Scientific American Medicine DOI 10.2310/7900.1208 01/17 © 2017 Decker Intellectual Properties Inc pulmonary and critical care medicine CHRONIC OBSTRUCTIVE PULMONARY DISEASE Andrew J. Schissler, MD, George Washko, MD, and Carolyn E. Come, MD, MPH* Definition Chronic obstructive pulmonary disease (COPD) is a disease state characterized by airflow obstruction that is not fully reversible. The airflow limitation is usually progressive and associated with an enhanced inflammatory response of the lungs to noxious particles and gases. 1 In most cases, the nox- ious particles and gases derive from tobacco smoking. This definition emphasizes the importance of airflow obstruction, which is determined using spirometry, in the diagnosis of COPD. Of note, unlike earlier definitions of COPD, the cur- rent definition does not mention “emphysema” or “chronic bronchitis.” Spirometry involves a forced expiratory maneuver after the patient has inhaled so as to achieve total lung capacity (TLC) [see Figure 1]. The volume of air exhaled in the first second of this maneuver is the forced expiratory volume in 1 second (FEV 1 ), and the total volume of air exhaled during the maneuver is the forced vital capacity (FVC). Airflow obstruction is defined in terms of a reduction in the ratio of the FEV 1 to the FVC; a ratio below 0.7 (commonly referred to as 70%, although it does not refer to a percentage of a predicted value) is often used to indicate significant airflow obstruction. The use of this fixed ratio to define airflow obstruction has been criticized 2 because the predicted values for the FEV 1 /FVC ratio normally decline with age, and a substantial number of normal elderly persons have an FEV 1 / FVC ratio of less than 0.7. The use of percentage of predicted values or lower limits of normal for FEV 1 /FVC ratio can overcome the limitations of a fixed threshold for defining an abnormal FEV 1 /FVC ratio. Airflow obstruction in COPD is caused by emphysema and airway disease. Emphysema is defined as abnormal, permanent enlargement of air spaces distal to the terminal bronchiole, accompanied by destruction of the alveolar walls [see Pathology, below]. The airway disease in COPD that results in airflow obstruction occurs principally in small air- ways (ie, those with an internal diameter of less than 2 mm). Chronic bronchitis, a condition of the large airways, is com- mon in persons with COPD. Chronic bronchitis is defined as a cough productive of sputum on most days for at least 3 months a year for at least 2 consecutive years and for which there is no other explanation. Like COPD, chronic bronchitis is often associated with cigarette smoking; however, individ- uals with chronic bronchitis but without airflow obstruction are not classified as having COPD. Whereas previous recommendations for management of COPD were based solely on the severity of the airflow obstruction, the most recent Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines recommend combined assessment using symptoms, breathlessness, spi- rometric classification, and risk of exacerbation to evaluate patients [see Figure 2] and guide treatment [see Table 1]. To assess symptoms, the GOLD guidelines recommend using the COPD Assessment Test (CAT) (http://www.cateston- line.org), a validated eight-item measure of impairment with scores ranging from 0 to 40. 3,4 The GOLD guidelines state that a CAT score of 10 or higher should prompt consid- eration of further treatment. 5 Spirometric classification is based on the degree of airflow limitation on postbronchodi- lator FEV 1 in the setting of a reduced FEV 1 /FVC ratio. 1 Grade 1 (mild) is defined as an FEV 1 value of greater than or equal to 80% of predicted; because of the limitations of using a fixed FEV 1 /FVC ratio to define airflow obstruction using the GOLD criteria, it is likely that many normal individuals would fit this definition of mild COPD. Grade 2 (moderate) is defined as an FEV 1 value between 50 and 80% of pre- dicted. Grade 3 (severe) is defined as an FEV 1 value between 30 and 50% predicted. Patients with FEV 1 values below 30% of the predicted value are categorized as grade 4 (ie, very severe). If only prebronchodilator spirometry is obtained, GOLD grades are assigned with less certainty. 6 The risk of exacerbation is based on a history of previously treated exacerbations, which has been shown to be the best predic- tor of having frequent exacerbations. 7 Epidemiology prevalence COPD is a leading cause of disability and death in the United States and worldwide. 8,9 Precise estimates of COPD prevalence are not available and likely differ by region and smoking rates. During 2007–2010, the National Health and Nutrition Examination Survey (NHANES) obtained pre- bronchodilator spirometry data on a nationally representa- tive sample of US adults and postbronchodilator pulmonary lung function data for the subset of adults with airflow lim- itation. 10 These data demonstrate an estimated overall COPD prevalence between 10 and 20% among adults in the United States between the ages of 40 and 79 depending on which diagnostic criteria were applied (fixed ratio or lower limit of normal) and whether pre- or postbronchodilator values were used. Similarly, a retrospective longitudinal cohort study in Canada published in 2011 determined that about one in four individuals is likely to receive medical attention for a physician diagnosis of COPD. 11 Recent data from nationally representative data sources suggest declines in the age-adjusted prevalence of COPD from 1999 to 2011, likely due to declines in smoking rates. Indeed, the preva- lence of smoking in 2010 was approximately one half of that in 1965 for both men and women. 12,13 *The authors and editors gratefully acknowledge the contributions of the previous authors, Robert M. Senior, MD, and Edwin K. Silverman, MD, PhD, to the develop- ment and writing of this review.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Scientific American MedicineDOI 10.2310/7900.1208
01/17
© 2017 Decker Intellectual Properties Inc
pulmonary and critical care medicine
C H R O N I C O B S T R U C T I V E P U L M O N A R Y D I S E A S E
Andrew J. Schissler, MD, George Washko, MD, and Carolyn E. Come, MD, MPH*
Definition
Chronic obstructive pulmonary disease (COPD) is a disease state characterized by airflow obstruction that is not fully reversible. The airflow limitation is usually progressive and associated with an enhanced inflammatory response of the lungs to noxious particles and gases.1 In most cases, the nox-ious particles and gases derive from tobacco smoking. This definition emphasizes the importance of airflow obstruction, which is determined using spirometry, in the diagnosis of COPD. Of note, unlike earlier definitions of COPD, the cur-rent definition does not mention “emphysema” or “chronic bronchitis.”
Spirometry involves a forced expiratory maneuver after the patient has inhaled so as to achieve total lung capacity (TLC) [see Figure 1]. The volume of air exhaled in the first second of this maneuver is the forced expiratory volume in 1 second (FEV1), and the total volume of air exhaled during the maneuver is the forced vital capacity (FVC). Airflow obstruction is defined in terms of a reduction in the ratio of the FEV1 to the FVC; a ratio below 0.7 (commonly referred to as 70%, although it does not refer to a percentage of a predicted value) is often used to indicate significant airflow obstruction. The use of this fixed ratio to define airflow obstruction has been criticized2 because the predicted values for the FEV1/FVC ratio normally decline with age, and a substantial number of normal elderly persons have an FEV1/FVC ratio of less than 0.7. The use of percentage of predicted values or lower limits of normal for FEV1/FVC ratio can overcome the limitations of a fixed threshold for defining an abnormal FEV1/FVC ratio.
Airflow obstruction in COPD is caused by emphysema and airway disease. Emphysema is defined as abnormal, permanent enlargement of air spaces distal to the terminal bronchiole, accompanied by destruction of the alveolar walls [see Pathology, below]. The airway disease in COPD that results in airflow obstruction occurs principally in small air-ways (ie, those with an internal diameter of less than 2 mm).
Chronic bronchitis, a condition of the large airways, is com-mon in persons with COPD. Chronic bronchitis is defined as a cough productive of sputum on most days for at least 3 months a year for at least 2 consecutive years and for which there is no other explanation. Like COPD, chronic bronchitis is often associated with cigarette smoking; however, individ-uals with chronic bronchitis but without airflow obstruction are not classified as having COPD.
Whereas previous recommendations for management of COPD were based solely on the severity of the airflow obstruction, the most recent Global Initiative for Chronic
Obstructive Lung Disease (GOLD) guidelines recommend combined assessment using symptoms, breathlessness, spi-rometric classification, and risk of exacerbation to evaluate patients [see Figure 2] and guide treatment [see Table 1]. To assess symptoms, the GOLD guidelines recommend using the COPD Assessment Test (CAT) (http://www.cateston-line.org), a validated eight-item measure of impairment with scores ranging from 0 to 40.3,4 The GOLD guidelines state that a CAT score of 10 or higher should prompt consid-eration of further treatment.5 Spirometric classification is based on the degree of airflow limitation on postbronchodi-lator FEV1 in the setting of a reduced FEV1/FVC ratio.1 Grade 1 (mild) is defined as an FEV1 value of greater than or equal to 80% of predicted; because of the limitations of using a fixed FEV1/FVC ratio to define airflow obstruction using the GOLD criteria, it is likely that many normal individuals would fit this definition of mild COPD. Grade 2 (moderate) is defined as an FEV1 value between 50 and 80% of pre-dicted. Grade 3 (severe) is defined as an FEV1 value between 30 and 50% predicted. Patients with FEV1 values below 30% of the predicted value are categorized as grade 4 (ie, very severe). If only prebronchodilator spirometry is obtained, GOLD grades are assigned with less certainty.6 The risk of exacerbation is based on a history of previously treated exacerbations, which has been shown to be the best predic-tor of having frequent exacerbations.7
Epidemiology
prevalence
COPD is a leading cause of disability and death in the United States and worldwide.8,9 Precise estimates of COPD prevalence are not available and likely differ by region and smoking rates. During 2007–2010, the National Health and Nutrition Examination Survey (NHANES) obtained pre-bronchodilator spirometry data on a nationally representa-tive sample of US adults and postbronchodilator pulmonary lung function data for the subset of adults with airflow lim-itation.10 These data demonstrate an estimated overall COPD prevalence between 10 and 20% among adults in the United States between the ages of 40 and 79 depending on which diagnostic criteria were applied (fixed ratio or lower limit of normal) and whether pre- or postbronchodilator values were used. Similarly, a retrospective longitudinal cohort study in Canada published in 2011 determined that about one in four individuals is likely to receive medical attention for a physician diagnosis of COPD.11 Recent data from nationally representative data sources suggest declines in the age-adjusted prevalence of COPD from 1999 to 2011, likely due to declines in smoking rates. Indeed, the preva-lence of smoking in 2010 was approximately one half of that in 1965 for both men and women.12,13
* The authors and editors gratefully acknowledge the contributions of the previous authors, Robert M. Senior, MD, and Edwin K. Silverman, MD, PhD, to the develop-ment and writing of this review.

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 2
gender effects
Historically, men have had higher rates of COPD because of higher rates of cigarette smoking when compared to women. During the 1960s, however, smoking rates in women significantly increased, and several decades after these increases, the rates of COPD in women began climbing rap-idly. Currently, some experts suggest that women may have increased susceptibility to COPD compared with men, although this position is controversial. In 2000, more women than men died from COPD in the United States, due in part to a higher percentage of women in the population.14 Among patients with severe, early-onset COPD, a marked female
predominance has been reported.15,16 It is worth noting that health care providers are more likely to diagnose COPD in males than in females. This may be in part related to differ-ent clinical presentations; women are more apt to report dyspnea, whereas men are more likely to report increased phlegm production.14
racial and ethnic differences
Although COPD prevalence has historically been highest in whites living in the United States and Europe, the rates of COPD have increased dramatically in populations in Asia and Africa as smoking rates have increased in those regions.17
Figure 1 Spirometry test results in a normal patient (a and b) and one with severe chronic obstructive pulmonary disease (COPD) (c and d); the two patients are of the same age, gender, and height. Readings were taken before administration of a bronchodilator (black lines) and afterward (blue lines). Flow-volume loops (a and c) reveal reduced forced vital capacity (FVC) with coving in the COPD patient; the squares represent pre-dicted normal values. Volume-time curves (b and d) demonstrate reduced forced expiratory volume in 1 second (FEV1) and prolonged exhalation without a plateau in the COPD patient.
Prebronchodilator
8a b
c d
4
6
2
0
10
6
8
4
2
00 41 2 3
Volume (L)
Flow
(L/s
)
61410 2 4 6 8 10 12
2 4 6 8 10 12
Time (s)
FEV1 FVC
Vol
ume
(L)
8
4
6
2
0
10
6
8
4
2
00 41 2 3
Volume (L)
Flow
(L/s
)
61410
Time (s)
FEV1 FVC
Vol
ume
(L)
Postbronchodilator
Prebronchodilator Postbronchodilator

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 3
In a comparison of 80 African-American COPD patients and 80 white COPD patients with airflow obstruction of similar severity, African Americans had fewer pack-years of smoking (44 ± 23 in African Americans versus 66 ± 31 in whites).18 A review of available data on racial disparities in COPD found similar results.19 There is also evidence to suggest racial differ-ences in the extent of emphysema and airway remodeling in COPD.20 COPD will continue to be a major international cause of morbidity and mortality in white, Asian, and African pop-ulations for decades to come.21 There is evidence to suggest that Hispanic subjects may be at lower risk for COPD and lung function decline.22 Data are still being amassed on racial dis-parities in susceptibility, treatment, and outcomes.22
Etiology
Normally, pulmonary function follows a predictable tra-jectory across a person’s life span: FEV1 values increase steadily with growth during childhood and adolescence, plateau in early adulthood, and then gradually decline with advancing age. Environmental and familial factors can influ-ence this trajectory; for example, childhood respiratory ill-nesses and airway hyperresponsiveness may result in lower FEV1 values.
Departure from the normal trajectory can lead to COPD by one of three paths: (1) a normal rate of decline in FEV1
after a reduced growth phase (ie, poor lung development); (2) early initiation of FEV1 decline after normal growth, with a shortened plateau phase; or (3) accelerated decline in FEV1 after normal growth. Environmental and genetic factors, together with their interactions, likely can decrease the max-imal level of pulmonary function achieved or increase the rate of decline in FEV1.23
cigarette smoking
Cigarette smoking is the major environmental risk factor for the development of COPD. The effects of cigarette smok-ing on the decline in pulmonary function depend on the intensity of the smoke exposure (typically assessed as the number of pack-years, where one pack-year is defined as smoking one pack of cigarettes a day for 1 year), the timing of this exposure during growth and development in child-hood and adolescence, and the maximally attained level of pulmonary function. There is a dose-response relation between smoking intensity as assessed by pack-years and average level of reduction in FEV1 [see Figure 3].24 The per-centage of smokers with significant airflow obstruction (ie, FEV1 of less than 80% predicted) increases with increas-ing pack-years. Although there is clearly an increased risk with more intensive smoking exposure, only a minority of smokers develop severe COPD. Even among very heavy smokers, many individuals continue to have normal FEV1
Figure 2 Global Initiative for Chronic Obstructive Lung Disease (GOLD) Combined COPD Assessment using symptoms of breathlessness, spirometric classification, and risk of exacerbation to group patients with chronic obstructive pulmonary disease (COPD) into one of four catego-ries (A, B, C, or D).1 CAT = COPD Assessment Test; FEV1 = forced expiratory volume in 1 second.
GOLD 4: Very Severe
C D
A
CAT < 10 CAT ≥ 10
Breathlessness Symptoms
B
GOLD 3: Severe
GOLD 2: Moderate
GOLD 1: MildG
OLD
Cla
ssif
icat
ion
ofA
irfl
ow L
imit
atio
n
Exacerbation History
FEV1 < 30% predicted
30% ≤ FEV1 < 50% predicted
50% ≤ FEV1 < 80% predicted
≥ 2 exacerbations(or ë 1 exacerbationleading to hospital
admission)
≤ 1 exacerbation (notleading to hospital
admission)
FEV1 ≥ 80% predicted
Table 1 Initial Pharmacologic Management of COPDPatient Group First Choice Alternate Choice
A Short-acting anticholinergic as needed or short-acting beta2 agonist as needed
Long-acting anticholinergic or long-acting beta2 agonist or short-acting beta2 agonist and short-acting anticholinergic
B Long-acting anticholinergic or long-acting beta2 agonist
Long-acting anticholinergic and long-acting beta2 agonist
C Inhaled corticosteroid and long-acting beta2 agonist or long-acting anticholinergic
Long-acting anticholinergic and long-acting beta2 agonist or long-acting anticholinergic and phosphodiesterase-4 inhibitor or long-acting beta2 agonist and phosphodiesterase-4 inhibitor
D Inhaled corticosteroid and long-acting beta2 agonist and/or long-acting anticholinergic
Inhaled corticosteroid and long-acting beta2 agonist and long-acting anticholinergic or inhaled corticosteroid and long-acting beta2 agonist and phosphodiesterase-4 inhibitor or long-acting anticholinergic and long-acting beta2 agonist or long-acting anticholinergic and phosphodiesterase-4 inhibitor
COPD = chronic obstructive pulmonary disease.

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 4
values. An analysis of longitudinal spirometric data from 4,391 individuals in the Framingham Offspring Cohort con-firmed the negative impact of ongoing cigarette smoking on lung function decline and the marked variability in response to smoking.25 Of interest, this study demonstrated that smoking cessation under age 30 led to normalization of the subsequent rate of lung function decline, whereas smoking cessation after age 40 was associated, on average, with a persistently
elevated rate of lung function decline. Notably, the preva-lence of smoking has been declining significantly among both men and women.13 Although the use of conventional cigarettes is decreasing, the use of electronic cigarettes is growing, including among those who were previously nev-er-smokers. Data on the long-term safety of electronic ciga-rettes are still being gathered.26
other risk factors
Surveys of the general population have found that a sig-nificant percentage of persons with airflow obstruction are lifelong nonsmokers.27,28 It is uncertain how many of these nonsmokers have COPD; however, the risk of severe COPD is markedly lower in persons who have never smoked than it is in current smokers or ex-smokers. Rare genetic syn-dromes (see below) can lead to the development of severe COPD in nonsmokers.
Lower respiratory tract infections in childhood appear to be associated with lower FEV1 and FVC levels in adult-hood.29,30 However, such childhood infections do not appear to be associated with an accelerated decline in lung function during adulthood.31 Airway responsiveness (ie, the tendency for bronchoconstriction to occur in response to stimuli such as methacholine) is observed in many COPD patients. Whether airway responsiveness was a cause or an effect of COPD was previously unclear; however, multiple longitudi-nal studies have demonstrated that airway responsiveness is a strong predictor of subsequent decline in pulmonary func-tion. Thus, airway responsiveness is a risk factor for COPD.32
An increased risk of COPD has been posited for a variety of occupations, including gold mining, coal mining, and work in cotton textile industries.33 Similarly, one study in Asia demonstrated that exposure to biomass fuels or dusty jobs among patients with COPD was associated with more frequent symptoms and severe airflow limitation.34 It remains unclear if occupational exposures are major risk factors for the development of COPD in nonsmokers. Increased air pol-lution has been associated with reduced lung function in children and increased exacerbation rates in COPD subjects; however, it remains unproven if air pollution is a significant risk factor for the development of COPD.35 Exposure to sec-ondhand cigarette smoke in childhood can affect the maxi-mal level of lung function attained, and secondhand smoke exposure may be a risk factor for COPD. Exposure to bio-mass fuel sources for cooking, including wood and crop res-idues, has been shown to be a risk factor for COPD and chronic bronchitis in nonsmoking women in developing countries.35 HIV infection can increase the risk of emphy-sema, particularly in smokers.36
genetics
α1-Antitrypsin Deficiency
α1-Antitrypsin (AAT) deficiency was discovered in Swe-den in the early 1960s and remains the most clearly defined genetic risk factor for COPD.37,38 AAT is a protease inhibitor that is encoded by the SERPINA1 (also known as protease inhibitor [PI]) gene. Most individuals carry two copies of the M allele at the SERPINA1 locus and have normal AAT levels. The S allele of SERPINA1 is associated with slightly reduced AAT levels, and the Z allele is associated with
Figure 3 In a general population sample, the mean forced expiratory volume in 1 second (FEV1) (expressed as a percentage of the predicted value) decreases with increasing smoking intensity (expressed as pack-years).16 However, many heavy cigarette smokers continue to have FEV1 values within the normal range. This variable response to smoking suggests that genetic and other environmental influences are involved in susceptibility to chronic obstructive pulmonary disease.
30
20
10
0
0–20 Pack-Years (578)30
20
10
0
21–40 Pack-Years (271)30
20
10
0
41–60 Pack-Years (154)30
20
10
0
61+ Pack-Years (100)30
20
10
0
40 60 80 100 120 140 160
Forced Expiratory Volume in 1 Second(FEV1) (% of predicted)
0 Pack-Years (945)
Median
– 1 Standard Deviation Mean + 1 Standard Deviation
Popu
lati
on (%
of t
otal
und
er s
tudy
)

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 5
markedly reduced AAT levels; the prevalence of these vari-ants is above 1% in most white populations. Much less com-monly, individuals inherit null alleles, which results in the complete absence of AAT production. Null alleles may result from any of multiple genetic alterations. The condi-tion of having two Z alleles or one Z and one null allele is referred to as PI Z because only the Z protein is observed within the bloodstream of such persons. This represents the most common type of severe AAT deficiency.
Three main types of clinical tests are used to detect AAT deficiency. The serum level of the protein, often measured by nephelometry (a technique used to determine the level of sev-eral blood plasma proteins), is a commonly performed screen-ing test. Patients with serum AAT levels below the normal range (typically below 80 mg/dL or 15 µM) should undergo testing to determine the type of AAT that they have inherited. This can be done by assessing the protein in serum or plasma with isoelectric focusing (referred to as the AAT phenotype) or the genetic variants in DNA (referred to as the AAT genotype). Because genotype testing is not available for the full range of null variants, AAT phenotyping is still commonly performed.
AAT deficiency is found in approximately 1 to 2% of COPD patients. Population screening studies in the United States indicate that the prevalence of individuals with AAT defi-ciency is between one in 2,857 and one in 5,097. On the basis of a population of about 320 million, there are approximately 100,000 individuals with AAT deficiency in the United States; however, the vast majority of them have not been diagnosed. For that reason, the natural history of lung disease (as well as liver disease, which also develops in a subset of PI Z individ-uals) in AAT deficiency is uncertain. In recognition of the marked underdiagnosis of AAT deficiency, AAT testing has been recommended for all patients who have COPD or who have asthma with chronic airflow obstruction.37
Despite the uncertainty in the natural history of AAT defi-ciency, it is well established that cigarette smoking is a major risk factor for early-onset COPD in persons with the PI Z phenotype. Nonetheless, the development of COPD in smok-ers with the PI Z phenotype is not absolute. Among non-smokers with PI Z, marked variability has been noted in the development of airflow obstruction.39 Reported risk factors for a more severe pulmonary course in AAT deficiency include male gender, asthma (especially in childhood), pneu-monia, and occupational exposures to respiratory irritants.40
The risk of lung disease in heterozygous individuals with the PI MZ phenotype remains controversial. A meta-analysis found that in case-control studies, a higher rate of PI MZ was observed in COPD patients than in normal control subjects.41 In population-based studies, however, similar FEV1 levels have been found in persons with PI MZ and those with PI MM. A subset of PI MZ individuals may be at increased risk for COPD as a result of other genetic and environmental risk factors, although this has not been proven. Greater risk of COPD in PI SZ individuals has been more definitively estab-lished,42 although the COPD risk associated with PI SZ is lower than that with PI Z. Individuals with the PI MS pheno-type do not appear to be at increased risk for COPD.
Other Genetic Syndromes
In addition to AAT deficiency, several very rare syndromes have been associated with COPD. Cutis laxa, a dermatologic
condition marked by loose and inelastic skin, is often asso-ciated with the development of emphysema in childhood or adolescence. Cutis laxa is etiologically heterogeneous; muta-tions in several genes, including those that code for elastin and fibulin-5, have been identified as causes. Hypocomple-mentemic urticarial vasculitis is a syndrome of unknown etiology that often includes COPD.43 Although lung blebs have been reported in other connective tissue disorders, such as Marfan syndrome and Ehlers-Danlos syndrome, the risk of COPD in these syndromes is unclear.44
Common Genetic Risk Factors
Among patients with COPD that does not have a specific known genetic cause, familial clustering of both low pulmo-nary function values and COPD cases has been demon-strated.45,46 One study of 22,422 Danish and 27,668 Swedish twin pairs estimated that approximately 60% of the individ-ual susceptibility to COPD could be explained by genetic factors.47 Thus, it is likely that additional genetic factors increase COPD risk in a subset of smokers. Genetic variants in several genomic regions (including chromosomes 4q22, 4q31, 15q25, and 14q32) have been associated with COPD at genome-wide significance levels in genome-wide association studies.48–51 Additional genome-wide significant loci have been associated with the development of severe COPD.51
A functional genetic variant upstream from hedgehog inter-acting protein (HHIP) has been found in the genomic region associated with COPD on chromosome 4q31. Several novel COPD susceptibility genes have been supported by animal models of emphysema.52,53 A better understanding of the genetic determinants of COPD has the potential to provide insights into the heterogeneity of this disease state and guide prevention, diagnosis, and treatment.
Pathogenesis
inflammation
COPD is a complex disease.54,55 Many pathogenetic mecha-nisms appear to be involved: (1) inflammation, (2) immunity, (3) oxidative stress, (4) apoptosis and senescence, and (5) turn-over of extracellular matrix (ECM).56–58 Epigenetic changes also occur.59,60 Research on genetically altered mice has been invalu-able in uncovering and investigating mechanisms of COPD,61 but the extent to which the mechanisms cited above are oper-ative in an individual COPD patient is not easily discerned. It is likely that the relative importance of the mechanisms var-ies between patients and at different grades in the course of the disease in an individual patient. Host factors specifically implicated in emphysema are shown here [see Figure 4].
The definition of COPD states that inflammation is central to the pathogenesis of COPD.1 Indeed, inflammation in COPD lungs is evident from a variety of sources, including surgical specimens, postmortem tissue, lung scans with iso-topes that label neutrophils, bronchoalveolar lavage fluid, sputum, and volatile products in exhaled breath. The sever-ity of airway inflammation by histopathologic examination correlates with the physiologic severity of the disease. In mouse smoking models, the inflammatory response and the degree of emphysema correlate. Many factors, as diverse as adiponectin and aryl hydrocarbon hydrolase, appear to be

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 6
involved in promoting or protecting against the inflamma-tion associated with smoke exposure.62,63 If inflammation is prevented or reduced in smoking models of emphysema, as, for example, by drugs such as the phosphodiesterase-4 inhibitor roflumilast [see Long-Term Care, below], there is a reduction in the development of emphysema.64
According to the inflammation paradigm of COPD patho-genesis, smoking and other types of inhaled irritants activate
resident macrophages and structural cells of the lungs to release factors that recruit and activate monocytes, lympho-cytes, dendritic cells, neutrophils, and eosinophils. These inflammatory cells, in turn, recruit other inflammator y cells; release oxidants, proteinases, and cytokines that promote apoptosis or senescence of lung cells; damage the lung ECM; and adversely affect its repair. In former smokers, high numbers of inflammatory cells in the lungs and other evi-dence of lung inflammation may be found years after smok-ing cessation,65 which suggests that the inflammation in COPD is self-perpetuating.
Although inflammatory cells and their products are clearly key in COPD, several points about inflammation and COPD are notable: (1) structural cells of the lungs and airways may trigger inflammation by producing proinflammatory factors66,67; (2) peptides derived from ECM components, especially elastin and collagens, have proinflammatory activity68,69; and (3) inflammation is not a feature of all exper-imental models of emphysema.70
immunity
Studies of both patients and experimental models implicate innate and adaptive immunity in the pathogenesis of COPD.71,72 Among many observations that associate immunity with COPD and with emphysema in mice are the following: (1) B cells, and CD4+ and CD8+ T cells, accumulate in alveolar, airway, and perivascular sites in COPD lungs; (2) lymphoid follicles are present in bronchiolar walls, at perivascular sites, and in alve-olar tissue in COPD, and their quantity correlates with airflow obstruction65; (3) antielastin antibodies are present in COPD bronchoalveolar lavage fluid and stimulate T cell prolifera-tion73; (4) smoke-induced emphysema does not develop in mice that lack CD4+ and CD8+ cells74; and (5) transfer of lung T cells from smoke-exposed mice to mice that have not been exposed to smoke drives a variety of COPD-like features in the recipient mice.75 A three-stage scheme of increasing immune responses coinciding with increasing severity of COPD has been proposed: in stage 1, which occurs in virtually all smok-ers, there is injury of pulmonary epithelium with release of proinflammatory mediators; in stage 2, there is intrapulmo-nary proliferation and maturation of dendritic cells and T cells; and in stage 3, there is augmentation of stage 2 and the appear-ance of antibodies to lung cells and other components of lung tissue.72,76 Autoimmunity to elastin has been reported,77 but this is not a consistent finding.78
proteinase-antiproteinase imbalance
Ever since the discovery in the 1960s of AAT deficiency associated with early-onset emphysema and the production of emphysema in experimental animals with elastolytic enzymes, proteinases have been considered key to the mech-anism of emphysema. In the intervening years, other mech-anisms of emphysema pathogenesis have been uncovered, most notably apoptosis and oxidant stress. But proteinase- antiproteinase imbalance continues to prevail as an import-ant mechanism, and within this paradigm, there has been marked growth of information about the types and cellular sources of proteinases, the induction and regulation of pro-teinases, and the substrates attacked by proteinases.79
Increased levels of several proteases from both inflamma-tory cells and epithelial cells have been found to be increased
Figure 4 The pathogenesis of emphysema from smoking. Smoking stimulates resident cells to release factors that recruit inflammatory cells to the lungs. The various inflammatory cells that accumulate in the periph-eral tissues of the lungs release proteinases and oxidants that damage or degrade extracellular matrix in the walls of alveoli, alveolar ducts, and respiratory bronchioles. In addition, agents in smoke and those released by inflammatory cells inactivate proteinase inhibitors, such as α1-antitrypsin, and cause senescence and apoptosis of lung cells that produce extracellu-lar matrix. Products of the damaged extracellular matrix, such as peptides of degraded elastin, are chemotactic for inflammatory cells; thus, degrada-tion of the extracellular matrix may lead to a feedback loop that perpetu-ates inflammation. These matrix-derived products may also elicit immune responses that lead to destruction of extracellular matrix. COPD = chronic obstructive pulmonary disease; IL-8 = interleukin-8.
Cigarette Smokeplus Host Factors
Chemoattractants(Leukotriene B4 ,
IL-8)
Antioxidants Antiproteases
Proteases(Cathepsins,
MatrixMetallo-
proteinases,Cathepsin G,NeutrophilElastase)
Oxidantsand FreeRadicals
EpithelialCell and
Matrix Injury
ActivatedMacrophage
ActivatedNeutrophil
Stimulates
Inhibits
IncreasedMucus
Emphysema
COPD
Chronic Bronchitis
H2O2

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 7
in patients with COPD. Among these, neutrophil elastase and its main inhibitor, AAT, and elastic fibers have most strongly supported the proteinase-antiproteinase hypothesis of emphysema. Matrix metalloproteinases (MMPs) have sub-sequently also been associated with emphysema.80 Many MMPs are found in emphysematous lung tissue and in bron-choalveolar lavage fluid along with alveolar macrophages from patients with emphysema. Macrophage MMP increases are also found in early-onset emphysema associated with HIV.81 MMPs are not inhibited by AAT; they are inhibited by proteins called tissue inhibitors of metalloproteinases. Just as the spectrum of proteinases has grown beyond neutrophil elastase, the focus on elastic fibers as the key component of the lung ECM in emphysema has been expanded to include other matrix components, notably collagens.82
Three aspects of proteinases and COPD should be noted: (1) in contrast to emphysema, relatively little is known about proteinase-antiproteinase imbalance in COPD airways; (2) besides affecting lung elastin and other matrix components, proteinases process cytokines involved in the inflammatory and immune responses in COPD; and (3) the apparently sim-ple model of emphysema caused by putting elastases in the lungs elicits, and requires, a complex response.83,84
oxidants
An increased burden of oxidants from smoke and inflam-matory cells is present in the lungs of cigarette smokers.85 Genes involved in responding to oxidant stress are affected in smokers’ airway epithelium, and the effects are exagger-ated in those with COPD.86 Similar types of changes are seen in experimental smoke-induced emphysema.87 Emphysema-tous tissue shows oxidative modifications of DNA.88
Oxidants promote inflammation and proteinase expression via several intracellular signaling pathways, including mitogen- activated protein kinases and nuclear factor κB. Oxidants can induce apoptosis, activate MMPs, and affect ECM directly as oxidative modification of tropoelastin, the building block of elastin, prevents its assembly into elastic fibers.89 Antioxidant mechanisms are crucial in resistance to emphysema develop-ment from smoke exposure. Mice lacking nuclear factor E2–related factor (Nrf-2), a transcription factor that promotes expression of antioxidants, develop emphysema in response to cigarette smoke, whereas mice with the same genetic back-ground except having normal Nrf-2 are resistant to emphy-sema from the same smoke exposure.90 Considering the importance of oxidants in COPD, there is interest in identify-ing ingestible compounds with antioxidant properties that protect against smoke-induced emphysema.91,92
apoptosis and senescence
Emphysematous human lung contains apoptotic and senescent cells, which are not seen in normal lungs.93,94 Cig-arette smoke can induce apoptosis in vitro. Emphysema develops in animal models of lung cell apoptosis induced by reducing vascular endothelial growth factor (VEGF) or his-tone deacetylase.70 Ceramide, a signaling sphingolipid, has been implicated in apoptosis in emphysema.95 Notably, inflammation is not present in models of emphysema caused by apoptosis via reductions in lung VEGF or histone deacety-lase.70 Senescent lung cells promote tissue injury through release of proinflammatory cytokines and proteinases and
lead to alveolar cellular loss via apoptosis.96 Cells from severe emphysema lungs are more easily induced to a senes-cent phenotype with cigarette smoke extract than cells from normal lungs. This difference between emphysema cells and normal cells correlates with levels of Werner syndrome pro-tein, which are low in emphysema cells.97
degradation and synthesis of lung ecm
The ECM of the lung parenchyma and airways can be changed markedly in COPD showing parenchymal spaces devoid of tissue and small airways that have fibrotic walls encroaching or even obliterating the airway lumina. Although emphysematous human lung parenchyma may look depleted of ECM and essentially nonviable, surprisingly, the quantity of collagen and elastin content of the remaining lung tissue may be increased above normal,98 and elastin synthesis is active by messenger RNA analysis even in lungs removed for lung transplantation for severe COPD.99 The response of the lung ECM in animal models of smoke exposure indicates that changes in airways, pulmonary blood vessels, and parenchyma ECM do not occur in the same time frame in each of these tissue compartments, and drugs to protect against changes do not necessarily affect the ECM in each compartment in the same way.100
Pathophysiology
Irreversibly reduced maximal expiratory airflow, most commonly measured as the FEV1 with a reduced FEV1/FVC ratio, is the defining physiologic feature of COPD.101 Other characteristic physiologic features include an increased residual volume (RV), increased functional residual capacity (FRC), increased residual volume to TLC ratio (RV/TLC), decreased inspiratory capacity (IC), maldistribution of ven-tilation, and ventilation-perfusion (V/Q) mismatching. The TLC may be normal or increased, whereas the carbon mon-oxide diffusing capacity (Dlco) may be normal or decreased.102
airflow obstruction
Unlike patients with asthma, in whom inhaled bronchodi-lators typically produce large improvements in airflow, patients with COPD commonly have only modest responses to inhaled bronchodilators (ie, increases of < 10% of the predicted FEV1 value).103 Inspiratory flow in patients with COPD may be relatively well preserved, even when FEV1 is markedly reduced. The abnormalities of expiratory flow seen in COPD are readily apparent from flow-volume curves: in mild COPD, the lower part of the expiratory limb of the flow-volume curve is “scooped out,” reflecting abnor-mally increased narrowing of the airways at lung volumes near the RV; in moderate and advanced COPD, decreased flow is seen over the entire expiratory limb [see Figure 1]. Two types of alterations of lung physiology account for reduced maximal expiratory airflow in COPD: reduced lung elastic recoil and increased airway resistance. Both of these alterations are common in individuals with COPD.
maldistribution of ventilation and v/q mismatching
Maldistribution of ventilation and V/Q mismatching occur in COPD as a result of the heterogeneous nature of the

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 8
underlying pathology in the airways and lung parenchyma. The heterogeneity of the distribution of ventilation can be detected by nitrogen washout during breathing of 100% oxygen and by radioisotopic ventilation scanning with xenon-133 (133Xe).
In the past, it was common to try to categorize patients with COPD as either “pink puffers” or “blue bloaters.” The distinction was based on the idea that there are two fun-damentally different patterns of V/Q abnormality among patients with COPD. In so-called pink puffers, the TLC is large, the Dlco is low, and the resting arterial oxygen tension (Pao2) is normal or near-normal. In these individuals, a sub-stantial percentage of ventilation is distributed to high V/Q regions (so-called dead space). In contrast, in so-called blue bloaters, TLC and Dlco are normal, but hypoxemia is pres-ent as a result of substantial perfusion of pulmonary blood into low V/Q regions (so-called shuntlike regions). This clas-sification has largely been discarded because most patients with COPD are not readily sorted into one type or the other.
hyperinflation
Hyperinflation, a supraphysiologic increase in lung vol-umes, is typical of moderate or more advanced COPD.104 Hyperinflation causes the diaphragm to be displaced into a flattened position, which makes it less effective. In addition, hyperinflation puts the thoracic cage at a mechanical disad-vantage. The net effect of these changes is to increase the work of breathing, to diminish the capacity for exercise, and to increase dyspnea. Typically, hyperinflation becomes worse with exercise because the increase in the respiratory rate lim-its the time available for the lung to empty between inhala-tions. This additional hyperinflation, designated dynamic hyperinflation, adds to the load on the inspiratory muscles while further reducing their mechanical advantage. The IC is commonly reduced in patients with COPD, and the reduc-tion appears to have prognostic significance that is indepen-dent of FEV1. In patients with moderate to severe COPD, survival was found to be markedly shorter in those whose IC/TLC ratio was less than 25% compared with the patients whose IC/TLC ratio was greater than 25%, even though the individuals in the two groups had COPD of comparable severity, as determined on the basis of FEV1.105
dyspnea
Dyspnea is characteristic of moderate or more advanced COPD. Its onset is typically gradual, only slowly affecting daily activities and quality of life. Dyspnea is seldom a com-plaint until the FEV1 has fallen below about 60% of the pre-dicted value. However, the correlation between FEV1 and dyspnea is not strong. Some individuals with COPD are relatively free of dyspnea despite remarkably low levels of FEV1.106
The mechanisms of dyspnea in COPD are still not fully understood.107 Neural signals from the chest wall and air-ways appear to be involved. Increased effort of breathing, as the respiratory muscles are stressed to produce pressures approaching their maximum pressure-generating capacity, is thought to be a factor in producing dyspnea. Signals of so-called length-tension inappropriateness from the respira-tory muscles occurring as a result of hyperinflation are another factor. Abnormalities of arterial blood gases are not
a major factor except in acute situations. Oxygen therapy may decrease dyspnea by reducing ventilation, as well as through direct effects that are not associated with changes in ventilation. Recent studies point to abnormalities of perfu-sion of respiratory muscles as a factor in exercise limitation in COPD.108,109
exacerbations
The natural history of COPD often includes exacerbations, which are episodes of worsening respiratory symptoms, includ-ing increased cough, discolored sputum, and/or increased dyspnea. Exacerbations tend to become more frequent as COPD becomes more severe; however, some patients with very severe COPD never have exacerbations, whereas others with mild COPD have frequent exacerbations.110 Exacerba-tion frequency appears to be a relatively stable characteristic; in 2,138 COPD subjects in the Evaluation of COPD Longitu-dinally to Identify Predictive Surrogate End-points (ECLIPSE) study, the strongest predictor of future exacerbations was previous exacerbation frequency.7 Frequent exacerbators, with at least two episodes per year requiring treatment with antibiotics and/or systemic steroids, tended to have a similar exacerbation frequency during 3 years of follow-up. COPD exacerbations are a major cause of morbidity and mortality. The most frequent triggers of exacerbations are respiratory infections; viruses and bacteria (including Streptococcus pneu-moniae, Haemophilus influenzae, and Moraxella catarrhalis and, less often, atypical pathogens such as Chlamydia pneumoniae) are common causes. Although many COPD patients are chronically colonized with potentially pathogenic bacteria, changes in the bacterial strains are frequently associated with episodes of exacerbation.111 In addition to respiratory infec-tions, other triggers for exacerbations include congestive heart failure, drug effects, and nonadherence to medical treatment. Investigators in France reported finding pulmo-nary embolism in a surprisingly high percentage (25%) of patients with episodes of severe COPD exacerbation of unknown etiology; of note, this study excluded patients with evidence of lower respiratory tract infections, the most com-mon cause of COPD exacerbations.112
COPD exacerbations can lead to respiratory failure and death. COPD patients who recover from an exacerbation usually take several weeks to return to their previous level of pulmonary function.113 Frequent exacerbations may be associated with accelerated decline in pulmonary function among some COPD patients.114,115
Pathology
Multiple pathologic changes are found in the respiratory tract of patients with COPD.116,117 However, from the stand-point of increasing airway resistance and creating maldistri-bution of V/Q relations, the changes of importance are those affecting airways smaller than 2 mm in internal diameter (commonly called small airways) and the sites of alveolar gas exchange, which are respiratory bronchioles, alveolar ducts, and alveoli.
Pathologic changes may be apparent in the lungs of smok-ers before the FEV1 becomes abnormally low. The lungs of apparently healthy cigarette smokers as young as 25 years show accumulation of pigmented macro phages in respiratory
Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 9
bronchioles, often accompanied by edema, epithelial hyper-plasia, and fibrosis in adjacent bronchiolar and alveolar walls.
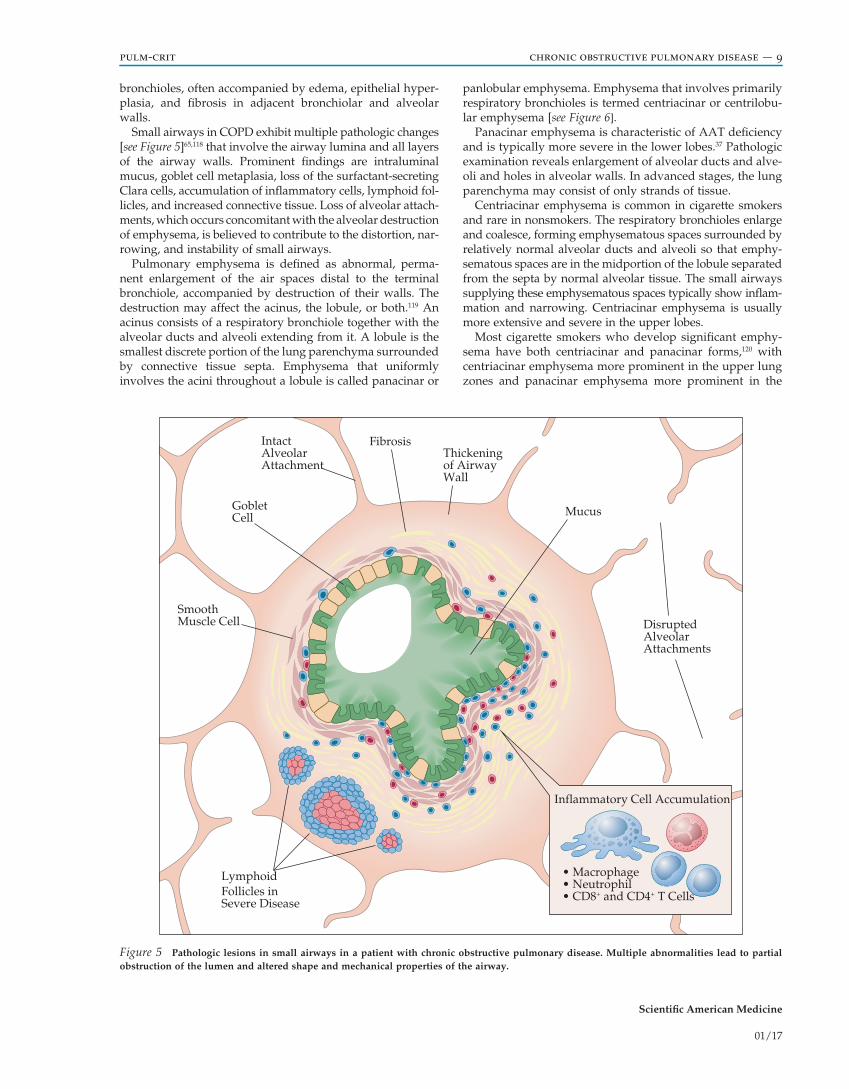
Small airways in COPD exhibit multiple pathologic changes [see Figure 5]65,118 that involve the airway lumina and all layers of the airway walls. Prominent findings are intraluminal mucus, goblet cell metaplasia, loss of the surfactant-secreting Clara cells, accumulation of inflammatory cells, lymphoid fol-licles, and increased connective tissue. Loss of alveolar attach-ments, which occurs concomitant with the alveolar destruction of emphysema, is believed to contribute to the distortion, nar-rowing, and instability of small airways.
Pulmonary emphysema is defined as abnormal, perma-nent enlargement of the air spaces distal to the terminal bronchiole, accompanied by destruction of their walls. The destruction may affect the acinus, the lobule, or both.119 An acinus consists of a respiratory bronchiole together with the alveolar ducts and alveoli extending from it. A lobule is the smallest discrete portion of the lung parenchyma surrounded by connective tissue septa. Emphysema that uniformly involves the acini throughout a lobule is called panacinar or
panlobular emphysema. Emphysema that involves primarily respiratory bronchioles is termed centriacinar or centrilobu-lar emphysema [see Figure 6].
Panacinar emphysema is characteristic of AAT deficiency and is typically more severe in the lower lobes.37 Pathologic examination reveals enlargement of alveolar ducts and alve-oli and holes in alveolar walls. In advanced stages, the lung parenchyma may consist of only strands of tissue.
Centriacinar emphysema is common in cigarette smokers and rare in nonsmokers. The respiratory bronchioles enlarge and coalesce, forming emphysematous spaces surrounded by relatively normal alveolar ducts and alveoli so that emphy-sematous spaces are in the midportion of the lobule separated from the septa by normal alveolar tissue. The small airways supplying these emphysematous spaces typically show inflam-mation and narrowing. Centriacinar emphysema is usually more extensive and severe in the upper lobes.
Most cigarette smokers who develop significant emphy-sema have both centriacinar and panacinar forms,120 with centriacinar emphysema more prominent in the upper lung zones and panacinar emphysema more prominent in the
Figure 5 Pathologic lesions in small airways in a patient with chronic obstructive pulmonary disease. Multiple abnormalities lead to partial obstruction of the lumen and altered shape and mechanical properties of the airway.
IntactAlveolarAttachment
Fibrosis
Mucus
Thickeningof AirwayWall
GobletCell
SmoothMuscle Cell
LymphoidFollicles inSevere Disease
DisruptedAlveolarAttachments
Inflammatory Cell Accumulation
• Macrophage• Neutrophil• CD8+ and CD4+ T Cells

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 10
lower lung zones. In advanced cases, it may not be possible to distinguish between these two forms of emphysema.
Physiologic-Pathologic Correlations
Maximal expiratory airflow from healthy lungs is limited primarily by the resistance of the central airways (ie, the tra-chea and the major bronchi) and the upper airways (ie, the larynx and the pharynx); it is limited to only a minor degree by the resistance of the small airways (defined as those having an internal diameter of 2 mm or less).121 As COPD develops, airflow resistance increases markedly. Almost all of this increase occurs in the small airways.122 GOLD classes of airflow obstruction correlate with pathologic changes in small air-ways, suggesting that the airflow obstruction in most patients with COPD is especially linked to abnormalities of small airways.65
As stated above [see Pathophysiology], an increased RV, an increased RV/TLC, a normal to increased FRC, and a normal to increased TLC are characteristic of COPD. The RV may be
up to severalfold larger than normal. As a result of an increased FRC, tidal breathing may take place at lung vol-umes 1 to 2 L above normal levels. An increased FRC pro-duces the advantage of enlarged airway diameter with greater radial support (which means less airway resistance) and increased driving pressure (ie, elastic recoil) for exhalation. However, an increased FRC increases the work of breathing because breathing has to overcome the decreased chest wall compliance at high lung volume. A reduction in Dlco rela-tive to lung volume has some correlation with emphysema. The presumed mechanism is a reduction of gas-exchanging surface area as a result of alveolar destruction.
There is considerable variability in the relation between the FEV1 and other physiologic abnormalities in COPD, but some generalizations can be made. The Pao2 usually remains near-normal until the FEV1 has decreased to below half of the predicted level. V/Q mismatching, rather than shunting of pulmonary blood flow, explains essentially all of the reduction in Pao2 that occurs in COPD. Accordingly, mod-estly elevated levels of inspired oxygen (eg, elevations of
Figure 6 In the top row, the acinar structure of normal lungs is compared with that of lungs in patients with centriacinar (centrilobular) emphy-sema or panacinar (panlobular) emphysema. The normal acinus has a clearly defined structure consisting of the terminal bronchiole (TB); first-, second-, and third-order respiratory bronchioles (RB1, RB2, and RB3, respectively); alveolar ducts (AD); and alveolar sacs (AS). In centriacinar emphysema, there is selective enlargement and destruction predominantly of the respiratory bronchioles. In contrast, panacinar emphysema is defined by universally enlarged and destroyed air spaces throughout the acinus. Patterns of lobular destruction are depicted in the center row. The normal pulmonary lobule is a macroscopic structure, the borders of which can be identified by the presence of connective tissue septa. In centriacinar emphysema, the predominant site of overdistention is in the center of the lobule, with relative sparing toward the periphery—hence the name centrilobular emphysema. In contrast, there are uniform destructive changes throughout the lobule in panlobular emphysema.
NormalA
cina
r Stru
ctur
eLo
bula
r Pat
tern
Centriacinar(Centrilobular) Emphysema
Panacinar(Panlobular) Emphysema
TBTBTB
RB1RB1 RB1
RB2RB2
RB2
RB3
RB3 RB3
AD ADAD
AS
Septum
AS AS + AS

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 11
24 to 30%) are usually effective in treating hypoxemia from COPD. If a patient with COPD has hypoxemia that is resis-tant to supplemental oxygen, the clinician should consider the possibility of additional problems, such as pulmonary emboli or pulmonary hypertension with right-to-left intrac-ardiac shunting.
The arterial carbon dioxide tension (Paco2) usually does not rise above normal in COPD until the FEV1 is less than about one fourth of the predicted value, and it may not do so even then. An elevation in the setting of only mild to moder-ate COPD raises questions of other causes of hypercapnia, such as muscular weakness or a depressed respiratory drive. The lack of a predictable relationship between the severity of airflow obstruction in COPD and the Paco2 appears to be related to differences in pulmonary mechanics, in the central control of ventilation, and in respiratory disturbances during sleep. Some patients have airflow obstruction primarily during expiration, whereas others exhibit increased resistance to air-flow during both expiration and inspiration. Another factor influencing the development of hypercapnia may be intrinsic properties of respiratory drive. Thus, patients in whom hypercapnia ultimately develops may have intrinsic defects that cause relatively depressed ventilatory responses to acute rises in Paco2, falls in Pao2, or both. The idea of an underlying, presumably genetic, basis for the risk of CO2 retention is sup-ported by the finding that healthy first-degree relatives of patients with COPD and chronic CO2 retention have blunted ventilatory responses to hypercapnia and hypoxia compared with first-degree relatives of patients with COPD but without chronic hypercapnia. Cor pulmonale and right ventricular failure occur as a complication of pulmonary hypertension attributable to COPD, but only with far-advanced disease that has caused chronic hypoxemia (Pao2 < 55 mm Hg) [see Pul-monary Hypertension, below].
Alveolar gas exchange during exercise is variable in COPD patients, a fact that reflects the different patterns of physiologic abnormalities seen among these patients. Some patients have abnormally high levels of ventilation for a given workload (expressed in terms of oxygen consump-tion), but, nevertheless, their Pao2 falls with exercise. The Dlco may remain low during exercise, reflecting insuffi-cient alveolar-capillary surface area available for gas exchange. Patients whose resting Dlco value is below 55% of the predicted normal value often experience oxygen desaturation with exercise. Other patients may have subnor-mal increases in ventilation for a given level of exertion (ie, a decrease in the ratio of minute ventilation to oxygen con-sumption) and exhibit a rise in Paco2 with exercise, yet the Pao2 may increase. This increase in Pao2 must reflect improved V/Q matching during exercise.
Diagnosis
According to the GOLD guidelines [see Definition, above], clinicians should consider the diagnosis of COPD in any patient who has dyspnea, chronic cough, or sputum produc-tion, as well as in any patient who has a strong family history of COPD or a history of exposure to risk factors for COPD, especially cigarette smoking.1 In developing countries, indoor air pollution from biomass heating and cooking over open fires is a COPD risk factor. Exposure to risk factors warrants
emphasis because spirometric abnormalities, by which a diagnosis of COPD is established, commonly antedate symp-toms or physical signs of COPD. However, there is no evidence that screening spirometry impacts management decisions or improves COPD outcomes. Therefore, the U.S. Preventive Services Task Force (USPSTF) and GOLD recommend against screening for COPD in asymptomatic adults.1,123
In essence, COPD is a diagnosis of exclusion [see Differen-tial Diagnosis, below]. A reduced FEV1 and a reduced FEV1/FVC in the absence of any other reason for airflow obstruc-tion are sufficient to warrant a strong suspicion of COPD. A history of long-term smoking and minimal or no response to an inhaled bronchodilator confirm the diagnosis. Occa-sionally, patients who have had symptoms of COPD for a long time will first seek medical attention because a new symptom, such as hemoptysis, has developed (perhaps as a result of a different disorder, such as lung cancer) or because their symptoms have worsened abruptly, such as occurs during a COPD exacerbation.
clinical manifestations
Chronic and progressive shortness of breath, often described as discomfort with activities formerly done without difficulty, is the hallmark symptom of COPD. Although exertional dys-pnea correlates in general with the degree of airflow obstruc-tion, wide variability among individuals makes it impossible to predict the extent of respiratory impairment on the basis of any single value or set of values of expiratory flow. Typically, only minimal dyspnea is experienced until the FEV1 falls below 60% of normal. As airflow obstruction progresses, dys-pnea develops with more modest levels of exertion. When the FEV1 drops below 35% of normal, patients commonly have dyspnea during activities of daily living, such as making the bed or bathing. Orthopnea is not a common complaint, although it may be reported in patients with advanced COPD, especially in those with chronic airway secretions. Patients with COPD may awaken after several hours of sleep with cough, shortness of breath, and chest congestion that mimic paroxysmal nocturnal dyspnea. Relief is obtained by cough-ing up secretions. A large deterioration in exercise tolerance or FEV1 within a few months or less should raise concern that more than COPD is present because COPD alone does not produce such rapid changes.
Chronic cough with or without sputum production may precede airflow limitation by years; these symptoms in a long-term smoker do not necessarily indicate COPD. Impor-tantly, the absence of cough and sputum in a long-term smoker should not be taken to mean that COPD is not present. Wheezing is a nonspecific finding that can occur transiently when mucus accumulates in airways, and some patients do experience wheezing attacks that mimic asthma with chest tightness. Anorexia and weight loss are often present in COPD and purport a worse prognosis. It is important to exclude other etiologies of weight loss, such as chronic infection and malignancy, before attributing this feature to COPD.124,125
physical examination findings
In early disease, the physical examination is likely to be normal. With more advanced COPD, the use of accessory muscles of ventilation may be evident at rest, as may noisy,

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 12
rapid breathing. On percussion of the chest, there may be hyperresonance over the lung fields, secondary to emphy-sema, and minimal movement of the diaphragm during deep breathing. Auscultation over the lungs during an FVC maneuver typically reveals prolonged exhalation (more than 6 seconds) and wheezes near the end of this effort. Decreased intensity of breath sounds, even during vigorous panting, is found in patients with advanced emphysema. A persistent localized wheeze on auscultation is not typical of COPD; rather, it raises suspicion of an obstructing lesion in an air-way. Likewise, clubbing of the digits is not a manifestation of COPD. Its recent appearance should prompt an investiga-tion for a coexisting lung cancer.126
laboratory tests
By definition, irreversible obstruction of airflow is the defining feature of COPD. Accordingly, pulmonary function tests that measure airflow, primarily the FEV1 and FEV1/FVC, are used for making a laboratory diagnosis of COPD. Radiologic studies of the chest may be suggestive or support-ive of the diagnosis but will not be diagnostic. Elevated levels of blood C-reactive protein and Clara cell secretory protein in the sputum have been associated with COPD.127,128 To date, there have been no biomarkers that are clinically used for the diagnosis of COPD or measurement of disease progression.
Pulmonary Function Tests
Spirometry can confirm the obstructive abnormality, quantify the severity of the obstruction, and provide some assessment of reversibility. Spirometry should be obtained postadministration of a short-acting inhaled bronchodilator. Once the diagnosis of COPD is established, pulmonary func-tion testing is useful to quantitatively monitor the course of the disease. In interpreting spirometry for possible COPD, focus is on the FEV1 and FEV1/FVC. For a number of years, the criteria set by GOLD were accepted, but it is now evi-dent that the cutoff value of 0.7 for FEV1/FVC by GOLD is too high for individuals over about age 45. Accordingly, among middle-aged and older individuals, the FEV1/FVC should be interpreted in relation to the lower limit of nor-mal.102,129 This precaution is particularly applicable for patients in GOLD category 1 or 2. Among such patients, a diagnosis of COPD should be based on the overall clinical picture and not solely on spirometry.
Blood Gas Studies and Oximetry
Oximetry and arterial blood gas measurements can be used to evaluate alveolar gas exchange at rest and during exertion. However, these tests usually give normal results in patients with COPD of GOLD grade 1 or 2, so they are sel-dom warranted at the time of diagnosis for COPD of this level of severity. In contrast, checking the arterial oxygen saturation by pulse oximetry is advised for patients in grade 3 or 4 or if there are clinical signs suggestive of right heart failure because abnormalities are common and may be an indication for therapy, most notably, supplemental oxygen.1 Checking oximetry during walking or some other exercise can be helpful in deciding on the need for supplemental oxygen during physical activity and for determining the amount of oxygen required. Oximeters may not be accurate in current smokers as many oximeters do not distinguish
between carboxyhemoglobin and oxyhemoglobin, so the oxy-gen saturation displayed is higher than the true level.130 Arterial blood gas determinations are advised for patients with periph-eral oxygen saturations less than 92% or if there are clinical signs suggestive of pulmonary failure or right heart failure.1
Radiographic Studies
Radiographic abnormalities on routine chest radiographs may be minimal, even in cases of advanced COPD.131 When correlations are made between radiographic and pathologic findings in advanced COPD, the results of chest radiography suggest a diagnosis of emphysema in fewer than half of the cases, even in patients with the highest emphysema scores pathologically. It is worth noting that radiographic studies can help elucidate alternative diagnoses such as pleural disease or bronchiectasis along with the presence of signifi-cant comorbidities (eg, cardiac disease, skeletal abnormali-ties, and mediastinal pathology).
Three types of radiographic abnormalities, when paired with the appropriate clinical history, suggest the diagnosis of emphysema. The first abnormality is arterial deficiency in the lung periphery: on the chest radiograph, narrowed or absent vessels in the lung periphery are associated with hyperlucency, usually in a symmetrical, bilateral distribu-tion. The second abnormality relates to hyperinflation and is evident in the standard posteroanterior and lateral chest radiographs, which are obtained at TLC; radiographic signs of hyperinflation in these films include a low position of the diaphragm (ie, at or below the seventh rib anteriorly), increased depth of the retrosternal air space, and a narrow, vertically oriented cardiac silhouette. Perhaps the most use-ful sign of hyperinflation is a flattening of the diaphragmatic contour and loss of the diaphragm’s normal domed appear-ance, especially as visualized on the lateral film. The third abnormality is bullous disease. The presence of a bulla, together with either of the other two radiographic findings, is virtually diagnostic of emphysema, although only a small percentage of patients with emphysema have bullae.
In smokers who have a chronic cough, with or without sputum production, and whose FEV1 is normal, a chest radiograph rarely exhibits abnormal findings consistent with COPD. On occasion, the diagnosis may be suspected because of visualization of thickened bronchial walls, partic-ularly in a parahilar bronchus viewed end-on. In some patients, bronchovascular markings at the lung bases may be accentuated—a pattern that has been dubbed the dirty chest of chronic bronchitis. A similar radiographic appear-ance has been referred to as the increased-markings pattern of emphysema, especially when increased vascular mark-ings are observed in the presence of pulmonary hyperten-sion and cor pulmonale.
Computed tomography (CT) of the chest is seldom neces-sary to establish the diagnosis of COPD. Occasionally, smokers who have normal spirometry findings but a dimin-ished Dlco have emphysema that can be detected only by high-resolution CT. In patients with irreversible airways obstruction who do not have a history of smoking, high- resolution CT may be helpful by revealing bronchiectasis, small nodular densities characteristic of diffuse panbronchiol-itis, or fine infiltrates associated with interstitial lung diseases. Chest CT scans are part of the routine evaluation for lung

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 13
volume reduction surgery (LVRS) and lung transplantation. Chest CT has become a valuable research tool for quantify-ing emphysema and various dimensions of the airways, such as wall thickness.132
Differential Diagnosis
The diagnosis of COPD should be considered in every patient with a history of exposure to risk factors for COPD, characteristic clinical manifestations of COPD (see above), or both. Generally, it is not difficult to diagnose COPD when the patient has a history of long-term smoking along with typical symptoms, especially shortness of breath. The FEV1 and FEV1/FVC will usually be abnormal in this setting. However, a number of conditions that cause chronic airflow obstruction might be considered in the absence of COPD risk factors [see Table 2]. It should be noted that not everyone who devel-ops COPD has been a smoker. Estimates are that 10 to 20% of cases of COPD occur in people who have never smoked. Asthma is the most common condition with which COPD may be confused. The distinction between asthma and COPD is not always possible, and, increasingly, these two conditions are recognized as often overlapping entities. The term asthma- COPD overlap syndrome (ACOS), a unique condition, has been used to describe these patients.133,134 Conditions other than COPD that cause chronic airways obstruction can usually be differentiated from COPD on the basis of the history and other clinical features. For example, cystic fibrosis typically begins in childhood and causes large quantities of purulent sputum. Panbronchiolitis occurs in middle-aged persons of Asian ethnicity, more often men; sinusitis typically precedes the onset of pulmonary symptoms.135 Constrictive (oblitera-tive) bronchiolitis typically occurs after toxic fume exposure, respiratory infection, in the setting of a connective disuse dis-order, or following transplantation (particularly lung, heart-lung, and bone marrow transplantations).136
Treatment
medical therapy
Preventive Care
Smoking cessation The most important treatment inter-vention for current cigarette smokers with COPD is complete
smoking cessation. The Lung Health Study demonstrated that complete smoking cessation reduced the accelerated decline in FEV1 that occurs in a subset of smokers.137 The earlier the age at smoking cessation, the greater the benefit because the rate of lung function decline will be normalized at an earlier stage of COPD. Reduction in the number of cigarettes smoked, without complete cessation, has not been convincingly shown to reduce COPD risk or disease progression.138 Although cig-arette smoking is the most widely studied risk factor for COPD, it is appropriate to recommend avoidance of smoking tobacco in other forms (eg, in cigars, pipes, or electronic ciga-rettes), as well as smoking nontobacco products. A simple verbal recommendation by a physician to quit smoking does provide some benefit in smoking cessation; organized smok-ing cessation classes provide increased benefit.139 A variety of pharmacologic measures, including nicotine replacement, can facilitate smoking cessation. In the Lung Health Study, the rates of smoking cessation were significantly higher with the combination of nicotine gum and intensive individual coun-seling than with usual care.
Nicotine replacement can be provided in various forms, including patches, gum, lozenges, nasal inhalers, and oral inhalers. Nicotine replacement by nasal inhalation provides a peak nicotine level more rapidly than other approaches, but none of the available nicotine delivery devices introduce nicotine into the bloodstream as rapidly as smoking a ciga-rette.139 There are insufficient data on the safety and efficacy of electronic cigarettes to presently support their use as ther-apy for smoking cessation.26 In addition to nicotine replace-ment, other pharmacologic approaches for smoking cessation include sustained-release bupropion, which may be particu-larly useful for COPD patients who also suffer from depres-sion.140 Combination treatment with both bupropion and nicotine replacement seems to be more effective than the individual medications. Varenicline, a partial nicotinic recep-tor antagonist, diminishes the response to nicotine provided by acute cigarette smoking and assists in smoking cessa-tion. Treatment with varenicline in combination with other smoking cessation medications is not currently recom-mended. In addition to counseling and pharmacologic treat-ment, a variety of other approaches for smoking cessation have been used, including hypnosis and acupuncture. The evidence for efficacy of these alternative approaches is less compelling.141
Vaccinations Because respiratory infections often trigger COPD exacerbations, vaccinations are promising approaches to limit the development of exacerbations. Influenza vaccina-tion has been shown to reduce influenza-related respiratory illnesses in COPD patients; such illnesses are a significant cause of COPD exacerbations.142 It is therefore recommended that all patients with COPD receive annual influenza vacci-nation.143 Although the evidence that pneumococcal vaccina-tion is beneficial in COPD patients is less compelling,144,145 it is reasonable to recommend that COPD patients receive this vaccine.146 Current guidelines from the Advisory Committee on Immunization Practices recommend administering the 23-valent pneumococcal polysaccharide vaccine (PPSV23) first to all patients under the age of 65. All patients with COPD should also receive a dose of the 13-valent pneumo-coccal conjugate vaccine (PCV13) at age 65 and older.147
Table 2 Differential Diagnosis of Chronic Obstructive Pulmonary Disease
AsthmaBronchiectasis (nonspecific and associated with ciliary
dyskinesia)Cystic fibrosisConstrictive (obliterative) bronchiolitisDiffuse panbronchiolitisMycobacterial infection (tuberculous and nontuberculous)Eosinophilic granulomaLymphangioleiomyomatosisAirway tumorsTracheal stenosisTracheobronchomalaciaTracheobronchomegalyScabbard trachea

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 14
Nutritional measures Many patients with severe COPD suffer from progressive weight loss, possibly related to increased work of breathing or the systemic inflammatory effects of COPD. Nutritional counseling and support to enable weight gain in these patients are appropriate. Alter-natively, other COPD patients have problems with obesity, often related to systemic corticosteroid use and inactivity. For these patients, nutrition counseling for weight loss is rec-ommended. Overall evidence suggests that a well-balanced diet in COPD patients is beneficial in mitigating metabolic and cardiovascular risks and possibly prevents pulmonary complications.148
Long-Term Care
Bronchodilators Although COPD is defined by irrevers-ible airflow obstruction, many COPD patients experience symptomatic or spirometric improvement with broncho-dilator medications.107 Bronchodilator responsiveness can be assessed with spirometry 15 to 30 minutes after administer-ing a short-acting bronchodilator (eg, two puffs of albuterol, which delivers 180 µg). Acute bronchodilator responsiveness varies substantially from day to day, however, so COPD patients who do not show an acute improvement in FEV1 on a particular day may still benefit from bronchodilator therapy.
In addition to improvement in FEV1, bronchodilators can also provide symptomatic relief by reducing dynamic lung hyperinflation at rest and with activity.
There are two major classes of inhaled bronchodilators: beta agonists and anticholinergic agents [see Table 3]. Within each of these classes, both short-acting and long-acting for-mulations are available [see Table 3]. Although the original beta agonists (eg, terbutaline) stimulated both beta1- and beta2-adrenergic receptors, currently used beta agonists are more selective for beta2 receptors, thus reducing the side effects of tachycardia and tremulousness. Bronchodilator treatment is given primarily for symptomatic relief; how-ever, several long-acting bronchodilator medications have been shown to reduce the frequency of exacerbation and related hospitalization (see below).
Bronchodilator therapy is typically prescribed in a step-wise fashion based on disease severity [see Figure 2 and Table 1] as assessed by spirometry, symptoms, and the risk of exac-erbation. For patients with mild disease, as-needed short- acting inhaled bronchodilators are appropriate, and either albuterol or ipratropium can be used. For symptomatic patients with moderate to severe COPD, regular use of a long-acting bronchodilator should be added; this can be in the form of either an anticholinergic agent (eg, tiotropium or glycopyrronium) or a beta agonist (eg, salmeterol, indacaterol,
Table 3 Commonly Used Inhaled Bronchodilators for COPD Treatment*Category Drug Formulation Maintenance Dosage Comment
Short-acting anticholinergics
Ipratropium bromide
MDI, 17 µg/puff Nebulizer
2 puffs q. 6 hr0.5 mg q. 6 hr
Use with caution in patients with narrow-angle glaucoma or prostatic hypertrophy
Use with caution in patients with narrow-angle glaucoma or prostatic hypertrophy
Short-acting beta2 agonists
Albuterol MDI, 90 µg/puff Nebulizer
2 puffs q. 4–6 hr 2.5 mg q. 6–8 hr
——
Levalbuterol MDI, 90 µg/puff Nebulizer
2 puffs q. 4–6 hr 0.63 mg q. 6–8 hr
May have fewer systemic side effects than albuterol
May have fewer systemic side effects than albuterol
Long-acting anticholinergics
Aclidinium DPI 400 µg/capsule 1 capsule q. 12 hr Similar precautions as for ipratropium
Glycopyrronium DPI, 15.6 µg/capsule 1 capsule q. 12 hr Similar precautions as for ipratropium
Tiotropium DPI, 18 µg/capsule 1 capsule inhaled q. 24 hr Similar precautions as for ipratropium
Umeclidinium DPI, 62.5 µg/capsule 1 capsule inhaled q. 24 hr Similar precautions as for ipratropium
Long-acting beta2 agonists
Salmeterol DPI, 50 µg/inhalation 1 capsule inhaled q. 12 hr Use in combination with inhaled corticosteroid in patients with underlying asthma
Formoterol DPI, 12 µg/capsuleNebulizer
1 capsule inhaled q. 12 hr20 µg q. 12 hr
Similar precautions as for salmeterolSimilar precautions as for salmeterol
Arformoterol Nebulizer 15 µg q. 12 hr Similar precautions as for salmeterol
Indacaterol DPI, 75 µg/capsule 1–4 capsules inhaled q. 24 hr
Similar precautions as for salmeterol
Olodaterol SMI, 50 µg/inhalation 5 µg q. 24 hr Similar precautions as for salmeterol
COPD = chronic obstructive pulmonary disease; DPI = dry-powder inhaler; MDI = metered-dose inhaler; SMI = soft mist inhaler. *Not all formulations are available in all countries; in some countries, other formulations may be available.

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 15
or formoterol). Patients with very severe COPD are typically treated with both long-acting anticholinergic agents and long-acting beta agonists. Such patients should also have short-acting bronchodilators available for as-needed rescue use. Many combination inhalers containing both long-acting beta agonists and long-acting anticholinergics are now avail-able [see Table 4].
Meta-analyses have demonstrated that long-acting formu-lations of anticholinergic agents and of beta agonists can reduce the frequency of COPD exacerbations.149,150 The Under-standing Potential Long-Term Impacts on Function with Tiotropium (UPLIFT) trial, a randomized, double-blind study comparing tiotropium with placebo in COPD patients over 4 years of follow-up, demonstrated a reduction in exacerba-tion frequency, increased FEV1 level, and improved health- related quality of life with tiotropium treatment; however, the rates of FEV1 decline and mortality were not significantly reduced by tiotropium.151 In a randomized, double-blind trial of 7,376 COPD subjects (Prevention of Exacerbations with Tiotropium in COPD [POET-COPD]), tiotropium was shown to reduce exacerbation frequency more effectively than sal-meterol; however, exacerbation frequency was low in both groups.152 A more recent trial that compared indacaterol and tiotropium and followed patients for 1 year similarly demon-strated a small but significant decrease in exacerbations in the tiotropium group compared with the indacaterol group.153 These results suggest that long-acting anticholinergic agents may be a better initial treatment option in moderate to severe COPD than long-acting beta agonists.
Multiple trials have demonstrated that long-acting bron-chodilators, used in combination with inhaled corticosteroids, can reduce the frequency of COPD exacerbations and may improve respiratory function. A randomized, double-blind, parallel-group study demonstrated that the addition of inhaled steroids to long-acting beta agonists provided a further reduc-tion in exacerbation frequency.154 Another large, randomized, double-blind trial, the TOwards a Revolution in COPD Health (TORCH) study, demonstrated that the combination of a long-acting beta agonist and inhaled steroid led to a reduction in the fre quency of COPD exacerbations, an increase in FEV1, and improved health-related quality of life compared with
placebo but did not conclusively demonstrate a reduction in mortality.155 The FLAME trial (Indacaterol–Glycopyrronium versus Salmeterol–Fluticasone for COPD), a recent multi-center, randomized, double-blind trial that followed patients for 1 year, compared indacaterol-glycopyrronium and salmeterol-fluticasone.156 It found that in patients with a his-tory of exacerbation during the previous year, indacaterol- glycopyrronium was more effective than salmeterol-fluticasone in preventing COPD exacerbations. These data suggest that it may be beneficial to initially treat with both a long-acting anticholinergic agent and a long-acting beta agonist before adding an inhaled steroid (see below for further discussion of inhaled corticosteroids).
Bronchodilator medications may be delivered by four types of devices: metered-dose inhalers (MDIs), dry-powder inhalers (DPIs), soft mist inhalers (SMIs), and nebulizers. A systematic review of randomized clinical trials found that although there is a relative paucity of data regarding bron-chodilator delivery devices in COPD patients, the efficacy of MDIs, DPIs, SMIs, and nebulizers appears to be similar.157,158 Therefore, clinicians should select a delivery device on the basis of the patient’s ability to use the device properly, the availability of devices for the preferred drug, the cost, and patient and physician preference. SMIs, which function by forcing a metered dose of drug solution via a soft mist gen-erated by the convergence of two fine jets of liquid, are sometimes useful in patients who have difficulty operating MDIs or DPIs. Nebulizers, which use either compressed air (jet nebulizers) or ultrasonic aerosolization, are useful in patients who have difficulty operating MDIs, DPIs, or SMIs but are often bulky and are not easily portable. Systemic absorption of beta2 agonists such as albuterol is greater with nebulized delivery than with MDI delivery, which can lead to greater increases in heart rate.156 MDIs and SMIs are not breath actuated and thus require excellent coordination for proper dose administration, whereas DPIs are typically breath actuated. If a spacer device is used with an MDI, the timing of inhalation is less important.
DPIs come in several forms; some require the patient to insert a capsule containing the dry powder into the device, whereas others prepare the dose automatically. All four
Table 4 Commonly Used Combination Inhalers for COPD Treatment*Category Drug Formulation Maintenance Dosage
Short-acting beta2 agonist plus anticholinergic
Albuterol/ipratropium Nebulizer 2.5 mg/0.5 mg 1 vial every 6 hr
Long-acting beta2 agonist plus anticholinergic
Indacaterol/glycopyrronium DPI 27.5 µg/15.6 µg 1–2 capsules inhaled twice daily
Olodaterol/tiotropium SMI 2.5 µg/2.5 µg 2 inhalations once daily
Vilanterol/umeclidinium DPI 25 µg/62.5 µg 1 inhalation once daily
Long-acting beta2 agonist plus corticosteroids
Formoterol/budesonide MDI 4.5 µg/160 µg 2 inhalations twice daily
Formoterol/mometasone↑ MDI 100 µg/5 µg 1–2 inhalations twice daily
Salmeterol/fluticasone DPI 50 µg/250 µg 1 inhalation twice daily
Vilanterol/fluticasone DPI 25 µg/100 µg 1 inhalation once daily
COPD = chronic obstructive pulmonary disease; DPI = dry-powder inhaler; MDI = metered-dose inhaler; SMI = soft mist inhaler. *Not all formulations are available in all countries; in some countries, other formulations may be available. ↑Not Food and Drug Administration approved for the treatment of COPD.

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 16
types of devices can be used in spontaneously breathing patients; DPIs cannot be used in intubated patients. There is no evidence that nebulizers are of greater benefit than prop-erly used DPIs, MDIs, or SMIs. Oral bronchodilator formu-lations (eg, tablets) of beta-agonist medications are available but tend to have greater side effects than inhaled formula-tions and should generally be avoided.
Inhaled corticosteroids Because inflammation is a central feature in the definition of COPD, inhaled corticosteroids have been extensively investigated in COPD. Inhaled steroids appear to have a beneficial effect in reducing the frequency of COPD exacerbations, especially in patients with severe COPD, and in reducing the decline in quality of life.159,160 Sev-eral large studies have investigated the hypothesis that inhaled corticosteroids reduce the rate of FEV1 decline in COPD patients.161,162 The studies suggest that corticosteroids may slow the rate of FEV1 decline, but long-term data are limited, and this remains controversial.159,163 The use of inhaled corticosteroids is not associated with improved mortality.164,165
Current treatment guidelines recommend inhaled steroids in combination with long-acting bronchodilators for COPD patients at high risk for exacerbations [see Figure 2 and Table 1].1 A number of combination long-acting bronchodilator and corticosteroids inhalers are now available [see Table 4]. Side effects are substantially less common with inhaled corticoste-roids than with systemic corticosteroids; however, the use of inhaled corticosteroids is associated with the development of oral candidiasis and hoarse voice and may be associated with an increase in the risk of cataracts and osteopenia. Although inhaled steroids appear to reduce COPD exacerbation rates, they are paradoxically associated with an increased rate of pneumonia, although inhaled steroids have not been associ-ated with increased mortality.165 It is worth noting that previ-ous data suggested that withdrawal from treatment with inhaled steroids may lead to exacerbations,166 but a more recent study demonstrated that steroids could be gradually withdrawn over a 12-week period without increasing the risk of exacerbations.163
Some COPD patients have features of asthma, including attacks of wheezing and sputum eosinophilia. Whether these patients have both asthma and COPD or ACOS is uncertain.133 It is reasonable to offer such patients a thera-peutic trial of inhaled steroids because inhaled steroids are effective treatments for asthma; in addition, one study found that treatment with an inhaled steroid led to a reduction in sputum eosinophilia and an improvement in FEV1.167
Oral corticosteroids Some COPD patients experience a modest improvement in FEV1 with a 2-week course of high-dose oral steroids. Long-term treatment with oral steroids has no proven benefit, however, and frequently results in significant adverse events.168 Consequently, oral corticoste-roid therapy should be avoided in patients with COPD except in the setting of acute exacerbations (see below).
Methylxanthines Although inhaled bronchodilators and inhaled steroids are the mainstays of pharmacologic therapy for COPD, theophylline, the most commonly used methylx-anthine, may be appropriate in selected patients.169 Theoph-ylline has traditionally been prescribed for its bronchodilator
properties, but this agent also has antiinflammatory effects; proposed mechanisms for its antiinflammatory action include its ability to activate histone deacetylation.170 Treatment with theophylline requires monitoring of serum levels. Although the standard therapeutic range is 10 to 20 mg/dL, symptom-atic benefit may be seen at serum levels below this range; these so-called subtherapeutic levels are associated with a reduction in the risk of toxic effects, most notably supraven-tricular arrhythmias. In patients with very severe COPD who remain symptomatic despite maximal inhaled bronchodilator and inhaled steroid treatment, a trial of theophylline is a rea-sonable consideration.169
Phosphodiesterase-4 inhibitors In 2011, roflumilast, a phosphodiesterase-4 inhibitor, was approved by the Food and Drug Administration (FDA) as a treatment to reduce COPD exacerbation frequency. Fabbri and colleagues reported two double-blind studies that compared adding roflumilast or placebo with either salmeterol (first study) or tiotropium (second study), with prebronchodilator FEV1 as the primary outcome.171 FEV1 improved significantly with roflumilast treatment in both studies; nausea, diarrhea, and weight loss were common side effects. Calverley and colleagues assessed the impact of roflumilast on the development of COPD exac-erbations requiring systemic steroid treatment in two trials of severe COPD subjects.172 Higher FEV1 values and lower exac-erbation rates were seen with roflumilast in both studies. Roflumilast appears to provide a therapeutic option for patients with persistently high exacerbation frequency despite treatment with long-acting anticholinergics, long-acting beta agonists, and inhaled steroid medications.173 Roflumilast should always be used in combination with at least one long-acting bronchodilator.1
Antibiotics Multiple studies have shown that continu-ous prophylactic macrolide use can reduce the frequency of COPD exacerbations. Concern remains, however, about the potential long-term side effects, including increased antibi-otic resistance.174–176 Current guidelines do not recommend the routine use of prophylactic antibiotic therapy in patients with COPD,1 although these may be of benefit in select patients who continue to have frequent exacerbations despite optimal medical therapy.
Vasodilators Pulmonary hypertension associated with COPD is associated with a worse prognosis.177 Despite this, both inhaled and systemic vasodilators have not shown bene-fit, likely because they can worsen V/Q mismatching and gas exchange.177–179 The routine use of vasodilators in pulmonary hypertension associated with COPD is not recommended.
Other medications Antitussives can be helpful for reliev-ing acute symptoms but are not recommended in the man-agement of stable COPD.1 Leukotriene antagonists, which are of benefit in some asthma patients, are of uncertain ben-efit in patients with COPD, and their routine use is not cur-rently recommended. Mucolytics (eg, mucokinetic, mucoregulator) have been evaluated in a number of studies and may produce a small reduction in acute execrations and a small effect on overall quality of life in some patients with COPD.180 The data on antioxidant agents (eg, ambroxol,

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 17
erdosteine, carbocysteine, iodinated glycerol, N-acetylcysteine) remain controversial, and their widespread use is not rec-ommended.181,182 There are no data to support the use of anti–tumor necrosis factor–α antibody (infliximab) in COPD; these agents may be harmful in COPD patients.183 Vitamin D is of uncertain benefit in patients with COPD; there is evi-dence that in patients with severe vitamin D deficiency at baseline, supplementation may reduce exacerbations.184 AAT augmentation therapy is not recommended for use in COPD that is unrelated to AAT deficiency [see AAT Deficiency, below].
Oxygen During COPD exacerbations, hypoxemia often worsens. Short-term supplemental oxygen therapy may be required during and immediately after such exacerbations. In severely hypoxemic COPD patients, long-term supple-mental oxygen therapy reduces mortality.185 In addition, supplemental oxygen can largely prevent the morbidity associated with chronic hypoxemia, which includes second-ary erythrocytosis, exercise limitation, pulmonary hyperten-sion, and impaired neuropsychiatric function.186,187
In general, COPD patients should receive long-term oxy-gen therapy if their resting Pao2 is 55 mm Hg or lower or if the peripheral oxygen saturation (Sao2) is below 88%. A rest-ing Pao2 threshold of 59 mm Hg or Sao2 of 88% is appropriate in patients with peripheral edema (a sign of cor pulmonale), hematocrit above 55%, or pulmonary hypertension. For patients who meet these criteria, supplemental oxygen used for at least 15 hours a day is beneficial; however, the reduction in mortality is greater if oxygen is used for 24 hours a day. Sup-plemental oxygen is administered at rates sufficient to main-tain the arterial oxygen saturation at 90% or higher. The rate of oxygen administration should be titrated to the individual patient’s needs as determined by pulse oximetry or arte-rial blood gas analysis. Supplemental oxygen requirements increase when such patients travel to high altitudes or by airplane.
The primary goal of supplemental oxygen therapy is to avoid hypoxemia. Supplemental oxygen can increase the retention of CO2, but maintaining oxygen saturation at slightly above 90% will minimize the risk of worsening hypercarbia.
The benefits of supplemental oxygen for patients with resting hypoxemia are clear. For patients with hypoxemia during exercise or with sleep, the benefits of supplemental oxygen are not well established185; however, supplemental oxygen is often considered for individuals whose oxygen saturation falls to 88% or less on ambulation or when sleep-ing at night. In addition, patients who experience moderate hypoxemia at sea level during rest may develop severe hypoxemia during air travel or at high altitude. Their need for supplemental oxygen can be assessed through the use of prediction equations.188 Testing patients with inhalation of hypoxic gas mixtures (15% oxygen) is theoretically ideal but impractical at most medical centers.
Oxygen for home use can be generated from ambient air using oxygen concentrators. Alternative sources of oxygen include cylinders of compressed gas or liquid oxygen. For freedom of movement, portable liquid oxygen tanks can pro-vide supplemental oxygen for several hours outside the home. Portable oxygen concentrators have also been developed and
are especially useful for air travel. The use of devices that release oxygen only during the inspiratory phase of the respiratory cycle conserves the oxygen and prolongs the time for which a particular tank can be used. Direct transtracheal administration can be considered, especially for patients requiring high supplemental oxygen concentrations; how-ever, clogging of these narrow catheters can be problematic.
Rehabilitation
Physical training programs, which often include treadmill walking and light weight lifting, significantly increase the exercise capacity of patients with severe COPD. Formal pul-monary rehabilitation programs contain an educational and a training component. Although exercise does not change spirometric values, it can lead to significant improvements in respiratory symptoms and functional capacity, as well as a reduction in the use of medical resources, including hospital-izations and emergency department visits.189 Whether there is additional, clinically important benefit from exercises that focus specifically on the inspiratory muscles remains unclear; however, a randomized controlled trial found that training with inspiratory pressure support significantly increased exercise capacity.190 In addition to general COPD manage-ment, pulmonary rehabilitation is routinely recommended before considering surgical options for patients with COPD, such as LVRS or lung transplantation.
Referral for pulmonary rehabilitation should be considered on the basis of respiratory symptoms and functional limita-tions rather than a specific FEV1 threshold. A randomized trial demonstrated that pulmonary rehabilitation following hospital admission for a COPD exacerbation led to lower rates of hospital readmission for an exacerbation over a 3-month period.191 Although the optimal duration of struc-tured pulmonary rehabilitation programs has not been deter-mined, it has been recommended that such programs should continue for at least 8 weeks.192 Subsequently, to avoid decon-ditioning, patients should continue to exercise; this may be done either in a monitored maintenance program or without direct supervision but with clearly defined recommendations for exercise. As with other forms of physical training, decon-ditioning occurs within a few weeks of cessation of the exer-cise program. Participation in maintenance rehabilitation programs or continuance of a defined set of exercises after leaving the program is essential to avoid deconditioning.
Exacerbations
Acute exacerbations of COPD typically involve increased dyspnea and cough, often with purulent sputum. Additiona l signs and symptoms can include wheezing, fatigue, fever, tachypnea, cyanosis, and mental status changes. Although respiratory infections are the most common trigger for COPD exacerbations, other triggers can also be involved [see Pathophysiology, Exacerbations, above]. Some complications of COPD, such as pneumothorax, can mimic COPD exacer-bations and need to be excluded. Distinctions between COPD exacerbations and other disease processes are more semantic. For example, some investigators classify pneumonia that is identified radiographically as distinct from COPD exacerba-tions. Similarly, pulmonary embolism may be viewed as a trigger for an exacerbation or as an alternative process other than an exacerbation. From a practical standpoint, it is

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 18
important to consider whether pneumothorax, pneumonia, congestive heart failure, cardiac arrhythmia, pulmonary embolism, or drug toxicity is present whenever a COPD patient develops worsening respiratory symptoms.
The most important initial therapeutic decision in exacer-bation management is whether emergent evaluation and possible hospitalization are required. Criteria for hospital admission include mental status changes, markedly increased shortness of breath, worsening hypoxemia or hypercarbia, and inadequate response to an intensified outpatient medical regimen. The more severe the COPD, the more likely the patient is to require emergent evaluation, especially if he or she has a history of respiratory failure requiring mechanical ventilation. Other factors to be considered regarding emer-gent evaluation and hospitalization include comorbid dis-eases, the availability of caregivers, and the distance to the medical center. For COPD patients without very severe exac-erbations, nurse-administered homecare programs may offer an alternative to hospitalization.1
Antibiotics Exacerbations are often triggered by respira-tory infections, which may be bacterial, viral, or both. In patients with a COPD exacerbation marked by increased dys-pnea, increased sputum production, and discolored sputum, a course of oral antibiotics is of proven benefit in reducing the duration of symptoms and risk of death.193 With advancing age, increased severity of disease, and comorbid conditions, the pathogens in COPD exacerbations are more likely to be resistant to antibiotics. Risk factors for resistance in bacterial pathogens include FEV1 of less than 50% of the predicted value, more than four exacerbations per year, and comorbid conditions such as cardiac disease.194 Patients without these risk factors can be treated with a second-generation macrolide, a second- or third-generation cephalosporin, ampicillin, doxy-cycline, or trimethoprim-sulfamethoxazole. In patients with any of the specified risk factors, many of the isolates of H. influ-enzae, M. catarrhalis, and S. pneumoniae are β-lactam resistant. In addition, organisms such as Klebsiella and Pseudomonas spe-cies become more common. For these patients, a quinolone or a β-lactam–β-lactamase inhibitor combination is recommended as first-line therapy. The local antibiotic resistance pattern should be considered in antibiotic selection for COPD exacer-bations. In addition, previous antibiotic treatment should be reviewed because exposure to a particular antibiotic class can increase the likelihood of antibiotic resistance.
Systemic corticosteroids A 2009 meta-analysis demon-strated that treatment with systemic corticosteroids (oral or parenteral) during COPD exacerbations led to fewer treat-ment failures, reduced duration of hospitalization, increased FEV1 values, and reduced dyspnea.195 However, mortality was not decreased. The Reduction in the Use of Corticoste-roids in Exacerbated COPD (REDUCE) trial, a recent multi-center, randomized, noninferiority trial, demonstrated that in patients presenting to the emergency department, 5-day treatment with systemic glucocorticoids was noninferior to a 14-day course with regard to reexacerbation within the subsequent 6 months.196 Current data support the contention that a 5-day course of oral corticosteroids, typically predni-sone 40 mg daily or an equivalent methylprednisolone dose, should be sufficient for treatment of most adults with acute
exacerbations of COPD.168 Although long-term use of sys-temic steroids increases the risk of infections, the short-term use of these agents for COPD exacerbations—many of which are triggered by infections—is reasonable as long as appro-priate antibiotic treatment is provided when bacterial infec-tion is suspected or in the setting of severe exacerbation.
Bronchodilators In acute exacerbations, both short-acting beta2 agonists (eg, albuterol) and short-acting anticholiner-gics (eg, ipratropium) are typically given. The initial dose of albuterol is four puffs every 4 hours by MDI or 2.5 mg every 4 hours by nebulizer. For patients who do not improve, the dosing interval can be shortened to every 2 hours. Ipratro-pium is given in a dosage of three or four puffs every 6 hours or, for severe exacerbations, five to eight puffs every 3 to 4 hours; alternatively, nebulized ipratropium can be admin-istered, often in combination with albuterol.
Delivery of short-acting bronchodilators by MDI and nebulizer should be equivalent, although greater systemic absorption occurs with nebulized treatment. Nebulized treat-ment is preferred for patients with extreme dyspnea or altered consciousness. There are no data suggesting that the addition of intravenous or oral theophylline to inhaled bronchodilator therapy is beneficial for acute COPD exacerbations. If a patient is already using theophylline, it can be continued. Monitoring of serum levels is necessary, however, because acute illness and other medications can influence theophyl-line levels and potentially precipitate theophylline toxicity.
Oxygen and mechanical ventilation COPD exacerba-tions are often associated with worsening of hypoxemia and hypercapnia. Oxygen supplementation should be provided to maintain oxygen saturation in the range of 88 to 92% to reduce the risk of worsening V/Q mismatching and hypercapnia while providing adequate tissue oxygenation. In patients with acute or acute-on-chronic respiratory acidosis, mechani-cal ventilation should be considered. Noninvasive positive pressure ventilation (NPPV) has been shown to reduce com-plications and mortality for patients with COPD compared with endotracheal intubation.1 NPPV can be given with either a nasal mask or a full face mask; the full face mask increases the risk of aspiration and limits com munication. Close atten-tion to patient comfort during the initiation of NPPV is asso-ciated with a higher success rate. Reasons for proceeding directly to endotracheal intubation include respiratory arrest, cardiovascular complications (eg, hypotension), difficulty in the management of respiratory secretions, and an inability to cooperate (eg, because of somnolence or agitation). If patients cannot tolerate NPPV or fail to improve, endotracheal intuba-tion and mechanical ventilation should be provided. When providing ventilatory support, care must be given to avoid auto-PEEP (ie, intrinsic positive end-expiratory pressure), which can result from inadequate exhalation times in patients with COPD. Extracorporeal carbon dioxide removal has been proposed for hypercapnic respiratory failure in COPD exac-erbations, but there are insufficient data to currently support its use as part of routine care.197
AAT Deficiency
In addition to receiving standard COPD treatment, patients with severe AAT deficiency (PI Z, PI Null-Null) should also

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 19
be considered for intravenous infusions of AAT (so-called augmentation therapy). AAT augmentation therapy is not indicated in PI MZ individuals, and the role of augmentation therapy in PI SZ subjects is controversial.38 The FDA has approved four commercial formulations of AAT augmenta-tion therapy; all are partially purified plasma preparations that are enriched for AAT. AAT augmentation therapy was approved by the FDA in the late 1980s on the basis of bio-chemical efficacy (ie, the demonstration of increased serum AAT levels and increased elastase inhibitory capacity in bron-choalveolar lavage fluid). Several observational studies and one recent multicenter, double-blind, randomized, paral-lel-group, placebo-controlled trial (Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency [RAPID]) have suggested that this therapy may reduce the rate of lung function decline in AAT-deficient patients who have moderate degrees of airflow obstruction.198–201 Because some AAT-deficient individuals will likely never develop COPD, augmentation therapy is not recommended for patients with normal lung function who show no radiographic evi-dence of emphysema. AAT-deficient patients with normal lung function but for whom there is radiographic evidence of emphysema represent a problematic group; individualized treatment decisions are required.
The FDA-approved regimen for augmentation therapy is a weekly intravenous infusion of 60 mg/kg; however, biweekly and monthly infusion regimens have been used in some cases, and other research of other regimens is ongoing.202 Augmentation therapy is generally well tolerated, with min-imal side effects, although there is a theoretical risk of ana-phylaxis in individuals with IgA deficiency. Peripheral intravenous access is typically used for as long as possible before considering permanent intravenous access (eg, an implanted infusion port). The processing of augmentation medications appears to destroy HIV and hepatitis viruses, and hepatitis B vaccination is no longer required before ini-tiating augmentation therapy. However, hepatitis A and B vaccinations are recommended for PI Z individuals as part of routine care. The possible transmission of Creutzfeldt-Jakob disease or undiscovered blood-borne pathogens through AAT augmentation therapy remains a concern, but no cases have as yet been reported.203
bronchoscopic treatment
Various bronchoscopic approaches to volume reduction are in development, including the use of valves and coils.204 Such approaches could provide simpler volume reduction procedures, with preliminary data suggesting potential improved exercise capacity. Further studies are needed to assess the role of these novel bronchoscopic approaches in the management of patients with advanced COPD and emphysema. These therapies are not currently available out-side of clinical studies.204–208
surgical treatment
Two surgical options are available for patients with very severe COPD: LVRS and lung transplantation. LVRS involves removal of the most severely affected emphysematous regions of the lung, which may be compressing less affected regions. After this operation was rediscovered by Cooper in the 1990s, it rapidly became widespread, with variable
results. The National Emphysema Treatment Trial (NETT) was a randomized controlled trial in which more than 1,200 severe emphysema patients were randomized to medical or surgical treatment.209 Both groups underwent pulmonary rehabilitation before randomization. Overall, LVRS did not have a significant effect on mortality, although exercise capacity did improve. The risk of mortality from LVRS was found to be significantly higher in patients who had an FEV1 of less than 20% of the predicted value and either a homo-geneous distribution of emphysema on a chest CT scan or a Dlco of less than 20% of the predicted value. In contrast, reduced mortality and an improvement in functional param-eters were seen in patients with upper lobe–predominant emphysema and low baseline exercise capacity (below 25 watts for women and below 40 watts for men on standard-ized exercise testing). Patients with upper lobe emphysema but with high exercise capacity had improved pulmonary function and quality of life but no improvement in survival. LVRS provided no compelling benefits for patients with non–upper lobe–predominant emphysema, although the subset of such patients who had low exercise capacity did report improved quality of life.
The operative mortality from LVRS is approximately 5%, but serious complications, including respiratory failure, pro-longed air leak, pneumonia, and cardiac morbidity, are much more common.210 Additionally, the benefits from LVRS diminish with time; after 5 years, only a small percentage of patients still have significantly improved pulmonary func-tion.211,212 The role of repeat LVRS after functional benefits have been lost is unclear. Because of the temporary benefit from LVRS, it can be used as a bridge to lung transplantation in young patients who have emphysema in a favorable ana-tomic distribution.211,213
Some patients have isolated large bullous lesions. If such patients have significant respiratory symptoms and func-tional impairment, surgical resection of these bullous lesions can lead to substantial improvement. Factors associated with greater functional improvement after bullectomy include bullae occupying at least one third of the hemithorax and a reduction in FEV1 below 50% of predicted.214 If the remaining lung parenchyma is relatively normal, these improvements can be long-standing. If there is substantial remaining emphy-sema, bullous lesions can recur.
In younger patients with very severe COPD (ie, an FEV1 of < 25% predicted), lung transplantation should be consid-ered.215 COPD patients with BODE scores of 7 or greater (see below) and other complications (eg, previous hospitalization with hypercapnia during a COPD exacerbation or pulmonary hypertension) can be considered for lung transplantation even if their FEV1 levels are less profoundly reduced.216 Despite concerns about overdistention of the remaining native lung, unilateral lung transplantation has been success-fully performed in patients with severe emphysema. In rare cases, lung volume reduction of the remaining native lung can be performed if overdistention does occur. In COPD patients, bilateral lung transplantation has been shown to increase survival compared with unilateral lung transplanta-tion in patients up to age 60 years,217 but the role of bilateral versus unilateral lung transplantation in COPD remains con-troversial. Even if patients have severe right heart dysfunc-tion, heart-lung transplantation is typically not required,

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 20
because the right ventricle recovers quickly after lung trans-plantation. Complications after lung transplantation include an increased risk of infections, malignancy, and a high rate of rejection (bronchiolitis obliterans); 5-year survival after lung transplantation is approximately 50%.218 Lung transplantation for COPD has not been proved to increase survival compared with continued medical therapy,219 but survivors may experi-ence improved quality of life. Patients with a history of COPD may be more susceptible to the development of lung cancer.220
Although priority for lung transplantation was previously determined by time spent on a waiting list, the guidelines for lung allocation by the United Network for Organ Shar-ing (UNOS) changed in 2005. Currently, a lung allocation score is calculated on the basis of the urgency for and likely benefit from lung transplantation, as assessed from the patient’s disease diagnosis, laboratory results, and other test results (https://optn.transplant.hrsa.gov/learn/professional- education/lung-allocation-system/). This lung allocation score, rather than waiting time, is used to prioritize patients for available organs.
Complications
copd as a systemic disease
There is increasing evidence that the inflammatory process in COPD has a systemic component: elevated levels of C- reactive protein have been observed in COPD patients.221 In addition, a variety of nonpulmonary illnesses are more com-mon in COPD patients, including coronary artery disease, hypertension, hyperlipidemia, diabetes, osteoporosis, and skeletal muscle dysfunction.222,223 There is also some evidence to suggest that COPD is associated with increased rates of mild cognitive impairment.224 It is difficult to separate the effects of cigarette smoking and reduced physical activity on these disease processes from the possible systemic impact of COPD related to inflammation and tissue hypoxia, but increased surveillance for these comorbid conditions is appropriate as they are likely to negatively impact survival.222 The marked weight loss that occurs in a subset of severe COPD patients, primarily from loss of skeletal muscle mass, may represent a manifestation of these systemic effects.
pulmonary hypertension
In a minority of COPD patients, secondary pulmonary hypertension develops. The elevations of pulmonary artery pressure in COPD tend to be mild to moderate and slowly progressive.225 However, the presence of pulmonary hyper-tension substantially increases the risk of mortality in COPD patients.226 Pulmonary hypertension can lead to the develop-ment of cor pulmonale, characterized by hypertrophy of the right ventricle and, ultimately, right ventricular failure, with associated peripheral edema and passive hepatic conges-tion. Cor pulmonale typically develops in advanced COPD; however, a population-based analysis in the Multi-Ethnic Study of Atherosclerosis (MESA) cohort demonstrated that both airflow obstruction assessed by spirometry and emphy-sema assessed quantitatively by chest CT scan were associ-ated with reductions in left ventricular stroke volume, left ventricular end-diastolic volume, and cardiac output in subjects ranging from normal lung function to moderate
COPD.227 Although pulmonary hypertension in COPD was originally believed to result purely from arterial hypertrophy in response to chronic hypoxic vasoconstriction, more recently, other mechanisms have been postulated, including inflamma-tion and direct effects of cigarette smoke components.226
Doppler echocardiography can be used to assess right ventricular function and to estimate pulmonary artery sys-tolic pressures noninvasively from a jet of tricuspid regurgi-tation. Unfortunately, estimates of pulmonary artery systolic pressure cannot be obtained with echocardiography in a substantial percentage of COPD subjects. Furthermore, in patients with advanced lung disease, echocardiographic estimates of pulmonary artery systolic pressure often indi-cate pulmonary hypertension when the patient has normal pulmonary artery pressures.228 Although right heart cathe-terization can provide more accurate assessment of pulmo-nary hypertension, it is invasive and thus is typically performed only in special circumstances, such as during evaluation for lung transplantation.
With appropriate supplemental oxygen therapy, pulmo-nary hypertension and cor pulmonale are substantially less likely to develop. However, oxygen therapy does not appear to reverse preexisting pulmonary hypertension.229 Systemic vasodilators, which are useful in the treatment of primary pulmonary arterial hypertension, may improve pulmo-nary hemodynamics in COPD but can also worsen arterial oxygenation by inhibiting hypoxic pulmonary vasoconstric-tion.230 Thus, systemic vasodilator treatment is not currently recommended for secondary pulmonary hypertension related to COPD [see Vasodilators, above].
lung cancer
Patients with COPD have a high risk of lung cancer. Whether this risk is simply related to the fact that many COPD patients have a history of cigarette smoking or reflects another shared determinant of COPD and lung cancer is currently unclear. The National Lung Screening Trial (NLST), a recent large, ran-domized trial, demonstrated that in high-risk patients, low-dose CT, compared with chest radiography, reduced both death from lung cancer and all-cause mortality.231 Currently, guidelines recommend offering low-dose CT screening to high-risk patients with significant tobacco histories, although providers should discuss the pros and cons of these tests, including a high risk of false-positive testing.232 Controversy remains over the cost-effectiveness of low-dose CT screening for lung cancer; risk-based approaches to screening have been proposed.233,234 Active smokers should be advised that screen-ing is not a substitute to tobacco cessation. Current studies are ongoing to develop biomarker sputum tests for early detection of lung cancer, but these are in the early stages of testing and not available for routine clinical use.
pneumothorax
COPD patients are at increased risk for pneumothorax.235 In patients with severe emphysema (especially bullous dis-ease), distinguishing between a bulla and pneumothorax radiographically can require careful examination. Pneumo-thoraces can be especially problematic in patients with severe COPD because of the reduced respiratory reserve in these patients. Tube thoracostomy is typically required to treat a spontaneous pneumothorax in a COPD patient.

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 21
reduce mortality in COPD are smoking cessation, oxygen therapy in severely hypoxemic patients, and LVRS in patients with upper lobe–predominant emphysema and low exercise capacity.
Financial Disclosures: George Washko, MD reports receiving consulting fees from GlaxoSmithKline, Boehringer Ingelheim, PulmonX, Genentech, and Janssen. Dr. Washko is a founding member of a consulting company focused on outcomes research in clinical trials. Dr. Washko’s spouse is an employee of Biogen. Andrew J. Schissler, MD, and Carolyn E. Come, MD, MPH, have no relevant financial relationships to disclose.
REFERENCE KEY
Review Clinical Trial Meta-analysis Guideline
References
1. Global Strategy for Diagnosis, Management, and Prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Available at: http://www.goldcopd.org (accessed October 5, 2016).
2. Hardie JA, Buist AS, Vollmer WM, et al. Risk of over- diagnosis of COPD in asymptomatic elderly never-smokers. Eur Respir J 2002;20:1117–22.
3. Jones PW. COPD Assessment Test–rationale, develop-ment, validation and performance. COPD 2013;10:269–71.
4. Gupta N, Pinto LM, Morogan A, Bourbeau J. The COPD assessment test: a systematic review. Eur Respir J 2014;44: 873–84.
5. Jones PW, Tabberer M, Chen WH. Creating scenarios of the impact of COPD and their relationship to COPD assessment test (CAT™) scores. BMC Pulm Med 2011;11:42.
6. Sterk PJ. Let’s not forget: the GOLD criteria for COPD are based on post-bronchodilator FEV1. Eur Respir J 2004;23: 497–8.
7. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exac-erbation in chronic obstructive pulmonary disease. N Engl J Med 2010;363:1128–38.
8. Kochanek KD, Xu J, Murphy SL, et al. Deaths: prelimi-nary data for 2008. Natl Vital Stat Rep 2011;59(10): 1–126.
9. Mannino DM. COPD: epidemiology, prevalence, morbid-ity and mortality, and disease heterogeneity. Chest 2002; 121:121S–6S.
10. Tilert T, Dillon C, Paulose-Ram R, et al. Estimating the U.S. prevalence of chronic obstructive pulmonary disease using pre- and post-bronchodilator spirometry: the National Health and Nutrition Examination Survey (NHANES) 2007–2010. Respir Res 2013;14:103.
11. Gershon AS, Warner L, Cascagnette P, et al. Lifetime risk of developing chronic obstructive pulmonary disease: a longitudinal population study. Lancet 2011;378:991–6.
12. Murray CJ, Lopez AD. Measuring the global burden of disease. N Engl J Med 2013;369:448–57.
13. Ford ES, Croft JB, Mannino DM, et al. COPD surveil-lance—United States, 1999–2011. Chest 2013;144:284–305.
14. Aryal S, Diaz-Guzman E, Mannino DM. COPD and gender differences: an update. Transl Res 2013;162:208–18.
15. Silverman EK, Weiss ST, Drazen JM, et al. Gender-related differences in severe, early-onset chronic obstructive pulmo-nary disease. Am J Respir Crit Care Med 2000;162:2152–8.
supraventricular arrhythmias
COPD patients are at increased risk for supraventricular arrhythmias. Some of this increased risk relates to medica-tion use. For example, theophylline use is associated with an increased risk of supraventricular arrhythmias, including multifocal atrial tachycardia, especially when drug levels exceed the relatively narrow therapeutic window. Beta2 ago-nists are not completely selective for beta2-adrenergic recep-tors; consequently, these agents can cause tachycardia and, in rare cases, cardiac arrhythmias.
anxiety and depression
The psychosocial impact of COPD can be devastating. Exercise limitation and fear of contracting respiratory infec-tions can be socially isolating. Diminished functional capac-ity and increased dependence on caregivers can be quite frustrating for patients. The ECLIPSE study found that 26% of COPD subjects met the criteria for depression, and depres-sion was more common with greater severity of airflow obstruction.236 A Veterans Affairs hospital survey in Texas found a very high prevalence of anxiety disorder, depres-sion, or both in outpatients with COPD, and only a minority of affected patients were receiving treatment for these psy-chiatric disorders.237 A large, multicenter, prospective trial of patients with COPD found that anxiety was prevalent in this population and had a strong association with increased risk of death.222 Anxiety often develops in response to dyspnea. Careful titration of benzodiazepines can alleviate these symp-toms, although caution must be exercised to avoid suppress-ing the respiratory drive. Depression is a common complication of COPD and is often undiagnosed. COPD patients should be screened for depression, and antidepressant therapy should be strongly considered when appropriate.238
Prognosis
COPD is a progressive condition. In most COPD patients, the FEV1 declines gradually, in an acceleration of the decline that normally occurs with advancing age; rarely, however, precipitous reductions in FEV1 values may occur. The risk of mortality from COPD is closely associated with the severity of FEV1 reduction. How long it takes for the FEV1 to fall to critical levels depends in part on how high the values were to begin with; this, in turn, is influenced by environmental and familial factors such as childhood respiratory illnesses, airway hyperresponsiveness, and intensity of exposure to cigarette smoke. Even after tobacco cessation, persistently high levels of inflammatory cells in the lungs of ex-smokers with COPD suggest that a self-perpetuating inflammatory process is present, which continues to lead to progressive emphysema and airway disease.
Airflow obstruction does not completely capture the mul-tifaceted nature of COPD severity, and current guidelines incorporate symptoms and the risk of exacerbation when making management decisions.1 The BODE index, which is a composite index including body mass index, airflow obstruction, dyspnea, and exercise tolerance, is a more accu-rate predictor of COPD mortality than FEV1 alone.239 Future genetic studies may provide insights into the heterogeneity of COPD and guide prevention, diagnosis, and treatment. The only therapeutic interventions that have been proven to

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 22
16. Foreman MG, Zhang L, Murphy J, et al. Early-onset chronic obstructive pulmonary disease is associated with female sex, maternal factors, and African American race in the COPDGene Study. Am J Respir Crit Care Med 2011;184:414–20.
17. Hurd S. The impact of COPD on lung health worldwide. Chest 2000;117:1S–4S.
18. Chatila WM, Wynkoop WA, Vance G, Criner GJ. Smoking patterns in African Americans and whites with advanced COPD. Chest 2004;125:15–21.
19. Dransfield MT, Bailey WC. COPD: racial disparities in sus-ceptibility, treatment, and outcomes. Clin Chest Med 2006;27:463–71.
20. Hansel NN, Washko GR, Foreman MG, et al. Racial differences in CT phenotypes in COPD. COPD 2013;10:20–7.
21. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006;3:e442.
22. Bruse S, Sood A, Petersen H, et al. New Mexican Hispanic smokers have lower odds of chronic obstructive pulmo-nary disease and less decline in lung function than non- Hispanic whites. Am J Respir Crit Care Med 2011;184: 1254–60.
23. Martinez FD. Early-life origins of chronic obstructive pul-monary disease. N Engl J Med 2016;375:871–8.
24. Burrows B, Knudson RJ, Cline MG, Lebowitz MD. Quan-titative relationships between cigarette smoking and ven-tilatory function. Am Rev Respir Dis 1977;115:195–205.
25. Kohansal R, Martinez-Camblor P, Agusti A, et al. The nat-ural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med 2009;180:3–10.
26. Dinakar C, O’Connor GT. The health effects of electronic cigarettes. N Engl J Med 2016;375:1372–81.
27. Coultas DB, Mapel D, Gagnon R, Lydick E. The health impact of undiagnosed airflow obstruction in a national sample of United States adults. Am J Respir Crit Care Med 2001;164:372–7.
28. Menezes AMB, Perez-Padilla R, Jardim JB, et al. Chronic obstructive pulmonary disease in five Latin American cities (the PLATINO study): a prevalence study. Lancet 2005;366:1875–81.
29. Shaheen SO, Barker DJ, Shiell AW, et al. The relationship between pneumonia in early childhood and impaired lung function in late adult life. Am J Respir Crit Care Med 1994;149:616–9.
30. Johnston IDA, Strachan DP, Anderson HR. Effect of pneu-monia and whooping cough in childhood on adult lung function. N Engl J Med 1998;338:581–7.
31. Marossy AE, Strachan DP, Rudnicka AR, Anderson HR. Childhood chest illness and the rate of decline of adult lung function between ages 35 and 45 years. Am J Respir Crit Care Med 2007;175:355–9.
32. O’Connor GT, Sparrow D, Weiss ST. A prospective longi-tudinal study of methacholine airway responsiveness as a predictor of pulmonary-function decline: the Normative Aging Study. Am J Respir Crit Care Med 1995;152: 87–92.
33. Balmes J, Becklake M, Blanc P, et al. American Thoracic Society statement: occupational contribution to the burden of airway disease. Am J Respir Crit Care Med 2003;167: 787–97.
34. Oh YM, Bhome AB, Boonsawat W, et al. Characteristics of stable chronic obstructive pulmonary disease patients in the pulmonology clinics of seven Asian cities. Int J Chron Obstruct Pulmon Dis 2013;8:31–9.
35. Eisner MD, Anthonisen N, Coultas D, et al. An official Amer-ican Thoracic Society public policy statement: novel risk fac-tors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010;182:693–718.
36. Diaz PT, King MA, Pacht ER, et al. Increased susceptibility to pulmonary emphysema among HIV-seropositive smok-ers. Ann Intern Med 2000;132:369-72.
37. American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: Standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003;168:818–900.
38. Silverman EK, Sandhaus RA. Clinical practice. Alpha 1-antitrypsin deficiency. N Engl J Med 2009;360:2749–57.
39. Piitulainen E, Tornling G, Eriksson S. Effect of age and occupational exposure to airway irritants on lung function in non-smoking individuals with alpha 1-antitrypsin defi-ciency (PiZZ). Thorax 1997;52:244–8.
40. DeMeo DL, Sandhaus RA, Barker AF, et al. Determinants of airflow obstruction in severe alpha-1-antitrypsin defi-ciency. Thorax 2007;62:806–13.
41. Hersh CP, Dahl M, Ly NP, et al. Chronic obstructive pul-monary disease in α1-antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax 2004;59:843–9.
42. Dahl M, Hersh CP, Ly NP, et al. The protease inhibitor PI*S allele and COPD: a meta-analysis. Eur Respir J 2005; 26:67–76.
43. Wisnieski JJ, Baer AN, Christensen J, et al. Hypocomplemen-temic urticarial vasculitis syndrome: clinical and serologic findings in 18 patients. Medicine (Baltimore) 1995;74:24–41.
44. Hersh CP, DeMeo D, Silverman EK. Chronic obstructive pulmonary disease. In: Silverman EK, Shapiro SD, Lomas DA, Weiss ST, editors. Respiratory Genetics. London: Hodder Arnold; 2005. p. 253–96.
45. McCloskey SC, Patel BD, Hinchliffe SJ, et al. Siblings of patients with severe chronic obstructive pulmonary dis-ease have a significant risk of airflow obstruction. Am J Respir Crit Care Med 2001;164:1419–24.
46. Silverman EK, Chapman HA, Drazen JM, et al. Genetic epide-miology of severe, early-onset chronic obstructive pulmonary disease—risk to relatives for airflow obstruction and chronic bronchitis. Am J Respir Crit Care Med 1998;157:1770–8.
47. Ingebrigtsen T, Thomsen SF, Vestbo J, et al. Genetic influ-ences on chronic obstructive pulmonary disease—a twin study. Respir Med 2010;104:1890–5.
48. Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet 2009;5:e1000421.
49. Wilk JB, Chen TH, Gottlieb DJ, et al. A genome-wide asso-ciation study of pulmonary function measures in the Framingham Heart Study. PLoS Genet 2009;5:e1000429.
50. Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmo-nary disease. Nat Genet 2010;42:200–2.
51. Cho MH, McDonald M-LN, Zhou X, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 23
association study and meta-analysis. Lancet Respir Med 2014;2:214–25.
52. Cho MH, Castaldi PJ, Hersh CP, et al. A genome-wide association study of emphysema and airway quantitative imaging phenotypes. Am J Respir Crit Care Med 2015;192: 559–69.
53. Castaldi PJ, Dy J, Ross J, et al. Cluster analysis in the COP-DGene study identifies subtypes of smokers with distinct patterns of airway disease and emphysema. Thorax 2014; 69:416–23.
54. MacNee W, Tuder RM. New paradigms in the pathogene-sis of chronic obstructive pulmonary disease I. Proc Am Thorac Soc 2009;6:527–31.
55. Pulmonary disease. In: How tobacco smoke causes disease: the biology and behavioral basis for smoking-attributable dis-ease. A report of the Surgeon General. Atlanta (GA): Centers for Disease Control and Prevention (US) 2010. p. 435–519.
56. Shapiro SD, Ingenito EP. The pathogenesis of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2005;32:367–72.
57. Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev 2004;56:515–48.
58. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003;22:672–88.
59. Qiu W, Baccarelli A, Carey VJ, et al. Variable DNA meth-ylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med 2012;185:373–81.
60. Ito K, Ito M, Elliott WM, et al. Decreased histone deacety-lase activity in chronic obstructive pulmonary disease. N Engl J Med 2005;352:1967–76.
61. Wright JL, Churg A. Animal models of cigarette smoke- induced chronic obstructive pulmonary disease. Expert Rev Respir Med 2010;4:723–34.
62. Baglole CJ, Maggirwar SB, Gasiewicz TA, et al. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-B family member RelB. J Biol Chem 2008;283:28944–57.
63. Miller M, Pham A, Cho JY, et al. Adiponectin-deficient mice are protected against tobacco-induced inflammation and increased emphysema. Am J Physiol Lung Cell Mol Physiol 2010;299:L834–42.
64. Martorana PA, Beume R, Lucattelli M, et al. Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smoke. Am J Respir Crit Care Med 2005;172:848–53.
65. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmo-nary disease. N Engl J Med 2004;350:2645–53.
66. Fuke S, Betsuyaku T, Nasuhara Y, et al. Chemokines in bronchiolar epithelium in the development of chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2004;31:405–12.
67. Churg A, Tai H, Coulthard T, et al. Cigarette smoke drives small airway remodeling by induction of growth factors in the airway wall. Am J Respir Crit Care Med 2006;174: 1327–34.
68. Houghton AM, Quintero PA, Perkins DL, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest 2006;116:753–9.
69. Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol 2008;40:1101–10.
70. Mizuno S, Yasuo M, Bogaard HJ, et al. Inhibition of his-tone deacetylase causes emphysema. Am J Physiol Lung Cell Mol Physiol 2011;300:L402–13.
71. Grumelli S, Corry DB, Song L-Z, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med 2004;1:e8.
72. Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med 2009;360:2445–54.
73. Low TB, Greene CM, O’Neill SJ, McElvaney NG. Quantifi-cation and evaluation of the role of antielastin autoantibod-ies in the emphysematous lung. Pulm Med 2011;2011:826160.
74. Maeno T, Houghton AM, Quintero PA, et al. CD8+ T cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol 2007;178: 8090–6.
75. Motz GT, Eppert BL, Wesselkamper SC, et al. Chronic cig-arette smoke exposure generates pathogenic T cells capa-ble of driving COPD-like disease in Rag2−/− mice. Am J Respir Crit Care Med 2010;181:1223–33.
76. Karayama M, Inui N, Suda T, et al. Antiendothelial cell antibodies in patients with COPD. Chest 2010;138:1303–8.
77. Lee S-H, Goswami S, Grudo A, et al. Antielastin autoim-munity in tobacco smoking–induced emphysema. Nat Med 2007;13:567–9.
78. Brandsma CA, Kerstjens HA, Geerlings M, et al. The search for autoantibodies against elastin, collagen and decorin in COPD. Eur Respir J 2011;37:1289–92.
79. Owen CA. Proteinases and oxidants as targets in the treat-ment of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005;2:373–85; discussion 394–5.
80. Greenlee KJ, Werb Z, Kheradmand F. Matrix metallopro-teinases in lung: multiple, multifarious, and multifaceted. Physiol Rev 2007;87:69–98.
81. Kaner RJ, Santiago F, Crystal RG. Up-regulation of alveolar macrophage matrix metalloproteinases in HIV1+ smokers with early emphysema. J Leukoc Biol 2009;86:913–22.
82. Braber S, Koelink PJ, Henricks PA, et al. Cigarette smoke- induced lung emphysema in mice is associated with prolyl endopeptidase, an enzyme involved in collagen break-down. Am J Physiol Lung Cell Mol Physiol 2010;300: L255–65.
83. Tasaka S, Inoue K, Miyamoto K, et al. Role of interleukin-6 in elastase-induced lung inflammatory changes in mice. Exp Lung Res 2010;36:362–72.
84. Shim YM, Paige M, Hanna H, et al. Role of LTB4 in the pathogenesis of elastase-induced murine pulmonary emphy-sema. Am J Physiol Lung Cell Mol Physiol 2010;299:L749–59.
85. MacNee W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005;2:50–60.
86. Pierrou S, Broberg P, O’Donnell RA, et al. Expression of genes involved in oxidative stress responses in airway epi-thelial cells of smokers with chronic obstructive pulmo-nary disease. Am J Respir Crit Care Med 2007;175:577–86.
87. Rangasamy T, Misra V, Zhen L, et al. Cigarette smoke- induced emphysema in A/J mice is associated with pul-monary oxidative stress, apoptosis of lung cells, and global

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 24
alterations in gene expression. Am J Physiol Lung Cell Mol Physiol 2009;296:L888–900.
88. Deslee G, Woods JC, Moore C, et al. Oxidative damage to nucleic acids in severe emphysema. Chest 2009;135:965–74.
89. Akhtar K, Broekelmann TJ, Miao M, et al. Oxidative and nitrosative modifications of tropoelastin prevent elastic fiber assembly in vitro. J Biol Chem 2010;285:37396–404.
90. Rangasamy T, Cho CY, Thimmulappa RK, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke–induced emphysema in mice. J Clin Invest 2004;114:1248–59.
91. Lanzetti M, Lopes AA, Ferreira TS, et al. Mate tea amelio-rates emphysema in cigarette smoke-exposed mice. Exp Lung Res 2011;37:246–57.
92. Suzuki M, Betsuyaku T, Ito Y, et al. Curcumin attenuates elastase- and cigarette smoke-induced pulmonary emphy-sema in mice. Am J Physiol Lung Cell Mol Physiol 2009;296: L614–23.
93. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med 2006;174:886–93.
94. Yokohori N, Aoshiba K, Nagai A. Respiratory Failure Research Group in Japan. Increased levels of cell death and proliferation in alveolar wall cells in patients with pul-monary emphysema. Chest 2004;125:626–32.
95. Petrache I, Medler TR, Richter AT, et al. Superoxide dis-mutase protects against apoptosis and alveolar enlarge-ment induced by ceramide. Am J Physiol Lung Cell Mol Physiol 2008;295:L44–53.
96. Aoshiba K, Nagai A. Senescence hypothesis for the patho-genetic mechanism of chronic obstructive pulmonary dis-ease. Proc Am Thorac Soc 2009;6:596–601.
97. Nyunoya T, Monick MM, Klingelhutz AL, et al. Cigarette smoke induces cellular senescence via Werner’s syndrome protein down-regulation. Am J Respir Crit Care Med 2009; 179:279–87.
98. Vlahovic G, Russell ML, Mercer RR, Crapo JD. Cellular and connective tissue changes in alveolar septal walls in emphysema. Am J Respir Crit Care Med 1999;160: 2086–92.
99. Deslee G, Woods JC, Moore CM, et al. Elastin expression in very severe human COPD. Eur Respir J 2009;34:324–31.
100. Churg A, Wang R, Wang X, et al. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax 2007;62:706–13.
101. Celli BR, MacNee W, Agusti A, et al. Standards for the diagnosis and treatment of patients with COPD: a sum-mary of the ATS/ERS position paper. Eur Respir J 2004;23: 932–46.
102. Bourdin A, Burgel PR, Chanez P, et al. Recent advances in COPD: pathophysiology, respiratory physiology and clin-ical aspects, including comorbidities. Eur Respir Rev 2009; 18:198–212.
103. Calverley PM, Burge PS, Spencer S, et al. Bronchodilator reversibility testing in chronic obstructive pulmonary dis-ease. Thorax 2003;58:659–64.
104. O’Donnell DE, Laveneziana P. The clinical importance of dynamic lung hyperinflation in COPD. COPD 2006;3:219–32.
105. Casanova C, Cote C, de Torres JP, et al. Inspiratory-to-total lung capacity ratio predicts mortality in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005;171:591–7.
106. Agusti A, Calverley PM, Celli B, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res 2010;11:122.
107. Mahler DA. Mechanisms and measurement of dyspnea in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2006;3:234–8.
108. West JB. The major limitation to exercise performance in COPD is inadequate energy supply to the respiratory and locomotor muscles vs. lower limb muscle dysfunction vs. dynamic hyperinflation. J Appl Physiol 2008;105:758–62.
109. Vogiatzis I, Athanasopoulos D, Habazettl H, et al. Inter-costal muscle blood flow limitation during exercise in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010;182:1105–13.
110. Donaldson GC, Wedzicha JA. COPD exacerbations - 1: epi-demiology. Thorax 2006;61:164–8.
111. Sethi S, Evans N, Grant BJB, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmo-nary disease. N Engl J Med 2002;347:465–71.
112. Tillie-Leblond I, Marquette CH, Perez T, et al. Pulmonary embolism in patients with unexplained exacerbation of chronic obstructive pulmonary disease: prevalence and risk factors. Ann Intern Med 2006;144:390–6.
113. Seemungal TA, Donaldson GC, Bhowmik A, et al. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000;161:1608–13.
114. Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002;57:847–52.
115. Dransfield MT, Kunisaki KM, Strand MJ, et al. Acute exac-erbations and lung function loss in smokers with and without COPD. Am J Respir Crit Care Med 2016 Aug 24. [Epub ahead of print]
116. Wright JL, Churg A. Pathologic features of chronic obstruc-tive pulmonary disease: diagnostic criteria and differential diagnosis. In: Fishman A, Elias J, Fishman J, et al., editors. Fishman’s Pulmonary Diseases and Disorders. 4th ed. New York: McGraw-Hill; 2008. p. 693–705.
117. Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol Mech Dis 2009;4:435–59.
118. Saetta M, Turato G, Baraldo S, et al. Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers with both symptoms of chronic bronchitis and chronic air-flow limitation. Am J Respir Crit Care Med 2000;161:1016–21.
119. Hogg JC, Senior RM. Chronic obstructive pulmonary disease—part 2: pathology and biochemistry of emphy-sema. Thorax 2002;57:830–4.
120. Kim WD, Eidelman DH, Izquierdo JL, et al. Centrilobular and panlobular emphysema in smokers: two distinct morphologic and functional entities. Am Rev Respir Dis 1991;144:1385–90.
121. Burgel PR. The role of small airways in obstructive airway diseases. Eur Respir Rev 2011;20:23–33.
122. Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 2004;364:709–21.
123. Guirguis-Blake JM, Senger CA, Webber EM, et al. Screen-ing for chronic obstructive pulmonary disease: evidence report and systematic review for the US Preventive Ser-vices Task Force. JAMA 2016;315:1378–93.
124. Schols AM, Slangen J, Volovics L, Wouters EF. Weight loss is a reversible factor in the prognosis of chronic

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 25
obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:1791–7.
125. Schols AMWJ, Soeters PB, Dingemans AM, et al. Preva-lence and characteristics of nutritional depletion in patients with stable COPD eligible for pulmonary rehabilitation. Am Rev Respir Dis 1993;147:1151–6.
126. Sarkar M, Mahesh DM, Madabhavi I. Digital clubbing. Lung India 2012;29:354–62.
127. Braido F, Riccio AM, Guerra L, et al. Clara cell 16 protein in COPD sputum: a marker of small airways damage? Respir Med 2007;101:2119–24.
128. Chen Y-WR, Leung JM, Sin DD. A systematic review of diagnostic biomarkers of COPD exacerbation. PloS One 2016;11:e0158843.
129. Miller MR, Quanjer PH, Swanney MP, et al. Interpreting lung function data using 80% predicted and fixed thresholds misclassifies more than 20% of patients. Chest 2011;139:52–9.
130. Ortega R, Hansen CJ, Elterman K, Woo A. Videos in clini-cal medicine—Pulse oximetry. N Engl J Med 2011;364:e33.
131. Müller NL, Coxson H. Chronic obstructive pulmonary dis-ease • 4: imaging the lungs in patients with chronic obstructive pulmonary disease. Thorax 2002;57:982–5.
132. Kim WJ, Silverman EK, Hoffman E, et al. CT metrics of airway disease and emphysema in severe COPD. Chest 2009;136:396–404.
133. Postma DS, Rabe KF. The asthma-COPD overlap syn-drome. N Engl J Med 2015;373:1241–9.
134. Behrendt CE. Mild and moderate-to-severe COPD in non-smokers. Chest 2005;128:1239–44.
135. Poletti V, Casoni G, Chilosi M, Zompatori M. Diffuse pan-bronchiolitis. Eur Respir J 2006;28:862–71.
136. Schlesinger C, Meyer CA, Veeraraghavan S, Koss MN. Constrictive (obliterative) bronchiolitis: diagnosis, etiology, and a critical review of the literature. Ann Diagn Pathol 1998;2:321–34.
137. Anthonisen NR, Connett JE, Murray RP. Smoking and lung function of Lung Health Study participants after 11 years. Am J Respir Crit Care Med 2002;166:675–9.
138. Simmons MS, Connett JE, Nides MA, et al. Smoking reduc-tion and the rate of decline in FEV(1): results from the Lung Health Study. Eur Respir J 2005;25:1011–7.
139. Rigotti NA. Clinical practice: Treatment of tobacco use and dependence. N Engl J Med 2002;346:506–12.
140. Wagena EJ, Knipschild PG, Huibers MJH, et al. Efficacy of bupropion and nortriptyline for smoking cessation among people at risk for or with chronic obstructive pulmonary disease. Arch Intern Med 2005;165:2286–92.
141. White AR, Rampes H, Liu JP, et al. Acupuncture and related interventions for smoking cessation. Cochrane Database Syst Rev 2014;(1):CD000009.
142. Wongsurakiat P, Maranetra KN, Wasi C, et al. Acute respi-ratory illness in patients with COPD and the effectiveness of influenza vaccination: a randomized controlled study. Chest 2004;125:2011–20.
143. Criner GJ, Bourbeau J, Diekemper RL, et al. Prevention of acute exacerbations of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest 2015;147:894–942.
144. Alfageme I, Vazquez R, Reyes N, et al. Clinical efficacy of anti-pneumococcal vaccination in patients with COPD. Thorax 2006;61:189–95.
145. Schenkein JG, Nahm MH, Dransfield MT. Pneumococcal vaccination for patients with COPD. Chest 2008;133:767–74.
146. Centers for Disease Control and Prevention (CDC). Use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine for adults with immunocompromising conditions: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2012;61:816–9.
147. Kobayashi M, Bennett NM, Gierke R, et al. Intervals between PCV13 and PPSV23 vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2015;64:944–7.
148. Schols AM, Ferreira IM, Franssen FM, et al. Nutritional assessment and therapy in COPD: a European Respiratory Society statement. Eur Respir J 2014;44:1504–20.
149. Barr RG, Bourbeau J, Camargo CA, Ram FS. Tiotropium for stable chronic obstructive pulmonary disease: a meta- analysis. Thorax 2006;61:854–62.
150. Stockley RA, Whitehead PJ, Williams MK. Improved out-comes in patients with chronic obstructive pulmonary disease treated with salmeterol compared with placebo/usual ther-apy: results of a meta-analysis. Respir Res 2006;7:147.
151. Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotro-pium in chronic obstructive pulmonary disease. N Engl J Med 2008;359:1543–54.
152. Vogelmeier C, Hederer B, Glaab T, et al. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med 2011;364:1093–103.
153. Decramer ML, Chapman KR, Dahl R, et al. Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE): a randomised, blinded, parallel-group study. Lancet Respir Med 2013;1:524–33.
154. Kardos P, Wencker M, Glaab T, Vogelmeier C. Impact of salmeterol/fluticasone propionate versus salmeterol on exacerbations in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007;175:144–9.
155. Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007;356:775–89.
156. Wedzicha JA, Banerji D, Chapman KR, et al. Indacaterol- glycopyrronium versus salmeterol-fluticasone for COPD. N Engl J Med 2016;374:2222–34.
157. Wise RA, Anzueto A, Cotton D, et al. Tiotropium Respimat Inhaler and the risk of death in COPD. N Engl J Med 2013; 369:1491–501.
158. Dolovich MB, Ahrens RC, Hess DR, et al. Device selection and outcomes of aerosol therapy: evidence-based guide-lines: American College of Chest Physicians/American College of Asthma, Allergy, and Immunology. Chest 2005; 127:335–71.
159. Yang IA, Clarke MS, Sim EH, Fong KM. Inhaled cortico-steroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012;(7):CD002991.
160. Jones PW, Willits LR, Burge PS, Calverley PMA. Disease severity and the effect of fluticasone propionate on chronic obstructive pulmonary disease exacerbations. Eur Respir J 2003;21:68–73.
161. Burge PS, Calverley PM, Jones PW, et al. Randomised, double blind, placebo controlled study of fluticasone pro-pionate in patients with moderate to severe chronic

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 26
obstructive pulmonary disease: the ISOLDE trial. BMJ 2000;320:1297–303.
162. Pauwels RA, Löfdahl C-G, Laitinen LA, et al. Long-term treatment with inhaled budesonide in persons with mild chronic obstructive pulmonary disease who continue smoking European Respiratory Society Study on Chronic Obstructive Pulmonary Disease. N Engl J Med 1999;340: 1948–53.
163. Magnussen H, Disse B, Rodriguez-Roisin R, et al. With-drawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med 2014;371:1285–94.
164. Vestbo J, Anderson JA, Brook RD, et al. Fluticasone furoate and vilanterol and survival in chronic obstructive pulmo-nary disease with heightened cardiovascular risk (SUMMIT): a double-blind randomised controlled trial. Lancet 2016;387:1817–26.
165. Festic E, Scanlon PD. Incident pneumonia and mortality in patients with chronic obstructive pulmonary disease: a double effect of inhaled corticosteroids? Am J Respir Crit Care Med 2015;191:141–8.
166. van der Valk P, Monninkhof E, van der Palen J, et al. Effect of discontinuation of inhaled corticosteroids in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;166:1358–63.
167. Leigh R, Pizzichini MM, Morris MM, et al. Stable COPD: pre-dicting benefit from high dose inhaled corticosteroid treat-ment. Eur Respir J 2006;27:964–71.
168. Walters JAE, Tan DJ, White CJ, Wood-Baker R. Different durations of corticosteroid therapy for exacerbations of chronic obstructive pulmonary disease. Cochrane Data-base Syst Rev 2014;(12):CD006897.
169. Ram FSF, Jones P, Jardim J, et al. Oral theophylline for chronic obstructive pulmonary disease. Cochrane Data-base Syst Rev 2002;(4):CD003902.
170. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflam-matory diseases. Lancet 2009;373:1905–17.
171. Fabbri LM, Calverley PMA, Izquierdo-Alonso JL, et al. Roflumilast in moderate-to-severe chronic obstructive pul-monary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 2009;374:695–703.
172. Calverley PMA, Rabe KF, Goehring U-M, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009;374:685–94.
173. Yu T, Fain K, Boyd CM, et al. Benefits and harms of roflu-milast in moderate to severe COPD. Thorax 2014;69:616–22.
174. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–98.
175. Seemungal TA, Wilkinson TM, Hurst JR, et al. Long-term erythromycin therapy is associated with decreased chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med 2008;178:1139–47.
176. Herath SC, Poole P. Prophylactic antibiotic therapy in chronic obstructive pulmonary disease (COPD). Cochrane Database Syst Rev 2013;(11):CD009764.
177. Adir Y, Harari S. Pulmonary hypertension associated with chronic obstructive lung disease and idiopathic pulmo-nary fibrosis. Curr Opin Pulm Med 2014;20:414–20.
178. Barberà JA, Roger N, Roca J, et al. Worsening of pulmo-nary gas exchange with nitric oxide inhalation in chronic obstructive pulmonary disease. Lancet 1996;347:436–40.
179. Blanco I, Santos S, Gea J, et al. Sildenafil to improve respi-ratory rehabilitation outcomes in COPD: a controlled trial. Eur Respir J 2013;42:982–92.
180. Poole P, Chong J, Cates CJ. Mucolytic agents versus pla-cebo for chronic bronchitis or chronic obstructive pulmo-nary disease. Cochrane Database Syst Rev 2015;(7):CD001287.
181. Zheng J-P, Kang J, Huang S-G, et al. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE Study): a randomised placebo-controlled study. Lancet 2008;371:2013–8.
182. Decramer M, Rutten-van Mölken M, Dekhuijzen PN, et al. Effects of N-acetylcysteine on outcomes in chronic obstruc-tive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomised placebo- controlled trial. Lancet 2005;365:1552–60.
183. Rennard SI, Fogarty C, Kelsen S, et al. The safety and effi-cacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007;175: 926–34.
184. Lehouck A, Mathieu C, Carremans C, et al. High doses of vitamin D to reduce exacerbations in chronic obstructive pulmonary disease. Ann Intern Med 2012;156:105–14.
185. Cranston JM, Crockett AJ, Moss JR, Alpers JH. Domiciliary oxygen for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2005;(4):CD001744.
186. Zielinski J, Tobiasz M, Hawrytkiewicz I, et al. Effects of long-term oxygen therapy on pulmonary hemodynamics in COPD patients: a 6-year prospective study. Chest 1998;113:65–70.
187. Stoller JK, Panos RJ, Krachman S, et al. Oxygen therapy for patients with COPD: current evidence and the long-term oxygen treatment trial. Chest 2010;138:179–87.
188. Johnson AO. Chronic obstructive pulmonary disease • 11: fitness to fly with COPD. Thorax 2003;58:729–32.
189. California Pulmonary Rehabilitation Collaborative Group. Effects of pulmonary rehabilitation on dyspnea, quality of life, and healthcare costs in California. J Cardiopulm Rehabil 2004;24:52–62.
190. van’t Hul A, Gosselink R, Hollander P, et al. Training with inspiratory pressure support in patients with severe COPD. Eur Respir J 2006;27:65–72.
191. Seymour JM, Moore L, Jolley CJ, et al. Outpatient pulmo-nary rehabilitation following acute exacerbations of COPD. Thorax 2010;65:423–8.
192. Troosters T, Casaburi R, Gosselink R, Decramer M. Pulmo-nary rehabilitation in chronic obstructive pulmonary dis-ease. Am J Respir Crit Care Med 2005;172:19–38.
193. Ram FS, Rodriguez-Roisin R, Granados-Navarrete A, et al. Antibiotics for exacerbations of chronic obstructive pulmo-nary disease. Cochrane Database Syst Rev 2006;(2): CD004403.
194. Martinez FJ, Anzueto A. Appropriate outpatient treatment of acute bacterial exacerbations of chronic bronchitis. Am J Med 2005;118 Suppl:39S-44S.
195. Walters JA, Gibson PG, Wood-Baker R, et al. Systemic cortico-steroids for acute exacerbations of chronic obstructive pulmo-nary disease. Cochrane Database Syst Rev 2009;(1):CD001288.
196. Leuppi JD, Schuetz P, Bingisser R, et al. Short-term vs con-ventional glucocorticoid therapy in acute exacerbations of chronic obstructive pulmonary disease: the REDUCE ran-domized clinical trial. JAMA 2013;309:2223–31.

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 27
197. Sklar MC, Beloncle F, Katsios CM, et al. Extracorporeal carbon dioxide removal in patients with chronic obstruc-tive pulmonary disease: a systematic review. Intensive Care Med 2015;41:1752–62.
198. Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double- blind, placebo-controlled trial. Lancet 2015;386:360–8.
199. Alpha 1-Antitrypsin Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998;158: 49–59.
200. Seersholm N, Wencker M, Banik N, et al. Does α1- antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary α1-antitrypsin deficiency? Eur Respir J 1997;10:2260–3.
201. Wencker M, Fuhrmann B, Konietzko N, Banik N. Longitu-dinal follow-up of patients with α1-protease inhibitor defi-ciency before and during therapy with IV α1-protease inhibitor. Chest 2001;119:737–44.
202. Sorrells S, Camprubi S, Griffin R, et al. SPARTA clinical trial design: exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiency. Respir Med 2015; 109:490–9.
203. Stoller JK, Fallat R, Schluchter MD, et al. Augmentation therapy with α1-antitrypsin: patterns of use and adverse events. Chest 2003;123:1425–34.
204. Sciurba FC, Ernst A, Herth FJ, et al. A randomized study of endobronchial valves for advanced emphysema. N Engl J Med 2010;363:1233–44.
205. Klooster K, ten Hacken NH, Hartman JE, et al. Endobron-chial valves for emphysema without interlobar collateral ventilation. N Engl J Med 2015;373:2325–35.
206. Sciurba FC, Criner GJ, Strange C, et al. Effect of endobron-chial coils vs usual care on exercise tolerance in patients with severe emphysema: the RENEW Randomized Clini-cal Trial. JAMA 2016;315:2178–89.
207. Davey C, Zoumot Z, Jordan S, et al. Bronchoscopic lung volume reduction with endobronchial valves for patients with heterogeneous emphysema and intact interlobar fis-sures (the BeLieVeR-HIFi study): a randomised controlled trial. Lancet 2015;386:1066–73.
208. Deslée G, Mal H, Dutau H, et al. Lung volume reduction coil treatment vs usual care in patients with severe emphy-sema: the REVOLENS Randomized Clinical Trial. JAMA 2016;315:175–84.
209. Fishman A, Martinez F, Naunheim K, et al. A randomized trial comparing lung-volume-reduction surgery with med-ical therapy for severe emphysema. N Engl J Med 2003;348: 2059–73.
210. Naunheim KS, Wood DE, Krasna MJ, et al. Predictors of operative mortality and cardiopulmonary morbidity in the National Emphysema Treatment Trial. J Thorac Cardio-vasc Surg 2006;131:43–53.
211. Tutic M, Lardinois D, Imfeld S, et al. Lung-volume reduc-tion surgery as an alternative or bridging procedure to lung transplantation. Ann Thorac Surg 2006;82:208–13.
212. Gelb AF, McKenna RJ Jr, Brenner M, et al. Lung function 5 yr after lung volume reduction surgery for emphysema. Am J Respir Crit Care Med 2001;163:1562–6.
213. Nathan SD, Edwards LB, Barnett SD, et al. Outcomes of COPD lung transplant recipients after lung volume reduc-tion surgery. Chest 2004;126:1569–74.
214. Snider GL. Reduction pneumoplasty for giant bullous emphysema: implications for surgical treatment of non-bullous emphysema. Chest 1996;109:540–8.
215. Patel N, Criner GJ. Transplantation in chronic obstructive pulmonary disease. COPD 2006;3:149–62.
216. Kreider M, Kotloff RM. Selection of candidates for lung transplantation. Proc Am Thorac Soc 2009;6:20–7.
217. Thabut G, Christie JD, Ravaud P, et al. Survival after bilat-eral versus single lung transplantation for patients with chronic obstructive pulmonary disease: a retrospective analysis of registry data. Lancet 2008;371:744–51.
218. Stehlik J, Edwards LB, Kucheryavaya AY, et al. The Regis-try of the International Society for Heart and Lung Trans-plantation: twenty-seventh official adult heart transplant report—2010. J Heart Lung Transplant 2010;29:1089–103.
219. Stavem K, Bjørtuft Ø, Borgan Ø, et al. Lung transplanta-tion in patients with chronic obstructive pulmonary disease in a national cohort is without obvious survival benefit. J Heart Lung Transplant 2006;25:75–84.
220. Lane CR, Tonelli A. Lung transplantation in chronic obstructive pulmonary disease: patient selection and spe-cial considerations. Int J Chron Obstruct Pulmon Dis 2015; 10:2137–46.
221. Pinto-Plata VM, Müllerova H, Toso JF, et al. C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax 2006;61:23–8.
222. Divo M, Cote C, de Torres JP, et al. Comorbidities and risk of mortality in patients with chronic obstructive pulmo-nary disease. Am J Respir Crit Care Med 2012;186:155–61.
223. Nussbaumer-Ochsner Y, Rabe KF. Systemic manifesta-tions of COPD. Chest 2011;139:165–73.
224. Singh B, Mielke MM, Parsaik AK, et al. A prospective study of chronic obstructive pulmonary disease and the risk for mild cognitive impairment. JAMA Neurol 2014;71: 581–8.
225. Wright JL, Levy RD, Churg A. Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax 2005;60:605–9.
226. Barberà JA, Peinado VI, Santos S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J 2003;21:892–905.
227. Barr RG, Bluemke DA, Ahmed FS, et al. Percent emphy-sema, airflow obstruction, and impaired left ventricular filling. N Engl J Med 2010;362:217–27.
228. Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardio-graphic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med 2003;167:735–40.
229. Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005;2:20–2.
230. Blanco I, Gimeno E, Munoz PA, et al. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hyperten-sion. Am J Respir Crit Care Med 2010;181:270–8.
231. National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality

Scientific American Medicine
01/17
pulm-crit chronic obstructive pulmonary disease — 28
with low-dose computed tomographic screening. N Engl J Med 2011;365:395–409.
232. Moyer VA, U.S. Preventive Services Task Force. Screening for lung cancer: U.S. Preventive Services Task Force rec-ommendation statement. Ann Intern Med 2014;160:330–8.
233. de-Torres JP, Marín JM, Casanova C, et al. Identification of COPD patients at high risk for lung cancer mortality using the COPD-LUCSS-DLCO. Chest 2016;149:936–42.
234. Katki HA, Kovalchik SA, Berg CD, et al. Development and validation of risk models to select ever-smokers for CT lung cancer screening. JAMA 2016;315:2300–11.
235. Guo Y, Xie C, Rodriguez RM, Light RW. Factors related to recurrence of spontaneous pneumothorax. Respirology 2005;10:378–84.
236. Hanania NA, Müllerova H, Locantore NW, et al. Determi-nants of depression in the ECLIPSE chronic obstructive pulmonary disease cohort. Am J Respir Crit Care Med 2011;183:604–11.
237. Kunik ME, Roundy K, Veazey C, et al. Surprisingly high prevalence of anxiety and depression in chronic breathing disorders. Chest 2005;127:1205–11.
238. Norwood R, Balkissoon R. Current perspectives on man-agement of co-morbid depression in COPD. COPD 2005;2: 185–93.
239. Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004;350:1005–12.
Acknowledgments
Figures 1, 5 Seward HungFigure 3 Al Miller Figure 4 Dana Burns-PizerFigure 6 Christine Kenney
Related Documents











