Protein structure determination & prediction

Protein structure determination & prediction
Dec 30, 2015
Protein structure determination & prediction. Tertiary protein structure: protein folding. Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2] Comparative modeling (based on homology) [3] Ab initio (de novo) prediction - PowerPoint PPT Presentation
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Protein structure determination & prediction

Tertiary protein structure: protein folding
Three main approaches:
[1] experimental determination (X-ray crystallography, NMR)
[2] Comparative modeling (based on homology)
[3] Ab initio (de novo) prediction (Dr. Ingo Ruczinski at JHSPH)

Experimental approaches to protein structure
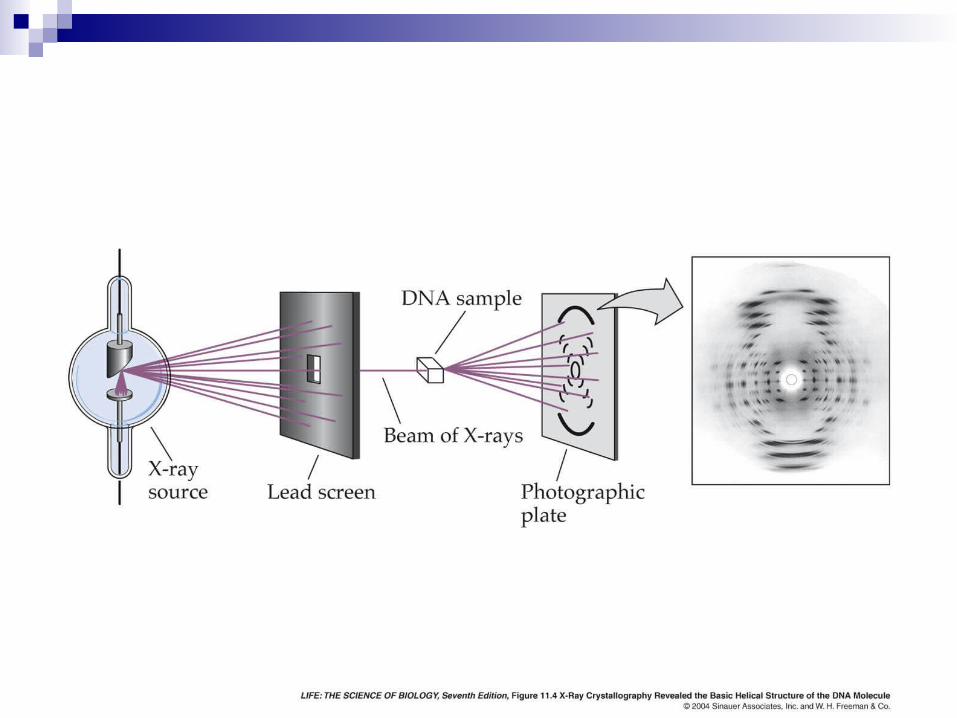
[1] X-ray crystallography-- Used to determine 80% of structures-- Requires high protein concentration-- Requires crystals-- Able to trace amino acid side chains-- Earliest structure solved was myoglobin
[2] NMR-- Magnetic field applied to proteins in solution-- Largest structures: 350 amino acids (40 kD)-- Does not require crystallization

Steps in obtaining a protein structure
Target selection
Obtain, characterize protein
Determine, refine, model the structure
Deposit in database

X-ray crystallographyhttp://en.wikipedia.org/wiki/X-ray_diffraction
Sperm Whale Myoglobin

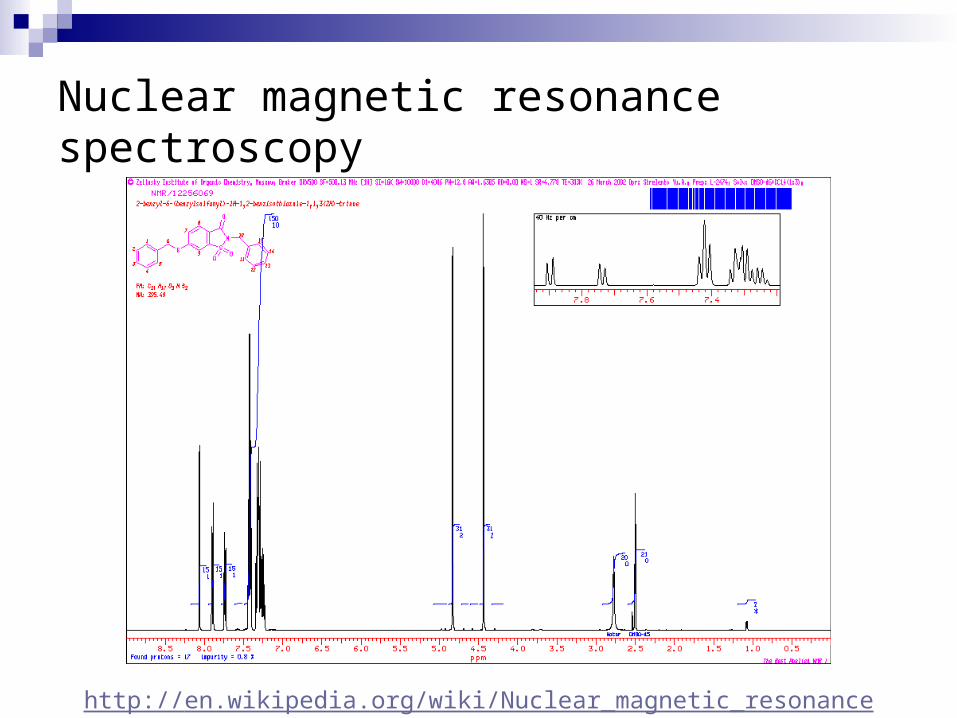
Nuclear magnetic resonance spectroscopy
http://en.wikipedia.org/wiki/Nuclear_magnetic_resonance

Article

Ab initio protein prediction
Starts with an attempt to derive secondary structure from the amino acid sequence Predicting the likelihood that a subsequence will fold into an
alpha-helix, beta-sheet, or coil, using physicochemical parameters or HMMs and ANNs
Able to accurately predict 3/4 of all local structures

Secondary structure prediction
Chou and Fasman (1974) developed an algorithmbased on the frequencies of amino acids found in helices, -sheets, and turns.
Proline: occurs at turns, but not in helices.
GOR (Garnier, Osguthorpe, Robson): related algorithm
Modern algorithms: use multiple sequence alignmentsand achieve higher success rate (about 70-75%)
Page 279-280



Fold recognition (structural profiles)
Attempts to find the best fit of a raw polypeptide sequence onto a library of known protein folds
A prediction of the secondary structure of the unknown is made and compared with the secondary structure of each member of the library of folds

Threading
Takes the fold recognition process a step further:Empirical-energy functions for residue pair
interactions are used to mount the unknown onto the putative backbone in the best possible manner
Related Documents






![PROTEIN STRUCTURE PREDICTION IIzhanglab.ccmb.med.umich.edu/papers/2007_7.pdfprotein structure determination [12].Another is to provide a complete library of solved protein structures](https://static.cupdf.com/doc/110x72/5f4f7883a1dae1214c7e89eb/protein-structure-prediction-protein-structure-determination-12another-is-to.jpg)




