J Med Genet 1997;34:63-72 Preparing for presymptomatic DNA testing for early onset Alzheimer's disease/cerebral haemorrhage and hereditary Pick disease A Tibben, M Stevens, G MW R de Wert, M F Niermeijer, C M van Duijn, J C van Swieten Department of Clinical Genetics, University Hospital Dijkzigt and Erasmus University, PO Box 1738, 3000 DR Rotterdam, The Netherlands A Tibben M F Niermeijer Department of Neurology, University Hospital Dijkzigt and Erasmus University, Rotterdam, The Netherlands M Stevens J C van Swieten Department of Medical Psychology and Psychotherapy, University Hospital Dijkzigt and Erasmus University, Rotterdam, The Netherlands A Tibben Department of Philosophy and Medical Ethics, University Hospital Dijkzigt and Erasmus University, Rotterdam, The Netherlands G M W R de Wert Department of Epidemiology and Biostatistics, University Hospital Dijkzigt and Erasmus University, Rotterdam, The Netherlands C M van Duijn Institute of Bioethics, Maastricht, The Netherlands G M W R de Wert Correspondence to: Dr Tibben. Received 13 March 1996 Revised version accepted for publication 9 August 1996 Abstract The acceptability of presymptomatic testing in 21 people at 50% risk for the APP-692 mutation causing presenile Alz- heimer's disease or cerebral haemorrhage resulting from cerebral amyloid an- giopathy (FAD-CH), and in 43 people at 50% risk for hereditary Pick disease (HPD) was assessed. Neither group differed in demographic variables. Thirty-nine people (64%) in the whole group would request presymptomatic testing if it were clinically available, although two-thirds did not yet feel ready to take it. The most important reasons in the HPD and FAD- CH group for taking the test were: to fur- ther basic research (42% and 47%, re- spectively), informing children (47% and 50%, respectively), future planning (29% and 47%, respectively), and relieving un- certainty (46% and 27%, respectively). The most commonly cited effect of an unfavourable test result concerned in- creasing problems for spouses (75% and 76%, respectively) and children (61% and 57%, respectively). Most respondents de- nied that an unfavourable result would have adverse effects on personal mood or relationship. One-third of all respondents favoured prenatal testing where one of the parents had an increased risk for HPD or FAD-CH. Participants would encourage their offspring to have the test before start- ing a relationship (35%) and before family planning (44%). Thirty-seven percent of the respondents would encourage their children to opt for prenatal diagnosis. People at risk for HPD were significantly more preoccupied with the occurrence of potential symptoms in themselves, com- pared with those at risk for FAD-CH, re- flecting the devastating impact that disinhibition in the affected patient has on the family. Our findings underline the need for adequate counselling and the availability of professional and community resources to deal with the impact of test results in subjects and their relatives. (J Med Genet 1997;34:63-72) Keywords: presymptomatic testing; Alzheimer's disease; Pick disease. An increasing number of neurodegenerative diseases have been defined at the molecular level in recent years, making it possible to determine the genotype precisely before the onset of symptoms. Presymptomatic testing programmes are available for Huntington's dis- ease (HD), hereditary cerebral haemorrhage with amyloid-Dutch type, inherited cerebral ataxia, myotonic dystrophy, and Alzheimer's disease. -6 For other autosomal dominant dis- orders, the genetic cause will be detected in the near, foreseeable future. Significant pro- gress has been made in unravelling the dy- namics of genes and their products.7 However, effective pharmacological or gene therapy for late onset neurodegenerative disorders is not expected to be available in the immediate fu- ture. In a collaborative programme on neuro- genetics in Rotterdam, two studies on early onset dementia are currently being carried out. The first study concerns a family with presenile Alzheimer's dementia and cerebral haem- orrhage (FAD-CH). FAD-CH is caused by a mutation in codon 692 of the gene for f- amyloid precursor protein (fAPP) on chro- mosome 21.8 Extracellular amyloid plaques, intraneuronal neurofibrillar tangles, and amy- loid angiopathy were found in the brains of two FAD-CH patients. Mutations in the ,APP gene account for less than 3% of disease onset before 65 years of age.8 In a second study, early onset Pick disease was found in three Dutch families with an autosomal dominant transmission pattern over five to seven generations. The typical clinical features in the patients with hereditary Pick disease (HPD) were disinhibition, stereotyped behaviour, roaming, and hyperorality. Frontal and temporal atrophy on CT scan supported the clinical diagnosis in eight, two, and five living patients in the three families, respectively. The diagnosis of HPD was confirmed in each family by pathological examination of the brain in one, 14, and 15 patients, respectively. Macroscopic examinations showed selective atrophy of the frontal cortex, non-specific changes (neuronal loss, spongiosis, gliosis), and ballooned cells. Evidence was found in these three families for linkage of HPD to chro- mosome 17.27 HPD and FAD-CH are primary degenerative diseases of the brain, with onset usually in the fourth to sixth decades of life.9-'2 Both conditions have an average age of onset be- tween 40 and 60 years of age. The course in both disorders is variable, with the development of profound dementia ranging from two to 10 years after diagnosis. No specific treatment is 63 group.bmj.com on July 15, 2011 - Published by jmg.bmj.com Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

J Med Genet 1997;34:63-72
Preparing for presymptomatic DNA testing forearly onset Alzheimer's disease/cerebralhaemorrhage and hereditary Pick disease
A Tibben, M Stevens, G M W R de Wert, M F Niermeijer, C M van Duijn,J C van Swieten
Department ofClinical Genetics,University HospitalDijkzigt and ErasmusUniversity, PO Box1738, 3000 DRRotterdam, TheNetherlandsA TibbenM F Niermeijer
Department ofNeurology, UniversityHospital Dijkzigt andErasmus University,Rotterdam, TheNetherlandsM StevensJ C van Swieten
Department ofMedical Psychologyand Psychotherapy,University HospitalDijkzigt and ErasmusUniversity,Rotterdam, TheNetherlandsA Tibben
Department ofPhilosophy andMedical Ethics,University HospitalDijkzigt and ErasmusUniversity,Rotterdam, TheNetherlandsG M W R de Wert
Department ofEpidemiology andBiostatistics,University HospitalDijkzigt and ErasmusUniversity,Rotterdam, TheNetherlandsC M van Duijn
Institute of Bioethics,Maastricht, TheNetherlandsG M W R de Wert
Correspondence to:Dr Tibben.
Received 13 March 1996Revised version accepted forpublication 9 August 1996
AbstractThe acceptability of presymptomatictesting in 21 people at 50% risk for theAPP-692 mutation causing presenile Alz-heimer's disease or cerebral haemorrhageresulting from cerebral amyloid an-giopathy (FAD-CH), and in 43 people at50% risk for hereditary Pick disease (HPD)was assessed. Neither group differedin demographic variables. Thirty-ninepeople (64%) in the whole group wouldrequest presymptomatic testing if it wereclinically available, although two-thirdsdid not yet feel ready to take it. The mostimportant reasons in the HPD and FAD-CH group for taking the test were: to fur-ther basic research (42% and 47%, re-spectively), informing children (47% and50%, respectively), future planning (29%and 47%, respectively), and relieving un-certainty (46% and 27%, respectively).The most commonly cited effect of anunfavourable test result concerned in-creasing problems for spouses (75% and76%, respectively) and children (61% and57%, respectively). Most respondents de-nied that an unfavourable result wouldhave adverse effects on personal mood orrelationship. One-third of all respondentsfavoured prenatal testing where one of theparents had an increased risk for HPD orFAD-CH. Participants would encouragetheir offspring to have the test before start-ing a relationship (35%) and before familyplanning (44%). Thirty-seven percent ofthe respondents would encourage theirchildren to opt for prenatal diagnosis.People at risk for HPD were significantlymore preoccupied with the occurrence ofpotential symptoms in themselves, com-pared with those at risk for FAD-CH, re-flecting the devastating impact thatdisinhibition in the affected patient hason the family. Our findings underline theneed for adequate counselling and theavailability ofprofessional and communityresources to deal with the impact of testresults in subjects and their relatives.(J Med Genet 1997;34:63-72)
Keywords: presymptomatic testing; Alzheimer's disease;Pick disease.
An increasing number of neurodegenerativediseases have been defined at the molecularlevel in recent years, making it possible to
determine the genotype precisely before theonset of symptoms. Presymptomatic testingprogrammes are available for Huntington's dis-ease (HD), hereditary cerebral haemorrhagewith amyloid-Dutch type, inherited cerebralataxia, myotonic dystrophy, and Alzheimer'sdisease. -6 For other autosomal dominant dis-orders, the genetic cause will be detected inthe near, foreseeable future. Significant pro-gress has been made in unravelling the dy-namics of genes and their products.7 However,effective pharmacological or gene therapy forlate onset neurodegenerative disorders is notexpected to be available in the immediate fu-ture.
In a collaborative programme on neuro-genetics in Rotterdam, two studies on earlyonset dementia are currently being carried out.The first study concerns a family with presenileAlzheimer's dementia and cerebral haem-orrhage (FAD-CH). FAD-CH is caused by amutation in codon 692 of the gene for f-amyloid precursor protein (fAPP) on chro-mosome 21.8 Extracellular amyloid plaques,intraneuronal neurofibrillar tangles, and amy-loid angiopathy were found in the brains oftwo FAD-CH patients. Mutations in the ,APPgene account for less than 3% of disease onsetbefore 65 years of age.8
In a second study, early onset Pick diseasewas found in three Dutch families with anautosomal dominant transmission pattern overfive to seven generations. The typical clinicalfeatures in the patients with hereditary Pickdisease (HPD) were disinhibition, stereotypedbehaviour, roaming, and hyperorality. Frontaland temporal atrophy on CT scan supportedthe clinical diagnosis in eight, two, and fiveliving patients in the three families, respectively.The diagnosis of HPD was confirmed in eachfamily by pathological examination of thebrain in one, 14, and 15 patients, respectively.Macroscopic examinations showed selectiveatrophy of the frontal cortex, non-specificchanges (neuronal loss, spongiosis, gliosis), andballooned cells. Evidence was found in thesethree families for linkage of HPD to chro-mosome 17.27HPD and FAD-CH are primary degenerative
diseases of the brain, with onset usually inthe fourth to sixth decades of life.9-'2 Bothconditions have an average age of onset be-tween 40 and 60 years of age. The course inboth disorders is variable, with the developmentof profound dementia ranging from two to 10years after diagnosis. No specific treatment is
63
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from

Tibben, Stevens, de Wert, Niermeijer, van Duijn, van Swieten
available, but the use of palliative treatmentsis now being explored.
Misdiagnosis of HPD or FAD-CH, such asdepression (FAD-CH) or manic states (HPD),may occur in the early stages of the disease andhas often led to unsuccessful psychotherapy ofcouples. The diagnosis of FAD-CH and HPDis psychologically devastating to the partner andhis/her offspring, who have seen the patient'sparent, sib, or another close relative becomeprogressively disabled. People sometimes in-correctly believe themselves to be at risk be-cause of symptom searching or preoccupationwith early signs. Often, "soft" signs, which arenot specific forFAD-CH or HPD, are perceivedas a precursor of the disease.
Genetic mutational or linkage analysis mayconfirm the diagnosis in patients with HPD orFAD-CH and could provide presymptomatictesting for at risk subjects. Presymptomatictesting for Huntington's disease is consideredas the paradigm for prediction programmes forother late onset neurodegenerative diseases andcancers'3 and should provide the experiencenecessary to improve pretest and post-testcounselling. Before the introduction of the pre-dictive test, attitudinal studies among those atrisk for Huntington's disease have shown thatthe commonly cited reasons for taking the pre-dictive test were the unbearable uncertainty,anticipating the future, and planning a family,but that an unfavourable result might also leadto depressive feelings and suicidal be-haviour. 14-6 These surveys indicated that themajority would make use of a predictive test ifit were available. With careful consideration ofthe ethical, clinical, and legal implications ofpresymptomatic testing for an incurable lateonset disease,'3 17-9 guidelines were carefullydeveloped and testing was carried out cau-tiously in research settings.20 To date, requestsfor the test have been far below the expectedrate, although in The Netherlands 150 peopleapply for it yearly, and about 15% of the es-timated cohort at 50% risk has received testresults.2' 22 Only a small amount of experiencehas been reported on testing for presenile de-mentia. In Sweden, one out of three peopletested at 50% risk for the APP 670/671 muta-tion was identified as a mutation carrier. Aftera one year period with depressive feelings andsuicidal thoughts, this subject could eventuallyhandle his situation. Intensive attention andcare at the genetics clinic was needed. Thenon-carriers had expressed their relief.23 Al-though the potential benefits of predictive test-ing may include relieving uncertainty andplanning the future, the acceptability of, andneed for, presymptomatic testing for early onsetdementia has not yet been established.2425
This study addresses the impact of ap-proaching families with a hereditary preseniledementia for genetic research with the aim ofestablishing a predictive testing programme, aswas done for Huntington's disease. We studiedthe ability to cope with being at risk for HPDor FAD-CH, the influence of the disease upona variety ofareas of life, and the attitude towardspresymptomatic testing. Guided by our clinicalexperience with these neurodegenerative dis-
orders, we expected similar attitudes as werefound in people at risk for Huntington's diseasein the Dutch presymptomatic testing pro-gramme.26 The results could be helpful for themedical-ethical evaluation of this and othergenetic research programmes, to establish asuitable, disease specific testing protocol, andto develop support strategies when testing doesbecome available.
Subjects and methodsClinical, genetic-epidemiological, and patho-logical research was conducted on FAD-CHand HPD in a collaborative programme inRotterdam, The Netherlands. DNA linkageand mutation studies were done in one familywith familial early onset FAD-CH8 and threefamilies with HPD.2728 Participants were at50% risk for HPD (n=43) or FAD-CH (n=21) and cooperated after fully informed con-sent. Participants had a general medical ex-amination, neurological examination, neuro-psychological testing, brain imaging (MRIscan), blood sampling, and were asked to par-ticipate in a clinical psychological assessmentand an attitudinal survey. The Medical EthicsCommittee of the University Hospital Dijkzigt,Rotterdam, approved the protocols. Par-ticipants were informed about the disease (MS,CMvD, JCvS), and received information aboutthe genetic pattern of familial FAD-CH orHPD. They were referred, when needed, tothe Clinical Genetics Department for furthergenetic counselling (MFN) or psychologicalsupport (AT).Forty-three out of 50 people at risk for HPD(86%) and 21 out of 26 people at risk forFAD-CH (81%) gave consent for the clinicalpsychological study which consisted of an indepth interview and administering psy-chological questionnaires that were completedat home. Questionnaires were returned withina week after the interview. Demographic data(gender, age, marital status, employment sta-tus, number of children, number of sibs, andlevel of education) were collected. An AttitudeQuestionnaire (AQ) was administered that con-sisted of questions on the following areas: ex-perience of the disorder, the age at which theperson learned about the heredity of HPD orFAD-CH, the subject's attitudes towards takingthe presymptomatic test, the expected outcomeofthe test, and attitudes toward prenatal testingand terminating a pregnancy in different cir-cumstances. Most questions had the responsecategories yes/agree, don't know, no/not agree.Questions on experience of the disease, impacton personal life, and reasons for and againstpredictive testing were open ended, for whichresponse categories were compiled to suit thecommon themes emerging from the answers.The estimated risk of inheriting the gene ordeveloping the disease was assessed using avisual analogue scale. People who consideredtaking a future predictive test when it becameclinically available answered questions aboutthe anticipated impact of either test results. TheAQ was adapted from the Dutch HuntingtonPresymptomatic Programme.26
64
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from
Presymptomatic DNA testing for Alzheimer's diseaselcerebral haemorrhage and Pick disease
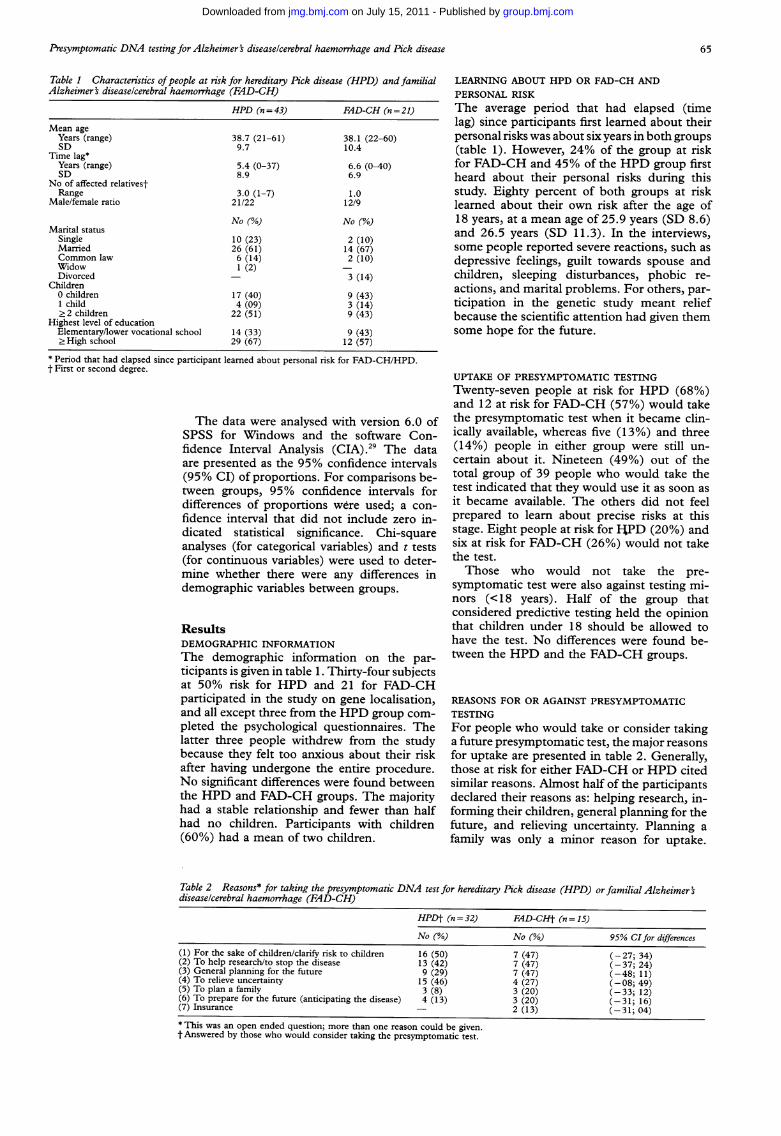
Table 1 Characteristics ofpeople at risk for hereditary Pick disease (HPD) and familialAlzheimer's diseaselcerebral haemorrhage (FAD-CH)
HPD (n = 43)
Mean ageYears (range)SD
Time lag*Years (range)SD
No of affected relativestRange
Male/female ratio
Marital statusSingleMarriedCommon lawWidowDivorced
Children0 children1 child.2 children
Highest level of educationElementary/lower vocational school> High school
38.7 (21-61)9.7
5.4 (0-37)8.9
3.0 (1-7)21/22
No (%)
10 (23)26 (61)6 (14)1 (2)
17 (40)4 (09)
22 (51)
14 (33)29 (67)
FAD-CH (n = 21)
38.1 (22-60)10.4
6.6 (0-40)6.9
1.012/9
No (%)
2 (10)14 (67)2 (10)
3 (14)
9 (43)3 (14)9 (43)9 (43)12 (57)
LEARNING ABOUT HPD OR FAD-CH ANDPERSONAL RISKThe average period that had elapsed (timelag) since participants first learned about theirpersonal risks was about six years in both groups(table 1). However, 24% of the group at riskfor FAD-CH and 45% of the HPD group firstheard about their personal risks during thisstudy. Eighty percent of both groups at risklearned about their own risk after the age of18 years, at a mean age of 25.9 years (SD 8.6)and 26.5 years (SD 11.3). In the interviews,some people reported severe reactions, such asdepressive feelings, guilt towards spouse andchildren, sleeping disturbances, phobic re-actions, and marital problems. For others, par-ticipation in the genetic study meant reliefbecause the scientific attention had given themsome hope for the future.
* Period that had elapsed since participant learned about personal risk for FAD-CH/HPD.t First or second degree.
The data were analysed with version 6.0 ofSPSS for Windows and the software Con-fidence Interval Analysis (CIA).29 The dataare presented as the 95% confidence intervals(95% CI) of proportions. For comparisons be-tween groups, 95% confidence intervals fordifferences of proportions were used; a con-fidence interval that did not include zero in-dicated statistical significance. Chi-squareanalyses (for categorical variables) and t tests(for continuous variables) were used to deter-mine whether there were any differences indemographic variables between groups.
ResultsDEMOGRAPHIC INFORMATIONThe demographic information on the par-ticipants is given in table 1. Thirty-four subjectsat 50% risk for HPD and 21 for FAD-CHparticipated in the study on gene localisation,and all except three from the HPD group com-pleted the psychological questionnaires. Thelatter three people withdrew from the studybecause they felt too anxious about their riskafter having undergone the entire procedure.No significant differences were found betweenthe HPD and FAD-CH groups. The majorityhad a stable relationship and fewer than halfhad no children. Participants with children(60%) had a mean of two children.
UPTAKE OF PRESYMPTOMATIC TESTINGTwenty-seven people at risk for HPD (68%)and 12 at risk for FAD-CH (57%) would takethe presymptomatic test when it became clin-ically available, whereas five (13%) and three(14%) people in either group were still un-certain about it. Nineteen (49%) out of thetotal group of 39 people who would take thetest indicated that they would use it as soon asit became available. The others did not feelprepared to learn about precise risks at thisstage. Eight people at risk for IJPD (20%) andsix at risk for FAD-CH (26%) would not takethe test.Those who would not take the pre-
symptomatic test were also against testing mi-nors (<18 years). Half of the group thatconsidered predictive testing held the opinionthat children under 18 should be allowed tohave the test. No differences were found be-tween the HPD and the FAD-CH groups.
REASONS FOR OR AGAINST PRESYMPTOMATICTESTINGFor people who would take or consider takinga future presymptomatic test, the major reasonsfor uptake are presented in table 2. Generally,those at risk for either FAD-CH or HPD citedsimilar reasons. Almost half of the participantsdeclared their reasons as: helping research, in-forming their children, general planning for thefuture, and relieving uncertainty. Planning afamily was only a minor reason for uptake.
Table 2 Reasons* for taking the presymptomatic DNA test for hereditary Pick disease (HPD) or familial Alzheimer'sdiseaselcerebral haemorrhage (FAD-CH)
HPDt (n =32) FAD-CHt (n =15)
No (%) No (%) 95% CIfor differences(1) For the sake of children/clarify risk to children 16 (50) 7 (47) (-27; 34)(2) To help research/to stop the disease 13 (42) 7 (47) (-37; 24)(3) General planning for the future 9 (29) 7 (47) (-48; 11)(4) To relieve uncertainty 15 (46) 4 (27) (-08; 49)(5) To plan a family 3 (8) 3 (20) (-33; 12)(6) To prepare for the future (anticipating the disease) 4 (13) 3 (20) (-31; 16)(7) Insurance - 2 (13) (-31; 04)* This was an open ended question; more than one reason could be given.t Answered by those who would consider taking the presymptomatic test.
65
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from

Tibben, Stevens, de Wert, Niermeijer, van Duijn, van Swieten
Table 3 Impact of hereditary Pick disease (HPD) or familial Alzheimer's diseaselcerebral haemorrhage (FAD-CH) onlife ofpeople at 50% risk
HPD (n = 40) FAD-CH (n=21)
No (Y.) No (%o) 95% CIfor differences
(1) Preoccupation with symptoms 24 (60) 3 (14) (24; 67)(2) Restriction in planning the future 21 (53) 7 (33) (-06; 45)(3) Anxiety, depression, uncertainty 24 (60) 6 (29) (07; 56)(4) Disturbance of family life 5 (13) 2 (10) (- 13; 19)(5) Positive influence - 4 (20) (-36; -02)(6) No influence 8 (20) 11 (52) (-57; -08)
Table 4 Most significant symptoms* of hereditary Pick disease (HPD or familial Alzheimers diseaselcerebralhaemorrhage (FAD-CH) as perceived and experienced by people at risk
HPD (n 40) FAD-CH (n 21)
No (0%) No (%O) 95% CIfor differences
(1) Change of personality 12 (30) 6 (29) (-23; 25)Disinhibition/restlessness 28 (70) 1 (5) (48; 82)Loss of insight and judgement 12 (30) 4 (19) (- 11; 33)Emotional lability/euphoria - 2 (10) (-22; 03)Lack of spontaneity/emotional unconcern 9 (23) 1 (5) (02; 34)Depressive episodes 4 (10) 3 (14) (-22; 13)Aggression 9 (23) 1 (5) (02; 34)
(2) Stereotyped behaviour 4 (10) 2 (10) (-15; 16)(3) Cognitive deterioration
Dysmnesia 17 (40) 7 (33) (-19; 32)Disorientation 4 (10) 6 (29) (-40; 03)Dysphasia 3 (7) 1 (5) (-09; 15)Dyspraxia 12 (30) 2 (10) (02; 39)
* In first or second degree affected relatives.
Two people would take the test for reasons ofinsurance. Considerations against pre-symptomatic testing were expressed by 28people out of the group that considered pre-dictive testing (60%). Fear of adverse effectssuch as anxiety and depression after an un-favourable result was mentioned by 24 of them(51%). Seven (15%) emphasised that an un-favourable result would overshadow their lifeentirely. One person feared that an un-favourable test result might lead to being re-fused insurance.
Fourteen subjects who would not take thetest (eight at risk for HPD (20%) and six atrisk for FAD-CH (26%)) cited as the mainreasons against testing the fear of unfavourabletest results and the inability to cope with suchan outcome, preoccupation with signs of theonset of the disease, and expected distortionof the current course of life. One person fearedpossible misuse by insurance companies. Inthis group only few reasons for testing weregiven: the responsibility to inform children (n =2), family planning (n = 2), and to help research(n = 2). No differences were found between theHPD group and the FAD-CH group.
IMPACT OF HPD/FAD-CH ON PERSONAL LIFE
The impact of the disease on personal life ispresented in table 3. Subjects at risk for HPDreported significantly more preoccupation withsymptoms than the FAD-CH-group. Morethan half (60%) of the HPD group said thatthe disease in their relatives had affected theirmood (anxiety, depression) and had led tofeelings of uncertainty. More than half (53%)of the HPD group felt restricted in makingplans for the future. Four had previously under-gone sterilisation to prevent transmission of thegene to their offspring. For FAD-CH, more
than half of those at risk (52%) said that thedisease had not influenced their personal life.Comparison of the most significant symp-
toms of HPD/FAD-CH in affected relatives asperceived by the participants (table 4) showedthat more than two-thirds (70%) of the HPDgroup mentioned the disinhibition and rest-lessness as the most significant symptoms. Insome cases, specific changes in oral/dietary be-haviour and sexual disinhibition was men-tioned. In the FAD-CH group at risk, themost significant symptoms were dysmnesia andpersonality changes.
EXPECTED EFFECTS OF AN INCREASED ORDECREASED RISK FOR HPD/FAD-CHPrevious feelings about getting the disease ornot were assessed with a visual analogue scale.Twenty-seven people at risk for HPD (68%)and 15 at risk for FAD-CH (71 %) thought thattheir personal risk was equal to or less than50%. Eighteen percent in the HPD group and10% in the FAD-CH group estimated their riskas higher than 70%. Anticipation of the effectsof being identified as a gene carrier did notdiffer between the groups and showed a highawareness of the increased burden for thespouse (table 5). Participants vividly com-mented on how their affected parent was notaware of the deterioration in the later stages ofthe disease, but that the healthy parent becameextremely burdened by the devastation causedby the disease and the difficult decisions tobe made regarding the patient. Twenty-threepercent of the HPD/FAD-CH group wereafraid of becoming depressed. Forty-nine per-cent of the HPD/FAD-CH groups had con-fidence that they could cope with anunfavourable test outcome, whereas 12% statedthat such a result would ruin their life.
66
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from
Presymptomatic DNA testing for Alzheimer's diseaselcerebral haemorrhage and Pick disease
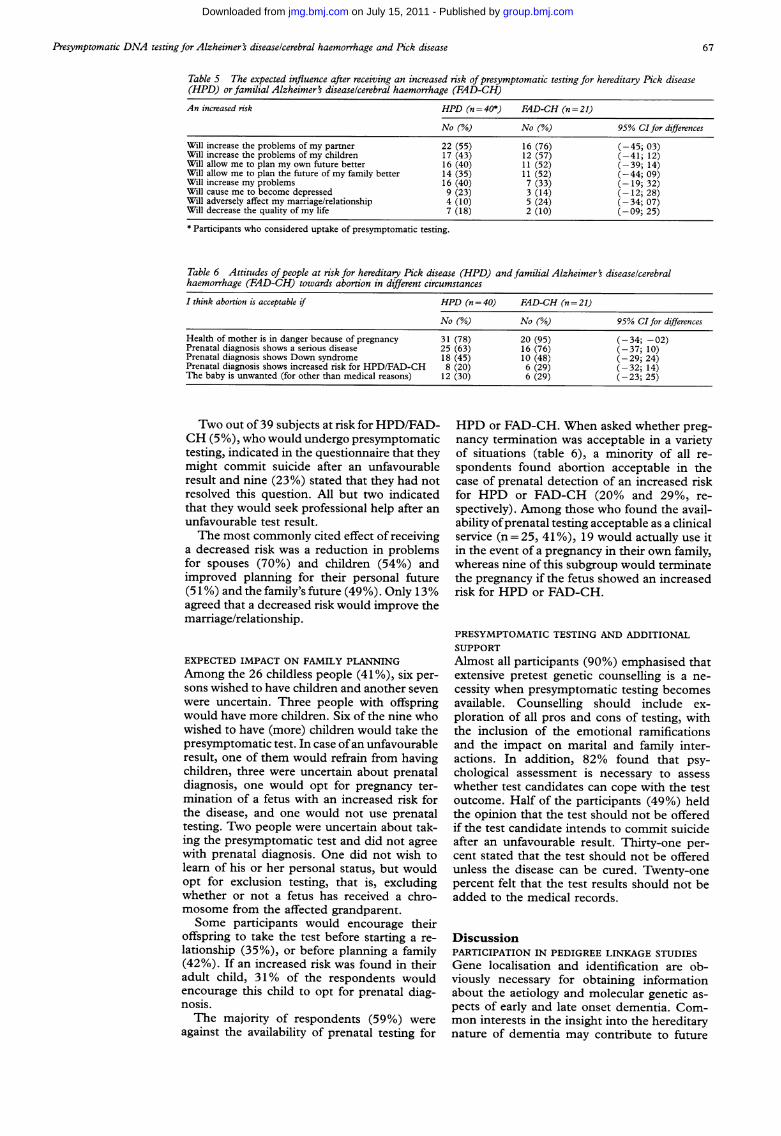
Table 5 The expected influence after receiving an increased risk ofpresymptomatic testing for hereditary Pick disease(HPD) or familial Alzheimer's diseaselcerebral haemorrhage (FAD-CH)An increased risk HPD (n = 40*) FAD-CH (n =21)
No (%) No (%) 95% CIfor differencesWill increase the problems of my partner 22 (55) 16 (76) (-45; 03)Will increase the problems of my children 17 (43) 12 (57) (-41; 12)Will allow me to plan my own future better 16 (40) 11(52) (-39; 14)Will allow me to plan the future of my family better 14 (35) 11(52) (-44; 09)Will increase my problems 16 (40) 7 (33) (-19; 32)Will cause me to become depressed 9 (23) 3 (14) (-12; 28)Will adversely affect my marriage/relationship 4 (10) 5 (24) (-34; 07)Will decrease the quality of my life 7 (18) 2 (10) (-09; 25)* Participants who considered uptake of presymptomatic testing.
Table 6 Attitudes ofpeople at risk for hereditary Pick disease (HPD) and familial Alzheimer' diseaselcerebralhaemorrhage (FAD-CH) towards abortion in different circumstances
I think abortion is acceptable if HPD (n = 40) FAD-CH (n = 21)
No (%) No (%) 95% CI for differences
Health of mother is in danger because of pregnancy 31 (78) 20 (95) (-34; -02)Prenatal diagnosis shows a serious disease 25 (63) 16 (76) (-37; 10)Prenatal diagnosis shows Down syndrome 18 (45) 10 (48) (-29; 24)Prenatal diagnosis shows increased risk for HPD/FAD-CH 8 (20) 6 (29) (-32; 14)The baby is unwanted (for other than medical reasons) 12 (30) 6 (29) (-23; 25)
Two out of 39 subjects at risk for HPD/FAD-CH (5%), who would undergo presymptomatictesting, indicated in the questionnaire that theymight commit suicide after an unfavourableresult and nine (23%) stated that they had notresolved this question. All but two indicatedthat they would seek professional help after anunfavourable test result.The most commonly cited effect of receiving
a decreased risk was a reduction in problemsfor spouses (70%) and children (54%) andimproved planning for their personal future(51 %) and the family's future (49%). Only 13%agreed that a decreased risk would improve themarriage/relationship.
EXPECTED IMPACT ON FAMILY PLANNINGAmong the 26 childless people (41%), six per-sons wished to have children and another sevenwere uncertain. Three people with offspringwould have more children. Six of the nine whowished to have (more) children would take thepresymptomatic test. In case ofan unfavourableresult, one of them would refrain from havingchildren, three were uncertain about prenataldiagnosis, one would opt for pregnancy ter-mination of a fetus with an increased risk forthe disease, and one would not use prenataltesting. Two people were uncertain about tak-ing the presymptomatic test and did not agreewith prenatal diagnosis. One did not wish tolearn of his or her personal status, but wouldopt for exclusion testing, that is, excludingwhether or not a fetus has received a chro-mosome from the affected grandparent.Some participants would encourage their
offspring to take the test before starting a re-lationship (35%), or before planning a family(42%). If an increased risk was found in theiradult child, 31% of the respondents wouldencourage this child to opt for prenatal diag-nosis.The majority of respondents (59%) were
against the availability of prenatal testing for
HPD or FAD-CH. When asked whether preg-nancy termination was acceptable in a varietyof situations (table 6), a minority of all re-spondents found abortion acceptable in thecase of prenatal detection of an increased riskfor HPD or FAD-CH (20% and 29%, re-spectively). Among those who found the avail-ability ofprenatal testing acceptable as a clinicalservice (n=25, 41%), 19 would actually use itin the event of a pregnancy in their own family,whereas nine of this subgroup would terminatethe pregnancy if the fetus showed an increasedrisk for HPD or FAD-CH.
PRESYMPTOMATIC TESTING AND ADDITIONALSUPPORTAlmost all participants (90%) emphasised thatextensive pretest genetic counselling is a ne-cessity when presymptomatic testing becomesavailable. Counselling should include ex-ploration of all pros and cons of testing, withthe inclusion of the emotional ramificationsand the impact on marital and family inter-actions. In addition, 82% found that psy-chological assessment is necessary to assesswhether test candidates can cope with the testoutcome. Half of the participants (49%) heldthe opinion that the test should not be offeredif the test candidate intends to commit suicideafter an unfavourable result. Thirty-one per-cent stated that the test should not be offeredunless the disease can be cured. Twenty-onepercent felt that the test results should not beadded to the medical records.
DiscussionPARTICIPATION IN PEDIGREE LINKAGE STUDIESGene localisation and identification are ob-viously necessary for obtaining informationabout the aetiology and molecular genetic as-pects of early and late onset dementia. Com-mon interests in the insight into the hereditarynature of dementia may contribute to future
67
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from

Tibben, Stevens, de Wert, Niermeijer, van Duijn, van Swieten
therapeutic interventions. Half of those at riskin this study mentioned "to help research" asan important motive for participation. Yet thepotential burden of participation in pedigreeand linkage studies is often underestimated byresearchers and medical specialists. Facing thethreat ofan appalling disease can cause a varietyof psychological, legal, and ethical problemsfor people at risk. In addition, family membersmay learn about their own risk for the first timethrough participation. This problem was oftenthe case for the groups at risk for HPD orFAD-CH. In the information sessions, manypeople did not fully understand all the ra-mifications ofbeing at risk. Ideas about hereditywere only vague and information previouslyobtained from professionals (neurologist, gen-eral practitioner) were often similarly unclear.Most of the participants were accordinglyshocked by the information about their ownrisks. Is it acceptable to recruit relatives forparticipation in research who may not evensuspect that the disease under investigation isgenetic, and that they may carry genes po-tentially harmful to them or their offspring?For some relatives the request for participationwas not ominous news because they suspectedthat the disease was hereditary. Other relativesmay have a positive attitude towards researchbecause genetic information may be relevant,for example, for reproductive decisions or in-forming their children. Refraining from con-ducting family studies leaves a family ignorantand might prevent members from knowing thepotential threat of personal risk. The moralprice of such a policy is that family membersare denied the possibility of anticipating theirfuture and making general decisions. Obtainingconsent, protecting privacy and confidentiality,and safeguarding divergent and conflicting in-trafamilial and intergenerational interests pres-ent moral challenges to the conduct of soundresearch.30 Our experience emphasises thatstrong collaboration of all disciplines (mo-lecular and clinical geneticist, neurologist,psychologist, medical ethicist, general prac-titioner) involved is a requirement for con-ducting genetic studies.Many people at risk for HPD were pre-
occupied with early symptoms in themselveswhich reflected anxiety and great concern abouttheir future, which was different for the FAD-CH group. The disinhibition/restlessness in theaffected parent and other affected relatives wasoften experienced as frightening and over-shadowed the lives of many of those at riskfor HPD. This fear affected their self-esteem,future prospects, and their relationship withspouses and relatives. Therefore, in the pro-gramming and institutional ethical review ofpedigree and linkage studies, attention must bepaid to the provision of genetic and psy-chological counselling. Also, familiarity withgenetic concepts in all medical disciplines be-comes essential and medical curricula mustmeet such requirements.
Localisation or finding of the gene oftenresults in the clinical application of predictivetesting programmes, given the experience withHuntington's disease, polyposis coli, and breast
and ovarian cancer. The predictive programmefor Huntington's disease was embedded in care-ful genetic counselling following the inter-national guidelines, and psychological followup.203132 Although the medical-ethical issuesand benefits of predictive testing are still underdebate, the widespread application of such test-ing as a clinical service proceeds for untreatablegenetic disorders. It is not known whether al-ternatives for solving the emotional and de-cision making problems in people at risk areoffered and can be sufficiently met in healthcare. Predictive programmes may be too easilyestablished as a result of finding a linkage ormutation, without proper ethical reflection orcontainment in a follow up research ex-perimental condition.Although genetic counselling often implies
being the devil's advocate by discouragingpeople at risk from undergoing the test fordiseases that have no outlook on treatment, theHuntington experience shows that applicantsfor the test are very determined to have testresults, even in those cases where other optionsof dealing with the threat might be preferable.Weighing the pros and cons of testing is even-tually a personal responsibility.
ACCEPTANCE OF PRESYMPTOMATIC TESTINGPROGRAMMESBoth FAD-CH and HPD are rare, devastatingdiseases, yet the majority of participants in thisstudy would take a presymptomatic test if itbecame available. As with results found in thoseat risk for Huntington's disease, many deniedthe potential untoward effects of becomingidentified as a gene carrier.263334 Preparationfor the future and worry about the spouse werethe main reasons for taking the test, in contrastto the HD group where family planning wasparamount.26 Because for the HPD/FAD-CHgroups the age ofonset is usually much later andthe risk increases dramatically as age advances,people at risk for HPD/FAD-CH mightconsider testing for general planning such asretirement, medical directives, and early diag-nosis and appropriate treatment.24The purpose of counselling is to safeguard
considerable deliberation. Half of the groupthat considered predictive testing found thattesting should also be accessible for minorsunder 18 years, which is similar to the opinionsof test candidates for Huntington's disease.26Yet the request of parents to test their childrenwho are minors should be rejected as this wouldviolate the child's right "not to know". It shouldbe safeguarded that the child can make anautonomous decision when he/she reaches theage of majority.35
Twenty-eight percent of those who con-sidered predictive testing would either commitor consider committing suicide after becomingidentified as a gene carrier, although they wouldseek professional support. This is similar toattitudinal studies in HD."6 Half of the par-ticipants thought that those who consideredcommitting suicide after an unfavourable resultshould not be given the test. This raises thequestion of what is good clinical practice. Ap-
68
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from

Presymptomatic DNA testing for Alzheimer's diseaselcerebral haemorrhage and Pick disease
plicants for the predictive test who are in shockor who are depressive, and who are con-sequently not able to make a well considered,autonomous choice, should not be given accessto the test or testing should be postponed.In all cases, extensive pretest counselling is aprerequisite, in which the pros and cons oftesting are explored and weighed. It should beinvestigated whether the suicidal intention isan indication of either a depressive state ofmind or of rational considerations. In addition,the counsellor can actively raise the issue ofpossible adverse reactions to unfavourable testresults, with the inclusion of suicide intentions.It should be noted that the experience withtesting for Huntington's disease has shown thatpeople at risk who are not able to cope withunfavourable test results exclude themselvesfrom testing (self-exclusion). What if a com-petent applicant expresses his intentions tocommit suicide after unfavourable results?Should access to the test be refused in suchcases? Such a policy has certain objections.First, prohibition oftesting ofthose consideringsuicide would lead to the concealment of sui-cidal intentions, as was experienced in the HDpresymptomatic testing programme. Second,unconditional refusal of access to testing wouldbe a violation of the principle of autonomy.This principle implies the professional respectfor the applicant's considerations regarding theconsequences of either test result. Moreover,refusal of access to the test has its moral pricebecause this would force test candidates toremain uncertain about their genetic status.Suicide is not immoral and the intention tocommit suicide in certain circumstances notunreasonable. Hence, suicide in case of anunfavourable test result is not a priori irrational.Consequently, it is, in our opinion, a priorimorally tenable to allow access to a futurepredictive test if an applicant expresses hisintention to commit suicide after unfavourabletest results. In conclusion, we recommend thatanxieties and expectancies regarding one's fatebe openly discussed. Testing may be postponedand additional support offered when needed. Itshould also be noted that, as clinical experiencewith Huntington's disease has shown, suicidemay become an option in the final stages ofthe disease, and not as a reaction to an increasedrisk test result or after onset of the first signsof the disorder.
People who are the first in a family to par-ticipate in genetic studies and presymptomatictesting programmes may assume the re-sponsibility for informing their offspring andrelatives about the new information. In thefamilies studied, the key person was often thepatient's spouse with whom the heredity of thedisease was first discussed and who consentedby proxy to testing the affected patient. Suchproxy consent is acceptable given the potentialinterests of children and other relatives withregard to certainty about personal risks, or therelevance of differential diagnosis. In addition,confirmation of diagnosis using DNA testingdoes not conflict with the demented patient'sinterests. It may be objected, however, thatthe potential interests of children and other
relatives disqualify them as proxy. Therefore,good medical and ethical practice requires closeconsideration and discussion whether personalinterests interfere with being a proxy.
Information on genetic studies may causeemotional upheaval in relatives who are in-formed and resentment against the informants.Informing and supporting people with this spe-cific mission about these family issues, whichare usually unexpected, may stimulate otherrelatives to appreciate the value of family stud-ies.The intended uptake of testing among the
HPD/FAD-CH groups is similar to the in-tended uptake in the HD groups at risk. Theactual uptake may be much lower given theHD experience,'6 which is illustrated by thefinding that only a minority wishes testing im-mediately upon availability. As in the HDstudies, participants at risk for HPD/FAD-CHemphasised the need for extensive pretest coun-selling and psychological assessment. Again,the group that requires predictive testing shouldalso be informed about alternative ways ofdealing with the issues that led to uptake ofthe test. Psychotherapy or behavioural therapymight help people to cope better with theiranxieties. Couples could be supported in ex-ploring other ways of dealing with their wishto have children. Predictive testing programmesseem to be subject to the "technological im-perative". Therefore, the counsellor should ap-proach applicants with full respect for theiropinions but must also play the role of thedevil's advocate when trying to discuss thepros and cons of testing and considerationof alternative coping strategies. However, thisrequires a closer collaboration of clinical gen-etics services and instititions of mental health.
Predictive testing for presenile dementia,such as Alzheimer's disease, should be under-taken only in the context of research protocols,using careful neurological and psychologicalassessments."4 Testing should not run un-noticed in a widespread clinical applicationwithout proper previous evaluation of such aservice. However, predictive testing is generallyconsidered, by both professionals and potentialusers, as a clinical service and not as a researchprotocol, with the consequence of a lack offollow up data, which hampers a thoroughmedical-ethical evaluation. Obviously, the rel-evance of mandatory research assessments forthe evaluation of predictive testing should beclarified. Consequently, the contribution to theimprovement of the clinical service must beconvincing. If these requirements are met,people should be encouraged to participate inresearch assessments, and to adhere to theprovisions of a research protocol. This may bean appropriate expectation by those offeringpresymptomatic testing for HPD or FAD-CHand HPD.One special issue that was not addressed by
the Alzheimer's study group,24 but which needsattention, concerns those at 25% risk, who areasymptomatic grandchildren of affected sub-jects. Identification of a person at 25% risk asa gene carrier identifies the unaffected, in-tervening, parent as a carrier of the disease
69
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from

Tibben, Stevens, de Wert, Niermeijer, van Duijn, van Swieten
mutation. Moreover, sibs would see their risksincrease to 50%. After the identification of theHD gene, recommendations for presympto-matic testing included a statement regardingthose at 25% risk. These recommendationsstated that extreme care should be exercisedwhen testing a person at risk would in-advertently provide information about anotherperson who has not requested the test. In suchcases, every effort should be made by the coun-sellors and the subjects concerned to providea satisfactory resolution of this conflict.20 Themajority of representatives from lay organ-isations favoured the opinion that if no con-sensus could be reached, the right of the personat 25% risk should have priority over the rightof the parent not to know. An important ar-gument is that planning a family may be themain reason for young adults to take the test,whereas their unaffected parents see theirchances of ever developing HD dramaticallydecrease after the age of 50. Those at risk forpresenile dementia are approaching the meanage of onset after 50 years of age, at a timewhen their children may start a family (three-quarters ofthe grandchildren in the study groupare older than 18 years). Thus, we expect moreconflict of interests compared with HD and,in line with the HD guidelines, every effortmust be made to solve such controversies bothat an individual and a family level. The seriousdilemma for the counsellor is whose rights andinterests should prevail. Should the counsellorgive priority to the applicant's right to know orshould he deny testing in order to protect aninvasion of the relatives' right not to know?Exclusion testing in a person at 25% risk maybe a solution, that is, excluding whether ornot one has received a chromosome from theaffected grandparent. Such an outcome doesnot change the risk of the parent at 50% risk.However, the initial conflict arises again if theapplicant's risk has increased to 50% and he/shewishes full certainty. Obviously, an unequivocalguideline is not compatible with the individualinterests of all parties involved. The coun-sellor's responsibility is to safeguard that alladvantages and disadvantages are discussedand weighed, with the inclusion of the impactof testing for relatives. Eventually, the test can-didate should decide and have the responsibilityfor his decisions. It is obvious that inheritedlate onset disorders should be considered as aproblem that may have affected the whole fam-ily for more than two generations. The familyis therefore a relevant clinical frame ofreferencefor the genetic counsellor and other health careprofessionals. 1837' 8
PRENATAL TESTINGWhen using the Dutch HD testing programme,one of the main aims was to obtain informationuseful for family planning.26 Family planningwas found to be less important in the presentsurvey of people at risk for HPD/FAD-CH.The majority were against the availability ofprenatal diagnosis as a clinical service. Littlemore than half of those who supported suchprovision would use it personally and even in
this group half of the respondents rejectedpregnancy termination if an increased risk inthe fetus was found. The eventual demandfor HD prenatal testing was lower than wasexpected from pretest attitudes, but was con-stant over time (unpublished data presented atthe 16th International Meeting of the WorldFederation of Neurology Research Group onHuntington's Disease, 1995). The expecteduse of prenatal diagnosis in HPD/FAD-CHmight be even lower. This expected uptakemight reflect feelings in the latter group thatonset of the disease is generally later.939 Theseattitudes reflect the painful and thoughtfulhandling of options by those at risk and makeclear the need for human compassion for peoplewho have experienced the tragedy ofthe diseasein their families. Hence, the individual requestfor prenatal diagnosis ought to be appreciated.However, access to prenatal diagnosis for un-treatable late onset disorders should be deniedto couples who would not consider a selectivepregnancy termination, in order to prevent theviolation of the future child's right not to know.In the present study, 10 out of 19 couples wouldopt for prenatal testing but not for selectiveabortion. This requires prudent counselling ofcouples before conception, if possible. Thismay be a task for the general practitioner fol-lowed by referral to a clinical genetics de-partment.
Preimplantation genetic diagnosis may be-come an alternative in the near future. Recently,it has been suggested that preimplantation gen-etic diagnosis could be used as a method toachieve prevention of untreatable, autosomaldominant late onset disorders in offspring with-out disclosure of parental genotype.40 Thecouple would be told only that their embryoswere tested, and that only apparently diseasefree embryos were replaced. No informationwould be given which might provide a basisfor inferring whether or not any embryos withthe mutation were identified. Hence, parentswould derive no direct or indirect informationabout their own genetic risk, while pre-implantation diagnosis could reduce the fetalrisk to zero. This option could be valuable forparents at risk who prefer not to know theirgenetic status. It remains to be seen, however,whether this is a realistic alternative. First, theburdens and risks of in vitro fertilisation shouldnot be underestimated. Furthermore, a con-dition would be to separate those involvedin the testing procedure and the counsellor,otherwise it may become impossible to protectthe parent's right not to know adequately.
GENETICS AND DISCRIMINATIONBoth employers and the health, life, pension,or disability insurance companies may dis-criminate against people known to have anincreased risk for cancer or neurodegenerativediseases.4" The Dutch debate on the person'sduty to reveal genetic information to insurancecompanies, and on exclusion from life in-surance of those at risk for HD and myotonicdystrophy, leads us to emphasise the potentialharm to carriers ofgenes for untreatable genetic
70
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from

Presymptomatic DNA testing for Alzheimer's diseaselcerebral haemorrhage and Pick disease
disorders with delayed onset. Our clinical ex-perience has taught us that most people at riskfor a variety of inherited late onset disordersare not aware of the risk of insurance andemployment discrimination or tend to under-estimate these issues. Some people have re-quested predictive testing in order to get accessto life insurance. This experience underlinesthe need for further discussion regarding theuse of genetic information by insurance com-panies and employers. We advocate that par-ticipants in genetic studies must be extensivelyinformed of the potential hazards, which maylead to withdrawal from the protocol, or todelay testing until arrangements have beenmade. At the time of our study, much mediaattention was paid to discrimination based ongenetic risks, which might explain why 20% ofthe participants held the opinion that test res-ults should not be added to the medical records.Local legislation should protect people with agenetic susceptibility so that those at risk feelfree to use the options of genetic testing, andscientists can proceed with research.41 Althoughlegal, ethical, biomedical, and psychosocial is-sues must be extensively addressed in pre- andpost-test counselling sessions, we are awarethat the informed consent remains un-satisfactory and has many limitations with re-gard to these issues.
GENETIC RESEARCH AND HEALTH CAREThe increasing number of diseases that can bepredicted by genetic testing (with far reachingconsequences) raises the question of how gen-etics services and other medical disciplines canmeet the need for careful pre- and post-testcounselling and additional support. Althoughthe need is acknowledged and emphasised inevery study, the planning and resources re-quired are rarely considered in most countries.This leaves the human aspect of genetics, suchas psychological support and evaluation stud-ies, dependent upon external, temporary fin-ancial support. Such lack of continuity inpatient care and research and dissemination ofclinical research findings may greatly endangerthe quality of genetic medicine in the future.Follow up care must provide proper and
consistent information and support about theeffects of test results on partner relationshipsand families. General practitioners must beproperly informed about the impact of beingat high risk on psychological well being. Healthcare providers must consider the complex psy-chological, ethical, and social issues in the ap-plication of presymptomatic testing. Theyshould be aware of their own feelings of help-lessness,42A4 and be careful not to consider thetest as the only option. They must be educatedon these issues in order to establish adequatesupport provisions.
ConclusionsThe major limitation of this study was therelatively small number of participants. An-other bias may be caused by the number ofsibs in the study as four different families were
involved. Therefore, the results must be con-sidered with caution. The group studied maynot be representative of the entire populationat risk for presenile dementia. Moreover, theresults, with the inclusion of the intention tohave predictive testing when available, mayhave been biased by the extensive psychologicalattention ofthe researchers that the participantsreceived. An important limitation is that thedata were obtained by means of self-report.The disadvantages of self-report data are wellknown and include possible social desirabilitybias. Therefore, qualitative studies using ob-servation and interview techniques and casestudies conducted by people who are able toobserve people at risk and their families ob-jectively can improve the understanding of theobservations which will consequently increasethe clinical significance.
The authors are very grateful to the people at risk and theirpartners who have participated in this study. They are alsograteful to Dr G C Beverstock for editing the manuscript.
1 Huntington's Disease Collaborative Research Group. Anovel gene containing a trinucleotide repeat that is ex-panded and unstable on Huntington's disease chro-mosomes. The Huntington's Disease CollaborativeResearch Group. Cell 1993;72:971-83.
2 Gispert S, Twells R, Orozco G, et al. Chromosomal as-signment of the second locus for autosomal dominantcerebellar ataxia (SCA2) to chromosome 12q23-24. 1. NatGenet 1993;4:295-9.
3 HaanJ, HardyjA, Roos RA. Hereditary cerebral hemorrhagewith amyloidosis-Dutch type: its importance for Alzheimerresearch. Trends Neurosci 1991;14:231-4.
4 Mullan M, Crawford F, Buchanan J. Technical feasibilityof genetic testing for Alzheimer's disease. Alzheimer DisAssoc Disord 1994;8:102-15.
5 Buxton J, Shelboume P, Davies J, et al. Detection of anunstable fragment of DNA specific to individuals withmyotonic dystrophy. Nature 1992;355:547-8.
6 Sherrington R, Rogaev EI, Liang Y, et al. Cloning of agene bearing missense mutations in early-onset familialAlzheimer's disease. Nature 1995;375:754-60.
7 Hoogeveen AT, Willemsen R, Meyer N, et al. Charac-terization and localization of the Huntington disease geneproduct. Hum Mol Genet 1993;2:2069-73.
8 Hendriks L, van Duijn CM, Cras P, et al. Presenile dementiaand cerebral haemorrhage linked to a mutation at codon692 of the beta-amyloid precursor protein gene. Nat Genet1992;1:218-21.
9 Heston LL, Mastri AR. Age at onset of Pick's and Alz-heimer's dementia: implications for diagnosis and re-search. J Gerontol 1982;37:422-4.
10 Gustafson L. Frontal lobe degeneration of non-Alzheimertype. II. Clinical picture and differential diagnosis. ArchGerontol Geriatr 1987;6:209-23.
11 Neary D, Snowden JS, Northen B, Goulding P. Dementiaof frontal lobe type. J Neurol Neurosurg Psychiatry 1988;51:353-61.
12 Mendez MF, Selwood A, Mastri AR, Frey W. Pick's diseaseversus Alzheimer's disease: a comparison of clinical char-acteristics. Neurology 1993;43:289-92.
13 Harper PS. Huntington disease and the abuse of genetics.Am J Hum Genet 1992;50:460-4.
14 Markel DS, Young AB, Penney JB. At risk person's attitudetoward presymptomatic testing in Huntington disease. AmJfMed Genet 1987;26:295-305.
15 Meissen GJ, Berchek RL. Intended use of predictive testingby those at risk for Huntington disease. Am JtMed Genet1987;26:283-93.
16 Kessler S, Field T, Worth L, Mosbarger H. Attitudes ofpersons at risk for Huntington disease toward predictivetesting. AmJMed Genet 1987;26:259-70.
17 Terrenoire G. Huntington's disease and the ethics of geneticprediction. J Med Ethics 1992;18:79-85.
18 Huggins M, Bloch M, Kanani S, et al. Ethical and legaldilemmas arising during predictive testing for adult-onsetdisease: the experience of Huntington disease. AmJ HumGenet 1990;47:4-12.
19 Hayden MR. Predictive testing for Huntington disease: arewe ready for widespread community implementation? AmJMed Genet 1991;40:515-17.
20 International Huntington Association and the World Fed-eration of Neurology Research Group on Huntington'sChorea. Guidelines for the molecular genetics predictivetest in Huntington's disease. JMed Genet 1994;31:555-9.
21 Quaid KA. Presymptomatic testing for Huntington's dis-ease: the U.S. experience. AmJ Med Genet 1992;51:1063.
22 Steenstraten IMvd, Tibben A, Roos RA, van de Kamp JJ,Niermeijer MF. Predictive testing for Huntington disease:nonparticipants compared with participants in the Dutchprogram. Am _THum Genet 1994;55:618-25.
71
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from

Tibben, Stevens, de Wert, Niermeijer, van Duijin, van Swieten
23 Lannfelt L, Axelman K, Lilius L, Basun H. Genetic coun-selling in a Swedish Alzheimer family with amyloid pre-cursor protein mutation. AmJ Hum Genet 1995;56:332-5.
24 Lennox A, Karlinsky H, Meschino W, Buchanan JA, PercyME, Berg JM. Molecular genetic predictive testing forAlzheimer's disease: deliberations and preliminary re-commendations. Alzheimer Dis Assoc Disord 1994;8: 126-47.
25 Karlinsky H, Sadovnick AD, Burgess MM, Langlois S,Hayden MR, Berg JM. Issues in molecular genetic testingof individuals with suspected early-onset familial Alz-heimer's disease. Alzheimer Dis Assoc Disord 1994;8: 116-25.
26 Tibben A, Frets PG, van de Kamp JJ, et al. PresymptomaticDNA-testing for Huntington disease: pretest attitudes andexpectations of applicants and their partners in the Dutchprogram. Am J7 Med Genet 1993;48:10-16.
27 Heutink P, van Swieten JC, Stevens M, et al. The gene forhereditary Pick's disease maps to chromosome 17q21-q22. Ann Neurol (in press).
28 Stevens M, van Duijn CM, Heutink P, et al. Hereditary Pick'sdisease. A comparison of clinical symptoms, radiologicalfeatures and neuropathological changes between threefamilies. (Submitted.)
29 Gardner MJ, Altman DG. Statistics with confidence. Belfast:The Universities Press, 1989.
30 Mac Kay CR. Discussion points to consider in researchrelated to the human genome. Hum Gene Ther 1993;4:477-95.
31 Wiggins S, Whyte P, Huggins M, et al. The psychologicalconsequences of predictive testing for Huntington's dis-ease. Canadian Collaborative Study of Predictive Testing.N Engl 3 Med 1992;327:1401-5.
32 Tibben A, Bannink EC, Timman RT, Duivenvoorden HJ.3-year follow-up after presymptomatic testing for Hun-tington disease in tested individuals and partners. HealthPsychol (in press).
33 Bloch M, Fahy M, Fox S, Hayden MR. Predictive testingfor Huntington disease. II. Demographic characteristics,life-style patterns, attitudes, and psychosocial assessments
ofthe first fifty-one test candidates. Anm_Med Genet 1989;32:217-24.
34 Meissen GJ, Mastromauro CA, Kiely DK, McNamara DS,Myers RH. Understanding the decision to take the pre-dictive test for Huntington disease. Amn 7 Med Genet 1991;39:404-10.
35 Feinberg J. The child's right to an open future. In: AikenW, LaFollette H, eds. Whose child? Childrens' rights, parentalauthority, and state power. Totowa, New Jersey: Littlefield,Adams & Co, 1980:124-53.
36 Quaid KA, Brandt J, Faden RR, Folstein SE. Knowledge,attitude, and the decision to be tested for Huntington'sdisease. Clin Genet 1989;36:431-8.
37 Kessler S. Forgotten person in the Huntingto; diseasefamily. AnmIMed Genet 1993;48:145-50.
38 Tibben A, Duivenvoorden HJ, Niermeijer MF, Vegter-vdVlis M, Roos RAC, Verhage F. Psychological effects ofpresymptomatic DNA testing for Huntington's disease inthe Dutch program. Psychosom Med 1994;56:526-32.
39 Roos RAC, Vegter-vd Vlis M, Hermans J, et al. Age at onsetin Huntington's disease: effect of line of inheritance andpatient's sex. 7 Med Genet 1991;28:515-19.
40 Schulman JD, Black SH, Handyside A, Nance WA. Pre-implantation genetic testing for Huntington disease andcertain other dominantly inherited disorders. Clin Genet1996;49:57-8.
41 Culliton BJ. Genes and discrimination. Nat Med 1995;1:385.
42 Martindale B. Huntington's chorea. Some psychodynamicsseen in those at risk and in the responses of helpingprofessions. Br
_ Psychiatry 1987;150:319-23.43 Thies U, Bockel B, Bochdalofsky V. Attitudes of neur-
ologists, psychiatrists, and psychotherapists towards pre-dictive testing for Huntington's disease in Germany. _7Med Genet 1993;30:1023-7.
44 Thomassen R, Tibben A, Niermeijer MF, van der Does E,van de Kamp JJ, Verhage F. Attitudes of Dutch generalpractitioners towards presymptomatic DNA-testing forHuntington disease. Clin Genet 1993;43:63-8.
72
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from

doi: 10.1136/jmg.34.1.63 1997 34: 63-72J Med Genet
A Tibben, M Stevens, G M de Wert, et al. hereditary Pick disease.disease/cerebral haemorrhage andfor early onset Alzheimer's Preparing for presymptomatic DNA testing
http://jmg.bmj.com/content/34/1/63Updated information and services can be found at:
These include:
References http://jmg.bmj.com/content/34/1/63#related-urls
Article cited in:
serviceEmail alerting
the box at the top right corner of the online article.Receive free email alerts when new articles cite this article. Sign up in
Notes
http://group.bmj.com/group/rights-licensing/permissionsTo request permissions go to:
http://journals.bmj.com/cgi/reprintformTo order reprints go to:
http://group.bmj.com/subscribe/To subscribe to BMJ go to:
group.bmj.com on July 15, 2011 - Published by jmg.bmj.comDownloaded from
Related Documents











