Recent Advances in the Chemistry of Parain£uenza-1 (Sendai) Virus Inhibitors Raffaele Saladino, 1 Umberto Ciambecchini, 1 Lucia Nencioni, 2 Anna Teresa Palamara 3 1 Agrobiological and Agrochemical Department, University of Tuscia, via San Camillo de Lellis snc, 00100, Viterbo, Italy 2 Department of Experimental Medicine and Biochemical Sciences, Faculty of Medicine, University of Rome ‘‘Tor Vergata,’’ Via Montpellier, 1, 00133, Rome, Italy 3 Institute of Microbiology, Faculty of Pharmacy, University of Rome ‘‘La Sapienza,’’ P.le Aldo Moro 5, 00185, Rome, Italy DOI 10.1002/med.10036 ! Abstract: Purine and pyrimidine derivatives, antioxidants, fusion inhibitors, statins, prostaglandins, antibiotic nucleosides, inhibitors of Ca 2 þ homeostasis, carbohydrate derivatives, antisense poly- nucleotides and chimeras, are described as inhibitors of parainfluenza-1 (Sendai) viral infections. ß 2003 Wiley Periodicals, Inc. Med Res Rev, 23 No. 4, 427–455, 2003 Key words: antivirals; synthesis; parainfluenza-1 (Sendai) virus; therapy 1. INTRODUCTION The family paramyxoviridae, which includes the parainfluenza, respiratory syncytial, measles and mumps viruses, cause disease in humans and animals. Two subfamilies have recently been distinguished: the paramyxovirinae, which comprises the genera respirovirus, rubulavirus, and morbillivirus, and the pneumovirinae, which includes the genera pneumovirus and metapneumo- virus. 1 The parainfluenza viruses (PIV) belong to the genus respirovirus and include four human types, hPIV1, hPIV2, hPIV3, and hPIV4, which were discovered between 1956 and 1960. The first isolated PIV was Sendai virus (SV), which is now classified as the murine counterpart of human PIV1, to which it is antigenetically related. Many of the basic biochemical and molecular biological properties of the paramyxoviruses have been identified through the study of SV. Respiratory syncytial Contract grant sponsor: Italian Ministry of Health; Contract grant sponsor: Italian National Research Council; Contract Grant number: CNRC007A72 _ 003. Correspondence to: Prof. Raffaele Saladino, Dipartimento A.B.A.C., Universita' della Tuscia, Via S. Camillo de Lellis, s.n.c., 00100, Viterbo, Italy. E-mail: [email protected] Medicinal Research Reviews, Vol. 23, No. 4, 427^ 455, 2003 ß 2003 Wiley Periodicals, Inc.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Recent Advances in the Chemistryof Parain£uenza-1 (Sendai)
Virus Inhibitors
Raffaele Saladino,1 Umberto Ciambecchini,1 Lucia Nencioni,2
Anna Teresa Palamara 3
1Agrobiological and Agrochemical Department, University of Tuscia, via San Camillo de Lellis snc,
00100, Viterbo, Italy2Department of Experimental Medicine and Biochemical Sciences, Faculty of
Medicine, University of Rome ‘‘Tor Vergata,’’ Via Montpellier, 1, 00133, Rome, Italy3Institute of Microbiology, Faculty of Pharmacy, University of Rome
‘‘La Sapienza,’’ P.le Aldo Moro 5, 00185, Rome, Italy
DOI 10.1002/med.10036
!
Abstract: Purine and pyrimidine derivatives, antioxidants, fusion inhibitors, statins, prostaglandins,
antibiotic nucleosides, inhibitors of Ca2þ homeostasis, carbohydrate derivatives, antisense poly-
nucleotides and chimeras, are described as inhibitors of parainfluenza-1 (Sendai) viral infections.
� 2003 Wiley Periodicals, Inc. Med Res Rev, 23 No. 4, 427–455, 2003
Key words: antivirals; synthesis; parainfluenza-1 (Sendai) virus; therapy
1 . I N T R O D U C T I O N
The family paramyxoviridae, which includes the parainfluenza, respiratory syncytial, measles
and mumps viruses, cause disease in humans and animals. Two subfamilies have recently been
distinguished: the paramyxovirinae, which comprises the genera respirovirus, rubulavirus, and
morbillivirus, and the pneumovirinae, which includes the genera pneumovirus and metapneumo-
virus.1 The parainfluenza viruses (PIV) belong to the genus respirovirus and include four human
types, hPIV1, hPIV2, hPIV3, and hPIV4, which were discovered between 1956 and 1960. The first
isolated PIVwas Sendai virus (SV), which is now classified as themurine counterpart of human PIV1,
to which it is antigenetically related. Many of the basic biochemical and molecular biological
properties of the paramyxoviruses have been identified through the study of SV. Respiratory syncytial
Contract grant sponsor: Italian Ministry of Health; Contract grant sponsor: Italian National Research Council; Contract
Grant number: CNRC007A72 _ 003.
Correspondence to: Prof. Raffaele Saladino, Dipartimento A.B.A.C., Universita' dellaTuscia, Via S. Camillo de Lellis, s.n.c., 00100,
Viterbo, Italy.E-mail: [email protected]
Medicinal Research Reviews, Vol. 23, No. 4, 427^455, 2003
� 2003 Wiley Periodicals, Inc.

virus (RSV), a member of the pneumovirus genus, shares many structural, pathogenic, epide-
miological and clinical features with the PIV, and for this reason, data on RSV are in many cases
relevant to our understanding of PIVas well.
The PIVare enveloped RNAviruses with linear, nonsegmented, negative-sense, single-stranded
genomes containing six to ten tandemly linked genes. The genome is enclosed within a helical
nucleocapsid containing nucleocapsid (NP), phospho- (P) and large polymerase complex (L)
proteins, which initiate intracellular virus replication. Protruding through the pleomorphic lipid-
containing envelope, the glycoproteins, hemagglutinin-neuraminidase (HN), and fusion protein (F),
mediate the virus’s entry into and exit from host cells. Another structural protein, the viral matrix
protein (M), is extremely important in virion architecture. It is found between the envelope and the
core and is released when the virus enters a host cell.
Parainfluenza virus replication occurs entirely within the cytoplasm of infected cells (Fig. 1).
It begins with transcription of the genomic RNA into 5 0-capped and 3 0-polyadenylated mRNAs.
Transcription occurs in a sequential manner, terminating and reinitiating at each gene junction.
The genome contains, in 3 0 to 5 0 order, the NP, P, M, F, HN, and L genes (named for the six
structural proteins they encode). After primary transcription and translation, when sufficient
amounts of unassembled NP are present, viral RNA synthesis becomes coupled to the concomitant
NP-encapsidation of the nascent (þ ) RNA chain. Under these conditions, viral RNA polymerase
ignores all the junctions, to produce an exact complementary antigenome chain. By a process known
as RNA editing, which involves the use of an overlapping open reading frame (ORF), the PIV P gene
also gives rise to multiple nonstructural protein species: the V protein and four carboxy-coterminal
proteins (C 0, C, Y1, and Y2) referred to collectively as the C proteins.
All of these proteins play functional roles in PIV replication and pathogenesis. The NP is a
58-kDa protein, and each of itsmonomers is associatedwith six nucleotides. It forms the nucleocapsid
in complex with RNA and the P protein, which represents the smaller subunit of RNA polymerase.
The P protein is a 68-kDa tetramer phosphorylated on one or two serine residues in the N-proximal
module. The third nucleocapsid-associated protein, the L protein, contains approximately 2200
amino acids and represents the major RNA polymerase subunit. Present in a small number of copies
per virion (about 20–40 in each virus particle), the L protein hasmultiple functions in RNA synthesis,
including catalytic activity, capping and methylation. The M protein, with a molecular weight of
about 40 kDa, is abundantly expressed within the virion. It interacts with both the viral envelope
and the nucleocapsid and is thought to play a role in virus maturation. The attachment protein, HN,
is an integral membrane glycoprotein with molecular weight of 72 kDa. It binds the virion to sialic
acid-containing receptors on the host cell surface (hemagglutinating activity), thus initiating the
process of infection. It also causes cleavage (neuraminidase activity) of sialic acid residues from
glycoproteins and glycolipids, which prevents self-aggregation of virus particles during their release
from infected cells. The second membrane glycoprotein, the F protein, is synthesized as an inactive
precursor (F0) that is post-translationally cleaved by a host cell protease to form the biologically active
molecule, which has a molecular weight of approximately 63 kDa. Following attachment, the F
protein mediates the fusion of the virion and host cell surface membranes. This fusion occurs at a
neutral pH and allows delivery of the nucleocapsid into the cell cytoplasm, where gene expression
begins. Later in infection, F protein is expressed on the surface of infected cells,mediating their fusion
with adjacent uninfected cells and allowing the virus to spread.2
As mentioned above, the P gene also encodes the nonstructural V and C proteins. The former
consists of a P module attached to a V carboxyl-terminal region containing seven highly conserved
cysteine residues that form zinc finger motifs and bind Zn2þ .3 The V protein is not necessary for viral
replication in tissue culture cells: its main purpose is the maintenance of high viral loads. This
function, which is involved in pathogenesis in vivo, has been mapped to carboxyl-terminal portion of
the V protein4 and is associated with its zinc-binding capacity.3 Its presence is associated with severe
pneumonia in mice.5 The C proteins, C 0, C, Y1, and Y2, are carboxyl-coterminal proteins consisting,
428 * SALADINO ET AL.
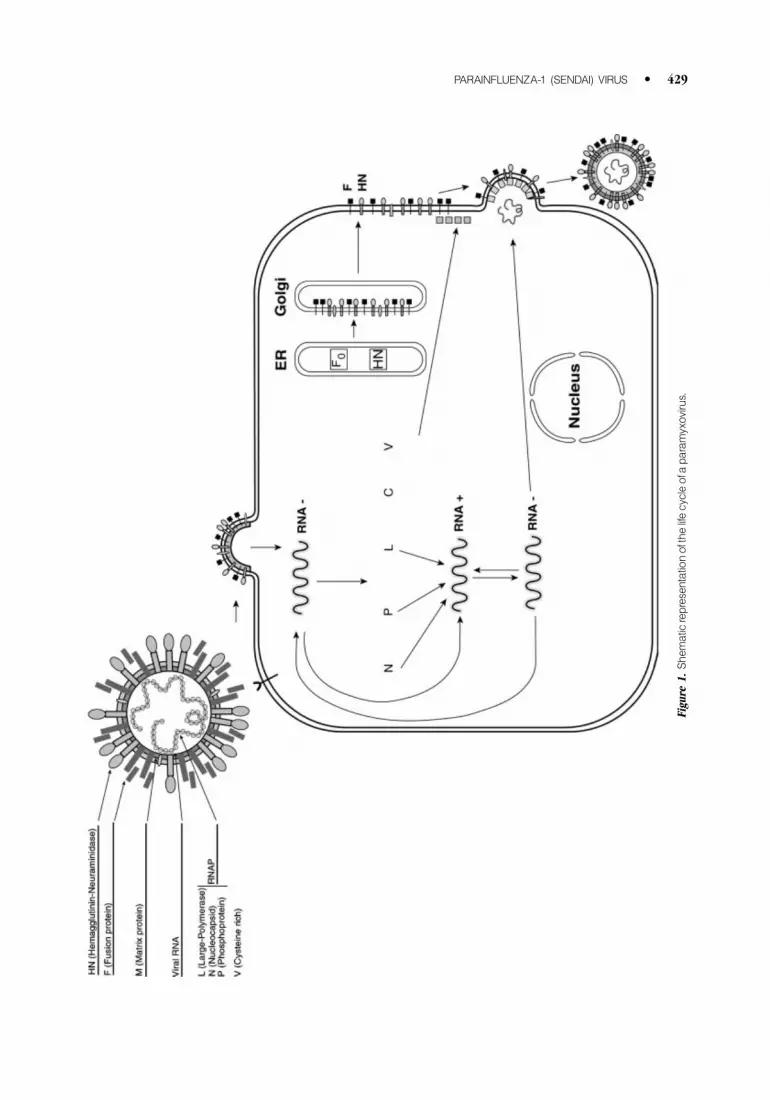
Figure
1.Shematic
representationofthelifecycleofa
paramyxovirus.
PARAINFLUENZA-1 (SENDAI) VIRUS * 429

in SV, of 215, 204, 181, and 175 amino acids, respectively. They are extremely versatile: they
counteract the antiviral action of interferons, inhibit viral RNA synthesis and are involved in virus
assembly. The C species is expressed in infected cells at a molar ratio several-fold higher than the
other three.1
A. Clinical Aspects and Pathogenesis
PIV and RSV have been known primarily as respiratory pathogens in young children, but now
they are also recognized as important pathogens in adults. Both replicate in the nasopharyngeal
epithelium after an incubation period of 2–8 days. They spread from cell to cell in the upper
respiratory tract, arriving in the lower airways 1–3 days later. The typical inflammation of RSV
bronchiolitis is characterized by necrosis of the bronchiolar epithelium and sloughing of the ciliated
epithelial cells of the small airways. Subsequently, lymphocytes migrate into affected tissues,
producing peribronchiolar infiltrates. The submucosal and adventitial tissues become edematous,
and secretions frommucous-producing cells increase. Plugs ofmucous, cellular debris, fibrin strands,
and DNA-like materials can occlude the smaller bronchioles and obstruct the flow in small airways.6
The clinical picture at this point has all the hallmarks of bronchiolitis: hyperinflation, atelectasis, and
wheezing.7
Among children, RSV infections account for 50–90% of hospitalizations for bronchiolitis,
5–40% of those for pneumonia and 10–30% of those for tracheobronchitis. PIV produce a similar
spectrum of respiratory illnesses but fewer hospitalizations. These infections generally involve the
upper respiratory tract, and 30–50% are complicated by otitis media. In about 15% of all cases,
however, PIV infections progress into the lower respiratory tract. Croup is the most typical clinical
manifestation of PIV infection (especially type 1) and is the principal cause of hospitalization in PIV-
infected children from 2–6 years of age. In older children and adults, PIVand RSV infections are not
as well recognized because other respiratory-tract infections can cause similar clinical manifesta-
tions.7 Some authors feel that there may be a pathogenic link between asthma and viral infections,
especially those caused by RSV. The inflammatory response elicited by asthmatic attacks is in fact
quite similar to that observed in viral infections. Furthermore, roughly half of the infants hospitalized
with RSV bronchiolitis later experience episodes of wheezing.
The number of patients subjected to intense immunosuppression for bone-marrow or solid-
organ transplantation is growing, and this increase has revealed another aspect of PIVand RSV, their
ability to cause opportunistic infections.8 Immunocompromised patients in transplantation units can
be exposed to these viruses through contact with staff members and/or visitors with mild upper
respiratory tract infections. Depending on the patient’s general condition and immune status, the type
of virus, and the time of exposurewith respect to the transplantation procedure, the results can be quite
severe, particularly with RSV.8
Many viruses, including the PIV, have evolved strategies to impede host defenses mediated by
interferon (IFN).9 Alpha/beta IFNs (IFN-a and IFN-b), the principal antiviral cytokines, act directlyon target cells to block viral replication.10Most of IFN’s antiviral effects require IFN-inducedmRNA
and protein synthesis. The complete signal transduction pathway from the IFN receptors to the
nucleus has been identified.11 IFN-a and IFN-b bind to heterodimeric IFN-a/b receptors consisting
of IFN-a receptor I and IFN-b receptor II. This interaction activates two cytoplasmic protein
tyrosine kinases, which consecutively phosphorylate tyrosine residues of the receptors. These
phosphotyrosines then bind to src homology 2 (SH2) domains of signal transducer and activator of
transcription (STAT) 1 and STAT2. The phosphorylation of these transducers leads to the formation
of heterodimers or homodimers through mutual SH2-domain-phosphotyrosine interactions. The
STAT1-STAT2 heterodimer associates with a third protein, interferon regulatory factor-9 (IRF9),
that allows DNA recognition. The result is an active transcriptional complex known as ISGF3 that
enhances transcription of target genes.
430 * SALADINO ET AL.

PIV neutralization of IFN-mediated cellular defenses seems to be related to the nonstructural
C and/or V proteins. The ability of SV to counteract the antiviral action of exogenous or endo-
genously produced IFNs is mediated by its C proteins,12 and it has recently been demonstrated that
the smallest of these, Y2, is as active as the C and Y1 proteins in this context.13 Recent data indicate
that the ISGF3 complex is the direct target of the anti-IFN strategies employed by certain negative-
stranded RNA viruses. The paramyxovirus, Simian virus 5, for example, provokes proteolytic
degradation of the STAT1 component of the complex, while hPIV2 preferentially degrades STAT2.14
Both of these effects aremediated by theviruses’Vproteins,15 andwhile the underlyingmechanism is
not entirely understood, it appears to involve subjugation of the host cell’s proteasome degradation
systems.15
B. Transmission
Most evidences indicate that PIV transmission depends upon direct contact with secretions,
fomites or large-particle aerosols. The major portals of entry for RSV infection are the eyes and nose;
the oral route is less permissive. Clinical observations suggest that PIV are transmitted similarly.
Parainfluenza virus type 1 has been recovered from air samples collected near infected patients.6
The high rates of primary infection and the frequency of reinfection suggest that these viruses spread
readily, that reinfected persons may be infectious and that a small inoculum is necessary to produce
an infection. Both viruses can survive for prolonged periods on skin, cloth, and other objects.
Fomites thus play an important role in the nosocomial diffusion of PIVand RSV, and hand washing
is essential in controlling infection.7
2 . C U R R E N T T H E R A P Y , N O V E L A N T I V I R A L A G E N T S A N D S T R A T E G I E S
The only antiviral agent currently licensed for treatment of RSV infections is aerosolized 1-b-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (Ribavirin, 1) (Fig. 2),16–21 a synthetic guanosine
analogue with broad-spectrum antiviral activity. Ribavirin prophylaxis is usually recommended for
children and adults undergoing bone-marrow transplantation.22
Ribavirin therapy for RSV infections is limited to treatment of infants. Improved oxygenation
and clinical scores and decreases in the levels of secretory mediators of inflammation associated
with severe bronchospasm have been observed in treated patients.23 However, its mode of action is
not well understood and it is also quite expensive. For these reasons, its use in RSV disease is the
object of continuing debate.24 Ribavirin exerts antiviral effects against PIV in vitro, and it has been
tested for treatment and prophylaxis of lower respiratory tract infections in immunocompromised
patients. Recently, Ramasamy and co-workers25 have described an efficient synthesis of triazole
L-ribofuranosyl nucleosides, comprising the lead compound (1) (Scheme 1). 1,2,3,5-tetra-O-acetyl-L-
ribose (3), easily prepared from L-ribose (2), was treated with methyl 1,2,4-triazole-3-carboxylate
to provide nucleosides (4) and (5). Treatment of (4) with ammonia at room temperature give (1) in90% yield.
Figure 2. Ribavirin (1).
PARAINFLUENZA-1 (SENDAI) VIRUS * 431

New antiviral agents being evaluated for RSVand PIV treatment include purine and pyrimidine
derivatives, antioxidants, fusion inhibitors, statins, prostaglandins, antibiotic nucleosides, inhibitors
of Ca2þ homeostasis, carbohydrate derivatives. Studies are also underway to evaluate antisense
polynucleotides and chimeras. Thus far, however, no antivirals for RSV have reached the clinical-trial
stage of testing.26
A. Pyrimidine and Purine Derivatives
Pyrimidines active against SV were prepared by oxidation of uracil derivatives with dimethy-
ldioxirane (DMD).27,28 The reaction proceeds through selective C-5,6 double bond oxidation to give
5,6-dihydro-5,6-oxiranyl uracils.29 As an example, when 1,3-dimethyl uracil (6), 1,3,5-trimethylur-
acil (13), and 1,3,6-trimethyluracil (14) were treated with DMD (1.0 M, acetone solution), the
corresponding 5,6-epoxides (7, 15, and 18) were obtained in good yields (Scheme 2) beside to cis- and
trans-diols (8, 9, 16, and 17) as side products.The reaction performed in the presence of nucleophiles (alcohols or amines) give 6-substituted-
5,6-dihydro-5-hydroxy uracils (10, 11, 22, 23, and 24) by ‘‘one-pot’’ ring-opening functionalizationof the epoxide moiety (Scheme 3).29
6-Substituted-5,6-dihydro uracils (6–24) were tested as antiviral agents. Among them trans-1,3-
dimethyl-5-hydroxy-6-ethylamino uracil (24) (Fig. 3) showed activity against SV, measured by
decreased haemagglutinin units (HAU).30
The dose-response effect of (24) is shown in Figure 4 (panel A). Concentrations of (24) lowerthan 1.0 mg/mL had no effect on HAU production. At higher concentrations, the inhibitions were
dose-dependent and reached maximum values (87.5% inhibition) at 100 mg/mL. The maximal
antiviral dose was not toxic for the cell.
Thiopurine and thiopyrimidine derivatives are also useful starting materials to synthesize new
SV inhibitors. The thioketo moiety may be easily functionalized by formation of the corresponding
sulphinic or persulphinic acid intermediates. As for example, treatment of 2-thiouracils,31a
pyrimidine-2-thione,31b 4-thiopyrimidine, and 6-thiopurine nucleosides31c with ozone afforded
Scheme 1.
432 * SALADINO ET AL.

desulfurized products or products of nucleophilic substitution depending on the experimental
conditions. The oxidation of the thioketo moiety with DMD is faster than other possible reactions.32
Thus, starting from thiopyrimidine or thiopurine nucleosides, alkoxy or alkylamino derivatives were
obtained in the presence of alcohols or amines as nucleophiles (Scheme 4).
The efficacy of this procedure is illustrated by the synthesis of 1-(b-D-arabinofuranosyl)pyrimidine derivatives with antiviral and antitumor properties (Scheme 5).
These compounds have been assayed for antiviral activity against parainfluenza 1 (Sendai)
virus,33 according with the HAU procedure.30 The 1-(b-D-arabinofuranosyl) derivative (27c) (Fig. 5)was found to inhibit virus replication at all the doses of the experiments. Unfortunately, toxic effects
have been found at dose of 100 mg mL�1 on uninfected cells.
In contrast to the extensive studies about 5-substitued pyrimidines, less attention has been
devoted to the 6-substituted isomers, probably because of the difficulty in their synthesis and
their supposed lack of biological activity. In the last few years, 6-substitued pyrimidines, for example
1-[(2-hydroxyethyl)methyl]-6-(phenylthio)thymine (HEPT)34a,b and 3,4-dihydro-2-alkoxy-6-ben-
zyl-4-oxopyrimidines (DABOs),35 showed potent and selective activity against human immunode-
ficiency virus type-1 (HIV-1). For this reason new synthetic procedures to obtain 6-substitued
pyrimidines are of great interest. Racemic and chiral 1,3-dimethyl-6-oxiranylpyrimidin-2,4-dione
(28a–b)36 showed remarkable anti-ASFV (African Swine Fever Virus) activity (Fig. 6).37,38
On the basis of these data, uracil and pyrimidinone derivatives (30a–g, 31a–c, 33a–e, 34a–j, 36,39a–c) bearing the oxiranyl moiety in the C-6 position of the pyrimidine ring were prepared by
selective metalation of 6-methyl-4(3H)-pyrimidinones39 and evaluated for their activity against
SV.40–43
Scheme 2.
Scheme 3.
PARAINFLUENZA-1 (SENDAI) VIRUS * 433
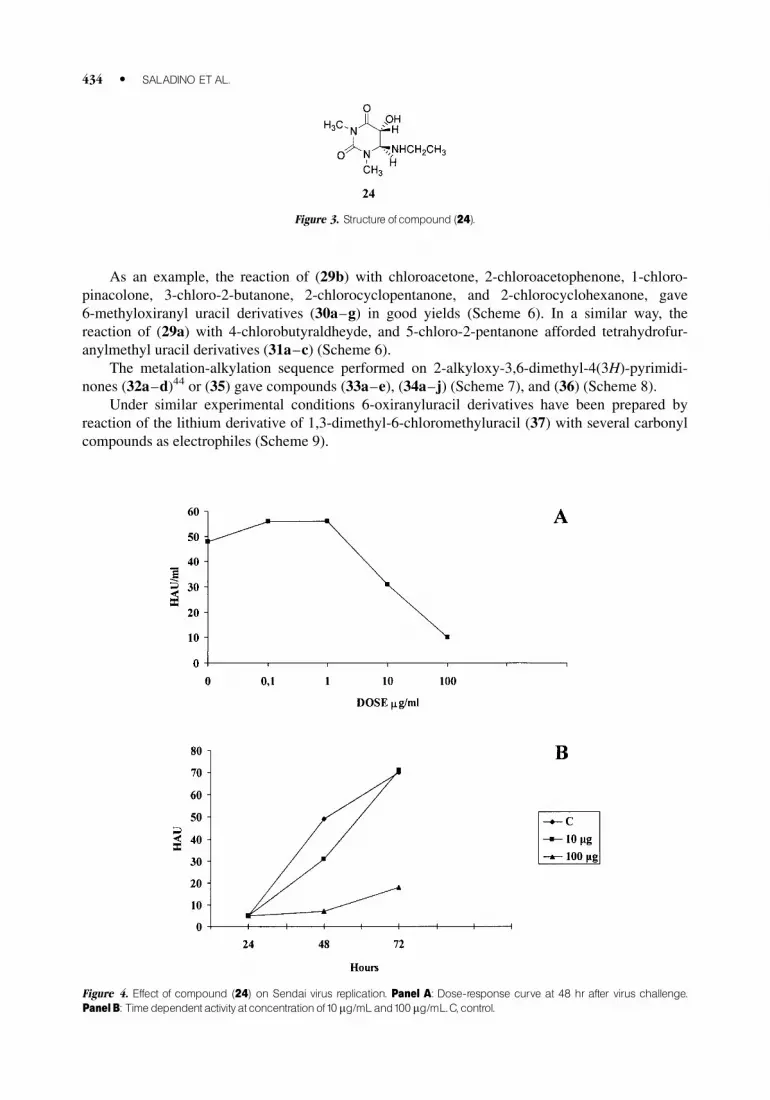
As an example, the reaction of (29b) with chloroacetone, 2-chloroacetophenone, 1-chloro-
pinacolone, 3-chloro-2-butanone, 2-chlorocyclopentanone, and 2-chlorocyclohexanone, gave
6-methyloxiranyl uracil derivatives (30a–g) in good yields (Scheme 6). In a similar way, the
reaction of (29a) with 4-chlorobutyraldheyde, and 5-chloro-2-pentanone afforded tetrahydrofur-
anylmethyl uracil derivatives (31a–c) (Scheme 6).
The metalation-alkylation sequence performed on 2-alkyloxy-3,6-dimethyl-4(3H)-pyrimidi-
nones (32a–d)44 or (35) gave compounds (33a–e), (34a–j) (Scheme 7), and (36) (Scheme 8).
Under similar experimental conditions 6-oxiranyluracil derivatives have been prepared by
reaction of the lithium derivative of 1,3-dimethyl-6-chloromethyluracil (37) with several carbonyl
compounds as electrophiles (Scheme 9).
Figure 3. Structure ofcompound (24).
Figure 4. Effect of compound (24) on Sendai virus replication. Panel A: Dose-response curve at 48 hr after virus challenge.
PanelB: Time dependentactivityat concentrationof10 mg/mLand100 mg/mL.C, control.
434 * SALADINO ET AL.

Most of the synthesized compounds showed inhibitory activity against SV. An ED50 lower
than micromolar was found in compounds (30a–c), (30f), (33e), and (39a–b), while most of the
remaining compounds were about 3 order of magnitude less potent (Table I). Moreover, (30b)presented a micromolar ED50 (1.1 mM) with a selectivity index of about 200.
On the basis of the results reported in Table I, the following structure-activity relationships
may be formulated: (i) The N,N-dimethyluracil scaffold, unusual in antiviral agents, along with
the 6-substitution on the uracil ring, seems to be an important property for active compounds. (ii) The
substitution pattern and the stereochemistry of the oxirane ring plays a role in modulating both
the activity and the toxic effect of the products. (iii) The substitution of the oxiranyl moiety with
a tetrahydrofuranyl ring resulted in less active agents, with the exception of (31b) which shows
an interesting selectivity index (about 200) as a consequence of a decreased toxic effect. (iv)
Unsubstituted derivatives (31a, 34h–j) show very low values of inhibitory activity and relevant
toxicity.
Computational studies,42 using the Catalyst software,43 have been performed to obtain a pharma-
cophore model describing the three dimensional structural properties for profitable interactions with
the binding site on SV. This computational approach has been applied to a set of 22 SV inhibitors
Scheme 4. Top:Method A:Dimethyldioxirane (DMD) 1.0 equiv.mol�1,CH2Cl2, 25�C.Method B:Dimethyldioxirane (DMD),1.0 equiv.
mol�1, dry alcohol-CH2Cl2(1:1 v/v), 25
�C. Method C: Dimethyldioxirane (DMD) 1.0 equiv. mol�1, amine, CH2Cl2, 25
�C. Bottom:Method B: R¼OR1; R1,:CH3,CH2CH3,CH2(CH2)2CH3.Method C:R¼NHR2; R2:CH3,CH2CH2CH3,C6H4Me-p.
Scheme 5.
PARAINFLUENZA-1 (SENDAI) VIRUS * 435

(Table I), chosen according to the Catalyst guideline. The resulting pharmacophore hypotheses
use chemical functions and their spatial location to explain the differences in inhibitory activity
within the training set. All but 1 of the 10 generated hypothesis have in common the presence of
three hydrogen bond acceptor groups (HBA) and one hydrophobic region (HY). Hypothesis 1,
characterized by the highest scoring and statistical parameters, has been chosen to represent the
‘‘pharmacophore model.’’ As a representative example, Figure 7 shows the best-fitted conformer of
(30b) into the pharmacophore model. The phenyl moiety fits within the region of the hydrophobic
group (HY), the carbonyl groups at the 2- and 4-position occupy two hydrogen bond regions (HBA2
and HBA3), and the oxirane oxygen is located in the third hydrogen bond acceptor region (HBA1) of
the hypothesis.
Deazapyrimidine nucleosides also show biological activity against SV. These derivatives
have extensive biochemical and medicinal applications.45,46 In particular, the effectiveness of 3-
deazauridine (deaza UR) (40) and 3-deazacytidine (deaza CT) (41) (Fig. 8) on replication of variousRNA viruses was measured by the extent of HAU production.47
The HAU experiments show that 3-deazacytidine (41) was more active than deaza UR (40).47
As illustrated in Figure 9, c.a. 100% inhibition in cytopathogenic effect (CPE) on chicken embryo
cells was obtained in the presence of deaza CT (41).A new synthesis48 of 3-deazapyrimidine derivatives (Scheme 10) has been proposed as a
synthetic alternative to previously reported methods49,50 which require dangerous materials such as
Figure 5. Structure ofcompound (27c).
Scheme 6.
436 * SALADINO ET AL.
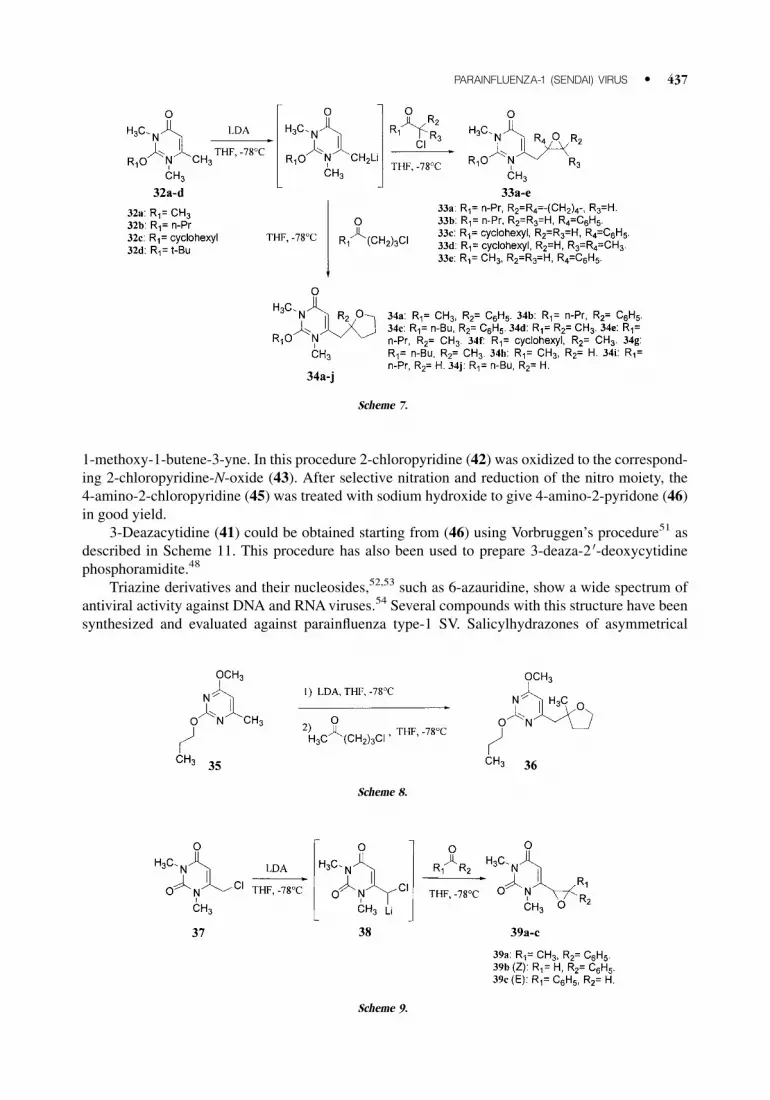
1-methoxy-1-butene-3-yne. In this procedure 2-chloropyridine (42) was oxidized to the correspond-ing 2-chloropyridine-N-oxide (43). After selective nitration and reduction of the nitro moiety, the
4-amino-2-chloropyridine (45) was treated with sodium hydroxide to give 4-amino-2-pyridone (46)in good yield.
3-Deazacytidine (41) could be obtained starting from (46) using Vorbruggen’s procedure51 as
described in Scheme 11. This procedure has also been used to prepare 3-deaza-2 0-deoxycytidinephosphoramidite.48
Triazine derivatives and their nucleosides,52,53 such as 6-azauridine, show a wide spectrum of
antiviral activity against DNA and RNAviruses.54 Several compounds with this structure have been
synthesized and evaluated against parainfluenza type-1 SV. Salicylhydrazones of asymmetrical
Scheme 7.
Scheme 8.
Scheme 9.
PARAINFLUENZA-1 (SENDAI) VIRUS * 437
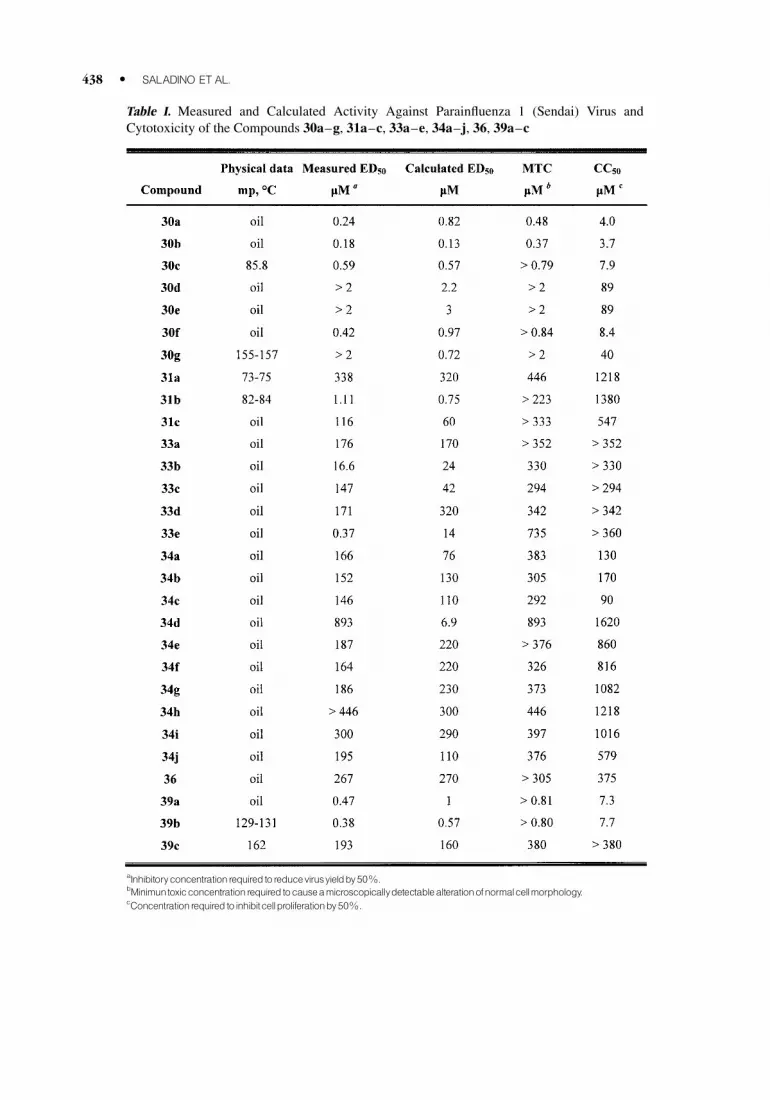
Table I. Measured and Calculated Activity Against Parainfluenza 1 (Sendai) Virus and
Cytotoxicity of the Compounds 30a–g, 31a–c, 33a–e, 34a–j, 36, 39a–c
aInhibitoryconcentrationrequiredtoreducevirusyieldby50%.
bMinimuntoxicconcentrationrequired tocauseamicroscopicallydetectablealterationofnormalcellmorphology.
cConcentrationrequired toinhibitcellproliferationby50%.
438 * SALADINO ET AL.

triazines (47–49) (Fig. 10) and the corresponding copper complexes show anti-SVactivity associated
to a superoxide radical scavenger activity.55
The inhibition of viral multiplication is probably due to the capture of oxygen radicals generated
after the contact of the virus with the host cell. The (47-Cu) complex was the most active compound
(Fig. 11).
Pseudonucleotidic derivatives of as-triazines (50–53) were also tested against SV (Fig. 12).56
B. Antioxidants
The pathogenesis of viral diseases depends not only on the characteristics of the infective agent, but
also on the metabolic state and defensive capacities of the host cell. Shifts in the intracellular redox
state towards pro-oxidant conditions have been observed during several viral infections in both invitro
and in vivo studies.57–60 Although, the role of the oxidative environment in viral replication is not
completely understood, there is reason to believe that the pro-oxidant state observed after infection is
important for completion of the life cycle of the virus.61–63
We have demonstrated that glutathione (GSH, 54) (Fig. 13), the major intracellular antioxidant,
can inhibit, in vitro and in vivo, the replication of SV, as well as that of other viruses with different
mechanisms of replication (herpes simplex-1 virus, human immunodeficiency virus type 1 (HIV-1))
(Fig. 14).30,64–67
Figure 6. Structures ofcompounds (28a^b).
Figure 7. Compound (30b) mapped into thepharmacophoremodel for Sendai virus inhibitors.
PARAINFLUENZA-1 (SENDAI) VIRUS * 439
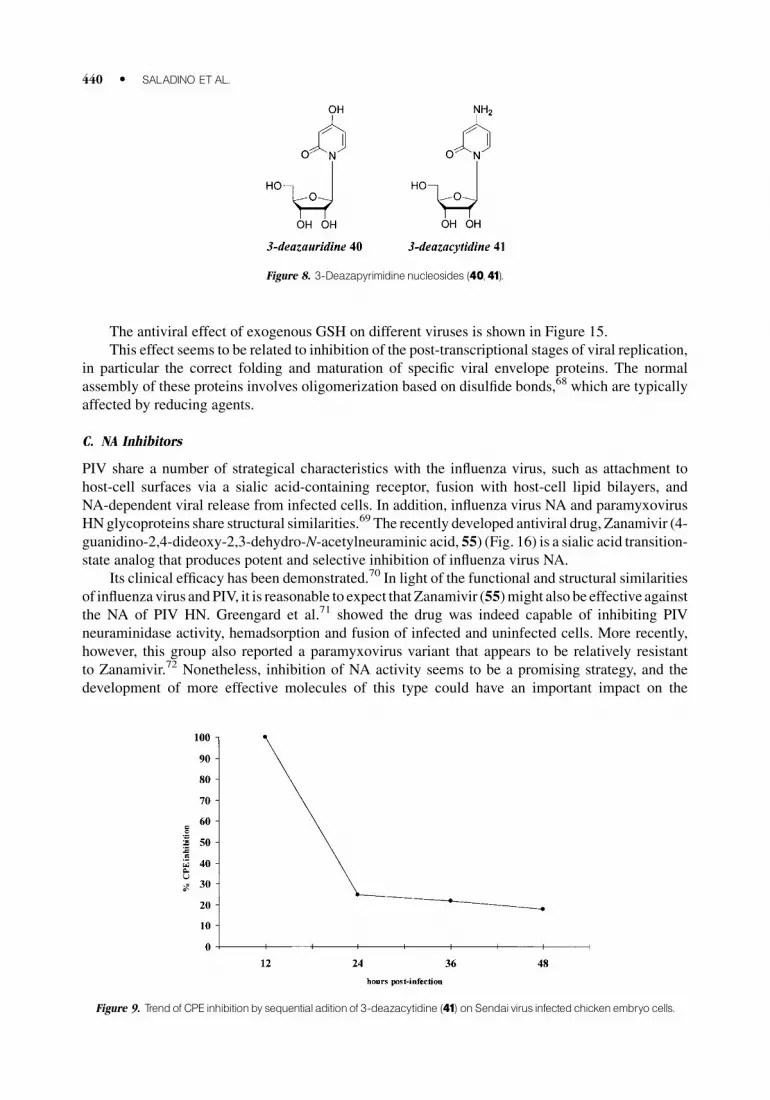
The antiviral effect of exogenous GSH on different viruses is shown in Figure 15.
This effect seems to be related to inhibition of the post-transcriptional stages of viral replication,
in particular the correct folding and maturation of specific viral envelope proteins. The normal
assembly of these proteins involves oligomerization based on disulfide bonds,68 which are typically
affected by reducing agents.
C. NA Inhibitors
PIV share a number of strategical characteristics with the influenza virus, such as attachment to
host-cell surfaces via a sialic acid-containing receptor, fusion with host-cell lipid bilayers, and
NA-dependent viral release from infected cells. In addition, influenza virus NA and paramyxovirus
HNglycoproteins share structural similarities.69 The recently developed antiviral drug, Zanamivir (4-
guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid, 55) (Fig. 16) is a sialic acid transition-state analog that produces potent and selective inhibition of influenza virus NA.
Its clinical efficacy has been demonstrated.70 In light of the functional and structural similarities
of influenza virus and PIV, it is reasonable to expect that Zanamivir (55)might also be effective against
the NA of PIV HN. Greengard et al.71 showed the drug was indeed capable of inhibiting PIV
neuraminidase activity, hemadsorption and fusion of infected and uninfected cells. More recently,
however, this group also reported a paramyxovirus variant that appears to be relatively resistant
to Zanamivir.72 Nonetheless, inhibition of NA activity seems to be a promising strategy, and the
development of more effective molecules of this type could have an important impact on the
Figure 8. 3-Deazapyrimidinenucleosides (40,41).
Figure 9. Trendof CPE inhibitionby sequential aditionof 3-deazacytidine (41) on Sendai virus infectedchickenembryo cells.
440 * SALADINO ET AL.

prophylaxis and treatment of PIV infections. An efficient synthesis of Zanamivir (55) has been
proposed73 starting from N-acetyl neuraminic acid. Other potent inhibitors of influenza
Neuraminidase are Tamiflu (Oseltamivir),74 the cyclopentane variant BCX-1812,75 and compounds
based on a pyrrolidine motif, such as A-315675.76
D. (HMG-CoA) Reductase Inhibitors
Lovastatin (56, Fig. 17) is used in the treatment of hypercholesterolemia. It inhibits hydro-
xymethylglutaryl coenzyme A (HMG-CoA) reductase, which plays an important role in cholesterol
biosynthesis.77
Since a branch of this pathway leads to the formation of isoprenyl groups, lovastatin has also
been used to study isoprenylation and membrane localization of proteins such as RhoA,78 a small
GTP-binding member of the Ras superfamily that is ubiquitously expressed in mammalian cells.
Interaction between the RSV F protein and RhoA has been demonstrated.79 RhoA activation, which
involves geranylgeranyltransferase-mediated isoprenylation at the protein’s carboxy terminus,80
leads to the production of several cytokines, such as interleukin (IL)-1-beta, IL-6, and IL-8, and
structural alterations of the cytoskeleton mediated by the organization of actin stress fibers and the
formation of focal adhesion plaques. RhoA activation has been demonstrated in RSV-infected cells.81
Gower et al.81 have suggested that RhoA-mediated signaling might be involved in various aspects of
RSV pathogenesis, including cell-to-cell fusion, induction of cytokine secretion, and airway
hyperreactivity. Indeed, they demonstrated lovastatin inhibits both in vitro and in vivo replication of
RSV and suggested that drugs like lovastatin that prevent isoprenylation or other pharmacological
approaches for preventing RhoA membrane localization might represent a novel approach to the
prevention of RSV-related disease in high risk groups.81 Lovastatin (56) is generally produced by a
fermentation process.82,83 However, total or semi-synthetic chemical routes have been consid-
ered.84,85 As an example, the preparation of lovastatin (56) and analogue compounds has been
reported86 via a regioselective enzymatic esterification of the commercially available diol (57) withthe (S)-(þ )-2-methylbutyric acid (58) (Scheme 12).
The reaction was performed using nylon-immobilized lipase from Candida Rugosa.
Scheme 10.
Scheme 11.
PARAINFLUENZA-1 (SENDAI) VIRUS * 441
E. Prostaglandins
Prostaglandins (PGs), a class of cyclic fatty acids, synthesized from polyunsatured precursors of the
phospholypids of the cell membrane, are involved in many biological and phatological phenomena
including regulation of cell cycle,87–89 immunization,90 inflammation,91 interferon action,92 and viral
replication. In the last 20 years more and more attention has been devoted on the antiproliferative and
antiviral property of PGs and related compounds. In particular, certain cyclopentenone prostaglandins
(PGAs and PGJs) have demonstarted potent antitumor effects in vitro and in vivo93–96 and antiviral
activity against awide range ofDNAandRNAviruses.97–100 In the case of SV, PGAs, both PGA1 (59)and PGA2 (60) were found to inhibit specifically the viral replication and to prevent persistent
infections, while PGs of the B, E, and F series were inactive (Fig. 18).101
The antiviral action of PGA1 (59) has been related to the selective block of viral protein synthesisoccurring at the translational level. Such a block has been associated to the synthesis of a 70-kDa heat
shock protein (HSP70) induced by PGA1(60) treatment in both uninfected and infected cells.102
PGJ2 (61), a dehydration product of PGD2, was also found to be a potent inhibitor of SV
replication at doses not toxic for uninfected cells.103
F. Inhibitors of Ca2þ -ATPase and Ca2þ -Perturbants
Treatment of cells with agents which interfere with Ca2þ -homeostasis has been shown to influence
the process of maturation and secretion of membrane proteins, being Ca2þ ions required in several
steps of the proteic pathway from endoplasmic reticulum (ER) to the plasma membrane.104
Thapsigargin (TG, 62) is a sequiterpene-g-lactone isolated from Thapsia garganica L.105,106that
Figure 10. Asimmetrical triazine derivatives (47^49).
Figure 11. The kinetics of the HA activity with virus cultivated in the absence of the complex compound 47-Cu (a) and with virus
incubated for 30minwith the complex conpound 47-Cu (b).
442 * SALADINO ET AL.

inhibits the ubiquitous sarcoplasmic and ER Ca2þ -dependent ATPases (SERCA’s) (Fig. 19).
Evaluation of its effects on SV infection in mouse fibroblast BALB3T3 cells demonstrated an
inhibition of virus replication due to a block in the transport of envelope proteins HN and F0 to host
cell membrane.107
Other inhibitors of Ca2þ pump such as Di-tert-butyl-hydroquinone (BHQ, 63) and cyclo-
piazonic acid (CPA, 64) and Ca2þ -ionophores such as Ionomycin (65) and Calcymicin (66) (Fig. 20)were found to inhibit SV by influencing the process of maturation of the glycoprotein HN.
G. Long Chain Fatty Acids
Although lipid bilayers are generally not considered to be involved inmembrane protein functionality,
the inactivation of viral infectivity has been reported by alteration of the lipid bilayer by various
means.108 In particular, treatment of SV with phospholipase A2 or B give reduction of its hemolytic
property.109 Of the different ways inwhich these phospholipases could inactivate the virus, the effects
of free fatty acids produced by the enzymes action have been evaluated in successive studies110 and
has been found that these compounds, especially cis-unsatured acids, are potent inhibitors of SV
induced hemolysis. Their action is ca. two orders of magnitude more potent than that of others
described membrane dissolving amphiphiles.111
H. Antiviral Glycosides
Several studies on the biological activity of carbohydrate derivatives as immunostimulant, antivirus or
interferon-inducing agents have been reported.112 Among them, many alkyl, aryl, or alkylaryl D-
glycopyranoside derivatives have been prepared and tested against enveloped viruses.113
The structure-activity relationships revealed that p-alkylphenyl-6-halogeno-6-deoxy-b-D-gluco-sides (67) (Fig. 21) were themost effective agents and the removal of aglyconmoiety resulted in a loss
of virucidal activity.114
When this series of compound was tested on SV, the p-(sec-butyl)-6-chloro-6-deoxy-b-D-glucopyranoside (68) results the most effective agent inhibiting the cell fusion capability and
infectivity without affecting hemagglutinating or neuramidase activities.114,115
Figure 13. Glutathione (GSH)54.
Figure 12. Structuresofcompounds (50^53).
PARAINFLUENZA-1 (SENDAI) VIRUS * 443
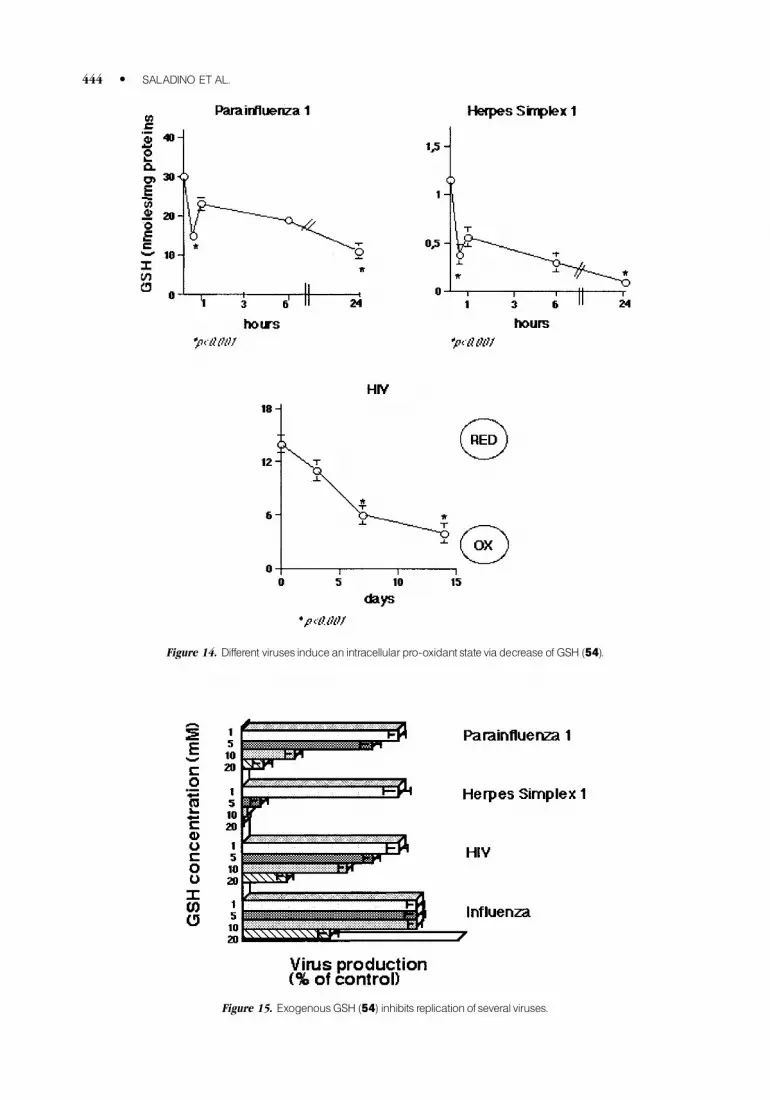
Figure 14. Different viruses inducean intracellular pro-oxidant state viadecrease of GSH (54).
Figure 15. ExogenousGSH (54) inhibits replicationof several viruses.
444 * SALADINO ET AL.

I. Antibiotic Nucleosides
Tunicamycins (TM, 69),116 a family of nucleosides isolated from Streptomyces lysosuperficus,
exhibit antibiotic and antiviral activity, inhibiting the biosynthesis of specific polysaccharides,
glycolipids, and glycoproteins. In particular, these antibiotics are involved in the enzyme inhibition of
processing of UDP glucose and UDP galactose derivatives. Structurally, the most characteristic
backbone is a sugar of 11 carbon atoms (undecose) formed by a furanose and a pyranose rings
separated by an ‘‘ethano’’ spacer. The uracil residue is attached to the ribose ring and an
additional carbohydrate residue, a D-2-deoxy-2-acetamidogalactose unit, is linked to the pyranose
moiety of the undecose (Fig. 22).
Several research groups have reported the effects of the TM on the growth of different viruses
such as Rous sarcoma virus,117a influenza virus,117b measles virus,117c vesicular stomatitis virus,117d
Sindbis virus.117e,118
SV replication is strongly inhibited byTM(69).118 Themechanismof the antiviral effect has been
related to an inhibition of HN and F0 envelope protein glycosilation.
J. Antisense Oligonucleotides
The first reported antiviral effects of antisense oligonucleotides were published in 1978 by Zamecnik
and Stephenson,116 who showed that Rous sarcoma virus replication could be inhibited by
oligonucleotides complementary to reiterated terminal sequences of the viral 35S RNA. Since then,
antisense oligonucleotides have been used against a variety of viruses, including negative-stranded
Figure 16. Zanamivir (55).
Figure 17. Lovastatin (56).
Scheme 12.
PARAINFLUENZA-1 (SENDAI) VIRUS * 445

RNA viruses (such as influenza virus, RSV, and rabies virus).119–121 Chimeric oligonucleotides
consisting of 2 0,5 0 oligoadenylate (2–5A) covalently linked to antisense (2–5A-antisense) have beendesigned to bind to the ubiquitous intracellular enzyme, endoribonuclease RNase L.122 Activation
of RNase L requires the presence of 2 0,5 0-oligoadenylates (2–5A), which are produced by cells afterexposure to IFN, and it results in the nonselective degradation of single-stranded RNA.123 The 2–5A-
antisense approach harnesses this enzyme to produce selective cleavage of individual RNA targets:
the antisense compound associates with complementary RNA sequences in the cell and activates
RNase to produce cleavage of the proximal RNA.
High levels of RNase have been demonstrated in human alveolar macrophages infected with
RSV,119 suggesting that this virus might be susceptible to the effects of 2–5A-antisense. The chimera
has been shown to produce potent inhibition of RSV replication in cultured human tracheal cells,119
and, more recently, dose-dependent reductions in nasal replication of the virus have been observed
following intranasal administration of a 2–5A-anti-RSV compound to African green monkeys.123
K. Chimeras
The cowpea mosaic virus (CPMV) has recently been developed as a biomolecular platform for the
display of heterologous peptide sequences. Khor et al.124 utilized a CPMV-chimera to create an
antiviral against measles virus (MV). This peptide sequence displayed on the CPMV platform
Figure 18. Antiviral prostaglandins (59^61).
Figure 19. Inhibitors of Ca2þ -ATPase (62^64).
446 * SALADINO ET AL.

corresponds to a portion of the MV binding site on the human MV receptor, CD46. The displayed
sequence retains its virus-binding activity and is capable of inhibiting viral entry in vitro and in vivo.
CPMV has also been used with promising results to produce vaccines against several viral
epitopes. In experimental animals, strongly protective antibody responses have been elicited
with chimeric particles expressing epitopes from HIV-1, human rhinovirus, canine parvovirus, foot-
and-mouth disease virus, mink enterititis virus, and the bacterium Staphylococcus aureus.125–128
CPMV-based antivirals, such as the one produced by Khor et al.,124 could also be co-administered
with vaccines to provide protection until full vaccine-induced immunity develops.
3 . I M M U N I Z A T I O N
The immune response to RSV and PIV is complex and not fully understood. Naturally acquired
immunity is neither complete nor durable, as reflected by the high rate of recurrent infections. These
factors have hindered attempts to produce effective vaccines, which must provide greater protection
than that afforded by natural infection and be effective during the early weeks of life.
The first RSVvaccine trials in the 1960s129were a dismal failure although important lessonswere
learned. The formalin-inactivated whole RSV vaccine used in these studies caused enhanced disease
in naive hosts following natural infection, and morbidity and mortality rates among vaccinated
children were greater than those of unvaccinated controls. Subsequent analysis revealed deficiencies
in the humoral response to this vaccine, compared to that elicited by natural infection, suggesting that
important surface glycoprotein epitopes had been selectively modified by formalin inactivation.
In addition, enhanced cell-mediated responses were observed in some of the vaccinated subjects.
These findings highlighted the importance of balance between the two components of immunity in
protection against RSV infection.
Later efforts focused on the development of attenuated vaccines. The first RSV vaccines
containing temperature-sensitive or cold-passaged mutants were effective in adults. In children,
Figure 20. Structuresof Ca2þ -ionophores (65^66).
Figure 21. Antiviral glycosides (67^68).
PARAINFLUENZA-1 (SENDAI) VIRUS * 447

however, they proved to be excessively virulent, too attenuated or unstable with reversion to wild-
type virus. More recent strategies have centered around the use of purified surface glycoproteins,
genomicmaterial, and synthetic peptides.With reverse genetics, a recombinant RSVhas been created
that expresses therapeutic levels of Interferon-g, and it seems to protect mice against reinfection
without inhibiting the immune response to vaccine.130
Attenuated PIV vaccines have been developed from both human and bovine strains.7 Bovine
PIV3, which is antigenically related to human PIV3, replicates poorly in humans and protects against
human PIV3 challenge.131 Reverse genetics has been use to produce an attenuated chimeric PIV1
containing internal proteins of PIV3 and surface glycoproteins F and HN of PIV1.132
4 . C O N C L U S I O N S A N D P E R S P E C T I V E S
Several new antiviral agents and therapeutic strategies against Parainfluenza virus infection are
actually under investigation. The ‘‘classical’’ approach is mainly focused on the synthesis of new
compounds characterized by high activity and selectivity in the inhibition of specific viral targets.
Among the pyrimidine derivatives, the synthesis of new 6-substituted uracils and 4-(3H)-
pyrimidinones, with an high degree of superimposition on the hydrophobic and hydrogen bond
acceptor regions present on the pharmacophore model, could be a relevant result.
Moreover, a ‘‘non-classical’’ approach based on the design of derivatives able to interfere with
cellular structures and/or metabolic pathways involved in supporting virus life-cycle (e.g., redox
state, cellular enzymes, transcription factors) appears to be a promising way to block virus replication
and to avoid the emergence of resistant virus strains.
A C K N O W L E D G M E N T S
Thisworkwas partially supported by ItalianMinistry ofHealth and ItalianNational ResearchCouncil
(CNRC007A72 _ 003) grants. INFM is acknowledged.
R E F E R E N C E S
1. Lamb RA, Kolakofsky D. Paramyxoviridae: The viruses and their replication. In: Fields BN, Knipe DM,Howley PM, editors. Virology 4th edn. Philadelphia: Lippincott-Raven Publishers; 2001. p 1305–1340.
2. Portner A. Parainfluenza viruses. In: Granoff A, Webster RG, editors. Encyclopaedia of virology. London,UK: Academic Press; 1999. p 1130–1140.
3. Huang C, Kiyotani K, Fujii Y, Fukuhara N, Kato A, Nagai Y, Yoshida T, Sakaguchi T. Involvement of thezinc-binding capacity of Sendai virus V protein in viral pathogenesis. J Virol 2000;74:7834–7841.
Figure 22. Tunicamycins (TM) (69).
448 * SALADINO ET AL.

4. Kato A, Kiyotani K, Sakai Y, Yoshida T, Shioda T, Nagai Y. Importance of the cysteine-rich carboxyl-terminal half of V protein for Sendai virus pathogenesis. J Virol 1997;71:7266–7272.
5. Kato A, Kiyotani K, Sakai Y, Yoshida T, Nagai Y. The paramyxovirus, Sendai virus, V protein encodes aluxury function required for viral pathogenesis. EMBO J 1997;16:578–587.
6. Piedra PA, Englund JA, Glezen WP. Respiratory syncytial virus and parainfluenza viruses. In:Richmann DD, Whitley RJ, Hayden FG, editors. Clinical virology. NY, USA: Churchill Livingstone;1997. p 787–819.
7. Hall CB. Respiratory syncytial virus and parainfluenza virus. N Eng J Med 2001;344:1917–1928.8. Whimbey E, Ghosh S. Respiratory Syncytial virus infections in immunocompromised adults. Curr Clin
Top Infect Dis 2000;20:232–255.9. Goodbourn S, Didcock L, Randall RE. Interferons: Cell signalling, immunemodulation, antiviral response
and virus countermeasures. J Gen Virol 2000;81:2341–2364.10. Isaacs A, Lindemann J. Virus interference. I. The interferons. Proc R Soc Lond B 1957;147:258–267.11. Horvath CM. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem Sci
2000;25:496–502.12. GarcinD, Latorre P,KolakofskyD. Sendai virus C proteins counteract the interferon-mediated induction of
an antiviral state. J Virol 1999;73:6559–6565.13. Kato A, Ohnishi Y, KohaseM, Saito S, TashiroM, Nagai Y. Y2, the smallest of the Sendai virus C proteins,
is fully capable of both counteracting the antiviral action of interferons and inhibiting viral RNA synthesis.J Virol 2001;75:3802–3810.
14. Parisien JP, Lau JF, Rodriguez JJ, Ulane CM,Horvath CM. Selective STAT protein degradation induced byParamyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signaltransduction. J Virol 2002;76:4190–4198.
15. Didcock L, Young DF, Goodbourn S, Randall RE. The V protein of Simian virus 5 inhibits interferonsignalling by targeting STAT1 for proteasome-mediated degradation. J Virol 1999;73:9928–9933.
16. Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowsky JT, Robins RK. Broad-spectrum antiviralactivity of Virazole: 1-b-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972;177:705–706.
17. Huffman JH, Sidwell RW, Khare GP, Witkowsky JT, Allen LB, Robins RK. In vitro effect of 1-b-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (Virazole) on deoxyribonucleic acid and ribonucleic acidviruses. Antimicr Agents Chemother 1973;3:235–241.
18. Walker JS, Stephen EL, Spertzel RO. Small-particle aerosols of antivirals compounds in treatment oftype A influenza pneumonia in mice. J Infect Dis 1976;133:suppl:A140–144.
19. Larson EW, Stephen EL, Walker JS. Therapeutic effects of small-particle aerosols of Ribavirin onParainfluenza (Sendai) Virus infections of mice. Antimicr Agents Chemother 1976;10:770–772.
20. Hall CB, McBride JT, Walsh EE, Bell DM, Gala CL, Hildreth S, Ten Hyck LG, Hall WJ. Aerosolizedribavirin treatment of infants with respiratory syncytial viral infection. N Engl J Med 1983;308:1443–1447.
21. Herzog KD, Long SS, McGuigan M, Fisher MC, Deforest A. Impact of treatment guidelines on use ofRibavirin. Am J Dis Child 1990;144:1001–1004.
22. McColl MD, Corser RB, Bremner J, Chopra R. Respiratory Syncytial virus infection in adult BMTrecipients: Effective therapy with short duration nebulised ribavirin. Bone Marrow Transplant 1998;21:423–425.
23. Welliver RC. Immunologic mechanisms of virus-induced wheezing and asthma. J Pedriatr 1999;135:14–20.
24. De Vincenzo JP. Therapy of respiratory syncytial virus infection. Pedriatr Infect Dis J 2000;19:786–790.25. Ramasamy KS, Tam RC, Bard J, Averett DR. Monocyclic L-nucleosides with type-1 cytokine-inducing
activity. J Med Chem 2000;43:1019–1028.26. Vujovic O, Mills J. Preventive and therapeutic strategies for Respiratory Syncytial virus infection.
Curr Opin Pharmacol 2001;1:497–503.27. Adam W, Curci R, Edwards JO. Dioxiranes: A new class of powerful oxidants. Acc Chem Res 1989;22:
205–211.28. Curci R. In: BaumstarkAL, editor. Advances in oxygenated process.Grenwich (CT): JAI Press; 1990.Vol. II.29. Saladino R, Bernini R, Crestini C, Mincione E, Bergamini A, Marini S, Palamara AT. Studies on the
chemistry of pyrimidine derivatives with dimethyldioxirane: Synthesis, cytotoxic effect and antiviralactivity of new 5,6-oxiranyl-5,6-dihydro and 5-hydroxy-5,6-dihydro-6-substitued uracil derivatives andpyrimidine nucleosides. Tetrahedron 1995;51:7561–7578.
30. Garaci E, Palamara AT, Di Francesco P, Favalli C, Ciriolo MR, Rotilio G. Glutathione inhibitsreplication and expression of viral proteins in cultured cells infected with Sendai Virus. Biochem BiophysRes Commun 1992;188:1090–1092.
PARAINFLUENZA-1 (SENDAI) VIRUS * 449

31. a Saladino R, Crestini C, Nicoletti R, Saladino R, Mincione E, Crestini C, Nicoletti R, Saladino R,Crestini C, Occhionero F, Nicoletti R. Ozonation of substituted 2-thiouracils and pyrimidine-2-thione.Tetrahedron Lett 1993;34:1631–1634. b Saladino R, Mincione E, Crestini C, Nicoletti R. Oxidation ofsubstituted 2-thiouracils and pyrimidine-2-thionewith ozone and 3,3-dimethyl-1,2-dioxirane. Tetrahedron1994;50:3259–3272. c Saladino R, Crestini C, Occhionero F, Nicoletti R. Ozonation of thionucleosides.A new chemical transformation of 4-thiouracil and 6-thioguanine nucleosides to cytosine and adenosinecounterparts. Tetrahedron 1995;51:3607–3616.
32. Saladino R, Mincione E, Crestini C, Mezzetti M. Transformation of thiopyrimidine and thiopurinenucleosides following oxidation of dimethyldioxirane. Tetrahedron 1996;52:6759–6780.
33. Saladino R, Mezzetti M, Mincione E, Savini P, Marini S, Palamara AT. Synthesis, cytotoxic effect andantiviral activity of 1-(b-D-arabinofuranosyl)-5-bromo-N4-subtituted cytosine and 1-(b-D-arabinofurano-syl)-5-bromo-4-methoxypyrimidin-2(1H)-one derivatives. Nucleosides Nucleotides 1999;18:2499–2510.
34. aMiyasaka T, Tanaka H, BabaM, Hayawaka H,Walter RT, Balzarini J, De Clercq E, Tanaka H, TakashimaH, UbasawaM, Hayawaka H, Sekiya K, Nitta I, BabaM, Shigeta CS,Walter RT, De Clercq E. A novel leadfor specific anti HIV-1 agents: 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine. J Med Chem1992;32:2507–2509. b Tanaka H, Takashima H, Ubasawa M, Hayawaka H, Sekiya K, Nitta I, Baba M,Shigeta CS, Walter RT, De Clercq E. Synthesis and antiviral activity of deoxy analogs of 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine (HEPT) as potent and selective anti HIV-1 agents. J MedChem 1992;35:4713–4719.
35. Artico M, Massa S, Mai A, Marongiu ME, Piras G, Tramontano E, La Colla P. 3,4-dihydro-2-alkoxy-6-benzyl-4-oxopyrimidines (DABOs): A new class of specific inhibitors of human immunodeficiency virustype 1. Antiviral Chem Chemother 1993;4:361–368.
36. Solladie G, Demailly G, Greck G. Reduction of b-hydroxy sulfoxides: Application to the synthesis ofoptically active epoxides. Tetrahedron Lett 1985;26:435–438.
37. Botta M, Saladino R, Gambacorta A, Nicoletti R. Research on antiviral agents. Enantiospecific synthesisof 1,3-dimethyl-6-oxiranylpyrimidin-2,4-dione with anti-ASFVactivity. Tetrahedron Asymmetry 1990;1:441–444.
38. Botta M, Fabrizi G, Lamba D, Saladino R. Structure of (þ )-(S)-1,3-dimethyl-6-oxiranyl-2,4-pyrimidinedione showing anti-ASFVactivity. Acta Cryst 1992;48:81–83.
39. BottaM, Saladino R, Gambacorta A, Nicoletti R. An unusual condensation of pyrimidinones: Synthesis ofbipyrimidinones and bipyrimidylmethane. Heterocycles 1991;32:1537–1545.
40. Botta M, Saladino R, Delle Monache G, Gentile G, Nicoletti R. Research on antiviral agents. Lithiation of6-methyluracil as a new and efficient entry to C(6)-substituted uracils. Heterocycles 1996;43:1687–1697.
41. Saladino R, Danti MC, Crestini C, Mincione E, Savini P, Marini S, Palamara AT, Botta M. A potent andselective inhibition of Parainfluenza 1 (Sendai) Virus by new 6-oxiranyl-, 6-methyloxiranyluracils, and4(3H)-pyrimidinone derivatives. Bioorg Med Chem Lett 1998;8:1833–1838.
42. Saladino R, Danti MC, Crestini C, Manetti F, Corelli F, Palamara AT, Garaci E, Botta M. Synthesis,biological evaluation, and pharmacophore generation of uracil, 4(3H)-pyrimidinone, and uridine deri-vatives as potent and selective inhibitors of Parainfluenza 1 (Sendai) Virus. J Med Chem 2001;44:4554–4562.
43. Catalyst 4.5 is from MSI Software. Scranton Road, MA.44. Botta M, De Angelis F, Finizia G, Gambacorta A, Nicoletti R. 6-Alkyl- and 5,6-dialkyl-2-methoxy-4(3H)-
pyrimidinones in the transformation of the pyrimidines. Conversion into 2-substituted amino- and4-chloropyrimidine derivatives. Synth Commun 1985;15:27–34.
45. Bloch A, Dutschman G, Currie BL, Robins RK, Robins MJ. Preparation and biological activity of various3-deazapyrmidines and related nucleosides. J Med Chem 1973;16:294–297.
46. a Ojha RP, Sanyal NK, McNamara DJ, Cook PD, Allen LB, Kehoe MJ, Holland CS, Teepe AG. Biologicalactivity of 3-deazauridine nucleoside analogue: a theoretical study. Int J Biol Macromol 1991;13:58–64. b
McNamara DJ, Cook PD, Allen LB, Kehoe MJ, Holland CS, Teepe AG. Synthesis, antitumor activity, andantiviral activity of 3-substituted 3-deazacytidines and 3-substituted 3-deazauridines. J Med Chem1990;33:2006–2011.
47. Khare GP, Sidwell RW, Huffman JH, Tolman RL, Robins RK. Inhibition of RNAVirus replication in vitroby 3-deazacytidine and 3-deazauridine. Proc Soc Exp Biol Med 1972;140:880–884.
48. Searls T,McLaughlinLW.Synthesis of the analogue nucleoside 3-Deaza-2 0-deoxycytidine and its templateactivity with DNA Polymerase. Tetrahedron 1999;55:11985–11996.
49. Currie BL, Robins RK, Robins MJ. The synthesis of 3-deazapyrimidine nucleosides related to uridine andcytidine and their derivatives. J Heterocyclic Chem 1970;7:323–329.
50. Cook D, Day RT, Robins RK. An improved synthesis of 3-deazacyclosine, 3-deazauracil, 3-deazacytidineand 3-deazauridine. J Heterocyclic Chem 1977;14:1295–1301.
450 * SALADINO ET AL.

51. a VorbruggenH,Niedballa U, VorbruggenH.A general synthesis of N-Glycosides. Synthesis of pyrimidinenucleosides. J Org Chem 1974;39:3654–3660. b Vorbruggen H. Adventures in Silicon-Organic chemistry.Acc Chem Res 1995;28:509–520.
52. Skoda P. Azapyrimidine nucleosides. In: Sartorelli AC, John OG, editors. Hundbuchder der experi-mentellen pharmakologie, Berlin: Springer-Verlag; 1975. vol. 38, p 348–372.
53. Cihak A, Vesely J, Skoda J. Azapyrimidine nucleosides: Metabolism and inhibitory mechanisms.Adv Enzyme Regul 1985;24:335–354.
54. a Liu MC, Luo MZ, Mozdziesz DE, Lin TS, Dutschman GE, Cheng YC, Sartorelli AC, Basnak I, Sun M,Hamor TA, Focher F, Verri A, Spadari S, Wroblowski B, Herdewijn P, Walker RT. Synthesis of 2 0-methylene-substituted 5-azapyrimidine, 6-azapyrimidine, and 3-deazaguanine nucleoside analogues aspotential antitumor/antiviral agents. Nucleosides & Nucleotides 1999;18:55–72. b Basnak I, Sun M,Hamor TA, Focher F, Verri A, Spadari S, Wroblowski B, Herdewijn P, Walker RT. Some 6-aza-5-substituted-2’-deoxyuridines show potent and selective inhibition of herpes simplex virus type 1 thymidinekinase. Nucleosides & Nucleotides 1998;17:187–206.
55. Zuivertz A, Tomas ST, Popescu A, Cristescu C, Tomas E. Effects of asimmetrical triazine copper complexon superoxide radical formation and on Sendai virus multiplication. Rev Roum Med Virol 1988;39:217–220.
56. Tomas E, Popescu A, Zuivertz A, Jucu V, Czabor F, Cristescu C. Study of the antiviral activity of some newclasses of As-triazine derivatives. Rev Rom J Virol 1995;46:51–56.
57. Ciriolo MR, Palamara AT, Incerpi S, Lafavia E, Bue MC, De Vito P, Garaci E, Rotilio G. Loss of GSH,oxidative stress, and decrease of intracellular pH as sequential steps in viral infection. J Biol Chem1997;272:2700–2708.
58. Garaci E, Palamara AT, Ciriolo MR, D’Agostini C, Ab del-Latif MS, Aquaro S, Lafavia E, Rotilio G.Intracellular GSH content and HIV replication in human macrophages. J Leukoc Biol 1997;62:54–59.
59. Boya P, De La Pena A, Beloqui O. Antioxidant status and glutathione metabolism in peripheral bloodmononuclear cells from patients with chronic hepatitis C. J Hepatol 1999;31:808–814.
60. Elbim C, Pillet S, Prevost MH. The role of phagocytes in HIV-related oxidative stress. J Clin Virol2001;20:99–109.
61. Mileva M, Tancheva L, Bakalova R, Galabov A, Savov V, Ribarov S. Effect of vitamin E on lipidperoxidation and liver monooxigenase activity in experimental influenza virus infection. Toxicol Lett2000;114:39–45.
62. Roederer M, Ela SW, Staal FJT. N-acetylcisteine: A new approach to anti-HIV therapy. AIDS Res HumRetroviruses 1992;8:209–217.
63. Palamara AT, Di Francesco P, Ciriolo M, Bue C, Rotilio G, Garaci E. Cocaine increases sendai virusreplication in cultured epithelial cells: Critical role of the intracellular redox status. Biochem Biophys ResCommun 1996;228:579–585.
64. PalamaraAT, PernoCF, CirioloMR,Dini L, Balestra E, D’Agostini C, Di Francesco P, Favalli C, Rotilio G,Garaci E. Evidence for antiviral activity of glutathione: In vitro inhibition of herpes simplex virus type 1replication. Antiviral Res 1995;27:237–253.
65. Palamara AT, Garaci E, Rotilio G, Ciriolo MR, Casabianca A, Fraternale A, Rossi L, Schiavano GF,Chiarantini L, Magnani M. Inhibition of murine AIDS by reduced glutathione. AIDS Res HumRetroviruses 1996;12:1373–1381.
66. Palamara AT, Perno CF, Aquaro S, Dini L, Bue C, Garaci E. Glutathione inhibits HIV replication byacting at late stages of virus life cycle. AIDS Res Hum Retroviruses 1996;12:1537–1541.
67. Nucci C, Palamara AT, Ciriolo MR, Nencioni L, Savini P, D’Agostini C, Rotilio G, Cerulli L, Garaci E.Imbalance in corneal redox state during Herpes Simplex virus 1-induced keratitis in rabbits. Effectivenessexogenous glutathione supply. Exp Eye Res 2000;70:215–220.
68. Vidal S, Mottet G, Kolakofsky D, Roux L. Addition of hgigh-mannose sugars must precede disulfidebondformation for proper folding of Sendai virus glycoproteins. J Virol 1989;63:892–900.
69. Colman PM, Hoyne PA, Lawrence MC. Sequence and structure alignement of paramyxovirushemagglutinin-neuraminidase with influenza virus neuraminidase. J Virol 1993;67:2972–2980.
70. Hayden FG, Osterhaus AD, Treanor JJ, Fleming DM, Aoki FY, Nicholson KG, Bohnen AM, Hirst HM,Keene O, Wightman K. Efficacy and safety of the neuraminidase inhibitor zanamivir in the treatmentof influenza virus infections. GG167 Influenza Study Group. N Engl J Med 1997;337:874–880.
71. Greengard O, Poltoratskaia N, Leikina E, Zimmemberg J, Moscona A. The anti-influenza virus agent 4-GU-DANA (zanamivir) inhibits cell fusionmediated by human parainfluenza virus and influenza virusHA.J Virol 2000;74:11108–11114.
72. Murrell MT, Porotto M, Greengard O, Poltoratskaia N, Moscona A. A single aminoacid alteration in theHuman Parainfluenza Virus type 3 hemagglutinin-neuraminidase glycoprotein confers resistance to
PARAINFLUENZA-1 (SENDAI) VIRUS * 451

the inhibitory effects of zanamivir on receptor binding and neuraminidase activity. J Virol 2001;75:6310–6320.
73. Chandrel M, Bamford MJ, Conroy R, Lamont B, Patel B, Patel VK, Steeples IP, Storer R, Weir NG,Wright M, Williamson C. Synthesis of the potent influenza neuraminidase inhibitor 4-guanidinoNeu5Ac2en. X-ray molecular structure of 5-acetamido-4-amino-2,6-anhydro-3,4,5,-trideoxy-D-erythro-L-gluco-nononic acid. J Chem Soc Perkin Trans 1 1995;9:1173–1180.
74. KimCU, LewW,WilliamsMA, LiuH, Zhang LJ, Swaminathan S, Bishofberger N, ChenMS,Mendel DB,Tai C, Laver WG, Stevens RC. Influenza neuraminidase inhibitors possessing a novel hydrophobicinteraction in the enzyme active syte: Design, synthesis, and structural analysis of carbocyclic sialic acidanologues with potent anti-influenza activity. J Am Chem Soc 1997;119:681–690.
75. Babu YS, Chand P, Bantia S, Kotian P, Dehghani A, El-Kattan Y, Lin TH, Hutchison TL, Elliott AJ, ParkerCD, Ananth SL, Horn LL, Laver GW, Montgomery JA. BCX-1812 (RWJ-270201): Discovery of a novel,highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drugdesign. J Med Chem 2000;43:3482–3486.
76. a Wang GT, Chen Y, Wang S, Gentles R, Sowin T, Kati W, Muchmore S, Giranda V, Stewart K, Sham H,Kempf D, Laver GW, Hanessian S, Bayrakdarian M, Luo X. Design, synthesis, and structural analysis ofinfluenza neuraminidase inhibitors containing pyrrolidine cores. J Med Chem 2001;44:1192–1201. b
Hanessian S, Bayrakdarian M, Luo X. Total synthesis of A-315675: a potent inhibitor of influenzaneuraminidase. J Am Chem Soc 2002;124:4716–4721.
77. Alberts A. HMG-CoA reductase inhibitors-the development. Atheroscler Rev 1988;18:123–129.78. Park HJ, Galper JB. 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors up-regulate transforming
growth factor-beta signaling in cultured heart cells via inhibition of geranylgeranylation of RhoAGTPase.Proc Natl Acad Sci USA 1999;96:11525–11530.
79. Pastey MK, Crowe JE, Graham BS. RhoA interacts with the fusion glycoprotein of Respiratory Syncytialvirus and facilitates virus-induced syncytium formation. J Virol 1999;73:7262–7270.
80. Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21rho proteins.J Biol Chem 1992;272:20033–20038.
81. Gower TL, Graham BS. Antiviral activity of Lovastatin against Respiratory Syncytial virus in vivo andin vitro. Antimicrob Agents Chemother 2001;45:1231–1237.
82. BucklandBK,GbewonyoK,Hallada T,KaplanL,Masurekar P.Novelmicrobial products formedicine andagriculture. In: Demain AL, Somkuti JC, Hunter-Cetera JC, Rossmoore HW, editors. Society for industrialmicrobiology. Amsterdam, the Netherlands: Elsevier Science Ltd; 1989. p 161–169.
83. Hajjaj H, Niederberger P, Duboc P. Lovastatin biosynthesis by Aspergillus terreus in a chemically definedmedium. Appl Environ Microbiol 2001;67:2596–2602.
84. HiramaM, IwashitaM. Total synthesis of (þ )-monacolinK (mevinolin). TetrahedronLett 1983;24:1811–1812.
85. Clive DLJ, Murthy KSK, Wee AGH, Prasad JS, Da Silva GVJ, Majewski M, Anderson PC, Evans CF,Haugen RD,Heerze LD, Barrie JR. Total synthesis of both (þ )-Compactin and (þ )-Mevinolin. A generalstrategy based on the use of a special titanium reagent for dicarbonyl coupling. J Am Chem Soc1990;112:3018–3028.
86. Yang F, Weber TW, Gainer JL, Carta G. Synthesis of Lovastatin with immobilized Candida rugosalipase in organic solvents: Effects of reaction conditions on initial rates. Biotechnol Bioeng 1997;56:671–680.
87. Narumiya S, Fukushima M. In: Honn KV, Marnett LJ, Nigam S, Walden T, Jr. editors. Eicosanoids andother bioactive lipids in cancer and radiation injury. Boston, MA: Kluwer Academic Publisher; 1989.p 439–448.
88. Forman BM, Tontonoz P, Chen J, Brun R, Spiegelman B, Evans RM. 15-Deoxy-D12,14-Prostaglandin J2is aligand for the adipocyte determination Factor PPARg. Cell 1995;83:803–812.
89. Kliewer JA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A Prostaglandin J2metabolitebinds peroxisome proliferator-activated receptor g and promotes adipocyte differentiation. Cell 1995;83:813–819.
90. Goodwin JS, Webb DR. Regulation of the immune response by prostaglandins. Clin Immun Immunopath1980;15:106–122.
91. Vane JR. In: Garaci E, Paoletti R, SantoroMG, editors. Prostaglandins in cancer research. Berlin: Springer-Verlag; 1987. p 12–28.
92. Pottathil R, Chandrabose KA, Cuatrecasas P, Lang PJ. Establishment of the interferon-mediated antiviralstate: Role of fatty acid cyclooxygenase. Proc Natl Acad Sci USA 1980;77:5437–5446.
93. Sasaki H, Fukushima M. Prostaglandins in the treatment of cancer. Anticancer Drugs 1994;5:131–138.
452 * SALADINO ET AL.

94. Fukushima M. In: Kabera JJ, editor. Role of lipids in cancer research. III. The pharmacological effect ofLipids. Chicago, IL: American Oil Chemist Society; 1989. p 215–221.
95. Garaci E, Paoletti R, Santoro MG. Prostaglandins in cancer research. Berlin: Springer-Verlag; 1987.96. FukushimaM, Kato T, Narumiya S, Mizushima Y, Sasaky H, Terashima Y, Nishiyama Y, Santoro MG. In:
Samuelsson B, Wong PYK, Sun FF, editors. Advances in prostaglandin, thromboxane, and leukotrieneresearch. New York: Raven Press; 1989. p 415–418.
97. Parker J, Ahrens PB, Ankel H. Antiviral effect of cyclopentenone prostaglandins on Vesicular StomatitisVirus replication. Antiviral Res 1995;26:83–96.
98. Conti C, Mastromarino P, Tomao P, Marco A, De Pica F, Santoro MG. Inhibition of Poliovirus replicationby prostaglandin A and J in human cells. Antimicr Agents Chemother 1996;40:367–372.
99. Rozera C, Carattoli A, Marco A, Amici C, Giorgi C, Santoro MG. Inhibition of HIV-1 replication bycyclopentenone prostaglandins in acutely infected human cells. J Clin Invest 1996;97:1795–1803.
100. O’Brien WJ, Taylor JL, Ankel H, Sitenga G. Assessment of antiviral activity, efficacy, and toxicityof prostaglandin A2 in a rabbit model of herpetic keratitis. Antimicr Agents Chemother 1996;40:2327–2331.
101. SantoroMG,Benedetto A, CarrubaG,Garaci E, Jaffe BM. ProstaglandinA compounds as antiviral agents.Science 1980;209:1032–1034.
102. Amici M, Giorgi C, Rossi A, Santoro MG. Selective inhibition of virus protein synthesis by prostaglandinA1: A translational block associated with HSP70 synthesis. J Virol 1994;68:6890–6899.
103. Santoro MG, Fukushima M, Amici C, Benedetto A. PGJ2, a new antiviral prostaglandin: Inhibition ofSendai virus replication and alteration of virus preotein synthesis. J Gen Virol 1987;68:1153–1158.
104. Lodish HF, Kong N. Perturbation of cellular calcium blocks exit of secretory proteins from roughendoplasmic reticulum. J Biol Chem 1990;265:10893–10899.
105. Rasmussen U, Christensen SB, Sandberg F. Thapsigargin and thapsigargicine, two new histamineliberators from Thapsia garganica. Acta Chem Suac 1978;15:133–140.
106. Christensen SB, Andersen A, Smitt UW. Sesquiterpenoids from Thapsia species and medicinal chemistryof the thapsigargins. Prog Chem Natl Prod 1997;71:129–167.
107. Ono A, Kawakita M. Transport of envelope proteins of Sendai Virus, HN and F0, is blocked at differentsteps by Thapsigargin and other perturbants to intracellular Ca2þ . J Biochem 1994;116:649–656.
108. Patzer EJ, Wagrer RR, Dubovi EJ. Viral membranes: Model systems for studying biological membranes.CRC Crit Rev Biochem 1979;7:165–217.
109. Barbanto-Brodano G, Possati L, La Placa M. Inactivation of polykaryocytogenic and hemolytic activitiesof Sendai virus by phospholipase B (lysolecithinase). J Virol 1971;8:796–800.
110. MacDonald RC, Dalle Ore V, MacDonald RI. Inhibition of Sendai virus-induced hemolysis by long chainfatty acids. Virology 1984;134:103–117.
111. Helenius A, Simons K. Solubilization of membranes by detergents. Biochim Biophs Acta 1975;415:29–79.
112. Arita H, Sugita K, Nomura A, Sato K, Kawanami J. Studies on antiviral glycosides. Synthesis andbiological evaluation of various phenyl glycosides. Carbohydrate Res 1978;62:143–154.
113. SugitaK, Arita H, SatoK,Kawanami J. Studies on antiviral glycosides.Mode of action for virucidal effectson Sendai virus. Bioch Bioph Acta 1979;552:404–412.
114. JizomotoH,AritaH, SugitaK,Kawanami J, SatoK,YuriyamaK. Studies on antiviral glycosides. Chemicalmodification of the envelope of Sendai virus. J Biochem 1980;88:995–999.
115. Arita H, SugitaK, SatoK,AmanoY,Kawanami J. Studies on antiviral glycosides. Formation of incompleteSendai virions in the presence of phenyl-6-chloro-6-deoxy-b-D-glucopyranoside. Chem Pharm Bull 1981;29:2928–2933.
116. Takasuki A, Arima K, Tamura G. Tunicamycin, a new antibiotic. Isolation and characterization ofTunicamycin. J Antibiot 1971;24:224–231.
117. a Diggelmann H, Nakamura K, Compans RW, Stallcup K, Fields BN, Gibson R, Schlesinger S, Kornfeld S,Leavitt R, Schlesinger S, Kornfeld S. Biosynthesis of an unglycosilated envelope glycoprotein ofRous sarcoma virus in presence of tunicamycin. J Virol 1979;30:799–804. b Nakamura K, Compans RW.Effects of glucosamine, 2-deoxy-D-glucose and tunicamycin on glycosilation, sulfation and assembly ofinfluenza virus glycoproteins. Virology 1978;84:303–319. c Stallcup K, Fields BN. The replication ofmeasles virus in presence of tunicamycin. Virology 1981;108:391–404. d Gibson R, Schlesinger S,Kornfeld S. The nonglycosylated glycoproteins of vesicular stomatitis virus containing nonglycosylated Gprotein. Cell 1978;13:671–679. e Leavitt R, Schlesinger S, Kornfeld S. Tunicamycin inhibits glycosilationand multiplication of sindbis and vesicular stomatitis virus. J Virol 1977;21:375–385.
118. NakamuraK,HommaM,Compans RW. Effect of tunicamycin on the replication of Sendai Virus. Virology1982;119:474–487.
PARAINFLUENZA-1 (SENDAI) VIRUS * 453

119. Cirino NM, Li G, Xiao W, Torrence PF, Silverman RH. Targeting RNA decay with 2 0-5 0 oligoadenylate-antisense in Respiratory Syncytial virus-infected cells. Proc Natl Acad Sci USA 1997;94:1937–1942.
120. Leiter JM,Agrawal S, Palese P, Zamecnik PC. Inhibition of Influenza virus replication by phosphorothioateoligodeoxynucleotides. Proc Natl Acad Sci USA 1990;87:3430–3434.
121. Fu ZF, Wickstrom E, Jiang M, Corisdeo S, Yang J, Dietzschold B, Koprowski H. Inhibition of rabies virusinfection by an oligodeoxynucleotide complementary to rabies virus genomic RNA. Antisense NucleicAcid Drug Dev 1996;6:87–93.
122. Torrence PF, Maitra RK, Lesiak K, Khamnei S, Zhou A, Silverman RH. Targeting RNA for degradationwith a (2 0-5 0)oligoadenylate-antisense chimera. Proc Natl Acad Sci USA 1993;90:1300–1304.
123. LeamanDW,LonganoFJ,Okicki JR, SoikeKF, Torrence PF, SilvermanRH,CramerH. Targeted therapy ofRespiratory Syncytial virus in African Green Monkeys by intranasally administered 2-5A antisense.Virology 2002;292:70–77.
124. Khor IW, Lin T, Langedijk PM, Johnson JE,Manchester M. Novel strategy for inhibiting viral entry by useof a cellular receptor-plant virus chimera. J Virol 2002;76:4412–4419.
125. McLainL,Durrani Z,Wisniewski LA, PortaC, LomonossoffGP,DimmockNJ. Stimulation of neutralizingantibodies to human immunodeficiency virus type 1 in three strains ofmice immunizedwith a 22 aminoacidpeptide of gp41 expressed on the surface of aplant virus. Vaccine 1996;14:799–810.
126. Porta C, Spall VE, Lin T, Johnson JE, Lomonossoff GP. The development of cowpea mosaic virus as apotential source of novel vaccines. Intervirology 1996;39:79–84.
127. Dalsgaard K, Uttenthal A, Jones TD, Xu F, Merryweather A, Hamilton W, Langeveld J, Boshuizen R,Kamstrup S, Lomonossoff G, Porta C,Vela C, Casal J,MeloenR, Rodgers P. Plant-derived vaccine protectstarget animals against a viral disease. Nat Biotechnol 1997;15:248–252.
128. Meloen RH, Hamilton WD, Casal JI, Dalsgaard K, Langeveld JP. Edible vaccines Vet Q 1998;20:S92–S95.
129. Kapikinian AA, Mitchell RH, Chanock RM, Shvedoff RA, Stewart CE. An epidemiologic study of alteredclinical reactivity to RSV infection in children previously vaccinated with an inactivated RSV vaccine.Am J Epidemiol 1969;89:405–412.
130. Bukreyev A, Whitehead SS, Bukreyeva N, Murphy BR, Collins PL. Interferon gamma expressed by arecombinant Respiratory Syncytial virus attenuates virus replication in mice without compromisingimmunogenicity. Proc Natl Acad Sci USA 1999;96:2367–2372.
131. MurphyBR,Collins PL.Current status of Respiratory Syncytial virus (RSV) and Parainfluenzavirus type 3(PIV3) vaccine development: Memorandum from a joint WHO/NIAIDmeeting. Bull World Health Organ1997;75:307–313.
132. Tao T, Skiadopolous MH, Durbin AP, Davoodi F, Collins PL, Murphy BR. A live attenuated chimericrecombinant Parainfluenza virus (PIV) encoding the internal proteins of PIV type 3 and the surfaceglycoproteins of PIV type 1 induces complete resistance to PIV1 challenge and partial resistance to PIV3challenge. Vaccine 1999;17:1100–1108.
Raffaele Saladino graduated in Chemistry at the University of Rome ‘‘La Sapienza,’’ Italy in 1989. He received
his Ph.D in 1993 at the University of Rome ‘‘La Sapienza’’ and carried out a period of postdoctoral research
with Prof. S. Hanessian (Universite de Montreal, Montreal, Quebec, Canada). He was appointed as research
assistant in 1992 at the University of Tuscia, Viterbo (Italy). In 2000, he was appointed as associate professor of
Organic Chemistry at the same university. His current research interests include the chemistry of natural
substances and the synthesis of new antiviral and antitumor agents.
Umberto Ciambecchini studied Medicinal Chemistry at the University of Rome ‘‘La Sapienza’’ and received
his degree in 1999. His thesis work was on 6-substituted pyrimidine derivatives. After a period, post lauream,
at University of Rome, focused on synthesis of new phosphoramidite nucleosides, he moved in 2000 to
University of Tuscia, Viterbo where he is currently engaged in synthesis of purine derivatives as antiviral
agents.
Lucia Nencioni graduated in Biology at the University of Florence in 1993. She received her Ph.D in 2002
at the University of Rome ‘‘Tor Vergata’’ under the direction of Prof. E. Garaci. Currently, she is carrying out
her post-doctoral research in the Department of Experimental Medicine and Biochemical Sciences of the
University of Rome ‘‘Tor Vergata.’’ Her research interests include the identification of molecular mechanisms
involved in the control of viral replication and apoptosis, and the study of antiviral agents.
454 * SALADINO ET AL.

Anna Teresa Palamara graduated in Medicine at the Catholic University of Rome, Italy in 1982. She started
her research activity in the Department of Experimental Medicine and Biochemical Sciences of the University
of Rome ‘‘Tor Vergata,’’ where she was appointed as research assistant in 1985. In 1998, she was appointed as
associate professor of Microbiology at the University of Naples ‘‘Federico II’’ and then full professor of
Microbiology at the University of Rome ‘‘La Sapienza’’ in 2001. Her current research interests include the
identification of the virus/host cell interaction involved in the control of viral replication and the study of new
antiviral therapeutic strategies.
PARAINFLUENZA-1 (SENDAI) VIRUS * 455
Related Documents











