Rhabdomyosarcoma of the Parotid Region Occurring in Childhood and Adolescence A Report from the Intergroup Rhabdomyosarcoma Study Group David O. Walterhouse, M.D. 1 Alberto S. Pappo, M.D. 2 K. Scott Baker, M.D. 3 David M. Parham, M.D. 4 James R. Anderson, Ph.D. 5 Sarah S. Donaldson, M.D. 6 Charles N. Paidas, M.D. 7 Richard B. Womer, M.D. 8 William M. Crist, M.D. 9 1 Department of Pediatrics, Children’s Memorial Hospital, Chicago, Illinois. 2 Department of Pediatrics, Hospital for Sick Chil- dren, Toronto, Ontario, Canada. 3 Department of Pediatrics, University of Minne- sota Hospital, Minneapolis, Minnesota. 4 Department of Pathology, Arkansas Children’s Hospital, Little Rock, Arkansas. 5 Department of Preventive and Societal Medicine, University of Nebraska Medical Center, Omaha, Nebraska. 6 Department of Radiation Oncology, Stanford Uni- versity Medical Center, Stanford, California. 7 Department of Pediatric Surgery, Johns Hopkins Hospital, Baltimore, Maryland. 8 Department of Pediatrics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania. 9 Dean, University of Missouri School of Medicine, Columbia, Missouri. Data presented at the American Society of Clinical Oncology Annual Meeting, New Orleans, Louisiana, May 19 –23, 2000. Supported in part by National Institutes of Health/ National Cancer Institute Grants CA24507 and CA72989. The authors prepared this article on behalf of the Intergroup Rhabdomyosarcoma Study Group (IRSG) representing the Children’s Cancer Group, the Pedi- atric Oncology Group, and the Intergroup Rhabdo- myosarcoma Statistical Office including: James R. Anderson, Ph.D., Richard J. Andrassy, M.D., Carola A. S. Arndt, M.D., K. Scott Baker, M.D., Frederic G. Barr, M.D., W. Archie Bleyer, M.D., Philip Breitfeld, M.D., John C. Breneman, M.D., Julia Bridge, M.D., Kenneth Brown, M.D., William M. Crist, M.D., Sarah S. Donaldson, M.D., Holcombe E. Grier, M.D., Doug- las Hawkins, M.D., Peter J. Houghton, Ph.D., Michael Link, M.D., Thom L. Lobe, M.D., Harold M. Maurer, M.D., William H. Meyer, M.D., Jeff Michalski, M.D., Sharon Murphy, M.D., Charles N. Paidas, M.D., Al- berto S. Pappo, M.D., David M. Parham, M.D., Ste- phen J. Qualman, M.D., R. Beverly Raney, M.D., Leslie Robison, Ph.D., Eric Sandler, M.D., Stephen Skapek, M.D., Lynn Smith, M.D., Poul H. B. Sorensen, M.D., Ph.D., Sheri Spunt, M.D., Lisa A. Teot, M.D., Timothy Triche, M.D., Ph.D., Teresa J. Vietti, M.D., David O. Walterhouse, M.D., Moody Wharam, M.D., Eugene S. Wiener, M.D., Suzanne Wolden, M.D., and Richard B. Womer, M.D. Address for correspondence: David O. Walter- house, M.D., Children’s Memorial Hospital, Divi- sion of Hematology Oncology, Box 30, 2300 Chil- dren’s Plaza, Chicago, IL 60614; Fax: (773) 880- 3223; E-mail: [email protected] Received October 8, 2000; revision received Au- gust 30, 2001; accepted September 8, 2001. BACKGROUND. Rhabdomyosarcoma (RMS) of the parotid region is rare and to the authors’ knowledge little information is available regarding the site of tumor origin, clinical presentation, and outcome in these patients. Therefore, the authors re- viewed the files of all patients with RMS of the parotid region who were registered on the Intergroup Rhabdomyosarcoma Studies (IRS) I–IV. METHODS. Patient charts and the Intergroup Rhabdomyosarcoma Study Group (IRSG) database were reviewed. RESULTS. Sixty-two patients presenting with a mass in the parotid region were identified. None of the tumors was localized exclusively to the parotid gland, so the primary site was referred to as the “parotid region.” The tumor invaded a para- meningeal site in 30 patients. These cases have been designated as parameningeal- parotid tumors to distinguish them from 32 cases that did not invade a parameni- ngeal site and were designated as nonparameningeal-parotid tumors. The majority of patients had Group III tumors in both the nonparameningeal-parotid and parameningeal-parotid subgroups. However, although there were 16 patients with Group I or II tumors in the nonparameningeal-parotid subgroup, no patients with Group I or II tumors were found in the parameningeal-parotid subgroup (P 0.001). Fifty-six of 62 patients (90%) received radiotherapy. The parameningeal primary site designation resulted in intensification of both chemotherapy and radiotherapy for patients with parameningeal-parotid RMS. The 5-year failure-free survival rate was 81% and the 5-year survival rate was 84%. There were no deaths reported among patients with Group I or II tumors. The 5-year failure-free survival did not appear to differ when comparing patients with parameningeal-parotid tumors with patients with nonparameningeal-parotid tumors (P 0.21). CONCLUSIONS. Treatment as defined by the IRS protocols has been reported to be highly effective for patients with RMS of the parotid region. Outcome for the more aggressively treated patients with parameningeal-parotid RMS appears similar to that for patients with nonparameningeal-parotid RMS. Cancer 2001;92:3135– 46. © 2001 American Cancer Society. KEYWORDS. chemotherapy, Intergroup Rhabdomyosarcoma Study Group (IRSG), parotid neoplasms, pediatric oncology, radiotherapy, rhabdomyosarcoma. 3135 © 2001 American Cancer Society DOI 10.1002/cncr.10172

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Rhabdomyosarcoma of the Parotid Region Occurringin Childhood and AdolescenceA Report from the Intergroup Rhabdomyosarcoma Study Group
David O. Walterhouse, M.D.1
Alberto S. Pappo, M.D.2
K. Scott Baker, M.D.3
David M. Parham, M.D.4
James R. Anderson, Ph.D.5
Sarah S. Donaldson, M.D.6
Charles N. Paidas, M.D.7
Richard B. Womer, M.D.8
William M. Crist, M.D.9
1 Department of Pediatrics, Children’s MemorialHospital, Chicago, Illinois.
2 Department of Pediatrics, Hospital for Sick Chil-dren, Toronto, Ontario, Canada.
3 Department of Pediatrics, University of Minne-sota Hospital, Minneapolis, Minnesota.
4 Department of Pathology, Arkansas Children’sHospital, Little Rock, Arkansas.
5 Department of Preventive and Societal Medicine,University of Nebraska Medical Center, Omaha,Nebraska.
6 Department of Radiation Oncology, Stanford Uni-versity Medical Center, Stanford, California.
7 Department of Pediatric Surgery, Johns HopkinsHospital, Baltimore, Maryland.
8 Department of Pediatrics, Children’s Hospital ofPhiladelphia, Philadelphia, Pennsylvania.
9 Dean, University of Missouri School of Medicine,Columbia, Missouri.
Data presented at the American Society of ClinicalOncology Annual Meeting, New Orleans, Louisiana,May 19–23, 2000.
Supported in part by National Institutes of Health/National Cancer Institute Grants CA24507 andCA72989.
The authors prepared this article on behalf of theIntergroup Rhabdomyosarcoma Study Group (IRSG)representing the Children’s Cancer Group, the Pedi-atric Oncology Group, and the Intergroup Rhabdo-myosarcoma Statistical Office including: James R.Anderson, Ph.D., Richard J. Andrassy, M.D., CarolaA. S. Arndt, M.D., K. Scott Baker, M.D., Frederic G.Barr, M.D., W. Archie Bleyer, M.D., Philip Breitfeld,
M.D., John C. Breneman, M.D., Julia Bridge, M.D.,Kenneth Brown, M.D., William M. Crist, M.D., SarahS. Donaldson, M.D., Holcombe E. Grier, M.D., Doug-las Hawkins, M.D., Peter J. Houghton, Ph.D., MichaelLink, M.D., Thom L. Lobe, M.D., Harold M. Maurer,M.D., William H. Meyer, M.D., Jeff Michalski, M.D.,Sharon Murphy, M.D., Charles N. Paidas, M.D., Al-berto S. Pappo, M.D., David M. Parham, M.D., Ste-phen J. Qualman, M.D., R. Beverly Raney, M.D.,Leslie Robison, Ph.D., Eric Sandler, M.D., StephenSkapek, M.D., Lynn Smith, M.D., Poul H. B. Sorensen,M.D., Ph.D., Sheri Spunt, M.D., Lisa A. Teot, M.D.,
Timothy Triche, M.D., Ph.D., Teresa J. Vietti, M.D.,David O. Walterhouse, M.D., Moody Wharam,M.D., Eugene S. Wiener, M.D., Suzanne Wolden,M.D., and Richard B. Womer, M.D.
Address for correspondence: David O. Walter-house, M.D., Children’s Memorial Hospital, Divi-sion of Hematology Oncology, Box 30, 2300 Chil-dren’s Plaza, Chicago, IL 60614; Fax: (773) 880-3223; E-mail: [email protected]
Received October 8, 2000; revision received Au-gust 30, 2001; accepted September 8, 2001.
BACKGROUND. Rhabdomyosarcoma (RMS) of the parotid region is rare and to the
authors’ knowledge little information is available regarding the site of tumor origin,
clinical presentation, and outcome in these patients. Therefore, the authors re-
viewed the files of all patients with RMS of the parotid region who were registered
on the Intergroup Rhabdomyosarcoma Studies (IRS) I–IV.
METHODS. Patient charts and the Intergroup Rhabdomyosarcoma Study Group
(IRSG) database were reviewed.
RESULTS. Sixty-two patients presenting with a mass in the parotid region were
identified. None of the tumors was localized exclusively to the parotid gland, so the
primary site was referred to as the “parotid region.” The tumor invaded a para-
meningeal site in 30 patients. These cases have been designated as parameningeal-
parotid tumors to distinguish them from 32 cases that did not invade a parameni-
ngeal site and were designated as nonparameningeal-parotid tumors. The majority
of patients had Group III tumors in both the nonparameningeal-parotid and
parameningeal-parotid subgroups. However, although there were 16 patients with
Group I or II tumors in the nonparameningeal-parotid subgroup, no patients with
Group I or II tumors were found in the parameningeal-parotid subgroup (P
� 0.001). Fifty-six of 62 patients (90%) received radiotherapy. The parameningeal
primary site designation resulted in intensification of both chemotherapy and
radiotherapy for patients with parameningeal-parotid RMS. The 5-year failure-free
survival rate was 81% and the 5-year survival rate was 84%. There were no deaths
reported among patients with Group I or II tumors. The 5-year failure-free survival
did not appear to differ when comparing patients with parameningeal-parotid
tumors with patients with nonparameningeal-parotid tumors (P � 0.21).
CONCLUSIONS. Treatment as defined by the IRS protocols has been reported to be
highly effective for patients with RMS of the parotid region. Outcome for the more
aggressively treated patients with parameningeal-parotid RMS appears similar to
that for patients with nonparameningeal-parotid RMS. Cancer 2001;92:3135– 46.
© 2001 American Cancer Society.
KEYWORDS. chemotherapy, Intergroup Rhabdomyosarcoma Study Group (IRSG),parotid neoplasms, pediatric oncology, radiotherapy, rhabdomyosarcoma.
3135
© 2001 American Cancer SocietyDOI 10.1002/cncr.10172

Malignant tumors of the parotid gland are reportedto be rare during childhood.1–3 Sarcomas account
for � 1.5% of malignant tumors of the parotid glandand controversy exists whether “parotid sarcomas”originate in the gland itself or invade it secondarily.4 –7
The prognosis for patients with sarcomas of the pa-rotid gland is correlated with histologic type, tumorsize, and resectability.4,5,7 Rhabdomyosarcoma (RMS)is one of the more common sarcomas of the parotidregion occurring during childhood and adolescence,but overall its incidence remains rare.4,7 Thus, infor-mation regarding the clinical presentation, manage-ment, and outcome for patients with RMS of the pa-rotid region has been based on series with smallnumbers of patients or case reports,1,2,4,5,7–10 reportsthat consider multiple histologic types of parotid tu-mors in both adults and children,1,4,5,11 or reports thatconsider patients with parotid region primary tumorstogether with patients with RMS of other head and neckregions.11–13 In the current study, we describe 62 pedi-atric patients with RMS of the parotid region who wereenrolled on Intergroup Rhabdomyosarcoma Studies(IRS) I–IV to better define the site of tumor origin, clinicalpresentation, therapy, and outcome in these patients.
MATERIALS AND METHODSSubjectsAll patients with RMS arising in the parotid region whowere registered on IRS-I (1972–1978), IRS-II (1978–1984),IRS-III (1984–1991), or IRS-IV (1991–1997) were includedin the current analysis. The parotid region was definedas lying immediately anterior and inferior to the ear andsurrounding both sides of the posterior aspect of theascending ramus of the mandible. Patients were in-cluded if a mass was present in the parotid region butinvaded into adjacent tissues including a parameningealsite. It is possible that some tumors invaded the parotidregion secondarily from a parameningeal or other headand neck site. Untreated patients, age � 21 years, with apathologically proven diagnosis of RMS, undifferenti-ated sarcoma, or extraosseous Ewing sarcoma (IRS-IIand IRS-III only) were eligible for the studies. Informedconsent was obtained for all participants at the time ofregistration on the study.
MethodsIndividual patient charts and the Intergroup Rhabdo-myosarcoma Study Group (IRSG) database were re-viewed for all cases. IRSG pretreatment stage was as-signed retrospectively for patients on IRS-I, IRS-II, orIRS-III based on primary tumor site, tumor size, tumorinvasiveness, regional lymph node involvement, and ev-idence of metastatic disease, but was assigned prospec-tively for those patients on IRS-IV (Table 1). The IRSG
pretreatment staging classification includes the parotidregion primary site as a favorable head and neck site inStage 1. However, a significant number of patients pre-senting with a mass in the parotid region had largetumors involving the infratemporal fossa or another sitedefined as parameningeal. Involvement of a parameni-ngeal site resulted in upstaging to Stage 2 or Stage 3 andmore aggressive treatment assignment as a paramenin-geal patient. Such patients were included in the currentanalysis and their primary tumor site was designated“parameningeal-parotid” to distinguish them from thepatients with “non-parameningeal-parotid” tumors thatdid not extend into a parameningeal site.
Excision of the primary tumor was attempted whenfeasible without causing a significant functional or cos-metic defect, and was limited to those patients withnonparameningeal-parotid tumors. IRSG group was de-termined for all patients based on the extent of diseaseafter the initial definitive surgical procedure prior to theinitiation of chemotherapy or radiotherapy (RT) (Table2). Pathologic material from all patients was reviewedcentrally by members of the IRSG pathology committee.
Patients received chemotherapy as assigned orwere randomized on IRS I–IV. Chemotherapy was as-signed based on group (IRS-I); group, primary tumorsite, and histology (IRS-II and IRS-III); or stage andprimary tumor site (IRS-IV) (Fig. 1).14 –17 Patients whowere eligible to receive less intensive chemotherapyregimens, which contained a two-drug combination ofvincristine and dactinomycin (VA), were those withGroup II tumors on IRS-I, Group I or Group II tumorson IRS-II, Group I tumors on IRS-III, or Group II orGroup III nonparameningeal-parotid tumors of favor-able histology (nonalveolar) on IRS-III. All other pa-tients received chemotherapy regimens containing atleast three drugs including an alkylating agent.
RT dose, volume, and schedule were determinedaccording to group, primary tumor site, age, and tumorsize (Fig. 1).14–17 RT records and adequacy of radiationdose were reviewed by the Quality Assurance ReviewCenter and confirmed by members of the IRSG Radio-therapy Committee. In addition, diagnostic radiographs,simulation films, and portal films were reviewed bymembers of the IRSG Radiotherapy Committee to eval-uate the adequacy of the treatment volume. In IRS-I theneed for RT in patients with Group I tumors was thesubject of a randomized trial. In IRS II–IV no routine RTwas given to Group I patients. In IRS-IV the value ofhyperfractionated versus conventionally fractionated RTwas examined in patients with Group III RMS. All otherpatients received conventionally fractionated treatment.After 1977, to prevent early central nervous system re-currence, patients with parameningeal RMS with high-risk features (evidence of intracranial extension of tu-
3136 CANCER December 15, 2001 / Volume 92 / Number 12

mor, base of skull erosion, or cranial nerve palsy) beganRT on Day 0 of treatment.18
DefinitionsComplete response (CR) was defined as the disappear-ance of all tumor for a minimum of 4 weeks. Partialresponse (PR) was defined as a � 50% decrease in thesize of all measurable lesions for a minimum of 4weeks. Stable disease (SD) was defined as a 25–50%decrease in the size of all measurable lesions for a min-
imum of 4 weeks. No response (NR) was defined as a� 25% decrease in tumor size. Progressive disease (PD)was defined as a � 25% increase in the size of measur-able lesions at any site and/or the appearance of newlesions. Relapse/recurrence was defined as the appear-ance of new lesions or the reappearance of old lesions.
Statistical AnalysisFailure-free survival (FFS) was measured from the timeof diagnosis to the time of PD, recurrence, or death (if
TABLE 1IRSG Pretreatment Staging Classification
Stage Sites T Classification Size N Classification M Classification
1 OrbitHead and neck
(Nonparameningeal)Genitourinary
(Nonbladder/nonprostate)
T1 or T2 A or B N0, N1 or Nx M0
2 Bladder/prostateExtremityCranial parameningealOther
T1 or T2 A N0 or Nx M0
3 Bladder/prostateExtremityCranial parameningealOther
T1 or T2 AB
N1
N0, N1 or Nx
M0
M0
4 All T1 or T2 A or B N0 or N1 M1
Definitions
Tumor invasiveness (T classification)T1 Confined to anatomic site of originT2 Extension and/or fixation to surrounding tissue
SizeA � 5 cm in greatest dimensionB � 5 cm in greatest dimension
Regional lymph nodes (N classification)N0 Regional lymph nodes not clinically involvedN1 Regional lymph nodes clinically involvedNx Status of regional lymph nodes unknown
Metastasis (M classification)M0 No distant metastasisM1 Metastasis present
TABLE 2IRSG Grouping System
Group I Localized disease, completely excised, regional lymph nodes not involvedGroup II Total macroscopic resection with evidence of regional spread
A. Macroscopically resected tumor with microscopic residual diseaseB. Regional disease with involved lymph nodes, completely resected with no microscopic residual diseaseC. Regional disease with involved lymph nodes, macroscopically resected, but with evidence of microscopic
residual disease and/or histologic involvement of the most distal regional lymph node in the dissectionGroup III Incomplete resection with macroscopic residual diseaseGroup IV Distant metastatic disease present at onset
IRSG: Intergroup Rhabdomyosarcoma Study Group.
Parotid Region Rhabdomyosarcoma/Walterhouse et al. 3137

FIGURE 1. Schema of therapy for (A-D) Intergroup Rhabdomyosarcoma Studies I–IV.

occurring prior to recurrence or PD). Patients who didnot fail were censored at their date of last contact. Sur-vival was measured from the time of diagnosis untildeath, or until the date of last contact if the patient wasalive at the time of last follow-up. All deaths were
counted as failures, regardless of whether they were dis-ease-related. Estimates of the time-to-event distribu-tions were calculated using the Kaplan–Meier method,and 95% confidence intervals (95% CI) for specific esti-mates of time-to-event distributions were calculated us-
FIGURE 1. (continued )
Parotid Region Rhabdomyosarcoma/Walterhouse et al. 3139

FIGURE 1. (continued )

ing the Greenwood formula for the variance of the esti-mates.19 Comparisons of outcome among patientsubsets were made using the log-rank test.20
RESULTSClinical Presentation and Pretreatment StageNone of the tumors was localized exclusively to the pa-rotid gland, so we refer to the primary site as the “parotidregion.” Sixty-two patients with tumors of the parotidregion were identified: 32 patients (52%) with nonpara-meningeal-parotid tumors and 30 patients (48%) withparameningeal-parotid tumors. These cases account for1.5% of all patients entered onto IRS I–IV. Fifty-one pa-tients (82%) were white, 6 patients (10%) were black, and5 patients (8%) were from other racial groups.
The duration of symptoms prior to the time ofpresentation ranged from 4 days to 17 weeks (median,5 weeks). All patients presented with a mass in theparotid region. The etiology of the mass often wasbelieved to be infectious and in 9 cases (15%), antibi-otics were administered before biopsy. The right pa-rotid region was affected in 33 patients (53%) and theleft was affected in 29 patients (47%). Preoperativefacial nerve palsy (9 patients; 15%) and/or pain (8patients; 13%) were present in 15 patients (24%).Other symptoms and physical findings included tris-mus (four patients), respiratory distress (two patients),dysfunction of cranial nerves V, VI, and XI (one pati-enteach), nasal congestion (one patient), dysphagia(one patient), and headache (one patient).
Table 3 lists patient characteristics by the invasionpattern of the tumor. The patient numbers were bal-anced among all studies. The median age of the pa-tients was 6 years. Patients with nonparameningeal-parotid tumors tended to be older than those patientswith parameningeal-parotid tumors (P � 0.03). Thir-teen patients (41%) with nonparameningeal-parotidtumors (median age, 6 years) were age � 10 yearscompared with 3 patients (10%) with parameningeal-parotid tumors (median age, 5 years). Patients withnonparameningeal-parotid tumors were somewhatmore likely to be male (P � 0.08) and have small (� 5cm) tumors (P � 0.07). The tumor invaded into tissuesadjacent to the parotid gland (T2) in all cases withsufficient data for evaluation.
By definition, all patients with Stage 1 tumors hadnonparameningeal-parotid RMS and all patients withStage 2 or Stage 3 tumors had parameningeal-parotidRMS. Parameningeal-parotid tumors demonstratedextension to the base of the skull, the infratemporalfossa, the nasopharynx, and the middle ear. Metastaticdisease was present in 5 cases (8%); metastatic sitesincluded the lungs (3 patients), bone (1 patient), andbone marrow (1 patient).
Group Assignment and HistologyClear surgical margins were obtained in only threepatients; all of whom were in the nonparamenin-geal-parotid subgroup. Facial nerve dysfunction wasdescribed for the first time postoperatively in 8 pa-tients (13%). In three of these patient the tumor wasresected macroscopically (1 Group I case and 2Group II cases) and in the remaining 5 patientsincisional biopsy (4 patients) or debulking surgery(1 patient) was performed. Although Group III caseswere most common in both the nonparameningeal-parotid and parameningeal-parotid subgroups, allGroup I and II cases were in the nonparameningeal-parotid subgroup (P � 0.001).
In addition to those patients with the embryonalor alveolar histologic subtype of RMS, there were twocases of undifferentiated sarcoma and one case ofextraosseous Ewing sarcoma in the nonparamenin-geal-parotid subgroup and two cases of undifferenti-ated sarcoma, one RMS of the spindle cell variant, andone RMS not otherwise specified in the paramenin-geal-parotid subgroup.
Chemotherapy and RTPatient characteristics affecting treatment regimen as-signment, chemotherapy regimen, RT dose, and out-come are listed for all patients in Table 4. Fourteen of30 patients (47%) with a localized nonparameningeal-parotid primary tumor received chemotherapy withVA. Only 1 of 27 patients (4%) with a localized para-meningeal-parotid primary tumor received VA. Thesingle patient receiving VA in the parameningeal-pa-rotid subgroup was treated according to protocol for anonparameningeal primary tumor site on IRS-III. Allremaining patients received at least three drugs in-cluding an alkylating agent.
Fifty-six patients received RT (90%). As specifiedper protocol, two patients with Group I tumors treatedon IRS-III did not receive RT.16 In three cases thetreating physicians elected not to give the protocol-specified RT. Radiation doses varied by study andgroup. RT was initiated on Day 0 of therapy in 13 ofthe 15 patients (87%) with parameningeal-parotidRMS who were considered to have high-risk para-meningeal features.18 Fifteen patients (50%) withparameningeal-parotid RMS did not have high-riskparameningeal features and received RT at the stan-dard time per protocol (Fig. 1).14 –17
Outcome and ToxicityForty-eight patients (77%) achieved a CR, 8 patients(13%) achieved a PR, 2 patients (3%) demonstrated NR,and 1 patient (2%) was found to have PD. A somewhat
Parotid Region Rhabdomyosarcoma/Walterhouse et al. 3141

TABLE 3Patient Characteristics Based on the Invasion Pattern of the Tumor
All patients IRSI–IV (n � 62)
Nonparameningeal-parotid (n � 32)
Parameningeal-parotid (n � 30)
StudyIRS-I 15 (24%) 9 (28%) 6 (20%)IRS-II 10 (16%) 5 (16%) 5 (17%)IRS-III 18 (29%) 12 (37%) 6 (20%)IRS-IV 19 (31%) 6 (19%) 13 (43%)
62 32 30P � 0.1
Age (yrs)�1 3 (5%) 1 (3%) 2 (7%)1–9 43 (69%) 18 (56%) 25 (83%)10–14 11 (18%) 8 (25%) 3 (10%)15–21 5 (8%) 5 (16%) 0
62 32 30P � 0.03
GenderMale 36 (58%) 22 (69%) 14 (47%)Female 26 (42%) 10 (31%) 16 (53%)
62 32 30P � 0.08
Tumor size (cm)�5 31 (50%) 19 (59%) 12 (40%)�5 29 (47%) 11 (34%) 18 (60%)Unknown 2 (3%) 2 (6%) 0
62 32 30P � 0.07
InvasivenessT1 0 0 0T2 59 (95%) 29 (91%) 30 (100%)Unknown 3 (5%) 3 (9%) 0
62 32 30Lymph node statusa
N0 24 (65%) 13 (72%) 11 (58%)N1 11 (30%) 5 (28%) 6 (32%)Nx 2 (5%) 0 2 (10%)
37 18 19P � 0.27
Stage1 30 (48%) 30 (94%) 02 6 (10%) 0 6 (20%)3 21 (34%) 0 21 (70%)4 5 (8%) 2 (6%) 3 (10%)
62 32 30P � 0.001
GroupI 3 (5%) 3 (9%) 0II 13 (21%) 13 (41%) 0III 41 (66%) 14 (44%) 27 (90%)IV 5 (8%) 2 (6%) 3 (10%)
62 32 30P � 0.001
HistologyEmbryonal 49 (79%) 26 (81%) 23 (77%)Alveolar 6 (10%) 3 (9%) 3 (10%)Other 7 (11%) 3 (9%) 4 (13%)
62 32 30
IRS: Intergroup Rhabdomyosarcoma Study.a Data available for Intergroup Rhabdomyosarcoma Studies III and IV only.
3142 CANCER December 15, 2001 / Volume 92 / Number 12

TABLE 4Patient Characteristics Affecting Treatment Assignment, Treatment, and Outcome for All Patients
Stage Group N/PMIRSno. Hist Chemotherapy
RT dose(Gy)
Response/recurrence Outcome
1 I N I Emb VAC 60.0 CR Alive1 I N III Emb VA None CR Alive1 I N III Other VA None CR Alive1 IIA N I Emb VA 52.7 CR Alive1 IIA N I Emb VA 50.0 CR Alive1 IIA N I Other VA 45.0 CR Alive1 IIA N II Emb VACD 45.0 CR Alive1 IIC N III Emb VA 41.4 CR Alive1 IIC N III Emb VA 41.4 CR Alive1 IIA N III Emb VA 41.4 CR Alive1 IIA N III Emb VA 52.1 CR/dis Alive1 IIC N III Emb VA 43.1 CR Alive1 IIA N III Alv VACDP 45.0 CR Alive1 IIA N IV Alv VIE 46.0 Unk Alive1 IIA N IV Emb VIE 48.2 CR Alive1 IIA N IV Alv VAI 41.4 CR Alive1 III N I Emb VAC 50.0 CR Alive1 III N I Emb VAC 48.0 CR Alive1 III N I Emb VAC 71.8 CR D(SM)1 III N I Emb VACD 46.8 CR Alive1 III N II Emb VAC 52.0 CR Alive1 III N II Emb VAC 44.9 CR Alive1 III N II Emb VAC None Unk/unk DOD1 III N III Other VA 48.8 CR Alive1 III N III Emb VA 50.4 CR/dis DOD1 III N III Emb VA 45.0 NR/loc DOD1 III N III Emb VA 50.4 CR Alive1 III N IV Emb VAI 41.4 CR Alive1 III N IV Emb VAI 54.0 CR Alive1 III N IV Emb VAI 56.8 PR Alive2 III PM I Emb VACD None NR/loc, dis DOD2 III PM II Emb VAC 44.0 CR Alive2 III PM II Emb VAC 46.8a PD DOD2 III PM II Emb VACD 52.2 CR Alive2 III PM III Emb VA 45.0 CR/loc DOD2 III PM IV Alv VAC 62.5 CR Alive3 III PM I Emb VAC 62.2a CR Alive3 III PM I Emb VACD 60.0 CR/loc, dis DOD3 III PM I Alv VACD 47.8a CR Alive3 III PM I Emb VACD 40.0 CR Alive3 III PM II Emb VAC 69.6 CR Alive3 III PM II Emb VACD 50.4 CR Alive3 III PM III Alv VACDP 45.9a CR Alive3 III PM III Emb VACDPE 45.0a PR/loc D( inf)3 III PM III Emb VACDPE 50.4 CR Alive3 III PM III Emb VACDPE 41.4a CR/dis DOD3 III PM IV Emb VAC 61.0a CR Alive3 III PM IV Emb VAC 50.4a CR Alive3 III PM IV Emb VAC 51.0a CR Alive3 III PM IV Emb VAC 55.2a PR Alive3 III PM IV Other VAC Unk Unk Alive3 III PM IV Other VAC 38.2 CR Alive3 III PM IV Emb VAI 59.4a CR Alive3 III PM IV Other VAI 60.9a PR Alive3 III PM IV Emb VAI 59.4 PR Alive3 III PM IV Emb VIE 53.2 CR Alive3 III PM IV Other VIE 62.9a PR/loc Alive4 IV PM I Emb VACD None PR/dis DOD4 IV N I Emb VACD 20.0 PR/dis DOD4 IV N II Emb VACD 50.5 CR Alive4 IV PM III Emb VACDP 48.6 CR Alive4 IV PM IV Emb VACDI 46.0 CR Alive
N: nonparameningeal; PM: parameningeal; Hist: histology; RT: radiotherapy; Gy: grays; Emb: embryonal; V: vincristine; A: dactinomycin; C: cyclophosphamide; CR: complete response; D: doxorubicin; dis: distant;
Alv: alveolar; P: cisplatin; I: ifosfamide; E: etoposide; Unk: unknown; D(SM): died of second malignancy; DOD: died of disease; NR: no response; loc: local; PR: partial response; PD: progressive disease; D(inf): died
of infection.a Indicates that radiotherapy was shifted to Day 0.
Parotid Region Rhabdomyosarcoma/Walterhouse et al. 3143
higher CR rate was noted for patients with nonparamen-ingeal-parotid tumors (93%) compared with patientswith parameningeal-parotid tumors (71%) (P � 0.05).
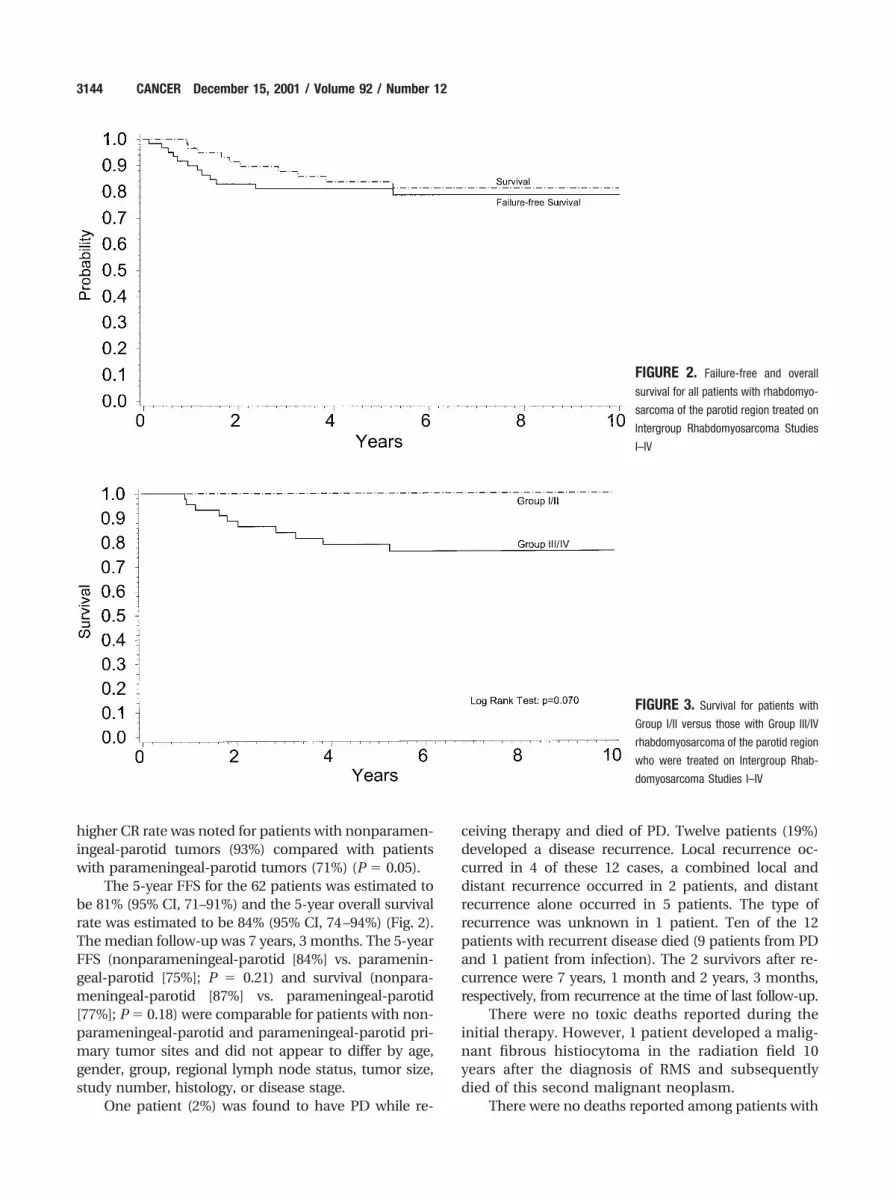
The 5-year FFS for the 62 patients was estimated tobe 81% (95% CI, 71–91%) and the 5-year overall survivalrate was estimated to be 84% (95% CI, 74–94%) (Fig. 2).The median follow-up was 7 years, 3 months. The 5-yearFFS (nonparameningeal-parotid [84%] vs. paramenin-geal-parotid [75%]; P � 0.21) and survival (nonpara-meningeal-parotid [87%] vs. parameningeal-parotid[77%]; P � 0.18) were comparable for patients with non-parameningeal-parotid and parameningeal-parotid pri-mary tumor sites and did not appear to differ by age,gender, group, regional lymph node status, tumor size,study number, histology, or disease stage.
One patient (2%) was found to have PD while re-
ceiving therapy and died of PD. Twelve patients (19%)developed a disease recurrence. Local recurrence oc-curred in 4 of these 12 cases, a combined local anddistant recurrence occurred in 2 patients, and distantrecurrence alone occurred in 5 patients. The type ofrecurrence was unknown in 1 patient. Ten of the 12patients with recurrent disease died (9 patients from PDand 1 patient from infection). The 2 survivors after re-currence were 7 years, 1 month and 2 years, 3 months,respectively, from recurrence at the time of last follow-up.
There were no toxic deaths reported during theinitial therapy. However, 1 patient developed a malig-nant fibrous histiocytoma in the radiation field 10years after the diagnosis of RMS and subsequentlydied of this second malignant neoplasm.
There were no deaths reported among patients with
FIGURE 3. Survival for patients with
Group I/II versus those with Group III/IV
rhabdomyosarcoma of the parotid region
who were treated on Intergroup Rhab-
domyosarcoma Studies I–IV
FIGURE 2. Failure-free and overall
survival for all patients with rhabdomyo-
sarcoma of the parotid region treated on
Intergroup Rhabdomyosarcoma Studies
I–IV
3144 CANCER December 15, 2001 / Volume 92 / Number 12

Group I or II tumors (Fig. 3). Three of five patients withGroup III tumors who were treated with VA plus RT diedof PD. The three patients with Group III or Group IVtumors who did not receive RT against protocol guide-lines died of PD. Three of 5 patients with Stage 4, GroupIV tumors were long-term survivors at the time of lastfollow-up (currently 11 years and 2 months, 10 years and2 months, and 7 years, respectively, from diagnosis).
DISCUSSIONIn the current study, we report the clinical presenta-tion and outcome for 62 children and adolescents withRMS of the parotid region who were treated on IRSI–IV. To our knowledge, this is the largest series ofpatients with RMS of the parotid region presented inthe literature to date.1,3–5,7–13
It has been debated whether sarcomas arise withinthe parotid gland itself or invade it secondarily fromsurrounding mesenchymal tissue.4–7 Although all pa-tients in the current series presented with a mass in theparotid region, it was not possible to identify any cases inwhich the tumor was localized only to the parenchymaof the parotid gland based on imaging studies, surgicalreports, and pathology specimens. Paraglandular tissuewas involved in all cases with sufficient data for analysis,including the smallest tumors, and the parotid gland wasnot involved by tumor in some cases as reported in thesurgical and pathology records. Although it is possiblethat some tumors originate in the parotid gland and thenextend into paraglandular tissue, overall these observa-tions suggest that the tumors usually originate in parag-landular tissue and subsequently invade the parotidgland, with the most appropriate designation being“RMS of the parotid region.”
The age and gender distribution of the patients inthe current study and the histologic subtype of thetumors were typical of RMS in general.14 –17 The ma-jority of the patients (76%) presented with a painlessmass in the parotid region without evidence of facialnerve dysfunction. Pain and facial nerve dysfunctionhave been described as characteristic features of ma-lignant tumors of the parotid gland.2,7 It is importantthat physicians be aware that pain or cranial nervedysfunction often do not occur in patients with RMSof the parotid region and further diagnostic evaluationof a mass in the parotid region should proceed with-out delay even in their absence. A delay in diagnosismay contribute to the possibility that the tumor mayinvade into a parameningeal site or may decrease thechance for a complete resection. RMS of the parotidregion occasionally may compress or invade othercranial nerves or obstruct nasal/respiratory passages.
The outcomes reported in the current series com-pare favorably with the outcomes for the case reports
and series that appear in the literature, although thestage/group of the patients in the literature has notalways been reported. In the three series in whichfollow-up data were available for patients with RMS ofthe parotid region, one of two patients died of diseasein one series, two of four patients with available fol-low-up data died in a second series, and seven of ninepatients died of disease in a third series, including onepatient with Group I RMS, four patients with Group IIIRMS, and four patients with Group IV RMS.4,5,7 Ourtreatment approach generally included conservativesurgical management; RT for patients with Group II,III, or IV RMS; and chemotherapy for all patients.
Excision of the primary tumor was attempted onlyin those patients in the nonparameningeal-parotidsubgroup and only if it could be accomplished withoutsignificant functional or cosmetic defects. Facial nervedysfunction was reported in the immediate postoper-ative period in some patients. Clinical or radiographicevidence of tumor involvement of regional lymphnodes was found in 31% of the patients entered ontoIRS-III or IRS-IV. Biopsy of enlarged lymph nodes wasundertaken for grouping, staging, and treatment pur-poses. Radical cervical lymph node dissection forthose patients with regional lymph node involvementis not recommended for patients with RMS.
There were no deaths reported among the 10 pa-tients with Stage 1, Group I or II RMS who received VAwith or without RT. However, one patient who diddevelop a recurrence was alive at last follow-up � 7years from the time of recurrence. There also were nodeaths reported among the six patients with Stage 1,Group I or II RMS who received chemotherapy includ-ing an alkylating agent plus RT. Whether some pa-tients with Stage 1, Group I or II tumors benefitedfrom the addition of the alkylating agent could not bedetermined from the current series because the num-ber of patients was small and all still were alive at thetime of last follow-up. Further refinement of therapyfor subsets of these patients will require evaluation ofthe larger number of patients who currently are beingtreated on the IRS-V low-risk protocol. On this proto-col, patients with Stage 1, Group I and Stage 1, GroupIIA (lymph node-negative) tumors of embryonal his-tology receive VA, whereas patients with Stage 1,Group IIB or IIC (lymph nodes clinically involved)tumors of embryonal histology receive VA plus cyclo-phosphamide (VAC). Patients with tumors of the alve-olar subtype receive treatment on the IRS-V interme-diate-risk protocol and are not eligible to receivetreatment with VA only.
It appears that therapy for the patients with Stage1, Group III tumors should include an alkylating agentin addition to VA plus RT. Eight of 10 patients with
Parotid Region Rhabdomyosarcoma/Walterhouse et al. 3145

Stage 1, Group III tumors who received chemotherapyincluding an alkylating agent were long-term survi-vors. One patient who did not receive RT died of PD,and a second patient died of a second malignancy thatdeveloped in the radiation field. VA does not appear tobe adequate for patients with Stage 1, Group III tu-mors because in the current study two of four patientswho received VA died of PD.
The parameningeal primary tumor site previouslyhas been shown to be unfavorable compared with pri-mary tumor sites such as the orbit and other nonpara-meningeal head and neck sites, including the nonpara-meningeal-parotid site.12 However, the outcome for theparameningeal-parotid subgroup in the current serieswas not significantly different from that for the nonpara-meningeal-parotid subgroup. The parameningeal desig-nation resulted in upstaging, intensification of chemo-therapy, and shifting RT to the beginning of thetreatment course in those patients with high-risk para-meningeal features. Approximately 47% of the patientswith localized tumors in the nonparameningeal-parotidsubgroup received the less intensive 2-drug combinationVA whereas only 3% of the patients with localized tu-mors in the parameningeal-parotid subgroup receivedVA. RT was shifted to the beginning of the treatmentcourse in 43% of the parameningeal-parotid patients,including 87% of those with high-risk parameningealfeatures.18 Therapy for patients in the parameningeal-parotid subgroup with Stage 2 or 3, Group III tumorsshould include VA plus an alkylating agent and RT. Thetiming of RT should be determined based on the pres-ence or absence of high-risk parameningeal features.
Three of the five patients with Stage 4, Group IVdisease were long-term survivors. Each of these patientswas age � 10 years and had embryonal RMS. Age � 10years and embryonal histology recently have been rec-ognized as favorable features for patients with metastaticRMS treated on the IRS-III and IRS-IV pilot studies.21 Along-term survival rate of approximately 50% has beenreported for this subset of patients.21 Intensive chemo-therapy with three drugs including an alkylating agentplus RT to the primary and metastatic tumor sites isrecommended for these patients. Optimal therapy forolder patients and those with unfavorable histologicsubtypes with metastatic RMS remains undefined andthe enrollment of patients on clinical trials is encouraged.
REFERENCES1. Marin VTW, Salmaso R, Onnis GL. Tumors of salivary
glands. Appl Pathol 1989;7:154 – 60.2. Schuller DE, McCabe BF. Salivary gland neoplasms in chil-
dren. Otolaryngol Clin North Am 1977;10:399 – 412.
3. Krolls SO, Trodahl JN, Boyers RC. Salivary gland lesions inchildren: a survey of 430 cases. Cancer 1972;30:459 – 69.
4. Luna MA, Tortoledo E, Ordonez NG, Frankenthaler RA, Bat-sakis JG. Primary sarcomas of the major salivary glands.Arch Otolaryngol Head Neck Surg 1991;117:302– 6.
5. Auclair PL, Langloss JM, Weiss SW, Corio RL. Sarcomas andsarcomatoid neoplasms of the major salivary gland regions.Cancer 1986;58:1305–15.
6. Kauffman SL, Stout AP. Tumors of the major salivary glandsin children. Cancer 1963;16:1317–31.
7. Rogers DA, Rao BN, Bowman L, Marina N, Fleming ID,Schropp KP, et al. Primary malignancy of the salivary glandin children. J Pediatr Surg 1994;29:44 –7.
8. Renick B, Clark RM, Feldman L. Embryonal rhabdomyosar-coma: presentation as a parotid gland mass. Oral Surg OralMed Oral Pathol 1988;65:575–9.
9. Pignataro L, Mariscotti C. Rhabdomyosarcoma of the parotidarea: a case study. Acta Otorhinolaryngol Ital 1990;10:193–7.
10. Daou RA, Schloss MD. Childhood rhabdomyosarcomas ofthe head and neck - 2 case reports on salivary glandular andparaglandular involvement. J Otolaryngol 1982;11:52– 6.
11. Wanebo HJ, Koness RJ, MacFarlane JK, Eilber FR, Byers RM,Elias EG, et al. Head and neck sarcoma: report of the headand neck sarcoma registry. Head Neck 1992;14:1–7.
12. Sutow WW, Lindberg RD, Gehan EA, Ragab AH, Raney RB,Ruymann F, et al. Three-year relapse-free survival rates inchildhood rhabdomyosarcoma of the head and neck. Cancer1982;49:2217–21.
13. Wharam MD, Foulkes MA, Lawrence W, Lindberg RD, Mau-rer HM, Newton WA, et al. Soft tissue sarcoma of the headand neck in childhood: non-orbital and non-parameningealsites. Cancer 1984;53:1016 –9.
14. Maurer HM, Beltangady M, Gehan EA, Crist W, HammondD, Hays DM, et al. The Intergroup RhabdomyosarcomaStudy-I: a final report. Cancer 1988;61:209 –20.
15. Maurer HM, Gehan EA, Beltangady M, Crist W, Dickman PS,Donaldson SS, et al. The Intergroup RhabdomyosarcomaStudy-II. Cancer 1993;71:1904 –22.
16. Crist W, Gehan EA, Ragab AH, Dickman PS, Donaldson SS,Fryer C, et al. The Third Intergroup RhabdomyosarcomaStudy. J Clin Oncol 1995;13:610 –30.
17. Baker KS, Anderson JR, Link MP, Grier HE, Qualman SJ,Maurer HM, et al. Benefit of intensified therapy for patientswith local or regional embryonal rhabdomyosarcoma: re-sults from the Intergroup Rhabdomyosarcoma Study IV.J Clin Oncol 2000;18:2427–34.
18. Raney BR, Tefft M, Newton WA, Ragab AH, Lawrence W,Gehan EA, et al. Improved prognosis with intensive treat-ment of children with cranial soft tissue sarcomas arising innon-orbital parameningeal sites. Cancer 1987;59:147–55.
19. Kaplan GL, Meier P. Nonparametric estimation from incom-plete observations. J Am Stat Assoc 1958;53:457– 81.
20. Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, HowardSV, et al. Design and analysis of randomized clinical trialsrequiring prolonged observation of each patient. II: analysisand examples. Br J Cancer 1977;35:1–39.
21. Anderson JR, Ruby E, Link M, Crist W, Maurer H. Identifi-cation of a favorable subset of patients (pts) with metastatic(MET) rhabdomyosarcoma (RMS): a report from the Inter-group Rhabdomyosarcoma Study Group (IRSG). Proc AmSoc Clin Oncol 1997;16:510a.
3146 CANCER December 15, 2001 / Volume 92 / Number 12
Related Documents











