Protein Kinase A Phosphorylation of Human Phosphodiesterase 3B Promotes 14-3-3 Protein Binding and Inhibits Phosphatase-catalyzed Inactivation * Received for publication, July 21, 2006, and in revised form, January 22, 2007 Published, JBC Papers in Press, January 25, 2007, DOI 10.1074/jbc.M606936200 Daniel Palmer ‡ , Sandra L. Jimmo ‡ , Daniel R. Raymond ‡ , Lindsay S. Wilson § , Rhonda L. Carter ‡ , and Donald H. Maurice ‡§1 From the ‡ Department of Pharmacology and Toxicology and § Department of Pathology and Molecular Medicine, Queen’s University, Kingston, Ontario K7L 3N6, Canada Recent studies confirm that intracellular cAMP concentra- tions are nonuniform and that localized subcellular cAMP hydrolysis by cyclic nucleotide phosphodiesterases (PDEs) is important in maintaining these cAMP compartments. Human phosphodiesterase 3B (HSPDE3B), a member of the PDE3 fam- ily of PDEs, represents the dominant particulate cAMP-PDE activity in many cell types, including adipocytes and cells of hematopoietic lineage. Although several previous reports have shown that phosphorylation of HSPDE3B by either protein kinase A (PKA) or protein kinase B (PKB) activates this enzyme, the mechanisms that allow cells to distinguish these two acti- vated forms of HSPDE3B are unknown. Here we report that PKA phosphorylates HSPDE3B at several distinct sites (Ser-73, Ser-296, and Ser-318), and we show that phosphorylation of HSPDE3B at Ser-318 activates this PDE and stimulates its inter- action with 14-3-3 proteins. In contrast, although PKB-cata- lyzed phosphorylation of HSPDE3B activates this enzyme, it does not promote 14-3-3 protein binding. Interestingly, we report that the PKA-phosphorylated, 14-3-3 protein-bound, form of HSPDE3B is protected from phosphatase-dependent dephosphorylation and inactivation. In contrast, PKA-phos- phorylated HSPDE3B that is not bound to 14-3-3 proteins is readily dephosphorylated and inactivated. Our data are pre- sented in the context that a selective interaction between PKA- activated HSPDE3B and 14-3-3 proteins represents a mecha- nism by which cells can protect this enzyme from deactivation. Moreover, we propose that this mechanism may allow cells to distinguish between PKA- and PKB-activated HSPDE3B. Cyclic AMP regulates a diverse array of cellular processes, including intermediary metabolism, vascular and visceral smooth muscle relaxation, hormonal secretion, cytoskeletal organization, as well as transcription, migration, proliferation, and apoptosis (reviewed in Refs.1–3). Although the elements regulating cAMP synthesis have been extensively studied (4, 5), the regulation of cyclic nucleotide phosphodiesterase (PDE) 2 - mediated hydrolysis of cAMP and its impact on cellular func- tions have only recently received considerable attention (6 –12). Based on their sequence homologies, substrate speci- ficities, and sensitivities to pharmacological inhibitors, mam- malian PDEs have been divided into 11 distinct enzyme families (6 –12). Two genes, phosphodiesterase 3A (PDE3A) and PDE3B, encode PDE3 family enzymes (6, 12). PDE3A mRNA is enriched in cells of the cardiovascular system and in oocytes, whereas PDE3B mRNA is abundant in adipocytes, hepatocytes, and cells of hematopoietic lineage (13). Full-length PDE3A and PDE3B contain two N-terminal hydrophobic regions (NHR1 and NHR2) that target these enzymes to the endoplasmic reticulum and perhaps the plasma membrane (13–16). Both PDE3A and PDE3B are substrates of protein kinase A (PKA) or protein kinase B (PKB), and activation of these kinases can result in phosphorylation-mediated activation of these enzymes in some cells (13–16). A consensus has emerged that protein-protein interactions play a central role in regulating cAMP-mediated signaling. Indeed, it is generally accepted that selective subcellular anchorage of PKA, through interaction with A-kinase anchor- ing proteins, allows selective coordination of PKA-dependent cellular events (17, 18). Subcellular targeting of certain PDEs also has emerged as a mechanism whereby these enzymes can coordinate various cellular effects of cAMP (17, 18). In this context, several individual variants of the phosphodiesterase 4 (PDE4) family of enzymes interact with proteins including A-kinase anchoring proteins, -arrestins, and receptor for acti- vated protein kinase C, and these interactions regulate PDE4 subcellular targeting and enzyme activity (17, 18). Although PDE3 activity can represent a significant fraction of total cAMP hydrolytic capacity in certain cell types (6, 11), little is known concerning how protein-protein interactions coordinate the activity and subcellular targeting of PDE3 enzymes. An HSPDE3B interaction with the insulin receptor in * This work was supported by Canadian Institutes for Health Research. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertise- ment” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. 1 Career Investigator of the Heart and Stroke Foundation of Ontario. To whom correspondence should be addressed: Botterell Hall, Rm. 229, Queen’s Uni- versity, Kingston, Ontario K7L 3N6, Canada. Tel.: 613-533-6000 (Ext. 75089); Fax: 613-533-6412; E-mail: [email protected]. 2 The abbreviations used are: PDE, phosphodiesterase; HSPDE3B, human cyclic nucleotide phosphodiesterase 3B; PKA, protein kinase A; PKB, pro- tein kinase B; GST, glutathione S-transferase; WT, wild type; DTT, dithio- threitol; IBMX, 3-isobutyl-1-methylxanthine; aa, amino acid; TEMED, N,N,N,N-tetramethylethylenediamine; PIPES, 1,4-piperazinediethanesul- fonic acid; PI3K, phosphoinositide 3-kinase ; PKC, protein kinase C; CIAP, calf intestinal alkaline phosphatase; C, catalytic; F/I, forskolin/IBMX; MA, membrane-associated; DN, dominant negative. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 282, NO. 13, pp. 9411–9419, March 30, 2007 © 2007 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 9411 at University of California, San Francisco on August 25, 2009 www.jbc.org Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Protein Kinase A Phosphorylation of HumanPhosphodiesterase 3B Promotes 14-3-3 Protein Bindingand Inhibits Phosphatase-catalyzed Inactivation*
Received for publication, July 21, 2006, and in revised form, January 22, 2007 Published, JBC Papers in Press, January 25, 2007, DOI 10.1074/jbc.M606936200
Daniel Palmer‡, Sandra L. Jimmo‡, Daniel R. Raymond‡, Lindsay S. Wilson§, Rhonda L. Carter‡,and Donald H. Maurice‡§1
From the ‡Department of Pharmacology and Toxicology and §Department of Pathology and Molecular Medicine,Queen’s University, Kingston, Ontario K7L 3N6, Canada
Recent studies confirm that intracellular cAMP concentra-tions are nonuniform and that localized subcellular cAMPhydrolysis by cyclic nucleotide phosphodiesterases (PDEs) isimportant in maintaining these cAMP compartments. Humanphosphodiesterase 3B (HSPDE3B), a member of the PDE3 fam-ily of PDEs, represents the dominant particulate cAMP-PDEactivity in many cell types, including adipocytes and cells ofhematopoietic lineage. Although several previous reports haveshown that phosphorylation of HSPDE3B by either proteinkinase A (PKA) or protein kinase B (PKB) activates this enzyme,the mechanisms that allow cells to distinguish these two acti-vated forms of HSPDE3B are unknown. Here we report thatPKA phosphorylates HSPDE3B at several distinct sites (Ser-73,Ser-296, and Ser-318), and we show that phosphorylation ofHSPDE3B at Ser-318 activates this PDE and stimulates its inter-action with 14-3-3 proteins. In contrast, although PKB-cata-lyzed phosphorylation of HSPDE3B activates this enzyme, itdoes not promote 14-3-3 protein binding. Interestingly, wereport that the PKA-phosphorylated, 14-3-3 protein-bound,form of HSPDE3B is protected from phosphatase-dependentdephosphorylation and inactivation. In contrast, PKA-phos-phorylated HSPDE3B that is not bound to 14-3-3 proteins isreadily dephosphorylated and inactivated. Our data are pre-sented in the context that a selective interaction between PKA-activated HSPDE3B and 14-3-3 proteins represents a mecha-nism by which cells can protect this enzyme from deactivation.Moreover, we propose that this mechanism may allow cells todistinguish between PKA- and PKB-activated HSPDE3B.
Cyclic AMP regulates a diverse array of cellular processes,including intermediary metabolism, vascular and visceralsmooth muscle relaxation, hormonal secretion, cytoskeletalorganization, as well as transcription, migration, proliferation,and apoptosis (reviewed in Refs.1–3). Although the elementsregulating cAMP synthesis have been extensively studied (4, 5),
the regulation of cyclic nucleotide phosphodiesterase (PDE)2-mediated hydrolysis of cAMP and its impact on cellular func-tions have only recently received considerable attention(6–12). Based on their sequence homologies, substrate speci-ficities, and sensitivities to pharmacological inhibitors, mam-malian PDEs have been divided into 11 distinct enzyme families(6–12).Two genes, phosphodiesterase 3A (PDE3A) and PDE3B,
encodePDE3 family enzymes (6, 12).PDE3AmRNA is enrichedin cells of the cardiovascular system and in oocytes, whereasPDE3BmRNA is abundant in adipocytes, hepatocytes, and cellsof hematopoietic lineage (13). Full-length PDE3A and PDE3Bcontain two N-terminal hydrophobic regions (NHR1 andNHR2) that target these enzymes to the endoplasmic reticulumand perhaps the plasma membrane (13–16). Both PDE3A andPDE3B are substrates of protein kinase A (PKA) or proteinkinase B (PKB), and activation of these kinases can result inphosphorylation-mediated activation of these enzymes in somecells (13–16).A consensus has emerged that protein-protein interactions
play a central role in regulating cAMP-mediated signaling.Indeed, it is generally accepted that selective subcellularanchorage of PKA, through interaction with A-kinase anchor-ing proteins, allows selective coordination of PKA-dependentcellular events (17, 18). Subcellular targeting of certain PDEsalso has emerged as a mechanism whereby these enzymes cancoordinate various cellular effects of cAMP (17, 18). In thiscontext, several individual variants of the phosphodiesterase 4(PDE4) family of enzymes interact with proteins includingA-kinase anchoring proteins, �-arrestins, and receptor for acti-vated protein kinase C, and these interactions regulate PDE4subcellular targeting and enzyme activity (17, 18).Although PDE3 activity can represent a significant fraction
of total cAMP hydrolytic capacity in certain cell types (6, 11),little is known concerning how protein-protein interactionscoordinate the activity and subcellular targeting of PDE3enzymes. AnHSPDE3B interaction with the insulin receptor in
* This work was supported by Canadian Institutes for Health Research. Thecosts of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked “advertise-ment” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1 Career Investigator of the Heart and Stroke Foundation of Ontario. To whomcorrespondence should be addressed: Botterell Hall, Rm. 229, Queen’s Uni-versity, Kingston, Ontario K7L 3N6, Canada. Tel.: 613-533-6000 (Ext. 75089);Fax: 613-533-6412; E-mail: [email protected].
2 The abbreviations used are: PDE, phosphodiesterase; HSPDE3B, humancyclic nucleotide phosphodiesterase 3B; PKA, protein kinase A; PKB, pro-tein kinase B; GST, glutathione S-transferase; WT, wild type; DTT, dithio-threitol; IBMX, 3-isobutyl-1-methylxanthine; aa, amino acid; TEMED,N,N,N�,N�-tetramethylethylenediamine; PIPES, 1,4-piperazinediethanesul-fonic acid; PI3K, phosphoinositide 3-kinase �; PKC, protein kinase C; CIAP,calf intestinal alkaline phosphatase; C, catalytic; F/I, forskolin/IBMX; MA,membrane-associated; DN, dominant negative.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 282, NO. 13, pp. 9411–9419, March 30, 2007© 2007 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 9411
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from

human adipocytes has been reported (19). Recently, rat adipo-cyte PDE3B was reported to interact with caveolin-1 (20) plac-ing this enzyme in lipid rafts in these cells (20, 21). Disruption ofan interaction between the murine PDE3B and phosphoinosit-ide 3-kinase � (PI3K�) (22), likely coordinated by one of itsregulatory subunits p87PIKAP (PI3K� adapter protein of 87 kDa)(23), reduced cardiomyocyte contractility (22, 23). Of moreimmediate relevance to the studies reported here, insulin wasreported previously to stimulate a PI3K-dependent interactionbetween murine PDE3B and 14-3-3� in 3T3-L1 adipocytes(24). Binding of 14-3-3 proteins to numerous proteins, usuallyfollowing their phosphorylation, allows 14-3-3 proteins to actas regulators, adaptors, or scaffolds for these proteins (25). APKC-dependent phosphorylation of HSPDE3A also stimulatesassociation of HSPDE3A with 14-3-3 proteins (26).In this study, we report that PKA-mediated phosphorylation
of HSPDE3B at Ser-318 activates this enzyme and promotes itsinteraction with 14-3-3 proteins. Although PKA is also shownto phosphorylate HSPDE3B at two other sites, Ser-73 and Ser-296, these events neither activated nor promoted HSPDE3Binteractions with 14-3-3 proteins. Although PKB activatedHSPDE3B, this kinase did not promote 14-3-3 protein bindingof HSPDE3B or influence the effects of PKA. Taken together,our data are consistent with the novel hypothesis that PKA-activated, but not PKB-activated, HSPDE3B interacts with14-3-3 proteins and that this selective protein-protein interac-tion protects the PKA-activated HSPDE3B from phosphatase-mediated deactivation in cells.
EXPERIMENTAL PROCEDURES
Two-hybrid Screen—Mass screening for protein-proteininteractions were carried out using amodification of the “inter-action trap” methodology described by Fields and Song (27)using the Y860 strain of yeast (Mat� ura3-1:URA3 lexAop-ADE2 leu2-3,112 his3-11,15 trp1-1 ade2-1 can1-100) (Dr. C.Boone, University of Toronto, Canada). Briefly, a murine brainexpression library in pACT (Clontech) that allowed expressionof mouse brain proteins as GAL4 fusions (Clontech) served as“prey.” “Bait” constructs were selected fragments of HSPDE3B(GenBankTM accession number NM-000922) in pEG202,which allowed expression of HSPDE3B-LexA fusion proteins.Primers for HSPDE3B fragment amplification were as follows:aa 2–90 (sense, 5�-ggggatccTGAGGAGGGACGAGCGA-GAC-3�; antisense, 5�-ctctcgagTTCCTCTTCATCTGCCTC-TTC-3�); aa 257–711 (sense, 5�-gcggatcCTGCTGGCCCTGG-GGTTGG-3�; antisense, 5�-gcctcgagTTCCAATAAACCAGT-GTC-3�); aa 712–1000 (sense, 5�-gcggatcCCCACTCAACAA-TTTATG-3�; antisense, 5�-gcctcgagACACAGGGGACCCAC-TATG-3�); and aa 978–1113 (sense, 5�-gcggatcCGTTCTTCT-CCTCAACTAGC-3�; antisense, 5�-cgctcgagTTCCTCTTCA-TCTGCCTCTTC-3�). Following transformation, yeast wasgrown in synthetic dropout (SD) media supplemented with tryp-tophan (72 mg/liter), adenine (110 mg/liter), leucine (327 mg/liter), and histidine (72 mg/liter) as appropriate for selection pur-poses. Positive clones were initially selected for growth in theabsence of adenine and then for the presence of LacZ activity.Plasmids were purified from yeast colonies that met both pheno-typic requirements, transformed into Escherichia coli (DH5�),
again tested inyeast, and identifiedbyDNAsequencing (CORTECDNA Service Laboratories, Kingston, Canada).Expression and Characterization of Glutathione S-Transfer-
ase Fusion Proteins—An amino-terminal FLAG-taggedHSPDE3B expression construct (HSPDE3B(AT)) was gener-ated by ligating an EcoRI-HSPDE3B-digested cDNA (base pairs1–1548 inclusively) into EcoRI-digested pCMVtag 2B vector. Acarboxyl-terminal FLAG-tagged HSPDE3B expression con-struct (HSPDE3B(CT)) was generated by ligation of a PstI-di-gested fragment ofHSPDE3B containing base pairs 1795–3269,inclusively into the PstI-digested pCMV tag 2C. For use in14-3-3 pulldown experiments, recombinant 14-3-3 (� or �) orHSPDE3B fragments were expressed as glutathione S-transfer-ase (GST) fusion proteins in E. coli. A pGEX-2T plasmid con-taining human 14-3-3� was a gift from Dr. Q. Medley (DanaFarber Cancer Institute, Harvard University), and the 14-3-3�-and HSPDE3B-GST fragments were expressed using pGEX-5X-3 (Amersham Biosciences). E. coli (BL21(DE)) expressionwas induced with 1 mM isopropyl 1-thio-�-D-galactopyrano-side at 30 °C for 3 h. Bacterial cell pellets were collected bycentrifugation (6000 � g, 15 min) and lysed by sonication inphosphate-buffered saline supplemented with 1% Triton-X-100, 0.02% sodium azide, 100 �M dithiothreitol (DTT), 10�g/ml lysozyme, 5 mM benzamidine, 1 mM EDTA, 2 �g/mlleupeptin, 5 �g/ml bestatin, 2 �g/ml aprotinin, 10 �g/ml anti-pain, 10�ME-64, 2�g/ml pepstatinA, and 0.1mMphenylmeth-ylsulfonyl fluoride. Filtered protein lysates were loaded onreduced glutathione (GSH)-Sepharose 4B (Amersham Bio-sciences), washed, and eluted with 5 mM free GSH in 10 mMTris, pH 8.0, and GSH was removed using of Amicon� Centri-con� centrifugation filters (Millipore). GST fusion proteinyields and purities were assessed by SDS-PAGE.Mammalian Cell Culture—HEK293T (herein 293T) or NIH
3T3 cells were cultured in Dulbecco’s modified Eagle’s mediumsupplemented with 10% fetal bovine serum, penicillin (100units/ml), streptomycin (100 �g/ml), 2 mM L-glutamine, and 1g/liter D-glucose.Heterologous Expression of HSPDE3B Constructs—A cDNA
encodingHSPDE3B (Dr. V. C.Manganiello, National Institutesof Health) was cloned into the mammalian expression vectorpCMV-Tag2C (Stratagene) using BamHI. This construct wasused to express HSPDE3B. In experiments in which HSPDE3Bwas expressed heterologously, transfections were carried outwith amounts of plasmid to limit overexpression of HSPDE3Bto levels not exceeding 4-fold those of endogenous HSPDE3B.With full-length HSPDE3B, levels of expression were deter-mined by PDE activity assays. Point mutations that allowedsubstitution of alanine (Ala) for serine (Ser) at positions 73, 295,296, or 318 within HSPDE3B were generated using the Quick-Change site-directed mutagenesis kit (Clontech) according tothe manufacturer’s protocol. Plasmids encoding wild type(WT),membrane-associated (MA), or dominant negative (DN)PKB were provided by Dr. D. Alessi (University of Dundee,Dundee, Scotland, UK). HSPDE3B constructs were transientlyexpressed in 293T or NIH 3T3 cells following transfectionusing FuGENE 6 (Roche Applied Science).In Vitro Phosphorylation of HSPDE3B—GST- or FLAG-
tagged proteins were purified by conventional approaches
14-3-3 Proteins and HSPDE3B Activation
9412 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from

using GSH-Sepharose orM2-agarose. HSPDE3B proteins wereincubated with recombinant PKA catalytic subunit or activatedrecombinant PKB� (Upstate Biotechnology, Inc., Lake Placid,NY) in a buffer without or with 200 �M ATP or 200 �M ATPsupplemented with [�-32P]ATP (10 �Ci per reaction; 3000Ci/mmol stock) for 1 h at 30 °C. For reference, reactions werecarried out with neither kinase nor ATP, with kinase or ATPalone, or with both ATP and kinase. At the completion of thesereactions, proteins were subjected to SDS-PAGE (10–12%gels).Treatment of Cells with Pharmacological Agents and Cell
Processing—Confluent cultures of 293T or NIH 3T3 cells wereincubated at 37 °C for 20 min in serum-free media containingforskolin (1–100 �M; Calbiochem), 3-isobutyl-1-methylxan-thine (IBMX; 10–100 �M; Sigma), or vehicle (Me2SO, 0.2% v/v;Fisher). When used, protein kinase inhibitors, H89, Bis-1,LY22945 (1–10 �M) (Sigma), were added 30 min prior to thetest agent. At the end of the incubation period, cells were eitherflash-frozen until use or immediately processed. Treated cellcultures were homogenized with a Tenbroeck tissue grinder ina lysis buffer composed of 50 mM Tris-HCl, pH 7.4, 150 mMNaCl, 5 mM MgCl2, 5 mM benzamidine, 100 mM DTT, 1 mMEDTA, 100 �M EGTA, 1% Triton X-100, 2 �g/ml leupeptin, 5�g/ml bestatin, 2 �g/ml aprotinin, 10 �g/ml antipain, 10 �ME-64, 2 �g/ml pepstatin A, 0.1 mM phenylmethylsulfonyl fluo-ride, 10 mM sodium orthovanadate, 10 mM sodium fluoride, 5mM sodium pyrophosphate, and 10 mM sodium glycerophos-phate. Cellular debris was removed by centrifugation (1,000 �g; 5 min), and cleared supernatants were used in the experi-ments. Protein concentrations were measured by the bicincho-ninic acid protein assay (Pierce).Precipitation of Cellular Proteins with Immobilized GST
Fusions orM2-Agarose—Interactions betweenHSPDE3B, or itsfragments or mutants, with GST-14-3-3 proteins were studiedusing precipitation by pulldownor immunoprecipitation assayswith GST-14-3-3�, GST-14-3-3�, or anti-FLAG(M2)-coupledagarose. For immobilized GST pulldown experiments, 50 �l(bed volume) of GSH-Sepharose-4B beads were saturated withGST, GST-14-3-3�, or GST-14-3-3� at 4 °C for 1 h, washed,and incubated for 16 h at 4 °C with cell extracts, prepared asdescribed above. For M2-agarose pulldowns, cell lysates weresimilarly incubated for 16 h at 4 °C. Following the incubations,Sepharose or agarose beads were centrifuged (6,000� g; 2 min)and washed repeatedly with 1% Triton X-100-containing lysisbuffer. cAMP PDE activities in pulldowns was measured fol-lowing resuspension of beads in lysis buffer (1:1 volume ratio)using an assay described previously (28). Cilostamide (1 �M) orRo20-1724 (10 �M) was used to inhibit PDE3 and PDE4 activi-ties, respectively (28). For immunoblotting, pellets were sus-pended in SDS-PAGE loading buffer. HSPDE3B, as well asother proteins of interest in these pellets, were detected usingseveral antisera, including a monoclonal antibody specific forHSPDE3B (281K; 1:3000 dilution), a monoclonal antisera spe-cific for PDE4D (1:4000) (all gifts from Drs. S. Wolda and V.Florio, ICOS Corp., Bothell, WA), or nonspecies selective anti-PDE3B polyclonal antibodies (sc-11835 and sc-11838; SantaCruz Biotechnology) as we described previously (29). Levels ofPKA-phosphorylated HSPDE3B in cells were detected using an
antiserum directed at phosphorylated PKA substrates (UpstateBiotechnology, Inc.). Detection of 14-3-3 proteins was carriedout by immunoblot analysis using antisera directed against14-3-3 isoforms (sc-629; pan-reactive; rabbit and goat poly-clonals; 1:1000; Santa Cruz Biotechnology).Statistical Analysis—Some of the data describing protein-
protein interactions are shown as representative immunoblots.In all cases, data consistent with that shown in the representa-tive immunoblot were obtained in at least three additionalseparate experiments. Numerical data are presented asmeans � S.E. and are from at least four independent experi-ments. Statistical differences were assessed using unpairedanalysis of variance, with a Tukey post hoc test, or unpairedStudent’s t test, as appropriate, with a value of p � 0.05 consid-ered statistically significant.Materials—Yeast and bacteria culture reagents were
obtained from Difco (yeast nitrogenous base without aminoacids), Fisher (peptone, tryptone, and dextrose), Qbiogene(complete supplement mixture �His �Leu �Ade �Trp), andSigma (adenine, hisitidine, leucine, tryptophan, and o-nitro-phenyl �-D-galactopyranoside). Restriction enzymes, TaqDNApolymerase, Superscript Moloney murine leukemia virus-re-verse transcriptase, isopropyl 1-thio-�-D-galactopyranoside,Dulbecco’s modified Eagle’s medium, RPMI 1640, antibiotic/antimycotic solution, trypsin-EDTA, bovine serum albumin,5-bromo-4-chloro-3-indolyl-�-D-galactopyranoside, and fetalbovine serum were from Invitrogen. Plasmid purification wasachieved using QIAprep spin columns, and MIDI columnswere from Qiagen. [3H]cAMP (25 Ci/mmol), 5�-[14C]AMP(590.4mCi/mmol), [�-32P]ATP (3000Ci/mmol), and enhancedchemiluminescence reagents (Western Lightning) were fromPerkinElmer Life Sciences. DTT, Triton X-100, EDTA, EGTA,PIPES, acrylamide, bisacrylamide, TEMED, and ammoniumpersulfate were purchased from ICN Biomedicals. Sodiumazide, Tris, trisodium citrate dihydrate, citric acid monohy-drate, HEPES, NaCl, and Tween 20were from Fisher. Benzami-dine-HCl, leupeptin hemisulfate, aprotinin, phenylmethylsul-fonyl fluoride, and G418 antibiotic were purchased fromCalbiochem. All other reagents, salts, and enzymes wereobtained from Sigma.
RESULTS
Identification of HSPDE3B-interacting Proteins—To identifypotential HSPDE3B-interacting proteins, we screened a yeasttwo-hybrid mouse brain cDNA library with HSPDE3B frag-ments as bait. “Bait-1” encoded amino acids 2–90, and “bait-2”encoded amino acids 257–711 of HSPDE3B, respectively. Nointeracting proteinswere isolatedwhenbait-2was used. In con-trast, multiple clones of a brain 14-3-3 protein, 14-3-3� (Gen-BankTM accession number NM-0011738), were isolated usingbait-1 (Fig. 1,A–D). Expressed alone in yeast, HSPDE3B “baits”or the 14-3-3� isolates did not promote adenine synthesis norLacZ activity (Fig. 1,A–D).When other fragments ofHSPDE3Bwere tested for their ability to interact with 14-3-3�, fragmentsencoding amino acids 257–711, 712–1000, or 978–1113 didnot possess such potential (not shown). Binding of 14-3-3 pro-teins usually requires that the binding partner be phosphoryla-ted (25). Consistent with the idea that phosphorylation could
14-3-3 Proteins and HSPDE3B Activation
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 9413
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from
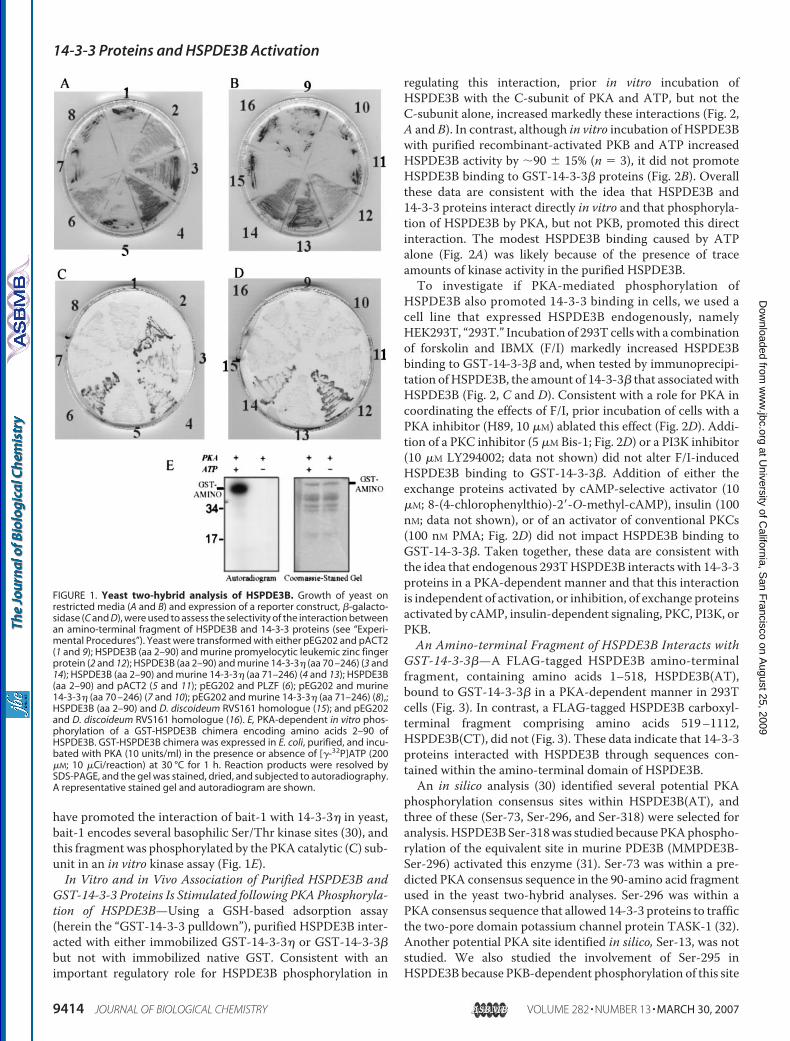
have promoted the interaction of bait-1 with 14-3-3� in yeast,bait-1 encodes several basophilic Ser/Thr kinase sites (30), andthis fragment was phosphorylated by the PKA catalytic (C) sub-unit in an in vitro kinase assay (Fig. 1E).In Vitro and in Vivo Association of Purified HSPDE3B and
GST-14-3-3 Proteins Is Stimulated following PKA Phosphoryla-tion of HSPDE3B—Using a GSH-based adsorption assay(herein the “GST-14-3-3 pulldown”), purified HSPDE3B inter-acted with either immobilized GST-14-3-3� or GST-14-3-3�but not with immobilized native GST. Consistent with animportant regulatory role for HSPDE3B phosphorylation in
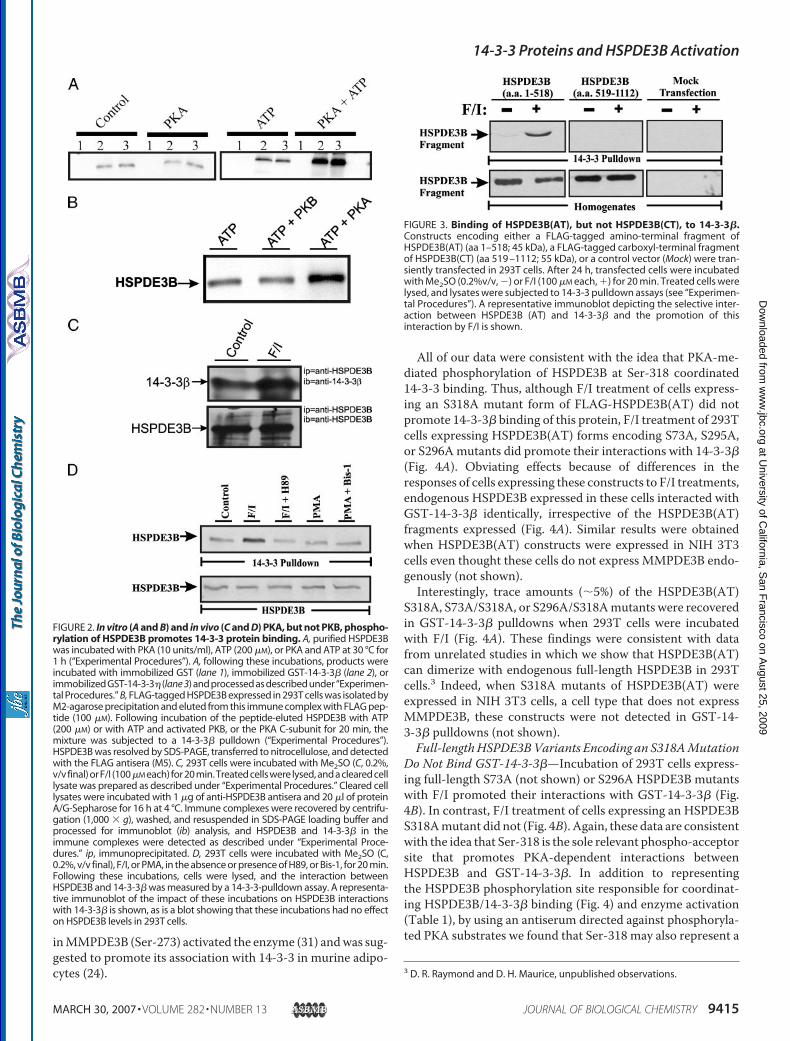
regulating this interaction, prior in vitro incubation ofHSPDE3B with the C-subunit of PKA and ATP, but not theC-subunit alone, increased markedly these interactions (Fig. 2,A and B). In contrast, although in vitro incubation of HSPDE3Bwith purified recombinant-activated PKB and ATP increasedHSPDE3B activity by �90 � 15% (n � 3), it did not promoteHSPDE3B binding to GST-14-3-3� proteins (Fig. 2B). Overallthese data are consistent with the idea that HSPDE3B and14-3-3 proteins interact directly in vitro and that phosphoryla-tion of HSPDE3B by PKA, but not PKB, promoted this directinteraction. The modest HSPDE3B binding caused by ATPalone (Fig. 2A) was likely because of the presence of traceamounts of kinase activity in the purified HSPDE3B.To investigate if PKA-mediated phosphorylation of
HSPDE3B also promoted 14-3-3 binding in cells, we used acell line that expressed HSPDE3B endogenously, namelyHEK293T, “293T.” Incubation of 293T cells with a combinationof forskolin and IBMX (F/I) markedly increased HSPDE3Bbinding to GST-14-3-3� and, when tested by immunoprecipi-tation ofHSPDE3B, the amount of 14-3-3� that associatedwithHSPDE3B (Fig. 2, C and D). Consistent with a role for PKA incoordinating the effects of F/I, prior incubation of cells with aPKA inhibitor (H89, 10 �M) ablated this effect (Fig. 2D). Addi-tion of a PKC inhibitor (5 �M Bis-1; Fig. 2D) or a PI3K inhibitor(10 �M LY294002; data not shown) did not alter F/I-inducedHSPDE3B binding to GST-14-3-3�. Addition of either theexchange proteins activated by cAMP-selective activator (10�M; 8-(4-chlorophenylthio)-2�-O-methyl-cAMP), insulin (100nM; data not shown), or of an activator of conventional PKCs(100 nM PMA; Fig. 2D) did not impact HSPDE3B binding toGST-14-3-3�. Taken together, these data are consistent withthe idea that endogenous 293THSPDE3B interacts with 14-3-3proteins in a PKA-dependent manner and that this interactionis independent of activation, or inhibition, of exchange proteinsactivated by cAMP, insulin-dependent signaling, PKC, PI3K, orPKB.An Amino-terminal Fragment of HSPDE3B Interacts with
GST-14-3-3�—A FLAG-tagged HSPDE3B amino-terminalfragment, containing amino acids 1–518, HSPDE3B(AT),bound to GST-14-3-3� in a PKA-dependent manner in 293Tcells (Fig. 3). In contrast, a FLAG-tagged HSPDE3B carboxyl-terminal fragment comprising amino acids 519–1112,HSPDE3B(CT), did not (Fig. 3). These data indicate that 14-3-3proteins interacted with HSPDE3B through sequences con-tained within the amino-terminal domain of HSPDE3B.An in silico analysis (30) identified several potential PKA
phosphorylation consensus sites within HSPDE3B(AT), andthree of these (Ser-73, Ser-296, and Ser-318) were selected foranalysis.HSPDE3BSer-318was studied because PKAphospho-rylation of the equivalent site in murine PDE3B (MMPDE3B-Ser-296) activated this enzyme (31). Ser-73 was within a pre-dicted PKA consensus sequence in the 90-amino acid fragmentused in the yeast two-hybrid analyses. Ser-296 was within aPKA consensus sequence that allowed 14-3-3 proteins to trafficthe two-pore domain potassium channel protein TASK-1 (32).Another potential PKA site identified in silico, Ser-13, was notstudied. We also studied the involvement of Ser-295 inHSPDE3B because PKB-dependent phosphorylation of this site
FIGURE 1. Yeast two-hybrid analysis of HSPDE3B. Growth of yeast onrestricted media (A and B) and expression of a reporter construct, �-galacto-sidase (C and D), were used to assess the selectivity of the interaction betweenan amino-terminal fragment of HSPDE3B and 14-3-3 proteins (see “Experi-mental Procedures”). Yeast were transformed with either pEG202 and pACT2(1 and 9); HSPDE3B (aa 2–90) and murine promyelocytic leukemic zinc fingerprotein (2 and 12); HSPDE3B (aa 2–90) and murine 14-3-3� (aa 70 –246) (3 and14); HSPDE3B (aa 2–90) and murine 14-3-3� (aa 71–246) (4 and 13); HSPDE3B(aa 2–90) and pACT2 (5 and 11); pEG202 and PLZF (6); pEG202 and murine14-3-3� (aa 70 –246) (7 and 10); pEG202 and murine 14-3-3� (aa 71–246) (8),;HSPDE3B (aa 2–90) and D. discoideum RVS161 homologue (15); and pEG202and D. discoideum RVS161 homologue (16). E, PKA-dependent in vitro phos-phorylation of a GST-HSPDE3B chimera encoding amino acids 2–90 ofHSPDE3B. GST-HSPDE3B chimera was expressed in E. coli, purified, and incu-bated with PKA (10 units/ml) in the presence or absence of [�-32P]ATP (200�M; 10 �Ci/reaction) at 30 °C for 1 h. Reaction products were resolved bySDS-PAGE, and the gel was stained, dried, and subjected to autoradiography.A representative stained gel and autoradiogram are shown.
14-3-3 Proteins and HSPDE3B Activation
9414 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from
inMMPDE3B (Ser-273) activated the enzyme (31) andwas sug-gested to promote its association with 14-3-3 in murine adipo-cytes (24).
All of our data were consistent with the idea that PKA-me-diated phosphorylation of HSPDE3B at Ser-318 coordinated14-3-3 binding. Thus, although F/I treatment of cells express-ing an S318A mutant form of FLAG-HSPDE3B(AT) did notpromote 14-3-3� binding of this protein, F/I treatment of 293Tcells expressing HSPDE3B(AT) forms encoding S73A, S295A,or S296Amutants did promote their interactions with 14-3-3�(Fig. 4A). Obviating effects because of differences in theresponses of cells expressing these constructs to F/I treatments,endogenous HSPDE3B expressed in these cells interacted withGST-14-3-3� identically, irrespective of the HSPDE3B(AT)fragments expressed (Fig. 4A). Similar results were obtainedwhen HSPDE3B(AT) constructs were expressed in NIH 3T3cells even thought these cells do not express MMPDE3B endo-genously (not shown).Interestingly, trace amounts (�5%) of the HSPDE3B(AT)
S318A, S73A/S318A, or S296A/S318Amutants were recoveredin GST-14-3-3� pulldowns when 293T cells were incubatedwith F/I (Fig. 4A). These findings were consistent with datafrom unrelated studies in which we show that HSPDE3B(AT)can dimerize with endogenous full-length HSPDE3B in 293Tcells.3 Indeed, when S318A mutants of HSPDE3B(AT) wereexpressed in NIH 3T3 cells, a cell type that does not expressMMPDE3B, these constructs were not detected in GST-14-3-3� pulldowns (not shown).Full-lengthHSPDE3BVariants Encoding an S318AMutation
Do Not Bind GST-14-3-3�—Incubation of 293T cells express-ing full-length S73A (not shown) or S296A HSPDE3B mutantswith F/I promoted their interactions with GST-14-3-3� (Fig.4B). In contrast, F/I treatment of cells expressing an HSPDE3BS318Amutant did not (Fig. 4B). Again, these data are consistentwith the idea that Ser-318 is the sole relevant phospho-acceptorsite that promotes PKA-dependent interactions betweenHSPDE3B and GST-14-3-3�. In addition to representingthe HSPDE3B phosphorylation site responsible for coordinat-ing HSPDE3B/14-3-3� binding (Fig. 4) and enzyme activation(Table 1), by using an antiserum directed against phosphoryla-ted PKA substrates we found that Ser-318 may also represent a
3 D. R. Raymond and D. H. Maurice, unpublished observations.
FIGURE 2. In vitro (A and B) and in vivo (C and D) PKA, but not PKB, phospho-rylation of HSPDE3B promotes 14-3-3 protein binding. A, purified HSPDE3Bwas incubated with PKA (10 units/ml), ATP (200 �M), or PKA and ATP at 30 °C for1 h (“Experimental Procedures”). A, following these incubations, products wereincubated with immobilized GST (lane 1), immobilized GST-14-3-3� (lane 2), orimmobilized GST-14-3-3� (lane 3) and processed as described under “Experimen-tal Procedures.” B, FLAG-tagged HSPDE3B expressed in 293T cells was isolated byM2-agarose precipitation and eluted from this immune complex with FLAG pep-tide (100 �M). Following incubation of the peptide-eluted HSPDE3B with ATP(200 �M) or with ATP and activated PKB, or the PKA C-subunit for 20 min, themixture was subjected to a 14-3-3� pulldown (“Experimental Procedures”).HSPDE3B was resolved by SDS-PAGE, transferred to nitrocellulose, and detectedwith the FLAG antisera (M5). C, 293T cells were incubated with Me2SO (C, 0.2%,v/v final) or F/I (100�M each) for 20 min. Treated cells were lysed, and a cleared celllysate was prepared as described under “Experimental Procedures.” Cleared celllysates were incubated with 1 �g of anti-HSPDE3B antisera and 20 �l of proteinA/G-Sepharose for 16 h at 4 °C. Immune complexes were recovered by centrifu-gation (1,000 � g), washed, and resuspended in SDS-PAGE loading buffer andprocessed for immunoblot (ib) analysis, and HSPDE3B and 14-3-3� in theimmune complexes were detected as described under “Experimental Proce-dures.” ip, immunoprecipitated. D, 293T cells were incubated with Me2SO (C,0.2%, v/v final), F/I, or PMA, in the absence or presence of H89, or Bis-1, for 20 min.Following these incubations, cells were lysed, and the interaction betweenHSPDE3B and 14-3-3� was measured by a 14-3-3-pulldown assay. A representa-tive immunoblot of the impact of these incubations on HSPDE3B interactionswith 14-3-3� is shown, as is a blot showing that these incubations had no effecton HSPDE3B levels in 293T cells.
FIGURE 3. Binding of HSPDE3B(AT), but not HSPDE3B(CT), to 14-3-3�.Constructs encoding either a FLAG-tagged amino-terminal fragment ofHSPDE3B(AT) (aa 1–518; 45 kDa), a FLAG-tagged carboxyl-terminal fragmentof HSPDE3B(CT) (aa 519 –1112; 55 kDa), or a control vector (Mock) were tran-siently transfected in 293T cells. After 24 h, transfected cells were incubatedwith Me2SO (0.2%v/v, �) or F/I (100 �M each, �) for 20 min. Treated cells werelysed, and lysates were subjected to 14-3-3 pulldown assays (see “Experimen-tal Procedures”). A representative immunoblot depicting the selective inter-action between HSPDE3B (AT) and 14-3-3� and the promotion of thisinteraction by F/I is shown.
14-3-3 Proteins and HSPDE3B Activation
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 9415
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from

major site of PKA phosphorylation of HSPDE3B in cells incu-bated with F/I (Fig. 5). Because it is currently unknown if thisantiserum reacts with similar affinity to all phosphorylationconsensus sequences, a more detailed analysis will be requiredto quantify the absolute levels of phosphate at each of thesesites. Although in silico analysis (30) identified other potentialPKA consensus sites within HSPDE3B, our data are consistentwith the idea that only Ser-318 was required for 14-3-3� bind-ing in 293T cells.PKB Does Not Promote HSPDE3B/14-3-3 Interactions—A
previous report proposed that an insulin-promoted, PKB-de-pendent phosphorylation ofMMPDE3B at Ser-279 and Ser-302(residues equivalent to Ser-295 and Ser-318 in HSPDE3B) acti-vated this enzyme and promoted its interactionwith 14-3-3� in
murine adipocytes (24). In marked contrast to this earlierreport, although our data unequivocally confirm that PKB canactivate HSPDE3B (see above), they are completely inconsis-tent with the idea that phosphorylation of HSPDE3B by PKB atSer-295, or any other site, promotes binding of this enzymewith 14-3-3�. Similarly, our data are inconsistent with the ideathat PKB-mediated actions on HSPDE3B alter the ability ofPKA phosphorylation at Ser-318 to promote 14-3-3� binding(Fig. 6). Indeed, expression ofwild type PKB (WTPKB), amem-brane-targeted and constitutively activated PKB (MA PKB), ora dominant negative and “kinase-dead” PKB (DN PKB) in 293Tcells did not alter either the basal levels of endogenousHSPDE3B binding to GST-14-3-3� in these cells nor the abilityof F/I treatment to promote this binding (Fig. 6). Consistentwith their state of activation, immunoblot analysis with a phos-pho-PKB antiserum indicated that a large fraction of the heter-ologously expressedwild type or activated PKBswere phospho-rylated and that the DNPKB, which is kinase-deadwas not (notshown). Similarly, because F/I treatment promoted S295A-HSPDE3B binding to 14-3-3� (Fig. 4A), it is also unlikely thatSer-295 was involved in coordinating the PKA-dependentbinding of HSPDE3B to 14-3-3� in these cells.Binding of the PKA Phosphorylated and Activated HSPDE3B
to 14-3-3� Alters Its Susceptibility to Phosphate-catalyzedDephosphorylation and Inactivation—Because our datashowed that PKA phosphorylation of Ser-318 in HSPDE3Bactivated this enzyme and promoted its binding to 14-3-3�, wehypothesized that this interactionmight alter the susceptibilityof this fraction of HSPDE3B to be dephosphorylated and inac-tivated by phosphatases. Our data are completely consistentwith this novel idea. Indeed, although a 20-min incubation ofM2-agarose-precipitated HSPDE3B from F/I-treated cells withcalf intestinal alkaline phosphatase (CIAP) resulted in substan-tial dephosphorylation (Fig. 7) and enzyme inactivation (Table2), this identical CIAP treatment of the PKA-activated, GST-14-3-3�-boundHSPDE3B did not result in substantial dephos-phorylation (Fig. 7) or enzyme inactivation (Table 2). BecausePKB phosphorylation of HSPDE3B did not promote GST-14-3-3� binding (Fig. 2), nor influence the extent of 14-3-3� bind-ing of this enzyme caused by F/I (Fig. 6), it is highly unlikely thatPKB-activated HSPDE3B would be similarly protected fromphosphatase-mediated dephosphorylation and inactivation. Ascheme depicting these concepts is presented in Fig. 8.
DISCUSSION
Many proteins involved in cellular signaling are promiscu-ous, regulating several distinct events simultaneously (33, 34).
FIGURE 4. A single HSPDE3B residue, Ser-318, coordinates the PKA-de-pendent interaction between HSPDE3B and 14-3-3�. A, plasmids encod-ing either a FLAG-tagged HSPDE3B(AT) or this same plasmid encodingHSPDE3B(AT) mutants, S73A, S295A, S296A, and S318A alone or in combina-tion, were each expressed in 293T cells. Cells were incubated either withMe2SO (0.2%, v/v final, �) or F/I (100 �M each, �) for 20 min. Treated cells werelysed, and lysates were subjected to 14-3-3 pulldown assays (see “Experimen-tal Procedures”). The top panel is an immunoblot in which anti-FLAG antibody(1:1000; M5, Sigma) was used to detect HSPDE3B(AT). Middle panel is animmunoblot in which M5 was used to determine the levels of expression ofthe HSPDE3B(AT) constructs in 293T cells. The lower panel is an immunoblot inwhich an anti-HSPDE3B antiserum (1:4000) was used to detect endogenous293T that had interacted with 14-3-3� in these cells. B, plasmids encodingFLAG-tagged point mutants of full-length HSPDE3B were each expressed in293T cells. These cells, and their lysates, were used in experiments identical tothose described in A. Immunoblots from two separate representative exper-iments are shown. Identical results were obtained in at least five individualsimilar experiments.
TABLE 1Impact of PKA-induced phosphorylation of HSPDE3B on enzyme activityNote that 293T cells were transiently transfected without or with a plasmid encoding the identified HSPDE3B variant. After 24 h, cells were incubated with vehicle (0.2%v/v Me2SO) or F/I for 20 min, lysed in lysis buffer supplemented with Ro20-1724 (10 �M), and processed for M2-agarose immunoprecipitation (ip) as described (seeExperimental Procedures). PDE3 activity was determined in 293T homogenates or in M2-agarose precipitates as described under Experimental Procedures. Data aremeans � S.E. from a representative experiment conducted in triplicate.
HSPDE3B Homogenate vehicle Homogenate F/I M2-agarose ip vehicle M2-agarose ip F/Ipmol/min/mg pmol/min/mg pmol/�l/min pmol/�l/min
Mock 36 � 3 50 � 2a 17 � 0 13 � 1S296A 93 � 5 125 � 2a 153 � 6 183 � 5aS318A 99 � 1 102 � 0 146 � 4 142 � 4S296/318A 90 � 2 93 � 2 101 � 12 106 � 7
a Data signify significant differences between vehicle and F/I-treated cells, p � 0.05. Similar data were obtained in three separate experiments.
14-3-3 Proteins and HSPDE3B Activation
9416 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from

Although the events that allow coordination of the multipleactions of signaling proteins are as yet poorly understood,dynamic regulation of protein phosphorylation has emerged asan important point of control (33, 34). In this study, we haveconfirmed that phosphorylation of HSPDE3B regulates thisenzyme activity in cells, and we have uncovered a mechanismthat may allow cells to discriminate between the PKA- and thePKB-activated populations of this enzyme.As described in the Introduction, cells express only one
HSPDE3B variant, and this enzyme can be targeted to boththe endoplasmic reticulum and, perhaps to a lesser extent, tothe plasma membrane of cells (21, 35–36). However, there iscurrently a paucity of information regarding the proteins withwhich these potentially separate populations of HSPDE3Bmight interact and on the impact such protein-protein interac-tionsmight have onHSPDE3B activity. Indeed, whereas PDE3Bin rodent or human adipocytes were reported to interact withthe insulin receptor and/or with caveolin-1, and the rodentenzyme was shown to interact with p87PIKAP in heart, theimpact of these events on PDE3 activity and on cellular events
regulated by the hydrolysis of cAMP by this enzyme are asyet poorly understood.Previously, the rat adipocyte PDE3B was reported to inter-
act with 14-3-3� in an insulin- and PI3K activation-depend-ent manner. Indeed, in this earlier study, it was suggestedthat phosphorylation of Ser-279 or Ser-302, sites equivalentto Ser-295 and Ser-318 in HSPDE3B, coordinated this inter-action (24). Herein we presented data demonstrating thatmembers of the 14-3-3 protein family of scaffolding proteins(14-3-3� or 14-3-3�) interact with HSPDE3B in human cells,and these interactions are markedly potentiated by incuba-tion of these cells with cAMP-elevating agents. Moreover,using a strategy of selective PKA inhibition, we identifiedPKA as the cAMP effector coordinating this cAMP-medi-ated effect. In addition to establishing that 14-3-3 proteinsinteracted with HSPDE3B in cells expressing these proteinsendogenously, and demonstrating that these events werecoordinated through cAMP-mediated activation of PKA, wealso made use of a combination of genetic screens and in situsite-directed mutagenesis studies to identify the HSPDE3Bresidue(s) coordinating this interaction. Our data identifytwo potential 14-3-3 protein-binding sites within HSPDE3B.First, based on our yeast two-hybrid analysis, we identified a14-3-3-binding domain within the first 90 amino acids ofHSPDE3B. Although this HSPDE3B domain containedpotential PKA phosphorylation consensus sites and wasphosphorylated by PKA in vitro, our mutagenesis studiesindicated that site(s) within this domain were unlikely to beinvolved in coordinating the increase in 14-3-3 protein bind-ing in mammalian cells in response to cAMP. More likely,based on the fact that this interaction was initially identifiedin yeast in which PKA had not been experimentally stimu-lated, this 14-3-3 protein interacting domain of HSPDE3Bmay be involved in coordinating basal levels of 14-3-3 pro-tein-HSPDE3B binding observed in our studies. This pro-posal is consistent with a basal level of 14-3-3 protein bind-
FIGURE 5. PKA phosphorylates HSPDE3B at several distinct sites. Plasmidsencoding a series of full-length HSPDE3B in which individual or combinationsof potential PKA sites had been mutated were transiently expressed in 293Tcells. After 24 h, transfected cells were incubated with Me2SO (0.2% v/v, �) orF/I (100 �M each, �) for 20 min. Treated cells were lysed, and lysates weresubjected to immunoprecipitation with M2-agarose. Immune complexeswere boiled, subjected to SDS-PAGE, and transferred to nitrocellulose forimmunoblot analysis using an antibody raised against phosphorylated PKAsubstrates (see “Experimental Procedures”). A, representative immunoblotdemonstrates that Ser3Ala mutations of individual or combinations of eachSer-73, Ser-296, and Ser-318 decreased phosphorylation of the FLAG-taggedHSPDE3B. B, immunoblot of HSPDE3B expressed in 293T cells transfectedwith the various plasmids. Similar results were obtained in three separateexperiments.
FIGURE 6. Increased, or antagonized, PKB activity has no impact on basalor PKA-mediated interactions between HSPDE3B and GST-14-3-3�. 293Tcells were transiently transfected with plasmids encoding either wild typePKB (Wt PKB), a membrane-targeted and constitutively activated PKB (MAPKB), or a dominant negative and kinase-dead PKB (DN PKB). After 24 h, trans-fected cells were incubated with Me2SO (0.2% v/v, �) or F/I (100 �M each, �)for 20 min. Following this incubation, cells were processed identically tothose described in the legend to Fig. 4. Upper panel depicts levels of associa-tion between endogenous 293T cell HSPDE3B and GST-14-3-3�, whereas themiddle and lower panels show the levels of expression of PKB, or HSPDE3B, inthe cell lysates used in this experiment, respectively. Similar results wereobtained in three separate experiments.
FIGURE 7. 14-3-3�-associated and PKA-phosphorylated HSPDE3B is pro-tected from CIAP-dependent dephosphorylation. 293T cells were tran-siently transfected with plasmids encoding FLAG-tagged HSPDE3B. After24 h, transfected cells were incubated with Me2SO (0.2% v/v, Control) or F/I(100 �M each) for 20 min. Following these incubations, cells were processedfor either M2-agarose or GST-14-3-3� pulldowns. In these experiments, 0.2mg of 293T cell lysate from each incubation was added either to M2-agarose(20-�l bed volume) or to 14-3-3-GST-coupled GSH-Sepharose (50-�l bed vol-ume) and processed as described (see “Experimental Procedures”). Followingthese pulldowns, resuspended pellets were equally divided such that half wasincubated with CIAP buffer and the other half incubated with this buffer sup-plemented with CIAP (1 unit) for 20 min at 37 °C. Reaction products wereprocessed for immunoblot analysis. Samples were first probed with an anti-body for phosphorylated PKA substrates (A) and subsequently with the M5anti-FLAG antiserum (B). Amounts of immunoreactive proteins were quanti-fied by densitometric analysis. Similar data were obtained in three separateexperiments.
14-3-3 Proteins and HSPDE3B Activation
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 9417
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from

ing with HSPDE3B in cells that was not sensitive to H89 orthe activation of PKB in our experiments. Examples of sim-ilar 14-3-3 interactions that were not dependent on proteinphosphorylation have been reported (37–38). Whetherphosphorylation of Ser-13 and Ser-73 in cells plays a role inorganizing cellular HSPDE3B complexes at a step not inves-tigated in our studies remains possible and will form thebasis of future work in our laboratory. From our site-directedmutagenesis studies we identified a dominant role for oneHSPDE3B residue, namely Ser-318, in coordinating thedirect, PKA-dependent, interaction between HSPDE3B and14-3-3 proteins. Indeed, binding of the S318A mutant ofHSPDE3B was not increased when cells were incubated withcAMP-elevating agents. Our data also rule out a direct rolefor phosphorylation of either of the other HSPDE3B PKAphospho-acceptor sites (Ser-73 and Ser-296) or the PKBphospho-acceptor residues (Ser-295) in coordinating PKA-dependent HSPDE3B binding to 14-3-3�. Indeed,HSPDE3B, in which multiple PKA sites were mutated, inter-acted with immobilized GST-14-3-3� in a manner consist-ent with the singular importance of Ser-318. Also, becausethe interaction between the S295A-HSPDE3B mutant usedin our studies and 14-3-3� was stimulated by activation ofPKA, this site is unlikely to have contributed to 14-3-3 bind-ing. Because expression of a constitutively activated PKB orof a kinase-dead dominant negative PKB had no impact on
basal or PKA-dependent HSPDE3B-14-3-3 protein binding,we suggest that HSPDE3B does not interact with 14-3-3 pro-teins in a PKB-dependent fashion in 293T cells. In unpub-lished data, we have similarly shown that PKB does not stim-ulate HSPDE3B binding to 14-3-3 proteins in either thepre-B acute lymphoblastic leukemia cell line (REH) or U937cells, a human cell line established from a diffuse histiocyticlymphoma that displays several monocytic characteristics.4Our data obviating the possible involvement of PKB in fos-
tering HSPDE3B binding to 14-3-3� are unequivocal. Indeed,we found no evidence to support a role for PKB in mediatingeither basal levels of HSPDE3B binding to 14-3-3� or in pro-moting association between these proteins in response toincreases in cellular cAMP. In relation to insulin-mediatedstimulation of RNPDE3B binding to 14-3-3� in adipocytes (24),it may be significant that insulin can activate adenylyl cyclase inseveral cell types and that this latter effect was PI3K-inhibitor-sensitive (39). Whether insulin stimulated RNPDE3B bindingto 14-3-3� in rat adipocytes through an insulin-mediated acti-vation of adenylyl cyclase, thus involving PKA, or whether adirect effect of PI3K was involved, which was not tested in ourstudy, remains to be established. Perhaps the use of a site-di-rectedmutation-based approach similar to that employed here,rather than the use of high concentrations of phosphorylatedpeptides, as was done in the earlier work (24), would be benefi-cial in future studies.To ourmind, themost interesting finding of this study relates
to our demonstration that the 14-3-3�-bound, PKA-activatedform of HSPDE3B was protected from phosphatase-catalyzedinactivation. Indeed, 14-3-3-bound HSPDE3B was less sensi-tive to phosphatase-based inactivation when compared withunbound PKA-activated HSPDE3B or with PKB-activatedHSPDE3B. Of course, the PKB-activated HSPDE3B was not14-3-3-bound. Indeed, we suggest that this difference in phos-phatase-catalyzed inactivation of the PKA- versus PKB-acti-vated forms of HSPDE3Bmay represent a novel mechanism bywhich cells can differentiate activation of HSPDE3B by thesetwo kinases (Fig. 8). Similarly, earlier works by others (40, 41)have shown that 14-3-3 protein binding to some, but not all,phosphorylated binding partners reduces their rate of dephos-phorylation. Ongoing studies in our laboratory are aimed atassessing the impact that 14-3-3 protein-mediated “protection”against dephosphorylation of the PKA-activated enzyme, butnot that activated by PKB, may have on intracellular cAMP
4 L. S. Wilson and D. H. Maurice, unpublished observations.
FIGURE 8. Model depicting HSPDE3B binding to 14-3-3 proteins and theimpact of this event on PKA- or PKB-mediated activation. A scheme ispresented that summarizes the data obtained in our analysis of the interac-tion of HSPDE3B and 14-3-3 proteins. The scheme depicts how PKA-phospho-rylated HSPDE3B is protected from phosphatase-mediated dephosphoryla-tion/inactivation compared with unbound PKA- or PKB-phosphorylatedHSPDE3B in cells.
TABLE 2Impact of phosphatase treatment on HSPE3B activityNote that 293T cells were transiently transfected with a plasmid encoding a FLAG-tagged HSPDE3B. After 24 h, cells were either treated with vehicle (0.2%Me2SO, v/v) orforskolin and IBMX (F/I, 100 �M each) for 20 min, rinsed, and lysed, and the lysates were used in M2-agarose precipitation, or GST-14-3-3� pulldown experiments (see“Experimental Procedures”). Pellets from the pulldownswere either incubatedwith nothing or 5 units/ml CIAP for 20min at 37 °C. Following these incubations, cAMPPDEactivity was determined using volumes of the reaction products from 14-3-3 pulldowns that gave roughly similar basal levels of activity.
AdditionsM2-agarose 14-3-3�
CIAP cAMP PDE activity % control CIAP cAMP PDE activity % controlpmol/min/�l pmol/min/�l
Vehicle � 38 � 4 � 14 � 2F/I � 49 � 1 29 � 81 � 6 479F/I � 33 � 3 �13 � 67 � 0 379
14-3-3 Proteins and HSPDE3B Activation
9418 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 13 • MARCH 30, 2007
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from

levels and the signaling events associated with HSPDE3B acti-vation in cells.
Acknowledgments—We thank Drs. V. C. Manganiello (NationalInstitutes of Health), R. J. Haslam (McMaster University, Hamilton,Canada), J. A. Beavo (University of Washington), V. Florio, S. Wolda(ICOS Corp.), and D. Alessi (University of Dundee, Scotland, UK) forprovision of reagents used in these studies.
REFERENCES1. Taylor, P., and Insel, P. A. (1988) in Principles of Drug Action: The Basis of
Pharmacology (Pratt, W. B., and Taylor, P., eds) 3rd Ed., pp. 144–164,Churchill Livingstone Inc., New York
2. De Cesare, D., Fimia, G.M., and Sassone-Corsi, P. (1999)Trends Biochem.Sci. 24, 281–285
3. Antoni, F. A. (2000) Front. Neuroendocrinol. 21, 103–1324. Marchese, A., George, S. R., Kolakowski, L. F., Lynch, K. R., and O’Dowd,
B. F. (1999) Trends Pharmacol. Sci. 20, 370–3755. Hanoune, J., and Defer, N. (2001) Annu. Rev. Pharmacol. Toxicol. 41,
145–1746. Manganiello, V. C., and Degerman, E. (1999) Thromb. Haemostasis 82,
407–4117. Soderling, S. H., and Beavo, J. A. (2000)Curr. Opin. Cell Biol. 12, 174–1798. Beavo, J. A., and Brunton, L. L. (2002)Nat. Rev. Mol. Cell Biol. 3, 710–7189. Houslay, M. D., and Adams, D. R. (2003) Biochem. J. 370, 1–1810. Conti, M., Richter,W., Mehats, C., Livera, G., Park, J. Y., and Jin, C. (2003)
J. Biol. Chem. 278, 5493–549611. Maurice, D. H., Palmer, D., Tilley, D. G., Dunkerley, H. A., Netherton, S. J.,
Raymond,D. R., El-Batarny,H. S., and Jimmo, S. L. (2003)Mol. Pharmacol.64, 533–546
12. Bender, A. T., and Beavo, J. A. (2006) Pharmacol. Rev. 58, 488–52013. Reinhardt, R. R., Chin, E., Zhou, J., Taira, M., Murata, T., Manganiello,
V. C., and Bondy, C. A. (1995) J. Clin. Investig. 95, 1528–1523814. Degerman, E., Belfrage, P., and Managaniello, V. C. (1997) J. Biol. Chem.
272, 6823–682615. Wechsler, J., Choi, Y. H., Krall, J., Ahmad, F., Manganiello, V. C., and
Movsesian, M. A. (2002) J. Biol. Chem. 277, 38072–3807816. Hambleton, R., Krall, J., Tikishvili, E., Honeggar, M., Ahmad, F., Manga-
niello, V. C., andMovsesian,M.A. (2005) J. Biol. Chem. 280, 39168–3917417. Dodge-Kafka, K. L., Langeberg, L., and Scott, J. D. (2006) Circ. Res. 98,
993–100118. McConnachie, G., Langeberg, L. K., and Scott, J. D. (2006) Trends Mol.
Med. 12, 317–323
19. Rondinone, C. M., Carvalho, E., Rahn, T., Manganiello, V. C., Degerman,E., and Smith, U. P. (2000) J. Biol. Chem. 275, 10093–10098
20. Zmuda-Trzebiatowska, E.,Manganiello, V., andDegerman, E. (2006)Cell.Signal. 19, 81–86
21. Nilsson, R., Ahmad, F., Sward, K., Andersson, U., Weston, M., Manga-niello, V., and Degerman, E. (2006) Cell. Signal. 18, 1713–1721
22. Patrucco, E., Notte, A., Barberis, L., Selvetella, G., Maffei, A., Brancaccio,M., Marengo, S., Russo, G., Azzolino, O., Rybalkin, S. D., Silengo, L., Al-truda, F., Wetzker, R., Wymann, M. P., Lembo, G., and Hirsch, E. (2004)Cell 118, 375–387
23. Voigt, P., Dorner, M. B., and Schaefer, M. (2006) J. Biol. Chem. 281,9977–9986
24. Onuma, H., Osawa, H., Yamada, K., Ogura, T., Tanabe, F., Granner, D. K.,and Makino, H. (2002) Diabetes 51, 3362–3367
25. Darling, D. L., Yingling, J., andWynshaw-Boris, A. (2005) Curr. Top. Dev.Biol. 68, 281–315
26. Pozuelo Rubio, M., Campbell, D. G., Morrice, N. A., and Mackintosh, C.(2005) Biochem. J. 392, 163–172
27. Fields, S., and Song, O. (1989) Nature 340, 245–24628. Rose, R. J., Liu, H., Palmer, D., andMaurice, D. H. (1997) Br. J. Pharmacol.
122, 233–24029. Liu, H., and Maurice, D. H. (1999) J. Biol. Chem. 274, 10557–1056530. Blom, N., Gammeltoft, S., and Brunak, S. (1999) J. Mol. Biol. 294,
1351–136231. Kitamura, T., Kitamura, Y., Kuroda, S., Hino, Y., Ando, M., Kotani, K.,
Konishi, H., Matsuzaki, H., Kikkawa, U., Ogawa, W., and Kasuga, M.(1999)Mol. Cell. Biol. 19, 6286–6296
32. Rajan, S., Preisig-Muller, R., Wischmeyer, E., Nehring, R., Hanley, P. J.,Renigunta, V., Musset, B., Schlichthorl, G., Derst, C., Karschin, A., andDaut, J. (2002) J. Physiol. (Lond.) 545, 13–26
33. Pawson, T., and Scott, J. D. (2005) Trends Biochem. Sci. 30, 286–29034. Seet, B. T., Dikic, I., Zhou,M.M., and Pawson, T. (2006)Nat. Rev.Mol. Cell
Biol. 7, 473–48335. Shakur, Y., Takeda, K., Kenan, Y., Yu, Z.-X., Rena, G., Brandt, D., Houslay,
M. D., Degerman, E., Ferrans, V. J., and Manganiello, V. C. (2000) J. Biol.Chem. 275, 38749–38761
36. Elbatarny, H. S., andMaurice, D. H. (2005)Am. J. Physiol. 289, E695–E70237. Feng, S., Christodoulides, N., Resendiz, J. C., Berndt, M. C., and Kroll,
M. H. (2000) Blood 95, 551–55738. Masters, S. C., Pederson, K. J., Zhang, L., Barbieri, J. T., and Fu, H. (1999)
Biochem. J. 38, 12499–1250439. Feldman, R. D. (1993) Br. J. Pharmacol. 110, 1640–164740. Chen, F., and Wagner, P. D. (1994) FEBS Lett. 347, 128–13241. Hutchins, J. R., Dikovskaya, D., and Clarke, P. R. (2002) FEBS Lett. 528,
267–271
14-3-3 Proteins and HSPDE3B Activation
MARCH 30, 2007 • VOLUME 282 • NUMBER 13 JOURNAL OF BIOLOGICAL CHEMISTRY 9419
at University of C
alifornia, San F
rancisco on August 25, 2009
ww
w.jbc.org
Dow
nloaded from
Related Documents











