
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript



MICROBIOLOGY RESEARCH ADVANCES
RETINOIC ACID INDUCIBLE-1
GENE (RAI1) AND CLINICAL
SUBTYPES OF SCHIZOPHRENIA
The exclusive license for this PDF is limited to personal website use only. No part of this digital document may be reproduced, stored in a retrieval system or transmitted commercially in any form or by any means. The publisher has taken reasonable care in the preparation of this digital document, but makes no expressed or implied warranty of any kind and assumes no responsibility for any errors or omissions. No liability is assumed for incidental or consequential damages in connection with or arising out of information contained herein. This digital document is sold with the clear understanding that the publisher is not engaged in rendering legal, medical or any other professional services.

MICROBIOLOGY RESEARCH
ADVANCES
Additional E-books in this series can be found on Nova‘s website
under the E-book tab.
CELL BIOLOGY
RESEARCH PROGRESS
Additional E-books in this series can be found on Nova‘s website
under the E-book tab.

MICROBIOLOGY RESEARCH ADVANCES
RETINOIC ACID INDUCIBLE-1
GENE (RAI1) AND CLINICAL
SUBTYPES OF SCHIZOPHRENIA
DUŠANKA SAVIĆ PAVIĆEVIĆ
MAJA IVKOVIĆ
JELENA KARANOVIĆ
GORAN BRAJUŠKOVIĆ
AND
STANKA ROMAC
———————————————
Nova Science Publishers, Inc.
New York

Copyright © 2012 by Nova Science Publishers, Inc.
All rights reserved. No part of this book may be reproduced, stored in a retrieval
system or transmitted in any form or by any means: electronic, electrostatic,
magnetic, tape, mechanical photocopying, recording or otherwise without the written
permission of the Publisher.
For permission to use material from this book please contact us:
Telephone 631-231-7269; Fax 631-231-8175
Web Site: http://www.novapublishers.com
NOTICE TO THE READER
The Publisher has taken reasonable care in the preparation of this book, but makes no
expressed or implied warranty of any kind and assumes no responsibility for any
errors or omissions. No liability is assumed for incidental or consequential damages
in connection with or arising out of information contained in this book. The Publisher
shall not be liable for any special, consequential, or exemplary damages resulting, in
whole or in part, from the readers‘ use of, or reliance upon, this material. Any parts of
this book based on government reports are so indicated and copyright is claimed for
those parts to the extent applicable to compilations of such works.
Independent verification should be sought for any data, advice or recommendations
contained in this book. In addition, no responsibility is assumed by the publisher for
any injury and/or damage to persons or property arising from any methods, products,
instructions, ideas or otherwise contained in this publication.
This publication is designed to provide accurate and authoritative information with
regard to the subject matter covered herein. It is sold with the clear understanding
that the Publisher is not engaged in rendering legal or any other professional services.
If legal or any other expert assistance is required, the services of a competent person
should be sought. FROM A DECLARATION OF PARTICIPANTS JOINTLY
ADOPTED BY A COMMITTEE OF THE AMERICAN BAR ASSOCIATION
AND A COMMITTEE OF PUBLISHERS.
LIBRARY OF CONGRESS CATALOGING-IN-PUBLICATION DATA
ISBN: 978-1-61942-471-5
Published by Nova Science Publishers, Inc. † New York

CONTENTS
Preface vii
Chapter 1 Introduction 1
Chapter 2 Methods 7
Chapter 3 Results 11
Chapter 4 Discussion 13
Acknowledgments 17
References 19
Index 27


PREFACE
Schizophrenia is a common neuropsychiatric disorder affecting
approximately 1% of the general population and displaying considerable
heterogeneity of symptoms, course and outcomes. It is generally
considered to be a neurodevelopmental disorder that ultimately affects
forebrain neurons and circuits. Retinoid signaling is involved in fetal
brain development, affecting patterning and neuronal differentiation, and
in the maintenance and regulation of neuronal plasticity in different areas
of the adult forebrain. Therefore, there may be a relationship between
retinoid signaling and perturbation involved in brain development and
neuronal plasticity in schizophrenia. Studies of the genes coding proteins
participating in retinoid metabolism and transport, retinoid signaling
pathway, as well as retinoic acid-responsive genes, are needed to
elucidate this relationship. To make contribution to this understudied
area, we present here our recent findings of a potential associations
between the retinoic acid inducible-1 (RAI1) gene and schizophrenic
patients of European descent.
RAI1 is retinoic acid-responsive gene expressed at high levels in the
neuronal and heart structures, and acts as transcription regulator with role
in neuronal development and differentiation, and neurobehavioral
regulation. It contains polymorphic CAG repeats coding for glutamine-
rich activation domain, involved in protein-protein interaction that can be
modulated by the number of repeats.
Our population-based case-control study showed that the number of
CAG repeats in the RAI1 gene ranged from 8 to 19 in the group of 115
unrelated patients, with the most frequent allele with 13 repeats, and

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. viii
from 10 to 19 in the group of 100 controls, with the most frequent allele
with 14 repeats. When alleles were divided as ≤13 and >13 repeats,
alleles ≤13 repeats appeared significantly more often in patient group
(57.6% patients vs. 47% controls; χ2=13813.0; p=.015).
Paranoid, disorganized, undifferentiated, and residual subtypes of
schizophrenia displayed similar allelic distributions (p=.208, Pearson‘s
chi-square test), which might be misleading as stratification of sample
reduced number of patients in each subgroup. When PANSS (Positive
and Negative Syndrome Scale) scores based stratification was analyzed,
significant association between alleles ≤13 repeats and positive forms of
schizophrenia was obtained (χ2=7.675; p=.021).
As patients with prominent delusions, hallucinations and/or
disorganized speech and behavior displayed significantly shorter CAG
repeat size compared to patients with predominant negative symptoms,
our finding suggests that polyglutamine polymorphism in RAI1 gene may
underlie processes at certain brain structures related to productive
symptomatology. Polyglutamine polymorphism may affect RAI1
interaction with partner proteins, modulating its function and thus
altering developmental and neuroplastic retinoid signaling in the brain.

Chapter 1
INTRODUCTION
Schizophrenia (MIM#181500) is a common neuropsychiatric
disorder displaying considerable heterogeneity in symptomatology,
etiopathology, neurobiology, treatment response, course, and outcomes.
The annual incidence of schizophrenia averages 15 per 100,000, the
point prevalence averages 4.5 per population of 1,000, and the risk of
developing the illness over one's lifetime averages 0.7% (Tandon et al.,
2008). Schizophrenia is characterized by an admixture of positive,
negative, cognitive, disorganization, psychomotor, and mood symptoms,
whose severity varies across patients and through the course of the illness
(Tandon et al., 2009). Positive symptoms are delusions, hallucinations
and other reality distortions, while negative ones includes impairments in
emotional experience and expression, loss of motivation, poverty of
speech, inability to experience pleasure, lack of initiative, lack of interest
and reduced social drive. Varying degrees of negative and cognitive
symptomatology often precede positive symptoms that usually begin in
adolescence or early adulthood. The illness is frequently a chronic and
relapsing with generally incomplete remissions, variable degrees of
functional impairment and social disability, frequent comorbid substance
abuse, and decreased longevity (Tandon et al., 2009).
The etiology of schizophrenia is heterogeneous and includes
complex interactions of many genetic and environmental risk factors
(Tienari et al., 2004; Tsuang et al., 2004). Schizophrenia is highly
heritable and genetic factors and gene-environmental interactions
contribute over 80% of the liability for developing illness (Tandon et al.,

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 2
2008). Despite the high degree of heritability, there is not a consensus
about genetic model for schizophrenia. According to the predominantly
accepted ―common disease‖ model, schizophrenia is a heterogeneous
polygenic/multifactorial disease with multiple common genetic
polymorphisms, each of which contributes a small effect to disease
susceptibility (Risch, 1990; Lichtermann et al., 2000). According another
model, schizophrenia is a highly heterogeneous genetic entity caused by
multiple, highly penetrant and individually very rare mutations that may
be specific to single cases or individual families (McClellan et al., 2007).
The third model postulates that epigenetic factors, heritable changes in
gene expression without DNA sequence variations, represent genetic
base of schizophrenia (Crow, 2007). The main epigenetic mechanisms
are DNA methylation and histone remodeling of chromatin structure.
Many environmental risk factors are linked to liability to develop
schizophrenia. They include both biological and psychosocial risk
factors: maternal infections and nutritional deficiency during the first and
early second trimester, obstetric and perinatal complications, older
paternal ages at conception, birth during late winter or early spring,
urbanicity and migration during the childhood period, cannabis use
during adolescence, social adversity and stressful life (Tandon et al.,
2008).
How genetic and environmental risk factors might interact to cause
schizophrenia and what neurobiological processes might mediate such
gene-gene, gene-environment, and environment-environment interactive
effects is not yet understood. There is a considerable body of evidence
for the "neurodevelopmental" model of schizophrenia, according which
illness represents the behavioral outcome of an aberration in
neurodevelopmental processes that ultimately affects forebrain neurons
and circuits, and begins long before the onset of clinical symptoms
(Murray and Lewis, 1987; Lewis and Levitt, 2002; Rapoport et al.,
2005). Reported abnormalities in neuronal migration and organization in
postmortem magnetic resonance imaging studies (Jakob and Beckmann,
1986) indicated early (pre- or perinatal) brain lesions involved in
schizophrenia, while reduced neuronal size and arborization (Selemon
and Goldman-Rakic, 1999) indicated extended time period of abnormal
neurodevelopment in schizophrenia. The later is supported by
longitudinal brain imaging studies that showed gray matter volume loss
was particularly striking in adolescence period of patients (Shenton et al.,

Introduction 3
2001) and appeared to be an exaggeration of the normal developmental
pattern (Giedd et al., 1996).
Retinoic acid, a derivate of vitamin A (retinol) that acts in the cells,
is involved in neurodevelopmental processes, affecting anteroposterior
patterning of the posterior hindbrain and the anterior spinal cord, and
neuronal differentiation (Maden, 2002). Certain aspects of forebrain
morphogenesis are also under control of retinoic acid (Schneider et al.,
2001). It has became evident that this molecular signal continues to play
a role in the adult, mediating neuronal plasticity in different areas of the
brain (e.g., hippocampus and components of the limbic system), normal
nigrostriatal functioning, nerve regeneration, and neuronal stem cell
production (Haskall et al., 2002; Madden, 2007).
The pathway of the retinoic acid synthesis and mechanism of its
action are referred as retinoid signaling or retinoid cascade (Maden,
2002: Maden 2007). Retinoic acid and other retinoids are obtained from
the diet in the form of retinyl esters or β-carotene. Lipoprotein lipase is
involved in reversible catalysis of retinyl esters to retinol. Circulating
retinol, bound to retinol-binding protein, is taken up by target cells.
Inside the cells retinol is converted to retinal by alcohol dehydrogenses,
and then to retinoic acid by retinealdehyde dehydrogenases. Synthesized
retinoic acid enters to nucleus and binds to nuclear retinoid receptors:
retinoic acid receptor (RARα, RARβ and RARγ) and retinoid X receptor
(RXRα, RXRβ and RXRγ) . Nuclear retinoid receptors are ligand-
activated transcription factors, which, as heterodimers, bind to retinoic
acid-responsive elements within the promoter/enhancer region of many
target genes, influencing their expression. The presence of a retinoic
acid-responsive elements has been identified, at least, in 27 genes, but it
is estimated that more than 500 genes are retinoic-acid responsive. Once
retinoic acid has activated nuclear retinoid receptors, it exits the nucleus
and is catabolized by one class of cytochrome P450 enzymes.
Accepting the "neurodevelopmental" model of schizophrenia and the
role of retinoic acid in neurodevelopment, maintenance and regulation of
neuronal plasticity in the adult brain, there may be a relationship between
retinoid signaling and disrupted neurodevelopment and neuronal
plasticity in some patients with schizophrenia. According to Goodman
(1998) there is three lines of evidence suggesting a close causal
relationship between deregulated retinoid pathway and pathophysiology
of schizophrenia. 1) Congenital anomalies similar to those caused by

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 4
retinoid dysfunction are found to schizophrenic patients and their
relatives (mental deficit, enlarged ventricles, agenesis of the corpus
callosum, microcephaly, and a variety of major and minor congenital
malformations, among which craniofacial and digital anomalies are
prominent) (Goodman, 1996). 2) Some of the genes coding proteins for
retinoid signaling are convergent loci to loci suggestively linked to
schizophrenia by genome-wide linkage studies (Moises et al, 1995;
Pulver et al., 1995; Ng et al., 2009). Potential relationship of retinoid
signaling genes and schizophrenia is supported by microarray analyses
and genome-wide association studies. Microarray analyses showed
decreased expression of retinaldehyde dehydrogenase 1A1 (ALDH1A1)
and albumin in schizophrenic patients (Goodman, 2005). Retinaldehyde
dehydrogenase 1A1 is normally strongly expressed in dopaminergic
neurons, while albumin is a ubiquitous serum transporter of retinoic acid
and some other free fatty acids. Genome-wide association studies
showed potential association of the genes for lipoprotein lipase (LPL)
and retinoic acid receptor β (RARB) with schizophrenia (GWAS, NIH).
3) Many retinoic acid-responsive genes can be connected to
neurotransmitter abnormalities in schizophrenia (Goodman, 1998) . For
example, retinoic acid may control the function of the dopaminergic
mesolimbic pathway, as there is a retinoic acid-response element within
the promoter of the D2 dopamine receptor gene (DRD2) (Samad et al.,
1997).
Retinoic acid inducible – 1 gene (RAI1) (ID#10743, MIM#607642)
is one of the retinoic acid-responsive genes, with the highest expression
in various brain regions and muscle heart (Tolouse et al., 2003;
GATExplorer). It is located on chromosome 17p11.2, contains six exons
and its promoter has binding sites for several regulatory proteins,
including a retinoic acid-responsive element, just upstream of exon 1
(Tolouse et al., 2003). RAI1 pre-mRNA undergoes alternative splicing,
giving 8 splice variants: 6 protein-coding transcripts and 2 processed
transcripts without protein coding potential (Ensembl). The RAI1 cDNA
characterized by Tolouse et al. (2003) was 7661 base pair long with an
open reading frame coding a 1906 amino-acid protein, with 79% identity
with its mouse ortholog Rai1 (ID#19377) (Imai et al., 1995).
Bioinformatics and comparative genomic analysis between human and
mouse orthologs revealed a zinc finger-like plant homeodomain at the C-
terminus that is conserved in the trithorax group of chromatin-based

Introduction 5
transcription regulators (Bi et al., 2004). This prediction suggested that
the RAI1 protein might be involved in transcription control as a part of a
multiprotein complex.
Mouse Rai1 gene is induced during neuronal differentiation of P19
embryonal carcinoma cells by retinoic acid, and is mainly expressed in
neuronal brain structures during development and adult life (Imai et al.,
1995). The deletion in 17p11.2 region and intragenic mutations in the
RAI1 gene is associated with Smith-Magenis syndrome (SMS,
MIM#182290), a mental retardation syndrome associated with various
congenital malformations, such as distinct craniofacial and skeletal
anomalies, and disrupted behavior including self-injurious behaviors and
sleep disturbance (Seranski et al., 2001; Edelman et al., 2007). All these
findings strongly suggest that retinoid signaling through RAI1 gene could
be involved in neurodevelopment, and its alternation may be related to
neurodevelopmental diseases.
RAI1 gene contains a polymorphic CAG repeats that code N-
terminal glutamine-rich activation domain (Seranski et al., 2001), an
important class of protein-protein interacting motifs (Tanese and Tjian,
1993; Gerber et al., 1994). Comparative genomic study of human, mouse
and rat showed that the RAI1 CAG repeats is one of the four rapidly
evolving coding triplet repeats in the human genome that are mainly
expressed in the brain (Huang et al., 2004). The number of the RAI1
CAG repeats varied up to 18 in humans, and is only 4 in the mouse
ortholog (Toulouse et al., 2003). Polyglutamine repeats are mainly found
in transcription regulators, suggesting they may be one of the main cause
for modulation of their activity, and thus result in subtle or overt genomic
effects (Gerber et al., 1994). CAG repeat polymorphism in the RAI1 gene
has been associated with the severity of schizophrenia and patient
response to neuroleptic medication (Jobber et al., 1999).
To make contribution to understudied relationship between retinoid
pathway and schizophrenia, we present here our recent findings of a
potential associations between the retinoic acid-responsive gene, RAI1
gene, and schizophrenic patients of European descent. We did
population-based case-control study with the aims to investigate a
potential association between the polymorphic CAG repeats in the RAI1
gene and schizophrenia, taking into account its clinical heterogeneity.


Chapter 2
METHODS
SUBJECTS
The clinical sample included 115 unrelated patients with
schizophrenia, 55 females and 60 males, treated at Institute for
Psychiatry, Clinical Centre of Serbia, Belgrade. All subjects were
inpatients, consecutively admitted to Institute from 2006 to 2008, due to
psychotic exacerbation. The patients were directly interviewed using the
Diagnostic Interview for Genetic Studies (DIGS) (Nurnberger et al.,
1994) and their medical records were comprehensively reviewed by a
research psychiatrist. Diagnosis was based on all available data,
according to DSM-IV criteria (American Psychiatry Association, 1994).
These criteria require the presence of psychotic symptoms for a
minimum period of one month and the exclusion of mood disorder,
substance use or other recognizable ―organic‖ etiological explanation for
psychotic symptomatology. Additionally, DSM-IV requires social
dysfunction and decline for a period of more than six months and the
exclusion of pervasive developmental disorder as an explanation for the
condition.
As schizophrenia displays considerably heterogeneity in clinical
manifestation, patients were stratified according the clinical features
defined by DSM-IV criteria (American Psychiatry Association, 1994)
and according the Positive and Negative Syndrome Scale (PANSS) (Kay
et al., 1988) .

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 8
In respect to clinical features defined by DSM-IV criteria, patients
were divided into 4 clinical subtypes: paranoid (n=37), disorganized
(n=25), undifferentiated (n=23), and residual (n=29). Paranoid type is
characterized by preoccupation with one or more systematized delusions
or presence of frequent hallucinations related to a single theme.
Disorganized type is associated with marked loosening of associations,
incoherence, grossly disorganized behavior, and flat or grossly
inappropriate affect. Undifferentiated type is considered when a patient
presents with psychotic symptoms that meet criteria for schizophrenia
but not for any specific subtype. Residual type is diagnosed by the
occurrence of at least one prior episode of florid phase of schizophrenia
(such as delusions, hallucinations, and disorganized thinking) with a
current clinical picture free from prominent psychotic symptoms, but
with minimal ‗residual‘ symptoms of the illness (principally cognitive
and negative symptoms).
Another stratification of patients included differentiation between the
patients with a predominantly positive (n=63) and a predominantly
negative (n=52) syndrome of schizophrenia by using the Positive and
Negative Syndrome Scale (PANSS) (Kay et al., 1988). PANSS is
designed to measure prevalence of positive, negative and general
psychopathology symptoms. Positive scale) includes delusions,
conceptual disorganization, hallucinatory behavior, excitement,
grandiosity and suspiciousness hostility, while negative scale) includes
blunted affect (lack of emotional reactivity), emotional withdrawal, poor
rapport, passive-pathetic social withdrawal, difficulty in abstract
thinking, lack of spontaneity and flow of conversation, and stereotyped
thinking (Kay et al., 1988). Patients who scored moderate or higher on at
least 3 of 7 positive items are considered as positive-type
schizophrenics), and those with the reverse pattern ("moderate" on at
least 3 negative items) as negative type). The ratings were performed in
the acute exacerbation phase, immediately after admission to hospital
treatment). Patients scored as a positive-type schizophrenics generally
showed prominent delusions, hallucinations and disorganized speech and
behavior
Controls were 100 unrelated healthy age-matched individuals, 50
males and 50 females, screened for DSM-IV axis 1 mental disorders
using the DIGS. All participating subjects were Caucasian from Serbia.
Informed consent was obtained from all participants, including the legal

Methods 9
guardians for incompetent patients. The study was approved by Ethic
Board of Clinical Center of Serbia.
DNA ANALYSES
Genomic DNA was extracted from peripheral blood samples using
Qiagen mini kit (QIAGEN, Germany), according to the manufacturer‘s
procedure. Polymerase chain reaction (PCR) was carried out using the
following primers RAI1/1: 5'- GCA GCG GGT CCA GAA TCT TC -3'
(forward) and RAI1/2: 5'- CAG TAG CCC TGG CCT TGC -3' (reverse)
in a total reaction volume of 12,5 µl. PCR reaction mix contained 100 ng
of genomic DNA, 1xTaq buffer +KCl -MgCl2 (pH 8.8) (Fermentas,
Germany), 1.5 mM MgCl2, 200 µM dNTPs, 0.5 µM of each primer, 0.6
µg/µl BSA and 0,03 U/µl of Taq polymerase (Fermentas, Germany).
PCR conditions consisted of a denaturation step at 96oC for 3 min,
followed by 30 cycles of denaturation at 94oC for 1 min, annealing at
60oC for 1 min and extension at 72
oC for 1 min, and a final extension at
72oC for 20 min.
The absolute number of the RAI1 CAG repeats for each sample was
identified on a silver stained 6% denaturing polyacrylamide gel. Samples
were compared with the alleles that were sequenced using BDT v.1.1. kit
(Applied Biosystems, CA, USA), and analyzed on an ABI 3100 Genetic
Analyzer (Applied Biosystems, CA, USA) with the instrument‘s
Sequencing Analysis Programs. To improve the possibility of detecting
possible expansions of the RAI1 CAG repeats we performed Southern
blot hybridization at 65 oC using a 3‘DIG-ddUTP-end labeled (CAG)12
probe.
STATISTICAL ANALYSIS
Differences in the RAI1 CAG repeat size between the patients and
controls were analyzed using Pearson‘s chi-square test.


Chapter 3
RESULTS
Patients with schizophrenia and healthy controls did not differ
according to age (p=0.23) and gender distribution (p=0.64). The mean
age of patients was 42.6±7.9, while the mean age of controls was
38.7±12.4. In patients‘ group, mean age at first psychiatric evaluation
was 21.8±2.8. Slightly over half of patients were assessed as having a
predominantly positive syndrome of schizophrenia according to PANSS
score.
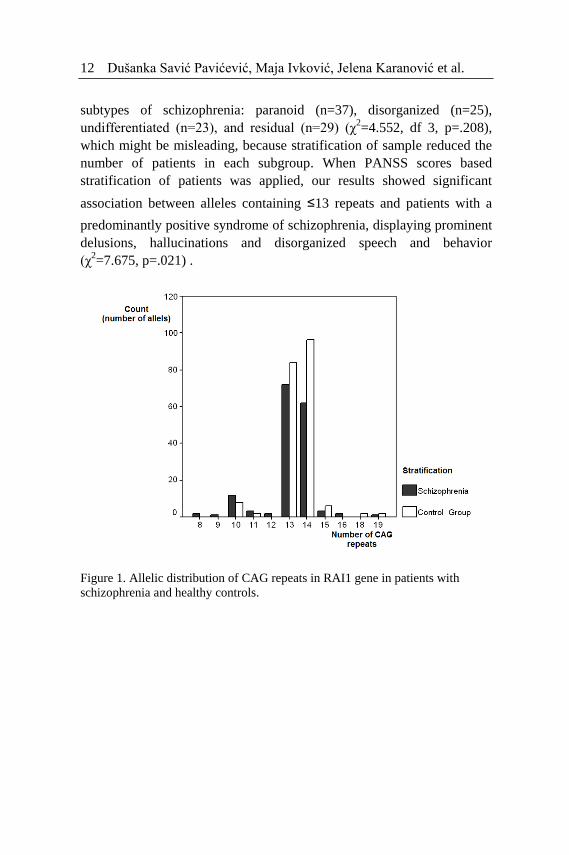
RAI1 CAG repeats allele distributions in patients with schizophrenia
and control groups are shown in Figure 1. The number of CAG repeats in
the RAI1 gene ranged from 8 to 19 in the patients with schizophrenia
(mean 12.86±1.41), with the most frequent one with 13 CAG repeats. In
control group, the number of CAG repeats ranged from 10 to 19 (mean
13.51±1.16), and the most frequent one was allele with 14 CAG repeats.
Alleles shorter than 10 repeats were observed only in the group of
patients with schizophrenia. Allele frequency distribution and
frequencies of individual alleles were not statistically significantly
different between patients and controls.
In order to see whether there is difference in an allele size interval
between patients and controls, data were dichotomized in intervals "less
or equal than" and "greater than" in respect to each allele size. When
alleles were divided as ≤13 and >13 repeats, it was shown that alleles
containing ≤13 repeats appeared significantly more often in patients
(57.6% patients vs. 47% controls; χ2=13813.0; p=.015) . There was no
association between the CAG repeat number and 4 different clinical
Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 12
subtypes of schizophrenia: paranoid (n=37), disorganized (n=25),
undifferentiated (n=23), and residual (n=29) (χ2=4.552, df 3, p=.208),
which might be misleading, because stratification of sample reduced the
number of patients in each subgroup. When PANSS scores based
stratification of patients was applied, our results showed significant
association between alleles containing ≤13 repeats and patients with a
predominantly positive syndrome of schizophrenia, displaying prominent
delusions, hallucinations and disorganized speech and behavior
(χ2=7.675, p=.021) .
Figure 1. Allelic distribution of CAG repeats in RAI1 gene in patients with
schizophrenia and healthy controls.

Chapter 4
DISCUSSION
One of the theories of pathogenesis proposes a neurodevelopmental
model for schizophrenia, with an early disruption in neuronal migration
or proliferation and late developmental derailments of peri-adolescent
process of synaptic pruning (Murray and Lewis, 1987; Lewis and Levitt,
2002; Rapoport et al., 2005). The model is supported by the occurrence
of specific neuronal loss and abnormal architecture in several brain
structures of schizophrenic patients. Retinoid signaling is involved in the
normal development of neuronal structures, including forebrain (Maden,
2002), and in the neuronal plasticity, maintenance and nerve regeneration
in the adult (Maden, 2007) . Deregulation of the retinoid signaling may
be an etiological factor of schizophrenia (Goodman, 1998), and studies
of the genes coding proteins participating in retinoid pathway, as well as
retinoid acid-responsive genes, are further needed to elucidate
relationship between retinoid signaling and schizophrenia. The
inducibility of RAI1 gene by retinoic-acid, its expression pattern with
highest level in a different brain structures, and its proposed role in the
neurodevelopmental and neurobehavioral processes, make this gene a
potential candidate for schizophrenia.
In our population-based case-control study we were interested in a
potential relationship between the polymorphic CAG repeats in the RAI1
gene and heterogeneous clinical manifestation of schizophrenia. Study
included 215 unrelated subjects of European descent (115 patients and
100 healthy controls) and showed significant association between shorter
alleles (≤13 CAG repeats) in RAI1 gene and schizophrenia. Different

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 14
subtypes of schizophrenia (paranoid, disorganized, undifferentiated, and
residual) displayed a similar allelic distributions, which might be
misleading, because stratification of sample reduced the number of
patients in each subgroup. Indeed, when patients were divided into larger
subgroups with a predominantly positive and predominantly negative
syndrome of schizophrenia, according to PANSS score, significant
difference was observed. Patients with prominent delusions,
hallucinations and disorganized speech and behavior displayed
significantly shorter alleles (≤13 CAG repeats) compared to patients with
a predominant negative syndrome of schizophrenia. According to our
study design, all patients were assessed in acute psychotic exacerbation.
This could be a limitation to our results, since PANSS score is not
constant parameter, and clinical manifestation could vary in same patient
through time. Nevertheless, studies repeatedly show negative symptoms
and cognitive deficit to be stable over time (Andreasen and Olson, 1982).
Considering the sample size and possible population stratification
bias, the obtained results should be taken with caution. However, our
findings could be important in terms of treatment and outcome, since a
positive syndrome of schizophrenia is generally better handled with
medication, and usually have more favorable prognosis than a negative
syndrome. Antipsychotic drugs are very effective mainly against positive
and disorganization symptoms (Mazure et al., 1992; Leucht et al., 2009;
Tandon et al., 2010), while are less successful in reducing negative
symptoms. They improve negative symptoms linked with positive ones,
and can worsen those negative symptoms associated with extrapyramidal
side effects (Stahl and Buckley, 2007). Additionally, response over the
first 2 to 4 weeks of antipsychotic therapy is highly predictive of long-
term response (Lambert et al., 2009; Kinon et al., 2010). The
responsiveness of a positive symptoms to antipsychotic drugs is linked to
a proposed underlying hyperactivity of the mesolimbic dopamine system
(Crow, 1980; Weinberger, 1987; Davis et al., 1991) as neuroleptics tend
to block D2 receptors in the brain's dopamine pathways (Snyder, 1989).
Previous study reported an association of the RAI1 CAG repeat
polymorphism with both the response to medication in schizophrenic
patients and the severity of the phenotype (Joober et al., 1999) .
Significantly shorter RAI1 CAG alleles were found in patients that
responded well to neuroleptic treatment, compared to non-responder and
controls. The group of neuroleptic responder had a later age of onset, and

Discussion 15
a better long-term outcome, as they were able to function autonomously
with only occasional supervision in domains of social and vocational
activities, compared to non-responder (Joober et al., 1999). To some
extent, our and Jobber and coworkers (1998) findings are in concordance
as disturbed dopamine neurotransmission is more frequently reported in
neuroleptic responder compared to non-responder (Mazure et al., 1991)
and, as mentioned above, a dopaminergic mesolimbic hyperactivity)
appears to underlie positive symptoms of schizophrenia (Crow, 1980;
Weinberger, 1987; Davis et al., 1991).
Our findings suggest that varying number of glutamine repeats in the
RAI1 gene may represent functional polymorphism related to a positive
syndrome of schizophrenia and, possibly, to underlying etiological
processes at certain brain structures). As it was suggested for some other
transcriptional regulators with homopolymeric stretches (Mitchell and
Tjian 1989; Perutz et al. 1994; Gerber et al., 1994), glutamine repeats can
mediate interaction of RAI1 protein with its partner protein and
variations in their number may alter the strength of that interactions. This
may slightly modulate RAI1 activity and transcriptional regulatory
network during the normal neurodevelopment, and thus contribute to a
positive syndrome of schizophrenia.
The relationship between RAI1 gene and disrupted forebrain
development is confirmed in patients suffering from Smith-Magensis
syndrome (Seranski et al., 2001). Comparison of the clinical
manifestation in patients with 17p11.2 deletion and intragenic mutations
in the RAI1 gene showed that haploinsufficiency of the RAI1 gene is
associated with craniofacial, behavioral, and neurologic signs and
symptoms of Smith-Magenis syndrome (Girirajan et al., 2005).
Moreover, experiments on animal models support RAI1 protein as a
transcriptional regulator involved in embryonic and postnatal
development, including neurologic and behavioral functions, in dose-
dependent manner. Most Rai1-/- mice died during gastrulation or
organogenesis, and survivors were growth retarded and displayed
malformations in both the craniofacial and the axial skeleton (Bi et al.,
2005). Obesity and craniofacial abnormalities were observed in Rai1+/-
mice (Bi et al., 2005). Transgenic mice with 4 and 6 copies of the Rai1
gene showed a dose-dependent exacerbation of the phenotype in mice
with higher Rai1 expression (Girirajan et al., 2008), The phenotype of
Rai1 transgenic mice included growth retardation, increased locomotor

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 16
activity, and severe neurologic deficits (impaired sensorimotor activity,
and abnormal anxiety-related behavior). According to a dose-dependent
effect of RAI1 gene on embryonic and postnatal development, it may be
speculated that polyglutamine polymorphism in RAI1 gene might be
related to more subtle functional alterations during development. Thus,
the varying number of coding CAG repeats in the RAI1 gene may
represent functional polymorphisam, which could slightly modify its
function in the normal neurodevelopment, and combined with other
genetic and/or environmental factors may predispose to a positive
syndrome of schizophrenia.
Although limited due to sample size and possible stratification bias,
our report, together with Jobber and coworkers' (1998) study, suggest
that short RAI1 CAG alleles might be related to clinical presentation,
treatment response, course and outcome in some schizophrenic patients.
Polyglutamine polymorphism in the RAI1 gene may affect interaction
between RAI1 and its partner proteins, modulating its activity as
transcription regulator to some extent. This may change retinoic-acid
response during neurodevelopment and neuronal plasticity, which may,
combined with other genetic and/or environmental factors, contribute to
a positive syndrome of schizophrenia. Further studies are needed to
elucidate possible relationship between retinoid signaling and
perturbation involved in brain development and neuronal plasticity in
schizophrenia. They may also reveal a new strategy for the treatment of
schizophrenia, as some retinoids has shown pharmacological successes in
dermatology and cancer (Thacher et al., 2000), and very recently a
beneficial effect in the antipsychotic treatment of schizophrenia patients
(Lerner et al., 2008). Study investigated the efficacy of augmentation of
bexarotene to ongoing antipsychotic treatment and showed significant
improvement on total PANSS score in chronic schizophrenia patients
who were stabilized on regular antipsychotic treatment (Lerner et al.,
2008).

ACKNOWLEDGEMENTS
This work was supported by grant #173016 funded by Serbian
Ministry of Education and Science.


REFERENCES
Alba, M.M. and Guigo, R., 2004. Comparative Analysis of Amino Acid
Repeats in Rodents and Humans. Genome Research. 14, 549-554.
American Psychiatric Association. Diagnostic and statistical manual of
mental disorders: DSM-IV, 4th ed. American Psychiatric
Association, Washington (DC), 1994.
Andreasen, N.C., Olson S, 1982. Negative versus positive schizophrenia:
definition and validation. Archives of Genetic Psychiatry 39, 789-
794.
Bi, W., Saifi, G.M., Shaw, C.J., Walz, K., Fonseca, P., Wilson, M.,
Potocki, L., Lupski, J.R., 2004. Mutations of RAI1, a PHD-
containing protein, in nondeletion patients with Smith-Magenis
syndrome. Human Genetics 115, 515-524.
Bi, W., Ohyama, T., Nakamura, H., Yan, J., Visvanathan, J., Justice, M.
J., Lupski, J. R., 2005. Inactivation of Rai1 in mice recapitulates
phenotypes observed in chromosome engineered mouse models for
Smith-Magenis syndrome. Human Molecular Genetics 14, 983-995.
Crow, T.J., 1980. Positive and negative schizophrenic symptoms and the
role of dopamine. British Journal of Psychiatry, 137, 383-386.
Crow, T.J., 2007. How and why genetic linkage has not solved the
problem of psychosis: review and hypothesis. American Journal of
Psychiatry 164, 13-21.
Davis, K.L., Kahn, R.S., Ko, G., Davidson, M., 1991. Dopamine in
schizophrenia: a review and reconceptualization. The American
Journal of Psychiatry 148, 1474–1486.
Edelman, E.A., Girirajan, S., Finucane, B., Patel, P.I., Lupski, J.R.,
Smith, A., Elsea, S.H., 2007. Gender, genotype, and phenotype

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 20
differences in Smith-Magenis syndrome: a meta-analysis of 105
cases. Clinical Genetics 71, 540-550.
Gerber, H.P., Seipel, K., Georgiev, O., Höfferer, M., Hug, M., Rusconi,
S., Schaffner, W., 1994. Transcriptional activation modulated by
homopolymeric glutamine and proline stretches. Science 263, 808-
811.
Giedd, J.N., Snell, J.W., Lange, N., Rajapakse, J.C., Casey, B.J., Kozuch,
P.L., Vaituzis, A.C., Vauss, Y.C., Hamburger, S.D., Kaysen, D.,
Rapoport, J.L.,1996. Quantitative magnetic resonance imaging of
human brain development: age 4-18. Cerebral Coretx 6, 551-560.
Girirajan, S., Elsas, L. J., II, Devriendt, K., Elsea, S. H., 2005. RAI1
variations in Smith-Magenis syndrome patients without 17p11.2
deletions. Journal Medical Genetics 42, 820-828.
Girirajan, S., Patel, N., Slager, R. E., Tokarz, M. E., Bucan, M., Wiley, J.
L., Elsea, S. H., 2008. How much is too much? Phenotypic
consequences of Rai1 overexpression in mice. European Journal of
Human Genetics 16, 941-954.
Goodman, A.B., 1996. Congenital anomalies in relatives of
schizophrenic probands may indicate a retinoid pathology.
Schizophrenia Research 19, 163-170.
Goodman, A.B., 1998. Three independent lines of evidence suggest
retinoids as causal to schizophrenia. Proceedings of the National
Academy of Sciences 95, 7240-7244.
Fiskerstrand, C.E., Lovejoy, E.A., Quinn, J.P., 1999. An intronic
polymorphic domain often associated with susceptibility to affective
disorders has allele dependent differential enhancer activity in
embryonic stem cells. Federation of European Biochemical Societies
Letters 458, 171-174.
Haskell, G.T., Maynard, T.M., Shatzmiller, R.A., Lamantia, A.S., 2002.
Retinoic acid signaling at sites of plasticity in the mature central
nervous system. Journal of Comparative Neurology 452, 228-241.
Huang, H., Winter, E.E., Wang, H., Weinstock, K.G., Xing, H.,
Goodstadt, L., Stenson P.D., Cooper D.N., Smith D., Albà M.M.,
Ponting C.P., Fechtel K., 2004. Evolutionary conservation and
selection of human disease gene orthologs in the rat and mouse
genomes. Genome Biology 5, R47.
Imai, Y., Suzuki, Y., Matsui, T., Tohyama, M., Wanaka, A., Takagi, T.,
1995. Cloning of a retinoic acid-induced gene, GT1, in the

References 21
embryonal carcinoma cell line P19: neuron-specific expression in the
mouse brain. Brain Research, Molecular Brain Research 31, 1-9.
Jakob, H. and Beckmann, H., 1986. Prenatal developmental disturbance
in the limbic allocortex in schizophrenics. Journal of Neuronal
Transmission 65, 303-326.
Joober, R., Benkelfat, C., Toulouse, A., Lafrenière, R.G., Lal, S., Ajroud.
S., Turecki G., Bloom D., Labelle A., Lalonde P., Alda M., Morgan
K., Palmour R., Rouleau G.A., 1999. Analysis of 14 CAG repeat-
containing genes in schizophrenia. American Journal of Medical
Genetics 88, 694-699.
Kay, S.R., Opler, L.A., Lindenmayer, J.P.,1988. Reliability and validity
of the positive and negative syndrome scale for schizophrenics.
Psychiatry Research 23, 99-110.
Karlin, S. and Burge, C., 1996. Trinucleotide repeats and long
homopeptides in genes and proteins associated with nervous system
disease and development. Proceedings of the National Academy of
Sciences 93,1560-1565.
Kinon, B.J., Chen, L., Ascher-Svanum, H., Stauffer, V.L., Kollack-
Walker, S., Zhou, W., Kapur, S., Kane, J.M., 2010. Early response
to antipsychotic drug therapy as a clinical marker of subsequent
response in the treatment of schizophrenia.
Neuropsychopharmacology 35, 581-590.
Lambert, M., Schimmelmann, B.G., Naber, D., Eich, F.X., Schulz, H.,
Huber, C.G., Karow, A., 2009. Early- and delayed antipsychotic
response and prediction of outcome in 528 severely impaired
patients with schizophrenia treated with amisulpride.
Pharmacopsychiatry 42, 277-283.
Lerner, V., Miodownik, C., Gibel, A., Kovalyonok, E., Shleifer, T.,
Goodman, A.B., Ritsner, M.S., 2008. Bexarotene as add-on to
antipsychotic treatment in schizophrenia patients: a pilot open-label
trial. Clinical Neuropharmacology 31, 25-33.
Leucht, S., Arbter, D., Engel, R.R., Kissling, W., Davis, J.M., 2009. How
effective are secondgeneration antipsychotic drugs? A meta-analysis
of placebo-controlled trials. Molecular Psychiatry 14, 429-447.
Lewis, D.A. and Levitt, P., 2002. Schizophrenia as a disorder of
neurodevelopment. Annual Review of Neuroscience 25, 409-432.

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 22
Lichtermann, D., Karbe, E., Maier, W., 2000. The genetic epidemiology
of schizophrenia and of schizophrenia spectrum disorders. European
Archives of Psychiatry and Clinical Neuroscience 250, 304-310.
Luo, T., Wagner, E., Grun, F., Drager, U.C., 2004. Retinoic acid
signaling in the brain marks formation of optic projections,
maturation of the dorsal telenchefalon, and function of limbic
system. Journal of Comparative Neurology 470, 297-316.
Maden, M., 2002. Retinoid signalling in the development of the central
nervous system. Nature Reviews Neuroscionce 3, 843-853.
Maden, M., 2007. Retionoic acid in the development, regeneration and
maintance of the nervous system. Nature Reviews Neuroscience 8,
755-765.
Mazure, C.M., Nelson, J.C., Jatlow, P.I., Bowers, M.B., 1991. Plasma
free homovanillic acid (HVA) as a predictor of clinical response in
acute psychosis. Biological Psychiatry 30, 475-482.
Mazure, C.M., Nelson, J.C., Jatlow, P.I., Bowers, M.B., 1992. Drug-
responsive symptoms during early neuroleptic treatment. Psychiatry
Research 41, 147-154.
McClellan, J., Susser, E., King, M.-C., 2007. Schizophrenia: a common
disease caused by multiple rare alleles. British Journal of Psychiatry
190, 194-199.
Mirnics, K., Middleton, F.A., Lewis, D.A. and Levitt, P., 2001. Analysis
of complex brain disorders with gene expression microarrays:
schizophrenia as a disease of the synapse. Trends in Neuroscience
24, 479-486.
Mitchell, P.J. and Tjian, R., 1989. Transcriptional regulation in
mammalian cells by sequence-specific DNA binding proteins.
Science 245, 371-378.
Moises, H.W., Yang, L., Kristbjarnarson, Wiese, H.C., Byerley, W.,
Macciardi, F., Arolt, V., Blackwood, D., Liu, X., Sjögren, B.,
Aschauer, H.N., Hwu, H.-G., Jang, K., Livesley, W.J., Kennedy,
J.L., Zoega, T., Ivarsson, O., Bui, M.-T., Yu, M.-H., Havsteen, B.,
Commenges, D., Weissenbach, J., Schwinger, E., Gottesman, I.I.,
Pakstis, A.J., Wetterberg, L., Kidd, K.K. and Helgason, T., 1995. An
international two−stage genome−wide search for schizophrenia
susceptibility genes. Nature Genetics 11, 321-324.

References 23
Murray, R.M., Lewis, S.W., 1987. Is schizophrenia a
neurodevelopmental disorder? [editorial]. British Medical Journal
(Clinical Research Ed.) 295, 681-682.
Ng, M.Y., Levinson, D.F., Faraone, S.V., Suarez, B.K., DeLisi, L.E.,
Arinami, T., Riley, B., Paunio, T., Pulver, A.E., Irmansyah,
Holmans, P.A., Escamilla, M., Wildenauer, D.B., Williams, N.M.,
Laurent, C., Mowry, B.J., Brzustowicz, L.M., Maziade, M., Sklar,
P., Garver, D.L., Abecasis, G.R., Lerer, B., Fallin, M.D., Gurling,
H.M., Gejman, P.V., Lindholm, E., Moises, H.W., Byerley, W.,
Wijsman, E.M., Forabosco, P., Tsuang, M.T., Hwu, H.G., Okazaki,
Y., Kendler, K.S., Wormley, B., Fanous, A., Walsh, D., O'Neill,
F.A., Peltonen, L., Nestadt, G., Lasseter, V.K., Liang, K.Y.,
Papadimitriou, G.M., Dikeos, D.G., Schwab, S.G., Owen, M.J.,
O'Donovan, M.C., Norton, N., Hare, E., Raventos, H., Nicolini, H.,
Albus, M., Maier, W., Nimgaonkar, V.L., Terenius, L., Mallet, J.,
Jay, M., Godard, S., Nertney, D., Alexander, M., Crowe, R.R.,
Silverman, J.M., Bassett, A.S., Roy, M.A., Mérette, C., Pato, C.N.,
Pato, M.T., Roos, J.L., Kohn, Y., Amann-Zalcenstein, D., Kalsi, G.,
McQuillin, A., Curtis, D., Brynjolfson, J., Sigmundsson, T.,
Petursson, H., Sanders, A.R., Duan, J., Jazin, E., Myles-Worsley, M.,
Karayiorgou, M., Lewis, C.M., 2009. Meta-analysis of 32 genome-
wide linkage studies of schizophrenia. Molecular Psychiatry 14, 774-
785.
Nurnberger, J.I. Jr, Blehar, M.C., Kaufmann, C.A., York-Cooler, C.,
Simpson, S.G., Harkavy-Friedman, J., Severe J.B., Malaspina D.,
Reich T., 1994. Diagnostic interview for genetic studies. Rationale,
unique features, and training. NIMH Genetics Initiative. Archives of
General Psychiatry 51, 849-859.
Perutz, M.F., Johnson, T., Suzuki, M., Finch, J.T., 1994. Glutamine
repeats as polar zippers: Their possible role in inherited
neurodegenerative diseases. Proceedings of the National Academy of
Sciences 91, 5355-5358.
Pulver, A.E., Lasseter, V.K., Kasch, L., Wolyniec, P., Nestadt, G.,
Blouin, J.L., Kimberland, M., Babb, R., Vourlis, S., Chen, H., et al.,
1995. Schizophrenia: a genome scan targets chromosomes 3p and 8p
as potential sites of susceptibility genes. American Journal of Human
Genetics 19, 252-260.

Dušanka Savić Pavićević, Maja Ivković, Jelena Karanović et al. 24
Risch, N., 1990. Linkage strategies for genetically complex traits. 1.
Multilocus models. American Journal of Human Genetics 46, 222-
228.
Samad, T.A., Krezel, W., Chambon, P., Borrelli, E, 1997. Regulation of
dopaminergic pathways by retinoids: activation of the D2 receptor
promoter by members of the retinoic acid receptor-retinoid X
receptor family. Proceedings of the National Academy of Sciences
94, 14349-14354.
Selemon, L.D. and Goldman-Rakic, P.S., 1999. The reduced neuropil
hypothesis: a circuit based model of schizophrenia. Biological
Psychiatry 45, 17-25.
Seranski, P., Hoff, C., Radelof, U., Hennig, S., Reinhardt, R., Schwartz,
C. E., Heiss, N. S., Poustka, A., 2001. RAI1 is a novel polyglutamine
encoding gene that is deleted in Smith-Magenis syndrome patients.
Gene 270, 69-76.
Shenton, M., Dickey, C.C., Frumin, M., McCarley, R.W., 2001. A
review of MRI findings in schizophrenia. Schizophrenia Research
49, 1-52.
Schneider, R.A., Hu, D., Rubenstein, J.L.R., Maden, M., Helms, J.A.,
2001. Local retinoid signaling coordinates forebrain and facial
morphogenesis by maintaining FGF8 and SHH. Development 128,
2755-2767.
Smith, D., Wagner, E., Koul, O., Mccafferty, P., Drager, U.C., 2001.
Retinoic acid synthesis for the developing telencephalon. Cerebral
Cortex 11, 894-905.
Snyder, S.H., 1989. Dopamine receptors, neuroleptics, and
schizophrenia. The American journal of psychiatry 138, 460–464.
Stahl, S.M., Buckley, P.F., 2007. Negative symptoms of schizophrenia: a
problem that will not go away. Acta Psychiatrica Scandinavica 115,
4-11.
Tanese, N. and Tjian, R., 1993. Coactivators and TAFs: A new class of
eukaryotic transcription factors that connect activators to the basal
machinery. Cold Spring Harbour Symposia on Quant Biology 58,
179-185.
Tandon, R., Keshavan, M.S. and Nasrallah, H.A., 2008. Schizophrenia,
"Just the Facts" What we know in 2008. 2. Epidemiology and
etiology. Schizophrenia Research 102, 1-18.

References 25
Tandon, R., Nasrallah, H.A. and Keshavan, M.S., 2009. Schizophrenia,
"Just the Facts" Clinical features and conceptualization.
Schizophrenia Research 110, 1-23.
Tandon, R., Nasrallah, H.A. and Keshavan, M.S., 2010. Schizophrenia,
―Just the Facts‖ 5. Treatment and prevention Past, present, and
future. Schizophrenia Research 122, 1-23.
Thacher, S.M., Vasudevan, J., Chandraratna, R.A., 2000. Therapeutic
applications for ligands of retinoid receptors. Current Pharmaceutical
Design 6, 25-58.
Tienari, P., Wynne, L.C., Sorri, A., et al., Lahti, I., Läksy K, Moring J,
Naarala, M., Nieminen, P., Wahlberg, K,E., 2004. Genotype-
environment interaction in schizophrenia spectrum disorders. Long-
term follow-up study of Finnish adoptees. British Journal of
Psychiatry 184, 216-222.
Toulouse, A., Rochefort, D., Roussel, J., Joober, R., Rouleau, G.A.,
2003. Molecular cloning and characterization of human RAI1, a
gene associated with schizophrenia. Genomics 82, 162-71.
Tsuang, M.T., Bar, J.L., Stone, W.S. and Faraone, S.V., 2004. Gene-
environment interactions in mental disorders. World Psychiatry 3,
73-83.
Wagner, E., Luo, T., Sakai, Y., Parada, L.F., 2006. Drager UC. Retinoic
acid delineates the topography of neuronal plasticity in postnatal
cerebral cortex. European Journal of Neuroscience 24, 329-340.
Weinberger, D.R., 1987. Implications of normal brain development for
the pathogenesis of schizophrenia. Archives of General Psychiatry
44, 660–669.
Database
GWAS, NIH: http://gwas.nih.gov/
Ensembl: www.ensembl.org/
GATExplorer: http://bioinfow.dep.usal.es/xgate/
OMIM: www.ncbi.nlm.nih.gov/omim
Gene: www.ncbi.nlm.nih.gov/gene


INDEX
A
antipsychotic drugs, 14
responsiveness of a positive
symptoms, 14
D
DNA analyses
determination of RAI1 CAG
repets number, 9
isolation of DNA, 9
PCR for RAI1 CAG repeats, 9
dopaminergic mesolimbic
hyperactivity, 14, 15
DSM-IV criteria, 7
G
glutamine rich activated domen, 5
N
nuclear retionid receptors, 3
P
polyglutamine repeats, 5, 15
population-based case-control study
of RAI1 and schizophrenia, 5, 13
association between positive and
negative type of schizophrenia,
12, 14
treatment and outcome
imortance, 14
association with clinical subtypes
of schizophrenia, 12, 14
association with schizophrenia, 11,
13
controls, 8
patients, 7
stratification, 7
positive and negative syndrome scale
(PANSS), 8
negative scale, 8
negative type patients, 8
positive scale, 8
positive type patients, 8
ratings of patients, 8

Index 28
R
Rai1 animal models
Rai1-/- mice, 15
Rai1+/- mice, 15
transgenic Rai1 mice, 15
RAI1 protein, 4
role in transcription, 5
relationship with schizophrenia,
15, 16
retinoic acid, 3
biological role, 3, 13
catabolism, 3
causal relationship with
schizophrenia, 3, 13
mechanism of action, 3
synthesis, 3
retinoic acid inducible 1 gene
(RAI1), 4
expression, 4
mRNA, 4
polyglutamine repeats, 5
polymorphic CAG repeats, 5
allele distribution, 11, 12
association with schizophrenia,
5, 14, 15, 16
potential candidate gene for
schizophreania, 13
retinoic acid responsive elements, 3
retinoic acid responsive genes, 3
in schizophrenia, 4
retinoid signaling, 3
retinoids, 3
bexarotene, 16
potentail antipsychotic treatment,
16
S
schizophrenia, 1
annual incidence, 1
clinical subtypes, 8
disorganized, 8
paranoid, 8
residual, 8
undifferentiated, 8
cognitive symptoms, 1
environmental risk factors, 2
etiology, 1
genetic models, 2
heritability, 1
negative symptoms, 1
neurodevelopmental model, 2, 13
positive and negative syndrome
scale (PANSS), 8
positive symptoms, 1
prevalence, 1
risk for illness, 1
Smith-Magenis syndrome, 5, 15
Related Documents











