RECOMBINANT DNA TECHNOLOGY DNA fragments from different sources can be joined together

RECOMBINANT DNA TECHNOLOGY DNA fragments from different sources can be joined together.
Dec 17, 2015
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

RECOMBINANT DNA TECHNOLOGY
DNA fragments from different sources can be joined together
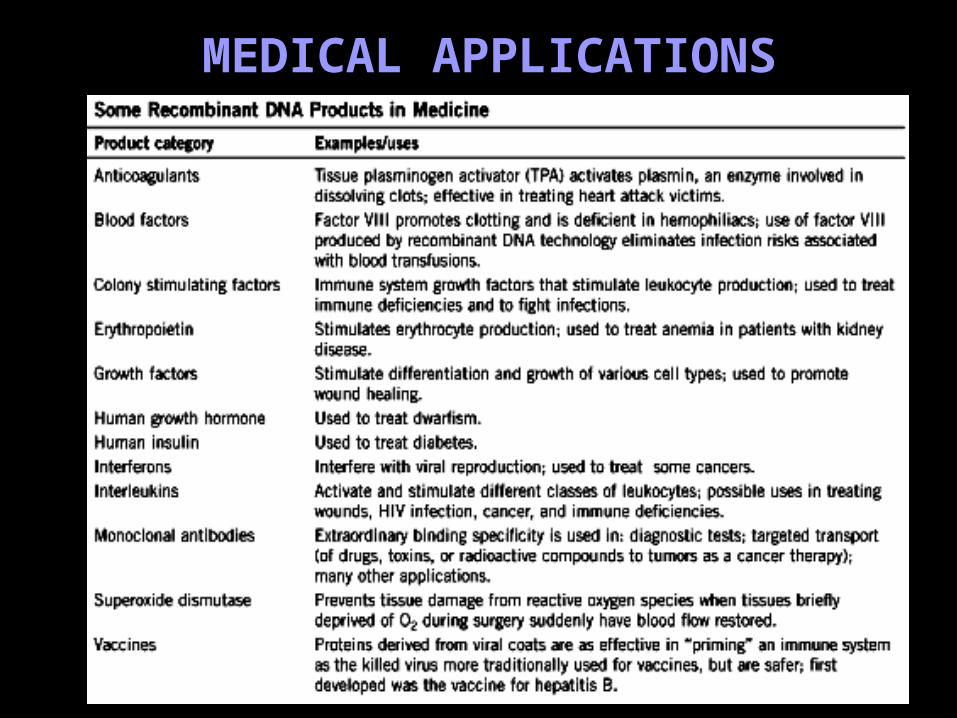
MEDICAL APPLICATIONS

The discovery of the restriction-modification system of bacteria led to the development of controlled and reproducible in vitro DNA cutting and ligation. This is the basic strategy of DNA cloning. The restriction-modification system is based on two enzymatic activities:
1. Restriction enzymes recognize specific DNA sequences and cut the double stranded DNA within or adjacent to the recognition site
The restriction endonucleases: In the „restriction endonucleases” expression, the „restriction" refers to their ability of the bacteria to break down the foreign DNA introduced by bacteriophage infection (therefore the host cell creates restrictive conditions for it).
2. DNA methylases modify the bacterial DNA with methyl groups within the sites recognized by their own restriction endonucleases, thus prevent them from cleaving their own DNA. The foreign DNA introduced into bacteria are not methylated on the right sites and thus will be degraded by these endonucleases.
THE RESTRICTION ENDONUCLEASES
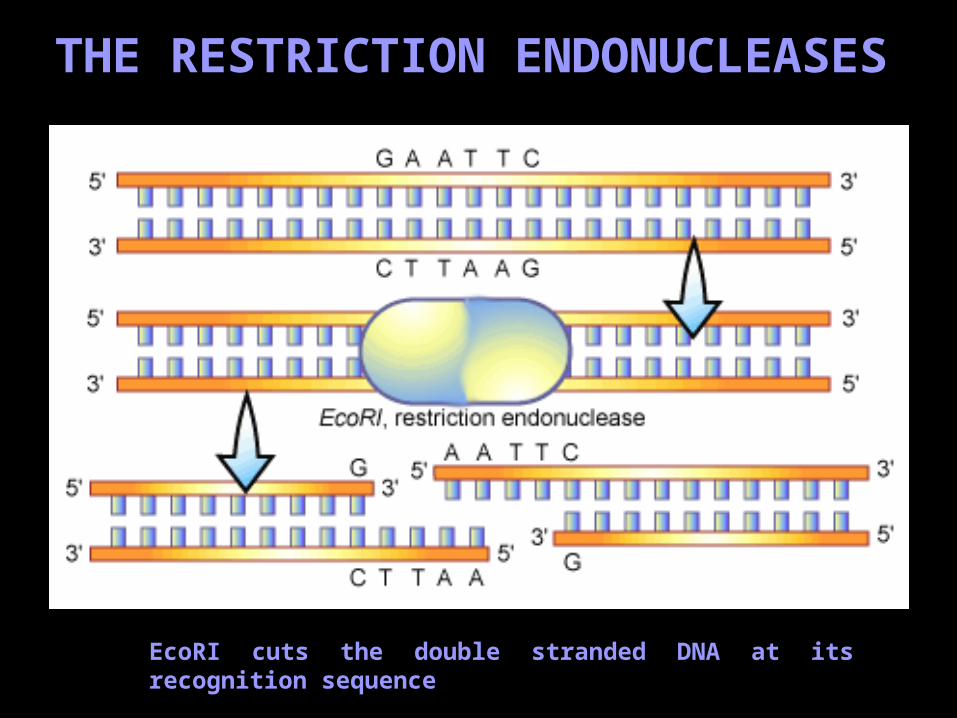
EcoRI cuts the double stranded DNA at its recognition sequence
THE RESTRICTION ENDONUCLEASES

THE RESTRICTION ENDONUCLEASES
blunt ends
sticky ends

Restriction endonucleases recognize specific DNA sequences of four to eight base
pairs (named as restriction sites) which are so-called palindroms: inverted
repetitive sequences, where the order of the bases is same in both strands in 5'-
>3' direction.
Many restriction enzymes make the cut leaving short, complementary single-
stranded tails on the ends of each fragment. These tails tend to adhere by base
pairing to any other complementary ends produced by the same restriction
enzyme, and they are thus often called sticky ends. Then the DNA ligase joins
permanently the two DNA fragments having homologous sticky ends.
Another group of restriction enzymes break DNA at the center of their recognition
sites and produce blunt-ended fragments. These ends, without single stranded
complementary tails, stick together with difficulty, but they have the advantage that
they can anneal to every other blunt end regardless to the enzyme that produced
it.
THE RESTRICTION ENDONUCLEASES

The agarose gel electrophoresis is a method for separation of DNA molecules according to their lenght (molecular weight). The DNA fragments are charged negatively and therefore they migrate in an electric field from the cathode towards the anode. The speed of their movement depends on:- the concentration of the gel which determines the size of the gel pores, - the size of the DNA molecule.- the conformation of the DNA molecule, - the composition of the electrolyte solution,- the electric field force.
Linear double-stranded DNA migrates through the gel at a rate that is inversely proportional to
the log10 of the number of base pairs. The size of any fragment can be established by
calibration with DNA molecules of known sizes (with so-called marker DNA). Staining of
these gels with dyes that attach to DNA makes it possible to visualize the separated fragments.
The most frequently used dye is ethidium bromide, a molecule that emits fluorescent light
when illuminated with UV light. DNA fragments can be purified from the gel for sequencing,
and cloning.
AGAROSE GEL ELECTROPHORESIS
Practical task: restriction digestion end gel electrophoresis of a plasmid
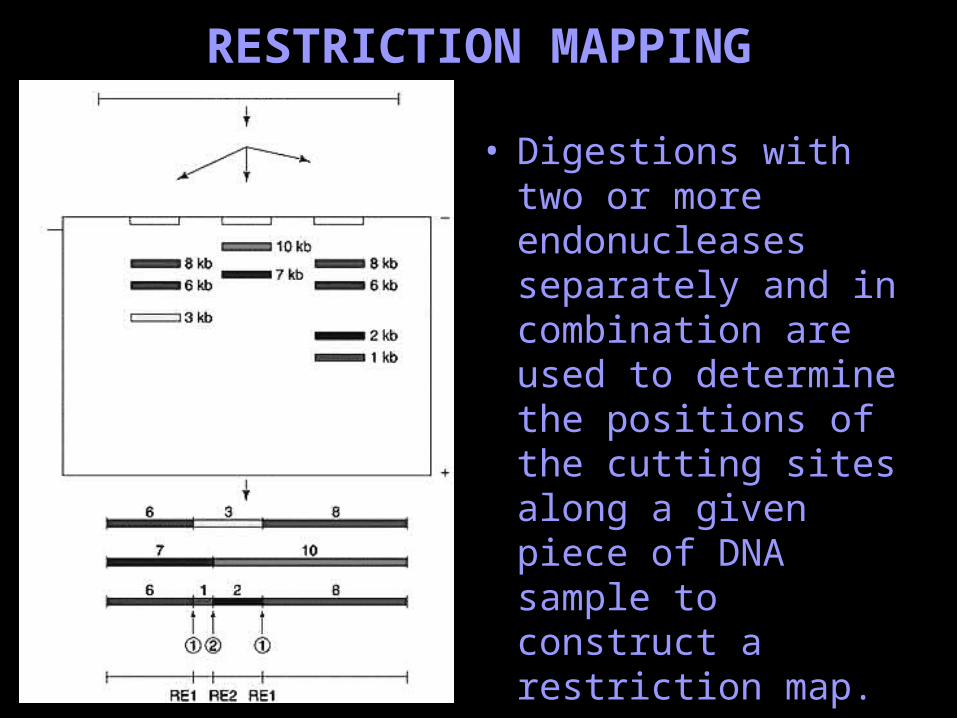
RESTRICTION MAPPING
• Digestions with two or more endonucleases separately and in combination are used to determine the positions of the cutting sites along a given piece of DNA sample to construct a restriction map.
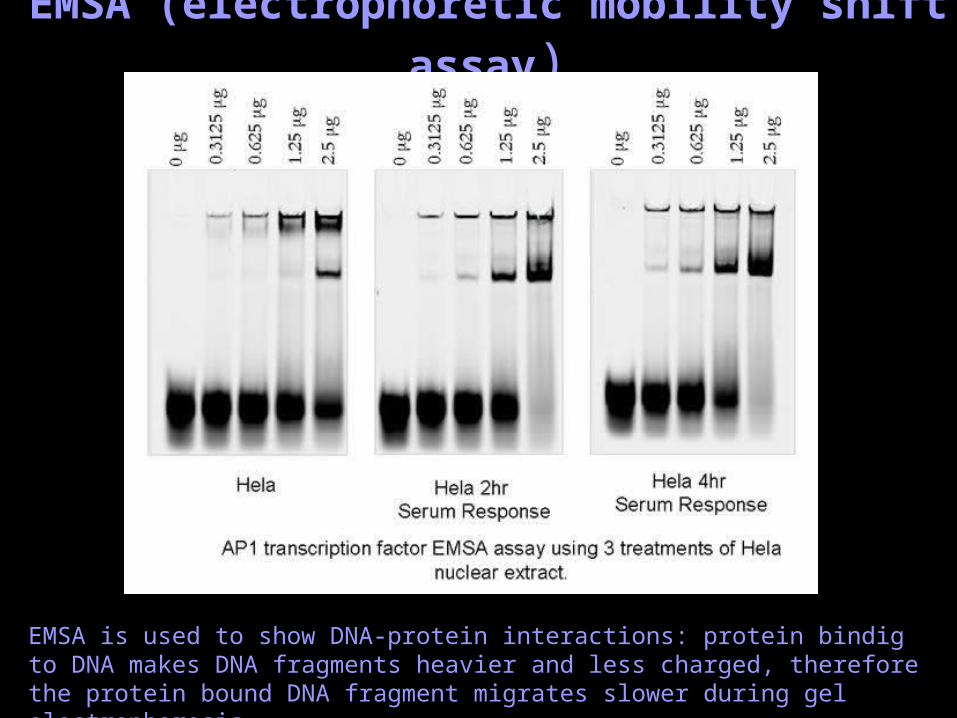
EMSA (electrophoretic mobility shift assay)
EMSA is used to show DNA-protein interactions: protein bindig to DNA makes DNA fragments heavier and less charged, therefore the protein bound DNA fragment migrates slower during gel electrophoresis.

FOOTPRINT ANALYSIS
Proteins bound to DNA protect it against random DNA breaking agents: bandless „windows” appear on the sequencing gel .

DNA CLONING
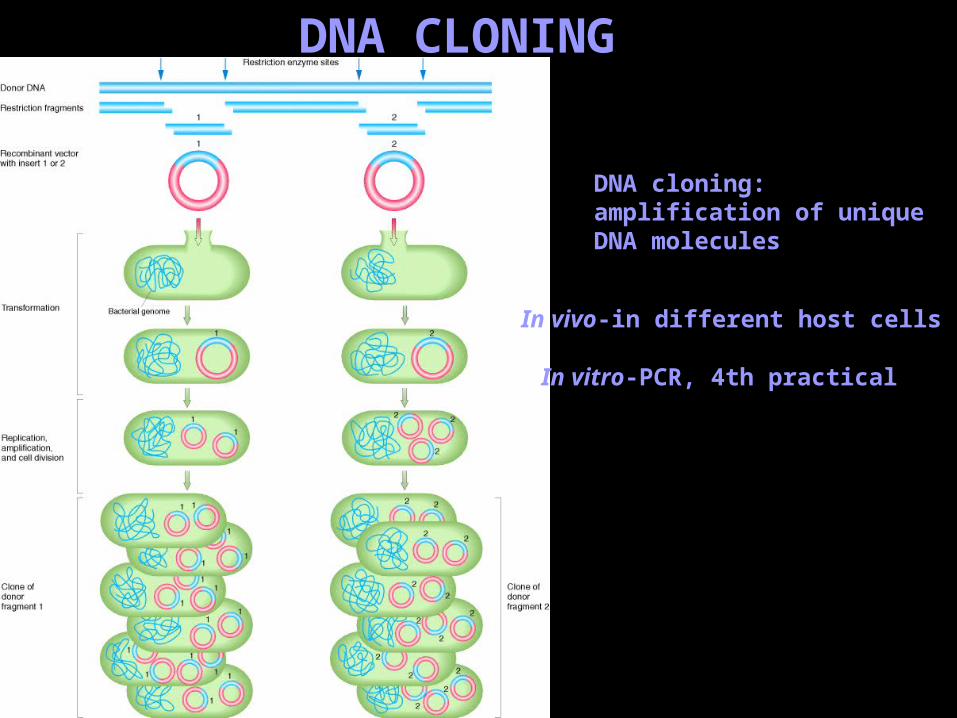
DNA CLONING
DNA cloning:amplification of unique DNA molecules
In vivo-in different host cells
In vitro-PCR, 4th practical
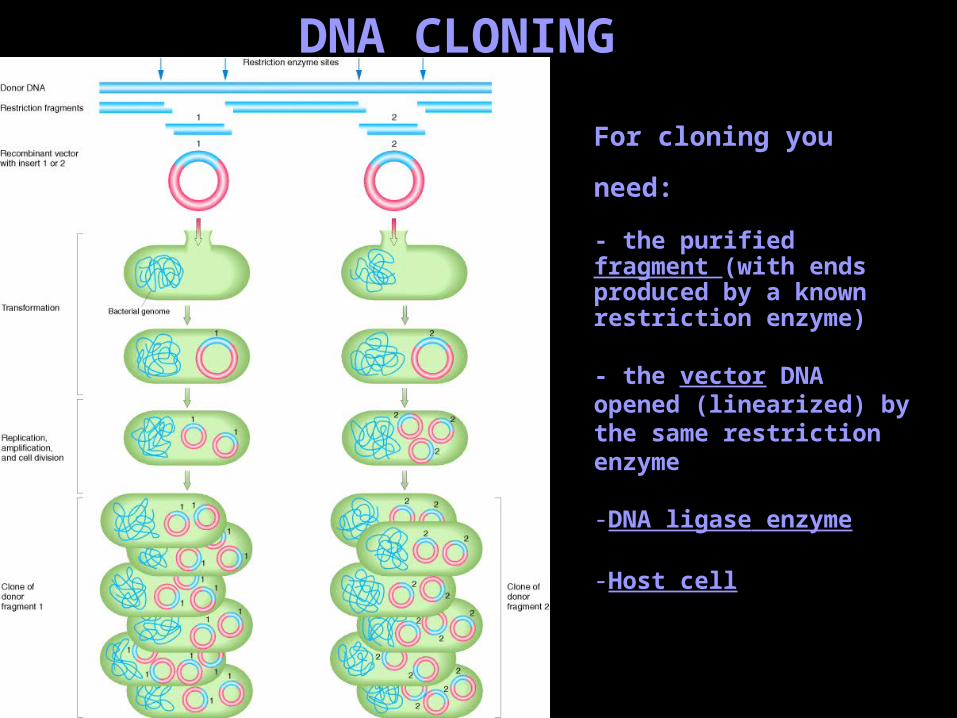
DNA CLONING
For cloning you need:
- the purified fragment (with ends produced by a known restriction enzyme)
- the vector DNA opened (linearized) by the same restriction enzyme
-DNA ligase enzyme
-Host cell

Escherichia coli is the "host cell" of the molecular biological laboratories.
For cloning we use non-pathogenic strains which are suitable for amplifying
foreign DNA in great amounts and in which a foreign DNA molecule is
stable. For these purposes, these strains are mutant in some genes coding for
the restriction-modification system and for recombination. Because of these
genetic modifications, the DNA molecules introduced from outside into these
cells will not be degraded by endonucleases and the absence of
recombination will results in the stability of the inserted DNA. E. coli has the
advantage that it is easy and cheap to sustain, it is growing very fast (1
division in every 20 minutes) and its genetic map is well-known. Besides,
Escherichia coli, many other prokaryotic and eukaryotic host cells are in use.
DNA CLONING
The host cell

The in vitro recombined DNA molecules can be introduced and maintained in host cells by the help of vectors. Vector DNAs are derived from prokaryotic or eukaryotic plasmids, viruses or transposons. After introducing these vectors by transformation or transfection into the host cells, they integrate into the host genome or they behave as an individual replicon within the cell. Vector DNAs are usually in vitro modified to be more convenient for molecular biological purposes. Thus they contain:- a replication origin (or sequences responsible for integrating into the host genome),- a gene responsible for selectivity (e.g. antibiotic resistance gene or requirement for a special nutrient) that makes it possible to distinguish the host cells containing the vector from those that do not contain it,- and unique restriction endonuclease sites for cloning that makes it possible to open the circular vector.There are vectors developed for special purposes, e.g. the so-called expression vectors that are suitable for not only the amplification of the inserted foreign DNA, but also for large-scale production of the protein coded by the insert.
DNA CLONINGVectors

Cloning vectors expression vectors
DNA CLONING

The DNA ligase enzyme catalyzes the formation of covalent phosphodiester bonds
between juxtaposed 5' phosphate and a 3'hydroxyl termini of the DNA ends kept
together by base pairing.
Ligation of sticky ends is simpler than that of blunt ends because the
complementary single-stranded tails can associate easier. In case of blunt ends,
higher concentration of ligase, insert and vector DNA can increase the ligation
efficiency. After incubation at the optimal temperature, the original linear DNA
(vector and foreign fragment), recircularized „empty" vectors, maybe circularized
foreign DNA and - hopefully - recombinant vectors containing the foreign insert are
present in the reaction mixture.
DNA CLONING
Ligation

During transformation, cells have to be able to take up exogenous DNA, they have to be competent. Experiments have shown that if the cells are exposed to high concentrations of bivalent cations (e.g. Ca2+), the cell membranes become leaky. The DNA to transform (or a few µl from the ligation reaction) has to be added to the cells kept on ice, a brief heat shock (42°C) is needed after, and these altogether cause the bacterial plasma membranes to admit foreign DNA.The bacterial suspension is then plated onto a solid medium, and after overnight incubation, single colonies can be seen on the plates. One colony consists of millions of genetically identical bacterial cell clones, derived from the division of one original cell. But how could we detect that this cell was transformed successfully or not?Since the transformation efficiency of linear DNA pieces is very low only the following possibilities should be distinguished:-the cell has not taken up any vector,-the cell has taken up only the „empty" and recircularized vector, - the circularized foreign DNA only,- the recombinant vector entered the cell. Members of the following vector family allow selection of these different cell clones.
DNA CLONINGTransformation into E. coli
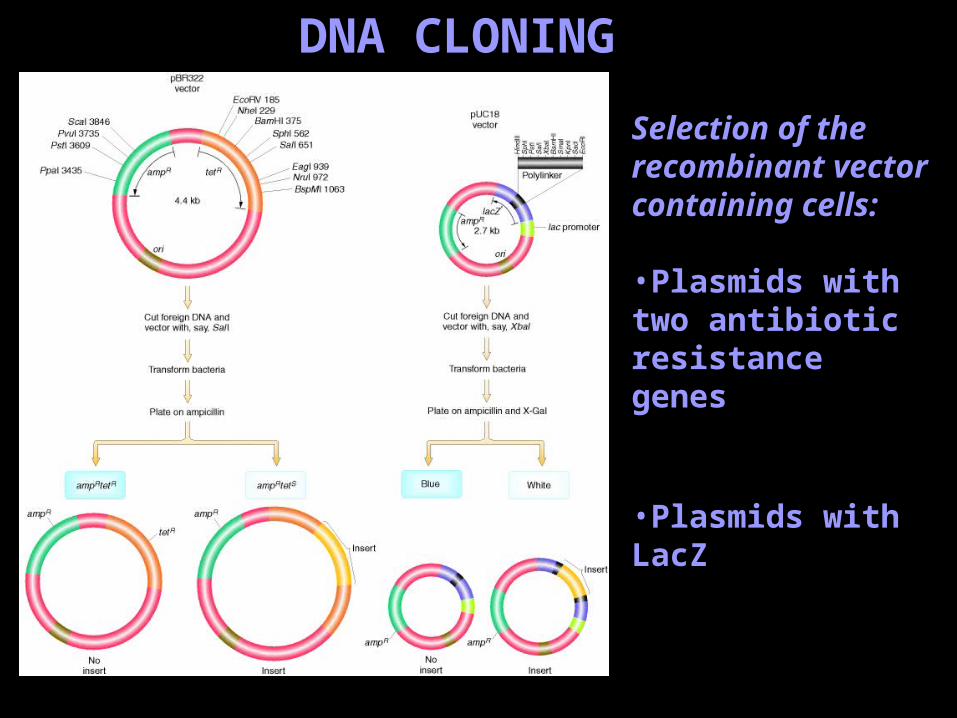
DNA CLONING
Selection of the recombinant vector containing cells:
•Plasmids with two antibiotic resistance genes
•Plasmids with LacZ
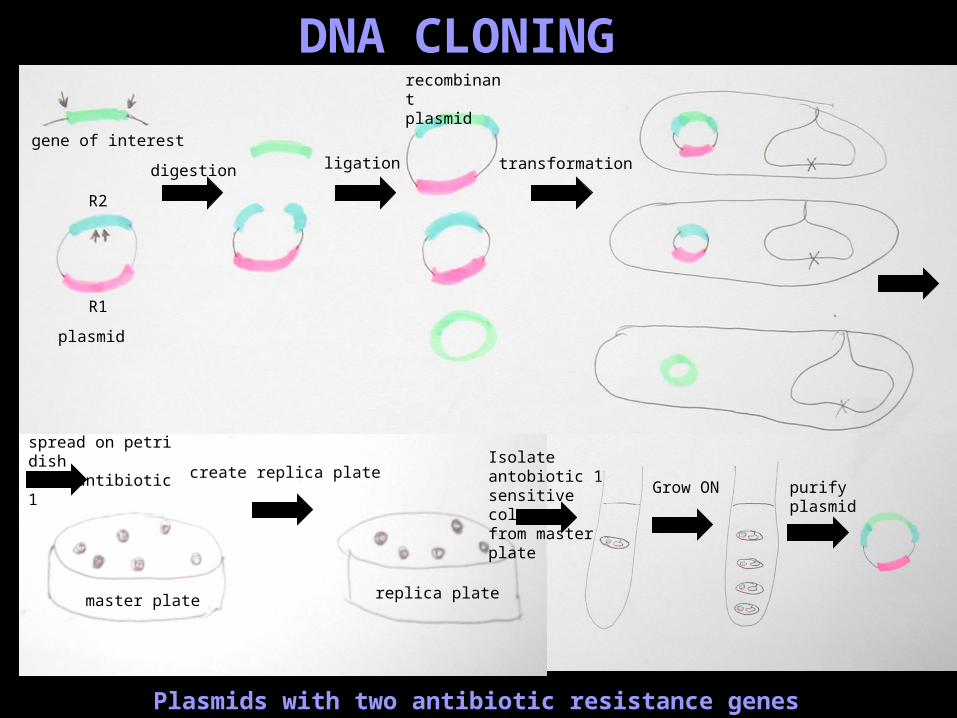
DNA CLONING
Plasmids with two antibiotic resistance genes
spread on petri dish with antibiotic 1
ligation
recombinant plasmid
transformationdigestion
master plate replica plate
create replica plate
R1
R2
Isolate antobiotic 1sensitive colony from master plate Grow ON purify
plasmid
plasmid
gene of interest
DNA CLONING
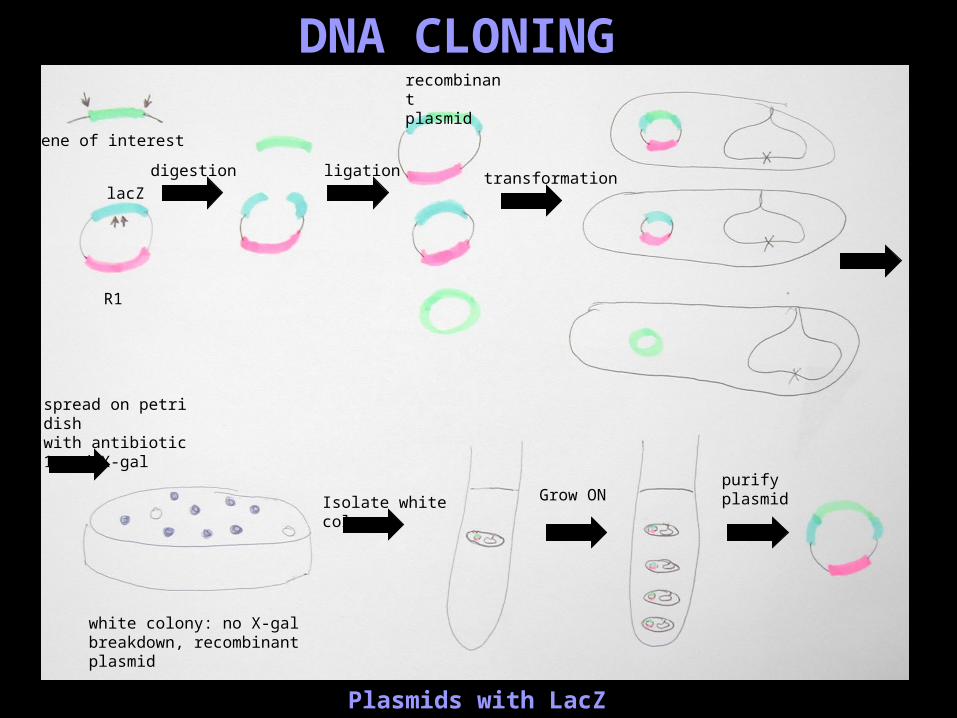
Plasmids with LacZ
gene of interest
R1
lacZ
digestion ligation
recombinant plasmid
transformation
spread on petri dish with antibiotic 1 and X-gal
Isolate white colony Grow ONpurify plasmid
white colony: no X-gal breakdown, recombinant plasmid

Restriction digestion of the pBluescript plasmid
1. Put together the following stuff in an eppendorf tube :
1 l DNA
1l 10X buffer
7 l distilled water
1 l Pvu I restriction enzime
2. Mix. centrifuge for 1-2 sec.
3. Incubate at 37°C for 1.5-2 hours
4. Prepare agarose gel, and run the digestion reaction
Practical task:
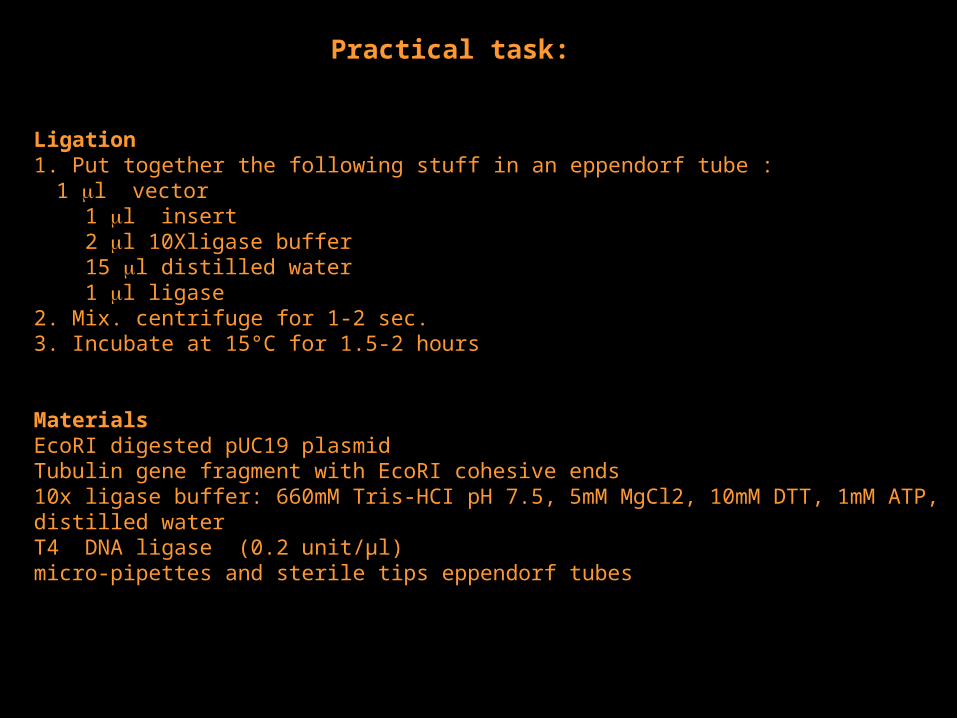
Ligation1. Put together the following stuff in an eppendorf tube : 1 l vector 1 l insert 2 l 10Xligase buffer 15 l distilled water 1 l ligase 2. Mix. centrifuge for 1-2 sec. 3. Incubate at 15°C for 1.5-2 hours
MaterialsEcoRI digested pUC19 plasmidTubulin gene fragment with EcoRI cohesive ends10x ligase buffer: 660mM Tris-HCI pH 7.5, 5mM MgCl2, 10mM DTT, 1mM ATP,distilled water T4 DNA ligase (0.2 unit/µl) micro-pipettes and sterile tips eppendorf tubes
Practical task:
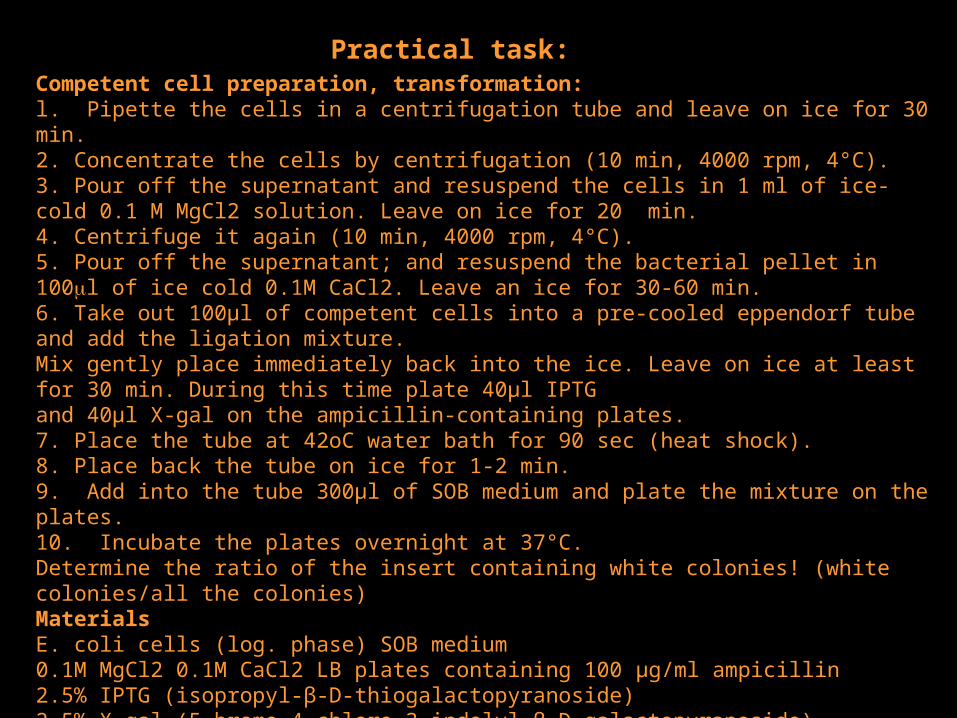
Competent cell preparation, transformation:l. Pipette the cells in a centrifugation tube and leave on ice for 30 min. 2. Concentrate the cells by centrifugation (10 min, 4000 rpm, 4°C).3. Pour off the supernatant and resuspend the cells in 1 ml of ice-cold 0.1 M MgCl2 solution. Leave on ice for 20 min.4. Centrifuge it again (10 min, 4000 rpm, 4°C). 5. Pour off the supernatant; and resuspend the bacterial pellet in 100l of ice cold 0.1M CaCl2. Leave an ice for 30-60 min.6. Take out 100µl of competent cells into a pre-cooled eppendorf tube and add the ligation mix ture. Mix gently place immediately back into the ice. Leave on ice at least for 30 min. During this time plate 40µl IPTG and 40µl X-gal on the ampicillin-containing plates.7. Place the tube at 42oC water bath for 90 sec (heat shock). 8. Place back the tube on ice for 1-2 min.9. Add into the tube 300µl of SOB medium and plate the mixture on the plates. 10. Incubate the plates overnight at 37°C.Determine the ratio of the insert containing white colonies! (white colonies/all the colonies)MaterialsE. coli cells (log. phase) SOB medium0.1M MgCl2 0.1M CaCl2 LB plates containing 100 µg/ml ampicillin2.5% IPTG (isopropyl-β-D-thiogalactopyranoside)2.5% X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) centrifuge and tubes42°C water bath thermostat at 37°C
Practical task:
Related Documents











