This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Role of adenosine A2A receptor signaling in the nicotine-evoked attenuation ofreflex cardiac sympathetic control
Mahmoud M. El-Mas ⁎, Sahar M. El-gowilly, Mohamed A. Fouda, Evan I. SaadDepartment of Pharmacology and Toxicology, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt
a b s t r a c ta r t i c l e i n f o
Article history:Received 22 January 2011Revised 18 April 2011Accepted 21 April 2011Available online 29 April 2011
Keywords:NicotineAdenosine receptorsArterial baroreceptorsCentral nervous systemCardiac autonomic controlRat
Baroreflex dysfunction contributes to increased cardiovascular risk in cigarette smokers. Given theimportance of adenosinergic pathways in baroreflex control, the hypothesis was tested that defectivecentral adenosinergic modulation of cardiac autonomic activity mediates the nicotine–baroreflexinteraction. Baroreflex curves relating changes in heart rate (HR) to increases or decreases in bloodpressure (BP) evoked by i.v. doses (1–16 μg/kg) of phenylephrine (PE) and sodium nitroprusside (SNP),respectively, were constructed in conscious rats; slopes of the curves were taken as measures of baroreflexsensitivity (BRS). Nicotine (25 and 100 μg/kg i.v.) dose-dependently reduced BRSSNP in contrast to no effecton BRSPE. BRSSNP was also attenuated after intracisternal (i.c.) administration of nicotine. Similar reductionsin BRSSNP were observed in rats pretreated with atropine or propranolol. The combined treatment withnicotine and atropine produced additive inhibitory effects on BRS, an effect that was not demonstratedupon concurrent exposure to nicotine and propranolol. BRSSNP was reduced in preparations treated with i.c.8-phenyltheophylline (8-PT, nonselective adenosine receptor antagonist), 8-(3-Chlorostyryl) caffeine(CSC, A2A antagonist), or VUF5574 (A3 antagonist). In contrast, BRSSNP was preserved after blockade of A1
(DPCPX) or A2B (alloxazine) receptors or inhibition of adenosine uptake by dipyridamole. CSC or 8-PTabrogated the BRSSNP depressant effect of nicotine whereas other adenosinergic antagonists were withouteffect. Together, nicotine preferentially impairs reflex tachycardia via disruption of adenosine A2A receptor-mediated facilitation of reflex cardiac sympathoexcitation. Clinically, the attenuation by nicotine ofcompensatory sympathoexcitation may be detrimental in conditions such as hypothalamic defenseresponse, posture changes, and ventricular rhythms.
© 2011 Elsevier Inc. All rights reserved.
Introduction
Cigarette smoking is a major risk factor for hypertension, athero-sclerosis, coronary heart disease, acute myocardial infarction, andsudden cardiac death (Barnoya and Glantz, 2005; Bullen, 2008). Thenicotine content of tobacco smoke is largely responsible for thedetrimental cardiovascular actions of cigarette smoking (Balakumarand Kaur, 2009). Cessation of smoking is associated with reducedcardiovascular morbidity and mortality (Bullen, 2008). Arterial barore-ceptor dysfunction, among several other factors, contributes toincreased vulnerability of smokers to cardiovascular risk (Mancia etal., 1997). Nicotine diminishes the baroreflex gain probably via reducingarterial compliance and stretch receptor responsiveness (Giannattasioet al., 1994). The direct interaction of nicotine with central mechanismsintegrating the baroreceptor input into autonomic responses is anotherplausible mechanism for the depressant action of nicotine on barore-flexes (Ashworth-Preece et al., 1998). A thirdmechanism is the ability of
nicotine to modify the effector responsiveness to reflex autonomicmodulation (Niedermaier et al., 1993). Contradictory data of no effect(Niedermaier et al., 1993) or even facilitation of baroreflex gain bynicotine has also been reported (Bennett and Richardson, 1984;Whitescarver et al., 1991). Differences in themethod used for baroreflexassessment, animal species, and dose and duration of nicotine regimenmight be responsible for discrepancies in the reported effect of nicotineon baroreflexes.
Adenosine has emerged in recent years as a major neuromodulatorin the central nervous system. Although some of the biological effects ofnicotine, e.g. behavioral (Wooters et al. 2009) and antinociception(Homayounfar et al., 2005), appear to be modulated by adenosinergicpathways, little or no information is available concerning the role ofadenosine receptors in the hemodynamic actions of nicotine. In a recentstudy, nicotine was found to decrease and increase the binding andprotein expression, respectively, of brainstem A2A receptors of sponta-neously hypertensive rats (de Matsumoto et al., 2010). It is not clear,however,whether these changes could explainnicotinehemodynamics.Evidence supports a modulatory role for central adenosine receptors onbaroreflex activity. For example, selective activation of A2A adenosinereceptors in the nucleus of the solitary tract (NTS), the first relay station
Toxicology and Applied Pharmacology 254 (2011) 229–237
⁎ Corresponding author. Fax: +20 3 487 3273.E-mail address: [email protected] (M.M. El-Mas).
0041-008X/$ – see front matter © 2011 Elsevier Inc. All rights reserved.doi:10.1016/j.taap.2011.04.014
Contents lists available at ScienceDirect
Toxicology and Applied Pharmacology
j ourna l homepage: www.e lsev ie r.com/ locate /ytaap

Author's personal copy
of baroreceptor information in the central nervous system (Zhang andMifflin, 2010), produces effects that are consistent with baroreceptorfacilitation such as decreases in BP, HR, and renal sympathetic nerveactivity (Barraco et al., 1991; Najem et al., 2006). Others reportedvariable effects for A2A adenosine receptors on baroreflex control ofsympathetic activity (Scislo and O'Leary, 1998; Ichinose et al., 2009).Selective activation of A1 adenosine receptors resets baroreflex controlof sympathetic neural activity to higher arterial pressure and reducessympathoexcitatory responses to unloading of arterial baroreceptors(Scislo et al., 2008).
The purpose of the current study was two-fold. To re-evaluate theeffect of nicotine on baroreflex gain and its autonomic modulation and,more importantly, to determine whether central neuronal pools ofadenosine receptors modulate the nicotine–baroreflex interaction.These aims were investigated by determining: (i) the dose-dependenteffect of acute nicotine onHR responses to baroreceptor loading (by PE)or unloading (by SNP) in absence and presence of atropine (muscarinicblocker) or propranolol (β-adrenergic blocker), and (ii) the impact ofpharmacologic maneuvers that selectively or nonselectively blockcentral adenosine receptors (A1, A2A, A2B, or A3) or elevate centraladenosine levels on the nicotine–baroreflex interaction. Notably, thestrategy of pharmacologic blockade of muscarinic or β-adrenergicblockadewas employed to determine the relative contributions of vagaland sympathetic autonomic components, respectively, to the baroreflexaction of nicotine.
Materials and methods
Male Wistar rats (200–250 g, Faculty of Pharmacy animal facility,Alexandria, Egypt) were used in the present study. All experimentswere performed in strict accordance with institutional animal careand use guidelines.
Intravascular cannulation. The method described in our previousstudies (El-Mas, 1998; El-Mas et al., 2009a, 2009b)was adopted. Briefly,rats were anesthetized with thiopental (50 mg kg−1, i.p.). The pawpinch reflex and tactile response were used for testing the adequacy ofanesthesia. Catheters (each consisted of a 5-cm polyethylene-10 tubingbonded to a 15-cm polyethylene-50 tubing) were placed in theabdominal aorta and vena cava via the femoral artery and vein formeasurement of BP and intravenous administration of drugs, respec-tively. The polyethylene-10 portion was used for the intravascularsegment of the catheter. The arterial catheter was connected to a bloodpressure transducer (Model P23XL, Astro-Med, Inc., West Warwick, RI,USA) that was attached through MLAC11 Grass adapter cable to acomputerized data acquisition system with LabChart-7 pro software(Power Lab 4/30, model ML866/P, AD Instruments, Bella Vista,Australia). HR was computed from BP waveforms and displayed onanother channel of the recording system.
Finally, the catheters were tunneled subcutaneously and exterior-ized at the back of the neck between the scapulae. The catheters wereflushed with heparin (0.2 ml, 100 U/ml) and plugged by stainless steelpins. Incisions were closed by surgical clips and swabbed withpovidone–iodine solution. Each rat received an intramuscular injectionof 60,000 U of penicillin G benzathine and penicillin G procaine in anaqueous suspension (Penicid) and was housed in a separate cage.Experiments started 2 days later in conscious freely moving rats.
Intracisternal cannulation (i.c.). Five days before starting the ex-periment (i.e. 3 days before intravascular cannulation), a stainlesssteel guide cannula was implanted into the cisterna magna underthiopental anesthesia (50 mg kg−1, i.p.) as described in our previousstudies (El-Mas and Abdel-Rahman, 1999; El-Mas et al., 2009a,2009b). A steel cannula (23G; Small Parts, Miami, FL, USA) waspassed between the occipital bone and the cerebellum so that itstip protruded into the cisterna magna. The cannula was secured in
place with dental acrylic cement (Glass ionomer, China). The guidecannula was considered patent when spontaneous outflow of cere-brospinal fluid was observed and by gross post mortem histolo-gical verification following injection of 5 μl of fast green dye (EMScience; Cherry Hill, NJ). After i.c. cannulation, rats were housedindividually.
Protocols and experimental groups
Effect of nicotine on reflex chronotropic responses. In this experiment,three groups of conscious male rats (n=6–8 each) were used toinvestigate the acute effect of nicotine (25 or 100 μg/kg i.v.) or equalvolume of saline (1 ml/kg) on baroreflex-mediated HR responses topressor or depressor responses caused by PE and SNP, respectively. Thesedoses of nicotine have been used in previous studies and produced dose-related hemodynamic effects (Marano etal., 1999). Each rat in a particulargroupwas employed in two experiments (2 and 4 days post intravascularcannulation). Baroreflex curves of PE or SNP were generated before and10min after i.v. administration of nicotine or saline. In the firstexperiment, 50% of rats in a particular group received PE and the other50% received SNP. In 100 μg/kg i.v. the second experiment, theadministration of PE and SNP was crossed over. For the determinationof plasma levels of cotinine, the main metabolic product of nicotine, twoblood samples (0.5 ml each) were drawn from each rat 10 and 30 minafter the administration of nicotine. The samples were centrifuged at800 g for 10 min and the plasma was aspirated and stored at −20 °C tillanalyzed for cotinine level by the chemiluminescent immunoassays(EURO/DPC Ltd, DPC Scandinavia, Mölmdal, Denmark) as reportedelsewhere (Andersen et al., 2009).
To determine whether the interaction of nicotine with reflextachycardic activity was SNP-specific, two more groups of rats(n=5 each) was used to determine the effect of nicotine (100 μg/kgi.v.) on reflex HR responses elicited by hydralazine (1 mg/kg i.v.). Peakchanges in MAP and HR caused by hydralazine in rats pretreated withsaline or nicotine were measured and used for the calculation ofBRShydralazine.
On the experiment day, the arterial catheter was connected to thepressure transducer and Power Lab data acquisition system formeasurement of BP and HR as mentioned above. A period of 30 minwas allowed at the beginning of the experiment for stabilization of BPand HR. For the generation of baroreflex curves, randomized i.v. doses(1–16 μg/kg) of PE or SNP were injected every 5 min as in ourprevious studies (El-Mas and Abdel-Rahman, 1997a; El-Mas et al.,2002). This time interval was adequate for BP and HR to regainbaseline levels. PE and SNP were dissolved in saline and the injectionvolume was kept constant at 0.05 ml/100 g body weight with a flushvolume of approximately 0.1 ml saline. The mean arterial pressure(MAP, diastolic plus one third pulse pressure) and HR values beforeand after PE or SNP administration were computed and the peakchanges in both variables (ΔMAP and ΔHR) were used for construc-tion of the baroreflex curves (El-Mas and Abdel-Rahman, 1997a;El-Mas et al., 2002).
Autonomic modulation of nicotine–baroreflex interaction. Because re-sults of the preceding experiment showed that nicotine reducedbaroreflex responsiveness to SNP and not PE, more studies wereundertaken to assess the relative contributions of sympathetic andvagal modalities, the two main efferent systems mediating the finalbaroreflex response (Glick and Braunwald, 1965; Coleman, 1980), tothe nicotine–BRSSNP interaction. Two groups of rats (n=6–8 each)were utilized to evaluate the effect of selective pharmacologic blockadeof cardiac vagal or sympathetic activity by atropine and propranolol(1 mg/kg each), respectively, on the BRSSNP response to subsequentlyadministered nicotine (100 μg/kg). After hemodynamic stabilization,rats were treated with atropine (n=6) or propranolol (n=8) andthis was followed 10 min later by the construction of the dose
230 M.M. El-Mas et al. / Toxicology and Applied Pharmacology 254 (2011) 229–237

Author's personal copy
response curve of SNP (1–16 μg/kg) as described earlier. Five minutesafter the last dose of SNP, rats were treated with nicotine (100 μg/kg)for another 10 min after which the dose–effect relationship of SNPwas re-established.
Central adenosinergic modulation of the nicotine–BRSSNP interaction.The issue whether central adenosine receptors mediate the depressantaction of nicotine on BRSSNP was investigated. A total of 7 groups of rats(n=6–8 each)were used to determine the effect of nicotine (100 μg/kgi.v.) on BRSSNP in rats pretreated intracisternally with 8-phenyltheophylline (8-PT, 20 μg/rat, nonselective adenosine receptorantagonist), DPCPX (6 μg/rat, selective A1 receptor antagonist), CSC(40 μg/rat, selective A2A receptor antagonist), alloxazine (40 μg/rat,selective A2B receptor antagonist), VUF5574 (2 μg/rat, selective A3
receptor antagonist), dipyridamole (150 μg/rat, adenosine uptakeinhibitor), or their vehicle (DMSO). The baroreflex curve of SNP (1–16 μg/kg) was established 10min after administration of each of theabove mentioned drugs. Nicotine was then injected intravenously andthe baroreflex curve of SNP was re-established 10 min later. Anothergroup of rats (n=7) was employed to investigate the effect of i.c.administration of adenosine (100 μg/rat) on BRSSNP. The doses ofadenosinergic drugs were selected based on reported studies (Stellaet al., 1998; Nassar and Abdel-Rahman, 2006; Yildirim and Marangoz,2007). Intracisternal injections (5 μl each) were made by inserting a30 G stainless steel injector cannula (Small Parts, Miami, FL, USA) intothe guide cannula with its tip protruding 1.0 mm below the tip of theguide cannula. Drugs were infused slowly over 3–4 min.
Because our findings showed that BRSSNP was reduced by CSC andVUF5574 in contrast to no effect for other adenosine receptorantagonists, two additional groups of rats (n=5 each) were used toevaluate the effect of CSC or VUF5574 on BRSPE. The baroreflex curveof PE (1–16 μg/kg) was established before and 10 min after admin-istration of CSC or VUF5574.
The possibility that nicotine alters baroreflex activity via directinteraction with central neurons was investigated. Three groups ofrats (n=6–8) were used to determine the effect of i.c. nicotine (2 or5 μg/rat) (Craft and Milholland, 1998) or equal volume of saline (5 μl)on the dose response curve of SNP (1–16 μg/kg).
Drugs
Phenylephrine hydrochloride, hydralazine hydrochloride, sodiumnitroprusside, atropine sulphate, propranolol hydrochloride, 8-phenyltheophylline, 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX),8-(3-Chlorostyryl) caffeine (CSC), alloxazine, N-(2-Methoxyphenyl)-N′-[2-(3-pyrindinyl)-4-quinazolinyl]-urea (VUF5574), dipyridamole(SigmaChemical Co., St. Louis,MO,U.S.A.), nicotine (Merck SchuchardtOHG, Hohenbrunn, Germany), thiopental (Thiopental, BiochemieGmbH, Vienna, Austria), povidone–iodine solution (Betadine, NilePharmaceutical Co., Cairo, Egypt) and Penicid (Cid Pharmaceutical Co.,Cairo, Egypt) were purchased from commercial vendors. All adenosi-nergic drugs were dissolved in DMSO whereas other drugs weredissolved in saline.
Statistical analysis
Values are expressed as mean±SEM. The relationship betweenchanges in MAP evoked by PE or NP and associated reciprocal changesin HR was assessed by regression analysis for individual animals asdescribed in our previous studies (El-Mas and Abdel-Rahman, 1997a;El-Mas et al., 2002). The regression coefficient (slope of the regressionline, BRSPE and BRSNP) expressed as beats/min/mmHgwas taken as anindex of baroreflex responsiveness. The Student's t-test was used inthe analysis of unpaired data. Analysis of variance (ANOVA) followedby a Newman–Keuls post-hoc analysis was used for multiplecomparisons with the level of significance set at Pb0.05.
Results
Autonomic modulation of the nicotine–baroreflex interaction
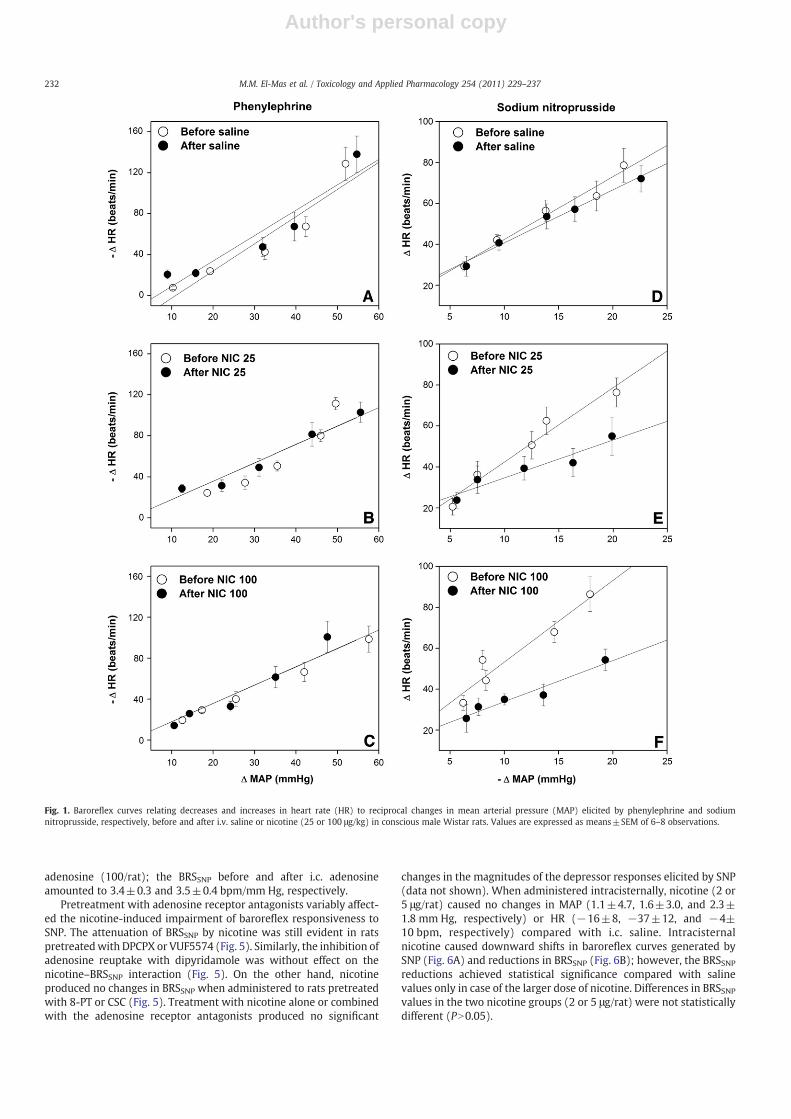
Baseline values of MAP (110±5, 112±4, and 104±8 mm Hg,respectively) and HR (385±13, 397±14, and 388±15 bpm,respectively) in conscious rats subsequently receiving saline ornicotine (25 or 100 μg/kg) were not statistically different. Plasmalevels of cotinine measured 10 and 30 min after the administrationof the 100 μg/kg dose of nicotine amounted to 96±9 and 115±13 ng/ml, respectively. Intravenous administration of nicotineproduced significant and dose-dependent increases in restingMAP (18±2 and 55±3 mm Hg, respectively) compared withsaline-treated values (2±2 mm Hg). MAP returned to pretreatmentlevels within 4–6 min of nicotine administration. HR was increasedand reduced by the lower and higher doses of nicotine, respectively.Fig. 1 depicts the effects of nicotine on baroreflex curves relatingthe baroreflex-mediated changes in HR to evoked increments anddecrements in BP. Compared with control (saline-treated) values,nicotine caused dose-dependent downward shifts in the SNP curves(Figs. 1D–F) in contrast to no effect on PE curves (Figs. 1A–C). Theslopes of the linear regression lines, which represented BRS, weresignificantly reduced by nicotine in case of SNP and not PE (Fig. 2).The reductions in BRSSNP caused by the 25 and 100 μg/kg doses ofnicotine amounted approximately to 40% and 50%, respectively.Nicotine (100 μg/kg) also significantly reduced the BRS (ΔHR/ΔMAP) tested by hydralazine (1 mg/kg) from −4.5±1.1 to −1.9±x0.8 bpm/mm Hg (Fig. 2). The administration of hydralazine to ratspretreated with nicotine or saline produced decreases and increasesin MAP and HR, respectively, that were maintained for at least30 min (data not shown).
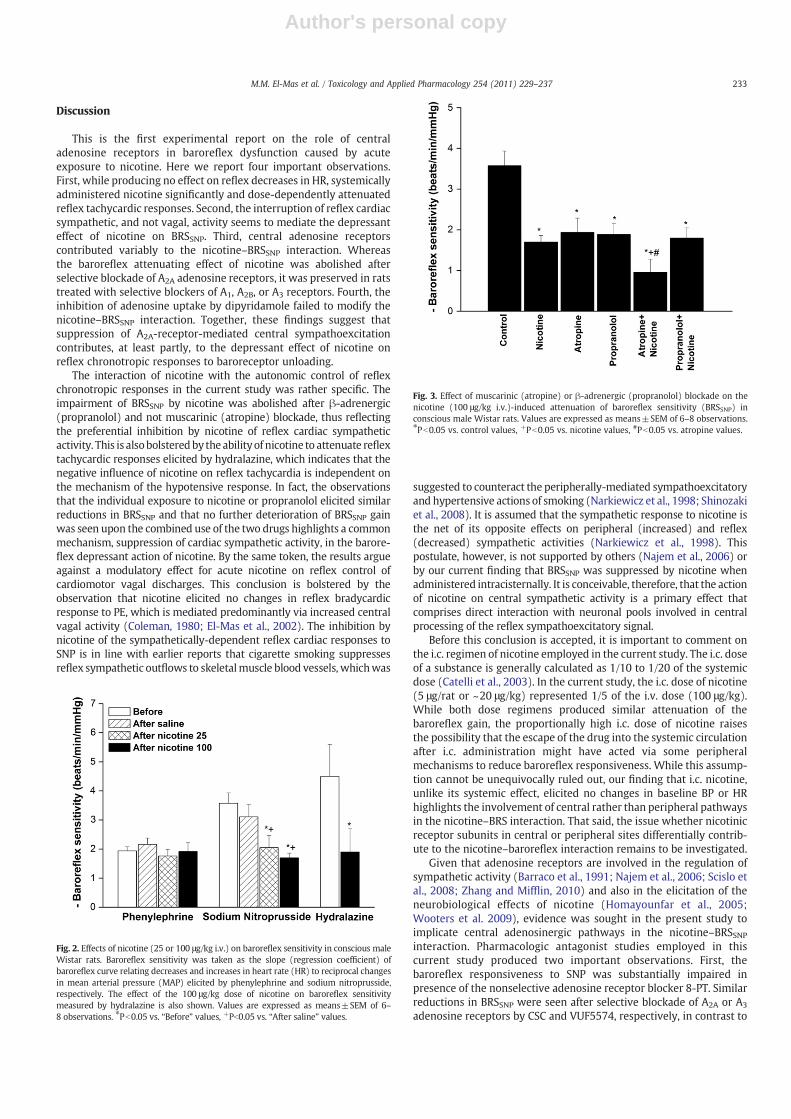
The effects of muscarinic or β-adrenergic blockade with atropineand propranolol, respectively, on the nicotine-induced impairment ofBRSSNP are shown in Fig. 3. The individual treatments with atropine orpropranolol (1 mg/kg each) caused a significant reduction in BRSSNPthat was similar in magnitude to that caused by the 100 μg/kg dose ofnicotine (Fig. 3). The subsequent administration of nicotine inatropine-treated rats elicited an additional reduction in BRSSNP(Fig. 3). In contrast, this exaggerated depressant effect on BRSSNPwas not observed in rats treated concurrently with nicotine pluspropranolol (Fig. 3).
Central adenosinergic modulation of the nicotine–BRSSNP interaction
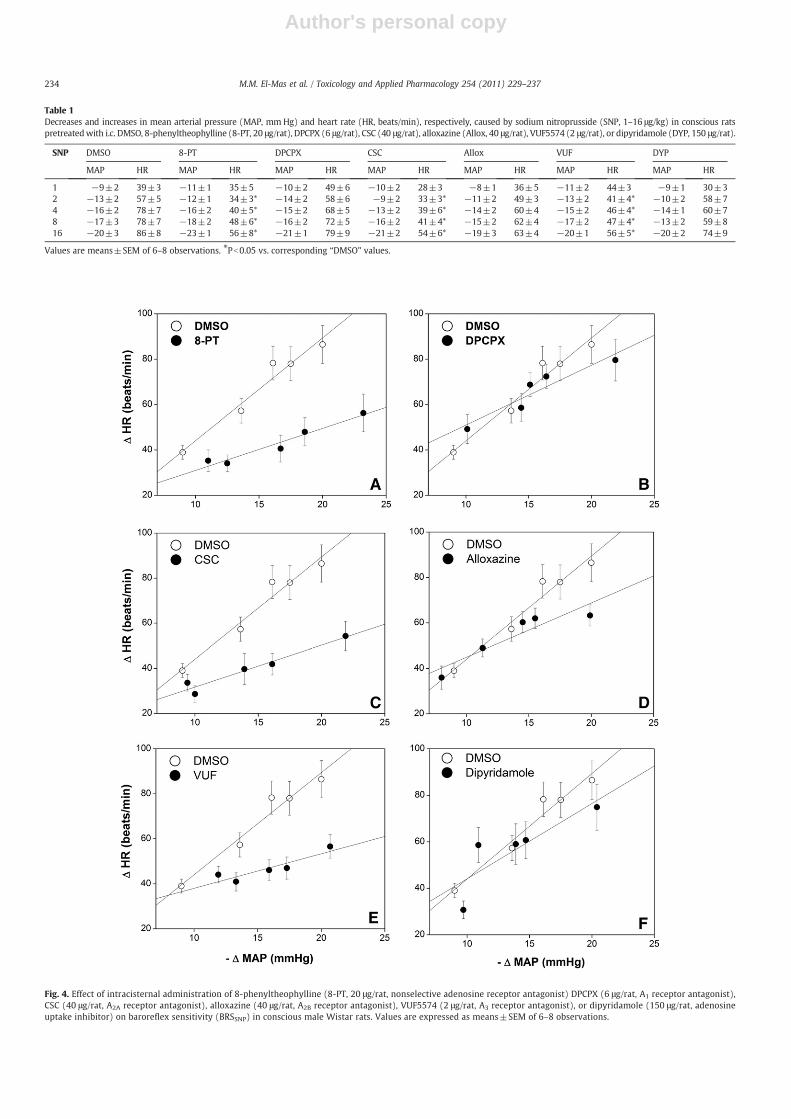
Table 1 and Figs. 4 and 5 illustrate the effects of selective ornonselective blockade of central adenosine receptors in absenceand presence of nicotine on baroreflex responsiveness measured bySNP. Compared with control (DMSO-treated) preparations, none ofthe adenosinergic antagonists significantly altered the depressorresponses elicited by SNP (Table 1). The nonselective blockade ofadenosine receptors (8-PT, 20 μg/rat) or selective blockade of A2A
(CSC, 40 μg/rat) or A3 receptors (VUF5574, 2 μg/rat) significantlyreduced reflex increases in HR caused by SNP (Table 1), causeddownward shift in the SNP baroreflex curves (Fig. 4), and decreasedthe slopes of the regression lines (BRSSNP, Fig. 5). The three drugsproduced comparable reductions in BRSSNP that amounted toapproximately 40%. On the other hand, BRSPE was significantlyreduced in rats treated with CSC (before, −1.5±0.3 bpm/mm Hg;after, −0.7±0.2 bpm/mm Hg) and not VUF5574 (before, −1.8±0.5 bpm/mm Hg; after, −1.6±0.3 bpm/mm Hg).
In contrast, the blockade of central A1 or A2B receptors by i.c.DPCPX (6 μg/rat) and alloxazine (40 μg/rat), respectively, or theinhibition of adenosine uptake by dipyridamole (150 μg/rat) failed tomodify reflex tachycardic responses to SNP (Table 1; Figs. 4, 5).Likewise, BRSSNP was not affected by the i.c. administration of
231M.M. El-Mas et al. / Toxicology and Applied Pharmacology 254 (2011) 229–237
Author's personal copy
adenosine (100/rat); the BRSSNP before and after i.c. adenosineamounted to 3.4±0.3 and 3.5±0.4 bpm/mm Hg, respectively.
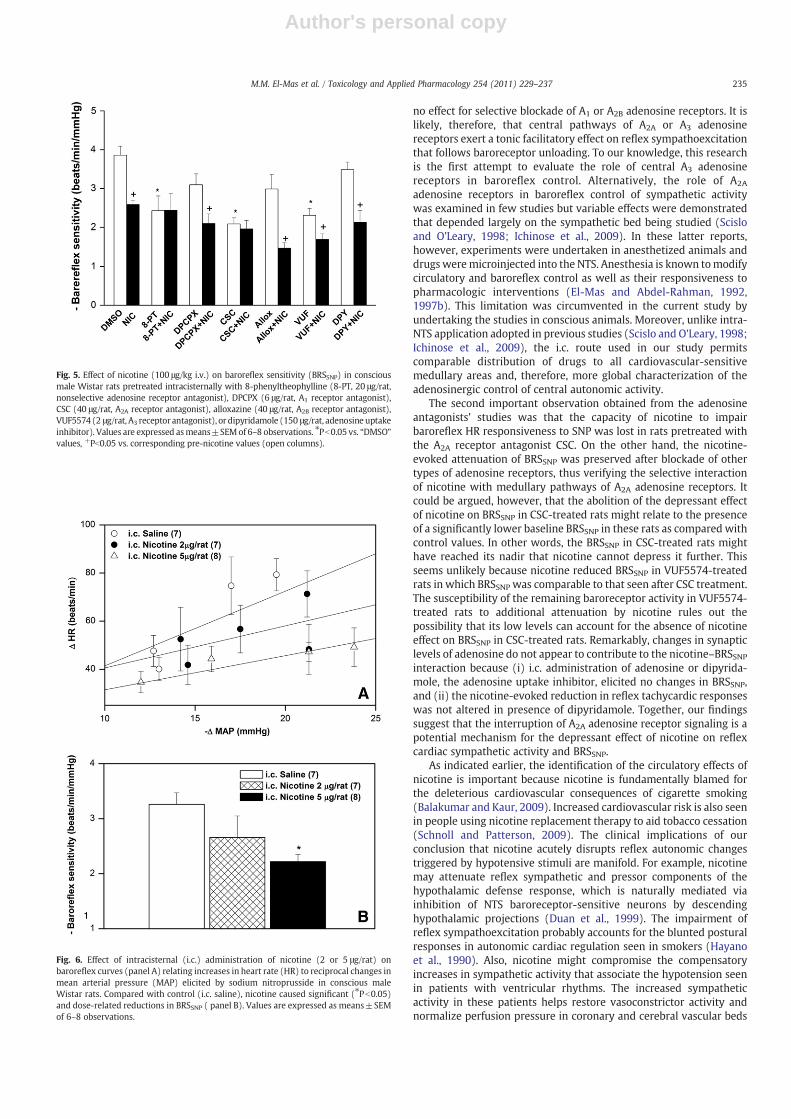
Pretreatment with adenosine receptor antagonists variably affect-ed the nicotine-induced impairment of baroreflex responsiveness toSNP. The attenuation of BRSSNP by nicotine was still evident in ratspretreatedwith DPCPX or VUF5574 (Fig. 5). Similarly, the inhibition ofadenosine reuptake with dipyridamole was without effect on thenicotine–BRSSNP interaction (Fig. 5). On the other hand, nicotineproduced no changes in BRSSNP when administered to rats pretreatedwith 8-PT or CSC (Fig. 5). Treatment with nicotine alone or combinedwith the adenosine receptor antagonists produced no significant
changes in the magnitudes of the depressor responses elicited by SNP(data not shown). When administered intracisternally, nicotine (2 or5 μg/rat) caused no changes in MAP (1.1±4.7, 1.6±3.0, and 2.3±1.8 mm Hg, respectively) or HR (−16±8, −37±12, and −4±10 bpm, respectively) compared with i.c. saline. Intracisternalnicotine caused downward shifts in baroreflex curves generated bySNP (Fig. 6A) and reductions in BRSSNP (Fig. 6B); however, the BRSSNPreductions achieved statistical significance compared with salinevalues only in case of the larger dose of nicotine. Differences in BRSSNPvalues in the two nicotine groups (2 or 5 μg/rat) were not statisticallydifferent (PN0.05).
Fig. 1. Baroreflex curves relating decreases and increases in heart rate (HR) to reciprocal changes in mean arterial pressure (MAP) elicited by phenylephrine and sodiumnitroprusside, respectively, before and after i.v. saline or nicotine (25 or 100 μg/kg) in conscious male Wistar rats. Values are expressed as means±SEM of 6–8 observations.
232 M.M. El-Mas et al. / Toxicology and Applied Pharmacology 254 (2011) 229–237
Author's personal copy
Discussion
This is the first experimental report on the role of centraladenosine receptors in baroreflex dysfunction caused by acuteexposure to nicotine. Here we report four important observations.First, while producing no effect on reflex decreases in HR, systemicallyadministered nicotine significantly and dose-dependently attenuatedreflex tachycardic responses. Second, the interruption of reflex cardiacsympathetic, and not vagal, activity seems to mediate the depressanteffect of nicotine on BRSSNP. Third, central adenosine receptorscontributed variably to the nicotine–BRSSNP interaction. Whereasthe baroreflex attenuating effect of nicotine was abolished afterselective blockade of A2A adenosine receptors, it was preserved in ratstreated with selective blockers of A1, A2B, or A3 receptors. Fourth, theinhibition of adenosine uptake by dipyridamole failed to modify thenicotine–BRSSNP interaction. Together, these findings suggest thatsuppression of A2A-receptor-mediated central sympathoexcitationcontributes, at least partly, to the depressant effect of nicotine onreflex chronotropic responses to baroreceptor unloading.
The interaction of nicotine with the autonomic control of reflexchronotropic responses in the current study was rather specific. Theimpairment of BRSSNP by nicotine was abolished after β-adrenergic(propranolol) and not muscarinic (atropine) blockade, thus reflectingthe preferential inhibition by nicotine of reflex cardiac sympatheticactivity. This is alsobolsteredby the abilityof nicotine to attenuate reflextachycardic responses elicited by hydralazine, which indicates that thenegative influence of nicotine on reflex tachycardia is independent onthe mechanism of the hypotensive response. In fact, the observationsthat the individual exposure to nicotine or propranolol elicited similarreductions in BRSSNP and that no further deterioration of BRSSNP gainwas seen upon the combined use of the two drugs highlights a commonmechanism, suppression of cardiac sympathetic activity, in the barore-flex depressant action of nicotine. By the same token, the results argueagainst a modulatory effect for acute nicotine on reflex control ofcardiomotor vagal discharges. This conclusion is bolstered by theobservation that nicotine elicited no changes in reflex bradycardicresponse to PE, which is mediated predominantly via increased centralvagal activity (Coleman, 1980; El-Mas et al., 2002). The inhibition bynicotine of the sympathetically-dependent reflex cardiac responses toSNP is in line with earlier reports that cigarette smoking suppressesreflex sympathetic outflows to skeletalmuscle blood vessels, whichwas
suggested to counteract the peripherally-mediated sympathoexcitatoryand hypertensive actions of smoking (Narkiewicz et al., 1998; Shinozakiet al., 2008). It is assumed that the sympathetic response to nicotine isthe net of its opposite effects on peripheral (increased) and reflex(decreased) sympathetic activities (Narkiewicz et al., 1998). Thispostulate, however, is not supported by others (Najem et al., 2006) orby our current finding that BRSSNP was suppressed by nicotine whenadministered intracisternally. It is conceivable, therefore, that the actionof nicotine on central sympathetic activity is a primary effect thatcomprises direct interaction with neuronal pools involved in centralprocessing of the reflex sympathoexcitatory signal.
Before this conclusion is accepted, it is important to comment onthe i.c. regimen of nicotine employed in the current study. The i.c. doseof a substance is generally calculated as 1/10 to 1/20 of the systemicdose (Catelli et al., 2003). In the current study, the i.c. dose of nicotine(5 μg/rat or ~20 μg/kg) represented 1/5 of the i.v. dose (100 μg/kg).While both dose regimens produced similar attenuation of thebaroreflex gain, the proportionally high i.c. dose of nicotine raisesthe possibility that the escape of the drug into the systemic circulationafter i.c. administration might have acted via some peripheralmechanisms to reduce baroreflex responsiveness. While this assump-tion cannot be unequivocally ruled out, our finding that i.c. nicotine,unlike its systemic effect, elicited no changes in baseline BP or HRhighlights the involvement of central rather than peripheral pathwaysin the nicotine–BRS interaction. That said, the issue whether nicotinicreceptor subunits in central or peripheral sites differentially contrib-ute to the nicotine–baroreflex interaction remains to be investigated.
Given that adenosine receptors are involved in the regulation ofsympathetic activity (Barraco et al., 1991; Najem et al., 2006; Scislo etal., 2008; Zhang and Mifflin, 2010) and also in the elicitation of theneurobiological effects of nicotine (Homayounfar et al., 2005;Wooters et al. 2009), evidence was sought in the present study toimplicate central adenosinergic pathways in the nicotine–BRSSNPinteraction. Pharmacologic antagonist studies employed in thiscurrent study produced two important observations. First, thebaroreflex responsiveness to SNP was substantially impaired inpresence of the nonselective adenosine receptor blocker 8-PT. Similarreductions in BRSSNP were seen after selective blockade of A2A or A3
adenosine receptors by CSC and VUF5574, respectively, in contrast to
Fig. 2. Effects of nicotine (25 or 100 μg/kg i.v.) on baroreflex sensitivity in conscious maleWistar rats. Baroreflex sensitivity was taken as the slope (regression coefficient) ofbaroreflex curve relating decreases and increases in heart rate (HR) to reciprocal changesin mean arterial pressure (MAP) elicited by phenylephrine and sodium nitroprusside,respectively. The effect of the 100 μg/kg dose of nicotine on baroreflex sensitivitymeasured by hydralazine is also shown. Values are expressed as means±SEM of 6–8 observations. ⁎Pb0.05 vs. “Before” values, +Pb0.05 vs. “After saline” values.
Fig. 3. Effect of muscarinic (atropine) or β-adrenergic (propranolol) blockade on thenicotine (100 μg/kg i.v.)-induced attenuation of baroreflex sensitivity (BRSSNP) inconscious male Wistar rats. Values are expressed as means±SEM of 6–8 observations.⁎Pb0.05 vs. control values, +Pb0.05 vs. nicotine values, #Pb0.05 vs. atropine values.
233M.M. El-Mas et al. / Toxicology and Applied Pharmacology 254 (2011) 229–237
Author's personal copy
Table 1Decreases and increases in mean arterial pressure (MAP, mm Hg) and heart rate (HR, beats/min), respectively, caused by sodium nitroprusside (SNP, 1–16 μg/kg) in conscious ratspretreatedwith i.c. DMSO, 8-phenyltheophylline (8-PT, 20 μg/rat), DPCPX (6 μg/rat), CSC (40 μg/rat), alloxazine (Allox, 40 μg/rat), VUF5574 (2 μg/rat), or dipyridamole (DYP, 150 μg/rat).
SNP DMSO 8-PT DPCPX CSC Allox VUF DYP
MAP HR MAP HR MAP HR MAP HR MAP HR MAP HR MAP HR
1 −9±2 39±3 −11±1 35±5 −10±2 49±6 −10±2 28±3 −8±1 36±5 −11±2 44±3 −9±1 30±32 −13±2 57±5 −12±1 34±3* −14±2 58±6 −9±2 33±3* −11±2 49±3 −13±2 41±4* −10±2 58±74 −16±2 78±7 −16±2 40±5* −15±2 68±5 −13±2 39±6* −14±2 60±4 −15±2 46±4* −14±1 60±78 −17±3 78±7 −18±2 48±6* −16±2 72±5 −16±2 41±4* −15±2 62±4 −17±2 47±4* −13±2 59±816 −20±3 86±8 −23±1 56±8* −21±1 79±9 −21±2 54±6* −19±3 63±4 −20±1 56±5* −20±2 74±9
Values are means±SEM of 6–8 observations. ⁎Pb0.05 vs. corresponding “DMSO” values.
Fig. 4. Effect of intracisternal administration of 8-phenyltheophylline (8-PT, 20 μg/rat, nonselective adenosine receptor antagonist) DPCPX (6 μg/rat, A1 receptor antagonist),CSC (40 μg/rat, A2A receptor antagonist), alloxazine (40 μg/rat, A2B receptor antagonist), VUF5574 (2 μg/rat, A3 receptor antagonist), or dipyridamole (150 μg/rat, adenosineuptake inhibitor) on baroreflex sensitivity (BRSSNP) in conscious male Wistar rats. Values are expressed as means±SEM of 6–8 observations.
234 M.M. El-Mas et al. / Toxicology and Applied Pharmacology 254 (2011) 229–237
Author's personal copy
no effect for selective blockade of A1 or A2B adenosine receptors. It islikely, therefore, that central pathways of A2A or A3 adenosinereceptors exert a tonic facilitatory effect on reflex sympathoexcitationthat follows baroreceptor unloading. To our knowledge, this researchis the first attempt to evaluate the role of central A3 adenosinereceptors in baroreflex control. Alternatively, the role of A2A
adenosine receptors in baroreflex control of sympathetic activitywas examined in few studies but variable effects were demonstratedthat depended largely on the sympathetic bed being studied (Scisloand O'Leary, 1998; Ichinose et al., 2009). In these latter reports,however, experiments were undertaken in anesthetized animals anddrugsweremicroinjected into the NTS. Anesthesia is known tomodifycirculatory and baroreflex control as well as their responsiveness topharmacologic interventions (El-Mas and Abdel-Rahman, 1992,1997b). This limitation was circumvented in the current study byundertaking the studies in conscious animals. Moreover, unlike intra-NTS application adopted in previous studies (Scislo and O'Leary, 1998;Ichinose et al., 2009), the i.c. route used in our study permitscomparable distribution of drugs to all cardiovascular-sensitivemedullary areas and, therefore, more global characterization of theadenosinergic control of central autonomic activity.
The second important observation obtained from the adenosineantagonists' studies was that the capacity of nicotine to impairbaroreflex HR responsiveness to SNP was lost in rats pretreated withthe A2A receptor antagonist CSC. On the other hand, the nicotine-evoked attenuation of BRSSNP was preserved after blockade of othertypes of adenosine receptors, thus verifying the selective interactionof nicotine with medullary pathways of A2A adenosine receptors. Itcould be argued, however, that the abolition of the depressant effectof nicotine on BRSSNP in CSC-treated rats might relate to the presenceof a significantly lower baseline BRSSNP in these rats as compared withcontrol values. In other words, the BRSSNP in CSC-treated rats mighthave reached its nadir that nicotine cannot depress it further. Thisseems unlikely because nicotine reduced BRSSNP in VUF5574-treatedrats in which BRSSNP was comparable to that seen after CSC treatment.The susceptibility of the remaining baroreceptor activity in VUF5574-treated rats to additional attenuation by nicotine rules out thepossibility that its low levels can account for the absence of nicotineeffect on BRSSNP in CSC-treated rats. Remarkably, changes in synapticlevels of adenosine do not appear to contribute to the nicotine–BRSSNPinteraction because (i) i.c. administration of adenosine or dipyrida-mole, the adenosine uptake inhibitor, elicited no changes in BRSSNP,and (ii) the nicotine-evoked reduction in reflex tachycardic responseswas not altered in presence of dipyridamole. Together, our findingssuggest that the interruption of A2A adenosine receptor signaling is apotential mechanism for the depressant effect of nicotine on reflexcardiac sympathetic activity and BRSSNP.
As indicated earlier, the identification of the circulatory effects ofnicotine is important because nicotine is fundamentally blamed forthe deleterious cardiovascular consequences of cigarette smoking(Balakumar and Kaur, 2009). Increased cardiovascular risk is also seenin people using nicotine replacement therapy to aid tobacco cessation(Schnoll and Patterson, 2009). The clinical implications of ourconclusion that nicotine acutely disrupts reflex autonomic changestriggered by hypotensive stimuli are manifold. For example, nicotinemay attenuate reflex sympathetic and pressor components of thehypothalamic defense response, which is naturally mediated viainhibition of NTS baroreceptor-sensitive neurons by descendinghypothalamic projections (Duan et al., 1999). The impairment ofreflex sympathoexcitation probably accounts for the blunted posturalresponses in autonomic cardiac regulation seen in smokers (Hayanoet al., 1990). Also, nicotine might compromise the compensatoryincreases in sympathetic activity that associate the hypotension seenin patients with ventricular rhythms. The increased sympatheticactivity in these patients helps restore vasoconstrictor activity andnormalize perfusion pressure in coronary and cerebral vascular beds
Fig. 5. Effect of nicotine (100 μg/kg i.v.) on baroreflex sensitivity (BRSSNP) in consciousmale Wistar rats pretreated intracisternally with 8-phenyltheophylline (8-PT, 20 μg/rat,nonselective adenosine receptor antagonist), DPCPX (6 μg/rat, A1 receptor antagonist),CSC (40 μg/rat, A2A receptor antagonist), alloxazine (40 μg/rat, A2B receptor antagonist),VUF5574 (2 μg/rat, A3 receptor antagonist), or dipyridamole (150 μg/rat, adenosineuptakeinhibitor). Values are expressed asmeans±SEMof 6–8 observations. ⁎Pb0.05 vs. “DMSO”values, +Pb0.05 vs. corresponding pre-nicotine values (open columns).
Fig. 6. Effect of intracisternal (i.c.) administration of nicotine (2 or 5 μg/rat) onbaroreflex curves (panel A) relating increases in heart rate (HR) to reciprocal changes inmean arterial pressure (MAP) elicited by sodium nitroprusside in conscious maleWistar rats. Compared with control (i.c. saline), nicotine caused significant (⁎Pb0.05)and dose-related reductions in BRSSNP ( panel B). Values are expressed as means±SEMof 6–8 observations.
235M.M. El-Mas et al. / Toxicology and Applied Pharmacology 254 (2011) 229–237

Author's personal copy
(Smith et al., 1999). Alternatively, the effect of nicotine in ventricularrhythmsmay be beneficial in the sense that reductions in sympatheticactivity could reduce the progression of ventricular tachycardia intofibrillation (Smith et al., 2010). Importantly, plasma levels of cotinine,the main metabolic product of nicotine, observed after administrationof the 100 μg/kg dose of nicotine amounted to ~100 ng/ml, which aresimilar to levels achieved in humans after moderate cigarette smoking(Shepperd et al., 2009). These cotinine levels, however, are far lessthan those observed during acute toxicity episodes (~0.9–1.4 μg/ml)that are associated with symptoms such as nausea, vomiting,headache and dizziness (Davies et al., 2001; Satora et al., 2009).
Remarkably, reported findings established critical modulatoryroles for autonomic and adenosinergic profiles in cardiovascularpathophysiology. Impairment of autonomic and baroreflex modalitiescontributes to hypertension, coronary artery disease and ischemicstroke (Doux and Yun, 2006; Kougias et al., 2010). On the other hand,adenosine diminishes myocardial injury following myocardial infarc-tion (Hodge et al., 1998) and A2A adenosine receptor activation guardsagainst atherosclerosis (McPherson et al., 2001) prevents progressionof atherosclerosis via upregulating proteins involved in reversecholesterol transport and prevention of foam cell transformation(Reiss et al., 2004). With this idea in mind, the issue whetheralterations in autonomic and adenosinergic pathways caused bynicotine contribute to the increased incidence of cardiovascular riskthat associates cigarette smoking remains to be investigated. Inaddition to nicotine, the roles of carbon monoxide and other volatilesubstances of the cigarette smoke in atherosclerosis and endotheliumdysfunction caused by smoking cannot be overlooked (ChellandCampbell et al., 2008).
In summary, our study provides new insights into cellularmechanisms of the nicotine–baroreflex interaction and its autonomicregulation. Nicotine dose-dependently impairs HR responsiveness tobaroreceptor unloading via inhibition of the A2A adenosine receptor-mediated control of reflex sympathoexcitatory outflow to cardiactissues. In support of this conclusion are the observations: (i) acutenicotine suppressed reflex tachycardic and not bradycardic responses,(ii) the inhibitory effect of nicotine on BRSSNP was abolished after β-adrenergic and notmuscarinic blockade, and (iii) selective blockade ofA2A adenosine receptors diminished reflex increases in HR andabolished BRSSNP attenuation caused by subsequently administerednicotine. This pattern of autonomic changes produced by nicotine islikely to influence compensatory sympathoexcitatory responses thatassociate conditions such as hypothalamic defense response, posturechanges, and ventricular rhythms.
Acknowledgment
This work was supported by the Faculty of Pharmacy, AlexandriaUniversity, Egypt.
References
Andersen, M.R., Simonsen, U., Uldbjerg, N., Aalkjær, C., Stender, S., 2009. Smokingcessation early in pregnancy and birth weight, length, head circumference, andendothelial nitric oxide synthase activity in umbilical and chorionic vessels. AnObservational Study of Healthy Singleton Pregnancies. Circulation 119, 857–864.
Ashworth-Preece, M., Jarrott, B., Lawrence, A.J., 1998. Nicotinic acetylcholine receptorsin the rat and primate nucleus tractus solitarius and on rat and human inferiorvagal (nodose) ganglia: evidence from in vivo microdialysis and [125I]alpha-bungarotoxin autoradiography. Neuroscience 83, 1113–1122.
Balakumar, P., Kaur, J., 2009. Is nicotine a key player or spectator in the induction andprogression of cardiovascular disorders? Pharmacol. Res. 60, 361–368.
Barnoya, J., Glantz, S.A., 2005. Cardiovascular effects of secondhand smoke: nearly aslarge as smoking. Circulation 111, 298–2684.
Barraco, R.A., el-Ridi, M.R., Ergene, E., Phillis, J.W., 1991. Adenosine receptor subtypes inthe brainstem mediate distinct cardiovascular response patterns. Brain Res. Bull.26, 59–84.
Bennett, C.H., Richardson, D.R., 1984. Effects of chronic tobacco smoke exposure onarterial blood pressure regulation. Am. J. Physiol. 247, H556–H562.
Bullen, C., 2008. Impact of tobacco smoking and smoking cessation on cardiovascularrisk and disease. Expert Rev. Cardiovasc. Ther. 6, 883–895.
Catelli, M., Feldman, J., Bousquet, P., Tibirica, E., 2003. Protective effects of centrallyacting sympathomodulatory drugs on myocardial ischemia induced by sympa-thetic overactivity in rabbits. Braz. J. Med. Biol. Res. 36, 85–95.
Chelland Campbell, S., Moffatt, R.J., Stamford, B.A., 2008. Smoking and smokingcessation: the relationship between cardiovascular disease and lipoproteinmetabolism: a review. Atherosclerosis 201, 225–235.
Coleman, T.G., 1980. Arterial baroreflex control of heart rate in the conscious rat. Am. J.Physiol. 238, H515–H520.
Craft, R.M., Milholland, R.B., 1998. Sex differences in cocaine- and nicotine-inducedantinociception in the rat. Brain Res. 809, 137–140.
Davies, P., Levy, S., Pahari, A., Martinez, D., 2001. Acute nicotine poisoning associatedwith a traditional remedy for eczema. Arch. Dis. Child. 85, 500–502.
deMatsumoto, J.P., de Ferrari, M.F., Fior-Chadi, D.R., 2010. Adenosine receptor type 2a isdifferently modulated by nicotine in dorsal brainstem cells of Wistar Kyoto andspontaneously hypertensive rats. J. Neural Transm. 117, 799–807.
Doux, J.D., Yun, A.J., 2006. The link between carotid artery disease and ischemic strokemay be partially attributable to autonomic dysfunction and failure of cerebrovas-cular autoregulation triggered by Darwinian maladaptation of the carotidbaroreceptors and chemoreceptors. Med. Hypotheses 66, 176–181.
Duan, Y.F., Kopin, I.J., Goldstein, D.S., 1999. Stimulation of the paraventricular nucleusmodulates firing of neurons in the nucleus of the solitary tract. Am. J. Physiol. 277,R403–R411.
El-Mas, M.M., 1998. Facilitation of reflex bradycardia does not contribute to the enhancedhypotensive effect of clonidine in aortic barodenervated rats. J. Cardiovasc. Pharmacol.31, 869–875.
El-Mas, M.M., Abdel-Rahman, A.A., 1992. Role of aortic baroreceptors in ethanol-induced impairment of baroreflex control of heart rate in conscious rats.J. Pharmacol. Exp. Ther. 262, 157–165.
El-Mas, M.M., Abdel-Rahman, A.A., 1997a. Aortic barodenervation upregulates α2-adrenoceptors in the nucleus tractus solitarius and rostral ventrolateral medulla:an autoradiographic study. Neuroscience 79, 581–590.
El-Mas, M.M., Abdel-Rahman, A.A., 1997b. Contrasting effects of urethane, ketamine,and thiopental anesthesia on ethanol–clonidine hemodynamic interaction. AlcoholExp Clin Res 21, 19–27.
El-Mas, M.M., Abdel-Rahman, A.A., 1999. Role of the sympathetic control of vascularresistance in ethanol–clonidine hemodynamic interaction in SHRs. J. Cardiovasc.Pharmacol. 34, 589–596.
El-Mas, M.M., Afify, E.A., Omar, A.G., Sharabi, F.M., 2002. Cyclosporine attenuates theautonomic modulation of reflex chronotropic responses in conscious rats. Can. J.Physiol. Pharmacol. 80, 766–776.
El-Mas, M.M., El-Gowelli, H.A., Ghazal, A.M., Harraz, O.F., Mohy El-Din, M.M., 2009a.Facilitation of central imidazoline I1-site/extracellular signal-regulated kinase/p38mitogen-activated protein kinase signalling mediates the hypotensive effect ofethanol in rats with acute renal failure. Br. J. Pharmacol. 158, 1629–1640.
El-Mas, M.M., Omar, A.G., Helmy, M.M., Mohy El-Din, M.M., 2009b. Interruption ofcentral neuronal pathway of imidazoline I1 receptor mediates the hypertensiveeffect of cyclosporine in rats. Brain Res. 1248, 96–106.
Giannattasio, C., Mangoni, A.A., Stella, M.L., Carugo, S., Grassi, G., Mancia, G., 1994. Acuteeffects of smoking on radial artery compliance in humans. J. Hypertens. 12,691–696.
Glick, G., Braunwald, E., 1965. Relative roles of the sympathetic and parasympatheticnervous system in the reflex control of heart rate. Circ. Res. 16, 363–375.
Hayano, J., Yamada, M., Sakakibara, Y., Fujinami, T., Yokoyama, K., Watanabe, Y., Takata,K., 1990. Short- and long-term effects of cigarette smoking on heart rate variability.Am. J. Cardiol. 65, 84–88.
Hodge, K.R., Bailey, J.K., Lobl, D.A., Laudon, A.B., Gibbons, R.J., 1998. Intravenousadenosine and lidocaine in patients with acute mycocardial infarction. Am. Heart J.136, 196–204.
Homayounfar, H., Jamali-Raeufy, N., Sahebgharani, M., Zarrindast, M.R., 2005.Adenosine receptor mediates nicotine-induced antinociception in formalin test.Pharmacol. Res. 51, 197–203.
Ichinose, T.K., O'Leary, D.S., Scislo, T.J., 2009. Activation of NTS A2a adenosine receptorsdifferentially resets baroreflex control of renal vs. adrenal sympathetic nerveactivity. Am. J. Physiol. Heart Circ. Physiol. 296, H1058–H1068.
Kougias, P., Weakley, S.M., Yao, Q., Lin, P.H., Chen, C., 2010. Arterial baroreceptors in themanagement of systemic hypertension. Med. Sci. Monit. 16, RA1–RA8.
Mancia, G., Groppelli, A., Di Rienzo, M., Castiglioni, P., Parati, G., 1997. Smoking impairsbaroreflex sensitivity in humans. Am. J. Physiol. 273, H1555–H1560.
Marano, G., Ramirez, A., Mori, I., Ferrari, A.U., 1999. Sympathectomy inhibits thevasoactive effects of nicotine in conscious rats. Cardiovasc. Res. 42, 201–205.
McPherson, J.A., Barringhaus, K.G., Bishop, G.G., Sanders, J.M., Rieger, J.M., Hesselbacher,S.E., Gimple, L.W., Powers, E.R., Macdonald, T., Sullivan, G., Linden, J., Sarembock, I.J.,2001. Adenosine A(2A) receptor stimulation reduces inflammation and neointimalgrowth in a murine carotid ligation model. Arterioscler. Thromb. Vasc. Biol. 21,791–796.
Najem, B., Houssière, A., Pathak, A., Janssen, C., Lemogoum, D., Xhaët, O., Cuylits, N., vande Borne, P., 2006. Acute cardiovascular and sympathetic effects of nicotinereplacement therapy. Hypertension 47, 1162–1167.
Narkiewicz, K., van de Borne, P.J., Hausberg, M., Cooley, R.L., Winniford, M.D., Davison,D.E., Somers, V.K., 1998. Cigarette smoking increases sympathetic outflow inhumans. Circulation 98, 528–534.
Nassar, N., Abdel-Rahman, A.A., 2006. Central adenosine signaling plays a key role incentrallymediated hypotension in conscious aortic barodenervated rats. J. Pharmacol.Exp. Ther. 318, 255–261.
236 M.M. El-Mas et al. / Toxicology and Applied Pharmacology 254 (2011) 229–237

Author's personal copy
Niedermaier, O.N., Smith, M.L., Beightol, L.A., Zukowska-Grojec, Z., Goldstein, D.S.,Eckberg, D.L., 1993. Influence of cigarette smoking on human autonomic function.Circulation 88, 562–571.
Reiss, A.B., Rahman, M.M., Chan, E.S., Montesinos, M.C., Awadallah, N.W., Cronstein,B.N., 2004. Adenosine A2A receptor occupancy stimulates expression of proteinsinvolved in reverse cholesterol transport and inhibits foam cell formation inmacrophages. J. Leukoc. Biol. 76, 727–734.
Satora, L., Goszcz, H., Gomółka, E., Biedroń, W., 2009. Green tobacco sickness in Poland.Pol. Arch. Med. Wewn. 119, 184–186.
Schnoll, R.A., Patterson, F., 2009. Sex heterogeneity in pharmacogenetic smokingcessation clinical trials. Drug Alcohol Depend. 104 (Suppl 1), S94–S99.
Scislo, T.J., O'Leary, D.S., 1998. Activation of A2a adenosine receptors in the nucleustractus solitarius inhibits renal but not lumbar sympathetic nerve activity. J. Auton.Nerv. Syst. 68, 145–152.
Scislo, T.J., Ichinose, T.K., O'Leary, D.S., 2008. Stimulation of NTS A1 adenosine receptorsdifferentially resets baroreflex control of regional sympathetic outputs. Am. J.Physiol. Heart Circ. Physiol. 294, H172–H182.
Shepperd, C.J., Eldridge, A.C., Mariner, D.C., McEwan, M., Errington, G., Dixon, M., 2009.A study to estimate and correlate cigarette smoke exposure in smokers in Germanyas determined by filter analysis and biomarkers of exposure. Regul. Toxicol.Pharmacol. 55, 97–109.
Shinozaki, N., Yuasa, T., Takata, S., 2008. Cigarette smoking augments sympatheticnerve activity in patients with coronary heart disease. Int. Heart J. 49, 261–272.
Smith, M.L., Joglar, J.A., Wasmund, S.L., Carlson, M.D., Welch, P.J., Hamdan, M.H., Quan,K., Page, R.L., 1999. Reflex control of sympathetic activity during simulatedventricular tachycardia in humans. Circulation 100, 628–634.
Smith, M.L., Hamdan, M.H., Wasmund, S.L., Kneip, C.F., Joglar, J.A., Page, R.L., 2010. High-frequency ventricular ectopy can increase sympathetic neural activity in humans.Heart Rhythm 7, 497–503.
Stella, L., de Novellis, V., Marabese, I., Berrino, L., Maione, S., Filippelli, A., Rossi, F., 1998.The role of A3 adenosine receptors in central regulation of arterial blood pressure.Br. J. Pharmacol. 125, 437–440.
Whitescarver, S.A., Roberts, A.M., Stremel, R.W., Jimenez, A.E., Passmore, J.C., 1991.Nicotine impairs reflex renal nerve and respiratory activity in deoxycorticosteroneacetate-salt rats. Hypertension 17, 179–186.
Wooters, T.E., Bevins, R.A., Bardo, M.T., 2009. Neuropharmacology of the interoceptivestimulus properties of nicotine. Curr. Drug Abuse Rev. 2, 243–255.
Yildirim, M., Marangoz, C., 2007. Anticonvulsant effects of focal and intracerebroven-tricular adenosine on penicillin-induced epileptiform activity in rats. Brain Res.1127, 193–200.
Zhang, W., Mifflin, S., 2010. Plasticity of GABAergic mechanisms within the nucleus ofthe solitary tract in hypertension. Hypertension 55, 201–206.
237M.M. El-Mas et al. / Toxicology and Applied Pharmacology 254 (2011) 229–237
Related Documents











