By Reza Nazari A thesis submitted in conformity with the requirements for the Degree of Philosophy Graduate Department of Laboratory Medicine and Pathobiology University of Toronto © Copyright by Reza Nazari (2008) RNA AND DNA INACTIVATION STRATEGIES TO PREVENT OR INHIBIT HIV-1 REPLICATION VIA GENE THERAPY

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

By
Reza Nazari
A thesis submitted in conformity with the requirements for the Degree of Philosophy
Graduate Department of Laboratory Medicine and Pathobiology University of Toronto
© Copyright by Reza Nazari (2008)
RNA AND DNA INACTIVATION
STRATEGIES TO PREVENT OR INHIBIT
HIV-1 REPLICATION VIA GENE THERAPY

RNA AND DNA INACTIVATION STRATEGIES TO
ON VIA
tly in use for
HIV/AIDS therapy, a number of gene therapy strategies have been designed as alternative
therapies. Most of these therapies target HIV RNA/proteins, which are subject to high rate of mutation,
resulting in escape mutants. Viral entry is mediated by CCR5 co-receptor in most routes of transmission. To
downregulate CCR5 as a gene therapy approach, we targeted seven unique sites within the CCR5 mRNA
by a multimeric hammerhead ribozyme, Rz1-7. Hammerhead ribozyme is a small RNA that cleaves a target
RNA upon binding to it. Expressing the Rz1-7 from HIV-1- and MSCV-based vectors in otherwise
susceptible cells inhibited replication of a CCR5-tropic strain of HIV-1 by 99-100%. The Rz1-7 will be tested
for inhibition of HIV-1 replication in the CD4+ T-lymphoid and myeloid progeny of transduced human
CD34+ hematopoietic progenitor stem cells.
It may be preferable to interfere HIV-1 life cycle at the DNA level since a one-time inactivation might
suffice to confer a complete and permanent inhibition of virus replication in the gene modified cells and
their progeny. This is what other strategies that target the HIV-1 RNA/protein can hardly offer. For this
purpose, group II introns, which are able to splice out and get incorporated into a specific DNA sequence,
PREVENT OR INHIBIT HIV-1 REPLICATI
GENE THERAPY
Reza Nazari A thesis submitted in conformity with the requirements for the Degree of Philosophy
Graduate Department of Laboratory Medicine and Pathobiology
University of Toronto
2008
AIDS is caused by a lentivirus, HIV-1. In addition to antiretroviral drugs that are curren
ii

can be designed/modified to gain novel DNA targeting specificities. As a novel approach, we have
examined whether insertion of a modified intron into an infectious HIV-1 clone at two sites within the
integrase domain of HIV-1 pol gene could inhibit virus replication. Intron insertion into the HIV-1 clone
was induced and mammalian cells were transfected with intron-inserted HIV-1 clones. Although similar
amounts of HIV-1 RNA, protein, and progeny virus were produced from the clones as from wild-type HIV-
1 provirus DNA, in the absence of a functional integrase, the HIV-1 reverse-transcribed DNA failed to
integrate and virus replication was aborted. These results demonstrate that modified group II introns can
confer complete inhibition of virus replication at the level of second round of infection. We are now
developing vectors to assess whether intron insertion can take place in mammalian cells.
iii

الَحكيمليم الَعم اِهللابْس
In the name of Allah,
The Knowing and The Wise
iv

Dedications and Acknowledgements
Praise to Allah for all the blessings!
To me, this thesis was more than a process of working on some projects and obtaining results, it
was a part of my life, in which I learned important lessons. I would like to briefly acknowledge
those individuals who have been with me during this challenging endeavor.
I dedicate this work to my wife, Parmis, who has my eternal gratitude for her love and patience. I
also dedicate this thesis to my parents, Mohammad and Mina, and my sister, Bita, who have
always provided me with their unconditional love, prayers, and support.
I am indebted to my supervisor, Dr. Sadhna Joshi, for her mentorship and guidance. I feel very
fortunate to have learned under her guidance, and I will always regard her as a close friend.
I greatly appreciate the advice and support given by my graduate advisory committee members,
Dr. Joe Minta and Dr. Stanley Read. I am also thankful to the department’s graduate
coordinators, Dr. Dittikavi Sarma and Dr. Harry Elsholtz for their kind support and wise advice.
My appreciations can never suffice for my closest friend, Dr. Masoud Ameli’s, innumerous
assistance and selfless sacrifices and dedications. His deep belief in Allah’s merci eased all my
difficulties.
Last but not least, I would like to extend my gratitude to my friends who helped and supported
me during all these years: My close friends: Dr. Sara Arab and Dr. Payman B. Bokaei; my
colleagues: Dr. Alka Arora, Darinka Sakac, Sabah Asad; and our undergraduate students: John
Kraft and Suraj Sharma.
v

Table of Contents
RNA and DNA Inactivation Strategies to Prevent or Inhibit HIV-1 Replication via Gene Therapy Abstract ii Dedications and Acknowledgements v Table of contents vi List of figures ix List of tables x List of abbreviations and short names xi Chapter 1: General Introduction 1 1.1. HIV-1 2
1.1.1. HIV-1 Life Cycle 4 1.1.2. Anti-HIV-1 Therapies 8 1.1.3. Anti-HIV-1 Gene Therapy Strategies 10
1.2. CCR5 as a Target for HIV-1 Therapy 11 1.2.1. HIV-1 Tropism and Co-receptor Utilization 11 1.2.2. Other HIV-1 Co-receptors 15 1.2.3. The Fusion Process 17 1.2.4. Dominance of CCR5 and Co-receptor Switch 17 1.2.5. Importance of CCR5 23 1.2.6. Receptor Downregulation and HIV-1 Therapy 24
1.2.6.1. Ligands and Intrakines 26 1.2.6.2. Anti-CCR5 Monoclonal Antibodies and Intrabodies 29 1.2.6.3. Zinc Finger-nucleases 31 1.2.6.4. RNA Interference 32 1.2.6.5. Antisense RNA 34 1.2.6.6. Ribozymes 35
1.2.7. Possible Consequences of CCR5 Inhibition 37 1.3. HIV-1 Gene Therapy at Pre-integration and Proviral DNA Levels 39
1.3.1. Integration Process 39 1.3.2. Gene Therapy Strategies to Target Components at Pre-integration and 41
Integration Steps 1.3.2.1. Targeting the Released Viral RNA 41 1.3.2.2. Targeting the Reverse Transcriptase 46 1.3.2.3. Targeting the Integrase 49 1.3.2.4. Targeting the Pre-integration Complex 50 1.3.2.5. Targeting the HIV-1 dsDNA or Integrated Proviral DNA 51
1.3.3. Group II Introns 51 1.3.4. Group II Intron as a Therapeutic Agent 57
1.4. Thesis Objectives 61
Chapter 2: Inhibition of HIV-1 Entry Using Vectors Expressing a Multimeric 63 Hammerhead Ribozyme Targeted Against the CCR5 mRNA
2.1. Abstract 64 2.2. Background 65 2.3. Materials and Methods 67
vi

2.3.1. Bacteria and Virus Strains and Cell Lines 67 2.3.2. Oligonucleotides and Primers 68 2.3.3. Plasmids 68 2.3.4. Construction of a Multimeric Hammerhead Ribozyme Targeted against 70
CCR5 mRNA 2.3.5. In vitro Cleavage Activity of Rz1-3 and Rz4-7 71 2.3.6. Vector Constructions 72 2.3.7. Transduction and Selection of Stable PM1 Transductants 73 2.3.8. PCR Analysis of Genomic DNA from Stable PM1 Transductants 74 2.3.9. RT-PCR Analysis of Total RNA from Stable PM1 Transductants 75 2.3.10.Immunoflowcytometry Analysis of Stable PM1 Transductants 75 2.3.11.HIV-1 Susceptibility of Stable PM1 Transductants 76 2.3.12.Progeny Virus Infectivity 77 2.3.13.PCR Analysis to Detect the HIV-1 Proviral DNA in the HIV-infected PM1 77
Transductants 2.4. Results 78
2.4.1. Design, Construction, and Activity of the Anti-CCR5 Multimeric 78 Hammerhead Ribozyme, Rz1-7
2.4.2. Oncoretroviral and Lentiviral Vectors Expressing Rz1-7 81 2.4.3. Development of Pools of Stable PM1 Transductants Expressing Rz1-7 81 2.4.4. Rz1-7-Mediated Downregulation of CCR5 mRNA 85 2.4.5. Rz1-7-Mediated Downregulation of the Surface CCR5 Co-receptor 85 2.4.6. HIV-1 Susceptibility of Pools of Stable PM1 Transductants Expressing Rz1-7 85 2.4.7. Infectivity of Progeny Viruses 90 2.4.8. PCR Analyses to Detect the Presence of HIV-1 Proviral DNA in Infected 90
PM1 Transductants 2.5. Discussion 92 2.6. Conclusion 95
Chapter 3: Exploring the Potential of Using Group II Introns to Inactivate HIV-1 97 3.1. Abstract 98 3.2. Introduction 99 3.3. Materials and Methods 100
3.3.1. Bacteria and Virus Strains and Cell Lines 100 3.3.2. Oligonucleotides and Primers 101 3.3.3. Plasmid Constructions 103 3.3.4. Intron Insertion Assay 103 3.3.5. Intron Insertion in HIV-1 Proviral DNA Clone and Purification of Intron- 104
inserted HIV-1 Proviral DNA Clones 3.3.6. Transfection of 293T Cells with Group II Intron-inserted HIV-1 Proviral 104
DNA Clones 3.3.7. RT-PCR Analysis to Detect Group II Intron-inserted HIV-1 RNA in 105
Transfected 293T Cells 3.3.8. Intracellular HIV-1 p24 and Progeny Virus Production from Transfected 105
293T Cells 3.3.9. RT-PCR Analysis to Detect Group II Intron-inserted HIV-1 RNA in the 106
Progeny Viruses Produced from Transfected 293T Cells 3.3.10.PM1 Cell Infection with Progeny Viruses Produced from the Transfected 106
293T Cells
vii

3.3.11.PCR Analyses to Detect Proviral DNA and Reverse-transcribed HIV-1 107 dsDNA in Infected PM1 Cells
3.3.12.Progeny Virus Production from Infected PM1 Cells 107 3.4. Results 108
3.4.1. Modified Group II Introns 108 3.4.2. Modified Group II Intron Insertion Frequencies 108 3.4.3. Group II Intron Insertion into an Infectious HIV-1 Proviral DNA Clone 111 3.4.4. Group II Intron-inserted HIV-1 Proviral DNA Replication in Mammalian Cells 115 3.4.5. Group II Intron-inserted Progeny Virus Replication during the Second Round 119
of Infection 3.5. Discussion 123
Chapter 4: General Discussion and Future Directions 126 4.1. Discussion and Thesis Summary 127 4.2. Future Directions 149 References 154
viii

List of Figures Figure 1.1. The HIV-1 schematic structure. 3
Figure 1.2. A schematic diagram of the HIV-1 LTRs and genes. 5
Figure 1.3. A schematic diagram of HIV-1 life cycle. 6
Figure 1.4- Schematic structure of CC chemokine receptor 5, CCR5. 14
Figure 1.5- Model for HIV-1 Entry. 18
Figure 1.6- A schematic diagram of the Ll.LtrB group II intron. 53
Figure 1.7- A schematic diagram of LtrA protein encoded by Ll.LtrB group II intron 54
Figure 1.8- Specifications of intron-insertion site. 56
Figure 1.9- Mechanism of Ll.LtrB intron splicing and the RecA-independent insertion 58 into the target DNA.
Figure 2.1- In vitro cleavage activity of the anti-CCR5 multimeric hammerhead 79 ribozyme, Rz1-7
Figure 2.2- Schematic diagrams of oncoretroviral and lentiviral vectors. 80 Figure 2.3- PCR analyses to determine the presence of vector DNA sequences in the 83
transduced PM1 cells.
Figure 2.4- RT-PCR analyses to determine the production of Rz1-7 RNA in transduced 84 PM1 cells.
Figure 2.5- RT-PCR analyses to determine CCR5 mRNA levels in transduced PM1 cells. 86
Figure 2.6- Immunoflowcytometry analyses to determine CCR5 co-receptor expression 87
level on the surface of untransduced and transduced PM1 cells.
Figure 2.7- Susceptibility of transduced PM1 cells to R5-tropic HIV-1 (Ba-L). 89
Figure 2.8- Assessment of HIV-1 proviral DNA from untransduced PM1 cells, as well 91 as from control or ribozyme vector-transduced PM1 cells challenged with HIV-1.
Figure 3.1- Structure of the group II intron-inserted HIV-1 proviral DNA, its transcripts 109 and the amino acid sequence of C-terminal region of the mutant Gag-Pol
Figure 3.2- Insertion frequencies of introns I4021s, I4021sIN, I4021sN, I4021s∆N, I4069s, I4069sIN, 112 I4069sN, and I4069s∆N.
Figure 3.3- Experimental scheme used to show insertion of I4021sN and I4069sN introns 113 into pHIV and isolation of the intron-inserted HIV-1 proviral DNA.
Figure 3.4- RT-PCR analysis for determining intron insertion. 116
Figure 3.5- HIV-1 p24 antigen detection in cell lysates and culture supernatants. 118
Figure 3.6- PCR analysis for determining intron insertion pHIV. 121
Figure 3.7- Progeny virus production by PM1 cells infected with the progeny virus 122 from pHIV-, pHIV-I4021sN-, or pHIV-I4069sN- transfected 293T cells.
ix

List of Tables Table 2.1- Oligonucleotides and primers sequences. 69
Table 3.1- Oligonucleotides and primers sequences. 102
Table 4.1- Summary of gene therapy approaches to downregulate CCR5 surface expression. 132
Table 4.2- Summary of gene therapy approaches to inhibit HIV-1 replication at pre- integration or integration stages. 141
x

List of Abbreviations and Short Names 3TC: Lamivudine
ABC: Abacavir succinate
ABT-378/r: Lopinavir
AIDS: acquired immunodeficiency syndrome
Ap: Ampicillin
APV: Amprenavir
ATP: adenosine triphosphate
AZT: azidothymidine
BAF: barrier-to-autointegration factor
CXCR4: CXC chemokines receptor 4
CCR5: CC chemokines receptor 5
cDNA: complementary deoxyribonucleic acid
Cm: Chloramphenicol
CMV: cytomegalo virus
CTL: cytotoxic T lymphocyte
CTP: cytidine triphosphate
d4T: Stavudine
dATP: deoxyadenosine 5'-triphosphate
dCTP: deoxycytidine 5'-triphosphate
ddI: Didanosine
ddC: Zalcitabine
dGTP: deoxyguanosine 5'-triphosphate
DMEM: Dulbecco's modified Eagle's medium DMSO: dimethyl sulfoxide
DNA: deoxyribonucleic acid
DNase: deoxyribonuclease
dNTP: deoxynucleoside 5'-triphosphate
DOHH: deoxyhypusine hydroxylase
dsDNA: double-stranded DNA
dTTP: deoxythymidine 5'-triphosphate
dUTP: deoxyuridine 5-triphosphate
DLV: Delaviridine
EBS: exon-binding site
EBV: Epstein Barr virus
xi

EF-1α: human elongation factor-1α
EFZ: Efavirenz
EGFP: enhanced green fluorescent protein
EMV: Emivirine
En: endonuclease
Env: envelop protein
ER: endoplasmic reticulum
E. coli: Escherichia coli
FACS: fluorescence-activated cell sorting
FDA: Food and Drug Administration
FTC: Emtricitabine
Gag: group antigen
GFP: green fluorescent protein
gp: glycoprotein
HAART: highly active anti-retroviral therapy
HIV-1: human immunodeficiency virus type 1
HIV-2: human immunodeficiency virus type 2
HLA-DR: human leukocyte antigen-DR
HMGA1: high-mobility group protein A1
HSA: heat-stable antigen marker
HS/PCs: hematopoietic stem/progenitor cells
IBS: intron-binding site
IDV: Indinavir
IEP: intron-encoded protein
IL: interleukin
imp: Importin
IN: integrase
IPTG: Isopropyl β-D-1-thiogalactopyranoside
IRES: internal ribosome entry site
Kn: Kanamycin
LEDGF: lens epithelium-derived growth factor
LTR: long terminal repeat
M-tropic: macrophage-tropic
MA: matrix protein
xii

MDM: monocyte-derived macrophage
MIP-1α: macrophage inflammatory protein-1α
MIP-1β: macrophage inflammatory protein-1β
MOI: multiplicity of infection
MoMuLV: moloney murine leukemia virus
mRNA: messenger ribonucleic acid
MSCV: mouse stem cell virus
Nef: negative factor
NFV: Nelfinavir mesylate
neo: neomycin phosphotransferase
NLS: nuclear localizing sequence
NNRTIs: non-nucleoside analogue RT inhibitors
NPC: nuclear pore complex
NRTIs: nucleoside/nucleotide analogue RT inhibitors
NSI: non-syncytium inducing
nt(s): nucleotide(s)
NVP: Nevirapine
ORF: open reading frame
PBL: peripheral blood lymphocyte
PBMC: peripheral blood mononuclear cell
PBS: primer binding site
PBS: phosphate buffered saline
PCR: polymerase chain reaction
PIC: pre-integration complex
Pol: polymerase
R: repeat sequence
RANTES: regulated on activation, normal T-cell expressed and secredted
Rev: regulator of expression viral proteins
RH: rapid-high RNA: ribonucleic acid
RNAi: interfering ribonucleic acid
RNase: ribonuclease
RNP: ribonucleoprotein
RPMI medium: Roswell Park Memorial Institute medium
RT: reverse transcriptase
xiii

RT-PCR: reverse transcription polymerase chain reaction
RTV: Ritonavir
Rz: ribozyme
scFv: single-chain variable fragment
SCID: severe combined immunodeficiency
SCID-hu: humanized severe combined immunodeficiency
SDS: sodium dodecyl sulfate
shRNA: short hairpin RNA
SI: syncytium inducing
siRNA: small interfering RNA
SIV: simian immunodeficiency virus
SL: slow-low
SQV: Saqinavir mesylate
+sssDNA: plus-strand strong stop DNA
T-tropic: T-cell-tropic
T-20: enfuvirtide, Fuzeon
TAR : transactivation responsive element
Tat: trans-activator of transcription
TCID50: 50% Tissue Culture Infective Dose
TDF: Tenofovir
Tet: Tetracycline
TPV: Tiparanavir
tRNA: transfer ribonucleic acid
TRTI : template analog RT inhibitor
U5: unique 5'
U3 : unique 3'
UTP: uridine 5'-triphosphate
VH: variable heavy
Vif: viral infectivity factor
VL: variable light
Vpr: viral protein R
Vpu: viral protein U
WHO: World Health Organization
WT: wild-type
xiv

ZDV: Zidovudine
ZFN: Zinc finger nuclease
xv

CHAPTER 1
GENERAL INTRODUCTION
1

The human immunodeficiency virus (HIV) was first identified in 19831,2. The complete
nucleotide sequence of the viral genome and the viral proteins were identified and characterized
soon after3,4. There are two types of HIV: HIV-1 and HIV-2. Both types are known to cause
acquired immunodeficiency syndrome (AIDS), however infection with HIV-1 is more common
worldwide.
Each year more than three million people die from AIDS. More than 25 million people have died
from AIDS since 1981. It is predicted that the number of infected people worldwide can rise to
90 million and 48 million will die by 2010. According to the surveillance report of the Public
Health Agency of Canada in April 2006, until the end of year 2005, 57,780 people were
diagnosed with HIV in Canada. According to the estimates from UNAIDS/WHO AIDS
Epidemic Update (December 2006), over 40 million people are living with HIV-1 worldwide
with the following distribution: Sub-Saharan Africa: 63%; Asia: 21%; North America, Western
and Central Europe: 5%; Eastern Europe and Central Asia: 4%; Latin America: 4%; North Africa
and Middle East: 1%; Caribbean: 0.8%; and Oceania: 0.2%. The overwhelming majority of
people with HIV-1 live in the developing world. The infection rate continues to rise in these
countries due to poverty, poor health care systems, and limited resources for prevention and care.
1.1. HIV-1
HIV-1 is a retrovirus of the lentivirus subfamily. Retroviruses (retro from Latin, `turning back`)
are RNA viruses that replicate via DNA intermediates using the viral reverse transcriptase (RT)
enzyme. The mature virion of HIV-1 has a diameter of approximately 100 nm (Figure 1.1). The
outer envelope, which is formed from the host cell membrane, is a lipid bilayer that contains host
cell proteins and spikes of the viral envelope glycoproteins (gp120 and gp41). Inside the lipid
2

Figure 1.1. The HIV-1 schematic structure. The virus is enveloped by a cell-originated lipid
bilayer membrane, containing HIV-1 surface and transmembrane glycoproteins. Matrix proteins
are arranged under the envelope. Capsid proteins enclose the virus core, which includes two plus
RNA strand genome, associated with nucleocapsid proteins. A tRNA3Lys and a reverse-
transcriptase are also bound to each of the RNA strands. The core also includes PR and IN
enzymes. Vpr, Vif, and p6 proteins are located in the space between matrix and the core. (From
Joshi & Joshi, 1996)5
3

bilayer are the internal structural capsid and core proteins, p17, p24, p7, and p6. These proteins
enclose two copies of the single-stranded 9-kb RNA genome and multiple RT molecules in the
center of the virus particle, which is called “core”. The virions also carry viral integrase (IN) and
protease (PR) enzymes inside the core. This core structure is often visible in thin sections viewed
by electron microscopy.
The HIV-1 genome contains at least nine different genes that encode 15 different proteins. Three
main genomic regions are: gag (coding for structural proteins), pol (coding for the viral enzymes
PR, RT and IN) and env (coding for the envelope glycoproteins) (Figure 1.2). The other HIV-1
gene products, tat, rev, vpr, and nef regulate the virus life cycle. The viral genome contains long
terminal repeat (LTRs) sequences at each end. The LTRs contain the viral promoter, as well as
binding sites for cellular proteins that are able to activate transcription. The HIV-1 genomic
RNA also contains signal and structural elements, which are involved in different stages of viral
life cycle, such as activation, transportation, packaging, and reverse-transcription5.
1.1.1. HIV-1 Life Cycle
HIV-1 begins the infection of a susceptible human host cell by binding to the CD4 receptor on
the cell surface. CD4+ is expressed on T lymphocytes, macrophages, microglial, dendritic and
Langerhans cells and makes them targets for HIV-1 infection. The CD4 transmembrane protein
along with one of two chemokine receptors, CCR5 and CXCR4, are necessary for viral
attachment and fusion to occur (Figure 1.3). During fusion, viral membranes fuse with target cell
membranes and HIV-1 matrix and capsid proteins eventually disassemble.
4

U3 U5R
5’ LTRgag
PR RT
pol
vif
vpr
tat vpu
rev
rev
tat
env
nef
U3 U5R
3’ LTRIIII IIIN
MA CA NC P6
gp120 gp41
Figure 1.2. A schematic diagram of the HIV-1 LTRs and genes. Like all retroviruses, HIV-1
genome encodes for gag, pol, and env. However, HIV-1 also contains six accessory gene
products (tat, rev, vif, vpu, vpr, and nef), some of which are essential for HIV replication and
reproduction.
5

Figure 1.3. A schematic diagram of HIV-1 life cycle. After binding and fusing to the surface of
the host cell, the HIV-1 core enters the cell. RT synthesizes dsDNA, which is transported to the
nucleus. Integration occurs and the resulting provirus expresses viral RNA that will synthesize
viral proteins using the host's ribosomes. New virions are created by assembly and budding
through the infected cell membrane and subsequent maturation due to the actions of PR. Adapted
from www.medspace.com
6

When the virus enters the cell, the genomic RNAs are released and undergo reverse transcription
into DNA by viral RT enzyme. Synthesis of proviral DNA includes several steps: binding of the
primer tRNA, synthesis of DNA from RNA by RT, degradation of viral RNA by the RNase H
domain of RT, and second strand DNA synthesis by RT.
The proviral DNA then enters the host cell nucleus, where it can be integrated into the genetic
material of the cell. In order for integration to occur in non-replicating cells, the HIV-1 pre-
integration complex (PIC) must be actively transported into the nucleus. A nuclear localization
signal (NLS) on the HIV-1 matrix protein, as well as other interactions with some cellular
proteins, have been reported to facilitate this transportation6. Viral protein R (VPR), an accessory
protein that appears to be incorporated in the virion, has also been reported to facilitate targeting
of HIV-1 PIC to the nucleus7.
After entry into the nucleus, HIV-1 double-stranded DNA (dsDNA) of the PIC undergoes
specific cleavages at the 5' and 3' termini and is integrated into the host DNA through the action
of the HIV-1 IN.
Although HIV-1 replicates extensively throughout all stages of infection8,9, latently infected cells
do exist10, which serve as HIV-1 reservoir in the body. The vast majority of virion production is
from the newly infected cells and not the activation of latently infected cells8,9.
In the cells where the virus is replicating, newly synthesized viral RNA is transported out of the
nucleus and translated (in the case of mRNA) or incorporated into new virions (in the case of
genomic RNA). The gag and pol genes are encoded out of frame on a single mRNA. Frame-
7

shifting during the translation of the viral gag-pol messenger RNA, therefore, is essential for the
production of pol gene products (PR, RT, and IN)11.
The viral proteins and genomic RNA then migrate to and accumulate on the cell membrane of
the releasing virions. The progeny virus buds from the cell membrane, and after release, undergo
the maturation process in which the pol-encoded PR cleaves the gag and gag-pol precursor into
matrix (MA, p17), capsid (CA, p24) and nucleocapsid (NC, p7) proteins12,13. Assembly of
infectious virions is also dependent on the action of cellular N-protein myristoyl transferase
(NMT), which adds myristic acid to the N-terminus of gag, gag-pol and nef precursors before the
virions bud14.
1.1.2. Anti-HIV-1 Therapies
To date, 22 FDA-approved antiretroviral agents are in use for HIV/AIDS therapy and several
others are currently in different stages of basic development and clinical trials. The year 2006
was the 10th anniversary of advent of highly active anti-retroviral therapy (HAART), which
significantly decreased the AIDS morbidity and mortality, and provided great hopes for the HIV-
positive individuals. The drugs that are used in HAART are developed to target viral proteins
RT, PR, IN and viral fusion compartments.
Anti-RT drugs include nucleoside/nucleotide analogue RT inhibitors (NRTIs) such as zidovudine
(ZDV, also called azidothymidine or AZT), didanosine (ddI), zalcitabine (ddC), stavudine (d4T),
lamivudine (3TC), abacavir succinate (ABC), emtricitabine (FTC), and tenofovir (TDF), and
non-nucleoside analogue RT inhibitors (NNRTIs) such as nevirapine (NVP), delaviridine (DLV),
efavirenz (EFZ), and emivirine (EMV). Anti-PR drugs include saquinavir mesylate (SQV),
ritonavir (RTV), indinavir (IDV), nelfinavir mesylate (NFV), amprenavir (APV), lopinavir
8

(ABT-378/r), tipranavir, and atazanavir. Anti-IN drugs consist of oligonucleotides, dinucleotides
and different kinds of chemical agents, such as pdCpI dicaffeoylquinic acids and 2,4-
dioxobutanoic acid analogous. The drugs that inhibit HIV-1 binding and fusion are T-20
(enfuvirtide, Fuzeon) and T-1249. These drugs are currently used as components of a multi-drug
cocktail in HAART15.
Other drugs are also being tested currently in different clinical trial phases, including four RT
inhibitors such as racivir (phase II), etravirine (phase III), rilpivirine (phase II), and BILR-355
BS (phase IIa), two PR inhibitors such as tiparanavir (TPV- phase II) and BMS 232,632 (phase
II), one IN inhibitor, MK-0518 (phase III), and three fusion inhibitors BMS-488043 (phase II),
NBD-556 and NBD-557 (both phase I)16. The drugs that target cellular factors include a
ribonucleotide reductase inhibitor (hydroxycarbamide (Hydrea), previously known as
hydroxyurea), cyclophilin A inhibitor (ciclosporin), deoxyhypusine hydroxylase (DOHH)
inhibitor (deferiprone), deoxyhypusine synthase inhibitor (CNI-1493, phase II), anti-CD4
monoclonal antibody (TNX-355), CCR5 inhibitors (maraviroc and vicriviroc, phase II), CXCR4
inhibitor (PRO 140, phase I)16.
In general, low intracellular permeability, drug toxicity, poor patient adherence to complicated
drug regimens, high mutation rate resulting in the emergence of drug-resistant isolates, and
persistence of viral reservoirs are the major obstacles facing current drug therapy. These
problems have led researchers to develop new drugs with novel mechanisms of action, as well as
alternative therapies such as gene therapy16,17.
9

1.1.3. Anti-HIV-1 Gene Therapy Strategies
A number of gene therapy strategies have been designed to inhibit HIV-1 replication. The
`therapeutic gene(s)` against identified key target(s) in HIV-1 or host cell were delivered to the
cell by a variety of vectors to interfere with various steps of HIV-1 life cycle15,18,19. Those
strategies that use interfering molecules against HIV-1 genes/RNAs/proteins, target highly
conserved regions in the virus. However, there is always a chance of escape mutations. So far,
many anti-HIV-1 genes that targeted HIV-1 proteins failed at different stages of trials. However,
it has been shown that by a combinational strategy that targets different stages of HIV-1 life
cycle, the effectiveness can be increased and the chances of generating escape mutants may be
minimized. Many examples of such combination therapies are described in reviews by Lamothe
and Joshi (2000)18 and Nielsen et al. (2005)20.
The anti-HIV-1 gene therapy strategies are categorized into two groups: protein-based and RNA-
based. The protein-based strategy includes intrakines, single-chain antibodies (intrabodies),
transdominant mutants, targeted/packageable nucleases, and Zinc finger-nucleases.
Transdominant RevM1021, gp41-disruptive peptide22, and anit-HIV-1 antibodies23 are among the
protein-based strategies that are currently at different stages of clinical trials. The RNA-based
approaches consist of antisense24,25, ribozymes26-28, RNA aptamers and decoys29-33, and RNAi
(shRNA and siRNA)34-37. Ribozyme against HIV tat and antisense RNA against HIV env are two
of the anti-HIV-1 RNA-bases therapeutic molecules that are currently in clinical trials23.
Another approach will be targeting a cellular factor that undergoes less diversity. CCR5 co-
receptor has such characteristic, because, as will be explained later, it is the only naturally
dispensable cellular molecule involved in HIV-1 infection. Downregulation of CCR5 expression
via gene therapy strategies has been attempted by several groups. Anti-CCR5 zinc-finger and
10

nucleases (protein-based strategies), and ribozyme and RNAi against CCR5 (RNA-bases
strategies) are now being tested in animals23.
The gene therapy strategies that interfere with HIV-1 RNA or proteins, mostly target the virus
life cycle at post-integration stages. While proviral DNA in infected cells continues producing
viral RNA and proteins, the therapeutic molecules must be expressed in the cells with the pace of
HIV-1 gene expression to be able to inhibit virus replication. Even if the virus replication is
inhibited, the originally infected cells are unable to function normally in the immune system.
Therefore, we believe that the best approach would be preventing viral entry to the cells via
downregulation of CCR5 expression or inactivating the proviral DNA in the infected cells by
insertion of a modified group II intron into specific sites of HIV-1 proviral DNA. In this
introduction, I will provide an in-depth review of the anti-HIV-1 gene therapy strategies that are
designed to downregulate or block CCR5 expression, and those that are designed to interfere
with pre-integration and integration process or to inactivate the proviral DNA.
1.2. CCR5 as a Target for HIV-1 Therapy
1.2.1. HIV-1 Tropism and Co-receptor Utilization
HIV-1 can infect various CD4+ human target cell types38. The viral isolates obtained from
peripheral blood of individuals, shortly after infection and during the asymptomatic phase, are
predominantly macrophage-tropic (M-tropic). As the infection progresses to AIDS, T-cell-tropic
(T-tropic) viruses can be isolated from many, but not all, patients.
The HIV-1 entry into the target cells starts by fusion between the viral envelope and the cell
membranes, which is initiated by a high-affinity binding of the viral envelope glycoprotein (Env)
11

to CD4 on the cell surface. However, CD4 is not the only cellular molecule involved in fusion, as
evidenced by studies showing that its expression alone on non-human cells does not render them
permissive to infection39.
Research focused on identification of the co-factors led to the discovery of the first HIV-1 co-
receptor by Feng et al. (1996)40. They showed that this co-receptor belongs to the superfamily of
the seven transmembrane G protein–coupled receptors. The co-receptor was called “Fusin” due
to its activity in HIV-1 Env-mediated fusion40. When Fusin and CD4 were co-expressed in non-
human cells, the cells could be infected by some of HIV-1 strains. In addition, it had been found
that anti-Fusin antibodies could inhibit infection of primary human CD4+ T lymphocytes.
However, Fusin could play roles in fusions and infections only when T-tropic HIV-1 strains were
used, not the M-tropic strains. Thus, Fusin was considered as the T-tropic HIV-1 co-receptor.
Also, Bleul et al. (1996) reported that infections of CD4+ T lymphocytes by the T-tropic HIV-1
strains were inhibited with use of stromal cell-derived factor-1 (SDF-1 or CXCL-12)41. Fusin
was later shown to be a receptor responding to SDF-141-43 and it was renamed CXCR4 as it
represented the fourth receptor for CXC chemokines. Chemokines (the abbreviation for
chemoattractant cytokines) are small proteins (92-125-amino acid long)44 produced during
immune activation, inflammation, and autoimmune diseases to induce a chemotactic migration
signal so that the cells migrate to the source of chemokine43.
The CC chemokines regulated on activation, normal T-cell expressed and secreted (RANTES or
CCL-5), macrophage inflammatory protein-1α (MIP-1α or CCL-4), and MIP-1β (CCL-4)45,
which are released mainly by CD8+ T lymphocytes, suppressed infection by M-tropic HIV-1
strains46. A receptor corresponding to these chemokines was identified by Samson et al. (1996),
Combadiere et al. (1996) and Raport et al. (1996)47-49. It was first designated CC CKR5 and was
12

later called CCR5 (Figure 1.4). CCR5 was shown to be the major co-receptor for M-tropic HIV-
1 strains50-54. It was also shown to be the only co-receptor used by the simian immunodeficiency
virus (SIV)55.
The identification of the chemokine receptors CCR5 and CXCR4 as HIV-1 co-receptors shed
light on the molecular basis of viral tropism and pathogenesis. CCR5 is expressed on the surface
of macrophages and CD4+ T-lymphocytes51, and is the common receptor for the M-tropic strains
that predominate during transmission56. Generally, strains that use this co- receptor cause the
majority of new infections47,57. In fact, the viruses that are transferred by infected persons can
replicate in both macrophages and primary CD4+ T-cells, but, neither will be able to infect T-
cell lines58-61, nor will form syncytia in T-cell lines. Therefore, the M-tropic strains were named
non-syncytium inducing (NSI) viruses. Considering the slow replication of the M-tropic viruses
in cell cultures, they were also called slow-low (SL) strains59. In the recent nomenclature, the M-
tropic strains are named R5-tropic, as they use CCR5 co-receptor.
Generally in about 50% individuals, about 4-5 years after the initial infection, viral strains evolve
to be able to infect T-cell lines, too40,62. Viral strains that utilize the CXCR4 co-receptor are
called X4-tropic. The virus evolution from R5-tropic to X4-tropic strain is correlated with
accelerated CD4+ T-cell decline and progression to AIDS63. Although sometimes the X4-tropic
strains loose their ability to replicate in macrophages, most of the time the primary isolates will
still be able to use both CCR5 and CXCR4 co-receptors and, therefore, they are called dual-
tropic or R5X4-tropic strains64.
13

HOOC
NH2
Extracellular Loops
Transmembrane Domains
Intracellular Loops
Figure 1.4. A schematic structure of CC chemokine receptor 5, CCR5, a 352-amino acid protein
encoded from chromosome 3p2149. Adapted from Lederman et al. (2006)65.
14

Viruses that can use CXCR4 and infect the T-cell lines have also been referred to as T-tropic,
syncytium-inducing (SI), or rapid-high (RH), respectively based on their ability to infect T-cells,
form syncytia in cultured T-cell lines, and rapid replication kinetics60.
The described nomenclature systems are sometimes confusing. For example, naming a virus
strain as macrophage (M)-tropic or T-cell (T)-tropic may be implied that the stain cannot infect
the other type of cells. Calling a strain as NSI may mean that the virus Env protein is deficient in
binding to the co-receptor. It may also be incorrectly understood that SI viruses are more
cytopathic and fusogenic than the NSI viruses, which we now know that this is not correct in
primary CD4+ T-cell cultures. It seems that this effect is more related to the type of cells than the
strain of virus. It has been shown that NSI viruses are able to form syncytia when infect the
CCR5-expressing cells lines. Therefore, for many instances, it is not appropriate to use terms
NSI, SL and M-tropic as synonyms; likewise for the terms SI, RH and T-tropic66.
To evolve to X4-tropic strains, R5-tropic viruses undergo mutations in the Env glycoprotein 120
(gp120), which are usually located in the V3-loop. It has been shown that the V3-loop of X4-
tropic HIV-1 Env protein is more basic than that of R5-tropic strains67,68. The X4-tropic strains
use the first and second extracellular loops of the CXCR4, which are noticeably more anionic
than the same domains of CCR569,70, therefore, an opposite-charge interaction may be involved
in the interaction of gp120 and CXCR4. In contrast, gp120 from the R5-tropic strains interacts
with the N-terminus of CCR5 co-receptor. Although the extracellular domains of CCR5 and
CXCR4 are less than 20% identical in amino acid sequence, R5X4-tropic strains are able to use
both co-receptors to efficiently enter the cells. The Env from the dual-tropic strains can interact
with the first and second extracellular loops of CXCR4 as well as the N-terminus of CCR570. It is
not clearly understood if the appearance of dual-tropic strains is an evolutionary step between
15

R5- to X4-tropic convergence or a final result of such phenomenon. However, it has been shown
that although R5X4-tropic HIV-1 strains are able to use both co-receptors, they prefer to use
CXCR4 to enter primary T-cells71.
1.2.2. Other HIV-1 Co-receptors
All HIV-1 strains examined so far use one or both of CCR5 and CXCR4 co-receptors66,72. In
addition to CCR5 and CXCR4, at least twelve other chemokine or chemokine receptor–like
orphan receptors have been detected to be involved in the cellular entry of one or more viral
strains. These include CCR2b53, CCR351,53,73, CCR874-76, CCR977, CXCR672, CX3CR1 (formerly
named CMKBRL1 or V28)78, GPR155, GPR15/BOB55,79, Apj77,80, US2881, ChemR2382, and
STRL33/Bonzo79,83.
There are discrepancies about the use of additional co-receptors. The most variable factor in
determining whether a given chemokine or orphan receptor can serve as an HIV-1 co-receptor is
the level of its expression. For example, when CCR3 is highly expressed in cells, most of the
tested X4-, R5-, or X4R5-tropic HIV-1 strains are able to infect the cells. However, when the
level of CCR3 expression is lower, only a few viral strains can infect the cells via this co-
receptor74. Another important factor is relative expression of the co-receptor compared to CD484.
It has been shown that low levels of CCR5 or CXCR4 can suffice virus entry when high level of
CD4 receptor is expressed on cell surface. In contrast, when the CD4 receptor level is low, high
levels of CCR5 or CXCR4 are required to allow virus entry84. However, viral entry via some
chemokine receptors is only shown to occur in vitro and there is not enough evidence of their
usage in vivo85.
16

It is known that the HIV-1 strains are very dependent on CD4 receptor to infect the target cell.
Only a few strains are found to be able to infect CD4-negative cells. However, presence of CD4
is not really crucial for some HIV-2 and SIV strains as they are able to use CCR5 or CXCR4 to
enter the target cells without need to CD466.
1.2.3. The Fusion Process
HIV-1 entry into target cells begins with interactions between viral Env gp120 and cell’s CD4
receptor. The HIV-1 envelope glycoproteins gp120 and gp41 trimers are non-covalently bound
together on the virus envelope (Figure 1.5)86,87. Gp120 consists of five variable loops (V1-V5)
and five constant domains (C1-C5), making an inner and an outer domain. A four-stranded
antiparallel β-sheet (bridging sheet) connects the domains to each other86. A highly conserved
groove at the boundary of the inner and outer domains and the bridging sheet of gp120 binds to
the first extracellular domain of the CD4 receptor88,89. Formation of the gp120-CD4 complex
causes conformational changes to the gp120 core structure that exposes a co-receptor-binding
site on gp120. This site contains a core of hydrophobic amino acids that is surrounded by
positively charged residues. Since the co-receptor is anchored in the host membrane, binding
moves the gp120 bridging sheet close to the target membrane89. The gp120-co-receptor
interaction causes additional conformational changes in the gp120-gp41 trimer that forces the
hydrophobic, glycine-rich fusion peptide region of gp41 to insert into the target cell’s plasma
membrane90.
1.2.4. Dominance of CCR5 and Co-receptor Switch
As mentioned before, CCR5 is the common co-receptor for R5-tropic HIV-1 strains that are
associated with transmission56,66,91. It is expressed on the surface of effector cells (e.g. T-cells,
17

Target cell
HIV virion
CD4
CCR5
b HIV virion a
gp41
gp120
Target cell
HIV virion
CCR5
CD4
c d
Target cell
HIV virion
Figure 1.5. Model for HIV-1 Entry. (a) the HIV-1 Env protein as a heterodimer consisting of
three gp120/gp41; (b) upon binding of cellular CD4 to gp120, the gp120 undergoes a
conformational change; (c) the altered gp120 now is capable of binding to the co-receptor, here
CCR5. The gp120-CCR5 interaction causes a conformational change in gp41, which enables it to
insert the hydrophobic parts into target cell membrane; (d) folding of gp41 trimer on itself brings
the membranes of virus and cell close together. Adapted from Lederman et al. (2006)65.
18

natural killer cells, and natural killer T-cells that produce inflammatory cytokines or destroy
infected cells)49,92,93, antigen presenting cells (e.g. monocytes, macrophages, and dendritic cells
that initiate immune responses)48,65,94-96, as well as the Langerhans cells97 and the mucosa of
rectum, colon, vagina, and cervix96,98. Viruses that use the CCR5 co-receptor cause the vast
majority of new infections57,99, are more frequently found in asymptomatic infected individuals,
and are involved in person-to-person and mother-to-child transmission100.
The co-receptor switch is not necessary for disease progression since X4-tropic strains are not
detected in 50% of individuals who develop AIDS101. However, the co-receptor switch is an
important factor contributing to HIV-1 pathogenesis since individuals, in which X4-tropic strains
dominate during the disease, show a faster decline in CD4+ T-cells and progress significantly
faster to AIDS than individuals who only bear the R5-tropic HIV-163.
There are three theories that explain the predominance of the R5-tropic virus in transmission.
Based on the “transmission-mutation hypothesis” the reasons for the dominance of R5-tropic
virus during or soon after transmission could be selection in favor of R5-tropic virus either in the
donor or the recipient101. The selection in donor can occur due to deferential distribution of R5-
and X4-tropic viruses in organs (such as the male genital tract) that are involved in transmission.
However, dominance of R5-tropic virus in semen could not be demonstrated102. CCR5 is also
thought to play an important role in selection in the recipient during early stages of transmission.
For instance, CCR5 is mainly expressed on the surface of intestinal epithelial cells. These cells
play an important role in infections via oral–genital contact (uncommon) and mother-to-child
transmissions. This may explain the reason of predominance of R5-tropic viruses when the
infection is transmitted through the above venues103,104. It has also been shown that among the
two types of viruses, mainly R5-tropics bind to dendritic cells and can be transported from the
19

mucosal tissues to lymph nodes by these cells105. On the other hand, mucosal surfaces express
high levels of SDF-1, causing the CXCR4 to be selectively downregulated on intestinal
lymphocytes103. However, we know that R5-tropic viruses form the majority of virus population
in patients that are infected through intravenous drug injection, blood transfusion, or sexual
intercourse91. Therefore, the predominance of R5-tropic virus occurs regardless of transmission
route106,107.
The “transmission-mutation hypothesis” also suggests that evolution in the virus population
during the infection results in co-receptor switch from CCR5 to CXCR4101. Neither selection of a
specific transmission route in the epithelial or mucosal tissue nor an advantage for predominance
of R5-tropic viruses in the competition for infecting the target cells is supported by other
observations. If competitive advantage is the reason of early-stage predominance of R5-tropic
strains, X4-tropic strains should predominate in infection of CCR5-deficient individuals. In
contrast, it has been shown that homozygous ccr5∆32/∆32 individuals are highly resistant to
HIV-infection108. Therefore, it was suggested that the predominance of R5-tropic strains may
occur because of replication of virus in some tissues, such as mucosa of the gastrointestinal tract,
in which higher levels of CCR5, but not CXCR4, are expressed63,109. However, late appearance
of X4-tropic virus strains is in contrast with this hypothesis110.
It is shown that, in most cases, for an R5-tropic strain to evolve to an X4-tropic type, only two
mutations are required. Considering the high mutation rate of HIV111 and the high production of
virus progeny (~1010–1012 virions every two days)112, appearance of X4-tropic strains must occur
earlier during infection68. It is known that switch to X4-tropic virus occurs only in ~50% of
individuals and at various times after infection. The reason Pastore et al. (2004)113 suggested for
20

these facts is that the intermediate mutants between R5-tropic and X4-tropic viruses have fitness
disadvantage and X4-tropic virus is fitter than R5-tropic virus.
Another hypothesis, the “immune-control hypothesis”, suggests that the patient’s immune
system recognizes the X4-tropic virus better than the R5-tropic virus and removes them at the
early stages of infection. By the time virus replication results in immune-system deterioration,
the selection against the X4-tropic virus is decreased101. Although the mathematical models
support this hypothesis, there is relatively little evidence to show such selective pressure
exists101. A specific immune mechanism that could lead to stronger selection pressure against
X4-tropic virus by the immune system is yet to be identified114.
The other hypothesis that tries to explain the predominance of R5-tropic virus at early stages of
infection and switch to X4-tropic virus later is called “target cell-based hypothesis”. We know
that CD4+ T-cells are the major HIV-1 target cells in vivo115. While high proportion of naïve
CD4+ T-cells expresses CXCR4, smaller fractions of memory CD4+ T-cells express both CCR5
and CXCR4116. This causes both R5- and X4-tropic viruses to have different target cell ranges,
which, however, may overlap117. During the infection, the CD4+ T-cell population changes in
several related manners: 1) in the peripheral blood, the number of memory CD4+ T-cells
increases and the number of naïve CD4+ T-cells decreases118. If we assume that the same
changes occur in the lymphatic system, this will cause the selection pressure to increase in the
favor of R5-tropic virus. 2) the number of proliferating naïve CD4+ T-cells at the early stages of
infection is lower than the number of proliferating memory CD4+ T-cells. This ratio will convert
at the later stage of infection in a way that the number of proliferating naïve CD4+ T-cells
increases to reach the level where the memory CD4+ T-cells used to be119. Such conversion
would increase the selection pressure in favor of X4-tropic virus.
21

In addition, when the immune system is activated upon infection, the expression of human
leukocyte antigen-DR (HLA-DR) increases, which results in proliferation of CCR5-expressing
CD4+ T-cells120. Higher number of CCR5-expressing CD4+ T-cells will increase the selection
pressure in favor of the R5-tropic virus. This may explain why in almost all cases of infection,
appearance of X4-tropic virus is delayed by years. More knowledge about the pathways of CD4+
T-cells differentiation and the CD4+ T-cell progeny’s susceptibility to R5- and X4-tropic virus is
required to approve this hypothesis114.
Other investigators focused on the kinetics and life cycle of HIV-1 in the infected target cells.
Rodrigo suggested that, during primary infection, there is a competition between R5- and X4-
tropic viruses121. This model hypothesizes that R5- and X4-tropic viruses would infect the same
cells. However, because X4-tropic virus causes syncytia and, therefore, has a higher
cytopathicity and shortens the lifespan of infected cells, it has a replicative disadvantage and R5-
tropic virus predominates.
Sometimes, actual findings support one or more of the above hypotheses. For instance, both
transmission-mutation hypothesis and the target-cell-based hypothesis are supported by the fact
that the X4-tropic viruses are subject to higher diversifying selection pressure than R5-tropic
viruses. However, other times, the findings can criticize the basis of a hypothesis. For example,
the fact that CCR5∆32 homozygous individuals are highly resistant to infection is in contrast to
the statement of the transmission-mutation hypothesis suggesting that predominance of R5-tropic
viruses at the early stages of infection is because of their competitive advantages over X4-tropic
viruses108.
22

1.2.5. Importance of CCR5
There are two reasons why CCR5 is an attractive target for HIV-1 therapy: first, the R5-tropic
HIV-1, which utilizes the CCR5 co-receptor, predominates during early infection. Second,
approximately 1% of Caucasians, who lack CCR5, are highly resistant to HIV-1 infection52,57,122.
In individuals heterozygous for the mutant allele, the infection is delayed123,124 and the plasma
viremia is lower52,122-124. The mutant allele of CCR5, which is present at a relatively high
frequency (allele frequency, 0.092 or ~10%) in Caucasians, but absent in black and Asian
populations, contains a 32-bp deletion (CCR5∆32) within the coding region57,99. This deletion
results in a frame-shift and generates a non-functional receptor that is truncated and cannot be
exported to the cell surface57. The membrane fusion or infection by R5- and R5X4-tropic HIV-1
strains does not occur in individuals who are homozygous for CCR5∆3299,108.
The reason progression to AIDS is delayed in CCR5 +/∆32 heterozygotes, is that the amount of
CCR5 protein on the cell surface of such individuals is lower than that expected from a simple
gene dosage effect92. This can be because of the transdominant effect of the ∆32 truncated
protein, which results in production of a deficient heterodimer that remains in the endoplasmic
reticulum99,125.
Rare cases of infection with X4-tropic strains have been reported in CCR5∆32
homozygotes56,126,127. This strongly indicates that CCR5 is the major co-receptor for HIV-1
transmission in vivo and it is not known why X4-tropic strains cannot inefficiently initiate
infection in a new host66. The fact that the R5-tropic strains are not able to infect stimulated
peripheral blood mononuclear cells (PBMCs) from CCR5∆32 homozygotes, also indicates that
receptors such as CCR2b and CCR3 do not play an important role in the process of the in vivo
virus entry into the primary target cells128.
23

1.2.6. Receptor Downregulation and HIV-1 Therapy
Interference with viral entry seems to be an optimistic approach to treat or prevent HIV-1
infection. Interfering molecules used for this purpose may target either HIV-1 or host cell
elements.
A small number of human monoclonal antibodies against the gp120 and gp41 are able to
neutralize HIV-1 infectivity and prevent entry129-131. However, using them in patients did not
show a prolonged inhibition132. Small molecules that block the CD4-binding site on gp120 have
also been developed. One of them, called BMS 378806 (Bristol-Myers Squibb, New York, NY)
was shown to have antiviral activity in vitro and is being developed to prevent mucosal HIV-1
transmission133. The first entry inhibitor approved for clinical use is a parenterally administered
peptide containing sequences of HIV-1 gp41. This peptide, called enfuvirtide or “Fuzeon”
(Roche/Trimeris, Basel, Switzerland/Durham, NC), blocks the zippering of gp41, which
normally brings the viral and cell membranes together to promote their fusion134.
Compared to the high variability of the viral Env protein, lack of variability in receptors and co-
receptors makes them better targets for therapeutic intervention. However, since there are several
receptors involved in the process of viral entry, it is likely that several different inhibitors will
have to be used in order to cover all possible means by which HIV-1 can enter.
Interference with the binding of CD4 and gp120 by using soluble CD4 was ineffective in early
studies135,136, but a polyvalent CD4-IgG fusion protein (PRO 542, Progenics Pharmaceuticals,
Tarrytown, NY) showed some antiviral activity in vivo137.
24

Complete and broad downregulation of CD4 or CXCR4 is possibly harmful to immune system
and immune cells maturation and homing. CXCR4 deficiency is lethal for mice embryos.
CXCR4-/- mice embryos were shown to have severe cardiac, neural, and hematopoietic
developmental defects. In adult mice, expression of CXCR4 and its interaction with SDF-1 is
required during homing and migration of hematopoietic progenitor cells, as well as cellular
positioning during thymic differentiation and immigration to the periphery43,138,139. On the
contrary, individuals carrying a non-functional CCR5 do not show any deficiency in their
immune system, have less susceptibility to HIV-1 infection, and the AIDS progression is delayed
in them140. Also, one study suggests that CCR5 downregulation in an HIV-2-infected cohort of
Senegalese women protected them from HIV-1 superinfection141. In addition, binding of β-
chemokines to CCR5 results in intracellular signal transduction and internalization of the co-
receptor, which prevents subsequent infection by HIV-1. For these reasons, CCR5 is an attractive
antiviral target and different approaches have been developed either to block the co-receptor
function or to decrease its expression on the cell surface.
To block the co-receptor function, ligands41,50,72,142-144 and anti-CCR5 monoclonal
antibodies92,145-147 were developed and reported with properties that may be attractive for anti-
HIV therapy, including reduced agonist activity, enhanced blocking of fusion/entry, and
improved selectivity for the desired co-receptor.
Among different CCR5-blocking approaches, CCR5 antagonists are the most advanced in their
clinical development and trials. However, investigation of some anti-CCR5 drugs, such as
aplaviroc and vicriviroc, was stopped at different stages of phase IIb and III trials due to
hepatotoxicity and inferior efficiency. For these reasons, many researchers, including ourselves,
have focused on gene therapy strategies to downregulate CCR5 expression.
25

Gene therapy strategies have been developed to inhibit either co-receptor synthesis or surface
expression. The anti-HIV-1 genes used to prevent surface expression include intrakines148-150 and
single-chain antibodies (or intrabodies)151 and those used to decrease co-receptor synthesis
include antisense RNAs152,153, ribozymes153-157, interfering RNAs (RNAi)35-37,158-163, and zinc
finger-nuclease164.
1.2.6.1. Ligands and Intrakines
The co-receptor blocking agents that seem useful are the natural chemokines that bind to and
inhibit fusion, entry and infection mediated by the corresponding co-receptors. Many
researchers, including Cocchi et al. (1995), Alkhatib et al. (1996), Dragic et al. (1996), Oravecz
(1996), Bleul et al. (1996), and Oberlin (1996) demonstrated inhibition of HIV-1 entry into
CD4+ T-cells and PBMCs, as well as monocytic and CD4+ T-cell lines by using ligands for
CCR5 (RANTES, MIP-1α, and MIP-1β)46,50,143,165 and CXCR4 (SDF-1)41,42. Therefore, it seems
that increasing expression of these ligands is part of host immune response to the infection46.
However, Yang et al. (1997) reported that a CCR5+/CD4+ human lymphoid cell line (PM1)
expressing RANTES and MIP1-α under the control of a cytomegalovirus (CMV) promoter failed
to grow166. In addition, there are conflicting reports by Schmidtmayerova et al. (1996), Coffey et
al. (1997), Simmons et al. (1997) and Kinter (1998) about the effects (enhancement, inhibition,
or no effect) of these ligands on HIV-1 replication in macrophages167-170. For example, in sharp
contrast to observed antiviral effects in T-cells, Schmidtmayerova et al. (1996) showed β-
chemokines stimulated the replication of primary HIV-1 strains in macrophages167. In the
presence of RANTES, Oravecz (1996) observed inhibition only in M-tropic isolates, and
RANTES did not inhibit virus replication in chronically infected PM1 cells or did not reduce
virus attachment to the cell membrane143. Tedla et al. (1996) demonstrated that β-chemokine
expression was strongly enhanced in lymph nodes of patients with HIV-1 disease171. Thus,
26

knowing that the primary physiologic role of β-chemokines is to direct the trafficking of
mononuclear cells to lymph nodes and sites of inflammation, cells recruited to lymph nodes are
likely to interact with β-chemokines before exposure to the virus171. However, studies to date
have examined the influence of β-chemokines only when added simultaneously with and/or after
HIV-1 infection. Accordingly, the effects of timing of exposure to β-chemokines on HIV-1
replication in monocytes and monocyte-derived macrophages (MDMs) were studied using an in
vitro system144. Kelly et al. (1998) reported that RANTES, MIP-1α and MIP-1β exposure
produced dichotomous effects on HIV-1 replication. When both monocytes and MDMs were
exposed to β-chemokines before infection, the HIV-1 replications were enhanced. By contrast,
addition of β-chemokines either simultaneously with or after HIV-1 infection inhibited
subsequent viral replication144. Acceleration of HIV-1 replication in the monocytes and MDMs
that are exposed to β-chemokines before infection suggests that mononuclear cells that are
exposed to β-chemokines during being recruited to the lymph nodes may be more susceptible to
HIV-1 infection. Also, note that high concentrations of β-chemokines used in some of these
studies may be well above those present in vivo46,165. The enhancing effects of β-chemokines on
HIV-1 replication observed in monocytes and MDMs were also observed in CD4+ T
lymphocytes. Furthermore, primary clinical isolates have also demonstrated similar results144.
Since interaction of β-chemokines with G protein-linked receptors stimulates the cell’s signaling
pathways, it may result in increased HIV-1 replication. The receptor signaling may upregulate a
nuclear transcription factor, such as κB, that may boost HIV-1 transcription172. Therefore,
developing an HIV-1/AIDS treatment based on using ligands cannot be successful unless the
entire role of β-chemokines during the disease is completely understood.
The chemokines can be converted to “intrakines”, which are intracellular chemokines that bind
to the chemokine receptors to block and decrease their surface expression. Intrakines can be
27

designed to contain KDEL (Lys-Asp-Glu-Leu) endoplasmic reticulum (ER) retention signal to
trap the bound protein in the ER during translation and/or recycling166.
Yang et al. (1997) expressed two intrakines, RANTES and MIP1-α containing KDEL, under the
control of a CMV promoter from a pCMV plasmid and a retroviral LNCX vector. The intrakines
decreased cell surface expression of CCR5 and syncytia formation, as well as R5-tropic HIV-1
replication in a T-lymphoid cell line (PM1)166. Since leakage of expressed intracellular
chemokines may induce signal transduction or inflammatory responses, Bai et al. (1998)
developed a deletion mutant (∆2-8) RANTES, which lacked amino acids 2-8. This intrakine, and
a KDEL-tagged derivative of it, ∆RANTES-KDEL, were cloned under the control of CMV
promoters in pRc/CMV plasmid and LNCX retroviral vector. ∆RANTES-KDEL demonstrated
the same efficiency as its ancestor in downregulating surface expression of CCR5, inhibiting
syncytia formation, R5-tropic HIV-1 replication, and desensitizing to chemotaxis in transduced
PM1 cells and peripheral blood lymphocytes (PBLs)148. In addition, to show the antigen-
stimulated function of ∆RANTES-KDEL-expressing lymphocytes, IL2 production and DNA
synthesis rate in these cells were assayed after exposure to tetanus toxoid as an antigen. These
cells demonstrated the same response as the untransduced control cells, suggesting that
∆RANTES-KDEL-expressing cells retained the basic biological functions in response to antigen
stimulation148.
Schroers et al. (2002) designed another intrakine, RANTES-SK, by adding a six amino acid ER
retaining sequence (Ser-Glu-Lys-Asp-Glu-Leu or SEKDEL) to the C-terminus. This intrakine
was cloned in LOX lentiviral vectors, expressing GFP (green fluorescent protein), under the
control of the human elongation factor-1α (EF-1α) promoter149. When PM1 cells were
transduced with LOX RANTES-SK vector particles, the surface expression of CCR5 was
28

reduced. Since RANTES binds to CCR1, CCR3, and CCR5, surface expression of CCR1 and
CCR3 were also downregulated. Cells were then challenged with R5-tropic HIV-1 (ADA,
SF162, and JRCSF) and X4-tropic HIV-1 (IIIB) at a multiplicity of infection (MOI) of 0.01 or
0.1. The HIV-1 p24 values obtained from infected RANTES-SK-expressing cells were much
lower than from control cells. Incomplete inhibition of virus infection might be due to residual
amounts of CCR5 molecules that were still expressed on cells surface. Using quantitative PCR
method, 44 copies of HIV-1 proviral DNA/ng genomic DNA were detected after 3 days in
infected RANTES-SK-expressing cells, compared to 743 copies/ng genomic DNA in control
cells. The HIV-1 proviral DNA copy number in RANTES-SK-expressing cells remained the
same during three weeks, while in control cells this number increased as high as 5277 copies/ng
genomic DNA149.
The disadvantages of using intrakines include off-target cellular effects and induction of an
inflammatory response. Besides, RANTES-intrakines disrupt expression of other RANTES
receptors, CCR1 and CCR3, whose normal expression during allergic reactions and
inflammatory responses are crucial for proper lymphocyte functions149.
1.2.6.2. Anti-CCR5 Monoclonal Antibodies and Intrabodies
Many anti-CCR5 monoclonal antibodies have also been made and tested, among which, some
were more successful at inhibiting HIV-1 entry. For example, the humanized monoclonal
antibody HGS004 developed and tested by Roschke et al. (2004)146 is in a phase I clinical trial93.
Another anti-CCR5 antibody, Pro-140, is a humanized monoclonal antibody developed by
Castagna et al. (2005)147 and results from a phase 1 clinical trial that was recently completed
showed a dose-dependent binding of Pro-140 to CCR5-expressing cells. At the highest
concentration tested, Pro-140 was shown to coat these cells for at least 60 days. A phase II study
29

in HIV-1 infected individuals is underway16. Binding of these agents to CCR5 initiates its
internalization, which is considered as an advantage: by intracellular arresting of the co-receptor,
there will be less chance for emergence of the drug-resistant HIV-1 isolates that are able to use
other regions of CCR5 to enter the cell. The only disadvantage is that the efficiency of the agent
is dependent on the extent and duration of receptor internalization.
An “intrabody” is an intracellularly expressed single-chain variable fragment (scFv) of antibody
against a specific protein173,174. Similar to intrakines, intrabodies can be designed to contain
KDEL signal to retain the target protein in ER173. Because of their high affinity, target
specificity, and stability in cellular environments173, intrabodies were also used to inhibit surface
CCR5 expression. Steinberger et al. (2000) produced an intrabody against N-terminal
extracellular domain of CCR5 for downregulation of CCR5 expression and inhibition of R5-
tropic HIV-1 infection151. To this end, the scFv was dimerized using a linker and tagged with
KDEL at the C-terminus. The resulting intrabody, ST6, was expected to be more efficient
because it had two CCR5 binding sites and two ER-retention signals. Steinberger et al. (2000)
expressed ST6 intrabody and RANTES-KDEL intrakine148 separately from pIB6 and pRAN
plasmids, respectively. ST6 was also expressed from Babe-Puro retroviral vector. To test the
efficiency of ST6 and RANTES-KDEL in downregulation of CCR5 surface expression, 293T
cells were first transfected with a CCR5-encoding plasmid (to produce high levels of CCR5) and
then with pIB6 or pRAN. Intracellular staining revealed that both intrakine and intrabody were
expressed equally. While ST6 resulted in a complete inhibition, RANTES-KDEL led to only a
slight reduction of CCR5 surface expression. Syncytia formation was also shown to be
completely inhibited by ST6, but only slightly by RANTES-KDEL. PM1 cells transduced with
the retroviral vector expressing ST6 showed a complete reduction of surface expression of CCR5
and inhibition of syncytia formation. These cells were also resistant to R5-tropic HIV-1 (SF162
30

and JR-CSF) infection at an MOI of 0.01 over the 10-day period of experiment151. Swan et al.
(2006) also showed that expression of ST6 intrabody from a lentiviral vector efficiently
disrupted surface expression of CCR5 in transduced primary CD4+ T-cells and macrophages
derived from transduced CD34+ cells175.
Cagnon et al. (2000) modified the scFv of an anti-CCR5 antibody (2C7) to contain KDEL and
then cloned it into an SV40-based vector under the control of CMV promoter to produce
pSV(2C7)176. Transduced PM1 cells and primary monocytes expressed 2C7 scFv for two months
and two weeks, respectively. When the monocytes were induced to differentiate into
macrophages, over 90% of the differentiated MDMs expressed 2C7 scFv. Surface CCR5
receptor was reduced 50-60% in transduced SupT1/CCR5 cells, PM1 cells, and MDMs. When
transduced SupT1/CCR5 and PM1 cells were challenged with 0.05-0.1 ng p24 equivalents of
HIV-1 (BaL), the infection was inhibited, but not completely. With 1 ng p24 equivalents of
virus, the infection was ~90% inhibited only when the cells were sequentially transduced with
SV(2C7) and SV(VCKA1), a vector expressing single hammerhead ribozyme against CCR5177.
SV(2C7)-transduced MDMs and microglial cells were partially resistant (20-50%) to 0.3 and 1
ng p24 equivalents of HIV-1 (BaL), respectively. The combination strategy showed a better
inhibition of virus infection. However, when SV(2C7)- and combined SV(2C7)/SV(VCKA1)-
transduced human monocytes were induced to differentiate to macrophages, only partial
inhibition of virus replication was observed at 1.5 ng p24 equivalent of HIV-1 (BaL).
1.2.6.3. Zinc Finger-nuclease
Proteins containing a Zinc finger (ZF) domain can bind with high affinities to specific DNA
sequences. Zinc finger nucleases (ZFNs) have been developed by fusing the non-specific
cleavage domain (N) of Fok I restriction enzyme to the ZF proteins, which can specifically bind
31

to an 18-bp target sequence within plant and mammalian genome. Upon binding the ZF domain
to target site, the nuclease cleaves the dsDNA in vitro73,178,179. Mani et al. (2005) combined and
fused the three ZF domains to the C-terminal 196 amino acids of Fok I restriction enzyme, which
constitute the Fok I cleavage domain, to develop a CCR5-specific ZFN to disrupt the ccr5 gene
at the DNA level164. A target site close to the start codon was chosen to target, so that only a
small peptide would be produced. The efficiency of this ZFN in downregulation of surface
expression of CCR5 is not reported yet.
1.2.6.4. RNA Interference
The endogenous or exogenous micro-RNAs and small interfering RNAs (siRNAs) control gene
expression, mRNA degradation and translation, as well as chromatin structure in eukaryotic
cells. All these pathways are referred to as RNAi180. The siRNAs, ranging in size from 19-24
nucleotides, can be targeted to any gene of interest. Silencing is performed by an inherent RNase
III-like endonuclease that uses specific siRNAs as triggers in cleaving target mRNAs181.
Martinez et al. (2002) showed that an siRNA, RNAR53i, corresponding to nucleotides +554 to
+572 of CCR5 relative to the start codon, conferred a 48% reduction of surface CCR5 expression
in transfected U87 cells that express CD4, CCR5, and CXCR4 receptors36. It also displayed a
33% inhibition of HIV-1 (BaL; MOIs between 0.03-0.24) entry as evaluated by intracellular p24
antigen detection 24 hrs post-infection, and a 79% inhibition of progeny virus production 48 hrs
post-infection. Therefore, the CCR5 mRNA cleavage was incomplete.
In order to further improve this strategy, Qin et al. (2003) designed a lentiviral FG12 vector to
express a CCR5 mRNA-specific siRNA, CCR5-siRNA (186), targeting nucleotides 186-204
within the CCR5 open reading frame37. CD4+ PBLs transduced with this vector showed >90%
32

reduction of surface CCR5 expression. By challenging the transduced CD4+ PBLs with a R5-
tropic HIV-1 reporter virus (expressing murine heat-stable antigen marker, HSA, instead of HIV-
1 Vpr gene) that could only undergo a single round of infection, more than 98% inhibition of
progeny virus production was observed. As expected, these cells were still susceptible to X4-
tropic HIV-137.
Anderson et al. (2003), Butticaz et al. (2003), Anderson et al. (2005) and Morris et al. (2006)
also investigated application of siRNAs in CCR5 downregulation159,160,163,182. Anderson et al.
(2003) designed a bispecific siRNA (with an 8-nucleotide spacer) to target both CCR5 and
CXCR4 mRNAs. The MAGI-CCR5 cells were transfected with the in vitro-transcribed
bispecific siRNA, which was shown to be processed in the cell to give rise to two 20-nt. long
monospecific products159. A 53% reduction of CCR5 expression was observed in the transfected
MAGI-CCR5 cells. Two days later, the cells were challenged with HIV-1 (BaL; MOI of 0.001),
and it was shown that there was inhibition of HIV-1 progeny virus production by ~95% at day 5
post-infection. However, when R5/X4 siRNA-transfected PBMCs were challenged with the
same virus (MOI of 0.001), only 32% inhibition of infection was observed on day 5 post-
infection. These results indicate that siRNA can be designed and assembled as multiple effector
motifs.
Anderson et al. (2005) co-expressed an anti-CXCR4 short hairpin183 that was expressed with the
anti-CCR5 siRNAs159 from a lentiviral vector, HIV-7-GFP-XHR, under the control of two
separate Pol-III promoters, U6 and H1, respectively163. In comparison to control cells, the
surface expression of CXCR4 and CCR5 co-receptors in transduced MAGI-CXCR4 and Ghost
R5 cells was reduced by 73% and 72%, respectively; however, the CCR5 mRNA was not
completely eliminated. When transduced cells were challenged with X4-tropic (NL4-3) or R5-
33

tropic (BaL) strains of HIV-1 (MOI of 0.01), over 90% reduction in progeny virus production
was observed with both cells on day 5 post-infection, as compared to untransduced or empty
vector-transduced cells. However, increased progeny virus production was detected on day 5-7
post-infection, probably because of the presence of cells that are untransduced and/or produce
low levels of siRNA. When PBMCs transduced with this vector were challenged with the same
HIV-1 strains, 33% reduction of p24 Ag inhibition was observed on days 3-7 post-infection163.
Lower levels of protection in PBMCs could have been due to the lower transduction efficiency.
Besides incomplete inhibition of HIV-1 replication, disadvantages of siRNA approach include
possibility of an interferon response and off-target gene regulation184-186.
1.2.6.5. Antisense RNA
Li et al. (2006) designed a 653-nt. long antisense RNA against the CCR5 mRNA (nts. 187-839
within the coding region) and cloned it into an adenovirus-based vector, to make pAd-
antisenseR5152. Inhibition of surface expression of CCR5 on U937 cells transduced with this
vector was 98.1%, compared to 13.8% from transduced cells expressing a sense RNA
corresponding to the same region. The amount of CCR5 mRNA in U937/Ad-antiR5 cells was
less than in control cells. No difference was seen in chemotactic activities responding to
RANTES in any of the cells. Compared to controls, when U937/Ad-antiR5 cells were challenged
with R5-tropic HIV-1 (CN97001; MOI of 0.01), ~55% inhibition of progeny virus production
was observed in 12 days post-infection. However, this antisense RNA possesses an ~87%
sequence homology to the CCR2a and CCR2b mRNAs, therefore, it may also inhibit the
function of these mRNAs, which may not be desired.
34

1.2.6.6. Ribozymes
Ribozymes are enzymatic RNA molecules that can be engineered to site-specifically cleave the
target RNAs187,188. The advantage of ribozymes over siRNA is that ribozymes have minimal
cellular toxicity and do not induce an interferon immune response154,189.
Qureshi et al. (2006)153 designed an anti-CCR5 ribozyme (CCR5Rz) against 13 nucleotides
flanking the second GUC (nts. 262-264) within the CCR5 mRNA. A 13-nt. long control
antisense RNA was also designed to bind to the same region153. These RNAs were expressed
from separate plasmids under control of a T7 promoter. When PBMCs were co-transfected with
either pCCR5Rz or pCCR5As, along with plasmids expressing the T7 RNA polymerase, CCR5
mRNA production was reduced by 95% in CCR5Rz-expressing cells, and by 80% in CCR5As-
expressing cells. The inhibition of surface CCR5 expression in PBMCs from different sources
varied from 50-90% between day 3-7 post-transfection. On day 5 post-transfection, the CCR5Rz-
or CCR5As-expressing PBMCs were challenged by 3 ng p24 equivalent of R5-tropic HIV-1
(SF162). On day 7 post-infection, progeny virus production was shown to be inhibited by 68%.
Another monomeric ribozyme targeted against nucleotide 23 within the CCR5 open reading
frame (ORF) was extensively studied. Cagnon et al. (2000)177 transfected HOS-CD4.CCR5 cell
line with a plasmid expressing this ribozyme and showed a 70% decrease in surface CCR5
expression, compared to a 50% decrease from a mutant ribozyme. However, both the active and
the mutant ribozymes conferred only a 1-3 days delay in R5-tropic HIV-1 (BaL; MOI of 0.001)
replication177. PM1 cells transduced with pBabe-Puro oncoretroviral vector expressing this
ribozyme conferred 70% (active ribozyme) vs 50% (mutant ribozyme) inhibition of BaL virus
replication (MOI of 0.02) on day 7 post-infection177. Bai et al. (2000) also used pG1Na
oncoretroviral vector expressing this ribozyme transduce CD34+ hematopoietic stem cells. The
35

differentiated macrophages showed inhibition of BaL virus replication (MOI of 0.02) on day 17
post-infection. However this inhibition was only slightly better than with the mutant ribozyme155.
Li M et al. (2005) also used an HIV-1-based vector, pHIV-7-GFP, expressing this ribozyme to
transduce primary T-cells and CD34+ stem cells190. The transduced primary T cells and the
monocytes differentiated in vitro from the transduced CD34+ cells, were challenged with the R5-
tropic HIV-1 (JR-FL; MOIs 0.01 and 0.05). A certain level of selective survival was observed,
compared to the control vector-expressing cells. Viral replication was reduced by 99% compared
to cells expressing the pHIV-7-GFP control vector, but still high amounts of progeny virus (1-10
ng/ml on day 7 and 500 ng/ml on day 28 post-infection) were produced190.
Ramezani et al. 199726 demonstrated that a multimeric ribozyme functions more efficiently than
a single (monomeric) ribozyme. Multimeric hammerhead ribozymes have an increased
probability of recognizing and cleaving at least one of the multiple target sites within the CCR5
mRNA. Therefore, Bai et al. (2001) designed a trimeric ribozyme against nucleotides 17, 153,
and 249 within the CCR5 ORF157. Oncoretroviral vectors, LN and MND, expressing this trimeric
ribozyme decreased CCR5 expression by 10-15% and conferred ~30% inhibition of R5 Env-
psuedotyped HIV-1 NL4-3 replication on day 4 post-infection157. Similar results were obtained
for inhibition of replication of HIV-1 (BaL; MOI of 0.001) in macrophages derived from
transduced CD34+ stem cells157.
The partial inhibition of HIV-1 replication observed using anti-CCR5 monomeric and trimeric
ribozymes could have been due to incomplete downregulation of surface CCR5 expression.
Therefore, to further improve this strategy, we designed a multimeric hammerhead ribozyme, Rz1-7,
which targets seven unique sites within the CCR5 mRNA. Rz1-7 was designed against nucleotides 17,
380, 390, 520, 556, 811, and 824 within the CCR5 ORF. These ribozymes were shown to effectively
36

cleave the CCR5 RNA in vitro191. An oncoretroviral vector (MGIN) and an HIV-1-based lentiviral
vector (HEG1) were used for the delivery and expression of Rz1-7. Rz1-7 expressed in the
transduced PM1 cells was shown to be active, since the CCR5 mRNA levels were found to be
decreased. As expected, high-levels of progeny virus were produced when the untransduced
cells, as well as MGIN-, MGIN-Rz1-7-, HEG1-, or HEG1-Rz1-7-transduced PM1 cells, were
challenged with the NL4-3 strain, suggesting that the cells were permissive to X4-tropic HIV-1
replication. Control untransduced PM1 cells and MGIN- or HEG1-transduced cells could be
infected by the R5-tropic HIV-1 (BaL; MOIs of 0.225, 0.675, and 2.025). However, when the
MGIN-Rz1-7-transduced cells were challenged with the BaL strain at all three MOIs, 99-100%
inhibition of progeny virus production was observed for the duration of the experiment (2-3
months post-infection). Inhibition of replication of the BaL strain was more prominent when the
multimeric ribozyme was expressed from the MGIN-Rz1-7 vector than from the HEG1-Rz1-7
vector. When the HEG1-Rz1-7-transduced cells were challenged with the BaL strain (MOIs of
0.225, 0.675, and 2.025), 80-99% inhibition of virus replication was observed for the duration of
the experiment (2 months post-infection). PCR analyses at different time intervals confirmed that
the inhibition of BaL virus replication in MGIN-Rz1-7- and HEG1-Rz1-7-transduced cells is at the
level of entry, as no or very little proviral DNA was detected by PCR.
1.2.7. Possible Consequences of CCR5 Inhibition
Although congenital deletion of CCR5 is well-tolerated, probably due to functions of other co-
receptors, use of mutated chemokines or antagonists to block CCR5 in a normal individual might
still result in induction of inflammatory responses or might block the trafficking and cellular
activation mediated by CCR5 in response to chemokines65. Also, as mentioned before, some
CCR5 inhibitors may increase the risk of opportunistic infections and malignancies in persons
who are already infected with HIV-1192,193. Recently, Glass et al. 2005194 showed that ∆32
37

homozygote individuals have a higher rate of mortality from a symptomatic infection with West
Nile virus.
38

1.3. HIV-1 Gene Therapy at Pre-integration and Proviral DNA
Levels
1.3.1. Integration Process
After fusion of HIV-1 envelop with cell membrane, the viral core enters the cytoplasm. Like all
known retroviruses, HIV-1 expresses RT, an enzyme that catalyzes the synthesis of proviral
linear DNA using the viral RNA as a template and a cellular tRNA3lys that binds to the primer
binding site (PBS) region located immediately 3' to the U5 region to serve as a primer. This
process consists of three catalytic steps: 1) synthesis of complementary DNA strand of the RNA
genome, resulting in a RNA-DNA hybrid; 2) removal of the RNA strand by ribonuclease H
(RNase H); and 3) simultaneous synthesis of a second DNA strand, complementary to the first
one. RT is an error-prone enzyme, introducing 3.4 × 10-5 mutations per base pair per cycle into
the HIV-1 genome20.
Following reverse-transcription, the PIC has to be quickly transported to the host cell’s nucleus,
but because the size of the PIC of most retroviruses is bigger than the nuclear pore complex
(NPC), the PIC cannot enter the nucleus of nondividing cells, such as macrophages and
microglia. To overcome this problem, HIV-1 and related lentiviruses, use a nuclear transport
machinery in a cell division-independent, ATP-dependent process195. Vpr, containing a nuclear
localization signal, is one of the viral proteins involved in transporting the PIC through NPC by
interacting directly with proteins in it196. A functional IN protein is not required for nuclear
import of the PIC197. The HIV-1 MA protein, is a component of the PIC, as well202. It facilitates
both nuclear import of the PIC (especially in non-dividing cells) and viral particle assembly with
the help of its two subcellular localization signals6,204.
39

Importin (imp) 7, a receptor for ribosomal proteins and histone H1, was found to be involved in
the process of importing the PIC198. A host transcriptional co-activator called lens epithelium-
derived growth factor (LEDGF/p75, also known as PSIP1), was shown to interact directly with
HIV-1 and other lentiviral IN protein in the PIC to localize it to the nucleus198, specifically into
the genes whose expression is regulated by LEDGF/p75199. This might be because LEDGF/p75
and, perhaps, other transcriptional factors make cellular DNA more accessible to the PIC200. It is
also shown that LEDGF/p75 protects IN from proteosomal degradation198. Another cellular
protein that directly and specifically binds to HIV-1 IN is the transcription factor Integrase
Interactor 1 (INI1/hSNF5), which is suggested to be required for the integration process and acts
as an ATP-dependent chromatin-remodeling protein20. It is shown that INI1/hSNF5 also
incorporates into the HIV-1 particles201. Two other cellular proteins were also identified in the
HIV-1 PIC: high-mobility group protein A1 (HMGA1) and barrier-to-autointegration factor
(BAF)202,203.
The imported dsDNA is integrated into the host chromosomal DNA by IN, which catalyzes
multiple steps, including DNA cutting and joining reactions: 1) the IN protein recognizes short
inverted repeats (ATT sites) at both ends of the proviral DNA, removes two nucleotides (AT)
from each 3’ end of the viral DNA, leaving recessed CA-OH’s at the 3’ end; 2) the IN cuts both
strands of target chromosomal DNA at a non-specific location with a 5-base distance, followed
by ligation between the viral processed 3’ end and host 5’ end; 3) IN fills the gap resulting in
production of a 5-bp duplication of host DNA at both ends on proviral DNA integration
site205,206.
IN is a protein encoded by the gag-pol polycistronic gene and is produced when the virion-
assembled gag-pol polyprotein is cleaved into individual components during viral maturation. IN
40

consists of three domains, a relatively highly conserved C-terminal domain, a central catalytic
core domain with a highly conserved D,D(35)E motif, which is required for the catalytic activity,
and an N-terminal zinc-binding domain (HHCC)201,206. In addition to its function in nuclear
localization of the PIC using its nuclear localization signals, IN plays roles in viral morphology,
particle formation, particle release, particle-associated RT activity, and infectivity201.
1.3.2. Gene Therapy Strategies to Target Components at Pre-Integration and
Integration Steps
As mentioned before, many drugs have been developed to target RT and IN, however, low
intracellular permeability, drug toxicity and high mutation rate resulting in emergence of drug-
resistant isolates, persistence of viral reservoirs, and poor patient adherence to complicated drug
regimens are the major obstacles faced with current drug therapy. These problems have led
researchers to develop new drugs with novel mechanisms of action, as well as alternative
therapies such as gene therapy15-17. A number of strategies have been developed to inhibit HIV-1
replication. Strategies designed to target the released viral RNA, RT, IN, PIC, and the integrated
proviral DNA are summarized below.
1.3.2.1. Targeting the Released Viral RNA
One of the potential targets for HIV-1 gene therapy is the viral RNA right after the viral core
enters the cytoplasm and before it has a chance to be reverse-transcribed into DNA. In a study by
Sarver et al. (1990), HeLa CD4+ cells allowing transient expression of a hammerhead ribozyme
against HIV-1 gag-coding region were >97% resistant to in-coming HIV-1 for 7 days post-
infection and the synthesized proviral DNA was 100-fold less than in control cells207. The
41

decreased amount of proviral DNA could be due to ribozyme-mediated cleavage of viral RNA
prior to reverse-transcription.
Ojwang et al. (1992) designed a hairpin ribozyme against a conserved site at position +111/112
relative to the transcription initiation site, within the U5 region of LTR of HIV-1 strain HXB2
mRNA208. HeLa cells allowing transient expression of ribozymes showed reductions in p24
protein (by 75%) and Tat activity (by 80%), suggesting the ribozyme efficiency in inhibition of
HIV-1 infection. This ribozyme was then expressed from a Moloney murine leukemia virus
(MoMuLV)-based LNL6 oncoretroviral vector, under the control of tRNAval, adenovirus VA1,
or β-actin promoters. Again, transient transfection of HeLa cells with these vectors and HIV-1
clones resulted in reduction of viral p24 protein. The best results were obtained when the
ribozyme was expressed under control of tRNAval promoter. Yu et al. (1993) reported that HeLa
cells allowing transient expression of this ribozyme inhibited progeny virus production by 95%
on day 2 post-transfection209. When Yamada et al. (1994) transduced a human T lymphoid Jurkat
cell line with the LNL6 oncoretroviral vector expressing the hairpin ribozyme under control of a
tRNAval promoter, virus replication was shown to be inhibited for up to 35 days post-infection
with HIV-1 (HXB2 strain). The efficiency of incoming virus to synthesize viral DNA was also
decreased by the ribozyme by approximately 50- to 100-fold210. Leavitt et al. (1994)
demonstrated that human PBLs transduced with this vector were also resistant for 10 days post-
infection to laboratory and clinical isolates of HIV-1211. Yu et al. (1995a) showed that
macrophages differentiated from transduced human fetal cord blood CD34+ cells expressed the
ribozyme and were resistant to infection by R5-tropic HIV-1 (BaL strain). The ribozyme
expressed by the promoter VA1 was more efficient (>95% inhibition for 10 days) than expressed
by the tRNAVal promoter (>90% inhibition for 6 days)212. Yu et al. (1995b) then designed
another hairpin ribozyme against the HIV-1 pol-coding region. This ribozyme was also
42

expressed from the LNL6 vector under the control of adenovirus VA1 promoter. This ribozyme
inhibited viral replication in stable CD34+-derived macrophage transductants till day 25 post-
infection. Inhibition by this ribozyme was slightly better than with the U5-ribozyme213. In a
preclinical study, Li et al. (1998) enriched and stimulated CD34+ cells from placental and
umbilical cord blood from ten newborns of HIV-1-positive mothers, and transduced them with
LNL6 vector expressing the U5-ribozyme under the control of the tRNAVal promoter. These cells
were then induced to differentiate and the progeny macrophage cells were challenged with HIV-
1 (BaL strain), or the HIV-1 isolate from the infant’s mother. Virus replication was significantly
inhibited for 20 to 35 days post-infection214. This study progressed to a phase-I clinical trial, in
which Wong-Staal et al. (1998) used MFT retroviral vector expressing the U5-ribozyme under
the control of the tRNAVal promoter215.
Westaway et al. (1995) fused an anti-HIV-1 hammerhead ribozyme targeted against the PBS
region of HIV-1 RNA to the 3'-terminus of tRNALys3 as it can be co-packaged with viral
genomes in newly synthesized virions216. This tRNA-ribozyme reduced the infectivity of
progeny virus by about 83%, probably by cleaving the viral genomic RNA. During the
subsequent round of infection, this tRNA-ribozyme also interrupted reverse-transcription by
competing with the host tRNALys3 for binding to RT/PBS and failed to prime the cDNA
synthesis due to presence of ribozyme at its 3’ end.
In a continuous study, Cohli et al. (1994) and Ding et al. (1998) demonstrated that production of
two antisense RNAs against Psi-gag-coding regions or U3-5’ untranslated region-gag-env-
coding regions in transduced MT4 cell line inhibited virus replication till day 30 and 78 post-
infection, respectively. These antisense RNAs were also able to inhibit HIV-1 replication in
PBLs for 21 days post-infection. Experiments revealed that both antisense RNAs co-packaged
43

into progeny virus and could interrupt virion RNA encapsidation and reverse-transcription
during the subsequent round of infection24,217.
Peng et al. (1997) also designed sense and antisense RNAs to hybridize to HIV-1 RNA or DNA
to inhibit reverse transcription. These include anti-RTn 1, a plus-sense RNA containing the RU5
sequence; anti-RTn 2, a plus-sense RNA containing the full U3RU5 sequence; anti-RTn 3, same
as anti-RTn 2 but with Sp1 and TATA box deleted, resulting in a defective HIV-1 promoter;
anti-RTn 4, a minus-sense RNA complementary to the plus-strand strong-stop DNA; and anti-
RTn 5, same as anti-RTn 4 along with a sequence complementary to the HIV-1 packaging
sequence218. The sense and antisense RNAs were expressed from a double-copy LSNP vector
under the control of the human tRNAmet promoter. Stably transduced Jurkat cells were
challenged with HIV-1 (NL4-3 strain; MOI of 0.01). While anti-RTn 1 enhanced viral infection
(for some unknown reason), no virus replication was observed in Jurkat cells expressing anti-
RTn 2, 3, 4, and 5 for 140 days post-infection.
Jacque et al. (2002) used siRNAs to destroy virion RNA before it has a chance to be reverse-
transcribed into cDNA34. Several 21-nt. long siRNA duplexes were directed against different
regions of the HIV-1 genome, including two siRNAs against the viral LTR, 5 siRNAs against the
vif-coding region, and 3 siRNAs against the nef-coding region. The siRNAs were co-transfected
with pHIVNL-GFP (containing a green fluorescent protein gene at the beginning of nef region) into
CD4+ MAGI cells. Compared to control cells that were only transfected with pHIVNL-GFP,
progeny virus production was reduced 30- to 50-fold in 24 h post-transfection. To investigate
whether siRNAs were able to specifically find and degrade the virion RNA, MAGI cells were
transfected with various siRNAs and infected with HIVNL-GFP 20 h later. No viral RNA could be
detected 1 h post-infection and no viral cDNA or integrated proviral DNA could be detected 36 h
44

post-infection. These results showed that HIV replication could be inhibited by degradation of
genomic HIV-1 RNA at the level of entry prior to reverse-transcription and provirus integration.
SiRNAs were also shown to be stable in cells because virus replication was inhibited when the
cells were challenged with HIVNL-GFP 20 h or 4 days after siRNA transfection.
SiRNAs were shown to inhibit HIV-1 replication not only before reverse-transcription but also
after transcription219. Capodici et al. (2002) designed two siRNAs, one targeting the gag-coding
region and the other targeting the 3’ LTR region. Infection of U87-CD4+-CXCR4+ and U87-
CD4+-CCR5+ cell lines with HIV-1 strains IIIB or BaL, respectively, and transfection with
either one of the two siRNAs on day 3 post-infection showed a 75 to 96% decrease in progeny
virus production for 3 days. Similar results were obtained using primary CD4+ T cells. Virus
replication was shown to be inhibited upon siRNA addition between day 2 and 5 post-infection.
Transfection of the infected cells with both gag and 3’-LTR siRNAs at one-half the
concentration of each resulted in better inhibition of viral replication, suggesting a synergistic
effect of targeting multiple sites. SiRNA can be protected against RNase A degradation by
incorporating 2’-F-dCTP and 2’-F-dUTP instead of CTP and UTP during transcription. Such
fluorine-derivatized RNA can be recognized by RTs, has the expected weight and structure by
mass spectroscopy, and can be delivered to cell by being directly added to serum without
complexing to lipofectin. Adding the gag-specific fluorine-derivatized siRNA to HIV-1-infected
primary CD4+ T cells culture on day 3 post-infection resulted in inhibition of HIV-1 replication
after 3 days. To determine whether viral infection could be inhibited after viral entry and before
reverse-transcription U87-CD4+-CXCR4+ and U87-CD4+-CCR5+ cells were transfected with
gag- or 3’-LTR-specific siRNA. One day later, cells were infected with HIV-1 strains IIIB and
BaL, respectively, and real-time quantitative PCR analysis for gag DNA of 24-hour cultured
cells demonstrated less gag DNA per cell. These results indicated that inhibition occurred before
45

the completion of reverse-transcription. The same results were obtained when primary CD4+ T
cells were treated with gag- or 3’-LTR-specific siRNA. Analysis with primers for 5’-negative
strand strong stop DNA gave similar results, suggesting that RNAi inhibited early and late stages
of reverse-transcription.
1.3.2.2. Targeting the Reverse Transcriptase
Maciejewski et al. (1995) cloned variable heavy (VH) and variable light (VL) gene fragments of
an anti-RT monoclonal antibody into an Epstein Barr virus (EBV)-based episomal eukaryotic
expression vector, pMEP4, to yield pMEP/VH and pMEP/VL220. This vector produces high levels
of protein in mammalian cells in the presence of cadmium. A lymphoid cell line MOLT-3 was
transfected with either of pMEP/VH and pMEP/VL, or with both of the vectors to express anti-RT
Fab. Stably transfected Fab-expressing cells were challenged by laboratory strains of HIV-1 IIIB
and RF, the clinical isolate HIV-1 571, and a low-passage isolate HIV-1 MN at MOI’s of 2. In
all cases, the infection was completely inhibited for 35 days post-infection. Quantitative PCR
analysis revealed a decrease of viral DNA in cells expressing anti-RT Fab to 1% of controls.
Shaheen et al. (1996) expressed an anti-RT intrabody (scFv fragment) from a MoMuLV-based
vector, SLXCMV, under the control of a CMV promoter221. Transduced SupT1-RT-SFv3 cells
were challenged with HIV-1 strains NL4-3 (MOI’s of 0.012 and 0.006) and R7-HXB2 (MOI’s of
0.01 and 0.001). Compared to controls, ~80 to 97% inhibition of progeny virus production was
observed 15 to 22 days post-infection. However, when cells were challenged with MOI of 1.0 of
viruses, no inhibition took place221. This study showed that intrabodies were able to reach the
target viral protein in the PIC.
46

Wu et al. (1993) produced an anti-RT monoclonal antibody, 1E8, that inhibited both RNA- and
DNA-dependent DNA polymerase, but had no effect on RNase H activity222. Strayer et al.
(2002) cloned the scFv of this and another anti-RT monoclonal antibody (RT#3)221 into an
SV40-based vector. SupT1 cells were individually or sequentially transduce with SV(1E8) and
SV(RT3) vectors223. When individually transduced cells were infected with 40 and 100 TCID50
(50% Tissue Culture Infective Dose) HIV-1 strain NL4-3, the progeny virus production was
highly inhibited. At 800 TCID50 HIV-1, both 1E8 and RT#3 failed to protect the individually
transduced cells, but in the cells, which were expressing both scFv’s, the infection was delayed
for about a week. In another effort, they transduced the cells with both SV(Aw) (an anti-IN scFv
cloned in SV40)224 and SV(RevM10) (a dominant-negative mutant of HIV-1 Rev21 cloned in
SV40) and showed that the cells were resistant to 800 TCID50 HIV-1. Transducing SupT1 cells
with SV(1E8) and SV(Aw) completely protected them from 2500 TCID50 HIV-1, made it the
best combination of scFv’s in one cell line.
Joshi et al. (2002) used anti-RT aptamers to inhibit HIV-1 replication. The anti-RT aptamers,
also referred to as template analog RT inhibitors (TRTI), are small RNA molecules that have
high affinity and specificity for HIV-1 RT and competitively inhibit its enzymatic activity in
vitro225. Ten aptamers were designed, out of which six (70.8,13, 70.15, 80.55,65, 70.28,
70.28t34, and 1.1) were chosen based on their binding constants and levels of RT inhibition in
vitro. Double-stranded fragments encoding different aptamer sequences were cloned into
pcDNA3.1 vector between two self-cleaving ribozymes under the control of the CMV promoter.
The flanking ribozymes were required to cleave and release the aptamers. Jurkat and 293T cells
were transfected by aptamer-expressing vectors separately and stable transfectants were selected.
When untransfected and transfected 293T cells were transfected with an infectious molecular
clone of HIV-1 strain R3B, they all produced the same amount of progeny virus. Immunoblot
47

analysis of viral proteins revealed that the assembly and maturation of the virus particles were
not affected. The authors assumed that the TRTI aptamers were co-packaged with the RT in the
progeny viruses. The infectivity of these viruses was reduced from 90 to 99% compared to the
control virus obtained from non-aptamer-expressing 293T cells. Out of six aptamers, the aptamer
70.8, which was the strongest RT-inhibitor in vitro, displayed the best (99%) reduction of HIV-1
infectivity. To determine the interference step during reverse-transcription, Jurkat cells were
infected with the TRTI-containing virion particles and the cells’ total genomic DNA were
analyzed 12 h post-infection. Synthesis of the earliest intermediate, minus-strand strong-stop
DNA, was not affected, but minus-strand transfer product and the formation of completed
proviral DNA was blocked by the aptamers 70.8,13 and 70.15. When stably transfected aptamer-
expressing Jurkat cells were infected with HIV-1 strain R3B at a low MOI (0.1), 23 to 96%
reduction of viral replication was observed for 22 days tested, 70.8,13 being the most effective
aptamer. Infection of the same Jurkat cells with HIV-1 R3B (MOI of 50) was significantly
inhibited with the three most effective aptamers, 70.8,13, 70.15, and 80.55,65. These three
aptamers also strongly suppressed the replication of a number of viral strains (MOI of 0.1)
resistant to NRTIs, NNRTIs or PR inhibitors225.
Surabhi et al. (2002) designed two other siRNAs (RT1 and RT2) against HIV-1 RT226. MAGI
cells were transfected with either of RT1 or RT2 siRNAs and 24 h later infected with DNase I-
treated HIV-1. Progeny virus production was inhibited by 90% on day 6 post-infection,
compared to the control cells. The inhibition was observed for at least 13 days. Western blot
analysis of whole-cell extracts from these cells demonstrated a specific decrease in the p51/p66
RT proteins following transfection of either of these RT siRNAs. Similar results were obtained
when Jurkat cells transfected with either of RT1 or RT2 siRNAs at a 20-time higher
concentration were infected with HIV-1.
48

1.3.2.3. Targeting the Integrase
Levy-Mintz et al. (1996) designed five anti-HIV-1 IN scFv antibodies against the zinc-finger-
like domain, core (catalytic) domain, and C-terminal nonspecific DNA binding domain227. The
scFvs were cloned under the control of a CMV promoter in the MoMuLV-based oncoretroviral
vector, pSLXCMV. The resulting vectors were used to transduce SupT1 cells. Stable
transductants were then challenged with HIV-1 strain NL4-3 at MOI’s of 0.04 and 0.06.
Compared to control vector-transduced and untransduced cells, low levels of HIV-1 p24 antigen
were detected in SupT1 IN scFv33 and scFv4 cells in a 22-day long experiment, while SupT1 IN
scFv12, scFv17 and scFv21 cells were susceptible to infection. The cytopathic effect (syncytia
formation) was also low and delayed in SupT1 IN scFv33 and scFv4 cells. It was also shown that
when two populations of SupT1 IN scFv33 and SupT1 IN scFv4 cells were mixed, infection by
HIV-1 (NL4-3) was approximately 95 to 98% inhibited. ScFv33/NU, a derivative of scFv33 that
contained the NLS of HIV-1 Tat protein between its VH and VL chains, was also made. When
SupT1 IN scFv33 and scFv33/NU cells were infected with HIV-1 (NL4-3), similar resistance
was observed, although the scFv33 seemed to be more efficient. The anti-IN scFv efficiency was
also tested in stimulated human PBMCs transduced with the retroviral vector expressing scFv33
or scFv33/NU. When these cells were challenged with HIV-1 strain NL4-3 at an MOI of 0.08,
about 92% inhibition was observed for 25 days post-infection.
BouHamdan et al. (1999) cloned the scFv33 in an SV40-based vector to obtain SV(Aw)224.
SupT1 cells transduced with this vector were challenged with two different doses of HIV-1
NL4–3 (0.05 pg and 0.5 pg p24 antigen equivalents). When lower amount of virus was used for
challenge, only very low levels of HIV-1 p24 antigen were detected in the supernatants. With
higher amount of virus, the syncytium formation was delayed for up to 18 days.
49

Goldstein et al. (2002) also injected an SV40-derived vector encoding IN#33 into the human
thymic grafts of thy/liv-SCID-hu mice228. Expression of IN#33 was shown to inhibit HIV-1 (800
TCID50) infection up to about 85% in two weeks after infection.
1.3.2.4. Targeting the Pre-integration Complex
As mentioned before, one of the cellular factors in importing the PIC is imp7. Fassati et al.
(2003) showed that applying an siRNA homologous to nucleotides 1392–1414 of human imp7
mRNA depleted the imp7 mRNA and resulted in 80-90% inhibition of HIV-1 replication198.
Inhibition of virus replication was only observed when HIV-1 infection occurred at an MOI of
0.01, and not when an MOI of >1 was used. The effect of depletion of imp9 on inhibition of
HIV-1 infection was also assessed. In cells transfected with an siRNA homologous to
nucleotides 527–547 of human imp9 mRNA, HIV-1 infection was partially reduced from 2% to
38% in three independent experiments.
Another cellular factor that can be potentially targeted is LEDGF/p75. RNAi-mediated depletion
of LEDGF/p75 resulted in instability and severe reduction of both cytoplasmic and nuclear HIV-
1 IN229. When Ciuffi et al. (2005) knocked down the LEDGF/p75 in Jurkat cells using short
hairpin (sh)RNA, the frequency of proviral DNA integration into genes regulated by
LEDGF/p75, as well as other transcription units, was partially reduced (6-10%), compared to
wild-type (WT) cells199. However, the frequency of integration into GC-rich DNA regions was
slightly increased.
Yung et al. (2004) showed that a transdominant mutant of INI1/hSNF5 (S6) that competes with
the WT protein for binding to IN, inhibited HIV-1 replication by 105 to 106 folds201.
50

To target HIV-1 MA protein, Levin et al. (1997) developed an intracellular Fab antibody against
a C-terminal epitope of MA from the Clade B HIV-1 Levin et al. (1997)230. Expression of
intrabody in the dividing CD4+ T-cells could inhibit HIV-1 infection. The progeny virions’
infectivity was also shown to be substantially reduced.
1.3.2.5. Targeting the HIV-1 dsDNA or Integrated Proviral DNA
To date, there are no reports of successful inactivation of HIV-1 dsDNA or integrated DNA. Our
attempt to test proviral DNA inactivation by targeting it with a mobile, retrotransposing RNA
element, called group II intron, is the first one of its type.
1.3.3. Group II Introns
Group II introns are found in both gram-negative and gram-positive bacteria231, the
mitochondrial and chloroplast genomes of lower eukaryotes and higher plants, but not in the
mitochondrial DNAs of metazoan animals nor in eukaryotic nuclear genes232.
Group II introns are mobile genetic elements that can insert themselves into the intronless allele
via a process called retrohoming. Mobility occurs when the spliced intron RNA reverse-splices
into a DNA site and is reverse-transcribed into dsDNA233. The initial study of group II intron by
Cech234 also showed that a self-splicing intron RNA could reverse-splice into an ectopic site
(similar to the original homing site) in dsDNA, resulting in retrotransposition.
A group II intron consists of a catalytic RNA and a multifunctional, intron-encoded protein (IEP)
with multiple structural and enzymatic activities. The intron RNA performs RNA splicing and
reverse-splicing (integration) reactions by its ribozyme activity and specific sequences, and IEP
facilitates these reactions by stabilizing the catalytically active RNA structure, DNA recognition
51

property, and its own enzymatic activities. Efficiency of this complex lets group II introns
retrohome (at the natural site) at a frequency of about 100%231 and retrotranspose (at an ectopic
site) at low frequencies (typically 10-4 to 10-5)235,236.
The intron of LtrB gene of Lactococcus lactis, also called Ll.LtrB intron, is a very well-studied
model group II intron, which unlike most of other bacterial group II introns, is mobile and can be
expressed at high levels in Escherichia coli. The Ll.LtrB intron RNA has a conserved secondary
structure consisting of six double-helical domains (DI-DVI) (Figure 1.6). Interaction of DI and
DV (containing the ribozyme motif) forms the catalytic core; DII and DIII are responsible for
RNA folding and catalytic efficiency; DIV encodes the intron’s IEP ORF, which can be provided
in trans for ribozyme activity; DVI contains a bulged A, which is necessary for intron
splicing238,239. In order for the splice junctions at the intron’s active site to be close together for
RNA splicing and reverse splicing reactions, the flanking 5’- and 3’-exon sequences base-pair
with three short sequences on intron RNA: are exon-binding site (EBS)1 and EBS2 in DI, which
base-pair with the 5’-exon sequences intron-binding site (IBS)1 and IBS2, and the sequence δ
adjacent to EBS1, which base pairs with δ’ made of the first three nucleotides of the 3’
exon232,240.
In vivo intron splicing requires RNA folding into a catalytically active structure that is facilitated
by the IEP, which binds specifically to the intron RNA to stabilize the active structure
(“maturase” activity)240,241.
The L. lactis IEP, named LtrA, consists of four domains: N-terminal reverse-transcriptase (RT),
maturase (X), DNA-binding (D), and DNA endonuclease (En) (Figure 1.7)242,243. The RT domain
binds the intron RNA, which will serve as a template for reverse transcription242, 243. Domain X
52

Figure 1.6. A schematic diagram of the Ll.LtrB group II intron, showing secondary structure of
double-helical domains (DI to DVI) and complementary intron and exon sequences. The ORF in
domain IV encodes the LtrA protein. EBS and IBS refer to exon- and intron-binding sites,
respectively. EBS1-IBS1, EBS2-IBS2, δ-δ’ interactions facilitates introns tertiary structure for
splicing and target DNA hybridization during insertion. Adapted from Guo et al. (2000)237.
53

N C
Z RT X D En
I II III IV V VI VII1 36 70 361 383 487 543 599
Figure 1.7. A schematic diagram of LtrA protein encoded by Ll.LtrB group II intron. Region Z
is characteristic of the RTs of non-LTR retroelements. The RT domain contains conserved motifs
I–VII. Domain X is an RNA binding domain with maturase activity, which is required for RNA
splicing. Other domains are D (DNA-binding) and En (DNA endonuclease). Key amino acids
flanking various domains are numbered. Adapted from San Filippo & Lambowitz (2002)244.
54

is an RNA binding domain with maturase activity that is required for RNA splicing242,243.
Although RNA splicing and synthesis of single-stranded DNA substrates occur independently
from the C-terminal D and En domains, they are required for intron mobility and prompting the
synthesis of dsDNA244. The En domain cleaves the second-strand DNA244.
The IEP is translated from unspliced precursor RNA. The interaction between the IEP and intron
RNA is crucial for all steps of intron splicing and mobility. It has been shown using the Ll.LtrB
that specific, high affinity binding of LtrA protein dimer to the DIV of intron RNA245 is
sufficient to initiate RNA splicing in vitro at physiologic Mg2+ concentrations241,246. LtrA also
interacts with intron’s catalytic core regions DI, DII, and DVI to fold the intron RNA into the
active structure241,245. This interaction is also crucial for (i) positioning the RT to initiate reverse-
transcription245, (ii) downregulation of translation by covering ribosome attachment site247,
which, in turn, prevents the accumulation of excess LtrA, and (iii) keeping ribosomes away from
the intron, which might, otherwise, prevent RNA splicing.
For reverse-splicing, first the ribonucleoprotein (RNP) complex binds nonspecifically to DNA
and then searches for a specific binding site. The binding site is located in the distal 5’ exon at
positions –24 to –13 corresponding to insertion site, where T–23, G–21 and A–20 seem to be the
most critical nucleotides. When the LtrA protein recognizes and binds to this location via major
groove interaction, it unwinds the dsDNA, enabling the EBS2, EBS1 and δ regions of the intron
RNA to base-pair to the IBS2, IBS1 and δ’ sequences of top-strand at positions –12 to +3
relative to the insertion site (Figure 1.8)231,233,248. While the protein is still associated with RNA,
it cleaves the bottom-strand of DNA (at +9) through interaction with a small number of bases in
the 3’ exon, especially T+5, and uses it as a primer to initiate reverse-transcription233,241,244.
55

Figure 1.8. Specifications of intron-insertion site. The most critical positions recognized by the
LtrA in the 5’ exon (T–23, G–21 and A–20) and 3’ exon (T+5) are indicated by squares. The
IBS2, IBS1 and δ’ sequences (positions –12 to +3) base-paired to intron RNA’s EBS2, EBS1
and δ sequences are shown. The black triangles indicate the positions where the dsDNA breaks.
The intron RNA inserts directly into the top-strand of DNA at the insertion site (IS) via reverse
splicing. Bottom-strand cleavage (CS) requires additional interaction between the LtrA and the 3'
exon. Adapted from Singh & Lambowitz (2001)233.
56

LtrA has very low efficiency DNA-dependent DNA polymerase activity. Therefore, second-
strand DNA synthesis might be performed by cellular machinery, suggesting that intron insertion
would require actively DNA replicating249.
Both retrohoming and retrotransposition of Ll.LtrB intron are Recombinase (Rec)A-independent
processes (Figure 1.9)231,250.
1.3.4. Group II Intron as a Therapeutic Agent
Since intron insertion into the DNA target site initiates by IEP recognition and the intron RNA
base-pairing, different DNA sites can be targeted for gene therapy purposes by modifying the
intron’s sequence237,248. Now, the target sequence can be scanned by computer programs to find
the best matches to the positions recognized by the IEP. These programs can be used to design
primers for modifying the intron’s EBS and δ sequences for insertion in the target sites251. An
advantage of using introns is that each gene can be targeted with multiple introns for each of its
available IEP-recognition sites. Efficient targeted gene disruptions have been shown already in
both gram-negative and gram-positive bacteria using Ll.LtrB intron251-254. In addition, the group
II introns can carry a gene, cloned in domain IV, into a specific chromosomal location252. This
property of Ll.LtrB intron was even more highlighted when it was shown that a drug-resistance
marker could be cloned in DIV in order to develop an intron-based vector243.
If group II introns can be modified and used to target genes in eukaryotes as efficiently as they
now do in bacteria, they would have potentially a broad range of gene therapy applications,
including gene disruption, gene repair/substitution, and gene insertion. To investigate the group
II abilities in gene repair, Jones III et al. used a modified Ll.LtrB group II intron to insert the WT
β-globin exons 2 and 3 into the second intron of a mutated β-globin gene in an E. coli system.
57

Second-strand synthesisand DNA repair
LtrA
Nicking of the DNA Strand by LtrA protein
Nicking of the DNA Strand by intron
Target recognition and hybridization
RNP Intron
LtrAIntronless allele
E1 E2
E2E1
LtrA
LtrA
LtrA
Intron
Intron releaseLtrA
Translation
Transcription and Translation of precursor
E1 E2 Ll.LtrB Intron
LtrA
Figure 1.9
Reverse transcription
Complete insertion
58

Figure 1.9. Mechanism of Ll.LtrB intron splicing and the RecA-independent insertion into the
target DNA. L1.LtrB (thick black line) is found in the LtrB gene (gray) between exons E1 and
E2. The intron encodes the LtrA protein (white), which facilitates self-splicing of the Ll.LtrB
intron (thin black line). The excised intron is in the form of a lariat that is bound to the LtrA
protein. This RNP complex targets an intronless target DNA. The RNA component recognizes
the IBS1, IBS2, and δ’ regions within the target DNA, nicks the DNA, and inserts itself (thin
black line) into the top strand of target dsDNA (gray). The LtrA protein (white) then cleaves the
opposite DNA strand and reverse transcribes the inserted intron RNA. Second-strand DNA
synthesis and DNA repair then complete the intron insertion into the intronless target DNA.
Adopted from Long et al. (2003)255.
59

Results revealed that approximately 2% of the mutant genes were repaired by this insertion and
could express RNAs that correspond to the WT β-globin transcript. The repaired gene also
expressed the full length β-globin protein compared to the truncated one from the mutant gene256.
This study demonstrated for the first time that a mutated mammalian gene can be repaired by
insertion of an intron carrying the correct exons in a bacterial model system and result in
expression of WT transcripts and proteins.
Another study to investigate intron insertion into a human gene and HIV-1 sequence has been
done by Dr. A. Lambowitz and his team237. They scanned CCR5 and HIV-1LA1 DNA sequences
and found 5 and 6 positions, respectively, that could be recognized by L. lactis IEP on sense and
antisense strands. The EBS-IBS and δ-δ‘ sequences of intron RNA were then modified to enable
it to base-pair with either of those positions. Each intron was cloned in a pACYC184 plasmid
while the LtrA ORF in domain IV was replaced by a T7 promoter and LtrA ORF was cloned
downstream of the intron, to make intron-donor pACD plasmids. To determine the introns
mobility, different CCR5 and HIV-1 DNA fragments were separately cloned in a recipient
plasmid immediately upstream of an intron-less TetR gene. Intron and LtrA expressions in E. coli
cells by a donor and a relevant recipient plasmid were induced by isopropyl β-D-1-
thiogalactopyranoside (IPTG). TetR cells were considered as cells in which the intron expression,
mobility and insertion had occurred. With this method, the insertion frequencies were measured,
which varied from 0.16-53% for CCR5-targeting introns and 3-66% for HIV-targeting introns.
To investigate whether the group II intron RNPs could function in human cells environment,
293T or CEM-T cells were co-transfected using liposome containing either of CCR5 nt. 332-
targeting or HIV-1 nt. 4069-targeting intron RNPs and their related recipient plasmids using
transfection. Note that the intron and LtrA protein were pre-expressed and bound to form RNP
prior to packing in the liposomes. PCR analysis on DNA of co-transfected cells revealed
60

occurrence of intron insertion. Note that the liposomes may have fused outside the cells before
delivering the material to the cell. Therefore, the insertion could have happened in a non-cellular
environment. Another question that one can ask about the study is that, while the ccr5 gene
exists in 293T cells, why did the researcher use a CCR5-expressing plasmid. Intron insertion into
the genomic ccr5 gene could be investigated by PCR analysis using Alu sequence-specific
primers.
1.4. Thesis Objectives
As mentioned before, one of the major reasons why most of the post-infection anti-HIV-1 gene
therapy strategies have not been successful to date is that they must inhibit and interfere with the
viral RNA/protein production and functions. Therefore, it is thought that the strategies to block
viral entry to the cell or to inactivate proviral DNA in the cells’ genome may be better
approaches. Since the viral genes undergo a high rate of mutation, human genes are more
constant targets for gene therapy. It is believed that among the human cells’ receptors, which
facilitate the virus entry, CCR5 is dispensable. The 10% of Caucasians, who carry this ∆32
allele, have a functional immune system and are resistant to HIV-1 infections. So far, the
ribozymes that are designed to downregulate CCR5 expression had better outcomes compared to
other therapeutic molecules. Therefore, I studied the inhibition of infection of an R5-tropic HIV-
1 strain by an anti-CCR5 multimeric hammerhead ribozyme.
The approach of inactivation of proviral DNA has not been well studied. Since Guo et al.
showed that 5 sites on HIV-1 proviral DNA can be targeted by modified group II introns,237 we
planned to investigate the feasibility of HIV-1 proviral DNA inactivation by further modified
group II introns and its effects on the virus infectivity.
61

Therefore, two objectives are pursued in this thesis:
1) Development and testing retroviral vectors expressing a 7-meric hammerhead ribozyme (Rz1-
7) against CCR5 mRNA. I will investigate whether transduction of mammalian cell lines with the
vectors expressing the Rz1-7 would inhibit ccr5 gene expression in these cells and their progeny,
rendering them non-permissive to HIV-1 infection and syncytia formation compared with the
untransduced HIV-1-infected cells.
2) Development a gene therapy strategy using a modified group II intron, which inhibits HIV-1
replication upon insertion within the HIV-1 provirus DNA and interfers with viral RNA/protein
production. I will develop a modified group II intron to insert into HIV-1 provirus DNA in the
IN domain of pol gene in order to inactivate the IN protein. Then I will investigate whether a) the
intron could target the full-length HIV-1 clone, b) further modifications to the intron to have a
selectable marker might alter the intron’s function and mobility, and c) the intron insertion would
inhibit HIV-1 replication.
62

CHAPTER 2
Inhibition of HIV-1 Entry Using Vectors Expressing a Multimeric Hammerhead Ribozyme Targeted against the
CCR5 mRNA
Reza Nazari1, Xue Zhong Ma1, Sadhna Joshi1,2,§
1Department of Laboratory Medicine and Pathobiology, Faculty of Medicine, University of Toronto, Toronto, Ontario M5S 3E2, Canada 2Department of Medical Genetics and Microbiology, Faculty of Medicine, University of Toronto, Toronto, Ontario M5S 3E2, Canada
§ Corresponding author
Email address:
This chapter has been modified from a published article in Journal of General Virology in 2008.
63

2.1. Abstract
HIV-1 entry is mediated by the specific interaction of the viral envelope glycoproteins with a cell
surface molecule, CD4, which serves as the primary receptor, and a chemokine receptor, CCR5
or CXCR4, which serves as a co-receptor. CCR5 appears to be required for all routes of
transmission. Therefore, research has been focused on downregulating CCR5 expression and/or
function.
Rz1-7 is a multimeric hammerhead ribozyme targeted against seven unique sites within the human
CCR5 mRNA. This multimeric ribozyme was shown to be active in vitro. It was inserted into a
mouse stem cell virus-based vector, as well as into an HIV-1-based vector so as to express the
multimeric ribozyme in a human CD4+ T lymphoid PM1 cell line. Stable transductants
expressed the virus-based ribozyme, which was further shown to be active since the CCR5
mRNA level decreased. High-levels of progeny virus were produced when the cells were
challenged with an X4-tropic HIV-1 strain, suggesting that cells were permissive to X4-tropic virus
replication. Replication of an R5-tropic HIV-1 strain was inhibited. Inhibition was more
prominent in cells transduced with the mouse stem cell virus-based vector than with the HIV-1-
based vector. When the PM1 cells transduced with the mouse virus-based vector were challenged
with the R5-tropic HIV-1 strain, 99-100% inhibition of progeny virus production was observed
for the duration of the experiment. Inhibition occurred at the level of viral entry, as no HIV-1
proviral DNA could be detected.
Our results demonstrate that the multimeric ribozyme can efficiently cleave the CCR5 mRNA and
prevent R5-tropic HIV-1 infection at the level of entry. Since this ribozyme confers excellent
64

inhibition of R5-tropic HIV-1 replication, we anticipate that it will be beneficial for HIV-1 gene
therapy.
2.2. Background
The anti-HIV-1 genes developed to date encode RNAs or proteins that interfere with the
functions of viral or cellular RNAs/proteins15,18,257. However, most of the strategies to prevent
infection, including several that are currently being evaluated in clinical trials, confer incomplete
inhibition. This would only delay disease progression and would not lead to the reconstitution of
an HIV-1 resistant immune system. Therefore, strategies are being developed to confer complete
inhibition of HIV-1 replication in the gene-modified cells15,190.
The best interference site to protect healthy cells from infection is at the level of entry. Hence,
strategies that inhibit viral entry are of particular interest. HIV-1 entry is mediated by the specific
interaction of the viral envelope glycoproteins with a cell surface molecule, CD4, which serves
as the primary receptor, and a chemokine receptor, CCR5 or CXCR4, which serves as a co-
receptor258. HIV-1 strains that utilize the CCR5 co-receptor are called R5-tropic strains and those
utilizing the CXCR4 co-receptor are called X4-tropic strains. To inhibit viral entry, either the
receptor or the co-receptor may be targeted. CD4 and CXCR4 cannot be downregulated because
of their critical roles in the immune function or cell maturation and homing138,259,260. However,
CCR5 appears not to play a major role in immune function. The ccr5 gene is polymorphic and a
mutant phenotype has been reported in the Caucasian population at a frequency of 1-2%57. The
defective ccr5 gene (∆32ccr5) in this population contains a 32-bp deletion; it gives rise to a
truncated ∆32CCR5 protein, which is not expressed on the cell surface. Individuals homozygous
for the defective ccr5 gene are resistant to HIV-1. Disease progression in heterozygotes is also
65

slower than in individuals with normal ccr5 alleles52,122,261. Resistance in ∆32CCR5
homozygotes implies that other co-receptors do not substitute CCR5 for infection by R5-tropic
HIV-1, which initiates transmission. Furthermore, CCR5 appears to be required for all routes of
transmission since ∆32CCR5 homozygous individuals, among hemophiliacs and intravenous
drug-users, are also protected from HIV-1 transmission57. ∆32CCR5 homozygotes that lack
CCR5 appear to be completely normal, implying that either CCR5 is not functionally critical or
other co-receptors can substitute for normal cell functions261. Therefore, research has been
focused on downregulating CCR5 expression and/or function15,18,262. A concern in using this
strategy to block HIV-1 entry was the potential of selective pressure on HIV-1 to use other co-
receptors, i.e. CCR1, CCR2b or CCR3, or to convert more rapidly towards X4-tropic HIV-1.
However, in vitro replication of two R5-tropic HIV-1 isolates in the presence of an anti-CCR5
monoclonal antibody did not convert the two isolates to use CCR1, CCR3, or CXCR4263.
Monomeric155,176,177,190,264 and trimeric157 hammerhead ribozymes were designed to target CCR5
mRNA and were tested for their ability to inhibit viral entry. A decrease in surface CCR5
expression was observed with ~70% inhibition of virus replication in the ribozyme-expressing
cells. Incomplete inhibition of HIV-1 replication may be due to the fact that only a portion of the
CCR5 mRNA was cleaved.
Since anti-HIV-1 multimeric hammerhead ribozymes targeted against HIV-1 RNA inhibited
HIV-1 replication better than monomeric ribozymes did18,26,28, we have developed and tested a
multimeric hammerhead ribozyme that targets seven unique sites within the CCR5 mRNA.
Oncoretroviral and lentiviral vectors were developed and used to express this multimeric
ribozyme. Stably transduced cells were then tested for CCR5 mRNA downregulation,
susceptibility to X4- and R5-tropic HIV-1, and absence of HIV-1 proviral DNA.
66

2.3. Materials and Methods
2.3.1. Bacteria and Virus Strains and Cell Lines
E. coli DH5α cells were obtained from GIBCO-BRL.
NL4-3 (Cat.# 2479) and BaL (Cat#. 510) were obtained from NIH AIDS Research and
Reference Reagent Program.
PA317, an adherent mouse packaging cell line (expressing MoMuLV gag, pol, and env genes)265
was obtained form ATCC (Cat.# CRL-9078) and used to obtain amphotropic retroviral vector
particles.
293T, an adherent human embryonic kidney cell line266, was obtained form Stanford Reference
(Cat.# S97-079) and used to produce lentiviral vector particles.
PM1267, a suspension human CD4+/CCR5+/CXCR4+-espressing T-lymphoid cell line, was
obtained from NIH AIDS Research and Reference Reagent Program and used for expression of
Rz1-7, producing stocks for HIV-1 strains NL4-3 and BaL, and challenged with the same HIV-1
strains.
The adherent 293T and PA317 cells were cultured in complete Dulbecco's modified Eagle's
medium (DMEM), and the PM1 cells in complete Roswell Park Memorial Institute medium
(RPMI-1640). Both media were supplemented with 10% cosmic calf serum (HyClone, USA),
1% antibiotic-antimycotic (Gibco-BRL, USA), and 1% L-Glutamine 200 mM (Gibco-BRL,
USA). Cells were incubated in a 5% CO2 incubator at 37°C. For freezing, media were removed
67

from cells by centrifugation at 600 × g for 5 min. Cell pellet was resuspended in cosmic calf serum
containing 10% dimethyl sulfoxide (DMSO), followed by 2 hr at -20°C and overnight at -70°C,
and stored in liquid nitrogen. To revive, frozen cells were melted at room temperature and mixed
with 3 ml fresh medium, followed by centrifugation as above. Cell pellet was resuspended in the
appropriate amount of fresh medium and transferred to flask or plate.
2.3.2. Oligonucleotides and Primers
All oligonucleotides and primers were synthesized by ACGT Corporation (Toronto, Canada). Long
oligonucleotides were cartridge-purified. Primers were received in powder form (0.2 µmole). See
table 2.1 for sequences of oligonucleotides and primers.
2.3.3. Plasmids
pGEM-7Zf(+) was purchased from Promega, USA. Mouse stem cell virus (MSCV)-based vector,
MGIN, containing the internal ribosome entry site (IRES) from encephalomyocarditis virus,
enhanced green fluorescent protein (egfp), and neomycin phosphotransferase (neo) genes was
obtained from Dr. R.G. Hawley268. pMD.G, expressing vesicular stomatitis virus membrane
glycoprotein (VSV-G) was obtained from Dr. J. C. Burns269. pCMV∆8.9, expressing HIV-1 gag,
gag-pol, tat and rev proteins, was obtained from Dr. L. Naldini270.
68

Table 2.1- Oligonucleotides and primers sequences
Name Sequence
β-actin-R 5’-caa-aca-tga-tct-ggg-tca-tct-tct-c-3’
β-actin-F 5’-gct-cgt-cgt-cga-caa-cgg-ctc-3’
EGFP-F 5’-tgg-tga-gca-agg-gcg-agg-a-3’
HEG1-R 5’-cta-aga-tct-aca-gct-gcc-3’
MGIN-R 5’-cag-tcg-act-acg-tag-cgg-3’
R5-Ti-F or R5-Ti-5’* 5’-ata-ggt-acc-tgg-ctg-tcg-tcc-atg -3’
R5-Ti-R or R5-Ti-3’* 5’-ggt-cca-acc-tgt-tag-agc-tac-tgc-3’
T7-Tat-F or Tat-5’* 5’-ata-tca-tat-gta-ata-cga-ctc-act-ata-ggg-cga-ata-ctt-ggg-cag-gag-tgg-aag-c-3’
Tat-R or Tat-3’* 5’-gat-cta-tgc-atg-agc-cag-3’
* Either name has been used in the text.
69

2.3.4. Construction of a Multimeric Hammerhead Ribozyme Targeted against CCR5 mRNA
Multimeric hammerhead ribozymes were designed to target CCR5 mRNA at nts. 17, 380, 390, 520,
556, 811, and 824. The ribozyme (Rz) target sites within CCR5 mRNA are as follows: Rz1 target
site: 5’-TCAAGTGTC17 AAGTCCAA-3’, Rz2 target site: 5’-CGATAGGTA380 -CCTGGCTG-
3’, Rz3 target site: 5’-CTGGCTGTC390 GTCCATGC-3’, Rz4 target site: 5’-AAGAAGGTC520 T
TCATTAC-3’, Rz5 target site: 5’-CATACAGTC556 AGTATCAA-3’, Rz6 target site: 5’-ATTG
CAGTA811 GCTCTAAC-3’, and Rz7 target site: 5’-TAACAGGT-T824 GGACCAAG-3’;
cleavage sites are indicated by arrows (Figure 2.1A).
The following oligonucleotides were designed to construct the multimeric ribozymes: Rz1F: 5’-
GCAGATCTAATCGCAAGGATCCTCAAGTGTTTCGTCCTCACGGACTCATCAGAAGTC
CAA-3’ (containing a BamH I site), Rz2R: 5’-CAGCCAGGCTGATGAGTCCGTGAGGACGAA
ACCTATCGTTGGACTTCTGATGAG-3’, Rz3R: 5’-ATCCATCTTGTTCCACCCGGATCGATG
CATGGACCTGATGAGTCCGTGAGGACGAAACAGCCAGGCTGATGAG 3’ (containing a
Cla I site), Rz4F: 5’-GCAGATCTAATCGCAAGGATCCAAGAAGGTTTCGTCCTCACGGAC
TCATCAGTTCATTAC-3’ (containing a BamH I site), Rz5R: 5’-TTGATACTCTGATGAGTCC
GTGAGGACGAAACTGTATGGTAATGAACTGATGAG-3’, Rz6R: 5’-GTTAGAGCCTGATG
AGTCCGTGAGGACGAAACTGCAATTTGATACTCT GATGAG-3’, and Rz7R: 5’-ATCCAT
CTTGTTCCACCCGGATCGATCTTGGTCCCTGATGAGTCCGTGAGGACGAAACCTGTTA
GAGCCTGATGAG-3’ (containing a Cla I site); the restriction sites are shown in italics, the
ribozyme flanking sequences that hybridize to the target sites are underlined, and the ribozyme
catalytic domains are in bold.
The oligonucleotides Rz1F (containing a BamH I site), Rz2R, and Rz3R (containing a Cla I site) were
joined by overlapping PCR271 to yield a 189-bp product, Rz1-3. This PCR product was digested with
70

BamH I and Cla I and cloned into the same sites in pGEM-7Zf(+) to yield pGEM-Rz1-3. Similarly,
oligonucleotides Rz4F (containing a BamH I site), Rz5R, Rz6R, and Rz7R (containing a Cla I site)
were joined by overlapping PCR271 to yield a 230-bp product, Rz4-7. This PCR product was digested
with BamH I and Cla I and cloned into the same sites in pGEM-7Zf(+) to yield pGEM-Rz4-7. Rz1-3
and Rz4-7 do not contain any nucleotides between individual ribozymes.
To obtain Rz1-7, pGEM-Rz4-7 was digested with BamH I and the 3’-CTAG-5’ overhang was partially
filled with the DNA polymerase I Klenow fragment (New England BioLabs, Canada) in the presence
of dGTP, dATP, and dTTP. This resulted in a 3’-G-5’ overhang. The DNA was then digested with
EcoR I, and the modified BamH I-EcoR I fragment containing the Rz4-7 was gel-purified. The
pGEM-Rz1-3 was digested with Cla I and the 5’-CG-3’ overhang was partially filled with the DNA
polymerase I Klenow fragment in the presence of dCTP to yield a 5’-C-3’overhang. This plasmid
was digested with EcoR I and used as the backbone to insert the modified BamH I-EcoR I fragment
containing Rz4-7. Note that the modified Cla I and BamH I sticky ends are compatible with each
other. The resulting plasmid was designated pGEM-Rz1-7.
2.3.5. In Vitro Cleavage Activity of Rz1-3 and Rz4-7
Rz1-3 and Rz4-7 RNAs were transcribed in vitro from pGEM-Rz1-3 and pGEM-Rz4-7, and the 32P-
labeled target CCR5 RNA was transcribed in vitro from a plasmid designated pc.CCR5, as
described27. Briefly, the pc.CCR5 plasmid was digested by Xba I. The linearized plasmid containing
the ccr5 gene was used as a transcription template for in vitro transcription reaction using T7 RNA
Polymerase kit (Invitrogen, USA) for 3 hr at 37°C. 20 µCi [α-32P]UTP was also added to the mixture
when labeled RNA was required. The produced RNAs were purified by running on a 5%
polyacrylamide, 7 M urea gel, followed by incubating the gel fragment containing the RNA in an
elution buffer, containing 2 M NH4Ac, 1% sodium dodecyl sulfate (SDS), and 25 µg tRNA for
71

overnight at 37°C. The eluted RNAs were precipitated using 1 ml 100% Ethanol, incubation on
ice for 15 min, centrifugation at maximum 15000 × g for 15 min, followed by air-drying and
dissolving in RNase-free water. The cleavage reactions were performed by mixing the ribozymes
and labeled CCR5 RNA at a 1:1 molar ratio followed by gel electrophoresis, as described27.
Briefly, Rz1-3 or Rz4-7 was mixed with the labeled CCR5 RNA in a buffer containing 40 mM Tris-
HCl (pH 8.0) and 10 mM NaCl. After 5 min of incubation at 65°C, the mixture was cooled stepwise
to 37°C, 13.3 mM MgCl2 was added, and the incubation continued for 30 min at 37°C. The cleavage
products were analyzed on a 5% polyacrylamide, 7 M urea gel.
2.3.6. Vector Constructions
The MGIN vector268 was previously modified in our laboratory to contain unique Csp45 I and
Bgl II sites downstream of the egfp gene28. The Csp45 I-BamH I 274-bp fragment of pGEM-Rz1-
7 was used to clone Rz1-7 into the modified MGIN vector at the Csp45 I and Bgl II sites, to obtain
MGIN-Rz1-7. The correct clone was identified and characterized by PCR and restriction enzyme
analyses (Figure 2.2).
To construct the HIV-1-based vectors, HEG1 and HEG1-Rz1-7, the Sfi I-EcoR I 5360-bp fragment of
SIN-EF-EGFP (containing the plasmid sequence, the HIV-1 5’ LTR, and the human EF-1α
promoter)272, the EcoR I-Not I fragment of MGIN (756 bp containing the egfp gene) or the same
digestion fragment of MGIN-Rz1-7 (1030 bp containing the egfp and Rz1-7 genes), and the Not I-
Sfi I 4100-bp fragment of the pHR’CMVLacZ vector (containing the HIV-1 3’ LTR and the
downstream sequences)270 were ligated using T4 DNA ligase (Invitrogen, USA). The correct clones
were identified and characterized by PCR and restriction enzyme analyses (Figure 2.2).
72

2.3.7. Transduction and Selection of Stable PM1 Transductants
PA317 was transfected with the MGIN or MGIN-Rz1-7 vector, as described27. Briefly, the
adherent cells were trypsinized and 4 × 106 of them were transferred to each 10 cm cell culture
plate containing 10 ml complete DMEM medium. Cells were incubated in a 37°C CO2 incubator
for 5-6 h until attached to the bottom of the plate. Alternatively, 2-3 × 106 cells were seeded in
late evening and incubated for 18 h. 15 µg of vector DNA (here MGIN or MGIN-Rz1-7), as well
as packaging constructs (where applicable), were mixed in up to 450 µl H2O by vortexing. 50 µl
ice-cold 2.5 M CaCl2 was added and vortexed briefly. 500 µl of ice-cold 2× HeBS (containing
0.283 M NaCl, 1.5 mM Na2HPO4.7H2O, 0.023 M HEPES, pH 7.05; filter-sterilized) was added
drop-wise while mixing with bubbles made by a transfer pipette and then mixed more by finger
tapping. The mixture was incubated at room temperature for 20 min and then added drop-wise to
the cell culture while rotating the plate in all directions. Cells were incubated in a 37°C CO2
incubator for over night. The medium was changed next morning. The transfected cells were
cultured for three weeks in medium containing 400 µg/ml G418 to select for stable transfectants.
These cells were used to collect vector particles, which were filtered through a 0.42 µm
Millipore filter and stored at -70°C until used.
To obtain lentiviral vector particles, the 293T cells were cotransfected with the following
plasmids: pCMV∆8.9 (5 µg), pMD.G (10 µg), and the HEG1 (15 µg) or HEG1-Rz1-7 (15 µg)
vectors, as described above. The vector particles were collected on day 3 post-transfection,
filtered and stored at -70°C until used.
The MGIN, MGIN-Rz1-7, HEG1, and HEG1-Rz1-7 vector particles were used to transduce PM1
cells as described previously28. Briefly, depending on the concentration of vector particles
73

determined by p24 Ag assays, 5 × 104 to 1 × 106 suspension cells were transduced. The cells were
mixed with appropriate amount of vector particles in the presence of 6-8 µg/ml of polybrene in a well
of a 12-well plate and left for 7-8 h in a 37°C CO2 incubator. Then 100-200 µl of fresh complete
RPMI-1640 medium was added to each well and cells were cultured for overnight. Next
morning, the supernatant was removed completely and fresh medium was added. Alternatively,
cells were mixed with vector particles in the presence of 6-8 µg/ml of polybrene in a 5-ml tube and
centrifuged at 450 × g for 2 h at 32°C. Then, cells were resuspended in the same mixture and
transferred to a well of a 12-well plate, where 100-200 µl fresh complete RPMI-1640 was added,
and cultured in a 37°C CO2 incubator for overnight. Next morning, the culture was centrifuged at
450 × g for 5 min and the cells’ pellet was resuspended in the same mixture of vector particles. On
the fourth day, the cells were resuspended in fresh media and cultured. The pools of green
fluorescent stable PM1 transductants were sorted twice by fluorescence-activated cell sorting
(FACS) machine at 2-3 month intervals and used in subsequent experiments.
2.3.8. PCR Analysis of Genomic DNA from Stable PM1 Transductants
Genomic DNA was extracted from the individual pools of stable PM1 transductants by phenol-
chloroform-isoamyl alcohol extraction protocol273. The EGFP-F and MGIN-R primer pair was
used to amplify the Rz1-7 gene from genomic DNA of the MGIN-Rz1-7-transduced cells. Another
primer pair, EGFP-F and HEG1-R, was used to amplify this gene from genomic DNA of the
HEG1-Rz1-7-transduced cells. Endogenous β-actin gene was amplified as control using the β-
actin-F and β-actin-R primer pair. PCRs were performed for 40 cycles (95°C for 1 min, 56°C for
1 min, and 72°C for 1.5 min), in a 5 µl reaction mixture containing 0.4 µM of each primer, 1×
PCR buffer, 100 µM of each dNTP, 0.5 µg DNA, and 2.5 units Taq DNA polymerase. The PCR
74

products were analyzed by 2% agarose gel electrophoresis along with the λ–Hind III DNA
marker (Gibco-BRL, USA).
2.3.9. RT-PCR Analysis of Total RNA from Stable PM1 Transductants
Total RNA was extracted from stable PM1 transductants using RNeasy Mini Kit (QIAGEN,
Canada). The RNA was incubated with RQI RNase-free DNase (Promega, USA) for 5 min at
37°C to degrade any residual DNA. To ensure that the DNase treatment was complete, the RNA
samples were analyzed by PCR. Reverse transcriptions were performed for 1 h at 37°C using the
MGIN-R, HEG1-R, β-actin-R, or R5-Ti-R primers and the RT-PCR kit (Invitrogen, USA). The
cDNAs were then PCR-amplified, as described above, using the EGFP-F/MGIN-R or EGFP-
F/HEG1-R primer pair. β-actin-F/β-actin-R or R5-Ti-F/R5-Ti-R primer pairs were used to
amplify a 353-bp or 456-pb long region within the β-actin or CCR5 ORF as endogenous PCR
controls. The RT-PCR products were analyzed on a 2% agarose gel along with the λ–Hind III
DNA marker (Gibco-BRL, USA).
2.3.10. Immunoflowcytometry analysis of stable PM1 transductants
Transduced and untransduced PM1 cells (2 × 106) were washed twice with 10 ml phosphate
buffered saline (PBS) and resuspended in 250 µl PBS containing 2% fetal calf serum (FCS)
(Hyclone, USA). Anti-CCR5 mouse IgG2aκ 2D7 monoclonal antibody (mAb; 2.5 µl)
(PharMingen, Canada) was added and the cells were incubated on ice in the dark for 30 min. The
cells were then washed twice with PBS and incubated in a similar manner with 2.5 µl
biotinylated goat anti-mouse IgG2a antibody (Southern Biotechnology Associates Inc., USA) and
then with 2.5 µl allophycocyanin (APC)-streptavidin conjugate. Finally, the cells were washed
twice with PBS, resuspended in 2 ml PBS containing 2 mM EDTA and 2% FCS, and analyzed
75

by flowcytometry. To remove background staining during flowcytometry, PM1 cells, which
were incubated in the presence of only one of the two antibodies or APC-streptavidin, were also
analyzed and the parameters were set to ignore these responses.
2.3.11. HIV-1 Susceptibility of Stable PM1 Transductants
Stable transductants were challenged with the BaL274 and NL4-3275 strains of HIV-1. To obtain
virus stocks, untransduced PM1 cells were infected with either the BaL or the NL4-3 strain and
the supernatants from day 10 to 14 were collected, filtered through a 0.42 µm Millipore filter,
and stored at -70°C.
To determine the MOI, the TCID50 was calculated as follows: 600,000 PM1 cells in 100 µl were
seeded in a 96-well plate in four repeats. BaL or NL4-3 viruses were diluted in series of 1:10,
1:100, 1:1000, 1:10,000, 1:100,000, 1:1,000,000, and added to each well. Cells were incubated
for 7 days and at day 7, samples were assayed for p24 Ag values. TCID50 was calculated by the
Karber, Reed and Muench method276 using the following formula: TCID50=L-d(S-0.5), where
L=log10 of last dilution that makes 100% infection, S=sum of positive wells that includes the
last row with 100% infection plus the following well, d=log10 of dilution factor. The TCID50
gives the number of infectious viral particles in the volume of inoculation. The MOI was
calculated using this formula: MOI=(V × NVP) / (1000 × NC), where V=volume of inoculated
virus, NVP=number of viral particles in that volume, and NC=number of cells initially used for
infection.
The pools of actively dividing stable PM1 transductants (6 × 105 cells in 1 ml) were each
inoculated with the BaL or NL4-3 strain at an MOI of 0.225, 0.675, or 2.025 for 3 h at room
temperature, with gentle shaking. The cells were then washed three times with phosphate
76

buffered saline (PBS), suspended in 2 ml complete RPMI-1640 medium, and cultured at 37°C. ~1
ml of each cell culture was collected every 3-4 days and replaced with 1 ml of fresh medium.
These aliquots were centrifuged at 450 × g for 5 min, and the cell pellets and supernatants were
both stored at -70°C. Experiments were stopped 2-3 months post-infection. The supernatants of
the frozen cultures were diluted as appropriate, and the amount of HIV-1 p24 antigen was
measured by the enzyme linked immunosorbent assay (ELISA) p24 Antigen EIA kit (Beckman
Coulter, USA).
2.3.12. Progeny Virus Infectivity
Progeny viruses produced from BaL virus-infected untransduced, as well as HEG1 or HEG1-
Rz1-7-transduced stable PM1 cells (MOI of 2.025) were collected from the supernatants from day
17 post-infection, filtered through a 0.42 µm Millipore filter, and stored at -70°C. The progeny
viruses (13 ng of HIV-1 p24 equivalent) were then used to inoculate actively dividing
untransduced PM1 cells (6 × 105 cells in 1 ml) for 3 h at room temperature, with gentle shaking.
The cells were then washed three times with PBS, suspended in 2 ml complete RPMI-1640
medium, and cultured at 37°C. ~1 ml of each cell culture was collected every 3-4 days and
replaced with 1 ml of fresh medium. These aliquots were centrifuged at 450 × g for 5 min, and
the supernatants were stored at -70°C. The amount of HIV-1 p24 antigen was measured by the
ELISA p24 Antigen EIA kit (Beckman Coulter, USA).
2.3.13. PCR Analysis to Detect the HIV-1 Proviral DNA in the HIV-infected PM1
Transductants
Genomic DNA was extracted from MGIN- and MGIN-Rz1-7-transduced frozen PM1 cell pellets
from day 4 and 43 post-infection. Genomic DNA was also extracted from HEG1- and HEG1-
Rz1-7-transduced frozen PM1 cell pellets on day 4 and day 29 post-infection. The T7-Tat-F
77

primer and the Tat-R primer were used for PCR to amplify a 424-bp region of the HIV-1 tat
gene. The R5-Ti-F/R5-Ti-R primer pair was used for PCR to amplify a 465-bp region of the
endogenous ccr5 gene. The PCRs were performed for 40 cycles (95°C for 15 sec, 53°C for 1
min, and 72°C for 45 sec) and the products analyzed by a 2% agarose gel electrophoresis.
2.4. Results
2.4.1. Design, Construction, and Activity of the Anti-CCR5 Multimeric Hammerhead
Ribozyme, Rz1-7
The multimeric hammerhead ribozyme was designed against seven unique sites at nucleotides 17,
380, 390, 520, 556, 811, and 824 within the human CCR5 open reading frame (ORF) (Figure
2.1A). These sites were not found anywhere else within the human genome. Overlapping PCRs
were performed using synthetic oligodeoxynucleotides to assemble Rz1-3 and Rz4-7, which were
cloned in pGEM-7Zf(+).
In vitro cleavage reactions were performed to determine the activity of cloned ribozymes. The
expected lengths of the products resulting from cleavage of a 989-nt. long 32P-labelled CCR5
target RNA by Rz1-3 or Rz4-7 are shown in Figure 2.1B. The in vitro cleavage products are shown
in Figure 2.1C. Rz1-3 cleaved the CCR5 RNA at three sites. Cleavage by Rz1 gave rise to 61- and
928-nt. long products, while cleavage by Rz2 or Rz3 resulted into 424- and 565-, or 434- and
555-nt. long products, respectively. Cleavage by Rz1 and Rz2 or Rz3 produced 363- and 565-, or
373- and 555-nt. long products, respectively, from the 928-nt. long fragment. Rz4-7 cleaved the
CCR5 target RNA at four sites. Cleavage by Rz4 should have given rise to 564- and 425-nt. long
products, which were not clearly detectable. Cleavage by Rz5 gave rise to 580- and 409-nt. long
products. Cleavage of the 409-nt. long product by Rz6 or Rz7 gave rise to 275 and 134, or to 288
78

Rz117 380
Rz2380
Rz3390 520
Rz4520 556
Rz5556
Rz6811 824
Rz7824
CCR5 ORF
a.
b.
121-134 b
275-288 b409 b580 b855-868 b
c.
928 b
363-373 b424-434 b555-565 b
61 b
CCR5 + Rz4 -7
CCR5+ Rz1 -3
CCR5
61 b
Rz2 Rz3
363 b 555 b
CCR5 RNA (989 b)
CCR5 RNA (989 b)
Rz1
Rz4 Rz5
Rz1-3 cleavage products:
10 b
Rz6 Rz7
Rz4-7 cleavage products:
424 b 565 b
555 b434 b
61 b 928 bRz1 cleavage products
Rz2 cleavage products
Rz3 cleavage products
Rz1, Rz2, Rz3cleavage products
564 b 121 b16 b 13 b275 b
564 b 425 b
580 b 409 b
855 b 134 b
868 b 121 b
Rz4, Rz5, Rz6, Rz7cleavage products
Rz4 cleavage products
Rz5 cleavage products
Rz6 cleavage products
Rz7 cleavage products
989 b
Rz117 380
Rz2380
Rz3390 520
Rz4520 556
Rz5556
Rz6811 824
Rz7824
CCR5 ORFCCR5 ORF
a.
b.
121-134 b
275-288 b409 b580 b855-868 b
c.
928 b
363-373 b424-434 b555-565 b
61 b
CCR5 + Rz4 -7
CCR5+ Rz1 -3
CCR5
61 b
Rz2 Rz3
363 b 555 b
CCR5 RNA (989 b)
CCR5 RNA (989 b)
Rz1
Rz4 Rz5
Rz1-3 cleavage products:
10 b
Rz6 Rz7
Rz4-7 cleavage products:
424 b 565 b
555 b434 b
61 b 928 bRz1 cleavage products
Rz2 cleavage products
Rz3 cleavage products
Rz1, Rz2, Rz3cleavage products
564 b 121 b16 b 13 b275 b564 b 121 b16 b 13 b275 b
564 b 425 b
580 b 409 b
855 b 134 b
868 b 121 b
Rz4, Rz5, Rz6, Rz7cleavage products
Rz4 cleavage products
Rz5 cleavage products
Rz6 cleavage products
Rz7 cleavage products
989 b
C B
A
Figure 2.1- In vitro cleavage activity of the anti-CCR5 multimeric hammerhead ribozyme,
Rz1-7: A) Rz1 to Rz7 cleavage sites are shown within the 1058-nt. long CCR5 ORF. B) The
expected lengths of the CCR5 RNA products upon cleavage by Rz1, Rz2, and/or Rz3 and Rz4,
Rz5, Rz6, and/or Rz7. C) In vitro cleavage activities of Rz1-3 and Rz4-7. The CCR5 target RNA
was incubated with the Rz1-3 or Rz4-7, which were transcribed in vitro from pGEM-Rz1-3 or
pGEM-Rz4-7. Rz1-3 cleavage products (lane 1), CCR5 RNA (lane 2), and Rz4-7 cleavage products
(lane 3) were analyzed on a 5% polyacrylamide, 7 M urea gel.
79

Cap (A)n
Cap (A)n
Cap (A)n
Cap(A)nCap(A)n
Cap (A)n
Cap(A)nCap(A)n
Cap (A)n
Cap (A)n
Cap (A)n
Cap(A)nCap(A)n
Cap (A)n
Cap(A)nCap(A)n
Figure 2.2- Schematic diagrams of oncoretroviral and lentiviral vectors: Schematic
diagrams of MGIN, MGIN-Rz1-7, HEG1, and HEG1-Rz1-7 vectors and their transcripts. MGIN
contains the egfp gene, an IRES element, and the neo gene. HEG1 contains the egfp gene under
control of the EF-1α promoter. The Rz1-7 gene in MGIN-Rz1-7 and HEG1-Rz1-7 vectors is cloned
downstream of the egfp gene.
80

and 121-nt. long products, respectively. These results show that Rz1-3 and Rz4-7 are active. These
ribozymes were then combined to yield Rz1-7. Rz1-7 contains the seven ribozymes with no
intercalated nucleotides, except for eight nucleotides between Rz3 and Rz4.
2.4.2. Oncoretroviral and Lentiviral Vectors Expressing Rz1-7
The mouse stem cell virus (MSCV)-based oncoretroviral vector, MGIN268, contains the
enhanced green fluorescence protein (egfp) gene, an internal ribosome entry site (IRES), and the
neomycin phosphotransferase (neo) gene (Figure 2.2). MGIN-Rz1-7 was engineered to express the
Rz1-7 gene, which was cloned between the egfp gene and the IRES element. In the MGIN and
MGIN-Rz1-7 vectors, the MSCV 5’ long terminal repeat (LTR) promoter allows constitutive
expression of a bicistronic vector RNA, which permits translation of the two ORFs, EFGP and
NEO.
An HIV-1-based HEG1 vector was designed to express the egfp gene under the control of an
internal human elongation factor-1α (EF-1α) promoter. The Rz1-7 gene was cloned downstream
of the egfp gene in the HEG1 vector to yield HEG1-Rz1-7 (Figure 2.2). In the HEG1 and HEG1-
Rz1-7 vectors, the EF-1α promoter allows constitutive expression of EGFP mRNA, whereas the
HIV-1 5’ LTR promoter allows inducible expression of vector RNA, which can also be spliced.
Rz1-7 is present on all the transcripts.
2.4.3. Development of Pools of Stable PM1 Transductants Expressing Rz1-7
Amphotropic MGIN, MGIN-Rz1-7, HEG1, and HEG1-Rz1-7 vector particles were each used to
transduce a PM1 cell line, which is a human CD4+ T lymphoid cell line. Pools of stable PM1
transductants were sorted twice at 2-3 month intervals by fluorescence activated cell sorter
81

(FACS) and used in subsequent experiments. The growth rates of transduced PM1 cells were
comparable to those of untransduced PM1 cells.
The presence of the Rz1-7 gene was confirmed by PCR analysis of genomic DNAs isolated from
various PM1 transductants (Figure 2.3) using the EGFP-F/MGIN-R and EGFP-F/HEG1-R
primer pairs. These forward and reverse primers were designed to hybridize to sequences
upstream and downstream of the ribozyme-cloning sites, respectively. No PCR product was
detected in the untransduced samples (Figure 2.3A and B, lane 1). As expected, by PCR using
the EGFP-F/MGIN-R primer pair, a 771-bp product was detected in the MGIN-transduced
sample, whereas a 1083-bp product was obtained from the MGIN-Rz1-7-transduced sample when
using the same primer pair (Figure 2.3A, lanes 2 and 3). Likewise, 834- and 1099-bp products
were detected in the HEG1- and HEG1-Rz1-7-transduced cells respectively, when the EGFP-
F/HEG1-R primer pair was used (Figure 2.3B, lanes 2 and 3). Genomic DNAs from the
untransduced and transduced cells were also analyzed using the β-actin-F/β-actin-R primer pair
to amplify a 474-bp region of the cellular β-actin gene (Figure 2.3A and B, lower panels).
Rz1-7 production was confirmed by RT-PCR analysis of total cellular RNA from various PM1
transductants. The RNA samples were treated with DNase and analyzed by PCR to ensure
absence of DNA contamination (results not shown). No RT-PCR product was obtained from the
untransduced cells (Figure 2.4A and B, lane 1). RT-PCR using the EGFP-F/MGIN-R primer pair
resulted in the amplification of a 771-bp product from the MGIN- transduced cells and a 1083-bp
product from the MGIN-Rz1-7-transduced cells (Figure 2.4A, lanes 2 and 3). RT-PCR using the
EGFP-F/HEG1-R primer pair resulted in the amplification of an 834-bp product from the HEG1-
transduced cells and a 1099-bp product from the HEG1-Rz1-7-transduced cells (Figure 2.4B,
lanes 2 and 3). RT-PCR analysis using the β-actin-F/β-actin-R primer pair (as a control) yielded
82

a.
b.1099 bp
834 bp
474 bp
1083 bp771 bp
474 bp
MGIN-Rz1-7 DNAMGIN DNA
β-actin gene
HEG1-Rz1-7 DNAHEG1 DNA
β-actin gene
1 2 3 4 5
1 2 3 4 5
a.
b.1099 bp
834 bp
474 bp
1083 bp771 bp
474 bp
MGIN-Rz1-7 DNAMGIN DNA
β-actin gene
HEG1-Rz1-7 DNAHEG1 DNA
β-actin gene
1 2 3 4 5
1 2 3 4 5
A
B
Figure 2.3- PCR analyses to determine the presence of vector DNA sequences in the
transduced PM1 cells: Genomic DNA was extracted from untransduced or transduced PM1
cells. A) PCR products obtained from the untransduced PM1 cells (lane 1), or MGIN (lane 2) or
MGIN-Rz1-7 (lane 3) vector-transduced PM1 cells using the EGFP-F/MGIN-R primer pair. PCR
products from MGIN (lane 4) and MGIN-Rz1-7 (lane 5) vectors were analyzed as controls. B)
PCR products obtained from untransduced PM1 cells (lane 1), or HEG1 (lane 2) or HEG1-Rz1-7
(lane 3) vector-transduced PM1 cells using the EGFP-F/HEG1-R primer pair. PCR products
from HEG1 (lane 4) and HEG1-Rz1-7 (lane 5) vectors were analyzed as controls. The
endogenous β-actin gene was PCR-amplified (lower panels) as a control using the β-actin-F/β-
actin-R primer pair. The PCR products were analyzed on a 2% agarose gel. The product sizes are
indicated on the left of the gels.
83

b.
a.1083 bp
771 bp
353 bp
1099 bp
834 bp
353 bp
MGIN-Rz1-7 RNAMGIN RNA
β-actin mRNA
HEG1-Rz1-7 RNA
HEG1 RNA
β-actin mRNA
1 2 3 4 5
1 2 3 4 5b.
a.1083 bp
771 bp
353 bp
1099 bp
834 bp
353 bp
MGIN-Rz1-7 RNAMGIN RNA
β-actin mRNA
HEG1-Rz1-7 RNA
HEG1 RNA
β-actin mRNA
1 2 3 4 5
1 2 3 4 5
A
B
Figure 2.4- RT-PCR analyses to determine the production of Rz1-7 RNA in transduced PM1
cells: Total RNA was extracted from untransduced or transduced PM1 cells. A) RT-PCR
products obtained from the untransduced PM1 cells (lane 1), or MGIN (lane 2) or MGIN-Rz1-7
(lane 3) vector-transduced PM1 cells using the EGFP-F/MGIN-R primer pair. PCR products
from MGIN (lane 4) and MGIN-Rz1-7 (lane 5) vectors were analyzed as controls. B) RT-PCR
products obtained from untransduced PM1 cells (lane 1), or HEG1 (lane 2) or HEG1-Rz1-7 (lane
3) vector-transduced PM1 cells using the EGFP-F/HEG1-R primer pair. PCR products from
HEG1 (lane 4) and HEG1-Rz1-7 (lane 5) vectors were analyzed as controls. The endogenous β-
actin mRNA was RT-PCR-amplified (lower panels) using the β-actin-F/β-actin-R primer pair.
The RT-PCR products were analyzed on a 2% agarose gel. The product sizes are indicated on the
left of the gels.
84

the expected 353-bp product corresponding to spliced β-actin mRNA in all untransduced and
transduced samples (Figure 2.4A and B, lower panels).
2.4.4. Rz1-7-mediated Downregulation of CCR5 mRNA
Downregulation of CCR5 mRNA in MGIN-Rz1-7- and HEG1-Rz1-7-transduced cells was
assessed by RT-PCR analysis. A 456-bp product, resulting from RT-PCR amplification of CCR5
mRNA, was detected in MGIN- and HEG1-transduced cells (Figure 2.5A and B, lane 1).
Absence of this band in MGIN-Rz1-7- and HEG1-Rz1-7-transduced cells (Figure 2.5A and B, lane
2) reveals that CCR5 mRNA was cleaved by the multimeric ribozyme.
Similar amounts of RNA were present in all samples as shown by RT-PCR amplification of
endogenous β-actin mRNA (Figure 2.5, lower panels).
2.4.5. Rz1-7-mediated Downregulation of the Surface CCR5 Co-receptor
To determine the level of expression of the CCR5 co-receptor on the surface of stable PM1
transductants lacking or expressing Rz1-7, cells were incubated with an anti-CCR5 mouse IgG2aκ
2D7 mAb, followed by a biotinylated goat anti-mouse IgG2a antibody and an APC-streptavidin
conjugate. The immunoflowcytometry results showed 90% and 99.6% downregulation of surface
CCR5 expression on PM1 cells expressing MGIN-Rz1-7 and HEG1-Rz1-7, respectively (Figure
2.6).
2.4.6. HIV-1 Susceptibility of Pools of Stable PM1 Transductants Expressing Rz1-7
Stable PM1 transductants lacking or expressing Rz1-7 were challenged with the BaL and NL4-3
strains of HIV-1. Progeny virus production was measured by determining the amount of HIV-1
85

a.
b.
CCR5 mRNA
1 2
353 bp
456 bp
353 bp
456 bp
β-actin mRNA
β-actin mRNA
CCR5 mRNA
1 2.
.
CCR5 mRNA
a
b1 2
353 bp
456 bp
353 bp
456 bp
β-actin mRNA
β-actin mRNA
CCR5 mRNA
1 2
B
A
Figure 2.5- RT-PCR analyses to determine CCR5 mRNA levels in transduced PM1 cells:
Total RNA was extracted from transduced PM1 cells. A) RT-PCR products obtained from the
MGIN (lane 1) or MGIN-Rz1-7 (lane 2) vector-transduced PM1 cells using the R5-Ti-F/R5-Ti-R
primer pair. B) RT-PCR products obtained from HEG1 (lane 1) or HEG1-Rz1-7 (lane 2) vector-
transduced PM1 cells using the R5-Ti-F/R5-Ti-R primer pair. The endogenous β-actin mRNA
was RT-PCR-amplified (lower panels) using the β-actin-F/β-actin-R primer pair. The RT-PCR
products were analyzed on a 2% agarose gel. The product sizes are indicated on the left of the
gels.
86

GIN PM1-MGIN-Rz1-7PM1-MPM1
PM1 PM1-HEG1 PM1-HEG1-Rz1-7
Figure 2.6- Immunoflowcytometry analyses to determine CCR5 co-receptor expression
level on the surface of untransduced and transduced PM1 cells: Untransduced PM1 cells as
well as MGIN, MGIN-Rz1-7, HEG1, and HEG1-Rz1-7 vector-transduced PM1 cells lacking or
expressing Rz1-7 were analyzed by immunoflowcytometry using a combined antibody system
containing anti-CCR5 mouse IgG2aκ 2D7 mAb, biotinylated goat anti-mouse IgG2a antibody, and
APC-streptavidin conjugate.
87

p24 present in the supernatants of infected cell cultures. Untransduced cells, as well as MGIN-,
MGIN-Rz1-7-, HEG1-, or HEG1-Rz1-7-transduced cells, challenged by the NL4-3 strain at a
multiplicity of infection (MOI) of 0.225 produced high amounts (>460 ng/ml) of HIV-1 p24 on
day 10 post-infection (results not shown). Likewise, both the untransduced cells and the cells
transduced with the MGIN or HEG1 vectors produced high amounts of progeny virus when
challenged by the BaL strain at an MOI of 0.225, 0.675, or 2.025 (Figure 2.7). Complete (100%)
inhibition of progeny virus production was observed up to day 87 (Figure 2.7A) or 66 (Figure
2.7B) post-infection in two independent experiments performed with the MGIN-Rz1-7-transduced
cells challenged by the BaL strain at an MOI of 0.225. Nearly total (~99%) inhibition of progeny
virus production was observed up to day 66 post-infection when the MGIN-Rz1-7-transduced
cells were challenged by the BaL strain at higher MOIs (0.675 and 2.025) (Figure 2.7B). When
HEG1-Rz1-7-transduced PM1 cells were cultured for a long period of time and then challenged
with the BaL strain at an MOI of 0.225, progeny virus production was delayed (up to day 29
post-infection) and strongly diminished (80% inhibition) up to day 60 post-infection (Figure
2.7C). However, nearly total (~99%) inhibition of progeny virus production was observed up to
day 66 post-infection when a fresh batch of HEG1-Rz1-7-transduced cells was challenged by the
BaL strain at three different MOIs (0.225, 0.675, and 2.025) (Figure 2.7D). The purpose of this
experiment was showing the resistance of Rz1-7-expressing cells in comparison with the cells,
which do not express it. The reproductivity of this experiment was tested by different
concentrations of virus rather than simple repeating with the same viral MOI.
2.4.7. Infectivity of Progeny Viruses
The infectivity of the progeny viruses from HEG1- or HEG1-Rz1-7-transduced cells, challenged
by the BaL strain at an MOI of 2.025, was assessed by infecting the untransduced PM1 cells. The
amount of progeny viruses produced on various days post-infection was measured by
88

a. b.
d.c.
0
100
200
300
400
500
600
0 10 20 30 40 50 60 70 80 90
Days post-infection
HIV
-1 p
24 (n
g/m
l)
0
200
400
600
800
1000
0 10 20 30 40 50 60
Days post-infection
HIV
-1 p
24 (n
g/m
l)
0
250
500
750
1000
1250
1500
1750
0 10 20 30 40 50 60 70
Days post-infection
HIV
-1 p
24 (n
g/m
l)
0
500
1000
1500
2000
2500
3000
0 10 20 30 40 50 60 70
Days post-infection
HIV
-1 p
24 (n
g/m
l)
a. b.
d.c.
0
100
200
300
400
500
600
0 10 20 30 40 50 60 70 80 90
Days post-infection
HIV
-1 p
24 (n
g/m
l)
0
200
400
600
800
1000
0 10 20 30 40 50 60
Days post-infection
HIV
-1 p
24 (n
g/m
l)
0
250
500
750
1000
1250
1500
1750
0 10 20 30 40 50 60 70
Days post-infection
HIV
-1 p
24 (n
g/m
l)
0
500
1000
1500
2000
2500
3000
0 10 20 30 40 50 60 70
Days post-infection
HIV
-1 p
24 (n
g/m
l)
D C
B A
Figure 2.7- Susceptibility of transduced PM1 cells to R5-tropic HIV-1 (BaL): Cells were
challenged with HIV-1 BaL at an MOI of 0.225, 0.675, or 2.025. HIV-1 p24 released in the
culture supernatant was determined (Y axis) at different time intervals (X axis). A) Progeny virus
production following HIV-1 infection (MOI of 0.225) of untransduced PM1 cells (×), PM1 cells
transduced with MGIN (open circles), or PM1 cells transduced with MGIN-Rz1-7 (closed
circles). B) Progeny virus production following HIV-1 infection of PM1 cells transduced with
MGIN (open symbols) or MGIN-Rz1-7 (closed symbols) at an MOI of 0.225 (circles), 0.675
(triangles), or 2.025 (rectangles). C) Progeny virus production following HIV-1 infection (MOI
of 0.225) of untransduced PM1 cells (×), PM1 cells transduced with HEG1 (open circles), or
PM1 cells transduced with HEG1-Rz1-7 (closed circles). D) Progeny virus production following
HIV-1 infection of PM1 cells transduced with HEG1 (open symbols) or HEG1-Rz1-7 (closed
symbols) at an MOI of 0.225 (circles), 0.675 (triangles), or 2.025 (rectangles).
89

determining the amount of HIV-1 p24 present in the supernatants of infected cell cultures.
Similar amounts of progeny viruses were produced from PM1 cells that were challenged with the
progeny viruses produced from the HEG1- (199 pg/ml) or HEG1-Rz1-7- (196 pg/ml) transduced
PM1 cells on day 14 post-infection.
2.4.8. PCR Analyses to Detect the Presence of HIV-1 Proviral DNA in Infected PM1
Transductants
PCR analyses of the genomic DNA of BaL and NL4-3 virus-infected untransduced and
transduced PM1 cells revealed the presence of HIV-1 proviral DNA in all cells that were
infected with the NL4-3 strain (Figure 2.8A and B, lanes 1, 3, and 5), as well as the untransduced
and the MGIN- or HEG1-transduced PM1 cells infected with the BaL strain (Figure 2.8A and B,
lanes 2 and 6). No HIV-1 proviral DNA could be detected at the two time points tested (day 4
and 43 post-infection) in the MGIN-Rz1-7-transduced PM1 cells (Figure 2.8A, lane 4), whereas a
faint band corresponding to this DNA was detectable by day 29 post-infection in the HEG1-Rz1-
7-transduced cells (Figure 2.8B, lane 4).
2.5. Discussion
CCR5 is an attractive candidate for HIV-1 gene therapy. A monomeric ribozyme targeted against
nt. 23 within the CCR5 ORF was extensively studied by Cagnon et al. (2000), Bai et al. (2000),
and Li et al. (2003). A cell line transfected with a plasmid expressing this ribozyme was shown
to lead to a 70% decrease in surface CCR5 expression, compared to a 50% decrease for a mutant
ribozyme. However, both the active and the mutant ribozymes conferred only a 1-3 day delay in
BaL virus replication177. PM1 cells transduced with an oncoretroviral vector expressing this
ribozyme conferred 70% (active ribozyme) vs 50% (mutant ribozyme) inhibition of BaL virus
replication on day 7 post-infection177. Another oncoretroviral vector expressing this ribozyme
90

a.
b.
Day 29
Day 4HIV-1 tat gene
424 bp
ccr5 gene Day 29 456 bp
Day 43
Day 4HIV-1 tat gene
424 bp
Day 43 456 bp
1 2 3 4 5 61 2 3 4 5 6
ccr5 gene
1 2 3 4 5 61 2 3 4 5 6
a.
b.
Day 29
Day 4HIV-1 tat gene
424 bp
ccr5 gene Day 29 456 bp
Day 43
Day 4HIV-1 tat gene
424 bp
Day 43 456 bp
1 2 3 4 5 61 2 3 4 5 6
ccr5 gene
1 2 3 4 5 61 2 3 4 5 6
B
A
Figure 2.8- Assessment of HIV-1 provirus DNA from untransduced PM1 cells, as well as
from control or ribozyme vector-transduced PM1 cells challenged with HIV-1: A)
Assessment of HIV-1 proviral DNA from untransduced PM1 cells (lanes 1 and 2), as well as
from MGIN-Rz1-7-transduced (lanes 3 and 4), and MGIN-transduced (lanes 5 and 6) PM1 cells
challenged with HIV-1 strains NL4-3 (lanes 1, 3, and 5) or BaL (lanes 2, 4, and 6). Genomic
DNA samples from day 4 and 43 post-infection (from the experiment described in Figure 2.7a)
were analyzed by PCR using the T7-Tat-F/Tat-R primer pair. B) Assessment of HIV-1 proviral
DNA from untransduced PM1 cells (lanes 1 and 2), as well as from HEG1-Rz1-7-transduced
(lanes 3 and 4) and HEG1-transduced (lanes 5 and 6) PM1 cells challenged with HIV-1 strains
NL4-3 (lanes 1, 3, and 5) or BaL (lanes 2, 4, and 6). Genomic DNA samples from day 4 and 29
post-infection (from the experiment described in Figure 2.7c) were analyzed by PCR using the
T7-Tat-F/Tat-R primer pair. As a control, a 456-bp region of the cellular ccr5 gene was PCR-
amplified (lower panels) using the R5-Ti-F/R5-Ti-R primer pair. The PCR products were
analyzed on a 2% agarose gel. The product sizes are indicated on the left of the gels.
91

was also used to transduce CD34+ hematopoietic stem cells. The differentiated macrophages
showed inhibition of BaL virus replication on day 17 post-infection. However this inhibition was
only slightly better than with the mutant ribozyme155. An HIV-1 based vector expressing this
ribozyme was also used to transduce primary T cells and CD34+ stem cells264. The transduced
primary T cells and the monocytes differentiated in vitro from the transduced CD34+ cells, were
challenged with the JR-FL strain. A certain level of selective survival was observed compared to
the control vector-expressing cells; inhibition of virus replication was not assessed264.
Multimeric hammerhead ribozymes have an increased probability of recognizing and cleaving at
least one of the multiple target sites within the CCR5 mRNA. Therefore, a trimeric ribozyme
was designed against nts. 17, 153, and 249 within the CCR5 ORF157. Oncoretroviral vectors
expressing this trimeric ribozyme decreased CCR5 expression by 10-15% and conferred ~30%
inhibition of R5-tropic HIV-1 replication on day 4 post-infection157. Similar results were
obtained for inhibition of HIV-1 replication in macrophages derived from transduced CD34+
stem cells157.
The partial inhibition of HIV-1 replication observed using anti-CCR5 monomeric and trimeric
ribozymes could have been due to incomplete downregulation of surface CCR5 expression.
Therefore, to further improve this strategy, we designed a multimeric hammerhead ribozyme, which
targets seven unique sites within the CCR5 mRNA. Rz1-7 was designed against nts. 17, 380, 390,
520, 556, 811, and 824 within the CCR5 ORF. These ribozymes were shown to effectively cleave
the CCR5 RNA in vitro (Figure 2.1).
Unlike oncoretroviral vectors, lentiviral vectors are able to pass through the nuclear envelope
into the cell nucleus, thereby allowing transduction of non-dividing cells. A lentiviral vector is
92

therefore more suitable for anti-HIV-1 gene delivery into human hematopoietic stem cells. We
have used an MSCV-based oncoretroviral (MGIN) vector and an HIV-1-based lentiviral (HEG1)
vector for delivery and expression of the multimeric hammerhead ribozyme Rz1-7 (Figure 2.2).
Transcription from the MGIN 5’ LTR promoter results in constitutive expression of a long
transcript containing the EGFP ORF, the IRES element, and the NEO ORF. The transcript
produced from MGIN-Rz1-7 also contains the Rz1-7 downstream of the EGFP ORF. In contrast,
the 5’ LTR promoter in the HEG1 and HEG1-Rz1-7 vectors is not expressed in the absence of
HIV-1 Tat protein. Therefore, an internal EF-1α promoter was used to allow strong constitutive
expression of a transcript containing the EGFP ORF or the EGFP ORF and the Rz1-7. CCR5
mRNA cleavage by the Rz1-7 would result in surface CCR5 downregulation, preventing HIV-1
entry.
RT-PCR analyses of total RNA extracted from PM1 cells transduced with MGIN-Rz1-7 or HEG1-
Rz1-7 showed that both vectors were able to express the Rz1-7 (Figure 2.4). The Rz1-7 expressed in
these cells was active, since the CCR5 mRNA (Figure 2.5) and surface CCR5 co-receptor
(Figure 2.6) levels decreased.
As expected, high-levels of progeny virus were produced when the untransduced cells, as well as
MGIN-, MGIN-Rz1-7-, HEG1-, or HEG1-Rz1-7-transduced PM1 cells, were challenged with the
NL4-3 strain, suggesting that the cells were permissive to X4-tropic HIV-1 replication. Control
untransduced PM1 cells and MGIN- or HEG1-transduced cells could be infected by the BaL
strain. However, when the MGIN-Rz1-7-transduced cells were challenged with the BaL strain
(MOIs of 0.225, 0.675, and 2.025), 99-100% inhibition of progeny virus production was
observed for the duration of the experiment (2-3 months post-infection) (Figure 2.7A and B).
Inhibition of replication of the BaL strain was more prominent when the multimeric ribozyme
93

was expressed from the MGIN-Rz1-7 vector than from the HEG1-Rz1-7 vector. When the HEG1-
Rz1-7-transduced cells were challenged with the BaL strain (MOIs of 0.225, 0.675, and 2.025),
90-99% inhibition of virus replication was observed for the duration of the experiment (2 months
post-infection) (Figure 2.7C and D).
In the progeny viruses released from the lentiviral vector-transduced PM1 cells, HEG1 and
HEG1-Rz1-7 vector RNAs could have been packaged with the HIV-1 RNA. The inclusion of Rz1-
7 was not believed to alter progeny virus infectivity since it is not targeted against HIV-1 RNA.
The infectivity of the progeny viruses produced from the BaL virus-infected HEG1 or HEG1-
Rz1-7-transduced PM1 cells was assessed by infecting untransduced PM1 cells. As expected,
similar amounts of progeny viruses were produced in both cases (196-199 pg/ml on day 14 post-
infection), suggesting that the progeny viruses produced from the control or ribozyme-expressing
cells were equally infectious.
PM1 cells have been shown to express CXCR4, CCR1, CCR3, CCR4 and CCR5 receptors277.
Therefore, significant inhibition of HIV-1 BaL virus replication in MGIN-Rz1-7 and HEG1-Rz1-7-
transduced cells (Figure 2.7B and D) indicates that the progeny viruses produced from these cells
do not correspond to escape viruses with altered tropism for CXCR4, CCR1, CCR3 or CCR4 co-
receptors.
PCR analyses at different time intervals confirmed that the inhibition of BaL virus replication in
MGIN-Rz1-7- and HEG1-Rz1-7-transduced cells is at the level of entry, as no or very little
proviral DNA was detected by PCR (Figure 2.8).
94

2.6. Conclusions
This study shows that Rz1-7-mediated downregulation of CCR5 mRNA results in almost
complete inhibition of R5-tropic HIV-1 entry and replication in MGIN-Rz1-7-transduced cells
that are permissive to X4-tropic HIV-1 replication. These results demonstrate the feasibility of a
multimeric hammerhead ribozyme-based strategy in successfully inhibiting HIV-1 entry. Vectors
expressing this multimeric hammerhead ribozyme will be challenged with all major clades of
HIV-1. These vectors will also be tested for inhibition of HIV-1 replication in transduced
peripheral blood mononuclear cells and in the in vitro differentiated progeny of transduced
CD34+ stem cells. Since this multimeric hammerhead ribozyme confers excellent inhibition of
R5-tropic HIV-1 replication up to an MOI of 2.025 (Figure 2.7B and D), we anticipate that it will
be beneficial for HIV-1 gene therapy.
List of Abbreviations
EF-1α, human elongation factor-1α; egfp, enhanced green fluorescence protein; FACS,
fluorescence activated cell sorter; IRES, internal ribosome entry site; LTR, long terminal repeat;
MOI, multiplicity of infection; MSCV, mouse stem cell virus; neo, neomycin
phosphotransferase; ORF, open reading frame; Rz, ribozyme.
Acknowledgements
This work was supported by grants from the Canadian Institutes of Health Research and the
Ontario HIV Treatment Network. R. N. is thankful to the Ontario HIV Treatment Network for a
doctoral fellowship. We thank Dr. R. G. Hawley for providing us with the MGIN and SIN-EF-
EGFP vectors and to Dr. D. Trono for the pHR’CMVLacZ, pCMV∆8.9, and pMD.G vectors. We
are grateful to Dr. A. L. Haenni for excellent scientific discussions and for critical proofreading
95

of this manuscript. We also thank Dr. A. Arora for the HEG1 vector construction and Dr. M.
Ameli for technical assistance. Flow cytometry experiments were performed by Ms. G. Knowles
at the Center for Cytometry and Scanning Microscopy (Sunnybrook Health Sciences Centre),
and FACS analyses were performed by Mr. W. Lee at the CIHR Group in Matrix Dynamics
(University of Toronto). The following reagents were obtained through the AIDS Research and
Reference Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 NL4-3 strain from Dr. A.
Adachi; HIV-1 BaL strain from Dr. S. Gartner; PM1 cell line from Dr. D. Richman, and
pc.CCR5 from Dr. N. Landau.
96

CHAPTER 3
Exploring the Potential of Using Group II Introns to Inactivate HIV-1
Reza Nazari1 and Sadhna Joshi1,2,*
1Department of Laboratory Medicine and Pathobiology, Faculty of Medicine, University of Toronto, Toronto, Ontario, M5S 3E2, Canada 2Department of Medical Genetics and Microbiology, Faculty of Medicine, University of Toronto, Toronto, Ontario, M5S 3E2, Canada
This chapter has been modified from a manuscript submitted to Journal of General Virology in 2008.
* To whom correspondence and reprint requests should be addressed. Sadhna Joshi, Department of Medical
Genetics and Microbiology, University of Toronto, 150 College St. # 212, Toronto Ontario M5S 3E2, Canada. Tel.
+ 416 978-2499; Fax +416 638-1459; Email: [email protected]
97

3.1. Abstract
Towards the development of a potential gene therapy strategy that could interrupt integration of
HIV-1 proviral DNA during the next round of infection, we have examined whether insertion of
a mobile group II intron into infectious HIV-1 proviral DNA could inhibit virus replication. We
have used introns targeted against two sites within the IN-coding region of the HIV-1 pol gene.
Intron insertion within this region should not interfere with viral RNA and protein synthesis;
however, the progeny virus produced is expected to be non-infectious. Intron insertion into an
infectious HIV-1 proviral DNA clone was allowed to occur in Escherichia coli. The intron-
inserted HIV-1 proviral DNA clones were then isolated and tested for virus replication in
mammalian cells. Similar amounts of HIV-1 RNA, protein, and progeny virus were produced
from HIV-1 proviral DNA as from intron-inserted HIV-1 proviral DNA. However, when the
progeny virus was tested for its infectivity, although the group II intron-inserted HIV-1 RNA
was packaged and reverse-transcribed, the DNA failed to integrate as expected in the absence of
a functional IN, and virus replication was aborted. These results demonstrate that group II introns
can inhibit HIV-1 replication and should be further exploited for HIV-1 gene therapy.
Key Words: HIV-1, gene therapy, interfering RNA, group II intron, Ll.LtrB
98

3.2. Introduction
A number of anti-HIV-1 gene therapy strategies have been developed that inhibit HIV-1
replication post-integration by interfering with the function of HIV-1 RNA and proteins18,278.
However for these strategies to be successful, inhibition must last for the life of the gene-
modified cells, as they all contain the proviral DNA. In contrast, a one-time intervention at the
HIV-1 DNA level might suffice to inhibit virus replication in the gene-modified cells and their
progeny.
Certain group II introns act as mobile genetic elements that not only can be spliced at the RNA
level, but with the help of a protein encoded by the introns, also can insert into specific DNA
target sites239,279,280. Recent advances led to the development of a Lactococcus lactis group II
intron (Ll.LtrB) with novel target DNA specificities248,252,255. Five Ll.LtrB intron insertion sites
have been identified within the HIV-1 DNA237. Intron insertion at two of these sites, located at
nts. 4021 and 4069 within the HIV-1 DNA (nt +1 denotes the first nucleotide of the RNA), was
shown to occur at high frequency in Escherichia coli. Liposomes were then used to deliver the
ribonucleoprotein complex containing the intron targeted against nt. 4069 and the intron-encoded
LtrA protein, as well as a plasmid containing the HIV-1 DNA target site, to a mammalian cell
line. Intron insertion within the target DNA was demonstrated by PCR, followed by DNA
sequencing237. This result demonstrates that the modified group II intron and the LtrA protein are
sufficient to allow intron insertion into mammalian cells.
Mobile group II introns offer the unique advantage of allowing the development of a gene
therapy strategy that could inhibit HIV-1 replication as a result of intron insertion within the
HIV-1 DNA. Intron insertion in the reverse-transcribed HIV-1 dsDNA or in the integrated HIV-1
99

proviral DNA may not only produce non-infectious virus that fails to infect a healthy cell, but
also rescue the HIV-1-infected cells. Furthermore, the reverse-transcribed HIV-1 dsDNA may be
more accessible for intron insertion than the integrated HIV-1 proviral DNA. Therefore, we
believe that HIV-1 represents a perfect model to assess the feasibility of developing group II
intron-based therapies (i.e. gene therapy). An important step towards the development of intron-
based HIV-1 gene therapy is to determine the extent of inhibition of virus replication following
intron insertion within the HIV-1 proviral DNA. Using Ll.LtrB-derived introns targeted against
nts. 4021 and 4069 within the IN-coding region of HIV-1 pol gene, we have tested whether
group II intron insertion within an infectious HIV-1 proviral DNA clone can inhibit virus
replication.
3.3. Materials and Methods
3.3.1. Bacteria and Viruses Strains and Cell Lines
E. coli DH5α was obtained from GIBCO-BRL. E. coli HMS174(DE3) was obtained from
Novagen, USA.
NL4-3 (Cat.# 2479) and BaL (Cat#. 510) were obtained from NIH AIDS Research and
Reference Reagent Program.
PA317, an adherent mouse packaging cell line (expressing MoMuLV gag, pol, and env genes)265
was obtained form ATCC (Cat.# CRL-9078) and used to obtain amphotropic retroviral vector
particles.
100

293T, an adherent human embryonic kidney cell line266, was obtained from Stanford Reference
(Cat.# S97-079) and used to produce lentiviral vector particles.
PM1267, a suspension human CD4+/CCR5+/CXCR4+ T-lymphoid cell line, was obtained from
NIH AIDS Research and Reference Reagent Program and used for expression of Rz1-7,
producing stocks for HIV-1 strains NL4-3 and BaL, and challenge with the same HIV-1 strains.
The adherent 293T and PA317 cells were cultured in complete Dulbecco's modified Eagle's
medium (DMEM), and the PM1 cells in complete Roswell Park Memorial Institute medium
(RPMI-1640). Both media were supplemented with 10% cosmic calf serum (HyClone, USA),
1% antibiotic-antimycotic (Gibco-BRL, USA), and 1% L-Glutamine 200 mM (Gibco-BRL,
USA). Cells were incubated in a 5% CO2 cabinet at 37°C. For freezing, media were removed from
cells by centrifugation at 600 × g for 5 min. Cell pellet was resuspended in cosmic calf serum
containing 10% dimethyl sulfoxide (DMSO), followed by 2 hr at -20°C and overnight at -70°C,
and stored in liquid nitrogen. To revive, frozen cells were thawed at room temperature and mixed
with 3 ml fresh medium, followed by centrifugation as above. Cell pellet was resuspended in the
appropriate amount of fresh medium and transferred to flask or plate.
3.3.2. Oligonucleotides and Primers
All oligonucleotides and primers were synthesized by ACGT Corporation (Toronto, CANADA).
Long oligonucleotides were cartridge-purified. Primers were received in powder form (0.2 µmole).
See table 3.1 for sequences of oligonucleotides and primers.
101

Table 3.1- Oligonucleotides and primers sequences
Name Sequence
β-actin-R 5’-caa-aca-tga-tct-ggg-tca-tct-tct-c-3’
β-actin-F 5’-gct-cgt-cgt-cga-caa-cgg-ctc-3’
DV-5’ 5’-gcc-gta-tac-tcc-gag-agg-3’
IIS-3’ 5’-agg-cgg-cct-taa-ctg-tag-3’
IIS-5’ 5’-atg-ggt-tgg-tca-gtg-ctg-3’
IIS-UP 5’-ttt-gca-gga-ttc-ggg-att-ag -3’
LTR-5’ 5’-gag-agc-tgc-atc-cgg-agt-ac-3’
LTR-3’ 5’-agg-caa-gct-tta-ttg-agg-ctt-aag-c-3’
R5-Ti-F or R5-Ti-5’* 5’-ata-ggt-acc-tgg-ctg-tcg-tcc-atg -3’
R5-Ti-R or R5-Ti-3’* 5’-ggt-cca-acc-tgt-tag-agc-tac-tgc-3’
T7MT Sense:
5’-ggg-ggg-tga-agc-ttc-tcg-agg-agc-tct-aat-acg-act-cac-tat-agg-gag-gtc-gac-gcc-atg-gag-gat-cca-ggg-ccc-tat-tta-aat-ggc-gcg-cca-gcg-gcc-gct-t-3’
T7MT Antisense:
5’-ggg-ggg-ggg-aat-tcc-tcg-agt-taa-tta-aca-cac-aaa-aaa-cca-aca-cac-aga-tgt-aat-gaa-aat-aaa-gat-att-tta-ttt-taa-tta-agc-ggc-cgc-tgg-cgc-gcc-a-3’
T7-Tat-F or Tat-5’* 5’-ata-tca-tat-gta-ata-cga-ctc-act-ata-ggg-cga-ata-ctt-ggg-cag-gag-tgg-aag-c-3’
Tat-R or Tat-3’* 5’-gat-cta-tgc-atg-agc-cag-3’
* Either name has been used in the text.
102

3.3.3. Plasmid Constructions
pACD-HIV1-4021s (pACD-I4021s), pACD-HIV1-4069s (pACD-I4069s), pACD-HIV1-54a (pACD-
I54a), pACD-HIV1-2654a (pACD-I2654a), and pBRR-HIV1(3805-4178s)/Tet plasmids237 were
kindly provided by Dr. A. Lambowitz. pACD-I4021sIN, pACD-I4069sIN, pACD-I4021sN, pACD-
I4069sN, pACD-I4021s∆N, and pACD-I4069s∆N were constructed as follows. A pair of single-stranded
oligonucleotides, T7MT sense and T7MT antisense, were designed to contain a T7 promoter, a
multiple cloning site and a transcription terminator (table 3.1). The single-stranded
oligonucleotides were converted to a double-strand fragment (T7MT cassette) by a single round
of PCR (95°C for 60 sec, 56°C for 60 sec, and 72°C for 300 sec). The PCR product was then
purified by agarose gel extraction method and were digested by Hind III and EcoR I restriction
enzymes. T7MT cassette was cloned into the EcoR I – Hind III digested pUC18 to produce pUC-
MCS. The internal ribosome entry site (IRES)268 and the neomycin phosphotransferase (neo) gene
were cloned in this plasmid. An Xho I – Xho I 1336-bp long fragment of pUC-MCS containing
the IRES element and the neo gene were then cloned at the Sal I sites within intron domain IV of
pACD-4021s and pACD-4069s to obtain pACD-I4021sIN and pACD-I4069sIN, respectively. Plasmids
pACD-I4021sN and pACD-I4069sN were obtained by replacing the Sal I – Bsp120 I fragment
containing the IRES element and the neo gene, by the Sal I – Bsp120 I 857-bp long fragment of
pMoTN281 containing the Shine-Dalgarno sequence and the neo gene. Deletion of an Eag I – Eag I
2107-bp long fragment from these plasmids then led to pACD-I4021s∆N and pACD-I4069s∆N.
3.3.4. Intron Insertion Assay
Intron insertion assay was performed as described elsewhere237. Briefly, E. coli HMS174(DE3)
cells were co-transformed with recipient pBRR-HIV1(3805-4178s)/Tet (ApR) and one of the
pACD-based donor plasmids (CmR). An ApRCmR colony, containing both the donor and the
recipient plasmids, was selected. Co-transformed bacteria were induced by culturing in the
103

presence of 0.1 mM isopropyl-β-D-thiogalactopyranosid (IPTG) for 1 hr to allow intron and LtrA
production, and intron insertion. Finally, the number of ApR and TetR colonies was determined and
used to calculate intron insertion frequencies by calculating the percentage of TetR colonies that
allow intron insertion over the total number of ApR colonies that do or do not allow intron
insertion.
3.3.5. Intron Insertion in HIV-1 Proviral DNA Clone and Purification of Intron-inserted HIV-
1 Proviral DNA Clones
E. coli HMS174(DE3) cells were co-transformed with recipient pHIV (an infectious HIV-1
proviral DNA clone, pNL4-3; ApR)275 and one of the two donors, pACD-I4021sN or pACD-I4069sN
(CmRKmR). A single ApRCmR colony was selected and cultured overnight in LB broth containing
Ap and Cm. The cells were then induced with 1 µM IPTG for 1 hr at 37°C, washed with LB broth,
and cultured overnight in LB broth containing Ap and Cm. Plasmid DNA was then extracted
using the QIAprep Spin Miniprep kit (Qiagen, Canada), and digested with Sac II to excise the
donor plasmids; pHIV, pHIV-I4021sN, and pHIV-I4069sN plasmids are insensitive to Sac II
digestion. The DNA was used to transform E. coli DH5α cells. The KmRApRCmS colonies were
identified as they contained pHIV-I4021sN or pHIV-I4069sN. Plasmid DNA from these colonies was
extracted and analyzed by restriction enzyme and PCR analyses.
3.3.6. Transfection of 293T Cells with Group II Intron-inserted HIV-1 Proviral DNA Clones
293T cells (4 × 106 cells/plate) were transfected with pHIV, pHIV-I4021sN, or pHIV-I4069sN, as
described before27. Briefly, the adherent cells were trypsinized and 4 × 106 of them were
transferred to each 10 cm cell culture plate containing 10 ml complete DMEM medium. Cells
were incubated in a 37°C CO2 incubator for 5-6 h till attached to the bottom of the plate. 15 µg
104

of the plasmid DNA were mixed in up to 450 µl H2O by vortexing. 50 µl ice-cold 2.5 M CaCl2
was added and vortexed briefly. 500 µl of ice-cold 2× HeBS (containing 0.283 M NaCl, 1.5 mM
Na2HPO4.7H2O, 0.023 M HEPES, pH 7.05; filter-sterilized) was added drop-wise while mixing
with bubbles made by a transfer pipette and then mixed more by finger tapping. The mixture was
incubated at room temperature for 20 min and then added drop-wise to the cell culture while
rotating the plate in all directions. Cells were incubated in a 37°C CO2 incubator for over night.
The medium was changed next morning. On day 4 post-transfection, the cells and the culture
supernatants were harvested and analyzed as described below.
3.3.7. RT-PCR Analysis to Detect Group II Intron-inserted HIV-1 RNA in Transfected 293T
Cells
Total RNA was extracted from the pHIV-, pHIV-I4021sN-, or pHIV-I4069sN-transfected 293T cells
using the RNeasy kit (Qiagen, Canada). The RNA was incubated with RQI RNase-free DNase
(Promega, USA) for 15 min at 37°C to degrade any residual DNA. To ensure that the DNase
treatment was complete, the RNA samples were analyzed by PCR. Reverse transcriptions were
performed for 1 hr at 37°C using the IIS-3’ primer and the RT-PCR kit (Invitrogen, USA). The
cDNAs were then PCR-amplified using IIS-5’, DV-5’, and IIS-3’ primers for 40 cycles (95°C
for 15 sec, 53°C for 1 min, and 72°C for 45 sec). The β-actin-F/β-actin-R primer pair was used
to amplify the endogenous β-actin gene. The RT-PCR products were analyzed on a 2% agarose
gel along with the λ–Hind III DNA marker (Gibco-BRL, USA).
3.3.8. Intracellular HIV-1 p24 and Progeny Virus Production from Transfected 293T Cells
To determine intracellular p24 concentrations, the transfected 293T cells were scraped from the
plates and washed three times with phosphate buffered saline (PBS). The cell pellets were
105

suspended in 0.5 ml of ice-cold lysing solution (containing Tris-Cl (pH 8.0) 50 mM, NaCl 150
mM, sodium azide 0.02%, phenylmethylsulfonyl fluoride 100 µg/ml, and NP-40 1%) and
incubated at 4°C for 20 min. The cell debris were then pelleted by centrifugation at 13000 × g for
2 min and the amount of intracellular p24 present in the supernatants was determined using the
enzyme linked immunosorbent assay (ELISA) p24 Antigen EIA kit (Beckman Coulter, USA)
Cell culture supernatants containing the progeny viruses were collected from the pHIV-, pHIV-
I4021sN-, or pHIV-I4069sN-transfected 293T cells. The amount of HIV-1 p24 antigen was measured
by the ELISA p24 Antigen EIA kit.
3.3.9. RT-PCR Analysis to Detect Group II Intron-inserted HIV-1 RNA in the Progeny
Viruses Produced from Transfected 293T Cells
Progeny viruses (280 µl) produced from the pHIV-, pHIV-I4021sN-, or pHIV-I4069sN-transfected
293T cells were used for virion RNA extraction by QIAamp Viral RNA minikit (Qiagen, Canada),
followed by DNase treatment as described above. Reverse transcriptions were performed for 1 h at
37°C using the IIS-3’ primer and the RT-PCR kit (Invitrogen, USA). The cDNAs were then
analyzed by PCR using IIS-Up/IIS-3’ primer pair to amplify a 613-bp region within the HIV-1
RNA, and the DV-5’/IIS-3’ primer pair to amplify a 275- or 227-bp region within the I4021sN- or
I4069sN-inserted HIV-1 RNA. PCRs were performed for 40 cycles (95°C for 60 sec, 50°C for 30
sec, and 72°C for 90 sec). The RT-PCR products were analyzed on a 2% agarose gel along with
the λ–Hind III DNA marker.
3.3.10. PM1 Cell Infection with Progeny Viruses Produced from the Transfected 293T Cells
PM1 cells were infected as described before282. Briefly, PM1 cells (6 × 105 cells in 1.5 ml) were
incubated for 1 hr at room temperature with ~10 ng p24 equivalent of progeny viruses from the
106

pHIV-, pHIV-I4021sN-, or pHIV-I4069sN-transfected 293T cells, respectively, followed by three
washes with PBS. The cells were then cultured in 2 ml complete RPMI in a 12-well plate. Every
3-4 days, ~1 ml of cultures was replaced by 1 ml fresh complete RPMI-1640. Samples were
centrifuged at 450 × g for 5 min, and the cells and supernatants were stored at -70°C.
3.3.11. PCR Analyses to Detect Proviral DNA and Reverse-transcribed HIV-1 dsDNA in
Infected PM1 Cells
Genomic DNAs were extracted using the DNeasy Tissue kit (Qiagen, Canada) from PM1 cells on
day 8 post-infection with progeny viruses from transfected 293T cells. PCRs were performed for
40 cycles (95°C for 15 sec, 53°C for 1 min, and 72°C for 45 sec) using the Tat-5’/Tat-3’ primer
pair to detect the HIV-1 sequence, and the R5-Ti-5’/R5-Ti-3’ primer pair to detect the
endogenous ccr5 gene. The PCR products were analyzed on a 2% agarose gel.
To detect the reverse-transcribed HIV-1 dsDNA, the progeny viruses (500 µl) from the pHIV,
pHIV-I4021sN-, and pHIV-I4069sN-transfected 293T cells were first treated with 30 units of DNase
for 30 min at 37°C283 and then used to inoculate PM1 cells. Genomic DNA was extracted 1 hr
post-inoculation using the DNeasy Tissue kit. PCR was performed using the LTR-5’ and LTR-
3’284 primer pair for 40 cycles (95°C for 1 min, 50°C for 30 sec, and 72°C for 30 sec). The PCR
products were analyzed on a 2% agarose gel.
3.3.12. Progeny Virus Production from Infected PM1 Cells
Cell culture supernatants collected from the pHIV-, pHIV-I4021sN-, or pHIV-I4069sN-infected PM1
cells were stored at -70°C. The amount of the HIV-1 p24 antigen was measured by the ELISA
p24 Antigen EIA kit.
107

3.4. Results
3.4.1. Modified Group II Introns
To investigate the potential utility of mobile group II introns in inhibiting HIV-1 replication, two
introns (I4021s and I4069s) were used that are derived from Ll.LtrB. These introns are 956-nt. long.
They are targeted against nts. 4021 or 4069 within the sense DNA strand of the IN-coding region
of the HIV-1 pol gene (Figure 3.1A).
Plasmids pACD-HIV1-4021s and pACD-HIV1-4069s237 (hereafter referred to as pACD-I4021s and
pACD-I4069s respectively) allow T7lac promoter-driven expression of RNA containing the I4021s and
I4069s introns. In these plasmids, the ltrA gene was deleted from its natural location within the
intron domain IV and cloned further downstream of the intron. Both plasmids were shown to
allow intron insertion at the respective HIV-1 DNA target sites in E. coli237.
We modified the pACD-I4021s and pACD-I4069s plasmids to confer a selectable (KmR) phenotype.
To this end, the T7 promoter and the neo gene were cloned in the intron domain IV. The
resulting plasmids were named pACD-I4021sN and pACD-I4069sN.
3.4.2. Modified Group II Intron Insertion Frequencies
To determine the insertion frequencies of the modified introns, E. coli cells were co-transformed
with the donor plasmid (pACD-I4021sN or pACD-I4069sN, allowing intron and LtrA protein
production and providing the T7 promoter) and the recipient plasmid (pBRR- HIV1(3805-
4178s)/Tet, providing the introns’ insertion sites and a promoterless TetR gene).
108

Figure 3.1
R
3’ LTRU3 U5R
3’ LTR
---ugg cug cua gau ugu aca cau uua gaa gga gug cgc cca gau agg gug uua agu caa gua guu uaa ------Trp Gln Leu Asp Cys Thr His Leu Glu Gly Val Arg Pro Asp Arg Val Leu Ser Gln Val Val
III VII II V
Cap (A)n
I4021sN intron-inserted 11.1kb HIV-1 RNA
C-terminus of mutant Gag-Pol
Intron domain I
---guu cau cua gcc agu gga uauagu gga uau aua gaa gca gug cgc cca gau agg gug uua agu caa gua guu uaa ------Val His Val Ala Ser Gly Tyr Ile Glu Ala Arg Pro Asp Arg Val Leu Ser Gln Val Val END
-terminus of mutant Gag-Pol (Processing of mutant Gag-Pol will give rise to 107 aa-long integrase)
Intron domain I
SD-neo
T7
U3 U5R
5’ LTRU3 U5R
5’ LTRpro RT
Group II intron-inserted integrase
poltatvpu
rev
rev
tat
env
I4021sN or I4069sN
U3 U5R
3’ LTRU3 U5R
3’ LTR
---ugg cug gau ugu aca cau uua gaa gga gug cgc cca gau agg gug uua agu caa gua guu uaa ---Trp Gln Leu Asp Cys His Leu Glu Gly Arg Pro Asp Val Leu Ser Gln Val Val
III VII II V
(A)n
-
Intron domain I
---guu cau cua gcc agu gga uau aua gaa gca gug cgc cca gau agg gug uua agu caa gua guu uaa ---Gly Ile Glu Ala Val Arg Pro Asp Val Val END
C- -
Intron domain I
neo
a
c
vif
vpr
gag
integrase domain)Group II intron--
Cap (A)n
integrase domain)--
integrase domain)---inserted 11.1 kb HIV-1 RNA Gag and mutant Gag-Pol (with a truncated -
Cap (A)n
Cap (A)n4-5 kb singly spliced HIV-1 RNAs Vif, Vpr, Vpu, and Env 4-
(A)n4-4-4-4-
Cap (A)n2 kb completely spliced HIV-1 RNAs- Tat, Rev, and Nef -
(A)n--------
b
(Processing of mutant Gag-Pol will give rise to 91 aa-long integrase)
END
nef
Group II intron-------inserted 11.1 kb HIV-1 RNA-
I4069sN intron-inserted 11.1kb HIV-1 RNA
R
3’ LTRU3 U5R
3’ LTR
---ugg cug cua gau ugu aca cau uua gaa gga gug cgc cca gau agg gug uua agu caa gua guu uaa ------Trp Gln Leu Asp Cys Thr His Leu Glu Gly Val Arg Pro Asp Arg Val Leu Ser Gln Val Val
III VII II V
Cap (A)n
I4021sN intron-inserted 11.1kb HIV-1 RNA
C-terminus of mutant Gag-Pol
Intron domain I
---guu cau cua gcc agu gga uauagu gga uau aua gaa gca gug cgc cca gau agg gug uua agu caa gua guu uaa ------Val His Val Ala Ser Gly Tyr Ile Glu Ala Arg Pro Asp Arg Val Leu Ser Gln Val Val END
-terminus of mutant Gag-Pol (Processing of mutant Gag-Pol will give rise to 107 aa-long integrase)
Intron domain I
SD-neo
T7
U3 U5R
5’ LTRU3 U5R
5’ LTRpro RT
Group II intron-inserted integrase
poltatvpu
rev
rev
tat
env
I4021sN or I4069sN
U3 U5R
3’ LTRU3 U5R
3’ LTR
---ugg cug gau ugu aca cau uua gaa gga gug cgc cca gau agg gug uua agu caa gua guu uaa ---Trp Gln Leu Asp Cys His Leu Glu Gly Arg Pro Asp Val Leu Ser Gln Val Val
III VII II V
(A)n
-
Intron domain I
---guu cau cua gcc agu gga uau aua gaa gca gug cgc cca gau agg gug uua agu caa gua guu uaa ---Gly Ile Glu Ala Val Arg Pro Asp Val Val END
C- -
Intron domain I
neo
a
c
vif
vpr
gag
integrase domain)Group II intron--
Cap (A)n
integrase domain)--
integrase domain)---inserted 11.1 kb HIV-1 RNA Gag and mutant Gag-Pol (with a truncated -
Cap (A)n
Cap (A)n4-5 kb singly spliced HIV-1 RNAs Vif, Vpr, Vpu, and Env 4-
(A)n4-4-4-4-
Cap (A)n2 kb completely spliced HIV-1 RNAs- Tat, Rev, and Nef -
(A)n--------
b
(Processing of mutant Gag-Pol will give rise to 91 aa-long integrase)
END
nef
Group II intron-------inserted 11.1 kb HIV-1 RNA-
I4069sN intron-inserted 11.1kb HIV-1 RNA
C
B
A
109

Figure 3.1- (A) Structure of the group II intron-inserted HIV-1 proviral DNA. Intron domains I,
II, III, V and VI are shown. Domain IV is modified to contain the T7 promoter and neo gene. (B)
Transcription from the U3 promoter in the HIV-1 5’ LTR generates primary 11.1 kb transcripts
containing the group II intron. Splicing of HIV-1 introns yields 4-5 and 2 kb RNAs, lacking
group II intron. Translation of the group II intron-inserted 11.1 kb HIV-1 RNA yields HIV-1 gag
and mutant gag-pol with a truncated IN domain. Translation of the 4-5 kb RNAs yields HIV-1
virion infectivity factor (Vif), virion protein R (Vpr), virion protein U (Vpu), and envelop protein
(Env). Translation of the 2 kb RNA yields HIV-1 trans-activator of transcription (Tat), regulator
of expression of virion proteins (Rev), and negative factor (Nef). (C) The HIV-1 RNA sequence
adjacent to the I4021sN and I4069sN insertion sites and the amino acid sequence of the C-terminal
region of the mutant gag-pol encoded by pHIV-I4021sN and pHIV-I4069sN, are shown. Processing of
mutant gag-pol produced from pHIV-I4021sN and pHIV-I4069sN gives rise to 91- and 107-aa long
IN, respectively.
110

The intron insertion frequencies were determined by calculating the percentage of colonies that
allow intron insertion over the total number of colonies that do or do not allow intron insertion.
Our results indicate that insertion frequencies of the modified introns that contain the neo gene
(I4021sN and I4069sN) are lower (Figure 3.2, samples 3 and 7) than those of the unmodified introns
(I4021s and I4069s) (Figure 3.2, samples 1 and 5). The decrease in insertion frequencies of the
modified introns, I4021sN and I4069sN, may have resulted from poor TetR gene expression because of
the extra distance created by the presence of neo gene between the T7 promoter and the TetR
gene. This observation is based on the fact that upon neo gene deletion, the insertion frequencies
of I4021s∆N and I4069s∆N introns returned to normal (Figure 3.2, samples 4 and 8), and that inclusion
of IRES element upstream of the neo gene in I4021sIN and I4069sIN reduced the insertion frequencies
even further (Figure 3.2, samples 2 and 6).
3.4.3. Group II Intron Insertion into an Infectious HIV-1 Proviral DNA Clone
Since intron insertion has not yet been demonstrated in mammalian chromosomes, we bypassed
this step by allowing intron insertion in an infectious HIV-1 proviral DNA clone in E. coli,
purifying the intron-inserted HIV-1 proviral DNA clones, and testing them for inhibition of virus
replication in mammalian cells. pACD-I4021sN and pACD-I4069sN were used to allow intron
insertion into an infectious HIV-1 proviral DNA clone in E. coli (Figure 3.3). To this end, the E.
coli cells were co-transformed with pACD-I4021sN or pACD-I4069sN along with pHIV (an infectious
HIV-1 proviral DNA clone, originally referred to as pNL4-3)275. Upon IPTG induction, pACD-
I4021sN and pACD-I4069sN would allow intron insertion at nts. 4021 and 4069 within the IN-coding
region of the HIV-1 pol gene. The intron-inserted pHIV, referred to as pHIV-I4021sN or pHIV-
I4069sN, respectively, would contain nts. 1-4021 or 1-4069 of the HIV-1 genome, the intron
domains I, II, and III, the T7 promoter and the neo gene, the intron domains V and VI, and the
rest of the HIV-1 genome (Figure 3.1A). Neo gene expression from the T7 promoter would
111

0
1020
3040
5060
7080
90
100
1 2 3 4 5 6 7 80
1020
3040
5060
7080
90
100
1 2 3 4 5 6 7 8
% In
tron
inse
rtio
n fr
eque
ncy
0
1020
3040
5060
7080
90
100
1 2 3 4 5 6 7 80
1020
3040
5060
7080
90
100
1 2 3 4 5 6 7 8
% In
tron
inse
rtio
n fr
eque
ncy
Figure 3.2- Insertion frequencies of introns I4021s, I4021sIN, I4021sN, I4021s∆N, I4069s, I4069sIN, I4069sN,
and I4069s∆N are presented (samples 1, 2, 3, 4, 5, 6, 7, and 8, respectively). The insertion
frequencies were determined by calculating the percentage of TetR colonies that allow intron-
insertion over the total number of ApR colonies that do or do not allow intron-insertion. Values
were normalized against those obtained for unmodified introns I4021s (66.4%) and I4069s (45.1%).
112

CmS
CmRKmR
pACD -I4021sN or pACD -I4069sN
VI
IPTG -induction
Isolate plasmid DNADigest donor plasmids with Sac II
(pHIV , pHIV -I4021sN, and pHIV-I4069sN are not digested)Transform DH5α cells, select ApRKmRCmS colonies
pHIV
ApR
pHIV -I4021sN or pHIV -I4069sN isolation and characterization
E. coliDH5α
E. coliHMS174 (DE3)
pHIV
E. coliHMS174 (DE3)
ApR ApRKmR
pHIV -I4021sN or pHIV -I4069sN
ApRKmR
pHIV -I4021sN or pHIV -I4069sN
CmRKmR
pACD -I4021sN or pACD -I4069sN
CmRKmR
or
CmRKmR
pACD -I4021sN or pACD -I4069sN
CmRKmR
pACD -I4021sN or pACD -I4069sN
Recipient5’ LTR 3’ LTR5’ LTR 3’ LTR
Sac IISac II Sac IISac II
LtrA
P PT7
I II V VIIIIE1 E2
DonorLtrA
T7
I II V VIIIIE1 E2
DonorLtrALtrA
T7 T7
I II V VIIIIE1 E2
T7Lac T7
I II V VIIIIE1 E2
Donor
SD-neoSD-neoSD-neoneo
LtrA
P PT7
I II V VIIIIE1 E2
DonorLtrA
T7
I II V VIIIIE1 E2
DonorLtrALtrA
T7 T7
I II V VIIIIE1 E2
T7LacT7
I II V VIIIIE1 E2
Donor
SD-neoSD-neoSD-neoneo
Recipient5’ LTR 3’ LTR5’ LTR 3’ LTR
PT7
I II V VIIII
T7
I II V VIIII
T7
I II V VIIII
T7
I II V VIIII SD-neoSD-neoSD-neoneo
5’ LTR 3’ LTR5’ LTR 3’ LTRIntron-inserted recipient
5’ LTR 3’ LTR5’ LTR 3’ LTRIntron-inserted recipient
PT7
I II V VIIII
T7
I II V VIIII
T7
I II V VIIII
T7
I II V VIIII SD-neoSD-neoSD-neoneo
CmS
CmRKmR
pACD -I4021sN or pACD -I4069sN
VI
IPTG -induction
Isolate plasmid DNADigest donor plasmids with Sac II
(pHIV , pHIV -I4021sN, and pHIV-I4069sN are not digested)Transform DH5α cells, select ApRKmRCmS colonies
pHIV
ApR
pHIV -I4021sN or pHIV -I4069sN isolation and characterization
E. coliDH5α
E. coliHMS174 (DE3)
pHIV
E. coliHMS174 (DE3)
ApR ApRKmR
pHIV -I4021sN or pHIV -I4069sN
ApRKmR
pHIV -I4021sN or pHIV -I4069sN
CmRKmR
pACD -I4021sN or pACD -I4069sN
CmRKmR
or
CmRKmR
pACD -I4021sN or pACD -I4069sN
CmRKmR
pACD -I4021sN or pACD -I4069sN
Recipient5’ LTR 3’ LTR5’ LTR 3’ LTR
Sac IISac II Sac IISac II
LtrA
P PT7
I II V VIIIIE1 E2
DonorLtrA
T7
I II V VIIIIE1 E2
DonorLtrALtrA
T7 T7
I II V VIIIIE1 E2
T7Lac T7
I II V VIIIIE1 E2
Donor
SD-neoSD-neoSD-neoneo
LtrA
P PT7
I II V VIIIIE1 E2
DonorLtrA
T7
I II V VIIIIE1 E2
DonorLtrALtrA
T7 T7
I II V VIIIIE1 E2
T7LacT7
I II V VIIIIE1 E2
Donor
SD-neoSD-neoSD-neoneo
Recipient5’ LTR 3’ LTR5’ LTR 3’ LTR
PT7
I II V VIIII
T7
I II V VIIII
T7
I II V VIIII
T7
I II V VIIII SD-neoSD-neoSD-neoneo
5’ LTR 3’ LTR5’ LTR 3’ LTR5’ LTR 3’ LTR5’ LTR 3’ LTRIntron-inserted recipient
5’ LTR 3’ LTR5’ LTR 3’ LTR5’ LTR 3’ LTR5’ LTR 3’ LTRIntron-inserted recipient
PT7
I II V VIIII
T7
I II V VIIII
T7
I II V VIIII
T7
I II V VIIII SD-neoSD-neoSD-neoneo
Figure 3.3
113

Figure 3.3- Experimental scheme used to show insertion of I4021sN and I4069sN introns into pHIV
(an infectious HIV-I proviral DNA clone) and isolation of the intron-inserted HIV-1 proviral
DNA clones (pHIV-I4021sN and pHIV-I4069sN). E. coli HMS174(DE3) cells were co-transformed
with the donor plasmid (pACD-I4021sN or pACD-I4069sN) and the recipient plasmid (pHIV). The
donor plasmids are shown to contain the intron domains I, II and III, the T7 promoter, neo gene,
and the intron domains V and VI. In these plasmids, the introns are flanked by the 5’ (E1) and 3’
(E2) HIV-1 exon sequences. These exons are required for the splicing of group II introns. IPTG-
induction allows intron and LtrA protein production and intron insertion into pHIV. Plasmids,
consisting of the donor (pACD-I4021sN or pACD-I4069sN), recipient (pHIV), and the intron-inserted
recipient (pHIV-I4021sN or pHIV-I4069sN), were extracted. They were digested with Sac II, which
excises pACD-I4021sN and pACD-I4069sN, but leaves the pHIV, pHIV-I4021sN, and pHIV-I4069sN
DNA intact. E. coli DH5α cells were then transformed with the resulting DNA and the
ApRKmRCmS colonies were selected. pHIV-I4021sN and pHIV-I4069sN were isolated from these
cells and characterized by restriction enzyme digestion and PCR analyses.
114

confer Km resistance. However, because pACD-I4021sN and pACD-I4069sN present in the co-
transformed cells would also confer Km resistance, Km selection at this stage cannot imply
intron insertion. We, therefore, used the following strategy to isolate pHIV-I4021sN and pHIV-
I4069sN (Figure 3.3). Plasmids were extracted from the IPTG-induced E. coli cells and digested
with Sac II, which excises pACD-I4021sN and pACD-I4069sN, but leaves both the intron-inserted
pHIV-I4021sN and pHIV-I4069sN, and the pHIV DNA intact. The E. coli cells were transformed with
the DNA after digestion with Sac II, and the ApRKmRCmS colonies containing pHIV-I4021sN or
pHIV-I4069sN were selected. Confirmation that the clones contained pHIV-I4021sN or pHIV-I4069sN
was obtained by restriction digestion and PCR analyses.
3.4.4. Group II Intron-inserted HIV-1 Proviral DNA Replication in Mammalian Cells
The intron-inserted HIV-1 proviral DNA clones, pHIV-I4021sN and pHIV-I4069sN, were first tested
for group II intron-inserted HIV-1 RNA production in mammalian cells. To this end, pHIV (as
positive control), pHIV-I4021sN, and pHIV-I4069sN were used to transfect human 293T cells. Total
RNA from these cells was extracted on day 4 post-transfection and analyzed by RT-PCR. As
expected, a 423-bp RT-PCR product was detected in the control pHIV-transfected 293T sample
(Figure 3.4A, lane 1). A 275-bp and a 227-bp RT-PCR product arising from group II intron-
inserted HIV-1 RNA was detected in pHIV-I4021sN- and pHIV-I4069sN-transfected 293T samples
(Figure 3.4A, lanes 2 and 3). Similar amounts of RNA were present in all samples analyzed by
RT-PCR (Figure 3.4B, lanes 1-3).
The pHIV-, pHIV-I4021sN-, and pHIV-I4069sN-transfected 293T cells were then tested for virus
replication. Similar amounts of gag protein were produced in these cells (Figure 3.5A, samples
1-3). The amount of progeny produced was also measured. Similar amounts of progeny viruses
were produced from cells transfected with pHIV (positive control), pHIV-I4021sN, or pHIV-I4069sN
115

1 2 3 4 5 6
-
-
-
-HIV 1 RNA 423 bp
275 bp227 bpinserted HIV 1
RNA
a
β actin mRNA 353 bpc
423 bp
275 bp227 bp
a 423 bp
275 bp227 bp
a
353 bpc
353 bpb
cHIV 1
virion RNA 613 bp
Group II intron 275 bp227 bpinserted HIV-1
virion RNA
275 bp227 bp275 bpd
-
-
-
-Group II intron
1 2 3 4 5 6
-
-
-
-HIV 1 RNA 423 bp
275 bp227 bpinserted HIV 1
RNA
a
β actin mRNA 353 bpc
423 bp
275 bp227 bp
a 423 bp
275 bp227 bp
a
353 bpc
353 bpb
cHIV 1
virion RNA 613 bp
Group II intron 275 bp227 bpinserted HIV-1
virion RNA
275 bp227 bp275 bpd
-
-
-
-Group II intron
D
C
B
A
Figure 3.4- RT-PCR analyses for determining intron insertions: (A) HIV-1 RNA or group II
intron-inserted HIV-1 RNA production in 293T cells transfected with pHIV (lane 1), pHIV-
I4021sN (lane 2), or pHIV-I4069sN (lane 3). Total RNA extracted from these cells on day 4 post-
transfection was analyzed by RT-PCR. RT-PCR amplification of the HIV-1 RNA by the IIS-
5’/IIS-3’ primer pair gave rise to a 423-bp product. This primer pair was designed to hybridize
with the HIV-1 RNA sequences that flank the intron insertion sites. RT-PCR amplification of the
I4021sN and I4069sN intron-inserted HIV-1 RNAs by the DV-5’/IIS-3’ primer pair gave rise to 275-
bp and 227-bp products, respectively. This primer pair was designed to hybridize within the
intron domain V and HIV-1 RNA further downstream of the intron insertion sites. PCR
amplification of pHIV DNA (lane 4) by the IIS-5’/IIS-3’ primer pair and of pHIV-I4021sN and
pHIV-I4069sN DNA (lanes 5 and 6) by the DV-5’/IIS-3’ primer pair was done in parallel. (B) The
endogenous β-actin mRNA from pHIV- (lane 1), pHIV-I4021sN- (lane 2), or pHIV-I4069sN- (lane 3)
transfected 293T cells was RT-PCR amplified as control. (C) HIV-1 RNA packaging in the
progeny viruses released from 293T cells transfected with pHIV (lane 1), pHIV-I4021sN (lane 2),
or pHIV-I4069sN (lane 3). Virion RNA extracted from these progeny viruses was analyzed by RT-
PCR using IIS-Up-5’ and IIS-3’ primers. PCR amplification of pHIV DNA (lane 4) by the IIS-
Up-5’/IIS-3’ primer pair was done in parallel. (D) Group II intron-inserted HIV-1 RNA
116

packaging in the progeny viruses released from 293T cells transfected with pHIV (lane 1),
pHIV-I4021sN (lane 2), or pHIV-I4069sN (lane 3). Virion RNA extracted from these progeny viruses
was analyzed by RT-PCR using DV-5’ and IIS-3’ primers. The RT-PCR products were analyzed
on 2% agarose gels. RT-PCR product sizes are indicated on the left of the gels. PCR
amplification of pHIV-I4021sN and pHIV-I4069sN DNA (lanes 5 and 6) by the DV-5’/IIS-3’ primer
pair was done in parallel.
117
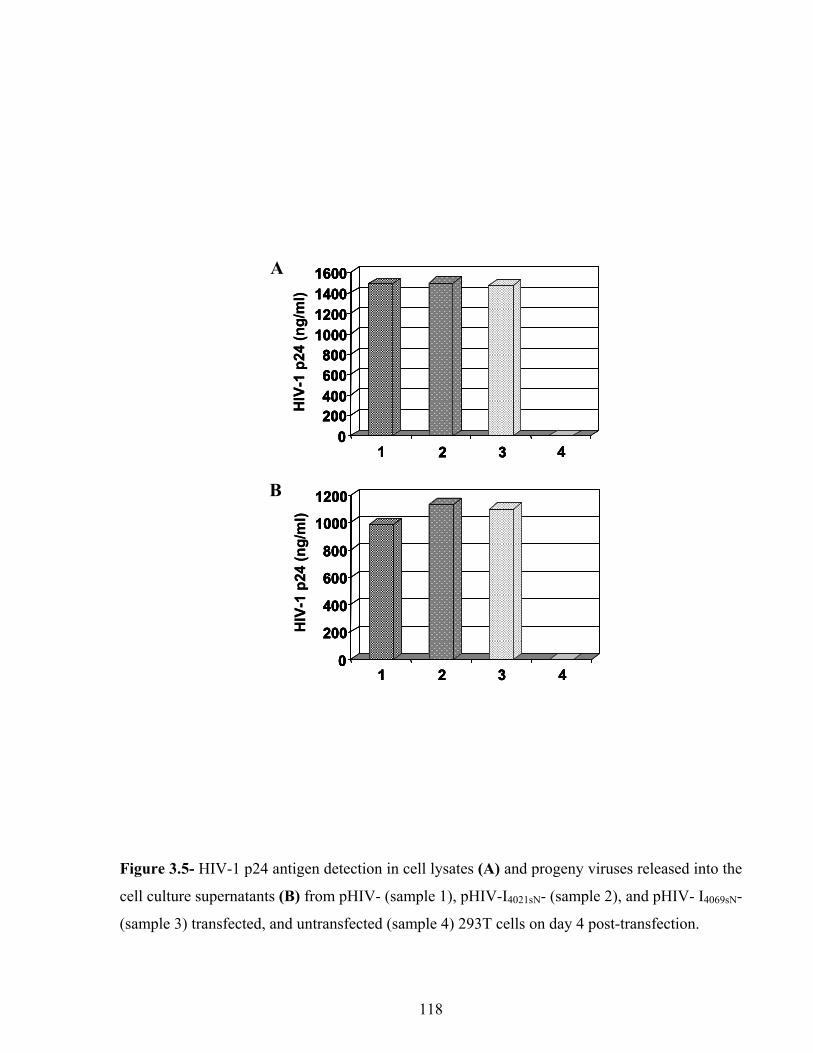
0200400600800
1000120014001600
1 4
HIV
-1 p
24 (n
g/m
l)
0200400600800
1000120014001600
4
0
200
400
600
800
1000
1200
1 2 3 4
HIV
-1 p
24 (n
g/m
l)
0
200
400
600
800
1000
1200
1 2 3 4
2 32 3
a
b
0200400600800
1000120014001600
1 4
HIV
-1 p
24 (n
g/m
l)
0200400600800
1000120014001600
4
0
200
400
600
800
1000
1200
1 2 3 4
HIV
-1 p
24 (n
g/m
l)
0
200
400
600
800
1000
1200
1 2 3 4
2 32 3
bB
aA
Figure 3.5- HIV-1 p24 antigen detection in cell lysates (A) and progeny viruses released into the
cell culture supernatants (B) from pHIV- (sample 1), pHIV-I4021sN- (sample 2), and pHIV- I4069sN-
(sample 3) transfected, and untransfected (sample 4) 293T cells on day 4 post-transfection.
118

(Figure 3.5B, samples 1-3). No gag protein or progeny virus could be detected in the
untransduced 293T samples (Figures 3.5A and B, sample 4).
Progeny viruses from pHIV-, pHIV-I4021sN-, and pHIV-I4069sN-transfected 293T cells were tested
for the HIV-1 RNA and group II intron-inserted HIV-1 RNA packaging. Virion RNA was
extracted from progeny viruses and analyzed by RT-PCR. When an HIV-1 RNA-specific IIS-
Up-5’/IIS-3’ primer pair was used, a 613-bp product was detected upon RT-PCR analysis of
virion RNA from the progeny of pHIV-transfected 293T cells (Figure 3.4C, lane 1). No RT-PCR
product could be detected when virion RNA from the progeny of pHIV-I4021sN- or pHIV-I4069sN-
transfected 293T cells was analyzed (Figure 3.4C, lanes 2 and 3). When a group II intron-
inserted HIV-1 RNA-specific DV-5’/IIS-3’ primer pair was used, 275- and 227-bp products were
detected upon RT-PCR analysis of virion RNA from the progeny of pHIV-I4021sN- and pHIV-
I4069sN-transfected 293T cells, respectively (Figure 3.4D, lanes 2 and 3). No RT-PCR product
could be detected when virion RNA from the progeny of pHIV-transfected 293T cells was
analyzed (Figure 3.4D, lane 1). These results demonstrate that the group II intron-inserted HIV-1
RNA was packaged.
3.4.5. Group II Intron-inserted Progeny Virus Replication during the Second Round of
Infection
The infectivity of the progeny viruses was assessed by infecting the human CD4+ T lymphoid
(PM1) cells. Since the group II intron-inserted HIV-1 RNA was packaged, reverse transcription
should have occurred. The presence of reverse-transcribed HIV-1 DNA was detected by PCR
analysis of the genomic DNA isolated 1 hr post-infection. A 220-bp product resulting from the
HIV-1 dsDNA was amplified from the genomic DNA of PM1 cells that were infected with the
progeny from pHIV- (positive control), pHIV-I4021sN-, or pHIV-I4069sN-transfected 293T cells
119

(Figure 3.6A, lanes 1-3). As expected, no PCR product was amplified from the genomic DNA
obtained from uninfected PM1 cells (Figure 3.6A, lane 4). We then tested for the presence of
integrated proviral DNA by PCR analysis of genomic DNA isolated on day 8 post-infection from
uninfected and infected PM1 cells. No proviral DNA could be detected in PM1 cells infected
with the progeny from pHIV-I4021sN- or pHIV-I4069sN-transfected 293T cells (Figure 3.6B, lanes 2
and 3). As expected, a 424-bp product resulting from HIV-1 proviral DNA amplification was
detected in control PM1 cells that were infected with the progeny from the pHIV-transfected
293T cells (Figure 3.6B, lane 1). No PCR product was detected in the uninfected PM1 sample
either (Figure 3.6B, lane 4). The amount of DNA analyzed by PCR was similar in all cases
(Figure 3.6C, lanes 1-4).
Infected PM1 cells were then tested for progeny virus production (Figure 3.7). As expected,
virus production, extensive cell death, and syncytia were observed in control PM1 cells that were
infected with the progeny virus from the pHIV-transfected 293T cells.
No virus was produced from PM1 cells infected with progeny virus from pHIV-I4021sN- or pHIV-
I4069sN-transfected 293T cells. These PM1 cells were healthy with no cell death, no syncytia, and
no progeny production for the length of the experiment (up to 62 days in two independent
experiments).
120

Reverse-transcribedHIV-1 ds DNA
HIV-1 provirus DNA
CCR5 DNA
220 bp
465 bpc
a
b
Reverse-transcribedHIV-1 ds DNA
HIV-1 provirus DNA
CCR5 DNA
220 bp
1 2 3 4 5
465 bp
424bp
c
a
b
c
a
b
Reverse-transcribedHIV-1 ds DNA
HIV-1 provirus DNA
CCR5 DNA
220 bp
465 bpc
a
b
Reverse-transcribedHIV-1 ds DNA
HIV-1 provirus DNA
CCR5 DNA
220 bp
1 2 3 4 5
465 bp
424bp
c
a
b
c
a
b
C
B
A
Figure 3.6- PCR analyses for determining intron inserted pHIV-1: (A) Detection of reverse-
transcribed HIV-1 dsDNA in PM1 cells infected with DNase-treated progeny from pHIV-
(positive control, lane 1), pHIV-I4021sN- (lane 2), or pHIV-I4069sN- (lane 3) transfected 293T cells.
One hr post-infection, the genomic DNA was extracted from the infected (lanes 1-3) or
uninfected (negative control, lane 4) PM1 cells. The DNA was analyzed by PCR using the HIV-
1 DNA specific LTR-5’/LTR-3’ primer pair to detect the HIV-1 and group II intron-inserted
HIV-1 dsDNA. PCR amplification of pHIV DNA (lane 5) by the LTR-5’/LTR-3’ primer pair
was done in parallel. (B) Detection of integrated proviral DNA in PM1 cells infected with
progeny from pHIV- (positive control; lane 1), pHIV-I4021sN- (lane 2), or pHIV-I4069sN- (lane 3)
transfected 293T cells. Genomic DNA was isolated from infected (day 8 post-infection; lanes 1-
3) and uninfected (lane 4) PM1 cells. They were analyzed by PCR using the HIV-1 Tat-5’/Tat-3’
primer pair to detect the presence of both HIV-1 and group II intron-inserted HIV-1 proviral
DNA. PCR amplification of pHIV DNA (lane 4) by the Tat-5’/Tat-3’ primer pair was done in
parallel. (C) The endogenous CCR5 DNA was amplified from PM1 cells infected with the
progeny from pHIV- (lane 1), pHIV-I4021sN- (lane 2), or pHIV-I4069sN- (lane 3) transfected 293T
cells. CCR5 DNA was also analyzed from uninfected (lane 4) PM1 cells. The PCR products
were analyzed on 2% agarose gels. PCR product sizes are indicated on the left of the gels.
121

0100200300400500600700
0 10 20 30 40 50 60 70
Days post-infection
HIV
-1 p
24 (n
g/m
l)
Figure 3.7- Progeny virus production by PM1 cells infected with the progeny virus from pHIV-
(— —), pHIV-I4021sN- (—▲—), or pHIV-I4069sN- (— —) transfected 293T cells. Virus
production from infected PM1 cells is reported until day 62 post-infection.
122

3.5. Discussion
In the present study, we have used HIV-1 as model to address a number of key questions
regarding the therapeutic application of a mobile group II intron. Two Ll.LtrB-derived introns
were used that are targeted against nts. 4021 or 4069 within the sense DNA strand of the IN-
coding region of the HIV-1 pol gene. pACD-I4021sN and pACD-I4069sN were engineered to express
introns that confer a selectable phenotype to the intron-inserted HIV-1 proviral DNA clones.
Insertion frequencies of the modified introns (I4021sN and I4069sN) were lower than those of the
unmodified introns or introns from which the neo gene had been deleted. These results indicate
that intron design (modifications within the intron domain IV) may directly (due to inadequate
RNA configuration) or indirectly (due to poor TetR gene expression) affect intron insertion
frequency.
pACD-I4021sN and pACD-I4069sN were used to allow intron insertion into an infectious HIV-1
proviral DNA clone in E. coli. The infectious HIV-1 proviral DNA clone (pHIV) and intron-
inserted HIV-1 proviral DNA clones (pHIV-I4021sN and pHIV-I4069sN) were then tested for virus
replication in 293T cells. Similar amounts of WT or group II intron-inserted HIV-1 RNA (Figure
3.4A), gag protein (Figure 3.5A), and progeny viruses (Figure 3.5B) were produced in all cases.
These results are in line with what we had anticipated. All viral RNAs and proteins produced in
pHIV-I4021sN- and pHIV-I4069sN-transfected 293T cells are expected to be WT, except for the
increased length of the group II intron-inserted HIV-1 RNA, and the gag-pol precursor that
would be truncated beyond the intron insertion site within the IN domain (Figure 3.1B).
Therefore, progeny viruses should have been produced in the pHIV-I4021sN- and pHIV-I4069sN-
transfected cells. However, these viruses were expected to be non-infectious because of the
increased length of the intron-inserted HIV-1 RNA (11.1 kb instead of 9.2 kb) that may prevent
123

encapsidation and/or the absence of a functional IN that may prevent integration even if the RNA
was packaged and reverse-transcribed. Since the insertion sites for the two introns are located in
the IN-coding region, a 91-aa long (aa 1-80 of the IN + 11 aa from the I4021sN intron) or a 107-aa
long (aa 1-96 of the IN + 11 aa from the I4069sN intron) truncated IN should be produced in the
pHIV-I4021sN- and pHIV-I4069sN-transfected 293T cells, respectively (Figure 3.1C); the full-length
IN is 298-aa long.
The intron-inserted HIV-1 RNAs were packaged in the progeny viruses released from the pHIV-
I4021sN- or pHIV-I4069sN-transfected 293T cells (Figure 3.4D). The infectivity of the progeny
viruses was then assessed by infecting the human CD4+ T lymphoid (PM1) cells. Although the
group II intron-inserted HIV-1 RNAs were reverse-transcribed (Figure 3.6A), no proviral DNA
could be detected (Figure 3.6B). The presence of reverse-transcribed HIV-I4021sN and HIV-I4069sN
dsDNA accompanied by the absence of integrated proviral DNA indicates that the reverse-
transcribed dsDNA could not integrate. These results suggest that the 91-aa and 107-aa long INs
(in the progeny from pHIV-I4021sN- and pHIV-I4069sN-transfected 293T cells) were non-functional.
Point mutations, insertions and deletions during HIV-1 reverse transcription285 could have
removed the group II introns and yielded a WT HIV-1 dsDNA. The absence of integrated
proviral DNA despite the formation of reverse-transcribed dsDNA further indicates that even if
these group II introns were deleted during reverse transcription, the resulting dsDNA could not
integrate due to the absence of a functional IN.
No virus was detected in PM1 cells infected with the progeny from pHIV-I4021sN- or pHIV-
I4069sN-transfected 293T cells (Figure 3.7). These PM1 cells were healthy with no cell death, no
syncytia, and no progeny production for the length of the experiment. PM1 cells are highly
permissive to HIV-1 replication. Therefore, if any WT HIV-1 had been produced as a result of
124

group II intron self-splicing in the pHIV-I4021sN- or pHIV-I4069sN-transfected 293T cells, or if
escape mutants had been generated during reverse transcription, progeny should have been
detected after two months. A concern with sense DNA targeting introns is that they may splice out
of the HIV-1 transcripts, enabling the group II intron-inserted HIV-1 proviral DNA to complete a
normal virus life cycle. Our results indicate that self-splicing of the particular introns used here is
not a concern for inhibition of HIV-1 replication.
In conclusion, we have shown here that the Ll.LtrB-derived I4021sN and I4069sN introns are capable
of conferring “complete” inhibition of HIV-1 replication at the intended step. These results
demonstrate that group II introns provide a powerful tool that should be further exploited for
HIV-1 gene therapy.
Acknowledgements
This work is supported by grants from the Canadian Institutes of Health Research and the
Ontario HIV Treatment Network. R.N. is thankful to the Ontario HIV Treatments Network for a
doctoral fellowship. We are grateful to Anne Lise Haenni (Institute Jacques Monod, Paris,
France) for critical proofreading of this manuscript. pACD-HIV1-4021s, pACD-HIV1-4069s,
and pBRR-HIV1(3805-4178s)/Tet were obtained from Alan Lambowitz (Institute for Cellular
and Molecular Biology, Austin, Texas). PM1 cell line and pNL4-3 were obtained through the
AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
125

CHAPTER 4
GENERAL DISCUSSION AND FUTURE DIRECTIONS
126

4.1. Discussion and Thesis Summary
To date, none of the anti-HIV drugs can eradicate the HIV-1 infection, no matter how early
treatment is initiated, or how long treatment is continued286. The reason is that the proviral DNA
persists in some cells. These cells, acting as viral reservoirs, can start virus production as soon as
the suppressive therapy stops. It has been shown that these cells, which are estimated to be about
0.05% of the total resting CD4+ T-cell population (~106 cells)287, are established early in HIV-1
infection288. Besides, genetic variations help the virus escape from the host immune system.
A mutation in CCR5, which results in a deletion of 32 nt. in the coding region, produces a non-
functional allele (CCR5∆32). About 10% of Caucasians carry this ∆32 allele and about 1% of
them are homozygotes. The ∆32/∆32 homozygotes are not usually susceptible to infection. R5-
tropic HIV-1 strains, which use CCR5 for entry, are favored for infection. Despite several
hypotheses that try to explain this phenomenon, the reason is not yet fully understood. Although
other CCR co-receptors, such as CCR1 and CCR3, can be used instead of CCR5 by some HIV-1
strains in vitro, there is no evidence of such infection and pathogenesis in vivo. The ∆32
heterozygotes are not completely protected, but progression to AIDS is slowed and delayed in
them108.
Once the infection is established, in 50% of patients, the viruses evolve from the R5-tropic strain
to an X4-tropic strain. The reason for such evolution is not understood, and its mechanism seems
to be mutations in viral glycoprotein gp120. The R5-tropic strains are less cytopathic in vitro and
have been shown to possess less replication capacity, killing, and syncytia formation, compared
to the X4-tropic strains. Therefore, shift from R5-tropic to X4-tropic results in a faster
progression to AIDS.
127

In infected individuals, one of the important keys that are involved in disease progression is
thought to be the activation of the immune system leading to production of inflammatory
cytokines and T-cell death/apoptosis289. HIV-1-infected macrophages may show abnormality in
function and production of cytokines, which may cause pathological and patho-physiological
problems. HIV-1 proviral DNA remains in the infected macrophage genome and the progeny
virus will be collected in and released by intracellular vesicles, without killing the cells or
producing high number of progeny virus, which makes the macrophages appropriate reservoirs
for virus. High levels of inflammatory cytokine production push both the infected and uninfected
cells to a pro-apoptotic state290.
During disease progression, some infected CD4+ T-cells produce high levels of viral proteins
and die after infection. A smaller number of infected T-cells, mainly the naïve and memory
CD4+ cells, will still carry the proviral DNA, but may remain as an HIV-1 reservoir and produce
little or no virus. The number of other uninfected CD4+ and CD8+ T-cells will also decline due
to apoptosis, which occurs either by cell-cell contact291 or by binding the HIV-1 Tat protein to
CD4 receptor of uninfected cell292. Also, the rate of CD4+ and CD8+ T-cell production will be
lowered by effects on bone marrow and/or thymus caused by the infection291. Hematopoiesis is
shown to be inhibited by secretion of certain hematopoietic factors, such as tumor growth factor-
α, from infected stromal fibroblasts and macrophages in bone marrow291.
As mentioned in the introduction, HIV-1 PIC, containing the reverse-transcribed dsDNA bound
to viral matrix protein p17, IN, RT, and Vpr, has the ability to integrate the HIV-1 DNA into the
chromosomes of non-dividing cells, which enables the HIV-1 to infect a wider number and type
of target cells. This ability is provided by Vpr functions, which, induces breaks in the nuclear
membrane293. The increase in numbers of such breaks may also induce cell death.
128

Increasing the amount of unintegrated DNA in re-infected cells294, as well as cytotoxic T
lymphocytes (CTL) attacks to the infected cells that express viral proteins295, are other reported
causes of infected cell death.
Nef is also an important HIV-1 protein involved in immunodeficiency and cell death, as well as
promoting more viral production. Although, by downregulating the surface CD4 expression, Nef
helps the infected cell escape being killed by CTL response, it enhances the expression of
apoptosis-inducing proteins, such as FasL and TNF-α, that induce apoptosis in uninfected cells
through a cell-cell contact296.
Knowing the above, if any therapeutic strategy is to be designed against HIV-1, in addition to
inhibition of viral replication, it must be able to stop cell death resulting from any of described
mechanisms. HIV-1 therapy strategies that focus on inhibiting proviral integration or disrupting
integrated viral DNA seem to be preferred because they would prevent viral gene activities.
Once no viral protein is produced, the infected cell may have a chance to remain and function in
the immune system. Our results demonstrated that group II introns, which are modified to target
integrated viral DNA, can be a good candidate for such strategy.
When HAART was developed in mid 1990s, its activity against HIV-1 was so promising that
people estimated the virus would be eliminated soon. Now, not only does the virus still infect
new patients, but also emergence of multi-drug resistant strains, HAART toxicity* and metabolic
* HIV/AIDS drug side effects may include appetite loss, nausea and vomiting, diarrhea, fatigue, gas and
bloating, headaches, nightmares and sleeping difficulties, hair loss, skin problems, muscles aches and
pains, sexual difficulties, liver toxicity, insulin resistance and diabetes, pancreatitis, kidney stones,
peripheral neuropathy, lose of bone density and osteonecrosis, body distortions, and cardiac
complications304.
129

complications are common problems in HIV/AIDS patients receiving HAART regimens. The
HAART therapy fails in about 50% of patients in less than two years286, and made the fatal
infection to become a chronic illness, which has increased the rate of transmission! According to
WHO estimates, 5 million people were diagnosed with new HIV-1 infection globally in 2005
and ~40,000 new infections occur yearly in the USA. Thus, other therapeutic approaches are
required to stop new infections along with treating the patients.
Since high levels of resistance to infection by HIV-1 was observed in CCR5∆32 homozygous
individuals, inhibiting viral entry through blockage/deletion of CCR5 may be the best possible
approach to eliminate and cure HIV-1 infection. This approach has many supporting reasons: 1)
CCR5 appearse to be dispensable. 2) viral shift to a more pathogenic X4-tropic strain is not a
concern anymore because it is known that R5-tropic strains alone can also produce AIDS.
Furthermore, resistance of a R5-tropic virus to a CCR5 inhibitor via changes in gp120 was
shown to result in a more efficient binding to CCR5 rather than a switching towards CXCR4
usage297.
Different classes of HIV-1 entry inhibitors, such as attachment inhibitors, CCR5 antagonists, and
fusion inhibitors have been developed. However, some anti-CCR5 drugs, such as aplaviroc and
vicriviroc, were ceased at different stages of phase IIb and III trials due to hepatotoxicity and
inferior efficiency. For these reasons, we and other researchers have focused on gene therapy
strategies to downregulate CCR5 expression. In table 4.1, I have summarized gene therapy
strategies that were developed and tested for downregulation of CCR5 expression. In addition to
their inefficiency in conferring complete CCR5 downregulation and durable inhibition of HIV-1
replication at high virus concentrations, each of them faced obstacles that slowed their
progression to clinical trials. For example, intrakines have off-target cellular effects that cause
130

immune system complications; intrabodies failed to protect the target cells when exposed to
higher virus concentrations; siRNAs were not durable and had off-target gene regulation effects;
and the antisense RNA was not efficient enough. Since ribozymes have not shown any off-target
gene regulation, cellular toxicity and immune system induction, they might be better candidate
tools for HIV-1 gene therapy. CCR5 downregulation by ribozymes is extensively studied.
Monomeric and multimeric ribozymes are designed to target CCR5 mRNA, however, none of
the studied monomeric or tirmeric ribozymes were completely efficient in CCR5 downregulation
and inhibition of HIV-1 replication. Therefore, we proposed that a multimeric ribozyme,
containing 7 ribozyme may function more efficiently.
131

Ref
.
Yan
g et
al.
(199
7)16
6
Schm
idtm
aye
rova
et
al.
(199
6)16
7
Tedl
a et
al.
(199
6)17
1
Kel
ly e
t al.
(199
8)14
4
Yan
g et
al.
(199
7)16
6
Dis
adva
ntag
e(s)
Into
lera
nce
to
ligan
ds o
ver-
ex
pres
sion
Tim
e of
add
ition
of
β-c
hem
okin
e is
im
porta
nt.
Upr
egul
atio
n of
tra
nscr
iptio
n fa
ctor
s via
G-
prot
ein
sign
alin
g is
a
fact
or15
3 .
Leak
age
of
intra
cellu
lar
chem
okin
es
indu
ces
infla
mm
ator
y re
spon
ses.
Res
ults
Cel
l gro
wth
failu
re
Stim
ulat
atio
n of
H
IV-1
repl
icat
ion
No
inhi
bitio
n or
vira
l at
tach
men
t
Bot
h en
hanc
emen
t an
d in
hibi
tion
of
infe
ctio
n
Dec
reas
e in
HIV
-1
repl
icat
ion
and
sync
ytia
form
atio
n
Vir
us
R5-
tropi
c H
IV-1
Cel
l(s)
PM1
Mac
roph
ages
Chr
onic
ally
in
fect
ed P
M1
Mon
ocyt
e,
MD
M a
nd
CD
4+ T
-cel
l
PM1
Vec
tor/
Pr
omot
er
pCM
V a
nd
LNC
X
(RV
)*
The
rape
utic
m
olec
ule
RA
NTE
S an
d M
IP1-α
RA
NTE
S
RA
NTE
S
RA
NTE
S M
IP1-α,
and
M
IP1-β
RA
NTE
S-K
DEL
and
M
IP1-α-
KD
EL
Tab
le 4
.1. S
umm
ary
of g
ene
ther
apy
appr
oach
es to
dow
nreg
ulat
e C
CR
5 su
rfac
e ex
pres
sion
.
Cla
ss o
f th
erap
eutic
m
olec
ule
Liga
nd
Mon
oclo
nal
ant
ibod
y * (R
V) =
retro
vira
l vec
tor;
(LV
) = le
ntiv
iral v
ecto
r; PI
= p
ost-i
nfec
tion;
(AV
) = a
deno
vira
l vec
tor;
Rz
= rib
ozym
e
132

Ref
.
Ros
chke
et
al.
(200
4)14
6
Cas
tagn
a et
al.
(200
5)16
,147
Stei
nber
ger e
t al.
(200
0)15
1
Swan
et a
l. (2
006b
)175
Cag
non
et
al.
(200
0)17
7
Dis
adva
ntag
e(s)
Pote
ncy
of a
gent
for
dura
tion
and
mag
nitu
de
of re
cept
or
inte
rnal
izat
ion.
Nee
ding
hig
h co
ncen
tratio
n, p
oten
cy
of a
gent
for d
urat
ion
and
mag
nitu
de o
f re
cept
or in
tern
aliz
atio
n.
Shor
t per
iod
of
inhi
bitio
n (1
0 da
ys P
I)
40%
inhi
bitio
n of
HIV
-1
repl
icat
ion
(day
20
PI)
20-5
0% in
hibi
tion
of
vira
l rep
licat
ion
(at 0
.3,
1 an
d 1.
5 ng
p24
of
BaL
)
Res
ults
Phas
e I c
linic
al tr
ial
Phas
e II
clin
ical
tria
l
Inhi
bitio
n of
CC
R5
expr
essi
on, s
yncy
tia
form
atio
n, a
nd H
IV-
1 in
fect
ion
Dis
rupt
ion
of C
CR
5 ex
pres
sion
50-6
0% re
duct
ion
of
CC
R5
expr
essi
on,
com
plet
e in
hibi
tion
of H
IV-1
repl
icat
ion
Vir
us
SF16
2 an
d JR
-CSF
(R
5) M
OI
of 0
.01
BaL
(1 n
g p2
4 eq
uiva
lent
)
BaL
(0.0
5-0.
1 ng
p24
eq
uiva
lent
)
Cel
l(s)
PM1
CD
4+ T
-cel
l an
d C
D34
+-de
rived
m
acro
phag
e
SupT
1,
PM1,
M
DM
s and
m
icro
glia
l
Vec
tor/
Pr
omot
er
Bab
e-Pu
ro
(RV
)
CA
D-R
5 (L
V)
SV40
-ba
sed
(RV
)/ C
MV
The
rape
utic
m
olec
ule
HG
S004
Pro-
140
ST6
(aga
inst
C
CR
5 N
-te
rmin
al)
ST6
(aga
inst
C
CR
5 N
-te
rmin
al)
2C7-
KD
EL
T
able
4.1
. con
tinue
d.
Cla
ss o
f th
erap
eutic
m
olec
ule
Mon
oclo
nal
antib
ody
Int
rabo
dy
* (R
V) =
retro
vira
l vec
tor;
(LV
) = le
ntiv
iral v
ecto
r; PI
= p
ost-i
nfec
tion;
(AV
) = a
deno
vira
l vec
tor;
Rz
= rib
ozym
e
133

Ref
.
Mar
tinez
et
al. (
2002
)36
Qin
et a
l. (2
003)
37
And
erso
n et
al.
(200
3)15
9
And
erso
n et
al.
(200
5)16
3
Li e
t al.
(200
6)15
2
Dis
adva
ntag
e(s)
79%
inhi
bitio
n of
HIV
-1
repl
icat
ion
on d
ay 2
PI
A d
efic
ient
HIV
-1 w
as
used
.
Onl
y 32
% in
hibi
tion
of
HIV
-1 re
plic
atio
n on
da
y 5
PI
Low
inhi
bitio
n of
C
CR
5 ex
pres
sion
and
on
ly 3
3% v
iral
repl
icat
ion
on d
ays 3
-7
PI
~55%
inhi
bitio
n of
vi
ral r
eplic
atio
n on
day
12
PI,
~87%
hom
olog
y to
CC
R2a
and
CC
R2b
m
RN
A
Res
ults
48%
redu
ctio
n of
C
CR
5 ex
pres
sion
90%
redu
ctio
n of
C
CR
5 ex
pres
sion
72%
redu
ctio
n of
C
CR
5 ex
pres
sion
98.1
% re
duct
ion
of
CC
R5
expr
essi
on
Vir
us
BaL
(MO
I 0.
03-0
.24)
Mod
ified
H
IV-1
, ex
pres
sing
H
SA
inst
ead
of
VPR
BaL
(MO
I of
0.0
01)
BaL
(MO
I of
0.0
1)
CN
9700
1 (R
5, M
OI
of 0
.01)
Cel
l(s)
U87
(e
xpre
ssin
g C
D4,
CC
R5
and
CX
CR
4)
CD
4+ P
BL
PBM
Cs
PBM
Cs
U93
7
Vec
tor/
Pr
omot
er
FG12
(LV
)
HIV
-7-
GFP
-XH
R
(LV
)/Pol
-II
I, U
6 an
d H
1
pAD
(AV
)
The
rape
utic
m
olec
ule
RN
AR
53i
(aga
inst
nt.
+554
to +
572
of C
CR
5)
siR
NA
(186
) (a
gain
st n
t. +1
86 to
+20
4 of
CC
R5)
Bis
peci
fic
siR
NA
(aga
inst
C
CR
5 an
d C
XC
R4)
siR
NA
(aga
inst
C
CR
5)15
9
653-
nt. l
ong
antis
ense
RN
A
(aga
inst
nts
. 18
7-83
9 of
C
CR
5-co
ding
re
gion
)
T
able
4.1
. con
tinue
d.
Cla
ss o
f th
erap
eutic
m
olec
ule
RN
Ai
(siR
NA
)
ant
isen
se
RN
A
* (R
V) =
retro
vira
l vec
tor;
(LV
) = le
ntiv
iral v
ecto
r; PI
= p
ost-i
nfec
tion;
(AV
) = a
deno
vira
l vec
tor;
Rz
= rib
ozym
e
134

Ref
.
Qur
eshi
et
al.
(200
6)15
3
Cag
non
et
al.
(200
0)17
7
Bai
et a
l. (2
000)
155
Li e
t al.
(200
5)19
0
Li e
t al.
(200
5)19
0
Bai
et a
l. (2
001)
157
Dis
adva
ntag
e(s)
68%
inhi
bitio
n of
vira
l re
plic
atio
n on
day
7 P
I
Mut
ant R
z sh
owed
the
sam
e ef
ficie
ncy
50%
inhi
bitio
n of
HIV
-1
repl
icat
ion
by m
utan
t R
z
Inhi
bitio
n w
as sl
ight
ly
bette
r tha
n ob
tain
ed b
y m
utan
t Rz
Hig
h m
ount
of p
roge
ny
viru
s pro
duct
ion
(1-1
0 ng
/ml o
n da
y 7
and
500
ng/m
l on
day
28 P
I)
30%
inhi
bitio
n of
HIV
-1
repl
icat
ion
on d
ay 4
PI
Res
ults
50-9
0% re
duct
ion
of
CC
R5
expr
essi
on
70%
redu
ctio
n of
C
CR
5 ex
pres
sion
, 1-
3 da
ys d
elay
in H
IV-
1 re
plic
atio
n
70%
inhi
bitio
n of
H
IV-1
repl
icat
ion
on
day
7 PI
Inhi
bitio
n of
HIV
-1
repl
icat
ion
on d
ay 1
7 PI
99%
inhi
bitio
n of
H
IV-1
repl
icat
ion
10-1
5% re
duct
ion
of
CC
R5
expr
essi
on
Vir
us
SF16
2 (3
ng
p4
equi
vale
nt)
BaL
(MO
I 0.
001)
BaL
(MO
I of
0.0
2)
BaL
(MO
I of
0.0
2)
JR-F
L (M
OIs
of
0.01
and
0.
05)
BaL
(MO
I of
0.0
01)
Cel
l(s)
PBM
Cs
HO
S-C
D4.
CC
R5
PM1
CD
34+-
deriv
ed
mac
roph
ages
T ce
lls a
nd
CD
34+-
deriv
ed
mac
roph
ages
CD
34+-
deriv
ed
mac
roph
ages
Vec
tor/
Pr
omot
er
Bab
e-Pu
ro
(RV
)
pG1N
a (R
V)
HIV
-7-
GFP
(LV
)
LN (R
V)
and
MN
D
(RV
)
The
rape
utic
m
olec
ule
Mon
omer
ic
CC
R5R
z*
(aga
inst
nts
. 26
2-26
4 of
C
CR
5 m
RN
A)
Mon
omer
ic R
z (a
gain
st n
ts. 2
3 of
CC
R5
mR
NA
)
Trim
eric
Rz
(aga
inst
nts
. 17
, 153
and
24
9 in
CC
R5
OR
F)
T
able
4.1
. con
tinue
d.
Cla
ss o
f th
erap
eutic
m
olec
ule
Rib
ozym
e
* (R
V) =
retro
vira
l vec
tor;
(LV
) = le
ntiv
iral v
ecto
r; PI
= p
ost-i
nfec
tion;
(AV
) = a
deno
vira
l vec
tor;
Rz
= rib
ozym
e
135

In our study, we investigated downregulation of the CCR5 expression by a heptameric
hammerhead ribozyme, containing 7 ribozymes that target nucleotides 17, 380, 390, 520, 556,
811, and 824 within the CCR5 ORF. These ribozymes were constructed in two sets: Set A,
containing Rz1, Rz2, and Rz3, and Set B containing Rz4, Rz5, Rz6, and Rz7. Each set of
multimeric ribozymes were tested for in vitro cleavage activity by providing with the CCR5
target RNA. The results revealed that the multimeric ribozymes were active (Figure 2.1). Then
the ribozymes of both sets were combined to make the heptameric hammerhead ribozyme, Rz1-7.
Rz1-7 was cloned into a MSCV-based oncoretroviral vector (MGIN), which contained egfp,
IRES, and neo, to produce MGIN-Rz1-7 vector. Constitutive activity of MGIN 5’ LTR results in
production of a long transcript containing the EGFP ORF, the Rz1-7, the IRES element, and the
Neo ORF. IRES facilitates protein expression from the Neo ORF as the second ORF on the same
transcript. Retroviral particles containing either MGIN or MGIN-Rz1-7 were produced by
transfection of an amphotropic packaging cell line, PA317, followed by G418 selection. The
viral particles were used to transduce CCR5+/CXCR4+/CD4+ human lymphoid cell line, PM1,
and the stably transduced cells were purified via FACS. The purification was performed two
times with 2-3 months interval. PCR analysis of stably transduced PM1 cells’ genomic DNA
showed the presence of the MGIN or MGIN-Rz1-7 vector DNAs (Figure 2.3A). RT-PCR analysis
on stably transduced PM1-MGIN and PM1-MGIN-Rz1-7 cells’ total RNA determined the
presence of vectors’ transcripts and expression of Rz1-7 (Figure 2.4A).
Lentiviral vectors are capable of transducing the resting cells by passing through the nuclear
envelope. Therefore, an HIV-1-based vector, HEG1, was constructed in our laboratory. It
contains the egfp gene under the control of the EF-1α promoter. Since HIV-1 5’ LTR promoter
depends on the HIV-1 Tat protein for activity, and this protein does not exist in uninfected cells,
136

EF-1α promoter serves as a strong internal promoter. I cloned the Rz1-7 gene downstream of the
egfp gene to construct HEG1-Rz1-7. Therefore, the transcript produced from EF-1α promoter will
contain both EGFP ORF and Rz1-7. Tat-dependent activity of HIV-1 5’ LTR promoter of the
HEG1-Rz1-7 vector in infected cells will also results in a long transcript containing the HIV-1 5’
sequence of gag gene and RRE element, the EGFP ORF and Rz1-7. HEG1 or HEG1-Rz1-7 vector
particles were used to transduce PM1 cells and the stable transductants were sorted by FACS.
The sorting was performed two times with 2-3 months interval. PCR analysis of stable
transductants’ genomic DNAs showed the presence of HEG1 and HEG1-Rz1-7 vectors DNAs
(Figure 2.3B). RT-PCR analysis of total RNA of PM1-HEG1-Rz1-7 cells also revealed the
expression of Rz1-7 in the cells (Figure 2.4B).
Total RNAs from the PM1-MGIN-Rz1-7 and PM1-HEG1-Rz1-7 cells were used to obtain Rz1-7
cDNAs via in vitro reverse-transcription reactions. These Rz1-7 cDNAs were used as templates to
produce Rz1-7 RNAs during in vitro transcription reactions. Cleavage activity of Rz1-7 originated
from PM1-MGIN-Rz1-7 and PM1-HEG1-Rz1-7 cells were demonstrated in vitro by its ability to
cleave in vitro-transcribed CCR5 mRNA.
RT-PCR analysis on both PM1-MGIN-Rz1-7 and PM1-HEG1-Rz1-7 cells’ total RNA also showed
that the CCR5 mRNA has been downregulated in the Rz-expressing cells (Figure 2.5A and B).
Untransduced PM1 cells, as well as MGIN-, MGIN-Rz1-7-, HEG1-, and HEG1-Rz1-7-transduced
PM1 cells became infected when challenged with NL4-3, an X4-tropic HIV-1 strain (Figure
2.7A and B). This confirms that the infection with an X4-tropic strain can still occur because this
strain uses CXCR4 co-receptor, which was not our target. Transducing the PM1 cells with the
vectors and expression of Rz1-7 did not alter the CXCR4 expression.
137

Untransduced PM1 cells, as well as MGIN- and HEG1-transduced PM1 cells were also infected
by BaL, an R5-tropic HIV-1 strain, indicating that these cells are susceptible to the R5-tropic
strains due to expression of CCR5 co-receptor on the cells’ surface (Figure 2.7A and B). This
experiment also showed that transduction of PM1 cells with empty MGIN and HEG1 vectors did
not affect the surface CCR5 expression. However, a high level of resistance was observed when
MGIN-Rz1-7- and HEG1-Rz1-7-transduced PM1 cells were challenged with BaL strain. The
challenging experiments lasted two to three months. Three different MOIs (0.225, 0.675, and
2.025) of BaL strain, which are quite high in comparison with other published
studies36,152,163,190,298, were used in our experiments. 99-100% inhibition of progeny virus
production was observed when PM1-MGIN-Rz1-7 cells were challenged with BaL strain at those
MOIs (Figure 2.6). In the case of PM1-HEG1-Rz1-7 cells, the inhibition was 80-99% (Figure
2.6). These levels of inhibitions are also among the highest reported inhibitions in all studies that
focused on inhibition at the level of virus entry36,37,152,153,157,163,176,177,190,298 (see table 4.1).
The incomplete inhibition in our study could have occurred for two reasons: first, the FACS
sorting did not result in a 100% purification of the EGFP-expressing cells, therefore, some
untransduced cells were still present in the sorted cells population. Second, the vectors
expression may have been silenced due to integration into the heterochromatin regions of some
host cells’ chromosomes, therefore, despite the presence of vector DNA in the host genome, the
Rz1-7 expression could have been downregulated due to silencing effects.
To confirm that the inhibitions were through blockade of R5 virus entry, PCR analysis were
performed on genomic DNAs extracted from infected PM1-MGIN and PM1-MGIN-Rz1-7 cells
on 4 and 43 days post-infection and from infected PM1-HEG1 and PM1-HEG1-Rz1-7 cells on 4
and 29 days post-infection. Although a tat gene fragment of HIV-1 proviral DNA was amplified
138

from infected PM1-MGIN and PM1-HEG1 cells’ DNAs, no HIV-1 proviral DNA could be
detected in infected PM1-MGIN-Rz1-7 and PM1-HEG1-Rz1-7 cells’ DNAs (Figure 2.5A and B).
To show that the resistance was not due to any effects on the virus infectivity, similar amount of
progeny virus produced on day 17 post-infection from the BaL virus-infected untransduced cells,
as well as HEG1- and HEG1-Rz1-7-transduced PM1 cells (at an MOI of 2.025) were used to
infect plain PM1 cells. On 14 days post-infection, similar amount of viral p24 was detected in
three cultures, which confirms the infectivity of the progeny viruses produced from control and
Rz1-7-expressing cells.
Since PM1 cells express variety of other chemokines receptors, such as CXCR4, CCR1, and
CCR3277, in addition to CCR5, the fact that the infection of an R5-tropic HIV-1 strain was
inhibited in the anti-CCR5 Rz1-7-expressing cells suggests that the inhibition was through
blockade of the entry and other chemokine receptors could not participate in infection with the
R5 strain. It can also be concluded that during two to three-month long experiments, the R5
strain did not evolve to gain the ability of using other chemokine receptors (i.e. CXCR4) for
infection.
Using the anti-CCR5 ribozyme, we were able to show that virus replication was inhibited at the
level of entry. However, one may ask what can be done if the virus still manages to enter the cell
or an X4-tropic virus infects the CXCR4-expressing cell.
The efficacy of drug and gene therapy strategies, which use interfering molecules that degrade
HIV-1 RNA or inactivate the downstream proteins, will depend on the levels of HIV-1
RNA/proteins expression. We believe that if, by any means, the virus manages to enter the cell
139

despite CCR5 downregulation, the best way to overcome the virus replication may be
inactivation of proviral DNA or blocking its integration to cells’ chromosomes. By doing this, no
viral RNA and protein would be produced and competition of therapeutic gene expression with
viral gene expression would not be necessary any more. Note that single or double LTR circles,
as well as noncircular forms, cannot express virus in vitro, unless integrated into the host
chromosome288. Therefore, we investigated whether an HIV-1-targeting group II intron can
insert into HIV-1 proviral DNA and inactivate the HIV-1 IN gene. Before analyzing our results, I
bring your attention to table 4.2 that summarizes some other gene therapy studies, which targeted
pre-integration and integration stages of HIV-1 life cycle. The gene therapy strategies that focus
on pre-integration and integration stages can be classified based on the molecules they target.
The targets include released viral RNA, RT, IN, PIC, HIV-1 dsDNA, and integrated proviral
DNA.
140

R
ef.
Sarv
er e
t al
. (1
990)
207
Yam
ada
et
al.
(199
4)21
0
Leav
itt e
t al
. (1
994)
211
Li e
t al.
(199
8)21
4
Yu
et a
l. (1
995a
)212
Yu
et a
l. (1
995b
)213
D
isad
vant
age(
s)
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
R
esul
ts
97%
resi
stan
ce to
in-
com
ing
HIV
-1 fo
r 7
days
PI*
95%
inhi
bitio
n of
pr
ogen
y vi
rus
prod
uctio
n til
l day
35
PI
Inhi
bitio
n of
pro
geny
vi
rus p
rodu
ctio
n fo
r 10
day
s PI
Inhi
bitio
n of
pro
geny
vi
rus p
rodu
ctio
n fo
r 20
-35
days
PI
95%
inhi
bitio
n of
pr
ogen
y vi
rus
prod
uctio
n fo
r 10
days
PI
Inhi
bitio
n of
pro
geny
vi
rus p
rodu
ctio
n til
l da
y 25
PI
V
irus
HX
B2
HX
B2
BaL
or
isol
ated
fr
om
patie
nts
BaL
C
ell(s
)
HeL
a C
D4+
Jurk
at
PBLs
CD
34+-
deriv
ed
mac
roph
ages
CD
34+-
deriv
ed
mac
roph
ages
CD
34+-
deriv
ed
mac
roph
ages
V
ecto
r/
Prom
oter
LNL6
(RV
)*/
tRN
AV
al
LNL6
(RV
) /tR
NA
Val
LNL6
(RV
) /tR
NA
Val
LNL6
(RV
) /V
A1
LNL6
(RV
) /V
A1
The
rape
uti
c m
olec
ule
HH
Rz*
ag
ains
t HIV
-1
gag-
codi
ng
regi
on
HPR
z*
agai
nst n
t. +1
11 a
t HIV
-1
LTR
HPR
z ag
ains
t H
IV-1
pol
-co
ding
regi
on
Tab
le 4
.2. S
umm
ary
of g
ene
ther
apy
appr
oach
es to
inhi
bit H
IV-1
repl
icat
ion
at p
re-in
tegr
atio
n or
inte
grat
ion
stag
es.
Tar
get
mol
ecul
e
Rel
ease
d vi
ral
RN
A
*HH
Rz
= ha
mm
erhe
ad ri
bozy
me;
HPR
z =
hairp
in ri
bozy
me;
PI =
pos
t-inf
ectio
n; R
V =
retro
vira
l vec
tor;
+sss
DN
A =
plu
s-st
rand
stro
ng
stop
DN
A
141

Ref
.
Wes
taw
ay
et a
l. (1
995)
216
Din
g et
al
. (1
998)
217
Din
g et
al
. (1
998)
217
Peng
et
al.
(199
7)21
8
Jacq
ue e
t al
. (2
002)
34
Cap
odic
i et
al.
(200
2)21
9
Dis
adva
ntag
e(s)
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
Shor
t-ter
m
inhi
bitio
n
Res
ults
83%
redu
ctio
n of
in
fect
ivity
of p
roge
ny
viru
s
Inhi
bitio
n of
pro
geny
vi
rus p
rodu
ctio
n fo
r 21
day
s PI
Inhi
bitio
n of
pro
geny
vi
rus p
rodu
ctio
n fo
r 21
day
s PI
Inhi
bitio
n of
vira
l re
plic
atio
n fo
r 140
da
ys P
I
30 to
50-
fold
re
duct
ion
of p
roge
ny
viru
s pro
duct
ion
on
day
2 PI
75-9
6% d
ecre
ase
in
prog
eny
viru
s pr
oduc
tion
for 3
day
s PI
Vir
us
NL4
-3 (3
0 ng
p24
eq
uiva
lent
)
NL4
-3 (3
0 ng
p24
eq
uiva
lent
)
NL4
-3
(MO
I of
0.01
)
pHIV
NL-
GFP
IIIB
and
B
aL
Cel
l(s)
PBLs
PBLs
Jurk
at
CD
4+ M
AG
I
U87
-CD
4+-
CX
CR
4+,
U87
-CD
4+-
CC
R5+
and
C
D4+
T c
ells
Vec
tor/
Pr
omot
er
Fuse
d to
tR
NA
Lys3
MO
TN a
nd
MO
TiN
(RV
)
MO
TN a
nd
MO
TiN
(RV
)
LSN
P (R
V)
/tRN
AM
et
The
rape
utic
m
olec
ule
HH
Rz*
ag
ains
t HIV
-1
PBS
regi
on
Ant
isen
se
RN
A a
gain
st
HIV
-1 ψ
-gag
Ant
isen
se
RN
A a
gain
st
HIV
-1 U
3-ga
g-en
v
Sens
e R
NA
ag
ains
t LTR
, se
nse
RN
A
agai
nst
+sss
DN
A*
siR
NA
s ag
ains
t LTR
, vi
f and
nef
siR
NA
s ag
ains
t LTR
an
d ga
g
Tab
le 4
.2. c
ontin
ued.
Tar
get
mol
ecul
e
Rel
ease
d vi
ral R
NA
*HH
Rz
= ha
mm
erhe
ad ri
bozy
me;
HPR
z =
hairp
in ri
bozy
me;
PI =
pos
t-inf
ectio
n; R
V =
retro
vira
l vec
tor;
+sss
DN
A =
plu
s-st
rand
stro
ng
stop
DN
A
142

Ref
.
Mac
ieje
ws
k et
al.
(199
5)22
0
Shah
een
et
al.
(199
6)22
1
Stra
yer e
t al
. (2
002)
223
Josh
i et a
l. (2
002)
225
Sura
bhi e
t al
. (2
002)
226
Dis
adva
ntag
e(s)
Shor
t-ter
m in
hibi
tion
Cha
lleng
ing
with
vi
ruse
s at M
OI o
f 1.0
sh
owed
no
inhi
bitio
n.
Cha
lleng
ing
with
vi
ruse
s at h
ighe
r TC
ID50
show
ed n
o in
hibi
tion
Shor
t-ter
m
inhi
bitio
n.
Shor
t-ter
m
inhi
bitio
n, Ju
rkat
ce
lls n
eede
d 20
-fol
d co
ncen
trate
d si
RN
As
for s
ame
inhi
bitio
n.
Res
ults
Com
plet
e in
hibi
tion
of v
iral r
eplic
atio
n fo
r 35
days
PI
80-9
7% in
hibi
tion
of
prog
eny
viru
s pr
oduc
tion
for 1
5-22
da
ys P
I
Hig
h in
hibi
tion
of
prog
eny
viru
s pr
oduc
tion
23-9
6% re
duct
ion
of
vira
l rep
licat
ion
for
22 d
ays P
I
90%
inhi
bitio
n of
pr
ogen
y vi
rus
prod
uctio
n fo
r 13
days
PI
Vir
us
IIIB
, RF,
57
1, a
nd
MN
(MO
Is
of 2
)
NL4
-3
(MO
Is o
f 0.
012
and
0.00
6) a
nd
R7-
HX
B2
(MO
Is o
f 0.
012
and
0.00
6)
NL4
-3 (4
0-10
0 TC
ID50
)
R3B
(MO
Is
of 0
.1 a
nd
50)
Cel
l(s)
MO
LT-3
ly
mph
oid
cell
line
SupT
1
SupT
1
Jurk
at a
nd
293T
MA
GI
Vec
tor/
Pr
omot
er
pMEP
4 (E
BV
-ba
sed
vect
or)
SLX
CM
V
(RV
)/CM
V
SV40
-bas
ed
(RV
)
pcD
NA
3.1/
CM
V
The
rape
utic
m
olec
ule
Ant
i-RT
Fab
Ant
i-RT
intra
body
Ant
i-RT
mA
b (1
E8)
Ant
i-RT
apta
mer
s
siR
NA
s ag
ains
t RT
Tab
le 4
.2. c
ontin
ued.
Tar
get
mol
ecul
e
Rev
erse
tra
nscr
ipta
se
*HH
Rz
= ha
mm
erhe
ad ri
bozy
me;
HPR
z =
hairp
in ri
bozy
me;
PI =
pos
t-inf
ectio
n; R
V =
retro
vira
l vec
tor;
+sss
DN
A =
plu
s-st
rand
stro
ng st
op
DN
A
143

Ref
.
Levy
-Min
tz
et a
l. (1
996)
227
Bou
Ham
dan
et a
l. (1
999)
224
Gol
dste
in e
t al
. (20
02)22
8
Fass
ati e
t al
. (20
03)19
8
Fass
ati e
t al
. (20
03)19
8
Dis
adva
ntag
e(s)
Shor
t-ter
m in
hibi
tion
Onl
y 18
day
s del
ay in
sy
ncyt
ia fo
rmat
ion
whe
n ch
alle
nged
with
th
e hi
gher
co
ncen
tratio
n of
viru
s
Shor
t-ter
m in
hibi
tion
No
inhi
bitio
n at
MO
Is
>1
Low
inhi
bitio
n
Res
ults
92%
inhi
bitio
n of
vi
ral r
eplic
atio
n fo
r 25
day
s PI
Dec
reas
e in
viru
s pr
oduc
tion
whe
n ch
alle
nged
with
the
low
er c
once
ntra
tion
of v
irus
85%
inhi
bitio
n of
vi
ral r
eplic
atio
n fo
r 14
day
s PI
80-9
0% in
hibi
tion
of
vira
l rep
licat
ion
(with
vi
ral M
OI o
f 0.0
1)
2-38
% in
hibi
tion
of
vira
l rep
licat
ion
Vir
us
NL4
-3
(MO
Is o
f 0.
08)
NL4
-3
(0.0
5 an
d 0.
5 pg
p24
eq
uiva
lent
)
NL4
-3 (8
00
TCID
50)
MO
Is o
f 0.
01 a
nd >
1
Cel
l(s)
PBM
Cs
SupT
1
Hum
an
thym
ic g
rafts
of
thy/
liv-
SCID
-hu
mic
e
NP2
, HeL
a,
HeL
a-C
D4
HeL
a
Vec
tor/
Pr
omot
er
SLX
CM
V
(RV
)/CM
V
SV40
-bas
ed
(RV
)
SV40
-bas
ed
(RV
)
The
rape
utic
m
olec
ule
Ant
i-IN
an
tibod
ie
(scF
v33
and
scFv
33/n
u)
Ant
i-IN
an
tibod
y (s
cFv3
3)
Ant
i-IN
an
tibod
y (s
cFv3
3)
siR
NA
aga
inst
hu
man
imp7
(n
ts. 1
392-
1414
)
siR
NA
aga
inst
hu
man
imp9
(n
ts. 5
27-5
47)
Tab
le 4
.2. c
ontin
ued.
Tar
get
mol
ecul
e
Inte
gras
e
Pre-
inte
grat
ion
com
plex
*HH
Rz
= ha
mm
erhe
ad ri
bozy
me;
HPR
z =
hairp
in ri
bozy
me;
PI =
pos
t-inf
ectio
n; R
V =
retro
vira
l vec
tor;
+sss
DN
A =
plu
s-st
rand
stro
ng st
op
DN
A
144

Ref
.
Ciu
ffi e
t al.
(200
5)19
9
Levi
n et
al.
(199
7)23
0
Dis
adva
ntag
e(s)
Incr
ease
of p
rovi
ral
inte
grat
ion
into
GC
-ric
h D
NA
regi
ons o
f hu
man
chr
omos
omes
Res
ults
6-10
% re
duct
ion
of
prov
iral i
nteg
ratio
n in
to L
EDG
F/p7
5-re
gula
ted
gene
s and
ot
her t
rans
crip
tion
units
Inhi
bitio
n of
vira
l re
plic
atio
n an
d re
duct
ion
of p
roge
ny
viru
s inf
ectiv
ity
Vir
us
Cel
l(s)
Jurk
at
Div
idin
g C
D4+
cel
ls
Vec
tor/
Pr
omot
er
The
rape
utic
m
olec
ule
shR
NA
ag
ains
t hum
an
LED
GF/
p75
Ani
t-MA
in
trabo
dy
Tab
le 4
.2. c
ontin
ued.
Tar
get
mol
ecul
e
Pre-
inte
grat
ion
com
plex
*H
HR
z =
ham
mer
head
ribo
zym
e; H
PRz
= ha
irpin
ribo
zym
e; P
I = p
ost-i
nfec
tion;
RV
= re
trovi
ral v
ecto
r; +s
ssD
NA
= p
lus-
stra
nd st
rong
stop
D
NA
145

As mentioned, despite the fact that the proviral DNA is thought to be the best gene therapy
candidate, it was not successfully targeted so far. This will highlight the importance and novelty
of our study of inactivating the HIV-1 proviral DNA by group II introns. We used the
Lactococcus lactis group II intron (Ll.LtrB) as a therapeutic tool that has an outstanding
advantage: since the Ll.LtrB group II intron RNP recognizes a 12-16 base pair region on the
target DNA site, its RNA sequence can be easily altered to base-pair with the flanking sequences
of the intron’s LtrA protein binding site. Previously, Dr. A. Lambowitz and his team had shown
that group II introns could be designed and modified to be able to insert into the HIV-1 proviral
DNA237. They cloned a ~370-bp cDNA fragment from nt. 3805 to 4178 (respect to nt. 1 of HIV-
1 RNA) of HIV-1 IN domain of pol region into the pBRR vector upstream of a promoter-less tetR
gene to make the pBRR-HIV1(3805-4178s)/Tet, a selectable ‘recipient’ plasmid. They also
cloned the introns targeting nts. 4021 and 4069 of HIV-1 provirus sense-strand DNA into the
pACD expression vector (donor) under the control of a T7 promoter to produce pACD-HIV1-
4021s and pACD-HIV1-4069s, respectively. The introns were modified to contain an internal T7
promoter in the intron’s domain IV. When E. coli cells were co-transformed with both donor and
recipient vectors, the introns were expressed (by induction with IPTG) and inserted into the
target sequence at a frequency of 66% and 63%, respectively. Since the promoter-less tetR gene
could be expressed by the intron’s internal T7 promoter, the frequency of insertion could be
measured by the ratio of TetR colonies to the total of TetR and TetS ones. However, the authors
did not assess the insertion and its frequency into a full-length HIV-1 clone. Because both intron
and target were cloned into bacterial vectors, to be able to test whether the intron-LtrA protein
(RNP) complex could function in human cellular environment, they co-transfected 293T cells
with liposomes carrying the pre-expressed RNP and the recipient vector, separately. The
insertion was detected by PCR analysis237. However, there is a doubt whether the fusion between
146

donor- and recipient-carrying liposomes did not occur outside the cell, which might result in a
better target accessibility for the intron.
In our study, we investigated if the intron can target the full-length HIV-1 clone, if further
modifications to the intron to have a selectable marker will alter the intron’s function and
insertion rate, and if the intron insertion will inhibit HIV-1 replication.
Both pACD-HIV1-4021s and pACD-HIV1-4069s plasmids were modified by cloning a
kanR/neoR gene in intron’s domain IV, downstream of the internal T7 promoter to produce
pACD-I4021sN and pACD-I4069sN (Figure 3.3). When E. coli cells were co-transformed with the
pBRR-HIV1(3805-4178s)/Tet recipient and each of the modified introns, we have shown that
upon IPTG induction, the insertions occurred, but at very lower frequencies compared to those of
unmodified introns (Figure 3.2). This might have happened because of either changes in the
tertiary structure of intron RNA due to cloning of the neoR gene in the intron or lower expression
of the tetR gene due to the additional distance made by cloning the neoR gene downstream of the
T7 promoter.
To investigate the insertion of modified introns into the full-length HIV-1 proviral DNA clone,
E. coli cells were co-transformed with pNL4-3 as the recipient and either of pACD-I4021sN or
pACD-I4069sN donor plasmids, followed by IPTG induction. The frequency of insertions could
not be measured because the recipient plasmid would not demonstrate any selective advantage
upon intron insertion. However, the insertions were detected by plasmid purification,
endonuclease digestions, and PCR analyses.
147

The intron-inserted HIV clones, pHIV-I4021sN and pHIV-I4069sN, were then tested for their ability
to produce progeny virus by transfecting the 293T cells. Since the sizes of the intron-inserted
HIV-1 RNA were about 2 kb larger than the wild-type RNA, we doubted whether or not they
could be packaged in the virions. It appeared that both pHIV-I4021sN and pHIV-I4069sN could
produce the same amount of progeny virus as the control pHIV-1-transfected 293T cells (Figure
3.5). However, the intron-containing progeny viruses were not infectious when PM1 cells were
challenged with them (Figure 3.7). This was anticipated because the intron insertion into the IN
domain would cause formation of an early stop codon and production of 88-aa or 96-aa long
truncated INs, which could not function normally in the integration process. This was proved by
PCR analysis on the infected PM1 cells’ DNAs. Although we could detect the HIV-1 cDNA
molecules in PM1 cells at 1 hr post-infection (meaning accomplishment of reverse-transcription
process), no HIV-1 proviral DNA could be detected 8 days post-infection. No sign of infection
was observed in a more than 2-month long experiment. The results were supported by high cell
viability and lack of syncytia formation in the cultures. The length of experiment was important
to show that the introns, which were inserted into the sense strand of target DNA, could not
splice out to produce wild-type HIV-1 RNA. This study showed that inactivation of IN by
specific introns could result in inhibition of HIV-1 infection in the second host cell, or as we call,
“the second round of infection”.
We also tried to test the anti-sense targeting introns, I54a and I2654a, which targeted the nucleotides
54 and 2654 on antisense strand of HIV-1 proviral DNA. We modified the introns to contain the
kanR/neoR gene, as well as a transcription terminator in the reverse orientation. The advantage of
presence of the terminator would be lack of viral RNA production in the first infected cell, which
would result in inhibition at “the first round of infection”. However, due to very low frequency
of insertion, this part of study could not be continued.
148

4.2. Future Directions
Gene therapy holds promise for prevention of HIV infection and control of established
infection. However, there were obstacles that made it progress slowly. The hematopoietic stem
cells that produce CD4+ cells needed to be identified and purified. Culture, transduction, and
induction of modified stem cells to the progeny cells must be established. Appropriate vectors
were needed to be developed with the following specifications: efficient and easy production,
safety, long-term and regulated gene expression, and specific cell targeting. It has been shown
that lentiviral vectors may cause gene silencing when the transduced cells are being cultured for
longer times. Also, integration of lentiviral vectors into various DNA sequences of the
mammalian cells’ genome, where they may be close to a growth-promoting gene, can cause
pathological disorders. An example is induction of leukemia in children treated for severe
combined immunodeficiency using retroviral vector-mediated gene transfer299. And, at last, but
not least, a therapeutic gene that confers complete resistance to HIV-1 needed to be developed.
In this project, I targeted the cellular CCR5 receptor, whose downregulation is strongly believed
to produce the most effective barrier against HIV-1 entry. I used multimeric hammerhead
ribozyme, which is proved to be one of the most efficient therapeutic molecule used in anti-
HIV-1 gene therapies. To deliver the anti-CCR5 Rz1-7 to the cells, I also used and modified a
self-inactivating HIV-1-based vector. Another self-inactivating lentiviral vector, SIN-EF-G-I300,
which contains an insulator to inhibit gene silencing and oncogene activation, has also been
used in our laboratory for cloning the anti-CCR5 Rz1-7 to produce SIN-EF-G-I-Rz1-7 vector.
Anti-HIV-1 ribozymes, Rz1-9 and Rz1-14, which were previously developed in our laboratory26,28,
were also cloned in this vector to obtain SIN-EF-G-I-Rz1-9 and SIN-EF-G-I-Rz1-14 vectors.
These clonings were done by Masoud Ameli and Xue Zhong Ma in our laboratory. I have
149

obtained vector particles for these vectors, however the titers were low and, therefore, high
concentration vector particles will be produced again to be able to obtain an appropriate
transduction frequency. Further experiments with three new vectors on hematopoietic stem cells
will be done in collaboration with Dr. J.J. Rossi (Division of Molecular Biology, City of Hope
National Medical Center, Duarte, CA, USA) and Dr. R. Akkina (Department of Pathology,
Colorado State University, CO, USA).
Considering the possibility that mutant HIV-1 strains may acquire the ability to infect the cells
with downregulated CCR5 via other co-receptors, a combination strategy has been thought as a
back-up. This would combine anti-CCR5 and anti-HIV-1 multimeric hammerhead ribozymes in
the SIN-EF-G-I vector. By this means, if the virus passes the first barrier at the level of entry and
enters the cell, the genomic or transcribed RNAs will be cleaved by the anti-HIV-1 ribozymes.
Cells will be transduced with the SIN-EF-G-I-Rz1-7-Rz1-9 vector particles and will be examined
for ribozymes expression and inhibition of viral entry and progeny virus production. The vector
particles will also be sent to our collaborators for further tests on hematopoietic stem cells.
In my second project, where inactivation of HIV-1 proviral DNA by modified group II introns
was tested, I showed that the intron insertion into the sense strand of the integrase domain of the
pol region could inhibit the HIV-1 infection. The inhibition of progeny virus replication from
the intron-inserted HIV-1 clones has been shown to occur in the second round of infection. It is
necessary, for gene therapy purposes, to inhibit the HIV-1 replication in the first round of
infection. Therefore, I also used another strategy, in which I modified the I54a and I2654a introns
that target the HIV-1 proviral DNA antisense strands at nts. 54 and 2654, respectively. The
modification included cloning the neoR gene and a mammalian transcription terminator in the
reverse orientation. When the intron inserts into the anti-sense strand of HIV-1 proviral DNA,
150

the terminator will be positioned in the forward orientation and will stop transcription.
Therefore, the transcription of HIV-1 RNA will be truncated, especially when intron integrates
after nt. 54, at the R region of the HIV LTR. However, I54a and I2654a intron-inserted HIV clones
could not be obtained due to the original low frequency of insertion at these sites. Further
modification to the intron’s EBS1, EBS2, and δ sequences, in order to obtain more sequence
match with the insertion site, may increase the insertion frequency.
Further modifications to the I4021s and I2654a introns were also done in our laboratory (by Masoud
Ameli and Zue Zhong Ma) to clone an anti-HIV-1 Rz1-926,28 into the introns. A ds-Red gene,
which expresses a red fluorescent protein, was cloned into the intron to facilitate sorting of
mammalian cells that allow intron insertion. Cloning the anti-CCR5 Rz1-7 into the introns is also
planned.
In the experiments reported in this thesis, intron insertions were allowed to take place in E. coli.
For future application of these introns in anti-HIV gene therapy, it is crucial to determine
whether intron expression and insertion can take place in mammalian cells. To this end, I have
recently shown that when 293T cells were co-transfected with I4021s and pBRR-HIV1(3805-
4178s)/Tet plasmids along with pCMV-T7Pol (a T7 RNA Polymerase-expressing plasmid)301, the
intron was expressed and inserted into the recipient plasmid in the mammalian cells. Experiments
are underway to determine the intron insertion frequency in mammalian cells. An ectopic I4021
intron insertion site was identified in the mammalian genome. Therefore, it will be ideal to
investigate whether intron insertion can take place in the chromosomal DNA in mammalian cells
that are co-transfected. If intron insertion at this site is detected, we will also determine whether
this takes place in all of the cells.
151

Intron insertion into the pHIV-1 clone in the co-transfected 293T cells with I4021s, pHIV-1 and
pCMV-T7Pol is planned. If successful, intron insertion into HIV-1 proviral DNA, which is
integrated into the cell line’s genome, will be investigated. However, for this purpose, the LtrA
protein will be modified to be codon-optimized and contain a nuclear localization signal, so that
it can be expressed in mammalian cells and be transported to the nucleus in its RNP form.
Following the successful demonstration of inhibition of HIV-1 replication through an intron
insertion approach, studies will be continued to determine the usefulness of this gene therapy
strategy in the inhibition of HIV-1 replication in transduced peripheral blood T lymphocytes
and in the progeny of transduced stem cells.
Inactivation of HIV-1 proviral DNA by introns has an advantage upon using retroviral or
lentiviral vectors for expressing other anti-HIV-1 genes: the Ll.LtrB group II intron RNP
recognizes a 12-16 base pairs on the target DNA site. Therefore, the insertion will be more
accurate compared to retroviral and lentiviral vectors, which usually integrate into the host
genome randomly and may cause unwanted gene disruption and malignancy.
The retrotransposition properties of group II introns are now being explored as potential tools in
genetic engineering and genomic analysis. The possibility of designing L. lactis group II intron
by modifying EBS–IBS and δ-δ’ regions to disrupt an unwanted or malfunction gene or to
introduce foreign sequences under the control of selected promoters raises a spectrum of
interesting applications in genetic engineering and gene therapy. In the future, efforts will have
to be focused on developing derivatives of the L. lactis mobile group II intron that can transfer a
wider variety of insert sequences without loss of repair efficiency. Moreover, development of
transfer and expression methods that allow for group II mobilization into mammalian
chromosomal DNA will be essential. The observation that L. lactis group II introns can mobilize
152

into plasmid DNA in mammalian cells is encouraging in this regard237. Moreover, the potential
utility of other group II introns and other mobile genetic elements for genetic repair should be
explored. Many mobile genetic elements exist naturally in the human genome302,303, suggesting
that if they can be controlled appropriately, they may become useful therapeutic agents for repair
of the mutant genetic instructions associated with many human maladies.
Although the development of HIV-1 gene therapy strategies from the laboratory design through
to the clinical trials, is an expensive process, the end result will be more beneficial. By the time
HIV-1 gene therapy strategies become a permanent solution, they can serve as a back-up to cope
with the current drug resistance or toxicity. In the future, when these therapies have been
developed, they can serve as the ultimate therapy in developed and developing countries.
153

REFERENCES
1. Barre-Sinoussi F., Chermann J. C., Rey F., Nugeyre M. T., Chamaret S., Gruest J., Dauguet C., Axler-Blin C., Vezinet-Brun F., Rouzioux C., Rozenbaum W. & Montagnier L. (1983) Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868-871.
2. Gallo R. C., Sarin P. S., Gelmann E. P., Robert-Guroff M., Richardson E., Kalyanaraman V. S., Mann D., Sidhu G. D., Stahl R. E., Zolla-Pazner S., Leibowitch J. & Popovic M. (1983) Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 220, 865-867.
3. Ratner L., Haseltine W., Patarca R., Livak K. J., Starcich B., Josephs S. F., Doran E. R., Rafalski J. A., Whitehorn E. A. & Baumeister K. (1985) Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 313, 277-284.
4. Wain-Hobson S., Sonigo P., Danos O., Cole S. & Alizon M. (1985) Nucleotide sequence of the AIDS virus, LAV. Cell 40, 9-17.
5. Joshi S., Joshi R. L. (1996) Molecular biology of human immunodeficiency virus type-1. Transfus. Sci. 17, 351-378.
6. Bukrinsky M. I., Haggerty S., Dempsey M. P., Sharova N., Adzhubel A., Spitz L., Lewis P., Goldfarb D., Emerman M. & Stevenson M. (1993) A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature 365, 666-669.
7. Bukrinsky M. I., Haffar O. K. (1997) HIV-1 nuclear import: in search of a leader. Front. Biosci. 2, d578-87.
8. Wei X., Ghosh S. K., Taylor M. E., Johnson V. A., Emini E. A., Deutsch P., Lifson J. D., Bonhoeffer S., Nowak M. A. & Hahn B. H. (1995) Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373, 117-122.
9. Ho D. D., Neumann A. U., Perelson A. S., Chen W., Leonard J. M. & Markowitz M. (1995) Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123-126.
10. Li X. D., Moore B. & Cloyd M. W. (1996) Gradual shutdown of virus production resulting in latency is the norm during the chronic phase of human immunodeficiency virus replication and differential rates and mechanisms of shutdown are determined by viral sequences. Virology 225, 196-212.
11. Jacks T., Power M. D., Masiarz F. R., Luciw P. A., Barr P. J. & Varmus H. E. (1988) Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 331, 280-283.
12. Kohl N. E., Emini E. A., Schleif W. A., Davis L. J., Heimbach J. C., Dixon R. A., Scolnick E. M. & Sigal I. S. (1988) Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. U. S. A. 85, 4686-4690.
13. Peng C., Ho B. K., Chang T. W. & Chang N. T. (1989) Role of human immunodeficiency virus type 1-specific protease in core protein maturation and viral infectivity. J. Virol. 63, 2550-2556.
154

14. Gottlinger H. G., Sodroski J. G. & Haseltine W. A. (1989) Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 86, 5781-5785.
15. Strayer D. S., Akkina R., Bunnell B. A., Dropulic B., Planelles V., Pomerantz R. J., Rossi J. J. & Zaia J. A. (2005) Current status of gene therapy strategies to treat HIV/AIDS. Mol. Ther. 11, 823-842.
16. Temesgen Z., Warnke D. & Kasten M. J. (2006) Current status of antiretroviral therapy. Expert Opin. Pharmacother. 7, 1541-1554.
17. Agrawal L., Lu X., Jin Q. & Alkhatib G. (2006) Anti-HIV therapy: Current and future directions. Curr. Pharm. Des. 12, 2031-2055.
18. Lamothe B., Joshi S. (2000) Current developments and future prospects for HIV gene therapy using interfering RNA-based strategies. Front. Biosci. 5, D527-55.
19. Singwi S., Joshi S. (2000) Potential nuclease-based strategies for HIV gene therapy. Front. Biosci. 5, D556-79.
20. Nielsen M. H., Pedersen F. S. & Kjems J. (2005) Molecular strategies to inhibit HIV-1 replication. Retrovirology 2, 10-30.
21. Bevec D., Dobrovnik M., Hauber J. & Bohnlein E. (1992) Inhibition of human immunodeficiency virus type 1 replication in human T cells by retroviral-mediated gene transfer of a dominant-negative Rev trans-activator. Proc. Natl. Acad. Sci. U. S. A. 89,
22. Fujii G., Horvath S., Woodward S., Eiserling F. & Eisenberg D. (1992) A molecular model for membrane fusion based on solution studies of an amphiphilic peptide from HIV gp41. Protein Sci. 1, 1454-1464.
23. Cohen J. (2007) Building an HIV-proof immune system. Science 317, 612-614.
24. Cohli H., Fan B., Joshi R. L., Ramezani A., Li X. & Joshi S. (1994) Inhibition of HIV-1 multiplication in a human CD4+ lymphocytic cell line expressing antisense and sense RNA molecules containing HIV-1 packaging signal and Rev response element(s). Antisense Res. Dev. 4, 19-26.
25. Veres G., Junker U., Baker J., Barske C., Kalfoglou C., Ilves H., Escaich S., Kaneshima H. & Bohnlein E. (1998) Comparative analyses of intracellularly expressed antisense RNAs as inhibitors of human immunodeficiency virus type 1 replication. J. Virol. 72, 1894-1901.
26. Ramezani A., Ding S. F. & Joshi S. (1997) Inhibition of HIV-1 replication by retroviral vectors expressing monomeric and multimeric hammerhead ribozymes. Gene Ther. 4, 861-867.
27. Ramezani A., Joshi S. (1996) Comparative analysis of five highly conserved target sites within the HIV-1 RNA for their susceptibility to hammerhead ribozyme-mediated cleavage in vitro and in vivo. Antisense Nucleic Acid Drug Dev. 6, 229-235.
28. Ramezani A., Ma X. Z., Nazari R. & Joshi S. (2002) Development and testing of retroviral vectors expressing multimeric hammerhead ribozymes targeted against all major clades of HIV-1. Front. Biosci. 7, a29-36.
29. Sullenger B. A., Gallardo H. F., Ungers G. E. & Gilboa E. (1990) Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell 63, 601-608.
155

30. Joshi S., Van Brunschot A., Asad S., van der Elst I., Read S. E. & Bernstein A. (1991) Inhibition of human immunodeficiency virus type 1 multiplication by antisense and sense RNA expression. J. Virol. 65, 5524-5530.
31. Bevec D., Volc-Platzer B., Zimmermann K., Dobrovnik M., Hauber J., Veres G. & Bohnlein E. (1994) Constitutive expression of chimeric neo-Rev response element transcripts suppresses HIV-1 replication in human CD4+ T lymphocytes. Hum. Gene Ther. 5, 193-201.
32. Sun L. Q., Pyati J., Smythe J., Wang L., Macpherson J., Gerlach W. & Symonds G. (1995) Resistance to human immunodeficiency virus type 1 infection conferred by transduction of human peripheral blood lymphocytes with ribozyme, antisense, or polymeric trans-activation response element constructs. Proc. Natl. Acad. Sci. U. S. A. 92, 7272-7276.
33. Bohjanen P. R., Colvin R. A., Puttaraju M., Been M. D. & Garcia-Blanco M. A. (1996) A small circular TAR RNA decoy specifically inhibits Tat-activated HIV-1 transcription. Nucleic Acids Res. 24, 3733-3738.
34. Jacque J. M., Triques K. & Stevenson M. (2002) Modulation of HIV-1 replication by RNA interference. Nature 418, 435-438.
35. Lee N. S., Dohjima T., Bauer G., Li H., Li M. J., Ehsani A., Salvaterra P. & Rossi J. (2002) Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat. Biotechnol. 20, 500-505.
36. Martinez M. A., Gutierrez A., Armand-Ugon M., Blanco J., Parera M., Gomez J., Clotet B. & Este J. A. (2002) Suppression of chemokine receptor expression by RNA interference allows for inhibition of HIV-1 replication. AIDS 16, 2385-2390.
37. Qin X. F., An D. S., Chen I. S. & Baltimore D. (2003) Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc. Natl. Acad. Sci. U. S. A. 100, 183-188.
38. Dragic T. (2001) An overview of the determinants of CCR5 and CXCR4 co-receptor function. J. Gen. Virol. 82, 1807-1814.
39. Berger E. A. (1997) HIV entry and tropism: the chemokine receptor connection. AIDS 11 Suppl A, S3-16.
40. Feng Y., Broder C. C., Kennedy P. E. & Berger E. A. (1996) HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272, 872-877.
41. Bleul C. C., Farzan M., Choe H., Parolin C., Clark-Lewis I., Sodroski J. & Springer T. A. (1996) The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382, 829-833.
42. Oberlin E., Amara A., Bachelerie F., Bessia C., Virelizier J. L., Arenzana-Seisdedos F., Schwartz O., Heard J. M., Clark-Lewis I., Legler D. F., Loetscher M., Baggiolini M. & Moser B. (1996) The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 382, 833-835.
43. Zaitseva M., Peden K. & Golding H. (2003) HIV coreceptors: role of structure, posttranslational modifications, and internalization in viral-cell fusion and as targets for entry inhibitors. Biochim. Biophys. Acta 1614, 51-61.
44. Baggiolini M., Dewald B. & Moser B. (1997) Human chemokines: an update. Annu. Rev. Immunol. 15, 675-705.
156

45. Moore J. P., Trkola A. & Dragic T. (1997) Co-receptors for HIV-1 entry. Curr. Opin. Immunol. 9, 551-562.
46. Cocchi F., DeVico A. L., Garzino-Demo A., Arya S. K., Gallo R. C. & Lusso P. (1995) Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 270, 1811-1815.
47. Samson M., Labbe O., Mollereau C., Vassart G. & Parmentier M. (1996a) Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry 35, 3362-3367.
48. Combadiere C., Ahuja S. K., Tiffany H. L. & Murphy P. M. (1996) Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1(alpha), MIP-1(beta), and RANTES. J. Leukoc. Biol. 60, 147-152.
49. Raport C. J., Gosling J., Schweickart V. L., Gray P. W. & Charo I. F. (1996) Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP-1beta, and MIP-1alpha. J. Biol. Chem. 271, 17161-17166.
50. Alkhatib G., Combadiere C., Broder C. C., Feng Y., Kennedy P. E., Murphy P. M. & Berger E. A. (1996) CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272, 1955-1958.
51. Choe H., Farzan M., Sun Y., Sullivan N., Rollins B., Ponath P. D., Wu L., Mackay C. R., LaRosa G., Newman W., Gerard N., Gerard C. & Sodroski J. (1996) The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85, 1135-1148.
52. Dean M., Carrington M., Winkler C., Huttley G. A., Smith M. W., Allikmets R., Goedert J. J., Buchbinder S. P., Vittinghoff E., Gomperts E., Donfield S., Vlahov D., Kaslow R., Saah A., Rinaldo C., Detels R. & O'Brien S. J. (1996) Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273, 1856-1862.
53. Doranz B. J., Rucker J., Yi Y., Smyth R. J., Samson M., Peiper S. C., Parmentier M., Collman R. G. & Doms R. W. (1996) A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85, 1149-1158.
54. Dragic T., Litwin V., Allaway G. P., Martin S. R., Huang Y., Nagashima K. A., Cayanan C., Maddon P. J., Koup R. A., Moore J. P. & Paxton W. A. (1996) HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381, 667-673.
55. Farzan M., Choe H., Martin K., Marcon L., Hofmann W., Karlsson G., Sun Y., Barrett P., Marchand N., Sullivan N., Gerard N., Gerard C. & Sodroski J. (1997) Two orphan seven-transmembrane segment receptors which are expressed in CD4-positive cells support simian immunodeficiency virus infection. J. Exp. Med. 186, 405-411.
56. O'Brien T. R., Winkler C., Dean M., Nelson J. A., Carrington M., Michael N. L. & White G. C.,2nd. (1997) HIV-1 infection in a man homozygous for CCR5 delta 32. Lancet 349, 1219.
57. Liu R., Paxton W. A., Choe S., Ceradini D., Martin S. R., Horuk R., MacDonald M. E., Stuhlmann H., Koup R. A. & Landau N. R. (1996) Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86, 367-377.
157

58. Roos M. T., Lange J. M., de Goede R. E., Coutinho R. A., Schellekens P. T., Miedema F. & Tersmette M. (1992) Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. J. Infect. Dis. 165, 427-432.
59. Schuitemaker H., Koot M., Kootstra N. A., Dercksen M. W., de Goede R. E., van Steenwijk R. P., Lange J. M., Schattenkerk J. K., Miedema F. & Tersmette M. (1992) Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 66, 1354-1360.
60. Connor R. I., Mohri H., Cao Y. & Ho D. D. (1993) Increased viral burden and cytopathicity correlate temporally with CD4+ T-lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected individuals. J. Virol. 67, 1772-1777.
61. Zhu T., Mo H., Wang N., Nam D. S., Cao Y., Koup R. A. & Ho D. D. (1993) Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261, 1179-1181.
62. Tersmette M., Lange J. M., de Goede R. E., de Wolf F., Eeftink-Schattenkerk J. K., Schellekens P. T., Coutinho R. A., Huisman J. G., Goudsmit J. & Miedema F. (1989) Association between biological properties of human immunodeficiency virus variants and risk for AIDS and AIDS mortality. Lancet 1, 983-985.
63. Berger E. A., Murphy P. M. & Farber J. M. (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17, 657-700.
64. Collman R., Balliet J. W., Gregory S. A., Friedman H., Kolson D. L., Nathanson N. & Srinivasan A. (1992) An infectious molecular clone of an unusual macrophage-tropic and highly cytopathic strain of human immunodeficiency virus type 1. J. Virol. 66, 7517-7521. .
65. Lederman M. M., Penn-Nicholson A., Cho M. & Mosier D. (2006a) Biology of CCR5 and its role in HIV infection and treatment. JAMA 296, 815-826.
66. Doms R.W. & Moore J.P. (1997) HIV-1 Co-receptor Use: A Molecular Window into Viral Tropism. Http://www.Hiv.Lanl.gov/content/hiv-dp/COMPENDIUM/1997/partIII/ doms.Pdf
67. Fouchier R. A., Groenink M., Kootstra N. A., Tersmette M., Huisman H. G., Miedema F. & Schuitemaker H. (1992) Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J. Virol. 66, 3183-3187.
68. De Jong J., Simon F., Van der Groen G., Baan E., Saragosti S., Brun-Vezinet F. & Goudsmit J. (1996) V3 loop sequence analysis of seven HIV type 1 group O isolates phenotyped in peripheral blood mononuclear cells and MT-2 cells. AIDS Res. Hum. Retroviruses 12, 1503-1507.
69. Brelot A., Heveker N., Pleskoff O., Sol N. & Alizon M. (1997) Role of the first and third extracellular domains of CXCR-4 in human immunodeficiency virus coreceptor activity. J. Virol. 71, 4744-4751.
70. Lu Z., Berson J. F., Chen Y., Turner J. D., Zhang T., Sharron M., Jenks M. H., Wang Z., Kim J., Rucker J., Hoxie J. A., Peiper S. C. & Doms R. W. (1997) Evolution of HIV-1 coreceptor usage through interactions with distinct CCR5 and CXCR4 domains. Proc. Natl. Acad. Sci. U. S. A. 94, 6426-6431.
158

71. Yi Y., Shaheen F. & Collman R. G. (2005) Preferential use of CXCR4 by R5X4 human immunodeficiency virus type 1 isolates for infection of primary lymphocytes. J. Virol. 79, 1480-1486.
72. Davis C. W., Doms R. W. (2004) HIV transmission: closing all the doors. J. Exp. Med. 199, 1037-1040.
73. Unutmaz D., KewalRamani V. N. & Littman D. R. (1998) G protein-coupled receptors in HIV and SIV entry: new perspectives on lentivirus-host interactions and on the utility of animal models. Semin. Immunol. 10, 225-236.
74. Rucker J., Edinger A. L., Sharron M., Samson M., Lee B., Berson J. F., Yi Y., Margulies B., Collman R. G., Doranz B. J., Parmentier M. & Doms R. W. (1997) Utilization of chemokine receptors, orphan receptors, and herpesvirus-encoded receptors by diverse human and simian immunodeficiency viruses. J. Virol. 71, 8999-9007.
75. Horuk R., Hesselgesser J., Zhou Y., Faulds D., Halks-Miller M., Harvey S., Taub D., Samson M., Parmentier M., Rucker J., Doranz B. J. & Doms R. W. (1998) The CC chemokine I-309 inhibits CCR8-dependent infection by diverse HIV-1 strains. J. Biol. Chem. 273, 386-391.
76. Jinno A., Shimizu N., Soda Y., Haraguchi Y., Kitamura T. & Hoshino H. (1998) Identification of the chemokine receptor TER1/CCR8 expressed in brain-derived cells and T cells as a new coreceptor for HIV-1 infection. Biochem. Biophys. Res. Commun. 243, 497-502.
77. Choe H., Farzan M., Konkel M., Martin K., Sun Y., Marcon L., Cayabyab M., Berman M., Dorf M. E., Gerard N., Gerard C. & Sodroski J. (1998) The orphan seven-transmembrane receptor apj supports the entry of primary T-cell-line-tropic and dualtropic human immunodeficiency virus type 1. J. Virol. 72, 6113-6118.
78. Combadiere C., Salzwedel K., Smith E. D., Tiffany H. L., Berger E. A. & Murphy P. M. (1998) Identification of CX3CR1. A chemotactic receptor for the human CX3C chemokine fractalkine and a fusion coreceptor for HIV-1. J. Biol. Chem. 273, 23799-23804.
79. Deng H. K., Unutmaz D., KewalRamani V. N. & Littman D. R. (1997) Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 388, 296-300.
80. Edinger A. L., Hoffman T. L., Sharron M., Lee B., Yi Y., Choe W., Kolson D. L., Mitrovic B., Zhou Y., Faulds D., Collman R. G., Hesselgesser J., Horuk R. & Doms R. W. (1998) An orphan seven-transmembrane domain receptor expressed widely in the brain functions as a coreceptor for human immunodeficiency virus type 1 and simian immunodeficiency virus. J. Virol. 72, 7934-7940.
81. Pleskoff O., Treboute C., Brelot A., Heveker N., Seman M. & Alizon M. (1997) Identification of a chemokine receptor encoded by human cytomegalovirus as a cofactor for HIV-1 entry. Science 276, 1874-1878.
82. Samson M., Edinger A. L., Stordeur P., Rucker J., Verhasselt V., Sharron M., Govaerts C., Mollereau C., Vassart G., Doms R. W. & Parmentier M. (1998) ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur. J. Immunol. 28, 1689-1700.
159

83. Liao F., Alkhatib G., Peden K. W., Sharma G., Berger E. A. & Farber J. M. (1997) STRL33, A novel chemokine receptor-like protein, functions as a fusion cofactor for both macrophage-tropic and T cell line-tropic HIV-1. J. Exp. Med. 185, 2015-2023.
84. Kozak S. L., Platt E. J., Madani N., Ferro F. E.,Jr, Peden K. & Kabat D. (1997) CD4, CXCR-4, and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J. Virol. 71, 873-882.
85. Collman R. G. (2003) HIV-1 Env-chemokine receptor interactions in primary human macrophages: entry and beyond. Res. Initiat. Treat. Action 8, 6-9.
86. Wyatt R., Kwong P. D., Desjardins E., Sweet R. W., Robinson J., Hendrickson W. A. & Sodroski J. G. (1998) The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393, 705-711.
87. Staropoli I., Chanel C., Girard M. & Altmeyer R. (2000) Processing, stability, and receptor binding properties of oligomeric envelope glycoprotein from a primary HIV-1 isolate. J. Biol. Chem. 275, 35137-35145.
88. Bour S., Geleziunas R. & Wainberg M. A. (1995) The human immunodeficiency virus type 1 (HIV-1) CD4 receptor and its central role in promotion of HIV-1 infection. Microbiol. Rev. 59, 63-93.
89. Kwong P. D., Wyatt R., Robinson J., Sweet R. W., Sodroski J. & Hendrickson W. A. (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648-659.
90. Chan D. C., Fass D., Berger J. M. & Kim P. S. (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell 89, 263-273.
91. van't Wout A. B., Kootstra N. A., Mulder-Kampinga G. A., Albrecht-van Lent N., Scherpbier H. J., Veenstra J., Boer K., Coutinho R. A., Miedema F. & Schuitemaker H. (1994) Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J. Clin. Invest. 94, 2060-2067.
92. Wu L., Paxton W. A., Kassam N., Ruffing N., Rottman J. B., Sullivan N., Choe H., Sodroski J., Newman W., Koup R. A. & Mackay C. R. (1997a) CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J. Exp. Med. 185, 1681-1691.
93. Lederman M. M., Offord R. E. & Hartley O. (2006b) Microbicides and other topical strategies to prevent vaginal transmission of HIV. Nat. Rev. Immunol. 6, 371-382.
94. Ayehunie S., Garcia-Zepeda E. A., Hoxie J. A., Horuk R., Kupper T. S., Luster A. D. & Ruprecht R. M. (1997) Human immunodeficiency virus-1 entry into purified blood dendritic cells through CC and CXC chemokine coreceptors. Blood 90, 1379-1386.
95. Rubbert A., Combadiere C., Ostrowski M., Arthos J., Dybul M., Machado E., Cohn M. A., Hoxie J. A., Murphy P. M., Fauci A. S. & Weissman D. (1998) Dendritic cells express multiple chemokine receptors used as coreceptors for HIV entry. J. Immunol. 160, 3933-3941.
96. Zhang L., He T., Talal A., Wang G., Frankel S. S. & Ho D. D. (1998) In vivo distribution of the human immunodeficiency virus/simian immunodeficiency virus coreceptors: CXCR4, CCR3, and CCR5. J. Virol. 72, 5035-5045.
160

97. Zaitseva M., Blauvelt A., Lee S., Lapham C. K., Klaus-Kovtun V., Mostowski H., Manischewitz J. & Golding H. (1997) Expression and function of CCR5 and CXCR4 on human Langerhans cells and macrophages: implications for HIV primary infection. Nat. Med. 3, 1369-1375.
98. Patterson B. K., Landay A., Andersson J., Brown C., Behbahani H., Jiyamapa D., Burki Z., Stanislawski D., Czerniewski M. A. & Garcia P. (1998) Repertoire of chemokine receptor expression in the female genital tract: implications for human immunodeficiency virus transmission. Am. J. Pathol. 153, 481-490.
99. Samson M., Libert F., Doranz B. J., Rucker J., Liesnard C., Farber C. M., Saragosti S., Lapoumeroulie C., Cognaux J., Forceille C., Muyldermans G., Verhofstede C., Burtonboy G., Georges M., Imai T., Rana S., Yi Y., Smyth R. J., Collman R. G., Doms R. W., Vassart G. & Parmentier M. (1996b) Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382, 722-725.
100. Ruibal-Ares B. H., Belmonte L., Bare P. C., Parodi C. M., Massud I. & de Bracco M. M. (2004) HIV-1 infection and chemokine receptor modulation. Curr. HIV. Res. 2, 39-50.
101. Regoes R. R., Bonhoeffer S. (2005) The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol. 13, 269-277.
102. Delwart E. L., Mullins J. I., Gupta P., Learn G. H.,Jr, Holodniy M., Katzenstein D., Walker B. D. & Singh M. K. (1998) Human immunodeficiency virus type 1 populations in blood and semen. J. Virol. 72, 617-623.
103. Agace W. W., Amara A., Roberts A. I., Pablos J. L., Thelen S., Uguccioni M., Li X. Y., Marsal J., Arenzana-Seisdedos F., Delaunay T., Ebert E. C., Moser B. & Parker C. M. (2000) Constitutive expression of stromal derived factor-1 by mucosal epithelia and its role in HIV transmission and propagation. Curr. Biol. 10, 325-328.
104. Meng G., Wei X., Wu X., Sellers M. T., Decker J. M., Moldoveanu Z., Orenstein J. M., Graham M. F., Kappes J. C., Mestecky J., Shaw G. M. & Smith P. D. (2002) Primary intestinal epithelial cells selectively transfer R5 HIV-1 to CCR5+ cells. Nat. Med. 8, 150-156.
105. Granelli-Piperno A., Delgado E., Finkel V., Paxton W. & Steinman R. M. (1998) Immature dendritic cells selectively replicate macrophagetropic (M-tropic) human immunodeficiency virus type 1, while mature cells efficiently transmit both M- and T-tropic virus to T cells. J. Virol. 72, 2733-2737.
106. Cornelissen M., Mulder-Kampinga G., Veenstra J., Zorgdrager F., Kuiken C., Hartman S., Dekker J., van der Hoek L., Sol C. & Coutinho R. (1995) Syncytium-inducing (SI) phenotype suppression at seroconversion after intramuscular inoculation of a non-syncytium-inducing/SI phenotypically mixed human immunodeficiency virus population. J. Virol. 69, 1810-1818.
107. Pratt R. D., Shapiro J. F., McKinney N., Kwok S. & Spector S. A. (1995) Virologic characterization of primary human immunodeficiency virus type 1 infection in a health care worker following needlestick injury. J. Infect. Dis. 172, 851-854.
108. Wilkinson D. A., Operskalski E. A., Busch M. P., Mosley J. W. & Koup R. A. (1998) A 32-bp deletion within the CCR5 locus protects against transmission of parenterally acquired human immunodeficiency virus but does not affect progression to AIDS-defining illness. J. Infect. Dis. 178, 1163-1166.
161

109. Veazey R., Lackner A. (2003) The mucosal immune system and HIV-1 infection. AIDS. Rev. 5, 245-252.
110. Holmes E. C. (2001) On the origin and evolution of the human immunodeficiency virus (HIV). Biol. Rev. Camb. Philos. Soc. 76, 239-254.
111. Mansky L. M., Temin H. M. (1995) Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69, 5087-5094.
112. Perelson A. S. (2002) Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2, 28-36.
113. Pastore C., Ramos A. & Mosier D. E. (2004) Intrinsic obstacles to human immunodeficiency virus type 1 coreceptor switching. J. Virol. 78, 7565-7574.
114. Seder R. A., Ahmed R. (2003) Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat. Immunol. 4, 835-842.
115. Zhang Z., Schuler T., Zupancic M., Wietgrefe S., Staskus K. A., Reimann K. A., Reinhart T. A., Rogan M., Cavert W., Miller C. J., Veazey R. S., Notermans D., Little S., Danner S. A., Richman D. D., Havlir D., Wong J., Jordan H. L., Schacker T. W., Racz P., Tenner-Racz K., Letvin N. L., Wolinsky S. & Haase A. T. (1999) Sexual transmission and propagation of SIV and HIV in resting and activated CD4+ T cells. Science 286, 1353-1357.
116. Lee B., Sharron M., Montaner L. J., Weissman D. & Doms R. W. (1999) Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc. Natl. Acad. Sci. U. S. A. 96, 5215-5220.
117. Bleul C. C., Wu L., Hoxie J. A., Springer T. A. & Mackay C. R. (1997) The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 94, 1925-1930.
118. Benito J. M., Zabay J. M., Gil J., Bermejo M., Escudero A., Sanchez E. & Fernandez-Cruz E. (1997) Quantitative alterations of the functionally distinct subsets of CD4 and CD8 T lymphocytes in asymptomatic HIV infection: changes in the expression of CD45RO, CD45RA, CD11b, CD38, HLA-DR, and CD25 antigens. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 14, 128-135.
119. Hazenberg M. D., Stuart J. W., Otto S. A., Borleffs J. C., Boucher C. A., de Boer R. J., Miedema F. & Hamann D. (2000) T-cell division in human immunodeficiency virus (HIV)-1 infection is mainly due to immune activation: a longitudinal analysis in patients before and during highly active antiretroviral therapy (HAART). Blood 95, 249-255.
120. Ostrowski M. A., Justement S. J., Catanzaro A., Hallahan C. A., Ehler L. A., Mizell S. B., Kumar P. N., Mican J. A., Chun T. W. & Fauci A. S. (1998) Expression of chemokine receptors CXCR4 and CCR5 in HIV-1-infected and uninfected individuals. J. Immunol. 161, 3195-3201.
121. Rodrigo A. G. (1997) Dynamics of syncytium-inducing and non-syncytium-inducing type 1 human immunodeficiency viruses during primary infection. AIDS Res. Hum. Retroviruses 13, 1447-1451.
122. Michael N. L., Louie L. G. & Sheppard H. W. (1997) CCR5-delta 32 gene deletion in HIV-1 infected patients. Lancet 350, 741-742.
162

123. Zimmerman P. A., Buckler-White A., Alkhatib G., Spalding T., Kubofcik J., Combadiere C., Weissman D., Cohen O., Rubbert A., Lam G., Vaccarezza M., Kennedy P. E., Kumaraswami V., Giorgi J. V., Detels R., Hunter J., Chopek M., Berger E. A., Fauci A. S., Nutman T. B. & Murphy P. M. (1997) Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol. Med. 3, 23-36.
124. Ioannidis J. P., Rosenberg P. S., Goedert J. J., Ashton L. J., Benfield T. L., Buchbinder S. P., Coutinho R. A., Eugen-Olsen J., Gallart T., Katzenstein T. L., Kostrikis L. G., Kuipers H., Louie L. G., Mallal S. A., Margolick J. B., Martinez O. P., Meyer L., Michael N. L., Operskalski E., Pantaleo G., Rizzardi G. P., Schuitemaker H., Sheppard H. W., Stewart G. J., Theodorou I. D., Ullum H., Vicenzi E., Vlahov D., Wilkinson D., Workman C., Zagury J. F., O'Brien T. R. & International Meta-Analysis of HIV Host Genetics. (2001) Effects of CCR5-Delta32, CCR2-64I, and SDF-1 3'A alleles on HIV-1 disease progression: An international meta-analysis of individual-patient data. Ann. Intern. Med. 135, 782-795.
125. Benkirane M., Jin D. Y., Chun R. F., Koup R. A. & Jeang K. T. (1997) Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32. J. Biol. Chem. 272, 30603-30606.
126. Balotta C., Bagnarelli P., Violin M., Ridolfo A. L., Zhou D., Berlusconi A., Corvasce S., Corbellino M., Clementi M., Clerici M., Moroni M. & Galli M. (1997) Homozygous delta 32 deletion of the CCR-5 chemokine receptor gene in an HIV-1-infected patient. AIDS 11, F67-71.
127. Sheppard H. W., Celum C., Michael N. L., O'Brien S., Dean M., Carrington M., Dondero D. & Buchbinder S. P. (2002) HIV-1 infection in individuals with the CCR5-Delta32/Delta32 genotype: acquisition of syncytium-inducing virus at seroconversion. J. Acquir. Immune Defic. Syndr. 29, 307-313.
128. Zhang L., Carruthers C. D., He T., Huang Y., Cao Y., Wang G., Hahn B. & Ho D. D. (1997) HIV type 1 subtypes, coreceptor usage, and CCR5 polymorphism. AIDS Res. Hum. Retroviruses 13, 1357-1366.
129. Muster T., Steindl F., Purtscher M., Trkola A., Klima A., Himmler G., Ruker F. & Katinger H. (1993) A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J. Virol. 67, 6642-6647.
130. Roben P., Moore J. P., Thali M., Sodroski J., Barbas C. F.,3rd & Burton D. R. (1994) Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J. Virol. 68, 4821-4828.
131. Zwick M. B., Labrijn A. F., Wang M., Spenlehauer C., Saphire E. O., Binley J. M., Moore J. P., Stiegler G., Katinger H., Burton D. R. & Parren P. W. (2001) Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 75, 10892-10905.
132. Trkola A., Kuster H., Rusert P., Joos B., Fischer M., Leemann C., Manrique A., Huber M., Rehr M., Oxenius A., Weber R., Stiegler G., Vcelar B., Katinger H., Aceto L. & Gunthard H. F. (2005) Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat. Med. 11, 615-622.
133. Moore P. L., Cilliers T. & Morris L. (2004) Predicted genotypic resistance to the novel entry inhibitor, BMS-378806, among HIV-1 isolates of subtypes A to G. AIDS 18, 2327-2330.
163

134. Matthews T., Salgo M., Greenberg M., Chung J., DeMasi R. & Bolognesi D. (2004) Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat. Rev. Drug Discov. 3, 215-225.
135. Husson R. N., Chung Y., Mordenti J., Butler K. M., Chen S., Duliege A. M., Brouwers P., Jarosinski P., Mueller B. U. & Ammann A. (1992) Phase I study of continuous-infusion soluble CD4 as a single agent and in combination with oral dideoxyinosine therapy in children with symptomatic human immunodeficiency virus infection. J. Pediatr. 121, 627-633.
136. Schacker T., Coombs R. W., Collier A. C., Zeh J. E., Fox I., Alam J., Nelson K., Eggert E. & Corey L. (1994) The effects of high-dose recombinant soluble CD4 on human immunodeficiency virus type 1 viremia. J. Infect. Dis. 169, 37-40.
137. Jacobson J. M., Israel R. J., Lowy I., Ostrow N. A., Vassilatos L. S., Barish M., Tran D. N., Sullivan B. M., Ketas T. J., O'Neill T. J., Nagashima K. A., Huang W., Petropoulos C. J., Moore J. P., Maddon P. J. & Olson W. C. (2004) Treatment of advanced human immunodeficiency virus type 1 disease with the viral entry inhibitor PRO 542. Antimicrob. Agents Chemother. 48, 423-429.
138. Zou Y. R., Kottmann A. H., Kuroda M., Taniuchi I. & Littman D. R. (1998) Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393, 595-599.
139. Plotkin J., Prockop S. E., Lepique A. & Petrie H. T. (2003) Critical role for CXCR4 signaling in progenitor localization and T cell differentiation in the postnatal thymus. J. Immunol. 171, 4521-4527.
140. O'Brien S. J., Moore J. P. (2000) The effect of genetic variation in chemokines and their receptors on HIV transmission and progression to AIDS. Immunol. Rev. 177, 99-111.
141. Shea A., Sarr D. A., Jones N., Penning L., Eisen G., Gueye-Ndiaye A., Mboup S., Kanki P. & Cao H. (2004) CCR5 receptor expression is down-regulated in HIV type 2 infection: implication for viral control and protection. AIDS Res. Hum. Retroviruses 20, 630-635.
142. Cocchi F., DeVico A. L., Garzino-Demo A., Cara A., Gallo R. C. & Lusso P. (1996) The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat. Med. 2, 1244-1247.
143. Oravecz T., Pall M. & Norcross M. A. (1996) Beta-chemokine inhibition of monocytotropic HIV-1 infection. Interference with a postbinding fusion step. J. Immunol. 157, 1329-1332.
144. Kelly M. D., Naif H. M., Adams S. L., Cunningham A. L. & Lloyd A. R. (1998) Dichotomous effects of beta-chemokines on HIV replication in monocytes and monocyte-derived macrophages. J. Immunol. 160, 3091-3095.
145. Wu L., LaRosa G., Kassam N., Gordon C. J., Heath H., Ruffing N., Chen H., Humblias J., Samson M., Parmentier M., Moore J. P. & Mackay C. R. (1997b) Interaction of chemokine receptor CCR5 with its ligands: multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J. Exp. Med. 186, 1373-1381.
146. Roschke V., Moore P. (2004) Characterization of novel human monoclonal antibodies that specifically antagonize CCR5 and block HIV-1 entry. 44 Th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). Abstract #2871, [Http://www.Hgsi.com/news/press/04-11-02_CCR5_ICAAC.Htm]
164

147. Castagna A., Biswas P., Beretta A. & Lazzarin A. (2005) The appealing story of HIV entry inhibitors : from discovery of biological mechanisms to drug development. Drugs 65, 879-904.
148. Bai X., Chen J. D., Yang A. G., Torti F. & Chen S. Y. (1998) Genetic co-inactivation of macrophage- and T-tropic HIV-1 chemokine coreceptors CCR-5 and CXCR-4 by intrakines. Gene Ther. 5, 984-994.
149. Schroers R., Davis C. M., Wagner H. J. & Chen S. Y. (2002) Lentiviral transduction of human T-lymphocytes with a RANTES intrakine inhibits human immunodeficiency virus type 1 infection. Gene Ther. 9, 889-897.
150. Luis Abad J., Gonzalez M. A., del Real G., Mira E., Manes S., Serrano F. & Bernad A. (2003) Novel interfering bifunctional molecules against the CCR5 coreceptor are efficient inhibitors of HIV-1 infection. Mol. Ther. 8, 475-484.
151. Steinberger P., Andris-Widhopf J., Buhler B., Torbett B. E. & Barbas C. F.,3rd. (2000) Functional deletion of the CCR5 receptor by intracellular immunization produces cells that are refractory to CCR5-dependent HIV-1 infection and cell fusion. Proc. Natl. Acad. Sci. U. S. A. 97, 805-810.
152. Li W., Yu M., Bai L., Bu D. & Xu X. (2006) Downregulation of CCR5 Expression on Cells by Recombinant Adenovirus Containing Antisense CCR5, a Possible Measure to Prevent HIV-1 From Entering Target Cells. J. Acquir. Immune Defic. Syndr. 43, 516-522.
153. Qureshi A., Zheng R., Parlett T., Shi X., Balaraman P., Cheloufi S., Murphy B., Guntermann C. & Eagles P. (2006) Gene silencing of HIV chemokine receptors using ribozymes and single-stranded antisense RNA. Biochem. J. 394, 511-518.
154. Rossi J. J. (1999) The application of ribozymes to HIV infection. Curr. Opin. Mol. Ther. 1, 316-322.
155. Bai J., Gorantla S., Banda N., Cagnon L., Rossi J. & Akkina R. (2000) Characterization of anti-CCR5 ribozyme-transduced CD34+ hematopoietic progenitor cells in vitro and in a SCID-hu mouse model in vivo. Mol. Ther. 1, 244-254.
156. Feng Y., Leavitt M., Tritz R., Duarte E., Kang D., Mamounas M., Gilles P., Wong-Staal F., Kennedy S., Merson J., Yu M. & Barber J. R. (2000) Inhibition of CCR5-dependent HIV-1 infection by hairpin ribozyme gene therapy against CC-chemokine receptor 5. Virology 276, 271-278.
157. Bai J., Rossi J. & Akkina R. (2001) Multivalent anti-CCR ribozymes for stem cell-based HIV type 1 gene therapy. AIDS Res. Hum. Retroviruses 17, 385-399.
158. Novina C. D., Murray M. F., Dykxhoorn D. M., Beresford P. J., Riess J., Lee S. K., Collman R. G., Lieberman J., Shankar P. & Sharp P. A. (2002) siRNA-directed inhibition of HIV-1 infection. Nat. Med. 8, 681-686.
159. Anderson J., Banerjea A. & Akkina R. (2003a) Bispecific short hairpin siRNA constructs targeted to CD4, CXCR4, and CCR5 confer HIV-1 resistance. Oligonucleotides 13, 303-312.
160. Butticaz C., Ciuffi A., Munoz M., Thomas J., Bridge A., Pebernard S., Iggo R., Meylan P. & Telenti A. (2003) Protection from HIV-1 infection of primary CD4 T cells by CCR5 silencing is effective for the full spectrum of CCR5 expression. Antivir Ther. 8, 373-377.
165

161. Cordelier P., Morse B. & Strayer D. S. (2003) Targeting CCR5 with siRNAs: using recombinant SV40-derived vectors to protect macrophages and microglia from R5-tropic HIV. Oligonucleotides 13, 281-294.
162. Pomerantz R. J. (2004) RNA interference:a potential novel therapeutic combating HIV-1 in the central nervous system. Arch. Immunol. Ther. Exp. (Warsz) 52, 401-409.
163. Anderson J., Akkina R. (2005) HIV-1 resistance conferred by siRNA cosuppression of CXCR4 and CCR5 coreceptors by a bispecific lentiviral vector. AIDS. Res. Ther. 2, 1.
164. Mani M., Kandavelou K., Dy F. J., Durai S. & Chandrasegaran S. (2005) Design, engineering, and characterization of zinc finger nucleases. Biochem. Biophys. Res. Commun. 335, 447-457.
165. Dragic T., Litwin V., Allaway G. P., Martin S. R., Huang Y., Nagashima K. A., Cayanan C., Maddon P. J., Koup R. A., Moore J. P. & Paxton W. A. (1996) HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381, 667-673.
166. Yang A. G., Bai X., Huang X. F., Yao C. & Chen S. (1997) Phenotypic knockout of HIV type 1 chemokine coreceptor CCR-5 by intrakines as potential therapeutic approach for HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 94, 11567-11572.
167. Schmidtmayerova H., Sherry B. & Bukrinsky M. (1996) Chemokines and HIV replication. Nature 382, 767.
168. Coffey M. J., Woffendin C., Phare S. M., Strieter R. M. & Markovitz D. M. (1997) RANTES inhibits HIV-1 replication in human peripheral blood monocytes and alveolar macrophages. Am. J. Physiol. 272, L1025-9.
169. Simmons G., Clapham P. R., Picard L., Offord R. E., Rosenkilde M. M., Schwartz T. W., Buser R., Wells T. N. & Proudfoot A. E. (1997) Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 276, 276-279.
170. Kinter A., Catanzaro A., Monaco J., Ruiz M., Justement J., Moir S., Arthos J., Oliva A., Ehler L., Mizell S., Jackson R., Ostrowski M., Hoxie J., Offord R. & Fauci A. S. (1998) CC-chemokines enhance the replication of T-tropic strains of HIV-1 in CD4(+) T cells: role of signal transduction. Proc. Natl. Acad. Sci. U. S. A. 95, 11880-11885.
171. Tedla N., Palladinetti P., Kelly M., Kumar R. K., DiGirolamo N., Chattophadhay U., Cooke B., Truskett P., Dwyer J., Wakefield D. & Lloyd A. (1996) Chemokines and T lymphocyte recruitment to lymph nodes in HIV infection. Am. J. Pathol. 148, 1367-1373.
172. Osborn L., Kunkel S. & Nabel G. J. (1989) Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. U. S. A. 86, 2336-2340.
173. Kontermann R. E. (2004) Intrabodies as therapeutic agents. Methods 34, 163-170.
174. Rondon I. J., Marasco W. A. (1997) Intracellular antibodies (intrabodies) for gene therapy of infectious diseases. Annu. Rev. Microbiol. 51, 257-283.
175. Swan C. H., Buhler B., Steinberger P., Tschan M. P., Barbas C. F.,3rd & Torbett B. E. (2006b) T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther. 13, 1480-1492.
176. Cordelier P., Kulkowsky J. W., Ko C., Matskevitch A. A., McKee H. J., Rossi J. J., Bouhamdan M., Pomerantz R. J., Kari G. & Strayer D. S. (2004) Protecting from R5-tropic
166

HIV: individual and combined effectiveness of a hammerhead ribozyme and a single-chain Fv antibody that targets CCR5. Gene Ther. 11, 1627-1637.
177. Cagnon L., Rossi J. J. (2000) Downregulation of the CCR5 beta-chemokine receptor and inhibition of HIV-1 infection by stable VA1-ribozyme chimeric transcripts. Antisense Nucleic Acid Drug Dev. 10, 251-261.
178. Kim Y. G., Cha J. & Chandrasegaran S. (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. U. S. A. 93, 1156-1160.
179. Alwin S., Gere M. B., Guhl E., Effertz K., Barbas C. F.,3rd, Segal D. J., Weitzman M. D. & Cathomen T. (2005) Custom zinc-finger nucleases for use in human cells. Mol. Ther. 12, 610-617.
180. Valencia-Sanchez M. A., Liu J., Hannon G. J. & Parker R. (2006) Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20, 515-524.
181. Sharp P. A. (2001) RNA interference--2001. Genes Dev. 15, 485-490.
182. Morris K. V., Rossi J. J. (2006) Lentiviral-mediated delivery of siRNAs for antiviral therapy. Gene Ther. 13, 553-558.
183. Anderson J., Banerjea A., Planelles V. & Akkina R. (2003b) Potent suppression of HIV type 1 infection by a short hairpin anti-CXCR4 siRNA. AIDS Res. Hum. Retroviruses 19, 699-706.
184. Jackson A. L., Linsley P. S. (2004) Noise amidst the silence: off-target effects of siRNAs? Trends Genet. 20, 521-524.
185. Hornung V., Guenthner-Biller M., Bourquin C., Ablasser A., Schlee M., Uematsu S., Noronha A., Manoharan M., Akira S., de Fougerolles A., Endres S. & Hartmann G. (2005) Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 11, 263-270.
186. Couzin J. (2004) Molecular biology. RNAi shows cracks in its armor. Science 306, 1124-1125.
187. Doherty E. A., Doudna J. A. (2001) Ribozyme structures and mechanisms. Annu. Rev. Biophys. Biomol. Struct. 30, 457-475.
188. Bagheri S., Kashani-Sabet M. (2004) Ribozymes in the age of molecular therapeutics. Curr. Mol. Med. 4, 489-506.
189. Shiota M., Sano M., Miyagishi M. & Taira K. (2004) Ribozymes: applications to functional analysis and gene discovery. J. Biochem. (Tokyo) 136, 133-147.
190. Li M. J., Kim J., Li S., Zaia J., Yee J. K., Anderson J., Akkina R. & Rossi J. J. (2005) Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 12, 900-909.
191. Nazari R., Joshi S. (2007) Inhibition of HIV-1 entry using oncoretroviral and lentiviral vectors expressing a multimeric hammerhead ribozyme targeted against seven unique sites within the CCR5 mRNA. J. Gen. Virol. (in press)
192. Dean M., Jacobson L. P., McFarlane G., Margolick J. B., Jenkins F. J., Howard O. M., Dong H. F., Goedert J. J., Buchbinder S., Gomperts E., Vlahov D., Oppenheim J. J., O'Brien S. J.
167

& Carrington M. (1999) Reduced risk of AIDS lymphoma in individuals heterozygous for the CCR5-delta32 mutation. Cancer Res. 59, 3561-3564.
193. Kenilworth N. J. (Oct. 27, 2005) Schering-Plough Discontinues Phase II Study of Vicriviroc in Treatment-Naive HIV Patients, Continues Phase II Study in Treatment-Experienced HIV Patients. PRNewswire-FirstCall (http://www.schering-plough.com/ schering_plough/news/release.jsp?releaseID=774673).
194. Glass W. G., Lim J. K., Cholera R., Pletnev A. G., Gao J. L. & Murphy P. M. (2005) Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 202, 1087-1098.
195. Goff S. P. (2001) Intracellular trafficking of retroviral genomes during the early phase of infection: viral exploitation of cellular pathways. J. Gene Med. 3, 517-528.
196. Heinzinger N. K., Bukinsky M. I., Haggerty S. A., Ragland A. M., Kewalramani V., Lee M. A., Gendelman H. E., Ratner L., Stevenson M. & Emerman M. (1994) The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl. Acad. Sci. U. S. A. 91, 7311-7315.
197. Bukrinsky M. I., Sharova N., Dempsey M. P., Stanwick T. L., Bukrinskaya A. G., Haggerty S. & Stevenson M. (1992) Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. U. S. A. 89, 6580-6584.
198. Fassati A., Gorlich D., Harrison I., Zaytseva L. & Mingot J. M. (2003) Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. EMBO J. 22, 3675-3685.
199. Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J. R. & Bushman F. (2005) A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 11, 1287-1289.
200. Ciuffi A., Bushman F. D. (2006) Retroviral DNA integration: HIV and the role of LEDGF/p75. Trends Genet. 22, 388-395.
201. Yung E., Sorin M., Wang E. J., Perumal S., Ott D. & Kalpana G. V. (2004) Specificity of interaction of INI1/hSNF5 with retroviral integrases and its functional significance. J. Virol. 78, 2222-2231.
202. Miller M. D., Farnet C. M. & Bushman F. D. (1997) Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J. Virol. 71, 5382-5390.
203. Lin C. W., Engelman A. (2003) The barrier-to-autointegration factor is a component of functional human immunodeficiency virus type 1 preintegration complexes. J. Virol. 77, 5030-5036.
204. von Schwedler U., Kornbluth R. S. & Trono D. (1994) The nuclear localization signal of the matrix protein of human immunodeficiency virus type 1 allows the establishment of infection in macrophages and quiescent T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 91, 6992-6996.
205. Coffin J. (1996) Retroviridae: the viruses and their replication. 1767-1830.
206. Craigie R. (2001) HIV integrase, a brief overview from chemistry to therapeutics. J. Biol. Chem. 276, 23213-23216.
207. Sarver N., Cantin E. M., Chang P. S., Zaia J. A., Ladne P. A., Stephens D. A. & Rossi J. J. (1990) Ribozymes as potential anti-HIV-1 therapeutic agents. Science 247, 1222-1225.
168

208. Ojwang J. O., Hampel A., Looney D. J., Wong-Staal F. & Rappaport J. (1992) Inhibition of human immunodeficiency virus type 1 expression by a hairpin ribozyme. Proc. Natl. Acad. Sci. U. S. A. 89, 10802-10806.
209. Yu M., Ojwang J., Yamada O., Hampel A., Rapapport J., Looney D. & Wong-Staal F. (1993) A hairpin ribozyme inhibits expression of diverse strains of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 90, 6340-6344.
210. Yamada O., Yu M., Yee J. K., Kraus G., Looney D. & Wong-Staal F. (1994) Intracellular immunization of human T cells with a hairpin ribozyme against human immunodeficiency virus type 1. Gene Ther. 1, 38-45.
211. Leavitt M. C., Yu M., Yamada O., Kraus G., Looney D., Poeschla E. & Wong-Staal F. (1994) Transfer of an anti-HIV-1 ribozyme gene into primary human lymphocytes. Hum. Gene Ther. 5, 1115-1120.
212. Yu M., Leavitt M. C., Maruyama M., Yamada O., Young D., Ho A. D. & Wong-Staal F. (1995a) Intracellular immunization of human fetal cord blood stem/progenitor cells with a ribozyme against human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 92, 699-703.
213. Yu M., Poeschla E., Yamada O., Degrandis P., Leavitt M. C., Heusch M., Yees J. K., Wong-Staal F. & Hampel A. (1995b) In vitro and in vivo characterization of a second functional hairpin ribozyme against HIV-1. Virology 206, 381-386.
214. Li X., Gervaix A., Kang D., Law P., Spector S. A., Ho A. D. & Wong-Staal F. (1998) Gene therapy targeting cord blood-derived CD34+ cells from HIV-exposed infants: preclinical studies. Gene Ther. 5, 233-239.
215. Wong-Staal F., Poeschla E. M. & Looney D. J. (1998) A controlled, Phase 1 clinical trial to evaluate the safety and effects in HIV-1 infected humans of autologous lymphocytes transduced with a ribozyme that cleaves HIV-1 RNA. Hum. Gene Ther. 9, 2407-2425.
216. Westaway S. K., Larson G. P., Li S., Zaia J. A. & Rossi J. J. (1995) A chimeric tRNA(Lys3)-ribozyme inhibits HIV replication following virion assembly. Nucleic Acids Symp. Ser. (33), 194-199.
217. Ding S. F., Noronha J. & Joshi S. (1998) Co-packaging of sense and antisense RNAs: a novel strategy for blocking HIV-1 replication. Nucleic Acids Res. 26, 3270-3278.
218. Peng H., Callison D. E., Li P. & Burrell C. J. (1997) Enhancement or inhibition of HIV-1 replication by intracellular expression of sense or antisense RNA targeted at different intermediates of reverse transcription. AIDS 11, 587-595.
219. Capodici J., Kariko K. & Weissman D. (2002) Inhibition of HIV-1 infection by small interfering RNA-mediated RNA interference. J. Immunol. 169, 5196-5201.
220. Maciejewski J. P., Weichold F. F., Young N. S., Cara A., Zella D., Reitz M. S.,Jr & Gallo R. C. (1995) Intracellular expression of antibody fragments directed against HIV reverse transcriptase prevents HIV infection in vitro. Nat. Med. 1, 667-673.
221. Shaheen F., Duan L., Zhu M., Bagasra O. & Pomerantz R. J. (1996) Targeting human immunodeficiency virus type 1 reverse transcriptase by intracellular expression of single-chain variable fragments to inhibit early stages of the viral life cycle. J. Virol. 70, 3392-3400.
169

222. Wu J., Amandoron E., Li X., Wainberg M. A. & Parniak M. A. (1993) Monoclonal antibody-mediated inhibition of HIV-1 reverse transcriptase polymerase activity. Interaction with a possible deoxynucleoside triphosphate binding domain. J. Biol. Chem. 268, 9980-9985.
223. Strayer D. S., Branco F., Landre J., BouHamdan M., Shaheen F. & Pomerantz R. J. (2002) Combination genetic therapy to inhibit HIV-1. Mol. Ther. 5, 33-41.
224. BouHamdan M., Duan L. X., Pomerantz R. J. & Strayer D. S. (1999) Inhibition of HIV-1 by an anti-integrase single-chain variable fragment (SFv): delivery by SV40 provides durable protection against HIV-1 and does not require selection. Gene Ther. 6, 660-666.
225. Joshi P., Prasad V. R. (2002) Potent inhibition of human immunodeficiency virus type 1 replication by template analog reverse transcriptase inhibitors derived by SELEX (systematic evolution of ligands by exponential enrichment). J. Virol. 76, 6545-6557.
226. Surabhi R. M., Gaynor R. B. (2002) RNA interference directed against viral and cellular targets inhibits human immunodeficiency Virus Type 1 replication. J. Virol. 76, 12963-12973.
227. Levy-Mintz P., Duan L., Zhang H., Hu B., Dornadula G., Zhu M., Kulkosky J., Bizub-Bender D., Skalka A. M. & Pomerantz R. J. (1996) Intracellular expression of single-chain variable fragments to inhibit early stages of the viral life cycle by targeting human immunodeficiency virus type 1 integrase. J. Virol. 70, 8821-8832.
228. Goldstein H., Pettoello-Mantovani M., Anderson C. M., Cordelier P., Pomerantz R. J. & Strayer D. S. (2002) Gene therapy using a simian virus 40-derived vector inhibits the development of in vivo human immunodeficiency virus type 1 infection of severe combined immunodeficiency mice implanted with human fetal thymic and liver tissue. J. Infect. Dis. 185, 1425-1430.
229. Llano M., Delgado S., Vanegas M. & Poeschla E. M. (2004) Lens epithelium-derived growth factor/p75 prevents proteasomal degradation of HIV-1 integrase. J. Biol. Chem. 279, 55570-55577.
230. Levin R., Mhashilkar A. M., Dorfman T., Bukovsky A., Zani C., Bagley J., Hinkula J., Niedrig M., Albert J., Wahren B., Gottlinger H. G. & Marasco W. A. (1997) Inhibition of early and late events of the HIV-1 replication cycle by cytoplasmic Fab intrabodies against the matrix protein, p17. Mol. Med. 3, 96-110.
231. Cousineau B., Smith D., Lawrence-Cavanagh S., Mueller J. E., Yang J., Mills D., Manias D., Dunny G., Lambowitz A. M. & Belfort M. (1998) Retrohoming of a bacterial group II intron: mobility via complete reverse splicing, independent of homologous DNA recombination. Cell 94, 451-462.
232. Lambowitz A. M., Zimmerly S. (2004) Mobile group II introns. Annu. Rev. Genet. 38, 1-35.
233. Singh N. N., Lambowitz A. M. (2001) Interaction of a group II intron ribonucleoprotein endonuclease with its DNA target site investigated by DNA footprinting and modification interference. J. Mol. Biol. 309, 361-386.
234. Cech T. R. (1985) Self-splicing RNA: implications for evolution. Int. Rev. Cytol. 93, 3-22.
235. Cousineau B., Lawrence S., Smith D. & Belfort M. (2000) Retrotransposition of a bacterial group II intron. Nature 404, 1018-1021.
170

236. Ichiyanagi K., Beauregard A., Lawrence S., Smith D., Cousineau B. & Belfort M. (2002) Retrotransposition of the Ll.LtrB group II intron proceeds predominantly via reverse splicing into DNA targets. Mol. Microbiol. 46, 1259-1272.
237. Guo H., Karberg M., Long M., Jones J. P.,3rd, Sullenger B. & Lambowitz A. M. (2000) Group II introns designed to insert into therapeutically relevant DNA target sites in human cells. Science 289, 452-457.
238. Peebles C. L., Zhang M., Perlman P. S. & Franzen J. S. (1995) Catalytically critical nucleotide in domain 5 of a group II intron. Proc. Natl. Acad. Sci. U. S. A. 92, 4422-4426.
239. Bonen L., Vogel J. (2001) The ins and outs of group II introns. Trends Genet. 17, 322-331.
240. Costa M., Michel F. (1999) Tight binding of the 5' exon to domain I of a group II self-splicing intron requires completion of the intron active site. EMBO J. 18, 1025-1037.
241. Matsuura M., Noah J. W. & Lambowitz A. M. (2001) Mechanism of maturase-promoted group II intron splicing. EMBO J. 20, 7259-7270.
242. Kennell J. C., Moran J. V., Perlman P. S., Butow R. A. & Lambowitz A. M. (1993) Reverse transcriptase activity associated with maturase-encoding group II introns in yeast mitochondria. Cell 73, 133-146.
243. Matsuura M., Saldanha R., Ma H., Wank H., Yang J., Mohr G., Cavanagh S., Dunny G. M., Belfort M. & Lambowitz A. M. (1997) A bacterial group II intron encoding reverse transcriptase, maturase, and DNA endonuclease activities: biochemical demonstration of maturase activity and insertion of new genetic information within the intron. Genes Dev. 11, 2910-2924.
244. San Filippo J., Lambowitz A. M. (2002) Characterization of the C-terminal DNA-binding/DNA endonuclease region of a group II intron-encoded protein. J. Mol. Biol. 324, 933-951.
245. Wank H., SanFilippo J., Singh R. N., Matsuura M. & Lambowitz A. M. (1999) A reverse transcriptase/maturase promotes splicing by binding at its own coding segment in a group II intron RNA. Mol. Cell 4, 239-250.
246. Saldanha R., Chen B., Wank H., Matsuura M., Edwards J. & Lambowitz A. M. (1999) RNA and protein catalysis in group II intron splicing and mobility reactions using purified components. Biochemistry 38, 9069-9083.
247. Singh R. N., Saldanha R. J., D'Souza L. M. & Lambowitz A. M. (2002) Binding of a group II intron-encoded reverse transcriptase/maturase to its high affinity intron RNA binding site involves sequence-specific recognition and autoregulates translation. J. Mol. Biol. 318, 287-303.
248. Mohr G., Smith D., Belfort M. & Lambowitz A. M. (2000) Rules for DNA target-site recognition by a lactococcal group II intron enable retargeting of the intron to specific DNA sequences. Genes Dev. 14, 559-573.
249. Zhong J., Lambowitz A. M. (2003a) Group II intron mobility using nascent strands at DNA replication forks to prime reverse transcription. EMBO J. 22, 4555-4565.
250. Eickbush T. H. (1999) Mobile introns: retrohoming by complete reverse splicing. Curr. Biol. 9, R11-4.
171

251. Perutka J., Wang W., Goerlitz D. & Lambowitz A. M. (2004) Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J. Mol. Biol. 336, 421-439.
252. Karberg M., Guo H., Zhong J., Coon R., Perutka J. & Lambowitz A. M. (2001) Group II introns as controllable gene targeting vectors for genetic manipulation of bacteria. Nat. Biotechnol. 19, 1162-1167.
253. Frazier C. L., San Filippo J., Lambowitz A. M. & Mills D. A. (2003) Genetic manipulation of Lactococcus lactis by using targeted group II introns: generation of stable insertions without selection. Appl. Environ. Microbiol. 69, 1121-1128.
254. Zhong J., Karberg M. & Lambowitz A. M. (2003b) Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Res. 31, 1656-1664.
255. Long M. B., Jones J. P.,3rd, Sullenger B. A. & Byun J. (2003) Ribozyme-mediated revision of RNA and DNA. J. Clin. Invest. 112, 312-318.
256. Jones J. P.,3rd, Kierlin M. N., Coon R. G., Perutka J., Lambowitz A. M. & Sullenger B. A. (2005) Retargeting mobile group II introns to repair mutant genes. Mol. Ther. 11, 687-694.
257. Lever A. M. (1995) Gene therapy for HIV infection. Br. Med. Bull. 51, 149-166.
258. Berger E. A., Doms R. W., Fenyo E. M., Korber B. T., Littman D. R., Moore J. P., Sattentau Q. J., Schuitemaker H., Sodroski J. & Weiss R. A. (1998) A new classification for HIV-1. Nature 391, 240.
259. Kawabata K., Ujikawa M., Egawa T., Kawamoto H., Tachibana K., Iizasa H., Katsura Y., Kishimoto T. & Nagasawa T. (1999) A cell-autonomous requirement for CXCR4 in long-term lymphoid and myeloid reconstitution. Proc. Natl. Acad. Sci. U. S. A. 96, 5663-5667.
260. Onai N., Zhang Y., Yoneyama H., Kitamura T., Ishikawa S. & Matsushima K. (2000) Impairment of lymphopoiesis and myelopoiesis in mice reconstituted with bone marrow-hematopoietic progenitor cells expressing SDF-1-intrakine. Blood 96, 2074-2080. .
261. Huang Y., Paxton W. A., Wolinsky S. M., Neumann A. U., Zhang L., He T., Kang S., Ceradini D., Jin Z., Yazdanbakhsh K., Kunstman K., Erickson D., Dragon E., Landau N. R., Phair J., Ho D. D. & Koup R. A. (1996) The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat. Med. 2, 1240-1243.
262. Rossi J. J. (2000) Ribozyme therapy for HIV infection. Adv. Drug Deliv. Rev. 44, 71-78.
263. Aarons E. J., Beddows S., Willingham T., Wu L. & Koup R. A. (2001) Adaptation to blockade of human immunodeficiency virus type 1 entry imposed by the anti-CCR5 monoclonal antibody 2D7. Virology 287, 382-390.
264. Li M. J., Bauer G., Michienzi A., Yee J. K., Lee N. S., Kim J., Li S., Castanotto D., Zaia J. & Rossi J. J. (2003) Inhibition of HIV-1 infection by lentiviral vectors expressing Pol III-promoted anti-HIV RNAs. Mol. Ther. 8, 196-206.
265. Miller A. D., Buttimore C. (1986) Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol. Cell. Biol. 6, 2895-2902.
266. DuBridge R. B., Tang P., Hsia H. C., Leong P. M., Miller J. H. & Calos M. P. (1987) Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 7, 379-387.
172

267. Lusso P., Cocchi F., Balotta C., Markham P. D., Louie A., Farci P., Pal R., Gallo R. C. & Reitz M. S.,Jr. (1995) Growth of macrophage-tropic and primary human immunodeficiency virus type 1 (HIV-1) isolates in a unique CD4+ T-cell clone (PM1): failure to downregulate CD4 and to interfere with cell-line-tropic HIV-1. J. Virol. 69, 3712-3720.
268. Cheng L., Du C., Murray D., Tong X., Zhang Y. A., Chen B. P. & Hawley R. G. (1997) A GFP reporter system to assess gene transfer and expression in human hematopoietic progenitor cells. Gene Ther. 4, 1013-1022.
269. Burns J. C., Friedmann T., Driever W., Burrascano M. & Yee J. K. (1993) Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. U. S. A. 90, 8033-8037.
270. Naldini L., Blomer U., Gage F. H., Trono D. & Verma I. M. (1996) Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. U. S. A. 93, 11382-11388.
271. Medina M. F., Joshi S. (1999) Design, characterization and testing of tRNA3Lys-based hammerhead ribozymes. Nucleic Acids Res. 27, 1698-1708.
272. Ramezani A., Hawley T. S. & Hawley R. G. (2000) Lentiviral vectors for enhanced gene expression in human hematopoietic cells. Mol. Ther. 2, 458-469.
273. Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Molecular cloning: A laboratory manual. 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
274. Gartner S., Markovits P., Markovitz D. M., Kaplan M. H., Gallo R. C. & Popovic M. (1986) The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 233, 215-219.
275. Adachi A., Gendelman H. E., Koenig S., Folks T., Willey R., Rabson A. & Martin M. A. (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59, 284-291.
276. Reed,L.J. and H. Muench. (1938) A simple method of estimating fifty percent endpoints. Am J Hyg 27, 493-497.
277. De Clercq E. (2000) Inhibition of HIV infection by bicyclams, highly potent and specific CXCR4 antagonists. Mol. Pharmacol. 57, 833-839.
278. Wolkowicz R., Nolan G. P. (2005) Gene therapy progress and prospects: novel gene therapy approaches for AIDS. Gene Ther. 12, 467-476.
279. Lambowitz A. M., Belfort M. (1993) Introns as mobile genetic elements. Annu. Rev. Biochem. 62, 587-622.
280. Martinez-Abarca F., Toro N. (2000) Group II introns in the bacterial world. Mol. Microbiol. 38, 917-926.
281. Magli M. C., Dick J. E., Huszar D., Bernstein A. & Phillips R. A. (1987) Modulation of gene expression in multiple hematopoietic cell lineages following retroviral vector gene transfer. Proc. Natl. Acad. Sci. U. S. A. 84, 789-793.
282. Liem S. E., Ramezani A., Li X. & Joshi S. (1993) The development and testing of retroviral vectors expressing trans-dominant mutants of HIV-1 proteins to confer anti-HIV-1 resistance. Hum. Gene Ther. 4, 625-634.
173

283. Zack J. A., Arrigo S. J., Weitsman S. R., Go A. S., Haislip A. & Chen I. S. (1990) HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61, 213-222.
284. Bokaei P. B., Ma X. Z., Sakac D. & Branch D. R. (2007) HIV-1 integration is inhibited by stimulation of the VPAC2 neuroendocrine receptor. Virology 362, 38-49.
285. Menendez-Arias L. (2002) Molecular basis of fidelity of DNA synthesis and nucleotide specificity of retroviral reverse transcriptases. Prog. Nucleic Acid Res. Mol. Biol. 71, 91-147.
286. Gallo R. C. (2002) Human retroviruses after 20 years: a perspective from the past and prospects for their future control. Immunol. Rev. 185, 236-265.
287. Adams M., Sharmeen L., Kimpton J., Romeo J. M., Garcia J. V., Peterlin B. M., Groudine M. & Emerman M. (1994) Cellular latency in human immunodeficiency virus-infected individuals with high CD4 levels can be detected by the presence of promoter-proximal transcripts. Proc. Natl. Acad. Sci. U. S. A. 91, 3862-3866.
288. Chun T. W., Carruth L., Finzi D., Shen X., DiGiuseppe J. A., Taylor H., Hermankova M., Chadwick K., Margolick J., Quinn T. C., Kuo Y. H., Brookmeyer R., Zeiger M. A., Barditch-Crovo P. & Siliciano R. F. (1997) Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387, 183-188.
289. Zagury D., Bernard J., Leonard R., Cheynier R., Feldman M., Sarin P. S. & Gallo R. C. (1986) Long-term cultures of HTLV-III--infected T cells: a model of cytopathology of T-cell depletion in AIDS. Science 231, 850-853.
290. Pantaleo G., Fauci A. S. (1996) Immunopathogenesis of HIV infection. Annu. Rev. Microbiol. 50, 825-854.
291. Roederer M., Dubs J. G., Anderson M. T., Raju P. A., Herzenberg L. A. & Herzenberg L. A. (1995) CD8 naive T cell counts decrease progressively in HIV-infected adults. J. Clin. Invest. 95, 2061-2066.
292. Westendorp M. O., Frank R., Ochsenbauer C., Stricker K., Dhein J., Walczak H., Debatin K. M. & Krammer P. H. (1995) Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375, 497-500.
293. De Noronha C. M., Sherman M. P., Lin H. W., Cavrois M. V., Moir R. D., Goldman R. D. & Greene W. C. (2001) Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science 294, 1105-1108.
294. Pauza C. D., Galindo J. E. & Richman D. D. (1990) Reinfection results in accumulation of unintegrated viral DNA in cytopathic and persistent human immunodeficiency virus type 1 infection of CEM cells. J. Exp. Med. 172, 1035-1042.
295. Xu X. N., Screaton G. R., Gotch F. M., Dong T., Tan R., Almond N., Walker B., Stebbings R., Kent K., Nagata S., Stott J. E. & McMichael A. J. (1997) Evasion of cytotoxic T lymphocyte (CTL) responses by nef-dependent induction of Fas ligand (CD95L) expression on simian immunodeficiency virus-infected cells. J. Exp. Med. 186, 7-16.
296. Geleziunas R., Xu W., Takeda K., Ichijo H. & Greene W. C. (2001) HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 410, 834-838.
174

297. Trkola A., Kuhmann S. E., Strizki J. M., Maxwell E., Ketas T., Morgan T., Pugach P., Xu S., Wojcik L., Tagat J., Palani A., Shapiro S., Clader J. W., McCombie S., Reyes G. R., Baroudy B. M. & Moore J. P. (2002) HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc. Natl. Acad. Sci. U. S. A. 99, 395-400.
298. Swan C. H., Torbett B. E. (2006a) Can gene delivery close the door to HIV-1 entry after escape? J. Med. Primatol. 35, 236-247.
299. Buchholz C. J., Cichutek K. (2006) Is it going to be SIN?: a European Society of Gene Therapy commentary. Phasing-out the clinical use of non self-inactivating murine leukemia virus vectors: initiative on hold. J. Gene Med. 8, 1274-1276.
300. Ramezani A., Hawley T. S. & Hawley R. G. (2003) Performance- and safety-enhanced lentiviral vectors containing the human interferon-beta scaffold attachment region and the chicken beta-globin insulator. Blood 101, 4717-4724.
301. Meyer-Ficca M. L., Meyer R. G., Kaiser H., Brack A. R., Kandolf R. & Kupper J. H. (2004) Comparative analysis of inducible expression systems in transient transfection studies. Anal. Biochem. 334, 9-19.
302. Prak E. T., Kazazian H. H.,Jr. (2000) Mobile elements and the human genome. Nat. Rev. Genet. 1, 134-144.
303. Bowen N. J., Jordan I. K. (2002) Transposable elements and the evolution of eukaryotic complexity. Curr. Issues Mol. Biol. 4, 65-76.
304. Lands L. (2002 (revised 2006)) A Practical Guide to HIV Drug Side Effects for People Living with HIV/AIDS.
175
Related Documents











