Proteasome inhibition induces stress kinase dependent transport deficits — Implications for Alzheimer's disease Lotta Agholme a,1 , Sangeeta Nath a , Jakob Domert a , Jan Marcusson b , Katarina Kågedal a , Martin Hallbeck a,c, ⁎ a Experimental Pathology, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, Linköping, Sweden b Geriatric, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, Linköping, Sweden c Department of Clinical Pathology, County Council of Östergötland, Linköping, Sweden abstract article info Article history: Received 20 August 2013 Revised 10 October 2013 Accepted 14 November 2013 Available online 21 November 2013 Keywords: Alzheimer's disease Proteasome dysfunction Tau phosphorylation Axonal transport ERK 1/2 c-Jun Alzheimer's disease (AD) is characterized by accumulation of two misfolded and aggregated proteins, β-amyloid and hyperphosphorylated tau. Both cellular systems responsible for clearance of misfolded and aggregated proteins, the lysosomal and the proteasomal, have been shown to be malfunctioning in the aged brain and more so in patients with neurodegenerative diseases, including AD. This malfunction could be contributing to β-amyloid and tau accumulation, eventually aggregating in plaques and tangles. We have investigated the impact of decreased proteasome activity on tau phosphorylation as well as on microtubule stability and transport. To do this, we used our recently developed neuronal model where human SH-SY5Y cells obtain neuronal morphology and function through differentiation. We found that exposure to low doses of the proteasome inhibitor MG-115 caused tau phosphorylation, microtubule destabilization and disturbed neuritic transport. Furthermore, reduced proteasome activity activated several proteins implicated in tau phosphorylation and AD pathology, including c-Jun N-terminal kinase, c-Jun and extracellular signal-regulated protein kinase (ERK) 1/2. Restoration of the mi- crotubule transport was achieved by inhibiting ERK 1/2 activation, and simultaneous inhibition of both ERK 1/2 and c-Jun reversed the proteasome inhibition-induced tau phosphorylation. Taken together, this study suggests that a decrease in proteasome activity can, through activation of c-Jun and ERK 1/2, result in several events relat- ed to neurodegenerative diseases. Restoration of proteasome activity or modulation of ERK 1/2 and c-Jun function can open new treatment possibilities against neurodegenerative diseases such as AD. © 2013 Elsevier Inc. All rights reserved. Introduction Alzheimer's disease (AD) is pathologically characterized by accumu- lation of amyloid-β (Aβ) in extracellular plaques, and tau in intracellular neurofibrillary tangles (Blennow et al., 2006). The familial forms of AD, representing about 1% of all cases, are caused by a number of mutations resulting in increased Aβ production. This in turn is thought to alter tau into a hyperphosphorylated form, more prone to aggregate into neuro- fibrillary tangles (Götz et al., 2011). The sporadic form is similarly characterized by Aβ and tau aggregates, but the cause of this has so far not been found. However, the progressive nature of AD has been explained by a possible transmission of Aβ oligomers (Hallbeck et al., 2013; Nath et al., 2012) or tau (Clavaguera et al., 2009). Plaques and tangles were for many years the focus of AD research, but are now thought to appear late in the disease process. Instead, mi- crotubule de-stabilization, obstruction of axonal transport and defects in synaptic function are believed to be early pathological events in AD (Stokin et al., 2005; Takahashi et al., 2002; Terry et al., 1991), and these events have all been shown to cause neuronal death (Perlson et al., 2010). The microtubule stability and subsequent transport along neurites are regulated by binding of tau to microtubules (Drewes et al., 1998). Tau regulates its binding to microtubules by phosphoryla- tion and de-phosphorylation, where hyperphosphorylation of tau causes it to completely detach from microtubules, resulting in its destabilization (Biernat and Mandelkow, 1999; Lindwall and Cole, 1984). More recently, it was also discovered that hyperphosphorylated tau interferes with proteins connecting cargo vesicles to microtubules, thus resulting in disturbance of axonal transport (Ittner et al., 2009). Glyco- gen synthase kinase (GSK) 3β and cyclin dependent kinase (CDK) 5 are implicated to be important for phosphorylation of tau in AD. In ad- dition, several other kinases, such as those belonging to the MAP kinase pathway have also been implicated in AD pathology (Churcher, 2006). All cells, including neurons, have two major systems for clearance of worn out, misfolded and aggregated proteins; the autophagy/lysosomal Molecular and Cellular Neuroscience 58 (2014) 29–39 Abbreviations: Aβ, β-amyloid; AD, Alzheimer's disease; APP, amyloid precursor protein; CDK, cyclin dependent kinase; ECM, extracellular matrix; ERK, extracellular signal-regulated protein kinase; GSK, glycogen synthase kinase; MSD, Meso Scale Discovery. ⁎ Corresponding author at: Pathology Building, Level 07, Linköping University Hospital, SE-58185 Linköping, Sweden. E-mail address: [email protected] (M. Hallbeck). 1 Present address: Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, The Sahlgrenska Academy at the University of Gothenburg, Gothenburg, Sweden. 1044-7431/$ – see front matter © 2013 Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.mcn.2013.11.001 Contents lists available at ScienceDirect Molecular and Cellular Neuroscience journal homepage: www.elsevier.com/locate/ymcne

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Molecular and Cellular Neuroscience 58 (2014) 29–39
Contents lists available at ScienceDirect
Molecular and Cellular Neuroscience
j ourna l homepage: www.e lsev ie r .com/ locate /ymcne
Proteasome inhibition induces stress kinase dependent transportdeficits — Implications for Alzheimer's disease
Lotta Agholme a,1, Sangeeta Nath a, Jakob Domert a, Jan Marcusson b, Katarina Kågedal a, Martin Hallbeck a,c,⁎a Experimental Pathology, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, Linköping, Swedenb Geriatric, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, Linköping, Swedenc Department of Clinical Pathology, County Council of Östergötland, Linköping, Sweden
Abbreviations: Aβ, β-amyloid; AD, Alzheimer's disprotein; CDK, cyclin dependent kinase; ECM, extracellsignal-regulatedprotein kinase; GSK, glycogen synthase kin⁎ Corresponding author at: Pathology Building, Level 07
SE-58185 Linköping, Sweden.E-mail address: [email protected] (M. Hallbeck).
1 Present address: Department of Psychiatry andNeuroscience and Physiology, The Sahlgrenska Academy aGothenburg, Sweden.
1044-7431/$ – see front matter © 2013 Elsevier Inc. All rihttp://dx.doi.org/10.1016/j.mcn.2013.11.001
a b s t r a c t
a r t i c l e i n f oArticle history:Received 20 August 2013Revised 10 October 2013Accepted 14 November 2013Available online 21 November 2013
Keywords:Alzheimer's diseaseProteasome dysfunctionTau phosphorylationAxonal transportERK 1/2c-Jun
Alzheimer's disease (AD) is characterized by accumulation of twomisfolded and aggregated proteins, β-amyloidand hyperphosphorylated tau. Both cellular systems responsible for clearance of misfolded and aggregatedproteins, the lysosomal and the proteasomal, have been shown to be malfunctioning in the aged brain andmore so in patients with neurodegenerative diseases, including AD. This malfunction could be contributing toβ-amyloid and tau accumulation, eventually aggregating inplaques and tangles.Wehave investigated the impactof decreased proteasome activity on tau phosphorylation aswell as onmicrotubule stability and transport. To dothis, we used our recently developed neuronal model where human SH-SY5Y cells obtain neuronal morphologyand function through differentiation. We found that exposure to low doses of the proteasome inhibitor MG-115caused tau phosphorylation, microtubule destabilization and disturbed neuritic transport. Furthermore, reducedproteasome activity activated several proteins implicated in tau phosphorylation and AD pathology, includingc-Jun N-terminal kinase, c-Jun and extracellular signal-regulated protein kinase (ERK) 1/2. Restoration of themi-crotubule transport was achieved by inhibiting ERK 1/2 activation, and simultaneous inhibition of both ERK 1/2and c-Jun reversed the proteasome inhibition-induced tau phosphorylation. Taken together, this study suggeststhat a decrease in proteasome activity can, through activation of c-Jun and ERK 1/2, result in several events relat-ed to neurodegenerative diseases. Restoration of proteasome activity ormodulation of ERK 1/2 and c-Jun functioncan open new treatment possibilities against neurodegenerative diseases such as AD.
© 2013 Elsevier Inc. All rights reserved.
Introduction
Alzheimer's disease (AD) is pathologically characterized by accumu-lation of amyloid-β (Aβ) in extracellular plaques, and tau in intracellularneurofibrillary tangles (Blennow et al., 2006). The familial forms of AD,representing about 1% of all cases, are caused by a number of mutationsresulting in increased Aβ production. This in turn is thought to alter tauinto a hyperphosphorylated form, more prone to aggregate into neuro-fibrillary tangles (Götz et al., 2011). The sporadic form is similarlycharacterized by Aβ and tau aggregates, but the cause of this has so farnot been found. However, the progressive nature of AD has beenexplained by a possible transmission of Aβ oligomers (Hallbeck et al.,2013; Nath et al., 2012) or tau (Clavaguera et al., 2009).
ease; APP, amyloid precursorular matrix; ERK, extracellularase;MSD,Meso ScaleDiscovery., Linköping University Hospital,
Neurochemistry, Institute oft the University of Gothenburg,
ghts reserved.
Plaques and tangles were for many years the focus of AD research,but are now thought to appear late in the disease process. Instead, mi-crotubule de-stabilization, obstruction of axonal transport and defectsin synaptic function are believed to be early pathological events in AD(Stokin et al., 2005; Takahashi et al., 2002; Terry et al., 1991), andthese events have all been shown to cause neuronal death (Perlsonet al., 2010). The microtubule stability and subsequent transport alongneurites are regulated by binding of tau to microtubules (Dreweset al., 1998). Tau regulates its binding to microtubules by phosphoryla-tion and de-phosphorylation, where hyperphosphorylation of taucauses it to completely detach from microtubules, resulting in itsdestabilization (Biernat and Mandelkow, 1999; Lindwall and Cole,1984).
More recently, it was also discovered that hyperphosphorylated tauinterferes with proteins connecting cargo vesicles to microtubules, thusresulting in disturbance of axonal transport (Ittner et al., 2009). Glyco-gen synthase kinase (GSK) 3β and cyclin dependent kinase (CDK) 5are implicated to be important for phosphorylation of tau in AD. In ad-dition, several other kinases, such as those belonging to theMAP kinasepathway have also been implicated in AD pathology (Churcher, 2006).
All cells, including neurons, have twomajor systems for clearance ofworn out, misfolded and aggregated proteins; the autophagy/lysosomal

30 L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
and the proteasomal systems. The lysosome, an acidic, protease-containing organelle, is generally responsible for degradation of long-lived proteins and organelles, such as worn out mitochondria. Theproteasome is a cytosolic, barrel shaped, protein complex. It is a key play-er in rapid degradation of partially folded or misfolded proteins, beingtagged for degradation through ubiquitination (Knecht et al., 2009). AsAD is pathologically characterized by deposits ofmisfolded and aggregat-ed proteins, disturbances of lysosomal and/or proteasomal function havebeen suggested to be involved in the pathogenesis. Indeed, several stud-ies suggest that disruption of both lysosomal and proteasomal functionsis present in AD brain (Nixon and Cataldo, 2006; Oddo, 2008), andthere is also evidence of defective ubiquitination, as well as accumulationof ubiquitinated proteins (López Salon et al., 2000; Perry et al., 1987).Degradation of tau has been shown to take place both in lysosomes andproteasomes (Grune et al., 2010; Wang et al., 2009), and aggregated tauand Aβ have been shown to inhibit proteasome function (Keck et al.,2003; Tseng et al., 2008). A decreased proteasome activity has alsobeen shown to cause increased Aβ generation (Agholme et al., 2012;Marambaud et al., 1997), possibly due to degradation of the C-terminalfragment of amyloid precursor protein (APP) by the proteasome(Nunan et al., 2001) or by increased levels of presenilin-1 upon protea-some inhibition (Marambaud et al., 1998).
Not only AD, but also other neurodegenerative diseases such asFrontotemporal Lobar Degeneration (FTLD) display tau pathology(Seltman and Matthews, 2012). Proteasome dysfunction has also beenrelated to other neurodegenerative diseases than AD, such as Parkinson'sdisease and Huntington's disease (Ross and Pickart, 2004; Valera et al.,2007). Taken together, there is much evidence pointing at the involve-ment of proteasome disturbance in AD and other neurodegenerative dis-eases. However, whether it contributes directly to disease progression,and if so how, is not known. Earlier studies investigating the effects ofproteasome inhibition on tau levels and phosphorylation have resultedin conflicting results (Brown et al., 2005; Delobel et al., 2005; Liu et al.,2009). In addition, it is not known if decreased proteasome functioncan affect microtubule stability and axonal transport.
In this study, the effects on tau phosphorylation,microtubule stabilityand axonal transport upon proteasome inhibition were investigated.Weused our recently developed neuronal model, enabling studies of tauphosphorylation and microtubule function in human cells (Agholmeet al., 2010). We found that proteasome inhibition caused an increasein tau phosphorylation and disruption of neuritic transport. This was,at least partly, due to activation of two kinases earlier implicated in AD;c-Jun and extracellular signal-regulated protein kinase (ERK) 1/2.
Results
Proteasome inhibition causes cell death
As reduced proteasome function has been implicated in AD (Kelleret al., 2000), proteasome inhibition was induced using low concentra-tions of MG-115, a reversible inhibitor of 20 and 26S proteasome sub-units. Exposure of ECM gel differentiated SH-SY5Y neuroblastoma cells(Agholme et al., 2010) to 0.1, 0.25 and 0.5 μM MG-115 for 24 and 48 hresulted in a dose- and time dependent reduction in proteasomeactivity (Fig. 1A). Exposure to 0.1 μM MG-115 resulted in a small, butsignificant decrease in cell viability after 24 h (Fig. 1B–C). Exposure to0.25 and 0.5 μM MG-115 resulted in a dose- and time dependent de-crease in cell viability (Fig. 1B–C). However, as the 24 hour incubationtime did not result in toxicity to the same extent as 48 hour incubation,the shorter time point was chosen for further experiments.
Proteasome inhibition disturbs vesicle transport along neurites
Disturbed axonal transport is believed to be a key event in early ADpathogenesis (Morfini et al., 2009). Therefore, vesicle transport inneurites, upon proteasome inhibition, was investigated using time-
lapse imaging. In this setting all vesicles, regardless of their origin,were included in the analysis. Exposure to 0.1 μMMG-115 significantlydecreased vesicle velocity compared to control, and the transport wasfurther decreased using 0.5 μMMG-115 (Fig. 2A–B). Vesicles in controlcells moved rapidly within neurites (Movie 1) whereas exposure to0.5 μM MG-115 resulted in almost complete halt of vesicles (Movie 2).Decrease in vesicle velocity was not due to increase in vesicle size, asthe changes in vesicle size did not correlate to the pattern of impairedtransport (Fig. 2C). Another sign of disturbed axonal transport is the re-traction of mitochondria from neurites (Ebneth et al., 1998). Localizationof mitochondria was investigated using fluorescent probe MitoTracker®for mitochondrial staining. MG-115 exposure decreased the number ofmitochondria in neurite dose dependently, being almost abolished uponexposure to 0.25 or 0.5 μMMG-115 (Fig. 2D, arrowheads). The decreasein mitochondria was not due to loss of MitoTracker® staining, as thestaining intensity in the cell soma remained unchanged, although thehigh density of mitochondria there made quantification impossible.When quantifying the number of mitochondria in neurites, a significantdecreasewas seenupon exposure to 0.1 or 0.25 μMMG-115, andwas fur-ther decreased using 0.5 μMMG-115 (Fig. 2E). These results indicate thata small reduction in proteasome activity is enough to disturb neuritictransport.
Proteasome inhibition affects microtubule stability
The above detected disturbance in neuritic transport could be a resultof destabilizedmicrotubules. Therefore, wewent further to investigate ifproteasome inhibition also caused a destabilization ofmicrotubule in oursystem. The appearance of “beaded” tubulin is one sign of microtubuledestabilization (Stokin et al., 2005). By immunochemistry we foundthat control cells displayed healthy, elongated tubulin-positive neurites(Fig. 3A, most left panel), and the majority of neurites in cells exposedto 0.1 μM MG-115 appeared healthy, with occasional beaded tubulin(Fig. 3A, second left panel; arrowheads). Higher concentrations ofMG-115 causeddisruption and beading of tubulin, aswell as loss of tubu-lin positive neurites, indicating that microtubules were destabilizedupon proteasome inhibition (Fig. 3A, right panels; arrowheads). Destabi-lization of microtubules is accompanied by two post-translational mod-ifications, de-acetylation and tyrosination. A decrease in acetylatedtubulin indicates de-stabilization of microtubules, as does increase intubulin tyrosination (Hempen and Brion, 1996; Wehland and Weber,1987). Western blot using acetylated- and tyrosinated tubulin-specificantibodies showed no change in the ratio of acetylated to totalα-tubulin, irrespective of MG-115 concentration (Fig. 3B–C). Surprising-ly, we detected a dose-dependent decrease in tubulin tyrosination uponMG-115 exposure, reaching significance using 0.25 and 0.5 μM (Fig. 3B–D). Thus, proteasome inhibition seems to induce microtubule destabili-zation without classical post-translational changes of tubulin.
Proteasome inhibition increases tau phosphorylation at severaldisease-relevant epitopes
Hyperphosphorylation of tau, causing detachment frommicrotubuleshas, for a long time, been suggested to cause microtubule destabilizationin AD and other neurodegenerative diseases (Li et al., 2007; Lindwalland Cole, 1984). Furthermore, hyperphosphorylation of tau is thoughtto impede axonal transport (Mudher et al., 2004). Phosphorylation atthe PHF1, AT8, and S422 epitope was investigated using western blot(Fig. 4A). Quantification using densitometry revealed no change in phos-phorylation at the PHF1 epitope (Fig. 4B). However, 0.25 and 0.5 μMMG-115 significantly increased phosphorylation at the AT8 epitope (Fig. 4C)and as low concentrations as 0.1 μMMG-115was enough to significantlyincrease phosphorylation at S422 (Fig. 4D). As western blot is a semiquantitative method, we also used the Meso Scale Discovery (MSD) sys-tem, a quantifiable antibody-based electrochemiluminescence method,to investigate tau phosphorylation. This system enabled simultaneous
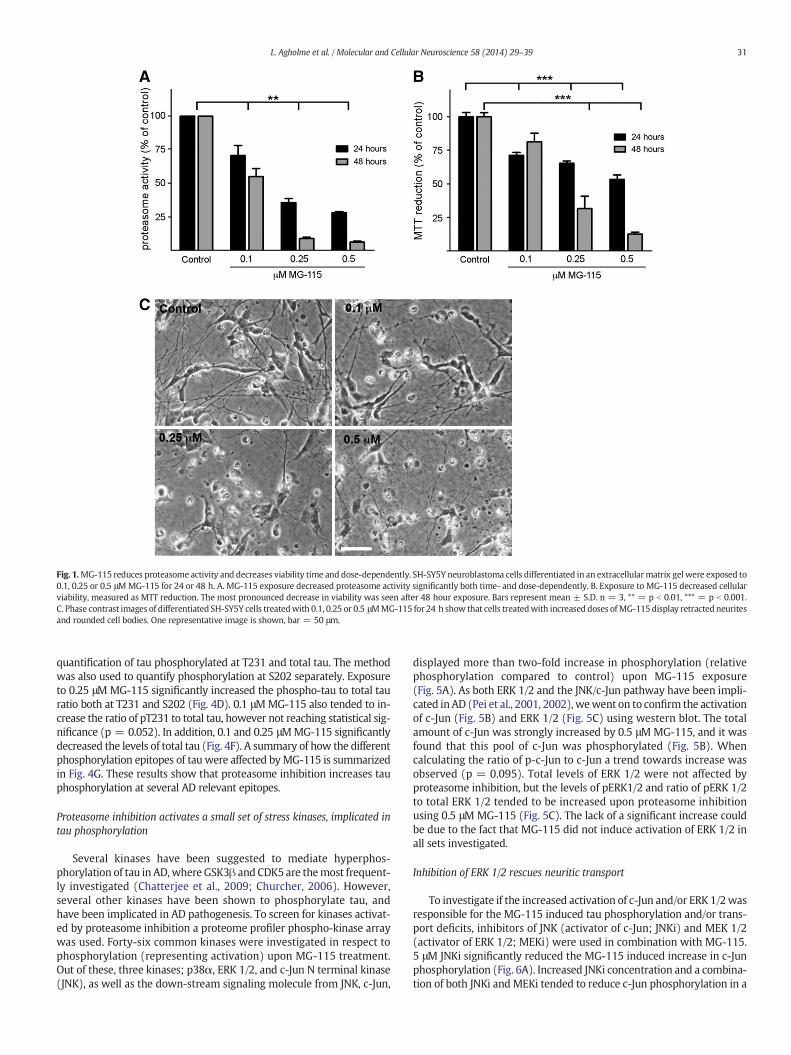
Fig. 1.MG-115 reduces proteasome activity and decreases viability time and dose-dependently. SH-SY5Y neuroblastoma cells differentiated in an extracellularmatrix gel were exposed to0.1, 0.25 or 0.5 μMMG-115 for 24 or 48 h. A. MG-115 exposure decreased proteasome activity significantly both time- and dose-dependently. B. Exposure to MG-115 decreased cellularviability, measured as MTT reduction. The most pronounced decrease in viability was seen after 48 hour exposure. Bars represent mean ± S.D. n = 3, ** = p b 0.01, *** = p b 0.001.C. Phase contrast images of differentiated SH-SY5Y cells treatedwith 0.1, 0.25 or 0.5 μMMG-115 for 24 h show that cells treatedwith increased doses ofMG-115display retracted neuritesand rounded cell bodies. One representative image is shown, bar = 50 μm.
31L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
quantification of tau phosphorylated at T231 and total tau. The methodwas also used to quantify phosphorylation at S202 separately. Exposureto 0.25 μM MG-115 significantly increased the phospho-tau to total tauratio both at T231 and S202 (Fig. 4D). 0.1 μMMG-115 also tended to in-crease the ratio of pT231 to total tau, however not reaching statistical sig-nificance (p = 0.052). In addition, 0.1 and 0.25 μMMG-115 significantlydecreased the levels of total tau (Fig. 4F). A summary of how the differentphosphorylation epitopes of tau were affected byMG-115 is summarizedin Fig. 4G. These results show that proteasome inhibition increases tauphosphorylation at several AD relevant epitopes.
Proteasome inhibition activates a small set of stress kinases, implicated intau phosphorylation
Several kinases have been suggested to mediate hyperphos-phorylation of tau in AD,where GSK3β and CDK5 are themost frequent-ly investigated (Chatterjee et al., 2009; Churcher, 2006). However,several other kinases have been shown to phosphorylate tau, andhave been implicated in AD pathogenesis. To screen for kinases activat-ed by proteasome inhibition a proteome profiler phospho-kinase arraywas used. Forty-six common kinases were investigated in respect tophosphorylation (representing activation) upon MG-115 treatment.Out of these, three kinases; p38α, ERK 1/2, and c-Jun N terminal kinase(JNK), as well as the down-stream signaling molecule from JNK, c-Jun,
displayed more than two-fold increase in phosphorylation (relativephosphorylation compared to control) upon MG-115 exposure(Fig. 5A). As both ERK 1/2 and the JNK/c-Jun pathway have been impli-cated inAD (Pei et al., 2001, 2002), wewent on to confirm the activationof c-Jun (Fig. 5B) and ERK 1/2 (Fig. 5C) using western blot. The totalamount of c-Jun was strongly increased by 0.5 μM MG-115, and it wasfound that this pool of c-Jun was phosphorylated (Fig. 5B). Whencalculating the ratio of p-c-Jun to c-Jun a trend towards increase wasobserved (p = 0.095). Total levels of ERK 1/2 were not affected byproteasome inhibition, but the levels of pERK1/2 and ratio of pERK 1/2to total ERK 1/2 tended to be increased upon proteasome inhibitionusing 0.5 μM MG-115 (Fig. 5C). The lack of a significant increase couldbe due to the fact that MG-115 did not induce activation of ERK 1/2 inall sets investigated.
Inhibition of ERK 1/2 rescues neuritic transport
To investigate if the increased activation of c-Jun and/or ERK 1/2wasresponsible for the MG-115 induced tau phosphorylation and/or trans-port deficits, inhibitors of JNK (activator of c-Jun; JNKi) and MEK 1/2(activator of ERK 1/2; MEKi) were used in combination with MG-115.5 μM JNKi significantly reduced the MG-115 induced increase in c-Junphosphorylation (Fig. 6A). Increased JNKi concentration and a combina-tion of both JNKi andMEKi tended to reduce c-Jun phosphorylation in a
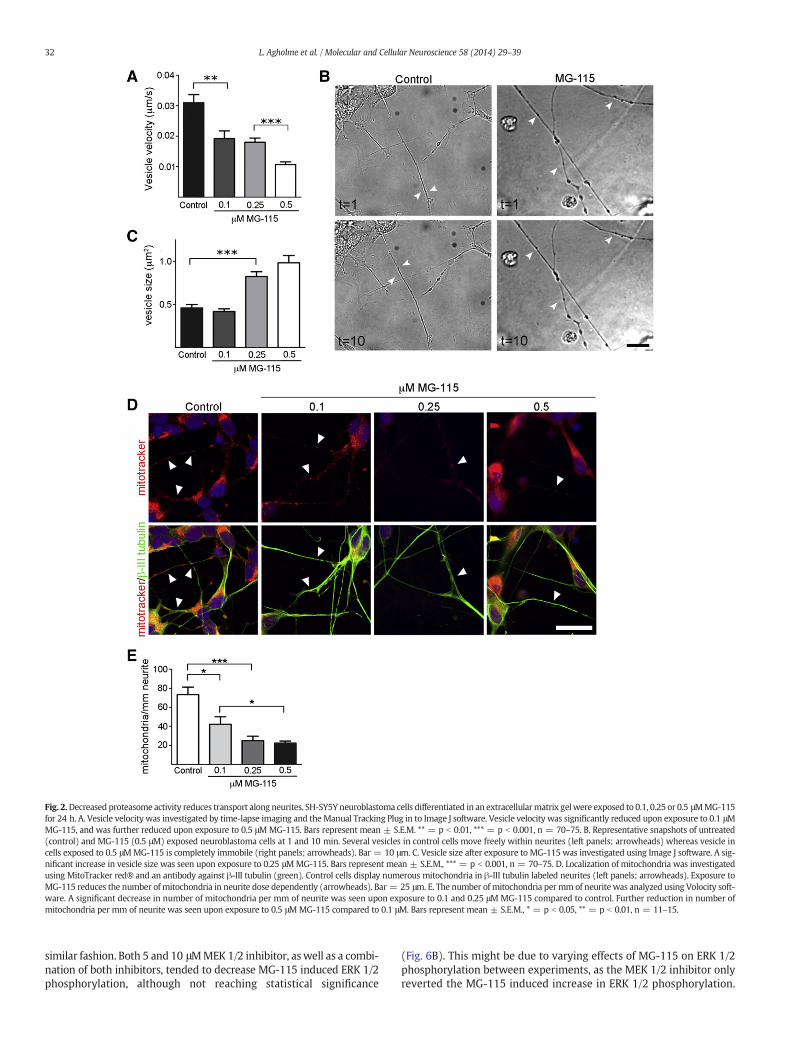
Fig. 2.Decreased proteasome activity reduces transport along neurites. SH-SY5Y neuroblastoma cells differentiated in an extracellularmatrix gelwere exposed to 0.1, 0.25 or 0.5 μMMG-115for 24 h. A. Vesicle velocity was investigated by time-lapse imaging and theManual Tracking Plug in to Image J software. Vesicle velocity was significantly reduced upon exposure to 0.1 μMMG-115, and was further reduced upon exposure to 0.5 μMMG-115. Bars represent mean ± S.E.M. ** = p b 0.01, *** = p b 0.001, n = 70–75. B. Representative snapshots of untreated(control) and MG-115 (0.5 μM) exposed neuroblastoma cells at 1 and 10 min. Several vesicles in control cells move freely within neurites (left panels; arrowheads) whereas vesicle incells exposed to 0.5 μMMG-115 is completely immobile (right panels; arrowheads). Bar = 10 μm. C. Vesicle size after exposure to MG-115 was investigated using Image J software. A sig-nificant increase in vesicle size was seen upon exposure to 0.25 μMMG-115. Bars represent mean ± S.E.M., *** = p b 0.001, n = 70–75. D. Localization of mitochondria was investigatedusing MitoTracker red® and an antibody against β-III tubulin (green). Control cells display numerous mitochondria in β-III tubulin labeled neurites (left panels; arrowheads). Exposure toMG-115 reduces the number of mitochondria in neurite dose dependently (arrowheads). Bar = 25 μm. E. The number of mitochondria permmof neurite was analyzed using Volocity soft-ware. A significant decrease in number of mitochondria per mm of neurite was seen upon exposure to 0.1 and 0.25 μM MG-115 compared to control. Further reduction in number ofmitochondria per mm of neurite was seen upon exposure to 0.5 μMMG-115 compared to 0.1 μM. Bars represent mean ± S.E.M., * = p b 0.05, ** = p b 0.01, n = 11–15.
32 L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
similar fashion. Both 5 and 10 μMMEK 1/2 inhibitor, aswell as a combi-nation of both inhibitors, tended to decrease MG-115 induced ERK 1/2phosphorylation, although not reaching statistical significance
(Fig. 6B). This might be due to varying effects of MG-115 on ERK 1/2phosphorylation between experiments, as the MEK 1/2 inhibitor onlyreverted the MG-115 induced increase in ERK 1/2 phosphorylation.
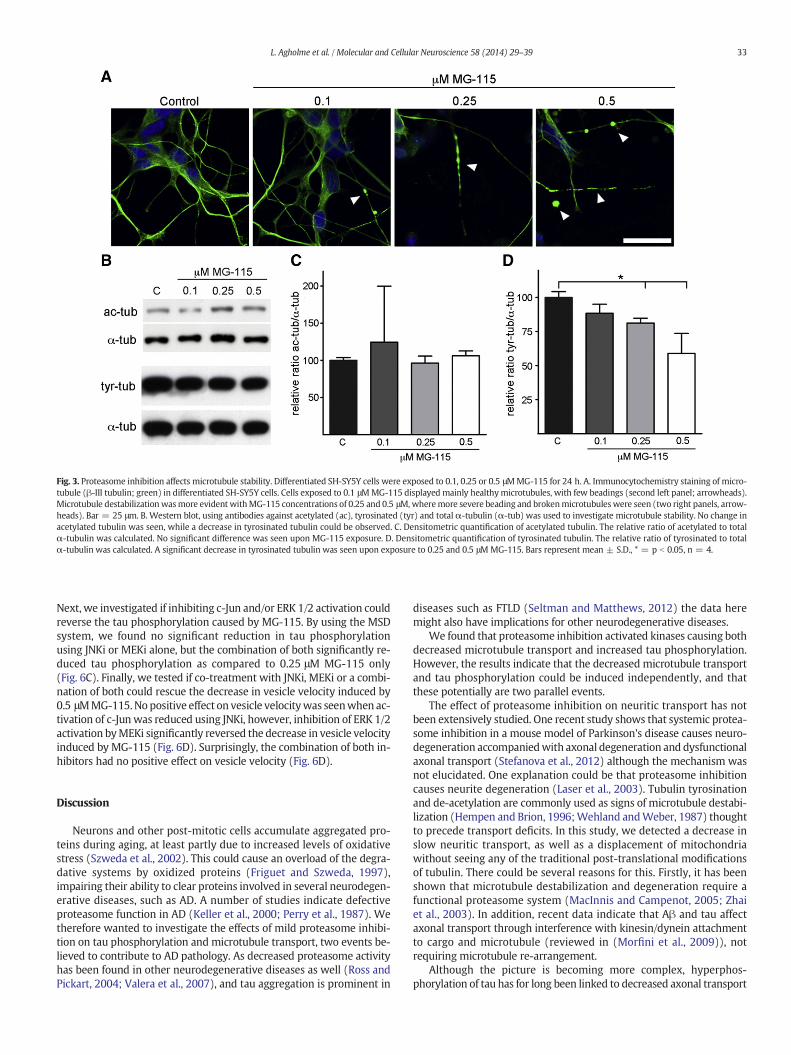
Fig. 3. Proteasome inhibition affects microtubule stability. Differentiated SH-SY5Y cells were exposed to 0.1, 0.25 or 0.5 μMMG-115 for 24 h. A. Immunocytochemistry staining of micro-tubule (β-III tubulin; green) in differentiated SH-SY5Y cells. Cells exposed to 0.1 μMMG-115 displayed mainly healthymicrotubules, with few beadings (second left panel; arrowheads).Microtubule destabilizationwasmore evident withMG-115 concentrations of 0.25 and 0.5 μM,wheremore severe beading and brokenmicrotubuleswere seen (two right panels, arrow-heads). Bar = 25 μm. B. Western blot, using antibodies against acetylated (ac), tyrosinated (tyr) and total α-tubulin (α-tub) was used to investigate microtubule stability. No change inacetylated tubulin was seen, while a decrease in tyrosinated tubulin could be observed. C. Densitometric quantification of acetylated tubulin. The relative ratio of acetylated to totalα-tubulin was calculated. No significant difference was seen upon MG-115 exposure. D. Densitometric quantification of tyrosinated tubulin. The relative ratio of tyrosinated to totalα-tubulin was calculated. A significant decrease in tyrosinated tubulin was seen upon exposure to 0.25 and 0.5 μM MG-115. Bars represent mean ± S.D., * = p b 0.05, n = 4.
33L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
Next, we investigated if inhibiting c-Jun and/or ERK 1/2 activation couldreverse the tau phosphorylation caused by MG-115. By using the MSDsystem, we found no significant reduction in tau phosphorylationusing JNKi or MEKi alone, but the combination of both significantly re-duced tau phosphorylation as compared to 0.25 μM MG-115 only(Fig. 6C). Finally, we tested if co-treatment with JNKi, MEKi or a combi-nation of both could rescue the decrease in vesicle velocity induced by0.5 μMMG-115.No positive effect on vesicle velocitywas seenwhen ac-tivation of c-Junwas reduced using JNKi, however, inhibition of ERK 1/2activation byMEKi significantly reversed the decrease in vesicle velocityinduced by MG-115 (Fig. 6D). Surprisingly, the combination of both in-hibitors had no positive effect on vesicle velocity (Fig. 6D).
Discussion
Neurons and other post-mitotic cells accumulate aggregated pro-teins during aging, at least partly due to increased levels of oxidativestress (Szweda et al., 2002). This could cause an overload of the degra-dative systems by oxidized proteins (Friguet and Szweda, 1997),impairing their ability to clear proteins involved in several neurodegen-erative diseases, such as AD. A number of studies indicate defectiveproteasome function in AD (Keller et al., 2000; Perry et al., 1987). Wetherefore wanted to investigate the effects of mild proteasome inhibi-tion on tau phosphorylation and microtubule transport, two events be-lieved to contribute to AD pathology. As decreased proteasome activityhas been found in other neurodegenerative diseases as well (Ross andPickart, 2004; Valera et al., 2007), and tau aggregation is prominent in
diseases such as FTLD (Seltman and Matthews, 2012) the data heremight also have implications for other neurodegenerative diseases.
We found that proteasome inhibition activated kinases causing bothdecreased microtubule transport and increased tau phosphorylation.However, the results indicate that the decreased microtubule transportand tau phosphorylation could be induced independently, and thatthese potentially are two parallel events.
The effect of proteasome inhibition on neuritic transport has notbeen extensively studied. One recent study shows that systemic protea-some inhibition in a mouse model of Parkinson's disease causes neuro-degeneration accompaniedwith axonal degeneration and dysfunctionalaxonal transport (Stefanova et al., 2012) although the mechanism wasnot elucidated. One explanation could be that proteasome inhibitioncauses neurite degeneration (Laser et al., 2003). Tubulin tyrosinationand de-acetylation are commonly used as signs of microtubule destabi-lization (Hempen and Brion, 1996;Wehland andWeber, 1987) thoughtto precede transport deficits. In this study, we detected a decrease inslow neuritic transport, as well as a displacement of mitochondriawithout seeing any of the traditional post-translational modificationsof tubulin. There could be several reasons for this. Firstly, it has beenshown that microtubule destabilization and degeneration require afunctional proteasome system (MacInnis and Campenot, 2005; Zhaiet al., 2003). In addition, recent data indicate that Aβ and tau affectaxonal transport through interference with kinesin/dynein attachmentto cargo and microtubule (reviewed in (Morfini et al., 2009)), notrequiring microtubule re-arrangement.
Although the picture is becoming more complex, hyperphos-phorylation of tau has for long been linked to decreased axonal transport
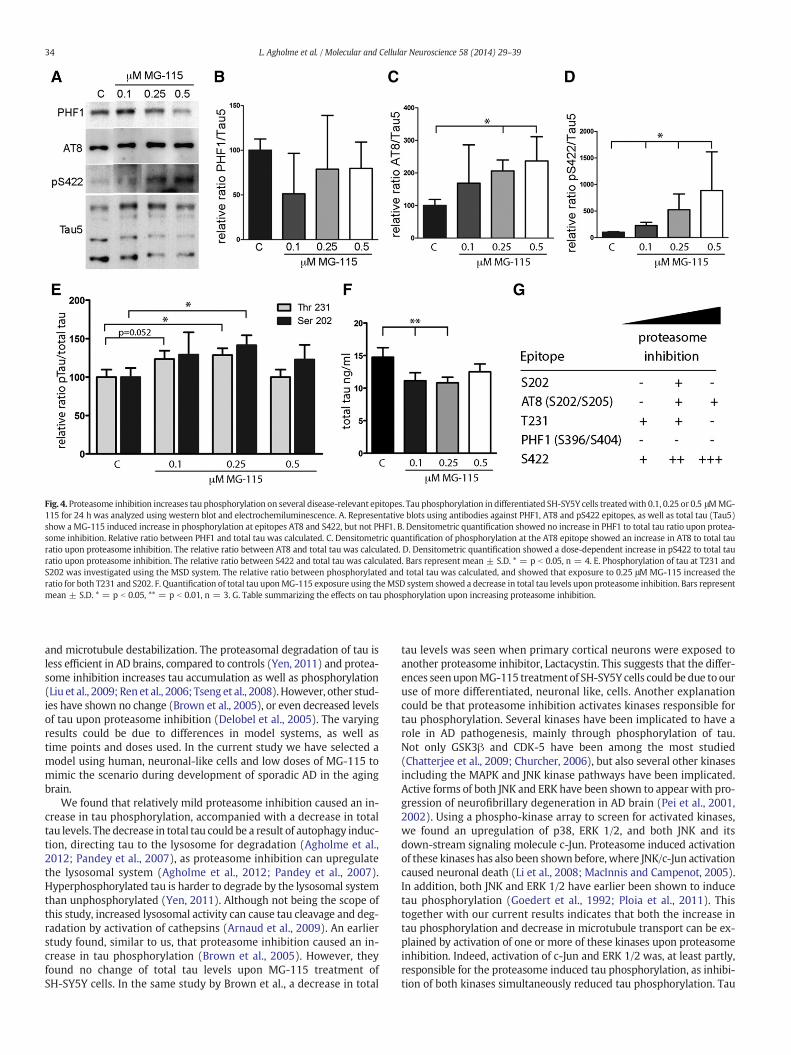
Fig. 4. Proteasome inhibition increases tau phosphorylation on several disease-relevant epitopes. Tau phosphorylation in differentiated SH-SY5Y cells treatedwith 0.1, 0.25 or 0.5 μMMG-115 for 24 h was analyzed using western blot and electrochemiluminescence. A. Representative blots using antibodies against PHF1, AT8 and pS422 epitopes, as well as total tau (Tau5)show aMG-115 induced increase in phosphorylation at epitopes AT8 and S422, but not PHF1. B. Densitometric quantification showed no increase in PHF1 to total tau ratio upon protea-some inhibition. Relative ratio between PHF1 and total tau was calculated. C. Densitometric quantification of phosphorylation at the AT8 epitope showed an increase in AT8 to total tauratio upon proteasome inhibition. The relative ratio between AT8 and total tau was calculated. D. Densitometric quantification showed a dose-dependent increase in pS422 to total tauratio upon proteasome inhibition. The relative ratio between S422 and total tau was calculated. Bars represent mean ± S.D. * = p b 0.05, n = 4. E. Phosphorylation of tau at T231 andS202 was investigated using the MSD system. The relative ratio between phosphorylated and total tau was calculated, and showed that exposure to 0.25 μM MG-115 increased theratio for both T231 and S202. F. Quantification of total tau uponMG-115 exposure using theMSD system showed a decrease in total tau levels upon proteasome inhibition. Bars representmean ± S.D. * = p b 0.05, ** = p b 0.01, n = 3. G. Table summarizing the effects on tau phosphorylation upon increasing proteasome inhibition.
34 L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
and microtubule destabilization. The proteasomal degradation of tau isless efficient in AD brains, compared to controls (Yen, 2011) and protea-some inhibition increases tau accumulation as well as phosphorylation(Liu et al., 2009; Ren et al., 2006; Tseng et al., 2008). However, other stud-ies have shown no change (Brown et al., 2005), or even decreased levelsof tau upon proteasome inhibition (Delobel et al., 2005). The varyingresults could be due to differences in model systems, as well astime points and doses used. In the current study we have selected amodel using human, neuronal-like cells and low doses of MG-115 tomimic the scenario during development of sporadic AD in the agingbrain.
We found that relatively mild proteasome inhibition caused an in-crease in tau phosphorylation, accompanied with a decrease in totaltau levels. The decrease in total tau could be a result of autophagy induc-tion, directing tau to the lysosome for degradation (Agholme et al.,2012; Pandey et al., 2007), as proteasome inhibition can upregulatethe lysosomal system (Agholme et al., 2012; Pandey et al., 2007).Hyperphosphorylated tau is harder to degrade by the lysosomal systemthan unphosphorylated (Yen, 2011). Although not being the scope ofthis study, increased lysosomal activity can cause tau cleavage and deg-radation by activation of cathepsins (Arnaud et al., 2009). An earlierstudy found, similar to us, that proteasome inhibition caused an in-crease in tau phosphorylation (Brown et al., 2005). However, theyfound no change of total tau levels upon MG-115 treatment ofSH-SY5Y cells. In the same study by Brown et al., a decrease in total
tau levels was seen when primary cortical neurons were exposed toanother proteasome inhibitor, Lactacystin. This suggests that the differ-ences seenuponMG-115 treatment of SH-SY5Y cells could be due to ouruse of more differentiated, neuronal like, cells. Another explanationcould be that proteasome inhibition activates kinases responsible fortau phosphorylation. Several kinases have been implicated to have arole in AD pathogenesis, mainly through phosphorylation of tau.Not only GSK3β and CDK-5 have been among the most studied(Chatterjee et al., 2009; Churcher, 2006), but also several other kinasesincluding the MAPK and JNK kinase pathways have been implicated.Active forms of both JNK and ERK have been shown to appear with pro-gression of neurofibrillary degeneration in AD brain (Pei et al., 2001,2002). Using a phospho-kinase array to screen for activated kinases,we found an upregulation of p38, ERK 1/2, and both JNK and itsdown-stream signaling molecule c-Jun. Proteasome induced activationof these kinases has also been shown before, where JNK/c-Jun activationcaused neuronal death (Li et al., 2008; MacInnis and Campenot, 2005).In addition, both JNK and ERK 1/2 have earlier been shown to inducetau phosphorylation (Goedert et al., 1992; Ploia et al., 2011). Thistogether with our current results indicates that both the increase intau phosphorylation and decrease in microtubule transport can be ex-plained by activation of one or more of these kinases upon proteasomeinhibition. Indeed, activation of c-Jun and ERK 1/2 was, at least partly,responsible for the proteasome induced tau phosphorylation, as inhibi-tion of both kinases simultaneously reduced tau phosphorylation. Tau
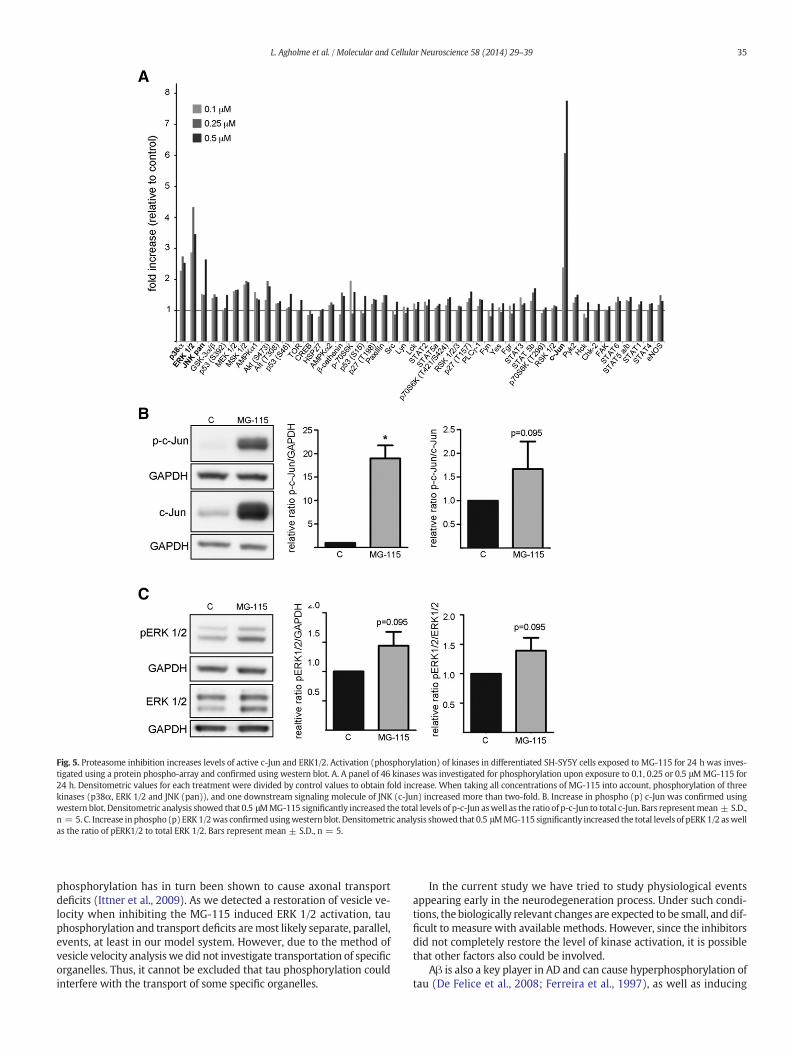
Fig. 5. Proteasome inhibition increases levels of active c-Jun and ERK1/2. Activation (phosphorylation) of kinases in differentiated SH-SY5Y cells exposed to MG-115 for 24 h was inves-tigated using a protein phospho-array and confirmed using western blot. A. A panel of 46 kinases was investigated for phosphorylation upon exposure to 0.1, 0.25 or 0.5 μMMG-115 for24 h. Densitometric values for each treatment were divided by control values to obtain fold increase. When taking all concentrations of MG-115 into account, phosphorylation of threekinases (p38α, ERK 1/2 and JNK (pan)), and one downstream signaling molecule of JNK (c-Jun) increased more than two-fold. B. Increase in phospho (p) c-Jun was confirmed usingwestern blot. Densitometric analysis showed that 0.5 μMMG-115 significantly increased the total levels of p-c-Jun aswell as the ratio of p-c-Jun to total c-Jun. Bars representmean ± S.D.,n = 5. C. Increase in phospho (p) ERK 1/2was confirmedusingwesternblot. Densitometric analysis showed that 0.5 μMMG-115 significantly increased the total levels of pERK 1/2 aswellas the ratio of pERK1/2 to total ERK 1/2. Bars represent mean ± S.D., n = 5.
35L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
phosphorylation has in turn been shown to cause axonal transportdeficits (Ittner et al., 2009). As we detected a restoration of vesicle ve-locity when inhibiting the MG-115 induced ERK 1/2 activation, tauphosphorylation and transport deficits aremost likely separate, parallel,events, at least in our model system. However, due to the method ofvesicle velocity analysiswe did not investigate transportation of specificorganelles. Thus, it cannot be excluded that tau phosphorylation couldinterfere with the transport of some specific organelles.
In the current study we have tried to study physiological eventsappearing early in the neurodegeneration process. Under such condi-tions, the biologically relevant changes are expected to be small, anddif-ficult to measure with available methods. However, since the inhibitorsdid not completely restore the level of kinase activation, it is possiblethat other factors also could be involved.
Aβ is also a key player in AD and can cause hyperphosphorylation oftau (De Felice et al., 2008; Ferreira et al., 1997), as well as inducing

Fig. 6. Inhibiting activation of ERK1/2 rescues proteasome induced transport deficits. Differentiated SH-SY5Y cellswere exposed toMG-115 in combinationwith 5 or 10 μMJNK inhibitor II(JNKi), 5 or 10 μMMEK inhibitor U0126 (MEKi), or a combination of 5 μM of each inhibitor (combo) for 24 h. A. The effect of 5 or 10 μM JNKi on MG-115 induced c-Jun phosphorylationwas investigated using western blot. Densitometric analysis showed that 5 μM JNKi significantly reduced the levels of phosphorylated (p) c-Jun. The same trendwas seen for 10 μM JNKiand a combination of both inhibitors. Bars representmean ± S.D., n = 4–5, * = p b 0.05. B. The effect of 5 or 10 μMMEKi onMG-115 induced ERK 1/2 phosphorylationwas investigatedusing western blot. Densitometric analysis showed that both 5 and 10 μM MEKi, as well as a combination of both inhibitors tended to reduce the levels of phosphorylated (p) ERK 1/2although not reaching statistical significance. Bars represent mean ± S.D., n = 5, * = p b 0.05. C. The effect of c-Jun and ERK 1/2 inhibition on MG-115 induced tau phosphorylationat the T231 epitope was investigated using the MSD system. 5 μM of either inhibitor was not enough to significantly reduce 0.25 MG-115 induced tau phosphorylation. A combinationof both inhibitors significantly reduced MG-115 induced tau phosphorylation. Bars represent mean ± S.D., n = 3, * = p b 0.05. D. The effect on vesicle velocity upon co-treatmentwith MG-115 and JNKi, MEKi or a combination was investigated by time-lapse imaging and the Manual Tracking Plug in to the Image J software. 5 μM MEKi significantly rescued thedecrease in vesicle velocity being induced by 0.5 μM MG-115. Bars represent mean ± S.E.M., n = 63–94, * = p b 0.05.
36 L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
microtubule dysfunction (Jin et al., 2011; King et al., 2006). We andothers have shown that proteasome inhibition can cause increasedlevels of intracellular and secreted Aβ (Agholme et al., 2012; Floodet al., 2005; Nunan et al., 2001). Among themany effects Aβ has on neu-ronal function and ADpathology, it has been shown that Aβ can activateboth the MEK/ERK and the JNK/c-Jun pathways (Viana et al., 2010;Young et al., 2009). Although not investigated in this study, it is there-fore possible that the proteasome inhibition induced kinase activationcould be regulated through Aβ, warranting further studies.
Conclusion
Decreased proteasome activity is seen in the aging brain and evenmore pronounced in the AD brain. This study proved a possible link be-tween the decrease in proteasome activity and downstream eventsleading to AD pathology, including deficits in axonal transport,
microtubule destabilization and tau phosphorylation. Activation ofstress kinases is indicated to be the link between these events. Thus,modulation of ERK 1/2 and c-Jun activation could therefore contributeto novel treatments against AD, as well as general improvement ofproteasome function.
Experimental methods
Cell culture and treatment
SH-SY5Y neuroblastoma cells (ECACC, Sigma Aldrich) were main-tained in MEM Glutamax (Invitrogen) supplemented with 10% fetalcalf serum (FCS; PAA Laboratories), 2 mM L-glutamine, 50 U/ml penicil-lin, and50 μg/ml streptomycin (all fromLonza), in a humidified chamberwith 5% CO2 at 37 °C. Medium was changed every 3–4 days, and cellswere split upon 80–90% confluence. Cells were pre-differentiated using

37L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
10 μM Retinoic acid (Sigma Aldrich) for 7 days and thereafter seeded inan extracellular matrix (ECM) gel and differentiated as described previ-ously (Agholme et al., 2010). After 10 days of differentiation, cells weretreated with 0.1, 0.25 or 0.5 μM MG-115 (stock solution 1 mM inDMSO; Calbiochem) for 24–48 h in serum free media. Control cellswere treated with DMSO alone. When applicable, cells were pre-treated with 5 or 10 μM JNK inhibitor II (#420119, Calbiochem) orMEK 1/2 inhibitor U0126 (Cell Signaling Technology) for 30 min, prioraddition of MG-115. At sample collection, the gel was disrupted usingdispase (2.2 U/ml in PBS; Invitrogen) for 60–90 min at 37 °C. Sampleswere thereafter collected on ice, washed once with PBS, and storedat−70 °C until further analysis.
Proteasome activity measurement
The chymotrypsin-like proteasomal activity was analyzed using thefluorescent proteasome substrate III (Calbiochem). Cells were lysed inreaction buffer containing 5 mM MgCl2, 0.5 mM ATP, 1% Triton, 20%Glycerol, 1 mM DTT and 0.1 mM substrate in 50 mM Tris–HCl, pH 8.8.An aliquot was removed for protein analysis, and the remaining samplewas incubated for 60 min at 37 °C. Thereafter, the reaction wasquenched by adding 10% SDS and 100 mM Tris–HCl (pH 9.0) at a ratioof 1:8. Released fluorescent AMC molecules were measured on a VictorWallac (PerkinElmer) using 380 nmexcitationwavelength and 460 nmemission wavelength. Fluorescent counts, correlating to proteasomeactivity were adjusted to protein concentration and calculated aspercentage of control.
Live cell imaging of vesicles and velocity analysis
Velocity of the vesicles in the neurites of the SH-SY5Y neuroblastomacells was analyzed after treatment with 0.1, 0.25 or 0.5 μM of MG-115.Time-lapse differential interference contrast (using Plan-Apochromat63×/1.40 oil DICM27 lens) images were collected using a Zeiss Axiovert200 M Fluorescence Microscope at every 30–60 s, with 1 s or shorterscan time, using the Axiovision 4.7 software. Cells were kept at 37 °C.Velocities were calculated with the help of Manual Tracking Plug in,and Image J software, where velocity was calculated by recording x andy coordinates during the time interval between two successive images.Both x and y pixel sizes are equal to 0.1 μmandvelocities are the averagemovements of 5–20 successive images. Area of each vesicle was alsocalculated selecting the pixel area using Image J software.
Immunocytochemistry
Immunocytochemistry was performed as described previously(Agholme et al., 2010), on cells maintained in the gel, plated onto4-well chamber slides (Thermo Fisher). When applicable, mitochondri-al staining was performed by incubating samples with MitoTracker®red (200 nM; Invitrogen) for 30 min at 37 °C prior fixation. Primary an-tibody used was neuron specific class III beta tubulin (1:2000; Abcam)and secondary antibody used was Goat anti-Rabbit Alexa 488 (1:400).Nuclei were stained with 1.5 μM ToPro3, and samples were mountedusing prolong gold antifade mounting media (all from Invitrogen). Im-ages were collected using a Nikon confocal microscope and the EZC1software (Nikon instruments), with a Plan Apo60×/1.40 lens, an eclipseC1 confocal unit and three lasers with a wavelengths of 488, 560 and630 nm respectively,
Mitochondria localization
Localization of mitochondria was performed using the Volocity soft-ware version 5.4.2 and the measurement tool. Mitochondria were se-lected based on fluorescence intensity and size, and all objects withinthe cell body or outside cells were removed manually. The neuritelengths were calculated using the line tool, and for each image the
number of mitochondria was divided by the total length of neurites.The data was presented as number of mitochondria per mm of neurite.
Quantification of tau phosphorylation by electrochemiluminescence linkedimmunoassay
For this analysis, 96-well 4-spotmultiplex (pTau T231/total tau) andsingleplex (pTau S202) were used (Meso Scale Discovery). Sampleswere lysed in MSD lysis buffer (150 mM NaCl, 20 mM Tris pH 7.5,1 mM EGTA, 1 mM EDTA, 1% Triton X-100) supplemented withphosphostop (Roche applied science) and protease inhibitor cocktail(Sigma Aldrich), and protein concentration was determined by the DCprotein assay (BioRad Laboratories). The assay was performed accord-ing to the manufacturer's instructions, and signals were measured ona SECTOR Imager 2400 reader (Meso Scale Discovery). For the T231epitope, the ratio of phosphate (U/ml) on T231 to total tau (ng/ml)was calculated. For S202, the ratio of signal from antibody specific forS202 (A.U.) to total tau (ng/ml) was calculated. Both were thereafterpresented as percentage of control.
Kinase phosphorylation assay
To investigate kinase activation (phosphorylation) upon protea-some inhibition, we used the Human-phospho-kinase antibody array(R&D systems). Cells were treated with 0.1, 0.25 and 0.5 μM MG-115for 24 h, and after gel disruption pooled together to increase cell num-ber to about 8× the amount normally used. The assay was then carriedout according to manufacturer's instructions. After protein determina-tion 155 μg of proteinwas added to eachmembrane. Detectionwas car-ried out using Amersham ECL western blotting reagent and hyperfilmECL (both from GE Healthcare). Densitometric analysis was carried outfor each dot using Image Gauge (Fujifilm), and the average of two wascalculated for each kinase. The relative ratio between control samplesand MG-115 treated samples was calculated.
Western blotting
Cells were lysed in buffer (62.5 mM Tris HCl pH 6.8, Glycerol, SDS)supplemented with protease inhibitor cocktail (Sigma Aldrich) andphosphostop (Roche Applied Science). Protein concentration was deter-mined using the DC protein assay (BioRad Laboratories). Samples wereadjusted to equal protein concentration before adding bromophenolblue and DTT to the samples. For each sample, 10 μg of proteinwas load-ed onto a 10 or 12% SDS gel (CBS Scientific Company). Proteins wereblotted onto a 0.45 μM nitrocellulose membrane (BioRad Laboratories),blocked in 5% bovine serum albumin in TBS-Tween 20, and incubatedwith the following; Tau5 1:2000 (Covance Research Products), AT81:1000 (Innogenetics), anti-pTau S422 1:1000 (Invitrogen), PHF11:1000 (a kind gift from Dr. P. Davies), anti-tyrosinated tubulin1:10000, anti-acetylated tubulin 1:10000 (both from Sigma Aldrich),anti-α-tubulin 1:10000 (DSHB, University of Iowa, IO, USA), anti c-Jun(1:1000), anti-phospho c-Jun (Ser63; 1:1000), anti-ERK 1/2 and anti-phospho ERK 1/2 (all from Cell Signaling Technology). Secondary anti-bodies used were HRP conjugated goat-anti mouse and goat-anti rabbit1:3000 (DAKO).
MTT viability assay
MTT salt (5 mg/ml in PBS; Calbiochem)was added to samples in 1:10dilution and incubated for 2 h at 37 °C. Media containing excess saltwere then removed and the gel was disrupted with dispase as describedabove. Produced formazan salt was collected by centrifugation at 3000 gfor 5 min, and dissolved in DMSO. Absorbance at 550 nmwasmeasuredon a Victor Wallac 1420 multilabel counter (PerkinElmer).

38 L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
Statistical analysis
Proteasome activity and viability results were analyzed using one-way ANOVA with Dunnett's post hoc test, using control samples asreference. Tau concentrations, vesicle velocity, and vesicle sizewere cal-culated using student's t-test. Statistical analysis of densitometric valuesobtained from western blot was performed using Mann–Whitney'snon-parametric test. All p-values less than 0.05 were considered statis-tically significant. All statistical calculations were performed with SPSSv. 16.0. All graphical presentation of data was prepared using GraphPadPrism v. 5.8.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.mcn.2013.11.001.
Funding
This work was funded by the Swedish Alzheimer's Foundation, theHans-Gabriel and Alice Trolle-Wachtmeister's Foundation for MedicalResearch, the Gustav V and Queen Victoria's Foundation, the SwedishDementia Foundation, the Linköping University Neurobiology Centerand the Östergötland County Council, Lions Research Fund and SwedishLundbeck Foundation. These funding sources had no involvement in theconduct of the research or in the preparation of the article.
Acknowledgments
We would like to thank Lisbeth Hjälle and Alexander Lamevski fortechnical assistance.
References
Agholme, L., Lindström, T., Kågedal, K., Marcusson, J., Hallbeck, M., 2010. An in vitromodelfor neuroscience: differentiation of SH-SY5Y cells into cells with morphological andbiochemical characteristics of mature neurons. J. Alzheimers Dis. 20, 1069–1082.
Agholme, L., Hallbeck, M., Benedikz, E., Marcusson, J., Kågedal, K., 2012. Amyloid-β secre-tion, generation, and lysosomal sequestration in response to proteasome inhibition:involvement of autophagy. J. Alzheimers Dis. 31, 343–358.
Arnaud, L.T., Myeku, N., Figueiredo-Pereira, M.E., 2009. Proteasome–caspase–cathepsinsequence leading to tau pathology induced by prostaglandin J2 in neuronal cells.J. Neurochem. 110, 328–342.
Biernat, J., Mandelkow, E.M., 1999. The development of cell processes induced by tau pro-tein requires phosphorylation of serine 262 and 356 in the repeat domain and isinhibited by phosphorylation in the proline-rich domains. Mol. Biol. Cell 10, 727–740.
Blennow, K., de Leon, M.J., Zetterberg, H., 2006. Alzheimer's disease. Lancet 368, 387–403.Brown, M.R., Bondada, V., Keller, J.N., Thorpe, J., Geddes, J.W., 2005. Proteasome or calpain
inhibition does not alter cellular tau levels in neuroblastoma cells or primaryneurons. J. Alzheimers Dis. 7, 15–24.
Chatterjee, S., Sang, T.K., Lawless, G.M., Jackson, G.R., 2009. Dissociation of tau toxicity andphosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum.Mol. Genet. 18, 164–177.
Churcher, I., 2006. Tau therapeutic strategies for the treatment of Alzheimer's disease.Curr. Top. Med. Chem. 6, 579–595.
Clavaguera, F., Bolmont, T., Crowther, R., Abramowski, D., Frank, S., Probst, A., Fraser, G.,Stalder, A., Beibel, M., Staufenbiel, M., Jucker, M., Goedert, M., Tolnay, M., 2009. Trans-mission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11,909–913.
De Felice, F.G., Wu, D., Lambert, M.P., Fernandez, S.J., Velasco, P.T., Lacor, P.N., Bigio, E.H.,Jerecic, J., Acton, P.J., Shughrue, P.J., Chen-Dodson, E., Kinney, G.G., Klein, W.L., 2008.Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβoligomers. Neurobiol. Aging 29, 1334–1347.
Delobel, P., Leroy, O., Hamdane, M., Sambo, A.V., Delacourte, A., Buee, L., 2005. Proteasomeinhibition and tau proteolysis: an unexpected regulation. FEBS Lett. 579, 1–5.
Drewes, G., Ebneth, A., Mandelkow, E.M., 1998. MAPs, MARKs and microtubule dynamics.Trends Biochem. Sci. 23, 307–311.
Ebneth, A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B., Mandelkow, E., 1998.Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles,mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease.J. Cell Biol. 143, 777–794.
Ferreira, A., Lu, Q., Orecchio, L., Kosik, K.S., 1997. Selective phosphorylation of adult tauisoforms in mature hippocampal neurons exposed to fibrillar Aβ. Mol. Cell. Neurosci.9, 220–234.
Flood, F., Murphy, S., Cowburn, R.F., Lannfelt, L., Walker, B., Johnston, J.A., 2005.Proteasome-mediated effects on amyloid precursor protein processing at theγ-secretase site. Biochem. J. 385, 545–550.
Friguet, B., Szweda, L.I., 1997. Inhibition of the multicatalytic proteinase (proteasome) by4-hydroxy-2-nonenal cross-linked protein. FEBS Lett. 405, 21–25.
Goedert, M., Cohen, E.S., Jakes, R., Cohen, P., 1992. p42 map kinase phosphorylation sitesin microtubule-associated protein tau are dephosphorylated by protein phosphatase2A1 implications for Alzheimer's disease. FEBS Lett. 312, 95–99.
Götz, J., Eckert, A., Matamales, M., Ittner, L., Liu, X., 2011. Modes of Aβ toxicity inAlzheimer's disease. Cell. Mol. Life Sci. 68, 3359–3375.
Grune, T., Botzen, D., Engels, M., Voss, P., Kaiser, B., Jung, T., Grimm, S., Ermak, G., Davies,K.J.A., 2010. Tau protein degradation is catalyzed by the ATP/ubiquitin-independent20S proteasome under normal cell conditions. Arch. Biochem. Biophys. 500, 181–188.
Hallbeck, M., Nath, S., Marcusson, J., 2013. Neuron-to-neuron transmission of neurode-generative pathology. Neuroscientist 19 (6), 560–566.
Hempen, B., Brion, J.P., 1996. Reduction of acetylated alpha-tubulin immunoreactivity inneurofibrillary tangle-bearing neurons in Alzheimer's disease. J. Neuropathol. Exp.Neurol. 55, 964–972.
Ittner, L.M., Ke, Y.D., Götz, J., 2009. Phosphorylated tau interacts with c-Jun N-terminalkinase-interacting protein 1 (JIP1) in Alzheimer disease. J. Biol. Chem. 284,20909–20916.
Jin, M., Shepardson, N., Yang, T., Chen, G., Walsh, D., Selkoe, D.J., 2011. Soluble amyloid β-protein dimers isolated fromAlzheimer cortex directly induce tau hyperphosphorylationand neuritic degeneration. Proc. Natl. Acad. Sci. U. S. A. 108, 5819–5824.
Keck, S., Nitsch, R., Grune, T., Ullrich, O., 2003. Proteasome inhibition by paired helicalfilament-tau in brains of patients with Alzheimer's disease. J. Neurochem. 85,115–122.
Keller, J.N., Hanni, K.B., Markesbery, W.R., 2000. Impaired proteasome function inAlzheimer's disease. J. Neurochem. 75, 436–439.
King, M.E., Kan, H.M., Baas, P.W., Erisir, A., Glabe, C.G., Bloom, G.S., 2006. Tau-dependentmicrotubule disassembly initiated by prefibrillar β-amyloid. J. Cell Biol. 175, 541–546.
Knecht, E., Aguado, C., Cárcel, J., Esteban, I., Esteve, J., Ghislat, G., Moruno, J., Vidal, J., Sáez,R., 2009. Intracellular protein degradation in mammalian cells: recent developments.Cell. Mol. Life Sci. 66, 2427–2443.
Laser, H., Mack, T.G.A., Wagner, D., Coleman, M.P., 2003. Proteasome inhibition arrestsneurite outgrowth and causes “Dying-back” degeneration in primary culture.J. Neurosci. Res. 74, 906–916.
Li, B., Chohan, M., Grundke-Iqbal, I., Iqbal, K., 2007. Disruption of microtubule network byAlzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 113, 501–511.
Li, X., Du, Y., Fan, X., Yang, D., Luo, G., Le, W., 2008. c-Jun N-terminal kinase mediateslactacystin-induced dopamine neuron degeneration. J. Neuropathol. Exp. Neurol.67, 933–944.
Lindwall, G., Cole, R.D., 1984. Phosphorylation affects the ability of tau protein to promotemicrotubule assembly. J. Biol. Chem. 259, 5301–5305.
Liu, Y.H., Wei, W., Yin, J., Liu, G.P., Wang, Q., Cao, F.Y., Wang, J.Z., 2009. Proteasome inhibi-tion increases tau accumulation independent of phosphorylation. Neurobiol. Aging30, 1949–1961.
López Salon, M., Morelli, L., Castaño, E.M., Soto, E.F., Pasquini, J.M., 2000. Defectiveubiquitination of cerebral proteins in Alzheimer's disease. J. Neurosci. Res. 62,302–310.
MacInnis, B.L., Campenot, R.B., 2005. Regulation of Wallerian degeneration and nervegrowth factor withdrawal-induced pruning of axons of sympathetic neurons by theproteasome and the MEK/Erk pathway. Mol. Cell. Neurosci. 28, 430–439.
Marambaud, P., Chevallier, N., Barelli, H., Wilk, S., Checler, F., 1997. Proteasome contrib-utes to the α-secretase pathway of amyloid precursor protein in human cells.J. Neurochem. 68, 698–703.
Marambaud, P., Ancolio, K., Lopez-Perez, E., Checler, F., 1998. Proteasome inhibitorsprevent the degradation of familial Alzheimer's disease-linked presenilin 1 andpotentiate A beta 42 recovery from human cells. Mol. Med. 4, 147–157.
Morfini, G.A., Burns, M., Binder, L.I., Kanaan, N.M., LaPointe, N., Bosco, D.A., Brown Jr., R.H.,Brown, H., Tiwari, A., Hayward, L., Edgar, J., Nave, K.-A., Garberrn, J., Atagi, Y., Song, Y.,Pigino, G., Brady, S.T., 2009. Axonal transport defects in neurodegenerative diseases.J. Neurosci. 29, 12776–12786.
Mudher, A., Shepherd, D., Newman, T.A., Mildren, P., Jukes, J.P., Squire, A., Mears, A.,Drummond, J.A., Berg, S., MacKay, D., Asuni, A.A., Bhat, R., Lovestone, S., 2004. GSK-3β inhibition reverses axonal transport defects and behavioural phenotypes inDrosophila. Mol. Psychiatry 9, 522–530.
Nath, S., Agholme, L., Kurudenkandy, F.R., Granseth, B., Marcusson, J., Hallbeck, M., 2012.Spreading of neurodegenerative pathology via neuron-to-neuron transmission ofβ-amyloid. J. Neurosci. 32, 8767–8777.
Nixon, R.A., Cataldo, A.M., 2006. Lysosomal system pathways: genes to neurodegenera-tion in Alzheimer's disease. J. Alzheimers Dis. 9, 277–289.
Nunan, J., Shearman, M.S., Checler, F., Cappai, R., Evin, G., Beyreuther, K., Masters, C.L.,Small, D.H., 2001. The C-terminal fragment of the Alzheimer's disease amyloidprotein precursor is degraded by a proteasome-dependent mechanism distinctfrom γ-secretase. Eur. J. Biochem. 268, 5329–5336.
Oddo, S., 2008. The ubiquitin–proteasome system in Alzheimer's disease. J. Cell. Mol. Med.12, 363–373.
Pandey, U.B., Nie, Z., Batlevi, Y., McCray, B.A., Ritson, G.P., Nedelsky, N.B., Schwartz, S.L.,DiProspero, N.A., Knight, M.A., Schuldiner, O., Padmanabhan, R., Hild, M., Berry, D.L.,Garza, D., Hubbert, C.C., Yao, T.P., Baehrecke, E.H., Taylor, J.P., 2007. HDAC6 rescuesneurodegeneration and provides an essential link between autophagy and the UPS.Nature 447, 859–863.
Pei, J.J., Braak, E., Braak, H., Grundke-Iqbal, I., Iqbal, K., Winblad, B., Cowburn, R.F., 2001.Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer'sdisease brains at different stages of neurofibrillary degeneration. J. Alzheimers Dis.3, 41–48.
Pei, J.J., Braak, H., An, W.L., Winblad, B., Cowburn, R.F., Iqbal, K., Grundke-Iqbal, I., 2002.Up-regulation of mitogen-activated protein kinases ERK1/2 andMEK1/2 is associatedwith the progression of neurofibrillary degeneration in Alzheimer's disease. BrainRes. Mol. Brain Res. 109, 45–55.

39L. Agholme et al. / Molecular and Cellular Neuroscience 58 (2014) 29–39
Perlson, E., Maday, S., Fu, M.M., Moughamian, A.J., Holzbaur, E.L.F., 2010. Retrogradeaxonal transport: pathways to cell death? Trends Neurosci. 33, 335–344.
Perry, G., Friedman, R., Shaw, G., Chau, V., 1987. Ubiquitin is detected in neurofibrillarytangles and senile plaque neurites of Alzheimer disease brains. Proc. Natl. Acad. Sci.U. S. A. 84, 3033–3036.
Ploia, C., Antoniou, X., Sclip, A., Grande, V., Cardinetti, D., Colombo, A., Canu, N., Benussi, L.,Ghidoni, R., Forloni, G., Borsello, T., 2011. JNK plays a key role in tau hyperphos-phorylation in Alzheimer's disease models. J. Alzheimers Dis. 26, 315–329.
Ren, Q.G., Liao, X.M., Wang, Z.F., Qu, Z.S., Wang, J.Z., 2006. The involvement of glycogensynthase kinase-3 and protein phosphatase-2A in lactacystin-induced tau accumula-tion. FEBS Lett. 580, 2503–2511.
Ross, C.A., Pickart, C.M., 2004. The ubiquitin–proteasome pathway in Parkinson's diseaseand other neurodegenerative diseases. Trends Cell Biol. 14, 703–711.
Seltman, R., Matthews, B., 2012. Frontotemporal Lobar Degeneration. CNS Drugs 26,841–870.
Stefanova, N., Kaufmann, W., Humpel, C., Poewe, W., Wenning, G., 2012. Systemic protea-some inhibition triggers neurodegeneration in a transgenic mouse model expressinghuman α-synuclein under oligodendrocyte promoter: implications for multiplesystem atrophy. Acta Neuropathol. 124, 51–65.
Stokin, G.B., Lillo, C., Falzone, T.L., Brusch, R.G., Rockenstein, E., Mount, S.L., Raman, R.,Davies, P., Masliah, E., Williams, D.S., Goldstein, L.S., 2005. Axonopathy andtransport deficits early in the pathogenesis of Alzheimer's disease. Science 307,1282–1288.
Szweda, P.A., Friguet, B., Szweda, L.I., 2002. Proteolysis, free radicals, and aging. Free Radic.Biol. Med. 33, 29–36.
Takahashi, R.H., Milner, T.A., Li, F., Nam, E.E., Edgar, M.A., Yamaguchi, H., Beal, M.F., Xu, H.,Greengard, P., Gouras, G.K., 2002. Intraneuronal Alzheimer Aβ42 accumulates in
multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161,1869–1879.
Terry, R.D., Masliah, E., Salmon, D.P., Butters, N., DeTeresa, R., Hill, R., Hansen, L.A., Katzman,R., 1991. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss isthe major correlate of cognitive impairment. Ann. Neurol. 30, 572–580.
Tseng, B.P., Green, K.N., Chan, J.L., Blurton-Jones, M., LaFerla, F.M., 2008. Aβ inhibits theproteasome and enhances amyloid and tau accumulation. Neurobiol. Aging 29,1607–1618.
Valera, A.G., Díaz-Hernández, M., Hernández, F., Lucas, J.J., 2007. Testing the possible inhi-bition of proteasome by direct interaction with ubiquitylated and aggregatedhuntingtin. Brain Res. Bull. 72, 121–123.
Viana, R.J.S., Ramalho, R.M., Nunes, A.F., Steer, C.J., Rodrigues, C.M.P., 2010. Modulation ofamyloid-β peptide-induced toxicity through inhibition of JNK nuclear localizationand caspase-2 activation. J. Alzheimers Dis. 22, 557–568.
Wang, Y., Martinez-Vicente, M., Kruger, U., Kaushik, S., Wong, E., Mandelkow, E.-M.,Cuervo, A.M., Mandelkow, E., 2009. Tau fragmentation, aggregation and clearance:the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170.
Wehland, J., Weber, K., 1987. Turnover of the carboxy-terminal tyrosine of alpha-tubulin andmeans of reaching elevated levels of detyrosination in living cells. J. Cell Sci. 88, 185–203.
Yen, S.S., 2011. Proteasome degradation of brain cytosolic tau in Alzheimer's disease. Int.J. Clin. Exp. Pathol. 4, 385–402.
Young, K.F., Pasternak, S.H., Rylett, R.J., 2009. Oligomeric aggregates of amyloid β peptide1–42 activate ERK/MAPK in SH-SY5Y cells via the α7 nicotinic receptor. Neurochem.Int. 55, 796–801.
Zhai, Q., Wang, J., Kim, A., Liu, Q., Watts, R., Hoopfer, E., Mitchison, T., Luo, L., He, Z., 2003.Involvement of the ubiquitin–proteasome system in the early stages of Walleriandegeneration. Neuron 39, 217–225.
Related Documents











