RNA Sequencing Peter Tsai Bioinformatics Institute, University of Auckland

RNA Sequencing Peter Tsai Bioinformatics Institute, University of Auckland.
Dec 18, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

RNA SequencingPeter Tsai
Bioinformatics Institute, University of Auckland

Study of transcriptomes Identify known genes, exons, splicing events, ncRNA,
miRNA Novel genes or transcripts Abundances of transcripts (quantitive expression) Differential expressed transcripts between different
conditions Reconstructing transcriptome.
What is RNA-seq?


General workflowRaw data
QC
Map to reference genome
De novo transcriptome
assembly
Estimate abundance
Normalisation
Differential expression
analysis
Require downstream annotation

Use FastQC, SolexQA Trim off low quality region, keep only proper-paired reads Most QC software assume normality, but in RNA-seq data
you will probably see none-normality You might see some duplicated reads, its probably due to
highly expressed gene. Specific reference mapping tool that can map across
splice junctions between exons, i.e. Tophat Specific de novo transcriptome assembly software for
reconstruction of transcriptomes from RNA-seq data, i.e. Trinity
Quality checks and mapping

The total number of reads mapped to a gene/transcript(Count data or raw counts or digital gene expression)
Complexity of using simple counts Sequencing depth: the higher the sequencing depth, the
higher the counts Gene length: Counts are proportional to the length of the
gene times mRNA expression level Counts distribution: difference on how counts are distributed
among samples.
Expression value in RNA-seq

RPKM (Mortazavi et al, 2008)
◦ Reads Per Kilobase of exon model per Million mapped reads FPKM (Mortazavi et al, 2010)
◦ Fragments Per Kilobase of exon model per Million mapped reads
◦ Paired-end RNA-Seq experiments produce two reads per fragment, but that doesn't necessarily mean that both reads will be mappable.
Normalisation
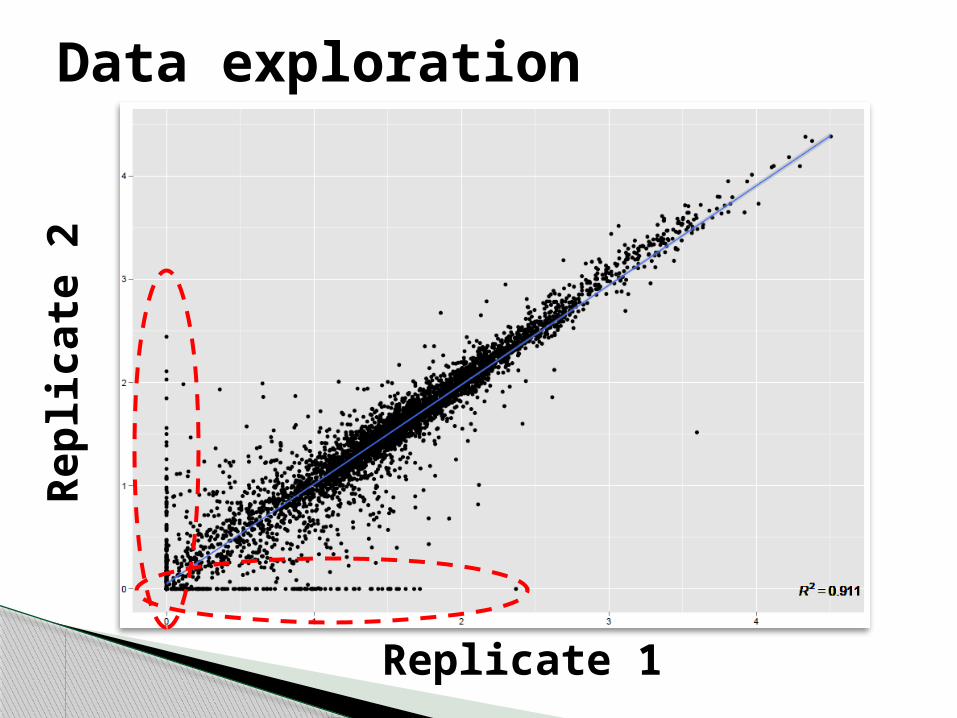
Data exploration
Replicate 1
Repl
icat
e 2

Gene.ID/Description logFC logCPM LR PValue FDR1 2.563086301 5.07961611 28.4599795 9.57E-08 2.72E-052 4.003686266 2.330395704 28.3288251 1.02E-07 2.72E-053 2.71372512 9.704651395 25.01930526 5.68E-07 0.0001006534 -2.052703196 3.402621025 21.11492168 4.33E-06 0.0005752875 1.95117636 4.438847349 19.21195535 1.17E-05 0.0012446516 2.465833373 12.20593577 10.91756889 0.000952565 0.0844607927 1.817858683 5.308092036 10.3738524 0.001278126 0.0971375538 1.577603322 6.556675456 9.690419768 0.001852312 0.1106877669 1.20515812 4.542565518 9.670466698 0.001872537 0.110687766
10 1.233090336 10.08249873 9.289827985 0.002304298 0.12258865211 1.120581944 12.14988136 7.710102379 0.005491264 0.26557748212 1.045292369 4.913492018 7.039209923 0.00797442 0.35027053713 1.089867189 3.885246135 6.912558621 0.008559242 0.35027053714 1.353955354 2.21406615 5.976193603 0.014500264 0.55101003615 1.049933686 3.281031472 5.737563572 0.016605812 0.58895279516 -1.032999983 1.480514873 4.712476717 0.029944481 0.99565399817 -1.313778857 4.325330722 4.169234925 0.041164384 0.99874210218 0.864451602 4.338668381 3.479808135 0.062121942 0.99874210219 -0.766266641 5.2972332 3.443865378 0.063486998 0.998742102

Up-regulated
Down-regulated

Set of external RNA transcripts with known concentration. Dynamic range and lower limit of detection Fold-change response Internal control, in order to measure against defined
performance criteria
ERCC spike-in control

The dynamic range can be measured as the difference between the highest and lowest concentration.
Measure of sensitivity, and it is defined as the lowest molar amount of ERCC transcript detected in each sample
Dynamic range and lower limit of detection

Fold-change response

Depends on a number of factors◦ Biological questions
Complexity of the organism Types of analysis Types of RNA, miRNA, lncRNA.
Literature search for similar work Pilot experiment
How much library depth is needed for RNA-seq?

Have 3 or more biological replicates Analysis your data with different normalisation
methods Perform data exploration Use a standard spike-in as internal control Validation with qPCR
Summary
Related Documents











