Restoration of Autophagy Alleviates Hepatic ER Stress and Impaired Insulin Signalling Transduction in High Fructose-Fed Male Mice Hao Wang, Ruo-Qiong Sun, Xiao-Yi Zeng, Xiu Zhou, Songpei Li, Eunjung Jo, Juan C. Molero, and Ji-Ming Ye Lipid Biology and Metabolic Disease Laboratory, Health Innovations Research Institute and School of Health Sciences, RMIT University, Melbourne, Victoria 3083, Australia High-carbohydrate (mainly fructose) consumption is a major dietary factor for hepatic insulin resistance, involving endoplasmic reticulum (ER) stress and lipid accumulation. Because autophagy has been implicated in ER stress, the present study investigated the role of autophagy in high- fructose (HFru) diet-induced hepatic ER stress and insulin resistance in male C57BL/6J mice. The results show that chronic HFru feeding induced glucose intolerance and impaired insulin signaling transduction in the liver, associated with ER stress and the accumulation of lipids. Intriguingly, hepatic autophagy was suppressed as a result of activation of mammalian target of rapamycin. The suppressed autophagy was detected within 6 hours after HFru feeding along with activation of both inositol-requiring enzyme 1 and protein kinase RNA-like endoplasmic reticulum kinase path- ways. These events occurred prior to lipid accumulation or lipogenesis and were sufficient to blunt insulin signaling transduction with activation of c-Jun N-terminal kinase/inhibitory-B kinase and serine phosphorylation of insulin receptor substrate 1. The stimulation of autophagy attenuated ER stress- and c-Jun N-terminal kinase/inhibitory-B kinase-associated impairment in insulin sig- naling transduction in a mammalian target of rapamycin -independent manner. Taken together, our data suggest that restoration of autophagy functions disrupted by fructose is able to alleviate ER stress and improve insulin signaling transduction. (Endocrinology 156: 169 –181, 2015) O verconsumption of carbohydrate (especially fruc- tose) is one of the major contributors to the current epidemic of the metabolic syndrome, including insulin re- sistance and fatty liver (1–3). Fructose is a main ingredient of high fructose corn syrup, a primary source of sweetener used in soft drinks and bakery items (4). Unlike other nutrients, fructose is almost entirely cleared at the first pass in the liver after absorption from the gut (3, 5). High- fructose ingestion can result in insulin resistance in the liver rapidly (within days) in humans (6, 7) and animals (8) before other tissues become insulin resistant. Compared with glucose, fructose also appears to be more deleterious to insulin action in both humans and animals (9 –11). Mechanistic studies have attributed fructose-induced insulin resistance to its strong lipogenic effect (12–14), which leads to ectopic lipid accumulation and consequen- tial interruption to insulin signaling transduction (15). Re- cently endoplasmic reticulum (ER) stress has emerged to be another important mechanism underlying insulin re- sistance (16 –18). For example, inositol-requiring enzyme ISSN Print 0013-7227 ISSN Online 1945-7170 Printed in U.S.A. Copyright © 2015 by the Endocrine Society Received June 6, 2014. Accepted October 16, 2014. First Published Online October 24, 2014 Abbreviations: ACC, acetyl-coenzyme A carboxylase; ATF6, activating transcription factor 6; Atg, autophagy-related gene; CH, chow; 4E-BP1, 4E-binding protein 1; eIF2, eukaryotic translation initiation factor 2; ER, endoplasmic reticulum; FAS, fatty acid synthase; Grp78, glucose-regulated protein 78; GSK3, glycogen synthase kinase-3; GTT, glucose tolerance test; HFru, high fructose; iAUC, incremental glucose area under the curve; IKK, inhibitory-B kinase; IRE1, inositol-requiring enzyme 1; IRS1, insulin receptor substrate 1; JNK, c-Jun N-ter- minal kinase; KHB, Krebs-Henseleit buffer; LC, light-chain; mSREBP1c, mature form of lipo- genic transcription factor SREBP1c; mTOR, mammalian target of rapamycin; PERK, protein kinase RNA-like endoplasmic reticulum kinase; p70 S6K, p70 S6 kinase; Rap, rapamycin; Rsv, resveratrol; SCD1, stearoyl coenzyme A desaturase 1; Spd, spermidine; SREBP-1, sterol regu- latory element-binding protein-1; sXBP1, spliced X-box-binding protein 1; UPR, unfolded pro- tein response; Veh, carrier; XBP1, X-box-binding protein 1. ENERGY BALANCE-OBESITY doi: 10.1210/en.2014-1454 Endocrinology, January 2015, 156(1):169 –181 endo.endojournals.org 169 The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Restoration of Autophagy Alleviates Hepatic ERStress and Impaired Insulin Signalling Transduction inHigh Fructose-Fed Male Mice
Hao Wang, Ruo-Qiong Sun, Xiao-Yi Zeng, Xiu Zhou, Songpei Li, Eunjung Jo,Juan C. Molero, and Ji-Ming Ye
Lipid Biology and Metabolic Disease Laboratory, Health Innovations Research Institute and School ofHealth Sciences, RMIT University, Melbourne, Victoria 3083, Australia
High-carbohydrate (mainly fructose) consumption is a major dietary factor for hepatic insulinresistance, involving endoplasmic reticulum (ER) stress and lipid accumulation. Because autophagyhas been implicated in ER stress, the present study investigated the role of autophagy in high-fructose (HFru) diet-induced hepatic ER stress and insulin resistance in male C57BL/6J mice. Theresults show that chronic HFru feeding induced glucose intolerance and impaired insulin signalingtransduction in the liver, associated with ER stress and the accumulation of lipids. Intriguingly,hepatic autophagy was suppressed as a result of activation of mammalian target of rapamycin. Thesuppressed autophagy was detected within 6 hours after HFru feeding along with activation ofboth inositol-requiring enzyme 1 and protein kinase RNA-like endoplasmic reticulum kinase path-ways. These events occurred prior to lipid accumulation or lipogenesis and were sufficient to bluntinsulin signaling transduction with activation of c-Jun N-terminal kinase/inhibitory-�B kinase andserine phosphorylation of insulin receptor substrate 1. The stimulation of autophagy attenuatedER stress- and c-Jun N-terminal kinase/inhibitory-�B kinase-associated impairment in insulin sig-naling transduction in a mammalian target of rapamycin -independent manner. Taken together,our data suggest that restoration of autophagy functions disrupted by fructose is able to alleviateER stress and improve insulin signaling transduction. (Endocrinology 156: 169–181, 2015)
Overconsumption of carbohydrate (especially fruc-tose) is one of the major contributors to the current
epidemic of the metabolic syndrome, including insulin re-sistance and fatty liver (1–3). Fructose is a main ingredientof high fructose corn syrup, a primary source of sweetenerused in soft drinks and bakery items (4). Unlike othernutrients, fructose is almost entirely cleared at the firstpass in the liver after absorption from the gut (3, 5). High-fructose ingestion can result in insulin resistance in theliver rapidly (within days) in humans (6, 7) and animals (8)
before other tissues become insulin resistant. Comparedwith glucose, fructose also appears to be more deleteriousto insulin action in both humans and animals (9–11).
Mechanistic studies have attributed fructose-inducedinsulin resistance to its strong lipogenic effect (12–14),which leads to ectopic lipid accumulation and consequen-tial interruption to insulin signaling transduction (15). Re-cently endoplasmic reticulum (ER) stress has emerged tobe another important mechanism underlying insulin re-sistance (16–18). For example, inositol-requiring enzyme
ISSN Print 0013-7227 ISSN Online 1945-7170Printed in U.S.A.Copyright © 2015 by the Endocrine SocietyReceived June 6, 2014. Accepted October 16, 2014.First Published Online October 24, 2014
Abbreviations: ACC, acetyl-coenzyme A carboxylase; ATF6�, activating transcription factor6�; Atg, autophagy-related gene; CH, chow; 4E-BP1, 4E-binding protein 1; eIF2�, eukaryotictranslation initiation factor 2�; ER, endoplasmic reticulum; FAS, fatty acid synthase; Grp78,glucose-regulated protein 78; GSK3�, glycogen synthase kinase-3�; GTT, glucose tolerancetest; HFru, high fructose; iAUC, incremental glucose area under the curve; IKK, inhibitory-�Bkinase; IRE1, inositol-requiring enzyme 1; IRS1, insulin receptor substrate 1; JNK, c-Jun N-ter-minal kinase; KHB, Krebs-Henseleit buffer; LC, light-chain; mSREBP1c, mature form of lipo-genic transcription factor SREBP1c; mTOR, mammalian target of rapamycin; PERK, proteinkinase RNA-like endoplasmic reticulum kinase; p70 S6K, p70 S6 kinase; Rap, rapamycin; Rsv,resveratrol; SCD1, stearoyl coenzyme A desaturase 1; Spd, spermidine; SREBP-1, sterol regu-latory element-binding protein-1; sXBP1, spliced X-box-binding protein 1; UPR, unfolded pro-tein response; Veh, carrier; XBP1, X-box-binding protein 1.
E N E R G Y B A L A N C E - O B E S I T Y
doi: 10.1210/en.2014-1454 Endocrinology, January 2015, 156(1):169–181 endo.endojournals.org 169
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

1 (IRE1) and protein kinase RNA-like endoplasmic retic-ulum kinase (PERK) branches of the unfolded protein re-sponse (UPR) pathways can activate c-Jun N-terminal ki-nase (JNK)/inhibitory-�B kinase (IKK), which have beensuggested to inhibit insulin signaling transduction byphosphorylation of insulin receptor substrate 1 (IRS1) atserine sites (16, 19–21). In addition, spliced X-box-bind-ing protein 1 (sXBP1), a downstream effector of IRE1, canalso promote lipogenesis and potentially lead to lipid-induced insulin resistance (22, 23).
Over the past decade, there has been increasing evidencesuggesting ER stress is closely associated with autophagy, adegradation process to remove protein aggregates and dam-aged organelles to maintain intracellular homeostasis (24–28). Autophagy was suggested to be an alternative machin-ery to dispose unfolded proteins apart from ER-associatedproteasomal degradation (24). A number of studies have in-dicated that autophagy can be activated in response to ERstressasaprotectivemechanism(25–28).Ontheotherhand,it hasalsobeen reported thatautophagyactivity is reduced inthe liver of high-fat-fed mice (29, 30) in association with ERstress and the genetic oblation of the autophagy-related gene(Atg)7 leads to hepatic ER stress and insulin resistance (30).Furthermore, the stimulation of autophagy in vivo by phar-macological agents can also reduce ER stress and apoptosisinpancreatic�-cells (31).Recent studies fromour laboratoryhave shown that ER stress is associated with HFru-inducedinsulin resistance (8, 32). However, the mechanism by whichhigh fructose (HFru) leads to ER stress is unknown. There-fore, the aim of current study is to investigate the role ofautophagy in ER stress and insulin resistance induced byHFru and the effects of autophagy modulation on these met-abolic events.
Materials and Methods
AnimalsMale C57BL/6J mice (10–12 wk) from the Animal Resources
Centre (Perth, Australia) were kept at 22 � 1°C on a 12-hourlight, 12-hour dark cycle. For chronic feeding studies, after 2weeks of acclimatization on chow (CH) diet (Rat and MouseCubes; Specialty Feeds; �70% carbohydrate, �10% fat, and�20% protein of caloric content), mice were fed ad libitum forup to 2 weeks with either a CH or a HFru diet (in-house prepared;35%fructose,35%starch,�10%fat,and�20%proteinofcaloriccontent) (32, 8). Twelve days after the diet intervention, intraperi-toneal (ip) glucose tolerance tests (GTTs;2.5gglucoseperkilogramof body weight) were performed on 5- to 7-hour fasted mice. Bloodglucose levels were monitored using a glucometer (AccuCheck Pro-forma Nano; Roche), and blood samples were collected at 0, 30,and60minutes forplasmainsulinmeasurementbyRIA(Millipore).At the end of 2 weeks, mice were fasted for 4–6 hours before culledfor tissue. Twenty minutes before the tissue collection, insulin (2
U/kg body weight) or vehicle (equivalent volume of saline) wasinjected (ip) to assess insulin signaling (8). Tissues of interest werecollected and freeze clamped immediately.
For acute feeding studies, after 2 weeks of acclimatization ona CH diet, the mice were fasted for 24 hours as the basal con-dition before being fed a CH or HFru diet for the periods indi-cated. Mice were killed at the end of each feeding time point andtissues of interest were freeze-clamped immediately. For drugtreatments, rapamycin (Rap; 4 mg/kg; Santa Cruz Biotechnol-ogy), spermidine (Spd; 50 mg/kg; Sigma), resveratrol (Rsv; 25mg/kg, Sigma), or carrier (Veh; 4% ethanol, 5% polyethyleneglycol 400, 5% Tween 80) was injected (ip) 1 hour prior to the6-hour feeding period (33–35). For the assessment of insulinsignaling in the liver, another cohort of mice was injected withinsulin (5 U/kg body weight, ip) along with glucose (3 g/kg bodyweight) to avoid hypoglycemia 40 minutes before the mice wereculled for tissues (32). All experiments were approved by theAnimal Ethics Committee of RMIT University.
Measurement of triglyceride levelsLiver samples were homogenized in chloroform-methanol
(2:1) solution before being subjected to an overnight lipid ex-traction with vigorous mixing. Two milliliters of 0.6% NaClwere then added, and the samples were centrifuged at 2000 rpmfor 10 minutes. The resulting organic phase was then transferredinto a fresh glass tube and dried with N2 at 45°C. The dried pelletwas dissolved in ethanol immediately prior to assay. Triglyceridelevels were measured using a Peridochrom triglyceride GPO-PAPkit (Roche Diagnostics).
Ex vivo studiesEx vivo studies were conducted as described previously (36).
Briefly, liver tissues were freshly collected from CH-fed mice andimmediately placed in ice-cold Krebs-Henseleit buffer (KHB; pH7.4, pregassed with carbogen). Tissues were then rinsed andchopped into 1- to 2-mm explants, followed by 30 minutes of pre-incubation at 37°C under continuous gassing with carbogen. Liverexplants were then incubated in KHB with or without 50 mM fruc-tose for 2 hours as indicated. At the end of the experiments, tissueswere pelleted and freeze clamped for immunoblot analysis.
Western blottingFreeze-clamped tissues were homogenized in ice-cold radio-
immunoprecipitation assay lysis buffer at pH 7.5 supplementedwith protease and phosphatase inhibitor cocktails. Protein levelsin the tissue homogenates were determined using the bicinchonnicacid method (Thermo Scientific) and resolved by SDS-PAGE. Pro-teins of interest were analyzed by imunoblotting using specific an-tibodies. Akt, phosphoSer473-Akt, glycogen synthase kinase-3�(GSK3�), phosphoSer9-GSK3�, acetyl-coenzyme A carboxylase(ACC), fatty acid synthase (FAS), stearoyl coenzyme A desaturase1 (SCD1), phosphoThr980-PERK, eukaryotic translation initiationfactor 2� (eIF2�), phosphoSer51-eIF2�, glucose-regulated protein(Grp) 78, mammalian target of rapamycin (mTOR), phosphoSer2448-mTOR, p70 S6 kinase (p70 S6K), phosphoThr389-p70 S6K,4E-binding protein 1 (4E-BP1), phosphoThr37/46-4E-BP1, au-tophagy-related gene (Atg)-7, Atg5, light-chain (LC) 3A/B, p62,JNK, phosphoThr183/Tyr185-JNK, c-Jun, phosphoSer73-cJun, IKK�,andphosphoSer176/180-IKK�/�werepurchasedfromCellSignaling.Sterol regulatory element-binding protein-1 (SREBP-1), X-box-
170 Wang et al Autophagy in Fructose-Induced Hepatic ER Stress Endocrinology, January 2015, 156(1):169–181
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

binding protein 1 (XBP1), and activating transcription factor6� (ATF6�) were purchased from Santa Cruz Biotechnology.IRE1 and phosphoSer724-IRE1 were purchased from Abcam.PhosphoSer307-IRS1 was purchased from Millipore. Proteins werevisualized using a ChemiDoc densitometer and quantified usingImage Lab software (Bio-Rad Laboratories).
Statistical analysisData are presented as means � SE. A one-way ANOVA was
used for comparison of the relevant groups unless indicated oth-erwise. When significant differences were found, the Tukey-Kramer multiple comparisons test was applied. Differences atP � .05 were considered to be statistically significant.
Results
HFru feeding induced glucose intolerance andimpaired hepatic insulin signaling transduction
HFru feeding for 2 weeks induced a significant increasein body weight gain (�2-fold, P � .05) compared with
CH-fed mice. This was accompanied with higher caloricintake (�30%, P � .01) and adiposity (�40%, P � .05)(Supplemental Table 1). Fasting blood glucose was alsosignificantly higher in HFru-fed mice, whereas no appar-ent differences in plasma insulin levels were observed.HFru feeding induced glucose intolerance, as indicated bya 30% (P � .05) increase in the incremental glucose areaunder the curve (iAUC) compared with CH-fed mice (Fig-ure 1, A and B). This effect was achieved without anysignificant change in the circulating levels of insulin duringthe glucose tolerance test (Figure 1C). We further exam-ined insulin-dependent phosphorylation of Akt and itssubstrate GSK3� in liver and skeletal muscle. Control CH-fed mice with insulin stimulation exhibited elevated phos-phorylation of Akt and GSK3� in liver by 14- and 3-fold,respectively (Figure 1D), whereas the HFru-fed mice elic-ited a 40%–50% reduction in insulin-stimulated phos-phorylation of Akt (P � .01) and GSK3� (P � .01). How-
Figure 1. HFru feeding induced insulin resistance. A, GTT was performed in 2 weeks HFru (f) or CH (E)-fed mice and glucose tolerance wasassessed by iAUC (B). Plasma insulin levels (C) during GTT were measured (8–12 mice per group). *, P � .05, **, P � .01 compared with CH. D,Liver insulin signaling transduction was determined by phosphorylation of Akt and GSK3� in response to insulin stimulation withimmnunoblotting. Two-way ANOVA was used to analyze insulin stimulated phosphorylation of Akt and GSK3� between diets (6–8 mice pergroup). *, P � .05, **, P � .01 compared with CH; ##, P � .01 compared with CH (�insulin).
doi: 10.1210/en.2014-1454 endo.endojournals.org 171
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

ever, we did not observe any significant difference in thephosphorylation of either protein in the skeletal muscle(Supplemental Figure 1A).
HFru feeding increased lipogenesis and ER stressin liver but not in muscle
HFru feeding for 2 weeks increased liver triglyceridessignificantly by greater than 2-fold compared with CHfeeding (Figure 2A). In agreement with this, the matureform of lipogenic transcription factor SREBP1c(mSREBP1c) was significantly elevated (3-fold, P � .01)(Figure 2B). The elevation of mSREBP1c also resulted inan up-regulation of its target lipogenic enzymes, includingACC (3-fold, P � .01), FAS (2-fold, P � .01), and SCD1(1.7-fold, P � .01) compared with CH-fed mice (Figure2B). HFru feeding did not alter the protein levels ofmSREBP1c, ACC, or SCD1 in skeletal muscle (Supple-mental Figure 1, B and C). HFru feeding also activated theIRE1 arm of the UPR pathway in liver, indicated by theincrease in phosphorylation of IRE1 (2.6-fold, P � .01)and the spliced form of its downstream target, XBP1 (1.6-fold, P � .01) (Figure 2C). Although we did not observeany significant changes in the phosphorylation of PERK inHFru-fed mice, there was a 1.8-fold increase in the phos-phorylation of eIF2�, a downstream of PERK. The levelsof ATF6, a marker of the third arm of the UPR pathways,were not affected by HFru feeding. In comparison, therewas no significant change in any of the measured markers
of the lipogenic and UPR pathwaysin skeletal muscle (Supplemental Fig-ure 1, B and D).
HFru feeding suppressedautophagy in liver but not inmuscle
We next examined whether au-tophagy was altered by HFru feedingin relation to ER stress. As shown inFigure 3A, Atg 5 and Atg 7, two pro-teins involved in the elongation of theautophagosome membrane were sig-nificantly reduced in HFu-fed miceby 12% and 27%, respectively. Au-tophagy activity is commonly assessedby the conversion of LC3II from LC3I(37), aprocessduringautophagosomeformation. Our results showed thatHFru feeding reduced LC3II to LC3Iratio in the liver by 30% (P � .05)compared with CH feeding. We fur-ther examined p62, an autophago-some protein cargo that directs target
proteins for degradation (37) and found a 30% (P � .05)increase in HFru-fed animals. Similar to the unaltered lipo-genic and UPR pathways in the skeletal muscle, the levels ofAtg7, LC3II, or p62 were not affected by HFru feeding (Sup-plemental Figure 1E).
HFru feeding activated mTOR pathways in liverbut not in muscle
Because mTOR has been reported to suppress au-tophagy (38), we next examined whether the suppressionof autophagy induced by HFru feeding was due to theactivation of mTOR. As shown in Figure 3B, HFru feedingindeed activated mTOR in the liver, indicated by a 70%increase in its phosphorylation compared with CH. Thiswas supported by the augment in the phosphorylation oftwo mTOR downstream targets, p70 S6K (2.5-fold, P �
.05) and 4E-BP1 (30%, P � .05). As expected, HFru feed-ing did not alter the phosphorylation of mTOR or p70 S6Kin skeletal muscle (Supplemental Figure 1F). To confirmthe direct effect of fructose on mTOR, we incubated liverexplants with fructose medium in the absence of aminoacid and insulin. Similar to what was observed in vivo,phosphorylation of both mTOR and p70 S6K was in-creased in liver explants after fructose treatment. Mean-while, the LC3II to LC3I ratio was reduced. These changeswere also accompanied with an elevation in phosphory-lated IRE1 and eIF2� (Figure 3C).
Figure 2. HFru feeding increased hepatic lipogenesis and ER stress. A, Triglyceride content wasdetermined in the liver of 2 weeks CH- and HFru-fed mice. Lipogenic proteins (B) mSREBP1c,ACC, FAS, SCD1, and ER stress markers (C) phosopho/total-IRE1, sXBP1, phospho-PERK,phospho/total-eIF2�, and ATF6 were measured with immunoblotting (8–12 mice per group).*, P � .05, **, P � .01 compared with CH.
172 Wang et al Autophagy in Fructose-Induced Hepatic ER Stress Endocrinology, January 2015, 156(1):169–181
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

Sequential changes in ER stress, lipogenesis, andautophagy during HFru feeding
Because changes in ER stress, lipogenesis, and au-tophagy were all apparent after 2 weeks of HFru feeding,we sought for shorter feeding period to determine whichof these events are manifested earlier. However, the cooc-
currence of these events was also observed, even after 1day of HFru feeding. This was supported by the increasedphosphorylation of mTOR and p70 S6K (2- to 4-fold),reduced LC3II to LC3I ratio (25%), increased level of p62(30%) (Supplemental Figure 2), and increased lipogene-sis-ER stress markers (data not shown). Therefore, a 2- to
Figure 3. HFru feeding suppressed autophagy and activated mTOR in the liver. A, After mice were fed a CH or HFru diet for 2 weeks, autophagywas assessed by autophagy proteins Atg7, Atg5, LC3II/LC3I, and p62 with immunoblotting. B, mTOR was determined by phosphorylation status ofmTOR and its downstream targets p70 S6K and 4E-BP1 with immunoblotting (8–12 mice per group). *, P � .05, **, P � .01 compared with CH.C, Liver explants were incubated in KHB without (control) or with 50 mM fructose (fructose) for 2 hours in the absence of insulin and amino acids.The effects of fructose on mTOR, autophagy, and ER stress were assessed by immunoblotting of the protein markers as indicated. *, P � .05compared with control (n � 6). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
doi: 10.1210/en.2014-1454 endo.endojournals.org 173
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

12 hour acute HFru feeding was used. To ensure the foodintake, mice were fasted for 24 hours (basal) prior to feed-ing. Our results suggest that the activation of mTOR andsuppression of autophagy were the first events triggered byHFru feeding, which can be detected as early as 2 hours.This was supported by the increase in the phosphorylationof mTOR and its downstream targets, p70 S6K and 4E-BP1 (2.6-fold, 7 fold, and 25%, respectively; all P � .05)(Figure 4, A–D). In contrast, the levels of the autophagyproteinAtg5inthe liverweresignificantlyreduced,evenafter2 hours of HFru feeding (20%, P � .01), and similar effectswere observed for Atg7 (Figure 4, F and G). Consistently, theratio of LC3II to LC3I was reduced by 60% (P � .01),whereas thep62 levelswere increasedby60%(P� .05)after2 hours of HFru feeding (Figure 4, H and I).
Notably, it was not until 6 hours of HFru feeding thatthe elevation of ER stress markers Grp78, phospho-IRE1,
and the spliced form of its downstream target XBP1 wasobserved (3.7-fold, 7.6-fold, and 44%, respectively, all P� .05) (Figure 5, A–D). Phospho-eIF2�, on the otherhand, increased after 2 hours of HFru feeding (88%, P �.01, Figure 5E). As for the lipogenic proteins, it was notuntil 12 hours of HFru feeding that a significant increasein ACC and FAS was observed (60%–80%, P � .05) alongwith a much greater up-regulation of SCD1 (30-fold, P �.01) (Figure 5, F and H–J). However, mSREBP1c proteinlevels and liver triglycerides remained unchangedthroughout this feeding period (Figure 5, F, G, and K).
Because 24 hours of fasting may affect these signalingpathways, we also compared nonfasted mice with 24-hour-fasted mice. There was no significant difference in the phos-phorylation of mTOR and p70 S6K between these 2 groups.Consistently, no difference in the LC3II to LC3I ratio or p62wasobserved (SupplementalFigure3A).Nonfastedmicedid
Figure 4. Acute HFru feeding activated mTOR and suppressed autophagy. Mice were acutely fed HFru diet for 2–12 hours after fasting (basal).Hepatic mTOR was determined by phosphorylation status of mTOR, p70 S6K, and 4E-BP1 (A) with densitometry using immunoblotting (B–D).Hepatic autophagy was assessed by autophagy proteins Atg7, Atg5, LC3II to LC3I ratio, and p62 (E) with densitometry using immunoblotting (F–I)(5–6 mice per group). *, P � .05, **, P � .01 compared with basal. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
174 Wang et al Autophagy in Fructose-Induced Hepatic ER Stress Endocrinology, January 2015, 156(1):169–181
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

not differ from the 24-hour-fasting group in either ER stressor inflammatory markers, except a greater Grp78 level (Sup-plemental Figure 3, B and C). We did observe higher lipo-genic protein expression in nonfasted mice, such as ACC andSCD1 (Supplemental Figure 3D).
Restoration of autophagy-alleviated ER stressinduced by HFru feeding
To investigate whether HFru-induced ER stress resultsfrom the suppression of autophagy, we treated HFru-fed
mice with autophagy inducers, both mTOR dependent(Rap) and mTOR independent (Spd and Rsv). As ex-pected,Rapreduced thephosphorylationofmTOR(30%,P � .01) and abolished the phosphorylation of p70 S6K(Figure 6, A–C). In contrast, neither Spd nor Rsv influ-enced the phosphorylation of these enzymes (Figure 6,A–C). Interestingly, all three agents restored autophagyactivity, as indicated by an increased LC3II to LC3I ratioand reduced p62 protein level (Figure 6, A, D, and E). Asa result, ER stress markers Grp78, phospho-IRE1, sXBP1,
Figure 5. HFru feeding led to ER stress and lipogenesis sequentially. Mice were acutely fed an HFru diet for 2–12 hours after fasting (basal).Hepatic ER stress and lipogenesis were measured by protein markers with immunoblotting: Grp78, phospho-IRE1, sXBP1, and phospho-eIF2� (A)with densitometry (B–E); mSREBP1c, ACC, FAS, and SCD1 (F) with densitometry (G–J). K, Triglyceride content in the liver was also assessed. 5–6mice per group. *, P � .05, **, P � .01 compared with basal. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
doi: 10.1210/en.2014-1454 endo.endojournals.org 175
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

and phospho-eIF2� were all significantly reduced (Figure6, F–J, all P � .05).
Restoration of autophagy abolished JNK/IKKactivation and improved insulin signalingtransduction
Both IRE1 and PERK have been reported to activateJNK/IKK to impair insulin signaling transduction by ser-ine-phosphorylating IRS1 (16, 19–21). As expected,HFru-fed mice exhibited increased phosphorylation ofIKK (2.4-fold, P � .01), JNK (43%, P � .01), and itsdownstream target cJun (5-fold, P � .01) (Figure 7, A–D).Consistently, serine phosphorylation levels of IRS1 weresignificantly elevated (40%, P � .01) (Figure 7E). This wasassociated with inhibition of insulin-stimulated phos-phorylation of Akt and GSK3� (by 40% and 30%, re-spectively, P � .01) (Figure 7, F and G). Moreover, treat-ment with Rap, Spd, and Rsv completely abolished theHFru-induced activation of JNK/IKK pathways and serinephosphorylation of IRS1 (Figure 7, A–E). As a result, theinsulin-stimulated phosphorylation of Akt was restoredafter the treatment of these autophagy drug inducers (allP � .05) (Figure 7F). A similar trend was also observed forGSK3� phosphorylation, although no statistical differ-ence was achieved (Figure 7G).
HFru diet showed more potent effect than CH dietin acute feeding
Feeding mice a CH diet after fasting has also been re-ported to induce ER stress (39). We therefore comparedmice fed HFru diet with those fed a CH diet for 6 hours.As shown in Supplemental Figure 4, there was a greaterthan 2-fold increase in mTOR and p70 S6K phosphory-lation in CH-fed mice compared with basal, but this effectwas more pronounced in HFru-fed mice (3.4-fold and 5.3-fold, respectively, P � .05 vs CH). Similarly, a 25% re-duction in the LC3II to LC3I ratio and a 50% increase inp62 were observed in CH-fed mice, whereas these changeswere more apparent in the HFru-fed mice (50% and2-fold, respectively, P � .05 vs CH). CH feeding elevatedonly Grp78 and phospho-IRE1 but not sXBP1 or phos-pho-eIF2�. HFru feeding increased all these ER stressmarkers. Its effects on Grp78 and phospho-IRE1 weremore potent than CH feeding (P � .05). No significantincrease in lipogenic proteins was observed with CHfeeding.
To examine whether the autophagy inducers can havesimilar effects on CH diet feeding, we treated mice withRap after fasting and fed mice a CH diet for 6 hours. Asshown in Supplemental Figure 5, Rap also normalizedmTOR and p70 S6K phosphorylation during CH feeding.Consequently, the LC3II to LC3I ratio and p62 levels were
restored. In contrast, there was a significant reduction inGrp78 after Rap treatment, although no suppression inphospho-IRE1 was observed. Because CH feeding did notalter the JNK/IKK signaling, there was no difference inphospho-JNK, phospho-IKK�/�, or phospho-IRS1 afterRap treatment. Consistently, the level of insulin stimu-lated phospho-Akt in Rap-treated mice was also compa-rable with CH-fed mice.
Discussion
The current study was set to investigate the role of au-tophagy in the development of ER stress and insulin re-sistance induced by HFru feeding. Several major findingshave emerged from this study. First, we found that HFrufeeding results in a marked suppression of autophagy inthe liver. To the best of our knowledge, this has not beenreported before. The suppression of autophagy is likely tobe mediated through the activation of mTOR, indepen-dent of insulin and amino acids. Second, HFru-inducedsuppression of autophagy may be a cause of ER stress andits resultant changes in JNK/IKK and insulin signaling cas-cades. These changes can occur in the absence of lipidaccumulation or the up-regulation of lipogenesis. Finally,restoration of autophagy by pharmacological agents isable to ameliorate ER stress and correct the associatedevents, irrespective of mTOR activity.
Dietary fructose is a major factor contributing to insu-lin resistance (1), which has been suggested to occur in theliver first (40). Clinical study in healthy subjects has shownbeing on a high-fructose diet for 6 days can lead to hepaticinsulin resistance (6, 7). In our current study, we alsoshowed that 2 weeks of HFru feeding in mice was suffi-cient to cause glucose intolerance with blunted insulin sig-naling in the liver but not in skeletal muscle (Figure 1 andSupplemental Figure 1). In contrast, we found both ERstress and increased lipogenesis in the liver but not in skel-etal muscle (Figure 2 and Supplemental Figure 1), suggest-ing ER stress and excess lipogenesis are potential mecha-nisms by which HFru feeding leads to hepatic insulinresistance.
Autophagy has been suggested to be triggered by ERstress to facilitate protein clearance (26). Intriguingly, ourstudy showed that autophagy was suppressed rather thanactivated in the presence of ER stress in the liver after micewere fed HFru for 2 weeks (Figure 3A). Similar observa-tions were also made in mice fed HFru for 2–12 hours(Figure 5) and liver explants incubated with fructose me-dium (Figure 3C). Recent studies from others have alsoshown hepatic ER stress is associated with suppression ofautophagy in high-fat fed and ob/ob mice (30), and dis-
176 Wang et al Autophagy in Fructose-Induced Hepatic ER Stress Endocrinology, January 2015, 156(1):169–181
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.
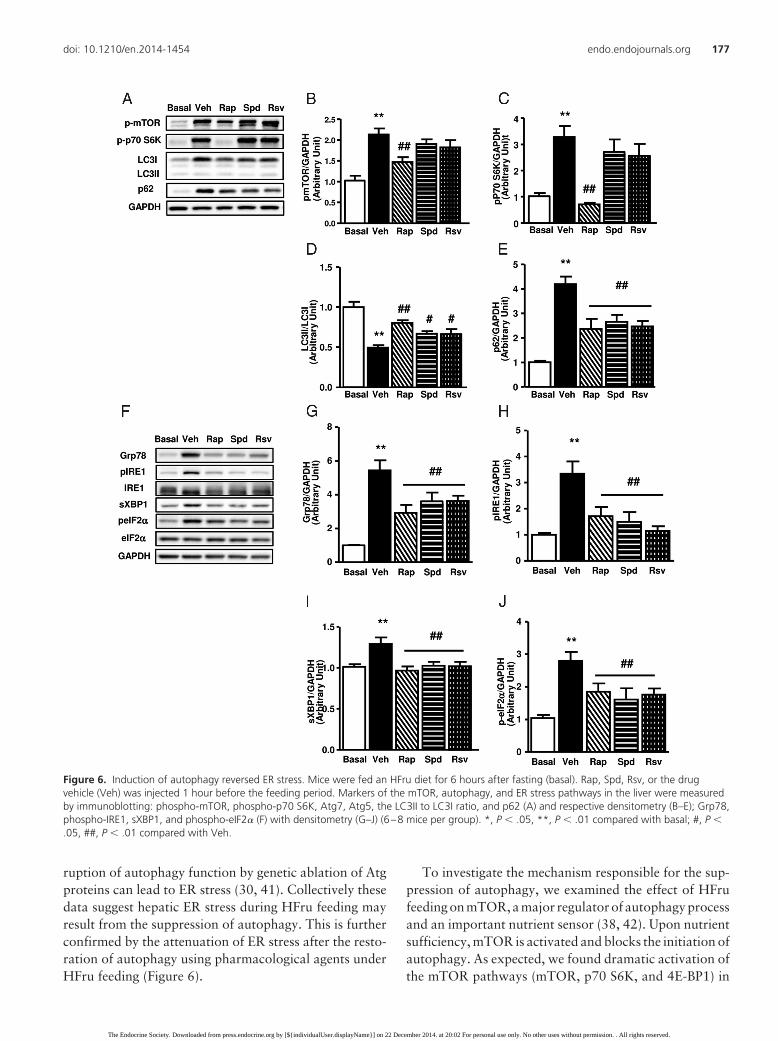
ruption of autophagy function by genetic ablation of Atgproteins can lead to ER stress (30, 41). Collectively thesedata suggest hepatic ER stress during HFru feeding mayresult from the suppression of autophagy. This is furtherconfirmed by the attenuation of ER stress after the resto-ration of autophagy using pharmacological agents underHFru feeding (Figure 6).
To investigate the mechanism responsible for the sup-pression of autophagy, we examined the effect of HFrufeeding on mTOR, a major regulator of autophagy processand an important nutrient sensor (38, 42). Upon nutrientsufficiency, mTOR is activated and blocks the initiation ofautophagy. As expected, we found dramatic activation ofthe mTOR pathways (mTOR, p70 S6K, and 4E-BP1) in
Figure 6. Induction of autophagy reversed ER stress. Mice were fed an HFru diet for 6 hours after fasting (basal). Rap, Spd, Rsv, or the drugvehicle (Veh) was injected 1 hour before the feeding period. Markers of the mTOR, autophagy, and ER stress pathways in the liver were measuredby immunoblotting: phospho-mTOR, phospho-p70 S6K, Atg7, Atg5, the LC3II to LC3I ratio, and p62 (A) and respective densitometry (B–E); Grp78,phospho-IRE1, sXBP1, and phospho-eIF2� (F) with densitometry (G–J) (6–8 mice per group). *, P � .05, **, P � .01 compared with basal; #, P �.05, ##, P � .01 compared with Veh.
doi: 10.1210/en.2014-1454 endo.endojournals.org 177
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

the liver after either chronic (Figure 3B) or acute (Figure 4)HFru feeding. Furthermore, inhibition of mTOR withrapamycin was able to relieve the suppression of au-tophagy induced by HFru (Figure 6). All these data suggestmTOR may be responsible for the observed changes inautophagy. mTOR can be stimulated by insulin and aminoacids (43, 44). However, plasma insulin levels and basalAkt/GSK3� phosphorylation in the liver were unalteredafter 2 weeks of HFru feeding despite overt activation ofmTOR at the same time (Supplemental Table 1 and Figure1D). Moreover, fructose also activated mTOR in liver ex-
plants in the absence of insulin and amino acids (Figure3C). These data indicate that fructose has direct effects onthe activation of mTOR in the liver. Consistent with un-altered ER stress or lipogenesis, no changes of autophagyor mTOR pathways were detected in muscle, suggestingthe changes induced by fructose are liver specific.
Although stimulation of autophagy through inhibitionof mTOR was able to ameliorate ER stress (Figure 6),mTOR itself has also been reported to affect ER stress andinsulin resistance (45, 46). To further confirm the role ofautophagy in HFru-induced ER stress, we examined the
Figure 7. Induction of autophagy improved insulin signaling transduction. Mice were fed an HFru diet for 6 hours after fasting (basal). Rap, Spd,Rsv, or drug vehicle (Veh) was injected 1 hour before the feeding period. The JNK/IKK-IRS1 signaling cascade was measured by immunoblotting.Shown are representative blots of phospho/total-JNK, phospho/total-cJun, phospho/total-IKK, and phospho-IRS1 (A) with densitometry (B–E). Inanother cohort of mice, insulin was injected and hepatic insulin signaling transduction was assessed by phosphorylation of Akt (F) and GSK3� (G)in response to insulin stimulation (6–8 mice per group). *, P � .05, **, P � .01 compared with basal; #, P � .05, ##, P � .01 compared with Veh.
178 Wang et al Autophagy in Fructose-Induced Hepatic ER Stress Endocrinology, January 2015, 156(1):169–181
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

impact of mTOR-independent induction of autophagy onER stress with HFru feeding, using the drug inducers, sper-midine and resveratrol (34, 35). The results showed thatboth drugs were able to restore autophagy and alleviateER stress without affecting mTOR activity (Figure 6). Fur-thermore, Bachar-Wikstrom et al (31) also demonstratedan attenuation of ER stress by stimulating autophagythrough mTOR inhibition, and this effect on ER stress wasabolished after cotreatment of autophagy inhibitors.These data together suggest that the role of autophagy inER stress is independent of mTOR.
ER stress and lipid accumulation have been revealed asimportant mechanisms for insulin resistance because bothcan lead to the serine phosphorylation of IRS and the se-quential disruption of insulin signaling transduction (47–49). With HFru feeding, lipids are mostly generatedthrough lipogenesis (50), a process promoted by ER stress(22, 23). Therefore, it is difficult to differentiate ER stressfrom its impact on lipogenesis toward insulin action. Inthis study, we found that both IRE1 and PERK signalingpathways were already activated after 6 hours of HFrufeeding (Figure 5). However, there was no significant in-crease in the levels of lipogenic proteins (eg, ACC, FAS,and SCD1) until 12 hour (Figure 5). Notably, 6 hours ofHFru feeding was sufficient to impair insulin signal trans-duction in the liver, as indicated by reduced phosphory-lation of Akt and GSK3� under insulin stimulation (Figure7, F and G). This impairment was accompanied by anactivation of the JNK/IKK pathways and the serine phos-phorylation of IRS1 (Figure 7, A–E). Moreover, when ERstress was alleviated by the stimulation of autophagy, boththe JNK/IKK and Akt/GSK3� signaling cascades were cor-rected (Figure 7). These results indicate that ER stress byitself is able to impair insulin signaling with HFru feeding,likely via the activation of JNK/IKK.
Some studies indicate that prolonged fasting can havesignificant impacts on metabolic status, which may affectsignaling pathways like lipogenesis in the liver (51, 52).Our results showed that apart from reduction in lipogenicenzymes, there was no significant difference in mTOR,autophagy, ER stress, or inflammatory makers after 24hours of fasting (Supplemental Figure 3). Interestingly,unaltered mTOR autophagy and UPR pathways have alsobeen reported in both liver tissues and cells after long-termfasting (53). These data indicate that the effects of HFrufeeding on mTOR autophagy, UPR, or inflammatorypathways are unlikely to be influenced by 24 hours offasting.
It may also be argued that changes in mTOR, au-tophagy, ER stress, and inflammatory pathways resultfrom refeeding per se rather than an HFru diet. To inves-tigate this possibility, we examined these signaling path-
ways after 6 hours of CH diet feeding. Consistent withearlier reports (39, 53), we also observed various degreesof changes in markers of mTOR, autophagy, and ER stresswith CH refeeding (Supplemental Figure 4). However, anHFru diet clearly showed an additional effect on thesepathways (especially mTOR autophagy) compared withthe CH diet, indicating its specific role apart from refeed-ing. Treatment with rapamycin in CH-refed mice is alsoable to normalize the mTOR autophagy signaling path-way and partially reverse ER stress. This further supportsour notion that the suppression of autophagy is a cause ofER stress (Supplemental Figure 5, A–H). Moreover, CHrefeeding does not activate the JNK/IKK signaling cas-cade. Consistently, no suppression in insulin-stimulatedphosphorylation of Akt was observed with CH refeeding(Supplemental Figure 5, I–L). These data together suggestthat the suppression of autophagy and the resultant ERstress are the underlying mechanisms of impaired insulinsignaling during HFru feeding.
In conclusion, this study provides the first evidence thatsuppressedautophagycontributes tohepaticERstress andinsulin resistance induced by HFru. Our results show thatthe restoration of the normal functions of autophagy isable to alleviate ER stress and improve insulin signal trans-duction. These findings also provide the scientific ratio-nale for targeting the autophagy pathway for the treat-ment of hepatic insulin resistance and its associatedmetabolic disorders.
Acknowledgments
We thank Dr Stanley M. H. Chan for the assistance with theanimal study.
Address all correspondence and requests for reprints to: Pro-fessor Ji-Ming Ye, Lipid Biology and Metabolic Disease Labo-ratory, Health Innovations Research Institute and School ofHealth Sciences, RMIT University, PO Box 71, Melbourne, Vic-toria 3083, Australia. E-mail: [email protected]; or Dr JuanC. Molero, Lipid Biology and Metabolic Disease Laboratory,Health Innovations Research Institute and School of Health Sci-ences, RMIT University, PO Box 71, Melbourne, Victoria 3083,Australia. E-mail: [email protected].
This work was supported by the National Health and Med-ical Research Council of Australia Program Grant 535921 (al-location to J.-M.Y.) and Australian Research Council Grant DP11010396 (to J.-M.Y.).
Disclosure Summary: The authors have nothing to disclose.
References
1. Dekker MJ, Su Q, Baker C, Rutledge AC, Adeli K. Fructose: a highlylipogenic nutrient implicated in insulin resistance, hepatic steatosis,
doi: 10.1210/en.2014-1454 endo.endojournals.org 179
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2010;299(5):E685–E694.
2. Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weightgain, and the insulin resistance syndrome. Am J Clin Nutr. 2002;76(5):911–922.
3. Tappy L, Le KA. Metabolic effects of fructose and the worldwideincrease in obesity. Physiol Rev. 2010;90(1):23–46.
4. Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructosecorn syrup in beverages may play a role in the epidemic of obesity.Am J Clin Nutr. 2004;79(4):537–543.
5. Bray GA. How bad is fructose? Am J Clin Nutr. 2007;86(4):895–896.
6. Lecoultre V, Carrel G, Egli L, et al. Coffee consumption attenuatesshort-term fructose-induced liver insulin resistance in healthy men.Am J Clin Nutr. 2014;99(2):268–275.
7. Faeh D, Minehira K, Schwarz JM, Periasamy R, Park S, Tappy L.Effect of fructose overfeeding and fish oil administration on hepaticde novo lipogenesis and insulin sensitivity in healthy men. Diabetes.2005;54(7):1907–1913.
8. Chan SM, Sun RQ, Zeng XY, et al. Activation of PPAR� ameliorateshepatic insulin resistance and steatosis in high fructose-fed micedespite increased endoplasmic reticulum stress. Diabetes. 2013;62(6):2095–2105.
9. Aeberli I, Hochuli M, Gerber PA, et al. Moderate amounts of fruc-tose consumption impair insulin sensitivity in healthy young men: arandomized controlled trial. Diabetes Care. 2013;36(1):150–156.
10. Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral ad-iposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119(5):1322–1334.
11. Thorburn AW, Storlien LH, Jenkins AB, Khouri S, Kraegen EW.Fructose-induced in vivo insulin resistance and elevated plasma tri-glyceride levels in rats. Am J Clin Nutr. 1989;49(6):1155–1163.
12. Samuel VT. Fructose induced lipogenesis: from sugar to fat to in-sulin resistance. Trends Endocrinol Metab. 2011;22(2):60–65.
13. Nagai Y, Yonemitsu S, Erion DM, et al. The role of peroxisomeproliferator-activated receptor gamma coactivator-1 � in the patho-genesis of fructose-induced insulin resistance. Cell Metab. 2009;9(3):252–264.
14. Le KA, Ith M, Kreis R, et al. Fructose overconsumption causes dys-lipidemia and ectopic lipid deposition in healthy subjects with andwithout a family history of type 2 diabetes. Am J Clin Nutr. 2009;89(6):1760–1765.
15. Samuel VT, Liu ZX, Qu XQ, et al. Mechanism of hepatic insulinresistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279(31):32345–32353.
16. Ozcan U, Cao Q, Yilmaz E, Hotamisligil GS, et al. Endoplasmicreticulum stress links obesity, insulin action, and type 2 diabetes.Science. 2004;306(5695):457–461.
17. Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic retic-ulum in hepatic lipid homeostasis and stress signaling. Cell Metab.2012;15(5):623–634.
18. Flamment M, Hajduch E, Ferre P, Foufelle F. New insights into ERstress-induced insulin resistance. Trends Endocrinol Metab. 2012;23(8):381–390.
19. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP,Ron D. Coupling of stress in the ER to activation of JNK proteinkinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666.
20. Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF.Phosphorylation of Ser307 in insulin receptor substrate-1 blocksinteractions with the insulin receptor and inhibits insulin action.J Biol Chem. 2002;277(2):1531–1537.
21. Liang SH, Zhang W, McGrath BC, Zhang P, Cavener DR. PERK(eIF2� kinase) is required to activate the stress-activated MAPKsand induce the expression of immediate-early genes upon disruptionof ER calcium homoeostasis. Biochem J. 2006;393(Pt 1):201–209.
22. Ning J, Hong T, Ward A, et al. Constitutive role for IRE1�-XBP1signaling pathway in the insulin-mediated hepatic lipogenic pro-gram. Endocrinology. 2011;152(6):2247–2255.
23. Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepaticlipogenesis by the transcription factor XBP1. Science. 2008;320(5882):1492–1496.
24. Ding WX, Yin XM. Sorting, recognition and activation of the mis-folded protein degradation pathways through macroautophagy andthe proteasome. Autophagy. 2008;4(2):141–150.
25. Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulatesAKT/TSC/mTOR pathway to enhance autophagy. Autophagy.2010;6(2):239–247.
26. Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulumstress triggers autophagy. J Biol Chem. 2006;281(40):30299–30304.
27. Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulumstress to autophagy by unfolded protein response and calcium. CellDeath Differ. 2007;14(9):1576–1582.
28. Qiu W, Zhang J, Dekker MJ, et al. Hepatic autophagy mediatesendoplasmic reticulum stress-induced degradation of misfoldedapolipoprotein B. Hepatology. 2011;53(5):1515–1525.
29. Liu HY, Han J, Cao SY, et al. Hepatic autophagy is suppressed in thepresence of insulin resistance and hyperinsulinemia: inhibition ofFoxO1-dependent expression of key autophagy genes by insulin.J Biol Chem. 2009;284(45):31484–31492.
30. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepaticautophagy in obesity promotes ER stress and causes insulin resis-tance. Cell Metab. 2010;11(6):467–478.
31. Bachar-Wikstrom E, Wikstrom JD, Ariav Y, et al. Stimulation ofautophagy improves endoplasmic reticulum stress-induced diabe-tes. Diabetes. 2013;62(4):1227–1237.
32. Ren LP, Chan SM, Zeng XY, et al. Differing endoplasmic reticulumstress response to excess lipogenesis versus lipid oversupply in re-lation to hepatic steatosis and insulin resistance. PLoS One. 2012;7(2):e30816.
33. Pfaffenbach KT, Nivala AM, Pagliassotti MJ, et al. Rapamycin in-hibits postprandial-mediated X-box-binding protein-1 splicing inrat liver. J Nutr. 2010;140(5):879–884.
34. Morselli E, Marino G, Bennetzen MV, et al. Spermidine and res-veratrol induce autophagy by distinct pathways converging on theacetylproteome. J Cell Biol. 2011;192(4):615–629.
35. Wang IF, Guo BS, Liu YC, et al. Autophagy activators rescue andalleviate pathogenesis of a mouse model with proteinopathies of theTAR DNA-binding protein 43. Proc Natl Acad Sci USA. 2012;109(37):15024–15029.
36. Molero JC, Jensen TE, Withers PC, et al. c-Cbl-deficient mice havereduced adiposity, higher energy expenditure, and improved periph-eral insulin action. J Clin Invest. 2004;114(9):1326–1333.
37. Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the useand interpretation of assays for monitoring autophagy. Autophagy.2012;8(4):445–544.
38. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulateautophagy through direct phosphorylation of Ulk1. Nat Cell Biol.2011;13(2):132–141.
39. Deng Y, Wang ZV, Tao C, et al. The Xbp1s/GalE axis links ER stressto postprandial hepatic metabolism. J Clin Invest. 2013;123(1):455–468.
40. Turner N, Kowalski GM, Leslie SJ, et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistancein mice by high-fat feeding. Diabetologia. 2013;56(7):1638–1648.
41. Li H, Zhou B, Xu L, et al. The reciprocal interaction between au-tophagic dysfunction and ER stress in adipose insulin resistance. CellCycle. 2014;13(4):565–579.
42. Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependentmTORC1 association with the ULK1-Atg13-FIP200 complex re-quired for autophagy. Mol Biol Cell. 2009;20(7):1981–1991.
43. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin
180 Wang et al Autophagy in Fructose-Induced Hepatic ER Stress Endocrinology, January 2015, 156(1):169–181
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.

signalling to mTOR mediated by the Akt/PKB substrate PRAS40.Nat Cell Biol. 2007;9(3):316–323.
44. Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bindraptor and mediate amino acid signaling to mTORC1. Science.2008;320(5882):1496–1501.
45. Ozcan U, Ozcan L, Yilmaz E, et al. Loss of the tuberous sclerosiscomplex tumor suppressors triggers the unfolded protein responseto regulate insulin signaling and apoptosis. Mol Cell. 2008;29(5):541–551.
46. Ueno M, Carvalheira JB, Tambascia RC, et al. Regulation of insulinsignalling by hyperinsulinaemia: role of IRS-1/2 serine phosphory-lation and the mTOR/p70 S6K pathway. Diabetologia. 2005;48(3):506–518.
47. Hotamisligil GS. Role of endoplasmic reticulum stress and c-JunNH2-terminal kinase pathways in inflammation and origin of obe-sity and diabetes. Diabetes. 2005;54(suppl 2):S73–S78.
48. Yu C, Chen Y, Cline GW, et al. Mechanism by which fatty acidsinhibit insulin activation of insulin receptor substrate-1 (IRS-1)-as-sociated phosphatidylinositol 3-kinase activity in muscle. J BiolChem. 2002;277(52):50230–50236.
49. Paz K, Hemi R, LeRoith D, et al. A molecular basis for insulinresistance. Elevated serine/threonine phosphorylation of IRS-1 andIRS-2 inhibits their binding to the juxtamembrane region of theinsulin receptor and impairs their ability to undergo insulin-inducedtyrosine phosphorylation. J Biol Chem. 1997;272(47):29911–29918.
50. Miyazaki M, Dobrzyn A, Man WC, et al. Stearoyl-CoA desaturase1 gene expression is necessary for fructose-mediated induction oflipogenic gene expression by sterol regulatory element-binding pro-tein-1c-dependent and -independent mechanisms. J Biol Chem.2004;279(24):25164–25171.
51. Hutchens TT, Van Bruggen JT, Cockburn RM, West ES. The effectof fasting upon tissue lipogenesis in the intact rat. J Biol Chem.1954;208(1):115–122.
52. Leveille GA. In vivo fatty acid and cholesterol synthesis in fasted andfasted-refed chicks. J Nutr. 1969;98(3):367–372.
53. Bae SH, Sung SH, OH SY, et al. Sestrins activate Nrf2 by promotingp62-dependent autophagic degradation of Keap1 and prevent oxi-dative liver damage. Cell Metab. 2013;17(1):73–84.
doi: 10.1210/en.2014-1454 endo.endojournals.org 181
The Endocrine Society. Downloaded from press.endocrine.org by [${individualUser.displayName}] on 22 December 2014. at 20:02 For personal use only. No other uses without permission. . All rights reserved.
Related Documents











