Reactivity Trends in the Oxidation of CO by Anionic Transition Metal Oxide Clusters † J. Ulises Reveles, ‡ Grant E. Johnson, § Shiv N. Khanna, ‡ and A. W. Castleman, Jr.* ,§ Department of Physics, Virginia Commonwealth UniVersity, Richmond, Virginia 23284-2000, and Departments of Chemistry and Physics, The PennsylVania State UniVersity, UniVersity Park, PennsylVania 16802 ReceiVed: June 23, 2009; ReVised Manuscript ReceiVed: September 27, 2009 Evidence obtained by experiments in the gas phase and density functional theory calculations indicates that differences in the CO binding energy to the transition metal site and the overall change in energy and spin multiplicity from reactants to products, underlie the variation in relative reactivity observed for the oxidation of CO by anionic transition metal oxide clusters. Experimental reactivity studies of MO 2 - and M 2 O 3 - (M ) Fe, Co, Ni, and Cu) reveal that anionic oxide clusters with the same number of atoms and stoichiometry but different elemental composition exhibit specific trends in relative oxidation reactivity with CO. The anionic MO 2 - and M 2 O 3 - clusters are found to be most reactive for M ) Fe and Cu and relatively less reactive for M ) Co and Ni. Theoretical electronic structure studies within the density functional theory framework indicate that the most reactive clusters either have relatively large initial binding energies of CO to the cluster which provide sufficient energy to overcome any subsequent barriers to oxidation, or that the reaction preserves the same overall spin multiplicity from reactants to products. Introduction Transition metal oxides are applied as both catalyst and catalyst-support materials for a variety of industrially relevant processes. 1,2 One reaction of particular importance to the abatement of environmental pollution is the oxidation of harmful carbon monoxide (CO) to more benign carbon dioxide (CO 2 ) in the presence of heterogeneous catalysts. 3 Oxides of the transition metals iron, 4-7 cobalt, 8,9 nickel, 10 and copper 11,12 have been found to be effective materials for promoting this reaction. These previous studies describe the preparation and catalytic behavior of bulk-phase transition metal oxides but have failed to identify the nature of the active sites, and to date, the molecular-level mechanisms of CO oxidation are not well understood. Gas-phase clusters are useful model systems that may be used to gain insight into the molecular-level details of heterogeneous catalytic reactions. 13,14 By avoiding the complications that result from different catalyst preparation methods, gas-phase cluster studies enable the fundamental behavior of catalyst materials to be studied with atomic-level precision. 15-17 Furthermore, mass selected studies provide insight into how factors such as size, stoichiometry, and ionic charge state influence reactivity. 18-25 Moreover, complete catalytic cycles have been observed in the gas phase, providing understanding of the fundamental mech- anisms of catalytic reactions. 26-33 In addition, due to their small size, clusters are amenable to treatment by high-level theoretical calculations which provide valuable insights into structure- reactivity relationships and the molecular-level mechanisms of catalytic reactions. 34 Concerning the influence of spin multiplicity on the efficiency of gas-phase ion molecule reactions, it is now recognized that a delicate interplay of electronic and kinetic energy, spin and spin-orbit coupling efficiency, as well as the electronic con- figuration determines the course and outcome of bond activation and in general the reactivity of transition metal complexes and the degree of spin forbiddeness of reactions, as discussed in an excellent review by Schwarz. 35 In addition, recent theoretical studies have characterized in detail the crossing points and possible spin inversion processes involved in the reaction of VO 2 + with ethylene, 36 ethane, 37 propene, 38 and propyne. 39 More- over, through a combination of gas-phase experiments and theoretical calculations, it was shown that the spin excitation energy 40 and overall spin conservation from reactants to products 41 directly determines the reactivity of anionic aluminum clusters with molecular oxygen. Similar effects were also observed in the etching of anionic aluminum hydride clusters by molecular oxygen. 42 From a fundamental point of view, the reaction of CO oxidation on bulk surfaces may be decomposed into two steps, namely (1) oxidation of CO by the metal oxide leaving a reduced oxide and (2) oxidation of the reduced oxide by oxygen. In a previous study one of the authors considered the oxidation of CO by Fe 2 O 3 surfaces. 43 The process involves oxidation of CO by the lattice oxygen in Fe 2 O 3 creating an oxygen vacancy. This vacancy is filled by an O 2 molecule to recreate the metal oxide. In this work, we focus on the first process, namely, the oxidation of CO by the metal oxide clusters. Previously, we systematically investigated the reactivity of CO with iron, 44-46 cobalt, 47 and nickel oxide 48 clusters. Through a combination of gas-phase experiments and theoretical calcula- tions, we determined that, out of a distribution of anionic iron oxide clusters containing one or two iron atoms and between one and six oxygen atoms, species with one more oxygen atom than iron atom (FeO 2 - and Fe 2 O 3 - ) are the most reactive toward the oxidation of CO. 45 The enhanced reactivity of these anionic clusters was found to result from their relatively low oxygen dissociation energy which makes the transfer of oxygen to CO energetically favorable and to the capacity of the clusters to geometrically rearrange which lowers the barriers to reaction. 45 In contrast to the anionic species, cationic iron oxide clusters containing one or two iron atoms and three or fewer oxygen † Part of the “Barbara J. Garrison Festschrift”. * Corresponding author. Tel: (814)-865-7242. Fax: (814)-865-5235. E-mail: [email protected]. ‡ Virginia Commonwealth University. § The Pennsylvania State University. J. Phys. Chem. C 2010, 114, 5438–5446 5438 10.1021/jp905895w 2010 American Chemical Society Published on Web 11/24/2009

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Reactivity Trends in the Oxidation of CO by Anionic Transition Metal Oxide Clusters†
J. Ulises Reveles,‡ Grant E. Johnson,§ Shiv N. Khanna,‡ and A. W. Castleman, Jr.*,§
Department of Physics, Virginia Commonwealth UniVersity, Richmond, Virginia 23284-2000, and Departmentsof Chemistry and Physics, The PennsylVania State UniVersity, UniVersity Park, PennsylVania 16802
ReceiVed: June 23, 2009; ReVised Manuscript ReceiVed: September 27, 2009
Evidence obtained by experiments in the gas phase and density functional theory calculations indicates thatdifferences in the CO binding energy to the transition metal site and the overall change in energy and spinmultiplicity from reactants to products, underlie the variation in relative reactivity observed for the oxidationof CO by anionic transition metal oxide clusters. Experimental reactivity studies of MO2
- and M2O3- (M )
Fe, Co, Ni, and Cu) reveal that anionic oxide clusters with the same number of atoms and stoichiometry butdifferent elemental composition exhibit specific trends in relative oxidation reactivity with CO. The anionicMO2
- and M2O3- clusters are found to be most reactive for M ) Fe and Cu and relatively less reactive for
M ) Co and Ni. Theoretical electronic structure studies within the density functional theory framework indicatethat the most reactive clusters either have relatively large initial binding energies of CO to the cluster whichprovide sufficient energy to overcome any subsequent barriers to oxidation, or that the reaction preserves thesame overall spin multiplicity from reactants to products.
Introduction
Transition metal oxides are applied as both catalyst andcatalyst-support materials for a variety of industrially relevantprocesses.1,2 One reaction of particular importance to theabatement of environmental pollution is the oxidation of harmfulcarbon monoxide (CO) to more benign carbon dioxide (CO2)in the presence of heterogeneous catalysts.3 Oxides of thetransition metals iron,4-7 cobalt,8,9 nickel,10 and copper11,12 havebeen found to be effective materials for promoting this reaction.These previous studies describe the preparation and catalyticbehavior of bulk-phase transition metal oxides but have failedto identify the nature of the active sites, and to date, themolecular-level mechanisms of CO oxidation are not wellunderstood.
Gas-phase clusters are useful model systems that may be usedto gain insight into the molecular-level details of heterogeneouscatalytic reactions.13,14 By avoiding the complications that resultfrom different catalyst preparation methods, gas-phase clusterstudies enable the fundamental behavior of catalyst materialsto be studied with atomic-level precision.15-17 Furthermore, massselected studies provide insight into how factors such as size,stoichiometry, and ionic charge state influence reactivity.18-25
Moreover, complete catalytic cycles have been observed in thegas phase, providing understanding of the fundamental mech-anisms of catalytic reactions.26-33 In addition, due to their smallsize, clusters are amenable to treatment by high-level theoreticalcalculations which provide valuable insights into structure-reactivity relationships and the molecular-level mechanisms ofcatalytic reactions.34
Concerning the influence of spin multiplicity on the efficiencyof gas-phase ion molecule reactions, it is now recognized thata delicate interplay of electronic and kinetic energy, spin andspin-orbit coupling efficiency, as well as the electronic con-
figuration determines the course and outcome of bond activationand in general the reactivity of transition metal complexes andthe degree of spin forbiddeness of reactions, as discussed in anexcellent review by Schwarz.35 In addition, recent theoreticalstudies have characterized in detail the crossing points andpossible spin inversion processes involved in the reaction ofVO2
+ with ethylene,36 ethane,37 propene,38 and propyne.39 More-over, through a combination of gas-phase experiments andtheoretical calculations, it was shown that the spin excitationenergy40 and overall spin conservation from reactants toproducts41 directly determines the reactivity of anionic aluminumclusters with molecular oxygen. Similar effects were alsoobserved in the etching of anionic aluminum hydride clustersby molecular oxygen.42
From a fundamental point of view, the reaction of COoxidation on bulk surfaces may be decomposed into two steps,namely (1) oxidation of CO by the metal oxide leaving a reducedoxide and (2) oxidation of the reduced oxide by oxygen. In aprevious study one of the authors considered the oxidation ofCO by Fe2O3 surfaces.43 The process involves oxidation of COby the lattice oxygen in Fe2O3 creating an oxygen vacancy. Thisvacancy is filled by an O2 molecule to recreate the metal oxide.In this work, we focus on the first process, namely, the oxidationof CO by the metal oxide clusters.
Previously, we systematically investigated the reactivity ofCO with iron,44-46 cobalt,47 and nickel oxide48 clusters. Througha combination of gas-phase experiments and theoretical calcula-tions, we determined that, out of a distribution of anionic ironoxide clusters containing one or two iron atoms and betweenone and six oxygen atoms, species with one more oxygen atomthan iron atom (FeO2
- and Fe2O3-) are the most reactive toward
the oxidation of CO.45 The enhanced reactivity of these anionicclusters was found to result from their relatively low oxygendissociation energy which makes the transfer of oxygen to COenergetically favorable and to the capacity of the clusters togeometrically rearrange which lowers the barriers to reaction.45
In contrast to the anionic species, cationic iron oxide clusterscontaining one or two iron atoms and three or fewer oxygen
† Part of the “Barbara J. Garrison Festschrift”.* Corresponding author. Tel: (814)-865-7242. Fax: (814)-865-5235.
E-mail: [email protected].‡ Virginia Commonwealth University.§ The Pennsylvania State University.
J. Phys. Chem. C 2010, 114, 5438–54465438
10.1021/jp905895w 2010 American Chemical SocietyPublished on Web 11/24/2009

atoms were found to be the most selective toward CO oxida-tion.46 Oxygen rich cationic iron oxide clusters exhibitedpredominately CO adsorption products.46 These trends inreactivity were explained by the variation in the calculated COand O2 binding energies with increasing oxygen content. Theinfluence of ionic charge state on the reactivity of iron oxideclusters of the same size and stoichiometry was also studiedrevealing that cationic FeO3
+ is far more reactive toward COoxidation than anionic FeO3
- due to the stronger binding ofCO and the weaker binding of atomic oxygen to the cationiccluster.44 Investigations of the interaction of anionic cobalt oxideclusters with CO revealed specific stoichiometries with enhancedreactivity toward oxidation (Co2O3
-, Co2O5-, Co3O5
-, andCo3O6
-).47 Similar to anionic iron oxides, these stoichiometriesof anionic cobalt oxide clusters were calculated to have relativelylower oxygen dissociation energies which makes the oxidationof CO thermodynamically favorable.47 Cationic cobalt oxides,in comparison, were observed to react predominately throughthe adsorption of CO onto the cluster accompanied by the lossof molecular O2 or cobalt oxide subunits.47 Similar behaviorwas observed experimentally for nickel oxide clusters: anionicspecies containing one more oxygen than nickel atom showedenhanced reactivity toward oxidation, and cationic clustersexhibited mainly CO adsorption products accompanied by theloss of molecular O2 or nickel oxide subunits.48
In this publication, we present results from our jointexperimental and theoretical investigation of the reactivity ofanionic metal oxide clusters with the same number of atomsand stoichiometry (MO2
- and M2O3-) with CO. We provide
evidence that when the elemental composition of the cluster ischanged (M ) Fe, Co, Ni, and Cu) variations are observed inthe experimentally measured rate constants for oxygen transferto CO. Theoretical calculations indicate that the variation inreactivity results from a combination of differences in the CObinding energy to the anionic cluster and the overall change inenergy and spin multiplicity from reactants to products. Ourresults suggest that a subtle interplay of energetics and spineffects determine the relative oxidation reactivity of differentanionic transition metal oxide clusters with CO.
Experimental Methods
The reactivity of anionic iron, cobalt, nickel, and copper oxideclusters with CO was studied using a guided-ion-beam massspectrometer described in detail in a previous publication.49
Briefly, metal oxide clusters were produced in a laser vaporiza-tion cluster source by pulsing oxygen seeded in helium (10%)into the plasma formed by ablating either an iron, cobalt, nickel,or copper rod with the second harmonic (532 nm) of a Nd:YAG laser. A 27 mm long conical expansion nozzle was usedto facilitate the clustering of these species. To prevent theundesired formation of hydroxo species, ultrahigh purity heliumand oxygen were used to create the expansion gas mixture.Furthermore, all of the gas transfer lines, which are constructedof stainless steel, were heated while under vacuum to desorbany residual water. The metal oxide clusters exit the sourceregion and are cooled through supersonic expansion intovacuum. During supersonic expansion the high pressure (13.2atm) expansion gas mixture passes through a narrow diameternozzle into vacuum. The random thermal energy of the clustersis thereby converted into directed kinetic energy of the molecularbeam. Consequently, the internal vibrational and rotationalenergy of the clusters is lowered through collisions with theHe carrier gas. The working pressure in the field free region ofthe source is between 5 to 9 × 10-5 Torr depending on the
length of time that the pulse valve is open per pulse.49 Thekinetic energy imparted to the cluster ions by the supersonicexpansion was determined previously, employing a retardingpotential analysis,49 to be approximately 1 eV in the laboratoryenergy frame (ELAB). Ideally, all clusters exiting the supersonicexpansion source have the same initial kinetic energy. The initialcenter-of-mass collision energy (ECM) was calculated for MO2
-
and M2O3- to be approximately 0.23 and 0.14 eV, respectively.
As subsequent collisions are expected to dissipate the initialenergy of a given cluster the values reported above serve toestablish an upper limit on the kinetic energy of the reactivecollisions.
After exiting the source region the clusters pass through a 3mm skimmer forming a collimated molecular beam and are thendirected into a quadrupole mass filter employing a set ofelectrostatic lenses. The quadrupole mass filter isolates clustersof a desired mass-to-charge ratio which are then passed into anoctopole collision cell. To maximize the intensity of a mass-selected cluster, the resolution of the first quadrupole mass filterwas adjusted to discriminate completely between adjacent metaloxides in the cluster distribution. Therefore, although care wastaken to prevent the formation of metal hydroxides in the laservaporization source, as mentioned above, we cannot excludethat a small percentage of the mass selected ions may consistof these species. Variable pressures of CO are introduced intothe octopole collision cell employing a low flow leak valve.The gas pressure is monitored using a MKS Baratron capaci-tance manometer. During reactivity experiments the sourceregion was grounded as was the electrostatic lens at the entranceto the octopole collision cell and the octopole rods, therebyensuring that no additional kinetic energy was imparted to thecluster ions in excess of that resulting from the supersonicexpansion (ELAB ) 1 eV). The reactivity studies were conductedat this kinetic energy because applying a retarding potential tothe octopole rods in order to slow down the cluster ions resultsin a significant reduction in overall signal intensity. Product ionsformed in the collision cell are mass analyzed by a secondquadrupole mass spectrometer. Again, to maximize the intensityof the product ions, the resolution of the second quadrupolemass spectrometer was adjusted to achieve the necessarydiscrimination between oxygen transfer products, which are thefocus of this publication. Finally, the ions are detected with achanneltron electron multiplier connected to a multichannelscalar card. The experimental data presented in the resultssection illustrate the change in normalized ion intensity withincreasing pressures of reactant gas. At higher gas pressures,therefore, the ratio of reactant ion intensity to total ion intensitybecomes smaller while the ratio of product ion intensity to totalion intensity becomes larger. We present the experimental dataas pressure dependent normalized ion intensities for the primaryreaction channels because the absolute intensities of differentmass selected reactant ions vary substantially between species.Product mass spectra at a specific reactant gas pressure wouldindicate the absolute intensity of both reactant and product ionsfor a given reaction but would not allow for a comparison ofthe relative reactivity of different cluster ions. Separate experi-ments were also conducted with inert N2 to verify that theproducts observed with CO are the result of a chemical reactionand not the products of collisional fragmentation.
Theoretical Calculations
The theoretical studies were performed using a linearcombination of atomic orbitals molecular orbital method withina gradient corrected density functional approach. The atomic
Oxidation of CO by Transition Metal Oxides J. Phys. Chem. C, Vol. 114, No. 12, 2010 5439
orbitals were expanded in a linear sum of Gaussian basis sets.The actual calculations were carried out within the densityfunctional theory (DFT) framework using the Naval ResearchLaboratory Molecular Orbital Library (NRLMOL) set of codesdeveloped by Pederson and co-workers.50-52 For these calcula-tions the PW91 generalized gradient approximation53 wasemployed. A 5s, 4p, and 3d basis set for the C and O atomsand 7s, 5p, and 4d basis for the Fe, Co, Ni, and Cu atoms wasused.51 In each case, the basis set was supplemented by a diffuseGaussian. These basis sets are roughly of triple to quadruple-�quality and have been shown to have negligible basis setsuperposition error.51 The transition states were located viaconstrained optimizations of selected bond lengths. All of themolecular geometries were plotted with the Schakal software.54
The benchmarks on the accuracy of the theoretical calculationson iron and cobalt oxides have been presented in previousreports.44-47 In the case of the nickel and copper oxide clusters,the benchmarks were established via calculations of the bondlengths, dissociation energies (DE), ionization energies (IE), andelectron affinities of CuxOy and NixOy clusters and theircomparison with the available corresponding experimentalvalues.55-67 These results are collected in Table 1. We foundthat the calculated bond lengths are in good agreement withexperiments, calculated DEs are higher than the experimentaldissociation energies, whereas the calculated IEs and EAspresented good agreement with respect to the experimentalvalues. The higher calculated DEs are known to occur in densityfunctional theory. Because the discussion is based on relativedifferences in the dissociation and binding energies, and alsothe theoretical studies consistently agree with the experimentalresults, we are confident about the conclusions based on thecurrent approach. Finally, we found good agreement in theground state spin configurations and geometries of the neutraland charged Cu2Oy (y ) 1-4) clusters, with respect to a previoustheoretical study.68
Results and Discussion
Anionic transition metal oxide clusters with two differentstoichiometries (MO2
- and M2O3-, M ) Fe, Co, Ni, and Cu)
were experimentally observed to exhibit dominant productsconsistent with the oxidation of CO to CO2 according to eq 1.
MO2-/M2O3
- + CO f MO-/M2O2- + CO2 (1)
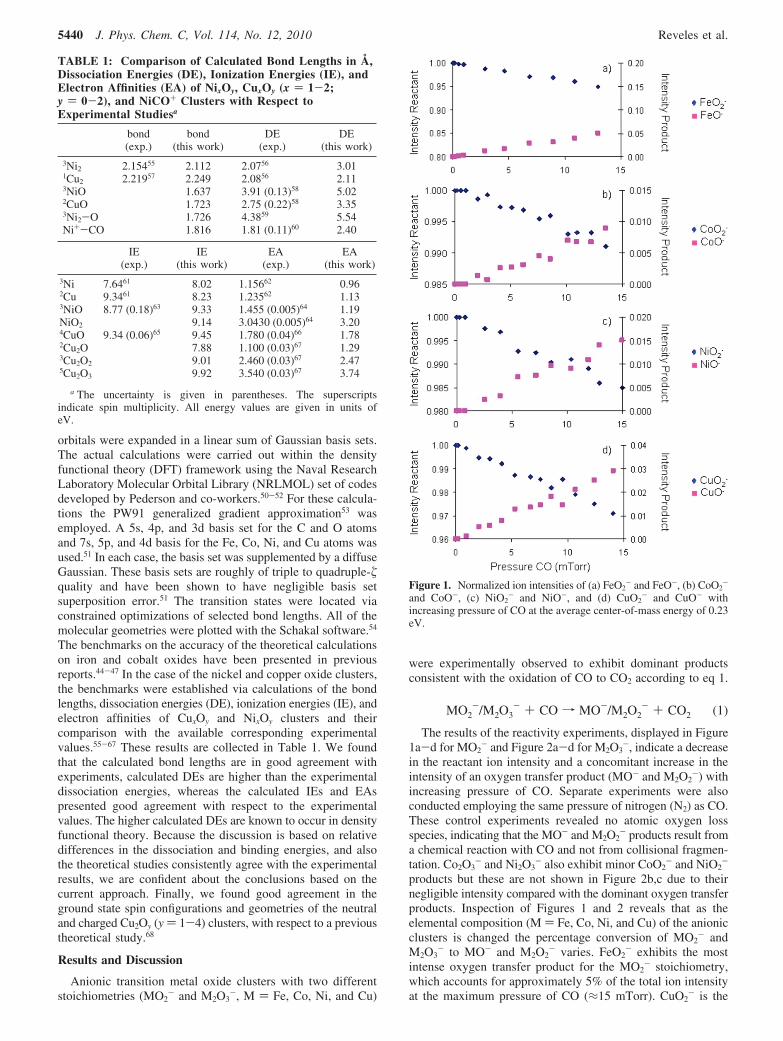
The results of the reactivity experiments, displayed in Figure1a-d for MO2
- and Figure 2a-d for M2O3-, indicate a decrease
in the reactant ion intensity and a concomitant increase in theintensity of an oxygen transfer product (MO- and M2O2
-) withincreasing pressure of CO. Separate experiments were alsoconducted employing the same pressure of nitrogen (N2) as CO.These control experiments revealed no atomic oxygen lossspecies, indicating that the MO- and M2O2
- products result froma chemical reaction with CO and not from collisional fragmen-tation. Co2O3
- and Ni2O3- also exhibit minor CoO2
- and NiO2-
products but these are not shown in Figure 2b,c due to theirnegligible intensity compared with the dominant oxygen transferproducts. Inspection of Figures 1 and 2 reveals that as theelemental composition (M ) Fe, Co, Ni, and Cu) of the anionicclusters is changed the percentage conversion of MO2
- andM2O3
- to MO- and M2O2- varies. FeO2
- exhibits the mostintense oxygen transfer product for the MO2
- stoichiometry,which accounts for approximately 5% of the total ion intensityat the maximum pressure of CO (≈15 mTorr). CuO2
- is the
TABLE 1: Comparison of Calculated Bond Lengths in Å,Dissociation Energies (DE), Ionization Energies (IE), andElectron Affinities (EA) of NixOy, CuxOy (x ) 1-2;y ) 0-2), and NiCO+ Clusters with Respect toExperimental Studiesa
bond(exp.)
bond(this work)
DE(exp.)
DE(this work)
3Ni2 2.15455 2.112 2.0756 3.011Cu2 2.21957 2.249 2.0856 2.113NiO 1.637 3.91 (0.13)58 5.022CuO 1.723 2.75 (0.22)58 3.353Ni2-O 1.726 4.3859 5.54Ni+-CO 1.816 1.81 (0.11)60 2.40
IE(exp.)
IE(this work)
EA(exp.)
EA(this work)
3Ni 7.6461 8.02 1.15662 0.962Cu 9.3461 8.23 1.23562 1.133NiO 8.77 (0.18)63 9.33 1.455 (0.005)64 1.19NiO2 9.14 3.0430 (0.005)64 3.204CuO 9.34 (0.06)65 9.45 1.780 (0.04)66 1.782Cu2O 7.88 1.100 (0.03)67 1.293Cu2O2 9.01 2.460 (0.03)67 2.475Cu2O3 9.92 3.540 (0.03)67 3.74
a The uncertainty is given in parentheses. The superscriptsindicate spin multiplicity. All energy values are given in units ofeV.
Figure 1. Normalized ion intensities of (a) FeO2- and FeO-, (b) CoO2
-
and CoO-, (c) NiO2- and NiO-, and (d) CuO2
- and CuO- withincreasing pressure of CO at the average center-of-mass energy of 0.23eV.
5440 J. Phys. Chem. C, Vol. 114, No. 12, 2010 Reveles et al.
second most reactive species with around 3% of the reactantion converted to products. NiO2
- and CoO2- are relatively less
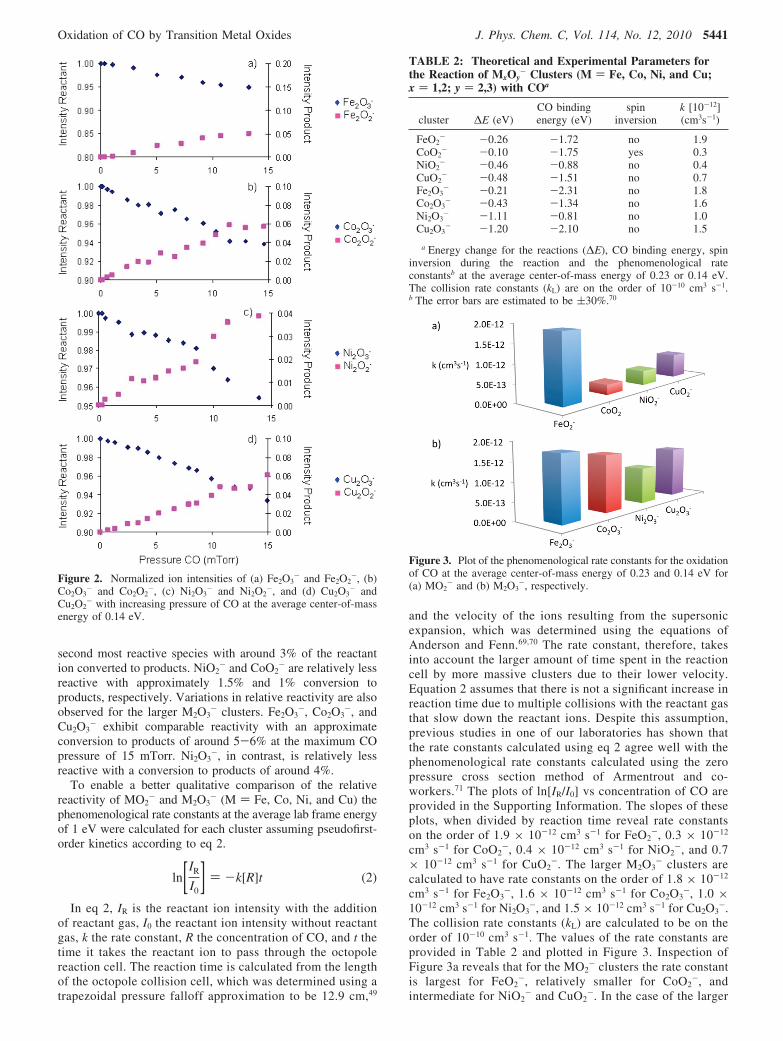
reactive with approximately 1.5% and 1% conversion toproducts, respectively. Variations in relative reactivity are alsoobserved for the larger M2O3
- clusters. Fe2O3-, Co2O3
-, andCu2O3
- exhibit comparable reactivity with an approximateconversion to products of around 5-6% at the maximum COpressure of 15 mTorr. Ni2O3
-, in contrast, is relatively lessreactive with a conversion to products of around 4%.
To enable a better qualitative comparison of the relativereactivity of MO2
- and M2O3- (M ) Fe, Co, Ni, and Cu) the
phenomenological rate constants at the average lab frame energyof 1 eV were calculated for each cluster assuming pseudofirst-order kinetics according to eq 2.
ln[IR
I0] ) -k[R]t (2)
In eq 2, IR is the reactant ion intensity with the additionof reactant gas, I0 the reactant ion intensity without reactantgas, k the rate constant, R the concentration of CO, and t thetime it takes the reactant ion to pass through the octopolereaction cell. The reaction time is calculated from the lengthof the octopole collision cell, which was determined using atrapezoidal pressure falloff approximation to be 12.9 cm,49
and the velocity of the ions resulting from the supersonicexpansion, which was determined using the equations ofAnderson and Fenn.69,70 The rate constant, therefore, takesinto account the larger amount of time spent in the reactioncell by more massive clusters due to their lower velocity.Equation 2 assumes that there is not a significant increase inreaction time due to multiple collisions with the reactant gasthat slow down the reactant ions. Despite this assumption,previous studies in one of our laboratories has shown thatthe rate constants calculated using eq 2 agree well with thephenomenological rate constants calculated using the zeropressure cross section method of Armentrout and co-workers.71 The plots of ln[IR/I0] vs concentration of CO areprovided in the Supporting Information. The slopes of theseplots, when divided by reaction time reveal rate constantson the order of 1.9 × 10-12 cm3 s-1 for FeO2
-, 0.3 × 10-12
cm3 s-1 for CoO2-, 0.4 × 10-12 cm3 s-1 for NiO2
-, and 0.7× 10-12 cm3 s-1 for CuO2
-. The larger M2O3- clusters are
calculated to have rate constants on the order of 1.8 × 10-12
cm3 s-1 for Fe2O3-, 1.6 × 10-12 cm3 s-1 for Co2O3
-, 1.0 ×10-12 cm3 s-1 for Ni2O3
-, and 1.5 × 10-12 cm3 s-1 for Cu2O3-.
The collision rate constants (kL) are calculated to be on theorder of 10-10 cm3 s-1. The values of the rate constants areprovided in Table 2 and plotted in Figure 3. Inspection ofFigure 3a reveals that for the MO2
- clusters the rate constantis largest for FeO2
-, relatively smaller for CoO2-, and
intermediate for NiO2- and CuO2
-. In the case of the larger
Figure 2. Normalized ion intensities of (a) Fe2O3- and Fe2O2
-, (b)Co2O3
- and Co2O2-, (c) Ni2O3
- and Ni2O2-, and (d) Cu2O3
- andCu2O2
- with increasing pressure of CO at the average center-of-massenergy of 0.14 eV.
TABLE 2: Theoretical and Experimental Parameters forthe Reaction of MxOy
- Clusters (M ) Fe, Co, Ni, and Cu;x ) 1,2; y ) 2,3) with COa
cluster ∆E (eV)CO bindingenergy (eV)
spininversion
k [10-12](cm3s-1)
FeO2- -0.26 -1.72 no 1.9
CoO2- -0.10 -1.75 yes 0.3
NiO2- -0.46 -0.88 no 0.4
CuO2- -0.48 -1.51 no 0.7
Fe2O3- -0.21 -2.31 no 1.8
Co2O3- -0.43 -1.34 no 1.6
Ni2O3- -1.11 -0.81 no 1.0
Cu2O3- -1.20 -2.10 no 1.5
a Energy change for the reactions (∆E), CO binding energy, spininversion during the reaction and the phenomenological rateconstantsb at the average center-of-mass energy of 0.23 or 0.14 eV.The collision rate constants (kL) are on the order of 10-10 cm3 s-1.b The error bars are estimated to be (30%.70
Figure 3. Plot of the phenomenological rate constants for the oxidationof CO at the average center-of-mass energy of 0.23 and 0.14 eV for(a) MO2
- and (b) M2O3-, respectively.
Oxidation of CO by Transition Metal Oxides J. Phys. Chem. C, Vol. 114, No. 12, 2010 5441
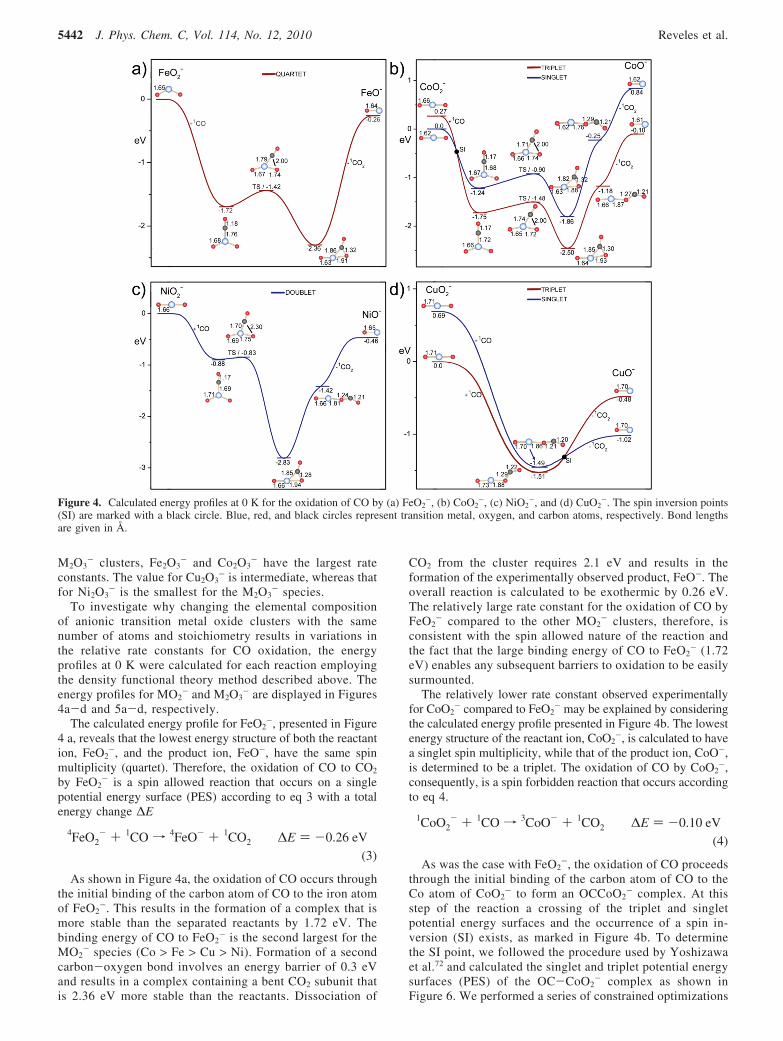
M2O3- clusters, Fe2O3
- and Co2O3- have the largest rate
constants. The value for Cu2O3- is intermediate, whereas that
for Ni2O3- is the smallest for the M2O3
- species.To investigate why changing the elemental composition
of anionic transition metal oxide clusters with the samenumber of atoms and stoichiometry results in variations inthe relative rate constants for CO oxidation, the energyprofiles at 0 K were calculated for each reaction employingthe density functional theory method described above. Theenergy profiles for MO2
- and M2O3- are displayed in Figures
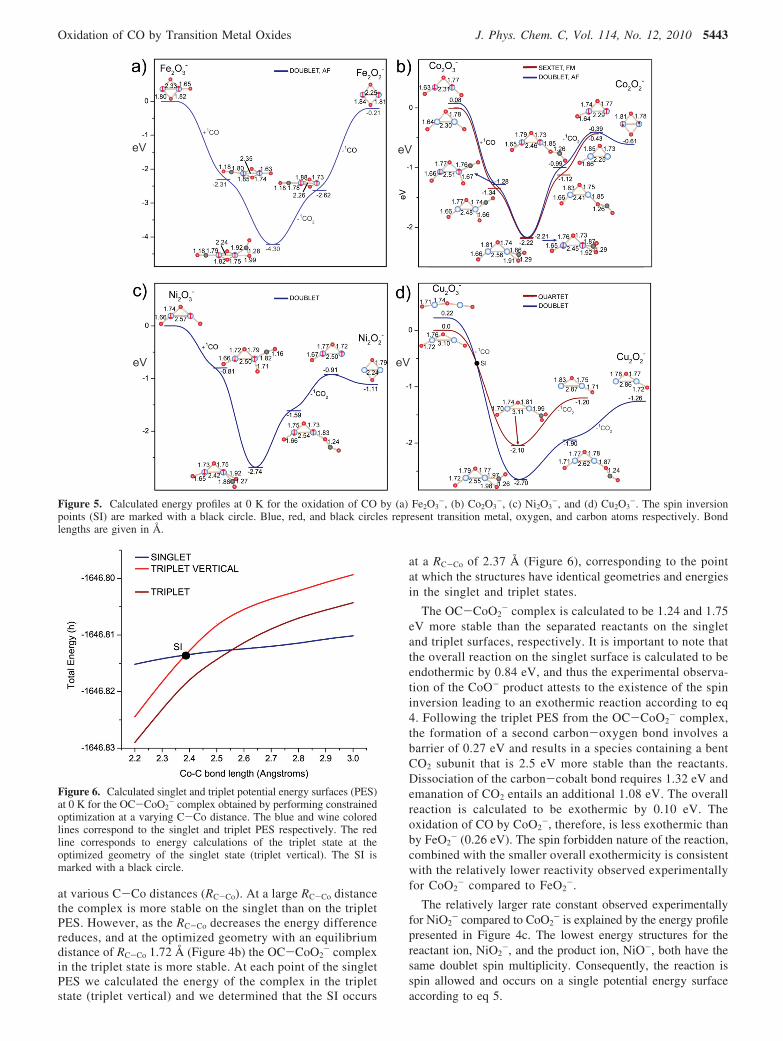
4a-d and 5a-d, respectively.The calculated energy profile for FeO2
-, presented in Figure4 a, reveals that the lowest energy structure of both the reactantion, FeO2
-, and the product ion, FeO-, have the same spinmultiplicity (quartet). Therefore, the oxidation of CO to CO2
by FeO2- is a spin allowed reaction that occurs on a single
potential energy surface (PES) according to eq 3 with a totalenergy change ∆E
4FeO2- + 1CO f 4FeO- + 1CO2 ∆E ) -0.26 eV
(3)
As shown in Figure 4a, the oxidation of CO occurs throughthe initial binding of the carbon atom of CO to the iron atomof FeO2
-. This results in the formation of a complex that ismore stable than the separated reactants by 1.72 eV. Thebinding energy of CO to FeO2
- is the second largest for theMO2
- species (Co > Fe > Cu > Ni). Formation of a secondcarbon-oxygen bond involves an energy barrier of 0.3 eVand results in a complex containing a bent CO2 subunit thatis 2.36 eV more stable than the reactants. Dissociation of
CO2 from the cluster requires 2.1 eV and results in theformation of the experimentally observed product, FeO-. Theoverall reaction is calculated to be exothermic by 0.26 eV.The relatively large rate constant for the oxidation of CO byFeO2
- compared to the other MO2- clusters, therefore, is
consistent with the spin allowed nature of the reaction andthe fact that the large binding energy of CO to FeO2
- (1.72eV) enables any subsequent barriers to oxidation to be easilysurmounted.
The relatively lower rate constant observed experimentallyfor CoO2
- compared to FeO2- may be explained by considering
the calculated energy profile presented in Figure 4b. The lowestenergy structure of the reactant ion, CoO2
-, is calculated to havea singlet spin multiplicity, while that of the product ion, CoO-,is determined to be a triplet. The oxidation of CO by CoO2
-,consequently, is a spin forbidden reaction that occurs accordingto eq 4.
1CoO2- + 1CO f 3CoO- + 1CO2 ∆E ) -0.10 eV
(4)
As was the case with FeO2-, the oxidation of CO proceeds
through the initial binding of the carbon atom of CO to theCo atom of CoO2
- to form an OCCoO2- complex. At this
step of the reaction a crossing of the triplet and singletpotential energy surfaces and the occurrence of a spin in-version (SI) exists, as marked in Figure 4b. To determinethe SI point, we followed the procedure used by Yoshizawaet al.72 and calculated the singlet and triplet potential energysurfaces (PES) of the OC-CoO2
- complex as shown inFigure 6. We performed a series of constrained optimizations
Figure 4. Calculated energy profiles at 0 K for the oxidation of CO by (a) FeO2-, (b) CoO2
-, (c) NiO2-, and (d) CuO2
-. The spin inversion points(SI) are marked with a black circle. Blue, red, and black circles represent transition metal, oxygen, and carbon atoms, respectively. Bond lengthsare given in Å.
5442 J. Phys. Chem. C, Vol. 114, No. 12, 2010 Reveles et al.
at various C-Co distances (RC-Co). At a large RC-Co distancethe complex is more stable on the singlet than on the tripletPES. However, as the RC-Co decreases the energy differencereduces, and at the optimized geometry with an equilibriumdistance of RC-Co 1.72 Å (Figure 4b) the OC-CoO2
- complexin the triplet state is more stable. At each point of the singletPES we calculated the energy of the complex in the tripletstate (triplet vertical) and we determined that the SI occurs
at a RC-Co of 2.37 Å (Figure 6), corresponding to the pointat which the structures have identical geometries and energiesin the singlet and triplet states.
The OC-CoO2- complex is calculated to be 1.24 and 1.75
eV more stable than the separated reactants on the singletand triplet surfaces, respectively. It is important to note thatthe overall reaction on the singlet surface is calculated to beendothermic by 0.84 eV, and thus the experimental observa-tion of the CoO- product attests to the existence of the spininversion leading to an exothermic reaction according to eq4. Following the triplet PES from the OC-CoO2
- complex,the formation of a second carbon-oxygen bond involves abarrier of 0.27 eV and results in a species containing a bentCO2 subunit that is 2.5 eV more stable than the reactants.Dissociation of the carbon-cobalt bond requires 1.32 eV andemanation of CO2 entails an additional 1.08 eV. The overallreaction is calculated to be exothermic by 0.10 eV. Theoxidation of CO by CoO2
-, therefore, is less exothermic thanby FeO2
- (0.26 eV). The spin forbidden nature of the reaction,combined with the smaller overall exothermicity is consistentwith the relatively lower reactivity observed experimentallyfor CoO2
- compared to FeO2-.
The relatively larger rate constant observed experimentallyfor NiO2
- compared to CoO2- is explained by the energy profile
presented in Figure 4c. The lowest energy structures for thereactant ion, NiO2
-, and the product ion, NiO-, both have thesame doublet spin multiplicity. Consequently, the reaction isspin allowed and occurs on a single potential energy surfaceaccording to eq 5.
Figure 5. Calculated energy profiles at 0 K for the oxidation of CO by (a) Fe2O3-, (b) Co2O3
-, (c) Ni2O3-, and (d) Cu2O3
-. The spin inversionpoints (SI) are marked with a black circle. Blue, red, and black circles represent transition metal, oxygen, and carbon atoms respectively. Bondlengths are given in Å.
Figure 6. Calculated singlet and triplet potential energy surfaces (PES)at 0 K for the OC-CoO2
- complex obtained by performing constrainedoptimization at a varying C-Co distance. The blue and wine coloredlines correspond to the singlet and triplet PES respectively. The redline corresponds to energy calculations of the triplet state at theoptimized geometry of the singlet state (triplet vertical). The SI ismarked with a black circle.
Oxidation of CO by Transition Metal Oxides J. Phys. Chem. C, Vol. 114, No. 12, 2010 5443

2NiO2- + 1CO f 2NiO- + 1CO2 ∆E ) -0.46 eV
(5)
Similar to the case for FeO2- and CoO2
-, the oxidationreaction commences through the exothermic (-0.88 eV) bindingof the carbon atom of CO to the Ni atom of NiO2
-. The bindingenergy of CO to NiO2
-, therefore, is much lower than to FeO2-
(1.72 eV) and CoO2- (1.75 eV). The formation of a second
carbon-oxygen bond entails a small barrier of 0.05 eV andresults in a complex containing a CO2 subunit that is 2.83 eVmore stable than the reactants. Dissociation of the carbon-nickelbond requires 1.41 eV and emanation of CO2 entails anadditional 0.96 eV and results in the experimentally observedproduct, NiO-. The relatively higher reactivity observed forNiO2
- compared to CoO2-, therefore, is consistent with the spin
allowed nature of the reaction. The fact that NiO2- is less
reactive than FeO2-, is consistent with the relatively lower CO
binding energy (NiO2- ) 0.88 eV, FeO2
- ) 1.72 eV).Consequently, less energy is gained through initial complexformation and is available to overcome the subsequent barriersto CO oxidation. Table 2 lists the overall energy changes, CObinding energies, spin inversion and phenomenological rateconstants for the reactions.
The calculated energy profile for the CuO2- cluster, which
has the second highest reactivity in the MO2- series after FeO2
-,is presented in Figure 4 d. The lowest energy structures forCuO2
- and CuO- have different spin multiplicity. Therefore,the oxidation of CO is a spin forbidden reaction that involvesa change in overall spin multiplicity according to eq 6.
3CuO2- + 1CO f 1CuO- + 1CO2 ∆E ) -1.02 eV
(6)
However, it should be noted that a spin allowed reaction wasalso determined to be possible resulting in an exothermic processaccording to eq 7:
3CuO2- + 1CO f 3CuO- + 1CO2 ∆E ) -0.48 eV
(7)
Thus, the violation of the spin conservation rule may beavoided if there exists an exothermic spin allowed path, asshown by Tanaka et al.73 for proton transfers reactions. In thecase of CuO2
-, the binding of the carbon atom of CO to the Cuatom of the 3CuO2
- cluster was calculated to be exothermic byonly 0.24 eV. Thus, an alternative reaction path was determinedto occur initiated by the direct binding of the carbon atom ofCO to an oxygen atom of CuO2
-. In this initial step the complexformed is 1.51 eV more stable than the reactants (Figure 4d),while a similar complex on the singlet surface lies 0.02 eVhigher in energy. At this stage of the energy profile a spininversion was determined to be possible during the CO2
emanation completing the reaction according to eq 6. The spinallowed oxidation of CO by CuO2
- is the most exothermicreaction (-0.48 eV) as compared with FeO2
- (0.26 eV), CoO2-
(0.10 eV), and NiO2- (0.46 eV). Oxidation of CO by CoO2
-
involves a spin inversion, and thus the spin allowed nature andthe greater overall exothermicity is consistent with CuO2
- beingtwo times more reactive than CoO2
- (Table 2). Finally, despitethe large exothermicity, CuO2
- may be less reactive than FeO2-
due to the possibility of the spin inversion that may slow downthe CuO2
- reaction, and the lower CO binding energy to CuO2-
(1.51 eV) compared to FeO2- (1.72 eV).
The larger M2O3- (M ) Fe, Co, Ni, and Cu) clusters also
exhibited measurable although smaller variations in reactivity
toward the oxidation of CO when their elemental compositionwas changed (Table 2). Similar to FeO2
- in the MO2- series,
the Fe2O3- cluster was observed experimentally to be the most
reactive M2O3- species. The calculated energy profile for Fe2O3
-
is displayed in Figure 5a. The oxidation of CO begins throughthe association of one CO molecule onto an iron atom of thecluster resulting in the formation of a complex that is 2.31 eVmore stable than the reactants. The association of the first COmolecule activates the terminal oxygen atom of the Fe2O3
-
cluster toward reaction with a second CO. Association of asecond CO produces an intermediate containing a CO2 subunitthat is 4.30 eV more stable than the reactants. Dissociation ofCO2 requires 1.68 eV of energy. Removal of the second COmolecule entails an additional 2.41 eV and results in theformation of the product ion, Fe2O2
-. Both the reactant ion,Fe2O3
-, and the product ion, Fe2O2-, are calculated to have
ground state structures with a doublet spin multiplicity andpresenting an antiferromagnetic (AF) coupling of the Fe sites.Therefore, the oxidation of CO by Fe2O3
- is a spin allowedprocess that occurs according to eq 8.
2Fe2O3- + 21CO f 2Fe2O2
- + 1CO2 + 1CO∆E ) -0.21 eV (8)
The relatively higher reactivity observed for Fe2O3- compared
to the other M2O3- (M ) Co, Ni, and Cu) clusters, therefore, is
consistent with the spin allowed nature of the reaction and thelarge amount of energy (4.30 eV) gained through the associationof two CO molecules onto the cluster.
The comparatively lower reactivity of Co2O3- in relation to
Fe2O3- may be understood from the calculated energy profile
presented in Figure 5 b. The most apparent difference betweenCo2O3
- and Fe2O3- is the geometry of the lowest energy
structures. Fe2O3- contains a four member ring structure with
two bridging oxygen atoms and one terminal oxygen atom.Co2O3
-, in contrast, has a three member ring structure with onlyone bridging oxygen atom and two terminal oxygen atoms. Thereactant Co2O3
- and the product ion Co2O2- are calculated to
have a sextet state with ferromagnetic (FM) coupling of the Cosites, and a doublet AF state, respectively. Therefore, theoxidation of CO by Co2O3
- is a spin forbidden process thatoccurs according to eq 9.
6Co2O3- + 1CO f 2Co2O2
- + 1CO2 ∆E ) -0.61 eV(9)
However, as in the case of CuO2- a spin allowed reaction
was determined to be possible according to eq 10:
6Co2O3- + 1CO f 6Co2O2
- + 1CO2 ∆E ) -0.43 eV(10)
The reaction begins through the exothermic (1.34 eV)association of CO onto one of the cobalt atoms of the clusterin the sextet spin state. The energetically low lying isomer ofthe association complex is found to lie 0.06 eV higher in energyin a doublet state. Both the sextet and doublet surfaces are foundto be very close along the reaction path (Figure 5b). Addition-ally, even though the ground state of Co2O3
- is a sextet (FM),the next isomer in a doublet (AF) state is only 0.08 eV higherin energy. Because of this small energy difference the spininversion is not to be expected to have an effect on the reactivityas strong as the one observed for the CoO2
- species, in whichthe next isomer that leads to a spin allowed path lies 0.27 eVabove the ground state (Figure 4b). These phenomena have beendiscussed previously as spin excitation energies and found to
5444 J. Phys. Chem. C, Vol. 114, No. 12, 2010 Reveles et al.

directly determine the reactivity of anionic aluminum andaluminum hydride clusters with oxygen.40
Coming back to the reaction path for Co2O3- with CO on
the sextet surface: after the formation of the CO associatedcomplex the formation of a carbon-oxygen bond is barrierlessand results in a species containing a CO2 subunit which is 2.22eV more stable than the reactants (Figure 5b). Dissociation ofthe carbon-cobalt bond requires 1.1 eV of energy and emana-tion of CO2 entails another 0.69 eV leading to the Co2O2
-
species with both a bridging and terminal oxygen atom. Overallthe Co2O3
- reaction is calculated to be exothermic by 0.43 eVas shown in Figure 5b. The relatively lower reactivity of Co2O3
-
compared to Fe2O3- is consistent with the different geometries
of the reactant ions that directly influences the CO bindingenergy. Specifically, Co2O3
-, due to the presence of a terminaloxygen atom on both cobalt atoms does not adsorb two COmolecules like Fe2O3
-. Therefore, the terminal oxygen atomsof the Co2O3
- cluster are less activated toward CO oxidationthan in Fe2O3
-.The reactivity of the Ni2O3
- cluster was found experimentallyto be the lowest among the M2O3
- series. The calculated reactionprofile is shown in Figure 5c. The reaction begins through theexothermic (0.81 eV) association of CO onto one of the nickelatoms of the cluster. This association energy is the smallestamong the series (Table 2) and is consistent with the relativelylower reactivity of Ni2O3
- with CO. Formation of a carbon-oxygen bond results in a complex containing a CO2 subunitthat is 2.74 eV more stable than the reactants. Dissociation ofthe Ni-C bond and emanation of CO2 are barrierless processesand result in two complexes that are 1.59 and 0.91 eV morestable than the reactants. Dissociation of the CO2 moleculerequires 0.68 eV of energy. Finally the Ni2O2
- cluster stabilizesforming a cyclic structure with an energy gain of 0.20 eV.
The reactivity of the Cu2O3- cluster was found to be relatively
higher with respect to Ni2O3- but slightly lower compared to
Fe2O3- and Co2O3
- as shown in Figure 3 and Table 2. Thecalculated energy profile of the Cu2O3
- reaction with CO isshown in Figure 5d. The Cu2O3
- and Cu2O2- clusters are
calculated to have a quartet and doublet spin multiplicity,respectively. Therefore, the oxidation of CO by Cu2O3
- is aspin forbidden process that occurs according to eq 11.
4Cu2O3- + 1CO f 2Cu2O2
- + 1CO2 ∆E ) -1.26 eV(11)
In addition to this reaction, a spin allowed reaction was alsodetermined to be possible according to eq 12:
4Cu2O3- + 1CO f 4Cu2O2
- + 1CO2 ∆E ) -1.20 eV(12)
In a previous DFT study the Cu2O3- structure was determined
to be a linear quartet state.68 In our study, however we foundtwo degenerate structures, the linear one and a ring structurewith a Cu-Cu bond length of 3.10 Å. We considered bothstructures in the calculation of the reaction path and found thatin both cases the complex formed after CO attachment is a ringstructure as shown in Figure 5d. The reaction begins throughthe exothermic (2.10 eV) association of CO onto one of thecopper atoms. A spin inversion process is determined to bepossible at this initial step. A CO association complex in whicha bent CO2 unit exists in the doublet state is found to be -2.70eV more stable than the reactants. From this species dissociationof the Cu-C bond and emanation of CO2 requires 0.80 and0.64 eV respectively. On the other hand, for the quartet surface
the emanation of CO2 requires 0.9 eV to produce the 4Cu2O2-
product. The oxidation of CO by Cu2O3- is calculated to be
the most exothermic reaction (1.20 eV) in the M2O3- series,
although the possible change in spin multiplicity is consistentwith the slightly slower reaction compared to Fe2O3
- andCo2O3
-.
Conclusion
Through a combination of experiments in the gas phase anddensity functional theory calculations, we provide evidence thatchanges in the initial binding energy of CO to the cluster, theoverall exothermicity of the reaction, and spin multiplicityunderlie the variations in relative reactivity observed for anionictransition metal oxide clusters toward the oxidation of CO. Ofall of the clusters considered here, the lowest reactivity isexhibited by CoO2
-. Theoretical studies for this cluster indicatethat the reaction path for the constrained spin multiplicity isendothermic and that the reaction will only proceed if there isa change in spin multiplicity. This illustrates the role of spin incontrolling the reactivity. The other important feature is theprogression in relative CO oxidation reactivity in going fromFe to Cu transition metal atoms in both series (MO2
- andM2O3
-). The Cu based clusters have higher reactivity than Nibased clusters and the theoretical studies show that the reactionpaths for Cu based clusters are qualitatively different from thosefor Fe, Co, and Ni transition metal based clusters. In general,clusters that are relatively more reactive (M ) Fe and Cu) areshown to have higher initial CO binding energies which providesufficient energy to overcome any subsequent barriers tooxidation, or to preserve the same overall spin multiplicity ingoing from reactants to products. In contrast, species that arecomparatively less reactive are shown to gain less energythrough the initial binding of CO or to undergo a change inspin multiplicity during oxidation.
Acknowledgment. J.U.R. and S.N.K. acknowledge supportfrom the Air Force Office of Scientific Research under theMURI Grant FA9550-08-1-0400. G.E.J. and A.W.C. gratefullyacknowledge the Department of Energy, Grant No. DE-FG02-92ER14258. Part of the calculations were performed on thecomputational equipment of DGSCA UNAM, particularly atthe super computer KanBalam.
Supporting Information Available: Plots of ln[Ir/Io] vsconcentration of CO. This material is available free of chargevia the Internet at http://pubs.acs.org.
References and Notes
(1) Fierro, J. L. G. Metal Oxides Chemistry and Applications; Taylor& Francis: London, 2006.
(2) Ertl, G.; Knozinger, H.; Weitkamp, J. Handbook of HeterogeneousCatalysis; Wiley-VCH: Weinheim, Germany, 1997.
(3) Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. J. Catal. 1989,115, 301.
(4) Yumura, T.; Amenomori, T.; Kagawa, Y.; Yoshizawa, K. J. Phys.Chem. A 2002, 106, 621.
(5) Lin, H.-Y.; Chen, Y.-W.; Wang, W.-J. J. Nanopart. Res 2005, 7,249.
(6) Li, P.; Miser, D. E.; Rabiei, S.; Yadav, R. T.; Hajaligol, M. R.Appl. Catal., B 2003, 43, 151.
(7) PalDey, S.; Gedevanishvili, S.; Zhang, W.; Rasouli, F. Appl. Catal.,B 2005, 56, 241.
(8) Wang, Y. Z.; Zhao, Y. X.; Gao, C. G.; Liu, D. S. Catal. Lett. 2007,116, 136.
(9) Lopes, I.; Davidson, A.; Thomas, C. Catal. Commun. 2007, 8, 2105.(10) Stoyanova, M.; Konova, P.; Nikolov, P.; Naydenov, A.; Chris-
toskova, S.; Mehandjiev, D. Chem. Eng. J. 2006, 122, 41.
Oxidation of CO by Transition Metal Oxides J. Phys. Chem. C, Vol. 114, No. 12, 2010 5445

(11) Li, D.; Liu, X.; Zhang, Q.; Wang, Y.; Wan, H. Catal. Lett. 2009,127, 377.
(12) Jin, L.-Y.; He, M.; Lu, J.-Q.; Luo, M.-F.; Fang, P.; Xie, Y.-L. Chin.J. Chem. Phys. 2007, 20, 582.
(13) Zemski, K. A.; Justes, D. R.; Castleman, A. W., Jr. J. Phys. Chem.B 2002, 106, 6136.
(14) Johnson, G. E.; Tyo, E. C.; Castleman, A. W., Jr. Proc. Natl. Acad.Sci 2008, 105, 18108.
(15) Schroder, D.; Schwarz, H. Angew. Chem., Int. Ed. 1995, 34, 1973.(16) Bohme, D. K.; Schwarz, H. Angew. Chem., Int. Ed. 2005, 44, 2336.(17) Kim, Y. D. Int. J. Mass. Spectrom 2004, 238, 17.(18) Feyel, S.; Schroder, D.; Schwarz, H. J. Phys. Chem. A 2006, 110,
2647.(19) Feyel, S.; Schroder, D.; Rozanska, X.; Sauer, J.; Schwarz, H. Angew.
Chem., Int. Ed. 2006, 45, 4677.(20) Feyel, S.; Dobler, J.; Schroder, D.; Sauer, J.; Schwarz, H. Angew.
Chem., Int. Ed. 2006, 45, 4681.(21) Feyel, S.; Scharfenberg, L.; Daniel, C.; Hartl, H.; Schroder, D.;
Schwarz, H. J. Phys. Chem. A 2007, 111, 3278.(22) Feyel, S.; Dobler, J.; Hockendorf, R.; Beyer, M. K.; Sauer, J.;
Schwarz, H. Angew. Chem. Int. Ed 2008, 47, 1946.(23) Dong, F.; Heinbuch, S.; Xie, Y.; Rocca, J. J.; Benstein, E. R.; Wang,
Z.-C.; Deng, K,; He, S.-G. J. Am. Chem. Soc. 2008, 130, 1932.(24) Justes, D. R.; Mitric, R.; Moore, N. A.; Bonacic-Koutecky, V.;
Castleman, A. W., Jr. J. Am. Chem. Soc. 2003, 125, 6289.(25) Johnson, G. E.; Mitric, R.; Tyo, E. C.; Bonacic-Koutecky, V.;
Castleman, A. W., Jr. J. Am. Chem. Soc. 2008, 130, 13912.(26) Kappes, M. M.; Staley, R. H. J. Am. Chem. Soc. 1981, 103, 1286.(27) Andersson, M.; Rosen, A. J. Chem. Phys. 2002, 117, 7051.(28) Schnabel, P.; Weil, K. G.; Irion, M. P. Angew. Chem., Int. Ed.
1992, 31, 636.(29) Socaciu, L. D.; Hagen, J.; Bernhardt, T. M.; Woste, L.; Heiz, U.;
Hakkinen, H.; Landman, U. J. Am. Chem. Soc. 2003, 125, 10437.(30) Bronstrup, M.; Schroder, D.; Kretzschmar, I.; Schwarz, H.; Harvey,
J. N. J. Am. Chem. Soc. 2001, 123, 142.(31) Shi, Y.; Ervin, K. M. J. Chem. Phys. 1998, 108, 1757.(32) Balaj, O. P.; Balteanu, I.; Ro�teuscher, T. T. J.; Beyer, M. K.;
Bondybey, V. E. Angew. Chem., Int. Ed. 2004, 43, 6519.(33) Siu, C.-K.; Reitmeier, S. J.; Balteanu, I.; Bondybey, V. E.; Beyer,
M. K. Eur. Phys. J. D 2007, 43, 189.(34) Cheng, M.-J.; Chenoweth, K.; Oxgaard, J.; van Duin, A.; Goddard,
W. A. J. Phys. Chem. C 2007, 111, 5115.(35) Schwarz, H. Int. J. Mass Spectrom. 2004, 75, 237.(36) Gracia, L.; Sambrano, J. R.; Safont, V. S.; Calatayud, M.; Beltran,
A.; Andres, J. J. Phys. Chem. A 2003, 107, 3107.(37) Gracia, L.; Andres, J.; Safont, V. S.; Beltran, A. Organometallics
2004, 23, 730.(38) Gracia, L.; Sambrano, J. R.; Andres, J.; Beltran, A. Organometallics
2006, 25, 1643.(39) Gracia, L.; Polo, V.; Sambrano, J. R.; Andres, J. J. Phys. Chem. A
2008, 112, 1808.(40) Reber, A. C.; Khanna, S. N.; Roach, P. J.; Woodward, W. H.;
Castleman, A. W., Jr. J. Am. Chem. Soc. 2007, 129, 16098.(41) Burgert, R.; Schnockel, H.; Grubisic, A.; Li, X.; Stokes, S. T.;
Bowen, K. H.; Gantefor, G. F.; Kiran, B.; Jena, P. Science 2008, 319, 438.(42) Roach, P. J.; Reber, A. C.; Woodward, W. H.; Khanna, S. N.;
Castleman, A. W., Jr. Proc. Natl. Acad. Sci. 2007, 104, 14565.(43) Kandalam, A. K.; Chatterjee, B.; Khanna, S. N.; Rao, B. K.; Jena,
P.; Reddy, B. V. Surf. Sci. 2007, 601, 4873.
(44) Reilly, N. M.; Reveles, J. U.; Johnson, G. E.; Khanna, S. N.;Castleman, A. W., Jr. Chem. Phys. Lett. 2007, 435, 295.
(45) Reilly, N. M.; Reveles, J. U.; Johnson, G. E.; Khanna, S. N.;Castleman, A. W., Jr. J. Phys. Chem. A 2007, 111, 4158.
(46) Reilly, N. M.; Reveles, J. U.; Johnson, G. E.; del Campo, J. M.;Khanna, S. N.; Koster, A. M.; Castleman, A. W., Jr. J. Phys. Chem. C2007, 111, 19086.
(47) Johnson, G. E.; Reveles, J. U.; Reilly, N. M.; Tyo, E. C.; Khanna,S. N.; Castleman, A. W., Jr. J. Phys. Chem. A 2008, 112, 11330.
(48) Johnson, G. E.; Reilly, N. M.; Castleman, A. W., Jr. Int. J. Mass.Spectrom 2009, 280, 93.
(49) Bell, R. C.; Zemski, K. A.; Justes, D. R.; Castleman, A. W., Jr.J. Chem. Phys. 2001, 114, 798.
(50) Pederson, M. R.; Jackson, K. A. Phys. ReV. B 1990, 41, 7453.(51) Jackson, K.; Pederson, M. R. Phys. ReV. B 1990, 42, 3276.(52) Porezag, D.; Pederson, M. R. Phys. ReV. A 1999, 60, 2840.(53) Perdew, J. P. In Electronic Structure of Solids; Ziesche, P., Eschrig,
H., Eds.; Akademie Verlag: Berlin, 1991.(54) Keller, E. Chem. Z. 1980, 14, 56. http://www.krist.uni-freiburg.de/
ki/Mitarbeiter/Keller/schakal.html.(55) Pinegar, J. C.; Langenberg, J. D.; Arrington, C. A.; Spain, E. M.;
Morse, M. D. J. Chem. Phys. 1995, 102, 666.(56) Morse, M. D.; Hansen, G. P.; Langridge-Smith, P. R. R.; Zheng,
L. S.; Geusic, M. E.; Michalopoulos, D. L.; Smalley, R. E. J. Chem. Phys.1984, 80, 5400.
(57) Ram, R. S.; Jarman, C. N.; Bernath, P. F. J. Mol. Spectrosc. 1992,156, 468.
(58) Pedley, J. B.; Marshall, E. M. J. Phys. Chem. Ref. Data 1983, 12,967.
(59) Goddard, W. A.; Walch, S. P.; Rappe, A. K.; Upton, T. H.; Melius,C. F. J. Vac. Sci. Technol. 1977, 14, 416.
(60) Khan, F. A.; Steele, D. L.; Armentrout, P. B. J. Phys. Chem. 1995,99, 7819.
(61) Sansonetti, J. E.; Martin, W. C.; Young, S. L. Handbook of BasicAtomic Spectroscopy Data (version 1.1), NIST Physical Data web site http://physics.nist.gov/PhysRefData/Handbook (October2004).
(62) Gas Phase Ion Chemistry; Bowers, M. T., Ed.; Academic Press:Orlando 1984; Vol. 3, Chapter 21, p 167; Chapter 22, p 213.
(63) Fisher, E. R.; Elkind, J. L.; Clemmer, D. E.; Georgiadis, R.; Loh,S. K.; Aristov, N.; Suderlin, L. S.; Armentrout, P. B. J. Chem. Phys. 1990,93, 2676.
(64) Ramond, T. M.; Davico, G. E.; Hellberg, F.; Svedberg, F.; Salen,P.; Soderqvist, P.; Lineberger, W. C. J. Mol. Spectrosc. 2002, 216, 1.
(65) Rodgers, M. T.; Walker, B.; Armentrout, P. B. Int. J. MassSpectrom. 1999, 182, 99.
(66) Wu, H.; Desai, S. R.; Wang, L. S. J. Phys. Chem. 1997, 101, 2103.(67) Wang, L. S.; Wu, H.; Desai, S. R.; Lou, L. Phys. ReV. B 1996, 53,
8028.(68) Dai, B.; Tian, L.; Yang, J. J. Chem. Phys. 2004, 120, 2746.(69) Anderson, J. B.; Fenn, J. B. Phys. Fluids 1965, 8, 780.(70) Moore, N. A.; Mitric, R.; Justes, D. R.; Bonacic-Koutecky, V.;
Castleman, A. W., Jr. J. Phys. Chem. B 2006, 110, 3015.(71) Ervin, K. M.; Armentrout, P. B. J. Chem. Phys. 1985, 83, 166.(72) Yoshizawa, K.; Shiota, Y.; Yamabe, T. J. Am. Chem. Soc. 1999,
121, 147.(73) Tanaka, K.; Betowski, L. D.; Mackay, G. I.; Bohme, D. K. J. Chem.
Phys. 1976, 65, 3203.
JP905895W
5446 J. Phys. Chem. C, Vol. 114, No. 12, 2010 Reveles et al.
Related Documents











