Rb Regulates Interactions between Hematopoietic Stem Cells and Their Bone Marrow Microenvironment Carl R. Walkley, 1 Jeremy M. Shea, 1 Natalie A. Sims, 2 Louise E. Purton, 3 and Stuart H. Orkin 1,4, * 1 Department of Pediatric Oncology, Dana-Farber Cancer Institute, Division of Hematology/Oncology and Stem Cell Program, Children’s Hospital Boston, Harvard Stem Cell Institute, Harvard Medical School, Boston, MA 02115, USA 2 St. Vincent’s Institute of Medical Research and Department of Medicine at St. Vincent’s Hospital, The University of Melbourne, Fitzroy, Victoria 3065, Australia 3 Center for Regenerative Medicine, Massachusetts General Hospital, Harvard Stem Cell Institute, Boston, MA 02114, USA 4 Howard Hughes Medical Institute, Boston, MA 02115, USA *Correspondence: [email protected] DOI 10.1016/j.cell.2007.03.055 SUMMARY Hematopoiesis is maintained by stem cells (HSCs) that undergo fate decisions by integrat- ing intrinsic and extrinsic signals, with the latter derived from the bone marrow (BM) microenvi- ronment. Cell-cycle regulation can modulate stem cell fate, but it is unknown whether this represents an intrinsic or extrinsic effector of fate decisions. We have investigated the role of the retinoblastoma protein (RB), a central regulator of the cell cycle, in hematopoiesis. Widespread inactivation of RB in the murine hematopoietic system resulted in profound myeloproliferation. HSCs were lost from the BM due to mobilization to extramedullary sites and differentiation. This phenotype was not intrinsic to HSCs, but, rather, was the con- sequence of an RB-dependent interaction between myeloid-derived cells and the micro- environment. These findings demonstrate that myeloproliferation may result from perturbed interactions between hematopoietic cells and the niche. Therefore, RB extrinsically regulates HSCs by maintaining the capacity of the BM to support normal hematopoiesis and HSCs. INTRODUCTION Under homeostatic conditions, the adult hematopoietic system is maintained by a small number of stem cells (HSCs) that reside in the bone marrow in a specialized microenvironment, termed the niche (Adams and Scad- den, 2006; Schofield, 1978). It is within the niche that HSCs undertake fate decisions, including differentiative divisions to generate progenitor cells and self-renewal divisions necessary to sustain HSCs throughout life. Both intrinsic and extrinsic cues are integrated within the niche to maintain effective control over HSCs, ensuring contribution to hematopoiesis without aberrant prolifera- tion (Fuchs et al., 2004; Moore and Lemischka, 2006). Whereas the majority of HSCs are in a slowly dividing state, termed relative quiescence, with a cell-division cycle in the mouse in the range of 2–4 weeks, progenitor cells exhibit rapid cycling (Bradford et al., 1997; Passegue et al., 2005). HSCs can also be stimulated to rapidly enter the cell cycle and contribute to hematopoiesis (Li and Johnson, 1994). In part, the dramatic contrast in cell-cycle status between stem and progenitor cells has led to the hypothesis that cell-cycle regulation plays a fundamentally important role in stem cell fate determination. Decisions to enter the cell cycle are regulated by the G 1 -S phase restriction point (Sherr and Roberts, 2004). The sequential phosphorylation and subsequent inactiva- tion of the retinoblastoma protein (RB) is an important part of this transition (Weinberg, 1995). RB is phosphorylated by cyclin-cyclin-dependent kinase (Cdk) complexes. Sev- eral negative regulators of Cdk activity have been studied in the context of HSC biology. Loss of the Cdk2 inhibitors p21 Cip1 and p27 Kip1 revealed a divergent role in HSC reg- ulation, with loss of p21 Cip1 resulting in a subtle increase in sensitivity to stress-induced exhaustion apparent in vivo after quaternary transplant (Cheng et al., 2000). Loss of p27 Kip1 resulted in a 2-fold increase in the number of long-term repopulating HSCs in addition to an enlarged progenitor compartment (Walkley et al., 2005). Loss of both Cdk4/6 inhibitors p16 Ink4a and p19 ARF revealed a small increase in serial transplant potential (Stepanova and Sorrentino, 2005), with a similar phenotype observed in p16 Ink4a single mutant HSCs (Janzen et al., 2006). Loss of p18 Ink4c resulted in increased HSC repopulation and frequency (Yuan et al., 2004). Collectively, these studies suggest that negative cell- cycle regulators that impact directly on RB-family protein function may influence HSC fate. It is indeterminate if Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1081

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Rb Regulates Interactions betweenHematopoietic Stem Cells and TheirBone Marrow MicroenvironmentCarl R. Walkley,1 Jeremy M. Shea,1 Natalie A. Sims,2 Louise E. Purton,3 and Stuart H. Orkin1,4,*1Department of Pediatric Oncology, Dana-Farber Cancer Institute, Division of Hematology/Oncology and Stem Cell Program,
Children’s Hospital Boston, Harvard Stem Cell Institute, Harvard Medical School, Boston, MA 02115, USA2St. Vincent’s Institute of Medical Research and Department of Medicine at St. Vincent’s Hospital, The University of Melbourne,
Fitzroy, Victoria 3065, Australia3Center for Regenerative Medicine, Massachusetts General Hospital, Harvard Stem Cell Institute, Boston, MA 02114, USA4Howard Hughes Medical Institute, Boston, MA 02115, USA
*Correspondence: [email protected] 10.1016/j.cell.2007.03.055
SUMMARY
Hematopoiesis is maintained by stem cells(HSCs) that undergo fate decisions by integrat-ing intrinsic and extrinsic signals, with the latterderived from the bone marrow (BM) microenvi-ronment. Cell-cycle regulation can modulatestem cell fate, but it is unknown whether thisrepresents an intrinsic or extrinsic effector offate decisions. We have investigated the roleof the retinoblastoma protein (RB), a centralregulator of the cell cycle, in hematopoiesis.Widespread inactivation of RB in the murinehematopoietic system resulted in profoundmyeloproliferation. HSCs were lost from theBM due to mobilization to extramedullary sitesand differentiation. This phenotype was notintrinsic to HSCs, but, rather, was the con-sequence of an RB-dependent interactionbetween myeloid-derived cells and the micro-environment. These findings demonstrate thatmyeloproliferation may result from perturbedinteractions between hematopoietic cells andthe niche. Therefore, RB extrinsically regulatesHSCs by maintaining the capacity of the BMto support normal hematopoiesis and HSCs.
INTRODUCTION
Under homeostatic conditions, the adult hematopoietic
system is maintained by a small number of stem cells
(HSCs) that reside in the bone marrow in a specialized
microenvironment, termed the niche (Adams and Scad-
den, 2006; Schofield, 1978). It is within the niche that
HSCs undertake fate decisions, including differentiative
divisions to generate progenitor cells and self-renewal
divisions necessary to sustain HSCs throughout life.
Both intrinsic and extrinsic cues are integrated within the
niche to maintain effective control over HSCs, ensuring
contribution to hematopoiesis without aberrant prolifera-
tion (Fuchs et al., 2004; Moore and Lemischka, 2006).
Whereas the majority of HSCs are in a slowly dividing
state, termed relative quiescence, with a cell-division
cycle in the mouse in the range of 2–4 weeks, progenitor
cells exhibit rapid cycling (Bradford et al., 1997; Passegue
et al., 2005). HSCs can also be stimulated to rapidly enter
the cell cycle and contribute to hematopoiesis (Li and
Johnson, 1994). In part, the dramatic contrast in cell-cycle
status between stem and progenitor cells has led to the
hypothesis that cell-cycle regulation plays a fundamentally
important role in stem cell fate determination.
Decisions to enter the cell cycle are regulated by the
G1-S phase restriction point (Sherr and Roberts, 2004).
The sequential phosphorylation and subsequent inactiva-
tion of the retinoblastoma protein (RB) is an important part
of this transition (Weinberg, 1995). RB is phosphorylated
by cyclin-cyclin-dependent kinase (Cdk) complexes. Sev-
eral negative regulators of Cdk activity have been studied
in the context of HSC biology. Loss of the Cdk2 inhibitors
p21Cip1 and p27Kip1 revealed a divergent role in HSC reg-
ulation, with loss of p21Cip1 resulting in a subtle increase in
sensitivity to stress-induced exhaustion apparent in vivo
after quaternary transplant (Cheng et al., 2000). Loss of
p27Kip1 resulted in a 2-fold increase in the number of
long-term repopulating HSCs in addition to an enlarged
progenitor compartment (Walkley et al., 2005). Loss of
both Cdk4/6 inhibitors p16Ink4a and p19ARF revealed
a small increase in serial transplant potential (Stepanova
and Sorrentino, 2005), with a similar phenotype observed
in p16Ink4a single mutant HSCs (Janzen et al., 2006). Loss
of p18Ink4c resulted in increased HSC repopulation and
frequency (Yuan et al., 2004).
Collectively, these studies suggest that negative cell-
cycle regulators that impact directly on RB-family protein
function may influence HSC fate. It is indeterminate if
Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1081

these phenotypes reflect intrinsic or extrinsic effects on
HSCs and hematopoiesis, as all studies to date have
utilized nonconditional mutant alleles that are not hemato-
poietic restricted in their effects. The analysis of HSCs
from germ-line deficient animals does not allow for the
clear delineation of intrinsic and extrinsic contribution to
the observed HSC phenotype. Such studies have largely
not accounted for effects on HSC genesis or potentially
defective niche support that affect HSCs prior to trans-
plantation analysis. While serial transplant studies are
suggestive of an intrinsic role for Cdkis in HSC biology,
they do not exclude a role for the environment from which
these cells were removed, necessitating analysis utilizing
hematopoietic restricted deletion. Indeed, a recent study
demonstrated that the p27Kip1�/� microenvironment me-
diates lymphoid expansion observed in the p27Kip1�/�
animals, possibly indicating that the HSC expansion
observed in p27Kip1�/� bone marrow is extrinsic in nature
(Chien et al., 2006; Walkley et al., 2005). This result sug-
gests that cell-cycle regulators may play a role in regulat-
ing the competence of the hematopoietic niche in addition
to intrinsic roles in HSC fate determination.
Recent studies have begun to characterize the adult
bone marrow niche (Schofield, 1978). Osteoblasts appear
to comprise an important component of the HSC niche, as
modulation of osteoblast number and function influences
hematopoiesis and HSC fate via extrinsic mechanisms
(Calvi et al., 2003; Visnjic et al., 2004; Zhang et al., 2003).
Additionally, numerous extrinsic factors modulate HSC
function. These factors include retinoic acid, extracellular
calcium, osteopontin, angiopoietins, and Notch ligands
(Adams et al., 2006; Arai et al., 2004; Purton et al., 2000;
Stier et al., 2005; Varnum-Finney et al., 1998; Zhang
et al., 2006a). Extrinsic regulation of homeostatic HSC
numbers can be dominant to intrinsic cues in vivo. For
example, HSCs engineered to overexpress HoxB4 expand
in vivo only to the level of normal HSCs despite markedly
enhanced in vitro self-renewal and proliferative capacity
(Krosl et al., 2003). Additionally, systemic factors con-
tained in the peripheral blood of young animals may
reactivate self-renewal-associated pathways in progeni-
tors of older animals, suggesting an important role for
extrinsic signaling in stem cell regulation (Conboy et al.,
2005). While these studies have begun to define the
bone marrow niche, little is currently known regarding
molecular regulators of the niche and their role in influenc-
ing HSC fate decisions. Regulatory interactions between
the hematopoietic cells and the nonhematopoietic-derived
microenvironment are largely unknown. Moreover, the
regulators of these potential interactions and how they
affect hematopoiesis and HSC function are unexplored.
Here we have utilized a conditional deletion strategy to
investigate the role of the RB in the regulation of adult HSC
fate. We found that widespread inactivation of Rb resulted
in the development of a myeloproliferative disease, char-
acterized by extramedullary hematopoiesis and mobiliza-
tion of primitive cells into the periphery. HSCs were lost
from the BM as a result of increased differentiation and
1082 Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc.
mobilization from the BM. The phenotype is not HSC
intrinsic, as it was not recapitulated upon inactivation of
RB in HSCs maintained in a wild-type environment (Walk-
ley and Orkin, 2006). Strikingly, however, concomitant
deletion of Rb from myeloid-derived cells and the micro-
environment generated the myeloproliferative disorder,
thereby demonstrating that RB is an essential regulator
of the interaction between myeloid-derived cells and the
BM microenvironment. Thus, RB extrinsically controls
HSCs by maintaining the competence of the BM to sup-
port normal HSCs and hematopoiesis.
RESULTS
Rb Deletion Leads to Myleoproliferation
Rb was inactivated in hematopoietic cells, including
HSCs, using the interferon-inducible Mx-Cre transgene
and pRbfl/fl animals (Kuhn et al., 1995; Sage et al., 2003;
Walkley and Orkin, 2006). We performed PCR on both
genomic DNA and cDNA from whole BM samples to
confirm Rb deletion (BM, Figures 1A and 1B). Rb was
quantitatively and stably deleted from hematopoietic
cells, and expression of the related p130 and p107 was
not altered as a result of Rb loss. Thus, with this condi-
tional system, we achieve specific loss of Rb without
compensatory gain of expression of other genes coding
for pocket proteins.
Analysis of the peripheral blood of control (Mx-Cre�pRbfl/fl,
pIpC injected) and RbD/D animals following pIpC treatment
revealed that RbD/D animals developed a mild but stable
anemia immediately following Rb deletion (C.R.W and
S.H.O, unpublished data) and by 6 weeks developed throm-
bocytosis (Figure 1C). By 4 weeks, RbD/D animals developed
pan-leukocytosis (Figure 1D) that was accompanied by
elevated levels of circulating progenitor cells, as determined
by in vitro colony-forming capacity (CFU-GEMM and CFU-
G/GM) and phenotypic staining (lin�c-Kit+Sca-1+, LKS+;
Figures 1E and 1F; Okada et al., 1992). Although leukocytosis
was apparent by 4 weeks post-Rb deletion, increased circu-
lating progenitors could be detected as early as 2 weeks after
pIpC (LKS+ increased 3.7-fold, p % 0.01, n = 7 per genotype;
CFU-GEMM increased 2.4-fold, p % 0.01, CFU-M/GM
increased3.9-fold,p%0.01,n=6pergenotype).Surprisingly,
the levels of circulating progenitors were comparable to those
achieved during pharmacologically induced mobilization of
stem and progenitors in the C57Bl/6 strain background
(Ghiaur et al., 2006). However this was a chronic, rather than
an acute, response in the RbD/D mutant.
BM cellularity was not initially altered; however, at 12
weeks post-pIpC it was increased by 40% in RbD/D
animals (Figure 2A). We observed the rapid development
of a myeloproliferative-like disease within the bone
marrow. The phenotype was fully penetrant and was char-
acterized by myeloid hyperplasia (predominantly neutro-
philia) and suppression of both B-lymphopoiesis and
erythropoiesis (Figures 2B, S1, and S2). Phenotypic
stem and primitive progenitor populations (LKS� and
LKS+) were increased significantly in the BM of RbD/D

Figure 1. Rapid Mobilization of Primitive Cells into the Peripheral Blood Following Deletion of Rb
(A) Genomic PCR on whole BM from control (Mx�pRbfl/fl) and Rb-deficient animals (Mx+pRbD/D) at 6 and 12 weeks post-pIpC.
(B) qRT-PCR for pRb, p130, and p107 on cDNA of control and Rb-deficient animals (n = 3 independent samples) 12 weeks post-pIpC.
(C) Platelets and (D) leukocytes in PB following Rb deletion (time 0 = final dose of pIpC); n R 4/time point; *p < 0.05.
(E) Day 12 CFU-GEMM and CFU-GM/M from the PB of at 12 weeks post-pIpC; n R 9/genotype; *p < 0.01. Value inside bars represents fold increase.
(F) FACS profile and mean number of Lin�c-Kit+Sca-1+ (LKS+) in the PB; n > 4/genotype; *p < 0.01. Methylcellulose plates from day 12 of culture. Data
expressed as mean ± SEM.
animals (Figure 2C); however, the number of pheno-
typic HSCs per femur was not significantly altered
(LKS+CD34�/lo; Osawa et al., 1996; Yang et al., 2005). In
addition, RbD/D animals exhibited striking changes in the
architecture of the bone, evidenced by loss of trabecular
bone (Figure 2D). Trabecular bone is thought to represent
an important niche for HSCs within the BM (Calvi et al.,
2003). Quantitative histomorphometric analysis of the
bone at 2 weeks post-pIpC, a time point correlating with
the presence of progenitors in the peripheral blood and
spleen, demonstrated a significant reduction in trabecular
volume as a proportion of total marrow volume, a �40%
reduction in the number of trabeculae, and a doubling of
the separation of trabeculae (Figures 2E–2H).
In parallel with BM myeloproiferation, RbD/D animals de-
veloped extensive extramedullary hematopoiesis. Spleen
weight increased rapidly by 5.5-fold relative to controls
due to expanded numbers of myeloid cells, megakaryo-
cytes, and erythroid cells (Figures 2I and 2J). B- and
T cell lymphopoiesis were present at comparable levels
in RbD/D and control spleens. Phenotypic stem and pro-
genitor populations (LKS+ and LKS�) increased progres-
sively in the spleens of RbD/D animals, and by 12 weeks
were increased 45- to 50-fold (Figures 2K, S3, and S4).
Splenic architecture was effaced as a result of myeloid
and erythroid elements (Figure 2J). Hematopoietic foci
were also observed in the liver but not in the kidney
(data not shown). Despite chronic myeloproliferation, no
hematopoietic tumors have developed during the lifespan
of RbD/D mutant animals. RbD/D animals survive for ap-
proximately 8 months post-pIpC; heterozygous animals
are normal (Figures S5 and S6). Eight-month-old RbD/D
animals present with a phenotype reminiscent of hemato-
poietic failure, characterized by a significant reduction in
Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1083

Figure 2. Myeloproliferation Following Rb Deletion
(A) Femoral cellularity, n R 3/genotype/time point.
(B) Number of cells of each lineage/femur at 12 weeks post-pIpC; Granulocytes CD11b+Gr-1+, Macrophages CD11b+F4/80+, Immature B lymphoid
IgM�B220+, Mature B lymphoid IgM+B220+, Mature Erythroid CD71�Ter119+, and Immature Erythroid CD71+Ter119+; n R 6/genotype; *p < 0.01.
(C) Number of phenotypic HSCs (LKS+CD34�/lo) and primitive progenitors/femur; 12 weeks post-pIpC; n R 5/genotype; *p < 0.05.
(D) Representative sections of tibiae at 12 weeks post-pIpC.
(E) Volume of marrow space occupied by bone (BV/TV); 2 weeks post-pIpC; n R 13/genotype; *p < 0.05.
(F) Trabecular number/mm; *p < 0.05.
(G) Separation of trabeculae; *p < 0.05.
(H) Representative longitudinal sections of tibiae stained with Von Kossa technique (mineralized bone stained black).
(I) Spleen cellularity; n R 3/genotype/time point; *p < 0.01.
1084 Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc.

spleen weight and replacement of BM by granulocytes;
however, pituitary tumors are also observed (Figure S5
and data not shown).
HSCs Are Lost from BM following Rb Deletion
RB and other negative cell-cycle regulators have been
postulated to play an important role in the regulation of
HSCs and in the subsequent hematopoiesis. However,
neither the myeloproliferative disease nor defective HSC
function was observed when Rb was deleted from HSCs
in the context of a wild-type microenvironment (Walkley
and Orkin, 2006). Given the striking phenotype we
observed when Rb was deleted from both hematopoietic
cells and the BM microenvironment, as occurs with
Mx-Cre (Zhang et al., 2003), we sought to determine the
consequences of RB loss on HSCs in these animals.
Within the BM we observed a significant increase in the
frequency of mature day 7 colony-forming cells but a de-
crease in the frequency of the more primitive in vivo day 12
colony-forming unit-spleen (CFU-S12, Figures 3A and 3B).
The numbers of both in vitro colony-forming cells (68-fold
increase in CFU-GEMM) and CFU-S8 in the spleen were
markedly increased (Figures 3C and 3D). As we observed
high levels of circulating progenitors and substantial levels
of progenitor activity in the spleen, we sought to determine
if latent HSC activity was also present in extramedullary
sites. Whole spleen cells from RbD/D animals (either 1 3
106 or 2 3 106) were transplanted with competitor whole
BM (2 3 105) into congenic recipients. At 17 weeks post-
transplant, significant multilineage repopulating activity
derived from the spleens of RbD/D animals was present,
demonstrating that functional HSCs were present in the
periphery of RbD/D mice (Figures 3E and 3F).
To determine the HSC content of the BM, we performed
limit-dilution competitive repopulation analysis with whole
BM from donor animals treated 12 weeks earlier with pIpC
(Purton et al., 2006; Szilvassy et al., 1990; Walkley et al.,
2005). Whole BM from control (Rbfl/fl) or Rb-deficient
(RbD/D, both CD45.2+) mice was mixed at varying doses
with a fixed number of competitor BM cells (CD45.1+/
CD45.2+) and transplanted into congenic recipient ani-
mals (CD45.1+). The frequency of long-term repopulating
HSCs in the RbD/D BM at 6 months posttransplant was re-
duced by 8-fold (p = 0.0005). When normalized to reflect
the increased cellularity of the RbD/D BM, this represents
a 5-fold decrease in the absolute number of HSCs per
femur (Figure 4A). Secondary transplantation demon-
strated that RbD/D HSCs were serially transplantable and
capable of stable multilineage contribution for at least
3 months. Importantly, we did not observe a progressive
decline in contribution from RbD/D HSCs to hematopoie-
sis, thereby demonstrating that self-renewal-mediated
maintenance of HSCs over time is not affected by the
absence of RB (Figure S7).
C
When 1000 freshly isolated lin�c-Kit+Sca-1+ cells were
competitively transplanted, RbD/D LKS+ displayed a
20-fold reduction in long-term repopulating potential on
a per-cell basis (Figure 4A). The reduced repopulating
potential of the RbD/D LKS+ fraction, despite a marked
increase in this phenotypic population observed in the
bone marrow (see Figure 2), demonstrates that the sur-
face phenotype of the cells does not faithfully reflect their
functional potential. We, and others, have previously
observed a lack of fidelity of phenotypic markers both in
mutant mice and following perturbation of homeostasis
in wild-type animals (Purton et al., 2006; Spangrude
et al., 1995; Tajima et al., 2000; Walkley et al., 2005).
HSCs may be lost from the BM for several reasons,
including a failed capacity of RbD/D HSCs to home and
engraft following transplantation, an increased rate of
apoptosis, or a mobilization/redistribution to extramedul-
lary sites. As cell-cycle status correlates with engraftment
capacity of HSCs (Gothot et al., 1998; Passegue et al.,
2005), we directly assessed the cell-cycle status of phe-
notypic RbD/D progenitors (LKS�), primitive progenitors
(LKS+), and HSCs (LKS+CD34�/lo). All three populations
displayed a comparable cell-cycle profile, with HSCs
from RbD/D displaying the same distribution of cells in
the G0/G1 or S phase of the cell cycle as control cells (fl/
fl: G0/G1 = 86.2 ± 5.7%; D/D: G0/G1 = 85.1 ± 2.4%, p =
0.85, fl/fl: S = 6.7 ± 1.9%; D/D: S = 7.8 ± 0.7%, p = 0.54,
n = 4 fl/fl, 7 D/D; expressed as mean ± SEM). There was
no difference in the rate of cell-cycle entry of LKS+ cells
between control and RbD/D cells as determined by
BrdU-incorporation rates at either 2 or 4 weeks post-
pIpC (Figure S9). Furthermore, analysis of the in vivo hom-
ing of RbD/D BM did not reveal a difference compared to
control BM at either 2 or 12 weeks post-pIpC (Figure 4B
and data not shown). RbD/D LKS+ cells exhibited
decreased apoptosis, as assessed by annexin-V staining,
at 2 weeks and exhibited normal levels at 12 weeks post-
pIpC compared to control LKS+ cells (Figure 4C). As we
had observed significantly increased progenitors and
HSCs in extramedullary sites, these data are consistent
with the loss of HSCs from the BM as a result of both
enhanced differentiation of HSCs within the BM and a
redistribution to extramedullary sites as a consequence
of the changes in the niche.
Myeloid-Restricted Inactivation of Rb Does
Not Result in Myeloproliferation
To determine the contribution of myeloid-derived cells
(granulocytes, macrophages, and osteoclasts) to the
myeloproliferation observed in Rb mutants, we generated
Lysozyme-M-Cre pRbfl/fl mice to achieve myeloid-
restricted deletion of Rb (Figure 5). Deletion of Rb with
Lys-M-Cre did not lead to myeloproliferation or extra-
medullary hematopoiesis, consistent with the results
(J) Number of cells of each lineage/spleen; 12 weeks post-pIpC; n R 6/genotype; *p < 0.01.
(K) Fold change in phenotypic LKS+ and LKS� in the spleen; n R 3/genotype/time point; p < 0.05.
(L) Representative spleen sections (12 weeks post-pIpC). Unless noted all data are expressed as mean ± SEM.
ell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1085

Figure 3. Increase in Progenitors in the
Bone Marrow and HSCs/Progenitors in
the Spleen of RbD/D Animals
(A) Number of day 7 CFC per femur; n R 5/ge-
notype/time point; *p < 0.05.
(B) Number of CFU-S12 from 1 3 105 whole BM
cells; n = 5/genotype.
(C) Splenic day 12 CFU-GEMM and CFU-GM/M
at 12 weeks post-pIpC; n R 9/genotype; *p <
0.01. Value inside bars is fold increase.
(D) CFU-S8/spleen; n = 5/genotype; p < 0.05.
(E) Percent PB chimerism at 17 weeks post-
transplant from either 1 3 106 or 2 3 106 whole
spleen cells from RbD/D animals 8 weeks post-
pIpC with 2 3 105 WT BM cells; n = 5/genotype.
(F) Lineage contribution of spleen-derived
HSCs at 17 weeks. Data are expressed as
mean ± SEM.
obtained by deletion of Rb from hematopoietic cells in
a wild-type environment (Figure 5A; Walkley and Orkin,
2006). We observed a subtle increase in the numbers of
granulocytes in the BM and slight reduction in erythroid
cells, but no change in either lymphoid or phenotypic
progenitor and HSC-enriched fractions (Figures 5A and
5B). Lineage distribution within the spleen or PB was
largely comparable to controls (Tables S1 and S2; data
not shown). Thus, deletion of Rb from myeloid-derived
populations does not recapitulate the phenotype observed
in the Mx-Cre model. Collectively, these results suggest
that RB may regulate HSCs and hematopoiesis in an
1086 Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc.
extrinsic manner, possibly through regulating the compe-
tence of the bone marrow niche.
An Rb-Dependent Interaction between
Myeloid-Derived Cells and the BM
Microenvironment Results in Myeloproliferation
We next sought to determine the relative contributions of
the hematopoietic cells and the nonhematopoietic (non-
transplantable) elements of the BM microenvironment
to the observed phenotype. Hematopoietic cells alone
were not capable of inducing either myeloproliferation or
the loss of HSCs from the BM that we observed in the

Figure 4. Loss of HSCs from the Bone
Marrow Following Rb Deletion
(A) HSC frequency and absolute number/
femur; n = 4–5 recipients/cell dose/genotype;
experiment was performed twice; data were
pooled from two independent experiments for
calculation of HSC frequency. Primary trans-
plant data are from 6 months posttransplant.
Secondary transplant data are from 3 months
posttransplant. One thousand freshly isolated
LKS+ were transplanted, and repopulating
unit (RU) was calculated 6 months posttrans-
plant; n = 5 recipients/cell dose/genotype.
(B) Number of CFDA-SE-labeled cells in the BM
of recipients 16 hr after injection; BM from 12
weeks post-pIpC; n = 5 recipients/genotype.
(C) Apoptotic cells in the LKS+ and LKS�populations; n = 9/genotype; p < 0.05. Data
are expressed as mean ± SEM.
Mx-CrepRbD/D model (Walkley and Orkin, 2006). Consis-
tent with this conclusion we did not observe myeloproli-
feration when previously excised RbD/D HSCs were
supported by a wild-type microenvironment, even at
high cell doses (described in Figure 3).
To ascertain if RB loss from the niche was responsible
for the myeloproliferation and loss of BM HSCs, reciprocal
transplants of wild-type hematopoietic cells into lethally
irradiated Mx-Cre�pRbfl/fl and Mx-Cre+pRbfl/fl recipients
were performed, and, following establishment of hemato-
poiesis, recipients were injected with pIpC to delete Rb
from the hematopoietic microenvironment. This strategy
was successfully used to demonstrate a role for BMP
receptor type 1 in the regulation of the HSC niche (Zhang
et al., 2003), and a similar approach demonstrated that
a RARg�/� microenvironment alone could induce myelo-
proliferation (Walkley et al., 2007 [this issue of Cell]). Fol-
lowing inactivation of Rb, recipients were monitored and
analyzed at 8 and 20 weeks posttransplant. We failed to
observe significant changes in hematological parameters
of these recipients (Figure 6A). The data show that loss of
RB uniquely in either hematopoietic cells or niche cells
alone is insufficient to account for the findings in
Mx-Cre+pRbD/D mice.
Bone homeostasis is maintained through balanced
activities of mesenchymal-derived osteoblasts and
myeloid-derived osteoclasts (Martin and Sims, 2005). As
we observed a rapid loss of trabecular bone following
Mx-Cre-mediated deletion, we quantitated the numbers
of osteoclasts present in the BM and spleen. The numbers
of osteoclasts in both the BM and spleen by 6 weeks post-
pIpC were markedly increased (Figures 5C and 5D).
Osteoclasts and macrophages derived from pIpC-treated
Mx-Cre mice showed efficient deletion of Rb, as did
osteoclasts and macrophages derived from Lysozyme-
M-Cre pRbfl/fl mice (Figure 5E). Osteoclasts have been
proposed to contribute to the release of HSCs from the
bone marrow during mobilization (Kollet et al., 2006).
Having observed significantly increased neutrophils and
monocytic-derived osteoclasts in the Mx-Cre+pRbD/D
model (Figure 5C), we hypothesized that deletion of Rb
from myeloid-derived cells together with an Rb-deficient
microenvironment might recapitulate the phenotype
observed in the Mx-Cre+pRbD/D model.
Hematopoietic cells from sex-mismatched Lysozyme-
M-Cre pRbfl/fl animals were transplanted into lethally
irradiated Mx-Cre�pRbfl/fl and Mx-Cre+pRbfl/fl recipients,
and, following establishment of hematopoiesis, recipi-
ents were injected with pIpC to delete Rb from the
BM microenvironment. Analysis of Y chromosome levels
by quantitative PCR on peripheral blood leukocytes
confirmed engraftment and high-level chimerism of all
recipients prior to pIpC and at the time of analysis
(data not shown). This transplant strategy results in Rb
deficiency in myeloid-derived cells (granulocytes, mac-
rophages, and osteoclasts) and an Rb-deficient niche.
In addition, RB expression is retained within the HSC
compartment.
We observed synergistic interaction between the pRbD/D
myeloid cells and the pRbD/D microenvironment (Figure 6).
Mx-Cre+pRbD/D recipients rapidly developed signs of
distress as early as 2 weeks after the completion of
Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1087
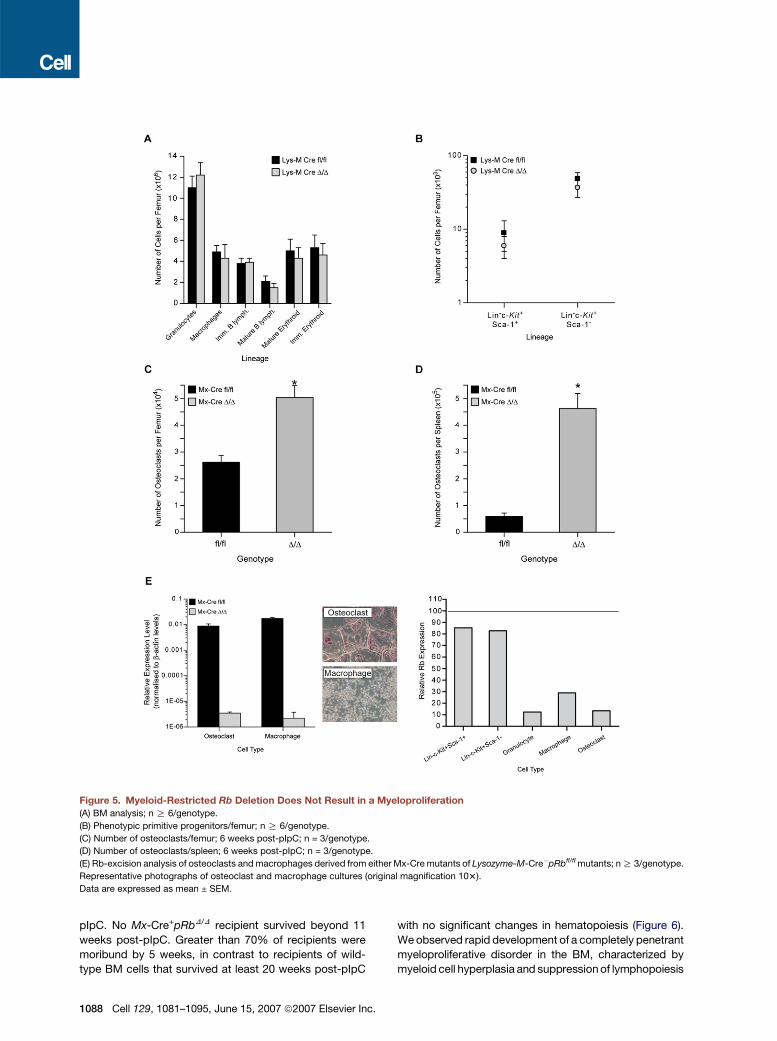
Figure 5. Myeloid-Restricted Rb Deletion Does Not Result in a Myeloproliferation
(A) BM analysis; n R 6/genotype.
(B) Phenotypic primitive progenitors/femur; n R 6/genotype.
(C) Number of osteoclasts/femur; 6 weeks post-pIpC; n = 3/genotype.
(D) Number of osteoclasts/spleen; 6 weeks post-pIpC; n = 3/genotype.
(E) Rb-excision analysis of osteoclasts and macrophages derived from either Mx-Cre mutants of Lysozyme-M-Cre�pRbfl/fl mutants; n R 3/genotype.
Representative photographs of osteoclast and macrophage cultures (original magnification 103).
Data are expressed as mean ± SEM.
pIpC. No Mx-Cre+pRbD/D recipient survived beyond 11
weeks post-pIpC. Greater than 70% of recipients were
moribund by 5 weeks, in contrast to recipients of wild-
type BM cells that survived at least 20 weeks post-pIpC
1088 Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc.
with no significant changes in hematopoiesis (Figure 6).
We observed rapid development of a completely penetrant
myeloproliferative disorder in the BM, characterized by
myeloid cell hyperplasia and suppression of lymphopoiesis

Figure 6. A pRb-Dependent Interaction between Myeloid Cells and the Bone Marrow Microenvironment Causes Myeloprolifera-
tion
(A) Wild-type BM was transplanted into unexcised Mx-Cre�pRbfl/fl and Mx-Cre+pRbfl/fl recipients, and 5 weeks posttransplant recipients received
pIpC. Recipients were analyzed at 8 (n = 3/genotype) and 20 weeks (n = 4/genotype) post-pIpC. Data are expressed as fold change of Mx+ recipients
compared to Mx�Cre�pRbfl/fl recipients (normalized to 100%). Data are shown from 8 weeks post-pIpC (comparable results at 20 weeks).
(B) Sex-mismatched Lysozyme-M-Cre+pRbfl/fl BM was transplanted into unexcised Mx-Cre�pRbfl/fl and Mx-Cre+pRbfl/fl recipients, and 5 weeks post-
transplant pIpC was administered. Data are expressed as fold change of Mx+ recipients compared to Mx-Cre�pRbfl/fl recipients (normalized to 100%).
y = approximate time of analysis post-pIpC; n = 11 Mx-Cre+ recipients in three independent experiments (two found dead 2 weeks post-pIpC); n = 10
Mx-Cre� control recipients.
(C) Comparison of spleen weights amongst groups.
(D) Splenic hematopoiesis in Mx-Cre+pRbfl/fl recipients of Lysozyme-M-Cre+pRbfl/fl bone marrow. Data are expressed as fold change compared to
Mx-Cre�pRbfl/fl recipients (normalized to 100%).
and erythropoiesis. Splenomegaly, accompanied by mye-
loid and erythroid hyperplasia and extramedullary hema-
topoiesis, was also observed, demonstrating a striking
similarity to the full Mx-Cre model (see Figure 2 and
Tables S1 and S2). The increased spleen size of Mx-
Cre� recipients of LysM-Cre pRbfl/fl BM can be accounted
for by the LysM-Cre pRbfl/fl BM itself, rather than a
contribution from the recipient environment (Tables S1
Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1089

and S2). These data demonstrate that the observed mye-
loproliferation is the consequence of an RB-dependent
interaction between myeloid-derived cells and the BM
microenvironment and that it develops independent of the
HSC RB status. Our data provide direct experimental
evidence that myeloproliferation may ensue from aberrant
interactions between myeloid-derived cells and the BM
microenvironment, revealing hematopoietic extrinsic con-
tribution to myeloproliferation.
DISCUSSION
We sought to determine the role RB plays in the regulation
of hematopoiesis and stem cell function. Recent studies
suggest that cell-cycle regulation is an important determi-
nant of stem cell fate; however, none have discriminated
between intrinsic or extrinsic contributions (Cheng et al.,
2000; Janzen et al., 2006; Walkley et al., 2005; Yuan
et al., 2004). RB was implicated as an important regulator
of stem cell maintenance in Arabidopsis; however, the
limitations of the experimental system did not allow for
the clear demonstration of a stem cell intrinsic role for
RB (Wildwater et al., 2005). Here we demonstrate that
RB extrinsically regulates HSCs by maintaining the com-
petence of the adult bone marrow to support HSCs and,
in turn, normal homeostatic hematopoiesis.
Rb and Stem Cell Self-Renewal
Understanding the regulation of cell cycle in stem cells is
important from several perspectives. Stem cells must en-
ter the cell cycle to self-renew; hence, induction of cycling
may be desirable to achieve HSC expansion. Engraftment
of transplanted HSCs is cell cycle dependent (Gothot
et al., 1998; Passegue et al., 2005). The slow cycling of
HSCs may spare them from acute toxicity (such as che-
motherapy) but may also prevent neoplastic cells from
eradication (Hodgson and Bradley, 1979; Lerner and
Harrison, 1990). Our understanding of the normal regula-
tion of self-renewal will also provide insight into tumori-
genesis, where self-renewal pathways are thought to be
active (Krivtsov et al., 2006).
The importance of cell-cycle regulation in HSC fate
decisions has been suggested by the analysis of animals
deficient in negative cell-cycle regulators such as
p21Cip1, p27Kip1, and p16INK4a/p19ARF (Cheng et al.,
2000; Stepanova and Sorrentino, 2005; Walkley et al.,
2005). However, these studies have not revealed if such
HSC defects are cell intrinsic or extrinsic in nature. The
‘‘Rb pathway’’ has also been implicated in phenotypes ob-
served in both the Bmi1�/� and ATM�/� HSCs (Ito et al.,
2004; Lessard and Sauvageau, 2003; Park et al., 2003).
Surprisingly, we did not observe an intrinsic requirement
for Rb in HSCs. If provided with a wild-type niche, RbD/D
HSCs contribute normally to multilineage hematopoiesis
and display serial transplant potential comparable to
wild-type HSCs. Furthermore, we failed to detect alter-
ations in numerous cell-cycle- or self-renewal-associated
genes in Rb-deficient HSCs and progenitors isolated from
1090 Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc.
both a wild-type and mutant microenvironment, consis-
tent with our interpretation that Rb is dispensable in the
HSCs (Figure S8). Our observations, taken together with
those from analysis of p27Kip1 mutant mice, reveal that
cell-cycle regulation is a novel extrinsic regulator of hema-
topoiesis (Chien et al., 2006; Walkley and Orkin, 2006). The
loss of BM HSCs in the Mx-Cre model is a secondary
consequence of the disrupted environment within the
BM and was not observed in the context of a wild-type
niche, demonstrating that myeloproliferative-like disor-
ders may deplete HSCs from the BM. A reanalysis of
cell-cycle mutants proposed to harbor HSC defects is
needed to clarify the intrinsic and extrinsic roles that
cell-cycle regulation plays in these phenotypes.
It has been documented that self-renewal of embryonic
stem cells occurs in an RB independent manner (Stead
et al., 2002); however, the RB pathway is thought to be
near universally targeted in human cancer cells (Hanahan
and Weinberg, 2000). We have described that self-renewal
of nontransformed HSCs occurs independent of RB, high-
lighting the important question of what role RB plays in the
regulation of the process of cellular self-renewal in both
normal and oncogenic settings. It may be that the require-
ment for RB and the RB pathway in self-renewal is devel-
opmentally and lineage dependent, with progenitor cells
having a greater dependence on RB for their division
than bona fide stem cells. One prediction of such a hypoth-
esis is that mutation of the RB pathway is of greater benefit
to a progenitor cell than a stem cell during tumor formation.
Rb and Hematopoiesis
Previous studies examining the role of RB in hematopoie-
sis have raised conflicting evidence regarding intrinsic and
extrinsic effects, particularly in erythropoiesis (Clark et al.,
2004; Iavarone et al., 2004; Spike et al., 2004; Whyatt
and Grosveld, 2002). Our study utilized compartment-
restricted somatic mutagenesis to analyze the role of
RB in adult hematopoiesis and HSCs. Compartment
restricted deletion enables a direct assessment of the
contribution of hematopoietic and nonhematopoietic cells
to the observed phenotype. We have not observed a
progressive failure of hematopoiesis as reported by Spike
et al. (2004) when RB-deficient HSCs were supported by
a wild-type environment, nor was this observed in a
separate study utilizing germline-deficient fetal liver
hematopoietic cells (Hu et al., 1997). In contrast to in vitro
findings (Iavarone et al., 2004), deletion of Rb from
myeloid-derived cells using Lys-M-Cre did not result in
anemia in vivo. Further studies utilizing lineage-restricted
deletion of Rb will be required to clarify the role of RB in
erythropoiesis.
Interactions between Hematopoietic Cells
and Their Microenvironment Regulate HSCs
and Hematopoiesis
The phenotype of Mx-Cre+pRbD/D animals is due to an
RB-dependent interaction between myeloid-derived cells
(most probably macrophages and osteoclasts) and the

Table 1. Summary of the Phenotype Observed Following Loss of Rb
Bone Marrow
Condition Hematopoietic CellsNiche /Microenvironment HSC Myeloid Lymphoid Erythroid
Mx-Cre pRbfl/fl model D/D D/D YYY [[[ Y 4
D/D HSC into WT nichea D/Db WT 4 [ 4 4
WT HSC into D/D niche WT D/Db 4 4 4 4
D/D myeloid cells D/D (myeloid) WT 4 4 4 4
D/D myeloid
cells into D/D niche
D/D (myeloid) D/Db [[[ Y Y
Spleen and Extramedullary Sites
Condition Hematopoietic Cells
Niche /
Microenvironment HSC / Progenitorsc Myeloid Lymphoid Erythroid
Mx-Cre pRbfl/fl model D/D D/D [[[ [[[ 4 [[[
D/D HSC into WT nichea D/Db WT 4 4 4 4
WT HSC into D/D niche WT D/Db 4 4 4 4
D/D myeloid cells D/D (myeloid) WT 4 4 4 [
D/D myeloid
cells into D/D niche
D/D (myeloid) D/Db [[[ [[[ 4 [[[
a Summary of data previously described (Walkley and Orkin, 2006).b Indicated compartment was nondeleted (Mx-Cre pRbfl/fl) at time of transplant and deleted 5 weeks after transplant with pIpC
injection.c HSC and Progenitors as determined by flow cytometry (Lin�c-Kit+Sca-1+ and Lin�c-Kit+Sca-1�) and in vitro progenitor analysis.
bone marrow microenvironment (summarized in Tables 1
and S1). Evidence supports a direct role for the bone mar-
row microenvironment, but we cannot entirely exclude
a contribution from other sites of Cre activity in the Mx-
Cre model. Myeloproliferation is generally considered to
be hematopoietic intrinsic, and evidence from the overex-
pression of activated kinase receptors in mouse models is
consistent with this view (Araki et al., 2004; Chan et al.,
2004; Le et al., 2004). In light of the data derived from
Mx-Cre+pRbD/D mice, the BM microenvironment may
play an active role in the promotion and/or maintenance
of myeloproliferative disorders. Additional studies are
required to define the cell(s) within the BM niche that are
responsible for this interaction. The BM microenvironment
is composed of numerous nonhematopoietic cell types
including osteoblasts, endothelial cells, adipocytes, and
nerve cells. Histomorphometry demonstrated a significant
disruption to bone homeostasis in the Rb-deficient
animals, correlating with the observed mobilization and
extramedullary hematopoiesis (Figures 2E–2H). Myelo-
proliferation in Mx-Cre+pRbD/D mice is dependent on con-
comitant deletion of Rb from both myeloid-derived cells
and the environment. In other situations, myeloprolifera-
tion may result directly from an aberrant niche and may
be independent on mutation(s) within hematopoietic cells
(Walkley et al., 2007).
Evidence of the role of stroma and the microenviron-
ment in oncogenesis is accumulating, notably from analy-
sis of solid tumors. Moreover, mathematical modeling of
tumor behavior predicts that the environment is a major
selective modifier of tumor morphology and phenotype
(Allinen et al., 2004; Anderson et al., 2006; Balkwill,
2004; Hill et al., 2005; Kurose et al., 2002; Oh et al.,
2004; St Croix et al., 2000). Somatic mutations divergent
from those found in the tumor have been identified in stro-
mal cells. In prostate cancer, results suggest that such
mutations may contribute nonautonomously to tumor
behavior (Hill et al., 2005; Kurose et al., 2002). Under-
standing the interactions between hematopoietic cells
and their microenvironment is directly relevant to hemato-
poietic disease. Mutations in the Rb pathway occur in
�75% of cases of multiple myeloma (Kramer et al.,
2002). Multiple myeloma clearly demonstrates that the
interaction of hematopoietic cells—in this case B cells—
and the BM microenvironment is a major contributor
to disease (Hideshima and Anderson, 2002; Mitsiades
et al., 2006). These studies have focused on mutations
present in established disease but have not addressed
the role of the microenvironment or stroma in the initiation
of the disease process.
In addition to our data focused on RB loss, the signifi-
cance of the hematopoietic microenvironment to disease
initiation has been suggested by recent studies. Mx-
Cre+PtenD/D mice (Pten-deficient hematopoietic cells
and microenvironment) develop rapid and aggressive
myeloproliferation that progresses to overt leukemia/
lymphoma in 4 to 5 weeks postdeletion (Yilmaz et al.,
2006; Zhang et al., 2006b). However, when Pten deletion
Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1091

was activated in the context of a wild-type BM microenvi-
ronment, phenotypic and functional HSCs were lost
without evidence of myeloproliferation or transformation
(Yilmaz et al., 2006). This striking result suggests that
PtenD/D hematopoietic cells alone are not intrinsically sus-
ceptible to myeloproliferation and subsequent malignant
transformation in the presence of a wild-type microenvi-
ronment. Mutations in PTEN have been reported in the
stroma of human breast tumors, suggesting a broader
role for this pathway in the microenvironment and stroma
of diverse organ systems (Kurose et al., 2002). Intriguingly
JunB, Bmi-1, and ATM, implicated in HSC regulation and
myeloproliferation, also have roles in regulating the bone
marrow microenvironment (Kenner et al., 2004; Oguro
et al., 2006; Passegue et al., 2004; Rasheed et al.,
2006). JunB-deficient mice develop severe osteopenia
due to intrinsic defects in osteoclasts and osteoblasts,
cellular constitutents of the HSC niche, while ATM
mutants develop osteoporosis as a result of defective
osteoblast differentiation. The contribution of these micro-
environmental defects to the HSC phenotypes in these
mutants has yet to be described. Such results demon-
strate the need for further analysis of the interaction
between the hematopoietic cells and their environment.
This reconsideration will further our understanding of
normal homeostatic hematopoiesis and the development
of hematopoietic disease.
Our finding that the myeloproliferative-like disorder in
the Rb mutants is the result of an interaction between
myeloid-derived cells and the bone marrow microenvi-
ronment, together with the microenvironment-induced
myeloproliferative-like disorder that develops in the
RARg�/�mice (Walkley et al., 2007), underscores a previ-
ously unrecognized role for the hematopoietic microenvi-
ronment in the development of myeloid disease. These
data further suggest that mutations within the hematopoi-
etic niche might also serve as initiating events in the devel-
opment of hematopoietic disease. In contrast to previous
reports of the importance of cell-cycle regulation in HSC
fate determination, we find scant evidence for an intrinsic
requirement for RB in HSCs and that, indeed, RB is a novel
extrinsic regulator of hematopoietic stem cells. As our
findings underscore, interactions between hematopoietic
cells and the bone marrow niche/microenvironment pro-
foundly affect hematopoietic homeostasis and the behav-
ior of HSCs.
EXPERIMENTAL PROCEDURES
A detailed version of the Experimental Procedures can be found in the
Supplemental Data.
Experimental Animals
pRbfl/fl mutant mice were generously provided by Dr. Tyler Jacks
(Massachusetts Institute of Technology, MA, USA; Sage et al., 2003).
Mx-Cre transgenic mice have been previously described (Kuhn et al.,
1995; Walkley and Orkin, 2006). Lys-M-Cre animals were purchased
from The Jackson Laboratory (Clausen et al., 1999). All experiments
1092 Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc.
were performed with approval of the respective Institute Animal Ethics
Committees (DFCI or CHB).
Flow Cytometry Analysis
All antibodies and clone numbers are listed in Supplemental Experi-
mental Procedures. Flow cytometry was performed on a FACSCalibur,
and sorting was performed on a FACS Aria; all data were analyzed
using Cell Quest Pro software (Becton Dickinson).
Progenitor Cells Assays: CFC and CFU-S
BM, spleen cells, and PB leukocytes were assessed for in vitro colony-
forming cell (CFC, defined as >50 cells/colony) potential at either day 7
CFC (DME/agar media) or day 12 CFC (IMDM/methylcellulose media)
as described in Supplemental Experimental Procedures. Colony-form-
ing unit-spleen (CFU-S) was performed using the CFU-S assay of Till
and McCulloch with both WBM (CFU-S day 12)- and spleen (CFU-S
day 8)-derived cells (Purton et al., 1999; Till and McCulloch, 1961).
Long-Term Repopulating Cell (HSC) Analysis
Limit-dilution competitive repopulation analysis was performed as
previously described (Purton et al., 2006; Szilvassy et al., 1990; Walk-
ley et al., 2005) using test cell doses of 5 3 104, 2 3 105, and 2 3 106
cells competed against 2 3 105 WT WBM (CD45.1+/CD45.2+). Four to
five recipients/cell dose/genotype/experiment were transplanted, and
the experiment was performed in duplicate. Recipients were analyzed
at 3 and 6 months posttransplant. HSC frequencies were calculated
using L-Calc software (StemCell Technologies Inc.) using Poisson sta-
tistical analysis (Taswell, 1981). Secondary recipients were analyzed at
3 months posttransplant. One thousand freshly isolated LKS+ were in-
jected with 2 3 105 competitor WBM into five recipients per genotype.
Whole spleen cells were isolated from RbD/D 8 weeks post-pIpC and
competitively transplanted (1 3 106 or 2 3 106) with 2 3 105 competitor
WBM into five recipients per genotype.
For transplant of WT or Lys-M-Cre+pRbfl/fl WBM into Mx-Cre+ or Mx-
Cre�pRbfl/fl recipients, recipients were transplanted with 3 3 106 WBM
from sex mismatched animals. Chimerism was confirmed either by
CD45.1/CD45.2 allele analysis or by Y chromosome qPCR as
indicated.
Analysis of Transplant Recipients
PB from each individual recipient was obtained from the retro-orbital
plexus at the indicated time points posttransplant and was analyzed
as described for chimerism and lineage contribution of test cells
(Purton et al., 2000; Walkley et al., 2005).
Statistical Analyses
Statistical analyses were performed using the paired and unpaired
Student’s t test. Calculation of HSC frequency was performed using
Poisson statistical analysis using L-Calc software (StemCell Technol-
ogies Inc.). Histomorphometric data was analyzed by ANOVA followed
by Fisher’s PLSD Test.
Supplemental Data
Supplemental Data include Experimental Procedures, References,
two tables, and nine figures and can be found with this article online
at http://www.cell.com/cgi/content/full/129/6/1081/DC1/.
ACKNOWLEDGMENTS
The authors thank Tyler Jacks for the generous provision of pRbfl/fl
mice and Ingrid Poulton for bone histology. We thank David Williams,
Kevin Shannon, Jack Martin, Hans Widlund, and Ernestina Schipani for
discussion, helpful suggestions, and critical comment; DFCI and
Children’s Animal Facility Staff for care of experimental animals;
John Daley and Suzan Lazo-Kallanian of DFCI HemNeo Flow facility
for assistance with FACS sorting; David Dombkowski for assistance

with FACS analysis; and DFCI/Harvard Cancer Centre Rodent Histol-
ogy Core.
This work was supported in part by a Center of Excellence in Molec-
ular Hematology Award from the NIH-NIDDK (S.H.O). C.R.W is a Spe-
cial Fellow of the Leukemia & Lymphoma Society, and S.H.O is an In-
vestigator of the Howard Hughes Medical Institute.
C.R.W designed and performed experiments, analyzed and inter-
preted data, and wrote the paper; J.M.S performed experiments;
N.A.S performed experiments and interpreted data; L.E.P analyzed
and interpreted data; S.H.O. analyzed and interpreted data and wrote
the paper.
Received: November 14, 2006
Revised: February 15, 2007
Accepted: March 29, 2007
Published: June 14, 2007
REFERENCES
Adams, G.B., and Scadden, D.T. (2006). The hematopoietic stem cell
in its place. Nat. Immunol. 7, 333–337.
Adams, G.B., Chabner, K.T., Alley, I.R., Olson, D.P., Szczepiorkowski,
Z.M., Poznansky, M.C., Kos, C.H., Pollak, M.R., Brown, E.M., and
Scadden, D.T. (2006). Stem cell engraftment at the endosteal niche
is specified by the calcium-sensing receptor. Nature 439, 599–603.
Allinen, M., Beroukhim, R., Cai, L., Brennan, C., Lahti-Domenici, J.,
Huang, H., Porter, D., Hu, M., Chin, L., Richardson, A., et al. (2004).
Molecular characterization of the tumor microenvironment in breast
cancer. Cancer Cell 6, 17–32.
Anderson, A.R.A., Weaver, A.M., Cummings, P.T., and Quaranta, V.
(2006). Tumor morphology and phenotypic evolution driven by selec-
tive pressure from the microenvironment. Cell 127, 905–915.
Arai, F., Hirao, A., Ohmura, M., Sato, H., Matsuoka, S., Takubo, K., Ito,
K., Koh, G.Y., and Suda, T. (2004). Tie2/angiopoietin-1 signaling regu-
lates hematopoietic stem cell quiescence in the bone marrow niche.
Cell 118, 149–161.
Araki, T., Mohi, M.G., Ismat, F.A., Bronson, R.T., Williams, I.R., Kutok,
J.L., Yang, W., Pao, L.I., Gilliland, D.G., Epstein, J.A., and Neel, B.G.
(2004). Mouse model of Noonan syndrome reveals cell type- and
gene dosage-dependent effects of Ptpn11 mutation. Nat. Med. 10,
849–857.
Balkwill, F. (2004). Cancer and the chemokine network. Nat. Rev.
Cancer 4, 540–550.
Bradford, G.B., Williams, B., Rossi, R., and Bertoncello, I. (1997). Qui-
escence, cycling, and turnover in the primitive hematopoietic stem cell
compartment. Exp. Hematol. 25, 445–453.
Calvi, L.M., Adams, G.B., Weibrecht, K.W., Weber, J.M., Olson, D.P.,
Knight, M.C., Martin, R.P., Schipani, E., Divieti, P., Bringhurst, F.R.,
et al. (2003). Osteoblastic cells regulate the haematopoietic stem cell
niche. Nature 425, 841–846.
Chan, I.T., Kutok, J.L., Williams, I.R., Cohen, S., Kelly, L., Shigematsu,
H., Johnson, L., Akashi, K., Tuveson, D.A., Jacks, T., and Gilliland,
D.G. (2004). Conditional expression of oncogenic K-ras from its en-
dogenous promoter induces a myeloproliferative disease. J. Clin. In-
vest. 113, 528–538.
Cheng, T., Rodrigues, N., Shen, H., Yang, Y., Dombkowski, D., Sykes,
M., and Scadden, D.T. (2000). Hematopoietic stem cell quiescence
maintained by p21cip1/waf1. Science 287, 1804–1808.
Chien, W.M., Rabin, S., Macias, E., Miliani de Marval, P.L., Garrison,
K., Orthel, J., Rodriguez-Puebla, M., and Fero, M.L. (2006). Genetic
mosaics reveal both cell-autonomous and cell-nonautonomous func-
tion of murine p27Kip1. Proc. Natl. Acad. Sci. USA 103, 4122–4127.
Clark, A.J., Doyle, K.M., and Humbert, P.O. (2004). Cell-intrinsic
requirement for pRb in erythropoiesis. Blood 104, 1324–1326.
Clausen, B.E., Burkhardt, C., Reith, W., Renkawitz, R., and Forster, I.
(1999). Conditional gene targeting in macrophages and granulocytes
using LysMcre mice. Transgenic Res. 8, 265–277.
Conboy, I.M., Conboy, M.J., Wagers, A.J., Girma, E.R., Weissman,
I.L., and Rando, T.A. (2005). Rejuvenation of aged progenitor cells by
exposure to a young systemic environment. Nature 433, 760–764.
Fuchs, E., Tumbar, T., and Guasch, G. (2004). Socializing with the
neighbors: stem cells and their niche. Cell 116, 769–778.
Ghiaur, G., Lee, A., Bailey, J., Cancelas, J.A., Zheng, Y., and Williams,
D.A. (2006). Inhibition of RhoA GTPase activity enhances hematopoi-
etic stem and progenitor cell proliferation and engraftment. Blood
108, 2087–2094.
Gothot, A., van der Loo, J.C., Clapp, D.W., and Srour, E.F. (1998). Cell
cycle-related changes in repopulating capacity of human mobilized
peripheral blood CD34(+) cells in non-obese diabetic/severe
combined immune-deficient mice. Blood 92, 2641–2649.
Hanahan, D., and Weinberg, R.A. (2000). The hallmarks of cancer. Cell
100, 57–70.
Hideshima, T., and Anderson, K.C. (2002). Molecular mechanisms of
novel therapeutic approaches for multiple myeloma. Nat. Rev. Cancer
2, 927–937.
Hill, R., Song, Y., Cardiff, R.D., and Van Dyke, T. (2005). Selective
evolution of stromal mesenchyme with p53 loss in response to epithe-
lial tumorigenesis. Cell 123, 1001–1011.
Hodgson, G.S., and Bradley, T.R. (1979). Properties of haematopoietic
stem cells surviving 5-fluorouracil treatment: evidence for a pre-CFU-S
cell? Nature 281, 381–382.
Hu, N., Gulley, M.L., Kung, J.T., and Lee, E.Y. (1997). Retinoblastoma
gene deficiency has mitogenic but not tumorigenic effects on erythro-
poiesis. Cancer Res. 57, 4123–4129.
Iavarone, A., King, E.R., Dai, X.M., Leone, G., Stanley, E.R., and Lasor-
ella, A. (2004). Retinoblastoma promotes definitive erythropoiesis by
repressing Id2 in fetal liver macrophages. Nature 432, 1040–1045.
Ito, K., Hirao, A., Arai, F., Matsuoka, S., Takubo, K., Hamaguchi, I., No-
miyama, K., Hosokawa, K., Sakurada, K., Nakagata, N., et al. (2004).
Regulation of oxidative stress by ATM is required for self-renewal of
haematopoietic stem cells. Nature 431, 997–1002.
Janzen, V., Forkert, R., Fleming, H.E., Saito, Y., Waring, M.T., Domb-
kowski, D.M., Cheng, T., DePinho, R.A., Sharpless, N.E., and Scad-
den, D.T. (2006). Stem-cell ageing modified by the cyclin-dependent
kinase inhibitor p16INK4a. Nature 443, 421–426.
Kenner, L., Hoebertz, A., Beil, T., Keon, N., Karreth, F., Eferl, R.,
Scheuch, H., Szremska, A., Amling, M., Schorpp-Kistner, M., et al.
(2004). Mice lacking JunB are osteopenic due to cell-autonomous os-
teoblast and osteoclast defects. J. Cell Biol. 164, 613–623.
Kollet, O., Dar, A., Shivtiel, S., Kalinkovich, A., Lapid, K., Sztainberg, Y.,
Tesio, M., Samstein, R.M., Goichberg, P., Spiegel, A., et al. (2006).
Osteoclasts degrade endosteal components and promote mobiliza-
tion of hematopoietic progenitor cells. Nat. Med. 12, 657–664.
Kramer, A., Schultheis, B., Bergmann, J., Willer, A., Hegenbart, U., Ho,
A.D., Goldschmidt, H., and Hehlmann, R. (2002). Alterations of the
cyclin D1/pRb/p16(INK4A) pathway in multiple myeloma. Leukemia
16, 1844–1851.
Krivtsov, A.V., Twomey, D., Feng, Z., Stubbs, M.C., Wang, Y., Faber,
J., Levine, J.E., Wang, J., Hahn, W.C., Gilliland, D.G., et al. (2006).
Transformation from committed progenitor to leukaemia stem cell
initiated by MLL-AF9. Nature 442, 818–822.
Krosl, J., Beslu, N., Mayotte, N., Humphries, R.K., and Sauvageau, G.
(2003). The competitive nature of HOXB4-transduced HSC is limited
by PBX1: the generation of ultra-competitive stem cells retaining full
differentiation potential. Immunity 18, 561–571.
Kuhn, R., Schwenk, F., Aguet, M., and Rajewsky, K. (1995). Inducible
gene targeting in mice. Science 269, 1427–1429.
Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1093

Kurose, K., Gilley, K., Matsumoto, S., Watson, P.H., Zhou, X.P., and
Eng, C. (2002). Frequent somatic mutations in PTEN and TP53 are
mutually exclusive in the stroma of breast carcinomas. Nat. Genet.
32, 355–357.
Le, D.T., Kong, N., Zhu, Y., Lauchle, J.O., Aiyigari, A., Braun, B.S.,
Wang, E., Kogan, S.C., Le Beau, M.M., Parada, L., and Shannon,
K.M. (2004). Somatic inactivation of Nf1 in hematopoietic cells results
in a progressive myeloproliferative disorder. Blood 103, 4243–4250.
Lerner, C., and Harrison, D.E. (1990). 5-Fluorouracil spares hemopoi-
etic stem cells responsible for long-term repopulation. Exp. Hematol.
18, 114–118.
Lessard, J., and Sauvageau, G. (2003). Bmi-1 determines the prolifer-
ative capacity of normal and leukaemic stem cells. Nature 423, 255–
260.
Li, C.L., and Johnson, G.R. (1994). Stem cell factor enhances the
survival but not the self-renewal of murine hematopoietic long-term
repopulating cells. Blood 84, 408–414.
Martin, T.J., and Sims, N.A. (2005). Osteoclast-derived activity in the
coupling of bone formation to resorption. Trends Mol. Med. 11, 76–81.
Mitsiades, C.S., Mitsiades, N.S., Munshi, N.C., Richardson, P.G., and
Anderson, K.C. (2006). The role of the bone microenvironment in the
pathophysiology and therapeutic management of multiple myeloma:
interplay of growth factors, their receptors and stromal interactions.
Eur. J. Cancer 42, 1564–1573.
Moore, K.A., and Lemischka, I.R. (2006). Stem cells and their niches.
Science 311, 1880–1885.
Oguro, H., Iwama, A., Morita, Y., Kamijo, T., van Lohuizen, M., and Na-
kauchi, H. (2006). Differential impact of Ink4a and Arf on hematopoietic
stem cells and their bone marrow microenvironment in Bmi1-deficient
mice. J. Exp. Med. 203, 2247–2253.
Oh, P., Li, Y., Yu, J., Durr, E., Krasinska, K.M., Carver, L.A., Testa, J.E.,
and Schnitzer, J.E. (2004). Subtractive proteomic mapping of the
endothelial surface in lung and solid tumours for tissue-specific ther-
apy. Nature 429, 629–635.
Okada, S., Nakauchi, H., Nagayoshi, K., Nishikawa, S., Miura, Y., and
Suda, T. (1992). In vivo and in vitro stem cell function of c-kit- and Sca-
1-positive murine hematopoietic cells. Blood 80, 3044–3050.
Osawa, M., Hanada, K., Hamada, H., and Nakauchi, H. (1996). Long-
term lymphohematopoietic reconstitution by a single CD34-low/nega-
tive hematopoietic stem cell. Science 273, 242–245.
Park, I.K., Qian, D., Kiel, M., Becker, M.W., Pihalja, M., Weissman, I.L.,
Morrison, S.J., and Clarke, M.F. (2003). Bmi-1 is required for mainte-
nance of adult self-renewing haematopoietic stem cells. Nature 423,
302–305.
Passegue, E., Wagner, E.F., and Weissman, I.L. (2004). JunB defi-
ciency leads to a myeloproliferative disorder arising from hematopoi-
etic stem cells. Cell 119, 431–443.
Passegue, E., Wagers, A.J., Giuriato, S., Anderson, W.C., and Weiss-
man, I.L. (2005). Global analysis of proliferation and cell cycle gene ex-
pression in the regulation of hematopoietic stem and progenitor cell
fates. J. Exp. Med. 202, 1599–1611.
Purton, L.E., Bernstein, I.D., and Collins, S.J. (1999). All-trans retinoic
acid delays the differentiation of primitive hematopoietic precursors
(lin-c-kit+Sca-1(+)) while enhancing the terminal maturation of com-
mitted granulocyte/monocyte progenitors. Blood 94, 483–495.
Purton, L.E., Bernstein, I.D., and Collins, S.J. (2000). All-trans retinoic
acid enhances the long-term repopulating activity of cultured hemato-
poietic stem cells. Blood 95, 470–477.
Purton, L.E., Dworkin, S., Olsen, G.H., Walkley, C.R., Fabb, S.A., Col-
lins, S.J., and Chambon, P. (2006). RAR{gamma} is critical for main-
taining a balance between hematopoietic stem cell self-renewal and
differentiation. J. Exp. Med. 203, 1283–1293.
1094 Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc.
Rasheed, N.,Wang, X., Niu, Q.T., Yeh, J., andLi,B. (2006).Atm-deficient
mice: an osteoporosis model with defective osteoblast differentiation
and increased osteoclastogenesis. Hum. Mol. Genet. 15, 1938–1948.
Sage, J., Miller, A.L., Perez-Mancera, P.A., Wysocki, J.M., and Jacks,
T. (2003). Acute mutation of retinoblastoma is sufficient for cell cycle
re-entry. Nature 424, 223–228.
Schofield, R. (1978). The relationship between the spleen colony-form-
ing cell and the haemopoietic stem cell. Blood Cells 4, 7–25.
Sherr, C.J., and Roberts, J.M. (2004). Living with or without cyclins and
cyclin-dependent kinases. Genes Dev. 18, 2699–2711.
Spangrude, G.J., Brooks, D.M., and Tumas, D.B. (1995). Long-term
repopulation of irradiated mice with limiting numbers of purified hema-
topoietic stem cells: in vivo expansion of stem cell phenotype but not
function. Blood 85, 1006–1016.
Spike, B.T., Dirlam, A., Dibling, B.C., Marvin, J., Williams, B.O., Jacks,
T., and Macleod, K.F. (2004). The Rb tumor suppressor is required for
stress erythropoiesis. EMBO J. 23, 4319–4329.
St Croix, B., Rago, C., Velculescu, V., Traverso, G., Romans, K.E.,
Montgomery, E., Lal, A., Riggins, G.J., Lengauer, C., Vogelstein, B.,
and Kinzler, K.W. (2000). Genes expressed in human tumor endothe-
lium. Science 289, 1197–1202.
Stead, E., White, J., Faast, R., Conn, S., Goldstone, S., Rathjen, J.,
Dhingra, U., Rathjen, P., Walker, D., and Dalton, S. (2002). Pluripotent
cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F ac-
tivities. Oncogene 21, 8320–8333.
Stepanova, L., and Sorrentino, B.P. (2005). A limited role for p16Ink4a
and p19Arf in the loss of hematopoietic stem cells during proliferative
stress. Blood 106, 827–832.
Stier, S., Ko, Y., Forkert, R., Lutz, C., Neuhaus, T., Grunewald, E.,
Cheng, T., Dombkowski, D., Calvi, L.M., Rittling, S.R., and Scadden,
D.T. (2005). Osteopontin is a hematopoietic stem cell niche compo-
nent that negatively regulates stem cell pool size. J. Exp. Med. 201,
1781–1791.
Szilvassy, S.J., Humphries, R.K., Lansdorp, P.M., Eaves, A.C., and
Eaves, C.J. (1990). Quantitative assay for totipotent reconstituting
hematopoietic stem cells by a competitive repopulation strategy.
Proc. Natl. Acad. Sci. USA 87, 8736–8740.
Tajima, F., Sato, T., Laver, J.H., and Ogawa, M. (2000). CD34 expres-
sion by murine hematopoietic stem cells mobilized by granulocyte col-
ony-stimulating factor. Blood 96, 1989–1993.
Taswell, C. (1981). Limiting dilution assays for the determination of im-
munocompetent cell frequencies. I. Data analysis. J. Immunol. 126,
1614–1619.
Till, J., and McCulloch, E. (1961). A direct measurement of the radiation
sensitivity of normal mouse bone marrow cells. Radiat. Res. 14, 213–
222.
Varnum-Finney, B., Purton, L.E., Yu, M., Brashem-Stein, C., Flowers,
D., Staats, S., Moore, K.A., Le Roux, I., Mann, R., Gray, G., et al.
(1998). The Notch ligand, Jagged-1, influences the development of
primitive hematopoietic precursor cells. Blood 91, 4084–4091.
Visnjic, D., Kalajzic, Z., Rowe, D.W., Katavic, V., Lorenzo, J., and
Aguila, H.L. (2004). Hematopoiesis is severely altered in mice with an
induced osteoblast deficiency. Blood 103, 3258–3264.
Walkley, C.R., Olsen, G.H., Dworkin, S., Fabb, S.A., Swann, J.,
McArthur, G.A., Westmoreland, S.V., Chambon, P., Scadden, D.T.,
and Purton, L.E. (2007). A microenvironment-induced myeloprolifera-
tive syndrome caused by Retinoic Acid Receptor g deficiency. Cell
129, this issue, 1097–1110.
Walkley, C.R., and Orkin, S.H. (2006). Rb is dispensable for self-
renewal and multilineage differentiation of adult hematopoietic stem
cells. Proc. Natl. Acad. Sci. USA 103, 9057–9062.
Walkley, C.R., Fero, M.L., Chien, W.M., Purton, L.E., and McArthur,
G.A. (2005). Negative cell-cycle regulators cooperatively control

self-renewal and differentiation of haematopoietic stem cells. Nat. Cell
Biol. 7, 172–178.
Weinberg, R.A. (1995). The retinoblastoma protein and cell cycle
control. Cell 81, 323–330.
Whyatt, D., and Grosveld, F. (2002). Cell-nonautonomous function of
the retinoblastoma tumour suppressor protein: new interpretations of
old phenotypes. EMBO Rep. 3, 130–135.
Wildwater, M., Campilho, A., Perez-Perez, J.M., Heidstra, R., Blilou, I.,
Korthout, H., Chatterjee, J., Mariconti, L., Gruissem, W., and Scheres,
B. (2005). The RETINOBLASTOMA-RELATED gene regulates stem cell
maintenance in Arabidopsis roots. Cell 123, 1337–1349.
Yang, L., Bryder, D., Adolfsson, J., Nygren, J., Mansson, R.,
Sigvardsson, M., and Jacobsen, S.E. (2005). Identification of Lin-
Sca1+kit+CD34+Flt3- short-term hematopoietic stem cells capable
of rapidly reconstituting and rescuing myeloablated recipients. Blood
105, 2717–2723.
Yilmaz, O.H., Valdez, R., Theisen, B.K., Guo, W., Ferguson, D.O., Wu,
H., and Morrison, S.J. (2006). Pten dependence distinguishes haema-
topoietic stem cells from leukaemia-initiating cells. Nature 441, 475–
482.
Yuan, Y., Shen, H., Franklin, D.S., Scadden, D.T., and Cheng, T. (2004).
In vivo self-renewing divisions of haematopoietic stem cells are in-
creased in the absence of the early G1-phase inhibitor, p18INK4C.
Nat. Cell Biol. 6, 436–442.
Zhang, C.C., Kaba, M., Ge, G., Xie, K., Tong, W., Hug, C., and Lodish,
H.F. (2006a). Angiopoietin-like proteins stimulate ex vivo expansion of
hematopoietic stem cells. Nat. Med. 12, 240–245.
Zhang, J., Niu, C., Ye, L., Huang, H., He, X., Tong, W.G., Ross, J.,
Haug, J., Johnson, T., Feng, J.Q., et al. (2003). Identification of the hae-
matopoietic stem cell niche and control of the niche size. Nature 425,
836–841.
Zhang, J., Grindley, J.C., Yin, T., Jayasinghe, S., He, X.C., Ross, J.T.,
Haug, J.S., Rupp, D., Porter-Westpfahl, K.S., Wiedemann, L.M., et al.
(2006b). PTEN maintains haematopoietic stem cells and acts in lineage
choice and leukaemia prevention. Nature 441, 518–522.
Cell 129, 1081–1095, June 15, 2007 ª2007 Elsevier Inc. 1095
Related Documents











