Review Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis Peter Ly 1, * and Don W. Cleveland 1, * Cancer genome sequencing has identified chromothripsis, a complex class of structural genomic rearrangements involving the apparent shattering of an individual chromosome into tens to hundreds of fragments. An initial error during mitosis, producing either chromosome mis-segregation into a micronu- cleus or chromatin bridge interconnecting two daughter cells, can trigger the catastrophic pulverization of the spatially isolated chromosome. The resultant chromosomal fragments are religated in random order by DNA double-strand break repair during the subsequent interphase. Chromothripsis scars the can- cer genome with localized DNA rearrangements that frequently generate exten- sive copy number alterations, oncogenic gene fusion products, and/or tumor suppressor gene inactivation. Here we review emerging mechanisms underly- ing chromothripsis with a focus on the contribution of cell division errors caused by centromere dysfunction. Hidden in Plain Sight: Chromothripsis in the Cancer Genome The karyotypes of cancer cells are often remarkably complex – littered not only with mutations but also small- and large-scale changes in both chromosome number and architecture. Copy number alterations in the form of whole-chromosome or segmental aneuploidy are present in most tumors, yet its role as a cause or consequence of cancer development remains under debate [1,2]. Structural aberrations and gross rearrangements alter the linear organization of chromosomes, and in some instances can directly drive tumorigenesis. The Philadelphia chromosome in chronic myelogenous leukemia is the classic example [3], involving a translo- cation between chromosomes 9 and 22 to generate an oncogenic gene fusion product that can be effectively targeted by clinical therapeutics [4]. Although it appears firmly established that tumorigenesis develops through the gradual and sequential accumulation of genetic and/or epigenetic changes [5], recent cancer genome sequencing efforts challenged this dogmatic view by identifying several new classes of mutational processes that form simultaneously in a single burst. Adapted from the age-old theory in evolution, the paradigm-shifting concept of punctuated equilibrium (see Glossary) has now been applied toward our understanding of cancer progression models, as recently described in the renewed context of pancreatic cancer [6]. These punctuated events currently include alterations such as kataegis (regions of clustered hypermutation) [7,8] and chromo- plexy (serial rearrangements linking multiple chromosomes) [9] the underlying bases of which are not well understood. Among the most striking examples of punctuated equilibrium is chromothripsis (a Greek neologism for ‘chromosome shattering’), in which tens to hundreds of structural Trends Chromothripsis is a catastrophic event in which one or a few chromosomes are shattered and stitched back together in random order, producing a derivative chromosome with com- plex rearrangements within a few cell cycles. Chromosome mis-segregation during cell division frequently produces small nuclear structures called micronuclei, which are prone to irreversible nuclear envelope disruption during interphase and impaired nucleocytoplasmic compartmentalization. Micronucleated chromosomes accu- mulate extensive DNA damage and are susceptible to shattering during the next mitosis, generating multiple, distinct DNA fragments. Chromosome fragments are reas- sembled by DNA double-strand break repair to form a derivative chromosome. Chromatin bridges trapped between daughter cells are attacked by a cyto- plasmic nuclease (three-prime repair exonuclease 1; TREX1) during inter- phase to generate DNA breaks and focal chromothripsis. 1 Ludwig Institute for Cancer Research, Department of Cellular and Molecular Medicine, University of California at San Diego, La Jolla, CA 92093, USA *Correspondence: [email protected] (P. Ly) and [email protected] (D.W. Cleveland). Trends in Cell Biology, December 2017, Vol. 27, No. 12 http://dx.doi.org/10.1016/j.tcb.2017.08.005 917 © 2017 Elsevier Ltd. All rights reserved.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ReviewRebuilding ChromosomesAfter Catastrophe: EmergingMechanisms of ChromothripsisPeter Ly1,* and Don W. Cleveland1,*
Cancer genome sequencing has identified chromothripsis, a complex class ofstructural genomic rearrangements involving the apparent shattering of anindividual chromosome into tens to hundreds of fragments. An initial errorduring mitosis, producing either chromosome mis-segregation into a micronu-cleus or chromatin bridge interconnecting two daughter cells, can trigger thecatastrophic pulverization of the spatially isolated chromosome. The resultantchromosomal fragments are religated in random order by DNA double-strandbreak repair during the subsequent interphase. Chromothripsis scars the can-cer genome with localized DNA rearrangements that frequently generate exten-sive copy number alterations, oncogenic gene fusion products, and/or tumorsuppressor gene inactivation. Here we review emerging mechanisms underly-ing chromothripsis with a focus on the contribution of cell division errorscaused by centromere dysfunction.
Hidden in Plain Sight: Chromothripsis in the Cancer GenomeThe karyotypes of cancer cells are often remarkably complex – littered not only with mutationsbut also small- and large-scale changes in both chromosome number and architecture. Copynumber alterations in the form of whole-chromosome or segmental aneuploidy are present inmost tumors, yet its role as a cause or consequence of cancer development remains underdebate [1,2]. Structural aberrations and gross rearrangements alter the linear organization ofchromosomes, and in some instances can directly drive tumorigenesis. The Philadelphiachromosome in chronic myelogenous leukemia is the classic example [3], involving a translo-cation between chromosomes 9 and 22 to generate an oncogenic gene fusion product that canbe effectively targeted by clinical therapeutics [4].
Although it appears firmly established that tumorigenesis develops through the gradual andsequential accumulation of genetic and/or epigenetic changes [5], recent cancer genomesequencing efforts challenged this dogmatic view by identifying several new classes ofmutational processes that form simultaneously in a single burst. Adapted from the age-oldtheory in evolution, the paradigm-shifting concept of punctuated equilibrium (see Glossary)has now been applied toward our understanding of cancer progression models, as recentlydescribed in the renewed context of pancreatic cancer [6]. These punctuated events currentlyinclude alterations such as kataegis (regions of clustered hypermutation) [7,8] and chromo-plexy (serial rearrangements linking multiple chromosomes) [9] � the underlying bases of whichare not well understood.
Among the most striking examples of punctuated equilibrium is chromothripsis (a Greekneologism for ‘chromosome shattering’), in which tens to hundreds of structural
TrendsChromothripsis is a catastrophic eventin which one or a few chromosomesare shattered and stitched backtogether in random order, producinga derivative chromosome with com-plex rearrangements within a few cellcycles.
Chromosome mis-segregation duringcell division frequently produces smallnuclear structures called micronuclei,which are prone to irreversible nuclearenvelope disruption during interphaseand impaired nucleocytoplasmiccompartmentalization.
Micronucleated chromosomes accu-mulate extensive DNA damage andare susceptible to shattering duringthe next mitosis, generating multiple,distinct DNA fragments.
Chromosome fragments are reas-sembled by DNA double-strand breakrepair to form a derivativechromosome.
Chromatin bridges trapped betweendaughter cells are attacked by a cyto-plasmic nuclease (three-prime repairexonuclease 1; TREX1) during inter-phase to generate DNA breaks andfocal chromothripsis.
1Ludwig Institute for CancerResearch, Department of Cellular andMolecular Medicine, University ofCalifornia at San Diego, La Jolla, CA92093, USA
*Correspondence:[email protected] (P. Ly) [email protected](D.W. Cleveland).
Trends in Cell Biology, December 2017, Vol. 27, No. 12 http://dx.doi.org/10.1016/j.tcb.2017.08.005 917© 2017 Elsevier Ltd. All rights reserved.

rearrangements are rapidly acquired within a short timeframe [10]. These complex rearrange-ments are curiously restricted to one or a few chromosomes, with breakpoints scattered acrossan entire chromosomal axis or clustered within a specific arm or region. Such alterations werepredicted to form de novo from massive DNA damage and repair events, leading to a derivativechromosome that shares little resemblance to its original configuration. The characteristicmutation signature for chromothripsis [11] has now been detected in a broad spectrum ofcancers, and of particular interest, at high frequency in specific tumor types – including bone,blood, and brain cancers [6,12–17]. Although the majority of chromothripsis cases occursomatically, inherited forms of germline chromothripsis have also been documented [18].Additionally, similar types of complex rearrangements have been reported in individuals withdevelopmental and/or cognitive disorders, which appear to be caused by DNA replicationerrors [19] in a process termed chromoanasynthesis. The catchall phrase chromoana-genesis (Greek for ‘to be reborn’) has been coined [20] to encompass all the possible types oflocalized and complex chromosomal rearrangements in human genomes irrespective of theirunderlying mechanism of formation.
If chromothripsis causes such drastic changes in chromosome structure and occurs frequentlyin some cancers, why had it not been discovered prior to 2011? Several types of large structuralrearrangements, such as gross translocations, can be easily identified by routine cytogeneticmethodologies involving chromosome-banding patterns or fluorescent in situ hybridization(FISH). However, submicroscopic structural variations in the range of kilobase to even mega-base rearrangements or more complex abnormalities, such as chromothripsis, can easilyescape recognition. Thus, initially missed by microscopy-based approaches, the recent adventof higher resolution next-generation DNA sequencing technologies enabled Stephens et al. [10]to identify and resolve the complexity of rearrangements characterizing chromothripsis incancer genomes.
The highly localized and complex nature of chromothripsis initially puzzled both cell biologistsand cancer geneticists, leading to a spectrum of proposed hypotheses for the underlyingmechanisms [10,20–23]. Considering that the rearrangements are often restricted to a singlechromosome, it was strongly suspected that the affected chromosome must have beenspatially isolated from the remaining genome, even if only for a transient period. DNA damagein the form of double-strand breaks (DSBs) resulting in chromosome breakage is also likelyinvolved followed by one or more mechanisms of error-prone DNA repair to produce theresulting rearrangements [10]. Many key aspects regarding the cellular mechanisms haveemerged over the last 5 years, including evidence supporting a role of cell division errors in theshattering of an initially mis-segregated chromosome [24–26]. In this review, we cover recentinsights into the mechanisms of chromothripsis with a particular focus on the role of mitoticerrors driven by centromere dysfunction. We also highlight a number of outstandingquestions.
Chromothripsis Driving TumorigenesisHow does chromothripsis contribute to cancer development? The simultaneous formation of amultiple alterations through chromothripsis can lead to the acquisition of one or more selectiveadvantages (Figure 1A). Because chromothripsis can result in both the loss of DNA segmentsand the formation of de novo rearrangements, two obvious culprits are the disruption of tumorsuppressor genes and the formation of oncogenic fusion products, respectively. Rearrange-ments formed between two normally distant loci may also juxtapose an active regulatoryelement (e.g., a promoter) adjacent or in close proximity to an otherwise repressed oncogene.Cancer genome sequencing efforts have indeed documented numerous examples of tumorsuppressor loss [10], gene fusion events [14], and perturbed regulatory elements [15,27]associated with chromothripsis in human malignancies (Box 1).
GlossaryAcentric: a chromosome, orfragment of a chromosome, thatlacks an active centromere.Centromere: a specialized region oneach chromosome designated forassembly of the kinetochore andwhose unique position is identifiedand maintained epigenetically.Chromoanagenesis: a catch-allterm that encompasses catastrophicmutational processes involving oneor a few chromosomes, independentof the precise mechanisms; includedhere are chromothripsis andchromoanasynthesis, which arisethrough distinct mechanisms.Chromoanasynthesis: complexrearrangements arising from thedefective replication of a single orfew chromosomes.Chromoplexy: a series of chained,complex rearrangements frequentlyinvolving five or more chromosomes.Chromothripsis: complexrearrangements arising from thecatastrophic shattering of a single orfew chromosomes.Dicentric: a chromosome harboringtwo active centromeres.Double minute (DM)chromosome: circular andreplication-competentextrachromosomal DNA elementsthat accumulate and amplify multiplegene copies that drive high levels ofexpression.Kataegis: clusters of focalhypermutation preferentially favoringcytosine substitutions.Micronucleus: small, nuclearstructures that encapsulate mis-segregated chromosomes and arespatially isolated from the mainnucleus.Micronuclear envelopedisruption: irreversible rupture of thenuclear envelope surrounding amicronucleus that causes abrupt lossof nucleocytoplasmic partitioning andterminates micronuclear function.Non-homologous end joining: amajor form of DNA DSB repairwhereby two damaged endsundergoes direct ligation.Punctuated equilibrium: a long-standing theory in evolutionarybiology with recent implications forcancer development, in particular therapid acquisition of a large number ofoncogenic alterations over a shorttimescale.Telomere: repetitive sequences thatprotect each of the terminal ends of
918 Trends in Cell Biology, December 2017, Vol. 27, No. 12

chromosomes from shorteningduring replication and detection bythe DNA damage response.
In certain cases [10,13,28], there is also an association between chromothripsis and geneamplification. These can be manifested as extrachromosomal DNA elements in the form ofdouble minute (DM) chromosomes, suggesting that the ends of one or more fragments canligate and circularize [25] into self-propagating entities. In the initial example from small-cell lungcancer, chromothripsis of chromosome 8 produced a megabase-long DM through the stitch-ing of 15 fragments that led to the amplification of the MYC oncogene [10]. These regions werelost from the reassembled derivative chromosome 8. Remarkably, such DMs have recentlybeen detected in nearly half of all human cancers [29] and can lead to exceptionally highexpression of the corresponding DM-located genes – typically oncogenes and/or genesconferring resistance to therapy [30]. That said, the frequency at which chromothripsis directlycontributes to the formation of DMs across varying cancer types has not been established.
Loss of the TP53 tumor suppressor gene, which can halt cell cycle progression in response toDNA damage, also appears to be a prerequisite event for chromothripsis [13]. Currentexperimental models for studying chromothripsis in human cells have thus far required deple-tion or inactivating mutations of p53 [24–26,31,32]. Bypassing the p53 checkpoint is thereforea critical step towards tolerating and overcoming the damage accompanying chromothripsis.Although most non-transformed cells experiencing chromothripsis probably do not survive, thecases exemplified in cancer genomes are likely rare exceptions that undergo clonal selectionand expansion towards cancer. Overall, one or a combination of these rearrangements, whichcollectively produce a hallmark mutation signature [11] for chromothripsis (Figure 1B), could fuelcancer development and tumor evolution through selective processes.
The Micronucleus RevisitedAt the exit of mitosis, nuclear lamins and pore complexes redeposit around newly segregatedchromosomal masses to encapsulate the genome within the nuclear envelope (NE), ultimatelyforming the cell nucleus. A chromosome that fails to correctly segregate to either of the twomitotic spindle poles, perhaps due to improper kinetochore–microtubule attachments, will
Box 1. Three Case Examples of Chromothripsis Driving Genomic Changes Associated with Cancerand Disease
Pancreatic cancer is thought to develop from an initiating clone that gradually accumulates oncogenic mutations overlong timescales to eventually acquire the capacity to disseminate to distant organs and become metastatic. However,the majority of pancreatic cancer patients are asymptomatic and not diagnosed until the tumor has reached themetastatic stage � an endpoint with extremely poor clinical outcome. This observation has challenged the conventionalstepwise paradigm of pancreatic cancer progression and instead raises an alternative model: the rapid acceleration ofearly pancreatic lesions toward metastatic disease occurs through punctuated events that simultaneously drive multipleoncogenic changes. Sequencing of 107 pancreatic cancer genomes revealed that, along with genome duplicationevents (polyploidy), chromothripsis affecting at least one chromosome was evident in two-thirds of examined cases [6].Multiple chromosomes characterized by the signatures of chromothripsis were also found, which concurrentlyinactivated several classic pancreatic driver genes, including CDKN2A, SMAD4, and TP53. Parallel events involvingpresumptive breakage–fusion–bridge cycles also triggered further genomic complexity, including the amplification ofmutated KRAS alleles.
Whole-genome sequencing analyses of nine supratentorial ependymomas, a type of brain and spinal cord tumor,strikingly revealed chromothripsis affecting chromosome 11 in all nine cases examined [14]. In eight cases, complexrearrangements generated a fusion of the oncogenic RELA gene with an uncharacterized C11orf95 gene, which arenormally located �2 megabases apart and separated by 73 genes on chromosome 11. Expression of RELA–C11orf95fusions in neural stem cells implanted into the cerebellum of mice resulted in a marked increase in brain tumors,providing evidence that gene fusion products created through chromothripsis can be highly oncogenic in nature.
In one remarkable medical case, a patient with a rare congenital immunodeficiency disorder called WHIM syndrome wasserendipitously cured through somatic chromothripsis of chromosome 2 [72]. These rearrangements resulted in theloss of a disease-causative, gain-of-function mutant allele of the CXCR4 gene within a hematopoietic stem cell clonethat was capable of repopulating the myeloid lineage and restoring normal neutrophil count.
Trends in Cell Biology, December 2017, Vol. 27, No. 12 919

produce a micro-NE assembled around the lagging chromosome. The resultant micronu-cleus spatially isolates one or sometimes few mis-segregated chromosomes into a smallnucleus-like structure that is positioned outside of the adjacent primary nucleus (Figure 2).Micronuclei are therefore a consequence of chromosome segregation errors during mitosis.
Accumulating evidence has established micronuclei as a unique source of genomic instabilityand DNA damage. The initial proposal for this emerged in 1968 when Kato and Sandbergdetermined that chromosomes in micronuclei undergo pulverization in mitosis after failing tocomplete DNA replication prior to mitotic entry [33]. Subsequent cell fusion studies betweencells in S phase and mitosis demonstrated that actively replicating chromosomes become
Figure 1. Genomic and Tumorigenic Consequences of Chromothripsis. (A) The shattering of an individualchromosome can produce tens to hundreds of acentric DNA fragments that persist as intermediates until they are religatedand stabilized by intrinsic DNA repair mechanisms. These fragments reassemble to form a scrambled, derivativechromosome containing multiple rearrangements (chromothripsis), become lost, and/or self-ligate into circular DNAstructures called double minutes. (B) Chromothriptic events can give rise to a characteristic mutation signature that hasbeen detected in a broad range of cancer genomes, including oscillating copy number states and complex patterns ofintrachromosomal rearrangements in apparently random fashion.
920 Trends in Cell Biology, December 2017, Vol. 27, No. 12

fragmented upon premature chromosome condensation caused by exposure to a mitoticcytoplasm [34].
More than 40 years later, the discovery of chromothripsis in 2011 [10] has reawakenedwidespread interest in what is now recognized as the distinct biology of micronuclei. Consistentwith the proposal by Kato and Sandberg, more recent evidence supports that micronuclearchromosomes do in fact acquire DNA damage and/or exhibit delayed replication kineticscompared to the main nucleus [24,35–37]. Additionally, elegant use of live-cell imagingcombined with single-cell DNA sequencing (a technique called Look-Seq) detected thepresence of multiple, aberrantly ligated DNA fragments in the daughter cells that arise froma previously micronucleated chromosome in a dividing mother cell [25]. These ligation eventsproduced complex patterns of localized rearrangements that are highly reminiscent of cancer-associated chromothripsis, although whether an actual derivative chromosome was producedhas not been established. Indeed, the experimental creation of a micronucleus-derived chro-mothriptic chromosome capable of stable transmission in subsequent cell cycles and thatinclude the defining features of a fully functional chromosome has not yet been accomplishedand represents a critical next step forward.
Similar mechanisms of chromosome shattering and religation also appear to be present ingenetic plant [38] and fission yeast [39] models of chromosome mis-segregation. In Arabi-dopsis models, apparent micronuclei have been reported to produce complex rearrangements[38] that are strikingly similar to chromothripsis observed in human cancer genomes, implicat-ing chromothripsis as a potentially conserved consequence of mitotic errors across evolution.Interestingly and along similar lines, protozoan ciliates such as Tetrahymena carry both amacronucleus and a micronucleus, the latter containing germline chromosomes that undergodeliberate DNA fragmentation and extensive rearrangements for subsequent conversion intothe macronucleus [40].
Sources of Micronuclear DNA DamageIn mammalian cells, why might micronuclear chromosomes acquire DNA damage in the form ofDSBs? As micronucleated cells progress through interphase (Figure 3), the micro-NE has a
Figure 2. Chromosome Segregation Errors during Mitotic Cell Division Can Entrap DNA within a Micro-nucleus. During mitosis, a chromosome that fails to congress, align, and/or form proper bipolar spindle microtubule–kinetochore attachments prior to anaphase onset can be left behind during the physical separation of the duplicatedgenome. A nuclear envelope assembles around the mis-segregated chromosome, subsequently forming a micronucleusat the exit of mitosis.
Trends in Cell Biology, December 2017, Vol. 27, No. 12 921
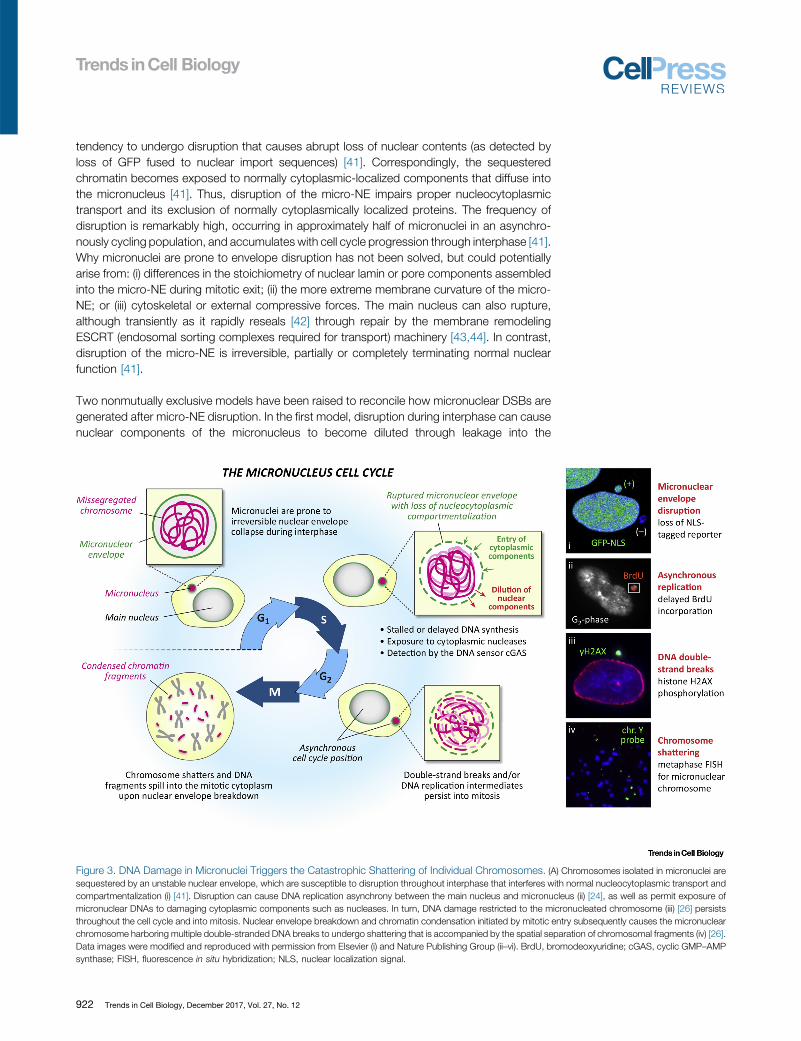
tendency to undergo disruption that causes abrupt loss of nuclear contents (as detected byloss of GFP fused to nuclear import sequences) [41]. Correspondingly, the sequesteredchromatin becomes exposed to normally cytoplasmic-localized components that diffuse intothe micronucleus [41]. Thus, disruption of the micro-NE impairs proper nucleocytoplasmictransport and its exclusion of normally cytoplasmically localized proteins. The frequency ofdisruption is remarkably high, occurring in approximately half of micronuclei in an asynchro-nously cycling population, and accumulates with cell cycle progression through interphase [41].Why micronuclei are prone to envelope disruption has not been solved, but could potentiallyarise from: (i) differences in the stoichiometry of nuclear lamin or pore components assembledinto the micro-NE during mitotic exit; (ii) the more extreme membrane curvature of the micro-NE; or (iii) cytoskeletal or external compressive forces. The main nucleus can also rupture,although transiently as it rapidly reseals [42] through repair by the membrane remodelingESCRT (endosomal sorting complexes required for transport) machinery [43,44]. In contrast,disruption of the micro-NE is irreversible, partially or completely terminating normal nuclearfunction [41].
Two nonmutually exclusive models have been raised to reconcile how micronuclear DSBs aregenerated after micro-NE disruption. In the first model, disruption during interphase can causenuclear components of the micronucleus to become diluted through leakage into the
Figure 3. DNA Damage in Micronuclei Triggers the Catastrophic Shattering of Individual Chromosomes. (A) Chromosomes isolated in micronuclei aresequestered by an unstable nuclear envelope, which are susceptible to disruption throughout interphase that interferes with normal nucleocytoplasmic transport andcompartmentalization (i) [41]. Disruption can cause DNA replication asynchrony between the main nucleus and micronucleus (ii) [24], as well as permit exposure ofmicronuclear DNAs to damaging cytoplasmic components such as nucleases. In turn, DNA damage restricted to the micronucleated chromosome (iii) [26] persiststhroughout the cell cycle and into mitosis. Nuclear envelope breakdown and chromatin condensation initiated by mitotic entry subsequently causes the micronuclearchromosome harboring multiple double-stranded DNA breaks to undergo shattering that is accompanied by the spatial separation of chromosomal fragments (iv) [26].Data images were modified and reproduced with permission from Elsevier (i) and Nature Publishing Group (ii–vi). BrdU, bromodeoxyuridine; cGAS, cyclic GMP–AMPsynthase; FISH, fluorescence in situ hybridization; NLS, nuclear localization signal.
922 Trends in Cell Biology, December 2017, Vol. 27, No. 12

cytoplasm. Disruption prior to or during S phase can delay active replication fork progression,slowing or completely stalling DNA synthesis [24,33,37,41], which itself may act as a source ofDNA damage owing to replication stress [45]. Disruption has been suggested to be a criticalstep for chromothriptic-like rearrangement signatures arising from micronuclei [25], whichaccumulate to peak frequency during S and G2 phases of the cell cycle [41]. The persistenceof a large number of unrepaired DNA DSBs and/or unresolved replication intermediates couldbe catastrophic upon the dramatic remodeling of chromatin organization that occurs duringmitosis, in particular the condensation of chromosomes that is required to facilitate the spatialseparation of DNA.
The second model involves aberrant exposure of the micronuclear chromosome to one or morepotentially damaging components from the cytoplasm. Upon disruption of the micro-NE,influxes of cytosol-localized proteins into the micronucleus have been observed (as detectedby the inclusion of GFP fused to nuclear export sequences). Recent evidence [43,44] suggeststhat transient NE rupture of the main nucleus triggers the recruitment of the cytosolic DNAsensor cyclic GMP–AMP synthase (cGAS) [46], which has evolved to detect foreign andpathogenic DNAs in the cytoplasm to activate the innate immune response [47]. Duringinterphase, cGAS can be rapidly detected at sites of NE rupture at the nuclear periphery,which is subsequently followed by the focal acquisition of DNA damage [43,44]. Similarrupturing events were reported to promote genomic copy number aberrations [48], and cGAShas been shown to associate with condensed chromosomes after NE breakdown duringmitosis [49]. Consistent with this, recent work confirmed that cGAS could indeed sensemicronuclear DNAs exposed to the cytoplasm following micro-NE disruption as cytosolicself-DNA [50], although the downstream consequences on the micronuclear chromosomeitself are unknown.
Harmful cytoplasmic components could include endo- or exonucleases whose localization istightly regulated or that are activated upon recognition of specific DNA intermediate structuresarising from replication stress. MUS81 is one potential nuclease given its role in inducing DSBsat replication stress-induced late-replicating loci, which activates DNA polymerase deltasubunit 3 (POLD3)-dependent mitotic DNA synthesis to safeguard against the formation ofultrafine anaphase bridges [51]. DNA synthesis errors during mitosis could be one source ofchromosomal rearrangements arising from micronuclei that undergo mitotic entry with partiallyreplicated DNA. Altogether, several mechanisms acting in parallel likely converge to promotethe massive DNA damage associated with chromothripsis in micronuclei.
One Centromere Too Few: Chromothripsis Driven by CentromereInactivationSeveral experimental approaches have been used to investigate the properties of micronucleiand the eventual fate of the encapsulated chromosome [52]. One widely used method togenerate micronuclei is through prolonged mitotic arrest using microtubule inhibitors, such asnocodazole, followed by release and subsequent mis-segregation of one or few randomchromosomes. An alternative experimental approach was recently developed involving theinactivation of a specific centromere (Box 2) to induce micronuclei containing a definedchromosome of interest (the human Y chromosome) – a strategy that has identified thetemporal sequence of chromothriptic events over several consecutive cell cycles [26].
Centromere inactivation initiates a series of events beginning with chromosome mis-segrega-tion into micronuclei at the end of the first cell cycle. DNA damage accumulates withinmicronuclei, triggering chromosome shattering during mitosis of the second cell cycle. Chro-mosome fragments are subsequently reassembled throughout interphase of the third cellcycle. Analysis of metaphase spreads by FISH has provided direct evidence supporting
Trends in Cell Biology, December 2017, Vol. 27, No. 12 923

micronucleus-mediated chromosome shattering, revealing >50 microscopic chromosomalfragments dispersed across the mitotic cytoplasm – all but one or a few of which lackedcentromeric DNA sequences [26]. Chromosome shattering is likely an intermediate stage forchromothripsis (Figure 1A) as the majority of acentric fragments would be unstable long-termand lost. With the exception of circularized extrachromosomal DMs, reassembly would berequired to stabilize acentric fragments to at least one fragment containing an active centro-mere to form a derivative chromosome that can be genetically inherited.
Bringing It All Back Home: Reassembly through DNA RepairMost DNA fragments produced by chromosome shattering are acentric [26], which alone areincapable of attaching to the mitotic spindle. The resulting fragments are therefore, at best,passively distributed, with likely asymmetric partitioning into newly formed daughter cells andreintegration into the main nucleus if they are in close proximity to either of the poleward-segregating chromosome masses (Figure 4A). The ends of these fragments are presumablyrecognized as DNA DSBs in the subsequent G1, thereby activating the DNA damage response(Figure 4B). Indeed, suppressing classical non-homologous end joining (c-NHEJ) (Box 3) bydepleting or inhibiting the activity of DNA ligase 4 (LIG4) or DNA-dependent protein kinase,catalytic subunit (DNA-PKcs) prevented micronuclei-derived fragments to undergo repair [26],evidence supporting that c-NHEJ is the predominant mechanism for reassembling chromo-somal fragments. LIG4 also appears to be required for repairing mis-segregated chromosomesin genetic plant models [38]. Chromosomal translocations in human cells are mediated primarilythrough c-NHEJ, although murine cells appear to favor alternative-EJ (alt-EJ) [53,54].
Figure 4. DNA Damage Repair Mechanisms Contributing to the Reassembly of Fragmented Chromosomes. (A) Chromosome fragments produced bychromothripsis spill into the mitotic cytoplasm and are subsequently incorporated into newly formed daughter cell nuclei at the exit of mitosis, possibly through thephysical tethering between fragments and/or onto intact, centromere-containing chromosomes. (B) In the next interphase, reintegrated fragments activate the DNAdamage response. In the absence of functional p53, DNA DSB repair ensues through error-prone non-homologous end joining, which directly links multiple fragmentstogether in a haphazard manner by ligation. The reassembled chromosome is characterized by extensive DNA rearrangements harboring de novo breakpoint junctionsthat carry the signatures of the underlying DNA repair mechanism. DNA-PK, DNA-dependent protein kinase; DNA-PKcs, DNA-PK, catalytic subunit; DSB, double-strand break; LIG4, DNA ligase 4; XRCC4, X-ray repair cross-complementary protein 4.
Box 2. Epigenetic Maintenance and Function of the Centromere
Centromeres are unique chromosomal loci that establish assembly of the kinetochore, a large multiprotein complex thatdirectly facilitates chromosome movement and segregation by attachment to spindle microtubules during mitosis.Although most human centromeres are found on repetitive a-satellite DNA sequences, these sequences are neithersufficient nor necessary for centromere formation, maintenance, or function, as evident by the discovery of mitoticallystable neocentromeres formed at distinct loci located on chromosome arms [73–75]. Instead, the position of eachcentromere is specified epigenetically by the histone H3 variant CENP-A [76], which self-templates its own propagationevery cell cycle (reviewed in [77]). These epigenetic mechanisms act to ensure that one, and strictly one, centromere isactive per chromosome to safeguard against genomic instability.
924 Trends in Cell Biology, December 2017, Vol. 27, No. 12

Sequence analysis of the breakpoint junctions from chromothriptic tumors [10] and experi-mental models of chromothripsis [25,38] has revealed that a large proportion of breakpoints(but not all, as discussed below) lack significant homology or microhomology � consistent withthe repair signature of c-NHEJ.
Although DNA DSBs accumulate in micronuclei following rupture in interphase, the resultantbreaks are likely not subjected to DNA repair as micro-NE disruption would cause dilution ofcomponents involved in theDNAdamage response [41]. Micronuclei with phosphorylated histonegH2AX, a marker of DNA DSBs, often failed to recruit or retain detectable levels of the DNA repairfactor 53BP1 [35]. As micronucleated cells enter mitosis, a fraction of micronuclei also seeminglyfail to disassemble the micro-NE and persist into the next interphase [24,55]. However, high-throughput sequencing of purified micronuclear DNAs demonstrated that extensive rearrange-ments do not accumulate to detectable levels within micronuclei [26], further indicating that mostreassembly events occur in the main nucleus following fragment reintegration.
Several lines of evidence suggest that nuclear ligation of fragmented DNA ensues rapidly withefficient repair kinetics. Analysis by Look-Seq of daughter cell pairs revealed that a highproportion of the fragments were ligated within �4 h after mitotic exit [25], most likely duringG1. In 3 out of 8 examples, fragments that were apparently lost from one daughter cell werefound ligated within the paired daughter cell [25], which reflects the oscillating regions of copynumber loss characteristic of chromothripsis [10,11]. Additionally, use of the Y centromere-specific inactivation strategy demonstrated that inhibiting c-NHEJ produced chromosomefragments that persist into the next mitosis [26], indicating that the duration of a single cellcycle is sufficient for complete (or nearly complete) fragment reassembly.
How chromosomal fragments are inherited between daughter cells during mitosis remains akey question. Spindle forces and motor proteins drive chromosome movement during mitosis,but these do not engage chromosome fragments lacking a centromere. In Drosophila neuro-blasts, acentric chromosomal fragments have been reported to partially segregate poleward[56] through kinetochore-independent microtubules and the chromokinesin Klp3a [57], whichshares similarity to human KIF4A. Alternative models include the topological linkage or tetheringof chromosome fragments to each other, as suggested by 5 out of 8 Look-Seq examples inwhich the majority of fragments were unequally distributed to a single daughter [25], or perhapsonto other chromosomes; a mechanism analogous to the proposal that extrachromosomal
Box 3. Repair Mechanisms for DNA DSBs
DNA DSBs are repaired by two primary mechanisms in mammalian cells. In the first, homologous recombination (HR)utilizes long tracts of homologous sequence as a template to repair DSBs and is most active during S and G2 phases ofthe cell cycle. In the second, NHEJ directly joins two DSBs together without use of long sequence homology and istherefore a more error-prone repair pathway.
There are at least two recognized subtypes of NHEJ that mechanistically operates through distinct components andpathways: classical NHEJ (c-NHEJ) and the less-characterized alt-EJ pathway (recently reviewed in [78,79]). c-NHEJfunctions through a signaling cascade involving DNA-PK, a heterotrimeric complex composed of DNA-PKcs, Ku70, andKu80 subunits, with direct end joining activity mediated by LIG4–XRCC4. Because end processing is minimal, repair byc-NHEJ can produce 0–4 bp of microhomology at junction sequences; most of which occurs by chance. In contrast,repair by alt-EJ does not require the components involved in c-NHEJ.
Microhomology-mediated end joining (MMEJ) is the major form of alt-EJ (reviewed in [80,81]). MMEJ requires an initialresection step mediated by the MRN (Mre11–Rad50–Nbs1) complex and CtIP (RBBP8), a step shared with HR,followed by a search for microhomologous sequences between the two resected ends. Once aligned, the ends aresubjected to fill-in synthesis by the DNA polymerase Polu and ligation by DNA ligase 3 (LIG3). Breakpoint junctionsrepaired by MMEJ contain scars of 3–8 bp of microhomology (and up to 20 bp) that are usually accompanied by smalldeletions.
Trends in Cell Biology, December 2017, Vol. 27, No. 12 925

DNAs (including DMs or viral episomes) could tether and segregate in trans with centromere-containing chromosomes [58–60]. Recent evidence suggests that the c-NHEJ componentsXRCC4–XLF (X-ray repair cross-complementing protein 4 and XRCC4-like factor) can physi-cally bridge two DSB ends prior to ligation [61]. Although DNA repair is normally suppressedthroughout mitosis to prevent telomere fusions [62,63], whether this or other tetheringmechanisms are involved in maintaining chromosome fragments in close spatial proximityuntil segregation and/or ligation remains unresolved.
One Centromere Too Many: Chromothripsis Driven by DicentricChromosomesIn certain instances, a single chromosome can harbor two active centromeres that are capableof attaching to the mitotic spindle. These dicentric chromosomes can be formed throughseveral mechanisms. A neocentromere can spontaneously form at a non-centromeric regionon the chromosome arm; an event that naturally occurs at a rare frequency through poorlydefined mechanisms (Box 2). Most often and perhaps by telomere shortening, a dicentric canbe produced by an end-to-end fusion event between the telomeres of two non-homologouschromosomes (chromosome-type fusion) or between sister chromatids (chromatid-typefusion). In the latter, a pseudodicentric chromosome is formed in which the sister centromeresare properly attached to the opposing spindle poles in mitosis but remain linked at the fusedend during chromatid separation at anaphase. Lastly, dicentric chromosomes can also resultfrom the fusion of two chromosome fragments that each contains an active centromere [64].Regardless of the mechanism of dicentric chromosome formation, a chromatin bridge is usuallyformed during mitosis that can persist until, or even long after, cytokinesis (Figure 5A).
Recent efforts identified that dicentric chromosomes developing into chromatin bridges duringlate mitosis could act as another source of chromothripsis [32]. Anaphase chromatin bridges werecreated using an established method to induce chromosome-type telomere fusions [65] throughexpression of a dominant-negative mutant of the telomere-associated shelterin componenttelomeric repeat-binding factor 2 (TRF2) [66]. Following the completion of an apparently normalcytokinesis, the chromatin bridge remains intact and interconnected between the two newlyformed daughter cells throughout early interphase. As the daughters migrate away from oneanother, the NEsurrounding the bridged DNA undergoes rupturingduring G1that isaccompaniedby the acquisition of the single-stranded DNA binding protein replication protein A (RPA). Thisrupture mediates access of the cytoplasmic 30 exonuclease TREX1 to the exposed and stretchedDNA, driving cleavage and ultimate resolution of the chromatin bridge (Figure 5B). Homozygousdeletions of TREX1, however, partially delays but does not completely prevent eventual bridgeresolution, suggesting possible roles of other nucleases and/or physical mechanisms responsiblefor the breakage of chromatin bridges. Sequencing of clones revealed chromothriptic-like rear-rangements involving the fused chromosomes [32] through an unidentified DNA repair mecha-nism. Consistent with a role for telomere fusions in chromothripsis, depletion of TRF2 to uncaptelomeric ends in non-cancerous cells followed by selection for partially transformed cells alsogenerated chromothriptic-like signatures [31]. Combinations of chromothripsis with breakage–fusion–bridge cycles (Figure 5C) can add another layer of complexity to the mutation signaturesassociated with cancer genomes [6,64,67].
How do chromothripsis and rearrangement patterns generated by chromatin bridges differfrom those arising through micronuclei? Whereas replication defects have been proposed to bean important component of the DNA damage associated with the micronucleus model,chromothripsis from telomere fusions likely do not require ongoing DNA synthesis. Rather,the fragmentation of the bridged DNA is dependent on its resolution by TREX1, whichfrequently occurs prior to S-phase entry [32]. Additionally, multiple examples from cancergenomes have been documented in which chromothriptic rearrangements were focally
926 Trends in Cell Biology, December 2017, Vol. 27, No. 12
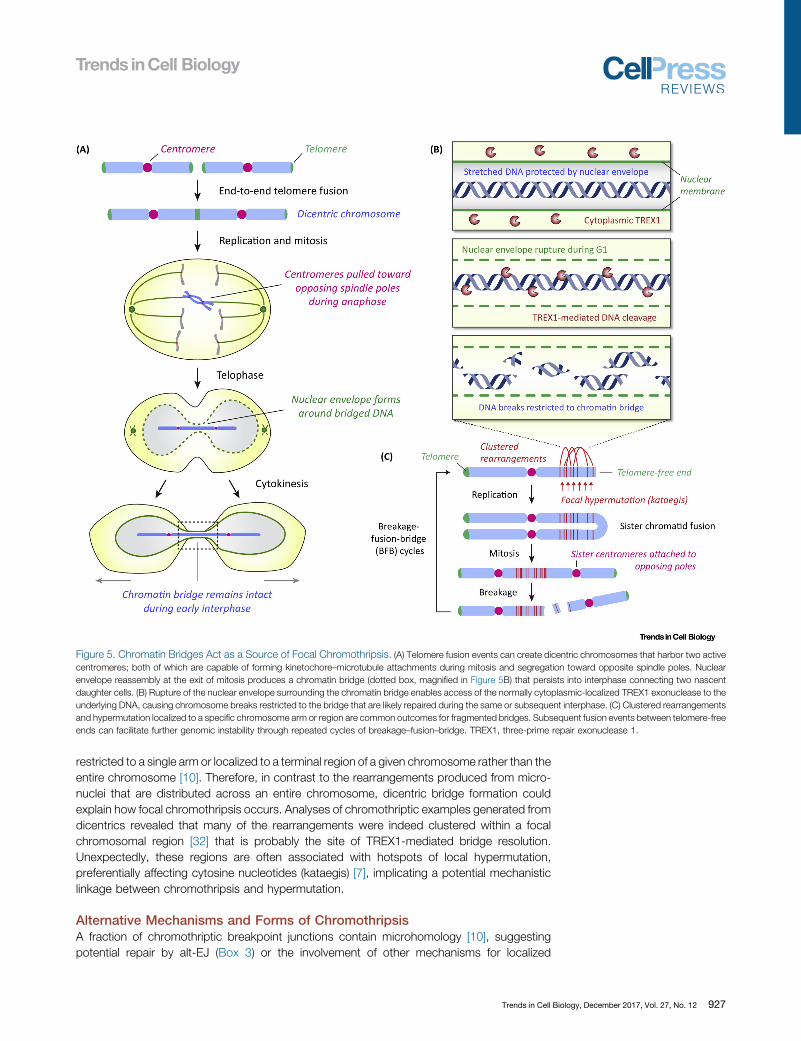
restricted to a single arm or localized to a terminal region of a given chromosome rather than theentire chromosome [10]. Therefore, in contrast to the rearrangements produced from micro-nuclei that are distributed across an entire chromosome, dicentric bridge formation couldexplain how focal chromothripsis occurs. Analyses of chromothriptic examples generated fromdicentrics revealed that many of the rearrangements were indeed clustered within a focalchromosomal region [32] that is probably the site of TREX1-mediated bridge resolution.Unexpectedly, these regions are often associated with hotspots of local hypermutation,preferentially affecting cytosine nucleotides (kataegis) [7], implicating a potential mechanisticlinkage between chromothripsis and hypermutation.
Alternative Mechanisms and Forms of ChromothripsisA fraction of chromothriptic breakpoint junctions contain microhomology [10], suggestingpotential repair by alt-EJ (Box 3) or the involvement of other mechanisms for localized
Figure 5. Chromatin Bridges Act as a Source of Focal Chromothripsis. (A) Telomere fusion events can create dicentric chromosomes that harbor two activecentromeres; both of which are capable of forming kinetochore–microtubule attachments during mitosis and segregation toward opposite spindle poles. Nuclearenvelope reassembly at the exit of mitosis produces a chromatin bridge (dotted box, magnified in Figure 5B) that persists into interphase connecting two nascentdaughter cells. (B) Rupture of the nuclear envelope surrounding the chromatin bridge enables access of the normally cytoplasmic-localized TREX1 exonuclease to theunderlying DNA, causing chromosome breaks restricted to the bridge that are likely repaired during the same or subsequent interphase. (C) Clustered rearrangementsand hypermutation localized to a specific chromosome arm or region are common outcomes for fragmented bridges. Subsequent fusion events between telomere-freeends can facilitate further genomic instability through repeated cycles of breakage–fusion–bridge. TREX1, three-prime repair exonuclease 1.
Trends in Cell Biology, December 2017, Vol. 27, No. 12 927

rearrangements that are independent of chromosome shattering events, such as chromoa-nasynthesis. Mechanisms that have been proposed to contribute to the complex structuralrearrangements defined by chromoanasynthesis include errors in DNA replication, mostnotably aberrant DNA template switching at stalled forks (called fork stalling and templateswitching, or FoSTeS) or collapsed/broken forks (called microhomology-mediated break-induced replication, or MMBIR) [68,69]. In individuals with inherited genomic disorders, iterativerounds of such replication-based events could produce catastrophic rearrangements that areaccompanied by templated insertions, duplications, and/or microhomology at the sequencejunctions [19]. Such complex rearrangements can be reminiscent of, but distinct from, chro-mothripsis through chromosome shattering and fragment religation [70,71]. Interestingly, a fewexamples of micronuclei-derived rearrangements also exhibited sequence features at junctionsthat share similarities with chromoanasynthesis, in particular small insertions and the presenceof microhomology [25], suggesting possible overlapping mechanisms between chromothripsisand chromoanasynthesis.
Concluding RemarksThe discovery of chromothripsis has advanced our understanding of the complexities associ-ated with cancer genomes, as well as opened exciting new avenues for research. Thedevelopment of novel cell biological tools combined with computational methods to examinethe fate and sequence characteristics of mis-segregated chromosomes has recently contrib-uted to defining the mechanisms underlying chromothripsis. Much remains to be determined(see Outstanding Questions); in particular, the exact causes of NE disruption and DNA damagein micronuclei. Key among these questions is what does it take to assemble a fully functionaland heritable human chromosome, and how is this achieved following catastrophic processessuch as the shattering of an individual chromosome? Whether other unidentified types ofpunctuated equilibrium-driven chromosomal alterations are present in cancer or diseaseremains to be seen, but advancements in DNA sequencing technologies will likely enablefurther discovery of more unexpected and hidden features of the human genome.
Disclaimer StatementThe authors declare no conflicts of interest.
AcknowledgmentsWe thank M. Hetzer, E. Hatch, and D. Pellman for sharing original data images. This work was supported by grants from
the National Institutes of Health (R35 GM122476 to D.W.C. and K99 CA218871 to P.L.) and the Hope Funds for Cancer
Research (HFCR-14-06-06 to P.L.). D.W.C. receives salary support from the Ludwig Institute for Cancer Research.
References1. Weaver, B.A. and Cleveland, D.W. (2008) The aneuploidy para-
dox in cell growth and tumorigenesis. Cancer Cell 14, 431–433
2. Sheltzer, J.M. and Amon, A. (2011) The aneuploidy paradox:costs and benefits of an incorrect karyotype. Trends Genet.27, 446–453
3. Nowell, P.C. and Hungerford, D.A. (1960) Minute chromosome inhuman chronic granulocytic leukemia. Science 132, 1497
4. Druker, B.J. et al. (2001) Efficacy and safety of a specific inhibitorof the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N.Engl. J. Med. 344, 1031–1037
5. Fearon, E.R. and Vogelstein, B. (1990) A genetic model forcolorectal tumorigenesis. Cell 61, 759–767
6. Notta, F. et al. (2016) A renewed model of pancreatic cancerevolution based on genomic rearrangement patterns. Nature538, 378–382
7. Nik-Zainal, S. et al. (2012) Mutational processes molding thegenomes of 21 breast cancers. Cell 149, 979–993
8. Alexandrov, L.B. et al. (2013) Signatures of mutational processesin human cancer. Nature 500, 415–421
9. Baca, S.C. et al. (2013) Punctuated evolution of prostate cancergenomes. Cell 153, 666–677
10. Stephens, P.J. et al. (2011) Massive genomic rearrangementacquired in a single catastrophic event during cancer develop-ment. Cell 144, 27–40
11. Korbel, J.O. and Campbell, P.J. (2013) Criteria for inference ofchromothripsis in cancer genomes. Cell 152, 1226–1236
12. Molenaar, J.J. et al. (2012) Sequencing of neuroblastoma iden-tifies chromothripsis and defects in neuritogenesis genes. Nature483, 589–593
13. Rausch, T. et al. (2012) Genome sequencing of pediatric medul-loblastoma links catastrophic DNA rearrangements with TP53mutations. Cell 148, 59–71
14. Parker, M. et al. (2014) C11orf95-RELA fusions drive oncogenicNF-kappaB signalling in ependymoma. Nature 506, 451–455
15. George, J. et al. (2015) Comprehensive genomic profiles of smallcell lung cancer. Nature 524, 47–53
16. Scarpa, A. et al. (2017) Whole-genome landscape of pancreaticneuroendocrine tumours. Nature 543, 65–71
Outstanding QuestionsWhat are the contributing factors thatpredispose micronuclei to undergonuclear envelope disruption duringinterphase?
What are the underlying sources ofDNA damage in a micronucleus follow-ing the disruption of its nuclearenvelope?
How are chromosomal fragments seg-regated between nascent daughtercells during mitosis, and are thesefragments topologically or physicallytethered to facilitate segregation enmasse?
Does the nuclear reintegration of chro-mosomal fragments activate the DNAdamage response to engage DNArepair, and if so, how are these frag-ments spatially positioned within theinterphase nucleus to promote efficientreassembly?
At what frequency do reassembledfragments form a fully functional chro-mosome that is capable of long-terminheritance?
Do chromothripsis and chromoana-synthesis share similar underlyingmechanisms?
What other types of complex DNAalterations caused by punctuatedevents exist in the genomes of individ-uals suffering from diseases ordisorders?
928 Trends in Cell Biology, December 2017, Vol. 27, No. 12

17. Fraser, M. et al. (2017) Genomic hallmarks of localized, non-indolent prostate cancer. Nature 541, 359–364
18. Kloosterman, W.P. et al. (2011) Chromothripsis as a mechanismdriving complex de novo structural rearrangements in the germ-line. Hum. Mol. Genet. 20, 1916–1924
19. Liu, P. et al. (2011) Chromosome catastrophes involve replicationmechanisms generating complex genomic rearrangements. Cell146, 889–903
20. Holland, A.J. and Cleveland, D.W. (2012) Chromoanagenesis andcancer: mechanisms and consequences of localized, complexchromosomal rearrangements. Nat. Med. 18, 1630–1638
21. Maher, C.A. and Wilson, R.K. (2012) Chromothripsis and humandisease: piecing together the shattering process. Cell 148, 29–32
22. Forment, J.V. et al. (2012) Chromothripsis and cancer: causesand consequences of chromosome shattering. Nat. Rev. Cancer12, 663–670
23. Jones, M.J. and Jallepalli, P.V. (2012) Chromothripsis: chromo-somes in crisis. Dev. Cell 23, 908–917
24. Crasta, K. et al. (2012) DNA breaks and chromosome pulveriza-tion from errors in mitosis. Nature 482, 53–58
25. Zhang, C.Z. et al. (2015) Chromothripsis from DNA damage inmicronuclei. Nature 522, 179–184
26. Ly, P. et al. (2017) Selective Y centromere inactivation triggerschromosome shattering in micronuclei and repair by non-homol-ogous end joining. Nat. Cell Biol. 19, 68–75
27. Peifer, M. et al. (2015) Telomerase activation by genomic rear-rangements in high-risk neuroblastoma. Nature 526, 700–704
28. Cheng, C. et al. (2016) Whole-genome sequencing revealsdiverse models of structural variations in esophageal squamouscell carcinoma. Am. J. Hum. Genet. 98, 256–274
29. Turner, K.M. et al. (2017) Extrachromosomal oncogene amplifi-cation drives tumour evolution and genetic heterogeneity. Nature543, 122–125
30. Nathanson, D.A. et al. (2014) Targeted therapy resistance medi-ated by dynamic regulation of extrachromosomal mutant EGFRDNA. Science 343, 72–76
31. Mardin, B.R. et al. (2015) A cell-based model system links chro-mothripsis with hyperploidy. Mol. Syst. Biol. 11, 828
32. Maciejowski, J. et al. (2015) Chromothripsis and kataegis inducedby telomere crisis. Cell 163, 1641–1654
33. Kato, H. and Sandberg, A.A. (1968) Chromosome pulverization inhuman cells with micronuclei. J. Natl. Cancer Inst. 40, 165–179
34. Johnson, R.T. and Rao, P.N. (1970) Mammalian cell fusion:induction of premature chromosome condensation in interphasenuclei. Nature 226, 717–722
35. Terradas, M. et al. (2009) DNA lesions sequestered in micronucleiinduce a local defective-damage response. DNA Repair (Amst.) 8,1225–1234
36. Xu, B. et al. (2011) Replication stress induces micronuclei com-prising of aggregated DNA double-strand breaks. PLoS One 6,e18618
37. Okamoto, A. et al. (2012) DNA replication occurs in all laminapositive micronuclei, but never in lamina negative micronuclei.Mutagenesis 27, 323–327
38. Tan, E.H. et al. (2015) Catastrophic chromosomal restructuringduring genome elimination in plants. eLife 4, e06516
39. Sabatinos, S.A. et al. (2015) Replication stress in early S phasegenerates apparent micronuclei and chromosome rearrange-ment in fission yeast. Mol. Biol. Cell 26, 3439–3450
40. Prescott, D.M. (1994) The DNA of ciliated protozoa. Microbiol.Rev. 58, 233–267
41. Hatch, E.M. et al. (2013) Catastrophic nuclear envelope collapsein cancer cell micronuclei. Cell 154, 47–60
42. Vargas, J.D. et al. (2012) Transient nuclear envelope rupturingduring interphase in human cancer cells. Nucleus 3, 88–100
43. Denais, C.M. et al. (2016) Nuclear envelope rupture and repairduring cancer cell migration. Science 352, 353–358
44. Raab, M. et al. (2016) ESCRT III repairs nuclear envelope rupturesduring cell migration to limit DNA damage and cell death. Science352, 359–362
45. Gaillard, H. et al. (2015) Replication stress and cancer. Nat. Rev.Cancer 15, 276–289
46. Sun, L. et al. (2013) Cyclic GMP-AMP synthase is a cytosolic DNAsensor that activates the type I interferon pathway. Science 339,786–791
47. Margolis, S.R. et al. (2017) Evolutionary origins of cGAS-STINGsignaling. Trends Immunol. Published online April 14, 2017.http://dx.doi.org/10.1016/j.it.2017.03.004
48. Irianto, J. et al. (2017) DNA damage follows repair factor depletionand portends genome variation in cancer cells after pore migra-tion. Curr. Biol. 27, 210–223
49. Yang, H. et al. (2017) cGAS is essential for cellular senescence.Proc. Natl. Acad. Sci. U. S. A. 114, E4612–E4620
50. Mackenzie, K.J. et al. (2017) cGAS surveillance of micronucleilinks genome instability to innate immunity. Nature 548, 461–465
51. Minocherhomji, S. et al. (2015) Replication stress activates DNArepair synthesis in mitosis. Nature 528, 286–290
52. Ly, P. and Cleveland, D.W. (2017) Interrogating cell division errorsusing random and chromosome-specific missegregationapproaches. Cell Cycle 16, 1252–1258
53. Ghezraoui, H. et al. (2014) Chromosomal translocations in humancells are generated by canonical nonhomologous end-joining.Mol. Cell 55, 829–842
54. Biehs, R. et al. (2017) DNA double-strand break resection occursduring non-homologous end joining in G1 but is distinct fromresection during homologous recombination. Mol. Cell 65 (671–684), e675
55. Huang, Y. et al. (2012) Lagging chromosomes entrapped inmicronuclei are not ‘lost’ by cells. Cell Res. 22, 932–935
56. Royou, A. et al. (2010) BubR1- and Polo-coated DNA tethersfacilitate poleward segregation of acentric chromatids. Cell 140,235–245
57. Karg, T. et al. (2017) The chromokinesin Klp3a and microtubulesfacilitate acentric chromosome segregation. J. Cell Biol. 216,1597–1608
58. Kanda, T. et al. (2001) Coupling of mitotic chromosome tetheringand replication competence in Epstein–Barr virus-based plas-mids. Mol. Cell Biol. 21, 3576–3588
59. Kanda, T. et al. (2001) Mitotic segregation of viral and cellularacentric extrachromosomal molecules by chromosome tethering.J. Cell Sci. 114, 49–58
60. Chiu, Y.F. et al. (2017) Kaposi’s sarcoma-associated herpesvirusstably clusters its genomes across generations to maintain itselfextrachromosomally. J. Cell Biol. Published online July 10, 2017.http://dx.doi.org/10.1083/jcb.201702013
61. Brouwer, I. et al. (2016) Sliding sleeves of XRCC4-XLF bridgeDNA and connect fragments of broken DNA. Nature 535, 566–569
62. Orthwein, A. et al. (2014) Mitosis inhibits DNA double-strandbreak repair to guard against telomere fusions. Science 344,189–193
63. Lee, D.H. et al. (2014) Dephosphorylation enables the recruitmentof 53BP1 to double-strand DNA breaks. Mol. Cell 54, 512–525
64. Garsed, D.W. et al. (2014) The architecture and evolution ofcancer neochromosomes. Cancer Cell 26, 653–667
65. van Steensel, B. et al. (1998) TRF2 protects human telomeresfrom end-to-end fusions. Cell 92, 401–413
66. de Lange, T. (2005) Shelterin: the protein complex that shapesand safeguards human telomeres. Genes Dev. 19, 2100–2110
67. Li, Y. et al. (2014) Constitutional and somatic rearrangement ofchromosome 21 in acute lymphoblastic leukaemia. Nature 508,98–102
68. Lee, J.A. et al. (2007) A DNA replication mechanism for generat-ing nonrecurrent rearrangements associated with genomic dis-orders. Cell 131, 1235–1247
69. Hastings, P.J. et al. (2009) A microhomology-mediated break-induced replication model for the origin of human copy numbervariation. PLoS Genet. 5, e1000327
70. Kloosterman, W.P. et al. (2012) Constitutional chromothripsisrearrangements involve clustered double-stranded DNA breaksand nonhomologous repair mechanisms. Cell Rep. 1, 648–655
Trends in Cell Biology, December 2017, Vol. 27, No. 12 929

71. Yang, L. et al. (2013) Diverse mechanisms of somatic structuralvariations in human cancer genomes. Cell 153, 919–929
72. McDermott, D.H. et al. (2015) Chromothriptic cure of WHIMsyndrome. Cell 160, 686–699
73. Voullaire, L.E. et al. (1993) A functional marker centromere with nodetectable alpha-satellite, satellite III, or CENP-B protein: activa-tion of a latent centromere? Am. J. Hum. Genet. 52, 1153–1163
74. Wandall, A. et al. (1998) A neocentromere on human chromo-some 3 without detectable alpha-satellite DNA forms morpho-logically normal kinetochores. Chromosoma 107, 359–365
75. Tyler-Smith, C. et al. (1999) Transmission of a fully functionalhuman neocentromere through three generations. Am. J.Hum. Genet. 64, 1440–1444
76. Fachinetti, D. et al. (2013) A two-step mechanism for epigeneticspecification of centromere identity and function. Nat. Cell Biol.15, 1056–1066
77. Black, B.E. and Cleveland, D.W. (2011) Epigenetic centromerepropagation and the nature of CENP-a nucleosomes. Cell 144,471–479
78. Ceccaldi, R. et al. (2016) Repair pathway choices andconsequences at the double-strand break. Trends Cell. Biol.26, 52–64
79. Chang, H.H.Y. et al. (2017) Non-homologous DNA end joiningand alternative pathways to double-strand break repair. Nat. Rev.Mol. Cell Biol. 18, 495–506
80. Ottaviani, D. et al. (2014) The role of microhomology in genomicstructural variation. Trends Genet. 30, 85–94
81. Sfeir, A. and Symington, L.S. (2015) Microhomology-mediatedend joining: a back-up survival mechanism or dedicated path-way? Trends Biochem. Sci. 40, 701–714
930 Trends in Cell Biology, December 2017, Vol. 27, No. 12
Related Documents











