of July 13, 2015. This information is current as Macrophages TLR Expression and Activity in Human Pulmonary Surfactant Protein A Regulates Crowther, Susheela Tridandapani and Larry S. Schlesinger Lisa N. Henning, Abul K. Azad, Kishore V. L. Parsa, Joy E. http://www.jimmunol.org/content/180/12/7847 doi: 10.4049/jimmunol.180.12.7847 2008; 180:7847-7858; ; J Immunol References http://www.jimmunol.org/content/180/12/7847.full#ref-list-1 , 43 of which you can access for free at: cites 82 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2008 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on July 13, 2015 http://www.jimmunol.org/ Downloaded from by guest on July 13, 2015 http://www.jimmunol.org/ Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

of July 13, 2015.This information is current as
MacrophagesTLR Expression and Activity in Human Pulmonary Surfactant Protein A Regulates
Crowther, Susheela Tridandapani and Larry S. SchlesingerLisa N. Henning, Abul K. Azad, Kishore V. L. Parsa, Joy E.
http://www.jimmunol.org/content/180/12/7847doi: 10.4049/jimmunol.180.12.7847
2008; 180:7847-7858; ;J Immunol
Referenceshttp://www.jimmunol.org/content/180/12/7847.full#ref-list-1
, 43 of which you can access for free at: cites 82 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2008 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on July 13, 2015
http://ww
w.jim
munol.org/
Dow
nloaded from

Pulmonary Surfactant Protein A Regulates TLR Expressionand Activity in Human Macrophages1
Lisa N. Henning,*† Abul K. Azad,*§¶ Kishore V. L. Parsa,*† Joy E. Crowther,*Susheela Tridandapani,*§¶ and Larry S. Schlesinger2*†‡§
The pulmonary innate immune system responds to various airborne microbes. Although its specificity is broad and based onthe recognition of pathogen-associated molecular patterns, it is uniquely regulated to limit inflammation and thereby preventdamage to the gas-exchanging alveoli. Macrophages, critical cell determinants of this system, recognize microbes throughpattern recognition receptors such as TLRs, which typically mediate proinflammatory responses. The lung collectin, sur-factant protein A (SP-A), has emerged as an important innate immune determinant that regulates microbe-macrophageinteractions in this environment. In this study, we report the basal and SP-A-induced transcriptional and posttranslationalregulation of TLR2 and TLR4 expression during the differentiation of primary human monocytes into macrophages. DespiteSP-A’s ability to up-regulate TLR2 expression on human macrophages, it dampens TLR2 and TLR4 signaling in these cells.SP-A decreases the phosphorylation of I�B�, a key regulator of NF-�B activity, and nuclear translocation of p65 which resultin diminished TNF-� secretion in response to TLR ligands. SP-A also reduces the phosphorylation of TLR signaling proteinsupstream of NF-�B, including members of the MAPK family. Finally, we report for the first time that SP-A decreases thephosphorylation of Akt, a major cell regulator of NF-�B and potentially MAPKs. These data identify a critical role for SP-Ain modulating the lung inflammatory response by regulating macrophage TLR activity. The Journal of Immunology, 2008,180: 7847–7858.
A lthough the majority of inhaled microbes and particu-lates are trapped and cleared by the upper airways of thelung, some organisms are able to travel further to the
terminal bronchioles and alveoli. The degree of inflammation ini-tiated in this site by a stimulus is tightly regulated by several el-ements of the innate immune system, as even moderate inflamma-tion could be harmful to the gas-exchanging alveolar structures (1).Two key components involved in this regulation are phagocytes,mainly alveolar macrophages (AM),3 and pulmonary surfactant.AM, which are bathed in surfactant, are the first professionalphagocytes to encounter, internalize, and degrade invading mi-crobes (2).
AM are considered alternatively activated, based upon theirunique biological attributes. These include a greater phagocyticpotential compared with other macrophages (2, 3) due to signifi-
cant expression and activity of PRRs, such as the mannose receptor(MR) and scavenger receptor A (SR-A) (4, 5); and immunoregu-lation through production of proinflammatory cytokines (e.g., suchas TNF-� and/or anti-inflammatory cytokines (e.g., TGF-�) (2, 3).They also produce less IL-1� (TNF-�), have a reduced oxidativeresponse to pathogens compared with blood monocytes (6–8), andserve as poor APCs (9), all of which are mechanisms that serve tocontrol alveolar inflammation.
TLRs are important cell-associated and intracellular PRRs.There are currently 12 identified mammalian TLRs, of whichTLR2 and TLR4 are among the most widely studied and are con-sidered the major transmembrane TLRs (10). TLR2 and TLR4play roles in initiating immune responses against pathogens. TLR2forms a heterodimer with TLR6 or TLR1 to recognize diacyl andtriacyl lipopeptides, respectively. TLR2 binds to zymosan, a par-ticle composed of yeast cell wall components; peptidoglycan, acomponent of the Gram-positive bacteria cell wall (11, 12), andPam3Cys-Ser-(Lys)4 hydrochloride (Pam3Cys), a lipohexapeptideanalog of the immunologically active N-terminal portion of bac-terial lipoprotein (13, 14). TLR4 associates with MD2 and CD14,enabling recognition of LPS, a major component of the Gram-negative cell wall, and heat shock proteins 60 and 70, which areubiquitously expressed (15–18).
Because an unrestricted inflammatory response induced by TLRsignaling can be potentially harmful to the host, this signaling istightly regulated, detecting the presence of certain microbial de-terminants and responding differently depending on the stimulus.The signaling cascade typically produces proinflammatory cyto-kines; however, activation through TLRs can also generate nega-tive regulators that inhibit a proinflammatory response (19).
Surfactant lipids and the surfactant-associated proteins A, B, C,and D are major components of pulmonary surfactant, which linesthe lung alveolus and serves to decrease surface tension at theair-liquid interface of the lung (20–22). Surfactant protein A
*Center for Microbial Interface Biology, †Division of Infectious Diseases, andDepartment of Medicine, and ‡Department of Molecular Virology, Immunology,and Medical Genetics, §Ohio State Biochemistry Program and ¶Dorothy M. DavisHeart and Lung Research Institute, Ohio State University, Columbus, OH 43210
Received for publication December 7, 2007. Accepted for publication April 4, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by National Institutes of Health Grant AI059639 (toL.S.S.).2 Address correspondence and reprint requests to Dr. Larry S. Schlesinger, Depart-ment of Medicine, Division of Infectious Diseases, Center for Microbial InterfaceBiology, Ohio State University, 460 West 12th Avenue, Biomedical Research Tower,Room 1004, Columbus, OH 43210. E-mail address: [email protected] Abbreviations used in this paper: AM, alveolar macrophage; MDM, monocyte-derived macrophage; MR, mannose receptor; SP-A, surfactant protein A; Pam3Cys,Pam3Cys-Ser-(Lys)4 hydrochloride; PRR, pattern recognition receptor; BAL, bron-choalveolar lavage; APP, alveolar proteinosis patient; MFI, mean fluorescence inten-sity; RCN, relative copy number; Ct, cycle threshold; SR-A, scavenger receptor A.
Copyright © 2008 by The American Association of Immunologists, Inc. 0022-1767/08/$2.00
The Journal of Immunology
www.jimmunol.org
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

(SP-A) is a large, multimeric protein with trimers that are assem-bled into an 18-mer protein through interactions between the col-lagen-like domains (23–25). It is classified as a collectin becauseeach monomer contains a linear collagen-like sequence, a shortlinking domain, and a globular Ca2�-dependent carbohydrate rec-ognition domain (CRD) (22).
SP-A has been implicated as a key component of the lung innateimmune response because it mediates host interactions with a va-riety of microbial pathogens (26) and regulates the nature of theinflammatory response. In this regard, SP-A can serve both as amicrobial opsonin and as a direct activator of macrophage func-tion. SP-A has been shown to up-regulate certain PRRs, such asSR-A and the MR, on macrophages (4, 5, 27). SP-A also reg-ulates TNF-� production, either up or down, depending onwhich receptor it binds on the cell surface and the activationstate of the cell (28). In human macrophages, SP-A down-reg-ulates oxidant production to stimuli by decreasing NADPH ox-idase activity (29).
Despite a large body of literature on the importance of TLR2and TLR4 in pathogen recognition and host response, there is lim-ited information about TLR2 and TLR4 expression on primaryhuman mononuclear phagocytes and the response of these cells toTLR agonists, particularly during monocyte differentiation intomacrophages. SP-A has emerged as a key regulator of the phago-cyte response in the alveolar compartment and contributes to thecharacteristic alternative activation state of the macrophages in thisenvironment. In this study, we report the discovery that SP-Adifferentially regulates the expression of TLR2 and TLR4 dur-ing primary human monocyte differentiation into macrophages.Despite up-regulation of TLR2 expression, SP-A markedly di-minishes the macrophage proinflammatory response generatedby both TLR2 and TLR4 agonists. The underlying mechanismis related to altered phosphorylation of a central regulator ofcellular function, Akt, as well as downstream intermediates inthe MAPK pathway and the activation of NF�B. These studiesunderscore the important role of SP-A in shaping the biology ofmacrophages in the lung alveoli.
Materials and MethodsBuffers, reagents, and media
Dulbecco’s PBS with and without Ca2� and Mg2� ions and RPMI 1640medium with L-glutamine (RPMI) were purchased from Invitrogen. PBFbuffer (PBS without Ca2� and Mg2� (Invitrogen), 5 mg/ml BSA (Sigma-Aldrich), and 10% heat-inactivated FBS (HyClone)) was used as a block-ing agent for confocal microscopy experiments. RHH medium (RPMI 1640plus 10 mM HEPES plus 0.4% human serum albumin) was used for cellculture experiments. Pam3Cys (Calbiochem) and Escherichia coli LPS(Sigma-Aldrich) were used for functional assays involving Western blot-ting and ELISAs.
Antibodies
Allophycocyanin-labeled anti-human TLR2 (clone T2.1), PE-labeledanti-human TLR4 (clone HTA125), and mouse IgG2a were purchasedfrom eBioscience for flow cytometry experiments. Unconjugated mouseanti-human TLR2 (clone TL2.1; Novus Biologicals), mouse anti-humanTLR4 (clone HTA125; Gene Tex), mouse IgG2a (clone 20102; R&DSystems), mouse anti-human MR (clone 19.2; BD Biosciences), andmouse IgG1 (R&D Systems) were used for confocal microscopy ex-periments in which Alexa Fluor 488-conjugated goat anti-mouse IgG(Molecular Probes and Invitrogen Detection Technologies) was used asa secondary Ab. Phosphorylated Akt (phospho-Akt Ser473), phosphor-ylated p38 (phospho-p38 MAPK Thr180/Tyr182), phosphorylated ERK(phospho-p44/42 MAPK Thr202/Tyr204), phosphorylated JNK (phos-pho-SAPK/JNK T183/Y185), phosphorylated I�B� (Ser32), and I�B�Abs were purchased from Cell Signaling Technology, while actin, totalAkt, goat anti-rabbit IgG-HRP (secondary Abs for primary Abs to phos-phorylated proteins), and donkey anti-goat IgG-HRP (actin and totalAkt secondary Ab) Abs were purchased from Santa Cruz Biotechnology
and used for Western blotting experiments. TNF-� ELISA kits werepurchased from R&D Systems.
SP-A proteins
The SP-A proteins used in this study were purified as previously de-scribed (4). In brief, bronchoalveolar lavage (BAL) from alveolar pro-teinosis patients (APP) was used to obtain APP-SP-A (4). Purity of theSP-A preparation was assessed by SDS-PAGE. Bacterial endotoxin lev-els were determined using the Limulus amebocyte lysate kit (BioWhit-taker). Endotoxin levels in SP-A preparations ranged from undetectableto 0.2 pg/�g protein. Two SP-A functional assays, oxidative burst andliposome aggregation (29, 30), were performed as quality controlexperiments.
Human monocytes and macrophages
Blood was obtained from healthy adult volunteers using an approvedprotocol by the Ohio State University Institutional Review Board.PBMC from single donors were isolated from heparinized blood onFicoll-Paque (Amersham Biosciences) and cultured in Teflon wells(Savillex) for 1 (monocytes) through 5 (monocyte-derived macrophages(MDMs)) days in the presence of RPMI 1640 medium containing 20%autologous serum (2.0 � 106 PBMC/ml) at 37°C (4). On the day of eachexperiment, PBMC were removed from Teflon wells and washed ex-tensively. Monocytes and MDMs were further purified by adherence intissue culture plates in some assays. Human AM were isolated fromBALs of healthy human donors as previously described (31) using anapproved protocol by the Ohio State University Institutional ReviewBoard. Briefly, the BAL was centrifuged (200 � g, 4oC, 10 min), su-pernatant removed, and the pellet resuspended in RPMI and washed twomore times with RPMI.
Flow cytometry
PBMC were incubated with APP-SP-A (10 �g/ml) or human serumalbumin (control) in Teflon wells for specific time periods. PBMC wereharvested from Teflon wells, centrifuged (200 � g, 4oC, 10 min), andresuspended in RPMI. After the cells were counted, they were centri-fuged, resuspended in FACS buffer (2% BSA), and incubated with al-lophycocyanin- or PE-conjugated Abs to TLR2 or TLR4 (20 �l/millioncells), respectively, for 20 min. PBMC incubated with the appropriateallophycocyanin- or PE-conjugated subtypic control mAb served asnegative controls.
For AM experiments, cells were counted, centrifuged, and resuspendedin FACS buffer. Two � 105 AM were incubated with TLR2, TLR4, orsubtype control Abs as described above for PBMC.
Ab-stained cells were fixed in 2% paraformaldehyde and 1 � 104 MDMor AM were analyzed for mean fluorescence intensity (MFI) and percent-age of positive cells (95/5% cutoff) using the BD FACSCalibur System(BD Biosciences). Macrophages were distinguished by the side scatter vsforward scatter (4), and the MFI due to nonspecific binding (subtypiccontrol Ab) was subtracted from each sample to obtain a specific MFI.The percent change in specific MFI in experiments with no treatment vsSP-A treatment were calculated as follows: (treated MFI � untreatedMFI)/untreated MFI) � 100. Triplicate samples in each experimentwere analyzed.
RNA isolation
One- to 5-day-old PBMC in Teflon wells were harvested and mononuclearphagocytes adhered to a 12-well tissue culture plate with 10% autologousserum (3 � 106 PBMC/ml). After washing away lymphocytes, the mono-cytes or MDMs (3 � 105 cells) were treated with SP-A (10 �g/ml) ormedium (untreated) over a time course. Finally, cells were lysed in TRIzol(Invitrogen) and total RNA was isolated by using the Qiagen RNeasy col-umn method. The Experion (Bio-Rad) was used to determine the RNAquality and quantity of each sample.
Real-time PCR
RNA (550 ng) was converted to cDNA by reverse transcriptase enzymeand real-time PCR was performed with a human TLR2, TLR4, or MRTaqMan gene expression kit (Applied Biosystems). TLR2, TLR4, andMR amplification were normalized to the �-actin housekeeping gene(�Ct). Relative copy number (RCN) and fold change were determined.RCN was calculated as follows: RCN � E��Ct � 100. E is the effi-ciency (2 � 100% efficiency) and �Ct � Ct(target) � Ct(reference)(32). Fold change was calculated as 2���Ct. ��Ct � �Ct (experimentalcell group) � �Ct (unstimulated cells). Duplicate samples were ana-lyzed in each experiment.
7848 SP-A REGULATES MACROPHAGE TLR ACTIVITY
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

Confocal microscopy
Two � 105 MDMs were adhered to a glass coverslip in each well of a24-well tissue culture plate for 2 h at 37oC, washed to remove lympho-cytes, and incubated with APP-SP-A (10 �g/ml) or RHH for 2 h at 37oC.The cells were washed, fixed with 2% paraformaldehyde (10 min at roomtemperature), and then incubated overnight in blocking buffer (PBF). Thecells were then stained with TLR2 (8 �g/ml), TLR4 (8 �g/ml), MR (1�g/ml), or the appropriate subtype (8 �g/ml or 1 �g/ml) control Ab for 1 hat room temperature, washed with PBF buffer, and counterstained with anAlexa Fluor 488-conjugated secondary Ab (1/500) for 1 h at room tem-perature. The cells were next washed with PBF buffer and coverslips weremounted on glass slides. Slides were viewed using a confocal, Zeiss scan-ning laser microscope and fluorescence intensity of each cell slice wasquantified using a pixel intensity measurement (NIH Image J program). Ananalytic box was placed on the cell membrane of each cell at four points (asrepresented on a clock at 12 a.m., 3 a.m., 6 a.m., 9 a.m.) and the MFI wascalculated as the mean of the MFI of all four points. The MFI was deter-mined for 20 cells per coverslip, with duplicate slides per experiment, anda minimum of two donors used for each treatment condition.
Western blotting
Three � 105 day 5 MDMs were adhered to wells of a 12-well plate for 2 hat 37oC, washed to remove lymphocytes, and repleted with RPMI contain-ing 10% autologous serum overnight at 37oC. APP-SP-A in RHH mediumwas added to appropriate wells for 10 min and then cells were washed withRHH and incubated for another 10 min at 37oC to allow for internalizationof all bound SP-A, as previously published (33). Dose (5–20 �g/ml) andtime (5 min to 2 h) course experiments were conducted initially to deter-mine the optimal concentration and time for SP-A incubation. Pam3Cys(100 or 500 ng/ml) or LPS (100 or 500 ng/ml) was added to appropriatewells for 5, 10, 15, or 30 min at 37oC, followed by the removal of mediumin preliminary experiments to determine the optimal time point for analy-sis. Lysis buffer (TN1 buffer: 50 mM Tris (pH 8.0), 10 mM EDTA, 10 mMNa4PO7, 10 mM NaF, 1% Triton X-100, and 125 mM NaCl, 10 mMNa3VO4, 10 �g/ml aprotinin, and 10 �g/ml leupeptin (34)) was added toeach well and the resultant lysates were added to Eppendorf tubes andincubated on ice, and then centrifuged at 13,000 � g for 10 min to removecell debris. The cleared lysates containing soluble proteins were collectedand protein content was calculated using a BCA kit (Pierce). Proteins were
FIGURE 1. Human monocytes and macrophages express TLR2 and TLR4. PBMC were isolated from human blood and incubated in Teflon wells withautologous serum. The monocytes differentiate into macrophages (MDM) by day 5. One (A and E)-, 3 (B and F)-, or 5 (C and G)-day-old PBMCs wereharvested and incubated with either an allophycocyanin-conjugated human TLR2 mAb (or an allophycocyanin-conjugated subtype control mAb) or aPE-conjugated human TLR4 mAb (or a PE-conjugated subtype control mAb). The stained cells were analyzed using flow cytometry by gating on themonocytes or macrophages (4), and a representative experiment is shown. The number in the top right corner represents the specific MFI (TLR2 or TLR4MFI � subtype control MFI). D and H, Bar graph with cumulative data (triplicate samples in each experiment; n � 5 for TLR2; n � 4 for TLR4). One-wayANOVA with post-Tukey test, ���, p � 0.005 compared with day 1 cells (�SEM).
FIGURE 2. TLR2 steady-state mRNA levels decrease, while TLR4 levels vary to only a small extent, as monocytes differentiate into macrophages. One-,3-, and 5- day-old PBMC in Teflon wells were harvested and adhered to a 12-well tissue culture plate in RPMI containing 10% autologous serum. Afterwashing away lymphocytes, the monocytes or MDMs were lysed in TRIzol and total RNA was isolated. mRNA was converted to cDNA and real-time PCRwas performed. TLR2 (A), TLR4 (B), and MR (C) amplification was normalized to the �-actin housekeeping gene and the RCN number was determined.A and B, Representative experiments (mean � SD, triplicate samples) and C is cumulative data (mean � SEM) (n � 6 (TLR2, TLR4); n � 4 (MR)). TheMR was used as a positive control as a macrophage marker. One-way ANOVA with post-Tukey test. �, p � 0.05 compared with day 1 monocytes. ���,p � 0.005 compared with day 1 monocytes.
7849The Journal of Immunology
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

separated by SDS-PAGE and transferred to nitrocellulose membranes. Fi-nally, the blots were blocked with 5% nonfat dry milk and probed with theappropriate primary (Abs to phosphorylated proteins 1/600; actin or totalAkt Ab 1/1000) and secondary HRP-conjugated secondary Abs (1/1000).The blots were developed using the ECL kit (Amersham Biosciences). TheImage J program was used to quantify band intensity. Background intensitywas subtracted from each sample and then fold change was determined asfollows: (treated sample band intensity/untreated sample band intensity).
ELISA
One � 105 day 5 MDMs were adhered to each well of a 96-well plate for2 h at 37oC, washed to remove lymphocytes, and then incubated with
RHH medium with APP-SP-A (10 �g/ml) or RHH for 2 h at 37oC. Cellswere washed with and resuspended in RHH and incubated for an ad-ditional 10 min at 37oC to allow for internalization of bound SP-A.Cells were then incubated with RHH alone, LPS (1 ng/ml), or Pam3Cys(10 ng/ml) for 1 h. Cell supernatants were collected and centrifuged at200 � g for 5 min to remove dead cells, and TNF-� was measured byELISA. Quantitative ELISAs were performed using the QuantikineELISA kits from R&D Systems according to the manufacturer’sinstructions.
NF-�B assay
Four � 105 day 5 MDMs were adhered to each well of a 12-well tissueculture plate for 2 h at 37oC, washed to remove lymphocytes, and thenincubated with APP-SP-A (10 �g/ml) or RHH medium for 10 min. Thecells were then washed with RHH and incubated for another 10 min at37oC to allow for internalization of all bound SP-A. Pam3Cys (500 ng/ml)or LPS (500 ng/ml) was added to appropriate wells for 1 h. Medium wasremoved and nuclear extracts from MDMs (two wells combined to make8 � 105 MDMs/sample) were prepared using the Transfactor extractionkit (BD Clontech). The nuclear lysates were assayed for the presence ofp65 in wells precoated with the DNA-binding consensus sequence byusing the colorimetric TransFactor kit (BD Clontech) and samples wereread at 655 nm. Untreated, SP-A plus LPS or Pam3Cys, and SP-Agroups were compared with LPS or Pam3Cys. LPS or Pam3Cys were setto 100% p65. Percent inhibition was determined as follows: ((experi-mental cell group � LPS or Pam3Cys cell group)/(LPS or Pam3Cys cellgroup) � 100); 100 minus percent inhibition � percent p65 comparedwith LPS or Pam3Cys.
Statistical analysis
An unpaired one-tailed Student’s t test was used to analyze differencesbetween two groups (e.g., with or without SP-A) and a one-way ANOVA
FIGURE 3. SP-A increases TLR2, but not TLR4, surface expression on MDMs. Five-day old PBMC in Teflon wells were incubated with or withoutSP-A (10 �g/ml) for 2 h (A and B). Following the same protocol as in Fig. 1, SP-A-treated and untreated MDMs were incubated with either a humanallophycocyanin-conjugated TLR2 mAb or allophycocyanin-conjugated subtype control mAb (A) or human PE-conjugated TLR4 mAb or PE-conjugatedsubtype control mAb (B). The stained cells were analyzed using flow cytometry by gating on the MDMs and the average of triplicate samples is shownin this experiment which is representative of n � 4 (A) and n � 5 (B). Five-day-old MDMs in monolayer culture on glass coverslips were incubated withor without SP-A (10 �g/ml) for 2 h. After washing, cells were fixed in paraformaldehyde (no permeabilization) and stained with mouse anti-human TLR2mAb, mouse anti-human TLR4 mAb, mouse anti-human MR mAb, or subtype control mAb followed by a secondary Alexa Fluor 488-conjugatedanti-mouse Ab. Glass coverslips were mounted and visualized by confocal microscopy. A representative experiment is shown in C (n � 3 (TLR2); n �2 (TLR4, MR)). Cumulative data are shown in D. A Student t test was performed. �, p � 0.05 relative to TLR2 expression on untreated MDMs. The MRwas used as a positive control.
Table I. TLR2 and TLR4 steady-state mRNA levels in humanmonocytes and macrophagesa
Receptorb
% Change RCNc Compared toDay 1 Monocytes
Day 3 Monocytes Day 5 MDMs
TLR2 �51 � 13d �65 � 10d
TLR4 68 � 40 �18 � 11MR 1193 � 415e 923 � 474e
a TLR2, TLR4, and MR mRNA levels were measured by real time PCR. Mean �SEM (n � 6 for TLR2 and TLR4 and n � 4 for MR).
b RNA was isolated and converted to cDNA and real-time PCR was performedusing TLR2, TLR4, and MR TaqMan gene expression kits.
c RCN � E��Ct � 100. E is the efficiency (2 � 100% efficiency) �Ct �Ct(target) � Ct(reference).
d p � 0.05 relative to day 1 monocytes.e p � 0.005 relative to day 1 monocytes.
7850 SP-A REGULATES MACROPHAGE TLR ACTIVITY
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

with the post-Tukey test was used to analyze differences among multipletest groups.
ResultsTLR2 surface protein expression decreases as human monocytesdifferentiate into macrophages, while TLR4 surface expressionremains unchanged
Studies of TLR2 and TLR4 expression and function have beenconducted largely in transfected murine cell lines or murine mac-rophages, with less data available in human monocytes and verylimited data in human macrophages. There are significant differ-ences in the structures of murine and human TLRs as well as intheir responsiveness to various stimuli, such as LPS (35–41).Therefore, we began our studies by determining the basal surfaceprotein expression of TLR2 and TLR4 by flow cytometry using anestablished model of human monocytes and MDMs (4). Such amodel allows us to assess changes in TLR expression during dif-ferentiation of cells from monocytes to macrophages in individualdonors. The results of a typical experiment are displayed as his-tograms (Fig. 1, A, B, C, and E–G) and cumulative data are pre-sented as bar graphs (Fig. 1, D and H). The data show that primaryhuman monocytes express the highest level of TLR2 and this ex-pression decreases when monocytes differentiate into macro-phages. Monocytes and macrophages express equivalent amountsof TLR4, indicating that TLR4 expression remains unchanged dur-ing differentiation. By flow cytometry, TLR2 and TLR4 were bothfound to be expressed on the surface of human alveolar macro-phages (data not shown).
TLR2, but not TLR4, mRNA expression decreases as humanmonocytes differentiate into macrophages
Previous data on TLR2 and TLR4 mRNA expression in mono-cytes/macrophages are inconsistent, which likely reflects differ-ences in cell type and activation state of the cells (38, 41–44). Weused real-time PCR to determine steady-state mRNA levels ofTLR2 and TLR4 in human monocytes and macrophages. The re-sults are displayed as bar graphs (Fig. 2) and cumulative data arein Table I. TLR2 mRNA expression is greatest in day 1 monocytesand steadily decreases thereafter, while TLR4 mRNA expressionchanges to a much lesser degree during differentiation into mac-rophages. Expression of MR mRNA was used as a positive controlbecause its expression is known to increase during monocyte dif-ferentiation into macrophages (4, 45).
FIGURE 4. SP-A regulates steady-state mRNA ex-pression of TLR2, but not TLR4, during monocyte dif-ferentiation into macrophages. SP-A (10 �g/ml) wasadded to 1-, 3-, and 5-day-old PBMC in selected Teflonwells for 1, 2, 6, or 20 h. PBMC were harvested andmonocytes/macrophages adhered to a 12-well tissueculture plate in autologous serum. After washing awaylymphocytes, the monocytes or MDMs were lysed inTRIzol and total RNA was isolated. mRNA was con-verted to cDNA and real-time PCR was performed.TLR2 and TLR4 mRNA amplification was normalizedto the �-actin housekeeping gene. The fold change wasdetermined by comparing SP-A-treated samples to sam-ples with no SP-A. Cumulative data are shown (�SEM)(TLR2: n � 6 (day 5, 20 h); n � 5 (day 5, 2 h); n � 4(day 1, 1, 2, 6, and 20 h; day 3, 1 and 2 h; day 5 1 and6 h); n � 2 (day 3, 6 and 20 h)) (TLR4: n � 6 (day 1,2 h); n � 5 (day 1, 1 and 20 h); n � 4 (day 1, 6 h); n �3 (day 3, 1 and 2 h; day 5, 2 and 20 h); n � 2 (day 3,6 h; day 5, 6 h); n � 1 (day 3, 20 h; day 5, 20 h)).Student t test was performed. �, p � 0.05 comparedwith day 1 monocytes.
Table II. Effect of SP-A on TNF-� secretion from human macrophagesa
Treatment Ligand Addedb % Decrease in TNF-�c n
SP-A None 0 3SP-A LPS 82 � 12d 3SP-A Pam3Cys 88 � 13d 3
a TNF-� secretion by MDMs was measured by ELISA.b Day 5 MDMs adhered to a 96-well tissue culture plate were incubated with or
without SP-A (10 �g/ml) for 2 h and then with or without the indicated TLR ligand.c Values shown represent percent change in TNF-� secretion � (treatment group
TNF-� secretion � control group TNF-� secretion)/control group secretion � 100).Control group is MDMs with no treatment or ligand added; mean TNF-� values(pg/ml) for LPS, Pam3Cys, SP-A plus LPS and SP-A plus Pam3Cys were 108, 108,19 and 13, respectively.
d p � 0.05 relative to TNF-� secretion by untreated MDMs.
7851The Journal of Immunology
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

SP-A up-regulates expression of TLR2, but not TLR4,on human macrophages
SP-A binds to one or more receptors on macrophages (22) whichtriggers signaling events that alter the biology of these cells (28,46, 47). Previous work from our laboratory has shown that SP-Aincreases MR expression on human MDMs, while another labo-ratory found that SP-A increases SR-A expression on rat macro-phages (4, 5); both receptors are known PRRs. The effect(s) ofSP-A on TLR expression and function in human macrophages hasnot been explored. We used flow cytometry to determine whetherSP-A regulated the basal levels of TLR2 and TLR4 surface ex-pression on MDMs. A typical experiment is displayed as histo-grams in Fig. 3, A (TLR2) and B (TLR4), with SP-A incubation for2 h. Incubation with SP-A for 1, 6, and 20 h showed equivalentresults (data not shown). Our results show that SP-A increasesTLR2, but not TLR4, expression on human macrophages. We nextsought to determine whether the increase in TLR2 expression isdue to a unique function of SP-A and/or SP-A-specific receptor
engagement or whether the function could be shared with otherstructurally related proteins such as the complement protein C1qwhich also shares some functional similarities with SP-A (28). Wedetermined that C1q does not regulate TLR2 expression (�8 �9%, mean � SD, n � 2).
To complement our data with SP-A, we used confocal micros-copy to visualize TLR surface expression. Consistent with the flowcytometry data, SP-A increased TLR2, but not TLR4, surface ex-pression on MDMs (Fig. 3, C and D). MR staining was used as apositive control because SP-A increases MR surface expression onMDMs (4).
SP-A regulates TLR2, but not TLR4, mRNA expression in adifferentiation-dependent manner
After determining that SP-A regulates TLR2, but not TLR4 surfaceprotein expression, we next examined whether SP-A regulatesTLR2 through a transcriptional mechanism. We used real-timePCR to examine whether SP-A regulates TLR2 mRNA expression
FIGURE 5. SP-A regulates the phosphorylation of I�B� in macrophages following the addition of TLR ligands. Five-day-old MDMs were adhered toa 12-well tissue culture plate, washed, and incubated overnight in autologous serum at 37oC. Cells were incubated with or without SP-A (10 �g/ml) for10 min. After washing, cells were incubated for an additional 10 min at 37oC to internalize any bound SP-A. LPS (10 min) or Pam3Cys (5 min) was addedto appropriate wells. MDMs were lysed and SDS-PAGE and Western blots were performed using pI�B� or actin (control) Abs. Representative Westernblots are shown (A and D) and bar graphs (B and E), generated by densitometric analysis, represent cumulative data (duplicate samples in eachexperiment; mean � SEM; n � 4: LPS, 10-min incubation, or n � 4: Pam3Cys, 5-min incubation). C and F, Five-day-old MDMs were adhered toa 12-well tissue culture plate, washed, and incubated overnight in autologous serum at 37oC. Cells were incubated with or without SP-A (10 �g/ml)for 10 min. After washing, cells were incubated for an additional 10 min at 37oC to internalize any bound SP-A. LPS (1 h) or Pam3Cys (1 h) wasadded to appropriate wells, medium was removed, and nuclear extracts were prepared. p65 nuclear translocation was measured according to themanufacturer of the colorimetric TransFactor kit (mean � SEM; n � 3). A Student t test was performed. �, p � 0.05 compared with LPS or Pam3Cys.
7852 SP-A REGULATES MACROPHAGE TLR ACTIVITY
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

in monocytes and macrophages. We examined SP-A regulation ofTLR4 mRNA expression for comparison. The results are displayedas bar graphs (Fig. 4). Although SP-A decreased TLR2 mRNAexpression in day 1 monocytes and increased its expression to asmall extent in day 3 monocytes; importantly, it did not regulateTLR2 mRNA expression in day 5 MDMs. In contrast to its effectson TLR2 mRNA levels, SP-A had very little effect on TLR4mRNA expression in monocytes and macrophages. Thus, the effectof SP-A on regulating TLR2 protein expression on macrophages(as opposed to monocytes), appears to be via a posttranslationalmechanism.
SP-A diminishes TNF-� secretion from macrophages stimulatedwith TLR2 or TLR4 ligands
Once we established that SP-A differentially regulates TLR2 andTLR4 expression on macrophages, we studied whether SP-A reg-ulates TLR function, because changes in expression do not alwayscorrelate with changes in function. When TLR2 or TLR4 is ac-tivated by an appropriate ligand, a signaling cascade is initiatedthat can result in the production of proinflammatory cytokines.We determined whether SP-A regulates the secretion of one ofthe major proinflammatory cytokines, TNF-�, in response toLPS (TLR4 ligand) or Pam3Cys (TLR2 ligand) by macro-phages. We found that SP-A markedly decreased TNF-� secre-tion by MDMs in the presence of either ligand (Table II). Thus,although SP-A regulates the expression of only TLR2, thesedata suggest that SP-A can alter the biological activity of bothTLR2 and TLR4.
SP-A regulates the phosphorylation of I�B� and the nucleartranslocation of p65 in macrophages stimulated with TLR2 orTLR4 ligands
Because SP-A regulates the biological outcome of TLR signalingpathways, we chose to study the effect of SP-A on the phosphor-ylation of specific protein kinases of these pathways, a prereq-uisite for kinase activity. We first examined whether SP-A reg-ulates the phosphorylation of the inhibitory protein I�B�. I�B�is a key regulator of NF-�B activation, because it binds to theNF-�B complex in the cytosol to prevent translocation ofNF-�B to the nucleus and subsequent transcription of NF-�B-regulated genes. After I�B� is phosphorylated, it is eventuallydegraded, which allows the NF-�B complex to translocate tothe nucleus (48, 49). As shown in Fig. 5, we found that SP-Adecreases the phosphorylation of I�B� in MDMs in response toTLR ligands. SP-A does not regulate the level of total I�B�protein in these cells (data not shown).
After determining that SP-A decreases the phosphorylation ofI�B�, we next examined whether SP-A directly regulates the nu-clear translocation of the NF-�B complex. We observed that SP-Asignificantly decreases the nuclear translocation of p65, a majorcomponent of the NF-�B complex, in the presence of TLR ligands(Fig. 5).
SP-A selectively regulates the phosphorylation of MAPKs inmacrophages stimulated with TLR2 or TLR4 ligands
A recent report has shown the ability of SP-A to activate SHP-1,a tyrosine phosphatase, and thereby decrease the phosphorylationof p38, one member of the MAPK family, in response to LPS (28).
FIGURE 6. SP-A selectively regu-lates the phosphorylation of MAPKsin macrophages following the addi-tion of TLR ligands. Following thesame protocol as in Fig. 5, treatedMDMs were lysed and SDS-PAGEand Western blots were performed us-ing pERK (A and D), pp38 (B and E),pJNK (C and F), or actin (control)Abs. Representative Western blotsare shown and bar graphs, generatedby densitometric analysis, representcumulative data (duplicate samples ineach experiment; mean � SEM; n �4 for pERK, LPS and Pam3Cys 15-min incubation; n � 4 for pp38, LPSand Pam3Cys 15-min incubation; andn � 3 for pJNK, LPS and Pam3Cys10-min incubation). A Student t testwas performed. �, p � 0.05 comparedwith LPS and Pam3Cys.
7853The Journal of Immunology
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
Thus, we sought to determine whether SP-A regulates the phos-phorylation of each of the MAPKs in the presence of TLR ligandsin human macrophages. These results would provide further in-sight as to whether SP-A directly regulates NF-�B activity orwhether the effect is upstream of this transcriptional complex. Wefound that while SP-A decreases the phosphorylation of p38 andERK, it does not affect the phosphorylation of JNK in MDMs, asassayed by Western blot analysis (Fig. 6).
SP-A regulates the phosphorylation of Akt in humanmacrophages stimulated with TLR2 or TLR4 ligands
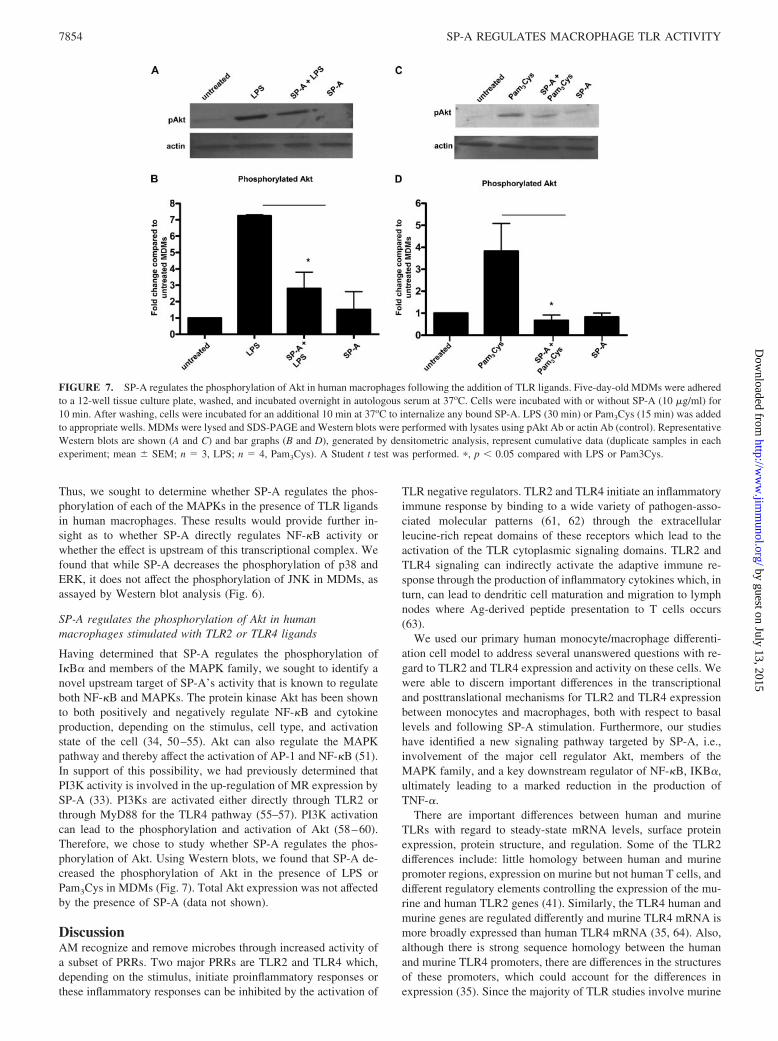
Having determined that SP-A regulates the phosphorylation ofI�B� and members of the MAPK family, we sought to identify anovel upstream target of SP-A’s activity that is known to regulateboth NF-�B and MAPKs. The protein kinase Akt has been shownto both positively and negatively regulate NF-�B and cytokineproduction, depending on the stimulus, cell type, and activationstate of the cell (34, 50–55). Akt can also regulate the MAPKpathway and thereby affect the activation of AP-1 and NF-�B (51).In support of this possibility, we had previously determined thatPI3K activity is involved in the up-regulation of MR expression bySP-A (33). PI3Ks are activated either directly through TLR2 orthrough MyD88 for the TLR4 pathway (55–57). PI3K activationcan lead to the phosphorylation and activation of Akt (58–60).Therefore, we chose to study whether SP-A regulates the phos-phorylation of Akt. Using Western blots, we found that SP-A de-creased the phosphorylation of Akt in the presence of LPS orPam3Cys in MDMs (Fig. 7). Total Akt expression was not affectedby the presence of SP-A (data not shown).
DiscussionAM recognize and remove microbes through increased activity ofa subset of PRRs. Two major PRRs are TLR2 and TLR4 which,depending on the stimulus, initiate proinflammatory responses orthese inflammatory responses can be inhibited by the activation of
TLR negative regulators. TLR2 and TLR4 initiate an inflammatoryimmune response by binding to a wide variety of pathogen-asso-ciated molecular patterns (61, 62) through the extracellularleucine-rich repeat domains of these receptors which lead to theactivation of the TLR cytoplasmic signaling domains. TLR2 andTLR4 signaling can indirectly activate the adaptive immune re-sponse through the production of inflammatory cytokines which, inturn, can lead to dendritic cell maturation and migration to lymphnodes where Ag-derived peptide presentation to T cells occurs(63).
We used our primary human monocyte/macrophage differenti-ation cell model to address several unanswered questions with re-gard to TLR2 and TLR4 expression and activity on these cells. Wewere able to discern important differences in the transcriptionaland posttranslational mechanisms for TLR2 and TLR4 expressionbetween monocytes and macrophages, both with respect to basallevels and following SP-A stimulation. Furthermore, our studieshave identified a new signaling pathway targeted by SP-A, i.e.,involvement of the major cell regulator Akt, members of theMAPK family, and a key downstream regulator of NF-�B, IKB�,ultimately leading to a marked reduction in the production ofTNF-�.
There are important differences between human and murineTLRs with regard to steady-state mRNA levels, surface proteinexpression, protein structure, and regulation. Some of the TLR2differences include: little homology between human and murinepromoter regions, expression on murine but not human T cells, anddifferent regulatory elements controlling the expression of the mu-rine and human TLR2 genes (41). Similarly, the TLR4 human andmurine genes are regulated differently and murine TLR4 mRNA ismore broadly expressed than human TLR4 mRNA (35, 64). Also,although there is strong sequence homology between the humanand murine TLR4 promoters, there are differences in the structuresof these promoters, which could account for the differences inexpression (35). Since the majority of TLR studies involve murine
FIGURE 7. SP-A regulates the phosphorylation of Akt in human macrophages following the addition of TLR ligands. Five-day-old MDMs were adheredto a 12-well tissue culture plate, washed, and incubated overnight in autologous serum at 37oC. Cells were incubated with or without SP-A (10 �g/ml) for10 min. After washing, cells were incubated for an additional 10 min at 37oC to internalize any bound SP-A. LPS (30 min) or Pam3Cys (15 min) was addedto appropriate wells. MDMs were lysed and SDS-PAGE and Western blots were performed with lysates using pAkt Ab or actin Ab (control). RepresentativeWestern blots are shown (A and C) and bar graphs (B and D), generated by densitometric analysis, represent cumulative data (duplicate samples in eachexperiment; mean � SEM; n � 3, LPS; n � 4, Pam3Cys). A Student t test was performed. �, p � 0.05 compared with LPS or Pam3Cys.
7854 SP-A REGULATES MACROPHAGE TLR ACTIVITY
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
cell lines and primary murine cells, we chose to focus our attentionon whether human monocytes and MDMs expressed TLR2and TLR4.
We observed the highest TLR2 surface protein and mRNA ex-pression in day 1 monocytes and this expression decreased asmonocytes differentiated into macrophages. Unlike TLR2, TLR4surface protein expression stayed constant and TLR4 mRNA ex-pression varied only slightly as monocytes differentiated into mac-rophages. The differences in basal surface protein expression andmRNA steady-state levels between TLR2 and TLR4 suggestedthat they may also be differentially regulated by SP-A. Our TLR2mRNA data are consistent with a previous finding by Haehnelet al. (41) where TLR2 mRNA expression (Northern blot analysis)in monocytes decreased after 24 h of adherence to a tissue cultureplate. Although others have shown TLR2 and TLR4 mRNA ex-pression in human monocytes or MDMs (38, 44, 65), these studiesdid not measure mRNA levels during monocyte differentiation intomacrophages. Our cell surface protein expression data are consis-tent with a previous report that showed more TLR2 and TLR4surface protein expression on human monocytes compared withhuman AM (66).
We found that while SP-A up-regulated TLR2 surface proteinexpression, it did not affect TLR4 surface expression on macro-phages. Interestingly, SP-A decreased TLR2 mRNA expression inday 1 monocytes, but increased to a small extent (not significant)mRNA expression in day 3 cells. Importantly, SP-A did not reg-ulate TLR2 mRNA expression in day 5 macrophages. These data
show that SP-A differentially regulates TLR2 mRNA expressionduring monocyte differentiation into macrophages. Due to the lackof TLR2 mRNA regulation by SP-A in day 5 MDMs, our datasupport that SP-A up-regulation of TLR2 surface expression onMDMs is most likely the result of a posttranslational mechanism,possibly by translocating intracellular pools of preformed TLR2 tothe cell surface. Confocal microscopy experiments using fixed andpermeabilized MDMs suggest that intracellular pools of TLR2 ex-ist (our unpublished observation). Our previous work indicated asimilar mechanism for SP-A’s up-regulation of the MR on mac-rophages (4) and there are reports that TLR2 and TLR4 can befound within intracellular vesicles (67, 68).
We examined the relationship between SP-A’s effects onTLR expression and its effects on TLR function because effectson expression and function do not always correlate. There arecontradictory data regarding the role of SP-A and TLRs in theinflammatory response. By using CHO cells and TLR4-deficient/wild-type mice, one group observed that SP-A can activate theNF-�B signaling pathway and up-regulate the synthesis of cyto-kines, such as TNF-�, by relying predominantly on a functionalTLR4 complex (69), while another group did not observe SP-Aactivation of NF-�B (70). Differences in these results may dependon the SP-A purification process used by the researchers. PurifiedSP-A used by the first group contained higher concentrations ofLPS compared with that of the second group. This difference inLPS contamination could result in the proinflammatory effects bySP-A which have been observed (71).
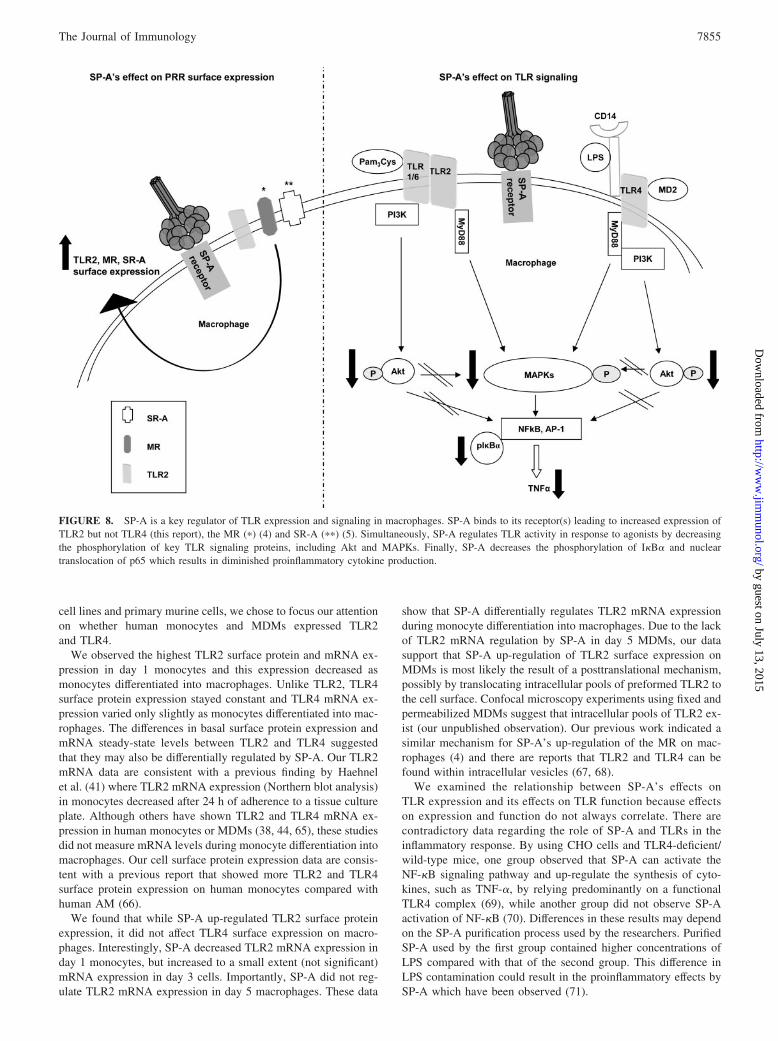
FIGURE 8. SP-A is a key regulator of TLR expression and signaling in macrophages. SP-A binds to its receptor(s) leading to increased expression ofTLR2 but not TLR4 (this report), the MR (�) (4) and SR-A (��) (5). Simultaneously, SP-A regulates TLR activity in response to agonists by decreasingthe phosphorylation of key TLR signaling proteins, including Akt and MAPKs. Finally, SP-A decreases the phosphorylation of I�B� and nucleartranslocation of p65 which results in diminished proinflammatory cytokine production.
7855The Journal of Immunology
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

We observed a significant reduction, but not a complete elimi-nation, of TNF-� secretion when macrophages were pretreatedwith SP-A and then treated with either the TLR4 ligand, LPS, orthe TLR2 ligand Pam3Cys. Our TLR4 data are consistent withprevious publications that showed that SP-A decreases TNF-� se-cretion in the presence of LPS (28, 72–74). Overall, these dataprovide further evidence for the role of SP-A in dampening theproinflammatory response in the lung environment.
Recently, SP-A was shown to directly associate with TLR2 andTLR4 proteins and thereby attenuate a proinflammatory responsein rat macrophages and murine cell lines (46, 75). In our studies,we used a protocol which allows for complete internalization ofSP-A in MDMs before adding the TLR ligands (33). Followingincubation with SP-A, we used specific mAbs to readily detect theTLRs (increased in the case of TLR2), indicating that surface TLRbinding sites were not occupied by SP-A and were free to bindother ligands. Thus, our studies point to a different mechanismindependent of TLR blockade by SP-A, whereby SP-A binds toone or more of its receptors, initiating a signaling cascade thatresults in altered expression and activity of the TLRs. Several po-tential SP-A receptors have been identified, including signal reg-ulatory protein �, calreticulin/CD91 (28) and SP-R210 (76, 77).
We found that SP-A significantly decreased the phosphorylationof I�B� in macrophages following the addition of the TLR2 andTLR4 ligands. Our data contradict previous findings by Wu et al.(70), which did not find an effect of SP-A on the phosphorylationof I�B� in the presence of LPS. The differences could be due to thelow concentration of LPS that was used to stimulate the cells andthe difference in cell type. Additionally, our data show that SP-Adirectly regulates the NF-�B complex by decreasing the nucleartranslocation of p65.
We found that SP-A decreases the phosphorylation of p38 andERK, but not JNK in the presence of TLR ligands. SP-A regulationof the phosphorylation of p38 in the presence of LPS are consistentwith previous findings by Gardai et al. (28) using murine RAWcells and human macrophages. However, they reported that SP-Aalso regulated the phosphorylation of JNK, although the data werenot shown. If RAW cells were used for their pJNK experiments,the differences in our findings could be explained by their use of amurine macrophage-like cell line vs our human MDMs. Our datasuggest that JNK is regulated by distinct mechanisms comparedwith p38 and ERK. Although the p38, ERK, and JNK pathwayscan be activated by unique upstream signaling molecules, there areoverlapping upstream and downstream kinases, which could ex-plain why we observed that SP-A regulated only two of the threeMAPKs (78).
We previously found that PI3K was involved in SP-A’s increasein MR expression on macrophages (33). Also, previous studieshave shown that when TLR2 or TLR4 is activated by appropriateligands, PI3Ks are activated (79). The activation of PI3K initiatesa signaling cascade that results in the phosphorylation and activa-tion of Akt. Akt is a kinase that is involved in numerous cellfunctions, one of which is regulation of NF-�B (80–82). The lit-erature provides evidence for a potential link among Akt, MAPKs,and the activation of AP-1 and NF-�B (51). We found that SP-Adecreased the phosphorylation of Akt in the presence of Pam3Cysand LPS, which is associated with the decrease in phosphorylationof MAPKs and I�B�, finally leading to a diminished proinflam-matory response.
Based on our previous work and current work and the literature,we propose a model of TLR modulation by SP-A (Fig. 8), whereSP-A binds to its macrophage receptor(s) and signals TLR2 intra-cellular pools to travel to the cell surface, which increases TLR2surface expression. Simultaneously, SP-A signals also regulate
TLR activity by diminishing proinflammatory cytokine productionas the result of a decrease in the phosphorylation of a key regulatorof NF-�B, I�B�, and a decrease in the nuclear translocation ofp65. SP-A down-regulates kinases upstream of I�B� by decreas-ing the phosphorylation of Akt and MAPKs in response to TLRligands. Therefore, while SP-A increases TLR2 expression, whichcan enhance pathogen recognition, it limits TLR signaling so thatthe lung is not damaged by an overreactive inflammatory response.
There is accumulating evidence that SP-A regulates the macro-phage surface expression and/or function of a subset of PRRs in-cluding the MR, SR-A, and, based on this report, TLR2 and TLR4,and controls the host inflammatory response and microbicidal ac-tivity against various pathogens and particulates routinely encoun-tered in the lung (Fig. 8). This is a critical function of SP-A thatpreserves the normal architecture of the alveolar environment andgas exchange and provides for effective microbial clearance andsterility of the alveolar environment.
AcknowledgmentsWe thank Dr. Mark Wewers for performing bronchoscopies to obtain BALfor the human alveolar macrophage experiments. Also, we thank Dr. Fran-cis McCormack and Dr. Bruce Trapnell for providing BAL from alveolarproteinosis patients for SP-A isolation and purification. We acknowledgethe support of two core facilities at the Ohio State University, the DorothyM. Davis Heart and Lung Research Institute Flow Cytometry Core Labo-ratory, and the Campus Microscopy and Imaging Facility.
DisclosuresThe authors have no financial conflict of interest.
References1. Zhang, P., W. R. Summer, G. J. Bagby, and S. Nelson. 2000. Innate immunity
and pulmonary host defense. Immunol. Rev. 173: 39–51.2. Fels, A., and Z. A. Cohn. 1986. The alveolar macrophage. J. Appl. Physiol. 60:
353–369.3. Lohmann-Matthes, M. L., C. Steinmuller, and G. Franke-Ullmann. 1994. Pul-
monary macrophages. Eur. Respir. J. 7: 1678–1689.4. Beharka, A. A., C. D. Gaynor, B. K. Kang, D. R. Voelker, F. X. McCormack, and
L. S. Schlesinger. 2002. Pulmonary surfactant protein A up-regulates activity ofthe mannose receptor, a pattern recognition receptor expressed on human mac-rophages. J. Immunol. 169: 3565–3573.
5. Kuronuma, K., H. Sano, K. Kato, K. Kudo, N. Hyakushima, S. Yokota,H. Takahashi, N. Fujii, H. Suzuki, T. Kodama, et al. 2004. Pulmonary surfactantprotein A augments the phagocytosis of Streptococcus pneumoniae by alveolarmacrophages through a casein kinase 2-dependent increase of cell surface local-ization of scavenger receptor A. J. Biol. Chem. 279: 21421–21430.
6. Wewers, M. D., S. I. Rennard, A. J. Hance, P. B. Bitterman, and R. G. Crystal.1984. Normal human alveolar macrophages obtained by bronchoalveolar lavagehave a limited capacity to release interleukin-1. J. Clin. Invest. 74: 2208–2218.
7. Balter, M. S., G. B. Toews, and M. Peters-Golden. 1989. Different patterns ofarachidonate metabolism in autologous human blood monocytes and alveolarmacrophages. J. Immunol. 142: 602–608.
8. Oren, R., A. E. Farnham, K. Saito, E. Milofsky, and M. L. Karnovsky. 1963.Metabolic patterns in three types of phagocytizing cells. J. Cell Biol. 17:487–501.
9. Lyons, C. R., E. J. Ball, G. B. Toews, J. C. Weissler, P. Stastny, andM. F. Lipscomb. 1986. Inability of human alveolar macrophages to stimulateresting T cells correlates with decreased antigen-specific T cell-macrophage bind-ing. J. Immunol. 137: 1173–1180.
10. Trinchieri, G., and A. Sher. 2007. Cooperation of Toll-like receptor signals ininnate immune defence. Nat. Rev. Immunol. 7: 179–180.
11. Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda,and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451.
12. Ozinsky, A., D. M. Underhill, J. D. Fontenot, A. M. Hajjar, K. D. Smith,C. B. Wilson, L. Schroeder, and A. Aderem. 2000. The repertoire for patternrecognition of pathogens by the innate immune system is defined by cooperationbetween toll-like receptors. Proc. Natl. Acad. Sci. USA 97: 13766–13771.
13. Hoffmann, P., K. H. Wiesmuller, J. Metzger, G. Jung, and W. G. Bessler. 1989.Induction of tumor cytotoxicity in murine bone marrow-derived macrophages bytwo synthetic lipopeptide analogues. Biol. Chem. Hoppe-Seyler 370: 575–582.
14. Muller, M. R., S. D. Pfannes, M. Ayoub, P. Hoffmann, W. G. Bessler, andK. Mittenbuhler. 2001. Immunostimulation by the synthetic lipopeptide P3CSK4:TLR4-independent activation of the ERK1/2 signal transduction pathway in mac-rophages. Immunology 103: 49–60.
7856 SP-A REGULATES MACROPHAGE TLR ACTIVITY
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

15. Poltorak, A., X. He, I. Smirnova, M. Y. Liu, H. C. Van, X. Du, D. Birdwell,E. Alejos, M. Silva, C. Galanos, M. Freudenberg, et al. 1998. Defective LPSsignaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science282: 2085–2088.
16. Vabulas, R. M., P. Ahmad-Nejad, S. Ghose, C. J. Kirschning, R. D. Issels, andH. Wagner. 2002. HSP70 as endogenous stimulus of the Toll/interleukin-1 re-ceptor signal pathway. J. Biol. Chem. 277: 15107–15112.
17. Ohashi, K., V. Burkart, S. Flohe, and H. Kolb. 2000. Cutting edge: heat shockprotein 60 is a putative endogenous ligand of the toll-like receptor-4 complex.J. Immunol. 164: 558–561.
18. Bulut, Y., E. Faure, L. Thomas, H. Karahashi, K. S. Michelsen, O. Equils,S. G. Morrison, R. P. Morrison, and M. Arditi. 2002. Chlamydial heat shockprotein 60 activates macrophages and endothelial cells through Toll-like receptor4 and MD2 in a MyD88-dependent pathway. J. Immunol. 168: 1435–1440.
19. Liew, F. Y., D. Xu, E. K. Brint, and L. A. O’Neill. 2005. Negative regulation oftoll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5: 446–458.
20. Pattle, R. E. 1955. Properties, function, and origin of the alveolar lining layer.Nature 175: 1125–1126.
21. Clements, J. A. 1957. Surface tension of lung extracts. Proc. Soc. Exp. Biol. Med.95: 170–172.
22. Wright, J. R. 2005. Immunoregulatory functions of surfactant proteins. Nat. Rev.Immunol. 5: 58–68.
23. Voss, T., H. Eistetter, K. P. Schafer, and J. Engel. 1988. Macromolecular orga-nization of natural and recombinant lung surfactant protein SP 28–36. J. Mol.Biol. 201: 219–227.
24. King, R., D. Simon, and P. M. Horowitz. 1989. Aspects of secondary and qua-ternary structure of surfactant protein A from canine lung. Biochim. Biophys.Acta 1001: 294–301.
25. Kuroki, Y., and D. R. Voelker. 1994. Pulmonary surfactant proteins. J. Biol.Chem. 269: 25943–25946.
26. Kuroki, Y., M. Takahashi, and C. Nishitani. 2007. Pulmonary collectins in innateimmunity of the lung. Cell. Microbiol. 9: 1871–1879.
27. Kudo, K., H. Sano, H. Takahashi, K. Kuronuma, S. Yokota, N. Fujii, K. Shimada,I. Yano, Y. Kumazawa, D. R. Voelker, et al. 2004. Pulmonary collectins enhancephagocytosis of Mycobacterium avium through increased activity of mannosereceptor. J. Immunol. 172: 7592–7602.
28. Gardai, S. J., Y. Q. Xiao, M. Dickinson, J. A. Nick, D. R. Voelker, K. E. Greene,and P. M. Henson. 2003. By binding SIRP� or calreticulin/CD91, lung collectinsact as dual function surveillance molecules to suppress or enhance inflammation.Cell 115: 13–23.
29. Crowther, J. E., V. K. Kutala, P. Kuppusamy, J. S. Ferguson, A. A. Beharka,J. L. Zweier, F. X. McCormack, and L. S. Schlesinger. 2004. Pulmonary surfac-tant protein a inhibits macrophage reactive oxygen intermediate production inresponse to stimuli by reducing NADPH oxidase activity. J. Immunol. 172:6866–6674.
30. Crowther, J. E., and L. S. Schlesinger. 2006. Endocytic pathway for surfactantprotein A in human macrophages: binding, clathrin-mediated uptake, and traf-ficking through the endolysosomal pathway. Am. J. Physiol. 290: L334–L342.
31. Gaynor, C. D., F. X. McCormack, D. R. Voelker, S. E. McGowan, andL. S. Schlesinger. 1995. Pulmonary surfactant protein A mediates enhancedphagocytosis of Mycobacterium tuberculosis by a direct interaction with humanmacrophages. J. Immunol. 155: 5343–5351.
32. Gavrilin, M. A., I. J. Bouakl, N. L. Knatz, M. D. Duncan, M. W. Hall, J. S. Gunn,and M. D. Wewers. 2006. Internalization and phagosome escape required forFrancisella to induce human monocyte IL-1� processing and release. Proc. Natl.Acad. Sci. USA 103: 141–146.
33. Beharka, A. A., J. E. Crowther, F. X. McCormack, G. M. Denning, J. Lees,E. Tibesar, and L. S. Schlesinger. 2005. Pulmonary surfactant protein a activatesa phosphatidylinositol 3-kinase/calcium signal transduction pathway in humanmacrophages: participation in the up-regulation of mannose receptor activity.J. Immunol. 175: 2227–2236.
34. Rajaram, M. V., L. P. Ganesan, K. V. Parsa, J. P. Butchar, J. S. Gunn, andS. Tridandapani. 2006. Akt/Protein kinase B modulates macrophage inflamma-tory response to Francisella infection and confers a survival advantage in mice.J. Immunol. 177: 6317–6324.
35. Lichtinger, M., R. Ingram, M. W. Hornef, C. Bonifer, and M. Rehli. 2007. Tran-scription factor PU.1 controls transcription start site positioning and alternativeTLR4 promoter usage. J. Biol. Chem. 282: 26874–26883.
36. Nomura, F., S. Akashi, Y. Sakao, S. Sato, T. Kawai, M. Matsumoto,K. Nakanishi, M. Kimoto, K. Miyake, K. Takeda, and S. Akira. 2000. Cuttingedge: endotoxin tolerance in mouse peritoneal macrophages correlates withdown-regulation of surface Toll-like receptor 4 expression. J. Immunol. 164:3476–3479.
37. Jiang, Q., S. Akashi, K. Miyake, and H. R. Petty. 2000. Lipopolysaccharideinduces physical proximity between CD14 and Toll-like receptor 4 (TLR4) priorto nuclear translocation of NF-�B. J. Immunol. 165: 3541–3544.
38. Siren, J., J. Pirhonen, I. Julkunen, and S. Matikainen. 2005. IFN-� regulatesTLR-dependent gene expression of IFN-�, IFN-�, IL-28, and IL-29. J. Immunol.174: 1932–1937.
39. Nahori, M. A., E. Fournie-Amazouz, N. S. Que-Gewirth, V. Balloy, M. Chignard,C. R. Raetz, I. Saint Girons, and C. Werts. 2005. Differential TLR recognition ofleptospiral lipid A and lipopolysaccharide in murine and human cells. J. Immu-nol. 175: 6022–6031.
40. Hajjar, A. M., R. K. Ernst, J. H. Tsai, C. B. Wilson, and S. I. Miller. 2002. HumanToll-like receptor 4 recognizes host-specific LPS modifications. Nat. Immunol. 3:354–359.
41. Haehnel, V., L. Schwarzfischer, M. J. Fenton, and M. Rehli. 2002. Transcrip-tional regulation of the human Toll-like receptor 2 gene in monocytes and mac-rophages. J. Immunol. 168: 5629–5637.
42. Miettinen, M., T. Sareneva, I. Julkunen, and S. Matikainen. 2001. IFNs activateToll-like receptor gene expression in viral infections. Genes Immun. 2: 349–355.
43. Visintin, A., A. Mazzoni, J. H. Spitzer, D. H. Wyllie, S. K. Dower, andD. M. Segal. 2001. Regulation of Toll-like receptors in human monocytes anddendritic cells. J. Immunol. 166: 249–255.
44. Xu, X. H., P. K. Shah, E. Faure, O. Equils, L. Thomas, M. C. Fishbein,D. Luthringer, X. P. Xu, T. B. Rajavashisth, J. Yano, et al. 2001. Toll-like re-ceptor-4 is expressed by macrophages in murine and human lipid-rich athero-sclerotic plaques and upregulated by oxidized LDL. Circulation 104: 3103–3108.
45. Stein, M., S. Keshav, N. Harris, and S. Gordon. 1992. Interleukin 4 potentlyenhances murine macrophage mannose receptor activity: a marker of alternativeimmunologic macrophage activation. J. Exp. Med. 176: 287–292.
46. Yamada, C., H. Sano, T. Shimizu, H. Mitsuzawa, C. Nishitani, T. Himi, andY. Kuroki. 2006. Surfactant protein A directly interacts with TLR4 and MD-2and regulates inflammatory cellular response: importance of supratrimeric oli-gomerization. J. Biol. Chem. 281: 21771–21780.
47. Sato, M., H. Sano, D. Iwaki, K. Kudo, M. Konishi, H. Takahashi, T. Takahashi,H. Imaizumi, Y. Asai, and Y. Kuroki. 2003. Direct binding of Toll-like receptor2 to zymosan, and zymosan-induced NF-�B activation and TNF-� secretion aredown-regulated by lung collectin surfactant protein A. J. Immunol. 171:417–425.
48. Ghosh, S., and M. Karin. 2002. Missing pieces in the NF-�B puzzle. Cell109(Suppl.): S81–S96.
49. Karin, M., and Y. Ben-Neriah. 2000. Phosphorylation meets ubiquitination: thecontrol of NF-�B activity. Annu. Rev. Immunol. 18: 621–663.
50. Fang, H., R. A. Pengal, X. Cao, L. P. Ganesan, M. D. Wewers, C. B. Marsh, andS. Tridandapani. 2004. Lipopolysaccharide-induced macrophage inflammatoryresponse is regulated by SHIP. J. Immunol. 173: 360–366.
51. Kao, S. J., H. C. Lei, C. T. Kuo, M. S. Chang, B. C. Chen, Y. C. Chang,W. T. Chiu, and C. H. Lin. 2005. Lipoteichoic acid induces nuclear factor-�Bactivation and nitric oxide synthase expression via phosphatidylinositol 3-kinase,Akt, and p38 MAPK in RAW 264.7 macrophages. Immunology 115: 366–374.
52. Islam, S., F. Hassan, M. M. Mu, H. Ito, N. Koide, I. Mori, T. Yoshida, andT. Yokochi. 2004. Piceatannol prevents lipopolysaccharide (LPS)-induced nitricoxide (NO) production and nuclear factor (NF)-�B activation by inhibitingI�kinase (IKK). Microbiol. Immunol. 48: 729–736.
53. Kim, H. G., B. Shrestha, S. Y. Lim, D. H. Yoon, W. C. Chang, D. J. Shin,S. K. Han, S. M. Park, J. H. Park, H. I. Park, et al. 2006. Cordycepin inhibitslipopolysaccharide-induced inflammation by the suppression of NF-�B throughAkt and p38 inhibition in RAW 264.7 macrophage cells. Eur. J. Pharmacol. 545:192–199.
54. Guha, M., and N. Mackman. 2002. The phosphatidylinositol 3-kinase-Akt path-way limits lipopolysaccharide activation of signaling pathways and expression ofinflammatory mediators in human monocytic cells. J. Biol. Chem. 277:32124–32132.
55. Arbibe, L., J. P. Mira, N. Teusch, L. Kline, M. Guha, N. Mackman,P. J. Godowski, R. J. Ulevitch, and U. G. Knaus. 2000. Toll-like receptor 2-me-diated NF-�B activation requires a Rac1-dependent pathway. Nat. Immunol. 1:533–540.
56. Koyasu, S. 2003. The role of PI3K in immune cells. Nat. Immunol. 4: 313–319.57. Ojaniemi, M., V. Glumoff, K. Harju, M. Liljeroos, K. Vuori, and M. Hallman.
2003. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediatedcytokine expression in mouse macrophages. Eur. J. Immunol. 33: 597–605.
58. Stokoe, D., L. R. Stephens, T. Copeland, P. R. Gaffney, C. B. Reese,G. F. Painter, A. B. Holmes, F. McCormick, and P. T. Hawkins. 1997. Dual roleof phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B.Science 277: 567–570.
59. Bellacosa, A., T. O. Chan, N. N. Ahmed, K. Datta, S. Malstrom, D. Stokoe,F. McCormick, J. Feng, and P. Tsichlis. 1998. Akt activation by growth factorsis a multiple-step process: the role of the PH domain. Oncogene 17: 313–325.
60. Toker, A., and L. C. Cantley. 1997. Signalling through the lipid products ofphosphoinositide-3-OH kinase. Nature 387: 673–676.
61. Janeway, C. A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu.Rev. Immunol. 20: 197–216.
62. Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteinslinking innate and acquired immunity. Nat. Immunol. 2: 675–680.
63. Pasare, C., and R. Medzhitov. 2004. Toll-like receptors: linking innate and adap-tive immunity. Microbes Infect. 6: 1382–1387.
64. Zarember, K. A., and P. J. Godowski. 2002. Tissue expression of human Toll-likereceptors and differential regulation of Toll-like receptor mRNAs in leukocytes inresponse to microbes, their products, and cytokines. J. Immunol. 168: 554–561.
65. Hornung, V., S. Rothenfusser, S. Britsch, A. Krug, B. Jahrsdorfer, T. Giese,S. Endres, and G. Hartmann. 2002. Quantitative expression of toll-like receptor1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells andsensitivity to CpG oligodeoxynucleotides. J. Immunol. 168: 4531–4537.
66. Droemann, D., T. Goldmann, T. Tiedje, P. Zabel, K. Dalhoff, and B. Schaaf.2005. Toll-like receptor 2 expression is decreased on alveolar macrophages incigarette smokers and COPD patients. Respir. Res. 6: 68.
67. Flo, T. H., O. Halaas, S. Torp, L. Ryan, E. Lien, B. Dybdahl, A. Sundan, andT. Espevik. 2001. Differential expression of Toll-like receptor 2 in human cells.J. Leukocyte Biol. 69: 474–481.
68. Latz, E., A. Visintin, E. Lien, K. A. Fitzgerald, B. G. Monks, E. A. Kurt-Jones,D. T. Golenbock, and T. Espevik. 2002. Lipopolysaccharide rapidly traffics toand from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex
7857The Journal of Immunology
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from

in a process that is distinct from the initiation of signal transduction. J. Biol.Chem. 277: 47834–47843.
69. Guillot, L., V. Balloy, F. X. McCormack, D. T. Golenbock, M. Chignard, andM. Si-Tahar. 2002. Cutting edge: The immunostimulatory activity of the lungsurfactant protein-A involves Toll-like receptor 4. J. Immunol. 168: 5989–5992.
70. Wu, Y., S. Adam, L. Hamann, H. Heine, A. J. Ulmer, U. Buwitt-Beckmann, andC. Stamme. 2004. Accumulation of inhibitory �B-� as a mechanism contributingto the anti-inflammatory effects of surfactant protein-A. Am. J Respir. Cell Mol.Biol. 31: 587–594.
71. Wright, J. R., D. F. Zlogar, J. C. Taylor, T. M. Zlogar, and C. I. Restrepo. 1999.Effects of endotoxin on surfactant protein A and D stimulation of NO productionby alveolar macrophages. Am. J. Physiol. 276: L650–L658.
72. Borron, P., J. C. McIntosh, T. R. Korfhagen, J. A. Whitsett, J. Taylor, andJ. R. Wright. 2000. Surfactant-associated protein A inhibits LPS-induced cyto-kine and nitric oxide production in vivo. Am. J. Physiol. 278: L840–L847.
73. McIntosh, J. C., S. Mervin-Blake, E. Conner, and J. R. Wright. 1996. Surfactantprotein A protects growing cells and reduces TNF-� activity from LPS-stimu-lated macrophages. Am. J. Physiol. 271: L310–L319.
74. Arias-Diaz, J., I. Garcia-Verdugo, C. Casals, N. Sanchez-Rico, E. Vara, andJ. L. Balibrea. 2000. Effect of surfactant protein A (SP-A) on the production ofcytokines by human pulmonary macrophages. Shock 14: 300–306.
75. Murakami, S., D. Iwaki, H. Mitsuzawa, H. Sano, H. Takahashi, D. R. Voelker,T. Akino, and Y. Kuroki. 2002. Surfactant protein A inhibits peptidoglycan-induced tumor necrosis factor-� secretion in U937 cells and alveolar macro-phages by direct interaction with toll-like receptor 2. J. Biol. Chem. 277:6830–6837.
76. Chroneos, Z. C., R. Abdolrasulnia, J. A. Whitsett, W. R. Rice, andV. L. Shepherd. 1996. Purification of a cell-surface receptor for surfactant proteinA. J. Biol. Chem. 271: 16375–16383.
77. Yang, C. H., J. Szeliga, J. Jordan, S. Faske, Z. Sever-Chroneos, B. Dorsett,R. E. Christian, R. E. Settlage, J. Shabanowitz, D. F. Hunt, et al. 2005. Identi-fication of the surfactant protein A receptor 210 as the unconventional myosin18A. J. Biol. Chem. 280: 34447–34457.
78. Guha, M., and N. Mackman. 2001. LPS induction of gene expression in humanmonocytes. Cell. Signal. 13: 85–94.
79. Akira, S., and K. Takeda. 2004. Toll-like receptor signaling. Nat. Rev. Immunol.4: 499–511.
80. Datta, S. R., A. Brunet, and M. E. Greenberg. 1999. Cellular survival: a play inthree Akts. Genes Dev. 13: 2905–2927.
81. Brazil, D. P., J. Park, and B. A. Hemmings. 2002. PKB binding proteins: gettingin on the Akt. Cell 111: 293–303.
82. Scheid, M. P., and J. R. Woodgett. 2001. PKB/AKT: functional insights fromgenetic models. Nat. Rev. Mol. Cell Biol. 2: 760–768.
7858 SP-A REGULATES MACROPHAGE TLR ACTIVITY
by guest on July 13, 2015http://w
ww
.jimm
unol.org/D
ownloaded from
Related Documents











