Rett Syndrome Research Rett Syndrome Research The Sydney Experience The Sydney Experience John Christodoulou John Christodoulou NSW Centre for Rett Syndrome Research NSW Centre for Rett Syndrome Research Western Sydney Genetics Program, Children’s Hospital at Western Sydney Genetics Program, Children’s Hospital at Westmead Westmead Disciplines of Paediatrics & Child Health and Medical Disciplines of Paediatrics & Child Health and Medical Genetics, Genetics, University of Sydney University of Sydney

Rett Syndrome Research The Sydney Experience John Christodoulou NSW Centre for Rett Syndrome Research Western Sydney Genetics Program, Children’s Hospital.
Dec 15, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Rett Syndrome ResearchRett Syndrome Research
The Sydney ExperienceThe Sydney Experience
John ChristodoulouJohn Christodoulou
NSW Centre for Rett Syndrome ResearchNSW Centre for Rett Syndrome Research
Western Sydney Genetics Program, Children’s Hospital at WestmeadWestern Sydney Genetics Program, Children’s Hospital at Westmead
Disciplines of Paediatrics & Child Health and Medical Genetics, Disciplines of Paediatrics & Child Health and Medical Genetics,
University of SydneyUniversity of Sydney

Presentation OutlinePresentation Outline
• Predicting disease severity by knowing Predicting disease severity by knowing thethe MECP2 MECP2 gene mutationgene mutation
• CDKL5CDKL5 – a second Rett syndrome gene – a second Rett syndrome gene
• hunt for MeCP2 targetshunt for MeCP2 targets

MECP2 MECP2 Mutation Studies:Mutation Studies:
phenotype-genotype correlationsphenotype-genotype correlations

Rett SyndromeRett Syndrome• first described by first described by Andreas Rett Andreas Rett in 1966in 1966
• almost exclusively affects femalesalmost exclusively affects females
• progressive loss ofprogressive loss of– intellectual functioningintellectual functioning
– fine and gross motor skillsfine and gross motor skills
• stereotypic hand movementsstereotypic hand movements
• 1:8,000 females by 15 yrs1:8,000 females by 15 yrs
• rarely familial recurrencesrarely familial recurrences
• most cases caused by mutations in most cases caused by mutations in MECP2MECP2

Clinical DiagnosisClinical Diagnosis• specific developmental profile based on a consistent specific developmental profile based on a consistent
constellation of clinical features constellation of clinical features (diagnosis is provisional < 3 yrs)(diagnosis is provisional < 3 yrs)
• diagnostic criteria developed and recently reviseddiagnostic criteria developed and recently revised
• classical and variant RTT phenotypesclassical and variant RTT phenotypes
Preserved Preserved SpeechSpeech
Forme Forme FrusteFruste
ClassicalClassicalCongenital Congenital RTTRTT
Late and Late and slow onset slow onset RTTRTT

Modified from Hagberg et al Eur J Paediatr Neurol 2002 (6) 293 - 297Modified from Hagberg et al Eur J Paediatr Neurol 2002 (6) 293 - 297

6 mutations identified in 21 sporadic classical cases6 mutations identified in 21 sporadic classical cases - 4 - 4 de novode novo missense mutations in methyl-binding domain (MBD) missense mutations in methyl-binding domain (MBD)
- 1 - 1 de novode novo frame-shift mutation in transcription repression domain (TRD) frame-shift mutation in transcription repression domain (TRD)
- 1 - 1 de novode novo nonsense mutation in TRD nonsense mutation in TRD
““Rett syndrome is caused by mutations in Rett syndrome is caused by mutations in X-linked X-linked MECP2MECP2, encoding methyl-CpG , encoding methyl-CpG
binding protein 2”binding protein 2”(Amir et al, Nature Genet 1999: 23; 185 - 188)(Amir et al, Nature Genet 1999: 23; 185 - 188)
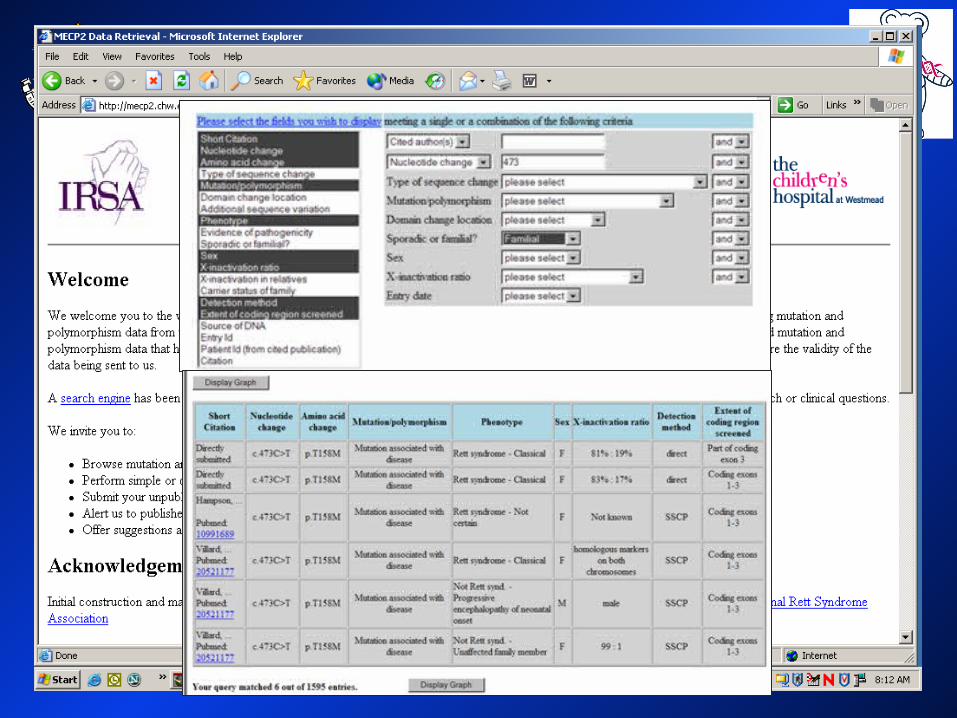

MECP2 MECP2 Mutations IdentifiedMutations Identified
> 270 different mutations to date> 270 different mutations to date > 3000 individuals> 3000 individuals
RettBASE: http://mecp2.chw.edu.auRettBASE: http://mecp2.chw.edu.au
Nucleotide change
Amino acid change
Cases reported
percentage
c.316C>T p.R106W 95 3.07
c.397C>T p.R133C 134 4.33
c.473C>T p.T158M 276 8.92
c.502C>T p.R168X 254 8.21c.763C>T p.R255X 222 7.17
c.808C>T p.R270X 198 6.40c.880C>T p.R294X 169 5.46
c.916C>T p.R306C 152 4.91
20 – 100 bp
deletions
various
truncations
203 6.5555%

• international study to examine clinical features of RTTinternational study to examine clinical features of RTT
• data are collected from 2 sourcesdata are collected from 2 sources– familiesfamilies
– cliniciansclinicians
• data are stored and compiled to produce an output data are stored and compiled to produce an output databasedatabase– this will be a searchable form in the futurethis will be a searchable form in the future
• both databases have been funded by IRSA (and now both databases have been funded by IRSA (and now the IRSF)the IRSF)
• development of clinical and mutation databases development of clinical and mutation databases
(J Child Neurol, 2003; Hum Mut, 2003)(J Child Neurol, 2003; Hum Mut, 2003)

Our Our MECP2MECP2 Mutation Studies Mutation Studies
• MECP2MECP2 mutation screening of a clinically well- mutation screening of a clinically well-characterised cohort of RTT patients characterised cohort of RTT patients (Am J Med Genet, 2003)(Am J Med Genet, 2003)
– pathogenic mutations in 74% of 234 patientspathogenic mutations in 74% of 234 patients
– (80% classical RTT patients, 70% atypical RTT patients)(80% classical RTT patients, 70% atypical RTT patients)
– truncation mutations clinically more severe than missense mutationstruncation mutations clinically more severe than missense mutations
– NLS & TRD mutations clinically more severe than MBD mutationsNLS & TRD mutations clinically more severe than MBD mutations
– higher proportion with skewing of X-inactivation Vs normal controlshigher proportion with skewing of X-inactivation Vs normal controls
• detailed evaluations of specific mutations detailed evaluations of specific mutations (J Med Genet, (J Med Genet, 2003; J Med Genet 2004; Brain Dev, 2005; Eur J Hum Genet, 2005; J Med 2003; J Med Genet 2004; Brain Dev, 2005; Eur J Hum Genet, 2005; J Med Genet, 2007)Genet, 2007)
– p.R133C mutation is milder; p.R270X most severep.R133C mutation is milder; p.R270X most severe
– 58% show unusual behaviours in the first 6 months of life58% show unusual behaviours in the first 6 months of life
– X-inactivation modulates disease severity of p.T158M & p.R168XX-inactivation modulates disease severity of p.T158M & p.R168X

Our Our MECP2MECP2 Mutation Studies Mutation Studies
• evaluation of clinical aspects ofevaluation of clinical aspects of RTT RTT (J Pediatr, (J Pediatr, 2005; J Pediatr, 2006; J Child Neurol, 2006; Eur J Pediatr Neurol, 2005; J Pediatr, 2006; J Child Neurol, 2006; Eur J Pediatr Neurol, 2008)2008)
– 78% survival by 25 yrs78% survival by 25 yrs
– 25% have seizures by 2 yr, 50% by 4 yr, 79% by 10 yr25% have seizures by 2 yr, 50% by 4 yr, 79% by 10 yr
– later onset of seizures with p.R294X vs p.R255Xlater onset of seizures with p.R294X vs p.R255X
– seizure rate highest 7 – 12 yr (lower with p.R294X, p.R255X, seizure rate highest 7 – 12 yr (lower with p.R294X, p.R255X, C-term)C-term)
– 75% have scoliosis by 13 yr (less likely if have p.R294X)75% have scoliosis by 13 yr (less likely if have p.R294X)
– 4 times more likely to have a fracture4 times more likely to have a fracture

Please participatePlease participate
• if you are not a current participant and you would like to take if you are not a current participant and you would like to take part in InterRett and the work practices pilot study just email part in InterRett and the work practices pilot study just email us….us….
• [email protected]@ichr.uwa.edu.au
• or visit our website or visit our website http://http://interrett.ichr.uwa.edu.auinterrett.ichr.uwa.edu.au
• encourage your doctor or the laboratory that did the testing to encourage your doctor or the laboratory that did the testing to submit their informationsubmit their information
• [email protected]@chw.edu.au
• or visit our website or visit our website http://http://mecp2.chw.edu.aumecp2.chw.edu.au

CDKL5CDKL5::
a new Rett Syndrome genea new Rett Syndrome gene

Clinical Summary Clinical Summary
Family 1Family 1
II:1II:1- clinically normal mother- clinically normal mother
II:1II:1 II:2II:2
III:5III:5III:1III:1 III:2III:2 III:3III:3 III:4III:4
III:1III:1- atypical (milder RTT)- atypical (milder RTT)- infantile spasms from 9 weeks- infantile spasms from 9 weeks
- - III:2III:2- autism & mild MR- autism & mild MR- never had seizures- never had seizures
III:3III:3- infantile spasms in the newborn period- infantile spasms in the newborn period- poor head control- poor head control- severe psychomotor retardation- severe psychomotor retardation- died age 16 yrs (unresponsive, frequent myoclonic jerks)- died age 16 yrs (unresponsive, frequent myoclonic jerks)
III:4III:4- clinically normal brother- clinically normal brother
III:5III:5- clinically normal sister- clinically normal sister
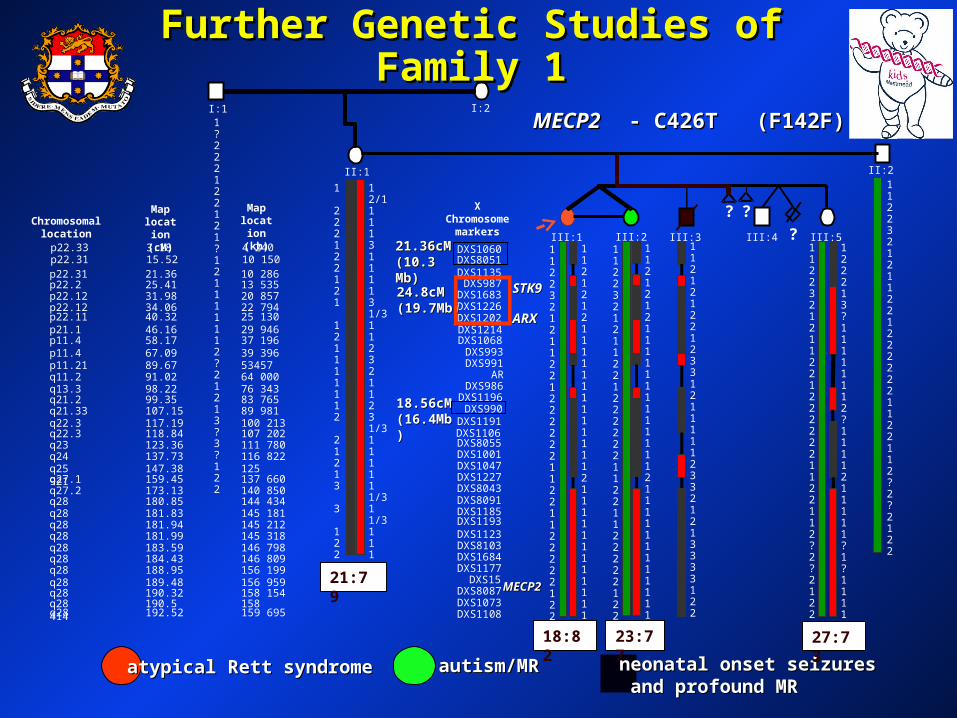
Further Genetic Studies of Family 1Further Genetic Studies of Family 1
atypical Rett syndromeatypical Rett syndrome autism/MRautism/MR neonatal onset seizuresneonatal onset seizures and profound MRand profound MR
DXS1193
DXS1060
DXS1135DXS8051
DXS987DXS1683DXS1226DXS1202
DXS993DXS991
ARDXS986
DXS1196DXS990
DXS1191DXS1106DXS8055DXS1001DXS1047DXS1227DXS8043DXS8091DXS1185
DXS1123DXS8103DXS1684DXS1177
DXS15DXS8087DXS1073DXS1108
I:2
II:2
III:3III:1 III:4
? ?
18.56cM18.56cM(16.4Mb)(16.4Mb)
MECP2MECP2
24.8cM24.8cM(19.7Mb)(19.7Mb)
21.36cM21.36cM(10.3 Mb)(10.3 Mb)
ARXARX
STK9STK9
II:1
III:5
I:1
?
21:79
18:82 23:77 27:73
DXS1214DXS1068
X Chromosome
markers
p22.33 3.18 4 240
p22.31 21.36 10 286p22.2 25.41 13 535
p22.12 34.06 22 794
p11.4 67.09 39 396p11.21 89.67 53457q11.2 91.02 64 000
q21.2 99.35 83 765q21.33 107.15 89 981
q22.3 118.84 107 202q23 123.36 111 780q24 137.73 116 822
q27.1 159.45 137 660q27.2 173.13 140 850
q28 181.83 145 181q28 181.94 145 212q28 181.99 145 318
q28 184.43 146 809q28 188.95 156 199
q28 190.32 158 154
q28 192.52 159 695
p22.31 15.52 10 150
p22.12 31.98 20 857
p22.11 40.32 25 130
p11.4 58.17 37 196p21.1 46.16 29 946
q13.3 98.22 76 343
q22.3 117.19 100 213
q25 147.38 125 921
q28 180.85 144 434
q28 183.59 146 798
q28 189.48 156 959
q28 190.5 158 414
Chromosomal location
Map location
(cM)
Map location
(kb)
11223212112212222221122112?2?2122
11223212112212222221122112?2?2122
122213?11111112?1111211111?1?1111
112121221233121111123321213333122
112121211111111111112111111111111
112232121122122222211221122222122
1
222122121
121111112
21213
3
122
12/11113111131/31123211231/3111111/311/3111
1?222122121?121111112?21213?3?122
112121211111111111112111111111111
112232121122122222211221122222122
III:2
MECP2 MECP2 - C426T (F142F)- C426T (F142F)
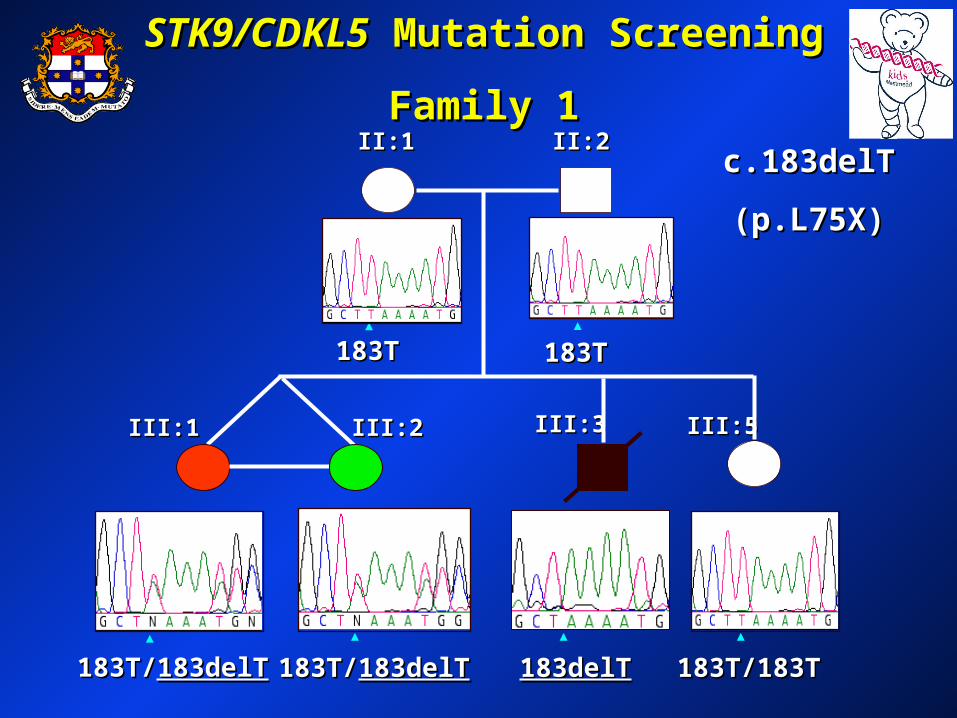
STK9/CDKL5STK9/CDKL5 Mutation Screening Mutation Screening
Family 1Family 1II:1II:1 II:2II:2
III:5III:5III:3III:3III:1III:1 III:2III:2
183T183T183T183T
183T/183T/183delT183delT 183T/183T/183delT183delT 183delT183delT 183T/183T183T/183T
c.183delTc.183delT
(p.L75X)(p.L75X)

Summary of currently known Summary of currently known CDKL5CDKL5 mutations mutations
66 55 44 33
1a1a11 1b1b 101022
ATGATG
1111 121233 44 55 66 77 88 99 1313 141415151616 1717 1818 1919 2020 2121
TelTel CenCen
CDKL5 gene
XLRS1 gene
IVS7-2A>GIVS7-2A>G
c.455G>Tc.455G>T
c.2635-2636delCTc.2635-2636delCT
c.838_847del10c.838_847del10
IVS11-2A>GIVS11-2A>G
IVS13-1G>AIVS13-1G>AIVS6-1G>TIVS6-1G>TIVS6-1G>CIVS6-1G>C
c.525A>Tc.525A>T
c.539C>Tc.539C>T
c.2343delGc.2343delG
IVS16+1G>CIVS16+1G>C
IVS16+1G>AIVS16+1G>A
c.2362_2366del5c.2362_2366del5del678_691del678_691ins683_673ins683_673
c.680T>Cc.680T>C
c.183delTc.183delTc.215T>Ac.215T>A
c.163-166delGAAAc.163-166delGAAAc.175C>Tc.175C>T

Further Mutation Screening Further Mutation Screening of CDKL5of CDKL5
• particular clinical phenotypeparticular clinical phenotype– Hanefeld variant –onset of severe seizures < 6 monthsHanefeld variant –onset of severe seizures < 6 months
• Archer et al ( J Med Genet 2006: 43; 729-734)Archer et al ( J Med Genet 2006: 43; 729-734)– 7 of 42 (17%) ♀ with seizures commencing < 6 months of age7 of 42 (17%) ♀ with seizures commencing < 6 months of age
– all with poor developmental progressall with poor developmental progress
– severe seizures mostly of myoclonic or infantile spasm typesevere seizures mostly of myoclonic or infantile spasm type
– few clinical signs suggestive of RTTfew clinical signs suggestive of RTT
– males rarely show males rarely show CDKL5CDKL5 mutations mutations
• our studies – 272 patients screened for mutations in the our studies – 272 patients screened for mutations in the CDKL5CDKL5 gene, incl. 89 RTT, 60 ISSX, 58 autism, 7 XLMR, gene, incl. 89 RTT, 60 ISSX, 58 autism, 7 XLMR, 58 others58 others– only 1 de novo missense mutation - c.586C>T (p.S196L)only 1 de novo missense mutation - c.586C>T (p.S196L)

Dapi CDKL5- Polyclonal
-Tubulin MergeUntransfected (HeLa)
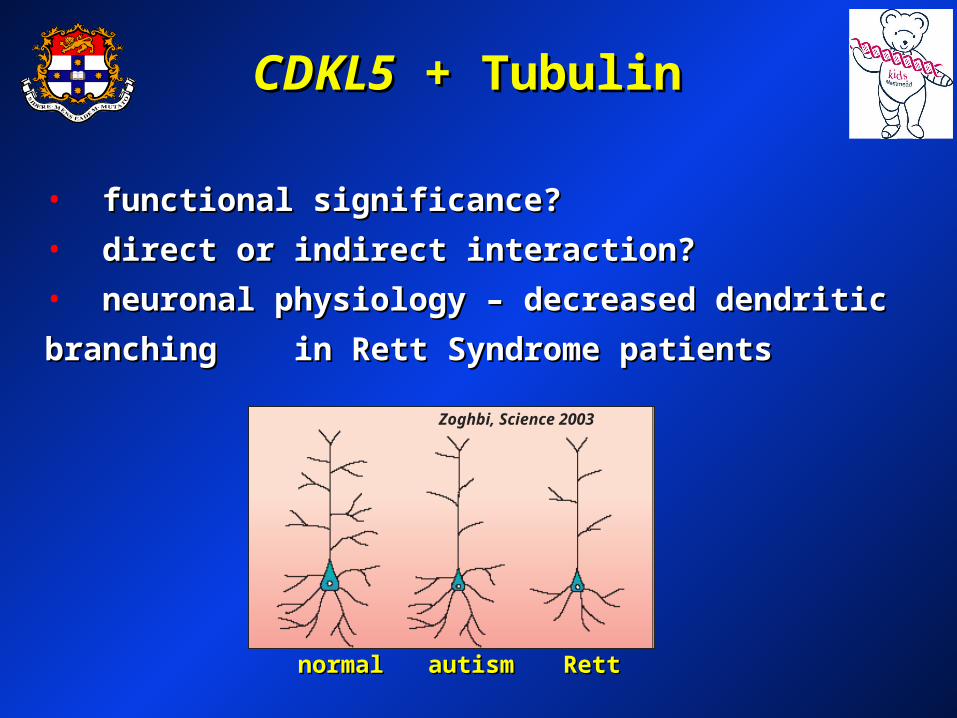
CDKL5CDKL5 + Tubulin + Tubulin
• functional significance?functional significance?
• direct or indirect interaction?direct or indirect interaction?
• neuronal physiology – decreased dendritic branching neuronal physiology – decreased dendritic branching
in Rett Syndrome patients in Rett Syndrome patients
normalnormal RettRett autismautism
Zoghbi, Science 2003

Identification of Specific MeCP2 Identification of Specific MeCP2
Downstream TargetsDownstream Targets

Neuropathology of RettNeuropathology of Rett• adult Rett brain ~ 900 grams (the adult Rett brain ~ 900 grams (the
same size as a non RTT 1 year old)same size as a non RTT 1 year old)
• regional volumetric lossregional volumetric loss
Control RettControl Rett
• small densely packed neurons with decreased dendritic small densely packed neurons with decreased dendritic branching branching (cerebral cortex, basal ganglia, hippocampus)(cerebral cortex, basal ganglia, hippocampus)
• occipital cortex escapes neuropathologyoccipital cortex escapes neuropathology
normalnormal RettRettautismautism
Zoghbi, Science 2003

-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
--------------aaaaUCGA------UCGA------
--------------aaaaUCGA------UCGA------
--------------aaaaUCGA------UCGA------
-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
--------------aaaaUCGA------UCGA------
--------------aaaaUCGA------UCGA------
--------------aaaaUCGA------UCGA------
Increased Increased ExpressionExpression
DecreasedDecreased ExpressionExpression
-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
-------AGCT--------------AGCT-------
--------------aaaaUCGA------UCGA------
--------------aaaaUCGA------UCGA------
UniformUniform ExpressionExpression
Label two mRNA Label two mRNA populations (populations (redred and and greengreen))
Bind labelled probe Bind labelled probe mRNAmRNA ssiimmuullttaanneeoouussllyy toto spotsspots
Created by Dan Catchpoole, CHW, 2001Created by Dan Catchpoole, CHW, 2001

Gene Expression StudiesGene Expression Studies• using mRNA from RTT patient brain samplesusing mRNA from RTT patient brain samples - compare regional expression patterns- compare regional expression patterns
- studies using frontal and occipital cerebral cortex- studies using frontal and occipital cerebral cortex
• microarray analysesmicroarray analysesExpression ProfilesExpression Profiles
increasedincreased
uniformuniform
reducedreduced

Gene NameGene Name FunctionFunction Median Median Fold Fold
ChangeChangeRTT FC:RTT OC/ RTT FC:RTT OC/ RTT FC: CON FCRTT FC: CON FC
(RNAi results)(RNAi results)
cytochrome c cytochrome c oxidase subunit Ioxidase subunit I
involved in energy involved in energy productionproduction
2.4/ 1.9 2.4/ 1.9
(4x)(4x)
clusterinclusterin control of cell survivalcontrol of cell survival 1.7/ 1.51.7/ 1.5
(3x)(3x)
guanine nucleotide guanine nucleotide binding protein binding protein
communication between communication between cellscells
2.5/ 1.82.5/ 1.8
(2x)(2x)
dynamin 1dynamin 1 communication between communication between brain cells brain cells
1.7/ 1.51.7/ 1.5
(3x)(3x)
Abnormal expression in Abnormal expression in Rett frontal cortexRett frontal cortex

Biology of Rett SyndromeBiology of Rett Syndrome
• functional abnormalities of energy production?functional abnormalities of energy production?– previous functional and structural studiesprevious functional and structural studies
• altered control of cell survival?altered control of cell survival?– increased sensitivity to agents that promote cell deathincreased sensitivity to agents that promote cell death
• abnormalities of communication between brain cells?abnormalities of communication between brain cells?– MeCP2 also found in synaptic regions, & shows punctate MeCP2 also found in synaptic regions, & shows punctate
cytoplasmic staining in COS-7 cells, WBC, fibroblasts and cytoplasmic staining in COS-7 cells, WBC, fibroblasts and PC12 cellsPC12 cells
plan to study these in more detail using our cell plan to study these in more detail using our cell culture model and mouse models at our disposalculture model and mouse models at our disposal

11stst Dimension Dimension Isoelectric Focusing Isoelectric Focusing EquilibrationEquilibration
22ndnd Dimension Dimension SDS- SDS-Polyacrylamide Gel Polyacrylamide Gel ElectrophoresisElectrophoresis
image Gelimage GelUsing the Typhoon MULTI LASER Using the Typhoon MULTI LASER ScannerScanner
Image AnalysisImage Analysis
Proteomic Study on Proteomic Study on Mecp2Mecp2
Mouse ModelMouse ModelDIGE (Differential Imaging GEL Electrophoresis)DIGE (Differential Imaging GEL Electrophoresis)

Overlaid ImageOverlaid Image
33 PHPH 10 10
250250
1515
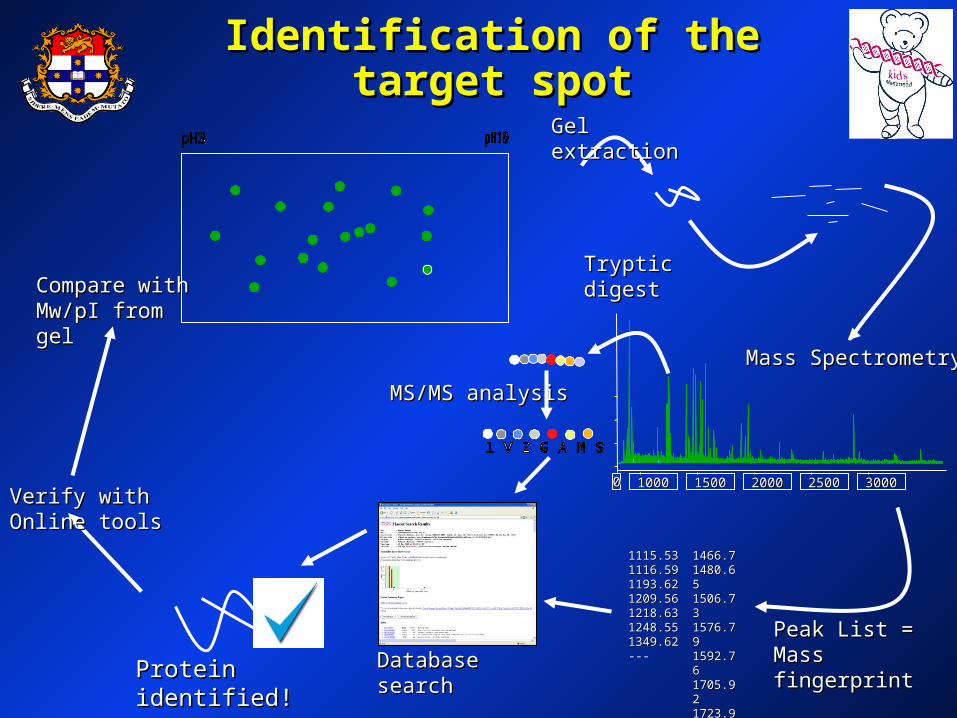
MS/MS analysisMS/MS analysis
Compare with Compare with Mw/pI from gelMw/pI from gel
Verify with Online Verify with Online toolstools
Tryptic digestTryptic digest
Gel extractionGel extraction
00 1000 1000 1500 1500 2000 2000 2500 2500 3000 3000
Mass SpectrometryMass Spectrometry
1115.531115.531116.591116.591193.621193.621209.561209.561218.631218.631248.551248.551349.621349.62------
1466.71466.71480.651480.651506.731506.731576.791576.791592.761592.761705.921705.921723.91723.9------
Peak List = Peak List = Mass fingerprintMass fingerprintDatabase searchDatabase searchProtein identified!Protein identified!
Identification of the target Identification of the target spotspot

ConclusionsConclusions
• the biological processes involved in RTT may in part the biological processes involved in RTT may in part be a consequence of abnormalities of:be a consequence of abnormalities of:
– energy productionenergy production
– cell survivalcell survival
– communication between brain cellscommunication between brain cells
• study of CDKL5/MeCP2 interactions will yield further study of CDKL5/MeCP2 interactions will yield further insights into RTT biologyinsights into RTT biology
• combination of clinical, combination of clinical, in vitroin vitro and animal model and animal model research is needed to answer questions relating to research is needed to answer questions relating to the biology of RTTthe biology of RTT
• the clinical - laboratory interface is critical to the clinical - laboratory interface is critical to translating research into clinical practicetranslating research into clinical practice

CollaboratorsCollaboratorsChildren’s Hospital at Westmead GroupChildren’s Hospital at Westmead Group
Current teamCurrent team Past teamPast teamRoksana ArmaniRoksana Armani Linda WeavingLinda WeavingBruce BennettsBruce Bennetts Alexandra BezlerAlexandra Bezler
Desiree Cloosterman Andrew GrimmDesiree Cloosterman Andrew Grimm Carolyn EllawayCarolyn Ellaway Joanne Gibson Joanne GibsonGladys HoGladys Ho Simon Hardwick Simon HardwickRania Kairouz-WabheRania Kairouz-Wabhe Hooshang LahootiHooshang Lahooti Vidya VasudevanVidya Vasudevan Abid Mohamedali Abid MohamedaliSarah Williamson Rose WhiteSarah Williamson Rose White
Children’s Medical Research InstituteChildren’s Medical Research Institute Patrick TamPatrick Tam
Gregory PelkaGregory Pelka Abid Mohamedali Abid Mohamedali
Phil RobinsonPhil Robinson
TVW Telethon Research Institute, PerthTVW Telethon Research Institute, PerthHelen Leonard & her ARSD teamHelen Leonard & her ARSD team
Westmead Millennium InstituteWestmead Millennium InstituteBarry Slobedman, Chris Bye & Josh SternBarry Slobedman, Chris Bye & Josh Stern
Baylor College of Medicine, HoustonBaylor College of Medicine, Houston
Huda ZoghbiHuda Zoghbi
Institute of Medical Genetics, Institute of Medical Genetics, University College of Medicine, CardiffUniversity College of Medicine, Cardiff
Angus Clarke, Hayley ArcherAngus Clarke, Hayley Archer
Women’s & Children’s Hospital, AdelaideWomen’s & Children’s Hospital, Adelaide Jozef GJozef Géécz, Kathie Friend & Olivia McKenziecz, Kathie Friend & Olivia McKenzie
West Australian Institute for Medical ResearchWest Australian Institute for Medical ResearchDavid Ravine & Alka SaxenaDavid Ravine & Alka Saxena

Funding AcknowledgementsFunding AcknowledgementsNHMRCNHMRC
International Rett Syndrome AssociationInternational Rett Syndrome Association
Rett Syndrome Research FoundationRett Syndrome Research Foundation
International Rett Syndrome FoundationInternational Rett Syndrome Foundation
Rotary Club of NarellanRotary Club of Narellan
CWA of NSWCWA of NSW
Rett Syndrome Australian Research Rett Syndrome Australian Research FundFund
Tissue Resource Centre, SydneyTissue Resource Centre, SydneyHarvard Brain BankHarvard Brain Bank


MethylatedMethylatedDNADNA
MeCP2MeCP2
AcAc
AcAc AcAc
AcAc
chromatin accessible & activechromatin accessible & active
Gene Silencing by Gene Silencing by Chromatin CondensationChromatin Condensation
chromatin condensed & inactivechromatin condensed & inactive
MeCP2MeCP2mSin3amSin3aHDACHDAC
MeCP2 binds to methyl-CpGsMeCP2 binds to methyl-CpGs
recruitment of mSin3a & recruitment of mSin3a & histone deacetylase (HDAC)histone deacetylase (HDAC)
HDACHDAC
mSin3amSin3a

Large Deletions in our RTT Large Deletions in our RTT PatientsPatients
teltel cencen
11 22 33 Exon 4Exon 4
MBDMBD TRDTRD 3’UTR3’UTR
11
~15 kb~15 kb
4.609 kb4.609 kb
~47 kb~47 kb
~37 kb~37 kb
~40 kb~40 kb
~65 kb~65 kb

CDKL5/STK9CDKL5/STK9
• novel, conserved serine/threonine kinasenovel, conserved serine/threonine kinase
• large gene of 23 exons with 2 alternative transcription large gene of 23 exons with 2 alternative transcription start sites generating two isoformsstart sites generating two isoforms
• CDKL5 protein localisation - cytoplasm/nucleus?CDKL5 protein localisation - cytoplasm/nucleus?
• wide tissue expression, including fetal and adult brainwide tissue expression, including fetal and adult brain

Unanswered Questions - CDKL5Unanswered Questions - CDKL5
• Does CDKL5 Does CDKL5 phosphorylate phosphorylate MeCP2 (and other MeCP2 (and other proteins)?proteins)?
POPO44
HDAC
mSin3amSin3a
MeCP2
HDAC
mSin3amSin3a
MeCP2
rat BDNF exon IIIrat BDNF exon III
rat BDNF exon IIIrat BDNF exon III
depolarizationdepolarization
Is CDKL5 the link??Is CDKL5 the link??

Unanswered QuestionsUnanswered Questions- CDKL5- CDKL5
• Does CDKL5 phosphorylate MeCP2 (and other proteins)?Does CDKL5 phosphorylate MeCP2 (and other proteins)?
• Do the different isoforms have different functions?Do the different isoforms have different functions?
• What is the developmental expression profile of What is the developmental expression profile of Cdkl5Cdkl5 in in mouse?mouse?
• Will mouse models for Will mouse models for Cdkl5Cdkl5 deficiency help us deficiency help us understand the biology of Rett syndrome?understand the biology of Rett syndrome?

Rett frontal cortexRett frontal cortex
Control occipital cortexControl occipital cortex
Rett occipital cortexRett occipital cortex
Control frontal cortexControl frontal cortex
Expression profilingExpression profiling
• cDNA microarrays with 19,000+ probe sequences cDNA microarrays with 19,000+ probe sequences (University Health Network, Ontario)(University Health Network, Ontario)
• 7 Rett and 7 control human frontal and occipital 7 Rett and 7 control human frontal and occipital corticescortices
• (a) Significance Analysis of Microarrays (a) Significance Analysis of Microarrays (modified t-test)(modified t-test)
• (b) >1.5 fold change, 5/7 biological replicates(b) >1.5 fold change, 5/7 biological replicates

Differentially expressed genesDifferentially expressed genes
13 UP13 UP
21 DOWN21 DOWN
14 unknown 14 unknown functionfunction
Rett frontal cortexRett frontal cortex
Control occipital cortexControl occipital cortex
Rett occipital cortexRett occipital cortex
Control frontal cortexControl frontal cortex
3434
1313
33
447 UP7 UP
6 DOWN6 DOWN
4 unknown 4 unknown functionfunction
Related Documents











