Pyrazinamide Resistance Is Caused by Two Distinct Mechanisms: Prevention of Coenzyme A Depletion and Loss of Virulence Factor Synthesis Pooja Gopal, † Michelle Yee, † Jickky Sarathy, † Jian Liang Low, † Jansy P. Sarathy, ‡ Firat Kaya, ‡ Ve ́ ronique Dartois, ‡ Martin Gengenbacher, † and Thomas Dick* ,† † Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Republic of Singapore ‡ Public Health Research Institute, RutgersNew Jersey Medical School, Newark, New Jersey 07103, United States * S Supporting Information ABSTRACT: Pyrazinamide (PZA) is a critical component of first- and second-line treatments of tuberculosis (TB), yet its mechanism of action largely remains an enigma. We carried out a genetic screen to isolate Mycobacterium bovis BCG mutants resistant to pyrazinoic acid (POA), the bioactive derivative of PZA, followed by whole genome sequencing of 26 POA resistant strains. Rather than finding mutations in the proposed candidate targets fatty acid synthase I and ribosomal protein S1, we found resistance conferring mutations in two pathways: missense mutations in aspartate decarboxylase panD, involved in the synthesis of the essential acyl carrier coenzyme A (CoA), and frameshift mutations in the vitro nonessential polyketide synthase genes mas and ppsA-E, involved in the synthesis of the virulence factor phthiocerol dimycocerosate (PDIM). Probing for cross resistance to two structural analogs of POA, nicotinic acid and benzoic acid, showed that the analogs share the PDIM- but not the CoA-related mechanism of action with POA. We demonstrated that POA depletes CoA in wild-type bacteria, which is prevented by mutations in panD. Sequencing 10 POA-resistant Mycobacterium tuberculosis H37Rv isolates confirmed the presence of at least 2 distinct mechanisms of resistance to the drug. The emergence of resistance through the loss of a virulence factor in vitro may explain the lack of clear molecular patterns in PZA-resistant clinical isolates, other than mutations in the prodrug-converting enzyme. The apparent interference of POA with virulence pathways may contribute to the drug’s excellent in vivo efficacy compared to its modest in vitro potency. KEYWORDS: tuberculosis, pyrazinamide, pyrazinoic acid, resistance, mechanism of action PZA is a critical component of the current first-line regimen for treating TB. Its inclusion in the regimen in the 1980s resulted in dramatically shortening the duration of therapy from 12 to 6 months; i.e., PZA is a key sterilizing drug. 1 The current 6-month regimen is still too lengthy to ensure compliance, not only affecting cure rates but also facilitating the development of drug resistance. Treatment shortening to 2 months or less is therefore a major goal in TB drug development. 2 Most new drug combinations currently in preclinical and clinical development include PZA, 3 although its mechanism of action remains ill- defined. Considering the clinically proven sterilizing activity of PZA, the identification of its target(s) may provide clues and effective approaches for the discovery of shortened chemo- therapeutic regimens. PZA is a prodrug that requires hydrolysis to the bioactive form, pyrazinoic acid (POA). Prodrug conversion is carried out by the bacterial amidase PncA, the inactivation of which causes resistance in vitro, 4 and also by host enzymes. 5 Until recently, it was believed that PZA/POA is active in vitro under acidic conditions only. Extracellular low pH was part of the suggested mechanism of action of the drug: POA, a low-molecular-weight carboxylic acid, was thought to act as an ionophore affecting membrane energetics and causing intracellular acidification. 6,7 On the basis of these in vitro findings, it was assumed that TB lesions must be acidic to allow PZA to be active in vivo. 1,8 In addition, two discrete molecular targets were suggested to be inhibited by the drug. POA was shown to block fatty acid synthesis, and fatty acid synthase I (FAS I) was suggested as the target of the drug. 9 FAS I as a direct target of POA was called into question by subsequent works. 10 Protein-binding studies found a large number of POA-binding partners, one of which was identified to be the 30S ribosomal S1/RpsA protein. Thus, by targeting RpsA, POA may impair trans−translation, a rescue mechanism that frees ribosomes stuck in translation. 11 However, gene sequencing of PZA-resistant clinical isolates did not support these biochemical findings. 12−14 Multiple investigators failed to isolate POA-resistant mutants in vitro on acidic agar, with Received: April 28, 2016 Article pubs.acs.org/journal/aidcbc © XXXX American Chemical Society A DOI: 10.1021/acsinfecdis.6b00070 ACS Infect. Dis. XXXX, XXX, XXX−XXX

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Pyrazinamide Resistance Is Caused by Two Distinct Mechanisms:Prevention of Coenzyme A Depletion and Loss of Virulence FactorSynthesisPooja Gopal,† Michelle Yee,† Jickky Sarathy,† Jian Liang Low,† Jansy P. Sarathy,‡ Firat Kaya,‡
Veronique Dartois,‡ Martin Gengenbacher,† and Thomas Dick*,†
†Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore,Republic of Singapore‡Public Health Research Institute, RutgersNew Jersey Medical School, Newark, New Jersey 07103, United States
*S Supporting Information
ABSTRACT: Pyrazinamide (PZA) is a critical component offirst- and second-line treatments of tuberculosis (TB), yet itsmechanism of action largely remains an enigma. We carriedout a genetic screen to isolate Mycobacterium bovis BCGmutants resistant to pyrazinoic acid (POA), the bioactivederivative of PZA, followed by whole genome sequencing of26 POA resistant strains. Rather than finding mutations in theproposed candidate targets fatty acid synthase I and ribosomalprotein S1, we found resistance conferring mutations in twopathways: missense mutations in aspartate decarboxylasepanD, involved in the synthesis of the essential acyl carrier coenzyme A (CoA), and frameshift mutations in the vitrononessential polyketide synthase genes mas and ppsA-E, involved in the synthesis of the virulence factor phthioceroldimycocerosate (PDIM). Probing for cross resistance to two structural analogs of POA, nicotinic acid and benzoic acid, showedthat the analogs share the PDIM- but not the CoA-related mechanism of action with POA. We demonstrated that POA depletesCoA in wild-type bacteria, which is prevented by mutations in panD. Sequencing 10 POA-resistant Mycobacterium tuberculosisH37Rv isolates confirmed the presence of at least 2 distinct mechanisms of resistance to the drug. The emergence of resistancethrough the loss of a virulence factor in vitro may explain the lack of clear molecular patterns in PZA-resistant clinical isolates,other than mutations in the prodrug-converting enzyme. The apparent interference of POA with virulence pathways maycontribute to the drug’s excellent in vivo efficacy compared to its modest in vitro potency.
KEYWORDS: tuberculosis, pyrazinamide, pyrazinoic acid, resistance, mechanism of action
PZA is a critical component of the current first-line regimen fortreating TB. Its inclusion in the regimen in the 1980s resulted indramatically shortening the duration of therapy from 12 to 6months; i.e., PZA is a key sterilizing drug.1 The current 6-monthregimen is still too lengthy to ensure compliance, not onlyaffecting cure rates but also facilitating the development of drugresistance. Treatment shortening to 2 months or less is thereforea major goal in TB drug development.2 Most new drugcombinations currently in preclinical and clinical developmentinclude PZA,3 although its mechanism of action remains ill-defined. Considering the clinically proven sterilizing activity ofPZA, the identification of its target(s) may provide clues andeffective approaches for the discovery of shortened chemo-therapeutic regimens.PZA is a prodrug that requires hydrolysis to the bioactive form,
pyrazinoic acid (POA). Prodrug conversion is carried out by thebacterial amidase PncA, the inactivation of which causesresistance in vitro,4 and also by host enzymes.5 Until recently,it was believed that PZA/POA is active in vitro under acidicconditions only. Extracellular low pH was part of the suggested
mechanism of action of the drug: POA, a low-molecular-weightcarboxylic acid, was thought to act as an ionophore affectingmembrane energetics and causing intracellular acidification.6,7
On the basis of these in vitro findings, it was assumed that TBlesions must be acidic to allow PZA to be active in vivo.1,8 Inaddition, two discrete molecular targets were suggested to beinhibited by the drug. POA was shown to block fatty acidsynthesis, and fatty acid synthase I (FAS I) was suggested as thetarget of the drug.9 FAS I as a direct target of POA was called intoquestion by subsequent works.10 Protein-binding studies found alarge number of POA-binding partners, one of which wasidentified to be the 30S ribosomal S1/RpsA protein. Thus, bytargeting RpsA, POA may impair trans−translation, a rescuemechanism that frees ribosomes stuck in translation.11 However,gene sequencing of PZA-resistant clinical isolates did not supportthese biochemical findings.12−14 Multiple investigators failed toisolate POA-resistant mutants in vitro on acidic agar, with
Received: April 28, 2016
Article
pubs.acs.org/journal/aidcbc
© XXXX American Chemical Society A DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX

mutations either in fatty acid synthase I, ribosomal protein RpsA,or any other target.9,15 During the course of this work, Zhang andcolleagues identified mutations in the aspartate decarboxylasePanD associated with PZA resistance16,17 and proposed thisenzyme as a direct target of POA. However, PanD as a target wascalled into question by Dillon et al.18
One of the major PZA conundrums is its very modest in vitropotency, contrasting with its excellent in vivo sterilizingactivity.3,8 A second major puzzle is that genetic analyses ofPZA-resistant clinical isolates do not yield any prominentclustering of PZA-associated resistance mutation patterns (otherthan those in the prodrug-activating pyrazinamidase).19−21 Thelack of obvious resistance epidemiology is in stark contrast toother tuberculosis drugs such as rifampicin, which inhibits RNApolymerase or the fluoroquinolones that target DNA gyrase. Forthose drugs, clinical resistance can be readily associated withmutations in the target enzymes, and the same mutations can beisolated in vitro.22 New genotype-based antibiotic-resistancepredicting software for TB such as Mykrobe Predictor excludePZA specifically because of the poor predictive value.23 The lackof clear-cut molecular resistance epidemiology or a transmissionpattern in clinical PZA-resistant isolates could indicate thatemerging resistant mutants may suffer from significant fitness orvirulence loss in vivo and therefore cannot be found in sputumisolates.22
Recently, the acid pH model for the mechanism of action ofPZA was called into question. First, it was shown (orrediscovered24) that PZA and POA also inhibit growth undernonacidic conditions in vitro.5,25 This was followed by the criticalfinding that POA does not act as a robust ionophore; i.e.,intracellular acidification as a major mechanism of action appearsunlikely.25 Finally, old reports from the 1930s26,27 wereconfirmed, showing that the necrotic core of TB lesions is notacidic.28−30
On the basis of these developments, we revisited a geneticapproach and successfully selected and isolated POA-resistantmutants on nearly neutral pH media rather than acidified agar todissect the mechanism of action of PZA. Phenotypic andmolecular characterization of these mutants suggests that POAresistance can be caused by two different mechanisms:prevention of depletion of an essential cofactor and interferencewith the synthesis of a cell-wall-associated virulence factor.
■ RESULTS
Missense Mutations in a CoA Synthesis Gene and Lossof Function Mutations in PDIM Synthesis Genes CausePOA Resistance. We previously showed that, in contrast tocommon belief, PZA and POA exert growth inhibitory activityagainst M. tuberculosis at nearly neutral pH 6.5.5 Here, we testedthe hypothesis that employing nearly neutral instead of acidicagar, as has been attempted unsuccessfully in the past, wouldenable the isolation of POA-resistant mutants.(Note that during the course of this work Zhang et al.17 and
Lanoix et al.31 showed the feasibility of this approach. See theDiscussion.) To facilitate this work, we usedMycobacterium bovisBCG as a biosafety level 2 compatible surrogate for M.tuberculosis. M. bovis BCG is naturally pyrazinamidase-negative(PncA−) because of a point mutation (C169G/His57Asp) in theamidase gene pncA and hence is not susceptible to PZA.4M. bovisBCG exhibits similar susceptibility to POA in vitro under bothnearly neutral (pH 6.5) and acidic (pH 5.8) conditions, withMIC50 values of 0.5−1 and 0.25 mM, respectively (1 mM POAcorresponds to 124 μg/mL).Pilot experiments to determine broth MICs using standard
(pH 6.5) growth media with and without glycerol surprisinglyshowed that glycerol hypersensitizes wild-type M. bovis BCG toPOA (Supporting Information, Figure S1A). Increasing theglycerol concentration from 0.01 to 0.8% resulted in a reductionof the MIC90 of POA from 4 to 1 mM (Supporting Information,Figure S1B). This shows that the POA susceptibility is influencedby the presence of glycerol, a phenomenon that we and othersobserved previously with other compound classes duringantimycobacterial drug screening.32,33 On the basis of theobserved glycerol effect, we used agar containing 0.5% glycerol oragar without glycerol, i.e., with glucose as themain carbon source,for resistant mutant selection. Four independent BCG cultureswere plated on the two agar types supplemented with 1, 2, or 4mM POA. POA-resistant colonies were observed at spontaneousresistance mutation frequencies of 10−4 to 10−5/CFU in thepresence of glycerol, but in the absence of glycerol, thefrequencies were 10−3 to 10−4/CFU. To confirm the resistance,a total of 26 colonies from both screens were restreaked on agarcontaining corresponding concentrations of POA. The resistanceof the 26 strains was further confirmed by the determination ofbroth MICs via serial dilution in broth with and without glycerol.
Table 1. Phenotypic and Genotypic Classes of POA-Resistant M. bovis BCG with Details for Five Representative Strains
phenotypicclass
POA resistance in 7H9broth without glycerol pH
6.5a
POA resistance in 7H9broth with glycerol pH
6.5agenotypicclass mutation types
representative M.bovis BCG strain
mutations inrepresentative strain
wt POAS MIC50= 1 POAS+ MIC50< 1 wt N.A. wt N.A.POA1 POAR++ MIC50> 4 POAR++ MIC50> 4 panD and
mas orppsA-E
panD: C-terminal missenseand mas or ppsA-E:Frameshift
POA1 [panD1mas-1]b
panD: T407G/Leu136Arg and mas:Ins4999G
POA2 POAR+ MIC50= 4 POAR+ MIC50= 2−4 panD panD: C-terminal missense POA2 [panD2]b panD: T395G/Leu132Arg
POA3c POAR MIC50= 2 POAS+ MIC50< 1 mas mas: Frameshift POA3A [mas-1]d mas: Ins4999GPOAR MIC50= 2 POAS+ MIC50< 1 ppsA-E ppsA-E: Frameshift POA3B [ppsA1
]dppsA: Ins4875A
POA4c POAR MIC50= 2 POAR+ MIC50= 4 glpK and masor ppsA-E
glpK: Frameshift and mas orppsA-E: Frameshift
POA4 [glpK1mas-1]b
glpK: Ins576G and mas:Ins4999G
aMIC50 values (expressed in mM) were obtained as a mean of technical replicates from independent experiments. Note that because of the verysmall molar mass of POA (124 g/mol), a concentration of 1 mM corresponds to ∼100 μg/mL. Resistance to POA at pH 6.5 was classified asfollows: POAS+/MIC50 < 1, POAS/MIC50 = 1, POAR/MIC50 = 2, POAR+/MIC50 = 2−4/4, and POAR++/MIC50 > 4. bM. bovis BCG POA1 [panD1mas-1], POA4 [glpK1 mas-1], and POA2 [panD2] were isolated from two independent cultures on POA agar with glycerol. cIsolated only on agarwithout glycerol (POA3) and on agar with glycerol (POA4), respectively. dM. bovis BCG POA3A [mas-1] and POA3B [ppsA1] were isolated fromthe same batch on POA agar without glycerol.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
B
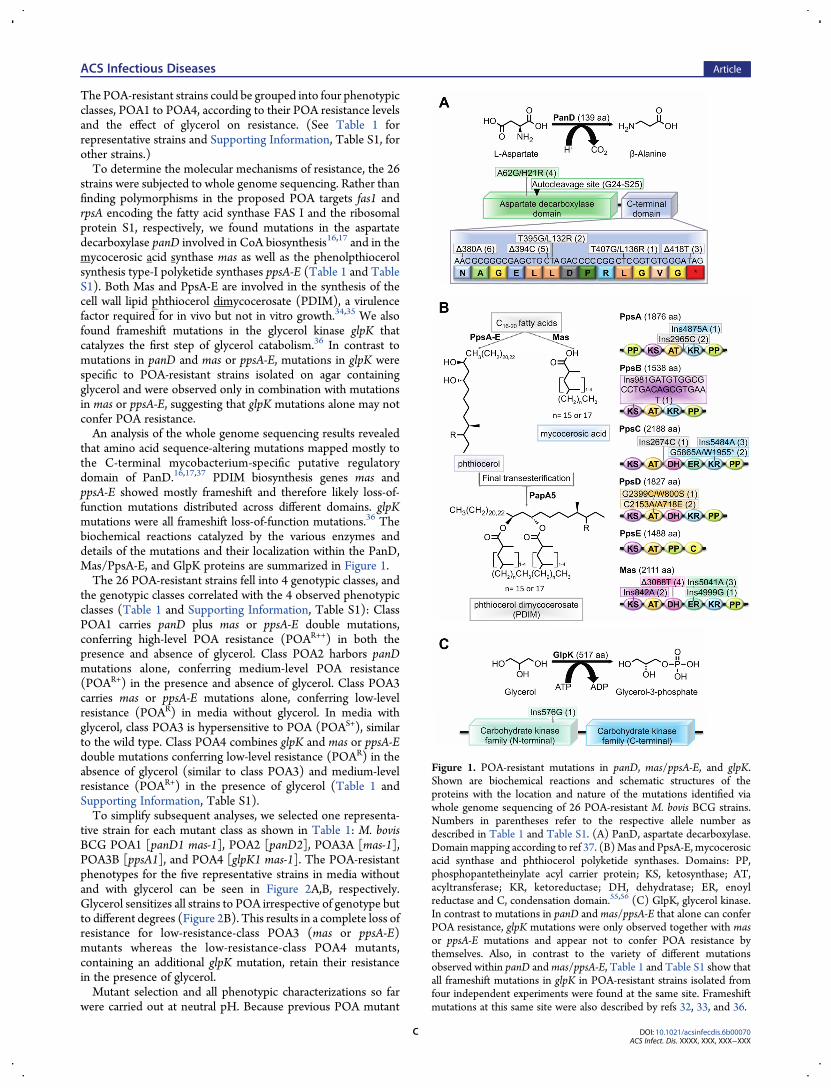
The POA-resistant strains could be grouped into four phenotypicclasses, POA1 to POA4, according to their POA resistance levelsand the effect of glycerol on resistance. (See Table 1 forrepresentative strains and Supporting Information, Table S1, forother strains.)To determine the molecular mechanisms of resistance, the 26
strains were subjected to whole genome sequencing. Rather thanfinding polymorphisms in the proposed POA targets fas1 andrpsA encoding the fatty acid synthase FAS I and the ribosomalprotein S1, respectively, we found mutations in the aspartatedecarboxylase panD involved in CoA biosynthesis16,17 and in themycocerosic acid synthase mas as well as the phenolpthiocerolsynthesis type-I polyketide synthases ppsA-E (Table 1 and TableS1). Both Mas and PpsA-E are involved in the synthesis of thecell wall lipid phthiocerol dimycocerosate (PDIM), a virulencefactor required for in vivo but not in vitro growth.34,35 We alsofound frameshift mutations in the glycerol kinase glpK thatcatalyzes the first step of glycerol catabolism.36 In contrast tomutations in panD and mas or ppsA-E, mutations in glpK werespecific to POA-resistant strains isolated on agar containingglycerol and were observed only in combination with mutationsin mas or ppsA-E, suggesting that glpK mutations alone may notconfer POA resistance.An analysis of the whole genome sequencing results revealed
that amino acid sequence-altering mutations mapped mostly tothe C-terminal mycobacterium-specific putative regulatorydomain of PanD.16,17,37 PDIM biosynthesis genes mas andppsA-E showed mostly frameshift and therefore likely loss-of-function mutations distributed across different domains. glpKmutations were all frameshift loss-of-function mutations.36 Thebiochemical reactions catalyzed by the various enzymes anddetails of the mutations and their localization within the PanD,Mas/PpsA-E, and GlpK proteins are summarized in Figure 1.The 26 POA-resistant strains fell into 4 genotypic classes, and
the genotypic classes correlated with the 4 observed phenotypicclasses (Table 1 and Supporting Information, Table S1): ClassPOA1 carries panD plus mas or ppsA-E double mutations,conferring high-level POA resistance (POAR++) in both thepresence and absence of glycerol. Class POA2 harbors panDmutations alone, conferring medium-level POA resistance(POAR+) in the presence and absence of glycerol. Class POA3carries mas or ppsA-E mutations alone, conferring low-levelresistance (POAR) in media without glycerol. In media withglycerol, class POA3 is hypersensitive to POA (POAS+), similarto the wild type. Class POA4 combines glpK and mas or ppsA-Edouble mutations conferring low-level resistance (POAR) in theabsence of glycerol (similar to class POA3) and medium-levelresistance (POAR+) in the presence of glycerol (Table 1 andSupporting Information, Table S1).To simplify subsequent analyses, we selected one representa-
tive strain for each mutant class as shown in Table 1: M. bovisBCG POA1 [panD1 mas-1], POA2 [panD2], POA3A [mas-1],POA3B [ppsA1], and POA4 [glpK1 mas-1]. The POA-resistantphenotypes for the five representative strains in media withoutand with glycerol can be seen in Figure 2A,B, respectively.Glycerol sensitizes all strains to POA irrespective of genotype butto different degrees (Figure 2B). This results in a complete loss ofresistance for low-resistance-class POA3 (mas or ppsA-E)mutants whereas the low-resistance-class POA4 mutants,containing an additional glpK mutation, retain their resistancein the presence of glycerol.Mutant selection and all phenotypic characterizations so far
were carried out at neutral pH. Because previous POA mutant
Figure 1. POA-resistant mutations in panD, mas/ppsA-E, and glpK.Shown are biochemical reactions and schematic structures of theproteins with the location and nature of the mutations identified viawhole genome sequencing of 26 POA-resistant M. bovis BCG strains.Numbers in parentheses refer to the respective allele number asdescribed in Table 1 and Table S1. (A) PanD, aspartate decarboxylase.Domain mapping according to ref 37. (B)Mas and PpsA-E, mycocerosicacid synthase and phthiocerol polyketide synthases. Domains: PP,phosphopantetheinylate acyl carrier protein; KS, ketosynthase; AT,acyltransferase; KR, ketoreductase; DH, dehydratase; ER, enoylreductase and C, condensation domain.55,56 (C) GlpK, glycerol kinase.In contrast to mutations in panD and mas/ppsA-E that alone can conferPOA resistance, glpK mutations were only observed together with masor ppsA-E mutations and appear not to confer POA resistance bythemselves. Also, in contrast to the variety of different mutationsobserved within panD and mas/ppsA-E, Table 1 and Table S1 show thatall frameshift mutations in glpK in POA-resistant strains isolated fromfour independent experiments were found at the same site. Frameshiftmutations at this same site were also described by refs 32, 33, and 36.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
C

selection experiments on acidic pH agar had failed to deliverresistant strains,9,15 we predicted that the representative POA-resistant strains would exhibit their resistance at neutral pH butnot at acidic pH. The results from broth dilution MICs carriedout at pH 5.8 (Figure 2C) indeed show that POA resistancemutations do not confer POA resistance under acidic pH, whichis consistent with the inability to isolate POA-resistant mutantson acidic pH agar.POA-Resistant Mutations Also Confer PZA Resistance.
In our studies, we directly used bioactive POA for the selection ofresistant mutants to avoid isolating pncA mutants that constitutethe vast majority of PZA resistance in vitro and in vivo.15 Todetermine whether POA resistance mutations confer resistanceto prodrug PZA, we introduced a functional copy of the pncAgene from M. tuberculosis H37Rv into the five representativemutants and wild-typeM. bovis BCG using an integrative plasmidconstruct. As expected, the introduction of M. tuberculosis pncAinto wild-type BCG rendered the previously PZA-resistant strainPZA-susceptible4 (Figure 2D). In contrast, the five representa-tive POA-resistant strains remained resistant to PZA aftercomplementation with functional pncA (Figure 2D). Thus, thePOA resistance conferring mutations also confers resistance toPZA. As observed previously with POA at acidic pH, the fiverepresentative strains complemented with functional pncA weresusceptible to PZA at acidic pH 5.8 (Figure 2E).To exclude the possibility that the identified POA/PZA-
resistant genotypes caused unspecific drug resistance, wemeasured the susceptibility of the five representative strains toisoniazid, rifampicin, streptomycin, and ciprofloxacin. The fivePOA/PZA-resistant strains were fully susceptible to the drugs,showing the sameMICs as the wild-type strain (data not shown).These results indicate that POA/PZA resistance conferring
mutations are drug-specific and do not cause general antibioticresistance.
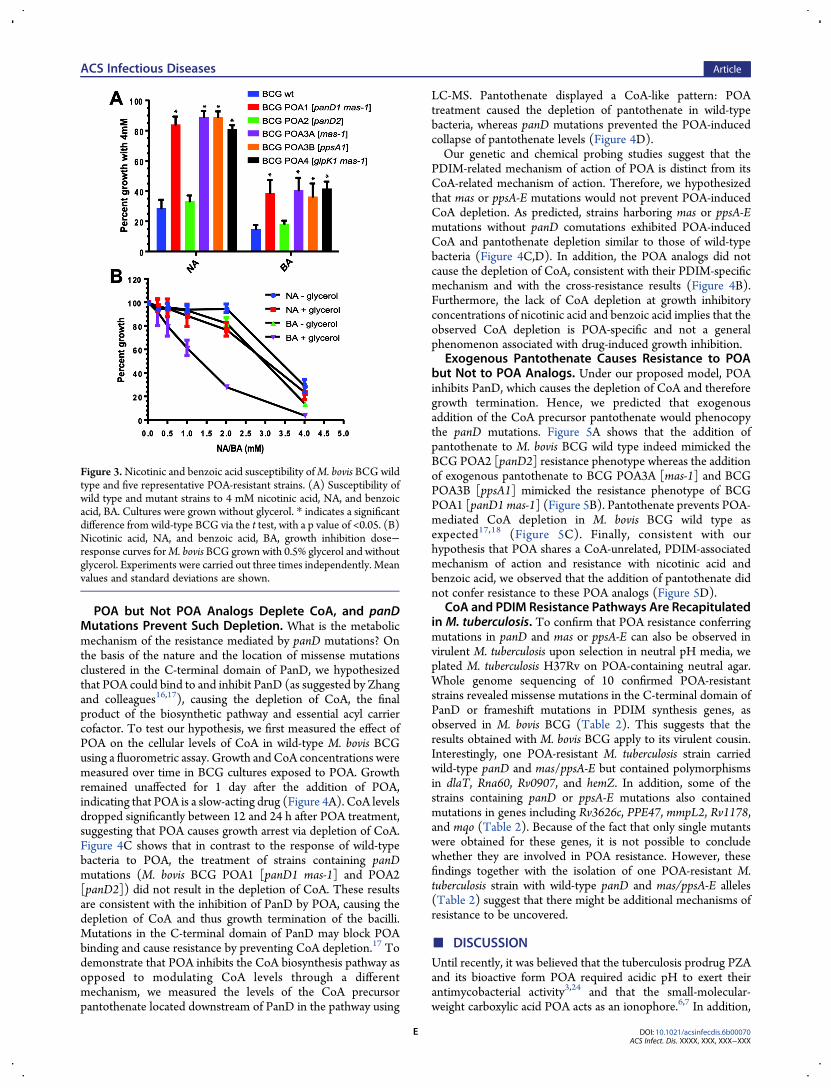
Mutations Affecting PDIM Synthesis but Not Muta-tions Affecting CoA Synthesis Confer Cross Resistance toPOA Analogs. Nicotinic acid and benzoic acid are twostructural analogs of POA, and both show antimycobacterialactivity,7 albeit with a lower potency than for POA (Figure 3B).Previous attempts to isolate mutants against these analogs onacidic agar were also unsuccessful.7 The genetic data abovesuggest that resistance to POA may be due to two distinctmechanisms involving either panD ormas/ppsA-Emutations, thelatter causing a lower level of resistance. To determine whetherthe analogs shared either of the two mechanisms of action withPOA, cross-resistance studies were carried out. The growth ofwild-type BCG was inhibited by nicotinic and benzoic acid asexpected (Figure 3A). POA-resistant strains containing mas orppsA-E mutations were also resistant to the analogs, whereaspanDmutations alone (POA2mutant class) did not confer cross-resistance (Figure 3A). The results indicate that the threecarboxylic acids share a PDIM synthesis-related mechanism ofaction whereas the CoA-related mechanism of action is unique toPOA. It is interesting that glycerol in themedium hypersensitizedM. bovis BCG to inhibition by benzoic acid and more weakly tonicotinic acid (Figure 3B), as was observed for POA.To confirm the role of mutations in PDIM synthesis, genes in
POA, and nicotinic/benzoic acid resistance, a functional copy ofthe mas gene, encoding the 2111 amino acid mycocerosic acidsynthase, was introduced into POA-resistant strainM. bovis BCGPOA3A [mas-1]. Upon complementation with functional mas,BCG POA3A [mas-1] lost its resistance to POA and reverted towild-type susceptibility (Supporting Information, Figure S2A).Figure S2B shows that mas complementation in BCG POA3Aalso restored susceptibility to nicotinic acid and benzoic acid.
Figure 2. Pyrazinoic acid and pyrazinamide susceptibility of M. bovis BCG wild type and five representative POA-resistant strains. Growth inhibitiondose−response curves are shown. (A−C) POA dose−response curves for BCG wild type and POA-resistant strains at neutral pH without (A) and with(B) glycerol (0.5%) and (C) at acidic pH 5.8 (without glycerol). (D, E) PZA dose−response curves for amidase pncA-complemented BCGwild type andPOA-resistant strains at neutral pH (D) and acidic pH 5.8 (E). Cultures in D and E were grown in glycerol-free media. Experiments were carried outthree times independently. Mean values and standard deviations are shown.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
D
POA but Not POA Analogs Deplete CoA, and panDMutations Prevent Such Depletion. What is the metabolicmechanism of the resistance mediated by panD mutations? Onthe basis of the nature and the location of missense mutationsclustered in the C-terminal domain of PanD, we hypothesizedthat POA could bind to and inhibit PanD (as suggested by Zhangand colleagues16,17), causing the depletion of CoA, the finalproduct of the biosynthetic pathway and essential acyl carriercofactor. To test our hypothesis, we first measured the effect ofPOA on the cellular levels of CoA in wild-type M. bovis BCGusing a fluorometric assay. Growth and CoA concentrations weremeasured over time in BCG cultures exposed to POA. Growthremained unaffected for 1 day after the addition of POA,indicating that POA is a slow-acting drug (Figure 4A). CoA levelsdropped significantly between 12 and 24 h after POA treatment,suggesting that POA causes growth arrest via depletion of CoA.Figure 4C shows that in contrast to the response of wild-typebacteria to POA, the treatment of strains containing panDmutations (M. bovis BCG POA1 [panD1 mas-1] and POA2[panD2]) did not result in the depletion of CoA. These resultsare consistent with the inhibition of PanD by POA, causing thedepletion of CoA and thus growth termination of the bacilli.Mutations in the C-terminal domain of PanD may block POAbinding and cause resistance by preventing CoA depletion.17 Todemonstrate that POA inhibits the CoA biosynthesis pathway asopposed to modulating CoA levels through a differentmechanism, we measured the levels of the CoA precursorpantothenate located downstream of PanD in the pathway using
LC-MS. Pantothenate displayed a CoA-like pattern: POAtreatment caused the depletion of pantothenate in wild-typebacteria, whereas panD mutations prevented the POA-inducedcollapse of pantothenate levels (Figure 4D).Our genetic and chemical probing studies suggest that the
PDIM-related mechanism of action of POA is distinct from itsCoA-related mechanism of action. Therefore, we hypothesizedthat mas or ppsA-E mutations would not prevent POA-inducedCoA depletion. As predicted, strains harboring mas or ppsA-Emutations without panD comutations exhibited POA-inducedCoA and pantothenate depletion similar to those of wild-typebacteria (Figure 4C,D). In addition, the POA analogs did notcause the depletion of CoA, consistent with their PDIM-specificmechanism and with the cross-resistance results (Figure 4B).Furthermore, the lack of CoA depletion at growth inhibitoryconcentrations of nicotinic acid and benzoic acid implies that theobserved CoA depletion is POA-specific and not a generalphenomenon associated with drug-induced growth inhibition.
Exogenous Pantothenate Causes Resistance to POAbut Not to POA Analogs. Under our proposed model, POAinhibits PanD, which causes the depletion of CoA and thereforegrowth termination. Hence, we predicted that exogenousaddition of the CoA precursor pantothenate would phenocopythe panD mutations. Figure 5A shows that the addition ofpantothenate to M. bovis BCG wild type indeed mimicked theBCG POA2 [panD2] resistance phenotype whereas the additionof exogenous pantothenate to BCG POA3A [mas-1] and BCGPOA3B [ppsA1] mimicked the resistance phenotype of BCGPOA1 [panD1 mas-1] (Figure 5B). Pantothenate prevents POA-mediated CoA depletion in M. bovis BCG wild type asexpected17,18 (Figure 5C). Finally, consistent with ourhypothesis that POA shares a CoA-unrelated, PDIM-associatedmechanism of action and resistance with nicotinic acid andbenzoic acid, we observed that the addition of pantothenate didnot confer resistance to these POA analogs (Figure 5D).
CoA and PDIM Resistance Pathways Are Recapitulatedin M. tuberculosis. To confirm that POA resistance conferringmutations in panD and mas or ppsA-E can also be observed invirulent M. tuberculosis upon selection in neutral pH media, weplated M. tuberculosis H37Rv on POA-containing neutral agar.Whole genome sequencing of 10 confirmed POA-resistantstrains revealed missense mutations in the C-terminal domain ofPanD or frameshift mutations in PDIM synthesis genes, asobserved in M. bovis BCG (Table 2). This suggests that theresults obtained with M. bovis BCG apply to its virulent cousin.Interestingly, one POA-resistant M. tuberculosis strain carriedwild-type panD and mas/ppsA-E but contained polymorphismsin dlaT, Rna60, Rv0907, and hemZ. In addition, some of thestrains containing panD or ppsA-E mutations also containedmutations in genes including Rv3626c, PPE47, mmpL2, Rv1178,and mqo (Table 2). Because of the fact that only single mutantswere obtained for these genes, it is not possible to concludewhether they are involved in POA resistance. However, thesefindings together with the isolation of one POA-resistant M.tuberculosis strain with wild-type panD and mas/ppsA-E alleles(Table 2) suggest that there might be additional mechanisms ofresistance to be uncovered.
■ DISCUSSIONUntil recently, it was believed that the tuberculosis prodrug PZAand its bioactive form POA required acidic pH to exert theirantimycobacterial activity3,24 and that the small-molecular-weight carboxylic acid POA acts as an ionophore.6,7 In addition,
Figure 3.Nicotinic and benzoic acid susceptibility ofM. bovis BCG wildtype and five representative POA-resistant strains. (A) Susceptibility ofwild type and mutant strains to 4 mM nicotinic acid, NA, and benzoicacid, BA. Cultures were grown without glycerol. * indicates a significantdifference from wild-type BCG via the t test, with a p value of <0.05. (B)Nicotinic acid, NA, and benzoic acid, BA, growth inhibition dose−response curves forM. bovis BCG grown with 0.5% glycerol and withoutglycerol. Experiments were carried out three times independently. Meanvalues and standard deviations are shown.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
E

the fatty acid synthase FAS I9 and the ribosomal protein S111
were proposed as two discrete molecular targets of POA.Attempts to isolate POA-resistant mutants of M. tuberculosis onacidified agar to determine the mechanism of action wereunsuccessful9,15 until recently.17,31 Furthermore, it was recentlyshown or rediscovered that the caseum of tuberculosis lesions isnot acidic,26−29 that the drug also works at neutral pH,5,25 andthat POA appears not to act as an ionophore as its majormechanism of action.25 On the basis of these new findings, werevisited the genetic approach using neutral instead of acidic agarfor the selection of resistant mutants and successfully isolatedPOA-resistant mutants in bothM. bovis BCG andM. tuberculosisH37Rv.The observed spontaneous resistance mutation frequencies
ranged, dependent on the specific selection medium, from 10−5
to 10−3/CFU. This is in the range of or is even higher than theresistance frequency for isoniazid (10−6 38) and is much higherthan the frequency reported for rifampicin (10−8 39). Thesimilarity between POA and isoniazid may be due to the fact thatin both cases mutations in multiple genes can cause resistanceand that, in some of the genes, loss-of-function mutations areallowed (see below). In contrast, rifampicin has only one majortarget that is essential; i.e., only very few resistance-causingpolymorphisms are tolerated.27,39
Unexpectedly, both FAS I and ribosomal protein S1 werefound to be intact from the 26 sequenced genomes of POA-
resistant M. bovis BCG and the 10 sequenced genomes of POA-resistant M. tuberculosis H37Rv. Rather, resistance to POAappears to be caused by (i) missense mutations in the aspartatedecarboxylase panD involved in the synthesis of the acyl carrierCoA and (ii) loss-of-function mutations in the polyketidesynthases mas and ppsA-E involved in the synthesis of the cell-envelope-associated virulence factor PDIM. The two mecha-nisms appear to be additive because strains carrying doublemutations in panD and mas or ppsA-E displayed a higher level ofresistance. Probing for cross resistance to two structural analogsof POA, nicotinic acid and benzoic acid, showed that the analogsshare the PDIM- but not the CoA-related mechanism of actionwith POA.To determine the molecular basis underlying the CoA-related
mechanism of resistance, we showed that POA, but not nicotinicor benzoic acid, depletes CoA in wild-type bacteria. This CoAdepletion is prevented by mutations in panD but not in mas orppsA-E. The exogenous addition of pantothenate copied thepanD phenotypes, conferring resistance to POA but not tonicotinic and benzoic acid, and prevented the POA-induceddepletion of intracellular CoA. Our finding that POA depletescellular CoA may explain the reported effect of PZA on fatty acidsynthesis. Jacobs and colleagues showed that PZA inhibits thesynthesis of palmitic acid,40 supporting the hypothesis that thedrug inhibits FAS I.9 However, Barry and colleagues providedevidence that this enzyme is not the direct target of the drug.10
Figure 4. Effect of pyrazinoic, nicotinic acid, and benzoic acid on CoA levels inM. bovis BCGwild type and five representative POA-resistant strains. (A)Effect of POA (4 mM) on the growth (red) and CoA level (purple with purple connecting line) ofM. bovis BCG wild type as a function of time. Blue:growth of drug-free control. Green with green connecting line: CoA levels of drug-free control. Growth on left y axis and CoA level on right y axis. *indicates a significant difference fromCoA levels of respective untreated samples at respective time points via the t test, with a p value of <0.05. (B) Effectof nicotinic acid, NA, and benzoic acid, BA (4 mM), on CoA level inM. bovis BCG wild type. * indicates a significant difference from untreated samplesvia the t test, with a p value of <0.05. (C) Effect of POA (4mM, striped bars) on the CoA level inM. bovisBCGwild type and POA-resistant strains. Filledbars: drug-free control. * indicates a significant difference from respective untreated samples via the t test, with a p value of <0.05. (D) Effect of POA onpantothenate level as determined via LC-MS. Solid bars: untreated control. Vertical striped bars: after treatment with 1 mM POA for 24 h . Horizontalstriped bars: after treatment with 4mMPOA for 24 h. * indicates a significant difference from respective untreated samples via the t test, with a p value of<0.05. (B−D) Measurements were taken after 24 h of treatment. Experiments were carried out three times independently. Mean values and standarddeviations are shown.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
F

This apparent contradiction may be explained by our finding thatPOA depletes the acyl carrier cofactor essential for fatty acidsynthesis: cessation of fatty acid synthesis upon drug exposuremay thus be an indirect effect of POA on CoA synthesis.Our findings that panD missense mutations confer POA
resistance, that POA depletes cellular CoA, and that POAresistance mutations in panD prevent CoA depletion areconsistent with and extend recent reports by Zhang andcolleagues made during the course of the current study.16,17
The authors attributed POA resistance to similar missensemutations in panD. Furthermore, they showed that POA inhibitsaspartate decarboxylase in vitro.17 Our collective data indicatethat PZA/POA acts via inhibition of aspartate decarboxylasePanD in the CoA biosynthetic pathway. This results in depletingthe pool of cellular cofactor essential in numerous pathways thatuse activated acyls, such as fatty acid biosynthesis, and bringsgrowth to a halt.In this work, we identified a secondmechanism of resistance to
PZA/POA involving PDIMs. PDIMs are cell-envelope-associ-ated polyketides, which are nonessential in vitro.41,42 However,PDIM knockout mutants are attenuated in mouse models ofinfection,34,35 indicating that these in vitro dispensable surfacemolecules are virulence factors required for growth and survivalin the host. If not required for in vitro growth, how are theyinvolved in POA-mediated growth inhibition? The finding thatall mas and ppsA-E POA resistance-conferring mutations were
frameshift events, causing a loss of function, may provide a clue. Itis conceivable that these polyketide synthases produce one orseveral toxic intermediates in the presence of POA (and nicotinicacid and benzoic acid). Loss-of-function mutations would theneliminate the generation of these toxic POA products and causeresistance. One may speculate that activated forms of these smallcarboxylic acids are incorporated into nascent PDIM polyketidesin place of the correct acyl groups and thus corrupt intracellularPDIM synthesis or transport processes to the outer cellenvelope.43 Loss-of-function mutations in PDIM synthesisgenes would then prevent this toxic effect and allow growth invitro in the presence of the drug and structural analogs.Interestingly, the newly identified PDIM-related mechanism
of resistance to PZA may shed light on two major puzzlesassociated with this old drug: the lack of clear resistanceepidemiology patterns in clinical isolates22 and the disconnectbetween PZA’s modest in vitro potency compared to its excellentin vivo activity1,44−46
For most TB drugs, a reasonable correlation exists between invitro and clinical resistance patterns, with the majority ofresistance mutations in targets or activating enzymes. For PZA-resistant clinical isolates, however, no such pattern has beenobserved other than a high frequency and diversity of pncAmutations, disabling the activation of the PZA prodrug.19−21 Ourfinding that the loss of the PDIM virulence factor causesresistance may partially explain this lack of clinically observed
Figure 5. Effect of pantothenate on pyrazinoic, nicotinic acid, and benzoic acid susceptibility and CoA level. (A) Effect of exogenous pantothenate on thePOA susceptibility ofM. bovis BCG wild type in comparison with the pantothenate-free susceptibility of POA-resistantM. bovis BCG harboring a panDmutation. (B) Effect of exogenous pantothenate on POA susceptibility of POA-resistantM. bovis BCG harboringmas or ppsAmutations in comparisonto the pantothenate-free susceptibility of POA-resistantM. bovis BCG harboring panD − mas double mutations. (C) Effect of exogenous pantothenateon the CoA level of POA-treatedM. bovis BCGwild type after 24 h of treatment. * indicates a significant difference from respective untreated samples viathe t test, with a p value of <0.05. (D) Lack of effect of exogenous pantothenate on nicotinic acid, NA, and benzoic acid, BA, susceptibility ofM. bovisBCG wild type. Pantothenate concentration: 0.1 mM. POA, NA, and BA concentrations: 4 mM. Experiments were carried out three timesindependently. Mean values and standard deviations are shown.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
G

resistance epidemiology. The in vitro isolated loss of functionmutations in mas and ppsA-E might occur in patients, but theassociated virulence cost is too high to sustain growth or survivalin lesions and sputum; therefore, these PZA-resistant strains arenot detected in clinical samples. Indeed, when we analyzed 1849genome-sequenced M. tuberculosis clinical isolates from thegenome-wide Mycobacterium tuberculosis variation (GMTV)database, we found none containing frameshift mutations inmas or ppsA-E genes.47 We also observed the same when welooked into sequences of XDR strains from Pakistan that werepncA wild type and resistant to PZA.48
Most TB drugs are potent antibacterials in vitro and displaysimilar activity in vivo. In contrast, PZA’s antibacterial activity invitro is modest at best, but the drug accelerates lesion sterilizationin vivo. Recently, it was suggested that the drug may act on hostfactors as an explanation of this disconnect.49 However, mouseinfection experiments by Grosset and colleagues suggest thathost factor interference does not play a significant role in the invivo action of the drug.50 The authors hypothesized that if PZAhas host-directed activity in addition to its antibacterial activitythen this should be detectable in mice infected with PncA−-,PZA-resistant organisms. However, no such activity could bedetected, pointing to a lack of host-directed activity. Todemonstrate that PZA does indeed act as an antibacterial invivo, the authors tested the efficacy of PZA in immune-deficientmice under the hypothesis that only antibacterial activity shouldand would be detected. Surprisingly, PZA was inactive inimmune-deficient athymic nude mice infected with PncA+ M.tuberculosis, where PZA cannot modulate the (absent) host’s
immune system. This very puzzling finding suggests that PZAworks neither via its antibacterial activity nor by modulating hostfactors in vivo. Our finding that PZA may act as an antivirulenceagent by corrupting PDIM synthesis may reconcile theseobservations. In standard M. tuberculosis infection models inwhich bacteria are PncA+ and the mouse immune system isintact, PZA exposure reduces the virulence of the bacteria, andefficacy is observed. The prevention of prodrug activation inPncA− bacilli growing in immune-competent mice preventsinterference with PDIM synthesis, resulting in no activity despitean intact immune system. Unmasking prodrug activation byinfecting immune-deficient mice with PncA+ bacteria does notrestore efficacy despite the corruption of PDIM synthesisbecause there is no immune system to take advantage of it.Interestingly, we observed that the addition of glycerol to
standard glucose-based growth media hypersensitizes mycobac-teria to POA. Consistent with this observation, low-level POAresistance of mas and ppsA-E mutants is exhibited only inglycerol-free media; in media containing glycerol, these strainsare POA-susceptible. Also consistent with glycerol-induced POAhypersensitization, strains containing mas or ppsA-E mutationswere resistant in the presence of glycerol when they coharboredloss-of-function mutations in glycerol kinase glpK, therebyblocking glycerol catabolism. These double mutants wereisolated only on medium containing glycerol. Loss-of-functionmutations in glpK seem to be insufficient to cause POA resistanceon their own because these were not observed in our geneticscreen. How glycerol hypersensitizes mycobacteria to POAremains an open question. Additionally, previous drug discoveryprograms and screens have identified compound classes thatexerted glycerol-dependent activity against M. tuberculosis,32,33
with ∼10% of the hits showing greater potency in glycerol-containing media.33 The observation that the presence ofglycerol increases the sensitivity of wild type and all mutantstrains (albeit to different degrees), not only to POA but also toPOA analogs nicotinic acid and benzoic acid (which interfereonly with POA’s PDIM but not the CoA-related mechanism),may indicate that glycerol catabolism creates a vulnerablemetabolic state of the bacterium in which bacilli show anincreased sensitivity to additional metabolic disturbances. Ourprevious observation that glycerol utilization induces methyl-glyoxal toxicity supports this notion.32 Whether glycerol’smethylglyoxal effect plays a role in the observed POAhypersensitization remains to be established. Whatever themechanistic basis for the observed glycerol effect on POAsensitivity may be, our data suggest that the results of clinicaldrug susceptibility testing are likely influenced by the presence orabsence of glycerol, which in addition to other assay variables51
may contribute to the often unreliable PZA susceptibility data.52
In conclusion, our genetic and phenotypic analyses, chemicalprobing with POA analogs, CoA measurements, and chemicalcomplementation studies with pantothenate suggest that PZA/POA has at least two distinct mechanisms of action andresistance. One involves the depletion of CoA, and resistance iscaused by mutations in panD, preventing CoA depletion. Theseresults confirm and extend recent findings suggesting PanD astarget for POA/PZA.16,17 A novel second mechanism involvesPDIM synthesis, where resistance is caused by loss-of-functionmutations in PDIM synthesizing genes mas and ppsA-E. Thefinding that the drug may interfere with the synthesis of avirulence factor could explain pyrazinamide’s excellent in vivoefficacy when compared to its modest in vitro potency, as well asthe lack of clear molecular resistance epidemiology of PZA.
Table 2. Sequence Polymorphisms of 10 Whole-Genome-Sequenced POA-Resistant M. tuberculosis H37Rv Strains
genotypicclass
M. tuberculosisH37Rv strain
mutations incandidate genes
mutations inadditional genes
panD POAR 1 [panD1]a panD: Δ380APOAR 2 [panD2]a panD: T395G/
Leu132ArgPOAR 3 [panD3]a panD: Δ416G Rv3626c: G710T/
Arg237LeuPOAR 4 [panD4]b panD: A349G/
Met117ValPPE47: Ins14G
POAR 5 [panD5]a panD: T407C/Leu136Pro
mmpL2: G2425A/Val809Ile
Rv1178: G395C/Arg132Pro
POAR 6 [panD6]b panD: G382T/Ala128Ser
mqo: A1325G/Asp442Gly
mas/ppsA-E
POAR 7 [ppsC1]a ppsC: Ins2674CPOAR 8 [ppsE1]a ppsE: G823A/
Gly275ArgRv3256c:Δ437TCGCG
POAR 9 [ppsA1]b ppsA: Ins491A Rv1178: G395C/Arg132Pro
others POAR 10a,c dlaT: Ins490ACRna60: Ins77CRv0907: C1190A/Thr397Lys
hemZ: G151A/Val51Ile
aM. tuberculosis H37Rv POAR 3 [panD3], POAR 10 and POAR 1[panD1], POAR 2 [panD2], POAR 5 [panD5], and POAR 7 [ppsC1]were isolated from two independent cultures on POA containing agarwithout glycerol. bM. tuberculosis H37Rv POAR 8 [ppsE1], POAR 9[ppsA1], POAR 4 [panD4], and POAR 6 [panD6] were isolated fromtwo independent cultures on POA agar with glycerol. cNo genotypegiven in strain name because multiple polymorphisms were observed.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
H

■ MATERIALS AND METHODS
Bacterial Strains, Culture Medium, and Chemicals. M.bovis BCG (ATCC 35734) and M. tuberculosis H37Rv (ATCC27294) strains were maintained in complete Middlebrook 7H9medium (BD Difco) supplemented with 0.05% (v/v) Tween 80(Sigma), 0.5% (v/v) glycerol (Fisher Scientific), and 10% (v/v)Middlebrook albumin-dextrose-catalase (BD Difco) at 37 °Cwith agitation at 80 rpm. Pyrazinamide, pyrazinoic acid, benzoicacid, and nicotinic acid were purchased from Sigma-Aldrich andwere freshly dissolved in 90%DMSO to a concentration of 0.5Mand sterilized using 0.2 μm PTFE membrane filters (AcrodiscPALL). Kanamycin and sodium pantothenate were obtainedfrom Sigma-Aldrich, dissolved in deionized water, and sterilizedusing 0.2 μm Minisart high-flow syringe filters (Sartorius) priorto adding to specific growth media under aseptic conditions. ThepH of broth and liquid media after the addition of POA/NA/BAwas checked using pH paper strips (Merck) and verified to bearound 6.5.Selection of Spontaneous Resistant Mutants and
Growth and Resistance Determination. CFU (104 to 106)from mid-log-phase cultures of M. bovis BCG (M. tuberculosis)was plated on complete 7H10 agar plates or 7H10 withoutglycerol containing 1, 2, or 4 mM POA and grown for 4 weeks.Colonies were picked from selection plates and restreaked onagar containing the same concentration of POA for colonypurification and to verify drug resistance. Isolated colonies werepicked from the restreak plates and expanded in 7H9 to anOD600of 0.7−0.8 and stored in 25% glycerol in 1 mL aliquots at−80 °C.This frozen stock was then used for the subsequent phenotypicand genetic characterization of mutants.Growth inhibitory activities of compounds against M. bovis
BCG were determined as described for M. tuberculosis in ref 5.The MIC90 and MIC50 reported here are those concentrations ofdrug that inhibit 90 and 50% of growth, respectively, as comparedto a drug-free control after 5 days of incubation. All MICs wereperformed in technical and biological replicates.Whole Genome Sequencing. Genomic DNA was isolated
from M. bovis BCG and M. tuberculosis H37Rv, wild type, andmutants as described.53 Library construction using a NEBNextUltra DNA library preparation kit (New England Biolabs), wholegenome sequencing on the Illumina MiSeq platform, andbioinformatics analyses was performed by AIT Biotech,Singapore. (Details are described in Supporting Information,Supplemental methods).pncA andmas Complementation Constructs. To restore
pyrazinamidase PncA activity in M. bovis BCG, which has anonfunctional endogenous pncA locus, the integrative vectorpMV306 was used.54 The M. tuberculosis pncA coding sequenceincluding the putative promoter was PCR amplified usingprimers pncA_F 5′ATGACACCTCTGTCACCGGACGGA3′and pncA_R 5′AAGCTTCAGGAGCTGCAAACCAACTCG-A3′ and inserted into pCR2.1TOPO by TOPO TA cloning (LifeTechnologies). The insert was subsequently excised using EcoRIand integrated into pMV306. The mas gene of M. bovis BCGincluding its putative promoter was amplified using PCR withspecific primers mas_F 5′TGGTACCGGTCGGATGTGA-TGTG3′ and mas_R 5′TATCTAGAATTCGACCGCTAT-GATGCC3′ and inserted into the KpnI/XbaI restriction sitesof pMV306. The integrity of the constructs was confirmed bycapillary sequencing (AIT Biotech, Singapore). The constructswere electroporated into M. bovis BCG strains and selected onKanamycin (25 μg/mL) containing 7H10 agar plates at 37 °C.
Kanamycin-resistant colonies were picked after 3 weeks,expanded with Kanamycin selection, and frozen at −80 °C asglycerol stock, which was subsequently characterized.
CoA Quantification. M. bovis BCG cultures were grown tomid-log phase in 1 L roller bottles (Corning) at 37 °C andpelleted in 50 mL tubes at 3200 rpm for 10 min. Pellets wereresuspended in the specific growth medium after adjusting to anOD600 of 0.2 and incubated with or without POA, nicotinic acid,or benzoic acid. At each time point for each treatment, thesamples were plated on 7H10 to determine CFU, and anequivalent of 2 × 109 CFU was pelleted by centrifugation. Thepellet was resuspended in 500 μL of phosphate-buffered saline(PBS) and homogenized by bead beating (Precellys 24homogenizer) at 6500 rpm three times for 30 s each. The lysatewas pelleted by centrifugation, and the supernatant wassubsequently used for analysis by a CoA assay kit (Sigma-AldrichMAK034) per the manufacturer’s instructions. The fluorescenceintensity was read on a Tecan Infinite M200 plate reader (λex =535 nm/λem = 587 nm), and the CoA quantity was determined innanomoles from a standard curve that was obtained for eachexperiment. These experiments were performed in technicalreplicates of independent biological replicates and normalizedper CFU.The lysate supernatant was also used for mass spectrometric
quantification of CoA and pantothenate, which was achieved byusing liquid chromatography coupled to mass spectrometry (LC-MS). (Details are described in Supporting Information,Supplemental methods.)
■ ASSOCIATED CONTENT*S Supporting InformationThe Supporting Information is available free of charge on theACS Publications website at DOI: 10.1021/acsinfecdis.6b00070.
Supplemental methods, phenotypic and genotypic classesof POA-resistant M. bovis BCG with details for anadditional 21 whole-genome-sequenced strains, glyceroleffect on POA susceptibility, and Mas complementation(PDF)
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected]. Phone/Fax: (65) 65166741/(65) 6776 6872.Author ContributionsP.G. and T.D. conceived the project, designed the strategy, andwrote themanuscript. P.G., M.Y., J.S., J.L.L., andM.G. carried outexperiments and analyses. J.P.S., F.K., and V.D. carried out themass spectrometric analyses.NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work was supported by the Singapore Ministry of Health’sNational Medical Research Council under its TranslationalClinical Research Flagship Grant (NMRC/TCR/011-NUHS/2014) and its Centre Grant MINE/Research core no. 4(NMRC/CG/013/2013) to T.D. and is part of the SingaporeProgramme of Research Investigating New Approaches toTreatment of Tuberculosis (SPRINT-TB; www.sprinttb.org),managed by Kristina Rutkute and led by Nick Paton. P.G.received a research scholarship from Yong Loo Lin School ofMedicine. We thank Sabai Phyu, the School of Medicine BSL3
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
I

core facility, for support; David Klinzing, AITBiotech, for wholegenome sequencing and genomics discussions; Claudia Setzerfor help with the initial BCGmutant screen; Joe Liu for help withplasmid construction; and Hayden Yeo for laboratory manage-ment and BSL-3 support.
■ ABBREVIATIONS
TB, tuberculosis; PZA, pyrazinamide; POA, pyrazinoic acid; NA,nicotinic acid; BA, benzoic acid; CoA, Coenzyme A; PDIM,phthiocerol dimycocerosate
■ REFERENCES(1) Mitchison, D. A. (1985) The action of antituberculosis drugs inshort-course chemotherapy. Tubercle 66, 219−225.(2) Zumla, A. I., Gillespie, S. H., Hoelscher, M., Philips, P. P. J., Cole, S.T., Abubakar, I., McHugh, T. D., Schito, M., Maeurer, M., and Nunn, A.J. (2014) New antituberculosis drugs, regimens, and adjunct therapies:needs, advances, and future prospects. Lancet Infect. Dis. 14, 327−340.(3) Zhang, Y., Shi, W., Zhang, W., and Mitchison, D. (2014)Mechanisms of Pyrazinamide Action and Resistance. Microbiol.Spectrum 2, 1−12.(4) Scorpio, A., and Zhang, Y. (1996) Mutations in pncA, a geneencoding pyrazinamidase/nicotinamidase, cause resistance to theantituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 2,662−667.(5) Via, L. E., Savic, R., Weiner, D. M., Zimmerman, M. D., Prideaux,B., Irwin, S. M., Lyon, E., O’Brien, P., Gopal, P., Eum, S., Lee, M., Lanoix,J.-P., Dutta, N. K., Shim, T., Cho, J. S., Kim, W., Karakousis, P. C.,Lenaerts, A., Nuermberger, E., Barry, C. E., and Dartois, V. (2015) Host-Mediated Bioactivation of Pyrazinamide: Implications for Efficacy,Resistance, and Therapeutic Alternatives. ACS Infect. Dis. 1, 203−214.(6) Zhang, Y., Scorpio, A., Nikaido, H., and Sun, Z. (1999) Role of AcidpH and Deficient Efflux of Pyrazinoic Acid in Unique Susceptibility ofMycobacterium tuberculosis to Pyrazinamide. J. Bacteriol. 181, 2044−2049.(7) Zhang, Y., Zhang, H., and Sun, Z. (2003) Susceptibility ofMycobacterium tuberculosis to weak acids. J. Antimicrob. Chemother. 52,56−60.(8) Zhang, Y., and Mitchison, D. (2003) The curious characteristics ofpyrazinamide: a review. Int. J. Tuberc Lung Dis 7, 6−21.(9) Zimhony, O., Cox, J. S., Welch, J. T., Vilcheze, C., and Jacobs, W. R.(2000) Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I(FASI) of Mycobacterium tuberculosis. Nat. Med. 6, 1043−1047.(10) Boshoff, H. I., Mizrahi, V., and Barry, C. E. (2002) Effects ofPyrazinamide on Fatty Acid Synthesis by Whole Mycobacterial Cellsand Purified Fatty Acid Synthase I. J. Bacteriol. 184, 2167−2172.(11) Shi, W., Zhang, X., Jiang, X., Yuan, H., Lee, J. S., Barry, C. E.,Wang, H., Zhang, W., and Zhang, Y. (2011) Pyrazinamide InhibitsTrans-Translation in Mycobacterium tuberculosis. Science 333, 1630−1632.(12) Alexander, D. C., Ma, J. H., Guthrie, J. L., Blair, J., Chedore, P., andJamieson, F. B. (2012) Gene Sequencing for Routine Verification ofPyrazinamide Resistance inMycobacterium tuberculosis: a Role for pncAbut Not rpsA. J. Clin Microbiol 50, 3726−3728.(13) Xia, Q., Zhao, L.-l., Li, F., Fan, Y.-m., Chen, Y.-y., Wu, B.-b., Liu,Z.-w., Pan, A.-z., and Zhu, M. (2015) Phenotypic and GenotypicCharacterization of Pyrazinamide Resistance amongMultidrug-resistantTuberculosis isolates in Zhejiang, China. Antimicrob. Agents Chemother.59, 1690−1695.(14) Akhmetova, A., Kozhamkulov, U., Bismilda, V., Chingissova, L.,Abildaev, T., Dymova, M., Filipenko, M., and Ramanculov, E. (2015)Mutations in the pncA and rpsA genes among 77 Mycobacteriumtuberculosis isolates in Kazakhstan. Int. J. Tuberc Lung Dis 19, 179−184.(15) Scorpio, A., Lindholm-Levy, P., Heifets, L., Gilman, R., Siddiqi, S.,Cynamon, M., and Zhang, Y. (1997) Characterization of pncAmutations in pyrazinamide-resistant Mycobacterium tuberculosis. Anti-microb. Agents Chemother. 41, 540−543.
(16) Zhang, S., Chen, J., Shi, W., Liu, W., Zhang, W., and Zhang, Y.(2013) Mutations in panD encoding aspartate decarboxylase areassociated with pyrazinamide resistance in Mycobacterium tuberculosis.Emerging Microbes Infect. 2, e34.(17) Shi, W., Chen, J., Feng, J., Cui, P., Zhang, S., Weng, X., Zhang, W.,and Zhang, Y. (2014) Aspartate decarboxylase (PanD) as a new target ofpyrazinamide in Mycobacterium tuberculosis. Emerging Microbes Infect. 3,e58.(18) Dillon, N. A., Peterson, N. D., Rosen, B. C., and Baughn, A. D.(2014) Pantothenate and Pantetheine Antagonize the AntitubercularActivity of Pyrazinamide. Antimicrob. Agents Chemother. 58, 7258−7263.(19) Stoffels, K., Mathys, V., Fauville-Dufaux, M., Wintjens, R., andBifani, P. (2012) Systematic Analysis of Pyrazinamide-ResistantSpontaneous Mutants and Clinical Isolates of Mycobacterium tuber-culosis. Antimicrob. Agents Chemother. 56, 5186−5193.(20) Casali, N., Nikolayevskyy, V., Balabanova, Y., Harris, S. R.,Ignatyeva, O., Kontsevaya, I., Corander, J., Bryant, J., Parkhill, J.,Nejentsev, S., Horstmann, R. D., Brown, T., and Drobniewski, F. (2014)Evolution and transmission of drug-resistant tuberculosis in a Russianpopulation. Nat. Genet. 46, 279−286.(21) Miotto, P., Cabibbe, A. M., Feuerriegel, S., Casali, N.,Drobniewski, F., Rodionova, Y., Bakonyte, D., Stakenas, P., Pimkina,E., Augustynowicz-Kopec, E., Degano, M., Ambrosi, A., Hoffner, S.,Mansjo, M., Werngren, J., Rusch-Gerdes, S., Niemann, S., and Cirillo, D.M. (2014) Mycobacterium tuberculosis Pyrazinamide ResistanceDeterminants: a Multicenter Study. mBio 5, e01819.(22) den Hertog, A. L., Sengstake, S., and Anthony, R. M. (2015)Pyrazinamide resistance in Mycobacterium tuberculosis fails to bite?Pathog. Dis. 73, 10.1093/femspd/ftv037.(23) Bradley, P., Gordon, N. C., Walker, T. M., Dunn, L., Heys, S.,Huang, B., Earle, S., Pankhurst, L. J., Anson, L., de Cesare, M., Piazza, P.,Votintseva, A. A., Golubchik, T., Wilson, D. J., Wyllie, D. H., Diel, R.,Niemann, S., Feuerriegel, S., Kohl, T. A., Ismail, N., Omar, S. V., Smith,E. G., Buck, D., McVean, G., Walker, A. S., Peto, T. E. A., Crook, D. W.,and Iqbal, Z. (2015) Rapid antibiotic-resistance predictions fromgenome sequence data for Staphylococcus aureus and Mycobacteriumtuberculosis. Nat. Commun. 6, 10063.(24) McDermott, W., and Tomsett, R. (1954) Activation ofpyrazinamide and nicotinamide in acidic environments in vitro. Am.Rev. Tuberc 70, 748−754.(25) Peterson, N. D., Rosen, B. C., Dillon, N. A., and Baughn, A. D.(2015) Uncoupling Environmental pH and Intrabacterial Acidificationfrom Pyrazinamide Susceptibility in Mycobacterium tuberculosis.Antimicrob. Agents Chemother. 59, 7320−7326.(26) Koller, F., and Leuthardt, F. (1934) Nekrose Und Autolyse.Bei-trag Zur Kenntnis Der Dystrophischen Verkalkung. Klin. Wochenschr.13, 1527−1529.(27) Gopal, P., and Dick, T. (2014) Reactive dirty fragments:implications for tuberculosis drug discovery. Curr. Opin. Microbiol. 21,7−12.(28) Lanoix, J.-P., Ioerger, T., Ormond, A., Kaya, F., Sacchettini, J.,Dartois, V., and Nuermberger, E. (2016) Selective Inactivity ofPyrazinamide against Tuberculosis in C3HeB/FeJ Mice Is BestExplained by Neutral pH of Caseum. Antimicrob. Agents Chemother.60, 735−743.(29) Lenaerts, A., Barry, C. E., and Dartois, V. (2015) Heterogeneity intuberculosis pathology, microenvironments and therapeutic responses.Immunol Rev. 264, 288−307.(30) Irwin, S. M., Prideaux, B., Lyon, E. R., Zimmerman, M. D., Brooks,E. J., Schrupp, C. A., Chen, C., Reichlen, M. J., Asay, B. C., Voskuil, M. I.,Nuermberger, E. L., Andries, K., Lyons, M. A., Dartois, V., and Lenaerts,A. J. (2016) Bedaquiline and Pyrazinamide Treatment Responses AreAffected by Pulmonary Lesion Heterogeneity in Mycobacteriumtuberculosis Infected C3HeB/FeJ. Mice. ACS Infect. Dis. 2, 251−267.(31) Lanoix, J.-P., Tasneen, R., O’Brien, P., Sarathy, J., Safi, H., Pinn,M., Alland, D., Dartois, V., and Nuermberger, E. (2016) High systemicexposure of pyrazinoic acid has limited anti-tuberculosis activity inmurine and rabbit models of tuberculosis. Antimicrob. Agents Chemother.60, 4197.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
J

(32) Pethe, K., Sequeira, P. C., Agarwalla, S., Rhee, K., Kuhen, K.,Phong, W. Y., Patel, V., Beer, D., Walker, J. R., Duraiswamy, J., Jiricek, J.,Keller, T. H., Chatterjee, A., Tan, M. P., Ujjini, M., Rao, S. P. S.,Camacho, L., Bifani, P., Mak, P. A., Ma, I., Barnes, S. W., Chen, Z.,Plouffe, D., Thayalan, P., Ng, S. H., Au, M., Lee, B. H., Tan, B. H.,Ravindran, S., Nanjundappa, M., Lin, X., Goh, A., Lakshminarayana, S.B., Shoen, C., Cynamon, M., Kreiswirth, B., Dartois, V., Peters, E. C.,Glynne, R., Brenner, S., and Dick, T. (2010) A chemical genetic screenin Mycobacterium tuberculosis identifies carbon-source-dependentgrowth inhibitors devoid of in vivo efficacy. Nat. Commun. 1, 1−8.(33) Stanley, S. A., Grant, S. S., Kawate, T., Iwase, N., Shimizu, M.,Wivagg, C., Silvis, M., Kazyanskaya, E., Aquadro, J., Golas, A., Fitzgerald,M., Dai, H., Zhang, L., and Hung, D. T. (2012) Identification of NovelInhibitors of M. tuberculosis Growth Using Whole Cell Based High-Throughput Screening. ACS Chem. Biol. 7, 1377−1384.(34) Cox, J. S., Chen, B., McNeil, M., and Jacobs, W. R. (1999)Complex lipid determines tissue-specific replication of Mycobacteriumtuberculosis in mice. Nature 402, 79−83.(35) Camacho, L. R., Ensergueix, D., Perez, E., Gicquel, B., andGuilhot, C. (1999) Identification of a virulence gene cluster ofMycobacterium tuberculosis by signature-tagged transposon mutagenesis.Mol. Microbiol. 34, 257−267.(36) Keating, L. A., Wheeler, P. R., Mansoor, H., Inwald, J. K., Dale, J.,Hewinson, R. G., and Gordon, S. V. (2005) The pyruvate requirementof some members of the Mycobacterium tuberculosis complex is due toan inactive pyruvate kinase: implications for in vivo growth. Mol.Microbiol. 56, 163−174.(37) Gopalan, G., Chopra, S., Ranganathan, A., and Swaminathan, K.(2006) Crystal structure of uncleaved L-aspartate-α-decarboxylase fromMycobacterium tuberculosis. Proteins: Struct., Funct., Genet. 65, 796−802.(38) Pansy, F., Stander, H., andDonovick, R. (1952) In vitro studies onisonicotinic acid hydrazide. Am. Rev. Tuberc. 65, 761−764.(39) Mariam, D. H., Mengistu, Y., Hoffner, S. E., and Andersson, D. I.(2004) Effect of rpoB Mutations Conferring Rifampin Resistance onFitness of Mycobacterium tuberculosis. Antimicrob. Agents Chemother.48, 1289−1294.(40) Zimhony, O., Vilcheze, C., Arai, M., Welch, J. T., and Jacobs, W. R.(2007) Pyrazinoic Acid and Its n-Propyl Ester Inhibit Fatty AcidSynthase Type I in Replicating Tubercle Bacilli. Antimicrob. AgentsChemother. 51, 752−754.(41) Azad, A. K., Sirakova, T. D., Rogers, L. M., and Kolattukudy, P. E.(1996) Targeted replacement of the mycocerosic acid synthase gene inMycobacterium bovis BCG produces a mutant that lacks mycosides.Proc. Natl. Acad. Sci. U. S. A. 93, 4787−4792.(42) Azad, A. K., Sirakova, T. D., Fernandes, N. D., and Kolattukudy, P.E. (1997) Gene Knockout Reveals a Novel Gene Cluster for theSynthesis of a Class of Cell Wall Lipids Unique to PathogenicMycobacteria. J. Biol. Chem. 272, 16741−16745.(43) Camacho, L. R., Constant, P., Raynaud, C., Laneelle, M.-A.,Triccas, J. A., Gicquel, B., Daffe, M., and Guilhot, C. (2001) Analysis ofthe Phthiocerol Dimycocerosate Locus ofMycobacterium tuberculosis:EVIDENCE THAT THIS LIPID IS INVOLVED IN THE CELLWALL PERMEABILITY BARRIER. J. Biol. Chem. 276, 19845−19854.(44) Tarshis, M. S., and Weed, W. A. J. (1953) Lack of significant invitro sensitivity of Mycobacterium tuberculosis to pyrazinamide onthree different solid media. Am. Rev. Tuberc. 67, 391−395.(45) Yeager, R. L., Munroe, W. G., and Dessau, F. I. (1952)Pyrazinamide (aldinamide) in the treatment of pulmonary tuberculosis.Am. Rev. Tuberc. 65, 523−546.(46) Malone, L., Schurr, A., Lindh, H., McKenzie, D., Kiser, J. S., andWilliams, J. H. (1952) The effect of pyrazinamide (aldinamide) onexperimental tuberculosis in mice. Am. Rev. Tuberc. 65, 511−518.(47) Chernyaeva, E. N., Shulgina, M. V., Rotkevich, M. S., Dobrynin, P.V., Simonov, S. A., Shitikov, E. A., Ischenko, D. S., Karpova, I. Y.,Kostryukova, E. S., Ilina, E. N., Govorun, V. M., Zhuravlev, V. Y.,Manicheva, O. A., Yablonsky, P. K., Isaeva, Y. D., Nosova, E. Y.,Mokrousov, I. V., Vyazovaya, A. A., Narvskaya, O. V., Lapidus, A. L., andO’Brien, S. J. (2014) Genome-wide Mycobacterium tuberculosis
variation (GMTV) database: a new tool for integrating sequencevariations and epidemiology. BMC Genomics 15, 1−8.(48) Ali, A., Hasan, Z., McNerney, R., XXMallard, K., Hill-Cawthorne,G., Coll, F., Nair, M., Pain, A., Clark, T. G., and Hasan, R. (2015) WholeGenome Sequencing Based Characterization of Extensively Drug-Resistant Mycobacterium tuberculosis Isolates from Pakistan. PLoS One10, e0117771.(49) Manca, C., Koo, M.-S., Peixoto, B., Fallows, D., Kaplan, G., andSubbian, S. (2013) Host Targeted Activity of Pyrazinamide inMycobacterium tuberculosis Infection. PLoS One 8, e74082.(50) Almeida, D. V., Tyagi, S., Li, S., Wallengren, K., Pym, A. S.,Ammerman, N. C., Bishai, W. R., and Grosset, J. H. (2014) RevisitingAnti-tuberculosis Activity of Pyrazinamide in Mice.Mycobac Dis 4, 145.(51) Zhang, Y., Permar, S., and Sun, Z. (2002) Conditions that mayaffect the results of susceptibility testing of Mycobacterium tuberculosisto pyrazinamide. J. Med. Microbiol. 51, 42−49.(52) Cui, Z., Wang, J., Lu, J., Huang, X., Zheng, R., and Hu, Z. (2013)Evaluation of Methods for Testing the Susceptibility of ClinicalMycobacterium tuberculosis Isolates to Pyrazinamide. J. Clin Microbiol51, 1374−1380.(53) Kaser, M., Ruf, M.-T., Hauser, J., Marsollier, L., and Pluschke, G.(2009) Optimized Method for Preparation of DNA from Pathogenicand Environmental Mycobacteria. Appl. Environ. Microbiol. 75, 414−418.(54) Stover, C. K., de la Cruz, V. F., Fuerst, T. R., Burlein, J. E., Benson,L. A., Bennett, L. T., Bansal, G. P., Young, J. F., Lee, M. H., Hatfull, G. F.,Snapper, S. B., Barletta, R. G., Jacobs, W. R., and Bloom, B. R. (1991)New use of BCG for recombinant vaccines. Nature 351, 456−460.(55) Jain, M., Petzold, C. J., Schelle, M. W., Leavell, M. D., Mougous, J.D., Bertozzi, C. R., Leary, J. A., and Cox, J. S. (2007) Lipidomics revealscontrol of Mycobacterium. Proc. Natl. Acad. Sci. U. S. A. 104, 5133.(56) Bachmann, B. O., and Ravel, J. (2009) Methods in Enzymology,Chapter 8, pp 181−217, Academic Press.
ACS Infectious Diseases Article
DOI: 10.1021/acsinfecdis.6b00070ACS Infect. Dis. XXXX, XXX, XXX−XXX
K
Related Documents











