Reduction of heart rate by chronic 1 -adrenoceptor blockade promotes growth of arterioles and preserves coronary perfusion reserve in postinfarcted heart Eduard I. Dedkov, 1 Lance P. Christensen, 1 Robert M. Weiss, 2,3,4 and Robert J. Tomanek 1,3 Departments of 1 Anatomy and Cell Biology and 2 Internal Medicine and 3 The Cardiovascular Center, University of Iowa Carver College of Medicine, and 4 Department of Veterans Affairs Medical Center, Iowa City, Iowa Submitted 10 October 2004; accepted in final form 26 January 2005 Dedkov, Eduard I., Lance P. Christensen, Robert M. Weiss, and Robert J. Tomanek. Reduction of heart rate by chronic 1- adrenoceptor blockade promotes growth of arterioles and preserves coronary perfusion reserve in postinfarcted heart. Am J Physiol Heart Circ Physiol 288: H2684 –H2693, 2005. First published January 28, 2005; doi:10.1152/ajpheart.01047.2004.—Adequate growth of coro- nary vasculature in the remaining left ventricular (LV) myocardium after myocardial infarction (post-MI) is a crucial factor for myocyte survival and performance. We previously demonstrated that post-MI coronary angiogenesis can be stimulated by bradycardia induced with the ATP-sensitive K channel antagonist alinidine. In this study, we tested the hypothesis that heart rate reduction with -blockade may also induce coronary growth in the post-MI heart. Transmural MI was induced in 12-mo-old male Sprague-Dawley rats by occlusion of the left anterior descending coronary artery. Bradycardia was induced by administration of the -adrenoceptor blocker atenolol (AT) via drink- ing water (30 mg/day). Three groups of rats were compared: 1) control/sham (C/SH), 2) MI, and 3) MI AT. In the MI AT rats, heart rate was consistently reduced by 25–28% compared with C/SH rats. At 4 wk after left anterior descending coronary ligation, infarct size was similar in MI and MI AT rats (67.1 and 61.5%, respec- tively), whereas a greater ventricular hypertrophy occurred in brady- cardic rats, as indicated by a higher ventricular weight-to-body weight ratio (3.4 0.1 vs. 2.8 0.1 mg/g in MI rats). Analysis of LV function revealed a smaller drop in ejection fraction in the MI AT than in the MI group (24 vs. 35%). Furthermore, in MI AT rats, maximal coronary conductance and coronary perfusion reserve were significantly improved compared with the MI group. The better myocardial perfusion indexes in MI AT rats were associated with a greater increase in arteriolar length density than in the MI group. Thus chronic reduction of heart rate induced with -selective block- ade promotes growth of coronary arterioles and, thereby, facilitates regional myocardial perfusion in post-MI hearts. myocardial infarction; angiogenesis; coronary circulation; resistance vessels; capillaries A LARGE TRANSMURAL MYOCARDIAL infarction (MI) of the left ventricle (LV) causes the sudden loss of a substantial number of cardiac myocytes, which leads to increased diastolic wall stress and chronic functional overload of the remaining portion of LV myocardium. As a result, the LV undergoes progressive structural remodeling, consisting of thinning and scarring of the infarcted region, chamber dilation, and eccentric hypertro- phy of the surviving portion of the LV myocardium (1, 27, 30). Because scar tissue is not capable of contracting, LV function after MI is entirely dependent on the hypertrophied portion of the surviving LV myocardium. However, to accommodate an increased O 2 demand in the surviving overloaded cardiac myocytes, an adaptation of the vascular bed is necessary. Two basic mechanisms allow the coronary vasculature to raise the myocardial oxygenation level as O 2 demand increases: 1) augmentation of blood flow via dila- tion of the existing resistance vessels and 2) increase in the number of arterioles and capillaries via angiogenesis. Cor- onary dilation and, consequently, increased coronary flow have been found under baseline conditions in the hypertro- phied LV myocardium of postinfarcted hearts (19, 20). This increased flow at rest, coupled with a decreased maximal coronary conductance, causes a reduction in coronary re- serve (19, 20). Such a decrease in coronary reserve in the remaining LV myocardium after infarction suggests an in- adequate compensatory angiogenesis (1). Therefore, the surviving LV myocardium of the postinfarcted heart re- mains vulnerable to the new episodes of ischemia. To minimize the risk of cardiac damage during enhanced cardiac work in a hypertrophied post-MI heart, angiogenic therapy may be beneficial. The principal goal of such therapy is to protect the surviving cardiac myocytes from O 2 deprivation by establishing a functional equilibrium be- tween O 2 demand and delivery. This can be accomplished by reduction of the myocyte O 2 demand via negative chro- notropic and inotropic actions or by augmentation of myo- cardial perfusion by means of coronary vascular growth. It appears that in clinical practice the effects described above might be easily achieved by a combined use of -blockers, a class of drugs designed to reduce heart rate and myocar- dial contractility via inhibition of sympathetic stimulation (26) and therapeutic angiogenesis (15, 38). In recent years, new evidence has emerged indicating that chronic -blockade in patients suffering from heart failure, including that caused by MI, improves LV dysfunction and reduces the risk of sudden death (21, 24). It has been suggested that one of the key cardioprotective roles of -blockers under such conditions is the characteristic decrease in heart rate, leading to improved myocardial blood flow due to a lengthened diastolic perfusion time (4). This is consistent with an earlier animal study that revealed that heart rate reduction associated with -blockers was beneficial for regional redistribution of blood flow, an effect that could be prevented by atrial pacing (32). In addition, it was shown that only those -blockers with -selectivity caused a more favorable redistribution of flow in ischemic myocardium (8). It has been known for at least two decades that bradycardia induced in animals by electrical pacing or pharmacological Address for reprint requests and other correspondence: E. I. Dedkov, Dept. of Anatomy and Cell Biology, Carver College of Medicine, 1-402 Bowen Science Bldg., Univ. of Iowa, Iowa City, IA 52242 (E-mail: eduard- [email protected]). The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. Am J Physiol Heart Circ Physiol 288: H2684 –H2693, 2005. First published January 28, 2005; doi:10.1152/ajpheart.01047.2004. http://www.ajpheart.org H2684

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Reduction of heart rate by chronic �1-adrenoceptor blockade promotes growthof arterioles and preserves coronary perfusion reserve in postinfarcted heart
Eduard I. Dedkov,1 Lance P. Christensen,1 Robert M. Weiss,2,3,4 and Robert J. Tomanek1,3
Departments of 1Anatomy and Cell Biology and 2Internal Medicine and 3The Cardiovascular Center, Universityof Iowa Carver College of Medicine, and 4Department of Veterans Affairs Medical Center, Iowa City, Iowa
Submitted 10 October 2004; accepted in final form 26 January 2005
Dedkov, Eduard I., Lance P. Christensen, Robert M. Weiss,and Robert J. Tomanek. Reduction of heart rate by chronic �1-adrenoceptor blockade promotes growth of arterioles and preservescoronary perfusion reserve in postinfarcted heart. Am J Physiol HeartCirc Physiol 288: H2684–H2693, 2005. First published January 28,2005; doi:10.1152/ajpheart.01047.2004.—Adequate growth of coro-nary vasculature in the remaining left ventricular (LV) myocardiumafter myocardial infarction (post-MI) is a crucial factor for myocytesurvival and performance. We previously demonstrated that post-MIcoronary angiogenesis can be stimulated by bradycardia induced withthe ATP-sensitive K� channel antagonist alinidine. In this study, wetested the hypothesis that heart rate reduction with �-blockade mayalso induce coronary growth in the post-MI heart. Transmural MI wasinduced in 12-mo-old male Sprague-Dawley rats by occlusion of theleft anterior descending coronary artery. Bradycardia was induced byadministration of the �-adrenoceptor blocker atenolol (AT) via drink-ing water (30 mg/day). Three groups of rats were compared: 1)control/sham (C/SH), 2) MI, and 3) MI � AT. In the MI � AT rats,heart rate was consistently reduced by 25–28% compared with C/SHrats. At 4 wk after left anterior descending coronary ligation, infarctsize was similar in MI and MI � AT rats (67.1 and 61.5%, respec-tively), whereas a greater ventricular hypertrophy occurred in brady-cardic rats, as indicated by a higher ventricular weight-to-body weightratio (3.4 � 0.1 vs. 2.8 � 0.1 mg/g in MI rats). Analysis of LVfunction revealed a smaller drop in ejection fraction in the MI � ATthan in the MI group (�24 vs. �35%). Furthermore, in MI � AT rats,maximal coronary conductance and coronary perfusion reserve weresignificantly improved compared with the MI group. The bettermyocardial perfusion indexes in MI � AT rats were associated witha greater increase in arteriolar length density than in the MI group.Thus chronic reduction of heart rate induced with �-selective block-ade promotes growth of coronary arterioles and, thereby, facilitatesregional myocardial perfusion in post-MI hearts.
myocardial infarction; angiogenesis; coronary circulation; resistancevessels; capillaries
A LARGE TRANSMURAL MYOCARDIAL infarction (MI) of the leftventricle (LV) causes the sudden loss of a substantial numberof cardiac myocytes, which leads to increased diastolic wallstress and chronic functional overload of the remaining portionof LV myocardium. As a result, the LV undergoes progressivestructural remodeling, consisting of thinning and scarring ofthe infarcted region, chamber dilation, and eccentric hypertro-phy of the surviving portion of the LV myocardium (1, 27, 30).
Because scar tissue is not capable of contracting, LVfunction after MI is entirely dependent on the hypertrophiedportion of the surviving LV myocardium. However, to
accommodate an increased O2 demand in the survivingoverloaded cardiac myocytes, an adaptation of the vascularbed is necessary. Two basic mechanisms allow the coronaryvasculature to raise the myocardial oxygenation level as O2
demand increases: 1) augmentation of blood flow via dila-tion of the existing resistance vessels and 2) increase in thenumber of arterioles and capillaries via angiogenesis. Cor-onary dilation and, consequently, increased coronary flowhave been found under baseline conditions in the hypertro-phied LV myocardium of postinfarcted hearts (19, 20). Thisincreased flow at rest, coupled with a decreased maximalcoronary conductance, causes a reduction in coronary re-serve (19, 20). Such a decrease in coronary reserve in theremaining LV myocardium after infarction suggests an in-adequate compensatory angiogenesis (1). Therefore, thesurviving LV myocardium of the postinfarcted heart re-mains vulnerable to the new episodes of ischemia.
To minimize the risk of cardiac damage during enhancedcardiac work in a hypertrophied post-MI heart, angiogenictherapy may be beneficial. The principal goal of suchtherapy is to protect the surviving cardiac myocytes from O2
deprivation by establishing a functional equilibrium be-tween O2 demand and delivery. This can be accomplishedby reduction of the myocyte O2 demand via negative chro-notropic and inotropic actions or by augmentation of myo-cardial perfusion by means of coronary vascular growth. Itappears that in clinical practice the effects described abovemight be easily achieved by a combined use of �-blockers,a class of drugs designed to reduce heart rate and myocar-dial contractility via inhibition of sympathetic stimulation(26) and therapeutic angiogenesis (15, 38).
In recent years, new evidence has emerged indicating thatchronic �-blockade in patients suffering from heart failure,including that caused by MI, improves LV dysfunction andreduces the risk of sudden death (21, 24). It has been suggestedthat one of the key cardioprotective roles of �-blockers undersuch conditions is the characteristic decrease in heart rate,leading to improved myocardial blood flow due to a lengtheneddiastolic perfusion time (4). This is consistent with an earlieranimal study that revealed that heart rate reduction associatedwith �-blockers was beneficial for regional redistribution ofblood flow, an effect that could be prevented by atrial pacing(32). In addition, it was shown that only those �-blockers with�-selectivity caused a more favorable redistribution of flow inischemic myocardium (8).
It has been known for at least two decades that bradycardiainduced in animals by electrical pacing or pharmacological
Address for reprint requests and other correspondence: E. I. Dedkov, Dept.of Anatomy and Cell Biology, Carver College of Medicine, 1-402 BowenScience Bldg., Univ. of Iowa, Iowa City, IA 52242 (E-mail: [email protected]).
The costs of publication of this article were defrayed in part by the paymentof page charges. The article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Am J Physiol Heart Circ Physiol 288: H2684–H2693, 2005.First published January 28, 2005; doi:10.1152/ajpheart.01047.2004.
http://www.ajpheart.orgH2684

agents stimulates angiogenesis in normal (5, 7, 17, 18, 40–42),hypertrophied (41), and infarcted hearts (22). Thus it has beenproposed that a chronic reduction of heart rate may be a way tostimulate angiogenesis in the heart after MI. Therefore, itseems reasonable to speculate that �-blockers, especially with�1-selectivity, may facilitate angiogenesis of the coronaryvasculature as a result of their negative chronotropic action onthe heart. In concert with this supposition, it was previouslydemonstrated that prolonged �-blockade with propranolol, anonselective �-blocker, could induce a significant increase incapillary density in the normal heart of young rabbits (35). Thepresent study is the first to address the issue of the potentialangiogenic effects of �1-selective blockade on the growth ofcoronary microvasculature in postinfarcted heart of middle-aged rats. We selected atenolol (AT), a �-selective adrenocep-tor blocker that is widely used in post-MI therapy, to test thehypothesis that 4 wk of reduction in heart rate by �-selectiveblockade can stimulate coronary angiogenesis, especially thegrowth of arterioles, in the surviving LV myocardium ofpostinfarcted heart and, thereby, can minimize the decline incoronary perfusion reserve (CPR).
MATERIALS AND METHODS
Animals. Male, 12-mo-old Sprague-Dawley rats (Harlan, Indianap-olis, IN) were housed under climate-controlled conditions and a12:12-h light-dark cycle and given standard rat chow and water adlibitum. All procedures were approved by the University of IowaAnimal Care and Use Committee and were in accordance with theregulations of the Animal Welfare Act of the National Institute ofHealth Guide for the Care and Use of Laboratory Animals.
Surgical procedure. Transmural MI was induced in animals byligation of the left anterior descending coronary artery (LAD) asdescribed in detail elsewhere (12, 22). Briefly, rats were anesthetizedwith a mixture of ketamine (100 mg/kg ip) and xylazine (10 mg/kg ip)and ventilated with a rodent ventilator (model 683, Harvard Appara-tus). The thoracic cavity was opened, the heart was exteriorized, andthe LAD was ligated near its origin with 5-0 Tycron suture. Insham-operated rats, the suture was pulled through the myocardium,but the LAD was not ligated. The heart was immediately interiorized,and the chest was closed in layers as the lungs were inflated. The ratswere observed until they awakened, at which time they were returnedto the Animal Care Unit. Standard postoperative care, includingantibiotics, was provided. The mortality rate was �20%, with mostdeaths occurring within the first 24 h.
�1-Blocker administration. AT powder (Sigma, St. Louis, MO)was dissolved in drinking water (1 mg/ml). Each rat received �30mg of AT per day based on the daily intake of drinking water,similar to that previously used in another laboratory to study theeffect of AT on LV remodeling after MI in rats (33). Heart rate wasmonitored daily in conscious, unrestrained rats with the use of
electrodes attached to an animal BioAmp differential amplifier anda PowerLab data-acquisition system (ADInstruments, Castle Hill,Australia). Only those rats that showed a reduction of heart rate�25% from the level detected in control rats (noted as brady-cardic) were processed for further evaluation.
Experimental design. At 24 h after surgery, the rats were examinedwith transthoracic echocardiography to estimate the size of the aki-netic (presumably ischemic) zone as well as to assess LV geometryand function. At 3 days after surgery, rats with a confirmed largeischemic zone (�30% of entire LV circumference; Table 1) wererandomly assigned to one of two groups: 1) MI only (n � 10) and 2)MI treated with AT (MI � AT, n � 8). AT administration began 3days after surgery, continued for 4 wk, and was terminated 24–48 hbefore the end of the experimental period, when echocardiographic(MI and MI � AT rats only) and myocardial perfusion analyses (allgroups) were performed. Because data from sham-operated (SH, n �6) and nonoperated control (C, n � 6) rats were similar, the twogroups were combined (C/SH).
At the end of the experimental period, the rats were anesthetizedand ventilated (see Surgical procedure) and surgically instrumented.Hemodynamics and regional myocardial perfusion (using the stableisotope-labeled microsphere technique) were measured at baselineand during maximal vasodilation induced by dipyridamole. Then, thehearts were arrested in diastole by infusion of lidocaine into the LV,excised, placed on a Langendorff apparatus, and fixed by perfusion(100 mmHg) with 4% phosphate-buffered paraformaldehyde (pH7.3–7.4).
After removal of the atria, the hearts were weighed and cuttransversely into five parallel, 2-mm-thick slices from apex to basewith a four-blade guillotine. In the MI hearts, ventricular sliceswith scar tissue were digitized, and images were used to estimateinfarct size. One medial slice from each heart was taken forimmunohistochemistry and morphometry. Anti-smooth muscle(SM) �-actin and anti-laminin immunostaining, as well as labelingwith BS-I lectin, was utilized to determine arteriolar length andcapillary numerical densities as well as nucleated cardiac myocytecross-sectional areas (CSAs). In all remaining slices, the rightventricular free wall was separated from the ring comprising theLV free wall and intraventricular septum. The myocardium fromthe septum and LV free wall was excised, weighed, and placed ina counting vial. In the MI hearts, myocardium from the LV freewall was excised under a dissecting microscope �1.5–2 mm fromthe edge of the scar tissue. Tissue samples, along with the refer-ence blood samples collected during myocardial blood flow mea-surements, were sent to the BioPhysics Assay Laboratory (Bio-PAL, Worchester, MA) for microsphere counting.
Echocardiographic analysis. Details of the echocardiographic pro-cedure have been described elsewhere (22, 45). Briefly, rats werelightly anesthetized with ketamine (50 mg/kg ip) and positionedsupine. The chest was shaved, and acoustic coupling gel was applied.Two-dimensional short- and long-axis images of the LV were ob-tained with a clinical echocardiograph (Sequoia model 256, Acuson,
Table 1. LV dimensions and function obtained 24 h and 4 wk after MI by transthoracic echocardiography
EDV/Mass, ml/g HR, beats/min SV, ml CO, ml/min EF, % IZ, %
MI24 h 0.83�0.06 450�14 0.35�0.04 148.8�23.2 39.4�2.9 39.7�2.74 wk 1.47�0.07c 403�11b 0.32�0.01 144.1�12.5 29.1�2.8b 45.2�1.9a
MI � AT24 h 0.79�0.07 416�22 0.29�0.02 116.9�6.9 41.3�5.3 33.2�2.54 wk 1.35�0.19b 334�9be 0.37�0.05a 134.5�15.3 33.9�3.1 39.3�2.6cd
Values are means � SE; n � 6. MI, myocardial infarction; EDV, end-diastolic volume; HR, heart rate; SV, stroke volume; CO, cardiac output; EF, ejectionfraction; IZ, ischemic (akinetic) zone. In the MI � AT group, echocardiographic analysis was done 24 h before �1-blocker [atenolol (AT)] administration and24 h after termination of AT treatment. aP � 0.05; bP � 0.01; cP � 0.001 vs. 24 h (paired t-test); dP � 0.05; eP � 0.001 vs. 4 wk MI (unpaired t-test).
H2685BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org

Mountain View, CA) equipped with an 8.0-MHz sector-array trans-ducer adapted for small rodents. Pulse-wave Doppler interrogation ofmitral inflow was used to calculate heart rate. Planimetric measure-ments were used to estimate the size of the ischemic zone thatdemonstrated systolic akinesis, and this region was expressed as apercentage of the entire LV. LV mass and volume were calculatedusing the area-length method. From these measurements, LV param-eters, including end-diastolic volume (EDV), volume-to-mass ratio,stroke volume, cardiac output, and ejection fraction, were calculated.
Hemodynamics and regional myocardial perfusion. Rats wereanesthetized with a mixture of ketamine (100 mg/kg ip) and xylazine(10 mg/kg ip) and ventilated with a Harvard rodent ventilator. Apolyethylene catheter (PE-50) attached to a blood pressure transducerwas inserted into the right femoral artery, and systemic hemodynamicparameters, including heart rate and systolic, diastolic, and meanarterial pressure, were recorded using a bridge amplifier and a Pow-erLab data-acquisition system equipped with Chart version 4 software(ADInstruments). Then, polyethylene catheters were inserted into theLV via the right common carotid artery, the left femoral artery, andthe left jugular vein, and LV systolic blood pressure and LV end-diastolic pressure were recorded.
Myocardial perfusion was determined using neutron-activated sta-ble isotope-labeled microspheres (B-series, BioPAL). For each flowdetermination, �1 106 microspheres were infused into the LV. Thecatheter was then flushed with 0.4 ml of heparinized saline (50 U/ml),and an arterial reference blood sample was withdrawn from the leftfemoral artery by a programmable syringe pump (model NE-1000,New Era Pump Systems, Farmingdale, NY) at a constant rate(0.2 ml/min). The microspheres labeled with three different iso-topes were used to determine myocardial perfusion once under thebaseline condition and twice after maximal coronary dilation.Maximal coronary dilation was induced by infusion of dipyridamole(6 mg �kg1 �min1 iv for 8 min) via the left jugular vein. The highestattained value was utilized as maximal flow. The mean arterialpressure was recorded via the right femoral artery during each micro-sphere infusion. Tissue samples from the heart, along with the refer-ence blood samples, were prepared for microsphere counting accord-ing to the manufacturer’s protocol (BioPAL).
From the measurements provided by BioPAL, myocardial bloodflow was computed according to a standard formula: (Cm/Cr) � Q̇r �100 g(ml �min1 �100 g1), where Cm is number of micro-spheres per gram of myocardium, Cr is number of microspheres inthe reference blood sample, and Q̇r is withdrawal rate of thereference blood sample (ml/min). On the basis of these data,regional coronary resistance was calculated by dividing systemicmean arterial pressure by regional myocardial blood flow. Finally,myocardial perfusion at baseline and after maximal coronarydilation was expressed as coronary conductance, which is thereverse of coronary resistance (ml �mmHg1 �min1 �100 g1).CPR was calculated as maximal coronary conductance divided bybaseline coronary conductance.
Estimation of infarct size. The procedure for measurements ofinfarct size was described previously (22, 45). Briefly, digital imagesfrom each ventricular slice of the hearts were analyzed with Image-Pro Plus software (Media Cybernetics, Silver Spring, MD). From eachslice, the lengths of LV free wall (obtained at a midtransmural point)and its portion occupied by scar tissue were measured, and the size ofthe scarred portion was estimated per slice as the ratio of scarredfraction to length of the LV free wall. Subsequently, the mean of suchratios was calculated for each heart. Infarct size was expressed as apercentage of the LV free wall.
Immunohistochemistry and immunofluorescence microscopy. Oneventricular slice from each heart was fixed in 4% paraformaldehyde in0.1 M PBS for 24 h at 4°C. The samples were washed overnight inPBS, cryoprotected in a graded sucrose series at 4°C, and then frozenin Tris-buffered saline (TBS)-tissue freezing medium (Triangle Bio-medical Sciences, Durham, NC) by isopentane precooled with dry ice.
Transverse 8.0-�m serial sections were cut with a cryostat andmounted on glass slides. The sections were incubated with 0.5%bovine serum albumin and labeled with primary antibodies. Theprimary antibodies used in this study were 1) monoclonal anti-smoothmuscle (SM) �-actin, clone 1A4 (1:600; Sigma) and 2) polyclonalantilaminin (1:30; Sigma). After they were labeled, the sections werewashed in PBS, incubated with 10% normal goat serum, and stainedwith secondary antibodies and/or BS-I lectin. The secondary antibod-ies used for visualization of primary antibodies were FITC- or Cy3-conjugated goat anti-mouse or goat anti-rabbit IgG (Jackson Immu-noResearch Laboratories, West Grove, PA). BS-I lectin was conju-gated with tetramethylrhodamine isothiocyanate (10 �g/ml; Sigma) orfluorescein (20 �g/ml; Vector Laboratories, Burlingame, CA). Forcontrols, primary antibodies were omitted or substituted with 10%normal goat serum. The sections were counterstained with 4�,6-diamidino-2-phenylindole (DAPI, 10 mg/ml) and mounted using theProLong antifade kit (Molecular Probes, Eugene, OR). Fluorescenceimages were captured on a computer using a microscope (EclipseE-600, Nikon) equipped with a digital camera (model DXM 1200,Nikon). Final images were prepared using Adobe Photoshop software(Adobe Systems, San Jose, CA).
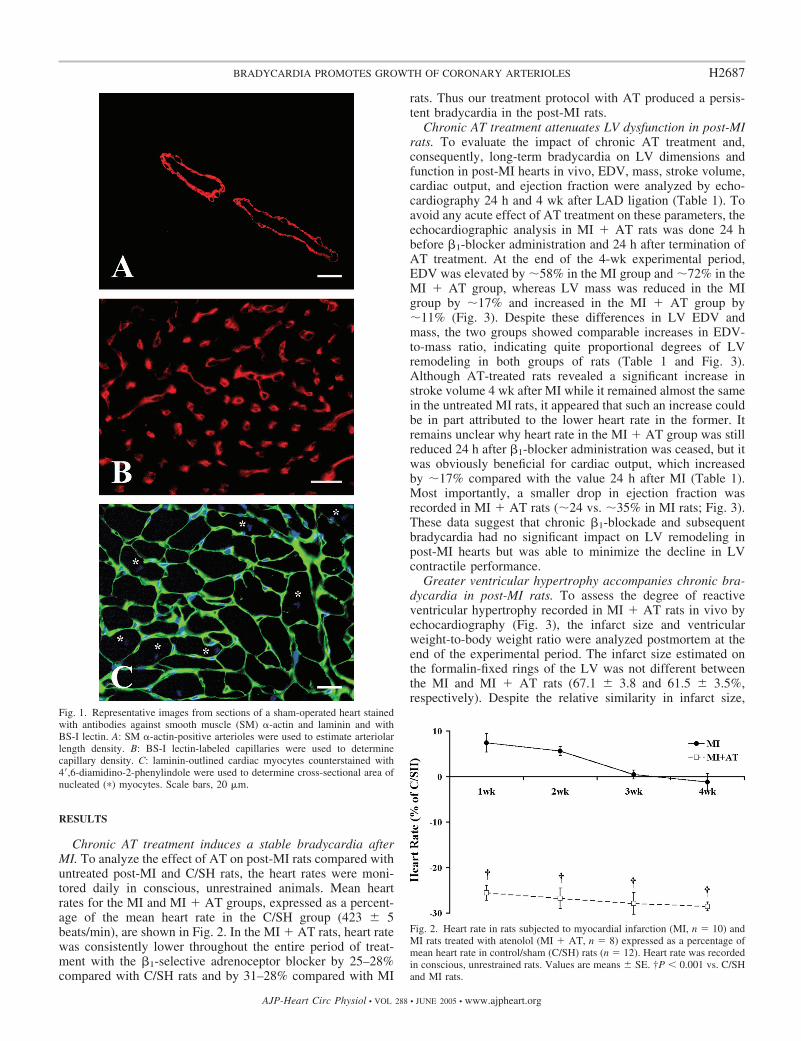
Morphometric and stereological analyses. The sections cut fromthe ventricular slices, taken from at least five hearts per group, werelabeled with anti-SM �-actin antibody (a marker of arteriolar SMcells; Fig. 1A), BS-I lectin (a marker of capillaries; Fig. 1B), and/oranti-laminin antibody counterstained with DAPI (used to outlinenucleated cardiac myocyte profiles; Fig. 1C) and then analyzed usingImage-Pro Plus software (Media Cybernetics).
To determine the length density of coronary arterioles (vessels withan uninterrupted SM �-actin-positive outline and an external diameterbetween 6 and 50 �m), the randomly selected regions of myocardiumin the LV free wall (excluding the area �2 mm from the edge of scartissue in post-MI hearts) and septum were digitized using low-powermagnification (2 objective), and their areas were measured. Theareas used in this analysis were �8–10.5 mm2 per region in eachheart. With the use of systematic scanning of these regions underhigh-power magnification (40 objective), every vessel profile (with-out respect to its sectioning plan) detected in the LV free wall andseptum was captured and their morphometric parameters were mea-sured. On the basis of these measurements, the length density (LV)was calculated as described previously (2): LV � (¥a/b)/N �N/A(�m/�m3), where a and b represent long and short axes, respectively,of individual arterioles, N is total number of arteriolar profiles, and Ais the total area in which arterioles were measured.
To calculate capillary numerical density (CD), the 7–10 randomlyselected optical fields from epimyocardium and endomyocardium ofthe LV free wall and the LV endomyocardial region of the septumwere digitized using high-power magnification. Only the areas thatdisplayed cross-sectioned capillary and myocyte profiles were se-lected for evaluation. The areas used in this analysis were �0.35–0.55mm2 per region in each heart. CD was calculated as N/A (counts/�m2), where N is number of cross-sectioned BS-I lectin-positivecapillary profiles and A is total area of the myocardium in whichcapillaries were counted.
To estimate cardiac myocyte CSA, the same regions used forcapillary counting were digitized on adjacent sections stained with ananti-laminin antibody and DAPI. Myocyte CSAs were determinedonly in those cells that showed a relatively circular shape and presenceof a nucleus. On average, �250–400 nucleated myocyte profiles perregion were analyzed in each heart.
Statistical analysis. Values are means � SE. One-way ANOVAfollowed by Bonferroni’s post test was used for multigroup compar-ison (C/SH, MI, and MI � AT). Paired t-test was used to compareintergroup differences, and an unpaired t-test was used to assess thedifference between two groups. P � 0.05 was selected to denotesignificant differences.
H2686 BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org
RESULTS
Chronic AT treatment induces a stable bradycardia afterMI. To analyze the effect of AT on post-MI rats compared withuntreated post-MI and C/SH rats, the heart rates were moni-tored daily in conscious, unrestrained animals. Mean heartrates for the MI and MI � AT groups, expressed as a percent-age of the mean heart rate in the C/SH group (423 � 5beats/min), are shown in Fig. 2. In the MI � AT rats, heart ratewas consistently lower throughout the entire period of treat-ment with the �1-selective adrenoceptor blocker by 25–28%compared with C/SH rats and by 31–28% compared with MI
rats. Thus our treatment protocol with AT produced a persis-tent bradycardia in the post-MI rats.
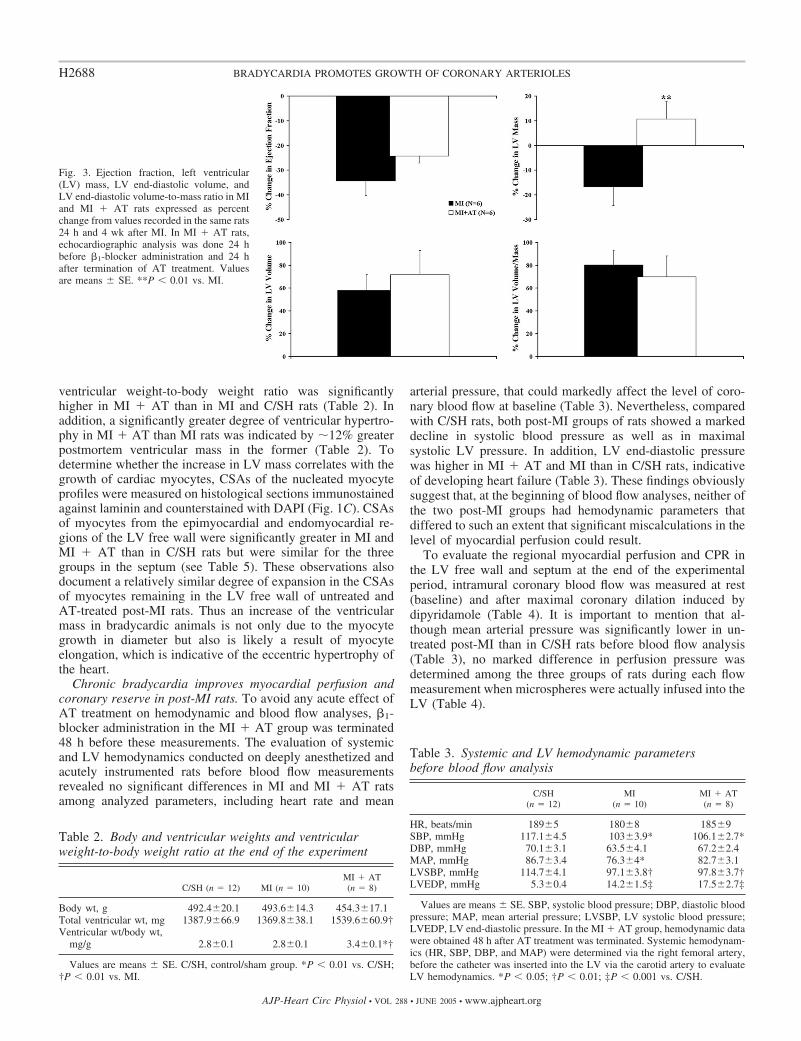
Chronic AT treatment attenuates LV dysfunction in post-MIrats. To evaluate the impact of chronic AT treatment and,consequently, long-term bradycardia on LV dimensions andfunction in post-MI hearts in vivo, EDV, mass, stroke volume,cardiac output, and ejection fraction were analyzed by echo-cardiography 24 h and 4 wk after LAD ligation (Table 1). Toavoid any acute effect of AT treatment on these parameters, theechocardiographic analysis in MI � AT rats was done 24 hbefore �1-blocker administration and 24 h after termination ofAT treatment. At the end of the 4-wk experimental period,EDV was elevated by �58% in the MI group and �72% in theMI � AT group, whereas LV mass was reduced in the MIgroup by �17% and increased in the MI � AT group by�11% (Fig. 3). Despite these differences in LV EDV andmass, the two groups showed comparable increases in EDV-to-mass ratio, indicating quite proportional degrees of LVremodeling in both groups of rats (Table 1 and Fig. 3).Although AT-treated rats revealed a significant increase instroke volume 4 wk after MI while it remained almost the samein the untreated MI rats, it appeared that such an increase couldbe in part attributed to the lower heart rate in the former. Itremains unclear why heart rate in the MI � AT group was stillreduced 24 h after �1-blocker administration was ceased, but itwas obviously beneficial for cardiac output, which increasedby �17% compared with the value 24 h after MI (Table 1).Most importantly, a smaller drop in ejection fraction wasrecorded in MI � AT rats (�24 vs. �35% in MI rats; Fig. 3).These data suggest that chronic �1-blockade and subsequentbradycardia had no significant impact on LV remodeling inpost-MI hearts but was able to minimize the decline in LVcontractile performance.
Greater ventricular hypertrophy accompanies chronic bra-dycardia in post-MI rats. To assess the degree of reactiveventricular hypertrophy recorded in MI � AT rats in vivo byechocardiography (Fig. 3), the infarct size and ventricularweight-to-body weight ratio were analyzed postmortem at theend of the experimental period. The infarct size estimated onthe formalin-fixed rings of the LV was not different betweenthe MI and MI � AT rats (67.1 � 3.8 and 61.5 � 3.5%,respectively). Despite the relative similarity in infarct size,
Fig. 2. Heart rate in rats subjected to myocardial infarction (MI, n � 10) andMI rats treated with atenolol (MI � AT, n � 8) expressed as a percentage ofmean heart rate in control/sham (C/SH) rats (n � 12). Heart rate was recordedin conscious, unrestrained rats. Values are means � SE. †P � 0.001 vs. C/SHand MI rats.
Fig. 1. Representative images from sections of a sham-operated heart stainedwith antibodies against smooth muscle (SM) �-actin and laminin and withBS-I lectin. A: SM �-actin-positive arterioles were used to estimate arteriolarlength density. B: BS-I lectin-labeled capillaries were used to determinecapillary density. C: laminin-outlined cardiac myocytes counterstained with4�,6-diamidino-2-phenylindole were used to determine cross-sectional area ofnucleated (�) myocytes. Scale bars, 20 �m.
H2687BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org
ventricular weight-to-body weight ratio was significantlyhigher in MI � AT than in MI and C/SH rats (Table 2). Inaddition, a significantly greater degree of ventricular hypertro-phy in MI � AT than MI rats was indicated by �12% greaterpostmortem ventricular mass in the former (Table 2). Todetermine whether the increase in LV mass correlates with thegrowth of cardiac myocytes, CSAs of the nucleated myocyteprofiles were measured on histological sections immunostainedagainst laminin and counterstained with DAPI (Fig. 1C). CSAsof myocytes from the epimyocardial and endomyocardial re-gions of the LV free wall were significantly greater in MI andMI � AT than in C/SH rats but were similar for the threegroups in the septum (see Table 5). These observations alsodocument a relatively similar degree of expansion in the CSAsof myocytes remaining in the LV free wall of untreated andAT-treated post-MI rats. Thus an increase of the ventricularmass in bradycardic animals is not only due to the myocytegrowth in diameter but also is likely a result of myocyteelongation, which is indicative of the eccentric hypertrophy ofthe heart.
Chronic bradycardia improves myocardial perfusion andcoronary reserve in post-MI rats. To avoid any acute effect ofAT treatment on hemodynamic and blood flow analyses, �1-blocker administration in the MI � AT group was terminated48 h before these measurements. The evaluation of systemicand LV hemodynamics conducted on deeply anesthetized andacutely instrumented rats before blood flow measurementsrevealed no significant differences in MI and MI � AT ratsamong analyzed parameters, including heart rate and mean
arterial pressure, that could markedly affect the level of coro-nary blood flow at baseline (Table 3). Nevertheless, comparedwith C/SH rats, both post-MI groups of rats showed a markeddecline in systolic blood pressure as well as in maximalsystolic LV pressure. In addition, LV end-diastolic pressurewas higher in MI � AT and MI than in C/SH rats, indicativeof developing heart failure (Table 3). These findings obviouslysuggest that, at the beginning of blood flow analyses, neither ofthe two post-MI groups had hemodynamic parameters thatdiffered to such an extent that significant miscalculations in thelevel of myocardial perfusion could result.
To evaluate the regional myocardial perfusion and CPR inthe LV free wall and septum at the end of the experimentalperiod, intramural coronary blood flow was measured at rest(baseline) and after maximal coronary dilation induced bydipyridamole (Table 4). It is important to mention that al-though mean arterial pressure was significantly lower in un-treated post-MI than in C/SH rats before blood flow analysis(Table 3), no marked difference in perfusion pressure wasdetermined among the three groups of rats during each flowmeasurement when microspheres were actually infused into theLV (Table 4).
Fig. 3. Ejection fraction, left ventricular(LV) mass, LV end-diastolic volume, andLV end-diastolic volume-to-mass ratio in MIand MI � AT rats expressed as percentchange from values recorded in the same rats24 h and 4 wk after MI. In MI � AT rats,echocardiographic analysis was done 24 hbefore �1-blocker administration and 24 hafter termination of AT treatment. Valuesare means � SE. **P � 0.01 vs. MI.
Table 2. Body and ventricular weights and ventricularweight-to-body weight ratio at the end of the experiment
C/SH (n � 12) MI (n � 10)MI � AT(n � 8)
Body wt, g 492.4�20.1 493.6�14.3 454.3�17.1Total ventricular wt, mg 1387.9�66.9 1369.8�38.1 1539.6�60.9†Ventricular wt/body wt,
mg/g 2.8�0.1 2.8�0.1 3.4�0.1*†
Values are means � SE. C/SH, control/sham group. *P � 0.01 vs. C/SH;†P � 0.01 vs. MI.
Table 3. Systemic and LV hemodynamic parametersbefore blood flow analysis
C/SH(n � 12)
MI(n � 10)
MI � AT(n � 8)
HR, beats/min 189�5 180�8 185�9SBP, mmHg 117.1�4.5 103�3.9* 106.1�2.7*DBP, mmHg 70.1�3.1 63.5�4.1 67.2�2.4MAP, mmHg 86.7�3.4 76.3�4* 82.7�3.1LVSBP, mmHg 114.7�4.1 97.1�3.8† 97.8�3.7†LVEDP, mmHg 5.3�0.4 14.2�1.5‡ 17.5�2.7‡
Values are means � SE. SBP, systolic blood pressure; DBP, diastolic bloodpressure; MAP, mean arterial pressure; LVSBP, LV systolic blood pressure;LVEDP, LV end-diastolic pressure. In the MI � AT group, hemodynamic datawere obtained 48 h after AT treatment was terminated. Systemic hemodynam-ics (HR, SBP, DBP, and MAP) were determined via the right femoral artery,before the catheter was inserted into the LV via the carotid artery to evaluateLV hemodynamics. *P � 0.05; †P � 0.01; ‡P � 0.001 vs. C/SH.
H2688 BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org
Because the level of blood flow within the vessels is depen-dent on vascular resistance and perfusion pressure, the use ofabsolute blood flow values to evaluate the changes in myocar-dial perfusion can lead to a major misinterpretation. Accord-ingly, we expressed myocardial perfusion at baseline and aftermaximal coronary dilation as coronary conductance. As aresult, we were able to accurately adjust each regional coronaryblood flow by corresponding perfusion pressure, because eachregional coronary resistance was calculated by dividing sys-temic mean arterial pressure by regional blood flow value.
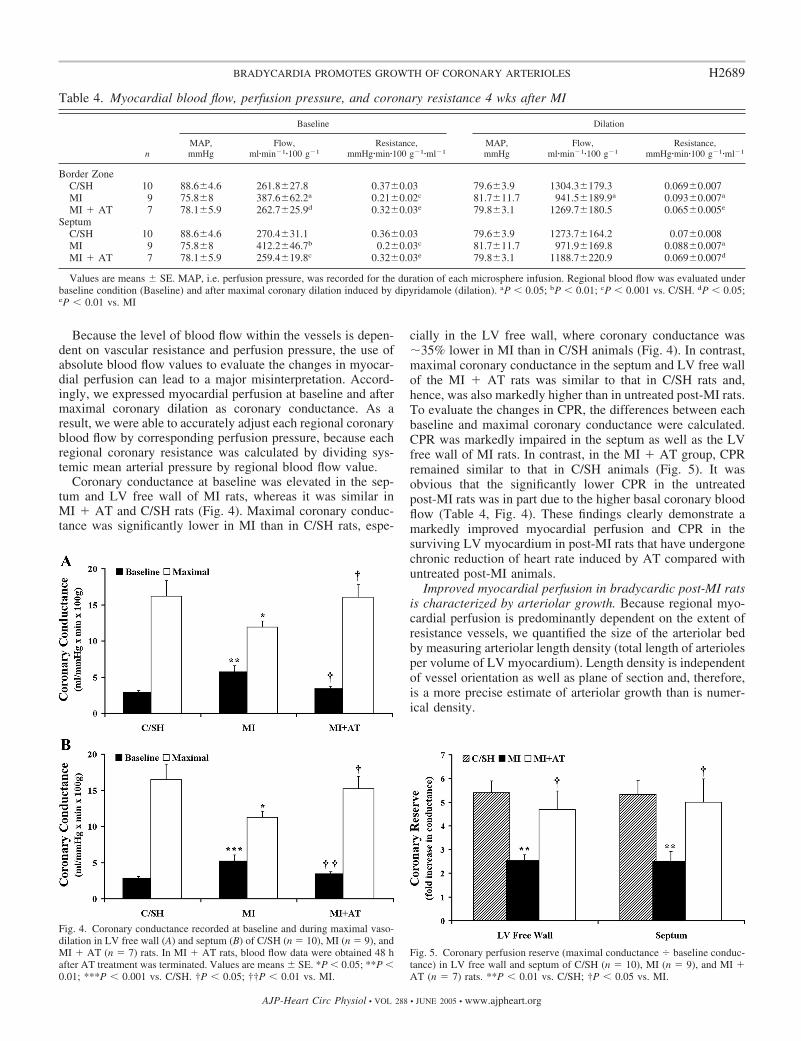
Coronary conductance at baseline was elevated in the sep-tum and LV free wall of MI rats, whereas it was similar inMI � AT and C/SH rats (Fig. 4). Maximal coronary conduc-tance was significantly lower in MI than in C/SH rats, espe-
cially in the LV free wall, where coronary conductance was�35% lower in MI than in C/SH animals (Fig. 4). In contrast,maximal coronary conductance in the septum and LV free wallof the MI � AT rats was similar to that in C/SH rats and,hence, was also markedly higher than in untreated post-MI rats.To evaluate the changes in CPR, the differences between eachbaseline and maximal coronary conductance were calculated.CPR was markedly impaired in the septum as well as the LVfree wall of MI rats. In contrast, in the MI � AT group, CPRremained similar to that in C/SH animals (Fig. 5). It wasobvious that the significantly lower CPR in the untreatedpost-MI rats was in part due to the higher basal coronary bloodflow (Table 4, Fig. 4). These findings clearly demonstrate amarkedly improved myocardial perfusion and CPR in thesurviving LV myocardium in post-MI rats that have undergonechronic reduction of heart rate induced by AT compared withuntreated post-MI animals.
Improved myocardial perfusion in bradycardic post-MI ratsis characterized by arteriolar growth. Because regional myo-cardial perfusion is predominantly dependent on the extent ofresistance vessels, we quantified the size of the arteriolar bedby measuring arteriolar length density (total length of arteriolesper volume of LV myocardium). Length density is independentof vessel orientation as well as plane of section and, therefore,is a more precise estimate of arteriolar growth than is numer-ical density.
Table 4. Myocardial blood flow, perfusion pressure, and coronary resistance 4 wks after MI
n
Baseline Dilation
MAP,mmHg
Flow,ml�min1�100 g1
Resistance,mmHg�min�100 g1�ml1
MAP,mmHg
Flow,ml�min1�100 g1
Resistance,mmHg�min�100 g1�ml1
Border ZoneC/SH 10 88.6�4.6 261.8�27.8 0.37�0.03 79.6�3.9 1304.3�179.3 0.069�0.007MI 9 75.8�8 387.6�62.2a 0.21�0.02c 81.7�11.7 941.5�189.9a 0.093�0.007a
MI � AT 7 78.1�5.9 262.7�25.9d 0.32�0.03e 79.8�3.1 1269.7�180.5 0.065�0.005e
SeptumC/SH 10 88.6�4.6 270.4�31.1 0.36�0.03 79.6�3.9 1273.7�164.2 0.07�0.008MI 9 75.8�8 412.2�46.7b 0.2�0.03c 81.7�11.7 971.9�169.8 0.088�0.007a
MI � AT 7 78.1�5.9 259.4�19.8c 0.32�0.03e 79.8�3.1 1188.7�220.9 0.069�0.007d
Values are means � SE. MAP, i.e. perfusion pressure, was recorded for the duration of each microsphere infusion. Regional blood flow was evaluated underbaseline condition (Baseline) and after maximal coronary dilation induced by dipyridamole (dilation). aP � 0.05; bP � 0.01; cP � 0.001 vs. C/SH. dP � 0.05;eP � 0.01 vs. MI
Fig. 4. Coronary conductance recorded at baseline and during maximal vaso-dilation in LV free wall (A) and septum (B) of C/SH (n � 10), MI (n � 9), andMI � AT (n � 7) rats. In MI � AT rats, blood flow data were obtained 48 hafter AT treatment was terminated. Values are means � SE. *P � 0.05; **P �0.01; ***P � 0.001 vs. C/SH. †P � 0.05; ††P � 0.01 vs. MI.
Fig. 5. Coronary perfusion reserve (maximal conductance baseline conduc-tance) in LV free wall and septum of C/SH (n � 10), MI (n � 9), and MI �AT (n � 7) rats. **P � 0.01 vs. C/SH; †P � 0.05 vs. MI.
H2689BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org
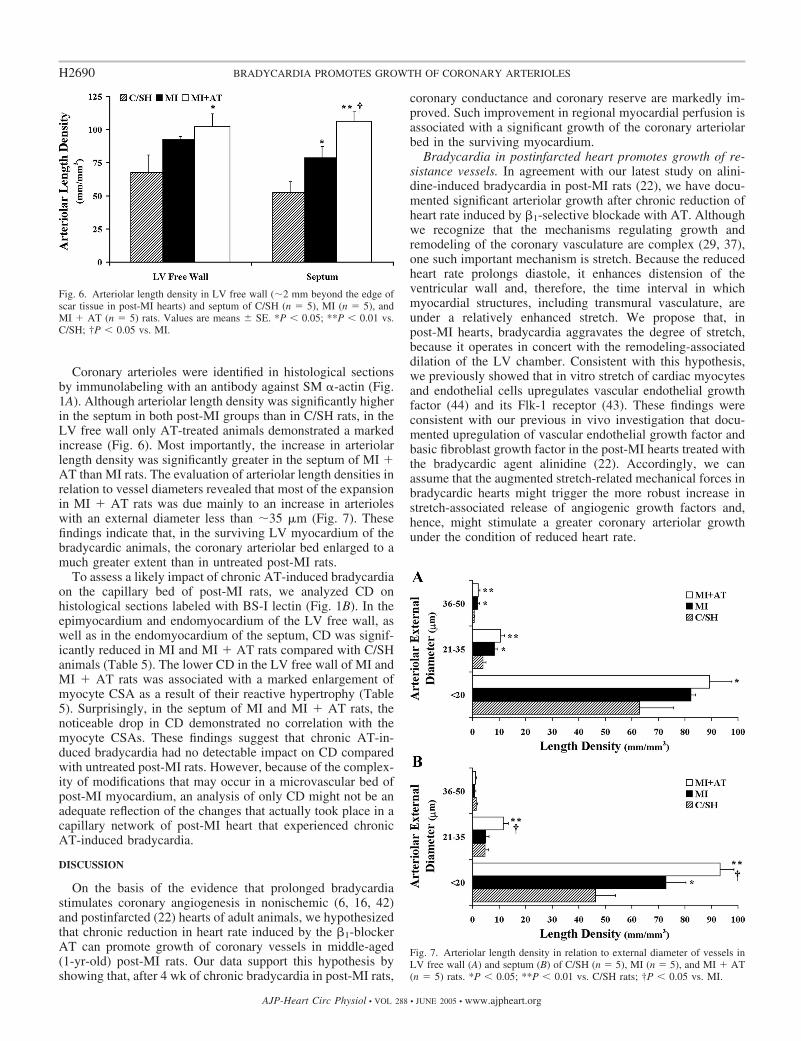
Coronary arterioles were identified in histological sectionsby immunolabeling with an antibody against SM �-actin (Fig.1A). Although arteriolar length density was significantly higherin the septum in both post-MI groups than in C/SH rats, in theLV free wall only AT-treated animals demonstrated a markedincrease (Fig. 6). Most importantly, the increase in arteriolarlength density was significantly greater in the septum of MI �AT than MI rats. The evaluation of arteriolar length densities inrelation to vessel diameters revealed that most of the expansionin MI � AT rats was due mainly to an increase in arterioleswith an external diameter less than �35 �m (Fig. 7). Thesefindings indicate that, in the surviving LV myocardium of thebradycardic animals, the coronary arteriolar bed enlarged to amuch greater extent than in untreated post-MI rats.
To assess a likely impact of chronic AT-induced bradycardiaon the capillary bed of post-MI rats, we analyzed CD onhistological sections labeled with BS-I lectin (Fig. 1B). In theepimyocardium and endomyocardium of the LV free wall, aswell as in the endomyocardium of the septum, CD was signif-icantly reduced in MI and MI � AT rats compared with C/SHanimals (Table 5). The lower CD in the LV free wall of MI andMI � AT rats was associated with a marked enlargement ofmyocyte CSA as a result of their reactive hypertrophy (Table5). Surprisingly, in the septum of MI and MI � AT rats, thenoticeable drop in CD demonstrated no correlation with themyocyte CSAs. These findings suggest that chronic AT-in-duced bradycardia had no detectable impact on CD comparedwith untreated post-MI rats. However, because of the complex-ity of modifications that may occur in a microvascular bed ofpost-MI myocardium, an analysis of only CD might not be anadequate reflection of the changes that actually took place in acapillary network of post-MI heart that experienced chronicAT-induced bradycardia.
DISCUSSION
On the basis of the evidence that prolonged bradycardiastimulates coronary angiogenesis in nonischemic (6, 16, 42)and postinfarcted (22) hearts of adult animals, we hypothesizedthat chronic reduction in heart rate induced by the �1-blockerAT can promote growth of coronary vessels in middle-aged(1-yr-old) post-MI rats. Our data support this hypothesis byshowing that, after 4 wk of chronic bradycardia in post-MI rats,
coronary conductance and coronary reserve are markedly im-proved. Such improvement in regional myocardial perfusion isassociated with a significant growth of the coronary arteriolarbed in the surviving myocardium.
Bradycardia in postinfarcted heart promotes growth of re-sistance vessels. In agreement with our latest study on alini-dine-induced bradycardia in post-MI rats (22), we have docu-mented significant arteriolar growth after chronic reduction ofheart rate induced by �1-selective blockade with AT. Althoughwe recognize that the mechanisms regulating growth andremodeling of the coronary vasculature are complex (29, 37),one such important mechanism is stretch. Because the reducedheart rate prolongs diastole, it enhances distension of theventricular wall and, therefore, the time interval in whichmyocardial structures, including transmural vasculature, areunder a relatively enhanced stretch. We propose that, inpost-MI hearts, bradycardia aggravates the degree of stretch,because it operates in concert with the remodeling-associateddilation of the LV chamber. Consistent with this hypothesis,we previously showed that in vitro stretch of cardiac myocytesand endothelial cells upregulates vascular endothelial growthfactor (44) and its Flk-1 receptor (43). These findings wereconsistent with our previous in vivo investigation that docu-mented upregulation of vascular endothelial growth factor andbasic fibroblast growth factor in the post-MI hearts treated withthe bradycardic agent alinidine (22). Accordingly, we canassume that the augmented stretch-related mechanical forces inbradycardic hearts might trigger the more robust increase instretch-associated release of angiogenic growth factors and,hence, might stimulate a greater coronary arteriolar growthunder the condition of reduced heart rate.
Fig. 7. Arteriolar length density in relation to external diameter of vessels inLV free wall (A) and septum (B) of C/SH (n � 5), MI (n � 5), and MI � AT(n � 5) rats. *P � 0.05; **P � 0.01 vs. C/SH rats; †P � 0.05 vs. MI.
Fig. 6. Arteriolar length density in LV free wall (�2 mm beyond the edge ofscar tissue in post-MI hearts) and septum of C/SH (n � 5), MI (n � 5), andMI � AT (n � 5) rats. Values are means � SE. *P � 0.05; **P � 0.01 vs.C/SH; †P � 0.05 vs. MI.
H2690 BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org

Because a marked compensatory hypertrophy of survivingcardiac myocytes in the LV develops in response to the largeintramural infarct (1, 30) as a result of myocyte overload, agrowth of resistance vessels (i.e., arterioles) must occur toprovide an adequate tissue perfusion. This supposition wasvalidated by the results of our present and previous (22)studies, which demonstrated a marked arteriolar growth inpost-MI hearts that underwent an eccentric hypertrophy. More-over, these data are in concert with our previous work, whichdocumented the induction of arteriolar development in volume-overloaded hearts, which also showed LV chamber remodelingtypical of eccentric hypertrophy (10). These findings suggestthat a greater development of the arteriolar bed in bradycardicpost-MI rats might, to some extent, be triggered by moreadvanced growth in the ventricular mass detected in MI � ATanimals.
In addition, we can propose that because of the evidence ofsome, more limited, arteriolar growth in the untreated post-MIrats, almost the same stimuli that triggered arteriolar growth inbradycardic heart, including mechanical stretch and myocar-dial hypertrophy, might also be effective in all post-MI hearts,only on more limited scale.
Most importantly, our present finding that the largest portionof the increase in arteriolar length density detected in post-MIhearts is due to formation of the smallest “terminal” arterioles(Fig. 7) is consistent with the concept that new arterioles mostlikely emerge from preexisting capillaries via capillary arterio-larization (28, 34, 39). Similar observations were previouslyreported in rats with MI that underwent pharmacological ther-apy (22, 45) or those subjected to volume overload (10).Furthermore, exercise training in swine also stimulated anincrease in the small arterioles in the heart (39). On the basis ofthese observations, we suggest that greater expansion of thecoronary arteriolar bed detected in MI � AT rats was in partdue to more advanced arteriolar transformation of preexistingcapillaries.
Is the cause of bradycardia and/or the type of experimentalmodel important for induction of capillary growth? In the lastfew decades, several lines of evidence demonstrated that bra-dycardia induced by electrical pacing (5, 17, 18, 40, 41) or bythe negative chronotropic agent alinidine (7, 42) stimulatesangiogenesis in the adult myocardium of normal or volume-overloaded hearts. The results from these studies demonstratedthat the long-term reduction in heart rate by 45% in rabbits (17,18, 40, 41), 34% in pigs (5), and 28% in rats (7) was associatedwith an increased CD or a higher capillary-to-fiber ratio than in
control animals. More recently, data from our laboratory alsorevealed that 3 wk of alinidine-induced bradycardia resulted inhigher capillary length density in the surviving LV myocar-dium of young post-MI rats (22).
In this study, AT consistently reduced heart rate in the12-mo-old post-MI rats by 25–28% compared with C/SH rats;thus the degree of bradycardia was similar to that achievedpreviously in younger (4-mo-old) post-MI rats by alinidine (7,22). Surprisingly, in contrast to these studies, the bradycardichearts of the MI � AT rats did not reveal any increase incapillary numerical density, even in the septum, where myo-cyte CSA enlargement was not detected. These contrastingdata raised the following question: Might the method of induc-ing bradycardia to some extent be accountable for the differ-ence between the present findings and data reported previ-ously? One possible explanation is that, in contrast to alinidine-induced bradycardia, treatment with the �1-blocker AT notonly lowers heart rate, it also markedly reduces myocardialcontractility (negative inotropy) as well as afterload (systemichypotension) and, therefore, decreases cardiac myocyte O2
consumption (11). Such a decrease in myocyte O2 consump-tion might inhibit capillary growth, because tissue PO2 is astrong determinant for induction and/or maintenance of capil-lary growth (9, 31). A second possible explanation is thatbecause AT administration reduces myocardial blood flow (3,13), it follows that shear stress and wall tension, the two vitalmechanical stimuli needed for the activation of endothelialcells and angiogenesis (6), would also be diminished within acapillary network during an entire period of �-blocker treat-ment. One additional explanation for the lack of capillarygrowth may be the use of 1-yr-old rats. A previous studyshowed a marked impact of advanced animal age on ability ofcapillaries to expand, in which 12 wk of exercise trainingstimulated capillary growth in 17-wk-old rats, whereas in twoolder age groups (31 and 94 wk) it did not (36). Finally, asnoted above, capillary-to-arteriolar transformation may, tosome extent, be responsible for the drop in capillary density inboth post-MI groups of rats.
It is also important to note that in our previous studies (22,42), where we showed a marked capillary growth in brady-cardic hearts, we used 1-�m-thick plastic sections and mea-sured the axial ratio of the capillary lumen. Such a methodpermitted computation of the capillary length density as analternative to the less precise numerical density reported in thisstudy. Because the degree of tissue shrinkage was much less incryosections and the numerical density did not use the capillary
Table 5. CD and CSA of nucleated cardiac myocytes
CD, counts/�m2 Myocyte CSA, �m2
LV free wall Septum LV free wall Septum
EpimyocardiumC/SH 3503.04�254.3 379.2�41.6MI 2651.7�173.3* 592.1�71.5*MI � AT 2721.8�138.2* 560.8�63.5*
EndomyocardiumC/SH 4204.1�245.8 4076.2�356.5 358.1�27.6 397.3�47.2MI 3405.4�156.4* 3404.3�63.2* 479.9�64.4* 391.1�39.8MI � AT 3240.6�224.1* 2984.5�147.3* 501.7�30.6† 416.1�44.3
Values are means � SE; n � 5. In the septum, capillary numerical density (CD) and myocyte cross-sectional area (CSA) were analyzed only on the side ofthe endomyocardium facing the LV. *P � 0.05; †P � 0.01 vs. C/SH.
H2691BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org

lumen axial ratio as a multiplier, it should not be a surprise thatcapillary density values demonstrated in our present study weresignificantly different from those reported previously. Regret-tably, the methodological limitation in this study has notpermitted a more comprehensive analysis of adaptations thatmight occur in the capillary bed of post-MI rats with AT-induced bradycardia.
Coronary conductance and vasodilator reserve are im-proved in post-MI hearts after chronic bradycardia. Previousstudies of myocardial perfusion in rat hearts with large trans-mural MI have shown significantly reduced maximal coronaryconductance and vasodilator reserve, especially in the regionwhere myocyte hypertrophy was evident (19, 20). Consistentwith these data, our present study has also documented mark-edly reduced levels of maximal coronary conductance andcoronary perfusion (or vasodilator) reserve (CPR) in untreatedpost-MI rats compared with C/SH animals. In contrast, post-MIrats that had undergone 4 wk of continuous bradycardia causedby chronic �1-selective blockade with AT revealed no declinein maximal coronary conductance and CPR compared withC/SH animals after withdrawal of the drug. Most importantly,both perfusion indexes were significantly higher in MI � ATthan in MI rats. This positive effect of chronic reduction inheart rate on myocardial perfusion was consistent with thatdocumented previously in our laboratory in a model of alini-dine-induced bradycardia in young post-MI rats (22).
Interestingly, our present findings as well as those of others(19, 20) indicate that a noticeable drop in CPR detected inuntreated post-MI rats was, to a certain extent, due to acombined effect of higher coronary blood flow at baseline andlower coronary blood flow after maximal dilation. In view ofthe fact that, during blood flow measurements in our experi-ments, hemodynamic parameters, such as heart rate and meanarterial pressure (or perfusion pressure), were comparableamong the three groups of rats at baseline as well as aftermaximal vasodilation, the alterations in coronary conductancemust be attributed to other factors, such as O2/metabolicdemand or the extent of arteriolar bed expansion.
The increase in basal myocardial perfusion in rats with largeMI is not surprising, because the surviving myocardium expe-riences an increased afterload (or wall stress), which increasesO2 demand in overloaded LV cardiac myocytes. In contrast,MI � AT rats demonstrated normal perfusion values at rest.The mechanism associated with the lower O2 demand inmyocytes of bradycardic animals after withdrawal of AT is notevident at this time.
Although noticeable growth of coronary arterioles was doc-umented in MI and MI � AT rats, the magnitude of arteriolarbed enlargement was less in MI than in MI � AT rats. Thus itis not unexpected that maximal coronary conductance was lessin the MI than in the MI � AT rats. Consistent with this resultis the fact that coronary minimal resistance was �43 and 27%higher in the LV free wall and septum, respectively, in the MIthan in the MI � AT rats (Table 4). Most importantly, becausemaximal coronary conductance was similar in MI � AT andC/SH rats, it can be proposed that the aggregate arteriolar CSAper volume of LV myocardium is similar in these two groups.These results and our previous data (22) indicate that arteriolardevelopment in bradycardic animals was able to fully preventa deficit in myocardial perfusion and, therefore, to avert animminent decline in CPR detected in untreated post-MI rats.
Nevertheless, the fact that arteriolar growth in post-MI heartsalways exceeded anticipated increases in maximal coronaryconductance suggests some functional inadequacy of the newlyformed vessels.
Could bradycardia in post-MI hearts minimize LV dysfunc-tion by improving myocardial perfusion? In recent years, it hasbecome more obvious that �-adrenoceptor blockade can reducethe symptoms of systolic dysfunction and diminish the risk ofsudden death in patients suffering from cardiovascular events,including MI and heart failure (4, 21, 24). Several differentmechanisms have been proposed to elucidate the beneficialeffect of �-blockers under such conditions (4, 23). Importantly,the most vital role of the �-blockers (including AT) in resto-ration of LV function was ascribed to reduction in heart rate(14, 25). For instance, studies on the ischemic myocardium ofconscious dogs revealed that AT-induced reduction in heartrate was associated with the concurrent improvement in coro-nary blood flow and contractile performance. Most impor-tantly, when electrical pacing was used concurrently with ATtreatment to increase heart rate to baseline levels, the improve-ment in blood flow as well as in cardiac function was elimi-nated (14, 32). In our present study, as well as in a previousstudy (22), we also found a dual beneficial role of chronicbradycardia 1) on LV contractile performance, as indicated byan attenuation of the drop in ejection fraction, and 2) oncoronary perfusion indexes, as indicated by normalized CPR.In conclusion, we suggest that the bradycardia-related devel-opment of the coronary arteriolar bed and, consequently, agreater myocardial perfusion may play an essential role in theamelioration of LV dysfunction in the postinfarcted heart.
Conclusion. Our data provide clear evidence that chronicbradycardia induced with �-selective blockade promotes agreater arteriolar growth in the surviving LV myocardium ofthe postinfarcted heart that is adequate to restore the compro-mised myocardial perfusion and coronary reserve.
ACKNOWLEDGMENTS
We thank Kathy Zimmerman for technical expertise.
GRANTS
This study was supported by National Heart, Lung, and Blood InstituteGrant RO1-HL-62587.
REFERENCES
1. Anversa P, Beghi C, Kikkawa Y, and Olivetti G. Myocardial infarctionin rats. Infarct size, myocyte hypertrophy, and capillary growth. Circ Res58: 26–37, 1986.
2. Anversa P and Capasso JM. Loss of intermediate-sized coronary arteriesand capillary proliferation after left ventricular failure in rats. Am J PhysiolHeart Circ Physiol 260: H1552–H1560, 1991.
3. Berdaux A, Bossier JR, and Giudicelli JF. Effects of atenolol onregional myocardial blood flow and ST segment elevation in the caninemyocardium. Br J Pharmacol 60: 433–439, 1977.
4. Bohm M and Maack C. Treatment of heart failure with �-blockers.Mechanisms and results. Basic Res Cardiol 95 Suppl I: I15–I24, 2000.
5. Brown MD, Davies MK, and Hudlicka O. The effect of long-termbradycardia on heart microvascular supply and performance. Cell Mol BiolRes 40: 137–142, 1994.
6. Brown MD and Hudlicka O. Angiogenesis in skeletal and cardiacmuscle: role of mechanical factors. In: The New Angiotherapy, edited byFan T-PD and Kohn EC. Totowa, NJ: Humana, 2002, p. 213–248.
7. Brown MD and Hudlicka O. Capillary growth in the heart. EXS 61:389–394, 1992.
8. Buck JD, Gross GJ, Warltier DC, Jolly SR, and Hardman HF.Comparative effects of cardioselective versus noncardioselective �-block-
H2692 BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org

ade on subendocardial blood flow and contractile function in ischemicmyocardium. Am J Cardiol 44: 657–663, 1979.
9. Carmeliet P and Collen D. Molecular basis of angiogenesis. Role ofVEGF and VE-cadherin. Ann NY Acad Sci 902: 249–264, 2000.
10. Chen Y, Torry RJ, Baumbach GL, and Tomanek RJ. Proportionalarteriolar growth accompanies cardiac hypertrophy induced by volumeoverload. Am J Physiol Heart Circ Physiol 267: H2132–H2137, 1994.
11. Colin P, Ghaleh B, Monnet X, Su J, Hittinger L, Giudicelli JF, andBerdeaux A. Contributions of heart rate and contractility to myocardialoxygen balance during exercise. Am J Physiol Heart Circ Physiol 284:H676–H682, 2003.
12. Fishbein MC, Maclean D, and Maroko PR. Experimental myocardialinfarction in the rat: qualitative and quantitative changes during pathologicevolution. Am J Pathol 90: 57–70, 1978.
13. Grover GJ, Tierney MA, and Weiss HR. �-Adrenoceptor control of themicrovascular reserve in rabbit myocardium. J Pharmacol Exp Ther 238:868–873, 1986.
14. Guth BD, Indolfi C, Heusch G, Seitelberger R, and Ross J Jr.Mechanisms of benefit in the ischemic myocardium due to heart ratereduction. Basic Res Cardiol 85 Suppl I: I57-I66, 1990.
15. Helisch A and Ware JA. Therapeutic angiogenesis for ischemic heartdisease. Adv Exp Med Biol 476: 327–350, 2000.
16. Hudlicka O, Egginton S, and Brown MD. Angiogenesis in the heart andskeletal muscles—models for capillary growth. In: Angiogenesis: Models,Modulators, and Clinical Applications, edited by Maragoudakis ME. NewYork: Plenum, 1998.
17. Hudlicka O, West D, Kumar S, el Khelly F, and Wright AJ. Cangrowth of capillaries in the heart and skeletal muscle be explained by thepresence of an angiogenic factor? Br J Exp Pathol 70: 237–246, 1989.
18. Hudlicka O, Wright AJ, Hoppeler H, and Uhlmann E. The effect ofchronic bradycardial pacing on the oxidative capacity in rabbit hearts.Respir Physiol 72: 1–12, 1988.
19. Kalkman EA, van Haren P, Saxena PR, and Schoemaker RG. Region-ally different vascular response to vasoactive substances in the remodelledinfarcted rat heart: aberrant vasculature in the infarct scar. J Mol CellCardiol 29: 1487–1497, 1997.
20. Karam R, Healy BP, and Wicker P. Coronary reserve is depressed inpostmyocardial infarction reactive cardiac hypertrophy. Circulation 81:238–246, 1990.
21. Kendall MJ. Clinical trial data on the cardioprotective effects of �-block-ade. Basic Res Cardiol 95, Suppl I: I25–I30, 2000.
22. Lei L, Zhou R, Zheng W, Christensen LP, Weiss RM, and TomanekRJ. Bradycardia induces angiogenesis, increases coronary reserve, andpreserves function of the postinfarcted heart. Circulation 110: 796–802,2004.
23. Lohse MJ, Engelhardt S, and Eschenhagen T. What is the role of�-adrenergic signaling in heart failure? Circ Res 93: 896–906, 2003.
24. Metra M, Nodari S, Parrinello G, Giubbini R, Manca C, and Dei CasL. Marked improvement in left ventricular ejection fraction during long-term �-blockade in patients with chronic heart failure: clinical correlatesand prognostic significance. Am Heart J 145: 292–299, 2003.
25. Nagatsu M, Spinale FG, Koide M, Tagawa H, DeFreitas G, Cooper GIV, and Carabello BA. Bradycardia and the role of �-blockade in theamelioration of left ventricular dysfunction. Circulation 101: 653–659,2000.
26. Norris RM. Therapeutic interventions in experimental infarction. I. Re-duction in myocardial oxygen and substrate demand. In: MyocardialInfarction: Its Presentation, Pathogenesis and Treatment. Edinburgh:Churchill Livingstone, 1982, p. 291–302.
27. Pfeffer JM, Pfeffer MA, Fletcher PJ, and Braunwald E. Progressiveventricular remodeling in rat with myocardial infarction. Am J PhysiolHeart Circ Physiol 260: H1406–H1414, 1991.
28. Price RJ, Owens GK, and Skalak TC. Immunohistochemical identifi-cation of arteriolar development using markers of smooth muscle differ-entiation. Evidence that capillary arterialization proceeds from terminalarterioles. Circ Res 75: 520–527, 1994.
29. Rakusan K. Coronary angiogenesis. From morphometry to molecularbiology and back. Ann NY Acad Sci 752: 257–266, 1995.
30. Rubin SA, Fishbein MC, and Swan HJ. Compensatory hypertrophy inthe heart after myocardial infarction in the rat. J Am Coll Cardiol 1:1435–1441, 1983.
31. Schelling ME, Meininger CJ, Hawker JR Jr, and Granger HJ. Venularendothelial cells from bovine heart. Am J Physiol Heart Circ Physiol 254:H1211–H1217, 1988.
32. Schulz R, Guth BD, and Heusch G. Pharmacological mechanisms toattenuate sympathetically induced myocardial ischemia. CardiovascDrugs Ther 3: 43–56, 1989.
33. Shimada K, Nishikimi T, Kawarabayashi T, Takeuchi K, and TakedaT. Effect of prolonged �-adrenergic blockade induced by atenolol on leftventricular remodeling after acute myocardial infarction in the rat. JpnHeart J 36: 81–89, 1995.
34. Skalak TC, Price RJ, and Zeller PJ. Where do new arterioles comefrom? Mechanical forces and microvessel adaptation. Microcirculation 5:91–94, 1998.
35. Tasgal J and Williams EM. The effect of prolonged propranolol admin-istration on myocardial transmural capillary density in young rabbits.J Physiol 315: 353–367, 1981.
36. Tomanek RJ. Effects of age and exercise on the extent of the myocardialcapillary bed. Anat Rec 167: 55–62, 1970.
37. Tomanek RJ and Torry RJ. Growth of the coronary vasculature inhypertrophy: mechanisms and model dependence. Cell Mol Biol Res 40:129–136, 1994.
38. Waltenberger J and Hombach V. Therapeutic angiogenesis for the heart.In: The New Angiotherapy, edited by Fan T-PD and Kohn EC. Totowa,NJ: Humana, 2002, p. 279–293.
39. White FC, Bloor CM, McKirnan MD, and Carroll SM. Exercisetraining in swine promotes growth of arteriolar bed and capillary angio-genesis in heart. J Appl Physiol 85: 1160–1168, 1998.
40. Wright AJ and Hudlicka O. Capillary growth and changes in heartperformance induced by chronic bradycardial pacing in the rabbit. CircRes 49: 469–478, 1981.
41. Wright AJ, Hudlicka O, and Brown MD. Beneficial effect of chronicbradycardial pacing on capillary growth and heart performance in volumeoverload heart hypertrophy. Circ Res 64: 1205–1212, 1989.
42. Zheng W, Brown MD, Brock TA, Bjercke RJ, and Tomanek RJ.Bradycardia-induced coronary angiogenesis is dependent on vascularendothelial growth factor. Circ Res 85: 192–198, 1999.
43. Zheng W, Christensen LP, and Tomanek RJ. Stretch induces upregu-lation of key tyrosine kinase receptors in microvascular cells. Am J PhysiolHeart Circ Physiol 287: H2739–H2745, 2004.
44. Zheng W, Seftor EA, Meininger CJ, Hendrix MJ, and Tomanek RJ.Mechanisms of coronary angiogenesis in response to stretch: role ofVEGF and TGF-�. Am J Physiol Heart Circ Physiol 280: H909–H917,2001.
45. Zheng W, Weiss RM, Wang X, Zhou R, Arlen AM, Lei L, LazartiguesE, and Tomanek RJ. DITPA stimulates arteriolar growth and modifiesmyocardial postinfarction remodeling. Am J Physiol Heart Circ Physiol286: H1994–H2000, 2004.
H2693BRADYCARDIA PROMOTES GROWTH OF CORONARY ARTERIOLES
AJP-Heart Circ Physiol • VOL 288 • JUNE 2005 • www.ajpheart.org
Related Documents











