ORIGINAL RESEARCH Properties, aromaticity, and substituents effects in poly nitro- and amino-substituted benzenes Irina V. Omelchenko • Oleg V. Shishkin • Leonid Gorb • Frances C. Hill • Jerzy Leszczynski Received: 11 December 2011 / Accepted: 4 February 2012 / Published online: 24 February 2012 Ó Springer Science+Business Media, LLC 2012 Abstract Geometrical parameters, aromaticity, and con- formational flexibility of the set of polysubstituted ben- zenes with different number and position of nitro and amino groups were calculated at the MP2/cc-pvdz level of theory. The key factor for structural and energetic changes has been identified. This is related to the presence of nitro and amino groups in vicinal positions that forms strong intramolecular resonance-assisted hydrogen bonds with a binding energy of 7–14 kcal/mol. Increasing number of such bonds facilitates a cooperative effect, inducing nota- ble changes in molecular geometry (particularly increasing bond alternation within H 2 N–C–C–NO 2 fragment and planarization of amino group), drastic increasing of con- formational flexibility and decreasing of aromaticity. In spite of well-known p-electron effects of nitro and amino substituents, influence of their push–pull interaction through aromatic moiety is negligible compared to the effect of the hydrogen bonding. That results in great dif- ference of the ortho-isomers as compared to meta- and para-isomers. Keywords Aromaticity Á Push–pull effect Á Intramolecular hydrogen bond Á Quantum chemical calculations Introduction Aromaticity is one of the most general and important concepts in organic chemistry [1]. This phenomenon also provides the background to understanding the influence of substituents on reactivity and stability of the very wide range of organic species containing aromatic and hetero- aromatic moieties. The numerous examples of such influ- ence are available in the literature elsewhere [2–4]. Among them, famous Hammett’s constants describing electronic strength of substituents were derived for aromatic mole- cules. Using this approach, it is very easy to understand that mechanism of such influence also including changes in aromaticity of benzene ring. Influenced by the Hammett equation, there have been numerous attempts to describe substituent effect on aro- matic moiety quantitatively [5]. The common way is the usage of some characteristic physicochemical properties (such as amino group basicity, catalytic activity, change of infrared, or NMR properties of substance) as an aromaticity measure [6]. Such characteristics are usually called aro- maticity indexes. Recently, aromaticity indexes were used to estimate substituent effect [7, 8]. Analysis of mono- substituted benzenes revealed that influence of a single Electronic supplementary material The online version of this article (doi:10.1007/s11224-012-9971-8) contains supplementary material, which is available to authorized users. I. V. Omelchenko (&) Á O. V. Shishkin STC’Institute for Single Crystals, National Academy of Sciences of Ukraine, 60 Lenina Ave, Kharkiv 61001, Ukraine e-mail: [email protected] O. V. Shishkin V.N. Karazin Kharkiv National University, 4 Svobody sq, Kharkiv 61077, Ukraine L. Gorb Badger Technical Services, LLC, Vicksburg, MS, USA F. C. Hill Á J. Leszczynski US Army ERDC, 3532 Manor Dr, Vicksburg, MS 39180, USA J. Leszczynski Interdisciplinary Center for Nanotoxicity, Department of Chemistry and Biochemistry, Jackson State University, P.O. Box 17910, 1325 Lynch Street, Jackson, MS 39217, USA 123 Struct Chem (2012) 23:1585–1597 DOI 10.1007/s11224-012-9971-8

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ORIGINAL RESEARCH
Properties, aromaticity, and substituents effects in polynitro- and amino-substituted benzenes
Irina V. Omelchenko • Oleg V. Shishkin •
Leonid Gorb • Frances C. Hill • Jerzy Leszczynski
Received: 11 December 2011 / Accepted: 4 February 2012 / Published online: 24 February 2012
� Springer Science+Business Media, LLC 2012
Abstract Geometrical parameters, aromaticity, and con-
formational flexibility of the set of polysubstituted ben-
zenes with different number and position of nitro and
amino groups were calculated at the MP2/cc-pvdz level of
theory. The key factor for structural and energetic changes
has been identified. This is related to the presence of nitro
and amino groups in vicinal positions that forms strong
intramolecular resonance-assisted hydrogen bonds with a
binding energy of 7–14 kcal/mol. Increasing number of
such bonds facilitates a cooperative effect, inducing nota-
ble changes in molecular geometry (particularly increasing
bond alternation within H2N–C–C–NO2 fragment and
planarization of amino group), drastic increasing of con-
formational flexibility and decreasing of aromaticity. In
spite of well-known p-electron effects of nitro and amino
substituents, influence of their push–pull interaction
through aromatic moiety is negligible compared to the
effect of the hydrogen bonding. That results in great dif-
ference of the ortho-isomers as compared to meta- and
para-isomers.
Keywords Aromaticity � Push–pull effect �Intramolecular hydrogen bond � Quantum chemical
calculations
Introduction
Aromaticity is one of the most general and important
concepts in organic chemistry [1]. This phenomenon also
provides the background to understanding the influence of
substituents on reactivity and stability of the very wide
range of organic species containing aromatic and hetero-
aromatic moieties. The numerous examples of such influ-
ence are available in the literature elsewhere [2–4]. Among
them, famous Hammett’s constants describing electronic
strength of substituents were derived for aromatic mole-
cules. Using this approach, it is very easy to understand
that mechanism of such influence also including changes in
aromaticity of benzene ring.
Influenced by the Hammett equation, there have been
numerous attempts to describe substituent effect on aro-
matic moiety quantitatively [5]. The common way is the
usage of some characteristic physicochemical properties
(such as amino group basicity, catalytic activity, change of
infrared, or NMR properties of substance) as an aromaticity
measure [6]. Such characteristics are usually called aro-
maticity indexes. Recently, aromaticity indexes were used
to estimate substituent effect [7, 8]. Analysis of mono-
substituted benzenes revealed that influence of a single
Electronic supplementary material The online version of thisarticle (doi:10.1007/s11224-012-9971-8) contains supplementarymaterial, which is available to authorized users.
I. V. Omelchenko (&) � O. V. Shishkin
STC’Institute for Single Crystals, National Academy of Sciences
of Ukraine, 60 Lenina Ave, Kharkiv 61001, Ukraine
e-mail: [email protected]
O. V. Shishkin
V.N. Karazin Kharkiv National University, 4 Svobody sq,
Kharkiv 61077, Ukraine
L. Gorb
Badger Technical Services, LLC, Vicksburg, MS, USA
F. C. Hill � J. Leszczynski
US Army ERDC, 3532 Manor Dr, Vicksburg, MS 39180, USA
J. Leszczynski
Interdisciplinary Center for Nanotoxicity, Department of
Chemistry and Biochemistry, Jackson State University,
P.O. Box 17910, 1325 Lynch Street, Jackson, MS 39217, USA
123
Struct Chem (2012) 23:1585–1597
DOI 10.1007/s11224-012-9971-8

group could be essential but aromaticity of central moiety
remains relatively high.
Much less attention has been paid to cooperative effect
of several substituents. Historically, such systems were
studied in view of transmission of some electronic effects
from one group to another through aromatic moiety
[6, 9]. This consideration was given mainly to disubsti-
tuted derivatives of benzene. It is usually discussed in
terms of push–pull interactions [10]. Substituents which
have an ability to donate (‘‘push’’) or withdraw (‘‘pull’’)
electrons from the p-system of aromatic ring are con-
sidered to interact through that ring in accordance with
internal resonance polarization of p-system, changing the
properties of aromatic ring. Unlike r-polarization, the
p-polarization depends strongly on the relative positions
of the substituents [6]. Appearance of substituents in
benzene ring always results in a decrease of aromaticity
[6]. Study of homodisubstituted benzenes also revealed
that electron-donating groups affects aromaticity much
stronger than electron-withdrawing [11] ones, in agree-
ment with the data for monosubstituted benzenes [8]. For
the molecules with different types of substituents, aro-
maticity as well as molecular geometry and some other
properties usually depends on their relative positions
[6, 12–16]. However, some studies show a negligible
distinction between position of isomers or even contro-
versial estimations of their influences by different aro-
maticity indices or molecular properties [9, 11, 17–20].
There is still lack of systematic study of heterosubstituted
systems with pronounced p-electronic effects by versatile
methods.
In the case of the presence of more than two substituents
situation becomes more complex, especially if they have
different or even opposite character of electronic effects.
Several ways for interaction between substituents are
possible including both electronic interactions through
aromatic p-system as well as non-bonding interactions
between vicinal groups.
Suitable models for investigation of cooperative effects
in the species possessing strong mesomeric effects of
opposite signs are provided by aminonitrobenzene deriva-
tives having different number and position of amino and
nitro groups. For such moieties, one could expect strong
interactions between substituents causing significant
changes of properties of aromatic system of benzene ring.
In addition some aminonitrobenzenes (e.g., triaminotrini-
trobenzene and its analogs) are objects of great practical
interest in fields of energetic materials [21, 22] and
molecular biochemistry [23, 24]. Nitroanilines and their
derivatives are of interest for photoelectronic spectroscopy
as model charge-transfer systems [25–27]. Also, they are
widely discussed in frameworks of different theoretical
methods—quantum chemical calculations as well as
molecular dynamic modeling [28–30]. Substituent effects
of nitro and amino groups on the specific molecular and
crystal properties, i.e., impact sensibility, decomposition
mechanism and its energetic barrier, biological activity,
photoelectronic properties, etc., are widely discussed in
such studies. However, there are much less studies focused
on aromaticity and cooperative substituent effects of amino
and nitro groups [11–13].
In this article, we study the dependence of structural
parameters of molecular system, aromaticity of benzene
ring, and its rigidity on the nature, amount, and the position
of substituents. The aminonitrobenzene derivatives having
different number and position of amino and nitro groups
are selected as the model systems to investigate coopera-
tive influence of substituents.
R4
R3
R2
R1
R6
R5
1
2
34
5
6
Methods of calculation
The structures of all examined molecules (Table 1) have
been optimized using the Møller–Plesset second-order
perturbation theory [31] with correlation-consistent double-
zeta basis set [32] (MP2/cc-pVDZ method). Normal
vibrational modes were founded by calculations of Hes-
sian, at the same level of theory. No negative eigenvalues
of Hessian were found for the studied species.
Aromaticity of molecules under study was described by
aromaticity indices [33]. All indices have restrictions in use
[34]; therefore, it is preferred to use several different
indices [35–38]. In this study, structural Bird (Ia) [39] and
HOMA [40] indices, and magnetic index NICS(1)zz [41]
were used. As for commonly used energetic indices, there
is no unambiguous scheme of calculation of aromatic sta-
bilization energies (ASE) for polysubstituted molecules
[34] using the most reliable homodesmotic reaction
scheme.
It was already demonstrated [8, 42–44] that aromaticity
of cyclic conjugated system is closely related with its
conformational flexibility. Energy of ring out-of-plane
deformation and other characteristics of ring flexibility
correlate well with aromaticity degree. Therefore, they also
may be used as aromaticity indices.
1586 Struct Chem (2012) 23:1585–1597
123

All aromaticity indices (Ia, NICS(1)zz, HOMA) were
calculated using MP2/cc-pVDZ level of theory. Gordy
bond order calculation scheme [45] with empirical con-
stants was applied to calculation of Ia index according to
the standard equation [33]. Nucleus-independent chemical
shifts were calculated in the point located by 1 A above the
center of the ring (NICS(1)zz) as it was recommended [46]
for obtaining more accurate data.
Conformational flexibility of rings was studied on the
same level of theory by scan of each of symmetry-inde-
pendent endocyclic torsion angles in the range ±30� with
5� steps. All remaining geometrical parameters were opti-
mized at every step of scan. For each molecule, the angle
with the smallest energy of deformation E(def) (difference
in energy between equilibrium and twisted on 30� geom-
etries) was chosen.
All calculations have been performed using the
Gaussian03 [47] and GAMESS US [48] program packages.
Electron density properties were studied using Bader’s
‘‘Atoms in Molecules’’ theory (AIM) with AIM2000 pro-
gram [49] Energy of intramolecular hydrogen bonds was
estimated following the Espinosa equation [50] based on
value of local potential energy in bond critical point for
interaction.
Results and discussion
Molecular structure
Homosubstituted molecules
Analysis of the structural parameters of homosubstituted
molecules reveals good agreement of the current data with
the previous results [8, 11, 15]. Presence of nitro substit-
uents leads to small elongation of the C–C bond lengths
through the whole ring (1.398–1.406 vs 1.394 A in ben-
zene) and to increasing of ipso bond angle (120.3–123.2).
Only the angle at the ‘‘central’’ NO2 group in the 1,2,3-
trinitrobenzene (123N) slightly decreases (118.0). As for
aminobenzenes, the geometric changes are more pro-
nounced but local elongation of bonds is limited to the
Ci–Co bonds (1.407–1.417 A). Ipso bond angles slightly
decrease (117.8�–119.8�). Thus, the homosubstitution
affects molecular structure of the benzene ring according to
the r-electronic effect model. Experimental data for the gas
phase reveal the same trend (see Tables 2, 3 and references
below), except the nitrobenzene case with notable alter-
nation of the bond length (1.375 A Ci–Co bonds, 1.403 A
Co–Cm, and 1.396 A Cm–Cp).
In the nitrobenzene, as well as in 1,3- and 1,4-dinitro-
benzenes, the nitro group tends to be coplanar with aromatic
ring through conjugation of their p-systems. 1,2-dinitro-
benzene and polynitrobenzenes with adjacent nitro groups
undergo notable steric repulsion that can be roughly esti-
mated by examination of torsion angle between planes of
base ring and substituent (Table 2). The maximum rotation
angle corresponds to ‘‘central’’ nitro group in 1,2,3-trini-
trobenzene that undergo the maximum repulsion. Rotation
of these nitro groups causes no evident structural effect on
the aromatic moiety (see Table S1 in Supplementary mate-
rial). However, one can note decreasing of the C–NO2 bond
length for adjacent nitro groups. In addition, big values of
rotation angles result in reducing the p-conjugation with
aromatic system. At the time, there is very weak but quite
clear trend for elongation of the C–N bond for substituents
located in meta-position, with respect to para-position.
As for aminobenzenes, C–NH2 bond length and pyramidality
of NH2 group increases in the ortho- and para-diaminobenzenes
in comparison with aniline and meta-diaminobenzene, and
in the ortho,para-triaminobenzene (124A) in comparison
Table 1 Molecules under consideration
Molecule R1 R2 R3 R4 R5 R6
Benzene H H H H H H
1N NO2 H H H H H
12N NO2 NO2 H H H H
13N NO2 H NO2 H H H
14N NO2 H H NO2 H H
123N NO2 NO2 NO2 H H H
135N NO2 H NO2 H NO2 H
1A NH2 H H H H H
1N2A NO2 NH2 H H H H
1N3A NO2 H NH2 H H H
1N4A NO2 H H NH2 H H
13N2A NO2 NH2 NO2 H H H
13N4A NO2 H NO2 NH2 H H
13N5A NO2 H NO2 H NH2 H
135N2A NO2 NH2 NO2 H NO2 H
12A NH2 NH2 H H H H
13A NH2 H NH2 H H H
14A NH2 H H NH2 H H
1N24A NO2 NH2 H NH2 H H
1N35A NO2 H NH2 H NH2 H
13N24A NO2 NH2 NO2 NH2 H H
13N25A NO2 NH2 NO2 H NH2 H
123N46A NO2 NO2 NO2 NH2 H NH2
135N24A NO2 NH2 NO2 NH2 NO2 H
124A NH2 NH2 H NH2 H H
135A NH2 H NH2 H NH2 H
1N246A NO2 NH2 H NH2 H NH2
13N246A NO2 NH2 NO2 NH2 H NH2
135N246A NO2 NH2 NO2 NH2 NO2 NH2
Struct Chem (2012) 23:1585–1597 1587
123

with meta,meta-isomer (135A) (Table 3). Adding extra
amino groups results in elongation of C–NH2 bond lengths in
all isomers as compared to aniline. Presence of amino groups
increases vicinal endocyclic C–C bond length up to
1.410–1.419 A (1.394 A for benzene) regardless of the rel-
ative substituents position. This effect is larger than analo-
gous increase predicted for any nitrobenzene derivatives
(1.398–1.406 A, Table S1 in Supplementary material).
Therefore, one may conclude that structure of the ben-
zene moiety is quite resistant to the perturbations caused by
the presence of several nitro groups and, in some less
extent, one or two amino groups (see Table S1 in Sup-
plementary material).
Isomeric nitroanilines
Molecules with both p-donor and p-acceptor substituents
are considered to reveal strong push–pull effect [10] that is
transmitted through aromatic system. It should change
molecular structure of ortho- and para-isomers according
to the quinoid canonical resonance structures with partial
charge transfer through p-system. Scheme 1 represents a
set of canonical structures for para-nitroaminobenzene.
Therefore, ortho- and para-nitroanilines 1N2A and 1N4A
are expected to reveal alternation in the benzene ring,
shortening of the C–NH2 and the C–NO2 bond lengths,
and flattening of the amino group, in contrast to meta-
nitroaniline 1N3A which does not share a push–pull effect
[10].
However, the results of calculation do not reveal sig-
nificant deformation of bond lengths within benzene ring in
para-isomer 1N4A as compared to meta-isomer 1N3A.
Slight changes in bond lengths may be explained by
superposition of the effects of nitro and amino groups
mentioned above (see Tables S1, S2 in Supplementary
material). There are only some shortening of the C–NH2
bond and flattening of amino group as compared to aniline.
These effects reflect conjugation interactions between
substituents (Table 4).
Table 2 Values of the C–N bond lengths (A) and the C–C–N–O
torsion angles (�) in nitrobenzenes
Molecule C–NO2 C–C–N–O
1N 1.483 0.0
Exp.a 1.492(1) 0.0(1)
12N 1.472 42.5
13N 1.487 0.0
14N 1.482 0.0
123N 1.477 (C1) 35.8
1.473 (C2) 62.4
135N 1.489 0.0
Identical values for symmetrically equivalent groups are omitteda Gas-phase electron diffraction and microwave spectroscopy
experiment, rs distances [51]
Table 3 Values of the C–N bond lengths (A), pyramidality of amino
group estimated as sum of bond angles centered at the nitrogen atom
(P
(NH2), �), and angle between planes of amino group and benzene
ring (u(NH2), �) in amino benzenes
C–NH2
P(NH2) u(NH)
1A 1.410 332.2 44.4
Exp.a 1.406(3) 44(4)
Exp.b 1.402(2) 37(2)
12A 1.416 328.7 54.6
13A 1.413 331.2 50.0
14A 1.417 329.5 51.3
Exp.c 1.422(2) 43(4)
124A 1.423 (C1) 326.8 57.0
1.415 (C2) 329 53.7
1.418 (C3) 329.2 51.8
135A 1.414 (C1, C5) 330.5 50.6
1.414 (C3) 330.9 50.3
Identical values for symmetrically equivalent groups are omitteda Gas-phase electron diffraction experiment, rg distances [52]b Gas-phase microwave and electronic spectroscopy experiment [53]c Gas-phase electron diffraction experiment, rg distances [54]
NH H
N+
OO
NH H
N+
OO
NH H
N+
OO ON
+O
NH H
ON
+O
NH H
ON
+O
NH H
ON
+O
NH H
+ +
+
+ +
-
- - +
+
Scheme 1 Canonical resonance structures for para-nitroaminobenzene (1N4A)
1588 Struct Chem (2012) 23:1585–1597
123

In ortho-isomer, the C–NH2 bond length is notably
shorter, and amino group is more flattened than in meta-
and para-isomers (Table 4). Nitro group in 1N2A is not
coplanar to benzene ring (Table 4) and the C–NO2 bond is
slightly shorter than in 1N3A and 1N4A. That is accom-
panied by alternation of endocyclic bond lengths (short-
ening of C(3)–C(4) and C(5)–C(6) bonds) and by
decreasing of the CC(NH2)C angle. Although such struc-
tural changes are typical for conjugative interaction as
well, no similar changes are observed in the para-nitro-
aniline structure. Thus, para- and meta-nitroanilines pos-
sess very close geometrical parameters that differ from
ortho-nitroaniline. This set of data does not match the
values expected due to push–pull effect but may be caused
by direct local interaction between vicinal substituents.
These results do not match experimental results obtained
from X-ray diffraction study of the crystalline state [56–59]
in which noticeable structural changes that correspond to
the push–pull effect are clearly observed in both ortho-
[56] and para- [57, 58] nitroanilines as compared to meta-
nitroaniline [59]. However, effect of the polar medium and
strong intermolecular hydrogen bonding that are observed
in crystals of all isomeric nitroanilines can affect properties
related to the p-electron system dramatically [60, 61].
These effects are a subject of particular study and therefore
crystal structure data as well as data obtained in polar
solvents cannot be a reference for molecules examined in
vacuum. Available gas-phase data exist only for para-
nitroaniline. Experimental C–NH2 and C–NO2 bond
lengths are some smaller than calculated (Table 4) but
precision of their determination is rather low, and only
averaged endocyclic bond length was given (rg(C–C) =
1.402(4) A). Thus, push–pull effect cannot be revealed
from these data.
Analysis of the electron density distribution using
Bader’s AIM theory demonstrated existence of the
N–H���O hydrogen bond between substituents bonding
(H���O 1.957 A, N–H���O 125.6�). Taking into account that
proton donating and withdrawing sites of this bond are
connected by benzene ring as resonant spout it is possible
to consider it [62–66] as resonance-assisted hydrogen bond
(RAHB) (Scheme 2). Considerable contribution of the
resonance structure C in the total structure of the 1N2A
molecule should cause shortening of the C–N bonds, flat-
tening of amino group mentioned above, and elongation of
the C1–C2 endocyclic bond which is 1.420 A. Taking into
account pyramidal configuration of amino group rotation of
the nitro group creates more favorable conditions for for-
mation of the N–H���O hydrogen bond. The N–O bond of the
nitro group of 1N2A that participates in RAHB is slightly
longer (1.240 A) than the other N–O bond (1.231 A). In all
other nitroanilines and homosubstituted molecules under
study, length of the N–O bonds of nitro groups are the same.
Therefore, some deformation of geometry of 1N2A may be
explained by the formation of intramolecular RAHB rather
than push–pull p-electronic effects.
Polyheterosubstituted molecules
Geometrical parameters of polynitroaminobenzenes vary in
a much more wide range than parameters of species dis-
cussed above. The specific structural feature of highly
substituted molecules with adjacent nitro and amino sub-
stituents is reflected by a minor non-planarity of benzene
rings in equilibrium geometry (endocyclic torsion angles
vary up to 6�, see Table 5). In the previous theoretical
studies of 2,4,6-triamino-1,3,5-trinitrobenzene (135N246A,
TATB), it was found that degree of non-planarity of mol-
ecule strongly depends on the level of theory and quality
of basis applied to chemical modeling [15, 29, 67–69].
High-quality calculations reveal non-planarity of TATB
Table 4 Values of the C–N bond lengths (A), pyramidality of amino
group estimated as sum of bond angles centered at the nitrogen atom
(P
(NH2), �) and the C–C–N–O torsion angles (�) in nitroanilines,
nitrobenzene, and aniline
Molecule C–NH2
P(NH2) C–NO2 C–C–N–O
1N – – 1.483 0.0
1A 1.410 332.2 – –
1N2A 1.387 341.3 1.473 16.1
1N3A 1.400 335.8 1.475 0.9
1N4A 1.400 336.2 1.481 0.3
Exp.a 1.36(3) (360) 1.47(1) (0)
a Gas-phase electron diffraction experiment, rg distances [55]. Values
in parenthesis were fixed during experiment analysis so they are non-
informative
N+
NH
OOH N
+
N+
O OH
H
N+
N
H
H
OO
A B C
Scheme 2 Resonance
structures proposed for
hydrogen bonding of ortho-
nitroaniline
Struct Chem (2012) 23:1585–1597 1589
123
molecule in equilibrium geometry that arises from an
extremely low energy of out-of-plane deformation of
molecule [12]. Experimental values of torsion angles [70]
measured by XRD method cannot be hold as reference due
to the strong influence of polar medium and intramolecular
interactions in crystal.
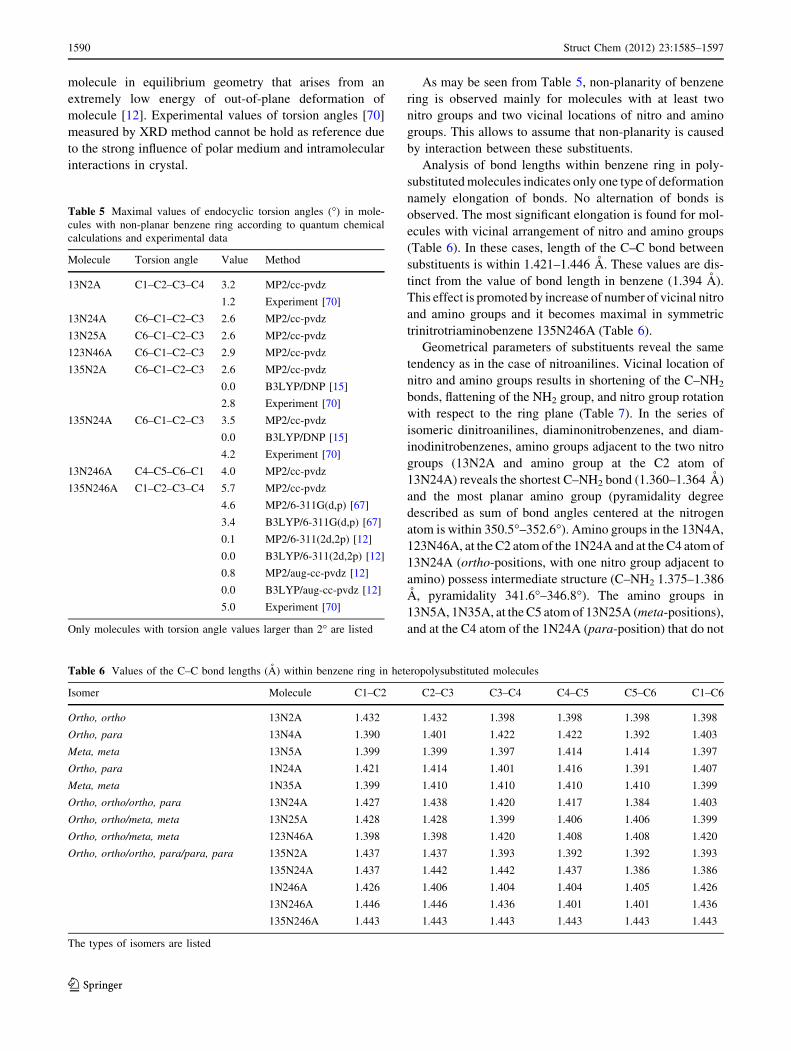
As may be seen from Table 5, non-planarity of benzene
ring is observed mainly for molecules with at least two
nitro groups and two vicinal locations of nitro and amino
groups. This allows to assume that non-planarity is caused
by interaction between these substituents.
Analysis of bond lengths within benzene ring in poly-
substituted molecules indicates only one type of deformation
namely elongation of bonds. No alternation of bonds is
observed. The most significant elongation is found for mol-
ecules with vicinal arrangement of nitro and amino groups
(Table 6). In these cases, length of the C–C bond between
substituents is within 1.421–1.446 A. These values are dis-
tinct from the value of bond length in benzene (1.394 A).
This effect is promoted by increase of number of vicinal nitro
and amino groups and it becomes maximal in symmetric
trinitrotriaminobenzene 135N246A (Table 6).
Geometrical parameters of substituents reveal the same
tendency as in the case of nitroanilines. Vicinal location of
nitro and amino groups results in shortening of the C–NH2
bonds, flattening of the NH2 group, and nitro group rotation
with respect to the ring plane (Table 7). In the series of
isomeric dinitroanilines, diaminonitrobenzenes, and diam-
inodinitrobenzenes, amino groups adjacent to the two nitro
groups (13N2A and amino group at the C2 atom of
13N24A) reveals the shortest C–NH2 bond (1.360–1.364 A)
and the most planar amino group (pyramidality degree
described as sum of bond angles centered at the nitrogen
atom is within 350.5�–352.6�). Amino groups in the 13N4A,
123N46A, at the C2 atom of the 1N24A and at the C4 atom of
13N24A (ortho-positions, with one nitro group adjacent to
amino) possess intermediate structure (C–NH2 1.375–1.386
A, pyramidality 341.6�–346.8�). The amino groups in
13N5A, 1N35A, at the C5 atom of 13N25A (meta-positions),
and at the C4 atom of the 1N24A (para-position) that do not
Table 5 Maximal values of endocyclic torsion angles (�) in mole-
cules with non-planar benzene ring according to quantum chemical
calculations and experimental data
Molecule Torsion angle Value Method
13N2A C1–C2–C3–C4 3.2 MP2/cc-pvdz
1.2 Experiment [70]
13N24A C6–C1–C2–C3 2.6 MP2/cc-pvdz
13N25A C6–C1–C2–C3 2.6 MP2/cc-pvdz
123N46A C6–C1–C2–C3 2.9 MP2/cc-pvdz
135N2A C6–C1–C2–C3 2.6 MP2/cc-pvdz
0.0 B3LYP/DNP [15]
2.8 Experiment [70]
135N24A C6–C1–C2–C3 3.5 MP2/cc-pvdz
0.0 B3LYP/DNP [15]
4.2 Experiment [70]
13N246A C4–C5–C6–C1 4.0 MP2/cc-pvdz
135N246A C1–C2–C3–C4 5.7 MP2/cc-pvdz
4.6 MP2/6-311G(d,p) [67]
3.4 B3LYP/6-311G(d,p) [67]
0.1 MP2/6-311(2d,2p) [12]
0.0 B3LYP/6-311(2d,2p) [12]
0.8 MP2/aug-cc-pvdz [12]
0.0 B3LYP/aug-cc-pvdz [12]
5.0 Experiment [70]
Only molecules with torsion angle values larger than 2� are listed
Table 6 Values of the C–C bond lengths (A) within benzene ring in heteropolysubstituted molecules
Isomer Molecule C1–C2 C2–C3 C3–C4 C4–C5 C5–C6 C1–C6
Ortho, ortho 13N2A 1.432 1.432 1.398 1.398 1.398 1.398
Ortho, para 13N4A 1.390 1.401 1.422 1.422 1.392 1.403
Meta, meta 13N5A 1.399 1.399 1.397 1.414 1.414 1.397
Ortho, para 1N24A 1.421 1.414 1.401 1.416 1.391 1.407
Meta, meta 1N35A 1.399 1.410 1.410 1.410 1.410 1.399
Ortho, ortho/ortho, para 13N24A 1.427 1.438 1.420 1.417 1.384 1.403
Ortho, ortho/meta, meta 13N25A 1.428 1.428 1.399 1.406 1.406 1.399
Ortho, ortho/meta, meta 123N46A 1.398 1.398 1.420 1.408 1.408 1.420
Ortho, ortho/ortho, para/para, para 135N2A 1.437 1.437 1.393 1.392 1.392 1.393
135N24A 1.437 1.442 1.442 1.437 1.386 1.386
1N246A 1.426 1.406 1.404 1.404 1.405 1.426
13N246A 1.446 1.446 1.436 1.401 1.401 1.436
135N246A 1.443 1.443 1.443 1.443 1.443 1.443
The types of isomers are listed
1590 Struct Chem (2012) 23:1585–1597
123

have adjacent nitro group reveals the longest C–NH2 bonds
(1.390–1.404 A) and the highest pyramidality (334.3�–
339.8�). The C–NO2 bond lengths depend on the presence
of adjacent amino group generally in the same way (the
shortest bond of 1.463 A is observed in the 13N24A, while
the longest bond of 1.485 A–in the 1N35A). Nitro group of
13N24A that has two vicinal amino groups undergo maxi-
mum rotation (29.6� deviation from the ring plane).
Molecular structures of trinitro- and triaminosubstituted
species (excluding 123N46A) agree with the above-men-
tioned trends. In general, the C–NH2 and the C–NO2 bonds
are shorter and amino group becomes more planar with the
increasing number of adjacent substituents. For example, the
C–NH2 bond length decreases from 1.364 A in 13N2A to
1.343 A in 13N246A, but it is much longer in the molecules
that do not contain adjacent nitro and amino groups (1.390 A
in 13N5A, 1.404 A in 1N35A). Correspondingly, the C–NO2
bond length decreases from 1.478 to 1.444 A (it is equal
1.485 A in 13N5A and 1.479 A in 1N35A), and amino group
becomes planar (pyramidality degree is 350.5� in 13N2A and
360.0� in 13N246A, and is much smaller in both meta,meta-
isomers—339.8� in 13N5A and 334.3� in 1N35A). The
remarkable exception is the 2,4,6-triaminonitrobenzene
(1N246A). The C–NH2 bonds 1.392–1.402 A and pyrami-
dality degree of amino groups 335.3�–338.4� are much clo-
ser to the values for mononitroanilines than for other
polysubstituted molecules. The molecular parameters of this
compound do not match the general trend in changing of
geometrical parameters with increasing number of amino
group. At the time, changes in the C–C bonds in 1N246A are
very close to those in 135N2A. Considerations of electron
density in the 1N246A molecule reveal that donating of the
extra electrons by amino groups into p-orbitals of the ben-
zene ring is energetically unfavorable for aromatic system.
Interestingly, this effect is compensated by electron-with-
drawing effect of the second and third nitro groups in all
other molecules.
As well as in mononitroanilines, direct interactions
between vicinal nitro and amino substituents play the
leading role in the all structural changes mentioned above.
Table 7 Values of the C–N bond lengths (A), pyramidality of amino groups estimated as sum of bond angles centered at the nitrogen atom
(P
(NH2), �) and the mean values of the two C–C–N–O torsion angles (�) for nitro groups in polynitroaminobenzenes
Isomer Molecule C–NH2
P(NH2) C–NO2 C–C–N–O
Ortho, ortho 13N2A 1.364 350.5 1.478 18.1
Ortho, para 13N4A 1.375 346.8 1.476 (C1) 0.4
1.476 (C3) 13.0
Meta, meta 13N5A 1.390 339.8 1.485 1.0
Ortho, para 1N24A 1.386 (C2) 341.6 (C2) 1.465 9.8
1.401 (C4) 335.6 (C4)
Meta, meta 1N35A 1.404 334.3 1.479 1.5
Ortho, ortho/ortho, para 13N24A 1.360 (C2) 352.6 (C2) 1.466 (C1) 1.5 (C1)
1.376 (C4) 345.3 (C4) 1.463 (C3) 29.6(C3)
Ortho, ortho/meta, meta 13N25A 1.377 (C2) 344.2 (C2) 1.478 (C1) 19.6 (C1)
1.400 (C5) 335.1 (C5) 1.478 (C3) 19.3 (C3)
Ortho, ortho/meta, meta 123N46A 1.381 342.1 1.465 (C1) 39.6 (C1)
1.475 (C2) 59.2 (C2)
Ortho, ortho/ortho, para/para, para 135N2A 1.350 358.0 1.482 (C1) 10.8 (C1)
1.482 (C3) 10.4 (C3)
1.477 (C5) 0.1 (C5)
135N24A 1.343 (C2) 359.6 (C2) 1.468 (C1) 2.0 (C1)
1.343 (C4) 359.8 (C4) 1.458 (C3) 16.4 (C3)
1.468 (C5) 3.8 (C5)
1N246A 1.392 (C2) 338.9 (C2) 1.454 27.9
1.402 (C4) 335.3 (C4)
1.392 (C6) 338.4 (C6)
13N246A 1.343 (C2) 360.0 (C2) 1.444 (C1) 7.0 (C1)
1.364 (C4) 352.7 (C4) 1.444 (C3) 6.5 (C3)
1.364 (C6) 352.5 (C6)
135N246A 1.336 360.0 1.445 11.2
Values are listed consequently from position 1 to position 6 of substituent. Identical values for symmetry equivalent groups are skipped
Struct Chem (2012) 23:1585–1597 1591
123

Repulsion of nitro groups affects them only slightly so
geometrical parameters of 123N46A with nitro group
rotated at almost 60� are close to the 13N25A (both
ortho,meta-isomers), but not to 135N24A (ortho,ortho-
isomer). Some minor changes can be referred to the push–
pull interaction with amino group in the para-position as
well. In the 135N2A, the C(5)–NO2 bond that does not
have adjacent substituents but has an amino group in the
para-position, is shorter than C(1)–NO2 and C(3)–NO2 by
0.005 A. In 13N4A, both C–NO2 bonds having amino
group in ortho- and para-positions are of the equal length.
No such effect was observed in the nitroanilines. Thus, in
polysubstituted molecules, C–NO2 bond length is little
more sensitive to the p-conjugation with amino group in
para-position. One may suppose that perturbative effect of
the intramolecular RAHB makes the aromatic system more
responsive to the push–pull effect. However, that does not
affect the endocyclic bond lengths of the benzene rings in
the corresponding molecules (see Table 6).
Thus, based on these data, it is possible to conclude that
effect of the p-electron interactions between substituents
through aromatic ring on the geometry of polysubstituted
nitroaminobenzenes is negligible as compared to direct
non-bonded interactions between vicinal substituents. The
character of changes in bond lengths indicates that this is
due to formation of resonance-assisted N–H���O hydrogen
bonds rather than to conjugation between substituents.
Lengths of the N–H���O–N hydrogen bonds vary from
1.957 A in 2-aminonitrobenzene 1N2A to 1.703 A in
135N246A (Table 8). Analysis of these molecules using
Bader’s AIM theory revealed existence of corresponding
(3; -1) bond critical points (BCP) for all N–H���O–N
contacts. Energy estimated from BCP properties indicates
rather strong bonding (Table 8).
The N–H���O bond strength grows noticeable with the
increase of the number of adjacent nitro and amino substit-
uents. For example, bond energy is about 8–9 kcal/mol in all
molecules with one to three corresponding bonds,
10–13 kcal/mol for molecules with four contacts (and
13N246A), and it is more than 14 kcal/mol in 135N246A
with six contacts. Therefore, such hydrogen bonding reveals
a cooperative effect [66, 71, 72] on the aromatic moiety.
Forming of the first bond disturbs the p-system and such
transformation facilitates its further alterations by other
RAHB. In addition, this makes benzene ring moiety more
sensible not only to the p-system disturbance but also to
r-electron perturbation, due to the polarization of the
r-skeleton by charges with opposite signs (Schemes 2, 3).
Extra elongation of the C–C bonds in 135N24A, 13N246A,
and 135N246A (see Table 8) provides the immediate
example.
Interestingly, AIM analysis revealed also the formation
of some C–H���O hydrogen bonds in 135N24A and
13N24A (Table 8). These bonds are significantly weaker
than the N–H���O–N bonds. However, formation of the
C–H���O bonds is observed only in molecules with at least
three N–H���O bonds.
In the extreme case of 135N246A, total energy of
hydrogen bonding is very high (about 87 kcal/mol) leading
to significant violation of structure of aromatic ring which
corresponds rather to derivative of cyclohexane with six
exocyclic double bonds than to hexasubstituted benzene
(Scheme 3). Probably, this significantly influences the
mechanism of decomposition of 135N246A. [73–78].
Conformational flexibility of benzene ring
Earlier it was demonstrated [42] that benzene and other six-
membered aromatic rings possess a notable conformational
flexibility. This property strongly depends on aromaticity
degree of cyclic conjugated system and presence of sub-
stituents. In particular, the presence of both p-electron-
donating and withdrawal groups in monosubstituted
derivatives of benzene results in increase of conformational
flexibility of ring due to decrease of aromaticity degree of
cyclic conjugated system [8].
In agreement with previous findings [8], energies of
benzene ring out-of-plane deformation in nitrobenzene 1N
and aniline 1A are smaller as compared to unsubstituted
benzene. Presence of several nitro groups results in a negli-
gible increase of flexibility, but existence of one or several
amino groups leads to more pronounced effect (Table 9).
Both these effects are non-additive: ring out-of-plane
deformation energy E(def) decreases by 0.67–0.98 kcal/mol
for nitro substituents and by 1.08–1.93 kcal/mol for amino
substituents and it does not depend on the number of
substituents.
Changes of the E(def) of the heterosubstituted molecules
clearly follow the revealed trends in structural changes.
The main factor that diminishes deformation energies is the
presence of vicinal nitro and amino groups that form
hydrogen bonds accompanied by perturbation of aromatic
system. Conformational flexibility of benzene ring in iso-
mers containing the N–H���O hydrogen bonds (for example,
1N2A, 1N24A) is always notably higher than for other
isomers (for example, 1N3A, 1N4A, 1N35A). Increase of
number of the N–H���O hydrogen bonds results in signifi-
cant decrease of E(def) which becomes minimal for
135N246A (less than 1 kcal/mol, Table 9). Thus, presence
of nitro and amino groups makes benzene ring very flexible
which does not match its aromatic character. This is
another evidence of significant violation of aromatic sys-
tem especially in trinitrotriaminobenzene 135N246A.
It should be noted that the value of ring out-of-plane
deformation energy is more sensitive to the electronic push–
pull effects than the molecular geometry of benzene ring.
1592 Struct Chem (2012) 23:1585–1597
123

The values of E(def) for 1N3A and 1N4A are slightly lower
than those for nitrobenzene and aniline (Table 9), in agree-
ment with polarization of p-system by push–pull effects.
It is known [42] that, for overwhelming majority of
simple aromatic molecules, including heterocycles, the
energy of ring out-of-plane deformation reveal strict har-
monic dependence on endocyclic torsion angle, at least in
the range from -30� to 30�. However, the results of cal-
culations demonstrate some deviations from that rule in
some of the highly substituted nitroaminobenzenes
Table 8 Geometrical param-
eters (H���O, A, D–H���O, �, O–N,
A) and AIM derived energy
(EAIM, kcal/mol) of
intramolecular hydrogen bonds
in nitroamino derivatives of
benzene
Molecule D–H���O–N H���O D–H���O O–N EAIM
1N2A N(10)–H���O(9)–N(7) 1.957 125.6 1.240 7.73
1N24A N(10)–H���O(9)–N(7) 1.934 126.8 1.242 8.09
1N246A N(10)–H���O(9)–N(7) 1.938 125.8 1.240 8.01
13N2A N(10)–H���O(9)–N(7) 1.893 129.3 1.240 8.69
N(10)–H���O(12)–N(11) 1.893 129.3 1.240 8.69
13N4A N(13)–H���O(12)–N(10) 1.943 125.6 1.239 7.91
13N246A N(10)–H���O(9)–N(7) 1.734 133.4 1.247 13.20
N(10)–H���O(12)–N(11) 1.733 133.5 1.247 13.22
N(14)–H���O(13)–N(11) 1.816 127.2 1.245 10.79
N(15)–H���O(8)–N(7) 1.817 127.2 1.245 10.76
135N2A N(10)–H���O(9)–N(7) 1.866 129.3 1.240 9.21
N(10)–H���O(12)–N(11) 1.863 129.4 1.240 9.26
135N24A N(10)–H���O(9)–N(7) 1.804 133.1 1.244 10.61
N(10)–H���O(12)–N(11) 1.797 128.8 1.242 11.10
N(14)–H���O(13)–N(11) 1.798 128.6 1.242 11.07
N(14)–H���O(16)–N(15) 1.805 133.0 1.244 10.59
C(6)–H���O(17)–N(15) 2.281 97.6 1.230 5.54
C(6)–H���O(8)–N(7) 2.280 97.6 1.230 5.53
135N246A N(10)–H���O(9)–N(7) 1.703 133.8 1.248 14.50
N(10)–H���O(12)–N(11) 1.705 133.6 1.248 14.38
N(14)–H���O(13)–N(11) 1.703 133.8 1.248 14.48
N(14)–H���O(16)–N(15) 1.704 133.6 1.248 14.40
N(18)–H���O(17)–N(15) 1.702 133.8 1.248 14.50
N(18)–H���O(8)–N(7) 1.704 133.6 1.248 14.40
13N24A N(10)–H���O(9)–N(7) 1.835 131.9 1.243 9.94
N(10)–H���O(12)–N(11) 1.888 128.0 1.239 8.77
N(14)–H���O(13)–N(11) 1.925 125.1 1.239 8.18
C(6)–H���O(8)–N(7) 2.281 98.3 1.232 5.36
13N25A N(10)–H���O(9)–N(7) 1.917 128.9 1.239 8.28
N(10)–H���O(12)–N(11) 1.916 128.9 1.239 8.28
123N46A N(16)–H���O(15)–N(13) 2.018 125.3 1.238 6.50
N(17)–H���O(8)–N(7) 2.018 125.3 1.238 6.50
N+
N+
O OH
H
Ν Η2
ΝΗ2
ΝΟ2Ο2Ν
N+
H
H
Ν Η2
ΝΗ2
ΝΟ2Ν+Ο
Ο
ΝΟ2
N+
H
H
Ν+
Ν+
Ν+
Ν+Ο
Ο
Ν+
Ο Ο
Η Η
Η
Η
Ο
Ο
Scheme 3 Some of resonance structures of TATB with single and multiple charge transfer that are stabilized by RAHB
Struct Chem (2012) 23:1585–1597 1593
123

(Table 9). Changes of endocyclic torsion angle in opposite
directions leads to different values of E(def). Asymmetrical
character of the PES in highly aromatic aniline and
polyaminobenzenes is caused by non-planar geometry of
the amino groups. Energy of non-bonded interactions of the
hydrogen atoms benzene ring with amino group is different
for out-of-plane deformations of the ring in the opposite
directions. Therefore, strict harmonic dependence is
maintained for both directions of PES scans (from -30� to
0� and from 0� to ?30�), but coefficients of these frag-
ments of parabolas are slightly different.
In molecules with two and more N–H���O hydrogen
bonds formed between vicinal nitro and amino groups, this
effect becomes considerably stronger because of the non-
coplanarity between the N–H���O hydrogen bonds and
benzene ring. Therefore, change of ring conformation
results in strengthening of hydrogen bonds during ring
out-of-plane deformation in one direction and weakening
of H bonds due to ring deformation in an opposite direc-
tion. This leads to notable asymmetry of potential energy
surface.
Aromaticity
Earlier, it was demonstrated [34] that appearance of any
substituent in benzene ring results in decrease of aroma-
ticity degree of cyclic conjugated system due to violation
of symmetry of p–p interactions within the ring. Therefore,
it is possible to expect that presence of several strong
p-electron-donating and withdrawing groups should sig-
nificantly decrease the aromaticity of benzene ring.
The results of present calculations demonstrate that
degree of aromaticity of molecules under study vary in quite
a wide range. It amounts to 8–96% for NICS(1)zz, 18–86%
for E(def), 74–100% for Ia, and 22–98% for HOMA, as
compared to benzene (Table 10). Some of molecules under
study, namely 2,4,6-triamino-1,3-dinitrobenzene 13N246A,
2,4-diamino-1,3,5-trinitrobenzene 135N24A, and 2,4,6-
triamino-1,3,5-trinitrobenzene 135N246A, possess extre-
mely low degree of aromaticity and may be considered even
as non-aromatic (Table 10).
Values of all aromaticity indices are quite consistent and
demonstrate reasonable correlation with each other
(Table 11). It is not a common result for aromatic systems,
e.g., monosubstituted benzenes and different heterocycles
[34, 35]. Correlation is observed in areas of both high and
low aromaticity. Therefore, it is possible to conclude that
molecules having high (small) values of all indices are
clearly aromatic (non-aromatic) without detailed discus-
sions regarding validity of the used index. As it was
demonstrated earlier [8], for highly aromatic systems,
structural indices are not very sensitive but the total cor-
relation is very good.
The remarkable result is that, in spite of above-men-
tioned features of potential energy surface for highly
substituted molecules, the values of ring out-of-plane
deformation energy describe changes of aromaticity degree
of the molecules under study in the agreement with general
trend. Moreover, correlation coefficients of E(def) with
other indices exceed 90% (Table 11). This confirms
applicability of ring out-of-plane deformation energy for
quantitative description of aromaticity even in the case of
quite complex molecules.
The Bird’s Ia index classifies all molecules as highly aro-
matic, while some of them possess clearly low degree of
aromaticity according to all other indices. This is well-known
limitation of the Bird’s index [79] for highly symmetrical
molecules. Therefore, 2,4,6-triamin-1,3-dinitrobenzene, 2,4-
diamino-1,3,5-trinitrobenzene and 2,4,6-triamin-1,3,5-trini-
trobenzene are outliers in all correlations. The HOMA index is
Table 9 Energies of ring out-of-plane deformations E(def) (kcal/mol)
upon change of the softest endocyclic torsion angle by ?30� (E?) and
-30� (E-) in molecules under consideration
Molecule Torsion angle E- E?
Benzene C1–C2–C4–C4 7.22 7.22
1N C5–C6–C1–C2 6.42 6.42
12N C1–C2–C4–C4 6.55 6.55
13N C2–C3–C4–C5 6.39 6.39
14N C5–C6–C1–C2 6.24 6.24
123N C3–C4–C5–C6 6.41 6.41
135N C1–C2–C4–C4 6.45 6.45
1A C5–C6–C1–C2 6.14 5.90
12A C1–C2–C4–C4 5.84 5.29
13A C1–C2–C4–C4 5.68 5.57
14A C1–C2–C4–C4 6.09 5.68
124A C1–C2–C4–C4 5.29 5.29
135A C1–C2–C4–C4 5.73 5.53
1N2A C5–C6–C1–C2 4.82 4.82
1N3A C3–C4–C5–C6 5.72 5.72
1N4A C1–C2–C4–C4 5.66 5.66
13N2A C6–C1–C2–C3 5.32 3.21
13N4A C4–C5–C6–C1 4.80 4.80
13N5A C3–C4–C5–C6 5.65 5.65
1N24A C6–C1–C2–C3 4.89 4.89
1N35A C2–C3–C4–C5 5.49 5.49
13N24A C6–C1–C2–C3 4.36 3.16
13N25A C6–C1–C2–C3 5.59 3.66
123N46A C3–C4–C5–C6 4.68 4.68
135N2A C3–C4–C5–C6 5.74 3.23
135N24A C5–C6–C1–C2 3.78 2.10
1N246A C3–C4–C5–C6 5.12 4.79
13N246A C6–C1–C2–C3 3.46 2.03
135N246A C3–C4–C5–C6 2.34 0.58
1594 Struct Chem (2012) 23:1585–1597
123

much more sensitive to structure deformation due to presence
of EN and GEO components while Ia index takes into con-
sideration only GEO part [40].
General trends in changes of aromaticity indices agree
well with the deformations of structure of benzene ring
mentioned above. Polysubstitution by amino groups affects
aromaticity stronger than polysubstitution by nitro groups,
and with no additive features of this effect (Table 10).
Steric repulsion of nitro groups causing their rotation does
not lead to considerable changes of aromaticity. Interest-
ingly, the values of aromaticity indices for sterically
strained 1,2,3-trinitrobenzene 123N and 1,2-dinitrobenzene
12N remains are quite high.
The most pronounced change of aromaticity is caused
by vicinal arrangement of nitro and amino groups.
Therefore, aromaticity of benzene ring in ortho-nitroaniline
1N2A is considerably lower as compared to meta- and
para-isomers 1N3A and 1N4A (Table 10). Aromaticity
Table 10 Aromaticity indices
of nitro-and amino-substituted
benzenes arranged by numbers
of nitro and amino substituents
Number of H bonds Molecule E(30), kcal/mol NICS(1)zz Ia HOMA
0 Benzene 7.22 -30.45 100.00 99.17
0 nitro groups
0 1A 5.90 -26.99 96.62 90.82
12A 5.29 -26.18 94.80 89.41
13A 5.57 -23.89 96.81 90.61
14A 5.68 -24.56 96.80 90.02
124A 5.29 -23.82 95.15 89.18
135A 5.53 -20.58 99.94 90.85
1 nitro group
0 1N 6.42 -29.10 97.78 93.73
1N3A 5.72 -25.46 94.48 91.57
1N4A 5.66 -25.71 93.88 92.22
1N35A 5.49 -21.81 94.73 90.84
1 1N2A 4.82 -24.99 90.59 87.83
1N24A 4.89 -20.97 90.07 86.84
1N246A 4.79 -16.36 89.93 82.63
2 nitro groups
0 12N 6.55 -27.72 98.03 94.48
13N 6.39 -28.84 97.34 95.67
14N 6.24 -27.69 99.52 95.17
13N5A 5.65 -24.25 92.34 92.33
1 13N4A 4.80 -23.88 87.46 88.32
2 13N2A 3.21 -22.49 84.60 81.94
13N25A 3.66 -20.49 87.90 82.65
3 13N24A 3.16 -16.32 82.43 73.53
4 13N246A 2.03 -8.25 80.31 49.57
3 nitro groups
0 123N 6.41 -27.01 98.32 95.29
135N 6.45 -28.74 100.00 97.60
2 123N46A 4.68 -17.63 91.24 87.00
135N2A 3.23 -21.27 79.75 79.25
4 135N24A 2.10 -11.65 74.28 54.71
6 135N246A 1.20 -2.46 99.79 21.52
Table 11 Correlation coefficients (%) between aromaticity indices
excluding outlying points (135N246A, 135N24A, 135N2A,
13N246A)
NICS(1)zz Ia HOMA
E(def) -91 93 (66) 92
NICS(1)zz -77 (-48) -93
Ia 91 (41)
Values in parenthesis correspond to all dataset including outlying
points
Struct Chem (2012) 23:1585–1597 1595
123

degree of the last two molecules is very similar, indicating
almost negligible push–pull effect.
Analysis of aromaticity degree of benzene ring in het-
eropolysubstituted molecules clearly indicates dependence
of aromaticity indices on number of intramolecular
N–H���O hydrogen bonds. For example, degree of varia-
tion of HOMA index stretches between 89.18–97.6 for
molecules without H bonds, 86.84–88.32 for molecules
with 1 H bond, 79.25–87.00 for molecules with 2 H
bonds, 73.53 for 13N24A with 3 H bonds, and
49.57–54.71 for molecules with 4 H bonds and only 21.52
for 135N246A containing 6 H bonds. This agrees well
with resonance-assisted character of hydrogen bonds
leading to specific deformation of geometry of benzene
ring mentioned above.
It is interesting to note that variation of aromaticity
degree within group of molecules with the same number of
intramolecular hydrogen bonds almost does not depend on
number of nitro and amino groups. For example, aroma-
ticity of benzene ring in 1N3A, 1N4A is very close to
aromaticity of 1N35A containing two amino groups or
13N5A containing two nitro groups. This also confirms
negligible influence of p–p interactions between substitu-
ents on aromaticity of cyclic conjugated system as com-
pared to formation of RAHB.
Conclusions
An analysis of structural properties and aromaticity of
nitro- and amino-substituted derivatives of benzene dem-
onstrates that the conjugation interactions between sub-
stituents (push–pull effect) is very small and almost does
not influence properties of investigated molecules. The
main factor governing molecular structure and aromaticity
of benzene ring is the formation of intramolecular N–H���Ohydrogen bonds between vicinal nitro and amino groups.
Resonance-assisted characteristics of these hydrogen bonds
result in significant elongation of the C–C bond between
nitro and amino groups accompanied by decrease of aro-
maticity of cyclic conjugated system and increase of con-
formational flexibility of ring. Degree of such deformations
depends on number of the N–H���O bonds. The most sig-
nificant deformation is observed in symmetric 1,3,5-trini-
tro-2,4,6-triaminobenzene (135N246A) in which structure
should be described as derivative of cyclohexane with six
exocyclic double bonds, rather than hexasubstituted ben-
zene. This is confirmed by very low values of aromaticity
indices and high conformational flexibility of benzene ring
corresponding to non-aromatic character of cyclic conju-
gated system. All other polysubstituted derivatives of
benzene have intermediate properties between nitroben-
zene or aniline and 135N246A.
Acknowledgments The use of trade, product, or firm names in this
report is for descriptive purposes only and does not imply endorse-
ment by the U.S. Government. Results in this study were funded and
obtained from research conducted under the Environmental Quality
Technology Program of the United States Army Corps of Engineers
by the USAERDC. Permission was granted by the Chief of Engineers
to publish this information. The findings of this report are not to be
construed as an official Department of the Army position unless so
designated by other authorized documents.
References
1. Krygowski TM, Cyranski MK (2001) Chem Rev 101:1385–1420
2. Schleyer PvR (2001) Chem Rev 101:1115–1117
3. Stock LM, Brown HC (1963) Adv Phys Org Chem 1:36–154
4. Hoggett JG, Moodie RB, Penton RB, Schofield K (1971)
Nitration and aromatic reactivity. Cambridge University Press,
Cambridge
5. Exner O, Bohm S (2002) J Org Chem 67:6320–6327
6. Krygowski TM, Stepien BT (2005) Chem Rev 105:3482–3512
7. Krygowski TM, Esmont K, Stepien BT, Cyranski MK, Poater J,
Sola M (2004) J Org Chem 69:6634–6640
8. Shishkin OV, Omelchenko IV, Krasovska MV, Zubatyuk RI,
Gorb L, Leszczynski J (2006) J Mol Struct 791:158–164
9. Krygowski TM, Stepien BT, Cyranski MK (2005) Int J Mol Sci
6:45–51
10. Smith MB, March J (2001) Advanced organic chemistry. Wiley,
New York
11. Krygowski TM, Dobrowolski MA, Zborowski K, Cyranski MK
(2006) J Phys Org Chem 19:889–895
12. Roszak S, Gee RH, Balasubramanian K, Fried LE (2003) Chem
Phys Lett 374:286
13. Lima CFRAC, Gomes LR, Santos LMNBF (2007) J Phys Chem
A 111:10598–10603
14. Krygowski TM, Palusiak M, Plonka A, Zachara-Horeglad JEJ
(2007) Phys Org Chem 20:297–306
15. Zhang C (2006) Chem Phys 324:547–555
16. Szatylowicz H, Krygowski TM, Hobza P (2007) J Phys Chem A
111:170–175
17. Borisenko VE, Krekov SA, MYu Fomenko, Koll A, Lipkovski PJ
(2008) Mol Struct 882:9–23
18. Gross KC, Seybold PG, Peralta-Inga Z, Murray JS, Politzer P
(2001) J Org Chem 66:6919–6925
19. Alonso M, Herradon B (2010) Phys Chem Chem Phys
12:1305–1317
20. Chermahini AN, Dabbagh HA, Teimouri A (2007) J Mol Struct
822:33–37
21. Badgujar DM, Talawar MB, Asthana SN, Mahulikar PP (2008)
J Hazard Mater 151:289–305
22. Keshavarz MH (2008) J Hazard Mater A 153:201–206
23. Takemura N, Shimizu H (1978) Mutat Res 54:256–257
24. Luther M (1990) Chemosphere 21:231–241
25. Levine BF (1976) Chem Phys Lett 37:516–520
26. Wolleben J, Testa AC (1977) J Phys Chem 81:429–431
27. in het Panhuis M, Munn RW, Popelier PLA (2004) J Chem Phys
120:11479–11486
28. Wang JX, Gong XD, Xiao HM (2009) Int J Quant Chem
109:1522–1530
29. Manaa MR, Gee RH, Fried LE (2002) J Phys Chem A
106:8806–8810
30. Gee RH, Roszak S, Balasubramanian K, Fried LE (2004) J Chem
Phys 120:7059–7066
31. Møller C, Plesset MS (1934) Phys Rev 46:618–622
1596 Struct Chem (2012) 23:1585–1597
123

32. Kendall RA, Dunning TH Jr, Harrison RJ (1992) J Chem Phys
96:6796–6806
33. Minkin VI, Glukhovtsev MN, Simkin BY (1994) Aromaticity and
antiaromaticity. Wiley, New York
34. Cyranski MK (2005) Chem Rev 105:3773–3811
35. Cyranski MK, Krygowski TM, Katritzky AR, Schleyer PvR
(2002) J Org Chem 67:1333–1338
36. Jug K, Oniciu DC, Katritzky AR (2001) Chem Rev 101:
1421–1449
37. Alonso M, Herradon BJ (2010) Comp Chem 31:917–928
38. Poater J, Duran M, Sola M, Silvi B (2005) Chem Rev 105:
3911–3947
39. Bird CW (1992) Tetrahedron 48:335–340
40. Cyranski MK, Krygowski TM (1999) Tetrahedron 55:6205–6210
41. Chen Z, Wannere CS, Corminboeuf C, Putcha R, Schleyer PvR
(2005) Chem Rev 105:3842–3888
42. Shishkin OV, KYu Pichugin, Gorb L, Leszczynski J (2002) J Mol
Struct 616:159–166
43. Zhigalko MV, Shishkin OV, Gorb L, Leszczynski J (2004) J Mol
Struct 693:153–159
44. Shishkin OV, Gorb L, Lesczynski J (2000) Chem Phys Lett
330:603–611
45. Gordy WJ (1947) J Chem Phys 15:305–310
46. Fallah-Bagher-Shaidaei H, Wannere CS, Corminboeuf C, Puchta
R, Schleyer PvR (2006) Org Lett 8:863–866
47. Gaussian 03, Revision C.01, Frisch MJ, Trucks GW, Schlegel
HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr,
Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi
J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N,
Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda
R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai
H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V,
Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O,
Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY,
Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski
VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK,
Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q,
Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G,
Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith
T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M,
Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, and
Pople JA (2004) Gaussian, Inc., Wallingford CT
48. Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS,
Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus
TL, Dupuis M, Montgomery JA (1993) J Comput Chem 14:
1347–1363
49. Bader RWF (1990) Atoms in molecules. A quantum theory.
Calendon Press, Oxford
50. Espinosa E, Molina E, Lecomte C (1998) Chem Phys Lett
285:170–173
51. Dorofeeva OV, Vishnevskiy YV, Vogt N, Vogt J, Khristenko LV,
Krasnoshchekov SV, Shishkov IF, Hargittai I, Vilkov LV (2007)
Struct Chem 18:739–753
52. Schultz G, Portalone G, Ramondo F, Domenicano A, Hargittai I
(1996) Struct Chem 7:59–71
53. Sinclair WE, Pratt DW (1996) J Chem Phys 105:7942–7956
54. Colapietro M, Domenicano A, Portalone G, Schultz G, Hargittai I
(1987) J Phys Chem 91:1728–1737
55. Sadova NI, Penionzhkevich NP, Vilkov LV (1976) J Struct Chem
(in Russian) 17:954–956
56. Zych T, Misiaszek T, Szostak MM (2007) Chem Phys 340:
260–272
57. Colapietro M, Domenicano A, Marciante C, Portalone G (1982)
Z Naturforsch 37B:1309–1311
58. Qian HY, Yin ZG, Jia J, Zhou N, Feng LQ (2006) Acta Crys-
tallogr 62E:o5048–o5049
59. Wojcik G, Holband J (2001) Acta Crystallogr 57B:346–352
60. Woodford JN, Pauley MA, Wang CH (1997) J Phys Chem
101B:1989–1992
61. Pappalardo RR, Marcos ES, Ruiz-Lopez MF, Rinaldi D, Rivail
JL (1993) J Am Chem Soc 115:3722–3730
62. Kovacs A, Szabo A, Hargittai I (2002) Acc Chem Res 35:
887–894
63. Chung G, Kwon O, Kwon Y (1997) J Phys Chem A 101:
4628–4632
64. Borisenko KB, Zauer K, Hargittai I (1996) J Phys Chem 100:
19303–19309
65. Gilli G, Belucci F, Ferretti V, Bertolesi V (1989) J Am Chem Soc
111:1023–1028
66. Sobczyk L, Grabowski SJ, Krygowski TM (2005) Chem Rev
105:3513–3560
67. Manaa MR, Fried LE (2001) J Phys Chem A 105:6765–6768
68. Rashid AN (2004) J Mol Struct 681:57–63
69. Cohen AJ, Mori-Sanchez P, Yang W (2008) Science 321:
792–794
70. Allen FH (2002) Acta Cryst B58:380–388
71. Fazli M, Raissi H, Chahkandi B, Aarabi M (2010) J Mol Struct
942:115–120
72. Wojtulewski S, Grabowski SJ (2003) J Mol Struct 621:285–291
73. Huanga Z, Chenb B, Gao G (2005) J Mol Struct 752:87–92
74. Kimmel AV, Sushko PV, Shluger AL, Kuklja MM (2008) J Phys
Chem A 112:4496–4500
75. Liu H, Zhao J, Ji G, Wei D, Gong Z (2006) Phys Lett A
358:63–69
76. Pravica M, Yulga B, Tkachev S, Liu Z (2009) J Phys Chem A
113:9133–9137
77. Wu C, Fried LE (2000) J Phys Chem A 104:6447–6452
78. Dobratz BM (1995) The insensitive high explosive triaminotri-
nitrobenzene (TATB): development and characterizations—1888
to 1994. Los Alamos National Laboratory, Los Alamos
79. Kotelevskii SI, Prezhdo OV (2001) Tetrahedron 57:5715–5729
Struct Chem (2012) 23:1585–1597 1597
123
Related Documents











