Lipid-InducedSignalingCausesReleaseofInflammatory
ExtracellularVesiclesFromHepatocytes.Gastroenterology.2016;150(4):956-67.
38. Jiang F, Chen Q, Wang W, Ling Y, Yan Y, Xia P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and
atherogenesisviamicroRNA-1.JHepatol.2020;72(1):156-66.
39. Antwi-Baffour S, Adjei J, Aryeh C, Kyeremeh R, Kyei F, SeiduMA. Understanding the biosynthesis of platelets-derived extracellular
vesicles.ImmunInflammDis.2015;3(3):133-40.
40.BalaphasA,MeyerJ,SadoulK,FontanaP,MorelP,Gonelle-GispertC,etal.PlateletsandPlatelet-DerivedExtracellularVesiclesinLiver
PhysiologyandDisease.HepatolCommun.2019;3(7):855-66.

27
41.SahaB,Momen-HeraviF,KodysK,SzaboG.MicroRNACargoofExtracellularVesiclesfromAlcohol-exposedMonocytesSignalsNaive
MonocytestoDifferentiateintoM2Macrophages.JBiolChem.2016;291(1):149-59.
42.LeeYA,WallaceMC,FriedmanSL.Pathobiologyofliverfibrosis:atranslationalsuccessstory.Gut.2015;64(5):830-41.
43.FriedmanSL.Hepaticstellatecells:protean,multifunctional,andenigmaticcellsoftheliver.PhysiolRev.2008;88(1):125-72.
44.HigashiT,FriedmanSL,HoshidaY.Hepaticstellatecellsaskeytargetinliverfibrosis.AdvDrugDelivRev.2017;121:27-42.
45.PoveroD,PaneraN,EguchiA,JohnsonCD,PapouchadoBG,deAraujoHorcelL,etal.Lipid-inducedhepatocyte-derivedextracellular
vesiclesregulatehepaticstellatecellviamicroRNAstargetingPPAR-gamma.CellMolGastroenterolHepatol.2015;1(6):646-63e4.
46.WangR,DingQ,YaqoobU,deAssuncaoTM,VermaVK,HirsovaP,etal.ExosomeAdherenceandInternalizationbyHepaticStellate
CellsTriggersSphingosine1-Phosphate-dependentMigration.JBiolChem.2015;290(52):30684-96.
47.ChenL,CharrierA,ZhouY,ChenR,YuB,AgarwalK,etal.EpigeneticregulationofconnectivetissuegrowthfactorbyMicroRNA-214
deliveryinexosomesfrommouseorhumanhepaticstellatecells.Hepatology.2014;59(3):1118-29.
48.CharrierA,ChenR,ChenL,KemperS,HattoriT,TakigawaM,etal.Exosomesmediateintercellulartransferofpro-fibrogenicconnective
tissuegrowthfactor(CCN2)betweenhepaticstellatecells,theprincipalfibroticcellsintheliver.Surgery.2014;156(3):548-55.
49.OwensAP,3rd,MackmanN.Microparticlesinhemostasisandthrombosis.CircRes.2011;108(10):1284-97.
50.VallaDC.Thrombosisandanticoagulationinliverdisease.Hepatology.2008;47(4):1384-93.
51.ChenL,BrennerDA,KisselevaT.CombattingFibrosis:Exosome-BasedTherapiesintheRegressionofLiverFibrosis.HepatolCommun.
2019;3(2):180-92.
52.ClaytonA,BuschmannD,ByrdJB,CarterDRF,ChengL,ComptonC,etal.SummaryoftheISEVworkshoponextracellularvesiclesas
diseasebiomarkers,heldinBirmingham,UK,duringDecember2017.JExtracellVesicles.2018;7(1):1473707.
53.ChoYE,SongBJ,AkbarM,BaekMC.Extracellularvesiclesaspotentialbiomarkersforalcohol-anddrug-inducedliverinjuryandtheir
therapeuticapplications.PharmacolTher.2018;187:180-94.
54. Arbelaiz A, Azkargorta M, Krawczyk M, Santos-Laso A, Lapitz A, Perugorria MJ, et al. Serum extracellular vesicles contain protein
biomarkersforprimarysclerosingcholangitisandcholangiocarcinoma.Hepatology.2017;66(4):1125-43.
55.MurphyDE,deJongOG,BrouwerM,WoodMJ,LavieuG,SchiffelersRM,etal.Extracellularvesicle-basedtherapeutics:naturalversus
engineeredtargetingandtrafficking.ExpMolMed.2019;51(3):32.
56.VillaF,QuartoR,TassoR.ExtracellularVesiclesasNatural,SafeandEfficientDrugDeliverySystems.Pharmaceutics.2019;11(11).
57.BalaphasA,MeyerJ,SadoulR,MorelP,Gonelle-GispertC,BuhlerLH.Extracellularvesicles:Futurediagnosticandtherapeutictoolsfor
liverdiseaseandregeneration.LiverInt.2019;39(10):1801-17.
58.BorrelliDA,YanksonK,ShuklaN,VilanilamG,TicerT,WolframJ.Extracellularvesicletherapeuticsforliverdisease.JControlRelease.
2018;273:86-98.
59.GaoJ,DongX,WangZ.Generation,purificationandengineeringofextracellularvesiclesandtheirbiomedicalapplications.Methods.
2019.
60.PatelDB, SantoroM,Born LJ, Fisher JP, Jay SM.Towards rationallydesignedbiomanufacturingof therapeutic extracellular vesicles:
impactofthebioproductionmicroenvironment.BiotechnolAdv.2018;36(8):2051-9.
61. Catalano M, O’Driscoll L. Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. Journal of Extracellular
Vesicles.2019(inpress).

28
62.KakazuE,MauerAS,YinM,MalhiH.Hepatocytesreleaseceramide-enrichedpro-inflammatoryextracellularvesiclesinanIRE1alpha-
dependentmanner.JLipidRes.2016;57(2):233-45.
63.PoveroD,EguchiA,NiesmanIR,AndronikouN,deMolleratduJeuX,MulyaA,etal.Lipid-inducedtoxicitystimulateshepatocytesto
releaseangiogenicmicroparticlesthatrequireVanin-1foruptakebyendothelialcells.SciSignal.2013;6(296):ra88.
64.IbrahimSH,HirsovaP,TomitaK,BronkSF,WerneburgNW,HarrisonSA,etal.Mixedlineagekinase3mediatesreleaseofC-X-Cmotif
ligand10-bearingchemotacticextracellularvesiclesfromlipotoxichepatocytes.Hepatology.2016;63(3):731-44.
65. Cannito S,Morello E, Bocca C, Foglia B, Benetti E, Novo E, et al.Microvesicles released from fat-laden cells promote activation of
hepatocellular NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS One.
2017;12(3):e0172575.
66.LiuXL,PanQ,CaoHX,XinFZ,ZhaoZH,YangRX,etal.LipotoxicHepatocyte-DerivedExosomalmiR-192-5pActivatesMacrophagesvia
Rictor/Akt/FoxO1SignalinginNAFLD.Hepatology.2019.
67.GuoQ,FurutaK, LucienF,GutierrezSanchez LH,HirsovaP,KrishnanA,etal. Integrinbeta1-enrichedextracellular vesiclesmediate
monocyteadhesionandpromoteliverinflammationinmurineNASH.JHepatol.2019;71(6):1193-205.
68.HernandezA,GengY, SepulvedaR, SolisN, Torres J,Arab JP, et al. ChemicalHypoxia inducespro-inflammatory signals in fat-laden
hepatocytes and contributes to cellular crosstalkwith Kupffer cells through extracellular vesicles. BBA -Molecular Basis of Disease (in
press).2020.
69.LeeYS,KimSY,KoE,LeeJH,YiHS,YooYJ,etal.Exosomesderivedfrompalmiticacid-treatedhepatocytesinducefibroticactivationof
hepaticstellatecells.SciRep.2017;7(1):3710.
70.ChenL,ChenR,KemperS,CharrierA,BrigstockDR.Suppressionof fibrogenicsignaling inhepaticstellatecellsbyTwist1-dependent
microRNA-214expression:RoleofexosomesinhorizontaltransferofTwist1.AmJPhysiolGastrointestLiverPhysiol.2015;309(6):G491-9.
71.SchuppanD,SurabattulaR,WangXY.DeterminantsoffibrosisprogressionandregressioninNASH.JHepatol.2018;68(2):238-50.
72.KoeckES, IordanskaiaT,SevillaS,FerranteSC,HubalMJ,FreishtatRJ,etal.Adipocyteexosomes induce transforminggrowth factor
betapathwaydysregulationinhepatocytes:anovelparadigmforobesity-relatedliverdisease.JSurgRes.2014;192(2):268-75.
73. Li J, LiuH,MauerAS, Lucien F, RaiterA, BandlaH, et al. Characterization of Cellular Sources andCirculating Levels of Extracellular
VesiclesinaDietaryMurineModelofNonalcoholicSteatohepatitis.HepatolCommun.2019;3(9):1235-49.
74.BanLA,ShackelNA,McLennanSV.ExtracellularVesicles:ANewFrontierinBiomarkerDiscoveryforNon-AlcoholicFattyLiverDisease.
IntJMolSci.2016;17(3):376.
75.KornekM,LynchM,MehtaSH,LaiM,ExleyM,AfdhalNH,etal.Circulatingmicroparticlesasdisease-specificbiomarkersofseverityof
inflammationinpatientswithhepatitisCornonalcoholicsteatohepatitis.Gastroenterology.2012;143(2):448-58.
76.WelshJA,ScorlettiE,CloughGF,EnglystNA,ByrneCD.Leukocyteextracellularvesicleconcentrationisinverselyassociatedwithliver
fibrosisseverityinNAFLD.JLeukocBiol.2018;104(3):631-9.
77.Momen-HeraviF,SzaboG.ExtracellularVesiclesandExosomes:BiologyandPathobiology.In:ARIASIM,ALTERHJ,BOYERJL,COHENDE,
SHAFRITZDA,THORGEIRSSONSS,etal.,editors.TheLiver:BiologyandPathobiology.Hoboken,NJ,USA:Wiley-Blackwell;2020.p.1022-8.
78.OharaM,OhnishiS,HosonoH,YamamotoK,YuyamaK,NakamuraH,etal.ExtracellularVesiclesfromAmnion-DerivedMesenchymal
StemCellsAmeliorateHepaticInflammationandFibrosisinRats.StemCellsInt.2018;2018:3212643.
79.BrunoS,PasquinoC,HerreraSanchezMB,TapparoM,FiglioliniF,GrangeC,etal.HLSC-DerivedExtracellularVesiclesAttenuateLiver
FibrosisandInflammationinaMurineModelofNon-alcoholicSteatohepatitis.MolTher.2019.

29
80. Rahman MA, Patters BJ, Kodidela S, Kumar S. Extracellular Vesicles: Intercellular Mediators in Alcohol-Induced Pathologies. J
NeuroimmunePharmacol.2019.
81. VermaVK, Li H,WangR,Hirsova P,MushrefM, Liu Y, et al. Alcohol stimulatesmacrophage activation through caspase-dependent
hepatocytederivedreleaseofCD40Lcontainingextracellularvesicles.Journalofhepatology.2016;64(3):651-60.
82. Momen-Heravi F, Saha B, Kodys K, Catalano D, Satishchandran A, Szabo G. Increased number of circulating exosomes and their
microRNAcargosarepotentialnovelbiomarkersinalcoholichepatitis.JTranslMed.2015;13:261.
83.BalaS,Petrasek J,MundkurS,CatalanoD, Levin I,Ward J, etal.CirculatingmicroRNAs inexosomes indicatehepatocyte injuryand
inflammationinalcoholic,drug-induced,andinflammatoryliverdiseases.Hepatology.2012;56(5):1946-57.
84.Momen-Heravi F, Bala S, Kodys K, Szabo G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific
miRNA-122andsensitizemonocytestoLPS.SciRep.2015;5:9991.
85.CsakT,BalaS,LippaiD,SatishchandranA,CatalanoD,KodysK,etal.microRNA-122regulateshypoxia-induciblefactor-1andvimentin
inhepatocytesandcorrelateswithfibrosisindiet-inducedsteatohepatitis.LiverInt.2015;35(2):532-41.
86.LiHD,DuXS,HuangHM,ChenX,YangY,HuangC,etal.NoncodingRNAsinalcoholicliverdisease.JCellPhysiol.2019.
87. Lamichhane TN, Leung CA, Douti LY, Jay SM. Ethanol Induces Enhanced Vascularization Bioactivity of Endothelial Cell-Derived
ExtracellularVesiclesviaRegulationofMicroRNAsandLongNon-CodingRNAs.SciRep.2017;7(1):13794.
88.Brandon-WarnerE,FeilenNA,CulbersonCR,FieldCO,deLemosAS,RussoMW,etal.ProcessingofmiR17-92ClusterinHepaticStellate
CellsPromotesHepaticFibrogenesisDuringAlcohol-InducedInjury.AlcoholClinExpRes.2016;40(7):1430-42.
89.ArabJP,VermaV,Martin-MateosR,SimonettoD,KamathPS,GoresGJ,etal.ExtracellularVesicleC16CeramideandS1PContent in
AlcoholicHepatitisCorrelateswithDiseaseSeverityandResolution.Gastroenterology.2018;154(6):S-1120.
90.SingalAK,ShahVH.TherapeuticStrategiesfortheTreatmentofAlcoholicHepatitis.Seminarsinliverdisease.2016;36(1):56-68.
91. Altamirano J,Miquel R, Katoonizadeh A, Abraldes JG, Duarte-Rojo A, Louvet A, et al. A histologic scoring system for prognosis of
patientswithalcoholichepatitis.Gastroenterology.2014;146(5):1231-9e1-6.
92.ArabJP,BarreraF,ArreseM.TheEvolvingRoleofLiverBiopsyinNon-alcoholicFattyLiverDisease.AnnHepatol.2018;17(6):899-902.
93.ArabJP,SehrawatTS,SimonettoDA,VermaVK,FengD,TangT,etal.AnOpenLabel,DoseEscalationStudyToAssessTheSafetyAnd
EfficacyOfIL-22AgonistF-652InPatientsWithAlcoholicHepatitis.Hepatology.2019.
Chapter3

30
CHEMICAL HYPOXIA INDUCES PRO-INFLAMMATORY SIGNALS IN FAT-LADEN HEPATOCYTES AND
CONTRIBUTESTOCELLULARCROSSTALKWITHKUPFFERCELLSTHROUGHEXTRACELLULARVESICLES
ABSTRACT
Background: Obstructive sleep apnea syndrome (OSAS), which is characterized by occurrence of
intermittent hypoxia (IH), is an aggravating factor of non-alcoholic fatty liver disease (NAFLD).We
investigatedtheeffectsofhypoxia inboth invitroandinvivomodelsofNAFLD.Methods:Primary
rathepatocytestreatedwithfreefattyacids(FFA)weresubjectedtochemicallyinducedhypoxia(CH)
usingthehypoxia-induciblefactor-1alpha(HIF-1α)stabilizercobaltchloride(CoCl2).Triglyceride(TG)
content, mitochondrial superoxide production, cell death rates, pro-inflammatory cytokines and
inflammasomecomponentsgeneexpressionandproteinlevelsofcleavedcaspase-1wereassessed.
Also,Kupffer cells (KC)were treatedwithconditionedmedium (CM)andextracellularvesicles (EV)
fromhypoxicfat-ladenhepatocytesandthecholinedeficientL-aminoaciddefined(CDAA)-fedmice
modelusedtoassesstheeffectsofIHonexperimentalNAFLDinvivo.Results:CHinduceda2-fold
increase in HIF-1α protein levels. Hepatocytes exposed to FFA and CoCl2 exhibited increased TG
content and higher cell death rates aswell as increasedmitochondrial superoxide production and
mRNAlevelsofpro-inflammatorycytokinesandofinflammasome-componentsinterleukin-1β,NLRP3
andASC.Proteinlevelsofcleavedcaspase-1increasedinCH-exposedhepatocytes.CMandEVfrom
hypoxic fat-laden hepatic cells evoked a pro-inflammatory phenotype in KC. Livers fromCDAA-fed
miceexposedtoIHexhibitedincreasedmRNAlevelsofpro-inflammatoryandinflammasomegenes
aswellasincreasedlevelsofcleavedcaspase-1.Conclusion:Hypoxiapromotesinflammatorysignals
including inflammasome/caspase-1 activation in fat-laden hepatocytes and contributes to cellular
crosstalkwithKupffercellsbyreleaseofEV.Thesemechanismsmayunderlietheaggravatingeffect
ofOSASonNAFLD.
1.-INTRODUCTION
Inrecentyears,obstructivesleepapneasyndrome(OSAS),acommonsleepdisordercharacterizedby
recurrentclosureoftheupperairwaysduringsleep,hasbeensuggestedtomodulatetheseverityof
differentmetabolicdisorders(1,2).ThehallmarkofOSASistheoccurrenceofintermittenthypoxia
(IH) leading to tissuehypoxiaandpromotingoxidative stress, inflammationandamyriadofmulti-
organ pathophysiological effects (3). Among conditions in which OSAS acts as both a potential
inducerandasanaggravatingfactorisnon-alcoholicfattyliverdisease(NAFLD),thecommonestliver
diseaseworldwide (4-6).NAFLDdescribesa clinicopathologicalentitydefinedby thepresenceofa
spectrumofhepatichistologicalchangesranginginseverityfromisolatedsteatosis(alsotermednon-
alcoholic fatty liver [NAFL]) tosteatohepatitis (namednon-alcoholicsteatohepatitis,NASH)through

31
to advanced fibrosis and cirrhosis (7, 8). Development of steatosis is closely linked to both
overweightandobesityaswellastoinsulinresistanceandtransitionfromisolatedsteatosistomore
advancedstagesofthediseaseoccursonlyinaminorityofNAFLDpatients.Multiplefactorsinfluence
NAFLD development and progression including individual’s genetic background, environmental
factorsandthepresenceofamyriadofcomorbiditiesincludingOSAS(6,9).
The main mechanisms of hepatocellular damage in NAFLD include those related to lipotoxicity,
mitochondrialdysfunction, formationof reactiveoxygenspecies,endoplasmic reticulumstressand
disturbed autophagy ultimately leading to hepatocyte injury and death that triggers hepatic
inflammation, hepatic stellate cell activation, and progressive fibrogenesis, thus driving disease
progression(8,10-12). Inaddition,recentstudieshaveshownthat inflammasomeactivationisalso
oneofthekeyeventsinNAFLDprogression(13-15).Specifically,theNOD-likereceptorPyrinDomain
Containing3 (NLRP3) inflammasomehasbeenrecognizedtobe involved intheprogressionof liver
damage in experimental in vitro (hepatocytes and immune liver cells) and in vivomodels ofNASH
(16-18)andalso inhuman studies (19).NLRP3 inflammasomemediates thematurationof inactive
pro-caspase-1 into active cleaved caspase-1, which cleaves gasdermin D (GSDMD) that in turn
determine the activation of the pro-inflammatory cytokines interleukin [IL]-1β and IL-18, which
amplifythepathologicalphenomenainNAFLDbypromotinginflammationandpyroptoticcelldeath
(20). Of note, the progression of damage in NAFLD also involves the participation of other liver-
residentcellssuchasmacrophagesorKupffercells(KC)aswellastherecruitmentof inflammatory
cellsfromtheperiphery(21-23).
WithregardtothepathophysiologicalconnectionsbetweenOSASandNAFLD,existingdataindicate
that OSASmay relate to both NAFLD occurrence as well as disease progression. Of note, several
studies have shown that IH is able to inducemetabolic alterations such as insulin resistance and
increased liver triglyceride (TG) accumulation as well as increased oxidative stress and increased
inflammation,whichare related tosteatosisdevelopmentandhepatocellulardamage, respectively
(24-26).HepaticTGaccumulationinthesettingofOSASmayresultfromhypoxia-relatedchangesin
lipid metabolic pathways such as a decrease in fatty acid β-oxidation and increased de novo
lipogenesis(5,27).Also,IHhasbeenshowntopromotepro-inflammatoryeffectsinanimalmodelsof
NAFLDmodulating inflammatory cytokineproduction (i.e. tumornecrosis factor-alpha [TNF-α] and
IL-6) (28,29).Althoughsomestudiessuggestarelevantroleofhypoxia, likelythrough inductionof
the hypoxia inducible factor 1 alpha (HIF-1α), in promoting hepatocellular cell death and the
generation of pro-inflammatory signals in several experimental settings (30), assessment of these
phenomenainthecontextofNAFLDhasbeenlessexplored(5,31).

32
In the present study, we aimed to assess the effects of hypoxia on cellular lipid accumulation,
hepatocellular death and pro-inflammatory signals including those related to the NLRP3
inflammasomeleadingtocaspase-1activationininvitroandinvivomodelsofNAFLD.Moreover,we
explored whether hypoxia modulates cellular crosstalk between hepatocytes and resident
macrophages (i.e. KCs) in the setting of NAFLD and if this crosstalk involves the release of
extracellularvesicles(EV)fromhepatocytes.Ofnote,EVhavebeenrecentlyrecognizedasplayingan
importantrole inamplifyingthe inflammatoryresponse inNASH(32,33)butfewdataexistonthe
potentialroleofhypoxiainmodulatingtheirrelease.Wefoundthathypoxiainduceshepatocellular
damageinfat-ladenhepatocytesthatinvolvesNLRP3inflammasome-associatedcaspase-1activation
and increasedmitochondrialsuperoxideproduction leadingto increasecelldeathratesbymultiple
mechanisms including apoptosis and pyroptosis. Also, hypoxia contributes to evoking a pro-
inflammatory response in Kupffer cells by a mechanism that involves EV release from fat-laden
hepatocytes.Theseobservationspartiallyclarifythemechanismsunderlyingtheaggravatingeffectof
OSASonNAFLD.
2.-MATERIALSANDMETHODS
2.1Animals
Specified pathogen-freemaleWistar rats (220–250 g; Charles River Laboratories Inc.,Wilmington,
MA,USA)andmaleC57bl6mice[purchasedfromJacksonLaboratories(BarHarbor,ME,USA)]were
used in the present study. Animals were housed under standard laboratory conditions with free
access to standard laboratory chowdiet andwater.All experimentswere carriedoutaccording to
the Dutch and Chilean laws on the welfare of laboratory animals and guidelines of the local
institutional animal care and use committees of the Pontificia Universidad Católica de Chile and
ethicscommitteeofUniversityofGroningenforcareanduseoflaboratoryanimals.Alleffortswere
madetominimizeanimalssufferingandtoreducethenumberofanimalsused.
2.2 Cell isolation techniques, culture conditions, cell surfacemarkers detection and use of liver-
derivedcelllines
ToconducttheexperimentsdescribedbelowweusedbothprimaryrathepatocytesandKC.Isolation
techniquesusedaredescribedelsewhere(34,35)andintheSupplementarymaterialfile.Detailsof
cultureconditionsandKCcellsurfacemarkersdetectionarealsoprovidedinthatsection.Thehuman
hepatocellular carcinoma cell line HepG2 (American Type Culture Collection [ATCC] HB-8065,
Manassas,VA,USA)wasused tocarryoutexperiments involvingEV isolationafter treatmentwith
FFA.Cultureconditionsaredescribedinthesupplementarymaterialfile.

33
2.3Treatmentwithfreefattyacidsandchemicalhypoxiainduction
Inordertoassess theeffectsofhypoxia in fat-laden livercells,hepatocyteswere incubatedwitha
mixtureoffreefattyacids(FFA)consistingofoleicacid(500μmol/L)andpalmiticacid(250μmol/L)
inanaqueoussolutionofBSAasdescribed(36).IncubationswerecarriedoutwithorwithoutCobalt
(II) chloride (CoCl2; Sigma Aldrich, Saint Louis, MO, USA) (200 μmol/L) for 24 hours to induce
chemicalhypoxia (37).CoCl2 isawell-knownhypoxia-mimeticagent, thatmimicshypoxia/ischemic
conditionsby stabilizationofhypoxia-inducible factorHIF-1α (38). Control cellswere treatedwith
BSA alone. To obtain the corresponding conditioned medium (CM), after treatment, hepatocyte
culturemediumwas replaced by FBS-freemedium for an additional 24 hours. CM from different
treatmentswereaddedtoKCfor24hoursinordertoexplorethepossibilitythathypoxiamodulates
hepatocytes-KCcellularcrosstalk.
2.4OilredOcellstainingandTGmeasurement
Primary rat hepatocytes cultured on glass cover slideswere treatedwith FFA and CoCl2 for 24 h.
Hepatocyteswerethenwashedwithphosphate-bufferedsaline(PBS)twice,andthenfixedwith4%
formalin for10minutes.Cellswerewashedwith60% isopropanol twiceandstainedwithoil-red-O
solution for 10minutes at room temperature. Then, cellswerewashedwith 60% isopropanol and
washedindistilledwaterfor5minutes.Thencellswereincubatedwithhematoxylin/eosinsolution
for 1minute andwashedwithdistilledwater for 5minutes. Slicesweremountedusing 1 dropof
glycerin-gelatin solution. Stained lipid droplets in cells were examined using a slide scanner,
NanozoomerTM (Hamamatsu Photonics K.K., Shizuoka, Japan). Intracellular TG content was
determined using TG Quantification Assay kit (ab65336, Abcam, Cambridge, UK) according to the
manufacturer’sinstructionsandnormalizedtoproteinconcentration.
2.5Apoptosismeasurement
Caspase-3 fluorometric assay was used to determine apoptosis induced by FFA and/or CoCl2 in
freshlyisolatedmousehepatocytes.Aftertreatment,hepatocyteswerescrapedandcelllysateswere
obtained by three cycles of freezing (−196°C) in liquid nitrogen and thawing (37°C) followed by
centrifugation for 5 minutes at 13,000g. Caspase-3 enzyme activity was assayed as described
previously(39).Arbitraryunitsoffluorescence(AUF)werequantifiedinaspectrofluorometeratan
excitationwavelengthof380nmandanemissionwavelengthof430nm.
2.6Assessmentofcelldeathassociatedtodisruptedcellularmembraneintegrity
SYTOXTMGreennucleicacidstain(Invitrogen,S7020,Carlsbad,CA,USA)wasusedtodeterminecell
deathinducedbyFFAand/orCoCl2inhepatocytes(40).Cellswereculturedin12-wellplates.After

34
treatment, diluted SYTOXGreen solution (1:40.000/HBSS)was added to the plates for at least 15
minutesat37°C,5%CO2.SYTOXTMGreenentersthecelluponlossofmembraneintegrityandbinds
toDNAactingasacounterstainthatcanbeanalyzedwhenexcitedat488nm.ALeicafluorescence
microscopewasusedat thatwavelengthof fordetectionof necrotic cells,whichwerequantified
using Image J software. Lactate dehydrogenase (LDH) release was assessed as described in the
Supplementary materials file. Additionally, we assessed the caspase-cleaved gasdermin-N domain
(GSDMD-N) to confirm pyroptotic cell death by Western blot to evaluate the occurrence of
pyroptoticcelldeath(19).
2.7Mitochondrialsuperoxidedetection
At the end of 24h-long incubations, hepatocyteswerewashed oncewithwarmHBSS followed by
incubation with 200 nmol/LMitoSOX™ RedMolecular Probes (InvitrogenTM, Carlsbad, CA,USA) in
William's E medium for 15 min at 37°C protected from light in order to detect mitochondrial
superoxideproduction(41).Then,cellswerewashedwithwarmHBSSandmountedontoglassslides
using DAPI staining solution (InvitrogenTM, Carlsbad, CA,USA). The fluorescence analyses were
immediatelyrecordedusingafluorescentmicroscopeatawavelengthof510/580nm(Ex/Em)bya
Leicamicroscope (LeicaMicrosystemsGmbH, �Wetzlar�,Germany).Quantificationof fluorescence in
microscopicimagesofMitoSOXwasperformedusingImageJsoftware.
2.8WesternBlotanalysesandantibodies
Protein levels expressionwere detected by total cell lysate subjected towestern blot as previous
described (42).The followingantibodieswereused:Monoclonalmouseanti-HIF1α1:1000 (Abcam,
USA); monoclonal mouse anti-GSDMDC1 1:500 (Santa Cruz Biotechnology, Dallas, TX, USA);
polyclonal rabbit anti-CASP-3 (Cell Signaling Technology, Leiden, The Netherlands); monoclonal
mouseanti-CASP11:500 (SantaCruzBiotechnology,Dallas,TX,USA);monoclonalmouseanti-CD81
1:1000(InvitrogenTM,Carlsbad,CA,USA)andmonoclonalrabbitanti-Bcl-21:1000(Abcam,UK)were
used in combination with appropriate peroxidase-conjugated secondary antibodies. Monoclonal
mouse anti-tubulin or actin were used as loading controls (Sigma, Life Sciences, Merck KGaA,
Darmstadt,German).Blotswereanalyzed inaChemiDocXRS system (Bio-Rad,Hercules,CA,USA).
ProteinbandintensitieswerequantifiedbyImageLabsoftware(BioRad,Hercules,CA,USA).
2.9RNAisolationandquantitativereal-timereversetranscriptionpolymerasechainreaction(qRT-
PCR)
Total RNA was isolated with TRI-reagent (Sigma-Aldrich, Saint Louis, MO, USA) according to the
manufacturer’s instructions. Quantification of RNA was measured with the Nanodrop

35
spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Reverse transcription (RT) was
performed using 2.5 µg of total RNA. Quantitative Real Time PCR (qRT-PCR) was carried out in a
StepOnePlus™(96-well)PCRSystem(AppliedBiosystems,Thermofisher, Waltham,MA,USA)using
TaqManmethodorSYBRGreenmethod.Thesequencesoftheprobesandprimersetsaredescribed
intheSupplementarymaterialsfile.mRNAlevelswerenormalizedtothehousekeepinggene18Sand
furthernormalizedtothemeanexpressionlevelofthecontrolgroup.Relativegeneexpressionwas
calculatedviathe2ddCT.
2.10Enzyme-linkedimmunosorbentassay(ELISA)ofInterleukin-1beta
Levels of pro-inflammatory cytokine of IL-1β in primary rat KC were assessed by the ELISA kit
(ab100768,Abcam,Cambridge,UK),accordingtothemanufacturer’sinstructions.
2.11EVisolationandcharacterization
EVwerecollectedfromculturemediaofHepG2cellsasdescribedpreviously(43)andsummarizedin
the Supplementary materials file. Nanoparticle tracking analysis (NTA) was performed using
NanoSightNS300instrumentation(Marvel,Egham,UK)thatusesbothlightscatteringandBrownian
motionanalysis fornanoparticlecharacterization.We furthercaharacterizedEVusingTransmission
electron microscopy. Sample preparation and details of these analysis are described in the
Supplementarymaterialsfile.
2.12TreatmentofKCcellswithandextracellularvesicles
RatprimaryKCwere incubatedwithFBS-freeWilliam'sEmediumandexposedto15μgofEVthat
wereisolatedfromHepG2cellstreatedwithCoCl2+FFAfor24h.After24hofEVtreatment,KCwere
harvestedtocontinuewithanalysesbyquantitativePCR.
2.13EffectsofintermittenthypoxiainexperimentalNASH
Animalexperimentswereapprovedbythe institutionalanimalcareandusecommittee(Comitéde
ética y bienestar Animal, Escuela de Medicina, Pontificia Universidad Católica de Chile, CEBA
100623003).MaleC57BL/6miceaged10weeksatthebeginningofthestudyanddividedintofour
experimental groups (n = 4–8) receiving either choline-deficient amino acid-defined (CDAA) diet
(Catalog#518753,DyetsInc.Bethlehem,PA)toinduceNASHorthecholine-supplementedL-amino
acid defined (CSAA, Catalog # 518754, Dyets Inc. Bethlehem, PA) diet as control for 22 weeks as
previouslydescribed(16,44).AnimalswereexposedtoIHornormoxia(chambers41x22x35cm,COY
labproducts™,GrassLake,MI,USA)duringthelast12weeksoftheexperimentalorcontrolfeeding
period.IHregimenconsistedin30events/hourofhypoxicexposuresfor8hour/dayduringtherest

36
cycle, between9 amand5pm. This cyclewas repeated7days aweek for 12 consecutiveweeks.
After ending the study, mice were anesthetized (ketamine 60 mg/kg plus xylazine 10 mg/kg
intraperitoneally) and then euthanized by exsanguination. Serum and liver tissue samples were
collected and processed or stored at −80 °C until analyzed. Gene expression and protein analyses
were carried out as described above. Positive control of hypoxia induction was evaluated by
measurementofhepaticgeneexpressionofHIF-1α.
2.14HistologicalStudies
Liversteatosiswasanalyzedonparaformaldehyde-fixedliversectionsstainedwithOilRed-Ostaining
that show lipiddeposits in red coloron frozen7 μm liver cryosections.Ablindedpathologist (J.T.)
assigneda score for steatosis, inflammationand fibrosis asdescribed (44). Scoresweregivenas it
follows: Steatosis: grade 0, none present; grade 1, steatosis of ≤25% of parenchyma; grade 2,
steatosisof26–50%ofparenchyma;grade3,steatosisof51–75%ofparenchyma;grade4,steatosis
of≥76%ofparenchymaandinflammation:grade0,noinflammatoryfoci;grade1,1–5inflammatory
foci per high power field; grade 2, >5 inflammatory foci/high power field. Fibrosis was estimated
qualitativelyinSiriusred-stainedsamples.
2.15Hepatictriglyceridedetermination
Hepatic triglyceride content (HTC)was using 40-50mg of homogenized liver tissue in 1.5ml of a
CHCl3-CH3OHmixture(2:1,v/v),followedbyaFolchextractiondescribedpreviously(16,31).
2.16StatisticalAnalyses
AnalyseswereperformedusingGraphPadsoftware(version5.03,GraphPadSoftwareInc.,CA,USA).
Statistical analyses were performed using one-way analysis of variance (ANOVA) with a post-hoc
Bonferronicorrectionwithmultiple-comparisontestorbyparametrict-testswhenneeded. Allthe
results are presented as a mean of at least 3 independent experiments ± SEM. Values were
representedas absolutenumber, normalizeddata respect to control or percentage for categorical
variables.Regardinginvitroassays, independentexperimentsmeansdifferentcellplatesharvested
fromatleastthreeanimals.Regardinginvivoassays,independentexperimentsmeansatleastthree
differentanimalsamples.AlltheresultswereanalyzedandplottedusingGraphPadsoftware(version
5.03,GraphPadSoftwareInc.,CA,USA).Statisticswithavalueofp<0.05wereconsideredsignificant.
3.-RESULTS
3.1HypoxiainducesHIF-1αandpromotestheincreaseoflipiddropletsinfat-ladenhepatocytes:To
investigatewhetherhypoxiaexacerbateslipotoxicityinan invitromodelofexperimentalNASH,we
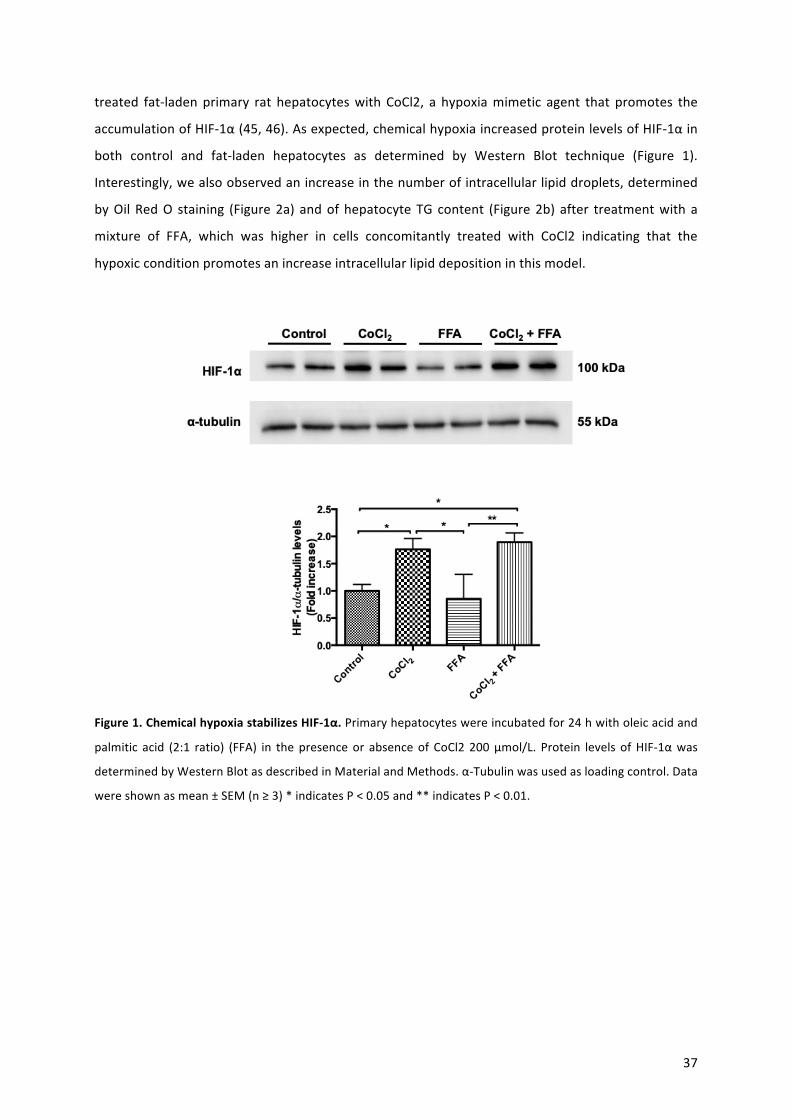
37
treated fat-ladenprimary rat hepatocyteswith CoCl2, a hypoxiamimetic agent that promotes the
accumulationofHIF-1α(45,46).Asexpected,chemicalhypoxiaincreasedproteinlevelsofHIF-1αin
both control and fat-laden hepatocytes as determined by Western Blot technique (Figure 1).
Interestingly,wealsoobservedanincreaseinthenumberofintracellularlipiddroplets,determined
byOilRedO staining (Figure2a) andofhepatocyteTG content (Figure2b) after treatmentwitha
mixture of FFA, which was higher in cells concomitantly treated with CoCl2 indicating that the
hypoxicconditionpromotesanincreaseintracellularlipiddepositioninthismodel.
Figure1.ChemicalhypoxiastabilizesHIF-1α.Primaryhepatocyteswereincubatedfor24hwitholeicacidand
palmitic acid (2:1 ratio) (FFA) in thepresenceor absenceof CoCl2 200μmol/L. Protein levels ofHIF-1αwas
determinedbyWesternBlotasdescribedinMaterialandMethods.α-Tubulinwasusedasloadingcontrol.Data
wereshownasmean±SEM(n≥3)*indicatesP<0.05and**indicatesP<0.01.

38
Figure 2. Steatosis in vitro: Free fatty acids and chemical hypoxia treatment increase the content of
triglycerides.Primaryhepatocyteswereincubatedfor24hwitholeicacidandpalmiticacid(2:1ratio)(FFA)in
thepresenceorabsenceofCoCl2200μmol/L.a)OilRedOstaining(scalebar:40μm)andb)Triglycerides(TG)
contentweremeasuredasdescribedinMaterialsandMethods.Datawereshownasmean±SEM(n≥3)***
indicatesP<0.005and****indicatesP<0.001.
3.2 Hypoxia increases apoptotic and pyroptotic cell death in fat-laden hepatocytes: To evaluate
whether the induction of chemical hypoxia exacerbates lipotoxic cell damage and death, we
determinedproteinlevelsandactivityofcaspase-3toassessapoptoticcelldeathandusedSYTOXTM
Green nucleic acid stain and LDH leakage to evaluate cell death associated to disrupted cellular
membraneintegrity.Additionally,weassessedthecaspase-cleavedgasdermin-Ndomain(GSDMD-N)
toevaluatepyroptoticcelldeath(19).WhileCoCl2didnotinfluencecleavedcaspase-3andcaspase3
activityinnormalhepatocytes,steatotichepatocytesundergoingchemicalhypoxiadisplayedatwo-
fold increase incleavedcaspase-3 (Figure3a)andsix-fold increase incaspase-3activity (Figure3b)
comparedtocontrolcells.Ofnote,treatmentwithamixtureofFFAaloneledtoatwo-foldincrease
(n.s.) in caspase 3 activity suggesting that chemical hypoxia exacerbates lipotoxicity and apoptotic
cell death in fat-laden hepatocytes. Also, using SYTOXTM Green nucleic acid stain (Figure 4a) and
measurementofcellularLDHleakage(Figure4b),wefoundthatchemicalhypoxiaalsoincreasedcell
death rate associated to losing plasmamembrane integrity of fat-laden hepatocytes compared to
control cells or cells treated only with FFA. Furthermore, protein levels of GSDMD-N, which is
considered a pyroptosis executor (19), increased three-fold in hypoxic fat-laden hepatocytes
comparedtocontrolcells(Figure4c).

39
Figure3.Chemicalhypoxiapromotesapoptoticcelldeathevaluatebya)cleavedCaspase-3andb)Caspase-3
activityinfat-ladenhepatocytes.Primaryhepatocyteswereincubatedfor24hwitholeicacidandpalmiticacid
(2:1ratio)(FFA) inthepresenceorabsenceofCoCl2200μmol/L. a)CleavedCaspase-3protein levelsandb)
Caspase-3activityweremeasuredasdescribedinMaterialsandMethods.Datawereshownasmean±SEM(n
≥3)*indicatesP<0.05;***indicatesP<0.005and****indicatesP<0.001.

40
Figure 4. Hypoxia promotes pyroptotic cell death
evaluate by a) SYTOX green, b) Lactate
dehydrogenase leakage and c) Gasdermin-N in fat-
laden hepatocytes. Primary hepatocytes were
incubated for 24h with oleic acid and palmitic acid
(2:1 ratio) (FFA) in thepresenceorabsenceofCoCl2
200 μmol/L. a) Sytox green (Representative images
andquantification),b)LDHleakagepercentageandc)
GSDMD-N protein levels determinated by Western
Blot were measured as described in Materials and
Methods.Datawereshownasmean±SEM(n≥3)*
indicatesP<0.05and**indicatesP<0.01.
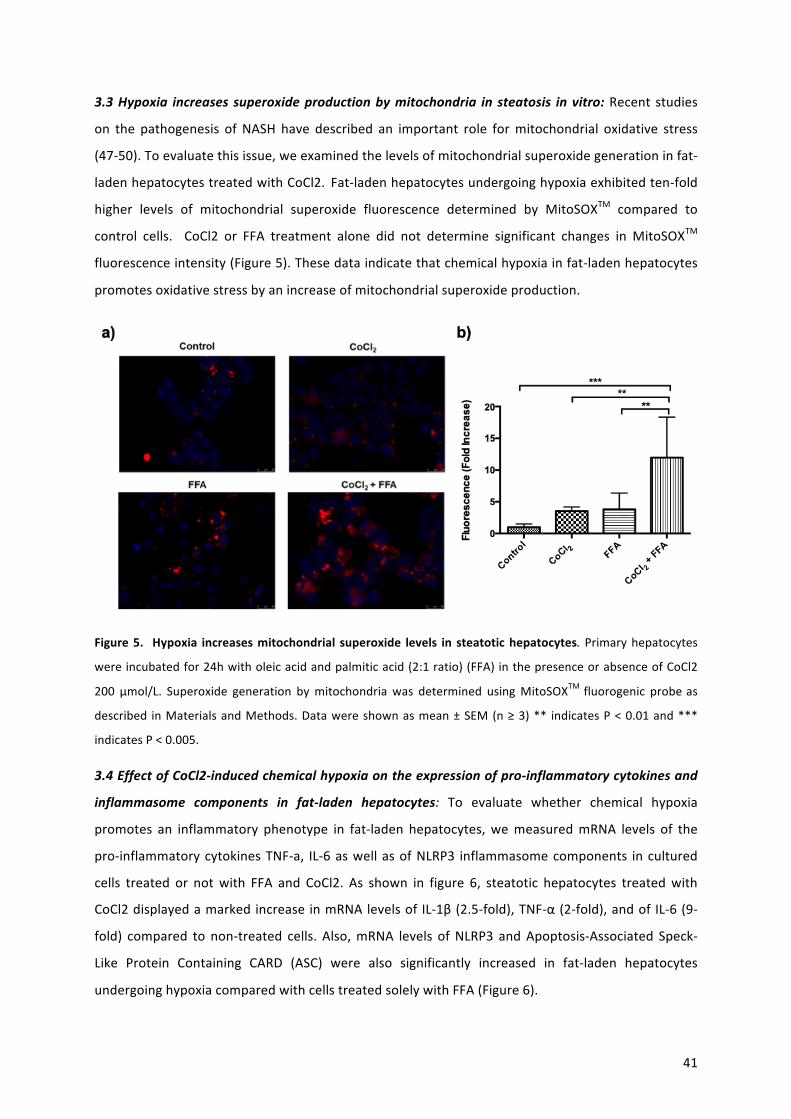
41
3.3Hypoxia increases superoxideproductionbymitochondria in steatosis invitro:Recent studies
on the pathogenesis of NASH have described an important role formitochondrial oxidative stress
(47-50).Toevaluatethisissue,weexaminedthelevelsofmitochondrialsuperoxidegenerationinfat-
ladenhepatocytestreatedwithCoCl2.Fat-ladenhepatocytesundergoinghypoxiaexhibitedten-fold
higher levels of mitochondrial superoxide fluorescence determined by MitoSOXTM compared to
control cells. CoCl2 or FFA treatment alone did not determine significant changes in MitoSOXTM
fluorescenceintensity(Figure5).Thesedataindicatethatchemicalhypoxiainfat-ladenhepatocytes
promotesoxidativestressbyanincreaseofmitochondrialsuperoxideproduction.
Figure5. Hypoxia increasesmitochondrial superoxide levels in steatotichepatocytes. Primaryhepatocytes
wereincubatedfor24hwitholeicacidandpalmiticacid(2:1ratio)(FFA)inthepresenceorabsenceofCoCl2
200 μmol/L. Superoxide generation bymitochondria was determined usingMitoSOXTM fluorogenic probe as
described inMaterialsandMethods.Datawereshownasmean±SEM(n≥3)** indicatesP<0.01and***
indicatesP<0.005.
3.4EffectofCoCl2-inducedchemicalhypoxiaontheexpressionofpro-inflammatorycytokinesand
inflammasome components in fat-laden hepatocytes: To evaluate whether chemical hypoxia
promotes an inflammatory phenotype in fat-laden hepatocytes,wemeasuredmRNA levels of the
pro-inflammatorycytokinesTNF-a, IL-6aswellasofNLRP3 inflammasomecomponents incultured
cells treated or notwith FFA and CoCl2. As shown in figure 6, steatotic hepatocytes treatedwith
CoCl2displayedamarkedincrease inmRNAlevelsof IL-1β(2.5-fold),TNF-α(2-fold),andof IL-6(9-
fold) compared to non-treated cells. Also,mRNA levels ofNLRP3 andApoptosis-Associated Speck-
Like Protein Containing CARD (ASC) were also significantly increased in fat-laden hepatocytes
undergoinghypoxiacomparedwithcellstreatedsolelywithFFA(Figure6).
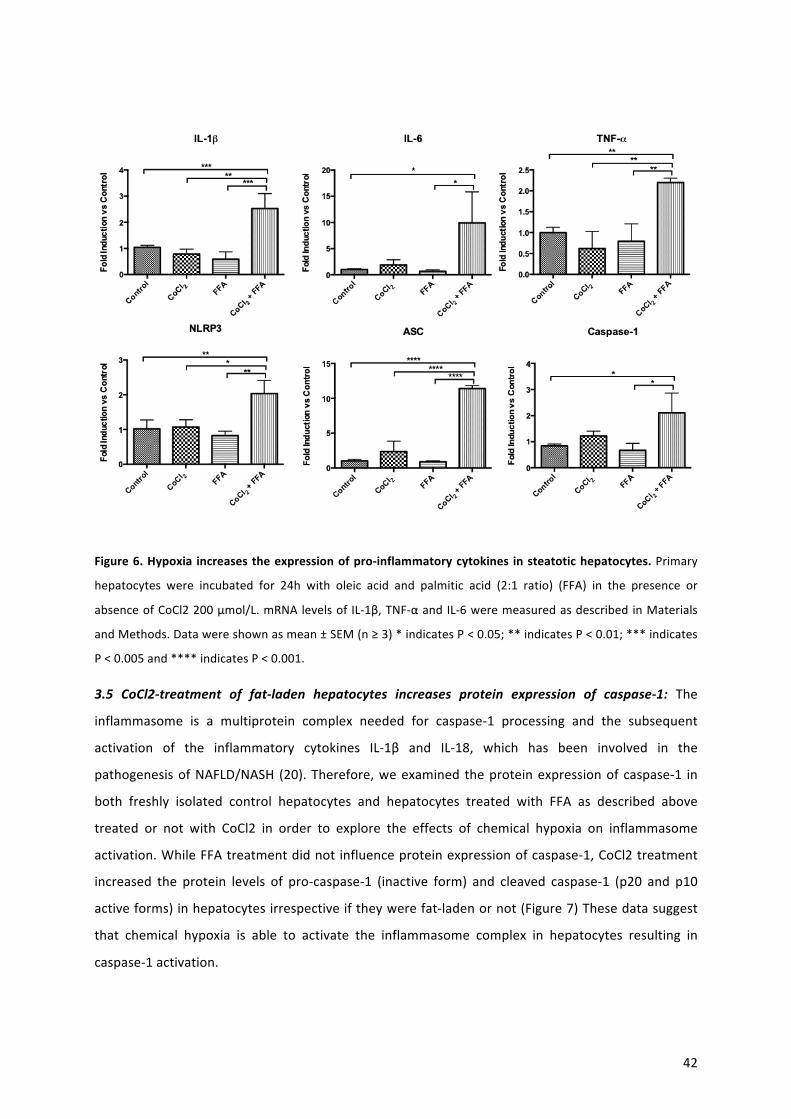
42
Figure6.Hypoxia increasestheexpressionofpro-inflammatorycytokines insteatotichepatocytes.Primary
hepatocytes were incubated for 24h with oleic acid and palmitic acid (2:1 ratio) (FFA) in the presence or
absenceofCoCl2200μmol/L.mRNAlevelsof IL-1β,TNF-αandIL-6weremeasuredasdescribedinMaterials
andMethods.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01;***indicates
P<0.005and****indicatesP<0.001.
3.5 CoCl2-treatment of fat-laden hepatocytes increases protein expression of caspase-1: The
inflammasome is a multiprotein complex needed for caspase-1 processing and the subsequent
activation of the inflammatory cytokines IL-1β and IL-18, which has been involved in the
pathogenesisofNAFLD/NASH (20).Therefore,weexamined theproteinexpressionofcaspase-1 in
both freshly isolated control hepatocytes and hepatocytes treated with FFA as described above
treated or not with CoCl2 in order to explore the effects of chemical hypoxia on inflammasome
activation.WhileFFAtreatmentdidnotinfluenceproteinexpressionofcaspase-1,CoCl2treatment
increased the protein levels of pro-caspase-1 (inactive form) and cleaved caspase-1 (p20 and p10
activeforms)inhepatocytesirrespectiveiftheywerefat-ladenornot(Figure7)Thesedatasuggest
that chemical hypoxia is able to activate the inflammasome complex in hepatocytes resulting in
caspase-1activation.
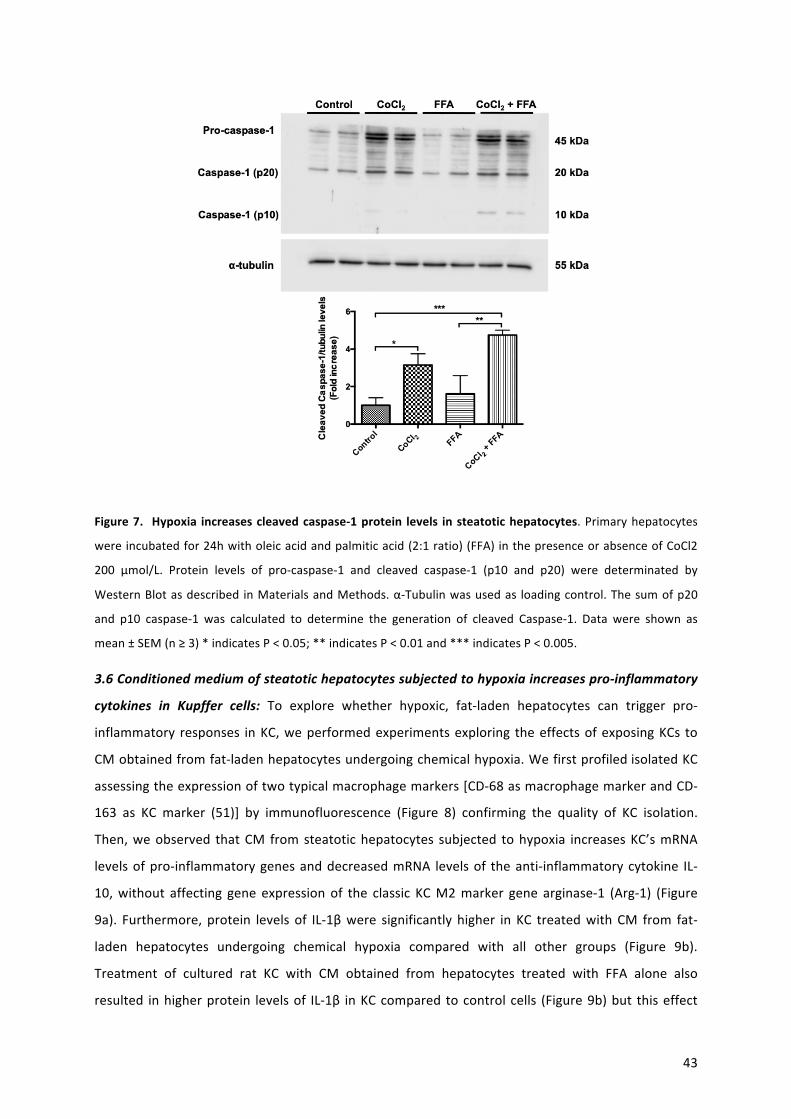
43
Figure7. Hypoxia increasescleavedcaspase-1protein levels insteatotichepatocytes.Primaryhepatocytes
wereincubatedfor24hwitholeicacidandpalmiticacid(2:1ratio)(FFA)inthepresenceorabsenceofCoCl2
200 μmol/L. Protein levels of pro-caspase-1 and cleaved caspase-1 (p10 and p20) were determinated by
WesternBlotasdescribed inMaterialsandMethods.α-Tubulinwasusedas loadingcontrol.Thesumofp20
and p10 caspase-1 was calculated to determine the generation of cleaved Caspase-1. Data were shown as
mean±SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01and***indicatesP<0.005.
3.6Conditionedmediumofsteatotichepatocytessubjectedtohypoxiaincreasespro-inflammatory
cytokines in Kupffer cells: To explore whether hypoxic, fat-laden hepatocytes can trigger pro-
inflammatory responses inKC,weperformedexperimentsexploring theeffectsofexposingKCs to
CMobtainedfromfat-ladenhepatocytesundergoingchemicalhypoxia.WefirstprofiledisolatedKC
assessingtheexpressionoftwotypicalmacrophagemarkers[CD-68asmacrophagemarkerandCD-
163 as KC marker (51)] by immunofluorescence (Figure 8) confirming the quality of KC isolation.
Then,weobserved thatCM fromsteatotichepatocytes subjected tohypoxia increasesKC’smRNA
levelsofpro-inflammatorygenesanddecreasedmRNA levelsof theanti-inflammatorycytokine IL-
10,without affecting gene expressionof the classic KCM2marker gene arginase-1 (Arg-1) (Figure
9a). Furthermore, protein levels of IL-1βwere significantly higher in KC treatedwithCM from fat-
laden hepatocytes undergoing chemical hypoxia compared with all other groups (Figure 9b).
Treatment of cultured rat KC with CM obtained from hepatocytes treated with FFA alone also
resulted inhigherprotein levelsof IL-1β inKCcompared tocontrolcells (Figure9b)but thiseffect
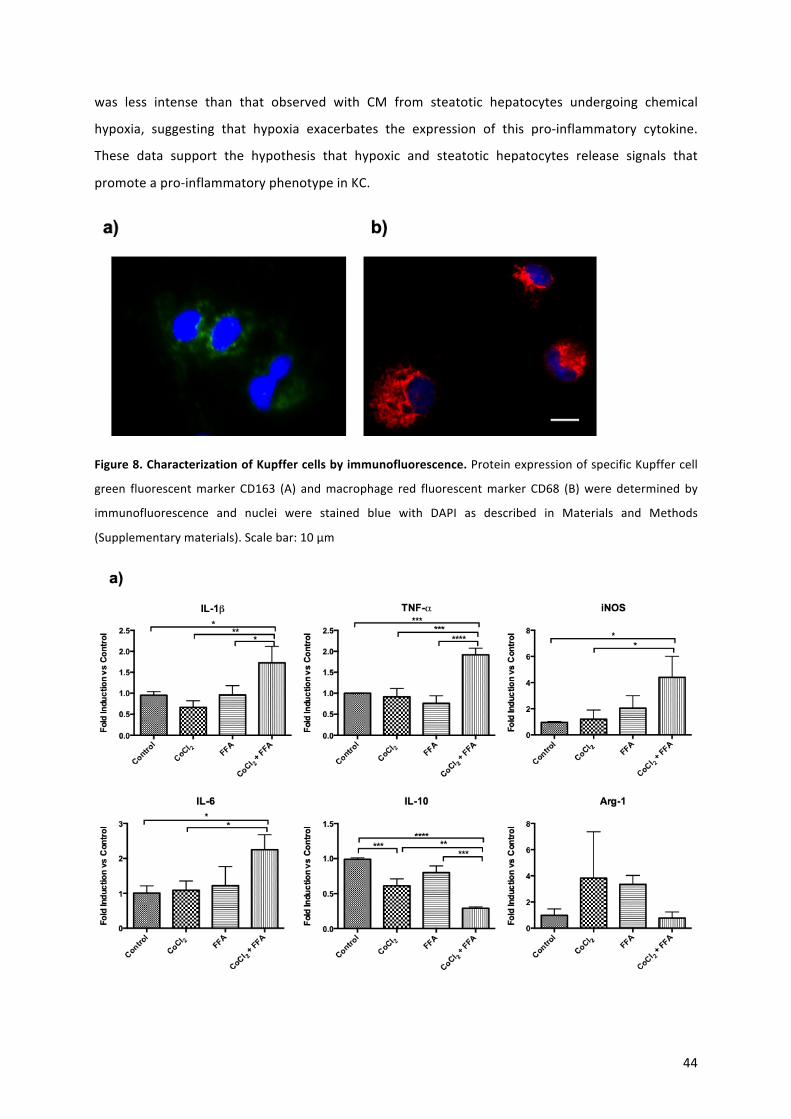
44
was less intense than that observed with CM from steatotic hepatocytes undergoing chemical
hypoxia, suggesting that hypoxia exacerbates the expression of this pro-inflammatory cytokine.
These data support the hypothesis that hypoxic and steatotic hepatocytes release signals that
promoteapro-inflammatoryphenotypeinKC.
Figure8.CharacterizationofKupffercellsbyimmunofluorescence.ProteinexpressionofspecificKupffercell
green fluorescentmarker CD163 (A) andmacrophage red fluorescentmarker CD68 (B)were determined by
immunofluorescence and nuclei were stained blue with DAPI as described in Materials and Methods
(Supplementarymaterials).Scalebar:10μm

45
Figure9.Conditionedmediumofsteatotichepatocytessubjectedtohypoxiaincreasesa)pro-inflammatory
genesand)IL-1βproteinlevelsinKupffercells.Culturemediumofprimaryhepatocyteswasreplaced24hours
aftertreatmentbyFFAandCoCl2-freemediumforanadditional24hours(conditionedmedium;CM).Kupffer
cellsweretreatedfor24hourswiththeCMfromhepatocytes.a)mRNAlevelsofIL-1β,TNF-α,iNOS,IL-6,IL-10
andArg-1 andb) IL-1βprotein levels by ELISAweremeasuredasdescribed inMaterials andMethods.Data
wereshownasmean±SEM(n≥3)* indicatesP<0.05;** indicatesP<0.01;*** indicatesP<0.005;****
indicatesP<0.001.
3.7Extracellularvesicles increased inCMfromfat-ladenhepatocellularcell lineexposedtoCoCl2
andpromotedpro-inflammatorygenesexpression inKC:Toexplore if EV released from fat-laden
hepatocytesundergoinghypoxiaplayaroleinpromotingapro-inflammatoryeffectsinKC,EVwere
collected from culture media obtained from cultured HepG2 cells. We first explored if CM from
HepG2 cells exposed to the different treatments evokedproinflammatory signals in KC. Results of
these experiments are shown in Supplementary figure 2. As shown in this figure, similar to the
effects observed in experiments involving hepatocytes, CMdid determine increased expression of
proinflammatorygenesinKC,adecreaseintheexpressionofIL-10andhadnoeffectonArginase-1
expression.We thenexplored theeffectsofHepG2-derivedEVonKCandcompared theobserved
effects with the effect of treatment with the EV-free fraction. We first characterized EV by
transmission electronmicroscopy aswell as byWestern blot detection of the presence of the EV
markerCD81andtheabsenceofthemitochondrialproteinBcl-2 inallEVfractions(Figure10aand
SupplementaryFigure1).Ofnote,thesizeandconcentrationofHepG2-derivedEVweredetermined
byNTA. Interestingly,we foundan increaseofEV inculturemedia fromfat-ladencellsexposed to
CoCl2(6.0x1011particles/mL)comparedtoculturemediafromcontrolcells(9.5x1010particles/mL)
(Figure10b).Asshowninthisfigure,theaveragesizeofEVobtainedfromculturemediaofcontrol
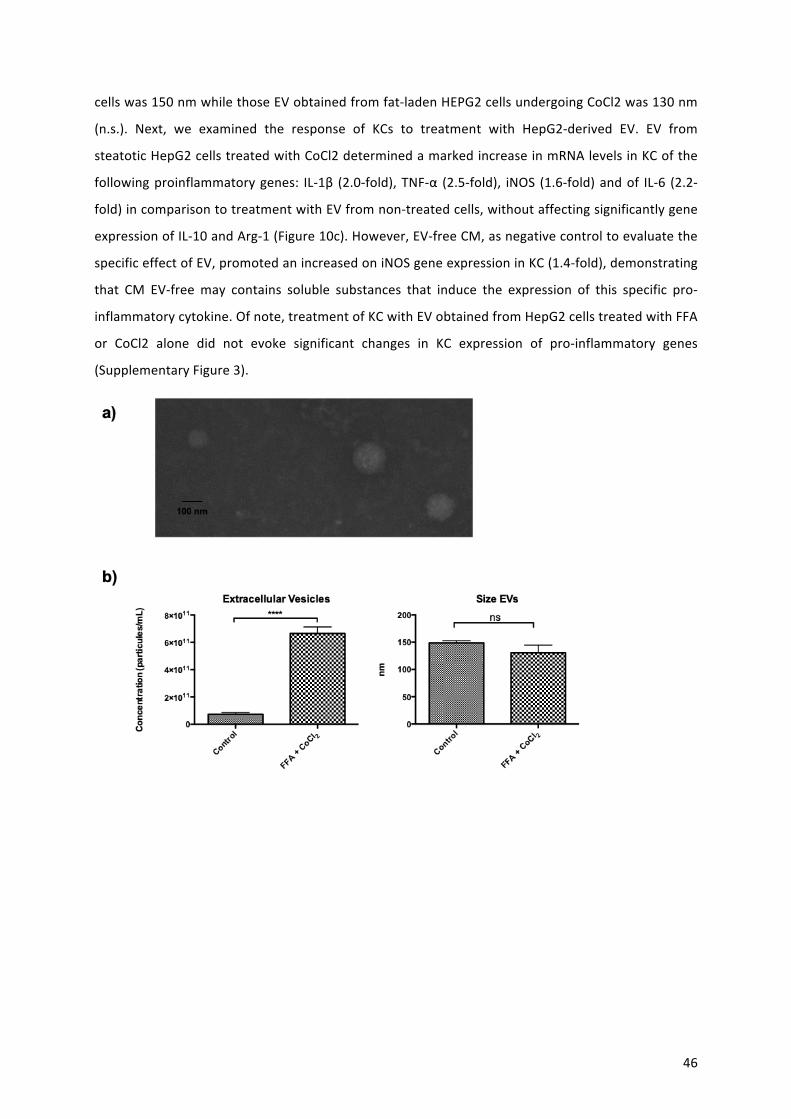
46
cellswas150nmwhilethoseEVobtainedfromfat-ladenHEPG2cellsundergoingCoCl2was130nm
(n.s.). Next, we examined the response of KCs to treatment with HepG2-derived EV. EV from
steatoticHepG2cellstreatedwithCoCl2determinedamarkedincreaseinmRNAlevelsinKCofthe
followingproinflammatorygenes: IL-1β (2.0-fold),TNF-α (2.5-fold), iNOS (1.6-fold)andof IL-6 (2.2-
fold)incomparisontotreatmentwithEVfromnon-treatedcells,withoutaffectingsignificantlygene
expressionofIL-10andArg-1(Figure10c).However,EV-freeCM,asnegativecontroltoevaluatethe
specificeffectofEV,promotedanincreasedoniNOSgeneexpressioninKC(1.4-fold),demonstrating
that CM EV-freemay contains soluble substances that induce the expression of this specific pro-
inflammatorycytokine.Ofnote,treatmentofKCwithEVobtainedfromHepG2cellstreatedwithFFA
or CoCl2 alone did not evoke significant changes in KC expression of pro-inflammatory genes
(SupplementaryFigure3).

47
Figure 10. EV obtained from culture media of fat-laden HepG2 cells exposed to CoCl2 increases pro-
inflammatory genes inKupfferCells. EVobtained fromculturemediaof fat-ladenHepG2 cells exposed to
CoCl2 increases pro-inflammatory genes in Kupffer Cells (KC). Presence of EV was confirmed by electron
microscopy (a) transmissionandquantifiedusingNanotrackinganalysis (b).Free fattyacids (FFA)andCoCl2
treatmentofHepG2cellsdeterminedan increase in thenumberof EV in culturedmediawithno significant
changesintheirsize.(c)KCweretreatedwithhypoxicandfat-ladenHepG2cells-derivedEV(15μg)andEV-
free culture media as negative control. mRNA levels of interleukin (IL)-1β, tumor necrosis factor TNF-α,
inducible nitric oxide synthase (iNOS), IL-6, IL-10 and Arginase−1 (Arg-1) were measured as described in
MaterialsandMethods.Dataisshownasmean±SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01;***
indicatesP<0.005;****indicatesP<0.001.
3.8 Intermittent hypoxia in an experimentalmodel of diet-induced non-alcoholic steatohepatitis
increasestheinflammatoryphenotype:Tovalidateourinvitrofindings,weinvestigatedtheeffectof
IH for12weeks inananimalmodel, theCDAAdiet-inducedNASH (16).Toevaluatesteatosis, liver
sections were stained with Oil red O staining (Figure 11a) (Abcam, Cambridge, UK). Steatosis,
inflammation and fibrosiswas gradedblindly by an experiencedpathologist (J.T.) according to the
NASscore.Specifically,theamountofsteatosis(percentageofhepatocytescontainingfatdroplets)
was scored as 0 (<5%), 1 (5–33%), 2 (>33–66%) and 3 (>66%). Although pathological scoring
suggestedahigherdegreeofsteatosisinanimalsonCDAAdietandexposedtohypoxiacomparedto
controlanimalsandcomparedtoanimalsonCDAAdietandnormoxicconditions(Figure11b),wedid
not observed significant differences in the hepatic TG levels between the CDAA-fed mice groups
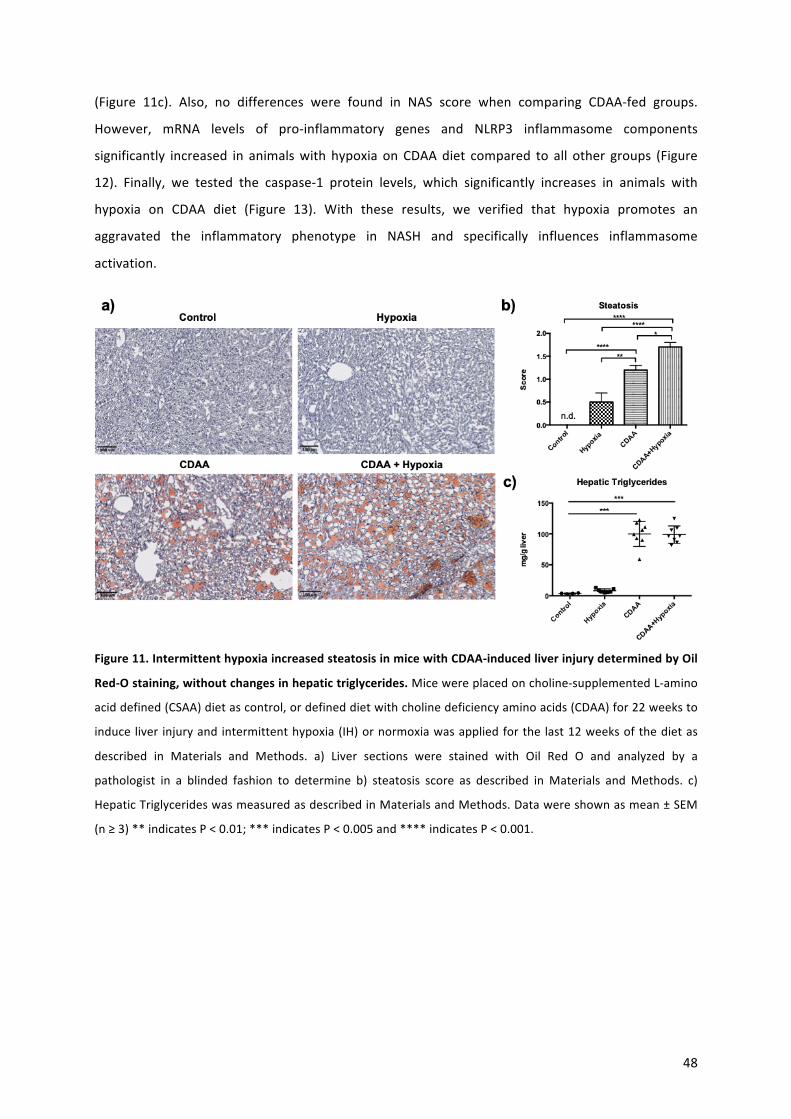
48
(Figure 11c). Also, no differences were found in NAS score when comparing CDAA-fed groups.
However, mRNA levels of pro-inflammatory genes and NLRP3 inflammasome components
significantly increased in animalswith hypoxia onCDAAdiet compared to all other groups (Figure
12). Finally, we tested the caspase-1 protein levels, which significantly increases in animals with
hypoxia on CDAA diet (Figure 13). With these results, we verified that hypoxia promotes an
aggravated the inflammatory phenotype in NASH and specifically influences inflammasome
activation.
Figure11.IntermittenthypoxiaincreasedsteatosisinmicewithCDAA-inducedliverinjurydeterminedbyOil
Red-Ostaining,withoutchangesinhepatictriglycerides.Micewereplacedoncholine-supplementedL-amino
aciddefined(CSAA)dietascontrol,ordefineddietwithcholinedeficiencyaminoacids(CDAA)for22weeksto
induce liver injuryandintermittenthypoxia(IH)ornormoxiawasappliedforthe last12weeksofthedietas
described in Materials and Methods. a) Liver sections were stained with Oil Red O and analyzed by a
pathologist in a blinded fashion to determine b) steatosis score as described inMaterials andMethods. c)
HepaticTriglycerideswasmeasuredasdescribedinMaterialsandMethods.Datawereshownasmean±SEM
(n≥3)**indicatesP<0.01;***indicatesP<0.005and****indicatesP<0.001.

49
Figure 12. Intermittent hypoxia in mice with CDAA-induced liver injury increases the expression of pro-
inflammatorycytokinesandinflammasomecomponents.Themicewereplacedoncholine-supplementedL-
aminoaciddefined (CSAA)dietascontrol,ordefineddietwithcholinedeficiencyaminoacids (CDAA) for22
weekstoinduceliverinjuryandintermittenthypoxia(IH)ornormoxiawasappliedforthelast12weeksofthe
dietasdescribedinMaterialsandMethods.mRNAlevelsofIL-1β,IL-18,NLRP3,caspase-1,IL-6andIFN-γwere
measuredasdescribedinMaterialsandMethods.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05;
**indicatesP<0.01;***indicatesP<0.005;****indicatesP<0.001.

50
Figura13.IntermittenthypoxiainmicewithCDAA-inducedliverinjuryincreasescleavedcaspase-1.Themice
wereplacedoncholine-supplementedL-aminoaciddefined(CSAA)dietascontrol,ordefineddietwithcholine
deficiencyaminoacids (CDAA) for22weeks to induce liver injuryand intermittenthypoxia (IH)ornormoxia
was applied for the last 12weeks of the diet as described inMaterials andMethods. Protein levels of pro-
caspase-1and cleaved caspase-1 (p10andp20)weredeterminedbyWesternBlot asdescribed inMaterials
andMethods.α-Tubulinwasusedasa loadingcontrol.Thesumofp20andp10caspase-1wascalculatedto
determinethegenerationofcleavedcaspase-1.Datawereshownasmean±SEM(n≥3)**indicatesP<0.01
and***indicatesP<0.005.
4.-DISCUSSION
NAFLD representsahighly frequent causeof liverdisease,which is closelyassociatedwithobesity
and insulin resistance (7, 52).Clinicalobservations suggest thatpatientswithOSAShavea greater
predispositiontodevelopNAFLDandNASH,theaggressiveformofNAFLD,whichischaracterizedby
the presence of necro-inflammatory and fibrotic phenomena in the liver (5). Several studies have
shown that OSAS, mainly through the occurrence of IH, can modulate hepatocellular damage by
triggeringproinflammatoryandprofibroticsignals(25,26),butpathwaysunderlyingthiseffectsare
ill-defined.Thepresent studyusedcellularmodelsofNAFLD/NASH toexplorepotential synergistic
interactionsbetweenhypoxiaandFFAexposure.Tothatend,thehypoxiamimeticagentCoCl2was
usedtotreatfat-ladenhepatocytes.Ourfindingssuggestthat indeedhypoxiadeterminesafurther
increaseincellularTGcontent inFFAtreatedhepatocytesaswellas increasedmRNAlevelsofpro-
inflammatory cytokines as well as those of the NLRP3 inflammasome components. These
phenomenawerealsoassociatedwithincreasedmitochondrialsuperoxidelevelsandincreasedrates
ofcelldeathduetoapoptosisanddisruptionofcellmembrane,likelypyroptosisassuggestedbythe
increase in GSDMD levels. Moreover, conditioned medium obtained from hypoxic fat-laden
hepatocytes promoted an inflammatory phenotype in KC, which is known to be decisive for the
amplificationofthepathologicalphenomenonofNASH(12,23).Additionally,inordertodetermineif
theobservedeffectsonKCcouldbemediatedbyEVreleasefromhepatocytes,weconductedfurther
experimentsinthehumanhepatomacelllineHepG2.Wechoosethisapproachforpracticalreasons
since using this cell line the EV isolation efficiency is high. Our results showed that treatment of
HepG2withFFAandCoCl2determinesanincreaseinthereleaseofEVtothemediumandthatthese
EValsoevokeapro-inflammatoryresponseinratKCinlinewithpreviousobservationsshowingthat
released EV from fat-laden hepatocytes can act in in macrophages and contribute to pro-
inflammatoryresponseinaparacrinefashion(15).
Finally,wefoundcorrelatesofourinvitroNASHmodelfindingsinananimalmodelofNASHinduced
by feeding a CDAA diet. In the latter experiments, we also confirmed that IH promotes NLRP3

51
inflammasome activation in the setting of NASH. Collectively taken, these findings indicate that
hypoxiamay influenceNAFLD development and progression by acting at different levels including
promotion of further lipid accumulation as well as enhancement of liver inflammation and
hepatocellulardeath.Ofnote,recentrodentstudiesindicatethathypoxiamaybealsolinkedtoliver
fibrosis, a key prognostic feature of NASH (8). In fact, Mersarwi et al. demonstrated that HIF-1α
deletioninhepatocytesprotectsagainstthedevelopmentofliverfibrosisinamousemodelofNAFLD
(53).ThisobservationaddstoourdataandsupporttheconceptthathypoxiacontributestoNAFLD
progressioninfluencingcriticalstepsofthedisease.
ExperimentaldataontheeffectsofhypoxiainNAFLDmodelsisscarceandmainlyrestrictedtowhole
animal studies (53, 54). No data is available on the effects of hypoxia in cellular models of
NAFLD/NASH (36). To assess this, we used the hypoxia mimetic compound CoCl2, which induces
chemical hypoxia by stabilizing HIF-1α and 2α (38). This model is well accepted and has several
advantagesoveralternativessuchasthehypoxiachamberoraCO2incubatorwithregulatedoxygen
levels,whichare lessavailable,mostcostlyandprovidea less stableexperimental conditions (38).
CoCl2 has been previously used in normal isolated hepatocytes (38, 45) but not in fat-laden cells
underlying the novelty of our approach. Of note, previous studies as well as experiments of our
laboratory on hepatocyte cell lines have demonstrated the non-toxic effect of CoCl2 at
concentrations not exceeding 400 μmol/L for 24 hours as previously described (55). On the other
hand, FFA treatment using combination of OA and PA effectively induced TG accumulation in the
absence of lipoapoptosis (56). In spite of these advantages and, although CoCl2-induced chemical
hypoxiaisasuitablemodeltoassesstheeffectsofhypoxiaincellularmodels,weacknowledgethat
thismodel reproduces only some of the effects generated by a decrease in oxygen supply,which
limitthegeneralizabilityofourresultsuntiltheyareconfirmedinothermodels.
The observed hypoxia-related increases in intracellular lipid droplets and TG content in fat-laden
hepatocytesisconsistentwithpreviousstudiesthatindicatethathypoxia,throughHIF-1α,promotes
an increase in lipid biosynthesis causing TG accumulation (57, 58).We did not observe amarked
increase in lipidcontent incellstreatedsolelywithCoCl2,whichmayrelateto lengthoftreatment
and experimental conditions.We also did not find an increase in the in vivo CDAA-feeding NASH
model.We think that thismaybe related to the fact that thisparticularmodeldetermines severe
steatosisandinflammation(59)andthatthereforeisdifficulttoobservesubtlechanges.
WithregardtotheeffectsofchemicalhypoxiaoncelldeathofFFAtreatedhepatocytes,qualitative
determination of apoptosis, necrosis and pyroptosis using determination of caspase-3 cleavage,
caspase-3activity,SYTOXgreenstaining,LDHreleaseandGSDMD-Nwerecarriedoutasperformed

52
inprevious studies (56,60).While, treatmentwithCoCl2or FFAalonedidnot inducea significant
increaseinapoptosiscomparedtocontrolconditions,CoCl2treatmentoffat-ladenhepatocytesdid
increase cleaved caspase-3 and caspase-3 activity reflecting ongoing apoptosis. The same
observationwasmaderegardingcelldeathassociatedtoadisruptedplasmamembrane,bySYTOX
GreenandLDH leakagemeasure, asan increasednumberofnecroticdeathcellswasobserved in
steatotichepatocytessubjectedtohypoxia.Giventhefactthathypoxiawasassociatedtoincreaseof
GSDMD cleavage and up-regulation of inflammasome components, it is likely that pyroptosis also
contributedtocelldeathoffat-ladenhepatocytesundergoingchemicalhypoxia.Ofnote,ithasbeen
suggested that pyroptosis may be an inflammatory link related to the progression from bland
steatosistoNASH,asNLRP3activation isnotpresent inanimalmodelof isolatedsteatosiswithout
inflammation(61).Additionally,weobservedasignificantincreaseinmitochondrialsuperoxidelevels
in the fat-laden hepatocytes exposed to chemical hypoxia. Interestingly, increased mitochondrial
superoxidegenerationhasbeencorrelatedtomitochondrialdysfunction,oxidativedamage,cellular
deathandpro-inflammatoryeffectsinNASH(49,62).Thus,thissynergisticeffectofhypoxiaandFFA
in hepatocytes with regard to mitochondrial superoxide generation, likely contributes to the
observedincreaseincelldeathwhenhepatocytesareexposedtobothstimuli.
Inadditiontotheobservedeffectsoncelldeathandoxidativestress,chemicalhypoxiaalsoinduced
theexpressionof several inflammatorycytokines, includingTNF-a, IL-6and IL-1β,aswell asof the
componentsoftheNLRP3inflammasome(caspase-1,NLRP3andASC),insteatotichepatocytes.The
NLRP3inflammasomeactivatespro-caspase-1intoactivecaspase-1(p10andp20cleavedcaspase-1)
that in turn cleaves pro-IL-1β into the mature form of IL-1β promoting amplification of liver
inflammation and cell death (12). In recent years, caspase-1has been studied extensively andhas
been shown to contribute to liver damage during the development of NASH (63, 64) and several
studieshaveshownprotectiveeffectsagainst liverdamagewhencaspase-1 is inhibited(63). Inour
study,weevaluatedtheproteinlevelsofcaspase-1(pro-caspase-1andactivecleaved-caspase-1)in
hepatocytes.Anincreaseinthetotallevelofpro-caspase-1wasobservedinhypoxichepatocytesand
hypoxichepatocytestreatedwithFFAcomparedtothecontrolconditions. Interestingly,cleaved-p-
20 andp-10, the active isoformsof caspase-1,were only detected in hypoxic hepatocytes treated
with FFA. These results are consistent with the observed up-regulation of inflammasome
components and similar to thoseobtainedbyotherauthors inprostateepithelial cellsundergoing
hypoxia, showing increased NLRP3 inflammasome activation as determined by increased cellular
levelsofcleavedcaspase-1(65).Takentogether,ourresultssuggestthattheinductionofhypoxiain
hepatocytes promotes susceptibility to damage by FFA, resulting in increased caspase-3mediated

53
apoptosis, (secondary) necrosis, pyroptosis and an inflammatory phenotype, characterized by
increasedexpressionofpro-inflammatorygenesandproteinlevelsofcaspase-1.
Kupffercells(KC)areinvolvedinthepathogenesisofvariousliverdiseases,includingsteatohepatitis
(66). However, their role in the context of hepatocellular damage due to hypoxia remains poorly
explored.KCarederivedfromcirculatingmonocytesand,onceestablishedintheliver,fulfillmultiple
functions related to the immune system. KC maintain constant paracrine communication with
neighboring hepatocytes, stellate cells and endothelial cells through the signals that they receive
from the extracellular environment (66). To verify the paracrine involvement of KCwith steatotic
hepatocytes in the context of hypoxia, KC were exposed to conditioned medium of hepatocytes
subjected to different treatments. We documented that conditioned medium from steatotic and
hypoxichepatocytesinducedapro-inflammatoryprofileinKC.Thesenovelresultscorrelatewellwith
recent studies showing micro-RNA-mediated cellular crosstalk between hepatocytes treated with
ethanolandmonocytes/macrophages(67).Asmentionedbefore,hepatocytesareabletomodulate
signaling pathways in macrophages, stellate cells and endothelial cells in a paracrine manner by
cytokines, micro-RNAs and EV (32). Our experiments involving isolation of EV from HEPG2 cells
strongly suggest that EV participate in the cellular communication between steatotic and hypoxic
hepatocytesandKC.Thisobservationsmayhavediagnosticandtherapeuticimplications(33).
In the present study, we also explored whether IH influences hepatic steatosis and the pro-
inflammatoryphenotypeinaninvivomodelofNASH.Tothisend,weassessedtheeffectsofhypoxia
for 22 weeks in CDAA-fed mice by evaluating liver histology and hepatic mRNA levels of pro-
inflammatorycytokines.Ourresultsshowedaworseningofhistologicalsteatosis inCDAA-fedmice
subjected to IHcompared to thoseanimals thatdidnotundergohypoxic conditions.However,we
didnotobserveacorrespondingincreaseinthepaticTGcontent.Wethinkthatthismayberelated
to the fact that this particularmodel determines severe steatosis and inflammation (59) and that
thereforeisdifficulttoobservesubtlechanges.
With regard to inflammatory markers, CDAA-fed mice exposed to IH exhibited an increase of all
inflammasome components (IL-1β, IL-18, NLRP3 and caspase-1) and other pro-inflammatory
cytokines (IL-6, IFN-γ). Also, active caspase-1 level protein increased in livers from CDAA-fedmice
exposedto IH,whichcorrelateswiththefindings inthe invitromodel.Theseobservationssupport
theconcept thathypoxiamaycontribute to liverdamage in thesettingofNAFLDbyactivating the
NLRP3inflammasome.ArecentstudyshowingthatIHactivatestheNLRP3inflammasomeinkidneys,
whichresultinprogressiverenalinjury(68)providessupporttothisnotion.Althoughnotexploredin

54
this study, thismight have implicationsnotonly for hepatocellular injurybut also for liver fibrosis
developmentandhepatocarcinogenesis(69,70).
In summary, our results in in vitro and in vivo models of NAFLD/NASH support the notion that
hypoxiaplaysanaggravatingroleinthesettingofexcessivelipidloadinlivercellsbyinfluencinglipid
deposition, hepatocellular death and pro-inflammatory signals, involving NLRP3 inflammasome
activation. Moreover, our novel findings demonstrate that hypoxia modulates cellular crosstalk
between steatotic hepatocytes and inflammatory cells (KCs). Future studies should focus on the
characterizationoftheextracellularenvironmentofhypoxicandsteatotichepatocytestoidentifythe
hypoxia-specificfactorsandthemechanismsofhepatocellulardamageatplayinNASH.
5.-SUPPLEMENTARYFIGURES
FigureS1.CharacterizationofHepG2-EV.EVwerecharacterizedinculturemediumfromcontrolHepG2cells
by detection of EV-positive marker CD81 and EV negative mitochondrial marker Bcl-2 by Western blot as
describedinMaterialandMethods.

55
Figure S2. Conditionedmediumof steatoticHepG2 cells subjected to hypoxia increases pro-inflammatory
genes in Kupffer cells. Culture medium of HepG2 cells was replaced 24 hours after treatment by FFA and
CoCl2-freemedium for an additional 24 hours (conditionedmedium; CM). Kupffer cellswere treated for 24
hourswiththeCMfromHepG2cells.mRNAlevelsofIL-1β,TNF-α,iNOS,IL-6,IL-10andArg-1weremeasured
as described in Materials and Methods. Data were shown as mean ± SEM (n ≥ 3) *indicates P < 0.05; **
indicatesP<0.01;***indicatesP<0.005.

56
Figure S3. EV obtained from culture media of fat-laden HepG2 cells exposed to CoCl2 increases
proinflammatoyrygenesinKupffercells.KCcellswereexposedto15μgofEVthatwereisolatedfromHepG2
cellsthathadbeentreatedwithCoCl2,FFAandCoCl2+FFAfor24h.mRNAlevelsofIL-1β,TNF-α,iNOS,IL-6,IL-
10andArg-1weremeasuredasdescribedinMaterialsandMethods.Datawereshownasmean±SEM(n≥3)
*indicatesP<0.05;***indicatesP<0.005;****indicatesP<0.001.
Figure S4. Intermittent hypoxia inmice increases the gene expression ofHIF-1α. Themicewere placed on
choline-supplemented L-amino acid defined (CSAA) diet as control, or defined diet with choline deficiency
aminoacids(CDAA)for22weekstoinduceliverinjuryandintermittenthypoxia(IH)ornormoxiawasapplied
forthelast12weeksofthedietasdescribedinMaterialsandMethods.mRNAlevelsofHIF-1αwasmeasured
as described inMaterials andMethods. Datawere shown asmean ± SEM (n ≥ 3) * indicates P < 0.05; ***
indicatesP<0.005;****indicatesP<0.001.

57
6.REFERENCES
1.Drager LF, LopesHF,Maki-NunesC,Trombetta IC,Toschi-DiasE,AlvesMJ,FragaRF,etal. The impactofobstructive sleepapneaon
metabolicandinflammatorymarkersinconsecutivepatientswithmetabolicsyndrome.PLoSOne2010;5:e12065.
2.Gaines J,VgontzasAN,Fernandez-Mendoza J,BixlerEO.Obstructivesleepapneaandthemetabolicsyndrome:Theroadtoclinically-
meaningfulphenotyping,improvedprognosis,andpersonalizedtreatment.SleepMedRev2018;42:211-219.
3.DempseyJA,VeaseySC,MorganBJ,O'DonnellCP.Pathophysiologyofsleepapnea.PhysiolRev2010;90:47-112.
4.AgrawalS,DusejaA,AggarwalA,DasA,MehtaM,DhimanRK,ChawlaY.Obstructivesleepapneaisanimportantpredictorofhepatic
fibrosisinpatientswithnonalcoholicfattyliverdiseaseinatertiarycarecenter.HepatolInt2015;9:283-291.
5.MesarwiOA,LoombaR,MalhotraA.ObstructiveSleepApnea,Hypoxia,andNonalcoholicFattyLiverDisease.AmJRespirCritCareMed
2019;199:830-841.
6.ParikhMP,GuptaNM,McCulloughAJ.ObstructiveSleepApneaandtheLiver.ClinLiverDis2019;23:363-382.
7.RinellaME.Nonalcoholicfattyliverdisease:asystematicreview.JAMA2015;313:2263-2273.
8.ArabJP,ArreseM,TraunerM.RecentInsightsintothePathogenesisofNonalcoholicFattyLiverDisease.AnnuRevPathol2018;13:321-
350.
9.ChalasaniN,YounossiZ,LavineJE,CharltonM,CusiK,RinellaM,HarrisonSA,etal.Thediagnosisandmanagementofnonalcoholicfatty
liverdisease:PracticeguidancefromtheAmericanAssociationfortheStudyofLiverDiseases.Hepatology2018;67:328-357.
10.NoureddinM,SanyalAJ.PathogenesisofNASH:TheImpactofMultiplePathways.CurrHepatolRep2018;17:350-360.
11. Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver
inflammation.Gut2018;67:963-972.
12. Schuster S, Cabrera D, ArreseM, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol
2018;15:349-364.
13.WreeA,McGeoughMD,PenaCA,SchlattjanM,LiH, InzaugaratME,MesserK,etal.NLRP3inflammasomeactivationisrequiredfor
fibrosisdevelopmentinNAFLD.JMolMed(Berl)2014;92:1069-1082.
14.WreeA, Eguchi A,McGeoughMD, Pena CA, JohnsonCD, CanbayA,HoffmanHM, et al.NLRP3 inflammasome activation results in
hepatocytepyroptosis,liverinflammation,andfibrosisinmice.Hepatology2014;59:898-910.
15. Cannito S, Morello E, Bocca C, Foglia B, Benetti E, Novo E, Chiazza F, et al. Microvesicles released from fat-laden cells promote
activationofhepatocellularNLRP3 inflammasome:Apro-inflammatory linkbetween lipotoxicity andnon-alcoholic steatohepatitis. PLoS
One2017;12:e0172575.
16. Cabrera D,Wree A, Povero D, Solis N, Hernandez A, PizarroM,Moshage H, et al. Andrographolide Ameliorates Inflammation and
FibrogenesisandAttenuatesInflammasomeActivationinExperimentalNon-AlcoholicSteatohepatitis.SciRep2017;7:3491.
17.CsakT,GanzM,PespisaJ,KodysK,DolganiucA,SzaboG.Fattyacidandendotoxinactivateinflammasomesinmousehepatocytesthat
releasedangersignalstostimulateimmunecells.Hepatology2011;54:133-144.
18.MridhaAR,WreeA,RobertsonAAB,YehMM,JohnsonCD,VanRooyenDM,HaczeyniF,etal.NLRP3inflammasomeblockadereduces
liverinflammationandfibrosisinexperimentalNASHinmice.JHepatol2017;66:1037-1046.
19.XuB, JiangM,ChuY,WangW,ChenD,LiX,ZhangZ,etal.GasderminDplaysakey roleasapyroptosisexecutorofnon-alcoholic
steatohepatitisinhumansandmice.JHepatol2018;68:773-782.

58
20.GuoH,CallawayJB,TingJP.Inflammasomes:mechanismofaction,roleindisease,andtherapeutics.NatMed2015;21:677-687.
21.Cha JY,KimDH,ChunKH.The roleofhepaticmacrophages innonalcoholic fatty liverdiseaseandnonalcoholic steatohepatitis. Lab
AnimRes2018;34:133-139.
22.YuY,LiuY,AnW,SongJ,ZhangY,ZhaoX.STING-mediated inflammation inKupffercellscontributestoprogressionofnonalcoholic
steatohepatitis.JClinInvest2019;129:546-555.
23.ArreseM,CabreraD,KalergisAM,FeldsteinAE.InnateImmunityandInflammationinNAFLD/NASH.DigDisSci2016;61:1294-1303.
24.DragerLF,LiJ,ReinkeC,Bevans-FontiS,JunJC,PolotskyVY.Intermittenthypoxiaexacerbatesmetaboliceffectsofdiet-inducedobesity.
Obesity(SilverSpring)2011;19:2167-2174.
25.SavranskyV,NanayakkaraA,ViveroA,LiJ,BevansS,SmithPL,TorbensonMS,etal.Chronicintermittenthypoxiapredisposestoliver
injury.Hepatology2007;45:1007-1013.
26.SavranskyV,ReinkeC, Jun J,Bevans-FontiS,NanayakkaraA, Li J,MyersAC,etal.Chronic intermittenthypoxiaandacetaminophen
inducesynergisticliverinjuryinmice.ExpPhysiol2009;94:228-239.
27.LiuY,MaZ,ZhaoC,WangY,WuG,XiaoJ,McClainCJ,etal.HIF-1alphaandHIF-2alphaarecriticallyinvolvedinhypoxia-inducedlipid
accumulationinhepatocytesthroughreducingPGC-1alpha-mediatedfattyacidbeta-oxidation.ToxicolLett2014;226:117-123.
28.Briancon-MarjolletA,MonneretD,HenriM,Joyeux-FaureM,TotosonP,CachotS,FaureP,etal.IntermittenthypoxiainobeseZucker
rats:cardiometabolicandinflammatoryeffects.ExpPhysiol2016;101:1432-1442.
29.KangHH,KimIK,LeeHI,JooH,LimJU,LeeJ,LeeSH,etal.Chronicintermittenthypoxiainducesliverfibrosisinmicewithdiet-induced
obesityviaTLR4/MyD88/MAPK/NF-kBsignalingpathways.BiochemBiophysResCommun2017;490:349-355.
30.JuC,ColganSP,EltzschigHK.Hypoxia-induciblefactorsasmoleculartargetsforliverdiseases.JMolMed(Berl)2016;94:613-627.
31.vanderGraaffD,KwantenWJ,FrancqueSM.Thepotentialroleofvascularalterationsandsubsequentimpairedliverbloodflowand
hepatichypoxiainthepathophysiologyofnon-alcoholicsteatohepatitis.MedHypotheses2019;122:188-197.
32.EguchiA,FeldsteinAE.Extracellularvesiclesinnon-alcoholicandalcoholicfattyliverdiseases.LiverRes2018;2:30-34.
33.MalhiH.Emergingroleofextracellularvesiclesinliverdiseases.AmJPhysiolGastrointestLiverPhysiol2019;317:G739-G749.
34.MoshageH,CasiniA,LieberCS.Acetaldehydeselectivelystimulatescollagenproductioninculturedratliverfat-storingcellsbutnotin
hepatocytes.Hepatology1990;12:511-518.
35.DambaT,ZhangM,Buist-HomanM,vanGoorH,FaberKN,MoshageH.Hydrogensulfidestimulatesactivationofhepaticstellatecells
throughincreasedcellularbio-energetics.NitricOxide2019;92:26-33.
36.Chavez-TapiaNC,RossoN,TiribelliC.Invitromodelsforthestudyofnon-alcoholicfattyliverdisease.CurrMedChem2011;18:1079-
1084.
37.PecoraroM,PintoA,PopoloA.InhibitionofConnexin43translocationonmitochondriaacceleratesCoCl2-inducedapoptoticresponse
inachemicalmodelofhypoxia.ToxicolInVitro2018;47:120-128.
38.Munoz-SanchezJ,Chanez-CardenasME.Theuseofcobaltchlorideasachemicalhypoxiamodel.JApplToxicol2019;39:556-570.
39.SchoemakerMH,CondedelaRosaL,Buist-HomanM,VrenkenTE,HavingaR,PoelstraK,HaismaHJ,etal.Tauroursodeoxycholicacid
protectsrathepatocytesfrombileacid-inducedapoptosisviaactivationofsurvivalpathways.Hepatology2004;39:1563-1573.
40.ChanLL,McCulleyKJ,KesselSL.AssessmentofCellViabilitywithSingle-,Dual-,andMulti-StainingMethodsUsing ImageCytometry.
MethodsMolBiol2017;1601:27-41.

59
41.WangG,MeminE,MuraliI,GaspersLD.Theeffectofchronicalcoholconsumptiononmitochondrialcalciumhandlinginhepatocytes.
BiochemJ2016;473:3903-3921.
42.Woudenberg-VrenkenTE,CondedelaRosaL,Buist-HomanM,FaberKN,MoshageH.Metforminprotectsrathepatocytesagainstbile
acid-inducedapoptosis.PLoSOne2013;8:e71773.
43.TheryC,AmigorenaS,RaposoG,ClaytonA. Isolationandcharacterizationofexosomesfromcellculturesupernatantsandbiological
fluids.CurrProtocCellBiol2006;Chapter3:Unit322.
44.PizarroM,SolisN,QuinteroP,BarreraF,CabreraD,Rojas-deSantiagoP,ArabJP,etal.Beneficialeffectsofmineralocorticoidreceptor
blockadeinexperimentalnon-alcoholicsteatohepatitis.LiverInt2015;35:2129-2138.
45.SuzukiE,MatsunagaT,AonumaA,SasakiT,NagataK,OhmoriS.Effectsofhypoxia-induciblefactor-1alphachemicalstabilizer,CoCl(2)
andhypoxiaongeneexpressionofCYP3Asinhumanfetallivercells.DrugMetabPharmacokinet2012;27:398-404.
46.WuD,YotndaP.Inductionandtestingofhypoxiaincellculture.JVisExp2011.
47.SunQ,GaoW,LoughranP,ShapiroR,FanJ,BilliarTR,ScottMJ.Caspase1activationisprotectiveagainsthepatocytecelldeathbyup-
regulatingbeclin1proteinandmitochondrialautophagyinthesettingofredoxstress.JBiolChem2013;288:15947-15958.
48.EgnatchikRA,LeamyAK,NoguchiY,ShiotaM,YoungJD.Palmitate-inducedactivationofmitochondrialmetabolismpromotesoxidative
stressandapoptosisinH4IIEC3rathepatocytes.Metabolism2014;63:283-295.
49.Garcia-RuizC,Fernandez-ChecaJC.MitochondrialOxidativeStressandAntioxidantsBalance inFattyLiverDisease.HepatolCommun
2018;2:1425-1439.
50.MasaroneM,RosatoV,DallioM,GravinaAG,AglittiA,LoguercioC,FedericoA,etal.RoleofOxidativeStress inPathophysiologyof
NonalcoholicFattyLiverDisease.OxidMedCellLongev2018;2018:9547613.
51.HeY,SadahiroT,NohSI,WangH,TodoT,ChaiNN,KleinAS,etal.Flowcytometric isolationandphenotypiccharacterizationoftwo
subsetsofED2(+)(CD163)hepaticmacrophagesinrats.HepatolRes2009;39:1208-1218.
52.YounossiZM.Non-alcoholicfattyliverdisease-Aglobalpublichealthperspective.JHepatol2019;70:531-544.
53. Mesarwi OA, Shin MK, Bevans-Fonti S, Schlesinger C, Shaw J, Polotsky VY. Hepatocyte Hypoxia Inducible Factor-1 Mediates the
DevelopmentofLiverFibrosisinaMouseModelofNonalcoholicFattyLiverDisease.PLoSOne2016;11:e0168572.
54. Chen J, Chen J, FuH, Li Y,Wang L, Luo S, LuH.Hypoxia exacerbates nonalcoholic fatty liver disease via theHIF-2alpha/PPARalpha
pathway.AmJPhysiolEndocrinolMetab2019;317:E710-E722.
55. Kong D, Zhang F, Shao J,Wu L, Zhang X, Chen L, Lu Y, et al. Curcumin inhibits cobalt chloride-induced epithelial-to-mesenchymal
transitionassociatedwithinterferencewithTGF-beta/Smadsignalinginhepatocytes.LabInvest2015;95:1234-1245.
56.RicchiM,OdoardiMR,CarulliL,AnzivinoC,BallestriS,PinettiA,FantoniLI,etal.Differentialeffectofoleicandpalmiticacidonlipid
accumulationandapoptosisinculturedhepatocytes.JGastroenterolHepatol2009;24:830-840.
57.MylonisI,SimosG,ParaskevaE.Hypoxia-InducibleFactorsandtheRegulationofLipidMetabolism.Cells2019;8.
58. Mylonis I, Sembongi H, Befani C, Liakos P, Siniossoglou S, Simos G. Hypoxia causes triglyceride accumulation by HIF-1-mediated
stimulationoflipin1expression.JCellSci2012;125:3485-3493.
59.JahnD,KircherS,HermannsHM,GeierA.AnimalmodelsofNAFLDfromahepatologist'spointofview.BiochimBiophysActaMolBasis
Dis2019;1865:943-953.
60.MoravcovaA,CervinkovaZ,KuceraO,MezeraV,RychtrmocD,LotkovaH.Theeffectofoleicandpalmiticacidoninductionofsteatosis
andcytotoxicityonrathepatocytesinprimaryculture.PhysiolRes2015;64Suppl5:S627-636.

60
61.WuJ,LinS,WanB,VelaniB,ZhuY.PyroptosisinLiverDisease:NewInsightsintoDiseaseMechanisms.AgingDis2019;10:1094-1108.
62.MariM,CaballeroF,ColellA,MoralesA,CaballeriaJ,FernandezA,EnrichC,etal.Mitochondrialfreecholesterolloadingsensitizesto
TNF-andFas-mediatedsteatohepatitis.CellMetab2006;4:185-198.
63. Dixon LJ, Flask CA, Papouchado BG, Feldstein AE, Nagy LE. Caspase-1 as a central regulator of high fat diet-induced non-alcoholic
steatohepatitis.PLoSOne2013;8:e56100.
64. TangY, CaoG,MinX,WangT, Sun S,DuX, ZhangW.CathepsinB inhibitionameliorates thenon-alcoholic steatohepatitis through
suppressingcaspase-1activation.JPhysiolBiochem2018;74:503-510.
65.PanchanathanR,LiuH,ChoubeyD.HypoxiaprimeshumannormalprostateepithelialcellsandcancercelllinesfortheNLRP3andAIM2
inflammasomeactivation.Oncotarget2016;7:28183-28194.
66.GuillotA,TackeF.LiverMacrophages:OldDogmasandNewInsights.HepatolCommun2019;3:730-743.
67. Momen-Heravi F, Saha B, Kodys K, Catalano D, Satishchandran A, Szabo G. Increased number of circulating exosomes and their
microRNAcargosarepotentialnovelbiomarkersinalcoholichepatitis.JTranslMed2015;13:261.
68.WuX,ChangSC,JinJ,GuW,LiS.NLRP3inflammasomemediateschronicintermittenthypoxia-inducedrenalinjuryimplicationofthe
microRNA-155/FOXO3asignalingpathway.JCellPhysiol2018;233:9404-9415.
69.AlegreF,PelegrinP,FeldsteinAE.InflammasomesinLiverFibrosis.SeminLiverDis2017;37:119-127.
70.WeiQ,MuK,LiT,ZhangY,YangZ,JiaX,ZhaoW,etal.DeregulationoftheNLRP3inflammasomeinhepaticparenchymalcellsduring
livercancerprogression.LabInvest2014;94:52-62.

61
Chapter4
EXTRACELLULARVESICLESDERIVEDFROMFATLADENHYPOXICHEPATOCYTESPROMOTEAPRO-
FIBROTICPHENOTYPEINSTELLATECELLS
ABSTRACT
Background:Transitionfromsteatosistonon-alcoholicsteatohepatitis(NASH)isakeyissueinNon-
alcoholicfattyliverdisease(NAFLD).Observationsinpatientswithobstructivesleepapneasyndrome
(OSAS),suggestthathypoxiacontributestoprogressiontoNASHandliverfibrosisandthereleaseof
extracellularvesicles(EV)byinjuredhepatocyteshasbeenimplicatedinNAFLDprogression.Aim:to
evaluatetheeffectsofhypoxiaonpro-fibroticresponseandthereleaseofEVinNAFLDandtoassess
cellularcrosstalkbetweenhepatocytesandhumanhepaticstellatecells(LX-2).Methods:HepG2cells
were treated with free fatty acids and subjected to chemically-induced hypoxia (CH) using the
hypoxia-inducible factor-1alpha (HIF-1α) stabilizer cobalt chloride (CoCl2). Lipid droplets, oxidative
stress, apoptosis and pro-fibrotic-associated genes were assessed. EV were isolated by
ultracentrifugation.LX-2cellsweretreatedwithEVfromhepatocytesandpro-fibrogenic-associated
markers were determined. CDAA-fed mice model was used to assess the effects of intermittent
hypoxia (IH) on experimental NASH.Results: CH-treatment in fat-laden HepG2 cells increased the
steatosis, oxidative stress, apoptosis, pro-fibrotic gene expression and increased the release of EV
compared to non-treated HepG2 cells. Treatment of LX2 cells with EV from fat-laden hypoxic
hepatocytesincreasedpro-fibroticmarkerscomparedtoEVfromnon-treatedhepatocytes.CDAA-fed
animalsexposedtoIHexhibitedincreasedfibrosisthatcorrelatedwithanincreaseofcirculatingEV.
Conclusion:Hypoxiapromoteshepatocellulardamageandpro-fibroticsignalingthatcorrelateswith
increasesofEVreleasefromhepatocytesandfromour invivomodelofNASH.EVfromhypoxicfat-
ladenhepatocytesevokepro-fibroticresponsesinLX-2cells.
1.-INTRODUCTION
Non-alcoholicfatty liverdisease(NAFLD)iscurrentlythemostcommonliverdiseaseandisamajor
globalhealthproblem(1).NAFLDischaracterizedbyfataccumulationintheliverwhichmayprogress
to hepatitis, cirrhosis and liver-relatedmorbidity andmortality (2). Recent evidence suggests that
saturated fatty acids (FFAs) contribute to the phenomenon of lipotoxicity and can trigger
inflammation in the liver and an abnormal wound-healing response or fibrosis leading to the
developmentofnon-alcoholicsteatohepatitis(NASH)(3-5).
ItisstillnotclearwhysomepatientswithfattyliverdevelopadvancedstagesofNASHmorerapidly
andseverelythanothers.ClinicalobservationsinpatientswhosufferfromObstructiveSleepApnea

62
syndrome (OSAS) have revealed that these patients have a higher risk of developingmore severe
NAFLD associated with significant liver damage (6,7). The pathophysiology of this condition is
characterizedbyintermittentairwayobstructionthataltersgasexchangeleadingtoperiodichypoxia
(8). Several studies have demonstrated that hypoxia inducesmetabolic alterations such as insulin
resistance, increased oxidative stress, increased liver triglyceride accumulation and increased
inflammation,hepatocellulardamageandfibrogenesis(9-14).
During hypoxia, the transcription factor hypoxia inducible factor 1 alpha (HIF-1α) is stabilized and
translocated to the nucleus to activate its target genes bymeans of binding ofHIF-1α toHypoxia
ResponsiveElements (HREs) located in the target genepromoters (15). These target genesmodify
hepatocytelipidmetabolismandalsoenergymetabolism,cellsurvival,inflammationandfibrosis(16-
18).InrodentmodelsofNASH,intermittenthypoxiawasshowntohaveapro-inflammatoryandpro-
fibroticeffectas indicatedby increased levelsofNF-κB-dependent inflammatorycytokines,suchas
TNF-α,IL-6andIL-1β,andanincreasedexpressionofcollagentypeIintheliver(20,21).Likewise,in
vitro studies using hepatocytes and stellate cells showed that HIF-1α canmodulate pro-fibrogenic
signaling(17,21-23),whichcouldbekeyforthedevelopmentandprogressionofNASH.
Theinvolvementofextracellularvesicles(EV)intheprogressionofNAFLDhasbeenstudiedrecently
in invitroand invivomodels (24-27).EVcorrespondtosmallparticles releasedafter the fusionof
multivesicularbodiesordirectlyfromthecellmembranewithasizeof50to150nm(28).EVplayan
important role in cellular communication innormalphysiological andpathophysiological situations
duetotheircontentofproteins,mRNAsand/orlipids(29).Thereareenoughevidencethatindicate
theinvolvementofEVinNASH(30,31),buttheirroleinthecontextofhypoxiaandeffectsonnon-
parenchymal cells, notably stellate cells, the principal cell type involved in matrix deposition in
fibrogenesis,remainstobeelucidated.
Therefore, the aim of this study was to test the hypothesis that hypoxia leads to hepatocellular
damagethatinvolvesreleaseofEVthathaveapro-fibroticeffectonstellatecells.Weusedboth in
vitroandinvivomodeltotestthishypothesis.
2.-MATERIALSANDMETHODS
2.1Animals
Animalexperimentswereapprovedbythe institutionalanimalcareandusecommittee(Comitéde
ética y bienestar Animal, Escuela de Medicina, Pontificia Universidad Católica de Chile, CEBA
100623003).MaleC57BL/6miceaged10weeksatthebeginningofthestudyanddividedintofour
experimental groups (n = 4-8) receiving either choline-deficient amino acid-defined (CDAA) diet

63
(Catalog#518753,DyetsInc.Bethlehem,PA)toinduceNASHorthecholine-supplementedL-amino
acid defined (CSAA, Catalog # 518754, Dyets Inc. Bethlehem, PA) diet as control for 22 weeks as
previouslydescribed(32,33).AnimalswereexposedtoIHornormoxia(chambers41x22x35cm,COY
labproducts™,GrassLake,MI,USA)duringthelast12weeksoftheexperimentalorcontrolfeeding
period.IHregimenconsistedof30events/hourofhypoxicexposuresfor8hour/dayduringtherest
cycle, between9 amand5pm. This cyclewas repeated7days aweek for 12 consecutiveweeks.
After ending the study, mice were anesthetized (ketamine 60 mg/kg plus xylazine 10 mg/kg
intraperitoneally) and then euthanized by exsanguination. Serum and liver tissue samples were
collectedandprocessedorstoredat−80 °Cuntilanalyzed.
2.2Histologicalstudies
Liver fibrosis was analyzed on paraformaldehyde-fixed liver sections stained with Sirius Red on
frozen7 μmlivercryosections. Stainingwasquantitatedbydigital imageanalysis(ImageJ,NIH,US)
aspreviouslydescribed(32).
2.3Cellcultureandtreatmentwithfreefattyacidsandchemicalhypoxiainduction
The human hepatocellular carcinoma cell line HepG2 (ATCC, USA) was cultured in Dulbecco’s
modifiedEaglemedium(1X) + GlutaMAX™-I(DMEM,10569010,Gibco)supplementedwith10%fetal
bovineserum(FBS;Gibco)and1%penicillin-streptomycin(Gibco)at37°Cand5%CO2.Allcellswere
platedinacellcultureplateatleast24h-36hbeforetreatment.Uponreaching80%confluence,the
cellswereincubatedwithamixtureoffreefattyacids(FFA)consistingofoleicacid(500μmol/L)and
palmiticacid(250μmol/L)inanaqueoussolutionofbovineserumalbumin(BSA)asdescribed(34).
IncubationswerecarriedoutwithorwithoutCobalt(II)chloride(CoCl2;SigmaAldrich)(200μmol/L)
for 24 hours to induce chemical hypoxia (35). CoCl2 is a well-known hypoxia-mimetic agent by
stabilizationofhypoxia-induciblefactor(HIF)-1α(36).ControlcellsweretreatedwithBSAalone.
2.4Westernblotanalyses
Cell lysateswere resolved onMini-PROTEAN® TGX Stain-Free™ Precast Gels (BioRad, UK, Oxford).
Semidry-blottingwasperformedusingTrans-BlotTurboMidiNitrocelluloseMembranewithTrans-
Blot Turbo System Transfer (BioRad). Ponceau S 0.1% w/v (Sigma) staining was used to confirm
protein transfer. Anti-HIF1α (610958, Biosciences), anti-CASP1 (SC-56036, Santa Cruz), anti-CD81
(10630D,Invitrogen),anti-Bcl-2(ab32370,Abcam),anti-TypeICollagen(1310-01;SouthernBiotech)
and anti-αSMA (A5228, Sigma)were used in combinationwith appropriate peroxidase-conjugated
secondaryantibodies.Tubulin(T9026,Sigma)oractin(A5228,Sigma)wereusedasloadingcontrols.

64
The blots were analyzed in a ChemiDoc XRS system (Bio-Rad). Protein band intensities were
quantifiedbyImageLabsoftware(BioRad).
2.5Nileredstaining
IntracellularlipiddropletsintheHepG2cellsweredetectedwithNileRedfluorescentprobe(N1142,
ThermoFisher).HepG2cellsweregrownin96-wellplatesandtreatedwithFFAand/orCoCl2.After
treatment,thecellswerewashedtwicewithPBSandstainedwithNileRedsolutionfor10 mininthe
dark. The cells were then washed twice with PBS and stained with Hoechst dye (33342, Thermo
Fisher).Thefluorescenceintensityofeachwellwasanalyzedwithexcitation/emissionwavelengthat
488nm/550nmusingamicroplatereader.Thefluorescenceimageswererecordedusinganinverted
fluorescencemicroscope
2.6Determinationofreactiveoxygenspecies
The intracellular generation of reactive oxygen species (ROS) in HepG2 cells was monitored with
DCFH-DAfluorescentprobe.HepG2cellsweregrownin96-wellplatesandtreatedwithFFAand/or
CoCl2. Cells were washed twice with PBS and incubated with the cell permeable reagent 2’,7’–
dichlorofluorescindiacetate(DCFDA;Abcam,USA)for45min.CellswerethenwashedtwicewithPBS
andthefluorescenceintensityofeachwellwasanalyzedwithexcitation/emissionwavelengthat495
nm/529nmusingamicroplatereader.
2.7Apoptosismeasurement
Caspase-3 fluorometric assay was used to determine apoptosis induced by FFA and/or CoCl2 in
HepG2cells.Aftertreatment,HepG2werescrapedandcelllysateswereobtainedbythreecyclesof
freezing(−80°C)andthawing(37°C)followedbycentrifugationfor5minutesat13,000g.Caspase-3
enzymeactivitywasassayedasdescribedpreviously (37).Thearbitraryunitsof fluorescence(AUF)
was quantified in a spectrofluorometer at an excitation wavelength of 380 nm and an emission
wavelengthof430nm.
2.8Assessmentofcelldeathassociatedtodisruptedcellularmembraneintegrity
SYTOX®Greennucleicacidstain(Invitrogen,S7020)wasusedtodeterminecelldeathinducedbyFFA
and/or CoCl2 in HepG2 (38). Cells were cultured in 12-well plates. After treatment, diluted Sytox
Green solution (1:40.000/PBS) was added to the cells for at least 15 minutes at 37°C, 5% CO2.
Necrotic cells have ruptured plasma membranes, allowing entrance of non-cell permeable Sytox
Greenintothecellsandbindingtonucleicacids.NecrosiswasdeterminedusingaLeicafluorescence
microscopeatawavelengthof488nm.

65
2.9RNAisolationandquantitativereal-timereversetranscriptionpolymerasechainreaction(qRT-
PCR)
HepG2cellswereharvestedoniceandwashedtwicewithice-coldPBS.TotalRNAwasisolatedwith
TRI-reagent (Sigma) according to the manufacturer’s instructions. Reverse transcription (RT) was
performedusing2.5 µgoftotalRNA,1XRTbuffer(500 mmol/lTris-HCl[pH8.3];500 mmol/lKCl;30
mmol/l MgCl2; 50 mmol/l DTT), 1 mmol/l deoxynucleotides triphosphate (dNTPs, Sigma), 10 ng/µl
random nanomers (Sigma), 0.6 U/µl RNaseOUT™ (Invitrogen) and 4 U/µl M-MLV reverse
transcriptase(Invitrogen) inafinalvolumeof50 µl.ThecDNAsynthesisprogramwas25°C/10min,
37°C/60minand95°C/5min.ComplementaryDNA(cDNA)wasdiluted20X innuclease-freewater.
Real-Time qPCR was carried out in a StepOnePlus™ (96-well) PCR System (Applied Biosystems,
Thermofisher)usingTaqManprobes.Thesequencesoftheprobesandprimersetsaredescribedin
Supplementary Material. For qPCR, 2X reaction buffer (dNTPs, HotGoldStar DNA polymerase, 5
mmol/LMgCl2) (Eurogentec,Belgium,Seraing),5µmol/Lfluorogenicprobeand50µmol/Lofsense
and antisense primers (Invitrogen)were used.mRNA levelswere normalized to the housekeeping
gene18Sandfurthernormalizedtothemeanexpressionlevelofthecontrolgroup.
2.10Extracellularvesiclesisolation
ForEVcollection,HepG2cellsweregrowninculturedishesof100mmtoobtain70mlofserum-free
conditionedmedium after different treatments for an additional 24 hours. EVwere isolated from
conditionedmediumbydifferential ultracentrifugation (UCFThermo-Sorvall 80wx+) according to a
modifiedpreviousprotocol(39).Atotalvolumeof70mlmediumpertreatmentwasdepletedofcells
andcelldebrisbyconsecutive,low-speedcentrifugations(2,000×gfor30minand12,000×gfor45
min).Theresultingsupernatantswerecarefullycollectedandcentrifugedfor70minat120,000×gat
4°C.PelletsfromthiscentrifugationstepwerewashedinPBS,pooledandcentrifugedagainfor60
minat 100,000× g at 4°C. For EV collection fromanimals, serumsampleswere reconstituted in a
totalvolumeof4.4mLandwerecentrifugatedat2,000×gfor30minand10,000×gfor30min.The
resulting supernatants were carefully collected and centrifuged for 70min at 120,000 × g at 4°C.
PelletsfromthiscentrifugationstepwerewashedinPBS,pooledandcentrifugedagainfor60minat
100,000×gat4°C.TheobtainedpelletsfromHepG2cellsorserumwereresuspendedinlysisbuffer
or PBS solution depending on subsequent experiments and stored in aliquots at -80 °C. Protein
concentrationinEVpelletweremeasuredusingBCAproteinassaykit(Pierce,Rockford,IL).
2.11Nanoparticletrackinganalysis
Concentration and size distribution of isolated EVwere assessed by nanoparticle tracking analysis
(NTA)usingNanoSightNS300 instrumentation (Marvel, Egham,UK). EV samplesweredilutedwith

66
PBS to a concentration of 108 to 109 particles/mL in a total volume of 1 ml. Each sample was
continuously run througha flow-cell top-plate setupat 18°Cusinga syringepump.At least three
videos of 20 seconds documenting Brownianmotion of nanoparticles were recorded and at least
1000ofcompletedtrackswereanalyzedbyNanoSightsoftware(NTAv3.2).
2.12Transmissionelectronmicroscopy
Isolated EV were fixed in 2% paraformaldehyde in 0.1M phosphate buffer overnight at 4°C. The
sampleswerethenplacedonFormvar/Carbon300Mesh(FCF300-CU,EMS)gridandairdriedfor10
min.Thegridswerefirstcontrastedwithuranyl-oxalatesolutionandthencontrastedandembedded
in amixtureof 4%uranyl acetate. ThegridswereairdriedandvisualizedwithaPhilips Tecnai 12
(Biotwin,Eindhoven,Netherlands)electronmicroscopeat80kV.Theimageswerecapturedwiththe
softwareItemOlympusSoftImagingSolutions(WindowsNT6.1).
2.13TreatmentofLX-2cellswithextracellularvesicles
ToinvestigatethecrosstalkbetweenEVfromHepG2andLX-2humanhepaticstellatecells,atypical
cell linetostudyhepaticfibrogenesis(40),LX-2cellsweretreatedwith isolatedEV.LX-2cellswere
culturedinDMEMsupplementedwith10%FBSand1%penicillin-streptomycinat37°Cand5%CO2.
All cellswere plated in a cell culture plate at least 24-36 h before treatment.Upon reaching 80%
confluence, thecellswere incubatedwithFBS-freemediumandexposed to15μgofEV thatwere
isolatedfromHepG2cellsthathadbeentreatedwithCoCl2,FFAandCoCl2+FFAfor24h.After24hof
EVtreatment,LX-2cellswereharvestedtocontinuewithanalysesbyquantitativePCR,WesternBlot
andimmunofluorescence.
2.14Immunofluorescencemicroscopy
LX-2 cellsweregrownonglass cover slipsplaced in12-wellplates.After treatment, culturemedia
wereremovedandcoverslipswerecarefullywashedtwicewithPBS.Cellswerethenfixedusinga4%
paraformaldehyde solution in PBS for 10 min at room temperature and washed twice with PBS.
Permeabilizationwasperformedby incubationof thesamples for10minutes in0.1%TritonX-100
(Sigma). The cells were washed twice with PBS and incubated with 2% BSA (Sigma) in PBS/0.1%
Tween 20 (Sigma) solution for 30 minutes to block non-specific binding sites. Goat anti-Type I
Collagen(1310-01;SouthernBiotech)wasusedatadilutionof1:200in2%BSA/PBSinahumidified
chamber for 1 hour at room temperature. Samples were subsequently washed twice with 2%
BSA/PBS.Finally,cellswereincubatedwithrabbitanti-goatAlexaFluor®568atadilutionof1:500in
2%BSA/PBSfor30minatroomtemperatureinthedark.SlidesweremountedwithProLongantifade

67
with DAPI (Molecular Probes) and images were evaluated using fluorescence microscopy and
analyzedbyLeicaALSAFSoftware(Leica).
2.15Statisticalanalyses
AnalyseswereperformedusingGraphPadsoftware(version5.03,GraphPadSoftwareInc.,CA,USA).
All results are presented as a mean of at least 3 independent experiments ± SEM or as absolute
number or percentage for categorical variables. The statistical significance of differences between
themeansof theexperimental groupswas evaluatedusingone-wayanalysis of variance (ANOVA)
with a post-hoc Bonferroni multiple-comparison test; P<0.05 was considered as statistically
significant.
3.-RESULTS
3.1 Chemical hypoxia increases lipid droplet content in fat laden hepatocytes: To investigate
whether hypoxia exacerbates lipid accumulation in an in vitro model of experimental NASH, we
treatedHepG2cellswithCoCl2,ahypoxiamimeticagentthatpromotestheaccumulationofHIF-1α.
HepG2 cells treatedwith CoCl2 showed a significant increase ofHIF-1α, independent of treatment
withFFA (Figure1).To inducesteatosis,HepG2cellsweretreatedwithamixtureofoleicacidand
palmitic acid (FFA) as described previously (41). Hepatocytes showed increased formation of lipid
droplets(Figure2a).Interestingly,asignificantincreaseinthecontentoflipiddropletswasobserved
inthesteatotichepatocytestreatedwiththechemicalinducerofhypoxia(Figure2b),indicatingthat
hypoxiaincreasessteatosisinthismodel.
Figure1.ChemicalhypoxiastabilizesHIF-1α.HepG2cellswereincubatedfor24hwitholeicacidandpalmitic
acid2:1 (FFA) in thepresenceorabsenceofCoCl2200μmol/L.Protein levelsofHIF-1αweredeterminedby

68
WesternBlotasdescribedinMaterialandMethods.α-Tubulinwasusedasloadingcontrol.Datawereshown
asmean±SEM(n≥3)****indicatesP<0.001.
Figure2.Hypoxia aggravates FFA-induced steatosis in vitro. HepG2 cellswere incubated for 24hwitholeic
acidandpalmitic acid2:1 (FFA) in thepresenceor absenceofCoCl2200μmol/L. Lipiddroplets contentwas
measuredby a) Fluorescence intensity andb) Fluorescence imagesofNileRedasdescribed inMaterial and
Methods.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05;****indicatesP<0.001.
3.2 Hypoxia increases oxidative stress and apoptotic cell death in fat laden hepatocytes: To
evaluatewhether the inductionofhypoxiaexacerbatesdamageby lipotoxicity,wedeterminedthe
generationof reactiveoxygen species (ROS)and theextentof celldeath.The increase inROSwas
evaluatedusingthefluorogenicdyeDCF.TheresultsindicatedasignificantincreaseinROSineachof
thetreatmentscomparedtothecontrol(Figure3).Furthermore,hypoxiaexacerbatestheincreasein
ROSinsteatoticHepG2cellscomparedtothehypoxicandFFAconditionsseparately.
Apoptoticcelldeathwasevaluatedbymeasuringtheactivityofcaspase-3.Theresultsdemonstrate
thathypoxiainsteatoticHepG2cellsinducesapoptoticlipotoxicitycomparedtocontrolcells.(Figure
4a).Hypoxiaorsteatosisalonedidnotinduceapoptoticcelldeath.Lipotoxicitywasalsoevaluatedby
Sytox Green assay to detect cell death associated to disrupted cellular membrane integrity. The
differenttreatmentsinHepG2cellsdidnotinduceanynecroticcelldeath(Figure4b).

69
Figure3.Hypoxia increasestheproductionofreactiveoxygenspecies insteatoticHepG2cells.HepG2cells
were incubated for 24hwith oleic acid and palmitic acid 2:1 (FFA) in the presence or absence of CoCl2 200
μmol/L. Reactive oxygen species (ROS) were measured as described in Material and Methods. Data were
shownasmean±SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01;***indicatesP<0.005;****indicates
P<0.001.
Figure4.Hypoxiaandsteatosisinvitropromoteapoptoticcelldeathbutdonotpromotenecroticcelldeath.
HepG2cellswere incubatedfor24hwitholeicacidandpalmiticacid2:1(FFA) inthepresenceorabsenceof
CoCl2200μmol/L.Caspase-3activityandnecrosisweremeasuredasdescribedinMaterialandMethods.Data
wereshownasmean±SEM(n≥3)*indicatesP<0.05
3.3Hypoxiainducestheexpressionofpro-fibroticcytokines:Toevaluatewhetherhypoxiapromotes
a profibrotic phenotype in fat laden HepG2 cells, we measured the mRNA levels of different
profibrotic cytokines.Wedemonstrate that chemical hypoxia in fat ladenhepatocytes significantly
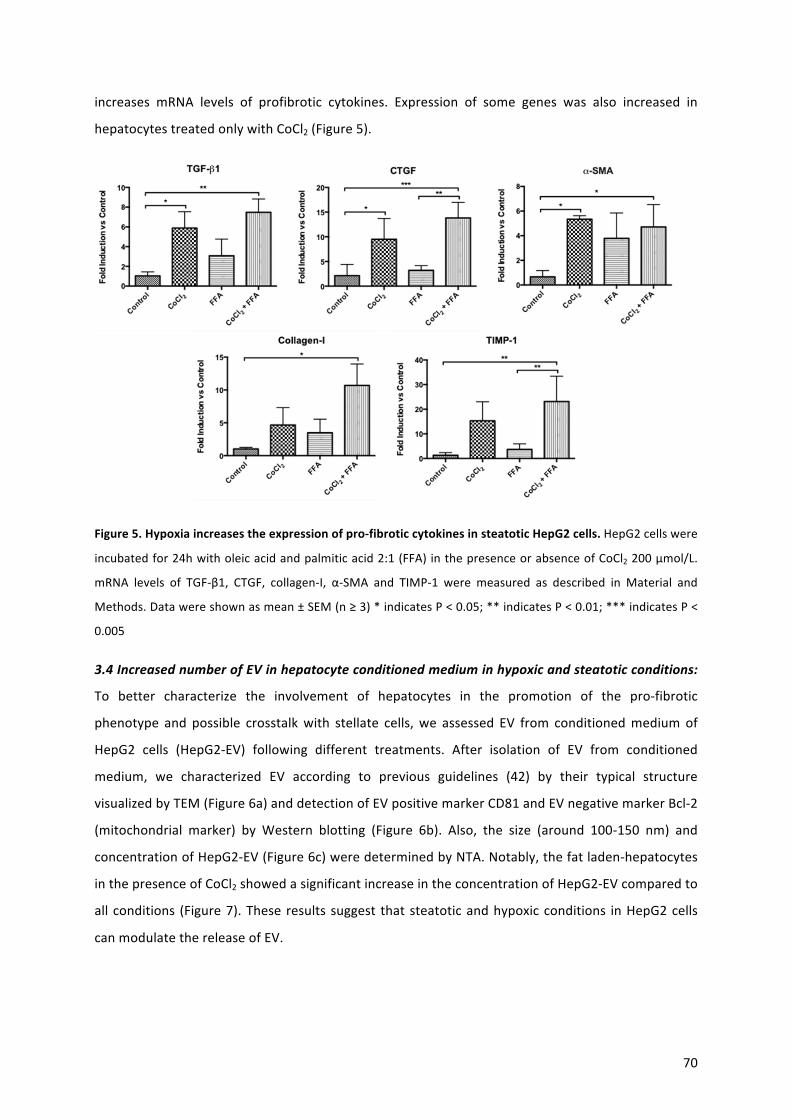
70
increases mRNA levels of profibrotic cytokines. Expression of some genes was also increased in
hepatocytestreatedonlywithCoCl2(Figure5).
Figure5.Hypoxiaincreasestheexpressionofpro-fibroticcytokinesinsteatoticHepG2cells.HepG2cellswere
incubatedfor24hwitholeicacidandpalmiticacid2:1(FFA)inthepresenceorabsenceofCoCl2200μmol/L.
mRNA levels of TGF-β1, CTGF, collagen-I, α-SMA and TIMP-1 were measured as described in Material and
Methods.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01;***indicatesP<
0.005
3.4IncreasednumberofEVinhepatocyteconditionedmediuminhypoxicandsteatoticconditions:
To better characterize the involvement of hepatocytes in the promotion of the pro-fibrotic
phenotype and possible crosstalkwith stellate cells,we assessed EV from conditionedmediumof
HepG2 cells (HepG2-EV) following different treatments. After isolation of EV from conditioned
medium, we characterized EV according to previous guidelines (42) by their typical structure
visualizedbyTEM(Figure6a)anddetectionofEVpositivemarkerCD81andEVnegativemarkerBcl-2
(mitochondrial marker) by Western blotting (Figure 6b). Also, the size (around 100-150 nm) and
concentrationofHepG2-EV(Figure6c)weredeterminedbyNTA.Notably,thefatladen-hepatocytes
inthepresenceofCoCl2showedasignificantincreaseintheconcentrationofHepG2-EVcomparedto
all conditions (Figure7).These results suggest thatsteatoticandhypoxicconditions inHepG2cells
canmodulatethereleaseofEV.
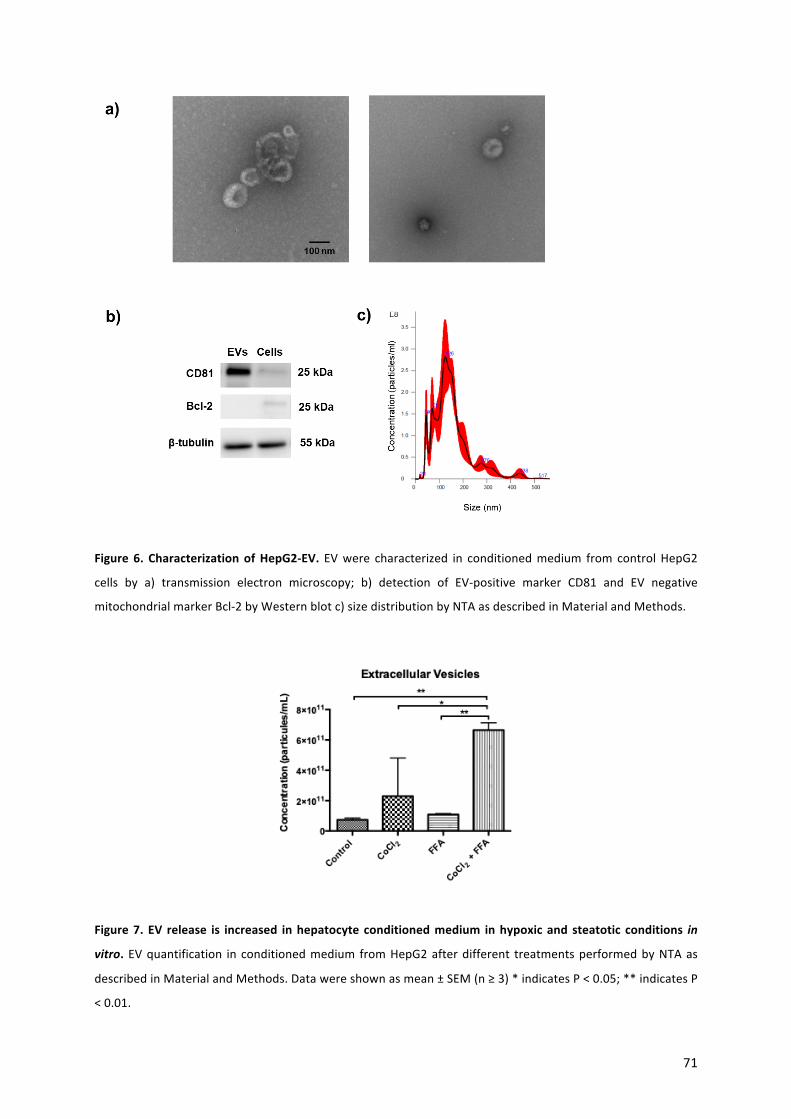
71
Figure 6. CharacterizationofHepG2-EV. EVwere characterized in conditionedmedium from controlHepG2
cells by a) transmission electron microscopy; b) detection of EV-positive marker CD81 and EV negative
mitochondrialmarkerBcl-2byWesternblotc)sizedistributionbyNTAasdescribedinMaterialandMethods.
Figure7. EV release is increased inhepatocyte conditionedmedium inhypoxicand steatotic conditions in
vitro. EVquantification in conditionedmedium fromHepG2afterdifferent treatmentsperformedbyNTAas
describedinMaterialandMethods.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05;**indicatesP
<0.01.
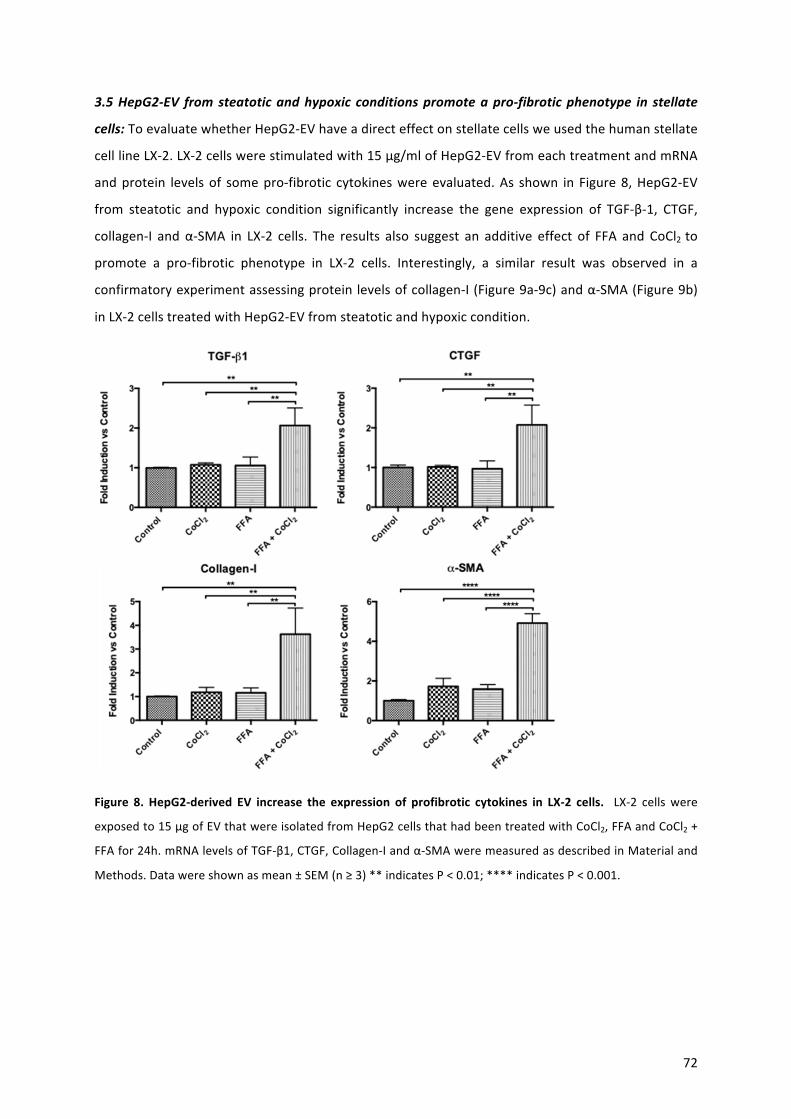
72
3.5HepG2-EV fromsteatoticandhypoxic conditionspromoteapro-fibroticphenotype in stellate
cells:ToevaluatewhetherHepG2-EVhaveadirecteffectonstellatecellsweusedthehumanstellate
celllineLX-2.LX-2cellswerestimulatedwith15μg/mlofHepG2-EVfromeachtreatmentandmRNA
andprotein levelsof somepro-fibrotic cytokineswereevaluated.As shown in Figure8,HepG2-EV
from steatotic and hypoxic condition significantly increase the gene expression of TGF-β-1, CTGF,
collagen-I and α-SMA in LX-2 cells. The results also suggest an additive effect of FFA and CoCl2 to
promote a pro-fibrotic phenotype in LX-2 cells. Interestingly, a similar result was observed in a
confirmatoryexperimentassessingproteinlevelsofcollagen-I(Figure9a-9c)andα-SMA(Figure9b)
inLX-2cellstreatedwithHepG2-EVfromsteatoticandhypoxiccondition.
Figure 8. HepG2-derived EV increase the expression of profibrotic cytokines in LX-2 cells. LX-2 cellswere
exposedto15μgofEVthatwereisolatedfromHepG2cellsthathadbeentreatedwithCoCl2,FFAandCoCl2+
FFAfor24h.mRNAlevelsofTGF-β1,CTGF,Collagen-Iandα-SMAweremeasuredasdescribedinMaterialand
Methods.Datawereshownasmean±SEM(n≥3)**indicatesP<0.01;****indicatesP<0.001.

73
a) b)
c)
Figure9.EVderivedfromsteatoticandhypoxicHepG2cellsincreasestheexpressionofprofibroticproteins
in stellate cells. LX-2 cellswereexposed to15μgof EV thatwere isolated fromHepG2 cells that hadbeen
treatedwithCoCl2,FFAandCoCl2+FFAfor24h.Proteinlevelsofa)Collagen-Iandb)α-SMAweredetermined
byWesternblotasdescribed inMaterialsandMethods.β-Actinwasusedas loadingcontrol. c) Intracellular
Collagen-I expressionwas determinedby immunofluorescence as described inMaterials andMethods.Data
wereshownasmean±SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01;***indicatesP<0.005.
.
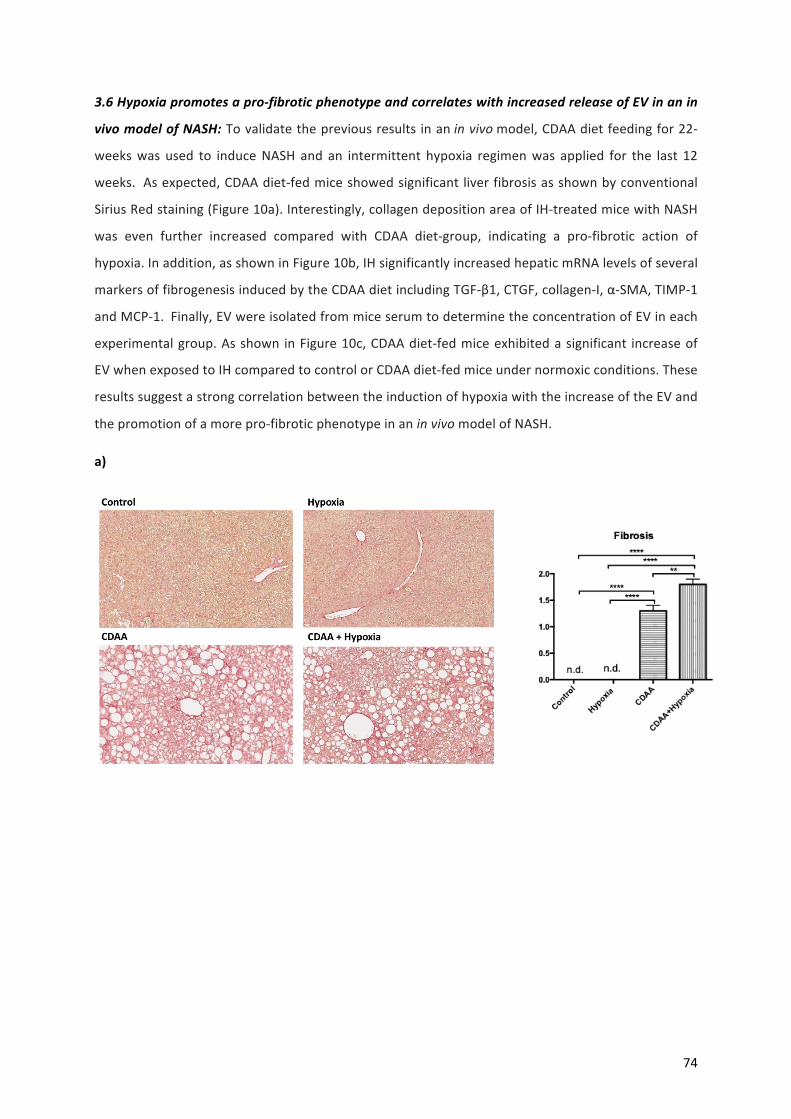
74
3.6Hypoxiapromotesapro-fibroticphenotypeandcorrelateswithincreasedreleaseofEVinanin
vivomodelofNASH:Tovalidatethepreviousresultsinaninvivomodel,CDAAdietfeedingfor22-
weekswas used to induceNASH and an intermittent hypoxia regimenwas applied for the last 12
weeks.Asexpected,CDAAdiet-fedmiceshowedsignificant liverfibrosisasshownbyconventional
SiriusRedstaining(Figure10a).Interestingly,collagendepositionareaofIH-treatedmicewithNASH
was even further increased compared with CDAA diet-group, indicating a pro-fibrotic action of
hypoxia.Inaddition,asshowninFigure10b,IHsignificantlyincreasedhepaticmRNAlevelsofseveral
markersoffibrogenesisinducedbytheCDAAdietincludingTGF-β1,CTGF,collagen-I,α-SMA,TIMP-1
andMCP-1.Finally,EVwereisolatedfrommiceserumtodeterminetheconcentrationofEVineach
experimentalgroup.Asshown inFigure10c,CDAAdiet-fedmiceexhibitedasignificant increaseof
EVwhenexposedtoIHcomparedtocontrolorCDAAdiet-fedmiceundernormoxicconditions.These
resultssuggestastrongcorrelationbetweentheinductionofhypoxiawiththeincreaseoftheEVand
thepromotionofamorepro-fibroticphenotypeinaninvivomodelofNASH.
a)
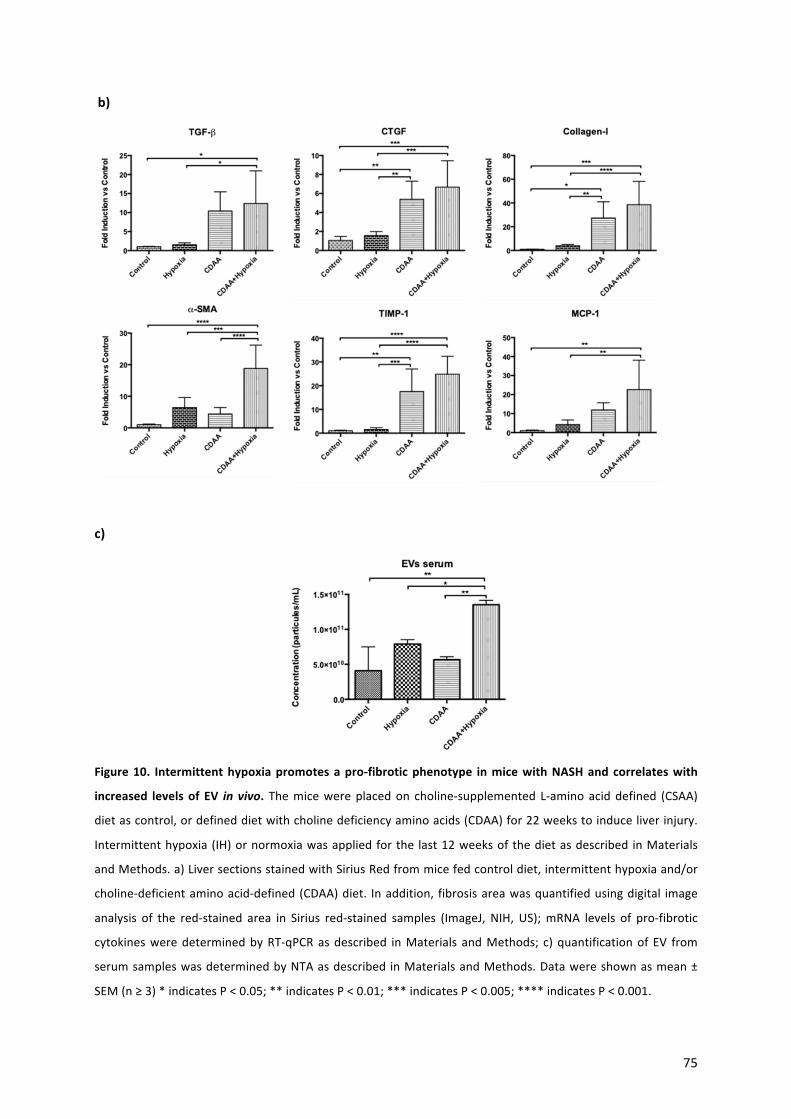
75
b)
c)
Figure10. Intermittenthypoxiapromotesapro-fibroticphenotype inmicewithNASHandcorrelateswith
increased levelsof EV in vivo. Themicewereplacedon choline-supplemented L-aminoaciddefined (CSAA)
dietascontrol,ordefineddietwithcholinedeficiencyaminoacids(CDAA)for22weekstoinduceliverinjury.
Intermittenthypoxia(IH)ornormoxiawasappliedforthe last12weeksofthedietasdescribed inMaterials
andMethods.a)LiversectionsstainedwithSiriusRedfrommicefedcontroldiet,intermittenthypoxiaand/or
choline-deficientaminoacid-defined(CDAA)diet. Inaddition, fibrosisareawasquantifiedusingdigital image
analysis of the red-stained area in Sirius red-stained samples (ImageJ, NIH, US);mRNA levels of pro-fibrotic
cytokinesweredeterminedbyRT-qPCRasdescribed inMaterials andMethods; c)quantificationofEV from
serumsampleswasdeterminedbyNTAasdescribedinMaterialsandMethods.Datawereshownasmean±
SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01;***indicatesP<0.005;****indicatesP<0.001.

76
4.-DISCUSSION
Inrecentyears, themechanismsunderlyingtheprogressionofNAFLD/NASHhavebeenthoroughly
studied. The early stages of NAFLD are characterized by fat accumulation in the liver resulting in
steatosisthatcanprogresstohepatocellulardamageandinflammationinaconditiontermedNASH
(2,5).Moreover,somepatientsdevelopliverfibrosisassociatingwithincreasedmortalityduetorisk
ofHCCdevelopment (43). Interestingly,clinicalstudieshave indicatedthatobstructivesleepapnea
(OSA) isanewpredictivefactorofhepatic fibrosis inpatientswithNAFLD(44-46).Recentresearch
hasprovidedevidenceregardingtheroleofhypoxia,ahallmarkofOSA,inthedevelopmentofliver
injuryandhepaticfibrosisinanimalmodelsofNAFLD(17,19,47).However,therearestillmajorgaps
inourunderstandingoftheprogressionofNASHtofibrosis,e.g.thecellularmechanismunderlying
thecrosstalkbetweenhepatocytesandstellatecells,theprincipalnon-parenchymalcellresponsible
for liver scar formation (48). In this study, we demonstrate that hypoxia, induced by a HIF-1α
chemical stabilizer in fat-laden hepatocytes, promotes hepatocellular damage, increases pro-
fibrogenicgeneexpressionand increasesthereleaseofextracellularvesicles (EV).Furthermore,EV
fromhypoxicandsteatotichepatocytespromotemRNAandproteinexpressionofimportantfibrosis
markersinstellatecells.
Inthepresentstudy,ourinvitromodelofsteatosiswaskeytotheanalysisofhypoxiainducedbythe
hypoxiamimeticCoCl2,aHIF-1stabilizer.Weusedthehumanhepatocytecell lineHepG2, inwhich
theeffectivenessofthehypoxiamimeticwasevaluatedthroughthequantificationofHIF-1αlevels,
which increaseddue to the intracellular stabilization,asdescribed inother invitro studies (49,50).
Weshowedthattreatmentwithpalmiticacid(PA)andoleicacid(OA)(1:2)increasedthecontentof
lipid droplets in hepatocytes in the absence of lipotoxicity. Previous studies have shown that OA
promotes steatosis inhepatocytes, both inprimaryhepatocyte cultureand inhepatomacell lines,
whilePAinducesacytotoxicresponse(51,52).Ourresultsareinlinewithapreviousreportusinga
combinationofboth fattyacids,usingahigherOAconcentration, thatpromotedananti-apoptotic
effect and triglyceride accumulation (51). Interestingly, the hypoxia mimetic CoCl2 increased the
contentoflipiddropletscomparedtoHepG2cellswithouthypoxia,asdeterminedviaquantification
ofNileRedfluorescence.Theseresultsaresupportedbyaninvivostudyindicatingthathypoxia,via
HIF-1α,promotesanincreaseinlipidbiosynthesisinsteatoticconditions(53).Hepatocytecelldeath
by apoptosis was measured using caspase-3 and as expected, treatment with FFA alone did not
inducecelldeath.However,HepG2cellsexposedtothecombinationofCoCl2andFFApresentedan
increaseof caspase-3 activity that correlatedwith an increase inoxidative stress compared to the
controlgroup.Unexpectedly,HepG2cellstreatedwithFFAsalsoincreasedROScomparedtoCoCl2
andcontrolgroups.A recent studydemonstrated thatOApreventsROSproduction inHepG2cells

77
treatedwith PA (54). Likewise, another study showed that FFA treatment of HepG2 cells induced
TNF-α generation, which is an importantmediator in hepatic steatohepatitis and liver injury (55).
Additionalstudiestofurtherdelineatethemechanism,suchasmeasurementofmitochondrialROS
production,remaintobeperformed.Wealsoanalyzedhepatocytecelldeathassociatedtodisrupted
cellular membrane integrity (necrosis) using SytoxGreen®. None of the treatments we applied
induced a significant increase in necrosis. The divergent results of apoptosis and necrosis can be
explainedbythefactthatdifferentstimulimayinduceddifferentmodesofcelldeathindifferentcell
types(56).Takentogether,ourfindingsindicatethathypoxiapromoteslipotoxicityasdeterminedby
theincreaseinhepatocellularapoptosisandoxidativestress.
To evaluate the pro-inflammatory effects of hypoxia in fat-laden hepatocytes, wemeasured gene
expression of pro-fibrotic cytokines. As expected and in accordancewith our previous results, the
conditionofhypoxiainsteatotichepatocytesincreasedtheexpressionofTGF-β1,CTGF,collagen-I,α-
SMAandTIMP-1.Moreover,wecheckedtheamountofEVreleasedintheculturemediumandwe
observed an increase after treatment of HepG2 cells with CoCl2 and FFA compared to all other
groups. EV are small structures surrounded by membrane and released from the cells to the
extracellularenvironment.Theyhaveapathophysiological role in variousdiseases, includingNASH
andotherchronicliverdiseases(30,31,57).WeperformedacharacterizationoftheisolatedEVfrom
HepG2 cells using positive and negativemarkers byWestern blotting and determinate EV size by
NTA.Finally,isolatedEVfromcontrolhepatocytesweredirectlyvisualizedusingTEM.
Previousstudieshavedemonstratedthatduringhepatocellulardamage,hepatocytesreleaseEVthat
modulatepro-inflammatoryandpro-fibroticsignaling innon-parenchymal livercells (25,58-60).To
confirmwhether EV from ourmodel of hypoxic and steatotic hepatocytes promote a pro-fibrotic
phenotypeinnon-parenchymallivercells,weusedthehumanstellatecelllineLX-2.EVderivedfrom
CoCl2-treatedfat-ladenhepatocytesincreasedgeneexpressionofpro-fibroticcytokinessuchasTGF-
β1, CTGF, collagen-I and α-SMA in LX-2 cells compared to all other groups. In addition, protein
expressionofcollagen-Iandα-SMAwasanalyzedinLX-2cellstreatedwithEVfromHepG2cells.Both
pro-fibrotic proteins increased when LX-2 cells were treated with EV derived from CoCl2-treated
steatotichepatocytes.ThisinterestingresultsuggeststhattheEV-cargoinourmodelisanimportant
topicforfutureanalysis.Recentstudiesthatsupportourfindingsshowthatpalmiticacidincreased
EV from hepatocytes and change their miRNA cargo inducing pro-fibrogenic signals to HSCs (60).
Anotherstudy,providingsimilarresults, indicatedthatEVfromlipotoxichepatocytesactivateHSCs
viamiRNA-128-3p(25).

78
The exact relation(s) between intermittent hypoxia, as hallmark of OSA, and NAFLD/NASH
progression,specificallyindevelopmentofliverfibrosis,remainincompletelyunderstood.Wefound
thatintermittenthypoxiapromotesapro-fibroticphenotypeinourCDAAdiet-fedanimalmodeland
increasescirculating levelsofEV.Previousstudieshavedemonstratedthat IHmimickingOSAalters
EVgenerationandreleaseaswellasEV-cargoandfunction,promotingpathophysiologyindifferent
in vivo models (61-63). This important finding correlates with our in vitro results and provide
evidence for a novel pro-fibroticmechanism, involving EV. Next challengewill be identified if the
mostofcirculatingEVarederivedfromliverorfromotherspecificcellsourceandidentifygeneand
proteinscargo,includingmembranesurfacecomposition.
In conclusion hypoxia promoted hepatocellular damage and increased pro-fibrotic genes that
correlates with increases of EV derived from hepatocytes and from our in vivo model of NASH.
Moreover, EV fromhypoxic fat-laden hepatocytes promoted a pro-fibrotic responses in LX-2 cells,
showinganovel cell to cell communication in thismodel. Finally,micemodelofNASHexposed to
intermittenthypoxiapresentedapro-fibroticphenotypethatcorrelatewithincreasedofcirculating
of EV. Futurework should focus on analysis to profile the genomic, transcriptomic, proteomic an
lipidomiccargo intoEV-derived fromcellsandbiological fluids fromrodentsmodelsunderhypoxia
conditionthatcouldbepromotesfibrosisprofileinthesettingofNAFLDandOSAS.
5.-REFERENCES
1.DrescherHK,Weiskirchen S,WeiskirchenR. Current Status in Testing forNonalcoholic Fatty LiverDisease (NAFLD) andNonalcoholic
Steatohepatitis(NASH).Cells2019;8.piiE845.
2.ArabJP,ArreseM,Trauner M.Recent insights intothepathogenesisofnon-alcoholic fatty liverdisease.AnnualReviewofPathology
MechanismsofDisease2018;13:321-350.
3.Trauner,M.;Arrese,M.;Wagner,M.Fattyliverandlipotoxicity.Biochim.Biophys.Acta2010;1801:299-310.
4.HirsovaP,IbrahimSH,GoresGJ,MalhiH.Lipotoxiclethalandsublethalstresssignalinginhepatocytes:relevancetoNASHpathogenesis.J
LipidRes2016;57:1758-1770.
5. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol
2018;15:349-364.
6.AgrawalS,DusejaA,AggarwalA,DasA,MehtaM,DhimanRK,ChawlaY.Obstructivesleepapneaisanimportantpredictorofhepatic
fibrosisinpatientswithnonalcoholicfattyliverdiseaseinatertiarycarecenter.HepatolInt2015;9:283-291.
7.MesarwiOA,LoombaR,MalhotraA.ObstructiveSleepApnea,Hypoxia,andNonalcoholicFattyLiverDisease.AmJRespirCritCareMed.
2019;199:830-841.
8.DempseyJA,VeaseySC,MorganBJ,O'DonnellCP.Pathophysiologyofsleepapnea.PhysiolRev2010;90:47-112.
9.SavranskyV,NanayakkaraA,ViveroA,LiJ,BevansS,SmithPL,TorbensonMS,etal.Chronic intermittenthypoxiapredisposestoliver
injury.Hepatology2007;45:1007-1013.

79
10.SavranskyV,ReinkeC, Jun J,Bevans-FontiS,NanayakkaraA, Li J,MyersAC,etal.Chronic intermittenthypoxiaandacetaminophen
inducesynergisticliverinjuryinmice.ExpPhysiol2009;94:228-239.
11. Drager LF, Lopes HF,Maki-Nunes C, Trombetta IC, Toschi-Dias E, AlvesMJ, Fraga RF, Jun JC, Negrão CE, Krieger EM, Polotsky VY,
Lorenzi-Filho G.The impact of obstructive sleep apnea onmetabolic and inflammatorymarkers in consecutive patients withmetabolic
syndrome.PLoSOne2010;5:e12065.
12.DragerLF,LiJ,ReinkeC,Bevans-FontiS,JunJC,PolotskyVY.Intermittenthypoxiaexacerbatesmetaboliceffectsofdiet-inducedobesity.
Obesity(SilverSpring)2011;19:2167-2174.
13. Paschetta E, Belci P, Alisi A, Liccardo D, Cutrera R, Musso G, Nobili V. OSAS-related inflammatory mechanisms of liver injury in
nonalcoholicfattyliverdisease.MediatorsInflamm2015;2015:815721.
14.ArchontogeorgisK,NenaE,PapanasN,RizzoM,etal.MetabolicSyndromeandVitaminDLevels inPatientswithObstructiveSleep
ApneaSyndrome.MetabSyndrRelatDisord2018;16:190-196.
15.NakayamaK.Cellularsignaltransductionofthehypoxiaresponse.JBiochem2009;146:757-65.
16.LiuY,MaZ,ZhaoC,WangY,WuG,XiaoJ,McClainCJ,etal.HIF-1alphaandHIF-2alphaarecriticallyinvolvedinhypoxia-inducedlipid
accumulationinhepatocytesthroughreducingPGC-1alpha-mediatedfattyacidbeta-oxidation.ToxicolLett2014;226:117-123.
17. Mesarwi OA, Shin MK, Bevans-Fonti S, Schlesinger C, Shaw J, Polotsky VY.Hepatocyte Hypoxia Inducible Factor-1 Mediates the
DevelopmentofLiverFibrosisinaMouseModelofNonalcoholicFattyLiverDisease.PLoSOne2016;11:e0168572.
18.Briancon-MarjolletA,MonneretD,HenriM,Joyeux-FaureM,TotosonP,CachotS,FaureP,etal.IntermittenthypoxiainobeseZucker
rats:cardiometabolicandinflammatoryeffects.ExpPhysiol2016;101:1432-1442.
19.KangHH,KimIK,LeeHI,JooH,LimJU,LeeJ,LeeSH,etal.Chronicintermittenthypoxiainducesliverfibrosisinmicewithdiet-induced
obesityviaTLR4/MyD88/MAPK/NF-kBsignalingpathways.BiochemBiophysResCommun2017;490:349-355.
20.WuW, LiW,Wei J,WangC, YaoY, et al. Chronic intermittenthypoxia accelerates liver fibrosis in ratswith combinedhypoxia and
nonalcoholicsteatohepatitisviaangiogenesisratherthanendoplasmicreticulumstress.ActaBiochimBiophysSin(Shanghai)2019;51:159-
1672.
21.ShiYF,FongCC,ZhangQ,CheungPY,TzangCH,WuRS,YangM.Hypoxia inducestheactivationofhumanhepaticstellatecellsLX-2
throughTGF-betasignalingpathway.FEBSLett2007;581:203-210.
22.CoppleBL,Bustamante JJ,WelchTP,KimND,Moon JO.Hypoxia-inducible factor-dependentproductionofprofibroticmediatorsby
hypoxichepatocytes.LiverInt2009;29:1010-21.
23.CoppleBL,BaiS,BurgoonLD,MoonJO.Hypoxia-induciblefactor-1alpharegulatestheexpressionofgenesinhypoxichepaticstellate
cellsimportantforcollagendepositionandangiogenesis.LiverInt2011;31:230-244.
24.PoveroD,EguchiA,LiH,JohnsonCD,PapouchadoBG,WreeA,MesserK,etal.Circulatingextracellularvesicleswithspecificproteome
andlivermicroRNAsarepotentialbiomarkersforliverinjuryinexperimentalfattyliverdisease.PLoSOne2014;9:e113651.
25.PoveroD,PaneraN,EguchiA, JohnsonCD,PapouchadoBG,deAraujoHorcelL,PinatelEM,etal. Lipid-inducedhepatocyte-derived
extracellularvesiclesregulatehepaticstellatecellviamicroRNAstargetingPPAR-gamma.CellMolGastroenterolHepatol2015;1:646-663.
26.HirsovaP,IbrahimSH,KrishnanA,VermaVK,BronkSF,WerneburgNW,CharltonMR,etal.Lipid-InducedSignalingCausesReleaseof
InflammatoryExtracellularVesiclesFromHepatocytes.Gastroenterology2016;150:956-967.
27. Cannito S,Morello E, Bocca C, Foglia B, Benetti E, Novo E, et al.Microvesicles released from fat-laden cells promote activation of
hepatocellular NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS One
2017;12:e0172575.

80
28.vanNielG,D'AngeloG,RaposoG.Sheddinglightonthecellbiologyofextracellularvesicles.NatRevMolCellBiol2018;19:213-228.
29. Devhare PB, Ray RB. Extracellular vesicles: Novelmediator for cell to cell communications in liver pathogenesis.Mol AspectsMed
2018;60:115-122.
30. Pradip B. Devhare, Ratna B. Ray. Extracellular vesicles: Novel mediator for cell to cell communications in liver pathogenesis. Mol
AspectsMed2018;60:115-122.
31.EguchiA,FeldsteinAE.Extracellularvesiclesinnon-alcoholicandalcoholicfattyliverdiseases.LiverRes2018;2:30-34.
32.PizarroM,SolisN,QuinteroP,BarreraF,CabreraD,Rojas-deSantiagoP,ArabJP,etal.Beneficialeffectsofmineralocorticoidreceptor
blockadeinexperimentalnonalcoholicsteatohepatitis.LiverInt2015;35:2129-2138.
33. Cabrera D,Wree A, Povero D, Solis N, Hernandez A, PizarroM,Moshage H, et al. Andrographolide Ameliorates Inflammation and
FibrogenesisandAttenuatesInflammasomeActivationinExperimentalNon-AlcoholicSteatohepatitis.SciRep2017;7:3491.
34.Chavez-TapiaNC,RossoN,TiribelliC.Invitromodelsforthestudyofnon-alcoholicfattyliverdisease.CurrMedChem2011;18:1079-
1084.
35.PecoraroM,PintoA,PopoloA.InhibitionofConnexin43translocationonmitochondriaacceleratesCoCl2-inducedapoptoticresponse
inachemicalmodelofhypoxia.ToxicolInVitro2018;47:120-128.
36.Munoz-SanchezJ,Chanez-CardenasME.Theuseofcobaltchlorideasachemicalhypoxiamodel.JApplToxicol2019;39:556-570.
37. SchoemakerMH,de laRosaLC,Buist-HomanM,etal. TauroursodeoxycholicAcidprotectsRathepatocytes fromBileAcid-induced
apoptosisviaactivationofsurvivalpathways.Hepatology2004;39:1563-1573.
38.ChanLL,McCulleyKJ,KesselSL.AssessmentofCellViabilitywithSingle-,Dual-,andMulti-StainingMethodsUsing ImageCytometry.
MethodsMolBiol2017;1601:27-41.
39.TheryC,AmigorenaS,RaposoG,ClaytonA. Isolationandcharacterizationofexosomesfromcellculturesupernatantsandbiological
fluids.CurrProtocCellBiol2006;3:22.
40.XuL,HuiAY,AlbanisE,ArthurMJ,O'ByrneSM,BlanerWS,MukherjeeP,FriedmanSL,EngFJ.Humanhepaticstellatecelllines,LX-1and
LX-2:newtoolsforanalysisofhepaticfibrosis.Gut2005;54:142-51.
41.FeldsteinAE,CanbayA,GuicciardiME,HiguchiH,BronkS,GoresG.Dietassociatedhepaticsteatosissensitizestofasmediated liver
injuryinmice.JHepatol2003;39:978-983.
42. Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, et al. Minimal information for studies of extracellular vesicles 2018
(MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J
ExtracellVesicles2018;7:1535750.
43.Vilar-GomezE,Calzadilla-BertotL,Wai-SunWongV,CastellanosM,Aller-de laFuenteR,etal.FibrosisSeverityasaDeterminantof
Cause-SpecificMortality in PatientsWith AdvancedNonalcoholic Fatty Liver Disease: AMulti-National Cohort Study. Gastroenterology.
2018;155:443-457.
44.MesarwiOA,ShinMK,DragerLF,Bevans-FontiS,JunJC,PutchaN,TorbensonMS,PedrosaRP,Lorenzi-FilhoG,SteeleKE,Schweitzer
MA,MagnusonTH,LidorAO,SchwartzAR,PolotskyVY.LysylOxidaseasaSerumBiomarkerofLiverFibrosisinPatientswithSevereObesity
andObstructiveSleepApnea.Sleep2015;38:1583-91.
45.ButtacavoliM,Gruttad'AuriaCI,OlivoM,VirdoneR,CastrogiovanniA,MazzucaE,MarottaAM,MarroneO,MadoniaS,BonsignoreMR.
Liver Steatosis and Fibrosis in OSA patients After Long-term CPAP Treatment: A Preliminary Ultrasound Study. Ultrasound Med Biol
2016;42:104-9.

81
46.MesarwiOA,LoombaR,MalhotraA.ObstructiveSleepApnea,Hypoxia,andNonalcoholicFattyLiverDisease.AmJRespirCritCareMed
2019;199:830-841.
47.FengSZ,TianJL,ZhangQ,WangH,SunN,ZhangY,ChenBY.Anexperimentalresearchonchronicintermittenthypoxialeadingtoliver
injury.SleepBreath2011;15:493-502.
48.FriedmanS.L.Hepaticstellatecells:protean,multifunctional,andenigmaticcellsoftheliver.PhysiolRev2008;88:125-172.
49.JeonYJ,SongKS,HanHJ,etal.Rosmarinicacidinhibitschemicalhypoxia-inducedcytotoxicityinprimaryculturedrathepatocytes.Arch
PharmRes2014;37:907-15.
50.Marin JJG, LozanoE,PerezMJ. LackofmitochondrialDNA impairs chemicalhypoxia-inducedautophagy in liver tumorcells through
ROS-AMPK-ULK1signalingdysregulationindependentlyofHIF-1α.FreeRadicBiolMed2016;101:71-84.
51. RicchiM,OdoardiMR, Carulli L, et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured
hepatocytes.JGastroenterolHepatol2009;24:830-40.
52.MoravcovaA,CervinkovaZ,KuceraO,MezeraV,RychtrmocD,LotkovaH.Theeffectofoleicandpalmiticacidoninductionofsteatosis
andcytotoxicityonrathepatocytesinprimaryculture.PhysiolRes2015;64:S627-636.
53.Li,J.,etal.,Intermittenthypoxiainduceshyperlipidemiainleanmice.CircRes2005;97:698-706.
54. ChenX, Li L, Liu X, LuoR, LiaoG, Li L, Liu J, Cheng J, Lu Y, ChenY.Oleic acid protects saturated fatty acidmediated lipotoxicity in
hepatocytesandratofnon-alcoholicsteatohepatitis.LifeSci2018;203:291-304.
55.FeldsteinAE,WerneburgNW,CanbayA,GuicciardiME,BronkSF,RydzewskiR,BurgartLJ,GoresGJ.Freefattyacidspromotehepatic
lipotoxicitybystimulatingTNF-alphaexpressionviaalysosomalpathway.Hepatology2004;40:185-94.
56.MalhiH,GoresGJ,LemastersJJ.Apoptosisandnecrosisintheliver:ataleoftwodeaths?Hepatology2006;43:S31-44.
57. Payance A, Silva-Junior G, Bissonnette J, et al. Hepatocyte Microvesicle Levels Improve Prediction of Mortality in Patients With
Cirrhosis.Hepatology2018;68:1508-1518.
58. PoveroD, Eguchi A, Niesman IR, AndronikouN, deMollerat du Jeu X,Mulya A, BerkM, LazicM, Thapaliya S, ParolaM, Patel HH,
Feldstein AE Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by
endothelialcells.SciSignal2013;6:ra88.
59.Momen-Heravi F, Bala S, Kodys K, Szabo G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific
miRNA-122andsensitizemonocytestoLPS.SciRep2015;5:9991.
60.LeeY,KimS,KoE,LeeJH,YiHS,YooY,JeJ,etal.Exosomesderivedfrompalmiticacid-treatedhepatocytesinducefibroticactivationof
hepaticstellatecells.SciRep2017;7:3710.
61.Almendros,I.,Y.Wang,L.Becker,F.E.Lennon,J.Zheng,B.R.Coats,K.S.Schoenfelt,A.Carreras,F.Hakim,S.X.Zhang,R.Farre,andD.
Gozal. Intermittenthypoxia-inducedchangesintumor-associatedmacrophagesandtumormalignancyinamousemodelofsleepapnea.
AmJRespirCritCareMed2014;189:593-601.
62.Luo,Y.,H.Y.Dong,B.Zhang,Z.Feng,Y.Liu,Y.Q.Gao,M.Q.Dong,andZ.C.Li.miR-29a-3pattenuateshypoxicpulmonaryhypertensionby
inhibitingpulmonaryadventitialfibroblastactivation.Hypertension2015;65:414-420.
63.Khalyfa,A.,R.Cortese,Z.Qiao,H.Ye,R.Bao, J.Andrade,andD.Gozal.Lategestational intermittenthypoxia inducesmetabolicand
epigeneticchangesinmaleadultoffspringmice.JPhysiol2017;595:2551-2568.

82
Chapter5
HYPOXIAINCREASEDCASPASE-1INEXTRACELLULARVESICLESDERIVEDFROMEXPERIMENTAL
NON-ALCOHOLICSTEATOHEPATITISMODELSANDPROMOTESINFLAMMASOMEACTIVATIONIN
KUPFFERCELLS
ABSTRACT
Background:Intercellularcrosstalkbetweenhepatocytesandnon-parenchymalcellsisanimportant
factorinthepathogenesisofnon-alcoholicfattyliverdisease(NAFLD)andthemoreaggressiveform
non-alcoholic steatohepatitis (NASH). Extracellular Vesicles (EV) plays an important role in
intercellular crosstalk in the context of liver diseases, including NAFLD. Clinical observations have
indicated that Obstructive sleep apnea (OSA) is an aggravating factor in NASH. Hypoxia is the
hallmarkofOSAS,however,whetherhypoxiamodulatesthecrosstalkbetweenhepatocytesandnon-
parenchymalKupffer cells (KCs)hasnotbeen investigateddeeplyyet.Aim: to investigatewhether
hypoxiamodulates hepatocellular damage and intercellular crosstalk in the context of NAFLD and
whetherEVandinflammasome/caspase-1areinvolvedinthismodulation.Methods:Hepatomacell
line HepG2 were treated with free fatty acids (FFAs) and chemical hypoxia (CH) was induced by
cobalt (II) chloride (CoCl2). Pro-inflammatory and inflammasome-related gene expressions were
assessedinhypoxicfat-ladenHepG2andinlivertissue.IntermittentHypoxia(IH)wasappliedtomice
fed a CDAA diet (NASH model). EV were isolated from conditioned medium (CM) or serum by
ultracentrifugation and characterized by nanoparticle tracking and electron microscopy. Cleaved
caspase-1and caspase-1activitywasmeasuredbyWesternblot andenzymatic assay respectively.
Results:CHinfat-ladenHepG2increasedexpressionofpro-inflammatoryandinflammasome-related
genescomparedtonon-treatedhepatocytes.Theseresultscorrelatedwiththe invivo results. The
silencing of HIF-1α using siRNA in HepG2 prevented the increased expression of
inflammasome-related genes in KCs. Treatment of KCs with EV from fat-laden hypoxic HepG2
increased pro-inflammatory and inflammasome-related gene expression in KCs compared to KC
treatedwithEV fromnon-treatedHepG2.Cleavedcaspase-1contentandactivityofcaspase-1was
increasedinEVfromhypoxicfat-ladenHepG2andfromserumofCDAA-fedanimalsexposedtoIH.
Conclusion:OurfindingsindicatethatCHpromotesinflammatoryphenotypeinfat-ladenHepG2and
theinflammasome-relatedgenesexpressioninKCsviaHIF-1α.CHinducesthereleaseofcaspase-1-
EVfromfat-ladenHepG2promotingtheinflammatoryphenotypeinKCs.ThesefindingssuggestEV
andtheircontent(caspase-1)asapotentialnovelbiomarkerinNAFLDandOSAS.

83
INTRODUCTION
Significant advances have been made in understanding the importance of intercellular
communication between hepatocytes and non-parenchymal cells in the pathogenesis of liver
diseases (1,2). In particular in the pathophysiology of non-alcoholic steatohepatitis (NASH), the
inflammatoryandprogressive formofnon-alcoholic fatty liverdisease (NAFLD),Kupffercells (KCs),
theliver-residentmacrophages,areconsideredtoplayakeyroleintheinflammatoryresponseand
diseaseprogression (2,3). Indeed, ithasbeendemonstrated thatKCactivationpromotessteatosis,
inflammationandfibrosisinNASH(4-7).AnearlyeventinthepathogenesisofNAFLDistheincreased
accumulation of lipids in the liver, in particular in hepatocytes. This lipid accumulation promotes
cytotoxic effects known as lipotoxicity, resulting in hepatocellular damage and triggering of
inflammatorysignaling(8-9).Emergingdatasuggestthat lipotoxichepatocytesreleaseextracellular
vesicles (EV) toneighboringtargetcells inanautocrine/paracrinemannerand in thiswaypromote
liver damage (10-11). EV are 50-150 nanometer-sized vesicles released after the fusion of
multivesicularbodiesordirectlyfromthecellmembrane(12).Theircargoisdiverseandmaycontain
proteins,variousRNAspeciesand/orlipids(13).SeveralstudieshaveindicatedtheimportanceofEV
inthecrosstalkbetweeninjuredhepatocytesandKCs(14-17).Moreover,increasedlevelsofEVhave
beenreportedinserumofmiceandhumanswithNASHandtheselevelscorrelatewithhistological
liverinflammationandmacrophageactivation(18-21).
Recent data have also indicated that injured hepatocytes release inflammasome components into
theextracellularenvironment,providingamechanismtospreadinflammasomesignalingtoadjacent
cells (22,23). The activation of the inflammasome promotes the maturation and release of pro-
inflammatoryinterleukinsviaactivationofcaspase-1.Theactivecaspase-1iscomposedofatetramer
containing two 20 kDa fragments and two 10 kDa fragments (24). The activation of caspase-1
initiatesanovelformofprogrammednecroticcelldeathnamedpyroptosis.Pyroptoticcellsrelease
proinflammatorycytokines like IL-1βand IL-18andthe intracellularenzyme lactatedehydrogenase
(LDH) via gasdermin-dependent pores in the plasma membrane (25,26). Recent studies have
indicated that caspase-1 activation plays an important role in inflammation and fibrosis in NASH
(27,28). A recent study reported the presence of caspase-1 in EV released from monocytes that
induced endothelial cell death in amodel of lung injury (29). However, the presence and role of
caspase-1inEVinthecontextofNASHhasnotbeenstudiedyet.
Emerging evidence from clinical studies indicates that intermittent hypoxia (IH), the principal
pathophysiological factor inobstructivesleepapnea(OSA),aggravatesNAFLD(30). Indeed,there is
substantialevidencefromrodentmodelsandhumansubjectsthatIHassociatedwithOSAincreases

84
hepatictriglyceride(TG)levelsandexacerbatesinflammationandfibrosisinNASH(31-35).Although
theprecisemolecularmechanismsunderlying theaggravatingeffectof IHonNASHhavenotbeen
elucidated yet, the activation of the transcription factor hypoxia inducible factor 1 alpha (HIF-1α)
appearstobeanimportantfactor.HepaticHIF-1αactivationincreasessteatosisandpromotespro-
inflammatoryandpro-fibroticsignalingpathways(35-38).
Taken together, there is strong evidence that hypoxia via HIF-1α and Inflammasome/Caspase-1
activationisanaggravatingfactorinNASH.However,itisstillnotfullyunderstoodwhethercaspase-
1participatesinhypoxia-inducedliverinjury.
InthepresentstudywehaveinvestigatedtheroleofhypoxiaandEVininvivoandinvitromodelsof
NASH.Wedemonstratethathypoxiapromotesinflammatoryphenotypeinfat-ladenHepG2andthe
inflammasome-related genes expression in KCs via HIF-1α. Moreover, CH induces the release of
caspase-1-EV from fat-laden HepG2 and promotes cellular crosstalk between hypoxic fat laden-
hepatocytesandKC.
2.-MATERIALSANDMETHODS
2.1Animals
Specified pathogen-freemaleWistar rats (220–250 g; Charles River Laboratories Inc.,Wilmington,
MA,USA)andmaleC57bl6mice[purchasedfromJacksonLaboratories(BarHarbor,ME)]wereused.
Animalswerehousedunderstandardlaboratoryconditionswithfreeaccesstostandardlaboratory
chowdietandwater.AllexperimentswerecarriedoutaccordingtotheDutchandChileanlawson
the welfare of laboratory animals and guidelines of the local institutional animal care and use
committeesofthePontificiaUniversidadCatólicadeChileandethicscommitteeoftheUniversityof
Groningenforcareanduseoflaboratoryanimals.Alleffortsweremadetominimizeanimalsuffering
andtoreducethenumberofanimalsused.
2.2Kupffercellisolation
Kupffercellswere isolated fromthenon-parenchymalcell fractionobtainedduring thehepatocyte
isolation. The supernatant was subjected to different low-speed centrifugations at 4°C and
resuspending the pellets in Hank’s Balanced Salt Solution (HBSS) (Gibco, California, USA)
supplemented with 0.3% bovine serum albumin (BSA). KC were purified using a 20% of density
gradient medium OptiPrepTM (Sigma-Aldrich). After different low-speed centrifugations, the final
pellet was dissolved in HBSS supplemented with 10% FBS to allow cells to attach. After the
attachment period, KC were cultured in William's E medium supplemented with 50 μg/ml
gentamycin,10%FBSand1%ofp/s/ffor18hoursuntilthestartoftheexperiments.

85
2.3HepG2hepatomacellline
The human hepatocellular carcinoma cell line HepG2 (ATCC, USA) was cultured in Dulbecco’s
modifiedEaglemedium(1X) + GlutaMAX™-I(DMEM,10569010,Gibco)supplementedwith10%fetal
bovineserum(FBS;Gibco)and1%penicillin-streptomycin(Gibco)at37°Cand5%CO2.Allcellswere
platedinacellcultureplateatleast24 h-36hbeforetreatmentuponreaching80%confluence.
2.4Treatmentwithfreefattyacidsandchemicalhypoxia
Inorder toassess theeffectsofhypoxiaon fat-laden liver cells,HepG2cellswas incubatedwitha
mixtureoffreefattyacids(FFA)consistingofoleicacid(500μmol/L)andpalmiticacid(250μmol/L)
inanaqueoussolutionofBSAasdescribed(41).IncubationswerecarriedoutwithorwithoutCobalt
(II)chloride(CoCl2;SigmaAldrich)(200μmol/L)for24hourstoinducechemicalhypoxia(40).CoCl2
isawell-knownhypoxia-mimeticagent, thatmimicshypoxia/ischemicconditionsbystabilizationof
hypoxia-induciblefactorHIF-1α(42).ControlcellsweretreatedwithBSAalone.
2.5EffectsofintermittenthypoxiainexperimentalNASH
Animalexperimentswereapprovedbythe institutionalanimalcareandusecommittee(Comitéde
ética y bienestar Animal, Escuela de Medicina, Pontificia Universidad Católica de Chile, CEBA
100623003).MaleC57BL/6miceaged10weeksatthebeginningofthestudyweredividedintofour
experimental groups (n = 4–8) receiving either choline-deficient amino acid-defined (CDAA) diet
(Catalog#518753,DyetsInc.Bethlehem,PA)toinduceNASHorthecholine-supplementedL-amino
acid defined (CSAA, Catalog # 518754, Dyets Inc. Bethlehem, PA) diet as control for 22 weeks as
previouslydescribed(43,44).AnimalswereexposedtoIHornormoxia(chambers41x22x35cm,COY
labproducts™,GrassLake,MI,USA)duringthelast12weeksoftheexperimentalorcontrolfeeding
period.IHregimenconsistedof30events/hourofhypoxicexposuresfor8hour/dayduringtherest
cycle,between9amand5pm.Thiscyclewasrepeated7daysaweekfor12consecutiveweeks.At
the end of the study, mice were anesthetized (ketamine 60 mg/kg plus xylazine 10 mg/kg
intraperitoneally) and then euthanized by exsanguination. Serum and liver tissue samples were
collected and processed or stored at −80 °C until analyzed. Gene expression and protein analyses
werecarriedoutasdescribedabove.
2.6WesternBlotanalyses
Protein lysateswerecollectedbyscrapingcells in lysisbuffer (HEPES25mmol/L,KAc150mmol/L,
EDTApH8.02mmol/L,NP-400.1%,NaF10mmol/L,PMSF50mmol/L,aprotinin1µg/µL,pepstatin1
µg/µL, leupeptin1µg/µL,DTT1mmol/L).Totalamountofprotein in lysateswasmeasuredbyBio-
Radproteinassay(Bio-Rad;Hercules,CA,USA).ForWesternblot,30-50µgproteinwasresolvedon

86
Mini-PROTEAN®TGXStain-Free™PrecastGels(BioRad,UK,Oxford).Semidry-blottingwasperformed
using Trans-Blot Turbo Midi Nitrocellulose Membrane with Trans-Blot Turbo System Transfer
(BioRad).PonceauS0.1%w/v(Sigma)stainingwasusedtoconfirmproteintransfer.anti-CASP-1(SC-
56036,SantaCruz),anti-CD81(10630D,Invitrogen),anti-CD63(sc-5275,SantaCruz)andanti-ASGPR1
(Novus, USA) were used in combination with appropriate peroxidase-conjugated secondary
antibodies.Tubulin(T9026,Sigma)oractin(A5228,Sigma)wereusedasloadingcontrols.Theblots
were analyzed in a ChemiDoc XRS system (Bio-Rad). Protein band intensities were quantified by
ImageLabsoftware(BioRad).
2.7RNAisolationandquantitativereal-timereversetranscriptionpolymerasechainreaction(qRT-
PCR)
Aftertreatment,HepG2cellsandrathepatocyteswereharvestedoniceandwashedtwicewithice-
cold PBS. Total RNA was isolated with TRI-reagent (Sigma) according to the manufacturer’s
instructions.Reversetranscription(RT)wasperformedusing2.5µgoftotalRNA,1XRTbuffer(500
mmol/l Tris-HCl [pH 8.3]; 500 mmol/l KCl; 30 mmol/l MgCl2; 50 mmol/l DTT), 1 mmol/l
deoxynucleotides triphosphate (dNTPs, Sigma), 10 ng/µl random nanomers (Sigma), 0.6 U/µl
RNaseOUT™(Invitrogen)and4U/µlM-MLVreversetranscriptase(Invitrogen)inafinalvolumeof50
µl.ThecDNAsynthesisprogramwas25°C/10min,37°C/60minand95°C/5min.ComplementaryDNA
(cDNA)wasdiluted20X innuclease-freewater.Real-TimeqPCRwascarriedout inaStepOnePlus™
(96-well) PCR System (Applied Biosystems, Thermofisher) using TaqMan probes. The sequences of
theprobesandprimer setsaredescribed inSupplementaryMaterial. ForqPCR,2X reactionbuffer
(dNTPs, HotGoldStar DNA polymerase, 5 mmol/l MgCl2) (Eurogentec, Belgium, Seraing), 5 µmol/l
fluorogenicprobeand50µmol/lofsenseandantisenseprimers(Invitrogen)wereused.mRNAlevels
werenormalizedtothehousekeepinggene18Sandfurthernormalizedtothemeanexpressionlevel
ofthecontrolgroup.RelativegeneexpressionwascalculatedusingtheΔCtmethod.Thesequences
oftheprobesandprimersetsaredescribedintheSupplementaryMaterial.
2.8TransfectionofHepG2cell linewithHIF-1α small interferingRNAor control small interfering
RNA
HepG2cellsweregrown toapproximately60%confluency in6-wellplatesbefore transfectionand
treatments. Small interfering RNA (esiRNA) was employed to silence HIF-1α in HepG2 cells
(EHU151981, Sigma-Aldrich) using standard Lipofectamine (Lipo3000, Invitrogen) in OPTI-MEM™ I
mediumatafinalconcentrationof100nmol/Laccordingtothemanufacturer’sprotocol.Cellswere
transfected with EGFP siRNA as control (EHUEGFP, Sigma-Aldrich). Experiments were performed
after 24 hours of transfection for 24 hours. The efficiency of transfection was determined by

87
quantitative RT-PCR and Western blot for HIF-1α (Supplementary Material). To obtain the
corresponding conditioned medium (CM), hepatocyte culture medium was replaced by FBS-free
mediumaftertreatmentforanadditional24hours.CMfromdifferenttreatmentswereaddedtoKC
for24hours.
2.9Extracellularvesiclesisolation
ForEVcollection,HepG2cellsweregrowninculturedishesof100mmtoobtain70mlofserum-free
conditioned media after 24 hours of treatment. EV were isolated from conditioned medium by
differential ultracentrifugation (UCF Thermo-Sorvall 80wx+) according to a modified protocol
describedpreviously(46).Atotalvolumeof70mlmediumpertreatmentwasdepletedofcellsand
cell debris by consecutive, low-speed centrifugations (2,000 × g for 30min and 12,000 × g for 45
min).Theresultingsupernatantswerecarefullycollectedandcentrifugedfor70minat120,000×gat
4°C.PelletsfromthiscentrifugationstepwerewashedinPBS,pooledandcentrifugedagainfor60
min at 100,000 × g at 4 °C. For EV collection from serum, samples were reconstituted in a total
volumeof4.4mLandcentrifugatedat2,000×gfor30minand10,000×gfor30min.Theresulting
supernatantswerecarefullycollectedandcentrifugedfor70minat120,000×gat4°C.Pelletsfrom
thiscentrifugationstepwerewashedinPBS,pooledandcentrifugedagainfor60minat100,000×g
at 4 °C. The obtained pellets fromHepG2 cells or serumwere resuspended in lysis buffer or PBS
dependingonsubsequentexperimentsandstoredinaliquotsat-80°C.ProteinfromHepG2-derived
EV or serum-derived EVwere obtained using the Total Exosome Protein Isolation kit (Invitrogen).
ProteinconcentrationinEVpelletsweremeasuredusingBCAproteinassaykit(Pierce,Rockford,IL).
2.10TreatmentofKupffercellswithextracellularvesicles
RatprimaryKupffercells(KCs)wereincubatedwithFBS-freeWilliam'sEmediumandexposedto15
μg of EV isolated fromHepG2 cells treatedwith CoCl2, FFA or CoCl2 + FFA for 24h. After 24h of
treatment,KCswereharvested.
2.11Caspase-1activity
Cell lysate, HepG2-derived EV or serum-derived EV were assayed for their ability to cleave a
fluorescentCASP-1substrate,followingthemanufacturer’sinstructions(Abcam,USA).
2.12Statisticalanalyses
AnalyseswereperformedusingGraphPadsoftware(version5.03,GraphPadSoftwareInc.,CA,USA).
All results are presented as a mean of at least 3 independent experiments ± SEM or as absolute
number or percentage for categorical variables. The statistical significance of differences between
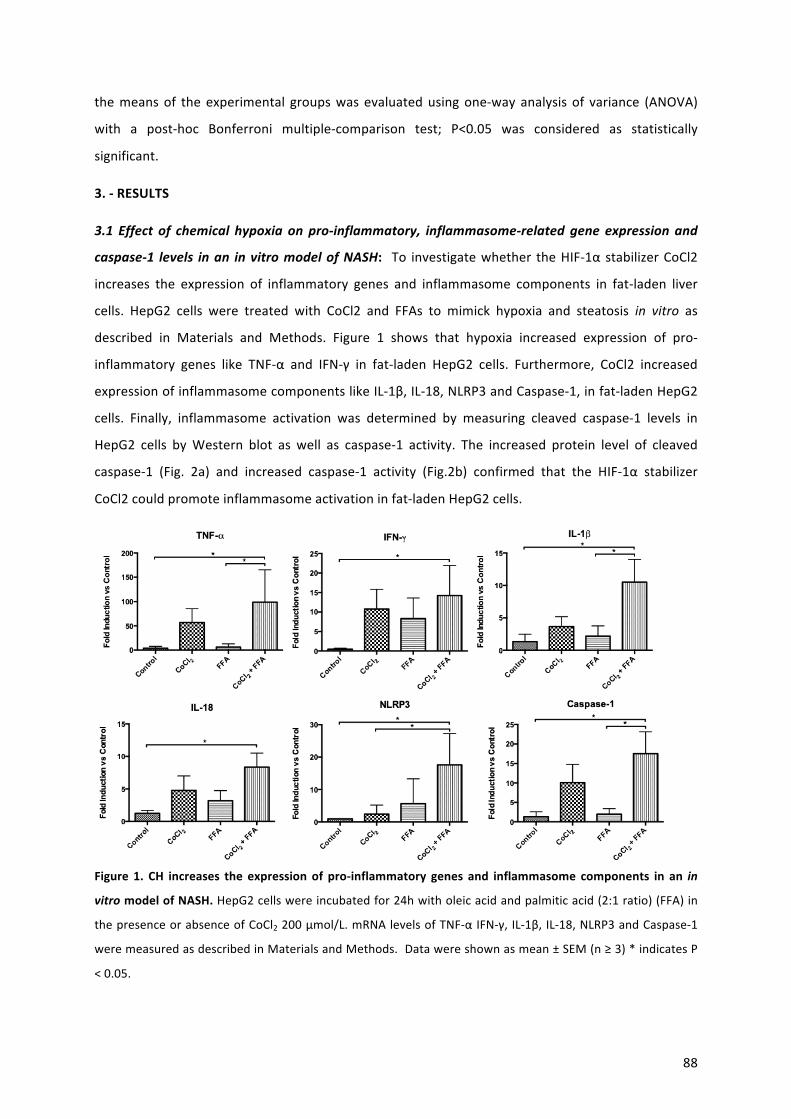
88
themeansof theexperimental groupswas evaluatedusingone-wayanalysis of variance (ANOVA)
with a post-hoc Bonferroni multiple-comparison test; P<0.05 was considered as statistically
significant.
3.-RESULTS
3.1 Effect of chemical hypoxia on pro-inflammatory, inflammasome-related gene expressionand
caspase-1 levels inan invitromodelofNASH: To investigatewhether theHIF-1αstabilizerCoCl2
increases the expression of inflammatory genes and inflammasome components in fat-laden liver
cells. HepG2 cells were treated with CoCl2 and FFAs to mimick hypoxia and steatosis in vitro as
described in Materials and Methods. Figure 1 shows that hypoxia increased expression of pro-
inflammatory genes like TNF-α and IFN-γ in fat-laden HepG2 cells. Furthermore, CoCl2 increased
expressionofinflammasomecomponentslikeIL-1β,IL-18,NLRP3andCaspase-1,infat-ladenHepG2
cells. Finally, inflammasome activation was determined by measuring cleaved caspase-1 levels in
HepG2 cells byWestern blot as well as caspase-1 activity. The increased protein level of cleaved
caspase-1 (Fig. 2a) and increased caspase-1 activity (Fig.2b) confirmed that the HIF-1α stabilizer
CoCl2couldpromoteinflammasomeactivationinfat-ladenHepG2cells.
Figure 1. CH increases the expressionof pro-inflammatory genes and inflammasome components in an in
vitromodelofNASH.HepG2cellswereincubatedfor24hwitholeicacidandpalmiticacid(2:1ratio)(FFA)in
thepresenceorabsenceofCoCl2200μmol/L.mRNAlevelsofTNF-αIFN-γ,IL-1β,IL-18,NLRP3andCaspase-1
weremeasuredasdescribedinMaterialsandMethods.Datawereshownasmean±SEM(n≥3)*indicatesP
<0.05.
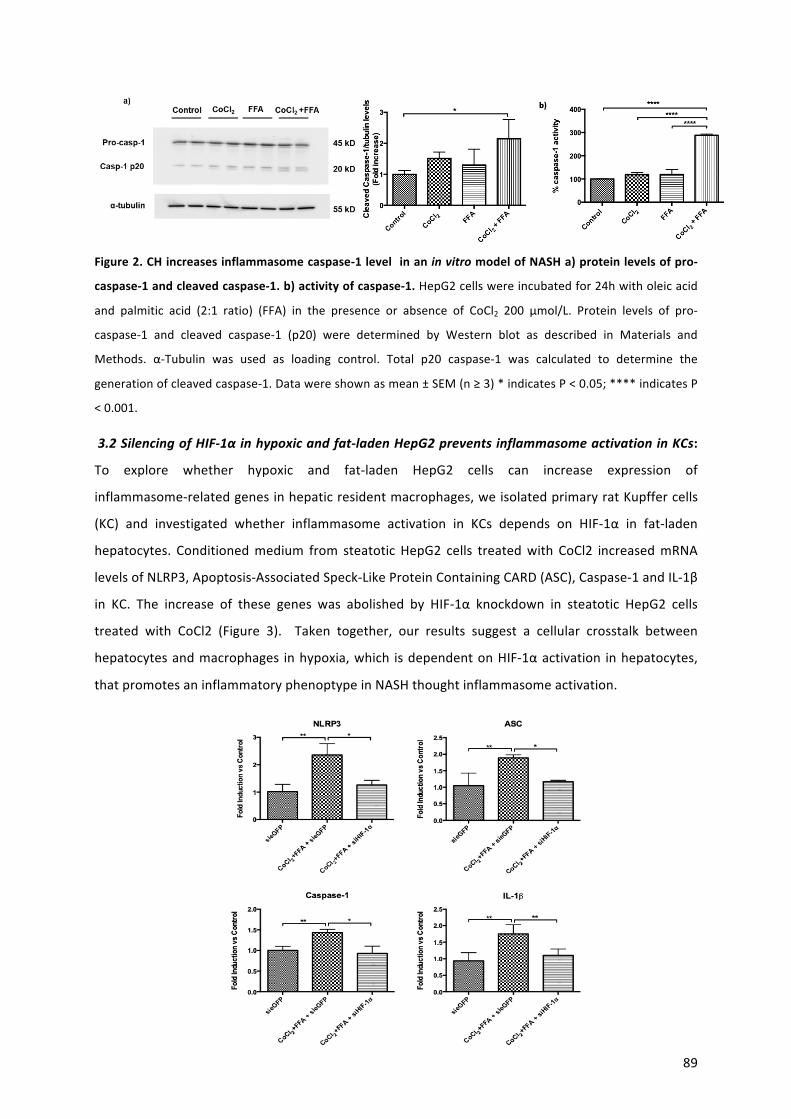
89
Figure2.CHincreasesinflammasomecaspase-1levelinaninvitromodelofNASHa)proteinlevelsofpro-
caspase-1andcleavedcaspase-1.b)activityofcaspase-1.HepG2cellswereincubatedfor24hwitholeicacid
and palmitic acid (2:1 ratio) (FFA) in the presence or absence of CoCl2 200 μmol/L. Protein levels of pro-
caspase-1 and cleaved caspase-1 (p20) were determined by Western blot as described in Materials and
Methods. α-Tubulin was used as loading control. Total p20 caspase-1 was calculated to determine the
generationofcleavedcaspase-1.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05;****indicatesP
<0.001.
3.2SilencingofHIF-1αinhypoxicandfat-ladenHepG2preventsinflammasomeactivationinKCs:
To explore whether hypoxic and fat-laden HepG2 cells can increase expression of
inflammasome-relatedgenesinhepaticresidentmacrophages,weisolatedprimaryratKupffercells
(KC) and investigated whether inflammasome activation in KCs depends on HIF-1α in fat-laden
hepatocytes. Conditionedmedium from steatoticHepG2 cells treatedwith CoCl2 increasedmRNA
levelsofNLRP3,Apoptosis-AssociatedSpeck-LikeProteinContainingCARD(ASC),Caspase-1andIL-1β
in KC. The increase of these genes was abolished by HIF-1α knockdown in steatotic HepG2 cells
treated with CoCl2 (Figure 3). Taken together, our results suggest a cellular crosstalk between
hepatocytesandmacrophages inhypoxia,which isdependentonHIF-1αactivation inhepatocytes,
thatpromotesaninflammatoryphenoptypeinNASHthoughtinflammasomeactivation.
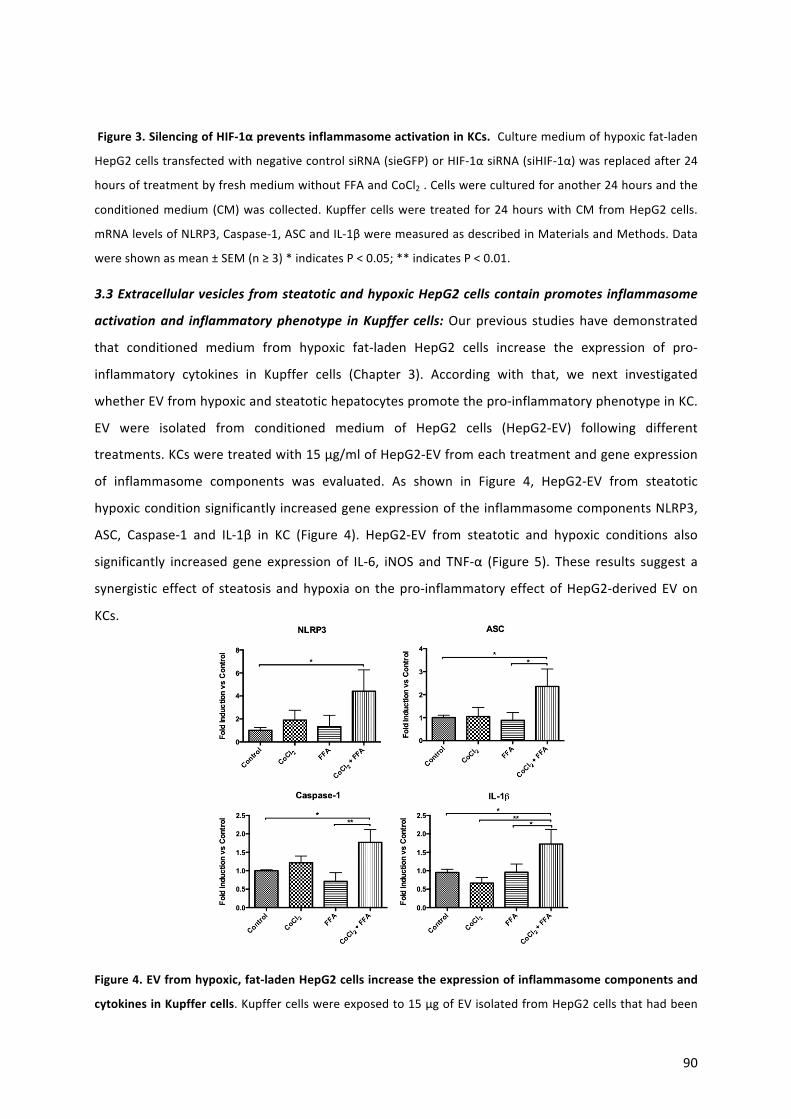
90
Figure3.SilencingofHIF-1αpreventsinflammasomeactivationinKCs.Culturemediumofhypoxicfat-laden
HepG2cellstransfectedwithnegativecontrolsiRNA(sieGFP)orHIF-1αsiRNA(siHIF-1α)wasreplacedafter24
hoursoftreatmentbyfreshmediumwithoutFFAandCoCl2.Cellswereculturedforanother24hoursandthe
conditionedmedium(CM)wascollected.Kupffercellswere treated for24hourswithCMfromHepG2cells.
mRNAlevelsofNLRP3,Caspase-1,ASCandIL-1βweremeasuredasdescribedinMaterialsandMethods.Data
wereshownasmean±SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01.
3.3ExtracellularvesiclesfromsteatoticandhypoxicHepG2cellscontainpromotesinflammasome
activationand inflammatoryphenotype inKupffer cells:Ourprevious studieshavedemonstrated
that conditioned medium from hypoxic fat-laden HepG2 cells increase the expression of pro-
inflammatory cytokines in Kupffer cells (Chapter 3). According with that, we next investigated
whetherEVfromhypoxicandsteatotichepatocytespromotethepro-inflammatoryphenotypeinKC.
EV were isolated from conditioned medium of HepG2 cells (HepG2-EV) following different
treatments.KCsweretreatedwith15μg/mlofHepG2-EVfromeachtreatmentandgeneexpression
of inflammasome components was evaluated. As shown in Figure 4, HepG2-EV from steatotic
hypoxicconditionsignificantlyincreasedgeneexpressionoftheinflammasomecomponentsNLRP3,
ASC, Caspase-1 and IL-1β in KC (Figure 4). HepG2-EV from steatotic and hypoxic conditions also
significantly increased gene expression of IL-6, iNOS and TNF-α (Figure 5). These results suggest a
synergisticeffectof steatosis andhypoxiaon thepro-inflammatoryeffectofHepG2-derivedEVon
KCs.
Figure4.EVfromhypoxic,fat-ladenHepG2cellsincreasetheexpressionofinflammasomecomponentsand
cytokinesinKupffercells.Kupffercellswereexposedto15μgofEVisolatedfromHepG2cellsthathadbeen
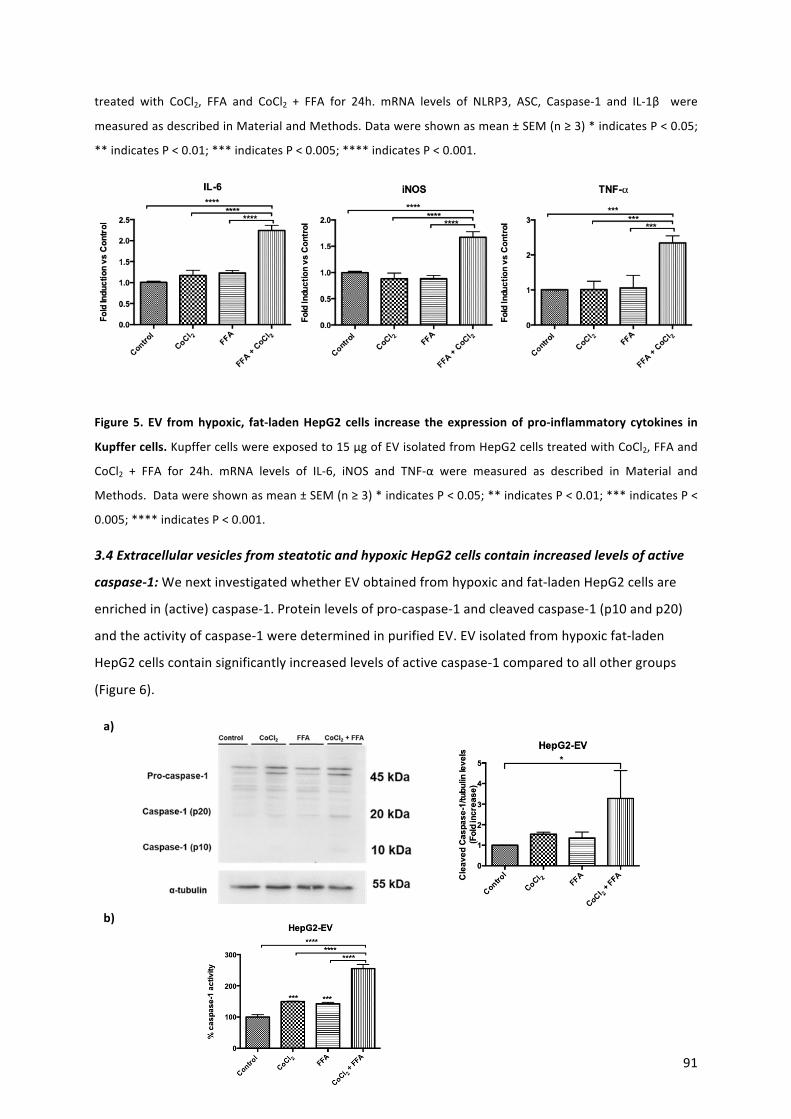
91
treated with CoCl2, FFA and CoCl2 + FFA for 24h. mRNA levels of NLRP3, ASC, Caspase-1 and IL-1β were
measuredasdescribedinMaterialandMethods.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05;
**indicatesP<0.01;***indicatesP<0.005;****indicatesP<0.001.
Figure5. EV fromhypoxic, fat-ladenHepG2cells increase theexpressionofpro-inflammatory cytokines in
Kupffercells.Kupffercellswereexposedto15μgofEVisolatedfromHepG2cellstreatedwithCoCl2,FFAand
CoCl2 + FFA for 24h. mRNA levels of IL-6, iNOS and TNF-α were measured as described in Material and
Methods.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05;**indicatesP<0.01;***indicatesP<
0.005;****indicatesP<0.001.
3.4ExtracellularvesiclesfromsteatoticandhypoxicHepG2cellscontainincreasedlevelsofactive
caspase-1:WenextinvestigatedwhetherEVobtainedfromhypoxicandfat-ladenHepG2cellsare
enrichedin(active)caspase-1.Proteinlevelsofpro-caspase-1andcleavedcaspase-1(p10andp20)
andtheactivityofcaspase-1weredeterminedinpurifiedEV.EVisolatedfromhypoxicfat-laden
HepG2cellscontainsignificantlyincreasedlevelsofactivecaspase-1comparedtoallothergroups
(Figure6).
a)
b)
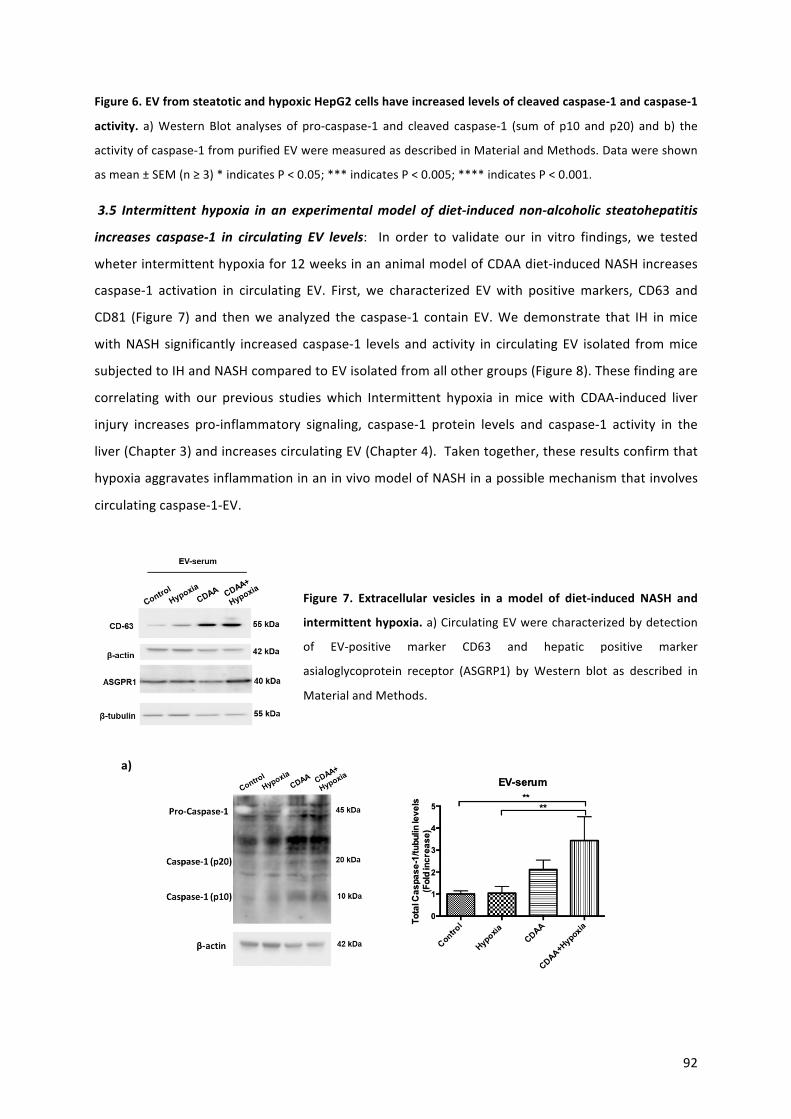
92
Figure6.EVfromsteatoticandhypoxicHepG2cellshaveincreasedlevelsofcleavedcaspase-1andcaspase-1
activity.a)Western Blot analyses of pro-caspase-1 and cleaved caspase-1 (sumof p10 and p20) and b) the
activityofcaspase-1frompurifiedEVweremeasuredasdescribedinMaterialandMethods.Datawereshown
asmean±SEM(n≥3)*indicatesP<0.05;***indicatesP<0.005;****indicatesP<0.001.
3.5 Intermittent hypoxia in an experimentalmodel of diet-induced non-alcoholic steatohepatitis
increases caspase-1 in circulating EV levels: In order to validate our in vitro findings, we tested
wheterintermittenthypoxiafor12weeksinananimalmodelofCDAAdiet-inducedNASHincreases
caspase-1 activation in circulating EV. First, we characterized EVwith positivemarkers, CD63 and
CD81 (Figure 7) and thenwe analyzed the caspase-1 contain EV.Wedemonstrate that IH inmice
withNASH significantly increased caspase-1 levels andactivity in circulatingEV isolated frommice
subjectedtoIHandNASHcomparedtoEVisolatedfromallothergroups(Figure8).Thesefindingare
correlatingwith our previous studies which Intermittent hypoxia inmicewith CDAA-induced liver
injury increases pro-inflammatory signaling, caspase-1 protein levels and caspase-1 activity in the
liver(Chapter3)andincreasescirculatingEV(Chapter4).Takentogether,theseresultsconfirmthat
hypoxiaaggravatesinflammationinaninvivomodelofNASHinapossiblemechanismthatinvolves
circulatingcaspase-1-EV.
Figure 7. Extracellular vesicles in a model of diet-induced NASH and
intermittenthypoxia.a)CirculatingEVwerecharacterizedbydetection
of EV-positive marker CD63 and hepatic positive marker
asialoglycoprotein receptor (ASGRP1) by Western blot as described in
MaterialandMethods.
a)

93
b)
Figure8. Extracellular vesicles fromCDAAdiet-fedmice subjected to IntermittentHypoxiaareenriched in
caspase-1.a)WesternBlotanalysisofpro-caspase-1andcleavedcaspase-1(p10andp20)andb)theactivityof
caspase-1 from purified circulating EV were measured as described in Materials and Methods. Data were
shownasmean±SEM(n≥3)**indicatesP<0.01;****indicatesP<0.001.
4.-DISCUSSION
Theprincipalfindingofthisstudyisthatextracellularvesciclesfromhepatocytesplayanimportant
roleinintercellularcommunicationinthecontextofNAFLD.Specifically,wehaveshownthatEVfrom
fat-laden and hypoxic hepatocytes induce a pro-inflammatiory phenotype in Kupffer cells, which
contributes to the initiation and progression of inflammation in NASH. The induction of this pro-
inflammatory phenotype is at least in part mediated by active caspase-1, and possibly other
componentsoftheinflammasome,containedwithinEV.
In recent years, different studies have provided evidence that hepatocyte injury induces a pro-
inflammatoryresponse inmacrophagesviathereleaseofhepatocyte-derivedEV,thuscontributing
to the development and progression of NAFLD (10, 47). The progression from simple steatosis to
NASH isnot completelyunderstood.TheonsetandprogressionofNAFLD fromsimple steatosis to
inflammation(NASH)ismostlikelytheresultofmultiplefactorsactinginparallelorsequentially(48).
Therecentclinicalobservationthatobstructivesleepapneasyndrome(OSAS) isapredictive factor
fortheprogressionandaggravationofNAFLD,promotedustoconsiderhypoxiathroughHIF-1α,the
hallmarkofOSAS,asan important trigger forhepatocellulardamage,exarcebating lipotoxicityand
inflammationandincreasingthereleaseofEV.Previously,wedemonstratedthatchemicalhypoxia,
mimicked by the HIF-1α stabilizer CoCl2, further increases hepatocellular TG content, pyroptosis-
induced cells death and mitochondrial ROS generation in FFA-treated primary hepatocytes and
HepG2 cells (10, 11). Now, we complement these finding, adding also that chemical hypoxia
increasesmRNA levelsof inflammasomecomponents inHepG2cells.These resultsare in linewith
our previous studies that hypoxia exacerbates apoptotic cell death determined by caspase-3 and
promotes disruption of cellular membranes in fat-laden hepatocytes (10). Interestingly, we

94
determinatedcellularcrosstalkmechanism-dependentofHIF-1alphainhypoxicfat-ladenHepG2cells
onexpressionof inflammasome-relatedgenes inKCs. Thisparacrineeffect is correlatedwithour
previous studies between hepatocytes with KCs and stellate cells (10, 11). Moreover, we
demonstratedanincreaseincaspase-1levelscontaininEVfromconditionedmediumobtainedfrom
hypoxicfat-ladenhepatocytes.TheseEVpromotedaninflammatoryphenotypeinKCsasevidenced
by increasedmRNA expression of inflammasome components in EV-treated Kupffer cells. Via this
mechanism,Caspase-1-EVmaycontributetotheinflammationassociatedwithNASH(12-49,50).The
inflammasome is an intracellularpro-inflammatory structure thatpromotes inflammatory signaling
and metabolic disruption (51). The activation of inflammasomes involves activation of caspase-1
which in turn cleaves pro-IL-1β into its active secreted form, thus amplifying the inflammatory
response(52). Interestingly,weobservedincreasedcaspase-1activity inEVfromfat-laden,hypoxic
hepatocytes.Inthecurrentstudywealsodemonstrateincreasedcaspase-1activityincirculatingEV
inmicewithNASHsubjectedtointermittenthypoxia(IH).Previousstudieshavedemonstratedthat
circulatingEVlevelsandinflammasomeactivationcorrelatestronglywithpathophysiologicalchanges
observedduringNASHdevelopment(4,27,28,53).Inourpreviousstudies,wealsoconfirmedthatIH
promotes NLRP3 inflammasome activation and pro-inflammatory and pro-fibrotic signaling in the
contextofNASH(10,11). In linewiththesepreviousresults,wehavenowdemonstrated increased
levelsofcirculatingEVinmicewithNASHandsubjectedtoIHandwealsodemonstratethattheseEV
containahigheractivityofcaspase-1comparedtocirculatingEVfromcontrolmice.Ourresultsare
supported by studies reporting that EV released by hepatocytes exposed to lipotoxic stress may
actively contribute to pro-inflammatory responses by activating macrophages in a NLRP3
inflammasome-dependent, paracrine pathway (54-56). In particular, a recent study demonstrated
the presence of the inflammasome components NLRP3, pro-caspase-1, pro-IL-1β and ASC in
macrophage-derivedEV(57).Takentogether,ourdataextendtheseobservationsbydemonstrating
thatEVfromhypoxicfat-ladenhepatocytes,andin invivomodelofhypoxiainexperimentalNASH,
containactivecaspase-1thatcouldinducesandextensepro-inflammatorysignaling.
Inconclusion,ourdataclearlyindicatetheimportanceofcrosstalkbetweenhepatocytesandKupffer
cells under steatotic and hypoxic conditions. Moreover, our results suggest that this crosstalk is
mediated via activated caspase-1 contained in EV and that this is essential for inflammasome
activationinmacrophages.Ourdataprovidenewinsightsinintercellularcommunicationandtherole
ofinflammasomesinhepatocyte-derivedEVinthecontextofNAFLD.
Our findingscouldhave implications in thedevelopmentofEV-basedbiomarkers,deliveryvehicles
andtherapeuticsforthetreatmentofliverdiseases.

95
5.-SUPPLEMENTARYFIGURES
FigureS1.SilencingofHIF-1α inHepG2cells . CulturemediumofhypoxicHepG2-transfectedwithnegative
control siRNA (sieGFP) orHIF-1αsiRNA (siHIF-1α).a)Protein levelsandb)mRNA levelsofHIF-1αdecreased
comparewithtreatmentofCoCl2 orCoCl2 +sieGFP.Valuesrepresentmean±standarddeviationfromthree
independentexperiments.Datawereshownasmean±SEM(n≥3)*indicatesP<0.05.
6.-REFERENCES
1.PetersKM,WilsonRB,BorradaileNM.Non-parenchymalhepaticcelllipotoxicityandthecoordinatedprogressionofnon-alcoholicfatty
liverdiseaseandatherosclerosis.CurrOpinLipidol.2018;29:417-422.
2.ChaJY,KimDH,ChunKH.Theroleofhepaticmacrophagesinnonalcoholicfattyliverdiseaseandnonalcoholicsteatohepatitis.LabAnim
Res.2018;34:133-139.
3.ArabJP,ArreseM,Trauner M.Recent insights intothepathogenesisofnon-alcoholic fatty liverdisease.AnnualReviewofPathology
MechanismsofDisease201813:1,321-350
4.CsakT,GanzM,PespisaJ,KodysK,DolganiucA,SzaboG.Fattyacidandendotoxinactivateinflammasomesinmousehepatocytesthat
releasedangersignalstostimulateimmunecells.Hepatology.2011;54:133-44.
5.PanJ,OuZ,CaiC,LiP,GongJ,RuanXZ,HeK.FattyacidactivatesNLRP3inflammasomesinmouseKupffercellsthroughmitochondrial
DNArelease.CellImmunol.2018;332:111-120
6. Tang Y, CaoG,Min X,Wang T, Sun S, Du X, ZhangW. Cathepsin B inhibition ameliorates the non-alcoholic steatohepatitis through
suppressingcaspase-1activation.JPhysiolBiochem.2018;74:503-510.
7.HoCM,HoSL,JengYM,LaiYS,ChenYH,LuSC,ChenHL,ChangPY,HuRH,LeePH.Accumulationoffreecholesterolandoxidizedlow-
densitylipoproteinisassociatedwithportalinflammationandfibrosisinnonalcoholicfattyliverdisease.JInflamm(Lond).2019;16:7.

96
8.Trauner,M.;Arrese,M.;Wagner,M.Fattyliverandlipotoxicity.Biochim.Biophys.Acta2010,1801,299–310
9.HirsovaP,IbrahimSH,GoresGJ,MalhiH.Lipotoxiclethalandsublethalstresssignalinginhepatocytes:relevancetoNASHpathogenesis.J
LipidRes.2016;57:1758-1770.
10.HernándezA,GengY,SepulvedaR,SolisN,TorresJ,ArabJP,BarreraF,CabreraD,MoshageH,ArreseM.Chemicalhypoxiainducespro-
inflammatorysignalsinfat-ladenhepatocytesandcontributestocellularcrosstalkwithKupffercells.BBA1866(2020)165753.
11.HernándezA,ReyesD,GengY,ArabJP,CabreraD,Buist-HomanM,ArreseM,MoshageH.Extracellularvesiclesderivedfromfat-laden
hepatocytesundergoingchemicalhypoxiapromoteapro-fibroticphenotypeinhepaticstellatecellsBBA1866(2020)165857
12.HernándezA,ArabJP,ReyesD,LapitzA,MoshageH,BañalesJM,ArreseM.ExtracellularvesiclesinNAFLD/ALD:frompathobiologyto
therapy.Cells9(2020)817
13.MalhiH.EmergingRoleofExtracellularVesiclesinLiverDiseases.AmJPhysiolGastrointestLiverPhysiol.2019;317:G739–G749.
14.Momen-Heravi F, Bala S, Kodys K, Szabo G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific
miRNA-122andsensitizemonocytestoLPS.SciRep.2015;5:9991.
15. Hirsova, P., Ibrahim, S.H., Krishnan, A. et al, Lipid-induced signaling causes release of inflammatory extracellular vesicles from
hepatocytes.Gastroenterology.2016;150:956–967
16. LiaoCY, SongMJ,Gao Y,MauerAS, RevzinA,MalhiH.Hepatocyte-Derived Lipotoxic ExtracellularVesicle Sphingosine 1-Phosphate
InducesMacrophageChemotaxis.FrontImmunol.2018;9:2980.
17.CannitoS,MorelloE,BoccaC,FogliaB,BenettiE,NovoE,ChiazzaF,RogazzoM,FantozziR,PoveroD,SuttiS,BugianesiE,FeldsteinAE,
AlbanoE,CollinoM,ParolaM.Microvesiclesreleasedfromfat-ladencellspromoteactivationofhepatocellularNLRP3 inflammasome:A
pro-inflammatorylinkbetweenlipotoxicityandnon-alcoholicsteatohepatitis.PLoSOne.2017;12:e0172575.
18.Kornek,M.,Lynch,M.,Mehta,S.H.etal.Circulatingmicroparticlesasdisease-specificbiomarkersofseverityofinflammationinpatients
withhepatitisCornonalcoholicsteatohepatitis.Gastroenterology.2012;143:448–458
19.PoveroD,PaneraN,EguchiA,JohnsonCD,PapouchadoBG,deAraujoHorcelL,PinatelEM,AlisiA,NobiliV,FeldsteinAE.Lipid-induced
hepatocyte-derived extracellular vesicles regulate hepatic stellate cell viamicroRNAs targeting PPAR-γ. CellMolGastroenterol Hepatol.
2015;1:646-663.e4.
20.EguchiA,LazaroRG,WangJ,KimJ,PoveroD,WillliamsB,HoSB,StärkelP,SchnablB,Ohno-MachadoL,TsukamotoH,FeldsteinAE.
ExtracellularvesiclesreleasedbyhepatocytesfromgastricinfusionmodelofalcoholicliverdiseasecontainaMicroRNAbarcodethatcanbe
detectedinblood.Hepatology.2017;65:475-490.
21.LiJ,LiuH,MauerAS,LucienF,RaiterA,BandlaH,MounajjedT,YinZ,GlaserKJ,YinM,MalhiH.CharacterizationofCellularSourcesand
CirculatingLevelsofExtracellularVesiclesinaDietaryMurineModelofNonalcoholicSteatohepatitis.HepatolCommun.201910;3:1235-
1249.
22.Baroja-MazoA,Martín-SánchezF,GomezAI.,etal.TheNLRP3inflammasomeisreleasedasaparticulatedangersignalthatamplifies
theinflammatoryresponse.NatImmunol2014;15:738-748
23.FranklinBS,BossallerL,DeNardoD.,etal.TheadaptorASChasextracellularand‘prionoid’activitiesthatpropagateinflammation.Nat
Immunol2014;15(08)727-737
24.Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, NaviaMA,MurckoMA, Chambers SP, Aldape RA, Raybuck SA. Structure and
mechanismofinterleukin-1betaconvertingenzyme.Nature.199428;370:270-5.
25.BeierJ,BanalesJ.Pyroptosis:AninflammatorylinkbetweenNAFLDandNASHwithpotentialtherapeuticimplications.JHepatol.2018;
68:643–645

97
26.LiaoJ,YangF,TangZ,YuW,HanQ,etal.InhibitionofCaspase-1-dependentpyroptosisattenuatescopper-inducedapoptosisinchicken
hepatocyte.EcotoxicolEnvironSaf.2019;174:110-119.
27. Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced
steatohepatitis.LabInvest.2012;92:713-23.
28.WreeA,EguchiA,McGeoughMD,PenaCA,JohnsonCD,CanbayA,HoffmanHM,FeldsteinAE.NLRP3inflammasomeactivationresults
inhepatocytepyroptosis,liverinflammation,andfibrosisinmice.Hepatology.2014;59:898-910.
29.MitraS,ExlineM,HabyarimanaF,GavrilinMA,BakerPJ,MastersSL,WewersMD,SarkarA1.MicroparticulateCaspase1Regulates
GasderminDandPulmonaryVascularEndothelialCellInjury.AmJRespirCellMolBiol.2018Jul;59(1):56-64.
30.MesarwiOA, LoombaR,MalhotraA.ObstructiveSleepApnea,Hypoxia,andNonalcoholic Fatty LiverDisease.Am JRespirCritCare
Med.2019Apr1;199(7):830-841
31.Qi JC,HuangJC,LinQC,ZhaoJM,LinX,ChenLD,HuangJF,ChenX.Relationshipbetweenobstructivesleepapneaandnonalcoholic
fattyliverdiseaseinnonobeseadults.SleepBreath2016;20:529-535.
32.PettaS,MarroneO,TorresD,ButtacavoliM,CammaC,DiMarcoV,LicataA,LoBueA,ParrinelloG,PintoA,SalvaggioA,Tuttolomondo
A,CraxiA,BonsignoreMR.ObstructiveSleepApnea IsAssociatedwithLiverDamageandAtherosclerosis inPatientswithNon-Alcoholic
FattyLiverDisease.PLoSOne2015;10:e0142210.
33. Paschetta E, Belci P, Alisi A, Liccardo D, Cutrera R, Musso G, Nobili V. OSAS-related inflammatory mechanisms of liver injury in
nonalcoholicfattyliverdisease.MediatorsInflamm.2015;2015:815721.
34.MesarwiOA,ShinMK,DragerLF,Bevans-FontiS, JunJC,PutchaN,TorbensonMS,PedrosaRP,Lorenzi-FilhoG,SteeleKE,Schweitzer
MA,MagnusonTH,LidorAO,SchwartzAR,PolotskyVY.LysylOxidaseasaSerumBiomarkerofLiverFibrosisinPatientswithSevereObesity
andObstructiveSleepApnea.Sleep2015;38:1583-1591.
35. Mesarwi OA, Shin MK, Bevans-Fonti S, Schlesinger C, Shaw J, Polotsky VY. Hepatocyte Hypoxia Inducible Factor-1 Mediates the
DevelopmentofLiverFibrosisinaMouseModelofNonalcoholicFattyLiverDisease.PLoSOne2016;11:e0168572.
36.LiuY,MaZ,ZhaoC,WangY,WuG,XiaoJ,McClainCJ,etal.HIF-1alphaandHIF-2alphaarecriticallyinvolvedinhypoxia-inducedlipid
accumulationinhepatocytesthroughreducingPGC-1alpha-mediatedfattyacidbeta-oxidation.ToxicolLett2014;226:117-123.
37.Briancon-MarjolletA,MonneretD,HenriM,Joyeux-FaureM,TotosonP,CachotS,FaureP,etal.IntermittenthypoxiainobeseZucker
rats:cardiometabolicandinflammatoryeffects.ExpPhysiol2016;101:1432-1442.
38.KangHH,KimIK,LeeHI,JooH,LimJU,LeeJ,LeeSH,etal.Chronicintermittenthypoxiainducesliverfibrosisinmicewithdiet-induced
obesityviaTLR4/MyD88/MAPK/NF-kBsignalingpathways.BiochemBiophysResCommun2017;490:349-355.
39.MoshageH,CasiniA,LieberCS.Acetaldehydeselectivelystimulatescollagenproductioninculturedratliverfat-storingcellsbutnotin
hepatocytes.Hepatology1990;12:511-518.
40.PecoraroM,PintoA,PopoloA.InhibitionofConnexin43translocationonmitochondriaacceleratesCoCl2-inducedapoptoticresponse
inachemicalmodelofhypoxia.ToxicolInVitro2018;47:120-128.
41.Chavez-TapiaNC,RossoN,TiribelliC.Invitromodelsforthestudyofnon-alcoholicfattyliverdisease.CurrMedChem2011;18:1079-
1084.
42.Munoz-SanchezJ,Chanez-CardenasME.Theuseofcobaltchlorideasachemicalhypoxiamodel.JApplToxicol2019;39:556-570.
43.PizarroM,SolisN,QuinteroP,BarreraF,CabreraD,Rojas-deSantiagoP,ArabJP,etal.Beneficialeffectsofmineralocorticoidreceptor
blockadeinexperimentalnon-alcoholicsteatohepatitis.LiverInt2015;35:2129-2138.

98
44. Cabrera D,Wree A, Povero D, Solis N, Hernandez A, PizarroM,Moshage H, et al. Andrographolide Ameliorates Inflammation and
FibrogenesisandAttenuatesInflammasomeActivationinExperimentalNon-AlcoholicSteatohepatitis.SciRep2017;7:3491.
45.Verhaag,E.M.,etal.,Hormesisincholestaticliverdisease;preconditioningwithlowbileacidconcentrationsprotectsagainstbileacid-
inducedtoxicity.PLoSOne,2016.11:p.e0149782.
46.TheryC,AmigorenaS,RaposoG,ClaytonA. Isolationandcharacterizationofexosomesfromcellculturesupernatantsandbiological
fluids.CurrProtocCellBiol2006;3:22.
47. Schattenberg JM, Lee M-S. Extracellular vesicles as messengers between hepatocytes and macrophages in non-alcoholic
steatohepatitis.Gastroenterology.2016;150:815±818.
48.TilgH,MoschenAR.Evolutionof inflammation innon-alcoholic fatty liverdisease: themultipleparallelhitshypothesis.Hepatology.
2010;52:1836±1846.
49.ArreseM,CabreraD,KalergisAM,FeldsteinAE.InnateImmunityandInflammationinNAFLD/NASH.DigDisSci2016;61:1294-1303.
50. Schuster S, Cabrera D, ArreseM, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol
2018;15:349-364
51.SzaboG,PetrasekJ.Inflammasomeactivationandfunctioninliverdisease.NatRevGastroenterolHepatol.2015;12:387–400.
52.KubesP,MehalWZ.Sterileinflammationintheliver.Gastroenterology.2012;143:1158–1172.
53.AmirMandCzajaM.Inflammasome-mediatedinflammationandfibrosis–itismorethanjusttheIL-1β.Hepatology.2018;67:479–481
54.Baroja-MazoA,Martin-SanchezF,GomezAI,MartinezCM,Amores-IniestaJ,CompanV,Barbera-CremadesM,YagueJ,Ruiz-OrtizE,
AntonJetal.TheNLRP3inflammasomeisreleasedasaparticulatedangersignalthatamplifiestheinflammatoryresponse.NatImmunol
2014;15:738–748
55.FranklinBS,BossallerL,DeNardoD,RatterJM,StutzA,EngelsG,BrenkerC,NordhoffM,MirandolaSR,Al-AmoudiAetal.Theadaptor
ASChasextracellularand“prionoid”activitiesthatpropagateinflammation.NatImmunol2014;15:727–73
56.WangL,FuH,NanayakkaraG,LiY,ShaoY,JohnsonC,ChengJ,YangWY,YangF,LavalleeMetal(2016)Novelextracellularandnuclear
caspase-1andinflammasomespropagateinflammationandregulategeneexpression:acomprehensivedatabaseminingstudy.JHematol
Oncol9:122
57. Lipinski S, Pfeuffer S, Arnold P, Treitz C, et al. Prdx4 limits caspase-1 activation and restricts inflammasome-mediated signaling by
extracellularvesicles.EMBOJ.2019;38:e101266.

99
Chapter6
1.-GENERALDISCUSSIONANDCONCLUSION
NAFLDreferstoaspectrumofhistologicalabnormalitiesintheliver,rangingfromisolatedsteatosis
tonon-alcoholicsteatohepatitis(NASH),theaggressiveformofNAFLD,whichischaracterizedbythe
presence of necro-inflammatory and fibrotic phenomena in the liver (1,2). Understanding the
transitionfromsimplesteatosistoNASHisakeyissueintheNAFLDfield.Currently,severalstudies
have shown thatOSAS,mainly via the occurrence of intermittent hypoxia (IH), predisposes to the
development of NASH (3). Recent studies have also demonstrated that hypoxia can aggravate
hepatocellular damage by triggering pro-inflammatory and pro-fibrotic signals (4,5). However, the
pathwaysunderlyingtheseeffectsarestill largelyunexplored.Thepresentdoctoralthesisproposes
an important role for extracellular vesicles in the hypoxia-induced exarcebation of hepatocellular
damage. We used in vitro models of NAFLD/NASH to explore whether hypoxia induces
hepatocellular damage, hepatic inflammation and fibrosis via mechanisms that involve cellular
crosstalkmediatedbyextracellularvesicles.
IntheChapter2,wereviewedthemostrecentfindingsconcerningtheroleofextracellularvesicles
(EV) inmediating autocrine and paracrine intercellular communication in both ALD andNAFLD as
wellastheirpotentialuseasbiomarkersofdiseaseseverityandprogression(15-17).Consideringour
resultsonthecrosstalkbetweenhepatocytesandnon-parenchymalcells,weproposearoleofEVin
intercellularcommunicationandtheaggravationofdamageinourNAFLDmodels
Theprincipal findings inChapter 3 suggest thathypoxiapromotes lipidsdroplet accumulation and
cell death via apoptosis, necrosis and pyroptosis in fat-laden primary rat hepatocytes. These
phenomena are associated with increased oxidative stress and inflammatory signals including
inflammasome/caspase-1 activation and contribute to cellular crosstalk between hepatocytes and
Kupffercells throughtEV,thusaggravatingthepathologicalphenomenaofNASH(6,7).Our invitro
findingscorrelatedwithour findings inananimalmodelofNASH inducedby feedingaCDAAdiet.
Ourresultsarealsoinlinewithpreviousstudiesthatindicatethathypoxia,viaHIF-1α,promoteslipid
accumulationinhepatocytesandcontributestoliver inflammationandfibrosis inrodentmodelsof
NAFLD (8-10).Our studiesare the first to investigate theeffectsofhypoxia inan invitromodelof
NAFLD/NASH that involves EV (11). Interestigly, we found a synergistic effect of hypoxia and fat
accumulationinhepatocytesontheincreasedexpressionofinflammasomecomponents,suggesting
that pyroptosis is the major form of cell death observed in these conditions. Moreover, we
investigatedtheeffectofhypoxia inan invivomicemodelofNASH(CDAAdiet). In linewithour in
vitroresults,intermittenthypoxiaincreasessteatosis,pro-inflammatorygeneexpressionandhepatic

100
inflammasome/caspase-1activationinmicewithNASH.Ithasbeenshownthatcaspase-1activation
contributestoliverdamageinNASH(12,13).
In Chapter 4 we demonstrated that hypoxia exarcebates hepatocellular damage in HepG2 cells.
Furthermore, hypoxia promotes pro-fibrotic signaling that correlates with increased release of EV
from hepatocytes in our experimental models of NASH. Morever, EV from hypoxic fat-laden
hepatocytesevokepro-fibroticresponsesinLX-2cells.Theseresultsareinagreementwithprevious
studies demonstrating that EV from lipotoxic hepatocytes induce pro-fibrogenic signals in stellate
cells(18,19).Furthermore,wedemonstratedincreasedlevelsofEVinserumfromCDAA-fedanimals
exposedtohypoxiaandthese increased levelscorrelatewithhistologicallydeterminedfibrosisand
pro-fibroticgeneexpressionintheliver.InChapter5weinvestigatedtheparacrineeffectofEVfrom
fat-ladenhypoxichepatocytesonKupffercellsthroughtCaspase-1likecargointoEV.Theexpression
ofpro-inflammatoryandinflammasome-relatedgeneswasincreasedinKupffercellstreatedwithEV
from hypoxic fat-laden hepatocytes compare to EV from non-treated hepatocytes, similar finding
thatwedescribesinChapter3.Theseresultscorrelatewellwithpreviousresultsindicatingcrosstalk
betweeninjuredhepatocytesandKupffercellsthatinvolvestheparticipationofEV(20-23).Wealso
performedexperiments to silenceHIF-1α inhypoxic fat-ladenHepG2 cells, to establish the roleof
HIF-1α in thepro-inflammatorycrosstalkbetweenhypoxic fat-ladenhepatocytes (HepG2cells)and
Kupffer cells. We demonstrated that conditioned medium from hypoxic fat-laden HepG2 cells
induced an increase in the expression of inflammasome-related genes in Kupffer cells via a
mechanismthatrequiresactivationofHIF-1α.Thesenovelresultsareinlinewitharecentstudythat
proposes a paracrine crosstalk between Kupffer cells and hepatocytes that promotes pro-
inflammatory signaling and leads to inflammatory injury (14). Taken together,we identify hypoxia
andHIF-1αactivationasapotentialaggravatingfactorinthedevelopmentofNASHinamechanism
thatinvolvescaspase-1/inflammasomeactivationandcellularcrosstalk.
Consideringthepro-infammatoryroleofEV(24-27),wealsoevaluatedthecontentofEV.Wecould
demonstratethepresenceofcleavedandactivecaspase-1inEVfromhypoxicfat-ladenhepatocytes
andEVfromserumofCDAA-fedmiceexposedtoIH.Collectively,thesenovelfindingsindicatethat
EV fromhypoxic fat-ladenhepatocytes inducepro-inflammatory andpro-fibrotic signals in Kupffer
cells and LX-2 cells, respectively via a mechanism that involves activation of caspase-1 and
inflammasomes.
In summary, this doctoral thesis identifies hypoxia as an aggravating factor in thepathogenesis of
NASH.ThisaggravatingeffectisdependentonintactHIF-1αsignalinginhepatocytesandintercellular
crosstalk between hepatocytes on the one hand and Kupffer cells and stellate cells on the other

101
hand. This crosstalk is dependentonextracellular vesicles released fromhepatocytes and involves
active caspase-1 and inflammasomes. We demonstrate that fat-laden hepatocytes are more
susceptible to the damaging effects of hypoxia than non-steatotic hepatocytes and that hypoxia
increasestheextentoffataccumulation,inflammationandfibrosisbothinvitroandinvivo.Further
characterizationofEVreleasedunderhypoxicconditionswillallowthedetaileddelineationoftheir
roleinthepromotionofsteatosis,inflammationandfibrosisinthecontextofNAFLDandOSAS.
Some limitations of our studies include the possible contamination of the EV fraction with
lipoproteinsduetosimilaritiesinsizeandcontamination,intheinvivostudies,withnon-hepaticEV.
Also,thesmallnumberofmicepergroupaswellastheuseofonly1modelofNASHarelimitations.
FuturestudiesshouldincludedifferentmodelsofNAFLD,e.g.highfatdietandalsohumanstudies,to
confirm our findings. Also, the cargo of EV should be further analyzed for the presence of
components,inadditiontoinflammasomecomponents,thatmaymodulatetheresponseofKupffer
cells and stellate cells to EV.Our findings couldhavepotential implications in thedevelopmentof
novel biomarkers that involve EV or selected components of EV, like caspase-1 and provide novel
therapeutictargetsforthetreatmentofNAFLDorliverdamageinOSAS.
2.-PERSPECTIVES
The information generated in this thesismay contribute to a better understanding of intercellular
communicationinthepathogenesisofNAFLDandthedevelopmentofnovel(EV-based)biomarkers
andtherapies.Itwillalsobeinterestingtoperformsimilarstudiesinotherliverdiseases.
AnexhaustivecharacterizationofthecontentofEVisolatedfrominvivoandinvitromodelsofNASH,
using genomic, proteomic and metabolomic techniques, will allow us to identify hypoxia-specific
mechanisms related to hepatocellular damage that occurs in NASH. In addition, our experimental
approach should be extended to clinical samples of patients with OSA and/or NASH, in order to
generate translational data. These studies should include characterization of EV obtained from
patients from blood or other bodily fluids such as urine or saliva. This non-invasive approachwill
allowtheidentificationandvalidationofnovelbiomarkersandimprovediagnosisandpredictionof
prognosis of NAFLD/NASH. Finally, non-pharmacological or pharmacological management of the
levelsofcirculatingEVcanbecomeanewtherapeutictargetinthecontextofNAFLDandOSAS.

102
3.-REFERENCES
1. YounossiZM,KoenigAB,AbdelatifD,FazelY,HenryL,WymerM.Globalepidemiologyofnonalcoholicfattyliverdisease-Meta-
analyticassessmentofprevalence,incidence,andoutcomes.Hepatology2016;64:73-84.
2. Arab JP, ArreseM, Trauner M. Recent insights into the pathogenesis of non-alcoholic fatty liver disease. Annual Review of
PathologyMechanismsofDisease201813:321-350
3. MesarwiOA,LoombaR,MalhotraA.ObstructiveSleepApnea,Hypoxia,andNonalcoholicFattyLiverDisease.AmJRespirCrit
CareMed.2019;199:830-841
4. SavranskyV,NanayakkaraA,ViveroA,LiJ,BevansS,SmithPL,TorbensonMS,etal.Chronicintermittenthypoxiapredisposesto
liverinjury.Hepatology2007;45:1007-1013.
5. Savransky V, Reinke C, Jun J, Bevans-Fonti S, Nanayakkara A, Li J, Myers AC, et al. Chronic intermittent hypoxia and
acetaminopheninducesynergisticliverinjuryinmice.ExpPhysiol2009;94:228-239.
6. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol
Hepatol2018;15:349-364.
7. ArreseM,CabreraD,KalergisAM,FeldsteinAE.InnateImmunityandInflammationinNAFLD/NASH.DigDisSci2016;61:1294-
1303.
8. MylonisI,SimosG,ParaskevaE.Hypoxia-InducibleFactorsandtheRegulationofLipidMetabolism.Cells2019;8.
9. MylonisI,SembongiH,BefaniC,LiakosP,SiniossoglouS,SimosG.HypoxiacausestriglycerideaccumulationbyHIF-1-mediated
stimulationoflipin1expression.JCellSci2012;125:3485-3493.
10. MesarwiOA,ShinMK,Bevans-FontiS,SchlesingerC,ShawJ,PolotskyVY.HepatocyteHypoxiaInducibleFactor-1Mediatesthe
DevelopmentofLiverFibrosisinaMouseModelofNonalcoholicFattyLiverDisease.PLoSOne2016;11:e0168572.
11. Chavez-Tapia NC, Rosso N, Tiribelli C. In vitro models for the study of non-alcoholic fatty liver disease. Curr Med Chem
2011;18:1079-1084.
12. DixonLJ,FlaskCA,PapouchadoBG,FeldsteinAE,NagyLE.Caspase-1asacentralregulatorofhighfatdiet-inducednon-alcoholic
steatohepatitis.PLoSOne2013;8:e56100.
13. Tang Y, Cao G,Min X,Wang T, Sun S, Du X, ZhangW. Cathepsin B inhibition ameliorates the non-alcoholic steatohepatitis
throughsuppressingcaspase-1activation.JPhysiolBiochem2018;74:503-510.
14. Zhang LY, Zhan DL, Chen YY,WangWH, He CY, Lin Y, Lin YC, Lin ZN. Aflatoxin B1 enhances pyroptosis of hepatocytes and
activationofKupffercellstopromoteliverinflammatoryinjuryviadephosphorylationofcyclooxygenase-2:aninvitro,exvivo
andinvivostudy.ArchToxicol.2019;14.
15. SzaboG,Momen-HeraviF.Extracellularvesicles in liverdiseaseandpotentialasbiomarkersandtherapeutictargets.NatRev
GastroenterolHepatol.2017;14(8):455-66.
16. EguchiA,FeldsteinAE.Extracellularvesiclesinnon-alcoholicandalcoholicfattyliverdiseases.LiverRes.2018;2(1):30-4.
17. UrbanSK,MocanT,SangerH,Lukacs-KornekV,KornekM.ExtracellularVesicles inLiverDiseases:Diagnostic,Prognostic,and
TherapeuticApplication.SeminLiverDis.2019;39(1):70-7.
18. PoveroD,PaneraN,EguchiA, JohnsonCD,PapouchadoBG,deAraujoHorcel L,PinatelEM,etal. Lipid-inducedhepatocyte-
derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-gamma. Cell Mol Gastroenterol
Hepatol2015;1:646-663.

103
19. LeeY, KimS, KoE, Lee JH, YiHS,YooY, Je J, et al. Exosomesderived frompalmitic acid-treatedhepatocytes induce fibrotic
activationofhepaticstellatecells.SciRep2017;7:3710.
20. Momen-Heravi F, Bala S, Kodys K, Szabo G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver
specificmiRNA-122andsensitizemonocytestoLPS.SciRep.2015;5:9991.
21. Hirsova,P., Ibrahim,S.H.,Krishnan,A.etal,Lipid-inducedsignalingcausesreleaseof inflammatoryextracellularvesicles from
hepatocytes.Gastroenterology.2016;150:956–967
22. Liao CY, Song MJ, Gao Y, Mauer AS, Revzin A, Malhi H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-
PhosphateInducesMacrophageChemotaxis.FrontImmunol.2018;9:2980.
23. Cannito S,Morello E, Bocca C, Foglia B, Benetti E,Novo E, Chiazza F, RogazzoM, Fantozzi R, PoveroD, Sutti S, Bugianesi E,
FeldsteinAE,AlbanoE,CollinoM,ParolaM.Microvesicles released from fat-ladencellspromoteactivationofhepatocellular
NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS One.
2017;12:e0172575.
24. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, Martinez CM, Amores-Iniesta J, Compan V, Barbera-Cremades M, Yague J,
Ruiz-OrtizE,AntonJetal.TheNLRP3inflammasomeisreleasedasaparticulatedangersignalthatamplifiestheinflammatory
response.NatImmunol2014;15:738–748
25. FranklinBS,BossallerL,DeNardoD,RatterJM,StutzA,EngelsG,BrenkerC,NordhoffM,MirandolaSR,Al-AmoudiAetal.The
adaptorASChasextracellularand“prionoid”activitiesthatpropagateinflammation.NatImmunol2014;15:727–73
26. WangL,FuH,NanayakkaraG,LiY,ShaoY,JohnsonC,ChengJ,YangWY,YangF,LavalleeMetal(2016)Novelextracellularand
nuclear caspase-1 and inflammasomes propagate inflammation and regulate gene expression: a comprehensive database
miningstudy.JHematolOncol9:122
27. LipinskiS,PfeufferS,ArnoldP,TreitzC,etal.Prdx4limitscaspase-1activationandrestrictsinflammasome-mediatedsignaling
byextracellularvesicles.EMBOJ.2019;38:e101266.

104
Appendices
EnglishSummary
Background:Nonalcoholic fatty liverdisease (NAFLD) isconsideredthemostcommon liverdisease
worldwide.Transitionfromsteatosistonon-alcoholicsteatohepatitis(NASH)isakeyissueinNAFLD.
Observations in patientswithobstructive sleep apnea syndrome (OSAS),which is characterizedby
occurrenceof intermittenthypoxia (IH),suggest thathypoxiacontributestodevelopNASH.Among
othermechanisms,releaseofextracellularvesicles(EV)byinjuredhepatocyteshasbeenimplicated
inNAFLDprogression.Aim: In this thesiswe aimed to investigate the roleof hypoxia condition in
modulation of steatosis and liver injury in both in vitro and in vivo models of NAFLD. Also we
evaluated the cellular crosstalk between hypoxic and steatotic hepatocytes and non-parenchymal
cellsthroughEV.Methods:PrimaryrathepatocytesandhepatomacelllineHepG2treatedwithfree
fatty acids (FFA) were subjected to chemically induced hypoxia (CH) using the hypoxia-inducible
factor-1alpha (HIF-1α)stabilizercobaltchloride (CoCl2).Triglyceride (TG)content,oxidativestress,
cell death rates, pro-inflammatory and pro-fibrotic cytokines, inflammasome components gene
expressionandproteinlevelsofcleavedcaspase-1wereassessed.Also,Kupffercells(KC)andhuman
stellate cells (LX-2) were treated with conditioned medium (CM) and EV from hypoxic fat-laden
hepatocytes. The choline deficient L-amino acid diet (CDAA)-fed mice model used to assess the
effects of IH on experimental NAFLD. Results: Hepatocytes exposed to FFA and CoCl2 exhibited
increased TG content and higher cell death rates aswell as increased, oxidative stress andmRNA
levelsofpro-inflammatory,pro-fibroticcytokinesandinflammasome-componentscomparedtonon-
treatedhepatocytes.Protein levelsof cleaved caspase-1 increased inCH-exposedhepatocytesand
from EV-hepatocytes. CM and EV from hypoxic fat-laden hepatic cells evoked a pro-inflammatory
and pro-fibrotic phenotype in KC and LX-2 respectively. Livers from CDAA-fedmice exposed to IH
exhibitedincreasedofsteatosis,portalinflammation,fibrosis,mRNAlevelsofpro-inflammatory,pro-
fibrotic and inflammasome genes as well as increased levels of cleaved caspase-1 that correlated
with an increase of circulating EV-caspase-1.Conclusion: Our findings in both in vivo and in vitro
models of NAFLD/NASH indicate that hypoxia may increase liver injury and promote disease
progression through amplification of inflammatory and fibrotic signals including
inflammasome/caspase-1 activation. Hypoxia also promotes the release of EV from hepatocytes
contributing to cellular crosstalkwith non-parenchymal cells by EV-caspase-1-relatedmechanisms.
TheseresultssuggestEVandtheircontent(caspase-1)asapotentialnovelbiomarkerinNAFLDand
OSAS. Further characterization of EV released under hypoxic conditions will allow the detailed
delineationof their role in thepromotionof steatosis, inflammation and fibrosis in the context of
NAFLDandOSAS.

105
Resumen
Antecedentes: El hígado graso no alcohólico (HGNA) es actualmente la enfermedad hepáticamás
prevalente en todo elmundo. Losmecanismos que subyacen a la transición de la esteatosis a la
esteatohepatitis no alcohólica (EHNA) no está del todo comprendida. Observaciones clínicas en
pacientesconelsíndromedelaapneaobstructivadelsueño(SAOS),caracterizadapor lapresencia
de hipoxia intermitente (HI), sugieren que esta condición contribuye al desarrollo de EHNA.
Recientemente, el rol de las vesículas extracelulares (EV) desde hepatocitos esteatóticos ha sido
estudiado en la progresión del daño en el HGNA. Objetivo general: Investigar si modelos
experimentalesdeEHNAsonmássusceptiblesalosefectosdehipoxia,ysitalcondiciónpromueveel
aumento de EV que señalizan a células no parenquimatosas. Métodos: Cultivo primario de
hepatocitosyuna líneacelularHepG2fuerontratadosconácidosgrasos libres(FFA)ysometidosa
hipoxia inducidaquímicamente (CH) usando clorurode cobalto (CoCl2). Se evaluóel contenidode
triglicéridos (TG), el estrés oxidativo, muerte celular, la expression génica de citoquinas pro-
inflamatorias,pro-fibróticas,ydeloscomponentesdelinflamasoma,incluídalacaspasa-1escindida.
Las células de Kupffer (KC) y las células estrelladas humanas (LX-2) se trataron con medio
condicionado (CM) y con EV aisladas de hepatocitos. El modelo de ratones alimentados con L-
aminoácidodefinidocondeficienciadecolina(CDAA)seusóparaevaluarlosefectosdeIHenHGNA
experimental.Resultados:LoshepatocitosexpuestosaFFAyCoCl2exhibieronunmayorcontenido
de TG, muerte celular, estrés oxidativo y un aumento en los niveles de la expression génica de
citoquinas proinflamatorias, pro-fibróticas y de los componentes de inflamasoma en comparación
con loshepatocitosnotratados.Losnivelesdecaspasa-1escindidaaumentaronen loshepatocitos
expuestos a CH, así comoen las EV de los hepatocitos hipóxicos y estetaóticos. El CM y elmayor
número de EV de las células hepáticas hipóxicas tratadas con FFA promovieron un fenotipo pro-
inflamatorio ypro fibróticoen las KC y LX-2 respectivamente. Loshígadosde ratones alimentados
conCDAAexpuestosaIHmostraronunaumentoenlaesteatosis,enlainflamaciónyfibrosisquese
correlacionó con un aumento de las EV circulantes con cargo de caspasa-1.Conclusión: Nuestros
hallazgosindicanquelosmodelosexperimentalesdeHGNAsonmássusceptiblesalosefectosdela
hipoxiaquesecorrelacionanconelaumentodelasEV.Lahipoxiapromovióseñalesinflamatoriasy
fibróticas, incluida la activación del inflamasoma/caspasa-1 en hepatocitos cargados de grasa y en
nuestromodeloinvivodeEHNA,contribuyendoaladiafoníacelularconcélulasnoparenquimatosas
por EV-caspasa-1. Estos resultadosproponena las EV, y su contenido (caspasa-1), comounnuevo
biomarcadorpotencialenHGNAySAOS.

106
Nederlandsesamenvatting
Achtergrond:Niet-alcoholische leververvetting (NAFLD)wordtwereldwijdbeschouwdalsdemeest
voorkomende leverziekte. Overgang van steatose naar niet-alcoholische steatohepatitis (NASH) is
een belangrijk probleem bij NAFLD. Waarnemingen bij patiënten met obstructief
slaapapneusyndroom(OSAS),datwordtgekenmerktdoorhetoptredenvanintermitterendehypoxie
(IH),suggererendathypoxiebijdraagtaandeontwikkelingvanNASH.Naastanderemechanismenis
de afgifte van extracellulaire blaasjes (EV) door beschadigde hepatocyten betrokken bij NAFLD-
progressie. Doel: In dit proefschrift wilden we de rol van hypoxie bij modulatie van steatose en
leverbeschadiging in zowel invitroals invivomodellenvanNAFLDonderzoeken.Ookevalueerden
wedecellulairesamenwerkingtussenhypoxischeensteatotischehepatocytenenniet-parenchymale
cellenviaEV.Methoden:PrimairerattenhepatocytenenhepatoomcellijnHepG2behandeldmetvrije
vetzuren (FFA)werdenonderworpenaan chemisch geïnduceerdehypoxie (CH)metbehulp vande
hypoxie-induceerbare factor-1 alfa (HIF-1α) stabilisator kobaltchloride (CoCl2). Triglyceride (TG) -
gehalte, oxidatieve stress, celsterftecijfers, pro-inflammatoire en pro-fibrotische cytokines,
genexpressie van ontstekingscomponenten en eiwitniveaus van gesplitst caspase-1 werden
beoordeeld. Ook werden Kupffer-cellen (KC) en humane stellaatcellen (LX-2) behandeld met
geconditioneerdmedium(CM)enEVvanmethypoxischevetgeladenhepatocyten.Hetmetcholine-
deficiënteL-aminozuurdieet(CDAA)gevoedemuizenmodeldatwerdgebruiktomdeeffectenvanIH
op experimentele NAFLD te beoordelen. Resultaten:Hepatocyten blootgesteld aan FFA en CoCl2
vertoonden een verhoogd TG-gehalte en hogere celsterftecijfers, evenals verhoogde oxidatieve
stress en mRNA-niveaus van pro-inflammatoire, pro-fibrotische cytokines en
inflammasoomcomponenten in vergelijking met niet-behandelde hepatocyten. Eiwitniveaus van
gesplitst caspase-1namen toe inCH-blootgesteldehepatocytenenvanEV-hepatocyten.CMenEV
van met hypoxisch vet geladen levercellen wekten respectievelijk een pro-inflammatoire en pro-
fibrotischefenotypeopinKCenLX-2.LeversvanCDAA-gevoedemuizendiewarenblootgesteldaan
IH vertoonden verhoogde steatose, portale ontsteking, fibrose, mRNA-niveaus van pro-
inflammatoire, pro-fibrotische en inflammasoomgenen, evenals verhoogde niveaus van gesplitst
caspase-1 die correleerden met een toename van circulerende EV-caspase-1. Conclusie: Onze
bevindingen in zowel in vivo als in vitro modellen van NAFLD/NASH geven aan dat hypoxie
leverbeschadiging kan vergroten en ziekteprogressie kan bevorderen door versterking van
inflammatoire en fibrotische signalen, waaronder de activering van inflammasoom/caspase-1.
HypoxiebevordertookdeafgiftevanEVuithepatocytendiebijdragenaancellulaireoverspraakmet
niet-parenchymalecellendoorEV-caspase-1-gerelateerdemechanismen.Dezeresultatensuggereren
EV en hun inhoud (caspase-1) als een potentiële nieuwe biomarker in NAFLD en OSAS. Verdere

107
karakterisering van EV die vrijkomt onder hypoxische omstandigheden zal de gedetailleerde
afbakening van hun rol bij de bevordering van steatose, ontsteking en fibrose in de context van
NAFLDenOSASmogelijkmaken.

108
ACKNOWLEDGEMENTS
ThisdoctoralthesiswassupportedbytheChileangovernmentthroughFondoNacionaldeDesarrollo
Científico y Tecnológico (FONDECYT 1150327 and 1119145) and PhD fellowship from Comisión
NacionalparalaInvestigaciónenCienciayTecnología(CONICYT)andVicerrectorateofResearchand
School of Medicine, PUC. Also, the support from the Abel Tasman Talent Program (ATTP) of the
GraduateSchoolofMedicalSciencesandtheUniversityMedicalCenterGroningen(UMCG)fromThe
Netherlandsisgratefullyacknowledged.

109
ABBREVIATIONS
OSAS:obstructivesleepapneasyndrome
IH:intermittenthypoxia
NAFLD:non-alcoholicfattyliverdisease
NAFL:non-alcoholicfattyliver
NASH:non-alcoholicsteatohepatitis
CDAA:choline-deficientaminoacid-defined
NLRP3:NOD-likereceptorPyrinDomainContaining3
GSDMD:gasderminD
KC:Kupffercells
TG:triglyceride
IL:interleukin
TNF-α:tumornecrosisfactor-alpha
HIF-1α:hypoxiainduciblefactor1alpha
EV:extracellularvesicles
FBS:fetalbovineserum
FFA:freefattyacids
CM:conditionedmedium
AUF:Arbitraryunitsoffluorescence
PBS:phosphate-bufferedsaline
LDH:Lactatedehydrogenase
NTA:nanoparticletrackinganalysis
ASC:Apoptosis-AssociatedSpeck-LikeProteinContainingCARD
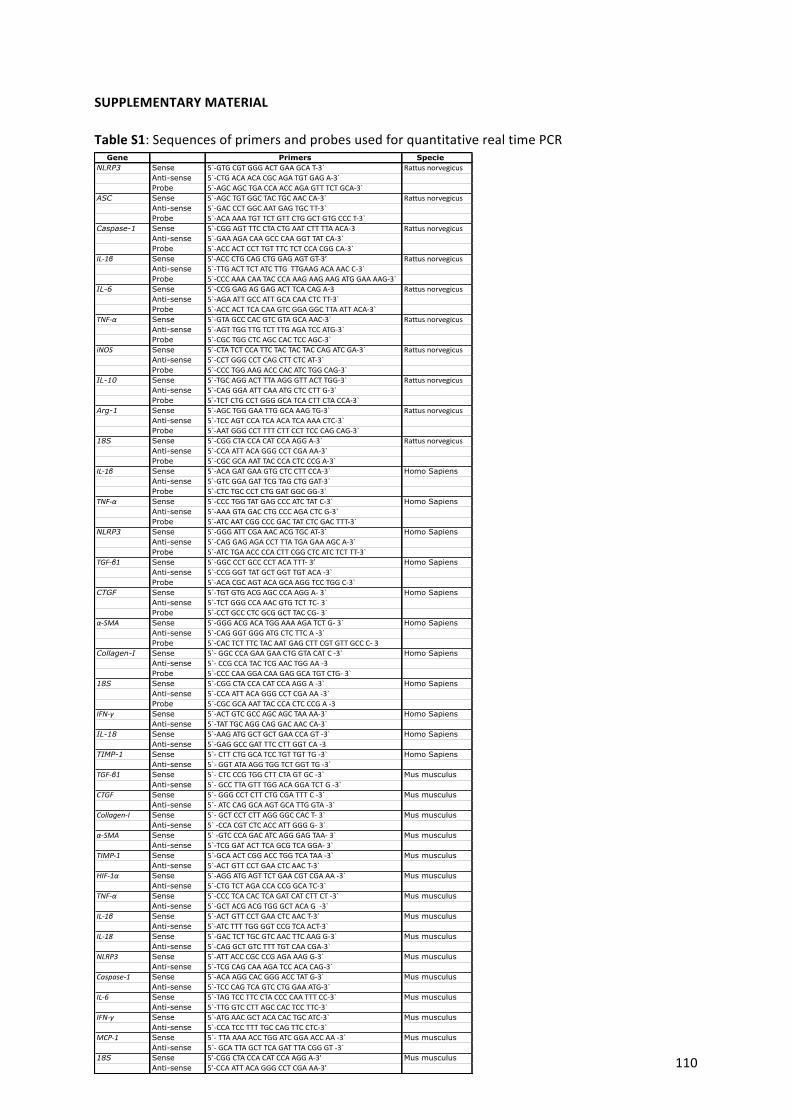
110
Gene Primers SpecieNLRP3 Sense 5`-GTGCGTGGGACTGAAGCAT-3` Rattusnorvegicus
Anti-sense 5`-CTGACAACACGCAGATGTGAGA-3`Probe 5`-AGCAGCTGACCAACCAGAGTTTCTGCA-3`
ASC Sense 5`-AGCTGTGGCTACTGCAACCA-3` RattusnorvegicusAnti-sense 5`-GACCCTGGCAATGAGTGCTT-3`Probe 5`-ACAAAATGTTCTGTTCTGGCTGTGCCCT-3`
Caspase-1 Sense 5`-CGGAGTTTCCTACTGAATCTTTTAACA-3 RattusnorvegicusAnti-sense 5`-GAAAGACAAGCCCAAGGTTATCA-3`Probe 5`-ACCACTCCTTGTTTCTCTCCACGGCA-3`
IL-1β Sense 5’-ACCCTGCAGCTGGAGAGTGT-3’ RattusnorvegicusAnti-sense 5`-TTGACTTCTATCTTGTTGAAGACAAACC-3`Probe 5`-CCCAAACAATACCCAAAGAAGAAGATGGAAAAG-3`
IL-6 Sense 5`-CCGGAGAGGAGACTTCACAGA-3 RattusnorvegicusAnti-sense 5`-AGAATTGCCATTGCACAACTCTT-3`Probe 5`-ACCACTTCACAAGTCGGAGGCTTAATTACA-3`
TNF-α Sense 5`-GTAGCCCACGTCGTAGCAAAC-3` RattusnorvegicusAnti-sense 5`-AGTTGGTTGTCTTTGAGATCCATG-3`Probe 5`-CGCTGGCTCAGCCACTCCAGC-3`
iNOS Sense 5`-CTATCTCCATTCTACTACTACCAGATCGA-3` RattusnorvegicusAnti-sense 5`-CCTGGGCCTCAGCTTCTCAT-3`Probe 5`-CCCTGGAAGACCCACATCTGGCAG-3`
IL-10 Sense 5`-TGCAGGACTTTAAGGGTTACTTGG-3` RattusnorvegicusAnti-sense 5`-CAGGGAATTCAAATGCTCCTTG-3`Probe 5`-TCTCTGCCTGGGGCATCACTTCTACCA-3`
Arg-1 Sense 5`-AGCTGGGAATTGGCAAAGTG-3` RattusnorvegicusAnti-sense 5`-TCCAGTCCATCAACATCAAAACTC-3`Probe 5`-AATGGGCCTTTTCTTCCTTCCCAGCAG-3`
18S Sense 5`-CGGCTACCACATCCAAGGA-3` RattusnorvegicusAnti-sense 5`-CCAATTACAGGGCCTCGAAA-3`Probe 5`-CGCGCAAATTACCCACTCCCGA-3`
IL-1β Sense 5`-ACAGATGAAGTGCTCCTTCCA-3`Homo SapiensAnti-sense 5`-GTCGGAGATTCGTAGCTGGAT-3`Probe 5`-CTCTGCCCTCTGGATGGCGG-3`
TNF-α Sense 5`-CCCTGGTATGAGCCCATCTATC-3`Homo SapiensAnti-sense 5`-AAAGTAGACCTGCCCAGACTCG-3`Probe 5`-ATCAATCGGCCCGACTATCTCGACTTT-3`
NLRP3 Sense 5`-GGGATTCGAAACACGTGCAT-3` Homo SapiensAnti-sense 5`-CAGGAGAGACCTTTATGAGAAAGCA-3`Probe 5`-ATCTGAACCCCACTTCGGCTCATCTCTTT-3`
TGF-β1 Sense 5`-GGCCCTGCCCCTACATTT-3’ Homo SapiensAnti-sense 5`-CCGGGTTATGCTGGTTGTACA-3`Probe 5`-ACACGCAGTACAGCAAGGTCCTGGC-3`
CTGF Sense 5`-TGTGTGACGAGCCCAAGGA-3` Homo SapiensAnti-sense 5`-TCTGGGCCAAACGTGTCTTC-3`Probe 5`-CCTGCCCTCGCGGCTTACCG-3`
α-SMA Sense 5`-GGGACGACATGGAAAAGATCTG-3` Homo SapiensAnti-sense 5`-CAGGGTGGGATGCTCTTCA-3`Probe 5`-CACTCTTTCTACAATGAGCTTCGTGTTGCCC-3
Collagen-I Sense 5`-GGCCCAGAAGAACTGGTACATC-3` Homo SapiensAnti-sense 5`-CCGCCATACTCGAACTGGAA-3Probe 5`-CCCCAAGGACAAGAGGCATGTCTG-3`
18S Sense 5`-CGGCTACCACATCCAAGGA-3` Homo SapiensAnti-sense 5`-CCAATTACAGGGCCTCGAAA-3`Probe 5`-CGCGCAAATTACCCACTCCCGA-3
IFN-γ Sense 5`-ACTGTCGCCAGCAGCTAAAA-3` Homo SapiensAnti-sense 5`-TATTGCAGGCAGGACAACCA-3`
IL-18 Sense 5`-AAGATGGCTGCTGAACCAGT-3` Homo SapiensAnti-sense 5`-GAGGCCGATTTCCTTGGTCA-3
TIMP-1 Sense 5`-CTTCTGGCATCCTGTTGTTG-3`Homo SapiensAnti-sense 5`-GGTATAAGGTGGTCTGGTTG-3`
TGF-β1 Sense 5`-CTCCCGTGGCTTCTAGTGC-3` Mus musculusAnti-sense 5`-GCCTTAGTTTGGACAGGATCTG-3`
CTGF Sense 5`-GGGCCTCTTCTGCGATTTC-3` Mus musculusAnti-sense 5`-ATCCAGGCAAGTGCATTGGTA-3`
Collagen-I Sense 5`-GCTCCTCTTAGGGGCCACT-3` Mus musculusAnti-sense 5`-CCACGTCTCACCATTGGGG-3`
α-SMA Sense 5`-GTCCCAGACATCAGGGAGTAA-3` Mus musculusAnti-sense 5`-TCGGATACTTCAGCGTCAGGA-3`
TIMP-1 Sense 5`-GCAACTCGGACCTGGTCATAA-3` Mus musculusAnti-sense 5`-ACTGTTCCTGAACTCAACT-3`
HIF-1α Sense 5`-AGGATGAGTTCTGAACGTCGAAA-3` Mus musculusAnti-sense 5`-CTGTCTAGACCACCGGCATC-3`
TNF-α Sense 5`-CCCTCACACTCAGATCATCTTCT-3`Mus musculusAnti-sense 5`-GCTACGACGTGGGCTACAG-3`
IL-1β Sense 5`-ACTGTTCCTGAACTCAACT-3` Mus musculusAnti-sense 5`-ATCTTTTGGGGTCCGTCAACT-3`
IL-18 Sense 5`-GACTCTTGCGTCAACTTCAAGG-3` Mus musculusAnti-sense 5`-CAGGCTGTCTTTTGTCAACGA-3`
NLRP3 Sense 5`-ATTACCCGCCCGAGAAAGG-3` Mus musculusAnti-sense 5`-TCGCAGCAAAGATCCACACAG-3`
Caspase-1 Sense 5`-ACAAGGCACGGGACCTATG-3` Mus musculusAnti-sense 5`-TCCCAGTCAGTCCTGGAAATG-3`
IL-6 Sense 5`-TAGTCCTTCCTACCCCAATTTCC-3` Mus musculusAnti-sense 5`-TTGGTCCTTAGCCACTCCTTC-3`
IFN-γ Sense 5`-ATGAACGCTACACACTGCATC-3` Mus musculusAnti-sense 5`-CCATCCTTTTGCCAGTTCCTC-3`
MCP-1 Sense 5`-TTAAAAACCTGGATCGGAACCAA-3` Mus musculusAnti-sense 5`-GCATTAGCTTCAGATTTACGGGT-3`
18S Sense 5’-CGGCTACCACATCCAAGGA-3’ Mus musculusAnti-sense 5’-CCAATTACAGGGCCTCGAAA-3’
SUPPLEMENTARYMATERIAL
TableS1:SequencesofprimersandprobesusedforquantitativerealtimePCR

111
LISTOFPUBLICATIONS
• HaoS,HernándezA,Quiroz-MuñozM,CéspedesC,VioC, FerreriN.ProstaglandinE2EP3
receptor downregulates COX-2 expression in themedullary thick ascending limb (mTAL)
inducedbyhypertonicNaCl.AmJPhysiolRenalPhysiol,2014;15:307
• CabreraD,WreeA,PoveroD,SolisN,HernándezA,PizarroM,MoshageH,TorresJ,Feldstein
A,Cabello-VerrugioC,BrandanE,BarreraF,ArabJ,ArreseM.Andrographolideameliorates
inflammation and fibrogenesis and attenuates inflammasome activation in experimental
non-alcoholicsteatohepatitis.ScientificReports,2017;7:3491
• ArreseM, Hernández A, Astete L, Estrada L, Cabello-Verrugio C, Cabrera D. TGF-beta and
Hepatocellular carcinoma: When a friend becomes an enemy Curr Protein Pept Sci.
2018;19:1172-1179.
• Geng Y , Hernández A, Oun A, Buist-Homan M, Blokzijl H, Faber KN, Dolga A, Moshage
Protective effect of metformin against palmitate-induced hepatic cell death. H. BBA
MolecularBasisofDisease,2020:1866(3):165621
• HernándezA,GengY,SepulvedaR,SolisN,TorresJ,ArabJP,BarreraF,CabreraD,Moshage
H,ArreseM.ChemicalHypoxia inducespro-inflammatorysignals infat-ladenhepatocytes
and contributes to cellular crosstalk with Kupffer cells. (BBA)Molecular Basis of Disease
1866,165753.2020
• HernándezA,ReyesD,ArabJP,ArreseM,MoshageH.ExtracellularVesiclesderivedfromfat
laden hypoxic hepatocytes promote a pro-fibrotic phenotype in stellate cells. (BBA)
MolecularBasisofDisease1866,165857.2020
• Hernández A, Arab JP, Reyes D, Lapitz A,MoshageH, Bañales JM, ArreseM.Extracellular
vesiclesinNAFLD/ALD:frompathobiologytotherapy.Cells9,817.2020
• MuñozN,ArreseM,HernándezA,JaraE,KalergisA,CabreraD.A.Mineralocorticoidreceptor
deficiencyinmyeloidcellsreducesliversteatosisbyimpairingactivationCD8+Tcells ina
NASHmousemodelLiverinternational.FrontImmunol563434.2020
• GengY, Serna S,HernándezA, Buist-HomanM,ArreseM,OlingaP, BlokzijH,MoshageH.
HepaticstellatecellsinducesaninflammatoryphenotypeinKupffercellsviathereleaseof
extracellularvesicles.JHEPReports.Summited2021
Related Documents











