HUMAN MUTATION 0,1^10, 2007 RESEARCH ARTICLE Progranulin Null Mutations in Both Sporadic and Familial Frontotemporal Dementia Isabelle Le Ber, 1–3 Julie van der Zee, 4,5 Didier Hannequin, 6 Ilse Gijselinck, 4,5 Dominique Campion, 6 Miche `le Puel, 7 Annie Laquerrie `re, 8 Tim De Pooter, 4,5 Agne `s Camuzat, 1 Marleen Van den Broeck, 4,5 Bruno Dubois, 2,3,9,10 Franc - ois Sellal, 11 Lucette Lacomblez, 2,10,12 Martine Vercelletto, 13 Catherine Thomas-Ante ´rion, 14 Bernard-Franc - ois Michel, 15 Ve ´ronique Golfier, 16 Mira Didic, 17 Franc - ois Salachas, 2,12 Charles Duyckaerts, 1,10,18 Marc Cruts, 4,5 Patrice Verpillat, 1 Christine Van Broeckhoven, 4,5 Alexis Brice, 1,2,10,19 and the French Research Network on FTD/FTD-MND 1 INSERM, UMR679, Paris, France; 2 Fe ´de ´ration des Maladies du Syste `me Nerveux, Groupe Pitie ´-Salpe ˆtrie `re, Assistance Publique-Ho ˆpitaux de Paris (AP-HP), Paris, France; 3 Centre de Neuropsychologie et du Langage, Groupe Pitie ´-Salpe ˆtrie `re, Assistance Publique-Ho ˆpitaux de Paris (AP- HP), Paris, France; 4 Neurodegenerative Brain Diseases Groupe, Department of Molecular Genetics, Flanders Interuniversity Institute for Biotechnology, Antwerpen, Belgium; 5 University of Antwerp, Antwerpen, Belgium; 6 INSERM U614 and De ´partement de Neurologie, Rouen University Hospital, Rouen, France; 7 Service de Neurologie, Centre Hospitalier Universitaire (CHU) Purpan, Toulouse, France; 8 Service de Neuropathologie, Rouen University Hospital, Rouen, France; 9 INSERM U610, Groupe Pitie ´-Salpe ˆtrie `re, Paris, France; 10 Universite Pierre et Marie Curie-Paris6, Institut Fe ´de ´ratif de Recherche Neurosciences Unite ´ Mixte S679, Faculte ´ de Me ´decine, Paris, France; 11 Service de Neurologie, Ho ˆpitaux Universitaires and INSERM U692, Strasbourg, France; 12 Service de Pharmacologie, Ho ˆpital Pitie ´-Salpe ˆtrie `re, Paris, France; 13 Service de Neurologie, Centre Hospitalier Universitaire (CHU) Guillaume et Rene ´ Lae ¨nnec, Nantes, France; 14 Service de Neurologie, Ho ˆpital de Bellevue, Saint-Etienne, France; 15 Unite ´ de Neuroge ´riatrie, Ho ˆpital Sainte-Marguerite, Marseille, France; 16 Service de Neurologie, Centre Hospitalier, Saint-Brieuc, France; 17 Service de Neurologie et Neuropsychologie, Ho ˆpital de la Timone Adultes, Marseille, France; 18 Laboratoire de Neuropathologie Escourolle, Ho ˆpital Pitie ´-Salpe ˆtrie `re, Paris, France; 19 De ´partement de Ge ´ne ´tique, Cytoge ´ne ´tique et Embryologie, Groupe Pitie ´- Salpe ˆtrie `re, Assistance Publique-Ho ˆpitaux de Paris (AP-HP), Paris, France Communicated by Jean-Louis Mandel Frontotemporal dementia (FTD) is the second most frequent type of neurodegenerative dementias. Mutations in the progranulin gene (GRN, PGRN) were recently identified in FTDU-17, an FTD subtype characterized by ubiquitin-immunoreactive inclusions and linkage to chromosome 17q21. We looked for PGRN mutations in a large series of 210 FTD patients (52 familial, 158 sporadic) to accurately evaluate the frequency of PGRN mutations in both sporadic and familial FTD, and FTD with associated motoneuron disease (FTD-MND), as well as to study the clinical phenotype of patients with a PGRN mutation. We identified nine novel PGRN null mutations in 10 index patients. The relative frequency of PGRN null mutations in FTD was 4.8% (10/210) and 12.8% (5/39) in pure familial forms. Interestingly, 5/158 (3.2%) apparently sporadic FTD patients carried a PGRN mutation, suggesting the possibility of de novo mutations or incomplete penetrance. In contrast, none of the 43 patients with FTD-MND had PGRN mutations, supporting that FTDU-17 and FTD-MND are genetically distinct. The clinical phenotype of PGRN mutation carriers was particular because of the wide range in onset age and the frequent occurrence of early apraxia (50%), visual hallucinations (30%), and parkinsonism (30%) during the course of the disease. This study supports that PGRN null mutations represent a more frequent cause of FTD than MAPT mutations (4.8% vs. 2.9%) but are not responsible for FTD-MND. It also demonstrates that half of the patients with a PGRN mutation in our series had no apparent family history of Published online inWiley InterScience (www.interscience.wiley.com). DOI 10.1002/humu.20520 Received12 October 2006; accepted revised manuscript 9 February 2007. Correspondence to: Pr. Alexis Brice, INSERM Unit 679, Ho Œ pital Pitie Ł -Salpe Œ trie Ø re, 47, boulevard de l’ho Œ pital, 75651 Paris cedex 13, France. E-mail: [email protected] Grant sponsors: Agence Nationale pour la Recherche; France-Alz- heimer Association; University of Antwerp; Interuniversity Attraction Poles Program P5/19 of the Belgian Science Policy O⁄ce; Interna- tional Alzheimer Research Foundation, Belgium; Zenith Award of the Alzheimer’s Association, USA; Institute for Science and Technology Flanders (IWT-F), Belgium;CRIC AP-HP; Grant number:01107; Grant sponsor: EU; Grant number: LSHMCT-2003-503330 (APOPIS); Grant sponsor: GIS-Institut des Maladies Rares; Grant numbers: A03081DS and APS03002DSA. The members of the French Research Network on FTD/FTD-MND are listed in Appendix A. Isabelle Le Ber and Julie van der Zee contributed equally to this work. r r 2007 WILEY-LISS, INC.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

HUMAN MUTATION 0,1^10,2007
RESEARCH ARTICLE
Progranulin Null Mutations in Both Sporadicand Familial Frontotemporal Dementia
Isabelle Le Ber,1–3 Julie van der Zee,4,5 Didier Hannequin,6 Ilse Gijselinck,4,5 Dominique Campion,6
Michele Puel,7 Annie Laquerriere,8 Tim De Pooter,4,5 Agnes Camuzat,1 Marleen Van den Broeck,4,5
Bruno Dubois,2,3,9,10 Franc-ois Sellal,11 Lucette Lacomblez,2,10,12 Martine Vercelletto,13
Catherine Thomas-Anterion,14 Bernard-Franc-ois Michel,15 Veronique Golfier,16 Mira Didic,17
Franc-ois Salachas,2,12 Charles Duyckaerts,1,10,18 Marc Cruts,4,5 Patrice Verpillat,1
Christine Van Broeckhoven,4,5 Alexis Brice,1,2,10,19� and the French Research Network on FTD/FTD-MND1INSERM, UMR679, Paris, France; 2Federation des Maladies du Systeme Nerveux, Groupe Pitie-Salpetriere, Assistance Publique-Hopitaux deParis (AP-HP), Paris, France; 3Centre de Neuropsychologie et du Langage, Groupe Pitie-Salpetriere, Assistance Publique-Hopitaux de Paris (AP-HP), Paris, France; 4Neurodegenerative Brain Diseases Groupe, Department of Molecular Genetics, Flanders Interuniversity Institute forBiotechnology, Antwerpen, Belgium; 5University of Antwerp, Antwerpen, Belgium; 6INSERM U614 and Departement de Neurologie, RouenUniversity Hospital, Rouen, France; 7Service de Neurologie, Centre Hospitalier Universitaire (CHU) Purpan, Toulouse, France; 8Service deNeuropathologie, Rouen University Hospital, Rouen, France; 9INSERM U610, Groupe Pitie-Salpetriere, Paris, France; 10Universite Pierre etMarie Curie-Paris6, Institut Federatif de Recherche Neurosciences Unite Mixte S679, Faculte de Medecine, Paris, France; 11Service de Neurologie,Hopitaux Universitaires and INSERM U692, Strasbourg, France; 12Service de Pharmacologie, Hopital Pitie-Salpetriere, Paris, France; 13Servicede Neurologie, Centre Hospitalier Universitaire (CHU) Guillaume et Rene Laennec, Nantes, France; 14Service de Neurologie, Hopital deBellevue, Saint-Etienne, France; 15Unite de Neurogeriatrie, Hopital Sainte-Marguerite, Marseille, France; 16Service de Neurologie, CentreHospitalier, Saint-Brieuc, France; 17Service de Neurologie et Neuropsychologie, Hopital de la Timone Adultes, Marseille, France; 18Laboratoire deNeuropathologie Escourolle, Hopital Pitie-Salpetriere, Paris, France; 19Departement de Genetique, Cytogenetique et Embryologie, Groupe Pitie-Salpetriere, Assistance Publique-Hopitaux de Paris (AP-HP), Paris, France
Communicated by Jean-Louis Mandel
Frontotemporal dementia (FTD) is the second most frequent type of neurodegenerative dementias. Mutationsin the progranulin gene (GRN, PGRN) were recently identified in FTDU-17, an FTD subtype characterized byubiquitin-immunoreactive inclusions and linkage to chromosome 17q21. We looked for PGRN mutations in alarge series of 210 FTD patients (52 familial, 158 sporadic) to accurately evaluate the frequency of PGRNmutations in both sporadic and familial FTD, and FTD with associated motoneuron disease (FTD-MND),as well as to study the clinical phenotype of patients with a PGRN mutation. We identified nine novel PGRNnull mutations in 10 index patients. The relative frequency of PGRN null mutations in FTD was 4.8% (10/210)and 12.8% (5/39) in pure familial forms. Interestingly, 5/158 (3.2%) apparently sporadic FTD patients carried aPGRN mutation, suggesting the possibility of de novo mutations or incomplete penetrance. In contrast, none ofthe 43 patients with FTD-MND had PGRN mutations, supporting that FTDU-17 and FTD-MND aregenetically distinct. The clinical phenotype of PGRN mutation carriers was particular because of the wide rangein onset age and the frequent occurrence of early apraxia (50%), visual hallucinations (30%), and parkinsonism(30%) during the course of the disease. This study supports that PGRN null mutations represent a morefrequent cause of FTD than MAPT mutations (4.8% vs. 2.9%) but are not responsible for FTD-MND. It alsodemonstrates that half of the patients with a PGRN mutation in our series had no apparent family history of
Published online inWiley InterScience (www.interscience.wiley.com).DOI10.1002/humu.20520
Received12October 2006; accepted revisedmanuscript 9 February2007.�Correspondence to: Pr. Alexis Brice, INSERM Unit 679, HoŒ pital
PitieŁ -SalpeŒ trieØ re, 47, boulevard de l’hoŒ pital, 75651 Paris cedex 13,France. E-mail: [email protected]
Grant sponsors: Agence Nationale pour la Recherche; France-Alz-heimer Association; University of Antwerp; Interuniversity AttractionPoles Program P5/19 of the Belgian Science Policy O⁄ce; Interna-tional Alzheimer Research Foundation, Belgium; Zenith Award of theAlzheimer’s Association, USA; Institute for Science and TechnologyFlanders (IWT-F), Belgium;CRICAP-HP;Grant number:01107;Grantsponsor: EU; Grant number: LSHMCT-2003-503330 (APOPIS);Grant sponsor: GIS-Institut des Maladies Rares; Grant numbers:A03081DS and APS03002DSA.
The members of the French Research Network on FTD/FTD-MNDare listed in Appendix A.
Isabelle Le Ber and Julie van der Zee contributed equally to thiswork.
rr 2007 WILEY-LISS, INC.

dementia. Taking this into account, genetic testing should be now considered more systematically, even inpatients without obvious familial history of FTD. Hum Mutat 0, 1–10, 2007. rr 2007 Wiley-Liss, Inc.
KEY WORDS: frontotemporal dementia; FTDU-17; ubiquitin-immunoreactive inclusions; GRN; PGRN; progranulin;TDP-43; TAR DNA-binding protein
INTRODUCTION
Frontotemporal dementia (FTD) is the second most frequenttype of neurodegenerative dementia after Alzheimer’s disease andis characterized by predominant and gradual behavioral changesassociated with language and affective disorders [Neary et al.,1998]. FTD results from severe neuronal degeneration of thefrontal and/or temporal cortices. In some patients, behavioralsymptoms are associated with a parkinsonian syndrome or withmotoneuron disease (MND).
FTD is inherited as an autosomal dominant trait in 20 to 50%of the patients. Familial forms are genetically heterogeneous.Mutations in the microtubule-associated protein tau gene(MAPT) were initially identified in families with FTD associatedwith parkinsonism (FTDP-17), in whom linkage to chromosome17 was previously demonstrated [Hutton et al., 1998; Poorkajet al., 1998; Spillantini et al., 1998]. So far, 39 mutations havebeen identified in 115 families worldwide (AD&FTD Mutationdatabase: www.molgen.ua.ac.be/FTDmutations) [Rademakerset al., 2004], which represents a relative frequency of 10 to 20%[Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998;Dumanchin et al., 1998]. The predominant histopathologicalchange in patients with MAPT mutations is usually widespreadtau pathology. Two other genes, valosin-containing protein (VCP)on the short arm of chromosome 9 [Watts et al., 2004], andcharged multivesicular body protein 2B (CHMP2B) on thepericentromeric region of chromosome 3 [Skibinski et al., 2005],are mutated in a smaller proportion of the autosomal dominantforms of FTD.
Recent genetic and pathological studies have shown that themajority of FTD patients do not have mutations in these genes,and lack tau pathology [Rademakers et al., 2004]. After theMAPT gene was identified, it became increasingly evident that asubset of families with conclusive linkage to the MAPT locus on17q21 did not carry mutations in this gene [Lendon et al., 1998;Rosso et al., 2001; Rademakers et al., 2002, 2004; Mackenzieet al., 2006; van der Zee et al., 2006]. Neuropathologically, thesepatients were characterized by the presence of neuronal ubiquitin-immunoreactive (ub-ir) cytoplasmic and nuclear inclusions[Rademakers et al., 2002; Mackenzie et al., 2006; van der Zeeet al., 2006]. Very recently, mutations in the progranulin gene(HUGO gene symbol GRN; also referred to as PGRN; MIM] 138945) were identified in families with FTD with ubiquitin-immunoreactive inclusions linked to chromosome 17 (FTDU-17)[Baker et al., 2006; Cruts et al., 2006]. The PGRN gene is locatedat the chromosomal region 17q21, 1.7 Mb centromeric of theMAPT gene. It codes for progranulin (PGRN, also known asacrogranin or epithelin precursor), a 593–amino acid glycoproteincomposed of 7.5 tandem repeats of a 12-cysteinyl-rich granulinmotif [Bateman and Benett, 1998], which is the commonprecursor of granulin proteins. PGRN is a widely expressedmultifunctional protein that may play a role in development,epithelial homeostasis, reproduction, and inflammation. PGRNalso modulates growth in a variety of cells and, when over-expressed, confers epithelial tumorigenicity [He and Bateman,2003]. In the central nervous system, PGRN is highly expressed inneurons of the cerebral cortex, particularly in granule cells of the
hippocampus and more intensively in Ammon’s horn and in thePurkinje cells of the cerebellum [Daniel et al., 2000].
To date, 31 PGRN mutations have been identified in FTDpatients worldwide (AD&FTD Mutation Database) [Baker et al.,2006; Cruts et al., 2006; Gass et al., 2006; Huey et al., 2006;Mukherjee et al., 2006; Masellis et al., 2006; Bronner et al., 2007].All these mutations were shown to lead to null alleles and a partialloss of functional PGRN. However, the phenotypes associated withthe PGRN mutations have not been extensively studied in a seriesof mutation carriers. To extend the mutation spectrum of thePGRN gene and more accurately evaluate its mutation frequencyand the associated phenotypes, we analyzed PGRN in a large seriesof 210 predominantly French FTD patients not known to belinked to chromosome 17.
MATERIALSANDMETHODSSubjects
DNA samples from 210 index patients with FTD were collectedin France through a French research network of neurologists, aftereach patient gave informed consent. The diagnosis of FTD wasbased on the Lund and Manchester criteria [Lund and ManchesterGroups, 1996]. Most patients originated from France (n 5 196).Others originated from Italy (n 5 5), the Caribbean islands(n 5 2), and one each from the Netherlands, Spain, Portugal,Morocco, Turkey, Greece, and Armenia. A total of 52 (25%)patients had a familial history of FTD, i.e., at least one first-degreerelative with disease from the FTD spectrum and 158 (75%) weresporadic. A MAPT mutation was found in six index patients[Dumanchin et al., 1998]. In the remaining 204 patients withoutMAPT mutations, the mean age at disease onset was 60.678.2years. In this subgroup of patients, parkinsonism was found in 25%of the patients and MND in 21%. This study was approved by theMedical Research Ethics Committee of Assistance Publique-Hopitaux de Paris (AP-HP) and the ethics committee of theSalpetriere Hospital.
A Yate’s corrected chi-squared test (for comparisons of smallgroups) was used to compare means age at onset and atexamination, disease duration at death, and phenotypic char-acteristics of the patients with PGRN mutations to a group ofpatients with MAPT mutations and to the patients without PGRNor MAPT mutations.
PGRN Sequencing Analysis
PGRN mutation analysis was performed in the 204 patientswithout MAPT mutations as well as in 175 neurologically healthycontrols of French origin (350 control chromosomes). For thePGRN exon 0 fragment containing the IVS013A4T(g.96239A4T, p.0) mutation, 90 additional French controls weresequenced (530 chromosomes in total). All PGRN exons,including the noncoding exon 0 and intron–exon boundaries weresequenced as described [Cruts et al., 2006]. Total genomic DNAwas prepared from peripheral blood according to standardprocedures. Exons and flanking regions were amplified by PCRon genomic DNA (20 ng) using previously described primers[Cruts et al., 2006]. Amplification products were purified with 1 U
2 HUMANMUTATION 0,1^10,2007
Human Mutation DOI 10.1002/humu

antarctic phosphatase (New England Biolabs, Ipswich, MA) and 1U exonuclease I (New England Biolabs) and sequenced in bothdirections using the BigDye Terminator Cycle Sequencing kit v3.1(Applied Biosystems, Foster City, CA) on an ABI3730 automatedsequencer (Applied Biosystems). Sequences were analyzed usingthe NovoSNP software package [Weckx et al., 2005]. STRgenotyping in Patients F001/001 and F329/001, who carriedthe same PGRN c.599_708del [p.Val200GlyfsX18] mutation, isdetailed in Supplementary Methods S1 (available online at: http://www.interscience.wiley.com/jpages/1053-1807/suppmat/).
Mutation nomenclature follows the HGVS recommendations.The nucleotides of genomic DNA variants were numbered relativeto the reverse complement of GenBank Accession NumberAC003043 and starting at nt 1. The nucleotides of the predictedRNA were numbered according to the largest PGRN transcript(GenBank Accession Number NM_002087.2), starting at thetranslation initiation codon. The amino acids of the predictedprotein were numbered according to the largest PGRN isoform(GenPept Accession Number NP_002078.1).
Neuropathological Evaluation
Brain autopsy was performed on Patient F171/002 carrying thePGRN mutation p.Gln401X. After fixation in 10% formaldehyde,tissue samples were taken from multiple representative areas of theright hemisphere, as well as from the brainstem and cerebellum.Seven micron sections were cut from paraffin-embedded blocksand stained with hematoxylin and eosin (HE), luxol-phloxin, andthe modified Bielchowski silver impregnation method. Immuno-histochemistry was carried out using antibodies directed againstubiquitin, TDP-43, alpha-synuclein, PHF-tau, glial fibrillary acidicprotein (GFAP), prion protein (PrP), phosphorylated neurofila-ments, and the macrophage marker CD68. Details on theimmunohistochemical procedures are given in SupplementaryMethods S2.
RESULTSPGRNMutation Analysis
We identified nine novel PGRN null mutations in 10 unrelatedand independently ascertained index patients (Table 1). Two werenonsense mutations (c.942C4A [p.Cys314X] and c.1201C4T[p.Gln401X]) and five were small insertions/deletions causingframeshifts (c.361delG [p.Val121TrpfsX135], c.384_387delTATG[p.Gln130SerXfsX125], c.468_474delCTGCTGT [p.Cys157LysfsX97], c.1095_1096delCT [p.Cys366fsX1], and c.1232_1233insGT [p.Ala412fsX1]). We also identified a mutation in the 50 splicedonor site of exon 6 (c.599_708del, IVS611G4A [p.Val200-GlyfsX18]) that caused skipping of exon 6, and one in the 50 splicedonor site of the noncoding exon 0 (g.96239A4T, IVS013A4T[p.0]) leading to IVS0 retention. One mutation, c.599_708del[p.Val200GlyfsX18], was found in the probands of two families(Families F001 and F329) that were ascertained independently.They shared a common haplotype of at least 6.8 Mb or 2.68 cMacross the PGRN locus (Supplementary Table S1), suggesting acommon ancestor in these families. The p.Gln401X mutationcosegregated with FTD in Family F171 (Fig. 1). Cosegregationwith the disease could not be verified for the other PGRNmutations as DNA samples of relatives were not available. Allmutations were identified in French patients; one of them was ofCaribbean origin (Family F587). Identified mutations were absentin 175 unrelated French control individuals, and the p.0 IVS01
3A>T mutation was absent in 90 additional French controlindividuals (530 control chromosomes in total). IVS013A>Twas
TABLE
1.PGRNMutations
Iden
ti¢ed
inFTDPatients
Patients
Age
atons
et(ran
gein
thefamily
)
Mutation
Alia
sGen
omea
Predicted
RNAb
Predicted
pro
tein
c
F587/0
06
58
IVS0
13A4T,
IVS0retention
g.962
39A4T
^p.0
F01
5/00
856(53^7
0)EX4
112
delG
g.10
1141
delG
c.36
1delG
p.Val12
1Trp
fsX13
5F52
1/01
045
EX4
135
_138
delTAGT
g.10
1164
_10
1167
delTAGT
c.38
4_3
87de
lTAGT
p.Gln13
0SerfsX12
5F583
/001
52EX5
16
_112
delC
TGCTG
Tg.10
1349
_101
355de
lCTGCTGT
c.468
_474
delC
TGCTGT
p.Cys15
7Ly
sfsX
97F00
1/00
152
IVS6
11G
4A,E
X6sk
ipping
g.10
1703
G4A
c.59
9_70
8de
lp.Val20
0GlyfsX18
F32
9/00
163
(63^7
2)IV
S6
11G
4A,E
X6sk
ipping
g.10
1703
G4A
c.59
9_70
8de
lp.Val20
0GlyfsX18
F52
4/00
172
(72^7
4)EX9
19C4A
g.10
2460C4A
c.94
2C4A
p.Cys
314X
F26
3/00
174
EX9
116
2_1
63de
lCT
g.10
2613
_102
614de
lCT
c.10
95_1
096de
lCT
p.Cys
366fsX1
F171
/001
57(54^6
2)EX10
122
C4T
g.10
2938
C4T
c.12
01C4T
p.Gln40
1XF05
7/00
761
EX10
153
_154insG
Tg.10
2969
_102
970insG
Tc.12
32_1
233insG
Tp.Ala41
2fsX1
a Num
beringrelative
tothereve
rseco
mplemen
tofG
enBan
kAcc
essionNum
ber
AC00
3043
andstartingat
nt1
.bNum
beringac
cordingto
thelarges
tPRGN
tran
script(Gen
Ban
kAcc
essionNum
berN
M_002
087.2)
andstartingat
tran
slationinitiationco
don.
c Num
beringac
cordingto
thelarges
tPGRN
isoform
(Gen
Pep
tAcc
essionNum
berN
P_00
2078
.1).Exo
nnu
mberingstarts
withno
ncoding¢rste
xonEX
0.EX,e
xon;
IVS,
intron.
HUMANMUTATION 0,1^10,2007 3
Human Mutation DOI 10.1002/humu

located in the splice donor site in intron 0, following thenoncoding exon 0 of the PGRN gene. We previously identified asimilar mutation in the splice donor site of exon 0 (IVS015G>C)in an extended Belgian founder family [Cruts et al., 2006; van derZee et al., 2006]. These mutations alter the canonical sequencerequired for U1 snRNP/50splice site base pairing [Malca et al.,2003] and are expected to reduce U1snRNP binding efficiencyleading to IVS0 retention in the mutant transcript. The nuclearretention signals remaining on the unspliced transcripts mightprevent the mutant mRNA from leaving the nucleus, marking itfor nuclear degradation. All other mutations lead to a prematuretermination codons and are predicted to be degraded by nonsense-mediated mRNA decay (NMD) also producing null alleles.
In addition to the PGRN null mutations, we also found sevenother sequence variants in the patients. Three were syno-nymous base substitutions (c.99C4T [p.Asp33], c.102C4T[p.Pro34], and c.1695C4T [p.Cys565]), and four missensevariants (c.743C4T [p.Pro248Leu], c.773G4A [p.Ser258Asn],c.1253G4A [p.Arg418Gln], and c.1294C4T [p.Arg432Cys]).Two variants (p.Asp33 and p.Arg418Gln) were also found incontrol chromosomes. Three missense variants (p.Pro248Leu,p.Ser258Asn, and p.Arg432Cys), however, were not present in theremaining patients or in the control individuals, indicating thatthey are either rare polymorphisms or possibly causative mutations[van der Zee et al., 2007].
Five index patients with PGRN null mutations had a positivefamily history of FTD (Table 2). The five other mutation carriershad no obvious familial history of neurological diseases (Fig. 2).In four of these patients, the family history was censured; i.e., atleast one parent had died before the age of 75 years. In three ofthese patients, both parents had survived past the age of 58 years,which is the mean age at disease onset in mutation carriers in ourseries. In the last patient (Patient F583/001), no relatives wereaffected and both parents were older than 75 when they died.Unfortunately, it was not possible to determine whether these fivepatients had de novo mutations, false paternity, or if the mutationthey carried presented with incomplete or age-dependantpenetrance, since DNA samples from the deceased parents werenot available. Nevertheless, age-dependant or incomplete pene-trance is likely given the variability in onset ages observed inFamilies F015 (range: 53–70 years) and F329 (range: 63–72 years)
(Table 2). In addition, the common founder haplotype in FamiliesF001 and F329 strongly support the hypothesis that there isincomplete penetrance and not a de novo mutation, at least inthis family.
This study establishes the relative frequency of PGRNmutations in FTD patients at 4.8% (10/210). The frequencyincreased to 9.6% (5/52) in familial forms including both FTD andFTD-MND, and 12.8% (5/39) in patients with familial FTDwithout MND. The PGRN mutation frequency was 3.2% (5/158)in nonfamilial patients. No patients with FTD-MND carriedPGRN mutations (0/43).
Phenotype
The mean age at onset in the 10 index patients with PGRNmutations was 58.779.1 years, and the mean disease durationat examination was 4.671.7 years (Table 3). Age at onset wasvariable between families (Families F521 and F524) as well aswithin families (Families F015 and F329) (Table 1), and rangedfrom 45 to 74 years. All probands but one (Patient F524/001) andtheir affected relatives were diagnosed with FTD at firstpresentation. Patient F524/001 had an atypical presentation withvisual hallucinations at onset and severe psychiatric disorders withparanoid ideas and delirium. Two patients had predominantlanguage disorders at onset, similar to or consistent with adiagnosis of progressive nonfluent aphasia in one case. Otherpatients had predominant behavioral disorders and personalitychanges. Additional signs developed during disease progression:visual hallucinations in 3 out of 10 patients (30%) andparkinsonism in 3 out of 10 patients (30%), one of whom hadreceived neuroleptic drugs. Interestingly, no patients developedMND. The rate of progression was variable, but most patients(5/10) developed apraxia early in the course of the disease. A totalof nine patients died after a mean disease duration of 6.772.2years (range: 4–11 years). The mean age at death was 65.4710.9years (range: 49–85 years). Frontal and/or temporal atrophy werefound in all patients that were investigated with brain CT scan orMRI, as well as brain hypoperfusion on SPECT in the same regions(Table 2).
Compared to MAPT mutation carriers, patients with a PGRNmutation had a later mean onset age (Po0.05) and morefrequently presented with visual hallucinations (Po0.01). Whencompared to the patients without known mutations they had morefrequent hallucinations (Po0.03) but were less frequentlyassociated with MND (Po0.035) (Table 3). Parkinsonismappeared to be more frequent in patients with a PGRN mutationthan in the two other groups, although this was not significant.
There were no correlations between the postulated conse-quences of the mutations and the phenotype. Patient F587/006,carrying the IVS013A4T (g.96239A4T) mutation leading tonuclear transcript degradation, had the same phenotype as theothers.
Neuropathology
Neuropathological examination was performed on Patient F171/002 carrying the PGRN mutation p.Gln401X. The unfixed brainweighed 990 g. External examination revealed diffuse atrophy,especially in the frontotemporal area (Fig. 3a). The substantianigra and the locus coeruleus were not macroscopically depig-mented. On coronal sections (Fig. 3b), cerebral atrophy was severeand diffuse, sparing only the occipital cortex and the hippocampalgyrus. The caudate nucleus was atrophic, as were the pallidum, theputamen, and the thalamus.
FIGURE 1. Segregation of the PGRN c.1201C4T (p.Gln401X)mutation with the disease in Family F171. A¡ected individualsare represented in black, una¡ected individuals in white. Pa-tients with history of dementia but not examined are noted ingray. Sampled individuals are numbered 001,002,003,004, and005 and their genotype is indicated below the symbol.The agesatonset (O), at death (D), and theagesofuna¡ected relatives areindicated when determined.The index patient is indicated by anarrow.
4 HUMANMUTATION 0,1^10,2007
Human Mutation DOI 10.1002/humu
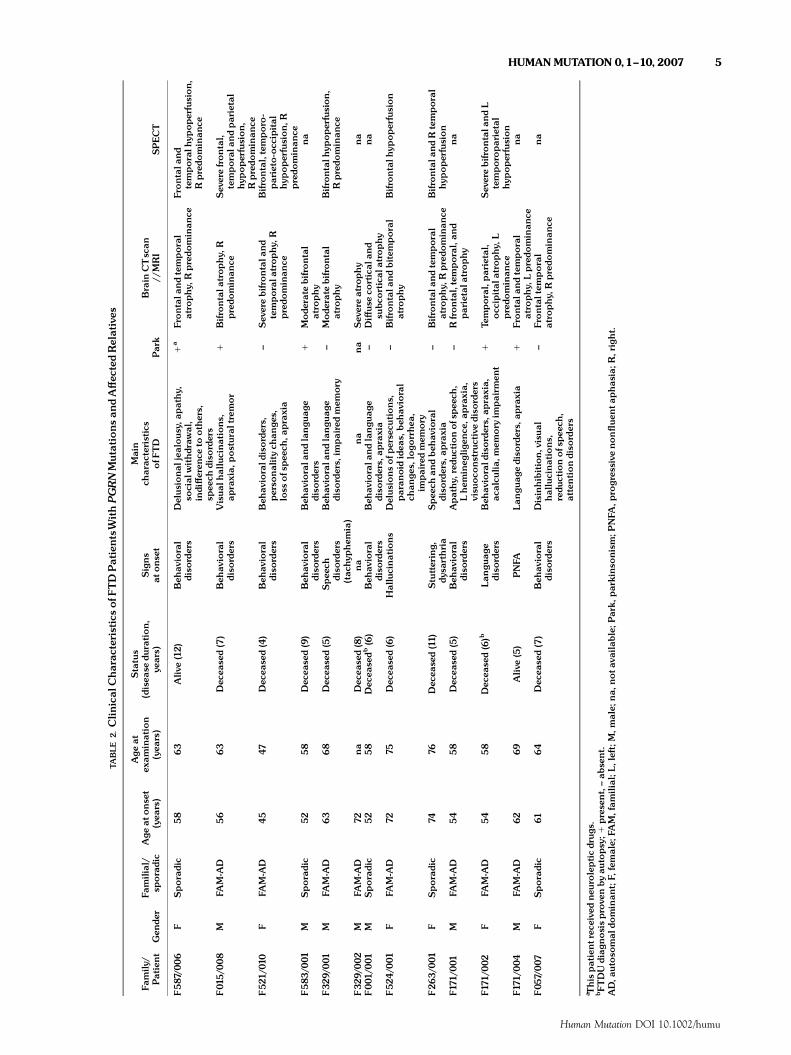
TABLE
2.Clin
ical
Charac
teristicsof
FTDPatientsWithPGRNMutations
andA¡ec
tedRelatives
Family
/Patient
Gen
der
Familial/
sporadic
Age
atonse
t(yea
rs)
Age
atex
amination
(yea
rs)
Status
(disea
sedu
ration,
years)
Signs
atons
et
Main
charac
teristics
ofF
TD
Park
Brain
CTscan
//MRI
SPECT
F587/0
06
FSporadic
58
63
Aliv
e(12)
Beh
avioral
diso
rders
Delus
ional
jealou
sy,a
pathy,
social
withdraw
al,
indi¡eren
ceto
others,
spee
chdiso
rders
1a
Frontala
ndtemporal
atro
phy,
Rpredo
minan
ceFrontala
ndtemporalh
ypoperfusion,
Rpredo
minan
ce
F01
5/00
8M
FAM-A
D56
63
Dec
ease
d(7)
Beh
avioral
diso
rders
Visual
hallucinations
,ap
raxia,
postur
altrem
or
1Bifro
ntala
trop
hy,
Rpredo
minan
ceSev
erefrontal,
temporala
ndparietal
hypoperfusion,
Rpredo
minan
ceF52
1/01
0F
FAM-A
D45
47Dec
eased(4)
Beh
avioral
diso
rders
Beh
aviorald
isorders,
perso
nalitych
ange
s,loss
ofs
peec
h,ap
raxia
^Sev
erebifrontala
ndtemporala
trop
hy,
Rpredo
minan
ce
Bifrontal,temporo-
parieto-occ
ipital
hypoperfusion,
Rpredo
minan
ceF583
/001
MSporadic
5258
Dec
ease
d(9)
Beh
avioral
diso
rders
Beh
aviorala
ndlangu
age
diso
rders
1Mode
rate
bifrontal
atro
phy
na
F32
9/001
MFA
M-A
D63
68
Dec
eased(5)
Spee
chdiso
rders
(tac
hyp
hem
ia)
Beh
aviorala
ndlangu
age
diso
rders,
impairedmem
ory
^Mode
rate
bifrontal
atro
phy
Bifrontalh
ypoperfusion,
Rpredo
minan
ce
F32
9/002
MFA
M-A
D72
na
Dec
eased(8)
na
na
na
Sev
ereatrophy
na
F00
1/00
1M
Sporadic
5258
Dec
easedb
(6)
Beh
avioral
diso
rders
Beh
aviorala
ndlangu
age
diso
rders,
apraxia
^Di¡us
eco
rtical
and
subc
ortical
atrophy
na
F52
4/001
FFA
M-A
D72
75Dec
eased(6)
Hallucinations
Delus
ionsofp
ersecu
tions,
paran
oididea
s,beh
avioral
chan
ges,
logo
rrhe
a,im
pairedmem
ory
^Bifro
ntala
ndbitem
poral
atro
phy
Bifrontalh
ypoperfusion
F26
3/00
1F
Sporadic
7476
Dec
eased(11)
Stuttering,
dysa
rthria
Spee
chan
dbeh
avioral
diso
rders,
apraxia
^Bifro
ntala
ndtemporal
atro
phy,
Rpredo
minan
ceBifrontala
ndRtemporal
hypoperfusion
F171
/001
MFA
M-A
D54
58
Dec
eased(5)
Beh
avioral
diso
rders
Apathy,
redu
ctionof
spee
ch,
Lhem
ineg
ligen
ce,a
praxia,
visu
ocons
truc
tive
diso
rders
^Rfrontal,temporal,an
dparietala
trophy
na
F171
/002
FFA
M-A
D54
58
Dec
ease
d(6)b
Lan
guag
ediso
rders
Beh
aviorald
isorders,
apraxia,
acalcu
lia,m
emory
impairm
ent
1Te
mporal,parietal,
occ
ipital
atrophy,
Lpredo
minan
ce
Sev
erebifrontala
ndL
temporoparietal
hypoperfusion
F171
/004
MFA
M-A
D62
69Aliv
e(5)
PNFA
Lan
guag
ediso
rders,
apraxia
1Frontala
ndtemporal
atro
phy,
Lpredo
minan
cena
F05
7/00
7F
Sporadic
6164
Dec
ease
d(7)
Beh
avioral
diso
rders
Disinhibition,
visu
alhallucinations,
redu
ctionofs
peec
h,attentiondiso
rders
^Frontaltem
poral
atro
phy,
Rpredo
minan
cena
a Thispatientrec
eive
dne
urolepticdrug
s.bFTDUdiag
nosisprove
nby
autopsy;1
prese
nt,^ab
sent.
AD,a
utoso
mal
dominan
t;F,
female;
FAM,fam
ilial;L,
left;M
,male;
na,
nota
vailab
le;P
ark,
parkinso
nism;P
NFA
,pro
gres
sive
non£ue
nta
phas
ia;R
,right.
HUMAN MUTATION 0,1^10,2007 5
Human Mutation DOI 10.1002/humu

On microscopic examination, almost all cortical structuresstudied were severely affected. Massive neuronal loss wasassociated with microvacuolization and sometimes macrovacuoli-zation of layers II and III in the frontotemporal areas (Fig. 3c), aswell as massive cortical astrogliosis. Severe neuronal loss was alsoobserved in the basal ganglia. The white matter displayed multiplefoci of myelin pallor, particularly around the vessels, whichremained unaffected (Fig. 3d). Extensive screening for Taupathology was entirely negative. No PrP, a-synuclein, or Taupathology was found, nor were Pick bodies, ballooned cells, Lewybodies, fibrillary tangles, neuritic plaques, amyloid vasculopathy, ordeposits. GFAP immunohistochemistry confirmed the presence ofprominent astrocytosis in the cerebral cortex (Fig. 3e). No skein orskein-like inclusions were labeled with antiubiquitin antisera in
the lower brainstem nuclei or in the anterior horns of the cervicalspinal cord. In contrast, ubiquitin pathology was severe in neuronsof the isocortex. Ubiquitin-positive inclusions were mainlyobserved in the vicinity of layers II and III. Well-defined roundor crescent-shaped intracytoplasmic inclusions (Fig. 3f) as well asmultiple ubiquitin-positive deposits were observed throughout theperikaryon. Intranuclear inclusions of variable sizes and shapeswere also seen, but the pathognomic ‘‘cat-eye’’ intranuclearinclusions were rare, and only found in the dentate gyrus andthe anterior frontal cortex. Some cortical neurons presented bothintracytoplasmic and ‘‘cat-eye’’ intranuclear inclusions (Fig. 3g).A dense ubiquitin-positive neuritic network was present in most ofthe cortical structures. The fibers were irregular in diameter,fragmented, and distorted. Finally, round oligodendroglial inclu-
FIGURE 2. Pedigrees of ‘‘sporadic’’patientswithPGRNmutations. A¡ected individuals are represented inblack,una¡ected individualsin white.The ages at onset (O), at death (D), and the ages of una¡ected relatives are indicated when determined.The index patientsare indicated by an arrow.
TABLE 3. Phenotypic Characteristics in PatientsWithPGRNMutations andThoseWithMAPTorWithout KnownMutation
PGRNmutations MAPTmutations Non-PGRN non-MAPT
General characteristicsNumber of patients 10a 9b 194c
Gender (male/female) 5/5 2/7 92/102Transmission (FAM/SPO) 50%/50% 100%/0% 24%/76%Mean age at onset, years7SD (range) 58.779.1 (45^74) 51.278.1� (32^61) 60.678.2 (30^82)Mean age at examination, years7SD (range) 63.078.6 (47^76) 55.678.4 (35^64) 66.277.3 (41^84)Meandisease duration at death, years7SD (n) 6.772.2 (9) 6.572.1 (2) 5.872.9 (58)
Main phenotypic characteristicsMotoneuron disease 0% 0% 23%�
Parkinsonism 30% 22% 19%Othermovement disorders 10% 22% 9%Oculomotor disorders 0% 0% 5%Hallucinations 30% 0%� 5%�
aTen index patients.bIncludes six index patients withMAPTmutation recruited in the present study, and three additional patients ascertained di¡erently.cIncludes 43 patients with FTD-MND.�P-value o0.05, comparisons between the patients withPGRNmutations and thosewithMAPTmutations or thosewith any mutation.FAM, familial transmission; n, number of patients; SD, standard deviations; SPO, sporadic patients.
6 HUMANMUTATION 0,1^10,2007
Human Mutation DOI 10.1002/humu

sions were observed in the cortex and in the superficial whitematter (Fig. 3h). TDP-43-positive cytoplasmic and intranuclearinclusions, as well as neuritic deposits, were found in both thefrontal cortex and dentate gyrus (Fig. 3i, j, and k).
DISCUSSION
In the present study, we analyzed a large series of FTD patientsfor mutations in the PGRN gene and identified nine novelmutations in 10 French patients; one of them was of Caribbeanorigin, ascertained independently. PGRN mutations were pre-viously identified in familial FTD patients from North America,Belgium, Sweden, United Kingdom, the Netherlands, and China,some of which had earlier published linkage to chromosome 17
[Baker et al., 2006; Cruts et al., 2006; Gass et al., 2006; Hueyet al., 2006; Masellis et al., 2006]. The most frequent mutationreported so far (g.96241G4C, IVS015G4C) was found in eightBelgian families and resulted from an ancestral founder effect[Cruts et al., 2006; van der Zee et al., 2006]. We did not detectthis mutation in our series.
As in previous studies, all mutations identified in this study werepredicted to result in null alleles. Except for the exon 0 splicedonor site mutation (IVS013A4T) that is predicted to lead tonuclear degradation of the unspliced transcript, all nonsense andframeshift mutations are expected to generate mutant mRNAtranscripts containing premature termination codons and there-fore be degraded by NMD. Previous studies of PGRN nonsenseand frameshift mutations have provided firm evidence for thedegradation of mutant mRNA by NMD [Baker et al., 2006; Cruts
FIGURE 3. Neuropathology of Patient F171/002 with the PGRN Gln401X mutation. A: Macroscopic view of the right hemisphereshowing di¡use atrophy, predominantly a¡ecting frontal and temporal lobes. B:Coronal section passing throughout themamillarybodies showing severe atrophy of the caudate and lenticular nuclei.C: Microscopic view of the super¢cial layer of the frontal cortex:macrovacuolizationof layers II and IIIwith severeneuronal loss andprominent reactiveastrogliosis (hematoxylinandeosin [HE],OM�100). D: Histological view of the frontal cortex and white matter: note di¡use myelin pallor (Luxol-Phloxin stain, OM �25).E:GFAP immunochemistry showing reactive astrogliosis in the cortex (arrow) and the sparing of white matter (OM �200). F:Cres-cent-shaped intracytoplasmic inclusions (OM �600).G: Dense intracytoplasmic inclusion (arrowhead) with typical ‘‘cat-eye’’ intra-nuclear inclusion (arrow) (OM �1000). H: Neuritic pathology in the frontal cortex: irregularly-shaped neuritic process (arrow) andubiquitin-positive oligodendroglial nuclear inclusions (arrowhead) (OM �600). I:TDP-43-positive inclusions in the dentate gyrus(OM �400). J:TDP43-positive neurites (arrow) in the frontal cortex (OM �400). K:TDP-43-positive nuclear inclusions (arrow) inthe frontal cortex (OM�400).
HUMAN MUTATION 0,1^10,2007 7
Human Mutation DOI 10.1002/humu

et al., 2006]. Therefore, partial loss of functional PGRN seeminglyleads to neurodegeneration through haploinsufficiency. PGRNmodulates growth of a variety of cells and is involved in multipleprocesses as development, epithelial homeostasis, reproduction,and inflammation [He and Bateman, 1999; He et al., 2003].PGRN is widely expressed throughout different tissues, includingthe nervous system, where it is abundantly expressed in corticalneurons, particularly in granule cells of the hippocampus, as well asin the Purkinje cells of the cerebellum [Daniel et al., 2000].Although the function of PGRN in the central nervous system isstill poorly understood, its presence seems to be crucial formaintaining neuronal survival in the cerebral cortex.
This study establishes the relative frequency of PGRN nullmutations in a predominantly French FTD patient sample at 4.8%(10/210) and 12.8% (5/39) in the familial subgroup with pureFTD. These frequencies are lower than those observed the Belgianpatient sample [Cruts et al., 2006], due to the high frequency of afounder mutation in the Belgian population. In addition, thePGRN mutation frequency (4.8%, 10/210) was 1.5-fold higherthan the frequency of MAPT mutations in the same totalpopulation patients (2.9%, 6/210). Unexpectedly, in the subgroupof familial FTD patients, the frequency of MAPT gene mutations(11%, 6/52) was slightly higher than the frequency of PGRN nullmutations (9.6%, 5/52). This is explained by the fact that half ofthe patients in our series with PGRN mutations had no familyhistory of the disease (5/10), whereas the patients with MAPTgene mutations all had a positive familial history.
More interesting is that half of the patients with a PGRNmutation (5/10 index patients) had no obvious family history ofdementia and were apparently sporadic. Accordingly, PGRNmutations accounted for 3.2% of nonfamilial FTD (5/158). Theparents of four sporadic FTD patients died after the age of 58years, the mean age at onset in our population. This raises thepossibility of de novo mutations that could not be investigated asDNA from the deceased parents was not available. However, theobservation of an 81-year-old asymptomatic obligate carrier in theBelgian PGRN founder family (Family DR8) has already demon-strated that incomplete or age-dependent penetrance can occur inFTDU-17 families [van der Zee et al., 2006; Cruts et al., 2006].This indicates that genetic testing should now be considered moresystematically, even in patients without obvious family history ofthe disease, and that an accurate estimation of penetrance isneeded for genetic counseling of at-risk individuals.
The mean age at onset of FTD in patients with a PGRNmutation was earlier than in other FTD patients with nomutations, but varied between families, ranging from 45 to 74years, as well as among affected relatives. In contrast to previousstudies [Baker et al., 2006; Cruts et al., 2006], onset after 70 yearof age was not uncommon in our patients (20%). Progressivenonfluent aphasia has been reported as a predominant initialsymptom in at least four families carrying PGRN mutations [vander Zee et al., 2006; Cruts et al., 2006; Mukherjee et al., 2006;Snowden et al., 2006]. In our series, the frequency of apredominant language disorders at onset was 20%. The clinicalphenotype was also unusual in our patients because of the highfrequency of visual hallucinations, not previously reported inmutation carriers, and the occurrence of apraxia early in thecourse of the disease. Visual hallucinations were much morefrequent (30%) in this series than usually admitted in FTD(Table 3). We emphasize that half of the patients developed earlyand severe apraxia in our series. Although only one PGRN familywith corticobasal degeneration (CBD) has been reported so far[Masellis et al., 2006], our study shows that PGRN mutation
should also be looked for in patients with a CBD phenotype.Disease progression with early instrumental dysfunction reflectsthe rapid progression of the neurodegenerative damage to theposterior cortical regions and suggests an aggressive pathologicalprocess. In this series, the frequency of parkinsonism was higher inpatients with mutations in PGRN than in patients with a MAPTmutation or in patients without an identified mutation. Patientswith PGRN mutations also had a later mean age at onset thanthose with MAPT mutations. Interestingly, none of the 43 indexpatients with associated MND carried a PGRN mutation. Thisstudy clearly confirms therefore that FTDU-17 and FTD-MNDare two genetically distinct diseases although they are bothneuropathologically characterized by the presence of ub-irinclusions.
Neuropathological examination of Patient F171/002 carryingthe PGRN p.Gln401X mutation showed ub-ir neuronal cytoplas-mic inclusions and neurites, associated with typical lentiform ub-ir‘‘cat-eye’’ intranuclear inclusions. These changes were similar tothe neuropathological characteristics described in other FTDU-17families [Rosso et al., 2001; Rademakers et al., 2002, 2004; vander Zee et al., 2006; Mackenzie et al., 2006]. Although PGRN ishighly expressed in Purkinje cells of the cerebellum, it is notablethat this structure appeared normal. TDP-43 was recentlyidentified as the major component of the ub-ir inclusions in bothFTLDU and FTD-MND [Neumann et al., 2006; Araı et al., 2006;Davidson et al., 2007]. TDP-43–positive cytoplasmic and nuclearinclusions and neurites were also present in our patient carryingthe PGRN mutation p.Gln401X. Since PGRN is not found in theub-ir inclusions [Baker et al., 2006, Cruts et al., 2006], thisobservation suggests an indirect relation between these twoproteins. It raises the question of the mechanism by which PGRNhaploinsufficiency may lead to TDP-43 aggregation.
In conclusion, this study provides convincing evidence thatmutations in PGRN are an important cause of dementia. Althoughthe exact biological function of PGRN in the central nervoussystem remains elusive, it appears to prevent neuronal death. Itremains to be determined why partial loss of functional PGRN, awidely expressed multifunctional protein, leads to a neurodegen-erative disease that predominantly affects the frontal and temporalcortices.
ACKNOWLEDGMENTS
We thank Dr. Merle Ruberg for her helpful suggestions on themanuscript (INSERM U679, Hopital Pitie-Salpetriere, Paris,France), Sophie Rivaud-Pechoux for the statistical analyses(INSERM U679, Hopital Pitie-Salpetriere, Paris, France), thepersonnel of the VIB Genetic Service Facility (www.vibgeneticservicefacility.be) for the genetic analyses of PGRN, and LydiaGuennec, Christiane Penet, Isabelle Lagroua, and ChristelleDussert at the DNA and cell bank (IFR070, Hopital Pitie-Salpetriere, Paris, France) for technical assistance. This study wassupported in part by Contrat de recherche et d’innovation clinique(CRIC) AP-HP (01107 to A.B.), Groupe d’interet scientifique(GIS)-Institut des Maladies Rares (A03081DS/APS03002DSA toA.B.), Agence Nationale pour la Recherche (to A.B.), France-Alzheimer Association (to A.B.), EU contract LSHMCT-2003-503330 APOPIS to A.B. and C.V.B.); and a Zenith Award of theAlzheimer’s Association, USA (to C.V.B.). I.L.B had a fellowshipfrom the France-Alzheimer Association; and J.v.d.Z. is holder of aPhD fellowship from the Institute for Science and Technology Flanders(IWT-F), Belgium. Scientific Research Flanders (FWO-F) provided aPhD fellowship to I.G. and a postdoctoral fellowship to M.C.
8 HUMANMUTATION 0,1^10,2007
Human Mutation DOI 10.1002/humu

APPENDIX A
The French Research Network on FTD/FTD-MND includes:Alexis Brice (Hopital Pitie-Salpetriere, Paris), Franc-oise Clerget-Darpoux (Hopital Paul-Brousse, Villejuif), Gilles Defer (CentreHospitalier Universitaire [CHU] Cote de Nacre, Caen), MiraDidic (CHU La Timone, Marseille), Claude Desnuelle (CHU,Nice), Bruno Dubois (Hopital Pitie-Salpetriere, Paris), CharlesDuyckaerts (Hopital Pitie-Salpetriere, Paris), Veronique Golfier(Centre Hospitalier [CH], Saint-Brieuc), Didier Hannequin(Rouen University Hospital), Lucette Lacomblez (Hopital Pitie-Salpetriere, Paris), Isabelle Le Ber (Hopital Pitie-Salpetriere,Paris), Bernard-Franc-ois Michel (CH Sainte-Marguerite, Mar-seille), Florence Pasquier (CHU Roger Salengro, Lille), CatherineThomas-Anterion (CHU Bellevue, Saint-Etienne), Michele Puel(CHU Purpan, Toulouse), Franc-ois Salachas (Pitie-Salpetriere,Paris), Franc-ois Sellal (Hopitaux Civils and INSERM U692,Strasbourg), Martine Vercelletto (CHU Laennec, Nantes), PatriceVerpillat (Hopital Pitie-Salpetriere, Paris), and William Camu(CHU Gui de Chauliac, Montpellier).
REFERENCES
Araı T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D,
Tsuchiya K, Yoshida M, Hashizume Y, Oda T. 2006. TDP-43 is a
component of ubiquitin-positive tau-negative inclusions in frontotem-
poral lobar degeneration and amyotrophic lateral sclerosis. Biochem
Biophys Res Commun 22;351:602–611.
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R,
Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S,
Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D,
Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R,
McGowan E, Mann D, Boeve B, Feldman H, Hutton M. 2006.
Mutations in progranulin cause tau-negative frontotemporal dementia
linked to chromosome 17. Nature 442:916–919.
Bateman A, Benett HPJ. 1998. Granulins: the structure and function of an
emerging family of growth factors. J Endocrinol 158:145–151.
Benson G. 1999. Tandem repeats finder: a program to analyze DNA
sequences. Nucleic Acids Res 27:573–580.
Bronner IF, Rizzu P, Seelaar H, van Mil SE, Anar B, Azmani A, Kaat LD,
Rosso S, Heutink P, van Swieten JC. 2007. Progranulin mutations in
Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet 15:
369–374.
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D,
Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C,
Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den
Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van
Broeckhoven C. 2006. Null mutations in progranulin cause ubiquitin-
positive frontotemporal dementia linked to chromosome 17q21. Nature
442:920–924.
Daniel R, Carmichael KP, Halper J, Bateman A. 2000. Cellular localization
of gene expression for progranulin. J Histochem Cytochem 48:999–1009.
Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary
D, Snowden JS, Mann DM. 2007. Ubiquitinated pathological lesions in
frontotemporal lobar degeneration contain the TAR DNA-binding
protein, TDP-43. Acta Neuropathol (Berl) Jan 12; [Epub ahead of print]
Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois
B, Saugier-Veber P, Martin C, Penet C, Charbonnier F, Agid Y, Frebourg
T, Brice A. 1998. Segregation of a missense mutation in the microtubule-
associated proteine tau gene with familial fronto-temporal dementia and
parkinsonism. Hum Mol Genet 7:1825–1829.
Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R,
Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-
Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio
E, Johnson N, Weintraub S, Mesulam M, White CL 3rd, Woodruff B,
Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, Rademakers
R. 2006. Mutations in progranulin are a major cause of ubiquitin-positive
frontotemporal lobar degeneration. Hum Mol Genet 15:2988–3001.
He Z, Bateman A. 1999. Progranulin gene expression regulates epithelial
cell growth and promotes tumor growth in vivo. Cancer Res 59:
3222–3229.
He Z, Bateman A. 2003. Progranulin (granulin-epithelin precursor, PC-
cell-derived growth factor, acrogranin) mediates tissue repair and
tumorigenesis. J Mol Med 81:600–612
He Z, Ong CH, Halper J, Bateman A. 2003. Progranulin is a mediator of
the wound response. Nat Med 9:225–229.
Huey ED, Grafman J, Wassermann EM, Pietrini P, Tierney MC, Ghetti B,
Spina S, Baker M, Hutton M, Elder JW, Berger SL, Heflin KA, Hardy J,
Momeni P. 2006. Characteristics of frontotemporal dementia patients
with a progranulin mutation. Ann Neurol 60:374–380.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H,
Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J,
Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de
Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM,
Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun
H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR,
Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd
D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann
D, Lynch T, Heutink P. 1998. Association of missense and 50-splice site
mutations in tau with the inherited dementia FTDP-17. Nature
393:702–705.
Lendon CL, Lynch T, Norton J, McKeel DW Jr, Busfield F, Craddock N,
Chakraverty S, Gopalakrishnan G, Shears SD, Grimmett W, Wilhelmsen
KC, Hansen L, Morris JC, Goate AM. 1998. Hereditary dysphasic
disinhibition dementia: a frontotemporal dementia linked to 17q21-22.
Neurology 50:1546–1555.
Lund and Manchester Groups. 1994. Consensus statement: clinical and
neuropathological criteria for fronto-temporal dementia. J Neurol
Neurosurg Psychiatry 57:416–418.
Mackenzie IR, Baker M, West G, Woulfe J, Qadi N, Gass J, Cannon A,
Adamson J, Feldman H, Lindholm C, Melquist S, Pettman R, Sadovnick
AD, Dwosh E, Whiteheart SW, Hutton M, Pickering-Brown SM.
2006. A family with tau-negative frontotemporal dementia and
neuronal intranuclear inclusions linked to chromosome 17. Brain 129:
853–867.
Malca H, Shomron N, Ast G. 2003. The U1 snRNP Base pairs with the 50
splice site within a penta-snRNP complex. Mol Cell Biol 23:3442–3455.
Masellis M, Momeni P, Meschino W, Heffner R, Elder J, Sato C, Liang Y, St
George-Hyslop P, Hardy J, Bilbao J, Black S, Rogaeva E. 2006. Novel
splicing mutation in the progranulin gene causing familial corticobasal
syndrome. Brain 129:3115–3123.
Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S,
Behrens MI, Hinrichs AL, Norton J, Levitch D, Taylor-Reinwald L,
Gitcho M, Tu PH, Tenenholz Grinberg L, Liscic RM, Armendariz J,
Morris JC, Goate AM. 2006. HDDD2 is a familial frontotemporal lobar
degeneration with ubiquitin-positive, tau-negative inclusions caused by a
missense mutation in the signal peptide of progranulin. Ann Neurol
60:314–322.
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman
M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J,
Benson DF. 1998. Frontotemporal lobar degeneration: a consensus on
clinical diagnostic criteria. Neurology 51:1546–1554.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou
TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller
BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA,
Trojanowski JQ, Lee VM. 2006. Ubiquitinated TDP-43 in frontotempor-
al lobar degeneration and amyotrophic lateral sclerosis. Science 314:
130–133.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L,
Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. 1998. Tau
is a candidate gene for chromosome 17 frontotemporal dementia. Ann
Neurol 43:815–825.
Rademakers R, Cruts M, Dermaut B, Sleegers K, Rosso SM, Van den
Broeck M, Backhovens H, van Swieten J, van Duijn CM, Van
Broeckhoven C. 2002. Tau negative frontal lobe dementia at 17q21:
significant fine mapping of the candidate region to a 4. 8 cM interval.
Mol Psychiatry 7:1064–1074.
HUMANMUTATION 0,1^10,2007 9
Human Mutation DOI 10.1002/humu

Rademakers R, Cruts M, Van Broeckhoven C. 2004. The role of tau
(MAPT) in frontotemporal dementia and related tauopathies. Human
Mutat 24:277–295.
Rosso SM, Kamphorst W, de Graaf B, Willemsen R, Ravid R, Niermeijer
MF, Spillantini MG, Heutink P, van Swieten JC. 2001. Familial
frontotemporal dementia with ubiquitin-positive inclusions is linked to
chromosome 17q21–22. Brain 124:1948–1957.
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL,
Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T,
Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA,
Gydesen S, Fisher EM, Collinge J. 2005. Mutations in the endosomal
ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat
Genet 37:806–808.
Snowden JS, Pickering-Brown SM, Mackenzie IR, Richardson AM, Varma
A, Neary D, Mann DM. 2006. Progranulin gene mutations associated
with frontotemporal dementia and progressive non-fluent aphasia.
Brain129:3091–3102.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A,
Ghetti B. 1998. Mutation in the tau gene in familial multiple system
tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:
7737–7741.
van der Zee J, Rademakers R, Engelborghs S, Gijselinck I, Bogaerts V,
Vandenberghe R, Santens P, Caekebeke J, De Pooter T, Peeters K, Lubke
U, Van den Broeck M, Martin JJ, Cruts M, De Deyn PP, Van
Broeckhoven C, Dermaut B. 2006. A Belgian ancestral haplotype
harbours a highly prevalent mutation for 17q21-linked tau-negative
FTLD. Brain 129:841–852.
van der Zee J, Le Ber I, Maurer-Stroh S, Engelborghs S, Gijselinck I,
Camuzat A, Brouwers N, Vandenberghe R, Sleegers K, Hannequin D,
Dermaut B, Schymkowitz J, Campion D, Santens P, Martin JJ, Lacomblez
L, De Pooter T, Peeters K, Mattheijssens M, Vercelletto M, Van den
Broeck M, Cruts M, De Deyn P, Rousseau F, Brice A, Van Broeckhoven
C. 2007. Mutations other than null mutations producing a pathogenic
loss of progranulin in frontotemporal dementia. Hum Mutat 28:416.
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D,
Pestronk A, Whyte MP, Kimonis VE. 2004. Inclusion body
myopathy associated with Paget disease of bone and frontotemporal
dementia is caused by mutant valosin-containing protein. Nat Genet
36:377–381.
Weckx S, Del-Favero J, Rademakers R, Claes L, Cruts M, De Jonghe P, Van
Broeckhoven C, De Rijk P. 2005. novoSNP, a novel computational tool
for sequence variation discovery. Genome Res 15:436–442.
10 HUMANMUTATION 0,1^10,2007
Human Mutation DOI 10.1002/humu
Related Documents











