Reactions of methylamines at the Si„100…-2Ã1 surface Collin Mui, George T. Wang, Stacey F. Bent, a) and Charles B. Musgrave b) Department of Chemical Engineering, Stanford University, Stanford, California 94305-5025 ~Received 28 December 2000; accepted 15 March 2001! We have investigated the room temperature adsorption of methylamine, dimethylamine and trimethylamine using density functional theory ~DFT! and multiple internal reflection Fourier transform infrared ~MIR-FTIR! spectroscopy. It was found that the reaction pathways of the amines resemble the precursor-mediated dissociative chemisorption of ammonia. Our calculations showed that although dissociation involving N–C bond cleavage is thermodynamically more favorable than the N–H dissociation pathway, the activation barrier for N–CH 3 dissociation is significantly higher than that for N–H dissociation. This leads to selective cleavage of N–H bonds in the surface reactions of methylamine and dimethylamine, while trapping trimethylamine in its molecularly chemisorbed state through the formation of a Si–N dative bond. We also identified the products of the reactions of the amines on the Si~100!-231 surface by surface IR studies, confirming the theoretical predictions. The selectivity observed in the surface chemistry of simple model amines is briefly discussed in the context of organic chemistry at semiconductor surfaces. © 2001 American Institute of Physics. @DOI: 10.1063/1.1370056# I. INTRODUCTION Reactions of organic molecules at semiconductor sur- faces are of fundamental and technological interest. Funda- mentally, the interaction between organic molecules and semiconductor surfaces is localized, a characteristic that makes the surface chemistry qualitatively different from catalytic reactions on metal surfaces. In particular, the unique structure of the Si~100!-231 surface has been shown to pro- vide a template of reactive sites for classical organic reac- tions, extending the principles of organic chemistry to cova- lent solid surfaces. Technologically, a chemically-modified silicon surface may serve as a dielectric or a lithography mask for ‘‘nanoelectronic devices.’’ Also, organic monolay- ers with known orientation and controllable properties on a semiconductor surface may have potential in applications such as chemical sensors, ‘‘DNA chips,’’ molecular elec- tronics and optoelectronic devices. The surface chemistry of organic molecules at semicon- ductor surfaces is closely related to the surface electronic structure. The Si~100!-231 surface consists of rows of dimers that result from surface reconstruction. 1,2 The bond- ing of a surface dimer involves a strong s-bond and a weak p-bond between the two Si atoms of the dimer. The strained geometry of the surface results in poor p-orbital overlap, and hence a weak p-bond between the two Si atoms in a dimer. The dimers on the surface are tilted in order to relieve strain, and the electronic structure of a tilted dimer has the proper- ties of both a weak p-bond and a zwitterion-like diradical. This unique surface electronic structure makes the Si~100!- 231 surface a template of reactive ‘‘organic reagent.’’ The p-bond character of the Si~100!-231 surface dimers has been explored previously through the reactivity towards unsaturated hydrocarbons. 3–6 It has been demonstrated that ethylene and acetylene react with the surface via di-s Si–C bond formation, also known as @212# cycloaddition. 7–16 Other unsaturated hydrocarbons with C5C double bonds are found to react in a similar fashion, some of them resulting in ordered organic monolayers on the surface. 17–22 Although @212# cycloaddition reactions are symmetry forbidden in or- ganic chemistry, it has been proposed that the symmetry rules are relaxed by the dynamic tilting of dimers at room temperature. 23–26 Conjugated unsaturated hydrocarbons such as 1,3-butadiene can react with the Si~100!-231 surface via @412# cycloaddition, more commonly known as the Diels– Alder reaction. 24,27–29 The Diels–Alder reaction is symmetry allowed, and there is direct theoretical evidence that it occurs on the Si~100!-231 surface with little or no activation barrier. 24,25 More complicated systems such as benzene show multiple binding configurations 30–36 and the conversion be- tween different binding states was observed by STM. 33,34 More recently, the reactivity of the Si~100!-231 surface towards unsaturated hydrocarbons that exhibit multiple cycloaddition reaction pathways, has also been studied and compared. 24–26,37–40 The generality of reactions involving surface p-bonds on group-IV semiconductors has also been demonstrated for other classes of molecules and other surfaces. Experimen- tally, it has been found that unsaturated organic molecules having heteroatom double bonds such as C5O, N5N and N5C5S react with the Si~100!-231 surface through cy- cloaddition reactions. 41–43 Recent theoretical calculations also investigated the possibility of cycloaddition reactions between several 1,3-dipolar organic molecules with the Si~100!-231 surface. 44 Furthermore, the ~100! surfaces of diamond and germanium consist of dimers as well, and their reactivity towards unsaturated hydrocarbons can be directly compared to Si~100!-231. It has been shown experimentally that cycloaddition reactions occur on both the Ge~100!-231 a! Electronic mail: [email protected] b! Electronic mail: [email protected] JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 22 8 JUNE 2001 10170 0021-9606/2001/114(22)/10170/11/$18.00 © 2001 American Institute of Physics Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

JOURNAL OF CHEMICAL PHYSICS VOLUME 114, NUMBER 22 8 JUNE 2001
Reactions of methylamines at the Si „100…-2Ã1 surfaceCollin Mui, George T. Wang, Stacey F. Bent,a) and Charles B. Musgraveb)
Department of Chemical Engineering, Stanford University, Stanford, California 94305-5025
~Received 28 December 2000; accepted 15 March 2001!
We have investigated the room temperature adsorption of methylamine, dimethylamine andtrimethylamine using density functional theory~DFT! and multiple internal reflection Fouriertransform infrared~MIR-FTIR! spectroscopy. It was found that the reaction pathways of the aminesresemble the precursor-mediated dissociative chemisorption of ammonia. Our calculations showedthat although dissociation involving N–C bond cleavage is thermodynamically more favorable thanthe N–H dissociation pathway, the activation barrier for N–CH3 dissociation is significantly higherthan that for N–H dissociation. This leads to selective cleavage of N–H bonds in the surfacereactions of methylamine and dimethylamine, while trapping trimethylamine in its molecularlychemisorbed state through the formation of a Si–N dative bond. We also identified the products ofthe reactions of the amines on the Si~100!-231 surface by surface IR studies, confirming thetheoretical predictions. The selectivity observed in the surface chemistry of simple model amines isbriefly discussed in the context of organic chemistry at semiconductor surfaces. ©2001 AmericanInstitute of Physics.@DOI: 10.1063/1.1370056#
udn
thmquo-avaedhy-
onc
onnf
ne
raie.
rd
at
in
r-etrymch
–yurs
ow-
pleand
foren-les
snsthe
eirctlyly
I. INTRODUCTION
Reactions of organic molecules at semiconductor sfaces are of fundamental and technological interest. Funmentally, the interaction between organic molecules asemiconductor surfaces is localized, a characteristicmakes the surface chemistry qualitatively different frocatalytic reactions on metal surfaces. In particular, the unistructure of the Si~100!-231 surface has been shown to prvide a template of reactive sites for classical organic retions, extending the principles of organic chemistry to colent solid surfaces. Technologically, a chemically-modifisilicon surface may serve as a dielectric or a lithograpmask for ‘‘nanoelectronic devices.’’ Also, organic monolaers with known orientation and controllable properties onsemiconductor surface may have potential in applicatisuch as chemical sensors, ‘‘DNA chips,’’ molecular eletronics and optoelectronic devices.
The surface chemistry of organic molecules at semicductor surfaces is closely related to the surface electrostructure. The Si~100!-231 surface consists of rows odimers that result from surface reconstruction.1,2 The bond-ing of a surface dimer involves a strongs-bond and a weakp-bond between the two Si atoms of the dimer. The straigeometry of the surface results in poorp-orbital overlap, andhence a weakp-bond between the two Si atoms in a dimeThe dimers on the surface are tilted in order to relieve strand the electronic structure of a tilted dimer has the propties of both a weakp-bond and a zwitterion-like diradicalThis unique surface electronic structure makes the Si~100!-231 surface a template of reactive ‘‘organic reagent.’’
Thep-bond character of the Si~100!-231 surface dimershas been explored previously through the reactivity towa
a!Electronic mail: [email protected]!Electronic mail: [email protected]
10170021-9606/2001/114(22)/10170/11/$18.00
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
r-a-dat
e
c--
y
as
-
-ic
d
.n,r-
s
unsaturated hydrocarbons.3–6 It has been demonstrated thethylene and acetylene react with the surface via di-s Si–Cbond formation, also known as@212# cycloaddition.7–16
Other unsaturated hydrocarbons with C5C double bonds arefound to react in a similar fashion, some of them resultingordered organic monolayers on the surface.17–22 Although@212# cycloaddition reactions are symmetry forbidden in oganic chemistry, it has been proposed that the symmrules are relaxed by the dynamic tilting of dimers at rootemperature.23–26Conjugated unsaturated hydrocarbons suas 1,3-butadiene can react with the Si~100!-231 surface via@412# cycloaddition, more commonly known as the DielsAlder reaction.24,27–29The Diels–Alder reaction is symmetrallowed, and there is direct theoretical evidence that it occon the Si~100!-231 surface with little or no activationbarrier.24,25More complicated systems such as benzene shmultiple binding configurations30–36 and the conversion between different binding states was observed by STM.33,34
More recently, the reactivity of the Si~100!-231 surfacetowards unsaturated hydrocarbons that exhibit multicycloaddition reaction pathways, has also been studiedcompared.24–26,37–40
The generality of reactions involving surfacep-bonds ongroup-IV semiconductors has also been demonstratedother classes of molecules and other surfaces. Experimtally, it has been found that unsaturated organic molecuhaving heteroatom double bonds such as C5O, N5N andN5C5S react with the Si~100!-231 surface through cy-cloaddition reactions.41–43 Recent theoretical calculationalso investigated the possibility of cycloaddition reactiobetween several 1,3-dipolar organic molecules withSi~100!-231 surface.44 Furthermore, the~100! surfaces ofdiamond and germanium consist of dimers as well, and threactivity towards unsaturated hydrocarbons can be direcompared to Si~100!-231. It has been shown experimentalthat cycloaddition reactions occur on both the Ge~100!-231
0 © 2001 American Institute of Physics
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

als
ouna
Tgar
enfir
H
thS
w
tiftosr
s,
nsspf
e,1iav
wikw
fosry
th-
willim-gech
rsors aty
reted
tinglay-
sur-s to
m-lcu-tial
fre-erore-ute-H
cal-trarorsree-r-
odeaceve.993enttheac-.pti-
ad-b-asisronicg
ribeinbe
YPby
10171J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Reactions of methylamines at the Si(100)-231 surface
and diamond~100!-231 surfaces.45–48 The possibility ofcycloaddition reactions on the surface of SiGe alloys hasbeen investigated theoretically.49
Although it is now established that thep-bonds of theSi~100!-231 surface are reactive towards a wide rangeorganic molecules, there has been less investigation intolizing the zwitterion-like diradical character of the silicodimers. The tilting of dimers on the surface results incharge separation between the two Si atoms in a dimer.down atom of the tilted dimer has a slight positive charrelative to the up atom, introducing some zwitterionic chacteristics into the weakp-bond of a dimer. The interactionof ammonia~NH3! with the Si~100!-231 surface provides anexample of a zwitterionic-type reaction. Previous experimtal and theoretical studies have shown that ammoniaadsorbs molecularly onto the Si~100!-231 surface into anNH3(a) precursor state, followed by scission of an N–bond to produce surface NH2(a) and H(a) species, as shownin reaction~1!.50–54 The formation of the NH3(a) precursorstate involves the interaction between the lone pair ofNH3 molecule and the electron-deficient down atom of thedimer, and the molecular adsorption is essentially a Leacid-base reaction.
Reaction~1!
There have also been previous studies on the interacof amines with the Si~100!-231 surface. The reaction oaniline was found to occur via N–H dissociation similarammonia, resulting in nonbridge bonded surface specie55
Hamers and coworkers proposed that 3-pyrroline adsoonto the Si~100!-231 surface via two competing pathwaynamely@212# cycloaddition and N–H dissociation.56,57 Zhuand coworkers found that the reaction of several orgaamines with the Si~100!-231 monochloride surface produceclose packed molecular assemblies of bridged–bondedcies on the surface.58,59 The surface chemistry o1,1-dimethylhydrazine,60 dimethylamine61 and pyrrole62
have also been investigated using several experimental tniques. It was proposed that the reaction of 1dimethylhydrazine involves both N–N and N–H dissoction, whereas dimethylamine and pyrrole dissociatesN–CH3 and N–H bond cleavage, respectively.
In this paper, we explore the reactivity of the Si~100!-231 surface towards several simple model amines. Wediscuss the surface chemistry in terms of the zwitterion-ldiradical character of the surface dimers. Specifically,investigate the reaction of methylamine@NH2CH3#, dimethy-lamine @NH~CH3!2# and trimethylamine@N~CH3!3# at theSi~100!-231 surface. We have calculated the energeticsthe adsorption and dissociation mechanisms of the aminethe Si~100!-231 surface using density functional theo~DFT! and cluster models of the surface~Sec. IV A!. Mul-tiple internal reflection Fourier transform infrared~MIR-FTIR! spectroscopy was used to identify the products ofsurface reactions~Sec. IV B!. We find that the amines un
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
o
fti-
hee-
-st
ei
is
on
.bs
ic
e-
ch---ia
illee
ron
e
dergo similar surface reactions to those of ammonia. Weshow that the surface dissociation of methylamine and dethylamine occurs preferentially via N–H bond cleavaover N–CH3 bond cleavage. Hence trimethylamine, whihas no N–H bonds, reacts with the Si~100!-231 surface toform a molecularly adsorbed species trapped in the precustate. Finally, we shall discuss the reactions of the aminethe Si~100!-231 in the context of surface modification borganic attachment.
II. THEORETICAL APPROACH
The electronic structure calculations in this work abased on DFT63,64 with Gaussian basis sets as implemenin theGAUSSIAN98suite of programs.65 The Si~100!-231 sur-face is modeled by a Si9H12 one-dimer cluster.24,54 TheSi9H12 cluster consists of two surface Si atoms representhe surface dimer, and seven Si atoms representing threeers of subsurface bulk Si. The dangling bonds of the subface Si atoms are terminated by twelve hydrogen atommaintain theirsp3 hybridization. All structures are fully op-timized without geometrical constraints on the clusters. Symetry restrictions are applied where appropriate. We calated the structures of the stationary points on the potenenergy surface at the BLYP/6-31G* level of theory.66,67 Allcalculated minima and transition states are verified byquency calculations at the same level of theory to have zand only one imaginary frequency, respectively. In the fquency calculations, the clusters are terminated with derium atoms in order to prevent coupling between bulk Si–normal modes and the surface Si–H frequencies in theculated vibrational spectra. All calculated vibrational specreported are scaled to minimize the known systematic erobserved in frequency calculations and for the best agment with experiments. Specifically, an empirically detemined scale factor is used to correct each stretching mtype: 0.964 for C–H stretching modes of dissociated surfproducts; 0.951 for C–H stretching vibrations of datibonded precursor states; 0.977 for Si–H stretches; and 0for N–H stretches. It has been shown that using differcorrections for different types of vibrations can increaseaccuracy of DFT frequency calculations, and the scale ftors used in this work are consistent with the literature68
Single point energy calculations are performed on the omized structures at the B3LYP level of theory67,69 with asplit basis set scheme, in which the 6-31111G** basis setwas used on the surface Si dimer atoms and the aminesorbate, and the 6-31G* basis set was used on all the susurface Si atoms and the terminating H atoms. The split bset scheme serves to enhance the accuracy of the electstructure of the chemically active atoms while minimizincomputational costs. The 6-31111G** triple-z basis setplus polarization and diffuse functions was used to descthe chemically active atoms such that the orbitals activethe bond breaking and bond forming processes wouldaccurately described during the surface reactions. All B3Lsingle point energies reported are correctedBLYP/6-31G* zero-point energies.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

m
v
eer
hanatinit
-
ner
foe
e
ismn
-
udsed
cuthhucretiv
dieoocte
th
forxo-ionalln of
ajare-
ofener-
cu-andcal/
el,nd
28
aventothee 4
era-
6.4hethe-thatande
theocal
in
re-ms
areses
10172 J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Mui et al.
III. EXPERIMENTAL DETAILS
All experiments were performed in an ultrahigh vacuu~UHV! chamber with a base pressure of less than 7.53 10210 Torr. The details of the experimental apparatus habeen described previously.70 Briefly, the chamber isequipped with an unshielded quadrupole mass spectromand an ion gun for surface sputtering. Infrared data wcollected in multiple internal reflection~MIR! mode using aFourier transform infrared~FTIR! spectrometer with aliquid-nitrogen-cooled, narrow-band HgCdTe detector. Tunpolarized beam from the FTIR spectrometer entersexits the UHV chamber by means of two KBr windowsright angles to each other. Polarized IR spectra were obtaby inserting a wire-grid polarizer in the IR path beforeenters the detector. A Si~100! single crystal sample of trapezoidal geometry~1 mm320 mm350 mm, 45° bevelededges, Harrick! is heated by a resistive tungsten heater acooled by heat exchange with a liquid nitrogen cold fingThe sample surface was cleaned by sputtering with Ar1 ionsat room temperature followed by annealing to 1100 Kone minute. This surface preparation procedure routinproduces a smooth Si~100!-231 surface, as verified by thpresence of sharp monohydride IR features at 2097 cm21
and 2086 cm21 after dosing the surface with disilane@Si2H6#~gas, purity 99.998%, Voltaix! while the sample is coolingfrom 900 K to 650 K. The back side of the crystal, whichnot cleaned by sputtering, was covered with a 0.127 mthick molybdenum plate to prevent molecular adsorption asurface reaction. Methylamine@NH2CH3# ~gas, purity99.51%, Matheson!, dimethylamine@NH~CH3!2# ~gas, pu-rity 991%, Aldrich! and trimethylamine@N~CH3!3# ~gas, pu-rity 99.51%, Matheson! were used without further purification, and their purities were verified usingin situ massspectrometry. Exposures are reported in units of Langm~L!, where 1 L51026 Torr s. All exposures were performeby filling the chamber with the compound of interest for apressure and time. The pressures have not been correcteion gauge sensitivities.
IV. RESULTS
A. Density functional theory calculations
1. Ammonia
In order to show that our theoretical approach is acrate, we calculated the reaction path of ammonia onSi~100!-231 surface, and compared our results to recent toretical and experimental studies. We optimized the strtures at the critical points on the potential energy surfaThe resulting energies along with the optimized structuare shown in Fig. 1. Our results confirmed that dissociachemisorption of ammonia onto the Si~100!-231 surface isprecursor mediated. Ammonia first forms a molecularly asorbed state through a dative bond to the electron deficdown Si atom of a silicon dimer. The adsorption energythis process was found to be 19.9 kcal/mol. Dissociationammonia then occurs through a transition state with an avation barrier of 13.6 kcal/mol relative to the adsorbed stalosing a hydrogen atom to the neighboring Si atom ofsame dimer and resulting in a NH2 group and a H atom
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
e
tere
ed
ed
d.
rly
d
ir
tfor
-e
e-c-e.se
-ntff
ti-,
e
chemisorbed on the dimer. The overall reaction energythe dissociative chemisorption process is 52.4 kcal/mol ethermic. Since the energy of the hydrogen-loss transitstate is 9.2 kcal/mol below the vacuum level, the overprocess is not activated, and hence the surface reactioammonia at Si~100! results in the formation of NH2(a) andH(a) species.
Our results are consistent with previous studies. Widjand Musgrave have performed a detailed analysis on theliability of DFT and cluster models, as well as the effectsbasis sets and subsurface constraints on the calculatedgetics of ammonia reaction on the Si~100!-231 surface. Us-ing a three dimer cluster model, the energies of the molelarly adsorbed state, the hydrogen-loss transition statethe hydrogen-dissociated state were calculated to be 26 kmol, 6 kcal/mol and 51 kcal/mol below the vacuum levrespectively.71 Using periodic slab models of the surface athe PBE generalized gradient approximation~GGA!, Lee andKang calculated the energies of the three states to bekcal/mol, 12 kcal/mol and 46 kcal/mol, respectively.53 Usingmolecular beam techniques, Takaoka and Kusunoki hshown that the dissociative adsorption of ammonia oSi~100!-231 is precursor-mediated, and they measuredenergy of the hydrogen-dissociation transition state to bkcal/mol below the vacuum level.52 The energy of NH3(g)recombinative desorption from the surface NH2(a) and H(a)species has been determined to be 47 kcal/mol by tempture programmed desorption~TPD! measurements.51 Ourcalculated values of 19.9 kcal/mol for the adsorbed state,kcal/mol for the transition state and 52.4 kcal/mol for tdissociated state are consistent with all of these previousoretical and experimental results. This agreement showsthe theoretical approach we used is generally accuratereliable in calculating the reaction of amines with thSi~100!-231 surface. The larger discrepancy observed inenergy of the molecularly adsorbed state is due to a nonleffect not included in the one dimer model, as discusseddetail by Widjaja and Musgrave.71
FIG. 1. Calculated reaction path and optimized structures for ammoniaaction on the Si9H12 one-dimer cluster. In the diagram, the large white atoare Si, the small white atoms are H and the gray atom is N. All energieswith respect to the vacuum level in kcal/mol. The numbers in parentheare energies at the BLYP/6-31G* level.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
r-n-e-ercie
avtl
theowc9onre
-Cth
hat
owneHd ame
aennotossme
of
e
ic.d
ateel.edndge-e-
e-en isco-ge-
de-asof
ss eadtTheh is
10173J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Reactions of methylamines at the Si(100)-231 surface
2. Methylamine
The initial adsorption of methylamine involves the inteaction of the N-lone pair of methylamine with the electrodeficient down Si atom of a silicon dimer, forming a dativbonded molecular adsorption state. The adsorption enwas calculated to be 24.1 kcal/mol. There are two dissotion pathways possible for molecular chemisorption of mthylamine: either N–H bond or N–C bond cleavage. Cleage of the N–H bond results in the formation of a covalenbound NHCH3(a) species and an H(a) at the other Si atomof the same dimer, while N–C bond cleavage results information of NH2(a) and CH3(a) species on the dimer. Thcalculated energies for the N–H cleavage pathway are shin Fig. 2. The overall reaction for N–H dissociation is unativated, since the energy of the H-loss transition state iskcal/mol below the vacuum level. The overall H-dissociatiprocess was found to be 53.2 kcal/mol exothermic. Theaction path for N–CH3 dissociation is shown in Fig. 3. According to our calculations, the activation barrier for N–cleavage is 18.0 kcal/mol above the vacuum level andoverall reaction is 68.8 kcal/mol exothermic.
FIG. 2. Calculated reaction path and optimized structures for N–H diciation of methylamine on the Si9H12 one-dimer cluster.
FIG. 3. Calculated reaction path and optimized structures for N–CH3 dis-sociation of methylamine on the Si one-dimer cluster.
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
gya---y
e
n-.0
-
e
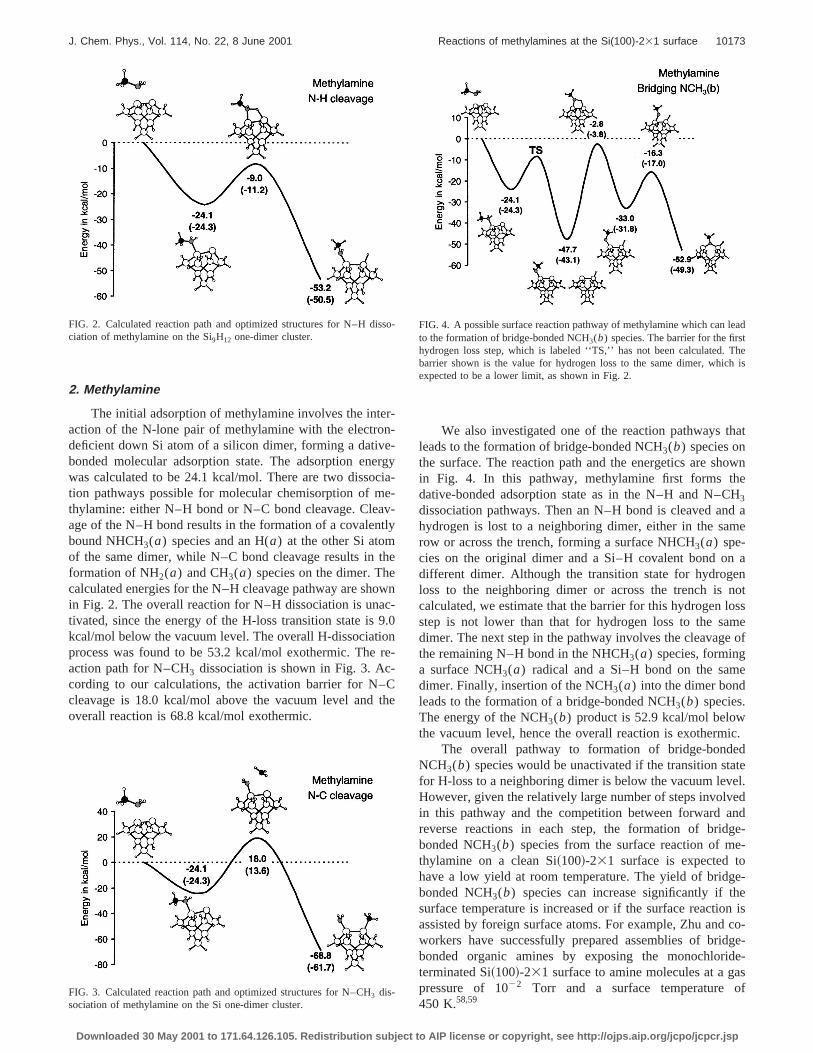
We also investigated one of the reaction pathways tleads to the formation of bridge-bonded NCH3(b) species onthe surface. The reaction path and the energetics are shin Fig. 4. In this pathway, methylamine first forms thdative-bonded adsorption state as in the N–H and N–C3
dissociation pathways. Then an N–H bond is cleaved anhydrogen is lost to a neighboring dimer, either in the sarow or across the trench, forming a surface NHCH3(a) spe-cies on the original dimer and a Si–H covalent bond ondifferent dimer. Although the transition state for hydrogloss to the neighboring dimer or across the trench iscalculated, we estimate that the barrier for this hydrogen lstep is not lower than that for hydrogen loss to the sadimer. The next step in the pathway involves the cleavagethe remaining N–H bond in the NHCH3(a) species, forminga surface NCH3(a) radical and a Si–H bond on the samdimer. Finally, insertion of the NCH3(a) into the dimer bondleads to the formation of a bridge-bonded NCH3(b) species.The energy of the NCH3(b) product is 52.9 kcal/mol belowthe vacuum level, hence the overall reaction is exotherm
The overall pathway to formation of bridge-bondeNCH3(b) species would be unactivated if the transition stfor H-loss to a neighboring dimer is below the vacuum levHowever, given the relatively large number of steps involvin this pathway and the competition between forward areverse reactions in each step, the formation of bridbonded NCH3(b) species from the surface reaction of mthylamine on a clean Si~100!-231 surface is expected tohave a low yield at room temperature. The yield of bridgbonded NCH3(b) species can increase significantly if thsurface temperature is increased or if the surface reactioassisted by foreign surface atoms. For example, Zhu andworkers have successfully prepared assemblies of bridbonded organic amines by exposing the monochloriterminated Si~100!-231 surface to amine molecules at a gpressure of 1022 Torr and a surface temperature450 K.58,59
o-FIG. 4. A possible surface reaction pathway of methylamine which can lto the formation of bridge-bonded NCH3(b) species. The barrier for the firshydrogen loss step, which is labeled ‘‘TS,’’ has not been calculated.barrier shown is the value for hydrogen loss to the same dimer, whicexpected to be a lower limit, as shown in Fig. 2.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ththa
heumith
lye
ci-5
th
oa
fom
1ivCg
orm-e
um
onifi
hy-
nin
y-.ineia,rstor-y-onrgybe
–Cfaceeer,l/essre-
inon
–
H
H
10174 J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Mui et al.
According to our calculations, the H dissociation paway of adsorbed methylamine is less exothermic thanN–C bond cleavage pathway. However, the H-loss pathwis not activated while the overall activation energy of tCH3 dissociation pathway is significantly above the vaculevel. Thus, we predict that the reaction of methylamine wa silicon dimer will result in the formation of a covalentbound NHCH3(a) species and a Si–H bond on the samdimer.
3. Dimethylamine
Dimethylamine undergoes precursor-mediated dissotive chemisorption at the Si~100!-231 surface, and the reaction pathways are similar to that of methylamine. Figureshows the energetics of the H dissociation pathway ofdimethylamine surface reaction. Initial adsorption of dimethylamine onto the silicon dimer results in the formationa stable dative-bonded molecularly adsorbed state withadsorption energy of 24.8 kcal/mol. The transition stateN–H dissociation is located 9.3 kcal/mol below the vacuulevel, and the formation of surface N~CH3!2(a) and H(a)species is exothermic, with an overall reaction energy of 5kcal/mol with respect to the vacuum level. The alternatdissociation pathway of dimethylamine, which involves N–bond cleavage, is shown in Fig. 6. We find that followininitial dimethylamine adsorption, the activation barrier fN–CH3 dissociation is 19.3 kcal/mol above the vacuulevel. Dissociation of CH3 from dimethylamine leads to surface NHCH3(a) and CH3(a) species and the energy of thdissociation products is 65.6 kcal/mol below the vaculevel.
Since the overall activation energy of N–H dissociatiis below the vacuum level while N–C cleavage has a signcant overall barrier, we expect that the reaction of dimetlamine on the Si~100!-231 surface involves N–H dissociation and produces N~CH3!2(a) and H(a). Although theformation of a surface CH3(a) from N–C bond cleavage ismore thermodynamically favorable than N–H dissociatiosurface reaction kinetics play an important role in determing the final reaction products.
FIG. 5. The calculated reaction path and optimized structures for Ndissociation of dimethylamine on the Si one-dimer cluster.
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
-ey
a-
e-fnr
.7e
--
,-
4. Trimethylamine
Since N–H dissociation is not possible for trimethlamine, only the N–CH3 dissociation pathway is studiedThe schematic potential energy curve for the trimethylamsurface reaction is shown in Fig. 7. Similar to ammonmethylamine and dimethylamine initial adsorption, the fistep of the trimethylamine surface reaction involves the fmation of a dative bond between the N-lone pair of trimethlamine and the electron-deficient down Si atom of the silicdimer. The adsorption process is exothermic and the eneof the stable molecularly chemisorbed state was found to23.4 kcal/mol below the vacuum level. Cleavage of the Nbond from this adsorbed state was found to produce surN~CH3!2(a) and CH3(a) species, and the energy of thproducts is 62.3 kcal/mol below the vacuum level. Howevthe transition state for CH3 dissociation is located 21.7 kcamol above the vacuum level, making the dissociation prockinetically unfavorable at room temperature. Hence, we pdict that trimethylamine will be trapped on the Si~100!-231surface in the initial molecularly adsorbed state, resultingthe formation of molecularly chemisorbed trimethylaminethe surface at room temperature.
H
FIG. 6. The calculated reaction path and optimized structures for N–C3
dissociation of dimethylamine on the Si one-dimer cluster.
FIG. 7. The calculated reaction path and optimized structures for N–C3
dissociation of trimethylamine on the Si one-dimer cluster.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
-
0
Ka-a
10175J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Reactions of methylamines at the Si(100)-231 surface
FIG. 8. Infrared spectra of amines adsorbed on the Si~100!-231 surface:~a!10 L of methylamine adsorbed at 30K; ~b! 100 L of dimethylamine ad-sorbed at 300 K;~c! 100 L of trim-ethylamine adsorbed at 300 K;~d!multilayers of methylamine at 100 K;~e! multilayers of dimethylamine at100 K; ~f! multilayers of trimethyl-amine at 100 K. The spectra at 300have been corrected for baseline instbilities. The low temperature spectrare all scaled.
ac
aidhth
rfsta
netc
re
in
ro
s,brb
u-eeubes
is
rs
od-im-ity
fre-ese
r isacey-ni-
i–Na
lvech
thetodw
of
ineethethe
B. Surface infrared spectroscopy
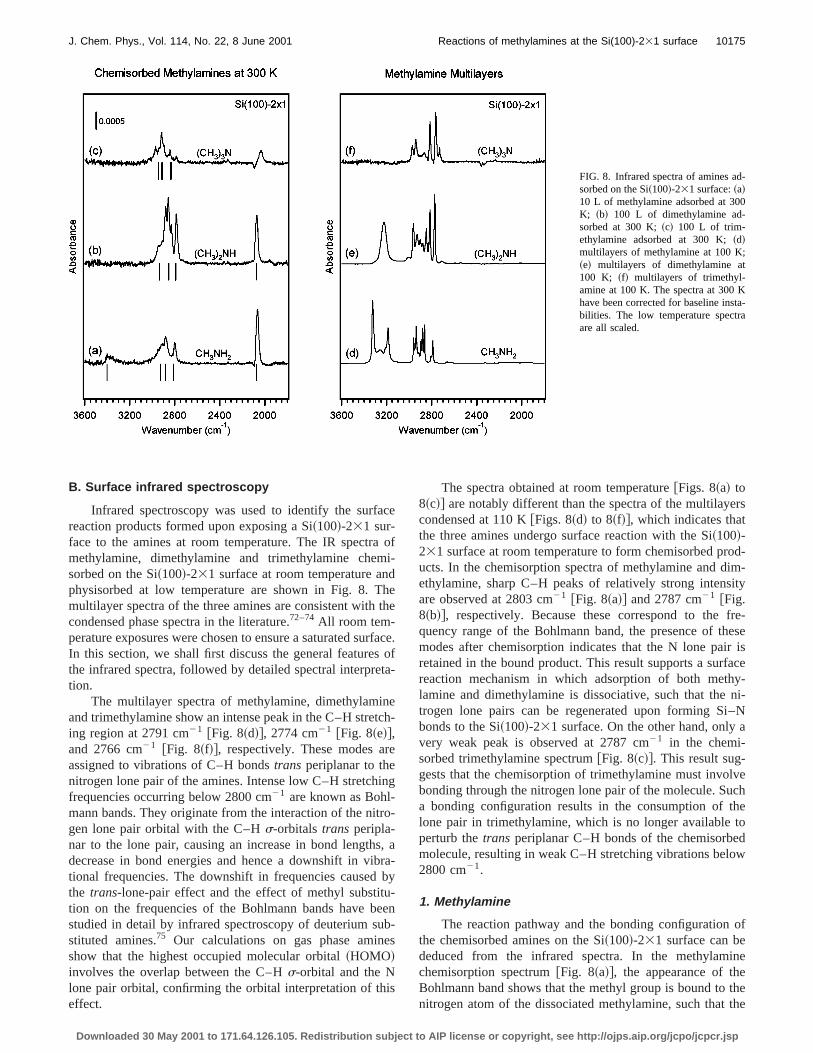
Infrared spectroscopy was used to identify the surfreaction products formed upon exposing a Si~100!-231 sur-face to the amines at room temperature. The IR spectrmethylamine, dimethylamine and trimethylamine chemsorbed on the Si~100!-231 surface at room temperature anphysisorbed at low temperature are shown in Fig. 8. Tmultilayer spectra of the three amines are consistent withcondensed phase spectra in the literature.72–74All room tem-perature exposures were chosen to ensure a saturated suIn this section, we shall first discuss the general featurethe infrared spectra, followed by detailed spectral interpretion.
The multilayer spectra of methylamine, dimethylamiand trimethylamine show an intense peak in the C–H streing region at 2791 cm21 @Fig. 8~d!#, 2774 cm21 @Fig. 8~e!#,and 2766 cm21 @Fig. 8~f!#, respectively. These modes aassigned to vibrations of C–H bondstrans periplanar to thenitrogen lone pair of the amines. Intense low C–H stretchfrequencies occurring below 2800 cm21 are known as Bohl-mann bands. They originate from the interaction of the nitgen lone pair orbital with the C–Hs-orbitals trans peripla-nar to the lone pair, causing an increase in bond lengthdecrease in bond energies and hence a downshift in vitional frequencies. The downshift in frequencies causedthe trans-lone-pair effect and the effect of methyl substittion on the frequencies of the Bohlmann bands have bstudied in detail by infrared spectroscopy of deuterium sstituted amines.75 Our calculations on gas phase aminshow that the highest occupied molecular orbital~HOMO!involves the overlap between the C–Hs-orbital and the Nlone pair orbital, confirming the orbital interpretation of theffect.
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
e
of-
ee
ace.of-
h-
g
-
aa-y
n-
The spectra obtained at room temperature@Figs. 8~a! to8~c!# are notably different than the spectra of the multilayecondensed at 110 K@Figs. 8~d! to 8~f!#, which indicates thatthe three amines undergo surface reaction with the Si~100!-231 surface at room temperature to form chemisorbed pructs. In the chemisorption spectra of methylamine and dethylamine, sharp C–H peaks of relatively strong intensare observed at 2803 cm21 @Fig. 8~a!# and 2787 cm21 @Fig.8~b!#, respectively. Because these correspond to thequency range of the Bohlmann band, the presence of thmodes after chemisorption indicates that the N lone pairetained in the bound product. This result supports a surfreaction mechanism in which adsorption of both methlamine and dimethylamine is dissociative, such that thetrogen lone pairs can be regenerated upon forming Sbonds to the Si~100!-231 surface. On the other hand, onlyvery weak peak is observed at 2787 cm21 in the chemi-sorbed trimethylamine spectrum@Fig. 8~c!#. This result sug-gests that the chemisorption of trimethylamine must invobonding through the nitrogen lone pair of the molecule. Sua bonding configuration results in the consumption oflone pair in trimethylamine, which is no longer availableperturb thetrans periplanar C–H bonds of the chemisorbemolecule, resulting in weak C–H stretching vibrations belo2800 cm21.
1. Methylamine
The reaction pathway and the bonding configurationthe chemisorbed amines on the Si~100!-231 surface can bededuced from the infrared spectra. In the methylamchemisorption spectrum@Fig. 8~a!#, the appearance of thBohlmann band shows that the methyl group is bound tonitrogen atom of the dissociated methylamine, such that
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ch3y-innioaferbci
in
a
h0
mifjo
tenaielse
bs
ct
ou
anoir
rly.ilietmrose
-
t the
byd thed-
a-
ine
ad-nts.enltser-ce
rmthe
ro-thenme-
–Hineinguldthy-–Hine
ate0%g,
lkybet-the–Hare
ach-
velytetionwocia-a-eof
ithi–Hhy-im-
10176 J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Mui et al.
nitrogen lone pair is able to interact with atrans periplanarC–H bond. Furthermore, the observation of a Si–H streting mode at 2066 cm21 and a N–H stretching mode at 340cm21 indicates that the principal pathway for the methlamine surface reaction occurs by N–H dissociation, leadto the formation of NHCH3(a) species and a Si–H bond othe surface. This is consistent with our theoretical predictthat N–H dissociation is the kinetically favored pathwayter molecular adsorption of methylamine on the Si dimFurthermore, a comparison of the experimental chemisorspectrum to the calculated frequencies of the N–H dissotion product of methylamine shows good [email protected]~a!#, which provides additional evidence to support thisterpretation.
2. Dimethylamine
The IR spectrum of dimethylamine multilayers hasbroad, strong N–H stretching mode at 3225 cm21 but showsno absorption features in the Si–H stretching region~2000cm21 to 2200 cm21!, as expected@Fig. 8~e!#. Upon chemi-sorption at room temperature, the N–H stretching modecompletely disappeared and a Si–H mode appears at 2cm21 @Fig. 8~b!#. These spectral changes indicate that diethylamine undergoes N–H dissociation upon a reaction wthe Si~100!-231 surface, which results in the formation oN~CH3!2(a) species and surface Si–H bonds as the mareaction products. The principal dissociation pathway demined by IR spectroscopy is consistent with our calculatiowhich predict that N–H dissociation is the preferred pathwfor dimethylamine surface reaction. Calculated frequencfor the N–H dissociated product of dimethylamine are ashown below the IR spectrum, and we find excellent agrment between theory and experiment@Fig. 8~b!#. We note,however, that two of the strong C–H stretching peaks~forexample, 2832 cm21 and 2880 cm21! observed in the IRspectrum of chemisorbed dimethylamine are not predictedtheory. Although we are not certain about the origin of thefeatures, we believe that they may originate from effesuch as Fermi resonance, overtones of CH3 deformationmodes or combination bands, which are not included incalculations.
3. Trimethylamine
As discussed above, the attenuation of the Bohlmband in the trimethylamine multilayer spectrum upon expsure at room temperature indicates the loss of the lone pathe chemisorbed product@Figs. 8~c! and 8~f!#. This resultsuggests that the majority of trimethylamine is moleculaadsorbed onto the Si~100!-231 surface through its lone pairThe bonding can be understood as follows. Buckling of scon dimers on the surface results in a charge asymmbetween the two atoms of the dimer. The down Si atowhich has a slight positive charge, can react with the nitgen lone pair of trimethylamine through a Lewis acid-bainteraction to form a molecularly adsorbed trimethylaminOur calculations show that N–CH3 dissociation has an acti
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
-
g
n-.eda-
-
as70-th
rr-s,ys
oe-
yes
r
n-in
-ry,-e.
vation barrier significantly above the vacuum level~Fig. 7!,and this barrier causes the surface reaction to terminate amolecularly adsorbed dative-bonded state.
This molecular adsorption model is further supportedthe close agreement between the observed IR spectra ancalculated vibrational frequencies for the molecularly asorbed precursor state, shown in Fig. 8~c!. Interestingly, thechemisorption spectrum of trimethylamine exhibits a derivtive shaped Si–H feature, with a negative peak at 2091 cm21
and a positive peak at 2035 cm21. A slight negative dip isalso observed at the same frequency in the methylamchemisorption spectrum@Fig. 8~a!#. This derivative featuremay originate from trace contaminants in the chambersorbed onto the surface before or during the experimeUpon adsorption of trimethylamine, the adsorbed hydrogis perturbed, causing a downshift in frequency which resuin the characteristic derivative shaped Si–H feature. Nevtheless, we can not entirely rule out the possibility of traC–H dissociation upon trimethylamine adsorption to fosurface Si–H species, particularly at defect sites onSi~100!-231 surface.
4. Saturation coverage of the amines
Analysis of the peak intensities in the IR spectra pvides further support of our proposed bonding model foramines at the Si~100!-231 surface. We shall first focus othe C–H stretching region in the spectra of chemisorbedthylamine and dimethylamine@Figs. 8~a! and 8~b!#. Assum-ing the transition dipole moments of the methyl groups Cstretching in chemisorbed methylamine and dimethylamare approximately the same, the integrated C–H stretchintensity in the chemisorbed dimethylamine spectrum shobe twice the corresponding area in the chemisorbed melamine spectrum. The integrated intensities of the Cstretching regions in the methylamine and dimethylamspectra are 0.11 and 0.19, respectively. Hence we estimthat the saturation coverage of dimethylamine is about 1lower than that of methylamine. This result is not surprisinconsidering the fact that methylamine is slightly less buthan dimethylamine and thus might be expected to packter at the surface. However, given the potential error inintensity integration and the assumption that the Cstretching transition dipole moments of the two aminesequal, 10% difference is not statistically significant.
Analysis of the Si–H stretching intensities leads tosimilar conclusion. The integrated areas of the Si–H streting regions~2000 cm21 to 2200 cm21! in the methylamineand dimethylamine spectra are 0.050 and 0.047, respecti@Figs. 8~a! and 8~b!#. Since dimethylamine can only generaone surface Si–H due to N–H dissociation, the observathat the integrated Si–H stretching intensities of the tspectra are roughly equal implies that the principal dissotion pathway of methylamine must also involve the formtion of one Si–H bond. This result is only possible if thsurface reaction of methylamine occurs by the cleavageone N–H bond in the methylamine molecule, consistent wour proposed mechanism. By comparing the integrated Sintensities in the chemisorbed methylamine and dimetlamine spectra, we find that the saturation coverage of d
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ehid
i
th–Htio
mehyia-wcimt
im
astith
lebir
rodispchetlite
acCha––Hdreta
ndin
ththH
i–adns
isan-
H-
n-,e itda-iumf aSi
sityH
the
ar-
s a
s an SiatetheneateSi
on
10177J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Reactions of methylamines at the Si(100)-231 surface
ethylamine is about 5% lower than that of methylaminConsidering the accuracy of the intensity integration, trelative saturation coverage is consistent with the valuetermined from integrated C–H stretching intensities.
The integrated C–H stretching intensity in the chemsorbed trimethylamine spectrum is 0.0768@Fig. 8~c!#, whichcorresponds to an integrated area of 0.0256 per megroup. By comparison to the integrated intensity of the Cregion in chemisorbed methylamine spectrum, the saturacoverage of trimethylamine on the Si~100!-231 surface isestimated to be only 25% of the saturation coverage ofthylamine. There is noa priori reason to expect that thsaturation coverage of molecularly chemisorbed trimetlamine would be significantly lower than that of dissoctively chemisorbed methylamine and dimethylamine. Hoever, our calculations suggest that electronic effects induon the surface by the dative bond formed between the trethylamine lone pair and the Si dimer may decrease bothadsorption probability and the saturation coverage of trethylamine on the Si~100!-231 surface.
V. DISCUSSION
Our results show that the reactivity of organic functiongroups at semiconductor surfaces can be altered by subent groups. Specifically, we have demonstrated that melamine and dimethylamine react with the Si~100!-231 sur-face selectively through N–H dissociation, whitrimethylamine, in the absence of N–H bonds, reactsforming a dative bond with the surface through its lone paConsequently, these results suggest that the methyl gcan be used to protect the amine functional group fromsociation at the Si~100!-231 surface. This protecting grousynthetic strategy may be useful in controlling the attament of organic functional groups to semiconductor surfacFor example, we have successfully demonstrated thatmethyl group can be used to protect the amine functionain pyrrolidine from undergoing N–H dissociativchemisorption.76
Our calculations show that although the surface retions of methylamine and dimethylamine involving N–bond cleavage are thermodynamically more favorable tN–H dissociation reactions, the activation energies for Nbond cleavage are significantly higher than those for Ndissociation. This general phenomenon can be understooexamining the geometry of the transition state structuFigure 9 shows the highest occupied molecular orbi~HOMO’s! of the transition states for N–H and N–CH3 dis-sociation of dimethylamine on a Si9H12 one-dimer cluster. Inthe transition state for the N–H dissociation [email protected]~a!#, the N–H bond length is 1.45 Å and the Si–H bolength is 1.83 Å. This means that the N–H bond lengththis transition state is 21% longer than the N–H bond inmolecularly adsorbed state, and the Si–H bond length intransition state is 29% longer than the Si–H bond in thedissociation state. On the other hand, the N–C and the Sbond lengths in the N–C bond cleavage transition state2.35 Å and 3.14 Å@Fig. 9~c!#, which are stretched 36% an63%, respectively, relative to the corresponding N–C aSi–C bonds in the reactant and the product. In other word
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
.se-
-
yl
n
e-
-
-ed-
he-
ltu-y-
y.up-
-s.hey
-
nC
bys.ls
ee
Cre
d, a
significant fraction of the Si–H bond formation energyavailable for N–H bond cleavage in the hydrogen loss trsition state, while the opposite is true for the CH3 loss tran-sition state. This results in the kinetic selectivity of N–cleavage over N–CH3 dissociation of methylamine and dimethylamine on the Si~100!-231 surface.
The selectivity of N–H versus N–CH3 dissociation canbe further visualized by orbital overlap analysis of the trasition states~Fig. 9!. Upon initial adsorption of the aminethe up Si atom in the dimer becomes electron rich sincreceives electron density from the amine lone pair. Thetive bonded state can be viewed as a quaternary ammonion. The N–H dissociation pathway involves the transfer oproton from the ammonium ion to the electron rich upatom through a Lewis acid-base reaction@Figs. 9~a! and9~b!#. The highest occupied molecular orbital~HOMO! ofthe N–H cleavage transition state shows that electron denfrom the Lewis-basic up Si atom interacts with the N–s-bond from the back side of the H atom. The N–Hs-bondbreaking process is facilitated by electron donation fromup Si atom to the N–Hs-antibonding orbital. Hence theN–H dissociation process has a relatively low activation brier.
The N–CH3 dissociation pathway can be considered anucleophilic substitution reaction@Figs. 9~c! and 9~d!#. Inthis case, the electron rich up Si atom of the dimer acts anucleophile, and the amine molecule attached to the dowatom is the leaving group. Inspection of the transition stHOMO shows that the nucleophilic up Si atom attacksmethyl group from its front side, where the leaving amigroup is bonded. The orbital overlap in the transition stresembles an insertion of electron density from the up
FIG. 9. Orbital interpretation of the N–H and N–C dissociation transitistates of dimethylamine:~a! Highest occupied molecular orbital~HOMO!diagram of the N–H dissociation transition state;~b! orbital interaction dur-ing N–H dissociation;~c! HOMO diagram of the N–CH3 dissociation tran-sition state;~d! orbital interaction during N–CH3 dissociation.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

istatryinonsiios
en
etiohyexinnthH
cuhee
ndnec
isdit
enio
tiohy
othoeioeearg
ento atu-
ine
he
-
re-uctsasedhatce.
ontion
ationthy-t. Ifm-er-ingndl in
he–Hine
y-
the
heyedSi
on,m-ndaceablekeries
llyex-
ayb-m-e-
ge-
hy-
the.
10178 J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Mui et al.
atom into the N–C antibonding orbital. This interactionsymmetry forbidden. On the other hand, the back side atof the methyl group by the up Si atom, which is symmeallowed, is energetically not favorable due to steric hdrance of the methyl hydrogens. Furthermore, the N–C bhas to be stretched in order to accommodate the frontnucleophilic attack. As a result, the energy of the transitstate is raised and hence the N–CH3 dissociation process haa relatively high activation barrier.
Table I shows a comparison of calculated reaction engies and activation barriers of the amine surface reactioThe reaction energies and barriers shown are all relativthe molecularly adsorbed state. We find that the activabarriers of N–H and N–C dissociation increases with metsubstitution, and the dissociation reactions become lessthermic when the number of methyl groups in the amincreases. As discussed above, the adsorbed state caviewed as a quaternary ammonium ion. The trends incalculated energetics can therefore be explained by N–1
and N–CH31 bond strengths of the amines. We have cal
lated the proton and methyl carbocation affinities of tamines at the same level of theory used in the surface chistry calculations:
A– H1→A:1H1, ~2!
A– CH31→A:1CH3
1, ~3!
where ‘‘A’’ represents a tetravalent ammonium ion a‘‘ A: ’ ’ represents a trivalent amine leaving group with a lopair. The proton affinity of the amine is the energy of reation ~2!, while the methyl carbocation affinity of the aminethe energy of reaction~3!. We find that both the proton anthe methyl carbocation affinities of the amines increase wthe number of methyl groups present in the amine. This trexplains the lower energy of reaction as methyl substitutincreases for both N–H and N–CH3 dissociation, since theenergy required to detach a proton or methyl carbocafrom the quaternary ammonium ion increases with metsubstitution.
The calculated trends in activation energies are also csistent with the Hammond postulate, which states thatactivation energy decreases when the reaction is mexothermic.77 For N–H dissociation, the reaction energy dcreases with more methyl substitutions, while the activatbarrier increases from ammonia to dimethylamine. Insption of the hydrogen-loss transition state structures revthat the transition state occurs later on the potential ene
TABLE I. Comparison of surface reaction energetics with bond strengThe energies are relative to the molecular chemisorbed precursor statenergies are in kcal/mol.
Si9H12 cluster NH3 NH2CH3 NH~CH3!2 N~CH3!3
H1 affinity 202.6 213.5 220.4 224.7CH3
1 affinity 101.5 112.2 118.1 120.3H-loss barrier 13.6 14.9 15.5H dissociation 232.5 229.3 227.0CH3-loss barrier 41.8 44.0 45.1CH3 dissociation 244.9 240.9 238.8
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
ck
-d
den
r-s.tonlo-
ebe
e
-
m-
-
hdn
nl
n-ere-nc-lsy
surface from ammonia to dimethylamine. In general, whthe reaction is exothermic, a later transition state leadshigher activation energy according to the Hammond poslate. Similar observations can be made for the N–CH3 dis-sociation transition states of methylamine, dimethylamand trimethylamine.
Mulcahy et al. have performed detailed studies on treaction of dimethylamine on the Si~100!-231 surface usingAuger electron spectroscopy~AES! and temperature programmed desorption~TPD! techniques.61 They observed dif-ferent desorption products in the low-dose and high-dosegimes, and proposed that the surface reaction prodformed are coverage dependent. They also proposed, bon their TPD results and thermodynamic arguments, tN–CH3 dissociation of dimethylamine occurs at the surfaHowever, our IR studies show no coverage dependencethe spectra, and our results show that the surface reacpathway of dimethylamine at the Si~100!-231 surface is ki-netically, not thermodynamically, controlled. We note thalthough dimethylamine forms a single surface reactproduct, the subsequent thermal decomposition of dimelamine, as probed in TPD, may be coverage dependenthis is the case, while the IR spectra obtained at room teperature will not show coverage dependence, different thmal desorption products may be observed in TPD, dependon the initial dimethylamine coverage. Further coverage atemperature dependent surface IR studies will be usefuelucidating the surface chemistry of amines at the Si~100!-231 surface.
Additional insights can be gained from examining tSi–H stretching modes of the reaction products. The Sipeak positions in the IR spectra of chemisorbed methylamand dimethylamine~2066 cm21 and 2070 cm21! are signifi-cantly downshifted from the well studied silicon monohdride vibrations at 2087 cm21 and 2099 cm21. Similar shiftsin Si–H stretching vibrations have been observed forreaction of ammonia on the Si~100!-231 surface.78 Thedown shifts are also predicted by our calculations, and toriginate from the electronic effects of the nitrogen adsorbonto the Si atom next to the Si–H bond on the samedimer. Since nitrogen is more electronegative than silicthe nitrogen lone pair of chemisorbed methylamine or diethylamine withdraws electron density from the Si–H boon the neighboring atom of the same dimer upon a surfreaction. This effect decreases the electron density availto form the surface Si–H covalent bonds, resulting in weaSi–H bonds and downshifts in Si–H stretching frequenccompared to the silicon monohydride surface.
The formation of a bridge-bonded NCH3(b) specieswith both Si atoms of a dimer was also explored theoreticaand is discussed above. However, we found little or noperimental evidence for the formation of the NCH3(b) spe-cies on the surface. Although a bridge-bonded product mgive rise to C–H stretching frequencies similar to the oserved spectrum, the N–H stretching mode would be copletely absent in the spectrum since both N–H bonds in mthylamine must be cleaved before the N atom can be bridbonded to two Si atoms to form NCH3(b). This is at oddswith the measured IR spectrum for chemisorbed met
s.All
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ia
one
–Hreae
ec
emn
noc
toxo
kcngtha
tiors
thth
stg
o
thust
onhe
rm
ingonthoreatisheinhythafagsuth
hed toionand
ple--
he-h a
tionolec-ionheacethe
D.uldgcialn.ceille
M.
tes,
.
. B
en-
n-
em.
re,
e
10179J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Reactions of methylamines at the Si(100)-231 surface
lamine. Furthermore, the integrated Si–H stretch intensitythe chemisorbed methylamine and dimethylamine spectrasimilar @Figs. 8~a! and 8~b!#, which suggests that only one Hatom is detached from methylamine upon chemisorptiThe retention of a N–H stretch upon chemisorption of mthylamine together with the fact that the integrated Sistretch intensity of methylamine and dimethylamine asimilar does not support the formation of bridge-bondNCH3(b) species as the principal surface reaction pathwOf course, it is possible that the amount of bridge-bondNCH3(b) species formed on the surface is below our dettion limit.
Our IR studies also show that the saturation coveragtrimethylamine is low compared to methylamine and diethylamine. This can be explained by the nonlocal electroeffects induced on the Si~100!-231 surface during the trim-ethylamine molecular chemisorption process. Widjaja aMusgrave have performed detailed studies on the nonleffects in the initial adsorption of ammonia onto the Si~100!-231 surface.71 They found that the adsorption of ammoniaform the dative bonded precursor state is 20 kcal/mol ethermic using a one-dimer cluster to model the Si~100!-231surface. However, the adsorption energy increases to 26mol when a three-dimer cluster, in which two neighboridimers along the same dimer row are included, is used incalculations. They explained the cluster-size-dependentsorption energy in terms of charge transfer after adsorpfrom the lone pair of ammonia to the neighboring dimealong the dimer row after the adsorption. In contrast,calculated energies of the H-loss transition state andH-dissociation state were found to be independent of clusize, which indicates that charge transfer effects are negible in this reaction pathway.
We expect similar nonlocal effects in the adsorptionalkyl amines onto the Si~100!-231 surface. We anticipateour calculated adsorption energies for the formation ofdative bonded species to increase when a three-dimer clis used to model the surface, and charge transfer fromlone pair to neighboring dimers to occur upon adsorptiThe charge transfer effect is particularly important in tcase of trimethylamine, since its reaction path is trappedthe dative bonded molecular chemisorption state. On foing the dative bond to the Si~100!-231 surface, electronstransfer from the trimethylamine lone pair to the neighbordimers along the dimer row. This redistribution of electrdensity on the surface decreases the electrophilicity ofdown Si atoms in the two neighboring dimers. Adsorptiontrimethylamine onto the less electrophilic Si atoms maysult in less exothermic chemisorption reactions, which clead to less tightly bound adsorbed products. The desorprate of these weakly adsorbed trimethylamine moleculeroom temperature will be much higher compared to ottrimethylamine molecules that are not influenced by theduced electronic effect from neighboring adsorbed trimetlamine. As a consequence, only the strongly bound trimelamine molecules can remain adsorbed onto the surfaceexposure and this may result in the low saturation coverobserved after the trimethylamine surface reaction. Theface reactions of methylamine and dimethylamine, on
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
nre
.-
edy.d-
of-ic
dal
-
al/
ed-n
ee
erli-
f
eterhe.
in-
ef-nonatr--
y-terer-e
other hand, involve dissociation after initial adsorption. Tbinding energies of the dissociated products are expectebe less affected by nonlocal effects of the initial adsorptprocess. This results in strongly bound surface productshence higher saturation coverages.
VI. CONCLUSION
We have studied the surface chemistry of three simmodel amines at the Si~100!-231 surface by density functional theory~DFT! calculations and multiple internal reflection Fourier transform infrared~MIR-FTIR! spectroscopy.We find that methylamine and dimethylamine react with tsurface primarily via N–H dissociation, while trimethylamine is trapped at the molecular adsorbed state througdative bond to the surface. We also find that the saturacoverage of trimethylamine is low relative to the other twamines, and we have explained this observation by the etronic effects induced on the surface from the adsorptprocess. Finally, we provided an orbital interpretation of tselectivity of dissociation pathways observed in the surfreactions, as well as the effect of methyl substituents oncalculated reaction energetics.
ACKNOWLEDGMENTS
We would like to thank Professor Christopher E.Chidsey for insightful discussion and suggestions. We wolike to thank Alan Sonnenfeld and Dr. Christopher A. Klufor experimental assistance. C.B.M. acknowledges finansupport from LSI Logic and the Charles Powell FoundatioS.F.B. acknowledges funding from the National ScienFoundation and the Beckman Foundation. S.F.B is a CamDreyfus Teacher–Scholar.
1C. B. Duke, Chem. Rev.96, 1237~1996!.2J. A. Kubby and J. J. Boland, Surf. Sci. Rep.26, 61 ~1996!.3R. A. Wolkow, Annu. Rev. Phys. Chem.50, 413 ~1999!.4R. J. Hamers, S. K. Coulter, M. D. Ellison, J. S. Hovis, D. F. Padowitz,P. Schwartz, C. M. Greenlief, and J. N. Russell, Acc. Chem. Res.33, 617~2000!.
5S. F. Bent, Surf. Sci.~submitted!.6S. F. Bent, J. Phys. Chem. B~to be submitted!.7J. Yoshinobu, H. Tsuda, M. Onchi, and M. Nishijima, J. Chem. Phys.87,7332 ~1987!.
8M. Nishijima, J. Yoshinobu, H. Tsuda, and M. Onchi, Surf. Sci.192, 383~1987!.
9C. C. Cheng, R. M. Wallace, P. A. Taylor, W. J. Choyke, and J. T. YaJ. Appl. Phys.67, 3693~1990!.
10A. J. Mayne, A. R. Avery, J. Knall, T. S. Jones, G. A. D. Briggs, and WH. Weinberg, Surf. Sci.284, 247 ~1993!.
11H. B. Liu and R. J. Hamers, J. Am. Chem. Soc.119, 7593~1997!.12L. Li, C. Tindall, O. Takaoka, Y. Hasegawa, and T. Sakurai, Phys. Rev
56, 4648~1997!.13U. Birkenheuer, U. Gutdeutsch, N. Rosch, A. Fink, S. Gokhale, D. M
zel, P. Trischberger, and W. Widdra, J. Chem. Phys.108, 9868~1998!.14F. Matsui, H. W. Yeom, I. Matsuda, and T. Ohta, Phys. Rev. B62, 5036
~2000!.15D. C. Sorescu and K. D. Jordan, J. Phys. Chem. B104, 8259~2000!.16S. H. Xu, M. Keeffe, Y. Yang, C. Chen, M. Yu, G. J. Lapeyre, E. Rote
berg, J. Denlinger, and J. T. Yates, Phys. Rev. Lett.84, 939 ~2000!.17R. J. Hamers, J. S. Hovis, S. Lee, H. B. Liu, and J. Shan, J. Phys. Ch
B 101, 1489~1997!.18G. C. Abeln, S. Y. Lee, J. W. Lyding, D. S. Thompson, and J. S. Moo
Appl. Phys. Lett.70, 2747~1997!.19G. P. Lopinski, D. J. Moffatt, D. D. Wayner, and R. A. Wolkow, Natur
392, 909 ~1998!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

ys
bu
.
J
Phy
ci.
W
.
ui,
J.
. B
oc
, J
ell,
lief,
,
tes,
l. B
m.
ett.
. B
10180 J. Chem. Phys., Vol. 114, No. 22, 8 June 2001 Mui et al.
20F. Jolly, F. Bournel, F. Rochet, G. Dufour, F. Sirotti, and A. Taleb, PhRev. B60, 2930~1999!.
21R. Akiyama, T. Matsumoto, and T. Kawai, Phys. Rev. B62, 2034~2000!.22K. Hamaguchi, S. Machida, K. Mukai, Y. Yamashita, and J. Yoshino
Phys. Rev. B62, 7576~2000!.23Q. Liu and R. Hoffmann, J. Am. Chem. Soc.117, 4082~1995!.24R. Konecny and D. J. Doren, Surf. Sci.417, 169 ~1998!.25C. H. Choi and M. S. Gordon, J. Am. Chem. Soc.121, 11311~1999!.26G. P. Lopinski, D. J. Moffatt, D. D. M. Wayner, and R. A. Wolkow, J
Am. Chem. Soc.122, 3548~2000!.27R. Konecny and D. J. Doren, J. Am. Chem. Soc.119, 11098~1997!.28A. V. Teplyakov, M. J. Kong, and S. F. Bent, J. Am. Chem. Soc.119,
11100~1997!.29A. V. Teplyakov, M. J. Kong, and S. F. Bent, J. Chem. Phys.108, 4599
~1998!.30Y. Taguchi, M. Fujisawa, T. Takaoka, T. Okada, and M. Nishijima,
Chem. Phys.95, 6870~1991!.31S. Gokhale, P. Trischberger, D. Menzel, W. Widdra, H. Droge, H.
Steinruck, U. Birkenheuer, U. Gutdeutsch, and N. Rosch, J. Chem. P108, 5554~1998!.
32M. J. Kong, A. V. Teplyakov, J. G. Lyubovitsky, and S. F. Bent, Surf. S411, 286 ~1998!.
33B. Borovsky, M. Krueger, and E. Ganz, Phys. Rev. B57, R4269~1998!.34G. P. Lopinski, D. J. Moffatt, and R. A. Wolkow, Chem. Phys. Lett.282,
305 ~1998!.35P. L. Silvestrelli, F. Ancilotto, and F. Toigo, Phys. Rev. B62, 1596
~2000!.36M. Staufer, U. Birkenheuer, T. Belling, F. Nortemann, N. Rosch,
Widdra, K. L. Kostov, T. Moritz, and D. Menzel, J. Chem. Phys.112,2498 ~2000!.
37J. S. Hovis, H. B. Liu, and R. J. Hamers, J. Phys. Chem. B102, 6873~1998!.
38G. T. Wang, C. Mui, C. B. Musgrave, and S. F. Bent, J. Phys. Chem103, 6803~1999!.
39M. J. Kong, A. V. Teplyakov, J. Jagmohan, J. G. Lyubovitsky, C. Mand S. F. Bent, J. Phys. Chem. B104, 3000~2000!.
40M. P. Schwartz, M. D. Ellison, S. K. Coulter, J. S. Hovis, and R.Hamers, J. Am. Chem. Soc.122, 8529~2000!.
41J. L. Armstrong, E. D. Pylant, and J. M. White, J. Vac. Sci. Technol. A16,123 ~1998!.
42M. D. Ellison, J. S. Hovis, H. B. Liu, and R. J. Hamers, J. Phys. Chem102, 8510~1998!.
43M. D. Ellison and R. J. Hamers, J. Phys. Chem. B103, 6243~1999!.44J. A. Barriocanal and D. J. Doren, J. Vac. Sci. Technol. A18, 1959
~2000!.45A. V. Teplyakov, P. Lal, Y. A. Noah, and S. F. Bent, J. Am. Chem. S
120, 7377~1998!.46G. T. Wang, S. F. Bent, J. N. Russell, J. E. Butler, and M. P. D’Evelyn
Am. Chem. Soc.122, 744 ~2000!.
Downloaded 30 May 2001 to 171.64.126.105. Redistribution subject to A
.
,
.
.s.
.
B
.
.
47J. S. Hovis, S. K. Coulter, R. J. Hamers, M. P. D’Evelyn, J. N. Russand J. E. Butler, J. Am. Chem. Soc.122, 732 ~2000!.
48S. W. Lee, J. S. Hovis, S. K. Coulter, R. J. Hamers, and C. M. GreenSurf. Sci.462, 6 ~2000!.
49C. Mui, S. F. Bent, and C. B. Musgrave, J. Phys. Chem. A104, 2457~2000!.
50M. Fujisawa, Y. Taguchi, Y. Kuwahara, M. Onchi, and M. NishijimaPhys. Rev. B39, 12 918~1989!.
51M. J. Dresser, P. A. Taylor, R. M. Wallace, W. J. Choyke, and J. T. YaSurf. Sci.218, 75 ~1989!.
52T. Takaoka and I. Kusunoki, Surf. Sci.413, 30 ~1998!.53S. H. Lee and M. H. Kang, Phys. Rev. B58, 4903~1998!.54Y. Widjaja, M. M. Mysinger, and C. B. Musgrave, J. Phys. Chem. B104,
2527 ~2000!.55R. M. Rummel and C. Ziegler, Surf. Sci.418, 303 ~1998!.56J. S. Hovis, S. Lee, H. B. Liu, and R. J. Hamers, J. Vac. Sci. Techno
15, 1153~1997!.57H. B. Liu and R. J. Hamers, Surf. Sci.416, 354 ~1998!.58W. F. Bergerson, J. A. Mulder, R. P. Hsung, and X. Y. Zhu, J. Am. Che
Soc.121, 454 ~1999!.59X. Y. Zhu, J. A. Mulder, and W. F. Bergerson, Langmuir15, 8147~1999!.60D. W. Robinson and J. W. Rogers, Appl. Surf. Sci.152, 85 ~1999!.61C. P. A. Mulcahy, A. J. Carman, and S. M. Casey, Surf. Sci.459, 1
~2000!.62M. H. Qiao, Y. Cao, J. F. Deng, and G. Q. Xu, Chem. Phys. Lett.325, 508
~2000!.63P. Hohenberg and W. Kohn, Phys. Rev. B136, 864 ~1964!.64W. Kohn and L. J. Sham, Phys. Rev. A140, 1133~1965!.65M. J. Frisch, G. W. Trucks, H. B. Schlegelet al., GAUSSIAN 98, Revision
A.5, Gaussian, Inc., Pittsburgh, PA, 1998.66A. D. Becke, Phys. Rev. A38, 3098~1988!.67C. T. Lee, W. T. Yang, and R. G. Parr, Phys. Rev. B37, 785 ~1988!.68G. Rauhut and P. Pulay, J. Phys. Chem.99, 3093~1995!.69A. D. Becke, J. Chem. Phys.98, 5648~1993!.70M. J. Kong, K. S. Lee, J. Lyubovitsky, and S. F. Bent, Chem. Phys. L
263, 1 ~1996!.71Y. Widjaja and C. B. Musgrave, Surf. Sci.469, 9 ~2000!.72J. R. Durig, S. F. Bush, and F. G. Baglin, J. Chem. Phys.49, 2106~1968!.73M. J. Buttler and D. C. McKean, Spectrochim. Acta21, 465 ~1965!.74T. D. Goldfarb and B. N. Khare, J. Chem. Phys.46, 3379~1967!.75D. C. McKean and I. A. Ellis, J. Mol. Struct.29, 81 ~1975!.76G. T. Wang, C. Mui, C. B. Musgrave, and S. F. Bent, J. Phys. Chem
105, 3295~2001!.77T. H. Lowry and K. S. Richardson,Mechanism and Theory in Organic
Chemistry, 3rd ed.~Harper Collins, New York, 1987!.78K. T. Queeney, Y. J. Chabal, and K. Raghavachari, Phys. Rev. Lett.86,
1046 ~2001!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
Related Documents











