Microelectronics Journal 39 (2008) 190–201 Prototyping bio-nanorobots using molecular dynamics simulation and virtual reality Mustapha Hamdi a , Antoine Ferreira a, , Gaurav Sharma b , Constantinos Mavroidis b, a Laboratoire Vision et Robotique, ENSI Bourges-University of Orle´ans, 18000 Bourges, France b Computational Bionano Robotics Laboratory, Northeastern University, Boston, MA-02120, USA Received 4 March 2006; received in revised form 1 December 2006; accepted 5 December 2006 Available online 5 February 2007 Abstract This paper presents a molecular mechanics study using a molecular dynamics software (NAMD) coupled to virtual reality (VR) techniques for intuitive bio-nanorobotic prototyping. Using simulated bio-nano environments in VR, the operator can design and characterize through physical simulation and 3D visualization the behavior of protein-based components and structures. The main novelty of the proposed simulations is based on the characterization of stiffness performances of passive joints-based proteins (a-helix deca-alanine, repressor of primer protein and immunoglobulin protein) and active joints-based viral protein motor (VPL) in their native environment. Their use as elementary bio-nanorobotic components are also simulated and the results discussed. r 2007 Elsevier Ltd. All rights reserved. Keywords: Bio-nanorobotics; Molecular dynamics; Computational simulation; Virtual reality; Nanotechnology; Proteins; Biomotors 1. Introduction Recent advances in understanding how biomolecular motors work has raised the possibility that they might find applications as protein-based nanomachines. For example, they could be used as molecule-sized robots that are able to apply forces and manipulate objects in the nanoscale world. Thus biomolecular motors could form the basis of bottom-up approaches for constructing active structuring and maintenance at the nanometer scale. Protein is the most versatile of the natural bio-nanomachines. As an example, the most familiar motor is the protein myosin [1] or dynein [2] which moves along filaments, formed through the protein actin, to drive the contraction of muscles. In addition, there are compliance devices such as spring-like proteins called fibronectin [3] and vorticellids [4], as well as synthetic contractile plant polymers [5] which can act as compliant joints in molecule-size robotic systems. The idea is to use biomolecular motors as the actuators of such bio- nanorobots, where the structural elements are carbon nanotubes (CNTs), while the joints are formed by appropriately designed biological spring elements [6]. To achieve these long-term goals, prototyping tools based on molecular dynamics (MD) simulators should be developed in order to understand the molecular mechanics of proteins and develop dynamic and kinematic models to study their performances and control aspects. The ability to visualize the atom-to-atom interaction in real-time and see the results in a fully immersive 3D environment is an additional feature of such simulations [7–9]. Virtual reality (VR) technology is applied here, which not only provides immersive visualization but also gives an added function- ality of CAD-based design, simulation, navigation and interactive manipulation of molecular graphical objects. Using virtual biological environments, the operator can design and characterize through physical simulation the behavior of bio-nanorobots. Adding haptic interaction, the operator can explore and prevent the problems of molecular robots in their native environment. Based on VR technology and MD simulators, our long-term goal is to prototype virtually bio-nanorobotic systems and control ARTICLE IN PRESS www.elsevier.com/locate/mejo 0026-2692/$ - see front matter r 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.mejo.2006.12.003 Corresponding author. Tel.: +33 2 4848 4079; fax: +33 2 4848 4050. Corresponding author. Tel.: +1 617 373 4121; fax: +1 617 373 2921. E-mail addresses: [email protected] (A. Ferreira), [email protected] (C. Mavroidis).

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ARTICLE IN PRESS
0026-2692/$ - se
doi:10.1016/j.m
�Correspond��CorresponE-mail addr
Microelectronics Journal 39 (2008) 190–201
www.elsevier.com/locate/mejo
Prototyping bio-nanorobots using molecular dynamics simulation andvirtual reality
Mustapha Hamdia, Antoine Ferreiraa,�, Gaurav Sharmab, Constantinos Mavroidisb,��
aLaboratoire Vision et Robotique, ENSI Bourges-University of Orleans, 18000 Bourges, FrancebComputational Bionano Robotics Laboratory, Northeastern University, Boston, MA-02120, USA
Received 4 March 2006; received in revised form 1 December 2006; accepted 5 December 2006
Available online 5 February 2007
Abstract
This paper presents a molecular mechanics study using a molecular dynamics software (NAMD) coupled to virtual reality (VR)
techniques for intuitive bio-nanorobotic prototyping. Using simulated bio-nano environments in VR, the operator can design and
characterize through physical simulation and 3D visualization the behavior of protein-based components and structures. The main
novelty of the proposed simulations is based on the characterization of stiffness performances of passive joints-based proteins (a-helixdeca-alanine, repressor of primer protein and immunoglobulin protein) and active joints-based viral protein motor (VPL) in their native
environment. Their use as elementary bio-nanorobotic components are also simulated and the results discussed.
r 2007 Elsevier Ltd. All rights reserved.
Keywords: Bio-nanorobotics; Molecular dynamics; Computational simulation; Virtual reality; Nanotechnology; Proteins; Biomotors
1. Introduction
Recent advances in understanding how biomolecularmotors work has raised the possibility that they might findapplications as protein-based nanomachines. For example,they could be used as molecule-sized robots that are able toapply forces and manipulate objects in the nanoscaleworld. Thus biomolecular motors could form the basis ofbottom-up approaches for constructing active structuringand maintenance at the nanometer scale. Protein is themost versatile of the natural bio-nanomachines. As anexample, the most familiar motor is the protein myosin [1]or dynein [2] which moves along filaments, formed throughthe protein actin, to drive the contraction of muscles. Inaddition, there are compliance devices such as spring-likeproteins called fibronectin [3] and vorticellids [4], as well assynthetic contractile plant polymers [5] which can act ascompliant joints in molecule-size robotic systems. The idea
e front matter r 2007 Elsevier Ltd. All rights reserved.
ejo.2006.12.003
ing author. Tel.: +332 4848 4079; fax: +33 2 4848 4050.
ding author. Tel.: +1 617 373 4121; fax: +1 617 373 2921.
esses: [email protected] (A. Ferreira),
.edu (C. Mavroidis).
is to use biomolecular motors as the actuators of such bio-nanorobots, where the structural elements are carbonnanotubes (CNTs), while the joints are formed byappropriately designed biological spring elements [6].To achieve these long-term goals, prototyping tools
based on molecular dynamics (MD) simulators should bedeveloped in order to understand the molecular mechanicsof proteins and develop dynamic and kinematic models tostudy their performances and control aspects. The abilityto visualize the atom-to-atom interaction in real-time andsee the results in a fully immersive 3D environment is anadditional feature of such simulations [7–9]. Virtual reality(VR) technology is applied here, which not only providesimmersive visualization but also gives an added function-ality of CAD-based design, simulation, navigation andinteractive manipulation of molecular graphical objects.Using virtual biological environments, the operator candesign and characterize through physical simulation thebehavior of bio-nanorobots. Adding haptic interaction, theoperator can explore and prevent the problems ofmolecular robots in their native environment. Based onVR technology and MD simulators, our long-term goal isto prototype virtually bio-nanorobotic systems and control

ARTICLE IN PRESSM. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201 191
their movements in their biological environment [10]. Inthis work, we consider real-time force-feedback technologyfor improving the methodology of studies of folding andunfolding proteins acting as passive or active joints inmolecule-size robots. We therefore decided to begin ourinvestigations by simulating the forces involved undervarious external mechanical stress (stretching, contraction,shearing, bending) to predict the type of force spectra andirreversible work that may be expected from single-molecule protein manipulation experiments.
These recent years, many mechanical proteins composedof multiple, individually folded protein domains have beenstudied using steered molecular dynamics (SMD) [11]. Theavailable SMD tool has provided the basis for a cascade ofstudies on the unique role of proteins as passive springsproviding restoring force either in shortened or stretchedconfigurations: SMD simulates force-induced unfolding byapplying force to the two termini of a protein module.Many SMD studies have been reported on the elasticityand structural properties of specific proteins, such as: giantmuscle protein titin I1-I27 [12–15], extra cellular matrixprotein fibronectin FN-III [16–18] or tropocollagen mole-cule [19]. Understanding the molecular mechanics behavior
Molecularsimulation
Quantum molec
Haptics M
Initial coordinates
bonds, charge…
VMD
Pdb Swiss
Conformation,trajectories,
Temperature
Force and temperature
feedback.
IMD
Molecular parameters
dihedrales...
IME Module
MD Module
files: Angles, bonds,
(pdb files), mass,
Fig. 1. Basic concept of virtual environment and ha
and control parameters of such proteins is an importantchallenge for interfacing protein nanostructures to struc-tural elements [20] such as single walled CNTs [21] orhighly conductive nanowires [22].The paper is organized as follows: Section 2 presents the
architecture of virtual MD simulator using haptics andVR. Section 3 presents the assembly of a protein-basedrobotic structure. Then, simulations of different active/passive proteins are analyzed in Section 4.
2. MD simulation using haptics and VR
2.1. CAD-design methodology using computational steering
Considering the above mentioned research issues, wedecided to adopt VR technologies coupled to computationalMD and quantum dynamics (QD) to simulate, analyze,model, and visualize the complex architecture and interac-tions of bio-nanorobotic components. Real-time explorationis what attracts researchers to develop man–machine interfaces for nanoscale manipulation that usehaptic display technologies. The developed simulationsystem presented in Fig. 1 permits manipulation, connection
dynamics (NAMD2) ular dynamics
odule
Initial coordinates
bonds, charge…
VegaZZ
PSFGen
Nanorobot structure : Initialcoordinates, mass, bonds,charges…
3D molecular assembly andcontrol (temperature, pH…)
Analyse
tools:
Matlab
Cluster
12 paralell PC
(pdb files), mass,
ptics technology for bio-nanorobot simulation.

ARTICLE IN PRESSM. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201192
and assembly of bio-nanorobotic components in MDsimulations with real-time VR devices such as stereo glasses,3D trackers, force-feedback devices and 3D graphicaldisplay.
The adopted CAD-design methodology is based oncomputational steering techniques where the user is able todesign or modify a simulation interactively in a virtualenvironment during run-time. It gives a tremendousadvantage over post-simulation visualization and analysisof results. In run-time steering the user does not have towait till the end of the simulation to see the results of hismodifications, instead he can immediately see the result ofthe interactively changed parameters giving him anopportunity to detect and modify them to steer thesimulation to a desired output. In computational steeringthe user can steer MD simulation by applying externalforces into the computations. These external forces canhelp a complex molecular nanorobotic system overcome apotential energy barrier and can even steer the system to anew geometric conformation for further analysis. Thespecific tasks are:
1.
To connect and assemble interactively the molecularcomponents of the bionanorobotic structure through adata glove (Ascension Technologies CyberGlove18interface) which can be displayed in the virtual environ-ment as a mapping of hand. It is composed of fullyenclosed fiber optic bend sensors that provides up to 22high-accuracy joint-angle measurements allowing tocapture all assembly hand postures.2.
To virtually steer the protein molecules using a hapticdevice (SensAble Technologies PHANToM Desktopinterface). The haptic device is virtually bound to anatom of a conformationally stable subset of themolecule. This system would allow applying stretching,shearing and bending mechanical constraints on theprotein.3.
To distinguish possible conformational paths, feel theenergy barriers associated with each path and choose theoptimal path/strategy.4.
To control the interaction mechanism in a solvent.Protein folding and unfolding can be controlled byassessing the influence of pH value, temperature,electron density or salt concentration.2.2. Software architecture
The software architecture is composed of three softwaremodules: (i) a haptic device module controlled by acomputer that generates the force environment, (ii) aninteractive molecular environment (IME) module formanipulation, attachment and assembly of nanocompo-nents in 3D virtual world and (iii) a MD module fordetermining the effects of force application.
As shown in Fig. 1, steering forces are issued from thehaptic device which measures a user’s hand position andexerts a precisely controlled force on the hand in order to
apply different mechanical constraints, force and energyfields on the virtual model for prototype bio-nanoroboticprototyping. The haptics module sends command data(position, velocity, force) and receives force-feedbacksignals from the IME module.The main role of the IME module is to gather the
nanorobotic components (proteins, DNA, CNT, graphite)in a one 3D representative space called visual moleculardynamics (VMD) [23]. VMD software is a molecularvisualization program for displaying, animating andanalyzing large biomolecular systems using 3D graphicsand built-in scripting. The VRPN software package [24]was used to connect VMD to the PHANToM Desktopdevice. The Data Glove input device is also supported byVRPN. The user manipulates in an intuitive way theattachment and assembly of the different molecularcomponents within VMD environment. As the linkersand/or connectors between multi-domain proteins are notalways clear during complex assemblies, Pdbswiss softwareallows the analysis of structural protein and/or nanos-tructure misalignments (rotation, translation, orientation).When the 3D structures are aligned, the molecularattachment between the different components is achievedvia VegaZZ software [25]. Finally, the psfgen tools generatea molecular structure file (:psf or :pdb format) associated tothe CAD-designed bio-nanorobotic structure: initial co-ordinates, force field parameters, structure solvation andionization.These data are sent to the MD module through a
communication protocol named interactive moleculardynamics (IMD) [26]. The IMD interface consists of asmall set of C-callable functions which can be adapted toany MD and/or visualization program. Periodically,VMD checks for a coordinate set from the MD program,updates the representation geometry if a new coordinateset was received, redraws the screen, and updates therestraint position of the haptic device. If the userhas applied a force through the haptic device, VMDroutes this force to NAMD2, which then integrates theforce into the equations of motion for the molecule.Updates to the haptic restraint point are made when VMDreceives a new coordinate set from NAMD2 software;while awaiting an update, the haptic server applies smoothfeedback forces based on the most recent restraint pointposition.The main program of the computational is NAMD2
[27], a fast, scalable, program which implements thepopular CHARMM27 force field [28] for MD. Theabsolute speed of NAMD2 is an essential ingredient of aresponsive interactive system. It runs on a parallel clustercomposed of 10 Pentium IV-4.1GHz personal computers.NAMD’s scalability enables us to use the computationalmodule to study large bio-nanorobotic structures as well assmall ones, given enough computational power. Thesecomputational resources allow the simulation of systemscontaining several thousand atoms at a speed sufficient forinteraction (�400Hz).
ARTICLE IN PRESSM. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201 193
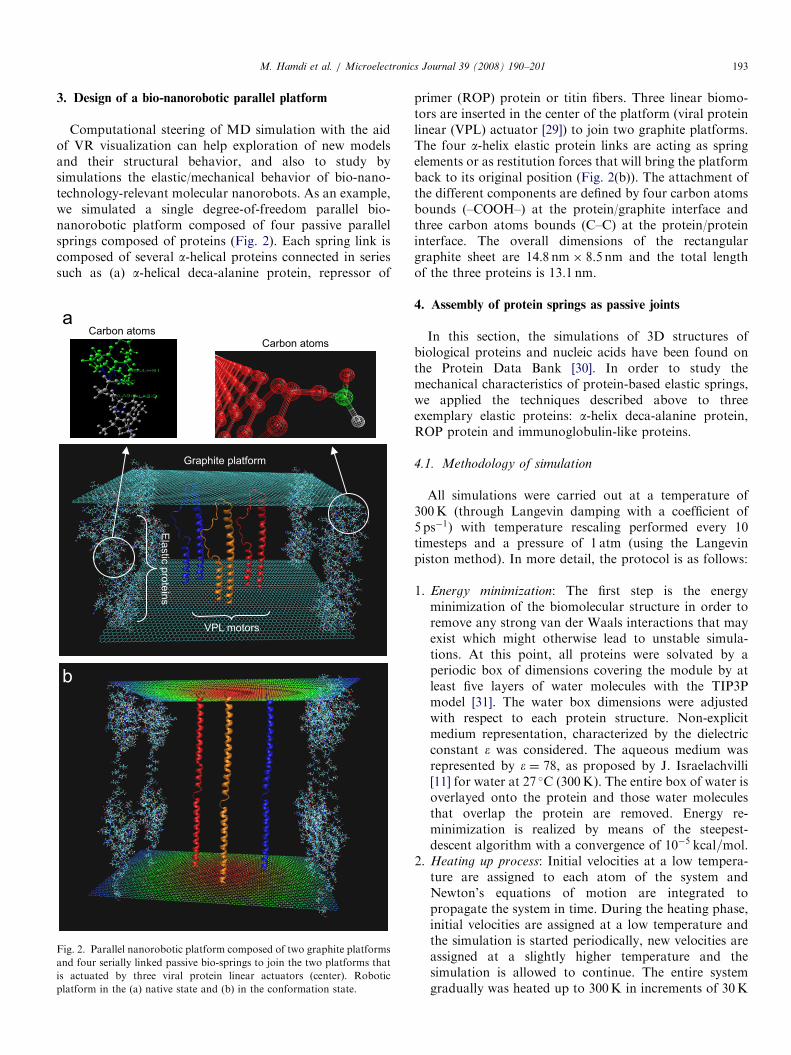
3. Design of a bio-nanorobotic parallel platform
Computational steering of MD simulation with the aidof VR visualization can help exploration of new modelsand their structural behavior, and also to study bysimulations the elastic/mechanical behavior of bio-nano-technology-relevant molecular nanorobots. As an example,we simulated a single degree-of-freedom parallel bio-nanorobotic platform composed of four passive parallelsprings composed of proteins (Fig. 2). Each spring link iscomposed of several a-helical proteins connected in seriessuch as (a) a-helical deca-alanine protein, repressor of
Graphite platform
Ela
stic
pro
tein
sVPL motors
Carbon atomsCarbon atoms
a
b
Fig. 2. Parallel nanorobotic platform composed of two graphite platforms
and four serially linked passive bio-springs to join the two platforms that
is actuated by three viral protein linear actuators (center). Robotic
platform in the (a) native state and (b) in the conformation state.
primer (ROP) protein or titin fibers. Three linear biomo-tors are inserted in the center of the platform (viral proteinlinear (VPL) actuator [29]) to join two graphite platforms.The four a-helix elastic protein links are acting as springelements or as restitution forces that will bring the platformback to its original position (Fig. 2(b)). The attachment ofthe different components are defined by four carbon atomsbounds (–COOH–) at the protein/graphite interface andthree carbon atoms bounds (C–C) at the protein/proteininterface. The overall dimensions of the rectangulargraphite sheet are 14:8 nm� 8:5 nm and the total lengthof the three proteins is 13.1 nm.
4. Assembly of protein springs as passive joints
In this section, the simulations of 3D structures ofbiological proteins and nucleic acids have been found onthe Protein Data Bank [30]. In order to study themechanical characteristics of protein-based elastic springs,we applied the techniques described above to threeexemplary elastic proteins: a-helix deca-alanine protein,ROP protein and immunoglobulin-like proteins.
4.1. Methodology of simulation
All simulations were carried out at a temperature of300K (through Langevin damping with a coefficient of5 ps�1) with temperature rescaling performed every 10timesteps and a pressure of 1 atm (using the Langevinpiston method). In more detail, the protocol is as follows:
1.
Energy minimization: The first step is the energyminimization of the biomolecular structure in order toremove any strong van der Waals interactions that mayexist which might otherwise lead to unstable simula-tions. At this point, all proteins were solvated by aperiodic box of dimensions covering the module by atleast five layers of water molecules with the TIP3Pmodel [31]. The water box dimensions were adjustedwith respect to each protein structure. Non-explicitmedium representation, characterized by the dielectricconstant � was considered. The aqueous medium wasrepresented by � ¼ 78, as proposed by J. Israelachvilli[11] for water at 27 �C (300K). The entire box of water isoverlayed onto the protein and those water moleculesthat overlap the protein are removed. Energy re-minimization is realized by means of the steepest-descent algorithm with a convergence of 10�5 kcal=mol.2.
Heating up process: Initial velocities at a low tempera-ture are assigned to each atom of the system andNewton’s equations of motion are integrated topropagate the system in time. During the heating phase,initial velocities are assigned at a low temperature andthe simulation is started periodically, new velocities areassigned at a slightly higher temperature and thesimulation is allowed to continue. The entire systemgradually was heated up to 300K in increments of 30K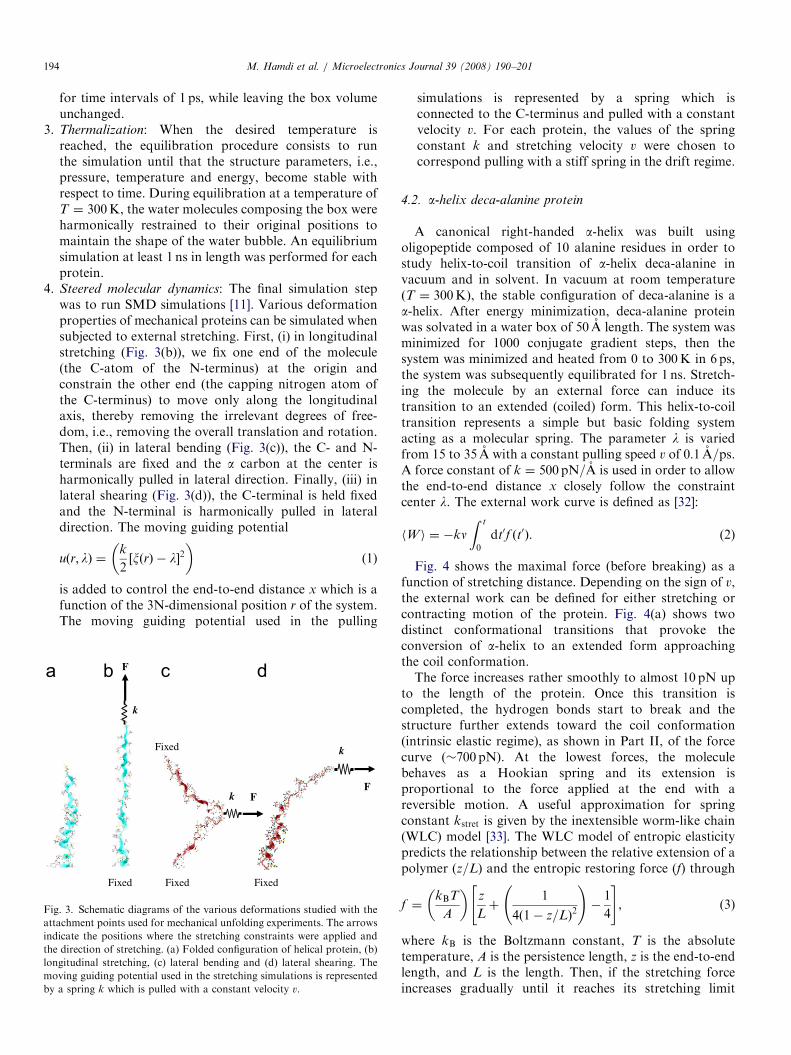
ARTICLE IN PRESS
a
Fig
att
ind
the
lon
mo
by
M. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201194
for time intervals of 1 ps, while leaving the box volumeunchanged.
3.
Thermalization: When the desired temperature isreached, the equilibration procedure consists to runthe simulation until that the structure parameters, i.e.,pressure, temperature and energy, become stable withrespect to time. During equilibration at a temperature ofT ¼ 300K, the water molecules composing the box wereharmonically restrained to their original positions tomaintain the shape of the water bubble. An equilibriumsimulation at least 1 ns in length was performed for eachprotein.4.
Steered molecular dynamics: The final simulation stepwas to run SMD simulations [11]. Various deformationproperties of mechanical proteins can be simulated whensubjected to external stretching. First, (i) in longitudinalstretching (Fig. 3(b)), we fix one end of the molecule(the C-atom of the N-terminus) at the origin andconstrain the other end (the capping nitrogen atom ofthe C-terminus) to move only along the longitudinalaxis, thereby removing the irrelevant degrees of free-dom, i.e., removing the overall translation and rotation.Then, (ii) in lateral bending (Fig. 3(c)), the C- and N-terminals are fixed and the a carbon at the center isharmonically pulled in lateral direction. Finally, (iii) inlateral shearing (Fig. 3(d)), the C-terminal is held fixedand the N-terminal is harmonically pulled in lateraldirection. The moving guiding potentialuðr; lÞ ¼k
2½xðrÞ � l�2
� �(1)
is added to control the end-to-end distance x which is afunction of the 3N-dimensional position r of the system.The moving guiding potential used in the pulling
FixedFixed
Fixed
Fixed
k
k
k
F
FF
b c d
. 3. Schematic diagrams of the various deformations studied with the
achment points used for mechanical unfolding experiments. The arrows
icate the positions where the stretching constraints were applied and
direction of stretching. (a) Folded configuration of helical protein, (b)
gitudinal stretching, (c) lateral bending and (d) lateral shearing. The
ving guiding potential used in the stretching simulations is represented
a spring k which is pulled with a constant velocity v.
simulations is represented by a spring which isconnected to the C-terminus and pulled with a constantvelocity v. For each protein, the values of the springconstant k and stretching velocity v were chosen tocorrespond pulling with a stiff spring in the drift regime.
4.2. a-helix deca-alanine protein
A canonical right-handed a-helix was built usingoligopeptide composed of 10 alanine residues in order tostudy helix-to-coil transition of a-helix deca-alanine invacuum and in solvent. In vacuum at room temperatureðT ¼ 300KÞ, the stable configuration of deca-alanine is aa-helix. After energy minimization, deca-alanine proteinwas solvated in a water box of 50 A length. The system wasminimized for 1000 conjugate gradient steps, then thesystem was minimized and heated from 0 to 300K in 6 ps,the system was subsequently equilibrated for 1 ns. Stretch-ing the molecule by an external force can induce itstransition to an extended (coiled) form. This helix-to-coiltransition represents a simple but basic folding systemacting as a molecular spring. The parameter l is variedfrom 15 to 35 A with a constant pulling speed v of 0:1 A=ps.A force constant of k ¼ 500 pN=A is used in order to allowthe end-to-end distance x closely follow the constraintcenter l. The external work curve is defined as [32]:
hW i ¼ �knZ t
0
dt0f ðt0Þ. (2)
Fig. 4 shows the maximal force (before breaking) as afunction of stretching distance. Depending on the sign of v,the external work can be defined for either stretching orcontracting motion of the protein. Fig. 4(a) shows twodistinct conformational transitions that provoke theconversion of a-helix to an extended form approachingthe coil conformation.The force increases rather smoothly to almost 10 pN up
to the length of the protein. Once this transition iscompleted, the hydrogen bonds start to break and thestructure further extends toward the coil conformation(intrinsic elastic regime), as shown in Part II, of the forcecurve (�700 pN). At the lowest forces, the moleculebehaves as a Hookian spring and its extension isproportional to the force applied at the end with areversible motion. A useful approximation for springconstant kstret is given by the inextensible worm-like chain(WLC) model [33]. The WLC model of entropic elasticitypredicts the relationship between the relative extension of apolymer (z=L) and the entropic restoring force (f) through
f ¼kBT
A
� �z
Lþ
1
4ð1� z=LÞ2
!�
1
4
" #, (3)
where kB is the Boltzmann constant, T is the absolutetemperature, A is the persistence length, z is the end-to-endlength, and L is the length. Then, if the stretching forceincreases gradually until it reaches its stretching limit
ARTICLE IN PRESS
Stretching distance (nm)
50
40
30
20
10
0
-10
Fo
rce
(p
N)
1 1.5 2 2.5 3 3.5 4 4.5 5
(I) (II) (III)
3.5
2.5
1.5
3
2
1
Dis
tan
ce
(n
m)
0 5 10 15 20 25
Time (pS)
stretching
relaxation
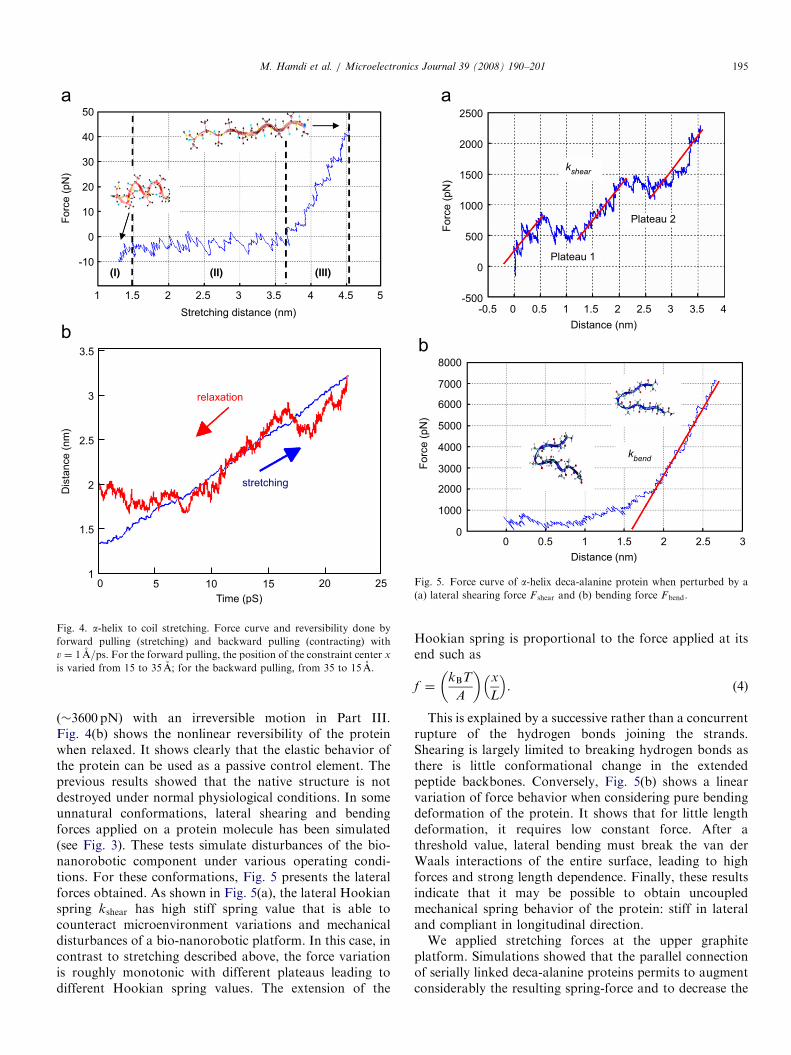
Fig. 4. a-helix to coil stretching. Force curve and reversibility done by
forward pulling (stretching) and backward pulling (contracting) with
v ¼ 1 A=ps. For the forward pulling, the position of the constraint center x
is varied from 15 to 35 A; for the backward pulling, from 35 to 15 A.
Distance (nm)
kbend
Forc
e (
pN
)
0 0.5 1 1.5 2 2.5 3
1000
2000
3000
4000
5000
6000
7000
8000
0
2500
2000
1500
1000
500
Fo
rce
(pN
)
0
-500-0.5 0.50 1 2 31.5 2.5 3.5 4
Distance (nm)
kshear
Plateau 2
Plateau 1
Fig. 5. Force curve of a-helix deca-alanine protein when perturbed by a
(a) lateral shearing force F shear and (b) bending force Fbend.
M. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201 195
(�3600 pN) with an irreversible motion in Part III.Fig. 4(b) shows the nonlinear reversibility of the proteinwhen relaxed. It shows clearly that the elastic behavior ofthe protein can be used as a passive control element. Theprevious results showed that the native structure is notdestroyed under normal physiological conditions. In someunnatural conformations, lateral shearing and bendingforces applied on a protein molecule has been simulated(see Fig. 3). These tests simulate disturbances of the bio-nanorobotic component under various operating condi-tions. For these conformations, Fig. 5 presents the lateralforces obtained. As shown in Fig. 5(a), the lateral Hookianspring kshear has high stiff spring value that is able tocounteract microenvironment variations and mechanicaldisturbances of a bio-nanorobotic platform. In this case, incontrast to stretching described above, the force variationis roughly monotonic with different plateaus leading todifferent Hookian spring values. The extension of the
Hookian spring is proportional to the force applied at itsend such as
f ¼kBT
A
� �x
L
� �. (4)
This is explained by a successive rather than a concurrentrupture of the hydrogen bonds joining the strands.Shearing is largely limited to breaking hydrogen bonds asthere is little conformational change in the extendedpeptide backbones. Conversely, Fig. 5(b) shows a linearvariation of force behavior when considering pure bendingdeformation of the protein. It shows that for little lengthdeformation, it requires low constant force. After athreshold value, lateral bending must break the van derWaals interactions of the entire surface, leading to highforces and strong length dependence. Finally, these resultsindicate that it may be possible to obtain uncoupledmechanical spring behavior of the protein: stiff in lateraland compliant in longitudinal direction.We applied stretching forces at the upper graphite
platform. Simulations showed that the parallel connectionof serially linked deca-alanine proteins permits to augmentconsiderably the resulting spring-force and to decrease the
ARTICLE IN PRESSM. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201196
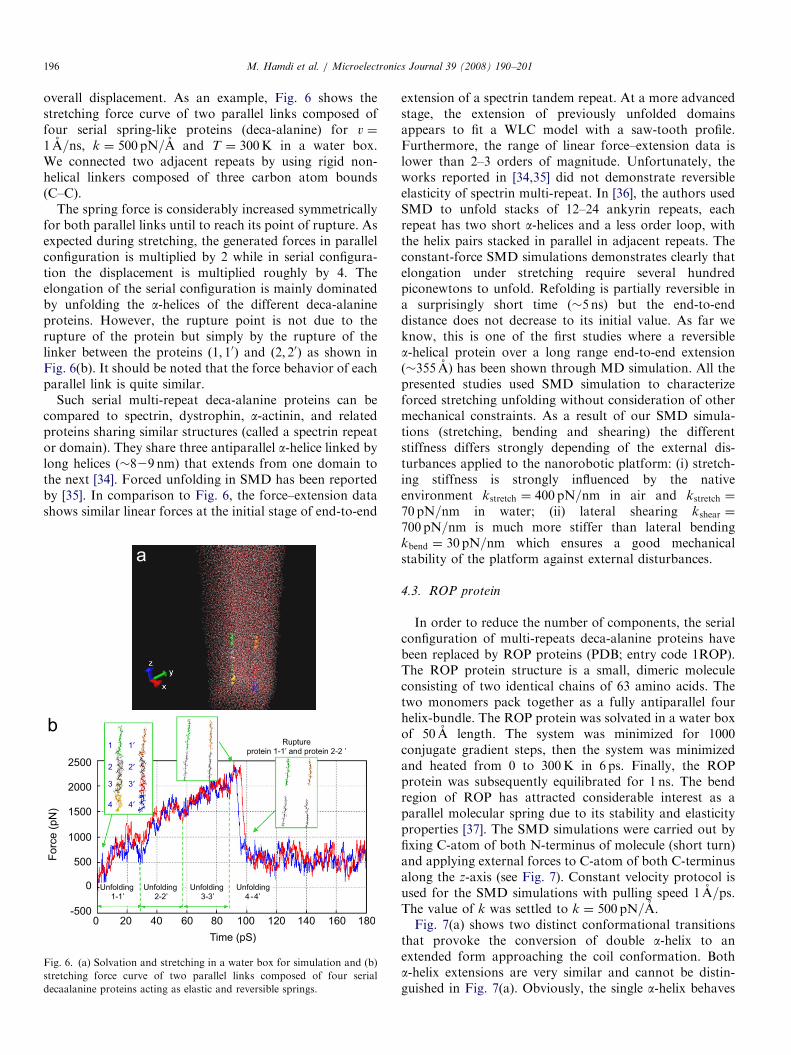
overall displacement. As an example, Fig. 6 shows thestretching force curve of two parallel links composed offour serial spring-like proteins (deca-alanine) for v ¼
1 A=ns, k ¼ 500 pN=A and T ¼ 300K in a water box.We connected two adjacent repeats by using rigid non-helical linkers composed of three carbon atom bounds(C–C).
The spring force is considerably increased symmetricallyfor both parallel links until to reach its point of rupture. Asexpected during stretching, the generated forces in parallelconfiguration is multiplied by 2 while in serial configura-tion the displacement is multiplied roughly by 4. Theelongation of the serial configuration is mainly dominatedby unfolding the a-helices of the different deca-alanineproteins. However, the rupture point is not due to therupture of the protein but simply by the rupture of thelinker between the proteins ð1; 10Þ and ð2; 20Þ as shown inFig. 6(b). It should be noted that the force behavior of eachparallel link is quite similar.
Such serial multi-repeat deca-alanine proteins can becompared to spectrin, dystrophin, a-actinin, and relatedproteins sharing similar structures (called a spectrin repeator domain). They share three antiparallel a-helice linked bylong helices (�829 nm) that extends from one domain tothe next [34]. Forced unfolding in SMD has been reportedby [35]. In comparison to Fig. 6, the force–extension datashows similar linear forces at the initial stage of end-to-end
Unfolding 3-3’
Unfolding 2-2’
Unfolding 1-1’
Unfolding 4 -4’
Ruptureprotein 1-1’ and protein 2-2 ’
Fo
rce
(p
N) 4
3
2
1
4′
3′
2′
1′
Time (pS)
2500
2000
1500
1000
500
-500
0
0 20 40 60 80 100 120 140 160 180
b
a
Fig. 6. (a) Solvation and stretching in a water box for simulation and (b)
stretching force curve of two parallel links composed of four serial
decaalanine proteins acting as elastic and reversible springs.
extension of a spectrin tandem repeat. At a more advancedstage, the extension of previously unfolded domainsappears to fit a WLC model with a saw-tooth profile.Furthermore, the range of linear force–extension data islower than 2–3 orders of magnitude. Unfortunately, theworks reported in [34,35] did not demonstrate reversibleelasticity of spectrin multi-repeat. In [36], the authors usedSMD to unfold stacks of 12–24 ankyrin repeats, eachrepeat has two short a-helices and a less order loop, withthe helix pairs stacked in parallel in adjacent repeats. Theconstant-force SMD simulations demonstrates clearly thatelongation under stretching require several hundredpiconewtons to unfold. Refolding is partially reversible ina surprisingly short time (�5 ns) but the end-to-enddistance does not decrease to its initial value. As far weknow, this is one of the first studies where a reversiblea-helical protein over a long range end-to-end extension(�355 A) has been shown through MD simulation. All thepresented studies used SMD simulation to characterizeforced stretching unfolding without consideration of othermechanical constraints. As a result of our SMD simula-tions (stretching, bending and shearing) the differentstiffness differs strongly depending of the external dis-turbances applied to the nanorobotic platform: (i) stretch-ing stiffness is strongly influenced by the nativeenvironment kstretch ¼ 400 pN=nm in air and kstretch ¼
70 pN=nm in water; (ii) lateral shearing kshear ¼
700 pN=nm is much more stiffer than lateral bendingkbend ¼ 30 pN=nm which ensures a good mechanicalstability of the platform against external disturbances.
4.3. ROP protein
In order to reduce the number of components, the serialconfiguration of multi-repeats deca-alanine proteins havebeen replaced by ROP proteins (PDB; entry code 1ROP).The ROP protein structure is a small, dimeric moleculeconsisting of two identical chains of 63 amino acids. Thetwo monomers pack together as a fully antiparallel fourhelix-bundle. The ROP protein was solvated in a water boxof 50 A length. The system was minimized for 1000conjugate gradient steps, then the system was minimizedand heated from 0 to 300K in 6 ps. Finally, the ROPprotein was subsequently equilibrated for 1 ns. The bendregion of ROP has attracted considerable interest as aparallel molecular spring due to its stability and elasticityproperties [37]. The SMD simulations were carried out byfixing C-atom of both N-terminus of molecule (short turn)and applying external forces to C-atom of both C-terminusalong the z-axis (see Fig. 7). Constant velocity protocol isused for the SMD simulations with pulling speed 1 A=ps.The value of k was settled to k ¼ 500 pN=A.Fig. 7(a) shows two distinct conformational transitions
that provoke the conversion of double a-helix to anextended form approaching the coil conformation. Botha-helix extensions are very similar and cannot be distin-guished in Fig. 7(a). Obviously, the single a-helix behaves
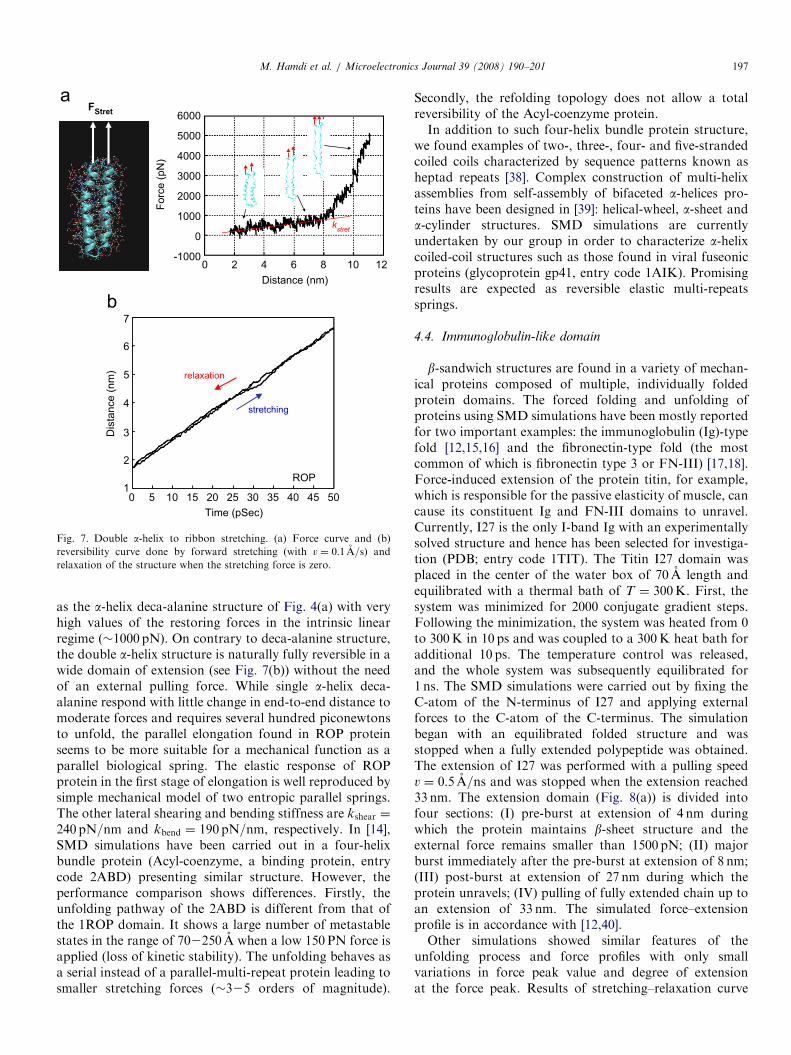
ARTICLE IN PRESS
kstret
FStret
relaxation
ROP
6000
5000
4000
3000
2000
1000
Forc
e (
pN
)
0
-10000 2 4 6 8 10 12
Distance (nm)
7
6
5
4
3
2
1
Dis
tance (
nm
)
stretching
0 5 10 15 20 25 30 35 40 45 50
Time (pSec)
Fig. 7. Double a-helix to ribbon stretching. (a) Force curve and (b)
reversibility curve done by forward stretching (with v ¼ 0:1 A=s) and
relaxation of the structure when the stretching force is zero.
M. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201 197
as the a-helix deca-alanine structure of Fig. 4(a) with veryhigh values of the restoring forces in the intrinsic linearregime (�1000 pN). On contrary to deca-alanine structure,the double a-helix structure is naturally fully reversible in awide domain of extension (see Fig. 7(b)) without the needof an external pulling force. While single a-helix deca-alanine respond with little change in end-to-end distance tomoderate forces and requires several hundred piconewtonsto unfold, the parallel elongation found in ROP proteinseems to be more suitable for a mechanical function as aparallel biological spring. The elastic response of ROPprotein in the first stage of elongation is well reproduced bysimple mechanical model of two entropic parallel springs.The other lateral shearing and bending stiffness are kshear ¼
240 pN=nm and kbend ¼ 190 pN=nm, respectively. In [14],SMD simulations have been carried out in a four-helixbundle protein (Acyl-coenzyme, a binding protein, entrycode 2ABD) presenting similar structure. However, theperformance comparison shows differences. Firstly, theunfolding pathway of the 2ABD is different from that ofthe 1ROP domain. It shows a large number of metastablestates in the range of 702250 A when a low 150 PN force isapplied (loss of kinetic stability). The unfolding behaves asa serial instead of a parallel-multi-repeat protein leading tosmaller stretching forces (�325 orders of magnitude).
Secondly, the refolding topology does not allow a totalreversibility of the Acyl-coenzyme protein.In addition to such four-helix bundle protein structure,
we found examples of two-, three-, four- and five-strandedcoiled coils characterized by sequence patterns known asheptad repeats [38]. Complex construction of multi-helixassemblies from self-assembly of bifaceted a-helices pro-teins have been designed in [39]: helical-wheel, a-sheet anda-cylinder structures. SMD simulations are currentlyundertaken by our group in order to characterize a-helixcoiled-coil structures such as those found in viral fuseonicproteins (glycoprotein gp41, entry code 1AIK). Promisingresults are expected as reversible elastic multi-repeatssprings.
4.4. Immunoglobulin-like domain
b-sandwich structures are found in a variety of mechan-ical proteins composed of multiple, individually foldedprotein domains. The forced folding and unfolding ofproteins using SMD simulations have been mostly reportedfor two important examples: the immunoglobulin (Ig)-typefold [12,15,16] and the fibronectin-type fold (the mostcommon of which is fibronectin type 3 or FN-III) [17,18].Force-induced extension of the protein titin, for example,which is responsible for the passive elasticity of muscle, cancause its constituent Ig and FN-III domains to unravel.Currently, I27 is the only I-band Ig with an experimentallysolved structure and hence has been selected for investiga-tion (PDB; entry code 1TIT). The Titin I27 domain wasplaced in the center of the water box of 70 A length andequilibrated with a thermal bath of T ¼ 300K. First, thesystem was minimized for 2000 conjugate gradient steps.Following the minimization, the system was heated from 0to 300K in 10 ps and was coupled to a 300K heat bath foradditional 10 ps. The temperature control was released,and the whole system was subsequently equilibrated for1 ns. The SMD simulations were carried out by fixing theC-atom of the N-terminus of I27 and applying externalforces to the C-atom of the C-terminus. The simulationbegan with an equilibrated folded structure and wasstopped when a fully extended polypeptide was obtained.The extension of I27 was performed with a pulling speedv ¼ 0:5 A=ns and was stopped when the extension reached33 nm. The extension domain (Fig. 8(a)) is divided intofour sections: (I) pre-burst at extension of 4 nm duringwhich the protein maintains b-sheet structure and theexternal force remains smaller than 1500 pN; (II) majorburst immediately after the pre-burst at extension of 8 nm;(III) post-burst at extension of 27 nm during which theprotein unravels; (IV) pulling of fully extended chain up toan extension of 33 nm. The simulated force–extensionprofile is in accordance with [12,40].Other simulations showed similar features of the
unfolding process and force profiles with only smallvariations in force peak value and degree of extensionat the force peak. Results of stretching–relaxation curve
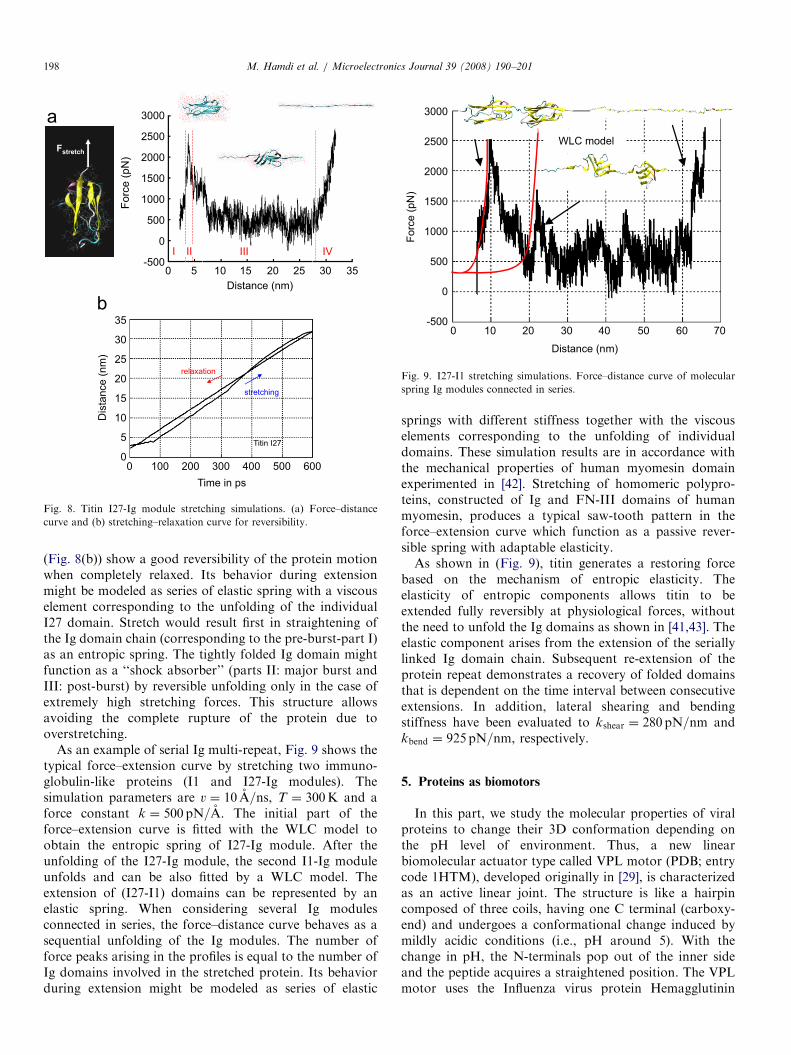
ARTICLE IN PRESS
I III IVII
Fstretch
Titin I27
stretching
relaxation
3000
2500
2000
1500
1000
500
-500
Distance (nm)
Fo
rce
(p
N)
0
0 5 10 15 20 25 30 35
35
30
25
20
15
10
5
0
Dis
tance (
nm
)
0 100 200 300 400 500 600
Time in ps
a
b
Fig. 8. Titin I27-Ig module stretching simulations. (a) Force–distance
curve and (b) stretching–relaxation curve for reversibility.
3000
2500
2000
1500
1000
Forc
e (
pN
)
500
0
-5000 10 20 30 40 50 60 70
Distance (nm)
WLC model
Fig. 9. I27-I1 stretching simulations. Force–distance curve of molecular
spring Ig modules connected in series.
M. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201198
(Fig. 8(b)) show a good reversibility of the protein motionwhen completely relaxed. Its behavior during extensionmight be modeled as series of elastic spring with a viscouselement corresponding to the unfolding of the individualI27 domain. Stretch would result first in straightening ofthe Ig domain chain (corresponding to the pre-burst-part I)as an entropic spring. The tightly folded Ig domain mightfunction as a ‘‘shock absorber’’ (parts II: major burst andIII: post-burst) by reversible unfolding only in the case ofextremely high stretching forces. This structure allowsavoiding the complete rupture of the protein due tooverstretching.
As an example of serial Ig multi-repeat, Fig. 9 shows thetypical force–extension curve by stretching two immuno-globulin-like proteins (I1 and I27-Ig modules). Thesimulation parameters are v ¼ 10 A=ns, T ¼ 300K and aforce constant k ¼ 500 pN=A. The initial part of theforce–extension curve is fitted with the WLC model toobtain the entropic spring of I27-Ig module. After theunfolding of the I27-Ig module, the second I1-Ig moduleunfolds and can be also fitted by a WLC model. Theextension of (I27-I1) domains can be represented by anelastic spring. When considering several Ig modulesconnected in series, the force–distance curve behaves as asequential unfolding of the Ig modules. The number offorce peaks arising in the profiles is equal to the number ofIg domains involved in the stretched protein. Its behaviorduring extension might be modeled as series of elastic
springs with different stiffness together with the viscouselements corresponding to the unfolding of individualdomains. These simulation results are in accordance withthe mechanical properties of human myomesin domainexperimented in [42]. Stretching of homomeric polypro-teins, constructed of Ig and FN-III domains of humanmyomesin, produces a typical saw-tooth pattern in theforce–extension curve which function as a passive rever-sible spring with adaptable elasticity.As shown in (Fig. 9), titin generates a restoring force
based on the mechanism of entropic elasticity. Theelasticity of entropic components allows titin to beextended fully reversibly at physiological forces, withoutthe need to unfold the Ig domains as shown in [41,43]. Theelastic component arises from the extension of the seriallylinked Ig domain chain. Subsequent re-extension of theprotein repeat demonstrates a recovery of folded domainsthat is dependent on the time interval between consecutiveextensions. In addition, lateral shearing and bendingstiffness have been evaluated to kshear ¼ 280 pN=nm andkbend ¼ 925 pN=nm, respectively.
5. Proteins as biomotors
In this part, we study the molecular properties of viralproteins to change their 3D conformation depending onthe pH level of environment. Thus, a new linearbiomolecular actuator type called VPL motor (PDB; entrycode 1HTM), developed originally in [29], is characterizedas an active linear joint. The structure is like a hairpincomposed of three coils, having one C terminal (carboxy-end) and undergoes a conformational change induced bymildly acidic conditions (i.e., pH around 5). With thechange in pH, the N-terminals pop out of the inner sideand the peptide acquires a straightened position. The VPLmotor uses the Influenza virus protein Hemagglutinin
ARTICLE IN PRESS
kstret
kshear
0 1 2 3 4 5 6 7 8
2500
2000
1500
1000
500
0
-500
kbend
0 1 2 3 4 5 6 7
1000
2000
3000
4000
5000
6000
7000
0
Distance (nm)
Forc
e (
pN
)F
orc
e (
pN
)F
orc
e (
pN
)
45004000350030002500200015001000500
-5000
2 3 4 5 6 7 8 9 10 11
Distance (nm)
M. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201 199
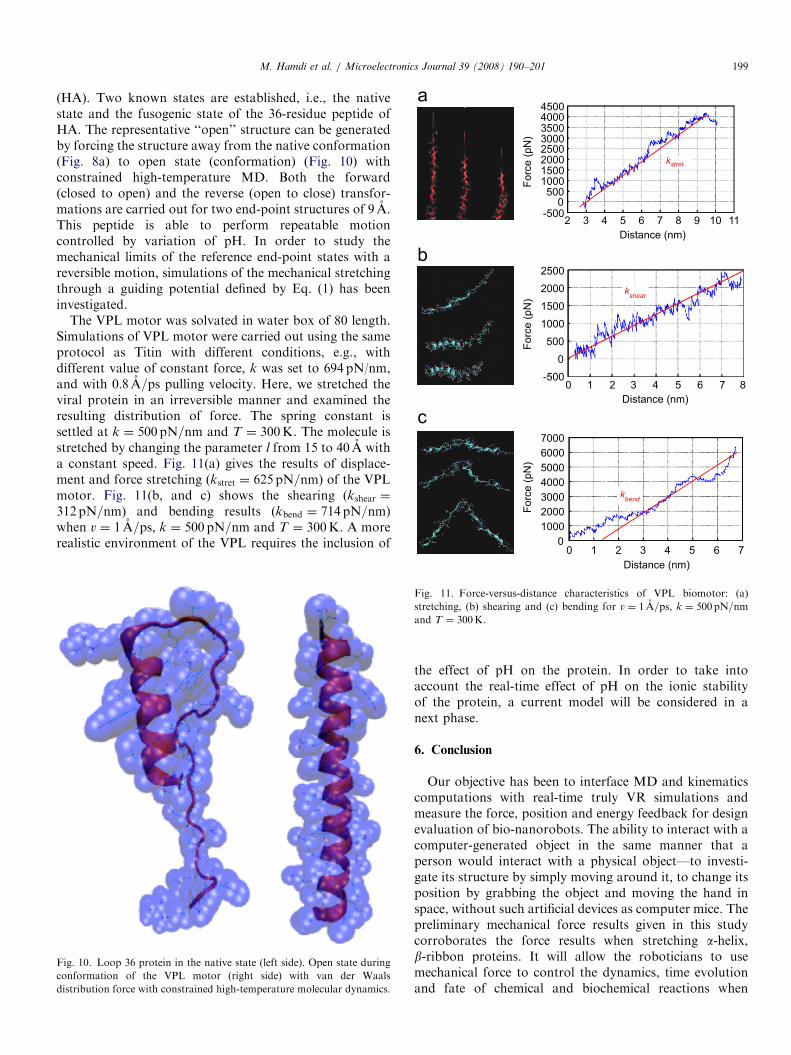
(HA). Two known states are established, i.e., the nativestate and the fusogenic state of the 36-residue peptide ofHA. The representative ‘‘open’’ structure can be generatedby forcing the structure away from the native conformation(Fig. 8a) to open state (conformation) (Fig. 10) withconstrained high-temperature MD. Both the forward(closed to open) and the reverse (open to close) transfor-mations are carried out for two end-point structures of 9 A.This peptide is able to perform repeatable motioncontrolled by variation of pH. In order to study themechanical limits of the reference end-point states with areversible motion, simulations of the mechanical stretchingthrough a guiding potential defined by Eq. (1) has beeninvestigated.
The VPL motor was solvated in water box of 80 length.Simulations of VPL motor were carried out using the sameprotocol as Titin with different conditions, e.g., withdifferent value of constant force, k was set to 694 pN/nm,and with 0:8 A=ps pulling velocity. Here, we stretched theviral protein in an irreversible manner and examined theresulting distribution of force. The spring constant issettled at k ¼ 500 pN=nm and T ¼ 300K. The molecule isstretched by changing the parameter l from 15 to 40 A witha constant speed. Fig. 11(a) gives the results of displace-ment and force stretching (kstret ¼ 625 pN=nm) of the VPLmotor. Fig. 11(b, and c) shows the shearing (kshear ¼
312 pN=nm) and bending results (kbend ¼ 714 pN=nm)when v ¼ 1 A=ps, k ¼ 500 pN=nm and T ¼ 300K. A morerealistic environment of the VPL requires the inclusion of
Fig. 10. Loop 36 protein in the native state (left side). Open state during
conformation of the VPL motor (right side) with van der Waals
distribution force with constrained high-temperature molecular dynamics.
Distance (nm)
Fig. 11. Force-versus-distance characteristics of VPL biomotor: (a)
stretching, (b) shearing and (c) bending for v ¼ 1 A=ps, k ¼ 500pN=nmand T ¼ 300K.
the effect of pH on the protein. In order to take intoaccount the real-time effect of pH on the ionic stabilityof the protein, a current model will be considered in anext phase.
6. Conclusion
Our objective has been to interface MD and kinematicscomputations with real-time truly VR simulations andmeasure the force, position and energy feedback for designevaluation of bio-nanorobots. The ability to interact with acomputer-generated object in the same manner that aperson would interact with a physical object—to investi-gate its structure by simply moving around it, to change itsposition by grabbing the object and moving the hand inspace, without such artificial devices as computer mice. Thepreliminary mechanical force results given in this studycorroborates the force results when stretching a-helix,b-ribbon proteins. It will allow the roboticians to usemechanical force to control the dynamics, time evolutionand fate of chemical and biochemical reactions when

ARTICLE IN PRESSM. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201200
connecting in series or parallel different bio-nanoroboticcomponents together. In the next step, we will integrate theexperimental and computational process by making apeptide-AFM-Phantom-VMD-NAMD system.
Acknowledgments
The authors would like to thank the NIH NationalResearch Resource in Molecular Graphics and Microscopyat the University of North Carolina at Chapel Hill,supported by the NIH National Center for ResearchResources and the NIH National Institute of BiomedicalImaging and Bioengineering for providing their help inNAMD2 and VRPN softwares. The work at NortheasternUniversity was supported by the National Science Founda-tion grant DMI-0422724. Any opinions, findings, conclu-sions or recommendations expressed in this publication arethose of the authors and do not necessarily reflect the viewsof the National Science Foundation.
References
[1] K. Kitamura, M. Tokunaga, A.H. Iwane, A. Yanada, A single
myosin head moves along an actin filament with rectangular steps of
5.3 nm, Nature 397 (1999) 129.
[2] C. Shingyoji, H. Higuchi, M. Yoshimura, E. Katayama, T. Yanagida,
Dynein arms are oscillating force generators, Nature 393 (2001)
711–714.
[3] C.D. Montegano, G.D. Bachand, Constructing nanomechanical
devices powered by biomolecular motors, Nanotechnology 10
(1999) 225–331.
[4] H.P. Erickson, Stretching single protein molecules: titin as a wired
spring, Science 276 (2003) 1090–1092.
[5] L. Mahadevan, P. Matsudaira, Mobility powered by supramolecular
springs and ratchets, Science J288 (2000) 95–99.
[6] M. Hamdi, G. Sharma, A. Ferreira, D. Mavroidis, Characterization
of protein based spring-like elastic joints for biorobotic applications,
in: IEEE International Conference on Robotics and Automation,
May 15–19, Orlando, FL, 2006, pp. 1794–1799.
[7] A. Ferreira, G. Sharma, D. Mavroidis, New trends in bio-
nanorobotics using virtual reality technologies, in: IEEE Interna-
tional Conference on Robotics and Biomimetics, June 30–July 3,
Hong Kong, 2005, pp. 89–94.
[8] H. Haase, J. Strassner, F. Dai, VR techniques for the investigation of
molecule data, in: Computers and Graphics, Special Issue on Virtual
Reality, Elsevier Science Ltd, Amsterdam, 1996, pp. 207–217.
[9] R.C. Drees, J. Pleiss, D. Roller, R.D. Schmid, Highly immersive
molecular modeling (HIMM): an architecture for the integration of
molecular modeling and virtual reality, Computer Science and
Biology, German Conference on Bioinformatics, Leipzig, Germany,
1996, pp. 190–192.
[10] M. Hamdi, G. Sharma, A. Ferreira, D. Mavroidis, Molecular
mechanics study on bio-nanorobotic components using force-feed-
back, in: IEEE International Conference on Robotics and Biomi-
metics, June 30–July 3, Hong Kong, 2005, pp. 105–110.
[11] B. Isralewitz, J. Baudry, J. Gullingsrud, D. Kosztin, K. Schulten,
Steered molecular dynamics investigations of protein function,
J. Mol. Graphic Model 19 (12) (2001) 13–25.
[12] H. Lu, B. Isralewitz, A. Kramer, V. Vogel, K. Schulten, Unfolding of
titin immunoglobulin domains by steered dynamics simulation,
Biophys. J. 75 (1998) 662–671.
[13] A. Minajeva, M. Kulke, J.M. Fernandez, W.A. Linke, Unfolding of
titin domains explains the viscoelastic behavior of skeletal micro-
fibrils, Biophys. J. 80 (2001) 1442–1451.
[14] E. Paci, M. Karplus, Unfolding proteins by external forces and
temperature: the importance of topology and energetics, Proc. Natl.
Acad. Sci. USA 97 (2000) 6521–6526.
[15] H. Granzier, S. Labeit, Cardiac titin: an adjustable multi-functional
spring, J. Physiol. 541 (2002) 335–342.
[16] H. Lu, K. Schulten, Steered molecular dynamics simulations
of force-induced protein domain unfolding, Proteins 35 (1999)
453–463.
[17] M. Gao, D. Graig, V. Vogel, K. Schulten, Identifying unfolding
intermediates of FN � III10 by steered molecular dynamics, Matrix
Biol. 232 (2002) 939–950.
[18] E. Paci, M. Karplus, Forced unfolding of fibronectin type 3 modules:
an analysis by biased molecular dynamics simulation, J. Mol. Biol.
288 (1999) 441–459.
[19] A.C. Lorenzo, E.R. Caffarena, Elastic properties, Young’s modulus
determination and structural stability of the tropocollagen molecule:
a computational study by steered molecular dynamics, J. Biomech. 38
(2005) 1527–1533.
[20] Y. Astier, H. Bayley, S. Howorka, Protein components for
nanodevices, Curr. Opin. Chem. Biol. 9 (2005) 576–584.
[21] S.G. Wang, R. Wang, P.J. Sellin, Q. Zhang, DNA biosensors based
on self-assembled carbon nanotubes, J. Mol. Graphics 14 (1996)
33–38.
[22] H. Yan, S.H. Park, G. Finkelstein, J.H. Reif, T.H. LaBean, DNA-
templated self-assembly of protein arrays and highly conductive
nanowires, Science 301 (2003) 1882–1883.
[23] W. Humphrey, A. Dalke, K. Schulten, VMD: visual molecular
dynamics, J. Mol. Graphics 14 (1996) 33–38.
[24] R.M. Taylor II, VRPN: virtual reality peripheral network, hhttp://
www.cs.unc.edu/Research/vrpni.
[25] hhttp://www.ddl.unimi.it/vega/i.
[26] J. Stone, J. Gullingstrud, P. Grayon, K. Schulten, A system for
interactive molecular dynamics simulation, in: 2001 ACM Sympo-
sium on Interactive 3D Graphics, ACM SIGGRAPH, New York,
2001, pp. 191–194.
[27] M. Nelson, W. Humphrey, A. Gursoy, A. Dalke, L. Kale, R.D.
Skeel, K. Schulten, NAMD—a parallel object-oriented molecular
dynamics program, Int. J. Supercomput. Appl. High. Perform.
Comput. 10 (1996) 251.
[28] A.D. MacKerell, D. Bshford, M. Bellott, R.L. Dunbrack, J.D.
Evanseck, M.J. Field, et al., All-atom empirical potential for
molecular modeling and dynamics studies of proteins, J. Phys. Chem.
B 102 (1998) 3586–3616.
[29] A. Dubey, C. Mavroidis, A. Thornton, K.P. Nikitczuk, M.L.
Yarmush, Viral protein linear (VPL) nano-actuators, in: IEEE
International Conference on Nanotechnology, San Francisco, Au-
gust, 2003, pp. 12–14.
[30] H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H.
Weissig, I.N. Shindyalov, P.E. Bourne, The protein data bank, Nucl.
Acid. Res. 28 (2000) 235–242.
[31] W.L. Jorgenson, J. Chandrasekhar, D. Madura, R.W. Impey,
Comparison of simple potential functions for simulating liquid
water, J. Chem. Phys. 79 (1983) 926–935.
[32] S. Park, F. Khalili-Araghi, E. Tajkhorshid, K. Schulten, Free energy
calculation from steered molecular dynamics simulations using
Jarzynski’s equality, J. Chem. Phys. 119 (6) (2003) 3559–3566.
[33] C. Bustamante, S.B. Smith, J. Liphardt, D. Smith, Single-molecule
studies of DNA mechanics, Struct. Biol. 10 (2000) 279–285.
[34] R. Law, P. Carl, S. Harper, P. Dalhaimer, D.W. Speicher, D.E.
Discher, Cooperativity in forced unfolding of tandem spectrin
repeats, Biophys. J. 84 (2003) 533–544.
[35] V. Ortiz, S.E. Nielsen, M.L. Klein, D.E. Discher, Unfolding a linker
between helical repeats, Biophys. J. 84 (2003) 533–544.
[36] M. Sotomayor, D.P. Corey, K. Schulten, In search of the hair-cell
spring: elastic properties of ankyrin and cadherin repeats, Structure
13 (2005) 669–682.
[37] H.P. Kresse, M. Czuayko, G. Nyakatura, G. Vriand, C. Sander, H.
Bloecker, Four-helix bundle topology re-engineered: monomeric Rop

ARTICLE IN PRESSM. Hamdi et al. / Microelectronics Journal 39 (2008) 190–201 201
protein variants with different loop arrangements, Protein Eng. 14
(11) (2001) 897–901.
[38] J. Walshaw, D.N. Woolfson, Extended knobs-into-holes packing in
classical and complex coiled-coil assemblies, J. Struct. Biol. 144
(2003) 349–361.
[39] J. Walshaw, D.N. Woolfson, Open-and-shut cases in coiled-coil
assembly: a-cylinders, Protein Sci. 10 (2001) 668–673.
[40] K. Trombitas, M. Greaser, S. Labeit, J.-P. Jin, M. Kellermayer, M.
Helmes, H. Granzier, Titin extensibility in situ: entropic elasticity of
permanently folded and permanently unfolded molecular segments,
J. Cell Biol. 140 (4) (1998) 853–859.
[41] R. Schoenauer, P. Bertoncini, G. Machaidze, U. Aebi, J.-C.
Perriard, M. Hegner, I. Agarkova, Myomesin is a mole-
cular spring with adaptable elasticity, J. Mol. Biol. 349 (2005)
367–379.
[42] A.A. Vazina, N.F. Lanina, D.G. Alexeev, W. Bras, I.P. Dolbnya, The
structural principles of multidomain organization of the giant
polypeptide chain of the muscle titin protein: SAXS/WAXS studies
during the stretching of oriented titin fibres, J. Struct. Biol. 155 (2)
(2006) 251–262.
[43] M. Gao, H. Lu, K. Schulten, Simulated refolding of stretched titin
immunoglobulin domain, Biophys. J. 81 (2001) 2268–2277.
Related Documents











