Protection Against Kainate Neurotoxicity by Ginsenosides: Attenuation of Convulsive Behavior, Mitochondrial Dysfunction, and Oxidative Stress Eun-Joo Shin, 1 Ji Hoon Jeong, 2 A-Young Kim, 3 Young Ho Koh, 3 Seung-Yeoul Nah, 4 Won-Ki Kim, 5 Kwang Ho Ko, 6 Hyun Ji Kim, 1 Myung-Bok Wie, 7 Yong Soo Kwon, 1 Yukio Yoneda, 8 and Hyoung-Chun Kim 1 * 1 Neuropsychopharmacology and Toxicology Program, College of Pharmacy, Kangwon National University, South Korea 2 Department of Pharmacology, College of Medicine, Chung-Ang University, Seoul, South Korea 3 Laboratory of Molecular Neurogenetics, ILSONG Institute of Life Science, Hallym University, Anyang, South Korea 4 Department of Physiology, College of Veterinary Medicine, Konkuk University, Seoul, South Korea 5 Department of Neuroscience, School of Medicine, Korea University, South Korea 6 Department of Pharmacology, College of Pharmacy, Seoul National University, Seoul, South Korea 7 School of Veterinary Medicine, Kangwon National University, Chunchon, South Korea 8 Laboratory of Molecular Pharmacology, Division of Pharmaceutical Sciences, Kanazawa University Graduate School of Natural Science and Technology, Kanazawa, Ishikawa, Japan We previously demonstrated that kainic acid (KA)-medi- ated mitochondrial oxidative stress contributed to hip- pocampal degeneration and that ginsenosides attenu- ated KA-induced neurotoxicity and neuronal degenera- tion. Here, we examined whether ginsenosides affected KA-induced mitochondrial dysfunction and oxidative stress in the rat hippocampus. Treatment with ginseno- sides attenuated KA-induced convulsive behavior dose- dependently. KA treatment increased lipid peroxidation and protein oxidation and decreased the reduced glutathione/oxidized glutathione (GSH/GSSG) ratio to a greater degree in the mitochondrial fraction than in the hippocampal homogenate. KA treatment resulted in decreased Mn-superoxide dismutase expression and diminished the mitochondrial membrane potential. Furthermore, KA treatment increased intramitochondrial Ca 21 and promoted ultrastructural degeneration in hip- pocampal mitochondria. Treatment with ginsenosides dose-dependently attenuated convulsive behavior and the KA-induced mitochondrial effects. Protection app- eared to be more evident in mitochondria than in tissue homogenates. Collectively, the results suggest that gin- senosides prevent KA-induced neurotoxicity by attenu- ating mitochondrial oxidative stress and mitochondrial dysfunction. V V C 2008 Wiley-Liss, Inc. Key words: GSH/GSSG; ultrastructural degeneration; hippocampus; Mn-superoxide dismutase; mitochondrial membrane potential Ginseng (Panax ginseng C.A. Meyer) is a well-known Asian medicinal plant that has long been used in folk medi- cine. Active constituents found in most ginseng species include ginsenosides, polysaccharides, peptides, polyacety- lenic alcohols, and fatty acids (Attele et al., 1999). The major component responsible for the actions of ginseng are the ginsenosides, which are also known as ginseng saponins (Yuan et al., 1998). Ginsenosides are a class of steroid-like compounds that are found exclusively in P. ginseng, and about 30 different forms have been isolated and chemically identified from ginseng root (Attele et al., 1999). Kainic acid (KA)-induced brain damage has been used as a model for temporal lobe epilepsy and excito- toxic neurodegenerative disorders (Sperk, 1994). Accu- mulating evidence indicates that hippocampal oxidative stress might be involved in KA-induced neurotoxicity in vivo (Floreani at al., 1997; Kim et al., 2000a, 2000b; Shin et al., 2008b) and in vitro (Shih et al., 2004; Kim et al., 2008). We previously demonstrated that ginseno- sides prevent KA-induced hippocampal degeneration in Contract grant sponsor: Brain Research Center from 21st Century Fron- tier Research Program funded by the Ministry of Science and Technol- ogy, Republic of Korea; Contract grant number: M103KV010013- 08K2201-01310. The first two authors contributed equally to this work. *Correspondence to: Hyoung-Chun Kim, PhD, Neuropsychopharmacol- ogy and Toxicology Program, College of Pharmacy, Kangwon National University, Chunchon 200-701, South Korea. E-mail: [email protected] Received 9 June 2008; Revised 9 July 2008; Accepted 11 July 2008 Published online 24 September 2008 in Wiley InterScience (www. interscience.wiley.com). DOI: 10.1002/jnr.21880 Journal of Neuroscience Research 87:710–722 (2009) ' 2008 Wiley-Liss, Inc.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Protection Against Kainate Neurotoxicityby Ginsenosides: Attenuation ofConvulsive Behavior, MitochondrialDysfunction, and Oxidative Stress
Eun-Joo Shin,1 Ji Hoon Jeong,2 A-Young Kim,3 Young Ho Koh,3
Seung-Yeoul Nah,4 Won-Ki Kim,5 Kwang Ho Ko,6 Hyun Ji Kim,1
Myung-Bok Wie,7 Yong Soo Kwon,1 Yukio Yoneda,8 and Hyoung-Chun Kim1*1Neuropsychopharmacology and Toxicology Program, College of Pharmacy,Kangwon National University, South Korea2Department of Pharmacology, College of Medicine, Chung-Ang University, Seoul, South Korea3Laboratory of Molecular Neurogenetics, ILSONG Institute of Life Science, Hallym University, Anyang,South Korea4Department of Physiology, College of Veterinary Medicine, Konkuk University, Seoul, South Korea5Department of Neuroscience, School of Medicine, Korea University, South Korea6Department of Pharmacology, College of Pharmacy, Seoul National University, Seoul, South Korea7School of Veterinary Medicine, Kangwon National University, Chunchon, South Korea8Laboratory of Molecular Pharmacology, Division of Pharmaceutical Sciences, Kanazawa UniversityGraduate School of Natural Science and Technology, Kanazawa, Ishikawa, Japan
We previously demonstrated that kainic acid (KA)-medi-ated mitochondrial oxidative stress contributed to hip-pocampal degeneration and that ginsenosides attenu-ated KA-induced neurotoxicity and neuronal degenera-tion. Here, we examined whether ginsenosides affectedKA-induced mitochondrial dysfunction and oxidativestress in the rat hippocampus. Treatment with ginseno-sides attenuated KA-induced convulsive behavior dose-dependently. KA treatment increased lipid peroxidationand protein oxidation and decreased the reducedglutathione/oxidized glutathione (GSH/GSSG) ratio to agreater degree in the mitochondrial fraction than inthe hippocampal homogenate. KA treatment resultedin decreased Mn-superoxide dismutase expressionand diminished the mitochondrial membrane potential.Furthermore, KA treatment increased intramitochondrialCa21 and promoted ultrastructural degeneration in hip-pocampal mitochondria. Treatment with ginsenosidesdose-dependently attenuated convulsive behavior andthe KA-induced mitochondrial effects. Protection app-eared to be more evident in mitochondria than in tissuehomogenates. Collectively, the results suggest that gin-senosides prevent KA-induced neurotoxicity by attenu-ating mitochondrial oxidative stress and mitochondrialdysfunction. VVC 2008 Wiley-Liss, Inc.
Key words: GSH/GSSG; ultrastructural degeneration;hippocampus; Mn-superoxide dismutase; mitochondrialmembrane potential
Ginseng (Panax ginseng C.A. Meyer) is a well-knownAsian medicinal plant that has long been used in folk medi-
cine. Active constituents found in most ginseng speciesinclude ginsenosides, polysaccharides, peptides, polyacety-lenic alcohols, and fatty acids (Attele et al., 1999). Themajor component responsible for the actions of ginseng arethe ginsenosides, which are also known as ginseng saponins(Yuan et al., 1998). Ginsenosides are a class of steroid-likecompounds that are found exclusively in P. ginseng, andabout 30 different forms have been isolated and chemicallyidentified from ginseng root (Attele et al., 1999).
Kainic acid (KA)-induced brain damage has beenused as a model for temporal lobe epilepsy and excito-toxic neurodegenerative disorders (Sperk, 1994). Accu-mulating evidence indicates that hippocampal oxidativestress might be involved in KA-induced neurotoxicity invivo (Floreani at al., 1997; Kim et al., 2000a, 2000b;Shin et al., 2008b) and in vitro (Shih et al., 2004; Kimet al., 2008). We previously demonstrated that ginseno-sides prevent KA-induced hippocampal degeneration in
Contract grant sponsor: Brain Research Center from 21st Century Fron-
tier Research Program funded by the Ministry of Science and Technol-
ogy, Republic of Korea; Contract grant number: M103KV010013-
08K2201-01310.
The first two authors contributed equally to this work.
*Correspondence to: Hyoung-Chun Kim, PhD, Neuropsychopharmacol-
ogy and Toxicology Program, College of Pharmacy, Kangwon National
University, Chunchon 200-701, South Korea.
E-mail: [email protected]
Received 9 June 2008; Revised 9 July 2008; Accepted 11 July 2008
Published online 24 September 2008 in Wiley InterScience (www.
interscience.wiley.com). DOI: 10.1002/jnr.21880
Journal of Neuroscience Research 87:710–722 (2009)
' 2008 Wiley-Liss, Inc.

rats (Lee et al., 2002). Furthermore, ginsenosides wereshown to attenuate neuronal degeneration induced bythe mitochondrial toxin 3-nitropropionic acid (Kimet al., 2005), and mitochondrial oxidative stress appearsto play a mechanistic role in neuronal degenerationinduced by KA (Kim et al., 2000a; Shin et al., 2008a).
To extend our understanding, we examined theeffect of ginsenosides in response to KA-induced mito-chondrial damage and oxidative stress in the rat hippo-campus. The data suggest that ginsenosides mediate therestoration of mitochondrial function after oxidativeinsults and that the attenuation of KA-induced mito-chondrial dysfunction is important for the neuroprotec-tion mediated by ginsenosides.
MATERIALS AND METHODS
Animals and Treatment
All animals were treated in accordance with theNational Institutes of Health Guide for the Humane Care andUse of Laboratory Animals. Male Sprague-Dawley rats (BioGenomic, Inc./Charles River Technology, Gapyung-Gun,Gyeonggi-Do, Korea) weighing �350 g were maintained ona 12-hr light/12-hr dark cycle and fed ad libitum. They wereallowed to adapt to these conditions for 2 weeks before exper-imentation. Ginsenosides were extracted and purified accord-ing to Namba et al. (1974) from a characterized saponin mix-ture from P. ginseng (supplied by the Korean Ginseng andTobacco Research Institute) that contained at least 11 glyco-sides as follows (%): Rb1 (18.26), Rb2 (9.07), Rc (9.65), Rd(8.24), Re (9.28), Rf (3.48), Rg1 (6.42), Rg2 (3.62), Rg3(4.70), Ro (3.82), Ra1 (2.91), and other minor ginsenosides(20.55) (Lee at al., 2002; Shin et al., 2005a). The chemicalname of each ginsenoside was shown in Table I.
Ginsenosides (50 or 100 mg/kg) were administered byintraperitoneal injection five times at 12-hr intervals. Controlrats received the same volume of saline. One hour after thelast administration of ginsenosides, KA (10 mg/kg) wasadministered intraperitoneally.
Seizure Activity and Sampling
We assessed wet dog shake behavior and seizure activityby means of an automated video tracking system (Noldus In-formation Technology, Wageningen, Netherlands). Seizure
activity was rated during a 4-hr period after the KA (10 mg/kg intraperitoneally [i.p.]) challenge, according to the scaledevised by Racine (1972): stage 1 (facial clonus), stage 2 (nod-ding), stage 3 (forelimb clonus), stage 4 (forelimb clonus withrearing), and stage 5 (rearing, jumping, and falling). Animalswere scored after having had three consecutive seizures at aparticular stage. A convulsion meter (CONVULS-1, Colum-bus Instruments, OH) was also used simultaneously. The sei-zure activity over a 60-min period was measured 2 hr afterthe KA treatment (Shin et al., 2008b).
Two days after the KA treatment, seizing rats wereanesthetized with pentobarbital (50 mg/kg) and perfusedtranscardially with saline (40 mL/100 g body weight) followedby 4% paraformaldehyde (80 mL/100 g body weight) via a140-mL syringe. The brains were removed and then cut into35-lm sections in the horizontal plane with a sliding micro-tome. The hippocampi of other seizing rats were dissected forevaluation of oxidative stress markers and mitochondrial dys-function 4 hr or 2 days after KA administration.
Mitochondrial Preparation
Isolated mitochondria were prepared as described previ-ously (Kwon et al., 2004) with slight modification (Xionget al., 1997). The rats were anesthetized with sodium pento-barbital (60 mg/kg) and perfused transcardially with 30 mL ofice-cold homogenization buffer (250 mM sucrose, 20 mMHEPES [N-[2-hydroxyethyl] piperazine-N0[2-ethanesulfonicacid]], 1 mM EDTA [ethylenediaminetetraacetic acid], pH7.2) 4 hr or 48 hr after KA. The animals were then decapi-tated, and their brain were dissected out and minced withscissors in 10 mL of ice-cold homogenization buffer. All sub-sequent steps were conducted at 48. The tissue was rinsedwith 10 mL of homogenization buffer and processed with atissue homogenizer. The resulting homogenate was centri-fuged for 10 min at 1300g. The supernatant was removed andcentrifuged at 10,000g for 10 min. The pellet was resuspendedin 30 mL of homogenization buffer with a homogenizer andcentrifuged for 10 min at 10,000g. The resulting pellet wasagain resuspended and rinsed in EDTA-free homogenizationbuffer. The mitochondrial pellet was then resuspended in250 mM sucrose solution at a final concentration of 10 mg/mLand placed on ice. The entire mitochondrial preparation pro-cedure required less than 1 hr to complete.
TABLE I. Chemical Name of Each Ginsenoside
Ginsenoside Chemical name
Rb1 Protopanaxadiol-3-[b-D-glucopyranosyl-(1?2)-b-D-glucopyranoside]-20-O-[b-D-glucopyranosyl-(1?6)-b-D-glucopyranoside]
Rb2 Protopanaxadiol-3-O-[b-D-glucopyranosyl-(1?2)-b-D-glucopyranoside]-20-O-[a-L-arabinopyranoside-(1?6)-b-D-glucopyanosie]
Rc Protopanaxadiol-3-O-[b-D-glucopyranosyl-(1?2)-b-D-glucopyranoside]-20-O-[a-L-arabinofuranoside-(1?6)-b-D-glucopyanosie]
Rd 20(S)-Protopanaxadiol-3-O-[b-D-glucopyranosyl-(1?2)-b-D-glucopyranoside]-20-O-[b-D-glucopyanosie]
Re Protopanaxtriol-6-O-[a-L-rhamnopyranosyl-(1?2)-b-D-glucopyranoside]-20-O-[b-D-glucopyanosie]
Rf Protopanaxatriol-6-O-[b-D-glucopyranosyl-(1?2)-b-D-glucopyranoside]
Rg1 Protopanaxa triol-6-O-[b-D-glucopyranoside]-20-O-[b-D-glucopyranoside]
Rg2 20(S)-Protopanaxatriol-6-O-[a-L-rhamnopyranosyl-(1?2)-b-D-lucopyranoside]
Rg3 20(S)-Protopanaxadiol-3-O-[b-D-glucopyranosyl-(1?2)-b-D-glucopyranoside]
Ro Oleanolic acid-3-O-[b-D-glucopyranosyl-(1?2)-b-D-glucopyranoside]-28-O-[b-D-glucopyanosie]ester
Ra1 Protopanaxadiol-3-O-[b-D-glucopyranosyl-(1?2)-b-D-glucopyranoside]-20-O-[b-D-xylopyranosyl-(1?4)-a-L-arabinopyranosyl-(1?6)-b-D-glucopyranoside]
Ginsenosides, Kainate, and Mitochondria 711
Journal of Neuroscience Research

Assessment of Mitochondrial TransmembranePotential (MTP)
MTP was measured as described previously (Bruce-Kel-ler et al., 1999) with the dye 5,50,6,60-tetrachloro-1,10,3,30-tet-raethylbenzimidazolycarbocyanine iodide (JC-1; MolecularProbes), which exists as a green fluorescent monomer at lowmembrane potential, but reversibly forms red fluorescent ‘‘J-aggregates’’ at polarized mitochondrial potentials. Briefly, ali-quots of 250 lg of isolated mitochondrial protein were sus-pended in respiration buffer (250 mM sucrose, 20 mMHEPES, 2 mM MgCl2, 2.5 mM inorganic phosphates [pH7.2], and 10 mM succinate (5 mM glutamate and 2.5 mMmaleate gave similar results in all paradigms]) in a final volumeof 200 lL. The energized mitochondria were then incubatedat 378 in the presence of 10 lM JC-1 for 30 min, after whichfluorescence was measured with a fluorometric plate reader.The relative amount of mitochondrial polarization per milli-gram of mitochondria was quantified by taking the ratio ofemission from 590 to 535 nm, respectively, with excitation at500 nm (Kwon et al., 2004).
Intramitochondrial Ca21 Levels
Intramitochondrial Ca21 levels were assessed asdescribed previously (Matton et al., 1998). Briefly, mitochon-drial fractions (250 lg) were incubated in the presence of theCa21 indicator rhod-2-AM (5 lM, Molecular Probes) for60 min at 378C and washed three times with Ca21-freeLocke’s solution. This reduced form of rhod-2-AM, is a col-orless, nonfluorescent dye that has a net positive charge,which promotes its sequestration into mitochondria. The dyeis then oxidized in the mitochondria where the AM ester iscleaved, trapping the dye in the mitochondria. Fluorescencewas quantified with a fluorometric plate reader with excitationand emission wavelengths of 549 and 581 nm, respectively.
Determination of Thiobarbituric Acid–reactiveSubstances (TBARS) and Protein Carbonyls
The amount of lipid peroxidation was determined bymeasuring the accumulation of TBARS in the tissue homoge-nate and mitochondrial fraction of the hippocampus (Fragaet al., 1988; Shin et al., 2008a). Samples were mixed with 0.1N HCl (2 mL), 10% phosphotungstic acid (0.3 mL), and0.67% 2-thiobarbituric acid (1 mL). The mixture was heatedfor 30 min in boiling water and extracted with n-butanol(5 mL). After a brief centrifugation, the absorption of the bu-tanol layer was measured at 535 nm (e 5 153 mM/cm) andthe results are expressed as pmol TBARS (malondialdehydeequivalent)/mg protein.
The extent of protein oxidation was assessed by meas-uring the content of protein carbonyl groups (Oliver et al.,1987). The homogenate and mitochondrial fractions wereadded to 10% trichloroacetic acid (TCA, 0.05 mL), and theprecipitated proteins were resuspended in 2,4-dinitrophenylhydrazine (0.05 mL), incubated for 1 hr at 378C, precipitatedagain with TCA, centrifuged, washed with ethanol:ethyl ace-tate (50:50), and dissolved in 6 mM guanidine hydrochloridein phosphate buffer (pH 6.5). The absorbance at 370 nm (e 521 mM/cm) was determined. Protein carbonyls are expressed
as pmol/mg protein. Protein was measured with the BCAprotein assay reagent (Pierce, Rockford, IL).
Determination of GSH and GSSG byHigh-performance Liquid Chromatography (HPLC)
GSH and GSSG were immediately measured (Kimet al., 2000b; Shin et al., 2005b, 2008a) from dissected tissuesby using a minor modification of the method described byReed et al. (1980). Briefly, each acidified supernatant of thehippocampal homogenate and mitochondrial fraction wasadded to the internal standard (1 mM cysteic acid) in 0.88 Miodoacetic acid. Excess sodium hydrogen carbonate was addedto the reaction to precipitate sodium perchlorate. Subse-quently, 0.5 mL of an alcoholic solution of 1.5% (v/v) 2,4-dinitrofluorobenzene was added to each sample, and the sam-ples were incubated for 4 hr. Diethyl ether (1.0 mL) wasadded, and the samples were shaken and centrifuged (20 min,2000g, room temperature). The residual aqueous phase con-taining derived glutathione was separated and analyzed byHPLC. The separation of 5-carboxymethyl glutathione wascarried out at room temperature with a flow rate of 1.2 mL/min. Chromatographic separation of derivatives was per-formed by injecting samples (10 lL) of the aqueous phaseonto a Spherisorb S-5 amino ODS column. Glutathione wassubsequently detected with an ultraviolet detector at 365 nm.Glutathione derivatives (GSH and GSSG) were quantified inrelation to the internal standard (cysteic acid).
Immunohistochemistry
Immunocytochemistry was performed as described pre-viously (Kim et al., 2000a, 2003). Sections were preincubatedwith 0.3% hydrogen peroxide in phosphate-buffered saline(PBS) for 30 min, with PBS containing 0.4% Triton X-100for 20 min, and then with 1% normal serum for 20 min. Sec-tions were then incubated for 48 hr at 48C with antibodiesagainst SOD-2 or c-Fos (1:1000 dilution; Santa Cruz Biotech.Inc., CA), followed by incubation with biotinylated secondaryantisera for 1 hr. The sections were immersed in a solutioncontaining an avidin-biotin-peroxidase complex (Vector Labo-ratories, Burlingame, CA) for 1 hr, and 3,30-diaminobenzidinewas used as the chromogen. The SOD-2 antibody has beendescribed previously (Kurobe et al., 1990; Kim et al., 2000a,2003; Kwon et al., 2004; Shin et al., 2008).
Western Blot Test
Hippocampi were homogenized and protein extractswere prepared as described previously (Kim et al., 2003). Pro-tein extracts (40 lg) were separated in 10% sodium dodecylsulfate–polyacrylamide gels and transferred onto membranes,and the resulting blots were blocked in PBS containing 3%skim milk for 30 min. Blots were incubated overnight at 48Cwith antibodies against SOD-2 (Kurobe et al., 1990; Kimet al., 2000a, 2003; Kwon et al., 2004; Shin et al., 2008a) orc-Fos (Shin et al., 2005c) at 1:1000 dilutions. After washing inPBS, the membranes were incubated with a biotinylated goatanti-rabbit antibody for 1 hr, followed by incubation in a so-lution containing an avidin-biotin horseradish-peroxidase con-jugate (Vectastatin kit, Vector) at room temperature. The blots
712 Shin et al.
Journal of Neuroscience Research

were then incubated in enhanced chemiluminescence reagent(Amersham, Arlington Heights, IL) and exposed to AmershamHyperfilm for 2 min. The intensities of the bands were quan-tified by laser densitometry.
Fluorescent Nissl Staining
At 2 days after KA injection, the mice were perfusedtranscardially with 50 mL of 50 mM PBS, followed by 100 mLof 4% paraformaldehyde in PBS, under pentobarbital anes-thesia. The brains were removed, postfixed for 24 hr in thesame fixative at 48C, and then immersed in 30% sucrose inPBS until they sank. The brains were cut into 35-lm transversefree-floating sections with a horizontal sliding microtome. Flu-orescent Nissl staining was performed with NeuroTrace 530/615 red fluorescent Nissl stain solution (N-21482, Invitrogen).Briefly, the sections were incubated in PBS containing 0.1%Triton X-100 (Sigma-Aldrich) for 10 min and then incubatedin a 100-fold dilution of NeuroTrace solution for 20 min. Thesections were then washed in PBS for 2 hr and mounted withan antifade agent (Fluoromount-G, Southern Biotech, Bir-mingham, AL). Digital images of fluorescent Nissl-stained neu-rons were acquired at 3003 magnification with a confocal laserscanning microscope (LSM 510 META, Carl Zeiss, Inc., Ober-kochen, Germany) (Shin et al., 2007, 2008a).
Fluoro-Jade B Staining
The brain sections were mounted on silane-coated glassslides and fully dried. The sections were rehydrated by immer-sion in 100% ethanol, 70% ethanol, and distilled water for1 min each. The slides were then transferred to a solution of0.06% potassium permanganate for 15 min on a horizontalshaker, rinsed for 1 min in distilled water, and then incubatedin a solution of 0.0004% Fluoro-Jade B (Chemicon Interna-tional, Inc., Temecula, CA) for 20 min at room temperature.The sections were rinsed 331 min each in distilled water,excess water was removed briefly, and the slides were fullydried. The slides were cleared by immersion in xylene for atleast 1 min before coverslips were emplaced. Digital imageswere acquired at 1003magnification with a confocal laserscanning microscope (LSM 510 META) with excitation at480 nm and emission at 525 nm (Schmued et al., 1997; Shinet al., 2008a).
Electron Microscopy
Specimens for morphologic assessments of mitochondrialdamage (Shin et al., 2008a) per hippocampal section were pre-pared as follows. A 2-mm-long cylindrical segment of theCA3 region of the hippocampus was prepared with a tissuechopper and dissecting scope immediately after killing the ani-mal. The segment was fixed in glutaraldehyde and osmic acidand then embedded in epoxy resin, following the routine pro-cedure of tissue preparation for electron microscopy.
Statistics
Statistical analyses were performed by one-way analysisof variance (ANOVA) with a post hoc Fisher’s protectedleast-significant difference (PLSD) test. A P value of <0.05was accepted as statistically significant.
RESULTS
Ginsenosides Protect Against KA-inducedConvulsive Behavior and Decrease c-FosImmunoreactivity
Animals receiving KA showed wet dog shakebehavior (202 6 20; Fig. 1A) and robust behavioral seiz-ures lasting 4–5 hr. Treatment with ginsenosides signifi-cantly reduced KA-induced wet dog shake number andseizure scores, as measured by Racine’s score, and com-pulsive impulses in a dose-dependent manner (Fig.1A,C). The onset of KA-induced seizures was delayedsignificantly by pretreatment with 100 mg/kg ginseno-sides (P < 0.01; Fig. 1B).
Expression of the proto-oncogene c-Fos has beenshown to increase rapidly and transiently in specific brainterritories during seizures (Dragunow and Robertson,1987); in that study, hippocampi exhibited spatial andtemporal expression of c-Fos that depended on the se-verity of the seizure. In our study, sections were proc-essed for c-Fos immunocytochemistry to examine thecellular response 4 hr after KA treatment. c-Fos–likeimmunoreactivity (c-Fos-IR) was minimally induced insaline-treated animals. Nuclear staining for c-Fos in thedentate gyrus was clearly visible 4 hr after KA. The c-Fos-IR was higher (P < 0.01) in the dentate gyrus thanin either the CA1 or CA3 region. KA-induced increasesin c-Fos-IR in CA1, CA3, and the dentate gyrus (P <0.01 for all regions) were significantly attenuated in adose-dependent manner by pretreatment with ginseno-sides (P < 0.01 for all regions; Fig. 1D).
Western blot analysis of c-Fos showed results con-sistent with those observed by immunocytochemistry.KA induced increases (P < 0.01) in c-Fos expression,which were attenuated dose-dependently in the presenceof ginsenosides (Fig. 1E).
Ginsenosides Attenuate KA-induced Increases inProtein Oxidation and Lipid Peroxidation in theHomogenate and Mitochondrial Fraction of theRat Hippocampus
Oxidative stress and mitochondrial dysfunctionhave been previously demonstrated to occur as a conse-quence of prolonged epileptic seizures and may play animportant role in seizure-induced brain damage (Lianget al., 2000). Our earlier study demonstrated thatincreases in lipid peroxidation and protein oxidation par-ticipated in the cascade of events leading to neuronaldamage after KA-induced seizures (Kim et al., 2000b;Shin et al., 2008a).
In the absence of KA, ginsenosides did not affectthe levels of protein oxidation (as assessed by proteincarbonyls) and lipid peroxidation (as assessed by thiobar-bituric acid reactive substances, TBARS) in the mito-chondrial fraction and homogenate (Fig. 2). These oxi-dative stress markers increased significantly 4 hr after KAtreatment in the mitochondrial fraction (P < 0.01) andhomogenate (protein carbonyls, P < 0.05; TBARS, P <0.01), and remained high in the mitochondrial fraction
Ginsenosides, Kainate, and Mitochondria 713
Journal of Neuroscience Research
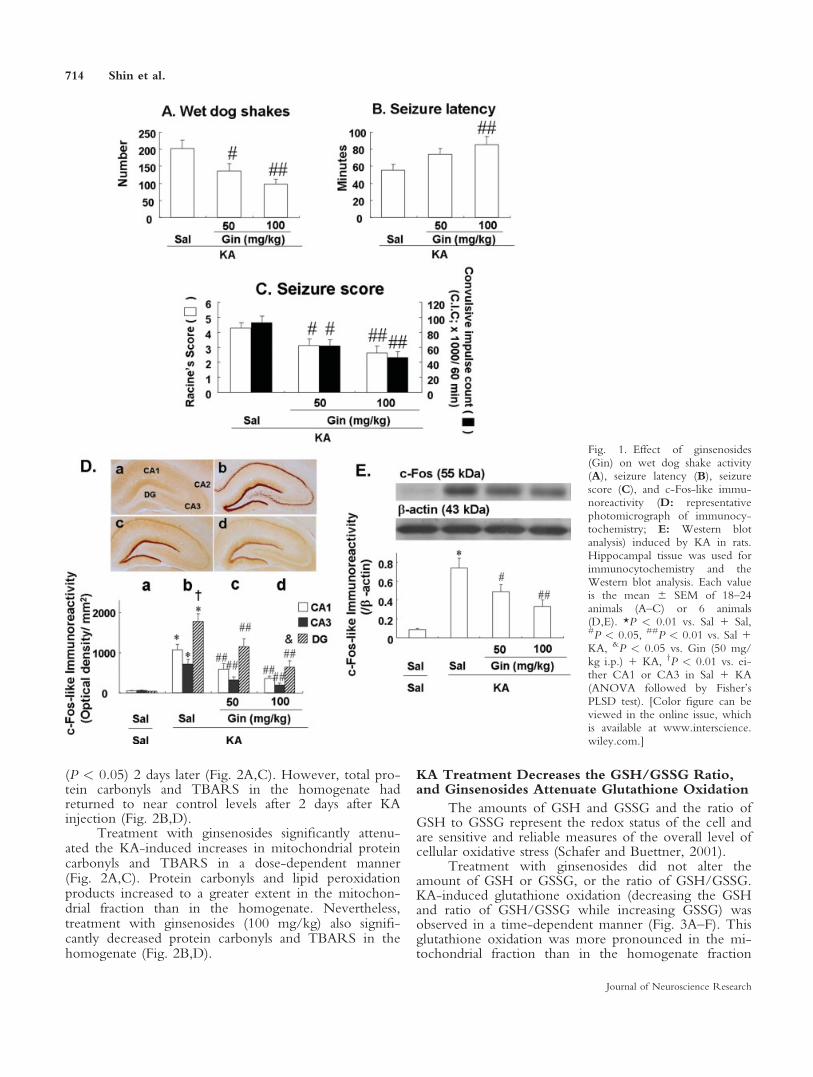
(P < 0.05) 2 days later (Fig. 2A,C). However, total pro-tein carbonyls and TBARS in the homogenate hadreturned to near control levels after 2 days after KAinjection (Fig. 2B,D).
Treatment with ginsenosides significantly attenu-ated the KA-induced increases in mitochondrial proteincarbonyls and TBARS in a dose-dependent manner(Fig. 2A,C). Protein carbonyls and lipid peroxidationproducts increased to a greater extent in the mitochon-drial fraction than in the homogenate. Nevertheless,treatment with ginsenosides (100 mg/kg) also signifi-cantly decreased protein carbonyls and TBARS in thehomogenate (Fig. 2B,D).
KA Treatment Decreases the GSH/GSSG Ratio,and Ginsenosides Attenuate Glutathione Oxidation
The amounts of GSH and GSSG and the ratio ofGSH to GSSG represent the redox status of the cell andare sensitive and reliable measures of the overall level ofcellular oxidative stress (Schafer and Buettner, 2001).
Treatment with ginsenosides did not alter theamount of GSH or GSSG, or the ratio of GSH/GSSG.KA-induced glutathione oxidation (decreasing the GSHand ratio of GSH/GSSG while increasing GSSG) wasobserved in a time-dependent manner (Fig. 3A–F). Thisglutathione oxidation was more pronounced in the mi-tochondrial fraction than in the homogenate fraction
Fig. 1. Effect of ginsenosides(Gin) on wet dog shake activity(A), seizure latency (B), seizurescore (C), and c-Fos-like immu-noreactivity (D: representativephotomicrograph of immunocy-tochemistry; E: Western blotanalysis) induced by KA in rats.Hippocampal tissue was used forimmunocytochemistry and theWestern blot analysis. Each valueis the mean 6 SEM of 18–24animals (A–C) or 6 animals(D,E). *P < 0.01 vs. Sal 1 Sal,#P < 0.05, ##P < 0.01 vs. Sal 1KA, &P < 0.05 vs. Gin (50 mg/kg i.p.) 1 KA, yP < 0.01 vs. ei-ther CA1 or CA3 in Sal 1 KA(ANOVA followed by Fisher’sPLSD test). [Color figure can beviewed in the online issue, whichis available at www.interscience.wiley.com.]
714 Shin et al.
Journal of Neuroscience Research
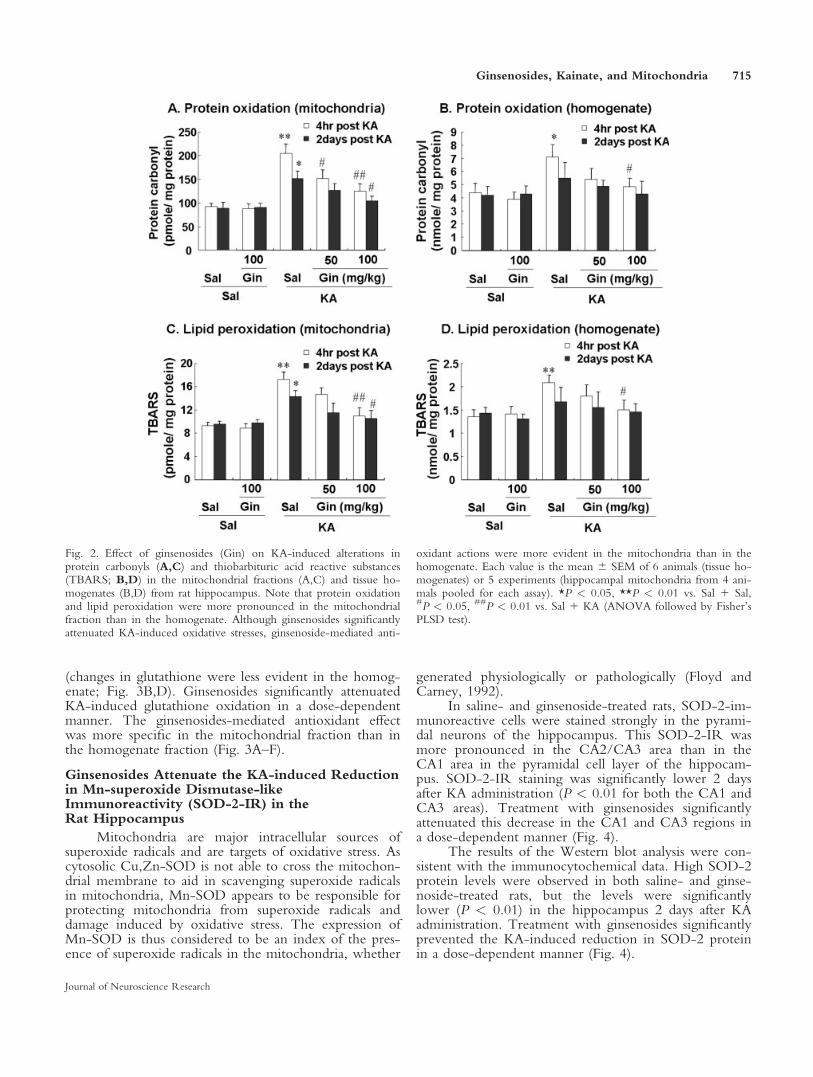
(changes in glutathione were less evident in the homog-enate; Fig. 3B,D). Ginsenosides significantly attenuatedKA-induced glutathione oxidation in a dose-dependentmanner. The ginsenosides-mediated antioxidant effectwas more specific in the mitochondrial fraction than inthe homogenate fraction (Fig. 3A–F).
Ginsenosides Attenuate the KA-induced Reductionin Mn-superoxide Dismutase-likeImmunoreactivity (SOD-2-IR) in theRat Hippocampus
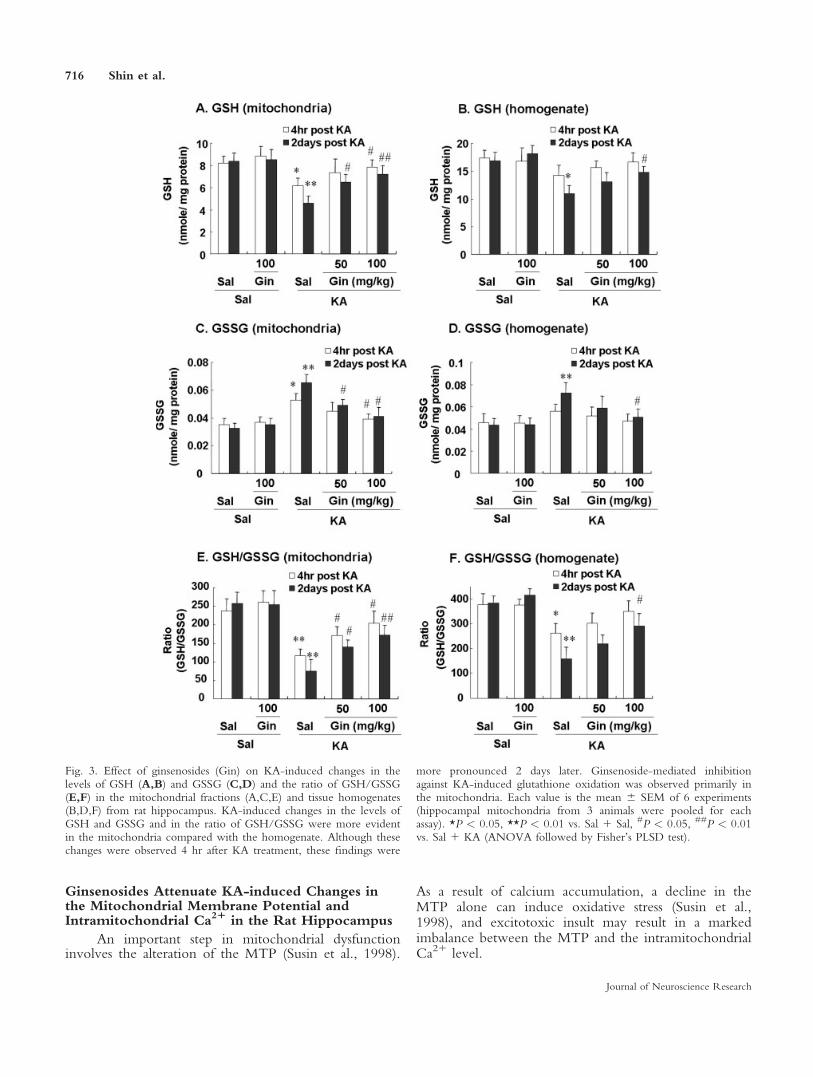
Mitochondria are major intracellular sources ofsuperoxide radicals and are targets of oxidative stress. Ascytosolic Cu,Zn-SOD is not able to cross the mitochon-drial membrane to aid in scavenging superoxide radicalsin mitochondria, Mn-SOD appears to be responsible forprotecting mitochondria from superoxide radicals anddamage induced by oxidative stress. The expression ofMn-SOD is thus considered to be an index of the pres-ence of superoxide radicals in the mitochondria, whether
generated physiologically or pathologically (Floyd andCarney, 1992).
In saline- and ginsenoside-treated rats, SOD-2-im-munoreactive cells were stained strongly in the pyrami-dal neurons of the hippocampus. This SOD-2-IR wasmore pronounced in the CA2/CA3 area than in theCA1 area in the pyramidal cell layer of the hippocam-pus. SOD-2-IR staining was significantly lower 2 daysafter KA administration (P < 0.01 for both the CA1 andCA3 areas). Treatment with ginsenosides significantlyattenuated this decrease in the CA1 and CA3 regions ina dose-dependent manner (Fig. 4).
The results of the Western blot analysis were con-sistent with the immunocytochemical data. High SOD-2protein levels were observed in both saline- and ginse-noside-treated rats, but the levels were significantlylower (P < 0.01) in the hippocampus 2 days after KAadministration. Treatment with ginsenosides significantlyprevented the KA-induced reduction in SOD-2 proteinin a dose-dependent manner (Fig. 4).
Fig. 2. Effect of ginsenosides (Gin) on KA-induced alterations inprotein carbonyls (A,C) and thiobarbituric acid reactive substances(TBARS; B,D) in the mitochondrial fractions (A,C) and tissue ho-mogenates (B,D) from rat hippocampus. Note that protein oxidationand lipid peroxidation were more pronounced in the mitochondrialfraction than in the homogenate. Although ginsenosides significantlyattenuated KA-induced oxidative stresses, ginsenoside-mediated anti-
oxidant actions were more evident in the mitochondria than in thehomogenate. Each value is the mean 6 SEM of 6 animals (tissue ho-mogenates) or 5 experiments (hippocampal mitochondria from 4 ani-mals pooled for each assay). *P < 0.05, **P < 0.01 vs. Sal 1 Sal,#P < 0.05, ##P < 0.01 vs. Sal 1 KA (ANOVA followed by Fisher’sPLSD test).
Ginsenosides, Kainate, and Mitochondria 715
Journal of Neuroscience Research
Ginsenosides Attenuate KA-induced Changes inthe Mitochondrial Membrane Potential andIntramitochondrial Ca21 in the Rat Hippocampus
An important step in mitochondrial dysfunctioninvolves the alteration of the MTP (Susin et al., 1998).
As a result of calcium accumulation, a decline in theMTP alone can induce oxidative stress (Susin et al.,1998), and excitotoxic insult may result in a markedimbalance between the MTP and the intramitochondrialCa21 level.
Fig. 3. Effect of ginsenosides (Gin) on KA-induced changes in thelevels of GSH (A,B) and GSSG (C,D) and the ratio of GSH/GSSG(E,F) in the mitochondrial fractions (A,C,E) and tissue homogenates(B,D,F) from rat hippocampus. KA-induced changes in the levels ofGSH and GSSG and in the ratio of GSH/GSSG were more evidentin the mitochondria compared with the homogenate. Although thesechanges were observed 4 hr after KA treatment, these findings were
more pronounced 2 days later. Ginsenoside-mediated inhibitionagainst KA-induced glutathione oxidation was observed primarily inthe mitochondria. Each value is the mean 6 SEM of 6 experiments(hippocampal mitochondria from 3 animals were pooled for eachassay). *P < 0.05, **P < 0.01 vs. Sal 1 Sal, #P < 0.05, ##P < 0.01vs. Sal 1 KA (ANOVA followed by Fisher’s PLSD test).
716 Shin et al.
Journal of Neuroscience Research
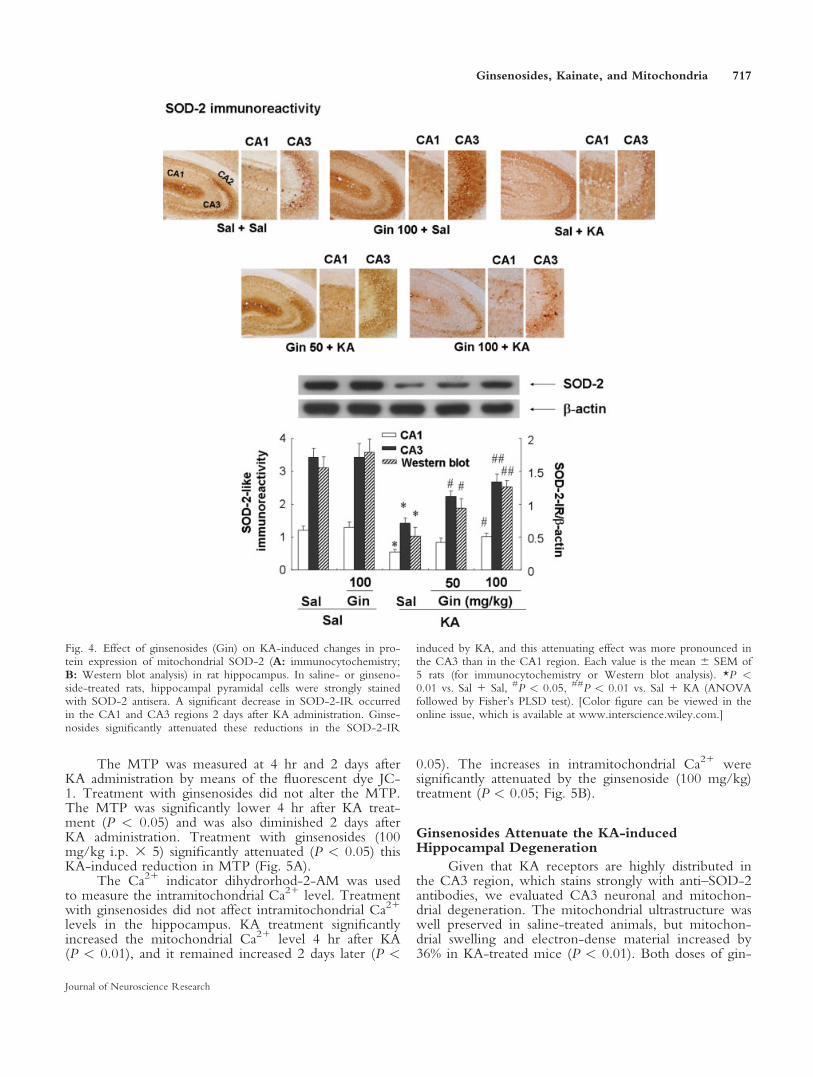
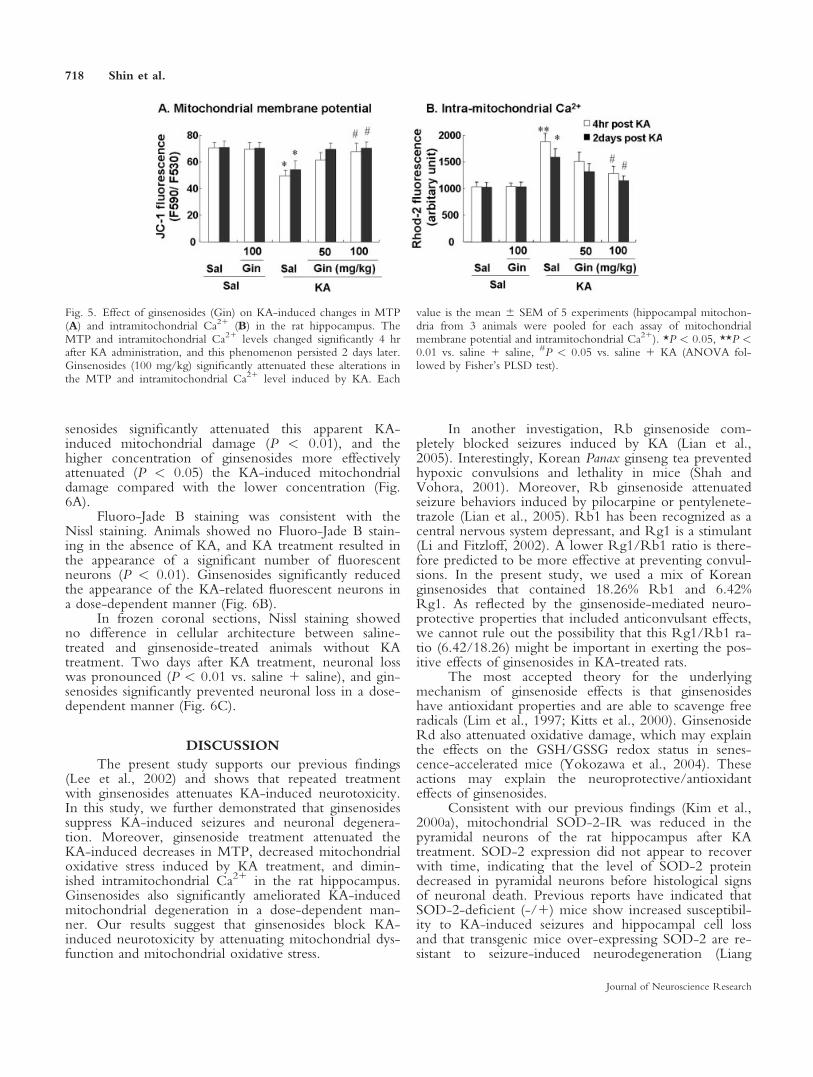
The MTP was measured at 4 hr and 2 days afterKA administration by means of the fluorescent dye JC-1. Treatment with ginsenosides did not alter the MTP.The MTP was significantly lower 4 hr after KA treat-ment (P < 0.05) and was also diminished 2 days afterKA administration. Treatment with ginsenosides (100mg/kg i.p. 3 5) significantly attenuated (P < 0.05) thisKA-induced reduction in MTP (Fig. 5A).
The Ca21 indicator dihydrorhod-2-AM was usedto measure the intramitochondrial Ca21 level. Treatmentwith ginsenosides did not affect intramitochondrial Ca21
levels in the hippocampus. KA treatment significantlyincreased the mitochondrial Ca21 level 4 hr after KA(P < 0.01), and it remained increased 2 days later (P <
0.05). The increases in intramitochondrial Ca21 weresignificantly attenuated by the ginsenoside (100 mg/kg)treatment (P < 0.05; Fig. 5B).
Ginsenosides Attenuate the KA-inducedHippocampal Degeneration
Given that KA receptors are highly distributed inthe CA3 region, which stains strongly with anti–SOD-2antibodies, we evaluated CA3 neuronal and mitochon-drial degeneration. The mitochondrial ultrastructure waswell preserved in saline-treated animals, but mitochon-drial swelling and electron-dense material increased by36% in KA-treated mice (P < 0.01). Both doses of gin-
Fig. 4. Effect of ginsenosides (Gin) on KA-induced changes in pro-tein expression of mitochondrial SOD-2 (A: immunocytochemistry;B: Western blot analysis) in rat hippocampus. In saline- or ginseno-side-treated rats, hippocampal pyramidal cells were strongly stainedwith SOD-2 antisera. A significant decrease in SOD-2-IR occurredin the CA1 and CA3 regions 2 days after KA administration. Ginse-nosides significantly attenuated these reductions in the SOD-2-IR
induced by KA, and this attenuating effect was more pronounced inthe CA3 than in the CA1 region. Each value is the mean 6 SEM of5 rats (for immunocytochemistry or Western blot analysis). *P <0.01 vs. Sal 1 Sal, #P < 0.05, ##P < 0.01 vs. Sal 1 KA (ANOVAfollowed by Fisher’s PLSD test). [Color figure can be viewed in theonline issue, which is available at www.interscience.wiley.com.]
Ginsenosides, Kainate, and Mitochondria 717
Journal of Neuroscience Research
senosides significantly attenuated this apparent KA-induced mitochondrial damage (P < 0.01), and thehigher concentration of ginsenosides more effectivelyattenuated (P < 0.05) the KA-induced mitochondrialdamage compared with the lower concentration (Fig.6A).
Fluoro-Jade B staining was consistent with theNissl staining. Animals showed no Fluoro-Jade B stain-ing in the absence of KA, and KA treatment resulted inthe appearance of a significant number of fluorescentneurons (P < 0.01). Ginsenosides significantly reducedthe appearance of the KA-related fluorescent neurons ina dose-dependent manner (Fig. 6B).
In frozen coronal sections, Nissl staining showedno difference in cellular architecture between saline-treated and ginsenoside-treated animals without KAtreatment. Two days after KA treatment, neuronal losswas pronounced (P < 0.01 vs. saline 1 saline), and gin-senosides significantly prevented neuronal loss in a dose-dependent manner (Fig. 6C).
DISCUSSION
The present study supports our previous findings(Lee et al., 2002) and shows that repeated treatmentwith ginsenosides attenuates KA-induced neurotoxicity.In this study, we further demonstrated that ginsenosidessuppress KA-induced seizures and neuronal degenera-tion. Moreover, ginsenoside treatment attenuated theKA-induced decreases in MTP, decreased mitochondrialoxidative stress induced by KA treatment, and dimin-ished intramitochondrial Ca21 in the rat hippocampus.Ginsenosides also significantly ameliorated KA-inducedmitochondrial degeneration in a dose-dependent man-ner. Our results suggest that ginsenosides block KA-induced neurotoxicity by attenuating mitochondrial dys-function and mitochondrial oxidative stress.
In another investigation, Rb ginsenoside com-pletely blocked seizures induced by KA (Lian et al.,2005). Interestingly, Korean Panax ginseng tea preventedhypoxic convulsions and lethality in mice (Shah andVohora, 2001). Moreover, Rb ginsenoside attenuatedseizure behaviors induced by pilocarpine or pentylenete-trazole (Lian et al., 2005). Rb1 has been recognized as acentral nervous system depressant, and Rg1 is a stimulant(Li and Fitzloff, 2002). A lower Rg1/Rb1 ratio is there-fore predicted to be more effective at preventing convul-sions. In the present study, we used a mix of Koreanginsenosides that contained 18.26% Rb1 and 6.42%Rg1. As reflected by the ginsenoside-mediated neuro-protective properties that included anticonvulsant effects,we cannot rule out the possibility that this Rg1/Rb1 ra-tio (6.42/18.26) might be important in exerting the pos-itive effects of ginsenosides in KA-treated rats.
The most accepted theory for the underlyingmechanism of ginsenoside effects is that ginsenosideshave antioxidant properties and are able to scavenge freeradicals (Lim et al., 1997; Kitts et al., 2000). GinsenosideRd also attenuated oxidative damage, which may explainthe effects on the GSH/GSSG redox status in senes-cence-accelerated mice (Yokozawa et al., 2004). Theseactions may explain the neuroprotective/antioxidanteffects of ginsenosides.
Consistent with our previous findings (Kim et al.,2000a), mitochondrial SOD-2-IR was reduced in thepyramidal neurons of the rat hippocampus after KAtreatment. SOD-2 expression did not appear to recoverwith time, indicating that the level of SOD-2 proteindecreased in pyramidal neurons before histological signsof neuronal death. Previous reports have indicated thatSOD-2-deficient (-/1) mice show increased susceptibil-ity to KA-induced seizures and hippocampal cell lossand that transgenic mice over-expressing SOD-2 are re-sistant to seizure-induced neurodegeneration (Liang
Fig. 5. Effect of ginsenosides (Gin) on KA-induced changes in MTP(A) and intramitochondrial Ca21 (B) in the rat hippocampus. TheMTP and intramitochondrial Ca21 levels changed significantly 4 hrafter KA administration, and this phenomenon persisted 2 days later.Ginsenosides (100 mg/kg) significantly attenuated these alterations inthe MTP and intramitochondrial Ca21 level induced by KA. Each
value is the mean 6 SEM of 5 experiments (hippocampal mitochon-dria from 3 animals were pooled for each assay of mitochondrialmembrane potential and intramitochondrial Ca21). *P < 0.05, **P <0.01 vs. saline 1 saline, #P < 0.05 vs. saline 1 KA (ANOVA fol-lowed by Fisher’s PLSD test).
718 Shin et al.
Journal of Neuroscience Research
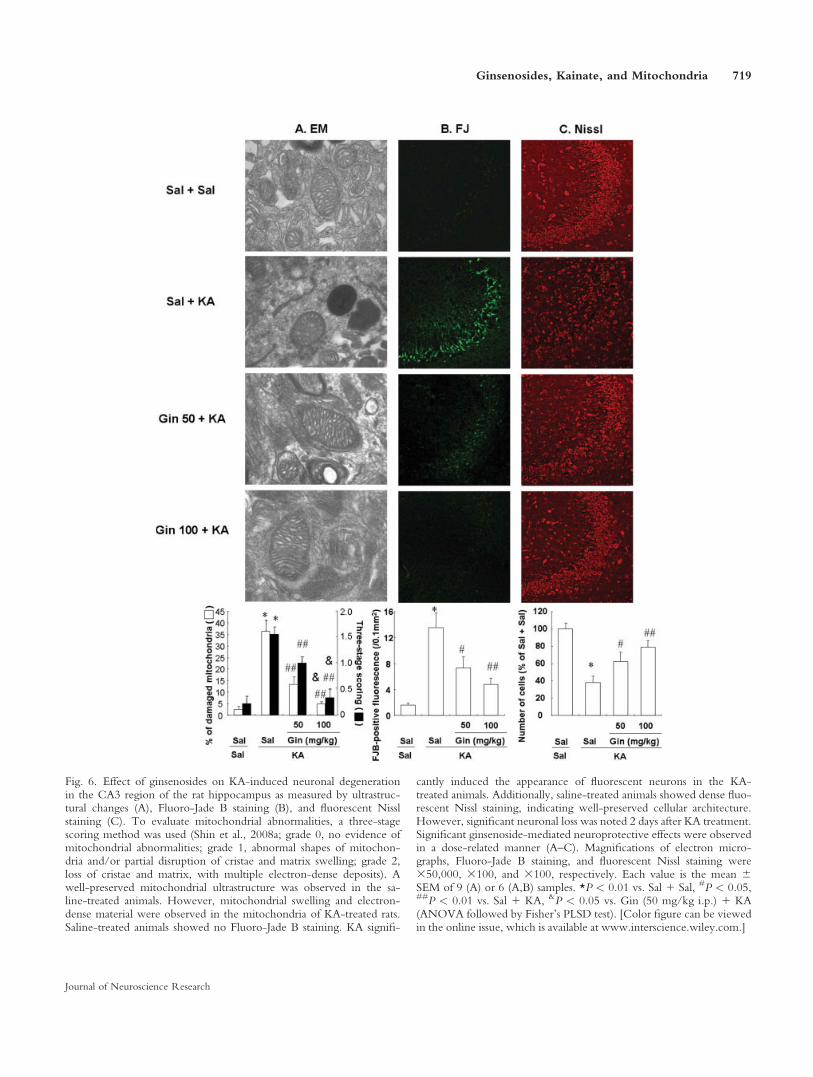
Fig. 6. Effect of ginsenosides on KA-induced neuronal degenerationin the CA3 region of the rat hippocampus as measured by ultrastruc-tural changes (A), Fluoro-Jade B staining (B), and fluorescent Nisslstaining (C). To evaluate mitochondrial abnormalities, a three-stagescoring method was used (Shin et al., 2008a; grade 0, no evidence ofmitochondrial abnormalities; grade 1, abnormal shapes of mitochon-dria and/or partial disruption of cristae and matrix swelling; grade 2,loss of cristae and matrix, with multiple electron-dense deposits). Awell-preserved mitochondrial ultrastructure was observed in the sa-line-treated animals. However, mitochondrial swelling and electron-dense material were observed in the mitochondria of KA-treated rats.Saline-treated animals showed no Fluoro-Jade B staining. KA signifi-
cantly induced the appearance of fluorescent neurons in the KA-treated animals. Additionally, saline-treated animals showed dense fluo-rescent Nissl staining, indicating well-preserved cellular architecture.However, significant neuronal loss was noted 2 days after KA treatment.Significant ginsenoside-mediated neuroprotective effects were observedin a dose-related manner (A–C). Magnifications of electron micro-graphs, Fluoro-Jade B staining, and fluorescent Nissl staining were350,000, 3100, and 3100, respectively. Each value is the mean 6SEM of 9 (A) or 6 (A,B) samples. *P < 0.01 vs. Sal 1 Sal, #P < 0.05,##P < 0.01 vs. Sal 1 KA, &P < 0.05 vs. Gin (50 mg/kg i.p.) 1 KA(ANOVA followed by Fisher’s PLSD test). [Color figure can be viewedin the online issue, which is available at www.interscience.wiley.com.]
Ginsenosides, Kainate, and Mitochondria 719
Journal of Neuroscience Research

et al., 2000). Moreover, we (Shin et al., 2008a) andothers (Liang and Patel, 2006) have demonstrated thatKA-induced oxidative stress was more evident in themitochondrial fraction than in the hippocampal homoge-nate. Thus, mitochondrial oxidative stress is thought tobe an important factor in epileptic brain damage.
In terms of histological evaluation, conventionalstaining techniques, such as hematoxylin and eosin andNissl staining, are technically simple and can be used toinfer degeneration on the basis of changes such as neuro-nal shrinkage, vacuolation, and hypochromatism. How-ever, these changes are not necessarily indicative of neu-ronal degeneration, but may instead be attributable toprocessing artifacts or nonlethal alterations in cellularmorphology. Fluoro-Jade is recognized as a more sensi-tive and definitive marker of neuronal degeneration thanconventional stains (Schmued et al., 1997).
Significant loss of hippocampal pyramidal cells wasobserved in the CA3 sector in this study. In the hippo-campus, the highest density of KA receptors is in theCA3 region, the area most severely damaged after KAadministration. Electron microscopic analysis showed sig-nificant mitochondrial deformity, with electron-densematerial in many fibers in the CA3 region. This materialwas probably mitochondrial degenerative debris. Thus,the possibility exists that ginsenosides could block activa-tion of the KA receptor, and then could contribute, atleast in part, to preventing mitochondrial dysfunction/degeneration activated by the KA receptor.
Accumulating evidence suggests that treatmentwith ginsenosides attenuates intracellular increases in
Ca21 by blocking various types of Ca21 channels suchas the L, N, and P/Q channels and by preventing depo-larization-induced Ca21 influx (Nah et al., 1995; Kimet al., 1998a; Rhim et al., 2002). Furthermore, ginseno-sides have been shown to inhibit receptor agonist-induced intracellular Ca21 mobilization (Jeong et al.,2004) and reduce glutamate/NMDA-mediated Ca21
influx in neurons (Kim et al., 1998b, 2002). Previousreports have suggested that the ginsenoside-mediated in-hibition of intracellular Ca21 influx could be the basis oftheir in vitro or in vivo protection against excitatoryamino acid- or neurotoxin-caused neuronal cell changes(Kim et al., 1998b; Lee et al., 2002; Liao et al., 2002).
Neurotoxins collapse the MTP, and lead toincreased oxidative stress due to calcium accumulation(Nicholls et al., 2003). Increased intracellular Ca21 pro-motes the accumulation of Ca21 within the mitochon-drial matrix when the total Ca21 uptake exceeds the totalCa21 efflux in the mitochondria (Nicholls and Aker-mann, 1982). Mitochondrial Ca21 overload may also leadto the uncoupling of mitochondrial electron transport andmay potentiate oxidative stress (Dykens, 1994). Decreasesin the mitochondrial membrane potential and increases inreactive oxygen species after KA treatment could bemediated by Ca21 entry through the kainate receptor.We speculate that ginsenosides might block Ca21 entry,at least in part, through the kainate receptor, given thatginsenosides primarily attenuated neuronal loss and mito-chondrial SOD-2 expression in the CA3 area.
In summary, the results of this study demonstratethat KA decreases the GSH/GSSG ratio, increases lipidperoxidation products and protein carbonyls, anddecreases SOD-2 expression in the rat hippocampus.Ginsenosides significantly prevented these KA-inducedchanges in oxidative stress markers. Specifically, augmen-tation of SOD-2 expression in ginsenoside-treatedanimals might contribute to the attenuation of the mito-chondrial structural damage and mitochondrial dys-function (i.e., altered membrane potential and intramito-chondrial Ca21) induced by KA (Fig. 7). To our knowl-edge, this is the first report showing that ginsenosidesprevent mitochondrial dysfunction, mitochondrialdegeneration, and oxidative stress induced by excitotoxicinsults in the rat brain. Further studies are required todefine the precise mechanism or mechanisms that con-vey ginsenoside neuronal protection.
ACKNOWLEDGMENTS
Equipment at the Institute of Pharmaceutical Sci-ence (Kangwon National University) was used for thisstudy. H.J.K. was supported by the BK 21 program.
REFERENCES
Attele AS, Wu JA, Yuan CS. 1999. Ginseng pharmacology: multiple
constituents and multiple actions. Biochem Pharmacol 58:1685–1693.
Bruce-Keller AJ, Geddes JW, Knapp PE, McFall RW, Keller JN, Holts-
berg FW, Parthasarathy S, Steiner SM, Mattson MP. 1999. Anti-death
properties of TNF against metabolic poisoning: mitochondrial stabiliza-
tion by MnSOD. J Neuroimmunol 93:53–71.
Fig. 7. Flow chart depicting our current working hypothesis on thepharmacological action of ginsenosides in response to KA insult inthe rat hippocampus.
720 Shin et al.
Journal of Neuroscience Research

Dragunow M, Robertson HA. 1987. Kindling stimulation induces c-fos
protein(s) in granule cells of the rat dentate gyrus. Nature 329:441–442.
Dykens JA. 1994. Isolated cerebral and cerebellar mitochondria produce
free radicals when exposed to elevated Ca21 and Na1: implications for
neurodegeneration. J. Neurodegeneration. J Neurochem 63:584–591.
Floreani M, Skaper SD, Facci L, Lipartiti M, Giusti P. 1997. Melatonin
maintains glutathione homeostasis in kainic acid–exposed rat brain tis-
sues. FASEB J 11:1309–1315.
Floyd RA, Carney JM. 1992. Free radical damage to protein and DNA:
mechanisms involved and relevant observations on brain undergoing
oxidative stress. Ann Neurol 32(Suppl):S22–S27.
Fraga CG, Leibovitz BE, Tappel AL. 1988. Lipid peroxidation measured
as thiobarbituric acid–reactive substances in tissue slices: characterization
and comparison with homogenates and microsomes. Free Radic Biol
Med 4:155–161.
Jeong SM, Lee JH, Kim S, Rhin H, Lee BH, Kim JH, Oh JH, Lee SM,
Nah SY. 2004. Ginseng saponins induce store-operated entry in Xeno-
pus oocytes. Br J Pharmacol 142:585–593.
Kim HS, Lee JH, Koo YS, Nah SY. 1998a. Effects of ginsenosides on
Ca21 channels and membrane capacitance in rat adrenal chromaffin
cells. Brain Res Bull 46:245–251.
Kim YC, Kim SR, Markelonis GJ, Oh TH. 1998b. Ginsenosides Rb1
and Rg3 protect cultured rat cortical cells from glutamate-induced neu-
rodegenertaion. J Neusosci Res 53:426–432.
Kim HC, Jhoo WK, Kim WK, Suh JH, Shin EJ, Kato K, Ko KH.
2000a. An immunocytochemical study of mitochondrial manganese-
superoxide dismutase in the rat hippocampus after kainate treatment.
Neurosci Lett 281:65–68.
Kim HC, Jhoo WK, Bing G, Shin EJ, Wie MB, Kim WK, Ko KH.
2000b. Phenidone prevents kainate-induced neurotoxicity via antioxi-
dant mechanisms. Brain Res 874:15–23.
Kim S, Ahn K, Oh TH, Nah SY, Rhim H. 2002. Inhibitory effect of
ginsenosides on NMDA receptor-mediated signals in rat hippocampal
neurons. Biochem Biophys Res Commun 296:247–254.
Kim HC, Yamada K, Olariu A, Tran MH, Mizuno M, Nakajima A,
Nagai T, Jhoo WK, Im DH, Shin EJ, Park SC, Kato K, Hjelle OP,
Ottersen OP, Mirault ME, Nabeshima T. 2003. Immunocytochemical
evidence that b-amyloid (1–42) impairs endogenous antioxidant systems
in vivo. Neuroscience 119:399–419.
Kim JH, Kim S, Yoon IS, Lee JH, Jang BJ, Jeong SM, Lee JH, Lee BH,
Han JS, Oh S, Kim HC, Park TK, Rhim H, Nah SY. 2005. Protective
effects of ginseng saponin on 3-nitropropionic acid–induced striatal
degeneration in rats. Neuropharmacology 48:743–756.
Kim EJ, Won R, Sohn JH, Chung MA, Nam TS, Lee HJ. 2008. Anti-
oxidant effect of ascorbic and dehydroascorbic acids in hippocampal
slice culture. Biochem Biophys Res Commun 366:8–14.
Kitts DD, Wijewickreme AN, Hu C. 2000. Antioxidant properties of a
North American ginseng extract. Mol Cell Biochem 203:1–10.
Kurobe N, Suzuki F, Kato K, Sato T. 1990. Sensitive immunoassay of
rat superoxide disnutase: concentration in the brain, liver, and kidney
are not affected by aging. Biomed Res 757:260–267.
Kwon YS, Ann HS, Nabeshima T, Shin EJ, Kim WK, Jhoo JH, Jhoo
WK, Wie MB, Kim YS, Jang KJ, Kim HC. 2004. Selegiline potentiates
the effects of EGb 761 in response to ischemic brain injury. Neuro-
chem Int 45:157–170.
Lee JH, Kim SR, Bae CS, Kim D, Hong H, Nah S. 2002. Protective
effect of ginsenosides, active ingredients of Panax ginseng, on kainic
acid–induced neurotoxicity in rat hippocampus. Neurosci Lett 325:
129–133.
Li W, Fitzloff JF. 2002. HPLC determination of ginsenosides content in
ginseng dietary supplements using ultraviolet detection. J Liquid Chro-
matogr Rel Technol 25:2485–2500.
Lian XY, Zhang ZZ, Stringer JL. 2005. Anticonvulsant activity of gin-
seng on seizures induced by chemical convulsants. Epilepsia 46:15–22.
Liang LP, Patel M. 2006. Seizure-induced changes in mitochondrial re-
dox status. Free Radic Biol Med 40:316–322.
Liang LP, Ho YS, Patel M. 2000. Mitochondrial superoxide production
in kainate-induced hippocampal damage. Neuroscience 101:563–570.
Liao B, Newmark H, Zhou R. 2002. Neuroprotective effects of ginseng
total saponin and ginsenosides Rb1 and Rg1 on spinal cord neurons in
vitro. Exp Neurol 173:224–234.
Lim JH, Wen TC, Matsuda S, Tanaka J, Maeda N, Peng H, Aburaya J,
Ishihara K, Sakanaka M. 1997. Protection of ischemic hippocampal
neurons by ginsenoside Rb1, a main ingredient of ginseng root. Neuro-
sci Res 28:191–200.
Matton MP, Keller JN, Begley JC. 1998. Evidence for synaptic apoptosis.
Exp Neurol 153:35–48.
Nah SY, Park HJ, McCleskey EW. 1995. A trace component of ginseng
that inhibits Ca21 channels through a pertussis toxin sensitive G pro-
tein. Proc Natl Acad Sci U S A 92:8739–8743.
Namba T, Yoshizaki T, Tominori K, Kobayashi K, Mitsui K, Hasse J.
1974. Fundamental studies on the evaluation of the crude drugs (I).
Planta Med 32:588–594.
Nicholls D, Akermann K. 1982. Mitochondrial calcium transport. Bio-
chim Biophys Acta 683:57–88.
Nicholls D, Vesce S, Kirk L, Chalmers S. 2003. Interactions between mi-
tochondrial bioenergetics and cytoplasmic calcium in cultured cerebellar
granule cells. Cell Calcium 34:407–424.
Oliver CN, Ahn BW, Moerman EJ, Goldstein S, Stadtman ER. 1987.
Age-related changes in oxidized proteins. J Biol Chem 262:5488–5491.
Racine RJ. 1972. Modification of seizure activity by electric stimulation:
II. Motor seizure. EEG Clin Neurophysiol 32:281–294.
Reed DJ, Babson JR, Beatty PW, Brodie AE, Ellis WW, Potter DW.
1980. High-performance liquid chromatography analysis of nanomole
levels of glutathione, glutathione disulfide, and related thiols and disul-
fides. Anal Biochem 106:55–62.
Rhim H, Kim H, Lee DY, Oh TH, Nah SY. 2002. Ginseng and ginse-
noside Rg3, a newly identified active ingredient of ginseng, modulate
Ca21 channel currents in rat sensory neurons. Eur J Pharmacol
436:151–158.
Schafer FQ, Buettner GR. 2001. Redox environment of the cell as
viewed through the redox state of the glutathione disulfide/glutathione
couple. Free Radic Biol Med 30:1191–1212.
Schmued LC, Albertson C, Slikker W Jr. 1997. Fluoro-Jade: a novel flu-
orochrome for the sensitive and reliable histochemical localization of
neuronal degeneration. Brain Res 751:37–46.
Shah ZA, Vohora SB. 2001. Protective effect of Korean ginseng tea on
hypoxic-convulsions and lethality in mice. Indian Drugs 38:181–182.
Shih YH, Chein YC, Wang JY, Fu YS. 2004. Ursolic acid protects hip-
pocampal neurons against kainate-induced excititoxity in rats. Neurosci
Lett 362:136–140.
Shin EJ, Nabeshima T, Suh HW, Jhoo WK, Oh KW, Lim YK, Kim
DS, Choi KH, Kim HC. 2005a. Ginsenosides attenuate methamphet-
amine-induced behavioral side effects in mice via activation of adeno-
sine A2A receptors: possible involvements of the striatal reduction in
AP-1 DNA binding activity and proenkephalin gene expression. Behav
Brain Res 158:143–157.
Shin EJ, Suh SK, Lim YK, Jhoo WK, Hjelle OP, Ottersen OP, Shin
CY, Ko KH, Kim WK, Kim DS, Chun W, Ali S, Kim HC. 2005b.
Ascorbate attenuates trimethyltin-induced oxidative burden and neuro-
nal degeneration in the rat hippocampus by maintaining glutathione ho-
meostasis. Neuroscience 133:715–727.
Shin EJ, Nah SY, Kim WK, Ko KH, Jhoo WK, Lim YK, Cha JY, Chen
CF, Kim HC. 2005c. The dextromethorphan analog dimemorfan
attenuates kainate-induced seizures via r1 receptor activation: compari-
son with the effects of dextromethorphan. Br J Pharmacol 144:908–
918.
Ginsenosides, Kainate, and Mitochondria 721
Journal of Neuroscience Research

Shin EJ, Chae JS, Jung ME, Bing G, Ko KH, Kim WK, Wie MB,
Cheon MA, Nah SY, Kim HC. 2007. Repeated intracerebroventricular
infusion of nicotine prevents kainate-induced neurotoxicity by activat-
ing the a7 nicotinic acetylcholine receptor. Epilepsy Res 73:292–298.
Shin EJ, Jeong JH, Bing G, Park ES, Chae JS, Yen TPH, Kim WK, Wie
MB, Jung BD, Kim HJ, Lee SY, Kim HC. 2008a. Kainate-induced mi-
tochondrial oxidative stress contributes to hippocampal degeneration in
senescence-accelerated mice. Cell Signal 20:645–658.
Shin EJ, Ko KH, Kim WK, Chae JS, Yen TP, Kim HJ, Wie MB, Kim
HC. 2008b. Role of glutathione peroxidase in the ontogeny of hippo-
campal oxidative stress and kainate seizure sensitivity in the genetically
epilepsy-prone rats. Neurochem Int 52:1134–1147.
Sperk G. 1994. Kainic acid seizures in the rat. Prog Neurobiol 42:1–32.
Susin SA, Zamzami N, Kroemer G. 1998. Mitochondria as regulators of
apoptosis: doubt no more. Biochim Biophys Acta 1366:151–165.
Xiong Y, Gu Q, Peterson PL, Muizelaar JP, Lee CP. 1997. Mitochon-
drial dysfunction and calcium perturbation induced by traumatic brain
injury. J Neurotrauma 14:23–34.
Yokozawa T, Satoh A, Cho EJ. 2004. Ginsenoside-Rd attenuates oxida-
tive damage related to aging in senescence-accelerated mice. J Pharm
Pharmacol 56:107–103.
Yuan CS, Wu JA, Lowell T, Gu M. 1998. Gut and brain effects of
American ginseng root on brainstem neuronal activities in rats. Am J
Chin Med 26:47–55.
722 Shin et al.
Journal of Neuroscience Research
Related Documents











