Reduced Corneal Thickness and Enlarged Anterior Chamber in a Novel ColVIIIa2 G257D Mutant Mouse Oliver Puk, 1 Claudia Dalke, 1,2 Julia Calzada-Wack, 3 Nafees Ahmad, 1 Matthias Klaften, 4 Sibylle Wagner, 4 Martin Hrabe ´ de Angelis, 4,5 and Jochen Graw 1 PURPOSE. The purpose of this study was the morphologic and genetic characterization of the novel eye size mutant Aca23 in the mouse. METHODS. The eyes of the mutants were characterized in vivo by optical low-coherence interferometry, Scheimpflug imag- ing, and funduscopy. Visual acuity was examined using a vir- tual optomotor system. Morphology was studied by histology, in situ hybridization, and immunohistochemistry. Linkage anal- ysis was performed using genomewide scans with single nu- cleotide polymorphisms and microsatellite markers. RESULTS. Aca23 is a new semidominant eye size mutant that was discovered in an ENU mutagenesis screen. The phenotype includes increased anterior chamber depths, extended axial lengths, and reduced thickness of corneal layers. Aca23 was mapped to chromosome 4. A G3 A point mutation was iden- tified at cDNA position 770 of Col8a2 encoding collagen VIII 2. The transition results in a G257D amino acid exchange affecting a highly conserved glycine residue in the collagenous domain. Proliferation of corneal endothelium, eye fundus, and visual acuity are not affected. CONCLUSIONS. The mouse mutant Aca23 described here offers the first point mutation of the Col8a2 gene in the mouse. The results of this study suggest that a functional collagen VIII 2 is essential for the correct assembly of the Descemet’s membrane and for corneal stability. Aca23 might be used as a novel model for keratoglobus. (Invest Ophthalmol Vis Sci. 2009;50: 5653–5661) DOI:10.1167/iovs.09-3550 M ore than 161 million people worldwide are blind or suffer from severe visual disorders. In most cases, pathologies of ocular tissues negatively affect visual properties. This in- cludes irregularities of different eye size parameters such as axial length of the entire eye globe (microphthalmia/macroph- thalmia), lens size (microphakia/macrophakia), or anterior chamber depth. Congenital eye size diseases very often occur in addition to further systemic abnormalities inside and outside the eye rather than as isolated pathologic phenotypes. The eye size-related disease with the highest frequency is microphthalmia (incidence 0.6 –2.3 per 10,000 births 1 ). A broad range of different syndromes associated with microph- thalmia have already been described. 2 However, only few genes have currently been identified to play an essential role in the development of this phenotype. SOX2 was found to be mutated in approximately 10% of patients with microphthal- mia/anophthalmia. 3–5 Other causative transcription factor genes include MITF, PAX6, RAX, CHX10, and OTX2 (for a recent review, see Verma and FitzPatrick 6 ). In contrast to microphthalmia, lens size abnormalities are usually rare diseases with an incidence between 1 per 10,000 births and a few dozen patients worldwide. Microphakia is part of the Warburg micro syndrome (WARBM1 7 ), Marfan’s syn- drome (MFS 8 ), rhizomelic chondrodysplasia calcificans punc- tata (RCDP 9 ), and ectopia lentis et pupillae (ELeP 10 ). The causative gene for each of these syndromes has been identified. RAB3GAP, encoding RAB3 GTPase activating protein, is af- fected in patients with WARBM1. 11 MFS cases result from alterations of the fibrillin-1 gene (FBN1 12 ). Furthermore, RCDP is caused by mutations in the PEX7 gene, 13–15 which codes for the peroxisomal type 2 targeting signal (PTS2) receptor. Congenital anterior chamber depth pathologies are typically observed in glaucomatous syndromes and corneal diseases. Abnormally large anterior chambers were described in patients with primary congenital glaucoma (buphthalmos 16 ). Buphthal- mos was demonstrated to be caused by mutations of the human cytochrome P4501B1 gene (CYP1B1 17 ). A shallow an- terior chamber is characteristic for autosomal dominant and recessive cornea plana (CNA1/CNA2) in which the cornea is opaque, flattened, and of a low refractive power. 18 –20 CNA2, but not CNA1, was identified to be caused by mutations in the keratocan-encoding gene KERA. 21–23 Furthermore, keratoglo- bus was identified in patients with posterior polymorphous corneal dystrophy (PPCD 24 ), a rare corneal endothelial disease caused by mutations in the transcription factor genes TCF8 25,26 and Zeb1, 27,28 and in the collagen type VIII gene COL8A2. 29,30 Even though various eye size-associated mutations have already been described, linkage studies point to a broad ge- netic heterogeneity with additional, not yet identified, loci and alleles. 31–33 The fact that causative mutations are still com- pletely unknown in many eye size associated-syndromes fur- ther indicates the necessity to search for additional mutations. To learn more about causative genes and alleles, we are cur- rently screening for novel abnormal eye size mouse models within a dominant ENU (N-ethyl-N-nitrosourea) mutagenesis program. 34 In this study, we present the first characterized eye size mutant line Aca23, which exhibits significantly larger anterior chamber depths, longer axes, and thinner corneas than determined for wild-type mice. 35 The causative mutation maps to chromosome 4 and represents a new missense muta- tion in Col8a2, encoding collagen type VIII. Funduscopic pa- rameters and visual properties are not altered. From the Institutes of 1 Developmental Genetics, 3 Pathology, and 4 Experimental Genetics, Helmholtz Center Munich, German Research Center for Environmental Health, Neuherberg, Germany; and the 5 De- partment of Experimental Genetics, Technical University Munich, Cen- ter of Life and Food Sciences, Freising-Weihenstephan, Germany. 2 Present affiliation: Institute of Radiation Biology, Helmholtz Cen- ter Munich, German Research Center for Environmental Health, Neu- herberg, Germany. Supported in part by European Community (EUMODIC) and sub- sequent National Genome Network Grants (NGFN, NGFN plus) LSHG- 2006-037188 and 01GS0850. Submitted for publication February 10, 2009; revised May 22, 2009; accepted August 20, 2009. Disclosure: O. Puk, None; C. Dalke, None; J. Calzada-Wack, None; N. Ahmad, None; M. Klaften, None; S. Wagner, None; M. Hrabe ´ de Angelis, None; J. Graw, None The publication costs of this article were defrayed in part by page charge payment. This article must therefore be marked “advertise- ment” in accordance with 18 U.S.C. §1734 solely to indicate this fact. Corresponding author: Oliver Puk, Institute of Developmental Genetics, Helmholtz Center Munich, German Research Center for Environmental Health, Ingolsta ¨dter Landstrasse 1, D-85764 Neuher- berg, Germany; [email protected]. Investigative Ophthalmology & Visual Science, December 2009, Vol. 50, No. 12 Copyright © Association for Research in Vision and Ophthalmology 5653

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Reduced Corneal Thickness and Enlarged AnteriorChamber in a Novel ColVIIIa2G257D Mutant Mouse
Oliver Puk,1 Claudia Dalke,1,2 Julia Calzada-Wack,3 Nafees Ahmad,1 Matthias Klaften,4
Sibylle Wagner,4 Martin Hrabe de Angelis,4,5 and Jochen Graw1
PURPOSE. The purpose of this study was the morphologic andgenetic characterization of the novel eye size mutant Aca23 inthe mouse.
METHODS. The eyes of the mutants were characterized in vivoby optical low-coherence interferometry, Scheimpflug imag-ing, and funduscopy. Visual acuity was examined using a vir-tual optomotor system. Morphology was studied by histology,in situ hybridization, and immunohistochemistry. Linkage anal-ysis was performed using genomewide scans with single nu-cleotide polymorphisms and microsatellite markers.
RESULTS. Aca23 is a new semidominant eye size mutant thatwas discovered in an ENU mutagenesis screen. The phenotypeincludes increased anterior chamber depths, extended axiallengths, and reduced thickness of corneal layers. Aca23 wasmapped to chromosome 4. A G3A point mutation was iden-tified at cDNA position 770 of Col8a2 encoding collagen VIII�2. The transition results in a G257D amino acid exchangeaffecting a highly conserved glycine residue in the collagenousdomain. Proliferation of corneal endothelium, eye fundus, andvisual acuity are not affected.
CONCLUSIONS. The mouse mutant Aca23 described here offersthe first point mutation of the Col8a2 gene in the mouse. Theresults of this study suggest that a functional collagen VIII �2 isessential for the correct assembly of the Descemet’s membraneand for corneal stability. Aca23 might be used as a novel modelfor keratoglobus. (Invest Ophthalmol Vis Sci. 2009;50:5653–5661) DOI:10.1167/iovs.09-3550
More than 161 million people worldwide are blind or sufferfrom severe visual disorders. In most cases, pathologies
of ocular tissues negatively affect visual properties. This in-cludes irregularities of different eye size parameters such asaxial length of the entire eye globe (microphthalmia/macroph-
thalmia), lens size (microphakia/macrophakia), or anteriorchamber depth. Congenital eye size diseases very often occurin addition to further systemic abnormalities inside and outsidethe eye rather than as isolated pathologic phenotypes.
The eye size-related disease with the highest frequency ismicrophthalmia (incidence 0.6–2.3 per 10,000 births1). Abroad range of different syndromes associated with microph-thalmia have already been described.2 However, only fewgenes have currently been identified to play an essential role inthe development of this phenotype. SOX2 was found to bemutated in approximately 10% of patients with microphthal-mia/anophthalmia.3–5 Other causative transcription factorgenes include MITF, PAX6, RAX, CHX10, and OTX2 (for arecent review, see Verma and FitzPatrick6).
In contrast to microphthalmia, lens size abnormalities areusually rare diseases with an incidence between 1 per 10,000births and a few dozen patients worldwide. Microphakia is partof the Warburg micro syndrome (WARBM17), Marfan’s syn-drome (MFS8), rhizomelic chondrodysplasia calcificans punc-tata (RCDP9), and ectopia lentis et pupillae (ELeP10). Thecausative gene for each of these syndromes has been identified.RAB3GAP, encoding RAB3 GTPase activating protein, is af-fected in patients with WARBM1.11 MFS cases result fromalterations of the fibrillin-1 gene (FBN112). Furthermore, RCDPis caused by mutations in the PEX7 gene,13–15 which codes forthe peroxisomal type 2 targeting signal (PTS2) receptor.
Congenital anterior chamber depth pathologies are typicallyobserved in glaucomatous syndromes and corneal diseases.Abnormally large anterior chambers were described in patientswith primary congenital glaucoma (buphthalmos16). Buphthal-mos was demonstrated to be caused by mutations of thehuman cytochrome P4501B1 gene (CYP1B117). A shallow an-terior chamber is characteristic for autosomal dominant andrecessive cornea plana (CNA1/CNA2) in which the cornea isopaque, flattened, and of a low refractive power.18–20 CNA2,but not CNA1, was identified to be caused by mutations in thekeratocan-encoding gene KERA.21–23 Furthermore, keratoglo-bus was identified in patients with posterior polymorphouscorneal dystrophy (PPCD24), a rare corneal endothelial diseasecaused by mutations in the transcription factor genes TCF825,26
and Zeb1,27,28 and in the collagen type VIII gene COL8A2.29,30
Even though various eye size-associated mutations havealready been described, linkage studies point to a broad ge-netic heterogeneity with additional, not yet identified, loci andalleles.31–33 The fact that causative mutations are still com-pletely unknown in many eye size associated-syndromes fur-ther indicates the necessity to search for additional mutations.To learn more about causative genes and alleles, we are cur-rently screening for novel abnormal eye size mouse modelswithin a dominant ENU (N-ethyl-N-nitrosourea) mutagenesisprogram.34 In this study, we present the first characterized eyesize mutant line Aca23, which exhibits significantly largeranterior chamber depths, longer axes, and thinner corneasthan determined for wild-type mice.35 The causative mutationmaps to chromosome 4 and represents a new missense muta-tion in Col8a2, encoding collagen type VIII. Funduscopic pa-rameters and visual properties are not altered.
From the Institutes of 1Developmental Genetics, 3Pathology, and4Experimental Genetics, Helmholtz Center Munich, German ResearchCenter for Environmental Health, Neuherberg, Germany; and the 5De-partment of Experimental Genetics, Technical University Munich, Cen-ter of Life and Food Sciences, Freising-Weihenstephan, Germany.
2Present affiliation: Institute of Radiation Biology, Helmholtz Cen-ter Munich, German Research Center for Environmental Health, Neu-herberg, Germany.
Supported in part by European Community (EUMODIC) and sub-sequent National Genome Network Grants (NGFN, NGFN plus) LSHG-2006-037188 and 01GS0850.
Submitted for publication February 10, 2009; revised May 22,2009; accepted August 20, 2009.
Disclosure: O. Puk, None; C. Dalke, None; J. Calzada-Wack,None; N. Ahmad, None; M. Klaften, None; S. Wagner, None; M.Hrabe de Angelis, None; J. Graw, None
The publication costs of this article were defrayed in part by pagecharge payment. This article must therefore be marked “advertise-ment” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Corresponding author: Oliver Puk, Institute of DevelopmentalGenetics, Helmholtz Center Munich, German Research Center forEnvironmental Health, Ingolstadter Landstrasse 1, D-85764 Neuher-berg, Germany; [email protected].
Investigative Ophthalmology & Visual Science, December 2009, Vol. 50, No. 12Copyright © Association for Research in Vision and Ophthalmology 5653

MATERIALS AND METHODS
Mice
Mice were kept under specific pathogen-free conditions at the Helm-holtz Center Munich. The use of animals was in accordance with theGerman Law of Animal Protection and the ARVO Statement for the Useof Animals in Ophthalmic and Vision Research. Male C57BL/6J micewere treated with ENU (80 mg/kg body weight applied by intraperi-toneal injection in three weekly intervals) at the age of 10 to 12 weeksaccording to Ehling et al.36 and were mated to untreated femaleC57BL/6J mice.34 The offspring of the ENU-treated mice were screenedat the age of 11 weeks for abnormalities of eye size. Mice withphenotypic deviations were tested for a dominant mode of inheritance.
Eye Size Determination
The eye sizes of the Aca23 mutants were examined at the age of 11weeks using optical low-coherence interferometry (ACMaster; Med-itec, Carl Zeiss, Jena, Germany). Briefly, each mouse was anesthetizedwith an intraperitoneal injection of ketamine (137 mg/kg) and xylazine(6.6 mg/kg). The anesthetized mouse was placed on a platform andoriented in an appropriate position using light signals from six infraredLEDs arranged in a circle that had to be placed in the center of thepupil. Central measurements of corneal thickness, anterior chamber
depth, lens thickness, and axial length as well as data evaluation wereperformed essentially as described.35,37
Funduscopy
Mice at the age of 9 months were administered 1% atropine to each eyeto dilate their pupils. The fundus examination was performed with anindirect ophthalmoscope (Heine Sigma 150K; Haag-Streit GmbH,Wedel, Germany) and a super field lens (Volk 90D; Haag-Streit GmbH).Digital fundus images were taken with an indirect ophthalmoscope(Heine Video Omega 2C; Haag-Streit GmbH) connected to a videograbber (VRmAVC; Dieter Mann GmbH, Mainaschaff, Germany) and a40-D or 60-D lens (Volk; Fronhauser GmbH, Unterhaching, Germany).The images were imported in an image-processing program (Photo-shop 10.0; Adobe, Unterschleissheim, Germany).
Scheimpflug Imaging
Scheimpflug images were taken from 3-month-old Aca23 mice using adigital camera system (Pentacam; Oculus GmbH, Wetzlar, Germany).Each mouse was anesthetized with an intraperitoneal injection ofketamine (137 mg/kg) and xylazine (6.6 mg/kg). For imaging, micewere positioned on a platform such that the vertical light slit (lightsource: LEDs, 475 nm) was orientated in the middle of the eyeball. Fineadjustment of the distance between eye and camera was undertaken
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0.8
1
0
3.13.23.33.43.53.63.73.83.9
An
teri
or
ch
am
ber
de
pth
ma
le (
mm
)
WT A/+ A/A
03.13.23.33.43.53.63.73.83.9
**
*
**
*
**
*
**
*
An
teri
or
ch
am
ber
de
pth
fe
ma
le (
mm
)a) b)A
xia
l le
ng
thm
ale
(m
m)
Ax
ial
len
gth
fe
ma
le (
mm
)
Co
rne
al
thic
kn
ess
m
ale
(µ
m)
Co
rne
al
thic
kn
ess
fe
ma
le (
µm
)
WT A/+ A/A0
20
40
60
80
100
120*
*
0
20
40
60
80
100
120*
***
0
0.2
0.4
0.6
0.8
1
0
0.2
0.4
0.6
0.8
1
0
3.13.23.33.43.53.63.73.83.9
An
teri
or
ch
am
ber
de
pth
ma
le (
mm
)
WT A/+ A/A
03.13.23.33.43.53.63.73.83.9
**
*
**
*
**
*
**
*
An
teri
or
ch
am
ber
de
pth
fe
ma
le (
mm
)a) b)A
xia
l le
ng
thm
ale
(m
m)
Ax
ial
len
gth
fe
ma
le (
mm
)
Co
rne
al
thic
kn
ess
m
ale
(µ
m)
Co
rne
al
thic
kn
ess
fe
ma
le (
µm
)
WT A/+ A/A0
20
40
60
80
100
120**
**
0
20
40
60
80
100
120*
**
0
20
40
60
80
100
120**
******
FIGURE 1. Eye size analysis. Mean anterior chamber depth and axial length were enlarged in Aca23�/� (A/�; hatched columns) and Aca23�/�
(A/A; white columns) compared with the C57BL/6J control (wild-type [WT]; black columns). Corneal thickness was reduced in the mutants. (a)Males. (b) Females. Significant differences are marked by asterisks (*P � 0.001; t-test).
5654 Puk et al. IOVS, December 2009, Vol. 50, No. 12

with the help of the provided software to guarantee optimal focus.Subsequently, measurements were taken manually.
Virtual Vision Test
Vision tests were performed between 9 AM and 4 PM using a virtualoptomotor system (Cerebral Mechanics, Lethbridge, AB, Canada), asdescribed previously.38 Briefly, a rotating cylinder covered with avertical sine wave grating was calculated and drawn in virtual three-dimensional space on four computer monitors facing to form a square.Visually unimpaired mice track the grating with reflexive head andneck movements (head-tracking). Visual acuity of the tested mice wasquantified by a stepwise increase of the spatial frequency (0.3, 0.319,0.342, 0.364, 0.386, 0.414, 0.439, 0.469, and 0.5 cyc/deg) until nooptomotor response could be detected. The highest spatial frequencythat the tested mouse could track was identified as the threshold forvisual acuity. To avoid habituation, rotation direction was changedseveral times during measurement. Rotation speed and contrast wereset to 12.0 d/s and 100%, respectively. Given that no significant thresh-old differences were observed between males and females (P � 0.05;calculated by Mann-Whitney U test), data of both sexes were com-bined. Thresholds of wild-type C57BL/6J, heterozygous individuals andhomozygous mice were compared using the Mann-Whitney U test.
Histology
Eyes of 3-month-old mice were histologically analyzed for cornea,retina, and optic nerve head pathologies. The eyes were fixed for 7days in Davidson solution and embedded in plastic medium (JB-4;Polyscience Inc., Eppelheim, Germany) according to the manufactur-er’s protocol. Sectioning was performed with an ultramicrotome(OMU3; Reichert-Jung, Walldorf, Germany). Serial transverse 3-�msections were cut with a glass knife and stained with methylene blueand basic fuchsin.
In Situ Hybridization
In situ hybridization of sections from postnatal day (P) 7 was per-formed essentially as described by Grimm et al.39 Briefly, the embryoswere fixed in paraformaldehyde and embedded (Jung Histowax; Cam-bridge Instruments, Nussloch, Germany). Sections (7–10 �m) were cutwith a microtome (RM-2065; Leica, Nussloch, Germany) and mountedonto slides. The sections were evaluated with a light microscope(Axioplan; Carl Zeiss). Images were acquired by means of a scanningcamera (AxioCam; Jenoptik, Jena, Germany) and imported into animage-processing program (Photoshop 10.0; Adobe, Unterschleis-sheim, Germany).
Immunohistochemistry
Immunohistochemistry was performed with 2-�m paraffin sectionsfrom eyes of 3-month-old mice on an automated immunostainer, asdescribed previously.40 Sections were incubated with a 1:800 dilutedprimary keratin 12 polyclonal antibody (TransGenic Inc., Kobe, Japan)and were counterstained with hematoxylin. Anti-rabbit biotinylatedIgG (Vector, Burlingame, Canada) was used as the secondary antibody.
Mapping
Heterozygous carriers (first generation) were mated to wild-typeC3HeB/FeJ mice, and the offspring (second generation) were back-crossed to wild-type C3HeB/FeJ mice. DNA was prepared from tail tipsof affected offspring of the third generation (G3), as described previ-ously.41 For genomewide linkage analysis, genotyping of a genome-wide mapping panel consisting of 149 single nucleotide polymor-phisms was performed using a MALDI-TOF (matrix-assisted laser/desorption ionization, time of flight analyzer) mass spectrometry high-throughput genotyping system supplied by Sequenom (MassExtend;San Diego, CA41). Fine mapping of the Aca23 mutation was performed
WT Aca23-/+A/+ A/A
CI
L
WT Aca23-/+A/+ A/A
CI
LFIGURE 2. Scheimpflug analysis. Ananterior globoid, keratoglobus-likeprotrusion was detected inAca23�/� (A/�) and Aca23�/�
(A/A) compared with the C57BL/6Jcontrol (wild-type [WT]). C, cornea;I, iris; L, lens.
D4Mit279
D4Mit338
D4Mit249
Aca23
D4Mit336
3 1
D4Mit73
D4Mit203
3 1 2
R= 3, n= 203; 1.48 ± 0.85
R= 1, n= 203; 0.49 ± 0.49
R= 3, n= 203; 1.48 ± 0.85
R= 1, n= 203; 0.49 ± 0.49
R= 2, n= 203; 0.99 ± 0.69
193
D4Mit279
D4Mit338
D4Mit249
Aca23
D4Mit336
3 1
D4Mit73
D4Mit203
3 1 2
R= 3, n= 203; 1.48 ± 0.85
R= 1, n= 203; 0.49 ± 0.49
R= 3, n= 203; 1.48 ± 0.85
R= 1, n= 203; 0.49 ± 0.49
R= 2, n= 203; 0.99 ± 0.69
193
FIGURE 3. Haplotype analysis of theAca23 mutation. Black boxes illus-trate the presence of two C3Hmarker alleles (recombination be-tween microsatellite marker andAca23); white boxes illustrate pres-ence of one copy of both alleles, C3Hand C57BL/6J (lack of recombina-tion). The number of G3 progenycarrying the particular recombina-tion pattern is given below theboxes. The total number of recombi-nation events between neighboringmarkers is shown to the right of theboxes, including the calculated rela-tive genetic distances (cM).
IOVS, December 2009, Vol. 50, No. 12 Novel ColVIIIa2 Mouse Mutant 5655
with the microsatellite markers D4Mit279, D4Mit338, D4Mit249,D4Mit73, D4Mit336, and D4Mit293.
PCR and Sequencing
For the molecular analysis, RNA was isolated at embryonic day (E) 12.5from heads of Aca23�/� or wild-type embryos (C57BL/6J) with theRNA-Bee kit (AMS Biotechnology, Abingdon, UK). RNA samples werereverse transcribed to cDNA using a T-primed first-strand kit (Ready-to-Go; GE Healthcare, Freiburg, Germany). Genomic DNA was isolatedfrom the tail tips of wild-type C57BL/6J mice or homozygous/heterozy-gous mutants according to standard procedures.
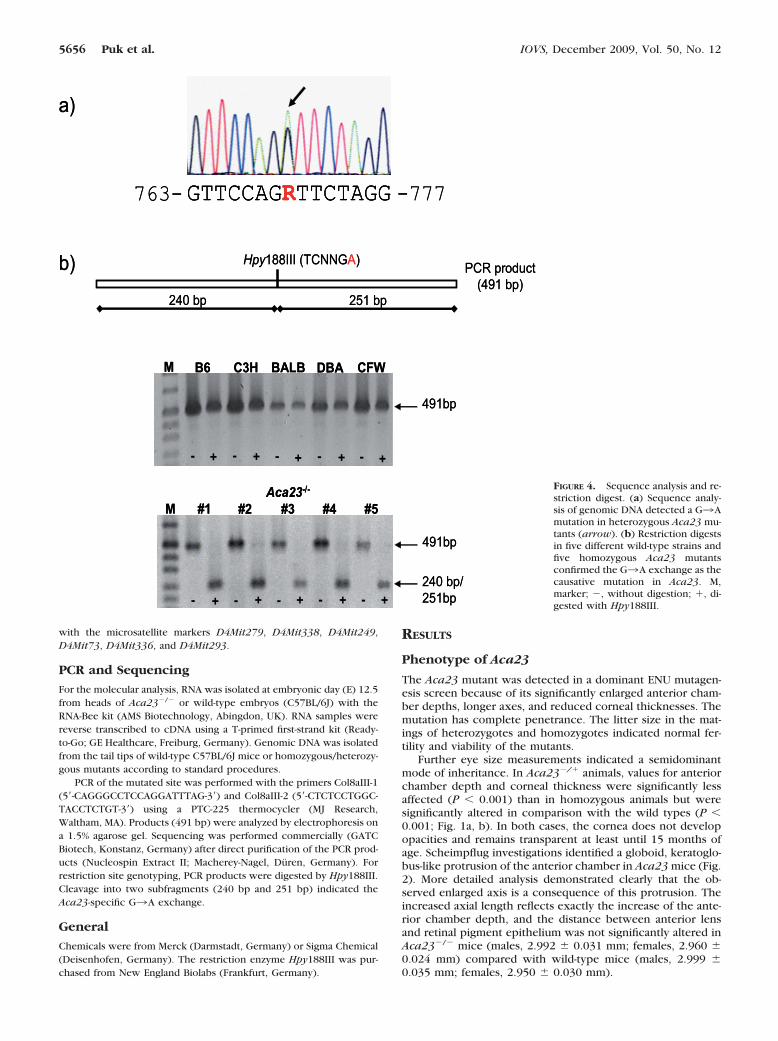
PCR of the mutated site was performed with the primers Col8aIII-1(5�-CAGGGCCTCCAGGATTTAG-3�) and Col8aIII-2 (5�-CTCTCCTGGC-TACCTCTGT-3�) using a PTC-225 thermocycler (MJ Research,Waltham, MA). Products (491 bp) were analyzed by electrophoresis ona 1.5% agarose gel. Sequencing was performed commercially (GATCBiotech, Konstanz, Germany) after direct purification of the PCR prod-ucts (Nucleospin Extract II; Macherey-Nagel, Duren, Germany). Forrestriction site genotyping, PCR products were digested by Hpy188III.Cleavage into two subfragments (240 bp and 251 bp) indicated theAca23-specific G3A exchange.
General
Chemicals were from Merck (Darmstadt, Germany) or Sigma Chemical(Deisenhofen, Germany). The restriction enzyme Hpy188III was pur-chased from New England Biolabs (Frankfurt, Germany).
RESULTS
Phenotype of Aca23
The Aca23 mutant was detected in a dominant ENU mutagen-esis screen because of its significantly enlarged anterior cham-ber depths, longer axes, and reduced corneal thicknesses. Themutation has complete penetrance. The litter size in the mat-ings of heterozygotes and homozygotes indicated normal fer-tility and viability of the mutants.
Further eye size measurements indicated a semidominantmode of inheritance. In Aca23�/� animals, values for anteriorchamber depth and corneal thickness were significantly lessaffected (P � 0.001) than in homozygous animals but weresignificantly altered in comparison with the wild types (P �0.001; Fig. 1a, b). In both cases, the cornea does not developopacities and remains transparent at least until 15 months ofage. Scheimpflug investigations identified a globoid, keratoglo-bus-like protrusion of the anterior chamber in Aca23 mice (Fig.2). More detailed analysis demonstrated clearly that the ob-served enlarged axis is a consequence of this protrusion. Theincreased axial length reflects exactly the increase of the ante-rior chamber depth, and the distance between anterior lensand retinal pigment epithelium was not significantly altered inAca23�/� mice (males, 2.992 � 0.031 mm; females, 2.960 �0.024 mm) compared with wild-type mice (males, 2.999 �0.035 mm; females, 2.950 � 0.030 mm).
GTTCCAGRTTCTAGG763- -777
a)
b) Hpy188III (TCNNGA)PCR product
(491 bp)240 bp 251 bp
M #1 #2 #3 #4 #5Aca23-/-
- - - - -+ + + + +
- - - - -
C3HB6 CFWBALB DBAM
+ + + + +
491bp
240 bp/251bp
491bp
GTTCCAGRTTCTAGGGTTCCAGRTTCTAGG763- -777
a)
b) Hpy188III (TCNNGA)PCR product
(491 bp)240 bp 251 bp
M #1 #2 #3 #4 #5Aca23-/-
- - - - -+ + + + +
- - - - -
C3HB6 CFWBALB DBAM
+ + + + +
491bp
240 bp/251bp
491bp
FIGURE 4. Sequence analysis and re-striction digest. (a) Sequence analy-sis of genomic DNA detected a G3Amutation in heterozygous Aca23 mu-tants (arrow). (b) Restriction digestsin five different wild-type strains andfive homozygous Aca23 mutantsconfirmed the G3A exchange as thecausative mutation in Aca23. M,marker; �, without digestion; �, di-gested with Hpy188III.
5656 Puk et al. IOVS, December 2009, Vol. 50, No. 12

Genotype of Aca23
We performed a genomewide linkage analysis by crossinghomozygous mutants on a C57BL/6J background (G1) to wild-type C3HeB/FeJ mice. Heterozygous mutants (G2) were back-crossed to C3HeB/FeJ mice. The SNP-based analysis of 73 G3offspring revealed linkage of the Aca23 phenotype to the distalpart of chromosome 4 within an interval spanning more than30 Mb (between rs3022989 and rs3023011; cytoband C6-D3).Further fine-mapping using 203 G3 mice located the Aca23mutation between the flanking microsatellite markersD4Mit249 and D4Mit73 (Fig. 3). Based on these data, thegenetic order was calculated (genetic distance � SD in paren-thesis): D4Mit249 (0.49 � 0.49 cM), Aca23 (1.48 � 0.85 cM),D4Mit73. The critical interval of approximately 1.0 Mb in-cludes 35 genes. Eighteen of them are predicted only becauseof the existence of ESTs, but they are not yet fully annotated.
Sequence analysis of the candidate gene Col8a2 in a het-erozygous Aca23 mouse detected a G3A exchange at cDNAposition 770 (counting the starting ATG as position 1; Fig. 4a).This transition generates an Hpy188III restriction site (TC-NNGA), which was confirmed in the genomic DNA of all testedhomozygous Aca23 mutants (n � 5). In contrast, all testedwild-type strains (C3H, C57BL/6, BALB/c, DBA, and CFW)lacked this restriction site, demonstrating that the observedexchange does not represent a general polymorphism (Fig.4b).
The 770 G3A transition is predicted to cause an exchangeof Gly to Asp at amino acid position 257 of the ColVIIIa2protein. This residue lies within the highly conserved collage-nous domain of ColVIIIa2 (position 137–532), which consistsof 136 (Gly-X-Y) units (X and Y � any amino acid). It repre-sents the first position of unit 45.
Histologic Consequences of Aca23
To investigate pathologic mechanisms initiated by the Aca23mutation, we performed histologic studies of the eyes of3-month-old mice. Structural alterations at the cellular level ofiris, trabecular meshwork, and lens were not found in Aca23mice (data not shown). In the cornea, we observed a reducedsize of epithelium, stroma, and Descemet’s membrane (Fig.5a). This was more pronounced in Aca23�/� than inAca23�/�. The corneal endothelium remained single-layeredin all tested eyes. In situ hybridizations revealed the expressionof Col8a2 in the Descemet’s membrane and the endotheliallayer of the wild-type cornea (Fig. 5b). The Col8a2 expressionpattern was not altered in heterozygous or homozygous indi-viduals.
To investigate the integrity of the corneal endothelium, weperformed keratin12 immunohistochemistry. In both wildtypes and mutants, expression of this epithelial marker waslimited to the superficial and suprabasal epithelial layers of theanterior cornea (Fig. 6). These data excluded a transformationof corneal endothelium into multilayered epithelial like cellsbecause it is associated with particular mutations in the humanCOL8A2 gene resulting in glaucoma from blocking of thetrabecular meshwork.
Detailed histology findings of the retina and optic nervehead, pathologies of which are often associated with glau-coma, were normal. The inner retinal layers were present, anddamage of the optic nerve fibers was not detectable in theretinal sections of the mutants (Fig 7a). Furthermore, a char-acteristic thick nerve fiber layer entered the optic nerve head(Fig. 7b). These findings were confirmed by ophthalmoscopicinvestigations of 9-month-old Aca23�/� and Aca23�/� ani-mals. All tested mutants revealed a homogenously pigmented
WT A/+ A/A
S
Ep
En
DM
a)
b)
WT A/+ A/A
S
Ep
En
DM
a)
b)
FIGURE 5. Histology of the cornea and in situ hybridization. (a) In3-month-old mice, thickness of corneal epithelium (Ep), stroma (S),and Descemet’s membrane (DM) is reduced in Aca23�/� (A/�; mid-dle) and Aca23�/� (A/A; right) compared with the C57BL/6J control(wild-type [WT]; left). En, endothelium. Scale bars, 10 �m. (b) Theexpression pattern of Col8a2 at P7 in the posterior cornea (arrow) ofAca23�/� (A/�) and Aca23�/� (A/A) is comparable to that inC57BL/6J control (wild-type [WT]). Signal intensity appears weaker inthe mutants. Scale bars, 20 �m.
S
En
S
Ep
En
WT A/+ A/A
S
En
S
Ep
En
WT A/+ A/AFIGURE 6. Immunohistochemistry. Corneas of wild-type (WT, left),Aca23�/� (A/�, middle), and Aca23�/� (A/A, right) were stainedwith the epithelial marker keratin 12. Keratin 12 expression wasrestricted to the corneal epithelium (arrow), excluding an epithelial-ization of corneal endothelium in Aca23. Ep, epithelium; S, stroma; En,endothelium. Scale bars, 20 �m.
IOVS, December 2009, Vol. 50, No. 12 Novel ColVIIIa2 Mouse Mutant 5657

fundus with a well-defined vessel pattern comparable to thewild-type fundus. Enlargement of the optic disc, typical forglaucoma, was not found (Fig. 7c).
Visual Acuity of Aca23
In humans, corneal protrusion is generally associated with strongmyopia. To study consequences of the anterior protrusion onvisual properties of Aca23 mice, vision tests with the virtualoptomotor system were performed with 2- to 8-month-old mice.The response of Aca23�/� and Aca23�/� mice clearly demon-
strated that visual properties were not affected. All tested miceresponded normally to the moving stripe pattern. A median spa-tial frequency threshold of x0.5 � 0.414 cyc/deg, a 25% quantileof x0.25 � 0.414 cyc/deg, and a 75% quantile of x0.75 � 0.439cyc/deg was observed for Aca23�/�. Aca23�/� individuals re-vealed a median threshold of x0.5 � 0.414 cyc/deg, a 25% quantileof x0.25 � 0.386 cyc/deg, and a 75% quantile of x0.75 � 0.414cyc/deg. These data did not differ significantly from the visualacuity of the C57BL/6J control (x0.5 � 0.414 cyc/deg; x0.25 �0.414 cyc/deg; x0.75 � 0.439 cyc/deg; Fig. 8).
WT
A/+
A/A
a) b) c)GCL
IPL
INL
OPL
ONL
IS/OS
RPE
GCL
IPL
INL
OPL
ONL
IS/OS
RPE
GCL
IPL
INLOPL
ONL
IS/OSRPE
N
WT
A/+
A/A
a) b) c)GCL
IPL
INL
OPL
ONL
IS/OS
RPE
GCL
IPL
INL
OPL
ONL
IS/OS
RPE
GCL
IPL
INLOPL
ONL
IS/OSRPE
N
FIGURE 7. Histology of the posterioreye and funduscopy. (a) Histology ofthe retina (3 months of age) revealedthe presence of all retinal layers inthe mutants excluding glaucomatousdamage. (b) Histology of the opticnerve head (3 months of age) con-firmed that an appropriate number ofnerve fibers enter the optic nerve inthe mutants (arrows). (c) Fundus ofAca23�/� (A/�), Aca23�/� (A/A),and C57BL/6J control (WT) at theage of 9 months. Optic disc struc-ture, blood vessel pattern, and pig-mentation are also not altered in themutants. GCL, ganglion cell layer;IPL, inner plexiform layer; INL, innernuclear layer; OPL, outer plexiformlayer; ONL, outer nuclear layer; IS/OS, inner segment/outer segment ofphotoreceptor layer; RPE, retinal pig-ment epithelium; N, optic nervehead. Arrows: nerve fiber layer enter-ing the optic nerve. Scale bars, 50�m.
0.364
0.386
Sp
atia
lfr
equ
enc
yth
resh
old
(cyc
/deg
)
0
0.3
0.414
0.439
0.469
0.5
A/+ A/AWT
0.342
0.319
0.364
0.386
Sp
atia
lfr
equ
enc
yth
resh
old
(cyc
/deg
)
0
0.3
0.414
0.439
0.469
0.5
A/+ A/AWT
0.342
0.319
FIGURE 8. Virtual vision test. Spatialfrequency thresholds were evaluatedfor C57BL/6J control (WT; black cir-cles), Aca23�/� (A/�; gray circles),and Aca23�/� (A/A; white circles)between 2 and 8 months of age. Eachspot represents the threshold of onetested mouse. Visual acuity is not sig-nificantly altered in the mutants(Mann-Whitney U test).
5658 Puk et al. IOVS, December 2009, Vol. 50, No. 12

DISCUSSION
We have established and characterized a novel mouse mutant,Aca23, with abnormalities of the anterior eye segment. Thephenotype includes larger anterior chambers and axes andthinner corneas. Histologic analysis confirmed a reduced sizeof corneal epithelium, stroma, and Descemet’s membrane. Thisputatively affects the resistance of the cornea to the naturalintraocular pressure, which might result in the observed ante-rior protrusion.
Genomewide linkage analysis and fine mapping placed theAca23 mutation on chromosome 4 within a 1.0-Mb intervalcontaining 35 coding genes. Two of them were described to beresponsible for ocular disorders: Col8a2 coding for a collagentype VIII protein29 and the neurochondrin gene Ncdn.42 Thelatter was excluded as a candidate because of its close distanceof approximately 0.05 Mb to the flanking marker D4Mit73,which did not fit to the calculated distance of 1.48 � 0.85 cMbetween D4Mit73 and Aca23. Concerning Col8a2, variousmissense mutations of the human COL8A2 gene were de-scribed to initiate posterior polymorphous corneal dystro-phies.29,30,43–45 Furthermore, targeted inactivation of Col8a1/Col8a2 has been reported to cause a thinning of cornealstroma and Descemet’s membrane in the mouse.46 Consideringthese data, it is likely that the observed missense mutation atcDNA-position 770 of Col8a2 represents the causative muta-tion of the Aca23 phenotype. Consequently, Aca23 is the firstdescribed pathogenic point mutation in the murine Col8a2gene. The affected glycine residue is highly conserved andhomologous to position 261 of the human COLVIIIA2. Mis-sense mutations at this position have not yet been identified inhumans with corneal endothelial dystrophies. Therefore,Aca23 demonstrates for the first time that this residue is es-sential for the functionality of ColVIIIa2. Moreover, this is thefirst described pathogenic mutation affecting the first positionof a (Gly-X-Y) collagen triple helix repeat subunit.
Type VIII collagen is a major component of specializedextracellular matrices such as sclera and skin47,48 (for a review,see Shuttleworth49). However, malformations seem to be re-stricted to the corneal layers in Aca23 animals. In the wild-typeDescemet’s membrane, the collagen type VIII molecules areassembled into a characteristic hexagonal lattice structure,50
which is formed by two distinct polypeptides, �1(VIII) and�2(VIII), encoded by the genes COL8A2 and COL8A1. A puta-tive misfolding of ColVIIIa2G257D could disrupt the latticestructure by preventing coordinated coassembly of both build-ing blocks. The result would be an irregular mosaic, in whichColVIIIa1 putatively predominates. Furthermore, incorpora-tion of ColVIIIa2G257D could be completely inhibited. Theobserved structural effects might be potentiated by an alteredexpression of the mutated Col8a2, which seems to be reducedin Aca23.
Various point mutations in the human collagen type VIII A2gene are associated with two distinct corneal dystrophies,FECD (OMIM 136800) and PPCD (OMIM 122000). Both formsshare many features including a thickened Descemet’s mem-brane caused by the secretion of a pathologic collagenous layerand an altered morphology of the corneal endothelium.44,45 Inparticular, endothelial wartlike guttate are typical for FECD.51
This was not found in Aca23. In PPCD, corneal endothelium isreplaced by aberrant epithelial-like cells,52 which might initiatesecondary glaucoma by extending into the anterior chamberand blocking the trabecular meshwork.29 However, our immu-nohistochemical and histologic data further excluded thesespecific symptoms. Aca23, therefore, does not seem to repre-sent a model for classical human FECD or PPCD. Rather, itcombines clinical features of keratoglobus, which is generallydefined by bilateral protrusions,53 with a yet undescribed cor-
neal pathology. A comparable combination has been reportedfor a human patient with keratoglobus with posterior polymor-phous dystrophy.24
The Aca23 phenotype described in this study resemblesocular pathologies of a Col8a2/Col8a1 null mutant, which wasalso reported to develop keratoglobus-like protrusions andirregularly thin corneal stromas and Descemet’s membranes.46
Stromal thinning was suggested to be initiated by an abnormalmigration of precursor cells during corneal development be-cause of the lacking collagen VIII �1 and �2.46 The fact thatColVIIIa2G257D causes comparable effects on stromal thicknessdemonstrates an essential role of ColVIIIa2 in these yet un-known mechanisms. Concerning the Descemet’s membrane,the reduced thickness observed in both mouse mutants is incontrast to the basal membrane thickening of human cornealdystrophies. Obviously, structural consequences of altered col-lagen VIII are not the same in the corneas of mice and humans.This might point to interspecies differences in collagen func-tion and corneal development. Further experiments are re-quired to understand these putatively alternative pathways.
Because the cornea contributes most of the focusing powerin the human eye, myopia is the most common refractivedisorder associated with anterior segment enlargement. How-ever, anterior chamber protrusion does not influence visualproperties in Aca23 mice. Even homozygous individuals re-sponded well in the virtual vision test and were still able toresolve spatial frequencies up to 0.469 cyc/deg. This furtherdemonstrates that a refractive error of a few diopters, as isexpected for Aca23, is less relevant for the vision of the mousebecause of a generally high refractive power of murine corneaand lens of approximately 500 diopters.54 Consequently,Aca23 would not have been detected as an eye-related mutantin a screening approach based on a vision test. This emphasizesthe benefits of the fast and reliable low-coherence interferom-etry technique for the establishment of eye size-associateddisease models.
CONCLUSION
We identified a new mouse mutant that represents a model forkeratoglobus. It is further characterized by a pathologic phe-notype of the corneal Descemet’s membrane. Molecular anal-ysis characterized the first point mutation in the murineCol8a2 gene encoding collagen type VIII �2. This mutationmight influence the assembly of the corneal basement mem-brane. Visual acuity is not altered by the resulting anteriorgloboid protrusion.
Acknowledgments
The authors thank Erika Burkle, Monika Stadler, Maria Kugler, JanEnke, and Jacqueline Muller for expert technical assistance and JackFavor for critical reading of the manuscript.
References
1. Lowry RB, Kohut R, Sibbald B, Rouleau J. Anophthalmia andmicrophthalmia in the Alberta Congenital Anomalies SurveillanceSystem. Can J Ophthalmol. 2004;40:38–44.
2. OMIM—Online Mendelian Inheritance in Man. McKusick-NathansInstitute of Genetic Medicine, Johns Hopkins University and Na-tional Center for BioTechnology Information. http://www.ncbi.n-lm.nih.gov/omim/. Accessed July 11, 2009.
3. Fantes J, Ragge NK, Lynch SA, et al. Mutations in SOX2 causeanophthalmia. Nat Genet. 2003;33:461–463.
4. Ragge NK, Lorenz B, Schneider A, et al. SOX2 anophthalmia syn-drome. Am J Med Genet A. 2005;135:1–7.
IOVS, December 2009, Vol. 50, No. 12 Novel ColVIIIa2 Mouse Mutant 5659

5. Bakrania P, Robinson DO, Bunyan DJ, et al. SOX2 anophthalmiasyndrome: 12 new cases demonstrating broader phenotype andhigh frequency of large gene deletions. Br J Ophthalmol. 2007;91:1471–1476.
6. Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Or-phanet J Rare Dis. 2007;2:47–55.
7. Warburg M, Sjo O, Fledelius HC, Pedersen SA. Autosomal recessivemicrocephaly, microcornea, congenital cataract, mental retarda-tion, optic atrophy, and hypogenitalism: micro syndrome. Am JDis Child. 1993;147:1309–1312.
8. Menez B. A case of Marfan’s syndrome in a black African. MedTrop. 1981;41:569–571.
9. Sanchez E, Munier F, Evequoz B, Marcoz JP, Balmer A. Ocularphenotype in a child with chondrodysplasia punctata, rhizomelicform. Klin Monatsbl Augenheilkd. 1997;210:329–331.
10. Luebbers JA, Goldberg MF, Herbst R, Hattenhauer J, MaumeneeAE. Iris transillumination and variable expression in ectopia lentiset pupillae. Am J Ophthalmol. 1977;83:647–656.
11. Aligianis IA, Johnson CA, Gissen P, et al. Mutations of the catalyticsubunit of RABGAP cause Warburg Micro syndrome. Nat Genet.2005;37:221–223.
12. Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome causedby a recurrent de novo missense mutation in the fibrillin gene.Nature. 1991;352:337–339.
13. Braverman N, Steel G, Obie C, et al. Human PEX7 encodes theperoxisomal PTS2 receptor and is responsible for rhizomelic chon-drodysplasia punctata. Nat Genet. 1997;15:369–376.
14. Motley AM, Hettema EH, Hogenhout EM, et al. Rhizomelic chon-drodysplasia punctata is a peroxisomal protein targeting diseasecaused by non-functional PTS2 receptor. Nat Genet. 1997;15:377–380.
15. Purdue PE, Zhang JW, Skoneczny M, Lazarow PB. Rhizomelicchondrodysplasia punctata is caused by deficiency of humanPEX7, a homologue of the yeast PTS2 receptor. Nat Genet. 1997;15:381–384.
16. Dickens CJ, Hoskins HD. Epidemiology and pathophysiology ofcongenital glaucoma. In: Ritch R, Shields BM, Krupin T, eds. TheGlaucomas. St. Louis: Mosby; 1996;729–738.
17. Stoilov I, Akarsu A, Sarfarazi M. Identification of three differenttruncating mutations in cytochrome P4501B1 (CYP1B1) as theprincipal cause of primary congenital glaucoma (Buphthalmos) infamilies linked to the GLC3A locus on chromosome 2p21. HumMol Genet. 1997;30:171–177.
18. Forsius H, Damsten M, Eriksson AW, Fellman J, Lindh S, Tah-vanainen E. Autosomal recessive cornea plana: a clinical and ge-netic study of 78 cases in Finland. Acta Ophthalmol Scand. 1998;76:196–203.
19. Sigler-Villanueva A, Tahvanainen E, Lindh S, Dieguez-Lucena J,Forsius H. Autosomal dominant cornea plana: clinical findings in aCuban family and a review of the literature. Ophthalmic Genet.1997;18:55–62.
20. Tahvanainen E, Forsius H, Karila E, et al. Cornea plana congenitagene assigned to the long arm of chromosome 12 by linkageanalysis. Genomics. 1995;26:290–293.
21. Pellegata NS, Dieguez-Lucena JL, Joensuu T, et al. Mutation inKERA, encoding keratocan, cause cornea plana. Nat Genet. 2000;25:91–95.
22. Lehmann OJ, El-ashry MF, Ebenezer MD, et al. A novel keratocanmutation causing autosomal recessive cornea plana. Invest Oph-thalmol Vis Sci. 2001;42:3118–3122.
23. Khan A, Al-Saif A, Kambouris M. A novel KERA mutation associatedwith autosomal recessive cornea plana. Ophthalmic Genet. 2004;25:147–152.
24. Harissi-Dagher M, Dana MR, Jurkunas UV. Keratoglobus in associ-ation with posterior polymorphous dystrophy. Cornea. 2007;26:1288–1291.
25. Krafchak CM, Pawar H, Moroi SE, et al. Mutations in TCF8 causeposterior polymorphous corneal dystrophy and ectopic expres-sion of COL4A3 by corneal endothelial cells. Am J Hum Genet.2005;77:694–708.
26. Aldave AJ, Yellore VS, Yu F, et al. Posterior polymorphous cornealdystrophy is associated with TCF8 gene mutations and abdominalhernia. Am J Med Genet A. 2007;143A:2549–2556.
27. Liskova P, Tuft SJ, Gwilliam R, et al. Novel mutations in the ZEB1gene identified in Czech and British patients with posterior poly-morphous corneal dystrophy. Hum Mutat. 2007;28:638.
28. Liu Y, Peng X, Tan J, Darling DS, Kaplan HJ, Dean DC. Zeb1 mutantmice as a model of posterior corneal dystrophy. Invest Ophthal-mol Vis Sci. 2008;49:1843–1849.
29. Biswas S, Munier FL, Yardley J, et al. Missense mutations inCOL8A2, the gene encoding the �2 chain of type VIII collagen,cause two forms of corneal endothelial dystrophy. Hum MolGenet. 2001;10:2415–2423.
30. Mok JW, Kim HS, Joo CK. Q455V mutation in COL8A2 is associ-ated with Fuchs’ corneal dystrophy in Korean patients. Eye. 2008;23:895–903.
31. Abdel-Salam GMH, Hassan NA, Kayed HF, Aligianis IA. Phenotypicvariability in Micro syndrome: report of new cases. Genet Counsel.2007;18:423–435.
32. Sarfarazi M, Akarsu AN, Hossain A, et al. Assignment of a locus(GLCA3) for primary congenital glaucoma (Buphthalmos) to 2p21and evidence for genetic heterogeneity. Genomics. 1995;30:171–177.
33. Tahvanainen E, Villanueva AS, Forsius H, Salo P, de la Chapelle A.Dominantly and recessively inherited cornea plana congenita mapto the same small region of chromosome 12. Genome Res. 1996;6:249–254.
34. Hrabe de Angelis M, Flaswinkel H, Fuchs H, et al. Genomewide,large-scale production of mutant mice by ENU mutagenesis. NatGenet. 2000;25:444–447.
35. Puk O, Dalke C, Favor J, Hrabe de Angelis M, Graw J. Variations ofeye size parameters among different strains of mice. Mamm Gen.2006;17:851–857.
36. Ehling UH, Charles DJ, Favor J, et al. Induction of gene mutationsin mice: the multiple endpoint approach. Mutat Res. 1985;150:393–401.
37. Schmucker C, Schaeffel F. In vivo biometry in the mouse eye withlow coherence interferometry. Vis Res. 2004;44:2445–2456.
38. Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantifica-tion of adult and developing mouse spatial vision using a virtualoptomotor system. Invest Ophthalmol Vis Sci. 2004;45:4611–4616.
39. Grimm C, Chatterjee B, Favor J, et al. Aphakia (ak), a mousemutation affecting early eye development: fine mapping, consid-eration of candidate genes, and altered Pax6 and Six3 expressionpattern. Dev Genet. 1998;23:299–316.
40. Kunder S, Calzada-Wack J, Holzlwimmer G, et al. A comprehensiveantibody panel for immunohistochemical analysis of formalin-fixed, paraffin-embedded hematopoietic neoplasms of mice: anal-ysis of mouse specific and human antibodies cross-reactive withmurine tissue. Toxicol Pathol. 2007;35:366–375.
41. Herbach N, Rathkolb B, Kemter E, et al. Dominant-negative effectsof a novel mutated Ins2 allele causes early-onset diabetes andsevere B-cell loss in Munich Ins2C95S mutant mice. Diabetes. 2007;56:1268–1276.
42. Hansen J, Floss T, Van Sloun P, et al. A large-scale, gene-drivenmutagenesis approach for the functional analysis of the mousegenome. Proc Natl Acad Sci USA. 2003;100:9918–9922.
43. Gottsch JD, Sundin OH, Liu SH, et al. Inheritance of a novelCOL8A2 mutation defines a distinct subtype of Fuchs cornealdystrophy. Invest Ophthalmol Vis Sci. 2005;46:1934–1939.
44. McCartney AC, Kirkness CM. Comparison between posterior poly-morphous dystrophy and congenital hereditary endothelial dystro-phy of the cornea. Eye. 1988;2:63–70.
45. Levy SG, Moss J, Sawada H, Dopping-Hepenstal PJ, McCartney AC.The composition of the wide-spaced collagen in normal and dis-eased Descemet’s membrane. Curr Eye Res. 1996;15:45–52.
46. Hopfer U, Fukai N, Hopfer H, et al. Targeted disruption of Col8a1and Col8a2 genes in mice leads to anterior segment abnormalitiesin the eye. FASEB J. 2005;19:1232–1244.
5660 Puk et al. IOVS, December 2009, Vol. 50, No. 12

47. Sage H, Bornstein P. Type VIII collagen. In: Mayne R, Burgeson RE,eds. Structure and Function of Collagen Types. New York: Aca-demic Press; 1990;173–193.
48. Kittelberger R, Davis PF, Flynn DW, Greenhill NS. Distribution oftype VIII collagen in tissues: an immunohistochemical study. Con-nect Tissue Res. 1990;24:303–318.
49. Shuttleworth CA. Type VIII collagen. Int J Biochem Cell Biol.1997;29:1145–1148.
50. Sawada H, Konomi H, Hirosawa K. Characterization of the colla-gen in the hexagonal lattice of Descemet’s membrane: its relationto type VIII collagen. J Cell Biol. 1990;110:219–227.
51. Bigar F. Specular microscopy of the corneal endothelium: opticalsolutions and clinical results. Dev Ophthalmol. 1982;6:1–94.
52. Levy SG, Moss J, Noble BA, McCartney AC. Early onset posteriorpolymorphous dystrophy. Arch Ophthalmol. 1996;114:1265–1268.
53. Baillif S, Garweg JG, Grange JD, Burillon C, Kodjikian L.Keratoglobus: review of the literature. J Fr Ophthalmol. 2005;28:1145–1149.
54. Schaffel F. The mouse as a model for myopia, and optics of its eye.In: Chalupa LM, Williams RW, eds. Eye, Retina, and Visual Systemof the Mouse. Boston: MIT Press; 2008;73–85.
IOVS, December 2009, Vol. 50, No. 12 Novel ColVIIIa2 Mouse Mutant 5661
Related Documents











