Molecular Cell Article Preventing Nonhomologous End Joining Suppresses DNA Repair Defects of Fanconi Anemia Adele Adamo, 1,6 Spencer J. Collis, 2,3,6 Carrie A. Adelman, 2 Nicola Silva, 1,4,5 Zuzana Horejsi, 2 Jordan D. Ward, 2 Enrique Martinez-Perez, 5 Simon J. Boulton, 2, * and Adriana La Volpe 1, * 1 Institute of Genetics and Biophysics ‘‘Adriano Buzzati-Traverso,’’ CNR, Via Pietro Castellino 111, 80131, Napoli, Italy 2 DNA Damage Response Laboratory, London Research Institute, Clare Hall, South Mimms, EN6 3LD, UK 3 Institute for Cancer Studies, University of Sheffield Medical School, Beech Hill Road, Sheffield, S10 2RX, UK 4 Department of Structural and Functional Biology, University of Naples, ‘‘Federico II,’’ Complesso di Monte S. Angelo, 80138, Napoli, Italy 5 Clinical Sciences Division, Imperial College, Du Cane Road, London, W12 0NN, UK 6 These authors contributed equally to this work *Correspondence: [email protected] (S.J.B.), [email protected] (A.L.V.) DOI 10.1016/j.molcel.2010.06.026 SUMMARY Fanconi anemia (FA) is a complex cancer suscepti- bility disorder associated with DNA repair defects and infertility, yet the precise function of the FA proteins in genome maintenance remains unclear. Here we report that C. elegans FANCD2 (fcd-2) is dispensable for normal meiotic recombination but is required in crossover defective mutants to prevent illegitimate repair of meiotic breaks by nonhomolo- gous end joining (NHEJ). In mitotic cells, we show that DNA repair defects of C. elegans fcd-2 mutants and FA-deficient human cells are significantly sup- pressed by eliminating NHEJ. Moreover, NHEJ factors are inappropriately recruited to sites of repli- cation stress in the absence of FANCD2. Our findings are consistent with the interpretation that FA results from the promiscuous action of NHEJ during DNA repair. We propose that a critical function of the FA pathway is to channel lesions into accurate, as opposed to error-prone, repair pathways. INTRODUCTION Fanconi anemia (FA) is a complex multigene disorder character- ized by severe genome instability, congenital abnormalities, acute myeloid leukemia, and/or bone marrow failure and cancer predisposition (D’Andrea and Grompe, 2003; Fanconi, 1967; Kennedy and D’Andrea, 2005). The hallmark of FA cells is exqui- site sensitivity to interstrand crosslinking (ICL) agents, indicative of a repair defect in response to agents that block the replication fork. The FA pathway is composed of at least 13 proteins corre- sponding to the complementation groups found mutated in FA patients: FANCA, B, C, D1, D2, E, F, G, I, J, L, M, and N (Moldovan and D’Andrea, 2009). In response to replication stress, the FA core complex (comprised of FANCA, B, C, E, F, G, L, and M) catalyzes the monoubiquitylation of the FANCD2/ FANCI heterodimer, which promotes its recruitment to damaged replication forks. Once recruited, FANCD2/FANCI colocalizes with BRCA2 (FANCD1), PALB2 (FANCN), and FANCJ in nuclear repair foci (Kennedy and D’Andrea, 2005; Wang, 2007). Since FANCJ, BRCA2, and PALB2 are essential for homologous recombination (HR) (Boulton, 2006; D’Andrea and Grompe, 2003; Martin et al., 2005; Petalcorin et al., 2006), it has been proposed that the FA pathway maintains genomic integrity by facilitating HR-mediated repair, but how this occurs remains unclear. The nematode C. elegans has emerged as a powerful system for the study of FA in the context of a whole organism (Youds et al., 2009). Similar to their vertebrate counterparts, mutants in C. elegans FCD-2 (FANCD2), FNCI-1 (FANCI), DOG-1 (FANCJ), and FCM-1 (FANCM) are exquisitely sensitive to ICL- inducing agents and display chromosomal aberrations and enhanced mutagenesis following treatment with these agents (Collis et al., 2006; Muzzini et al., 2008; Youds et al., 2009). At the molecular level, the C. elegans FCD-2 and FNCI-1 proteins are monoubiquitylated in response to replication stress, and this requires the replication stress checkpoint proteins ATL-1 (ATR), CHK-1 (CHK1), and RPA-1, as well as the FA core complex protein FCM-1 (Collis et al., 2006, 2007; Lee et al., 2010). Once activated, FCD-2 is recruited to nuclear DNA repair foci, demon- strating the conservation of the central elements of the FA pathway (Collis et al., 2006; Lee et al., 2010). The C. elegans germline also offers an excellent system to study the repair of double strand breaks (DSBs), which are phys- iologically generated by the topoisomerase-like protein SPO-11 during meiosis. Meiotic DSBs are preferentially repaired by HR using a parental homolog to form interhomolog crossovers that are essential for accurate chromosome segregation at the first meiotic division (Page and Hawley, 2003). In wild-type C. ele- gans, RAD-51 foci, which form at meiotic DSBs, arise during the late zygotene and early pachytene stages, and then rapidly decrease in number during pachytene stage as meiotic DSB repair progresses (Colaia ´ covo et al., 2003; Rinaldo et al., 2002). Both timely disappearance of RAD-51 foci at pachytene and establishment of crossovers require the presence of the mismatch repair related proteins MSH-4 and MSH-5 (Kelly et al., 2000; Zalevsky et al., 1999) and the assembly of the syn- aptonemal complex (SC), a proteinaceous structure that is Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc. 25

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Molecular Cell
Article
Preventing Nonhomologous End JoiningSuppresses DNA Repair Defects of Fanconi AnemiaAdele Adamo,1,6 Spencer J. Collis,2,3,6 Carrie A. Adelman,2 Nicola Silva,1,4,5 Zuzana Horejsi,2 Jordan D. Ward,2
Enrique Martinez-Perez,5 Simon J. Boulton,2,* and Adriana La Volpe1,*1Institute of Genetics and Biophysics ‘‘Adriano Buzzati-Traverso,’’ CNR, Via Pietro Castellino 111, 80131, Napoli, Italy2DNA Damage Response Laboratory, London Research Institute, Clare Hall, South Mimms, EN6 3LD, UK3Institute for Cancer Studies, University of Sheffield Medical School, Beech Hill Road, Sheffield, S10 2RX, UK4Department of Structural and Functional Biology, University of Naples, ‘‘Federico II,’’ Complesso di Monte S. Angelo, 80138, Napoli, Italy5Clinical Sciences Division, Imperial College, Du Cane Road, London, W12 0NN, UK6These authors contributed equally to this work*Correspondence: [email protected] (S.J.B.), [email protected] (A.L.V.)
DOI 10.1016/j.molcel.2010.06.026
SUMMARY
Fanconi anemia (FA) is a complex cancer suscepti-bility disorder associated with DNA repair defectsand infertility, yet the precise function of the FAproteins in genome maintenance remains unclear.Here we report that C. elegans FANCD2 (fcd-2) isdispensable for normal meiotic recombination butis required in crossover defective mutants to preventillegitimate repair of meiotic breaks by nonhomolo-gous end joining (NHEJ). In mitotic cells, we showthat DNA repair defects of C. elegans fcd-2 mutantsand FA-deficient human cells are significantly sup-pressed by eliminating NHEJ. Moreover, NHEJfactors are inappropriately recruited to sites of repli-cation stress in the absence of FANCD2. Our findingsare consistent with the interpretation that FA resultsfrom the promiscuous action of NHEJ during DNArepair. We propose that a critical function of the FApathway is to channel lesions into accurate, asopposed to error-prone, repair pathways.
INTRODUCTION
Fanconi anemia (FA) is a complex multigene disorder character-
ized by severe genome instability, congenital abnormalities,
acute myeloid leukemia, and/or bone marrow failure and cancer
predisposition (D’Andrea and Grompe, 2003; Fanconi, 1967;
Kennedy and D’Andrea, 2005). The hallmark of FA cells is exqui-
site sensitivity to interstrand crosslinking (ICL) agents, indicative
of a repair defect in response to agents that block the replication
fork. The FA pathway is composed of at least 13 proteins corre-
sponding to the complementation groups found mutated in FA
patients: FANCA, B, C, D1, D2, E, F, G, I, J, L, M, and N
(Moldovan and D’Andrea, 2009). In response to replication
stress, the FA core complex (comprised of FANCA, B, C, E, F,
G, L, and M) catalyzes the monoubiquitylation of the FANCD2/
FANCI heterodimer, which promotes its recruitment to damaged
replication forks. Once recruited, FANCD2/FANCI colocalizes
with BRCA2 (FANCD1), PALB2 (FANCN), and FANCJ in nuclear
repair foci (Kennedy and D’Andrea, 2005; Wang, 2007). Since
FANCJ, BRCA2, and PALB2 are essential for homologous
recombination (HR) (Boulton, 2006; D’Andrea and Grompe,
2003; Martin et al., 2005; Petalcorin et al., 2006), it has been
proposed that the FA pathway maintains genomic integrity by
facilitating HR-mediated repair, but how this occurs remains
unclear.
The nematode C. elegans has emerged as a powerful system
for the study of FA in the context of a whole organism (Youds
et al., 2009). Similar to their vertebrate counterparts, mutants
in C. elegans FCD-2 (FANCD2), FNCI-1 (FANCI), DOG-1
(FANCJ), and FCM-1 (FANCM) are exquisitely sensitive to ICL-
inducing agents and display chromosomal aberrations and
enhanced mutagenesis following treatment with these agents
(Collis et al., 2006; Muzzini et al., 2008; Youds et al., 2009).
At the molecular level, the C. elegans FCD-2 and FNCI-1 proteins
are monoubiquitylated in response to replication stress, and this
requires the replication stress checkpoint proteins ATL-1 (ATR),
CHK-1 (CHK1), and RPA-1, as well as the FA core complex
protein FCM-1 (Collis et al., 2006, 2007; Lee et al., 2010). Once
activated, FCD-2 is recruited to nuclear DNA repair foci, demon-
strating the conservation of the central elements of the FA
pathway (Collis et al., 2006; Lee et al., 2010).
The C. elegans germline also offers an excellent system to
study the repair of double strand breaks (DSBs), which are phys-
iologically generated by the topoisomerase-like protein SPO-11
during meiosis. Meiotic DSBs are preferentially repaired by HR
using a parental homolog to form interhomolog crossovers that
are essential for accurate chromosome segregation at the first
meiotic division (Page and Hawley, 2003). In wild-type C. ele-
gans, RAD-51 foci, which form at meiotic DSBs, arise during
the late zygotene and early pachytene stages, and then rapidly
decrease in number during pachytene stage as meiotic DSB
repair progresses (Colaiacovo et al., 2003; Rinaldo et al.,
2002). Both timely disappearance of RAD-51 foci at pachytene
and establishment of crossovers require the presence of the
mismatch repair related proteins MSH-4 and MSH-5 (Kelly
et al., 2000; Zalevsky et al., 1999) and the assembly of the syn-
aptonemal complex (SC), a proteinaceous structure that is
Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc. 25

Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
required to maintain homologous chromosomes in close align-
ment (Colaiacovo et al., 2003). In C. elegans, meiotic DSBs are
not a prerequisite for SC assembly (Dernburg et al., 1998), and
axis morphogenesis and SC formation are not required for
loading of the RAD-51 strand-exchange protein onto DSBs
(Colaiacovo et al., 2003; Rinaldo et al., 2002). These attributes
of C. elegans meiosis allow factors that affect synapsis to be
distinguished from those required for meiotic DSB repair. For
instance, despite a failure to form crossovers, the lack of chro-
mosome fragments in the oocytes of msh-4/5 and SC mutants
(syp-1 or syp-2) demonstrates that these mutants retain the
ability to repair meiotic DSBs. The repair observed in msh-4/5
and syp-1/2 mutants is most likely achieved by HR repair
between sister chromatids. Indeed, recent studies have estab-
lished that C. elegans BRCA1 (BRC-1), while being dispensable
for crossover formation between homologs, is essential for inter-
sister HR repair of meiotic DSBs in SC mutants (Adamo et al.,
2008).
Prompted by the infertility of FA patients (Alter, 1993) and the
presence of meiotic defects in FANCD2 knockout mice
(Houghtaling et al., 2003), we have investigated the meiotic
role of C. elegans FCD-2. Whereas FCD-2 is dispensable for
crossover recombination in normal meiosis, loss of FCD-2 in
syp-2 (SC defective) or msh-4 (crossover defective) mutants re-
sulted in erroneous repair of meiotic DSBs, a phenotype that
could be suppressed by deleting the DNA ligase component
lig-4. Strikingly, eliminating LIG-4 activity also suppressed the
ICL-induced defects of fcd-2 mutants to levels indistinguishable
from wild-type. Similarly, deletion or inhibition of NHEJ sup-
pressed the ICL sensitivity of FA-deficient mammalian cells.
Consistent with a role for the FA pathway in ensuring that DNA
lesions are repaired with high fidelity, cells lacking FANCD2
exhibited promiscuous recruitment of DNA-PKcs to sites of repli-
cation stress. Our findings reveal that the FA pathway
suppresses illegitimate repair of meiotic DSBs and ICL lesions
by NHEJ and that inhibition of NHEJ suppresses several FA
phenotypes that stem from defects in DNA repair.
RESULTS
Crossovers Occur Normally but Meiotic RAD-51 FociAre Elevated in fcd-2 MutantsAlthough FA patients and mouse models of FA have reduced
fertility, and FANCD2 has been previously implicated in DNA
repair processes, upon initial examination, no apparent meiotic
phenotype is detectable in C. elegans fcd-2 mutants. Both the
incidence of males (0.04%), reflecting the frequency of X chro-
mosome nondisjunction during meiosis, and the levels of
embryonic lethality (0.51%), indicative of missegregation of the
autosomes, appear normal in fcd-2 mutants (Collis et al., 2006;
Polanowska et al., 2006; this study). Homolog pairing, synapsis,
and chiasmata formation also occur as normal in fcd-2 mutant,
indicating that FCD-2 is dispensable for crossing over (Figures
1A and 1B). To confirm this, we assayed the frequency of recom-
bination in the fcd-2 mutant in two different chromosomal inter-
vals: (1) dpy-5 to unc-29 on chromosome I (a region with a low
level of recombination of about 1 cM/Mb), and (2) unc-60 to
dpy-11 on chromosome V (an interval with a relatively high
26 Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc.
recombination frequency of about 3.7 cM/Mb). In both of these
intervals the frequency of recombination observed in fcd-2
mutants (3.17% and 20.10%, respectively) was similar to that
observed in wild-type worms (Table S1). These data indicate
that fcd-2 is dispensable for crossover formation in meiosis.
Given the potential link between the FA pathway and HR-
mediated repair, we also analyzed fcd-2 mutant germlines for
HR repair events by immunostaining with anti-RAD-51 anti-
bodies. RAD-51 foci were elevated between the leptotene and
pachytene stages of meiotic prophase (Figure 2 and Table S2).
This result was confirmed in 11C3[fcd-2] worms (Figure S1),
which carry an extrachromosomal fcd-2 transgene array that
induces cosuppression of both itself and the endogenous fcd-
2 gene (Dernburg et al., 2000). In contrast, animals derived
from the 11C3 line that have lost the extrachromosomal array
(at a frequency of 43%) do not exhibit increased meiotic RAD-
51 foci (Figure S1). Analysis of RAD-51 foci in spo-11;fcd-2
double mutants, in which meiotic DSB formation is impaired,
revealed a very similar RAD-51 staining pattern to that of spo-
11 mutants. Thus, the increase in RAD-51 foci in fcd-2 mutants
might either represent an increase in DSB formation or most
likely reflects compromised repair of SPO-11-induced meiotic
DSBs, as previously suggested for other DNA repair genes
such as msh-4, msh-5, syp-2, or brc-1 (Colaiacovo et al., 2003,
Adamo et al., 2008). These results establish that loss of fcd-2
leads to accumulation of meiotic RAD-51 foci, which suggest
that FCD-2 is required for efficient conversion of HR intermedi-
ates into poststrand exchange products in meiosis.
FCD-2 Prevents Illegitimate Repair of Meiotic DSBsin Crossover Deficient MutantsThe accumulation of meiotic RAD-51 foci together with the wild-
type levels of crossovers in fcd-2 mutants is highly reminiscent of
the meiotic phenotype of brc-1 mutant (Adamo et al., 2008).
BRC-1 is required for the repair of meiotic DSBs using the sister
chromatid under circumstances where the homologs are
unavailable for repair, such as in SC mutants (syp-2) or in
mutants that lack the prointerhomolog crossover protein MSH-
4 (Adamo et al., 2008). Prompted by these observations, we
sought to determine if FCD-2 plays a similar role to BRC-1 in in-
tersister repair of meiotic DSBs. In wild-type diakinesis oocytes,
six DAPI-stained bodies, or bivalents (representing the six homo-
logs held together by chiasmata) are observed, whereas syp-2 or
him-14/MSH4 (hereafter called msh-4) mutant oocytes typically
contain 12 unpaired chromosomes (univalents) at diakinesis.
In contrast, cytological analysis revealed that the majority of
fcd-2; syp-2 double-mutant oocytes possessed between 6 and
12 DAPI-stained bodies (Figures 1A and 1B). This phenotype
contrasts with the chromosome fragmentation observed in
brc-1;syp-2 double mutants (Adamo et al., 2008). Furthermore,
fcd-2; msh-4 double mutant oocytes displayed between 4 and
12 DAPI-stained bodies (Figures 1A and 1B).
We reasoned that the presence of between 4 and 12 DAPI-
stained bodies in fcd-2; syp-2 and fcd-2;msh-4 double-mutant
oocytes may reflect noncovalent aggregation of nonhomologous
chromosomes, partial restoration of HR-mediated interhomolog
repair, or illegitimate repair between nonhomologous chromo-
somes. To discriminate between these possibilities we used
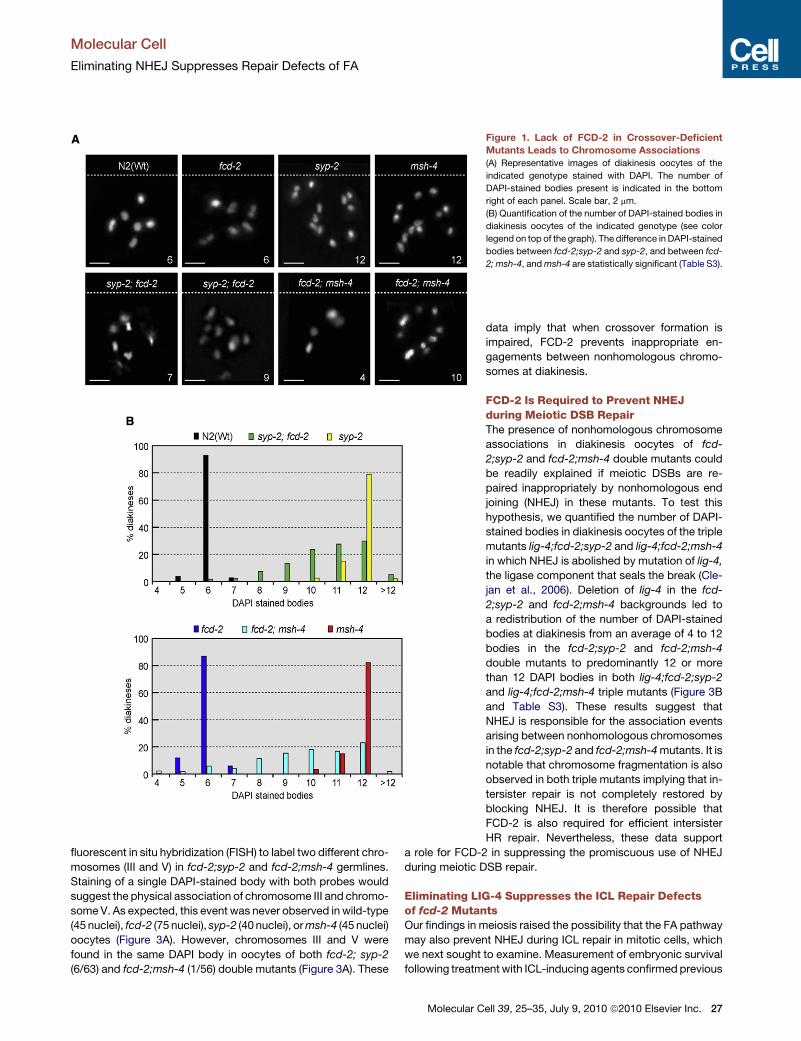
Figure 1. Lack of FCD-2 in Crossover-Deficient
Mutants Leads to Chromosome Associations
(A) Representative images of diakinesis oocytes of the
indicated genotype stained with DAPI. The number of
DAPI-stained bodies present is indicated in the bottom
right of each panel. Scale bar, 2 mm.
(B) Quantification of the number of DAPI-stained bodies in
diakinesis oocytes of the indicated genotype (see color
legend on top of the graph). The difference in DAPI-stained
bodies between fcd-2;syp-2 and syp-2, and between fcd-
2; msh-4, and msh-4 are statistically significant (Table S3).
Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
fluorescent in situ hybridization (FISH) to label two different chro-
mosomes (III and V) in fcd-2;syp-2 and fcd-2;msh-4 germlines.
Staining of a single DAPI-stained body with both probes would
suggest the physical association of chromosome III and chromo-
some V. As expected, this event was never observed in wild-type
(45 nuclei), fcd-2 (75 nuclei), syp-2 (40 nuclei), or msh-4 (45 nuclei)
oocytes (Figure 3A). However, chromosomes III and V were
found in the same DAPI body in oocytes of both fcd-2; syp-2
(6/63) and fcd-2;msh-4 (1/56) double mutants (Figure 3A). These
Molecular C
data imply that when crossover formation is
impaired, FCD-2 prevents inappropriate en-
gagements between nonhomologous chromo-
somes at diakinesis.
FCD-2 Is Required to Prevent NHEJduring Meiotic DSB RepairThe presence of nonhomologous chromosome
associations in diakinesis oocytes of fcd-
2;syp-2 and fcd-2;msh-4 double mutants could
be readily explained if meiotic DSBs are re-
paired inappropriately by nonhomologous end
joining (NHEJ) in these mutants. To test this
hypothesis, we quantified the number of DAPI-
stained bodies in diakinesis oocytes of the triple
mutants lig-4;fcd-2;syp-2 and lig-4;fcd-2;msh-4
in which NHEJ is abolished by mutation of lig-4,
the ligase component that seals the break (Cle-
jan et al., 2006). Deletion of lig-4 in the fcd-
2;syp-2 and fcd-2;msh-4 backgrounds led to
a redistribution of the number of DAPI-stained
bodies at diakinesis from an average of 4 to 12
bodies in the fcd-2;syp-2 and fcd-2;msh-4
double mutants to predominantly 12 or more
than 12 DAPI bodies in both lig-4;fcd-2;syp-2
and lig-4;fcd-2;msh-4 triple mutants (Figure 3B
and Table S3). These results suggest that
NHEJ is responsible for the association events
arising between nonhomologous chromosomes
in the fcd-2;syp-2 and fcd-2;msh-4 mutants. It is
notable that chromosome fragmentation is also
observed in both triple mutants implying that in-
tersister repair is not completely restored by
blocking NHEJ. It is therefore possible that
FCD-2 is also required for efficient intersister
HR repair. Nevertheless, these data support
a role for FCD-2 in suppressing the promiscuous use of NHEJ
during meiotic DSB repair.
Eliminating LIG-4 Suppresses the ICL Repair Defectsof fcd-2 MutantsOur findings in meiosis raised the possibility that the FA pathway
may also prevent NHEJ during ICL repair in mitotic cells, which
we next sought to examine. Measurement of embryonic survival
following treatment with ICL-inducing agents confirmed previous
ell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc. 27
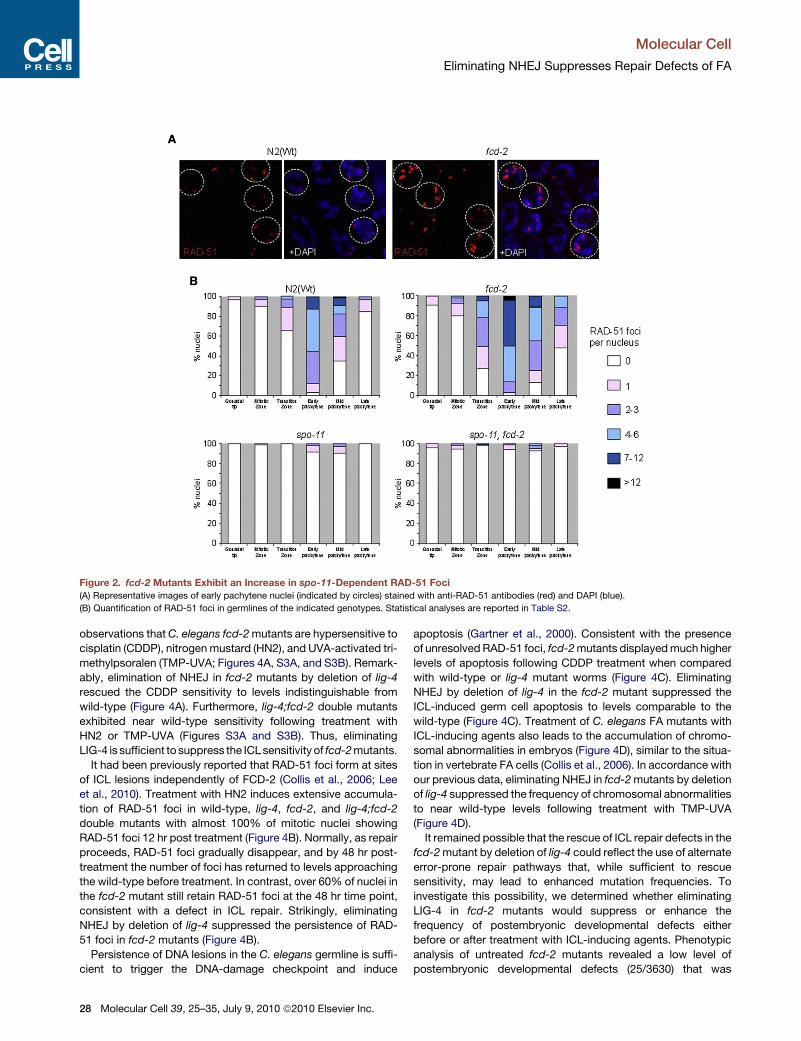
Figure 2. fcd-2 Mutants Exhibit an Increase in spo-11-Dependent RAD-51 Foci
(A) Representative images of early pachytene nuclei (indicated by circles) stained with anti-RAD-51 antibodies (red) and DAPI (blue).
(B) Quantification of RAD-51 foci in germlines of the indicated genotypes. Statistical analyses are reported in Table S2.
Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
observations that C. elegans fcd-2 mutants are hypersensitive to
cisplatin (CDDP), nitrogen mustard (HN2), and UVA-activated tri-
methylpsoralen (TMP-UVA; Figures 4A, S3A, and S3B). Remark-
ably, elimination of NHEJ in fcd-2 mutants by deletion of lig-4
rescued the CDDP sensitivity to levels indistinguishable from
wild-type (Figure 4A). Furthermore, lig-4;fcd-2 double mutants
exhibited near wild-type sensitivity following treatment with
HN2 or TMP-UVA (Figures S3A and S3B). Thus, eliminating
LIG-4 is sufficient to suppress the ICL sensitivity of fcd-2 mutants.
It had been previously reported that RAD-51 foci form at sites
of ICL lesions independently of FCD-2 (Collis et al., 2006; Lee
et al., 2010). Treatment with HN2 induces extensive accumula-
tion of RAD-51 foci in wild-type, lig-4, fcd-2, and lig-4;fcd-2
double mutants with almost 100% of mitotic nuclei showing
RAD-51 foci 12 hr post treatment (Figure 4B). Normally, as repair
proceeds, RAD-51 foci gradually disappear, and by 48 hr post-
treatment the number of foci has returned to levels approaching
the wild-type before treatment. In contrast, over 60% of nuclei in
the fcd-2 mutant still retain RAD-51 foci at the 48 hr time point,
consistent with a defect in ICL repair. Strikingly, eliminating
NHEJ by deletion of lig-4 suppressed the persistence of RAD-
51 foci in fcd-2 mutants (Figure 4B).
Persistence of DNA lesions in the C. elegans germline is suffi-
cient to trigger the DNA-damage checkpoint and induce
28 Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc.
apoptosis (Gartner et al., 2000). Consistent with the presence
of unresolved RAD-51 foci, fcd-2 mutants displayed much higher
levels of apoptosis following CDDP treatment when compared
with wild-type or lig-4 mutant worms (Figure 4C). Eliminating
NHEJ by deletion of lig-4 in the fcd-2 mutant suppressed the
ICL-induced germ cell apoptosis to levels comparable to the
wild-type (Figure 4C). Treatment of C. elegans FA mutants with
ICL-inducing agents also leads to the accumulation of chromo-
somal abnormalities in embryos (Figure 4D), similar to the situa-
tion in vertebrate FA cells (Collis et al., 2006). In accordance with
our previous data, eliminating NHEJ in fcd-2 mutants by deletion
of lig-4 suppressed the frequency of chromosomal abnormalities
to near wild-type levels following treatment with TMP-UVA
(Figure 4D).
It remained possible that the rescue of ICL repair defects in the
fcd-2 mutant by deletion of lig-4 could reflect the use of alternate
error-prone repair pathways that, while sufficient to rescue
sensitivity, may lead to enhanced mutation frequencies. To
investigate this possibility, we determined whether eliminating
LIG-4 in fcd-2 mutants would suppress or enhance the
frequency of postembryonic developmental defects either
before or after treatment with ICL-inducing agents. Phenotypic
analysis of untreated fcd-2 mutants revealed a low level of
postembryonic developmental defects (25/3630) that was
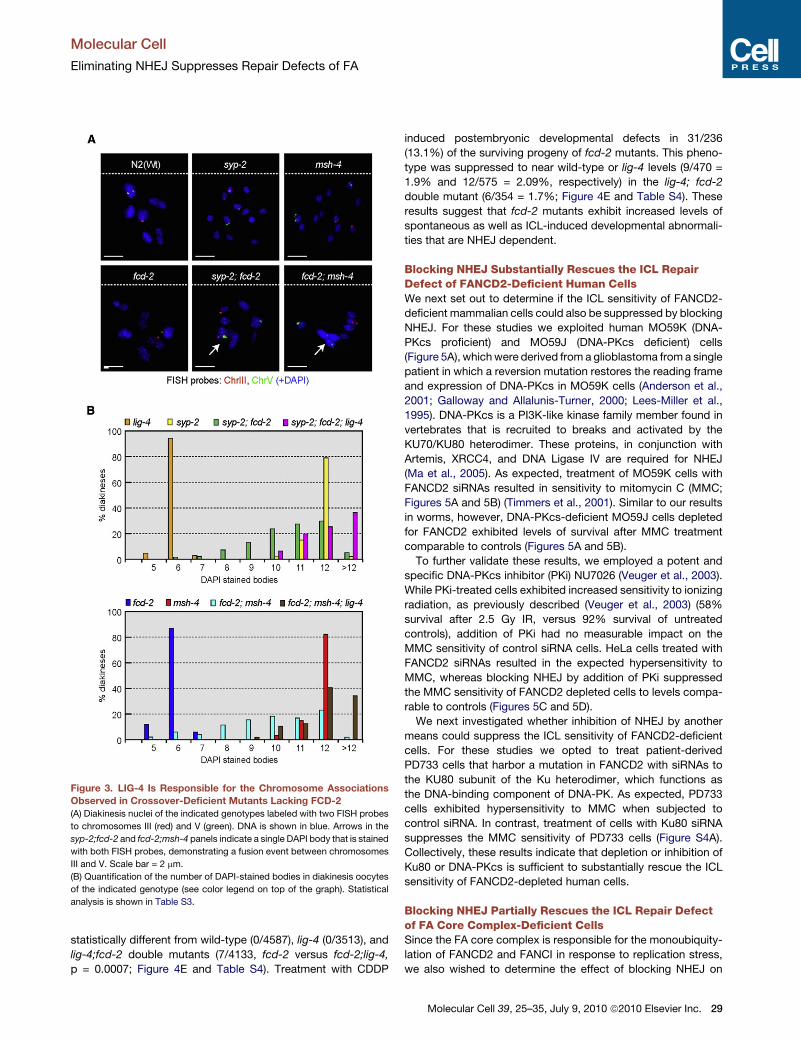
Figure 3. LIG-4 Is Responsible for the Chromosome Associations
Observed in Crossover-Deficient Mutants Lacking FCD-2
(A) Diakinesis nuclei of the indicated genotypes labeled with two FISH probes
to chromosomes III (red) and V (green). DNA is shown in blue. Arrows in the
syp-2;fcd-2 and fcd-2;msh-4 panels indicate a single DAPI body that is stained
with both FISH probes, demonstrating a fusion event between chromosomes
III and V. Scale bar = 2 mm.
(B) Quantification of the number of DAPI-stained bodies in diakinesis oocytes
of the indicated genotype (see color legend on top of the graph). Statistical
analysis is shown in Table S3.
Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
statistically different from wild-type (0/4587), lig-4 (0/3513), and
lig-4;fcd-2 double mutants (7/4133, fcd-2 versus fcd-2;lig-4,
p = 0.0007; Figure 4E and Table S4). Treatment with CDDP
induced postembryonic developmental defects in 31/236
(13.1%) of the surviving progeny of fcd-2 mutants. This pheno-
type was suppressed to near wild-type or lig-4 levels (9/470 =
1.9% and 12/575 = 2.09%, respectively) in the lig-4; fcd-2
double mutant (6/354 = 1.7%; Figure 4E and Table S4). These
results suggest that fcd-2 mutants exhibit increased levels of
spontaneous as well as ICL-induced developmental abnormali-
ties that are NHEJ dependent.
Blocking NHEJ Substantially Rescues the ICL RepairDefect of FANCD2-Deficient Human CellsWe next set out to determine if the ICL sensitivity of FANCD2-
deficient mammalian cells could also be suppressed by blocking
NHEJ. For these studies we exploited human MO59K (DNA-
PKcs proficient) and MO59J (DNA-PKcs deficient) cells
(Figure 5A), which were derived from a glioblastoma from a single
patient in which a reversion mutation restores the reading frame
and expression of DNA-PKcs in MO59K cells (Anderson et al.,
2001; Galloway and Allalunis-Turner, 2000; Lees-Miller et al.,
1995). DNA-PKcs is a PI3K-like kinase family member found in
vertebrates that is recruited to breaks and activated by the
KU70/KU80 heterodimer. These proteins, in conjunction with
Artemis, XRCC4, and DNA Ligase IV are required for NHEJ
(Ma et al., 2005). As expected, treatment of MO59K cells with
FANCD2 siRNAs resulted in sensitivity to mitomycin C (MMC;
Figures 5A and 5B) (Timmers et al., 2001). Similar to our results
in worms, however, DNA-PKcs-deficient MO59J cells depleted
for FANCD2 exhibited levels of survival after MMC treatment
comparable to controls (Figures 5A and 5B).
To further validate these results, we employed a potent and
specific DNA-PKcs inhibitor (PKi) NU7026 (Veuger et al., 2003).
While PKi-treated cells exhibited increased sensitivity to ionizing
radiation, as previously described (Veuger et al., 2003) (58%
survival after 2.5 Gy IR, versus 92% survival of untreated
controls), addition of PKi had no measurable impact on the
MMC sensitivity of control siRNA cells. HeLa cells treated with
FANCD2 siRNAs resulted in the expected hypersensitivity to
MMC, whereas blocking NHEJ by addition of PKi suppressed
the MMC sensitivity of FANCD2 depleted cells to levels compa-
rable to controls (Figures 5C and 5D).
We next investigated whether inhibition of NHEJ by another
means could suppress the ICL sensitivity of FANCD2-deficient
cells. For these studies we opted to treat patient-derived
PD733 cells that harbor a mutation in FANCD2 with siRNAs to
the KU80 subunit of the Ku heterodimer, which functions as
the DNA-binding component of DNA-PK. As expected, PD733
cells exhibited hypersensitivity to MMC when subjected to
control siRNA. In contrast, treatment of cells with Ku80 siRNA
suppresses the MMC sensitivity of PD733 cells (Figure S4A).
Collectively, these results indicate that depletion or inhibition of
Ku80 or DNA-PKcs is sufficient to substantially rescue the ICL
sensitivity of FANCD2-depleted human cells.
Blocking NHEJ Partially Rescues the ICL Repair Defectof FA Core Complex-Deficient CellsSince the FA core complex is responsible for the monoubiquity-
lation of FANCD2 and FANCI in response to replication stress,
we also wished to determine the effect of blocking NHEJ on
Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc. 29
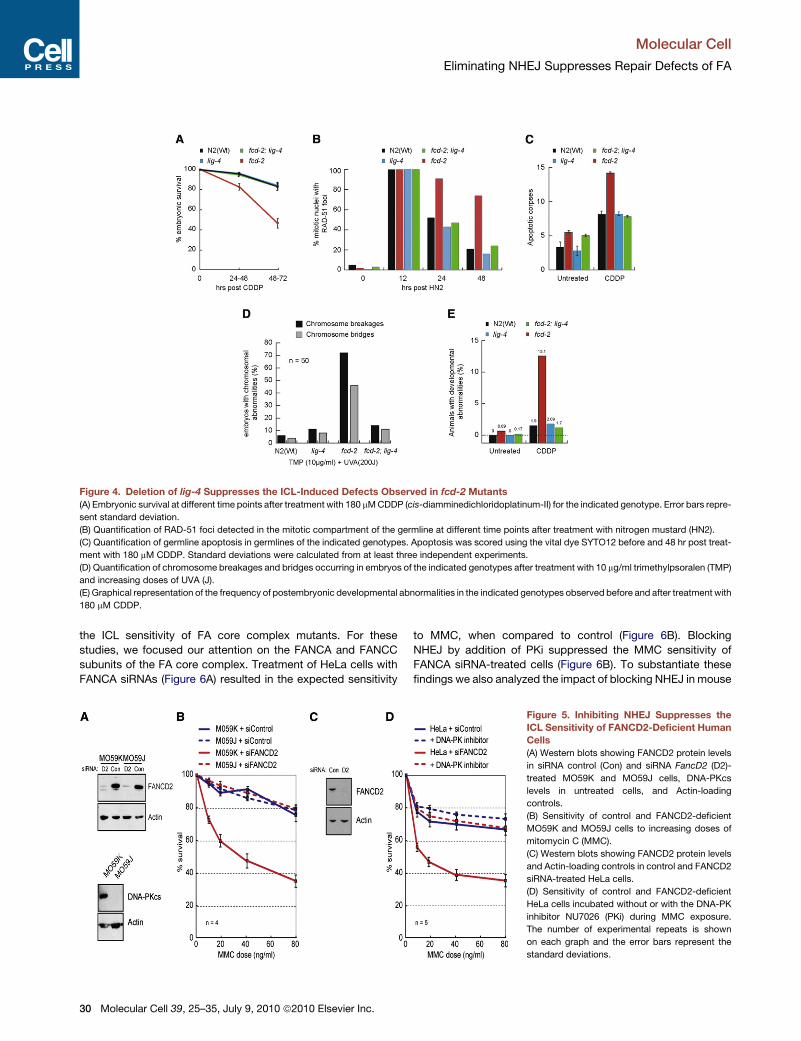
Figure 4. Deletion of lig-4 Suppresses the ICL-Induced Defects Observed in fcd-2 Mutants
(A) Embryonic survival at different time points after treatment with 180 mM CDDP (cis-diamminedichloridoplatinum-II) for the indicated genotype. Error bars repre-
sent standard deviation.
(B) Quantification of RAD-51 foci detected in the mitotic compartment of the germline at different time points after treatment with nitrogen mustard (HN2).
(C) Quantification of germline apoptosis in germlines of the indicated genotypes. Apoptosis was scored using the vital dye SYTO12 before and 48 hr post treat-
ment with 180 mM CDDP. Standard deviations were calculated from at least three independent experiments.
(D) Quantification of chromosome breakages and bridges occurring in embryos of the indicated genotypes after treatment with 10 mg/ml trimethylpsoralen (TMP)
and increasing doses of UVA (J).
(E) Graphical representation of the frequency of postembryonic developmental abnormalities in the indicated genotypes observed before and after treatment with
180 mM CDDP.
Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
the ICL sensitivity of FA core complex mutants. For these
studies, we focused our attention on the FANCA and FANCC
subunits of the FA core complex. Treatment of HeLa cells with
FANCA siRNAs (Figure 6A) resulted in the expected sensitivity
30 Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc.
to MMC, when compared to control (Figure 6B). Blocking
NHEJ by addition of PKi suppressed the MMC sensitivity of
FANCA siRNA-treated cells (Figure 6B). To substantiate these
findings we also analyzed the impact of blocking NHEJ in mouse
Figure 5. Inhibiting NHEJ Suppresses the
ICL Sensitivity of FANCD2-Deficient Human
Cells
(A) Western blots showing FANCD2 protein levels
in siRNA control (Con) and siRNA FancD2 (D2)-
treated MO59K and MO59J cells, DNA-PKcs
levels in untreated cells, and Actin-loading
controls.
(B) Sensitivity of control and FANCD2-deficient
MO59K and MO59J cells to increasing doses of
mitomycin C (MMC).
(C) Western blots showing FANCD2 protein levels
and Actin-loading controls in control and FANCD2
siRNA-treated HeLa cells.
(D) Sensitivity of control and FANCD2-deficient
HeLa cells incubated without or with the DNA-PK
inhibitor NU7026 (PKi) during MMC exposure.
The number of experimental repeats is shown
on each graph and the error bars represent the
standard deviations.
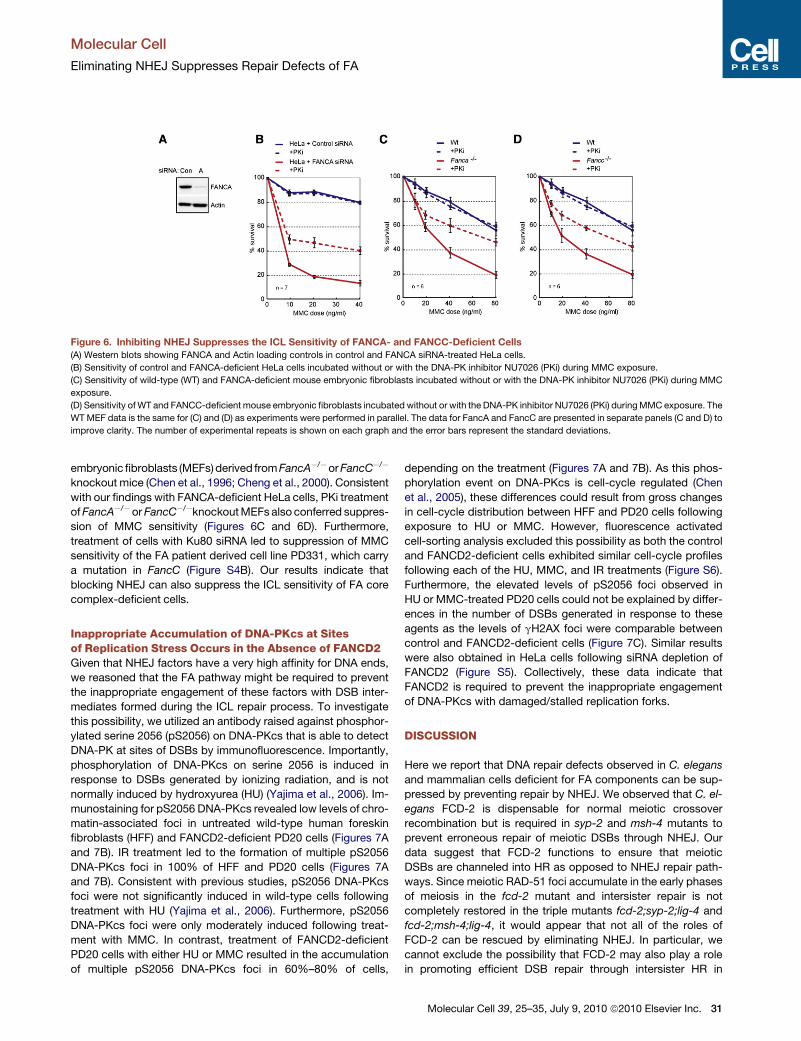
Figure 6. Inhibiting NHEJ Suppresses the ICL Sensitivity of FANCA- and FANCC-Deficient Cells
(A) Western blots showing FANCA and Actin loading controls in control and FANCA siRNA-treated HeLa cells.
(B) Sensitivity of control and FANCA-deficient HeLa cells incubated without or with the DNA-PK inhibitor NU7026 (PKi) during MMC exposure.
(C) Sensitivity of wild-type (WT) and FANCA-deficient mouse embryonic fibroblasts incubated without or with the DNA-PK inhibitor NU7026 (PKi) during MMC
exposure.
(D) Sensitivity of WT and FANCC-deficient mouse embryonic fibroblasts incubated without or with the DNA-PK inhibitor NU7026 (PKi) during MMC exposure. The
WT MEF data is the same for (C) and (D) as experiments were performed in parallel. The data for FancA and FancC are presented in separate panels (C and D) to
improve clarity. The number of experimental repeats is shown on each graph and the error bars represent the standard deviations.
Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
embryonic fibroblasts (MEFs) derived from FancA�/�or FancC�/�
knockout mice (Chen et al., 1996; Cheng et al., 2000). Consistent
with our findings with FANCA-deficient HeLa cells, PKi treatment
of FancA�/� or FancC�/�knockout MEFs also conferred suppres-
sion of MMC sensitivity (Figures 6C and 6D). Furthermore,
treatment of cells with Ku80 siRNA led to suppression of MMC
sensitivity of the FA patient derived cell line PD331, which carry
a mutation in FancC (Figure S4B). Our results indicate that
blocking NHEJ can also suppress the ICL sensitivity of FA core
complex-deficient cells.
Inappropriate Accumulation of DNA-PKcs at Sitesof Replication Stress Occurs in the Absence of FANCD2Given that NHEJ factors have a very high affinity for DNA ends,
we reasoned that the FA pathway might be required to prevent
the inappropriate engagement of these factors with DSB inter-
mediates formed during the ICL repair process. To investigate
this possibility, we utilized an antibody raised against phosphor-
ylated serine 2056 (pS2056) on DNA-PKcs that is able to detect
DNA-PK at sites of DSBs by immunofluorescence. Importantly,
phosphorylation of DNA-PKcs on serine 2056 is induced in
response to DSBs generated by ionizing radiation, and is not
normally induced by hydroxyurea (HU) (Yajima et al., 2006). Im-
munostaining for pS2056 DNA-PKcs revealed low levels of chro-
matin-associated foci in untreated wild-type human foreskin
fibroblasts (HFF) and FANCD2-deficient PD20 cells (Figures 7A
and 7B). IR treatment led to the formation of multiple pS2056
DNA-PKcs foci in 100% of HFF and PD20 cells (Figures 7A
and 7B). Consistent with previous studies, pS2056 DNA-PKcs
foci were not significantly induced in wild-type cells following
treatment with HU (Yajima et al., 2006). Furthermore, pS2056
DNA-PKcs foci were only moderately induced following treat-
ment with MMC. In contrast, treatment of FANCD2-deficient
PD20 cells with either HU or MMC resulted in the accumulation
of multiple pS2056 DNA-PKcs foci in 60%–80% of cells,
depending on the treatment (Figures 7A and 7B). As this phos-
phorylation event on DNA-PKcs is cell-cycle regulated (Chen
et al., 2005), these differences could result from gross changes
in cell-cycle distribution between HFF and PD20 cells following
exposure to HU or MMC. However, fluorescence activated
cell-sorting analysis excluded this possibility as both the control
and FANCD2-deficient cells exhibited similar cell-cycle profiles
following each of the HU, MMC, and IR treatments (Figure S6).
Furthermore, the elevated levels of pS2056 foci observed in
HU or MMC-treated PD20 cells could not be explained by differ-
ences in the number of DSBs generated in response to these
agents as the levels of gH2AX foci were comparable between
control and FANCD2-deficient cells (Figure 7C). Similar results
were also obtained in HeLa cells following siRNA depletion of
FANCD2 (Figure S5). Collectively, these data indicate that
FANCD2 is required to prevent the inappropriate engagement
of DNA-PKcs with damaged/stalled replication forks.
DISCUSSION
Here we report that DNA repair defects observed in C. elegans
and mammalian cells deficient for FA components can be sup-
pressed by preventing repair by NHEJ. We observed that C. el-
egans FCD-2 is dispensable for normal meiotic crossover
recombination but is required in syp-2 and msh-4 mutants to
prevent erroneous repair of meiotic DSBs through NHEJ. Our
data suggest that FCD-2 functions to ensure that meiotic
DSBs are channeled into HR as opposed to NHEJ repair path-
ways. Since meiotic RAD-51 foci accumulate in the early phases
of meiosis in the fcd-2 mutant and intersister repair is not
completely restored in the triple mutants fcd-2;syp-2;lig-4 and
fcd-2;msh-4;lig-4, it would appear that not all of the roles of
FCD-2 can be rescued by eliminating NHEJ. In particular, we
cannot exclude the possibility that FCD-2 may also play a role
in promoting efficient DSB repair through intersister HR in
Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc. 31
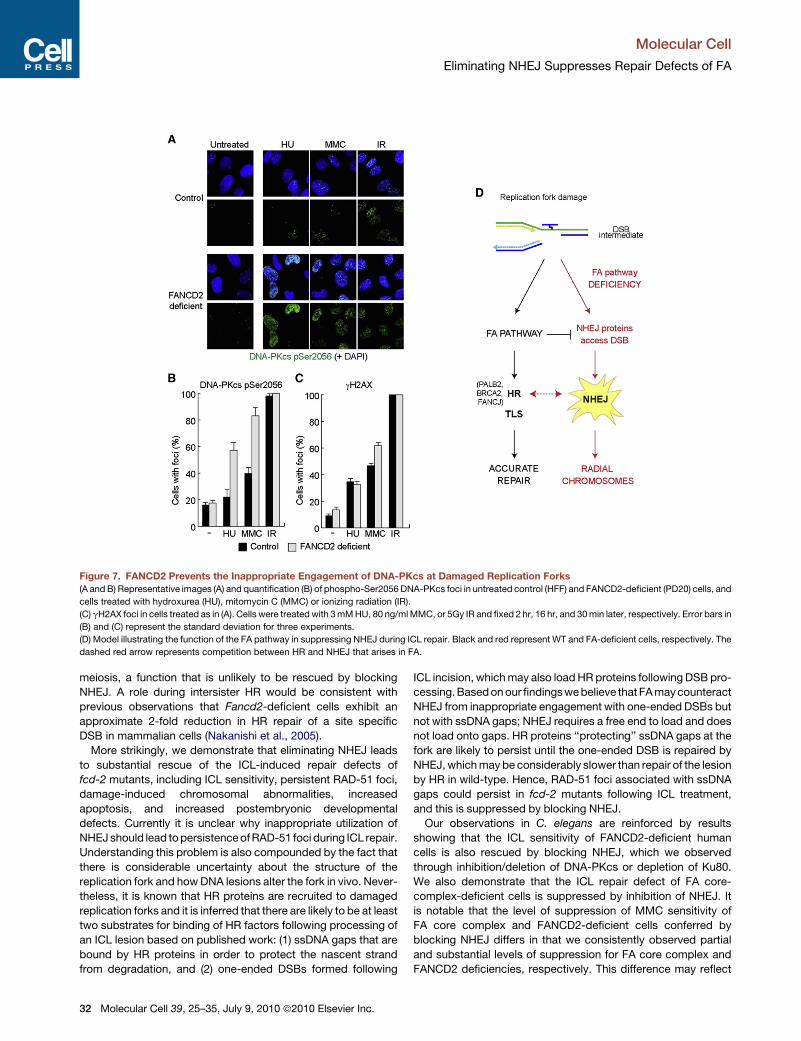
Figure 7. FANCD2 Prevents the Inappropriate Engagement of DNA-PKcs at Damaged Replication Forks
(A and B) Representative images (A) and quantification (B) of phospho-Ser2056 DNA-PKcs foci in untreated control (HFF) and FANCD2-deficient (PD20) cells, and
cells treated with hydroxurea (HU), mitomycin C (MMC) or ionizing radiation (IR).
(C) gH2AX foci in cells treated as in (A). Cells were treated with 3 mM HU, 80 ng/ml MMC, or 5Gy IR and fixed 2 hr, 16 hr, and 30 min later, respectively. Error bars in
(B) and (C) represent the standard deviation for three experiments.
(D) Model illustrating the function of the FA pathway in suppressing NHEJ during ICL repair. Black and red represent WT and FA-deficient cells, respectively. The
dashed red arrow represents competition between HR and NHEJ that arises in FA.
Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
meiosis, a function that is unlikely to be rescued by blocking
NHEJ. A role during intersister HR would be consistent with
previous observations that Fancd2-deficient cells exhibit an
approximate 2-fold reduction in HR repair of a site specific
DSB in mammalian cells (Nakanishi et al., 2005).
More strikingly, we demonstrate that eliminating NHEJ leads
to substantial rescue of the ICL-induced repair defects of
fcd-2 mutants, including ICL sensitivity, persistent RAD-51 foci,
damage-induced chromosomal abnormalities, increased
apoptosis, and increased postembryonic developmental
defects. Currently it is unclear why inappropriate utilization of
NHEJ should lead to persistence of RAD-51 foci during ICL repair.
Understanding this problem is also compounded by the fact that
there is considerable uncertainty about the structure of the
replication fork and how DNA lesions alter the fork in vivo. Never-
theless, it is known that HR proteins are recruited to damaged
replication forks and it is inferred that there are likely to be at least
two substrates for binding of HR factors following processing of
an ICL lesion based on published work: (1) ssDNA gaps that are
bound by HR proteins in order to protect the nascent strand
from degradation, and (2) one-ended DSBs formed following
32 Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc.
ICL incision, which may also load HR proteins following DSB pro-
cessing. Based on our findings we believe that FA may counteract
NHEJ from inappropriate engagement with one-ended DSBs but
not with ssDNA gaps; NHEJ requires a free end to load and does
not load onto gaps. HR proteins ‘‘protecting’’ ssDNA gaps at the
fork are likely to persist until the one-ended DSB is repaired by
NHEJ, which may be considerably slower than repair of the lesion
by HR in wild-type. Hence, RAD-51 foci associated with ssDNA
gaps could persist in fcd-2 mutants following ICL treatment,
and this is suppressed by blocking NHEJ.
Our observations in C. elegans are reinforced by results
showing that the ICL sensitivity of FANCD2-deficient human
cells is also rescued by blocking NHEJ, which we observed
through inhibition/deletion of DNA-PKcs or depletion of Ku80.
We also demonstrate that the ICL repair defect of FA core-
complex-deficient cells is suppressed by inhibition of NHEJ. It
is notable that the level of suppression of MMC sensitivity of
FA core complex and FANCD2-deficient cells conferred by
blocking NHEJ differs in that we consistently observed partial
and substantial levels of suppression for FA core complex and
FANCD2 deficiencies, respectively. This difference may reflect

Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
the fact that the FA core complex may perform additional roles in
ICL repair independent of its role in regulating FANCD2/FANCI,
such as ubiquitylaton of other factors important for ICL repair
(Matsushita et al., 2005). It is notable that suppression of ICL
repair defects by inhibition of NHEJ is not applicable to all FA
genes as the ICL sensitivity of dog-1 mutants (C. elegans FANCJ)
(Youds et al., 2008) was neither rescued nor enhanced by the lig-
4 mutation (Figure S3C). This may reflect the fact that DOG-1 has
functions independent of the FA pathway during replication fork
repair (Youds et al., 2008). Finally, we demonstrate that DNA-
PKcs is promiscuously activated and inappropriately recruited
to sites of replication stress in the absence of FANCD2 in human
cells. Collectively, our results lead us to propose that DNA repair
defects in FA cells arise as a consequence of the misuse of NHEJ
during lesion repair in S phase and in meiosis. This hypothesis
provides a plausible explanation for many of the observed FA
phenotypes (DNA-repair defects and meiotic problems) and is
consistent with the suppression of these phenotypes upon elim-
ination of the NHEJ pathway. Such a scenario is also supported
by cytological analysis of radial chromosomes in FA cells; over
99% of these events arise as a result of translocations between
nonhomologous chromosomes, which is a signature of repair by
NHEJ (Newell et al., 2004).
Our results raise the possibility that FA proteins act to regulate
the choice of DSB repair pathway, and we propose two potential
models that are consistent with published observations and our
current data: (1) FA proteins directly antagonize NHEJ by block-
ing or displacing NHEJ factors from DSB ends, and/or (2) FA
proteins coordinate the processing of meiotic DSBs and ICL
lesions to favor HR and disfavor NHEJ. Studies of the ICL repair
process have established that DSB intermediates are formed at
the replication fork following ICL incision. DSB ends are also
believed to arise as a result of replication fork regression, which
could be promoted by the helicase/translocase activities of
FANCM and/or BLM (Gari et al., 2008). Given that NHEJ factors
are present constitutively throughout the cell cycle and possess
a high affinity for DSB ends, one possible explanation for our
data is that FA proteins bind to and protect DSB intermediates
from engagement and repair by NHEJ. Supporting this idea,
in vitro DNA-binding assays indicate that FANCD2 is able to
interact with a number of DNA substrates including forked and
linear molecules containing double-stranded DNA ends (Park
et al., 2005; Roques et al., 2009). The hypothesis that FANCD2
physically occludes access of NHEJ factors to repair intermedi-
ates is consistent with our findings that eliminating NHEJ is suffi-
cient to substantially rescue FANCD2 deficiency in most, if not
all, assays and that DNA-PKcs engages inappropriately with
sites of replication stress in the absence of FANCD2. Engage-
ment of NHEJ factors with DSB intermediates during S phase
may inhibit the processing of DSBs required for loading of HR
factors while also promoting illegitimate repair (Figure 7D).
Our results are also consistent with an alternate model in which
the FA proteins function to coordinate processing of ICL lesions/
DSB intermediates in a way that would favor HR and disfavor
NHEJ. Indeed, it is known that the FA core complex associates
with the Blooms and MRN complexes (Meetei et al., 2003;
Nakanishi et al., 2002; Pichierri et al., 2002; Roques et al.,
2009), which are both implicated in DSB processing (Gravel
et al., 2008; Mimitou and Symington, 2008; Zhu et al., 2008).
Therefore, it is possible that the FA pathway may coordinate
BLM- and/or MRN-dependent end processing of DSB intermedi-
ates arising at damaged replication forks and meiotic DSBs. It is
also possible that the FA pathway may recruit other DSB end-
processing activities to promote HR. Ultimately, processing of
the DSB to produce a 30 single-stranded overhang would provide
an optimal substrate for binding by HR factors, while at the same
time reducing the affinity of NHEJ factors for these DNA ends. A
role for FCD-2/FANCD2 in coordinating ICL processing is also
supported by recent observations that FANCD2/FANCI-depleted
frog egg extracts are deficient for the initial incision of an ICL
lesion present within a synthetic DNA substrate, and this can
be rescued by addition of ubiquitylated FANCD2/FANCI
(Knipscheer et al., 2009). An essential role for FANCD2 in ICL inci-
sion is potentially difficult to reconcile with our findings that the
phenotypes resulting from FCD-2/FANCD2 deficiency can be
rescued by eliminating NHEJ. However, it is entirely possible
that in vivo, alternative nucleases process the ICL lesion inappro-
priately in the absence of FANCD2/FANCI and this produces
a substrate for NHEJ. This scenario is consistent with previous
observations and our findings (Figures 4 and 6) that DSBs are
formed at wild-type levels at ICL lesions in vivo in the absence
of FANCD2 (Rothfuss and Grompe, 2004). Moreover, structure-
specific nucleases such as MUS81, XPF, and SNM1A (PSO2)
have all been implicated in ICL processing in vivo and could
process the ICL lesion inappropriately in the absence of FANCD2
(Ciccia et al., 2008; Li et al., 2005). Additionally, a recent study has
shown that the unhooking of an ICL by XPF/ERCC1 is necessary
for the stable association of FANCD2 with damaged chromatin
in vivo, which questions the requirement for FANCD2 in initial
ICL incision (Bhagwat et al., 2009). Nevertheless, our data indi-
cate that blocking NHEJ is sufficient to allow the lesion to be
repaired effectively in the absence of FANCD2, whether or not
the ICL is inappropriately processed at the incision step.
Finally, it is important to mention that our results showing
that deletion or inhibition of Ku80 or DNA-PKcs is sufficient to
rescue the ICL sensitivity of FANCD2-deficient human cells was
not observed in FANCD2/DNA-PKcs double knockout MEFs
(Houghtaling et al., 2005). The most likely reason for this difference
is that human DNA-PKcs possesses additional cellular functions
that are not shared with its counterparts in other vertebrates, or
alternatively inhibition of DNA-PKcs has a different effect to dele-
tion of this kinase (Ruis et al., 2008). Irrespective of the reason for
thisdifference, our discovery that the defects inFANCD2-deficient
human cells can be substantially suppressed by blocking NHEJ
raises the possibility that DNA-PKcs inhibitors could be used in
a subset of FA patients to delay the onset of solid tumors, which
remains the most significant health issue in adult FA patients.
EXPERIMENTAL PROCEDURES
Strains
Strains were cultured according to Wood (1988), experiments were performed
at 20�C. The following C. elegans strains were provided by the Caenorhabditis
Genetics Center: wild-type strain Bristol N2. AV106 spo-11(ok79)IV/ nT1[unc-
?(n754)let-?](IV;V). AV276 syp-2(ok307)V/nT1[unc-?(n754)let-? qls50](IV;V).
AV308 him-14(it21)/mnCI, DR102 dpy-5(e61) unc-29(e403)I. BC3217
unc-60(m35) dpy-11(e224)V; sDp30 (V;X). RB873 lig-4(ok716) III. VC13 dog-1
Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc. 33

Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
(gk10) I. FX1298 fcd-2(tm1298) IV mutant (Collis et al., 2006) was generated
and provided by Shoehi Mitani of the National Bioresource Project for the
nematode, Japan.
Immunolocalization of RAD-51
The anti-RAD-51 antibody was described in Colaiacovo et al. (2003). DAPI
staining, immunostaining, and analysis of meiotic nuclei were performed using
whole-mount preparations of dissected gonads as described in Colaiacovo
et al. (2003). Quantitative analysis of RAD-51 foci was performed on Z series
of images acquired using a Leica DM6000 fluorescence microscope, Leica
DC 350 FX camera under control of Leica LAS AF 6000 software. Optical
sections were collected at 0.25 mm increments and a minimum of 100 nuclei
per zone of the germline was scored for each genotype. Quantitative analyses
of germline RAD-51 foci (Table S2) were analyzed with student’s t test for inde-
pendent samples. Distributions of numbers of DAPI-stained bodies in the
different genotypes were analyzed with the c2 test (Table S3).
C. elegans Treatment with ICL-Inducing Agents
Young adult worms were picked and individually cloned on freshly seeded
plates containing 180 mM cis-diamminedichloroplatinum(II) (CDDP, Sigma).
Each worm was transferred onto a fresh plate every 24 hr, and the eggs laid
after 48 and 72 hr were scored. The progeny were scored for embryonic
lethality (unhatched eggs) and for organisms with evident postembryonic
developmental defects under a dissecting microscope. Untreated controls
were singled out and screened at the same time points. Conditions for HN2
and TMP-UVA treatments were performed as previously described (Collis
et al., 2006; Youds et al., 2008).
Cell Culture
All cell culture, RNAi conditions, whole-cell extracts, and immunoblotting were
carried out as previously described (Collis et al., 2007). For survival assays,
cells were treated with 0–80ng/ml mitomycin C (MMC; Sigma). Three to four
days after damage, survival was assessed using MTT (Sigma). Cells were
treated with 0.5 mM DNA-PK inhibitor (NU7026; Tocris BioScience) or vehicle
control for 1–2 hr before damage induction. FancA�/� and FancC�/� MEFs
were derived from crosses with FancA+/� FancC+/� double heterozygous
mice. The control MEFs were derived from a wild-type littermate resulting
from the same cross. Six replicas were used per condition for each experi-
ment; the average of four to six independent experiments is shown.
Immunostaining for Phospho-DNA-PKcs and gH2AX
Cells were seeded in duplicate on coverslips and treated 24 hr later with either
3 mM HU, 80 ng/ml MMC, or 5Gy IR and fixed 2 hr, 16hr, 30 min, and 30 mins
later respectively. Cells were then stained for phospho-DNA-PKcs (Abcam;
ab18192, 1:500) and imaged using a DeltaVision microscope as previously
described (Collis et al., 2007). The presence of phospho-DNA-PKcs foci was
scored in over 200 cells per coverslip and the percentage of positive (foci-con-
taining) cells was calculated for each experiment. gH2AX staining (Upstate;
05-636, 1:1000) was carried out on cells treated identically to those processed
for phosho-DNA-PKcs staining.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures, four tables, and Supplemental
Experimental Procedures and can be found with this article online at
doi:10.1016/j.molcel.2010.06.026.
ACKNOWLEDGMENTS
The labs of S.J.B. and A.L.V. contributed equally to this work and the cofirst
and cocorresponding authors are positioned alphabetically. We would like to
thank Jillian Youds for critical reading of the manuscript. The lab of S.J.B. is
funded by Cancer Research UK, A.L.V. is funded by CNR and Regione Cam-
pania legge 5. N.S. was recipient of a predoctoral fellowship from MIUR (PhD
School Molecular Genetics and Medicine) and EMBO ST fellowship. The lab of
EM-P is funded by a BBSRC David Phillips Fellowship and the MRC. S.J.B. is
a Royal Society Wolfson Research Merit Award holder.
34 Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc.
Received: February 16, 2010
Revised: May 13, 2010
Accepted: June 9, 2010
Published online: July 1, 2010
REFERENCES
Adamo, A., Montemauri, P., Silva, N., Ward, J.D., Boulton, S.J., and La Volpe,
A. (2008). BRC-1 acts in the inter-sister pathway of meiotic double-strand
break repair. EMBO Rep. 9, 287–292.
Alter, B.P. (1993). Fanconi’s anaemia and its variability. Br. J. Haematol. 85,
9–14.
Anderson, C.W., Dunn, J.J., Freimuth, P.I., Galloway, A.M., and Allalunis-
Turner, M.J. (2001). Frameshift mutation in PRKDC, the gene for DNA-PKcs,
in the DNA repair-defective, human, glioma-derived cell line M059J. Radiat.
Res. 156, 2–9.
Bhagwat, N., Olsen, A.L., Wang, A.T., Hanada, K., Stuckert, P., Kanaar, R.,
D’Andrea, A., Niedernhofer, L.J., and McHugh, P.J. (2009). XPF-ERCC1
participates in the Fanconi anemia pathway of cross-link repair. Mol. Cell.
Biol. 29, 6427–6437.
Boulton, S.J. (2006). Cellular functions of the BRCA tumour-suppressor
proteins. Biochem. Soc. Trans. 34, 633–645.
Chen, M., Tomkins, D.J., Auerbach, W., McKerlie, C., Youssoufian, H., Liu, L.,
Gan, O., Carreau, M., Auerbach, A., Groves, T., et al. (1996). Inactivation of Fac
in mice produces inducible chromosomal instability and reduced fertility remi-
niscent of Fanconi anaemia. Nat. Genet. 12, 448–451.
Chen, B.P., Chan, D.W., Kobayashi, J., Burma, S., Asaithamby, A., Moroto-
mi-Yano, K., Botvinick, E., Qin, J., and Chen, D.J. (2005). Cell cycle depen-
dence of DNA-dependent protein kinase phosphorylation in response to
DNA double strand breaks. J. Biol. Chem. 280, 14709–14715.
Cheng, N.C., van de Vrugt, H.J., van der Valk, M.A., Oostra, A.B., Krimpenfort,
P., de Vries, Y., Joenje, H., Berns, A., and Arwert, F. (2000). Mice with
a targeted disruption of the Fanconi anemia homolog Fanca. Hum. Mol. Genet.
9, 1805–1811.
Ciccia, A., McDonald, N., and West, S.C. (2008). Structural and functional rela-
tionships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 77,
259–287.
Clejan, I., Boerckel, J., and Ahmed, S. (2006). Developmental modulation of
nonhomologous end joining in Caenorhabditis elegans. Genetics 173,
1301–1317.
Colaiacovo, M.P., MacQueen, A.J., Martinez-Perez, E., McDonald, K., Adamo,
A., La Volpe, A., and Villeneuve, A.M. (2003). Synaptonemal complex assembly
in C. elegans is dispensable for loading strand-exchange proteins but critical
for proper completion of recombination. Dev. Cell 5, 463–474.
Collis, S.J., Barber, L.J., Ward, J.D., Martin, J.S., and Boulton, S.J. (2006). C.
elegans FANCD2 responds to replication stress and functions in interstrand
cross-link repair. DNA Repair (Amst.) 5, 1398–1406.
Collis, S.J., Barber, L.J., Clark, A.J., Martin, J.S., Ward, J.D., and Boulton, S.J.
(2007). HCLK2 is essential for the mammalian S-phase checkpoint and
impacts on Chk1 stability. Nat. Cell Biol. 9, 391–401.
D’Andrea, A.D., and Grompe, M. (2003). The Fanconi anaemia/BRCA
pathway. Nat. Rev. Cancer 3, 23–34.
Dernburg, A.F., McDonald, K., Moulder, G., Barstead, R., Dresser, M., and
Villeneuve, A.M. (1998). Meiotic recombination in C. elegans initiates by
a conserved mechanism and is dispensable for homologous chromosome
synapsis. Cell 94, 387–398.
Dernburg, A.F., Zalevsky, J., Colaiacovo, M.P., and Villeneuve, A.M. (2000).
Transgene-mediated cosuppression in the C. elegans germ line. Genes Dev.
14, 1578–1583.
Fanconi, G. (1967). Familial constitutional panmyelocytopathy, Fanconi’s
anemia (F.A.). I. Clinical aspects. Semin. Hematol. 4, 233–240.

Molecular Cell
Eliminating NHEJ Suppresses Repair Defects of FA
Galloway, A.M., and Allalunis-Turner, J. (2000). cDNA expression array anal-
ysis of DNA repair genes in human glioma cells that lack or express DNA-
PK. Radiat. Res. 154, 609–615.
Gari, K., Decaillet, C., Delannoy, M., Wu, L., and Constantinou, A. (2008). Re-
modeling of DNA replication structures by the branch point translocase
FANCM. Proc. Natl. Acad. Sci. USA 105, 16107–16112.
Gartner, A., Milstein, S., Ahmed, S., Hodgkin, J., and Hengartner, M.O. (2000).
A conserved checkpoint pathway mediates DNA damage—induced apoptosis
and cell cycle arrest in C. elegans. Mol. Cell 5, 435–443.
Gravel, S., Chapman, J.R., Magill, C., and Jackson, S.P. (2008). DNA helicases
Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22,
2767–2772.
Houghtaling, S., Timmers, C., Noll, M., Finegold, M.J., Jones, S.N., Meyn,
M.S., and Grompe, M. (2003). Epithelial cancer in Fanconi anemia comple-
mentation group D2 (Fancd2) knockout mice. Genes Dev. 17, 2021–2035.
Houghtaling, S., Newell, A., Akkari, Y., Taniguchi, T., Olson, S., and Grompe,
M. (2005). Fancd2 functions in a double strand break repair pathway that is
distinct from non-homologous end joining. Hum. Mol. Genet. 14, 3027–3033.
Kelly, K.O., Dernburg, A.F., Stanfield, G.M., and Villeneuve, A.M. (2000). Cae-
norhabditis elegans msh-5 is required for both normal and radiation-induced
meiotic crossing over but not for completion of meiosis. Genetics 156,
617–630.
Kennedy, R.D., and D’Andrea, A.D. (2005). The Fanconi Anemia/BRCA
pathway: new faces in the crowd. Genes Dev. 19, 2925–2940.
Knipscheer, P., Raschle, M., Smogorzewska, A., Enoiu, M., Ho, T.V., Scharer,
O.D., Elledge, S.J., and Walter, J.C. (2009). The Fanconi anemia pathway
promotes replication-dependent DNA interstrand cross-link repair. Science
326, 1698–1701.
Lee, K.Y., Chung, K.Y., and Koo, H.S. (2010). The involvement of FANCM,
FANCI, and checkpoint proteins in the interstrand DNA crosslink repair
pathway is conserved in C. elegans. DNA Repair (Amst.) 9, 374–382.
Lees-Miller, S.P., Godbout, R., Chan, D.W., Weinfeld, M., Day, R.S., 3rd,
Barron, G.M., and Allalunis-Turner, J. (1995). Absence of p350 subunit of
DNA-activated protein kinase from a radiosensitive human cell line. Science
267, 1183–1185.
Li, X., Hejna, J., and Moses, R.E. (2005). The yeast Snm1 protein is a DNA
50-exonuclease. DNA Repair (Amst.) 4, 163–170.
Ma, Y., Pannicke, U., Lu, H., Niewolik, D., Schwarz, K., and Lieber, M.R. (2005).
The DNA-dependent protein kinase catalytic subunit phosphorylation sites in
human Artemis. J. Biol. Chem. 280, 33839–33846.
Martin, J.S., Winkelmann, N., Petalcorin, M.I., McIlwraith, M.J., and Boulton,
S.J. (2005). RAD-51-dependent and -independent roles of a Caenorhabditis
elegans BRCA2-related protein during DNA double-strand break repair.
Mol. Cell. Biol. 25, 3127–3139.
Matsushita, N., Kitao, H., Ishiai, M., Nagashima, N., Hirano, S., Okawa, K.,
Ohta, T., Yu, D.S., McHugh, P.J., Hickson, I.D., et al. (2005). A FancD2-mono-
ubiquitin fusion reveals hidden functions of Fanconi anemia core complex in
DNA repair. Mol. Cell 19, 841–847.
Meetei, A.R., Sechi, S., Wallisch, M., Yang, D., Young, M.K., Joenje, H., Hoat-
lin, M.E., and Wang, W. (2003). A multiprotein nuclear complex connects
Fanconi anemia and Bloom syndrome. Mol. Cell. Biol. 23, 3417–3426.
Mimitou, E.P., and Symington, L.S. (2008). Sae2, Exo1 and Sgs1 collaborate in
DNA double-strand break processing. Nature 455, 770–774.
Moldovan, G.L., and D’Andrea, A.D. (2009). How the fanconi anemia pathway
guards the genome. Annu. Rev. Genet. 43, 223–249.
Muzzini, D.M., Plevani, P., Boulton, S.J., Cassata, G., and Marini, F. (2008).
Caenorhabditis elegans POLQ-1 and HEL-308 function in two distinct DNA
interstrand cross-link repair pathways. DNA Repair (Amst.) 7, 941–950.
Nakanishi, K., Taniguchi, T., Ranganathan, V., New, H.V., Moreau, L.A., Stot-
sky, M., Mathew, C.G., Kastan, M.B., Weaver, D.T., and D’Andrea, A.D. (2002).
Interaction of FANCD2 and NBS1 in the DNA damage response. Nat. Cell Biol.
4, 913–920.
Nakanishi, K., Yang, Y.G., Pierce, A.J., Taniguchi, T., Digweed, M., D’Andrea,
A.D., Wang, Z.Q., and Jasin, M. (2005). Human Fanconi anemia monoubiquiti-
nation pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. USA
102, 1110–1115.
Newell, A.E., Akkari, Y.M., Torimaru, Y., Rosenthal, A., Reifsteck, C.A., Cox, B.,
Grompe, M., and Olson, S.B. (2004). Interstrand crosslink-induced radials
form between non-homologous chromosomes, but are absent in sex chromo-
somes. DNA Repair (Amst.) 3, 535–542.
Page, S.L., and Hawley, R.S. (2003). Chromosome choreography: the meiotic
ballet. Science 301, 785–789.
Park, W.H., Margossian, S., Horwitz, A.A., Simons, A.M., D’Andrea, A.D., and
Parvin, J.D. (2005). Direct DNA binding activity of the Fanconi anemia D2
protein. J. Biol. Chem. 280, 23593–23598.
Petalcorin, M.I., Sandall, J., Wigley, D.B., and Boulton, S.J. (2006). CeBRC-2
stimulates D-loop formation by RAD-51 and promotes DNA single-strand
annealing. J. Mol. Biol. 361, 231–242.
Pichierri, P., Averbeck, D., and Rosselli, F. (2002). DNA cross-link-dependent
RAD50/MRE11/NBS1 subnuclear assembly requires the Fanconi anemia C
protein. Hum. Mol. Genet. 11, 2531–2546.
Polanowska, J., Martin, J.S., Garcia-Muse, T., Petalcorin, M.I., and Boulton,
S.J. (2006). A conserved pathway to activate BRCA1-dependent ubiquitylation
at DNA damage sites. EMBO J. 25, 2178–2188.
Rinaldo, C., Bazzicalupo, P., Ederle, S., Hilliard, M., and La Volpe, A. (2002).
Roles for Caenorhabditis elegans rad-51 in meiosis and in resistance to
ionizing radiation during development. Genetics 160, 471–479.
Roques, C., Coulombe, Y., Delannoy, M., Vignard, J., Grossi, S., Brodeur, I.,
Rodrigue, A., Gautier, J., Stasiak, A.Z., Stasiak, A., et al. (2009). MRE11-
RAD50-NBS1 is a critical regulator of FANCD2 stability and function during
DNA double-strand break repair. EMBO J. 28, 2400–2413.
Rothfuss, A., and Grompe, M. (2004). Repair kinetics of genomic interstrand
DNA cross-links: evidence for DNA double-strand break-dependent activation
of the Fanconi anemia/BRCA pathway. Mol. Cell. Biol. 24, 123–134.
Ruis, B.L., Fattah, K.R., and Hendrickson, E.A. (2008). The catalytic subunit of
DNA-dependent protein kinase regulates proliferation, telomere length, and
genomic stability in human somatic cells. Mol. Cell. Biol. 28, 6182–6195.
Timmers, C., Taniguchi, T., Hejna, J., Reifsteck, C., Lucas, L., Bruun, D.,
Thayer, M., Cox, B., Olson, S., D’Andrea, A.D., et al. (2001). Positional cloning
of a novel Fanconi anemia gene, FANCD2. Mol. Cell 7, 241–248.
Veuger, S.J., Curtin, N.J., Richardson, C.J., Smith, G.C., and Durkacz, B.W.
(2003). Radiosensitization and DNA repair inhibition by the combined use of
novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) poly-
merase-1. Cancer Res. 63, 6008–6015.
Wang, W. (2007). Emergence of a DNA-damage response network consisting
of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 8, 735–748.
Wood, W. (1988). The nematode Caenorhabditis elegans (Cold Spring Harbor,
NY: Cold Spring Harbor Laboratory Press).
Yajima, H., Lee, K.J., and Chen, B.P. (2006). ATR-dependent phosphorylation
of DNA-dependent protein kinase catalytic subunit in response to UV-induced
replication stress. Mol. Cell. Biol. 26, 7520–7528.
Youds, J.L., Barber, L.J., Ward, J.D., Collis, S.J., O’Neil, N.J., Boulton, S.J.,
and Rose, A.M. (2008). DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ
homologue and functions in interstrand cross-link repair. Mol. Cell. Biol. 28,
1470–1479.
Youds, J.L., Barber, L.J., and Boulton, S.J. (2009). C. elegans: a model of Fan-
coni anemia and ICL repair. Mutat. Res. 668, 103–116.
Zalevsky, J., MacQueen, A.J., Duffy, J.B., Kemphues, K.J., and Villeneuve,
A.M. (1999). Crossing over during Caenorhabditis elegans meiosis requires
a conserved MutS-based pathway that is partially dispensable in budding
yeast. Genetics 153, 1271–1283.
Zhu, Z., Chung, W.H., Shim, E.Y., Lee, S.E., and Ira, G. (2008). Sgs1 helicase
and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell
134, 981–994.
Molecular Cell 39, 25–35, July 9, 2010 ª2010 Elsevier Inc. 35
Related Documents











