2004;64:2782-2792. Cancer Res S. Sianna Castillo, John Brognard, Pavel A. Petukhov, et al. Ether Lipid Analogues Cancer Cells by Rationally Designed Phosphatidylinositol Preferential Inhibition of Akt and Killing of Akt-Dependent Updated version http://cancerres.aacrjournals.org/content/64/8/2782 Access the most recent version of this article at: Cited Articles http://cancerres.aacrjournals.org/content/64/8/2782.full.html#ref-list-1 This article cites by 70 articles, 32 of which you can access for free at: Citing articles http://cancerres.aacrjournals.org/content/64/8/2782.full.html#related-urls This article has been cited by 24 HighWire-hosted articles. Access the articles at: E-mail alerts related to this article or journal. Sign up to receive free email-alerts Subscriptions Reprints and . [email protected] Department at To order reprints of this article or to subscribe to the journal, contact the AACR Publications Permissions . [email protected] Department at To request permission to re-use all or part of this article, contact the AACR Publications Research. on August 28, 2013. © 2004 American Association for Cancer cancerres.aacrjournals.org Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

2004;64:2782-2792. Cancer Res S. Sianna Castillo, John Brognard, Pavel A. Petukhov, et al. Ether Lipid AnaloguesCancer Cells by Rationally Designed Phosphatidylinositol Preferential Inhibition of Akt and Killing of Akt-Dependent
Updated version
http://cancerres.aacrjournals.org/content/64/8/2782
Access the most recent version of this article at:
Cited Articles
http://cancerres.aacrjournals.org/content/64/8/2782.full.html#ref-list-1
This article cites by 70 articles, 32 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/64/8/2782.full.html#related-urls
This article has been cited by 24 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

[CANCER RESEARCH 64, 2782–2792, April 15, 2004]
Preferential Inhibition of Akt and Killing of Akt-Dependent Cancer Cells byRationally Designed Phosphatidylinositol Ether Lipid AnaloguesS. Sianna Castillo,1 John Brognard,1 Pavel A. Petukhov,2 Chunyu Zhang,1 Junji Tsurutani,1 Courtney A. Granville,1
Min Li,2 Michael Jung,1 Kip A. West,1 Joell G. Gills,1 Alan P. Kozikowski,2 and Phillip A. Dennis1
1Cancer Therapeutics Branch, Center for Cancer Research, National Cancer Institute, Bethesda, Maryland; and 2Department of Medicinal Chemistry and Pharmacognosy,College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois
ABSTRACT
Activation of the PI3k/Akt pathway controls key cellular processes andcontributes to tumorigenesis in vivo, but investigation of the PI3k/Akt path-way has been limited by the lack of specific inhibitors directed against Akt. Todevelop Akt inhibitors, we used molecular modeling of the pleckstrin homol-ogy (PH) domain of Akt to guide synthesis of structurally modified phos-phatidylinositol ether lipid analogues (PIAs). Here, we characterize the bio-chemical and cellular effects of PIAs. Of 24 compounds tested, five PIAs withmodifications at two sites on the inositol ring inhibited Akt with IC50s < 5�M. Molecular modeling identified putative interactions of PIAs with thephosphoinositide-binding site in the PH domain of Akt, and growth factor-induced translocation of Akt to the plasma membrane was inhibited by PIAadministration. Inhibition of Akt occurred rapidly and was maintained forhours. PIAs decreased phosphorylation of many downstream targets of Aktwithout affecting upstream kinases, such as PI3k or phosphoinositide-de-pendent kinase-1, or members of other kinase pathways such as extracellularsignal-regulated kinase. Importantly, PIAs increased apoptosis 20–30-fold incancer cell lines with high levels of endogenous Akt activity but only 4–5-foldin cancer cell lines with low levels of Akt activity. These studies identify PIAsas effective Akt inhibitors, and provide proof of principle for targeting the PHdomain of Akt.
INTRODUCTION
The PI3k/Akt signal transduction pathway is activated by many typesof cellular stimuli or toxic insults and once active, can regulate funda-mental cellular functions such as transcription, translation, proliferation,growth, and survival (1, 2). The serine/threonine kinase Akt (or PKB) isa crucial kinase in this pathway. Akt was first described as the cellularhomologue of the product of the v-akt oncogene (3–5), and it has threeisoforms, Akt1, Akt2, and Akt3 (or PKB-�, PKB-�, and PKB-�). Acti-vation of all three isoforms is similar in that phosphorylation of two sites,one in the activation domain and one in the COOH-terminal hydrophobicmotif, are necessary for full activity. For Akt1, phosphorylation of Thr-308 in the activation domain by phosphoinositide-dependent kinase-1(PDK-1) is dependent on the products of PI3k, phosphatidylinositol 3,4bisphosphate (PIP2), and phosphatidylinositol 3,4,5 trisphosphate (PIP3;Refs. 1 and 6). Levels of PIP2 and PIP3 are controlled by the tumorsuppressor, dual-phosphatase PTEN, whose tumor suppressor function isdue to dephosphorylation of PIP2 and PIP3 at the 3� position (7). Themechanism of Ser-473 phosphorylation is less clear. Kinases potentiallyresponsible for Ser-473 phosphorylation include PDK-1 (8), integrin-linked kinase (ILK) or an ILK-associated kinase (9, 10), Akt itself (11),or a plasma membrane lipid raft-associated kinase (12). In addition, arecent study has suggested that tyrosine phosphorylation may also beimportant for Akt activation (13).
Once active, Akt exerts many cellular effects through the phosphoryl-ation of downstream substrates such as Bad, caspase 9, ASK1, andMDM2 that regulate the apoptotic machinery (14–18); the cdk inhibitorsp21 and p27 that control cell cycle progression (19–22); forkhead tran-
scription family members and inhibitor of nuclear factor-�B kinases thatcontrol gene expression (23, 24); the kinase glycogen synthase kinase(GSK)-3 that controls glucose metabolism, cell cycle, and apoptosis (25);and tuberin and the mammalian target of rapamycin that control proteintranslation (26–29). The cumulative effect of altering these cellularprocesses can promote transformation in vitro or tumorigenesis andtumor growth in vivo.
Many lines of evidence demonstrate that Akt is a critical player in thedevelopment, growth, and therapeutic resistance of cancers. First, studiesin transgenic mice that overexpress constitutively active Akt show thatAkt, in combination with other genetic alterations, contributes to tumorformation in many tissues (30–32). Tumor formation is also character-istic of PTEN heterozygous knockout mice that have increased levels ofactive Akt (2, 6). Second, activation of Akt is an early response tocarcinogen exposure in vitro and in vivo and may have a permissive rolefor the development of tobacco-related cancers (33). Third, active Akt hasbeen detected in over eight types of human cancers in vivo3 and has beenfunctionally linked with poor clinical outcomes (34–36). Fourth, Aktactivity promotes resistance to chemotherapy and radiation (37, 38).Collectively, these studies suggest that inhibiting the Akt pathway mighthave therapeutic value for patients with cancer, and they have formed thebasis for widespread efforts to develop approaches that inhibit Akt.
Despite the potential value of inhibiting Akt and concerted efforts byindustry and academia to develop Akt inhibitors, no small molecule Aktinhibitors exist. One approach has used molecular modeling of theinteraction of PIP2 with the pleckstrin homology (PH) domain of Akt toguide the synthesis of phosphatidylinositol ether lipid analogues (PIAs)that were designed to inhibit this interaction (39). The most effective PIAfrom this set, DPIEL, contained a single 3-deoxy substitution on theinositol ring and inhibited platelet-derived growth factor-induced Aktactivation and cell growth (40). To improve potency and metabolicstability, we recently modified the inositol ring to create 2-modified3-deoxy PIAs. Evaluation of these compounds showed that two 2-modified 3-deoxy PIAs were able to inhibit constitutive Akt phosphoryl-ation in cancer cell lines but that DPIEL was ineffective (41).
In this paper, we extend these studies and provide a biologicalanalysis of a series of chemically modified PIAs that bear differentsets of two substitutions on the inositol ring. We identify five PIAsthat inhibit Akt and downstream signaling without inhibiting otherkinases upstream of Akt, and we show that these compounds prefer-entially induce apoptosis in cancer cell lines with high levels of Aktactivity. Our data support the concept of selectively targeting Akt totreat cancer and identify PIAs as lead compounds for possible devel-opment as small molecule Akt inhibitors.
MATERIALS AND METHODS
Materials
The synthesis of the PIAs has been described previously (41). All antibodies(except anti-�-tubulin, anti-p85, and anti-cyclin D1) and the Akt kinase assay kitwere purchased from Cell Signaling Technologies (Beverly, MA). Anti �-tubulin
Received 5/28/03; revised 2/2/04; accepted 2/5/04.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
Requests for reprints: Phillip A. Dennis, Building 8, Room 5101, 8901 WisconsinAvenue, Bethesda, MD 20889. Phone (301) 496-0901; Fax (301) 496-0047; E-mail:[email protected].
3 S. Hewitt, X. Yang, S. Steinberg, S. Swain, and P. A. Dennis. In situ analysis of thePI3K/Akt pathway in normal and cancerous tissues on human multi-tissue arrays, sub-mitted for publication.
2782
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

was purchased from Sigma Chemical Co. (St. Louis, MO). Antibodies against thep85 subunit of PI3k (anti-p85) were obtained from Upstate Biotechnology (LakePlacid, NY). Antibodies to cyclin D1 were from Santa Cruz Biotechnology (SantaCruz, CA). Protease inhibitor mixture was obtained from Sigma Chemical Co.,and protein assay materials were from Bio-Rad (Hercules, CA). All cell culturereagents were purchased from Life Technologies, Inc. (Rockville, MD). Protranpure nitrocellulose membranes were purchased from Schleicher & Schuell (Das-sel, Germany). Adenoviruses containing �-galactosidase or myristolated-Akt1were generous gifts from Dr. Kenneth Walsh (Boston University, Boston, MA)and have been described previously (42).
Methods
Cell Culture. NSCLC lines were provided by H. Oie or Dr. F. Kaye atNCI/Navy Medical Oncology (Bethesda, MD). All cell lines were maintainedin 75-cm2 flasks in DMEM and supplemented with 10% (v/v) fetal bovineserum (FBS), 100 units/ml penicillin, and 100 �g/ml streptomycin. Cells wereincubated in a 37°C and 7.0% CO2 atmosphere incubator. The stock cultureswere split on a weekly basis at a 1:5 or 1:10 ratio. Breast cancer cell lines wereobtained from Dr. S. Lipkowitz at the NCI/Navy Medical Oncology and weremaintained in RPMI 1640 supplemented with 10% (v/v) FBS, 100 units/mlpenicillin, and 100 �g/ml streptomycin in an incubator calibrated to 37°C and6% CO2 in 75-cm2 flasks. Stock flasks were split on a weekly basis at a 1:4,1:10, or 1:20 ratio. BEAS2B cells were provided by Dr. Curtis C. Harris (NCI,Bethesda, MD) and were grown in LHC8 medium (BioSource, Camerillo, CA)supplemented with 100 units/ml penicillin in an incubator at 37°C and 3.5%CO2 in 75-cm2 flasks. Stock flasks were split on a weekly basis at a 1:4 ratio.
Pharmacological Treatments. NSCLC cells were plated 2–2.5 � 105
cells/well in 6- or 12-well plates in DMEM containing 10% FBS and incubatedfor 24 h. The medium was then changed to DMEM with 0.1% FBS, and thecells were incubated overnight. After overnight incubation, cells were treatedwith PIAs dissolved in DMSO (10 �M) for 2 h (immunoblotting/kinaseassays), 18 h (immunoblotting experiments), or 24 h (apoptosis studies). In allexperiments, DMSO was added to control samples and had no effect on Aktactivity. After incubation with PIAs, the cells were harvested for immunoblotanalysis or for assessment of apoptosis as described below. For dose responsestudies, NSCLC cells were plated 2–2.5 � 105 cells/well in 6-well plates. Afterattachment, the medium was changed to DMEM containing 0.1% FBS over-night. Cells were treated with the indicated doses of PIAs for 2 h, and the cellswere harvested for immunoblot analysis. Quantification of band density wasperformed using NIH Image software. Dose curves were obtained by quanti-fying ratios of P-Ser-473 to total Akt under each condition and setting the ratioof P-Ser-473/total Akt for DMSO-treated cells equal to 1.
Immunoblotting. Cell extracts were prepared by washing cells with PBS andadding 100 �l of 2� Laemmli sample buffer supplemented with 2 �l proteaseinhibitor mixture/well as described previously (43). Lysates were sonicated for15 s with a Vibra Cell sonicator. The protein yield was quantified using theBio-Rad Dc protein assay kit. Equivalent protein was loaded, and the lysates wereseparated by SDS-PAGE and then transferred to nitrocellulose membranes. Equiv-alent loading was confirmed by staining membranes with fast green as describedpreviously (44). The membranes were blocked for 1 h in blocking buffer (1�Tris-buffered saline, 5% milk, and 0.20% Tween 20) and placed in primaryantibody (1� Tris-buffered saline, 5% milk, 0.10% Tween 20, and 1:1000 anti-body) overnight at 4°C. Nitrocellulose membranes were washed three times inwash buffer (0.10% NP40, 0.10% Tween 20, and 1� Tris-buffered saline).Primary antibody was detected using horseradish peroxidase-linked goat anti-mouse or goat antirabbit IgG antibodies and visualized with the enhanced chemi-luminescent detection system (Super Signal; Pierce, Rockford, IL). Immunoblotexperiments were performed at least three times.
PI3k Assays. H1703 or H157 cells were plated at a density of 2 � 106
cells/100-mm dish. The following day, the medium was changed to DMEMcontaining 0.1% FBS. After overnight incubation, cells were treated with 10 �M
PIAs 5, 6, 7, 23, 24, 25, or DMSO control for 2 h. Cells were lysed in 1% NP40lysis buffer containing 137 mM NaCl, 20 mM Tris-HCl (pH 7.4), 1 mM CaCl2, 1mM MgCl2, 1% NP40, 1 mM phenylmethylsulfonyl fluoride, and 0.1 mM sodiumorthovanadate. Cellular extracts containing 900-1100 �g of protein were incubatedwith anti-p85 for 1 h at 4°C, followed by protein A-agarose beads (Santa CruzBiotechnology) for an additional 1 h at 4°C. The reaction to demonstrate PI3kactivity in PI3k immunoprecipitates was performed as described previously (45).
In brief, immunoprecipitates were incubated with kinase reaction buffer mix [10nM Tris-HCl (pH 7.4), 150 nM NaCl, 5 mM EDTA, and 0.1 mM sodium or-throvanadate added fresh], 100 mM MgCl2, 2 mg/ml phosphatidylinositol (AvantiPolar Lipids, Alabaster, AL), and 30 �Ci of [�-32P]ATP (Redivue ATP; Amer-sham Biosciences, Piscataway, NJ) for 10 min at 37°C. Reaction products wereseparated by TLC on 1% potassium oxalate-treated TLC plates (Whatman LK6DFsilica gel 60). 32P-labeled 3�-phosphoinositides were visualized by autoradiogra-phy. Kinase assays were repeated at least three times.
Akt Kinase Assays. Akt kinase assays of cells treated with PIAs wereperformed as described previously (38), using the manufacturer’s recommenda-tions (Cell Signaling Technologies) with modifications described below. Cellswere plated at a concentration of 2–4 � 105 and treated with PIAs as describedabove for 2 h. Cells were washed once with ice-cold PBS, and 200 �l of ice-coldlysis buffer [20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1%Triton X-100, 2.5 mM sodium PPi, 1 mM �-glycerol phosphate, 1 mM Na3VO4, 1mM phenylmethylsulfonyl fluoride, and 1 mM leupeptin] were added to the cellsfor 10 min. Lysates were cleared and allowed to immunoprecipitate for 2–3 h at4°C with anti-Akt antibody. Immunoprecipitates were washed twice with lysisbuffer and twice with kinase buffer. Kinase reaction was performed for 30 min at30°C in kinase buffer supplemented with 200 �M ATP and 1 �g of GSK-3�/�fusion protein. Reaction was terminated with 3� SDS buffer. The samples wereheated at 100°C for 5 min and loaded into a 12% SDS-polyacrylamide gel.Additional kinase assays were performed on Akt isolated from untreated H157cells as follows. H157 cells were grown in T-75 flasks until �80% confluent. Cellswere harvested with ice-cold lysis buffer, and cleared lysate was divided into threeequal volumes. Aliquots were immunoprecipitated for 2–3 h at 4°C with anti-Aktantibody. Immunoprecipitates were washed twice with lysis buffer and twice withkinase buffer and incubated with 10 �M PIA 7, PIA 5, or DMSO for 30 min atroom temperature. After incubation, kinase reactions were performed and pro-cessed as described above.
Apoptosis Assays. Cells were treated with PIAs for 18–24 h as describedabove. Floating cells were collected, and adherent cells were harvested bytrypsinization and then centrifuged at 1000 � g for 5 min. Cells were fixed inice-cold 70% methanol added dropwise and then incubated at �20°C for 30 min.Cells were centrifuged and incubated with propidium iodide (25 �g/ml) supple-mented with RNase A (30 �g/ml) for 30 min at room temperature. Quantificationof sub2N DNA was determined by flow cytometry analysis using a Becton-Dickinson FACSort and by manual gating using CellQuest software. Apoptosisexperiments were performed in triplicate and repeated at least three times.
“Washout” Experiments. Additional apoptosis assays were performed asdescribed above but with the following modifications. Cells were treated withPIAs for 2, 4, 6, or 24 h. After PIA treatment, media were changed to freshDMEM media with 0.1% FBS without PIAs for the remainder of the experi-ment. At 24 h, cells were collected and prepared for flow cytometric analysisas described above. Apoptosis experiments were performed in triplicate andrepeated at least three times.
Adenoviral Infections. BEAS2B cells were plated at a density of2.5 � 105 cells/well in 6-well plates. After attachment, cells were washed withPBS and infected with media containing adenoviral constructs for �-galacto-sidase or myristolated Akt (MOI 100) for 24 h. Infected cells were then treatedwith PIA 24 or DMSO, and cells were harvested and analyzed for apoptosis orfor immunoblot analysis as described above.
Translocation Assays. H157 or A549 cells were transfected with 2 �g ofgreen fluorescent protein (GFP)-Akt-PH construct using Fugene (Roche, In-dianapolis, IN) transfection reagent according to the manufacturer’s instruc-tions. Transfection efficiencies were �30% at 36 h, as assessed by fluorescentmicroscopy. Cells were placed into 0.1% FBS DMEM for 5 h and pretreatedor not with PIA5 (10 �M) for 2 h, with or without insulin-like growth factor I(IGF-I; 50 ng/ml) for 10 min. After IGF-I stimulation, cells were washed oncewith PBS and then fixed for 10 min at �20°C in a mixture of methanol:acetone(1:1). After fixation, GFP and 4�,6-diamidino-2-phenylindole fluorescence wasassessed using an Axioscope 2 microscope (Zeiss, Thornwood, NY).
Molecular Modeling. Docking and scoring studies of the interactions of PIAswith Akt were performed using a virtual screening package from Tripos (46). Forcalculation purposes, the long lipophilic tail of the PIAs was truncated leaving onlyglycerol fragments where the hydroxyl groups were substituted with methylgroups. Because of this simplification, the binding mode of inositol 1,3,4,5-tetraphosphate (IP4), the head group of PIP3, was compared with the bindingmodes of PIAs predicted by FlexX. The ligands PIA 5, PIA 6, PIA 23, PIA 24
2783
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

phosphorylated at positions 4 and 5, and PIA 25 phosphorylated at position 3ewere docked to the binding site of Akt and scored using FlexX (47, 48) and CScore(46) modules in Sybyl, respectively. The 30 best conformations generated afterrunning FlexX were clustered according to the pose of the inositol ring. The mostpopulated poses of PI analogues were saved and depicted.
RESULTS
Identification of PIAs That Inhibit Akt. Using molecular model-ing studies of the PH domain of Akt, a series of phosphatidylinositolanalogues were rationally designed, synthesized, and modified. Thechemical modifications of the PIAs are shown in Fig. 1A. Cell-basedassays were conducted to screen the effectiveness of the different PIAs ondecreasing Akt activation. Activation of Akt was assessed using phospho-specific antibodies directed against Ser-473 in immunoblotting experi-ments. These phospho-specific antibodies only recognize Akt in an activestate. Cancer cell lines with high levels of endogenous Akt activity thathave mutant PTEN (H157 and MB468) or wild-type PTEN (H1703)were treated for 2 h with PIAs (10 �M). PIA 7 serves as a negative controlbecause it is composed of the ether lipid backbone and lacks an inositolmoiety. As shown in representative immunoblots (Fig. 1B), a subset ofPIAs were able to decrease Akt phosphorylation without affecting totallevels of Akt in two lung cancer cell lines that differ in status of p53,K-ras, and PTEN (H1703 and H157; Ref. 38). The cumulative data fromscreening 24 PIAs in three cancer cell lines is shown in Fig. 2A. PIAs 5,6, 23, 24, and 25 exhibited the most complete and consistent inhibition ofAkt phosphorylation in the three cell lines tested. PIAs 10, 13, 15, and 16exhibited cell line-specific inhibition of Akt phosphorylation. Interest-ingly, the lead compound from an earlier generation of PIAs, DPIEL, wascompletely ineffective in these assays. Because PIAs 5, 6, 23, 24, and 25inhibited Akt phosphorylation in these three cell lines, they were chosenfor additional evaluation. The structures of these PIAs are shown inFig. 2B.
To confirm that inhibition of Akt phosphorylation was indicative ofinhibition of kinase activity and to show that PI3k activity was notaffected, in vitro kinase assays were performed (Fig. 2C). Similar tothe inhibition of Akt phosphorylation by PIAs observed in the immu-noblotting experiments, treatment of H1703 cells with PIA 5, 6, 23,24, and 25 decreased Akt phosphorylation that is indicative of Aktactivity, as determined by the decrease in phosphorylation of anexogenously added, GSK-3 peptide substrate (Fig. 2C, top left pan-els). PIAs 10 and 7 (the ether lipid moiety used as a negative control)did not inhibit Akt. When active Akt was immunoprecipitated fromuntreated H157 cells and subsequently incubated with PIAs 7 and 5,there was no inhibition of Akt activity (Fig. 2C, top right panels).These studies show that inhibition of Akt by PIAs requires intact cells.PI3k activity was not affected by PIAs 5, 6, 23, 24, 25, and 7 (Fig. 2C,lower panels), consistent with earlier observations that DPIEL did notinhibit PI3k (49). Similar results were also obtained when PI3k wasimmunoprecipitated using anti-p110 or antiphosphotyrosine antibod-ies (data not shown). Together, these studies indicate that PIAs 5, 6,23, 24, and 25 inhibits the phosphorylation and activity of Akt inintact cells without affecting PI3k.
Modeling of the Interaction of PIAs with the PH Domain ofAkt. To characterize how PIAs might interact with the PH domain ofAkt, molecular modeling of the head group of PIP3, IP4, and the biolog-ically active PIAs was performed (Fig. 2D). Previously, crystallographicstudies had shown that IP4 in the phosphoinositide-binding site of the PHdomain of Akt is positioned so that its 2-hydroxyl group is pointed insidethe binding cleft and that the network of the hydrogen bonds in thebinding site of Akt recognizes only three phosphate groups bound to theequatorial 1-, 3-, and 4-hydroxyl groups of the inositol ring and the axialhydroxyl group in position 2 (50, 51). Docking studies revealed that the
2-hydroxy substitutions in PIA5, PIA23, and PIA24 (Fig. 2D, panelsA–C) cannot be accommodated by the binding site unless they arepositioned outside of the binding groove. This sterically driven change infacial orientation of the inositol ring within the binding site leads torotation of the ring to maximize binding interactions between the polarphosphate groups and the complementary residues. For PIA6 and PIA25,from which a second hydroxyl group has been deleted, additional reori-entation of the inositol ring takes place to maximize hydrogen-bondinginteractions (Fig. 2D, panels D and E). Assuming that the X-ray structureof Akt cocrystallized with IP4 correctly represents the orientation of PIP2
and PIP3 when bound to Akt, our calculated binding results for the PIAssuggest that the orientation of their lipophilic side chains will be differentfrom that observed for natural substrates PIP2 or PIP3 when bound to Akt.Given that the binding of PIP3 to the PH domain of Akt causes aconformational change that allows Akt to be activated by PDK-1 (52), itis possible that PIAs inhibit Akt through changing the conformationalstate of the PH domain. One prediction from this model is that PIAsinhibit activation of Akt by inhibiting its translocation to the plasmamembrane.
Growth Factor-Stimulated Translocation of Akt Is Inhibited byPIA Treatment. To test whether PIAs inhibit Akt translocation, H157cells were transfected with a plasmid containing the PH domain of Aktfused to GFP (GFP-PH-Akt), and the cells were treated with IGF-I withour without pretreatment with PIA5 (Fig. 2E). In untreated cells, GFP-PH-Akt is predominantly cytoplasmic. When cells were treated withIGF-I, punctate membranous staining was observed, indicating translo-cation of Akt. In the presence of PIA5, IGF-I-induced translocation ofGFP-PH-Akt was inhibited. PIA5 treatment induced rounding of the cellsbut did not affect subcellular localization of GFP-PH-Akt. Similar inhi-bition of translocation of Akt was observed when these experiments wereperformed with A549 cells (data not shown).
PIAs Inhibit Akt in a Dose- and Time-Dependent Manner.Because the identification of PIAs 5, 6, 23, 24, and 25 as inhibitors ofAkt was determined at a given dose (10 �M) and time of exposure (2h), additional studies were conducted to determine the dose depend-ence and time dependence of Akt inhibition by PIAs. Fig. 3A showsthe dose response of H1703 cells treated with different concentrationsof PIAs 5, 6, and 24 for 2 h. Inhibition of Akt was virtually completeat 5 �M. The calculated IC50s for PIAs 5, 6, and 24 in H1703 cellswere 4.13, 4.28, and 2.49 �M, respectively (Fig. 3A, top panels).Similar dose-dependent responses were observed in the H157 cellstreated with PIAs 5, 6, and 24 (data not shown).
To determine the time course of Akt inhibition by PIAs, H1703 orH157 cells were treated with PIAs 5 and 6 (10 �M) for different times,and Akt phosphorylation was assessed (Fig. 3B). An inhibitor of PI3k,LY294002 (10 �M), was added as a positive control, and PIA 7, the etherlipid lacking an inositol ring, served as a negative control. In H1703 cells,PIAs 5 and 6 and LY294002 completely inhibited Akt phosphorylationwithin 15 min. Complete inhibition was maintained until the 2-h timepoint, when cells treated with PIAs 5 and 6 began to restore Aktphosphorylation. At 18 h, PIAs continued to inhibit Akt activation, butthe level of inhibition was less than that observed with LY294002.Time-dependent inhibition of Akt was also observed when H157 cellswere treated with PIA 5 or PIA 6 or LY294002. Although Akt phospho-rylation was decreased within 15 min after treatment with PIAs 5 and 6,LY294002 treatment completely attenuated Akt phosphorylation within15 min. After 2 h of incubation, PIAs 5 and 6 completely inhibited Aktphosphorylation. Interestingly, Akt phosphorylation was still attenuatedby PIAs 5 and 6 at 18 h, but Akt phosphorylation had returned to baselinein the LY294002-treated cells. The basis for the different kinetics ofinhibition by PIAs or LY294002 is unknown. The results of the dosedependence and time course experiments show that PIAs inhibit Aktwithin minutes at low micromolar concentrations.
2784
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

PIAs Selectively Inhibit Downstream Targets of the Akt Path-way. To determine the specificity of PIAs, activation state-specificantibodies against other kinases and downstream substrates were usedin immunoblotting experiments in H1703 and H157 cells. Theseexperiments evaluated the activation state of PDK-1, a kinase up-stream of Akt that activates Akt; nine downstream proteins whosephosphorylation is increased in response to Akt activation—tuberin,4EBP-1, p70S6K, AFX, FKHR, GSK-3, c-Raf, ASK-1, and MDM2;and a member of the mitogen-activated protein kinase superfamily,extracellular signal-regulated kinase (ERK), whose phosphorylation isnot affected by Akt activation or administration of LY294002 in thesecell lines (data not shown). Of the downstream substrates, tuberin,AFX, FKHR, GSK-3, c-Raf, ASK-1, and MDM2 are direct substratesof Akt, whereas 4EBP-1 and p70S6K are indirectly phosphorylated asa consequence of Akt activation.
Treatment of H1703 cells with PIAs 5, 6, 23, 24, and 25 altered theactivation of Akt and many downstream substrates (Fig. 4A), but did notalter phosphorylation of the upstream kinase, PDK-1, at a site necessaryfor PDK-1 activity. PIAs decreased Akt phosphorylation at Ser-473 andThr-308 without affecting levels of total Akt protein. Ser-473 phospho-rylation was inhibited to a greater extent than Thr-308 phosphorylation,and this may be related to better recovery of Thr-308 phosphorylation at18 h or to the fact that PDK-1, the Thr-308 kinase, is commonlyconstitutively active and is not inhibited by PIAs. Many downstream Aktsubstrates showed decreased phosphorylation after PIA treatment. Phos-phorylation of tuberin, 4EBP-1, and p70S6K, substrates that control
protein translation, was inhibited by PIA treatment, as was phosphoryl-ation of forkhead family members AFX and FKHR that control tran-scription. Phosphorylation of GSK-3 and c-Raf was attenuated by PIAs 5,6, 23, 24, and 25. Inhibition of ASK-1 phosphorylation was only ob-served with PIAs 23, 24, and 25 in H157 cells, but phosphorylation ofMDM2 was not affected by PIA treatment. To further demonstrateselectivity of PIAs, we assessed the phosphorylation status of a kinasedownstream of Ras, ERK, which is a member of the mitogen-activatedprotein kinase kinase superfamily. ERK phosphorylation was not inhib-ited by PIAs 23, 24, and 25 and was slightly increased by PIAs 5 and 6.Collectively, these data show that although no PIAs decreased PDK-1phosphorylation, PIAs inhibited the phosphorylation of Akt and of manysubstrates downstream of Akt.
Similar inhibitory effects of PIAs were observed in H157 cells. WhenPIAs were added to H157 cells, phosphorylation of Akt was diminished,but phosphorylation of PDK-1 was not (Fig. 4B). Downstream of Akt,PIAs inhibited phosphorylation of three substrates that control translation(tuberin, 4EBP-1, and p70S6K). The virtual complete inhibition ofp70S6K phosphorylation by PIAs 5, 6, 23, 24, and 25 in H157 cells is incontrast to the results observed in H1703 cells. Phosphorylation offorkhead family members was also decreased by PIA treatment, as wasphosphorylation of GSK-3 and c-Raf. Inhibition of ASK-1 phosphoryl-ation or MDM2 phosphorylation was not observed with PIA treatment inH157 cells. In general, PIAs 5 and 6 were overall more effective inhib-itors of downstream substrates in H157 cells. PIA-specific inhibition ofdownstream substrates was less commonly observed in H157 cells,
Fig. 1. Synthesis and screening of PIAs. A, synthesis and chemical modification of PIAs. The chemical modifications for individual sites on the inositol ring and ether lipid backboneare shown. B, representative immunoblots using activation state-specific, phospho-specific antibodies (P-S473) and antibodies against total Akt in H1703 and H157 cells treated withvarious PIAs (10 �M). Immunoblotting experiments were performed at least three times. Con, control.
2785
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

Fig. 2. Identification of PIAs that inhibit Akt. A, cumulativeresults of screening three cancer cell lines (MB468, H1703, andH157) with 24 PIAs (10 �M) in immunoblotting experiments. B,structures of PIAs that inhibited Akt phosphorylation in the threecancer cell lines tested. C, effect of PIAs on Akt or PI3k in vitrokinase activity. H1703 cells were treated with indicated PIAs (10 �M)or DMSO (C, control) for 2 h. Active Akt or PI3k was immunopre-cipitated using antibodies against phospho-Ser-473 or p85, respec-tively, as described. For Akt assays, phosphorylation of exogenousGSK-3 peptide was assessed using phospho-specific GSK-3 antibod-ies in immunoblotting experiments. Left panels show results fromH157 cells treated with PIAs. Right panels show effects of addingPIAs to kinase mixture after isolation of active Akt. Fast greenstaining shows equal amounts of protein in the lysates used forimmunoprecipitations, and Ser-473 Akt immunoblot shows thatequal amounts of active Akt were immunoprecipitated. For PI3kassays, phosphorylation of phosphatidylinositol was assessed usingTLC and autoradiography. Equivalent loading for Akt assays wasdemonstrated using fast green staining of cellular extracts beforeimmunoprecipitation. Equivalent loading for PI3k assays was shownby immunoblotting for p85 after immunoprecipitation with anti-p85antibodies. Experiments shown are representative of three independ-ent experiments. D, overlay of IP4 and PIA 5 (panel A), PIA 6 (panelB), PIA 23 (panel C), PIA 24 (panel D), and PIA 25 (panel E) in thePH binding site of Akt. IP4 is rendered by the green model. PIAs arerendered by ball-and-stick models, and key amino acids interactingwith IP and PIAs are rendered by stick model. Carbon atoms arecolored gray, hydrogen atoms are colored cyan, and nitrogen atomsare colored blue. E, translocation of GFP-PH-Akt. H157 cells trans-fected with GFP-PH-Akt were treated with or without IGF-I or PIA5. Translocation was assessed using fluorescent microscopy.
2786
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

although within the group of PIAs 23, 24, and 25, PIA 23 exerted thegreatest inhibition. Finally, PIAs 5 and 6 increased phosphorylation ofERK, but PIAs 23, 24, and 25 did not affect ERK phosphorylation.
Additional support for selective effects of PIAs on Akt and down-stream substrates was provided by analysis of two other cell lines withhigh endogenous levels of Akt activity (MB468 and ZR751). Whentested in these cells, PIAs 5, 6, 23, 24, and 25 uniformly inhibitedphosphorylation of Akt, p70S6K, 4EBP-1, and GSK-3, but not PDK-1(data not shown). Together, these studies show that PIAs 5, 6, 23, 24,and 25 selectively inhibit Akt activation and phosphorylation ofcomponents downstream of Akt without inhibiting ERK activation.
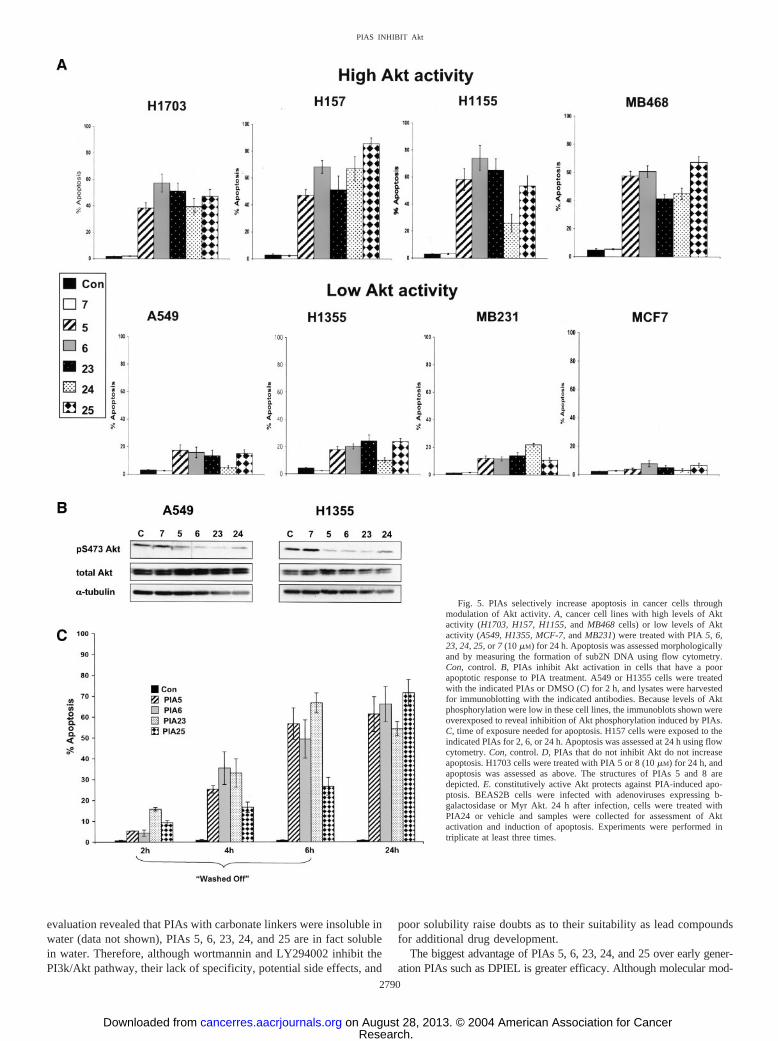
PIAs Preferentially Induce Apoptosis in Cell Lines with HighLevels of Active Akt. Because PIAs inhibited Akt downstream com-ponents in H157 or H1703 cells that have high levels of constitutivelyactive Akt that depend upon Akt for survival (37, 38), we assessedinduction of apoptosis by PIAs. For these experiments, we tested PIAs inlung cancer or breast cancer cell lines that maintain high (H1703,MB468, H157, and H1155) or low (H1355, A549, MCF-7, and MB231)endogenous levels of Akt activity under conditions of serum starvation(37, 38). Cells were treated with PIAs 5, 6, 23, 24, and 25 for 24 h, andapoptosis was assessed morphologically and by measuring the generationof sub2N DNA formation using flow cytometry. When measured quan-
titatively in cells with high levels of active Akt, apoptosis increased20–30-fold with the administration of PIAs 5, 6, 23, 24, and 25, ascompared with DMSO-treated cells or cells treated with PIA 7 (Fig. 5B).In contrast, PIAs were less effective in cell lines with low levels ofconstitutively active Akt, as manifest by fewer morphological changesand a less robust induction of apoptosis (4–5-fold). In general, individualPIAs induced apoptosis similarly, with the one exception of PIA 24 in theH1155 cells, where apoptosis caused by PIA 24 was approximatelyone-half of that induced by PIAs 5, 6, 23, and 25. Similar effects ofPIA-induced apoptosis were also observed when apoptosis was measuredusing an ELISA-based assay that measures histone release (data notshown). The fact that PIAs increased apoptosis by 4–5-fold in cell lineswith low levels of Akt activity probably reflects the fact that these celllines have low, but not absent, levels of active Akt (37, 38). Indeed, Fig.5B shows that PIA treatment does inhibit Akt activation in two cell lineswith low levels of endogenous Akt activity that exhibited less of anapoptotic response to PIA treatment (A549 and H1355 cells). Thissuggests that the ability of PIAs to induce apoptosis is related to thedependence of the cells on Akt rather than to the degree of inhibition of Akt.
Previous experiments from our laboratory using traditional cytotoxicchemotherapies have shown that the NSCLC and breast cancer cell linesused in these experiments typically require 24–48 h for apoptosis to
Fig. 3. Dose- and time-dependent inhibition of Akt by PIAs. A, dose-dependent inhibition of Akt. H1703 cells were treated with different doses of PIAs 5, 6, and 24 for 2 h.Phosphorylation of Akt was quantified relative to levels of total Akt using densitometry and NIH Image software. The ratio of phospho-Akt:total Akt was set to 1 for DMSO-treatedsamples. Ratios and SE were calculated for each experimental condition from three independent experiments. B, time-dependent inhibition of Akt. H1703 or H157 cells were treatedwith PIA 5, 6, or 7 or LY294002 (LY; 10 �M). C, control. Phosphorylation of Akt was assessed at the time points shown. �-Tubulin or fast green images were included as loadingcontrols. A representative experiment from three independent experiments is shown.
2787
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

occur (37, 38). To assess the time of exposure to PIAs that is needed tocommit these cells to apoptosis, H157 cells were treated with PIAs for 2,4, or 6 h, then the medium containing PIAs was removed, and apoptosiswas assessed 24 h after the initiation of the experiment (Fig. 5C). Smallincreases in apoptosis were observed after only 2 h of exposure. After 6 hof exposure, levels of apoptosis were similar to those observed forcontinuous 24 h of exposure for three of four PIAs tested. These studiesshow that PIAs cause an early commitment to apoptosis.
To demonstrate that the cytotoxicity of the PIAs was dependent uponthe ability of PIAs to inhibit Akt, we compared induction of apoptosis byPIAs 5 and 8 that are structurally identical (except for the linker group;Fig. 5D) but that differ greatly in ability to inhibit Akt (Fig. 2A). PIA 5contains a phosphate linker and inhibits Akt (Fig. 1B). PIA 8 contains acarbonate linker and did not inhibit Akt phosphorylation (Fig. 1B). Whenadministered to H157 cells, only PIA 5 was cytotoxic (Fig. 5C), therebycorrelating inhibition of Akt and induction of apoptosis. Collectively, theresults of these cellular experiments show that PIAs 5, 6, 23, 24, and 25increase apoptosis preferentially in cells lines with constitutively highlevels of Akt activity. Moreover, these data suggest that cytotoxicity ofPIAs correlates with the ability to inhibit Akt.
To confirm the role of Akt modulation in controlling the cytotoxiceffects of PIAs, BEAS2B lung epithelial cells were infected with adeno-viruses expressing a constitutively active form of Akt (MyrAkt) or�-galactosidase. (Of note, these studies were attempted with the NSCLCand breast cancer cell lines used above, but these cell lines were notinfectable with these adenoviruses.) Administration of PIA24 increasedapoptosis and decreased Akt phosphorylation in BEAS2B cells infectedwith �-galactosidase (Fig. 5E). In contrast, PIA24 was less cytotoxic andcaused less inhibition of Akt phosphorylation in BEAS2B cells infectedwith MyrAkt. These studies confirm the role of modulation of Aktactivation as the main biological effect underlying PIA cytotoxicity.
DISCUSSION
In the present study, we analyzed a series of modified PIAs for theirability to inhibit Akt activity and to induce apoptosis in cancer celllines with constitutively active Akt. Of the initial 24 compounds testedin our cell panel, PIAs 5, 6, 23, 24, and 25 markedly inhibited Akt at2 h. The common structural features of these effective PIAs were aphosphate linker between the ether side chain and the inositol ring andmodifications at positions 2 and 3 or 4 and 5 of the inositol ring. The2,3-modified analogues included: a 2-methoxy,3-deoxy analogue(PIA 5); a 2,3-di-deoxy analogue (PIA 6); a 2-isobutyoxy,3-deoxyanalogue (PIA 23); and a 2-cyclohexylmethoxy,3-deoxy analogue(PIA 24). The 4,5-modified analogue (PIA 25) was deoxygenated atthe 4- and 5-positions of the inositol ring. Interestingly, when thephosphate group linking the ether lipid component to the inositol ringof the active PIAs was replaced by a carbonate linker, the inhibitoryactivity of the PIAs was lost (e.g., compare PIAs 8 versus 5 or 9versus 6). Other PIAs with carbonate linkers that were ineffective atinhibiting Akt included PIAs 12, 14, and 15. Dimeric PIAs (PIAs 17,18, 19, 20, 21, and 22) were also ineffective at inhibiting Akt.However, some PIAs with phosphate linkers and different ring mod-ifications (PIAs 10, 13, and 16) were able to inhibit Akt in a cellline-specific manner. The basis for cell line specificity is presentlyunclear but may be partially related to differences in uptake of PIAsbecause when PIAs 10 and 16 were administered to H157 or H1703cells for 24 h (instead of 2 h), inhibition of Akt and induction ofapoptosis was observed (data not shown).
Inhibition of Akt by PIAs was dose dependent and time dependent.The IC50s for PIAs 5, 6, and 24 were in the low micromolar range, whichmight be achievable in vivo but is a concern regarding additional devel-opment of these compounds. When PIAs 5 and 6 were analyzed in H1703cells for time-dependent inhibition of Akt activation and were compared
Fig. 4. PIAs selectively inhibit Akt and downstream components. A, H1703 cells were treated with PIA 5, 6, 23, 24, 25, or 7 (10 �M) for 18 h and were harvested for immunoblottingexperiments with the phospho-specific and native antibodies shown. The positions of each protein within the PI3k/Akt pathway are indicated by Upstream, Akt, or Downstream, andthe involvement of individual downstream substrates in various cellular processes is to the right of the immunoblots. B, same as A, only H157 cells were used. Immunoblottingexperiments were performed at least three times. C, control.
2788
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

with a PI3k inhibitor, LY294002, similar rapid times of onset of inhibi-tion were observed (within 15 min), but the inhibition of Akt wasmaintained longer with LY294002 treatment. In contrast, the onset ofinhibition of Akt by PIAs 5 and 6 in H157 cells lagged inhibition byLY294002, but inhibition by PIAs 5 and 6 was maintained longer thanwith LY294002. These cell lines differ in many ways, but one relevantmolecular difference might be PTEN status. Although the activity ofPIAs is likely PTEN independent because PIAs can inhibit Akt phospho-rylation in cancer cells with wild-type PTEN (H1703) or mutant PTEN(H157 and MB468; Fig. 2A), the delayed onset of inhibition in cells withmutant PTEN suggests that PTEN status may determine the kinetics ofAkt inhibition by PIAs. Future studies will address this issue.
Inhibition of Akt phosphorylation and activity by PIAs was not due toinhibition of upstream kinases such as PI3k or PDK-1. The lack ofinhibition of PI3k by earlier PIAs such as DPIEL was observed in twoprior studies (40, 49). Additional evidence that PIAs did not inhibitPDK-1 includes the fact that PIAs did not inhibit the activity of purifiedPDK-1 in a high-throughput kinase assay (data not shown) and that PIAsdid not affect the phosphorylation of another PDK-1 substrate, proteinkinase C-�, at its PDK-1 site (T505; data not shown).
In contrast to the lack of inhibition of upstream components, PIAs 5,6, 23, 24, and 25 inhibited phosphorylation of downstream substrates thatcontrol key cellular functions. Differences in PIA-induced inhibition ofphosphorylation of downstream substrates might be related to differencesin stability or metabolism of the various PIAs, differences in kinetics ofAkt inhibition, or phosphorylation of these substrates by other kinasesthat are not inhibited by PIAs. The apparent greater inhibition of phos-phorylation of downstream substrates by PIAs compared with inhibitionof Akt phosphorylation might be related to the fact that some Aktconsensus sites in downstream substrates can also be phosphorylated byother kinases. If these “off-target” kinases were to be affected as anindirect result of PIA-induced Akt inhibition for 18 h, then one mightobserve greater inhibition of downstream substrates than Akt itself.Among substrates directly phosphorylated by Akt, GSK-3 phosphoryla-tion was decreased with PIA administration. Because GSK-3 activity canpromote apoptosis by altering glucose metabolism, inhibiting antiapop-totic molecules such as heat shock factor 1 and heat shock protein 70, aswell as inhibiting cell cycle regulatory molecules such as cyclin D1 andp21 (53), PIA-induced decreased GSK-3 phosphorylation could have anoverall effect of increasing or maintaining GSK-3 activity, inhibiting cellcycle progression, and/or promoting apoptosis. Of note, cyclinD1 proteinexpression was not altered by PIA administration in H157 or H1703 cells(data not shown). PIAs did not alter cell cycle distribution in the eight celllines tested in Fig. 5B, although LY294002 has been previously shown toinduce a G1-G0 arrest in these cell lines (37, 38).
Other direct Akt substrates that were inhibited by PIAs in H1703 andH157 cells included members of the Forkhead family of transcriptionfactors, AFX and FKHR, which regulate apoptosis through alteringtranscription of genes such as BIM, p27, and Fas ligand (54). PIAtreatment also decreased phosphorylation of three substrates downstreamof Akt that control the initiation phase of protein synthesis—tuberin,4EBP-1, and p70S6K. Tuberin is a direct substrate of Akt, and 4EBP-1and p70S6K are indirectly phosphorylated as a consequence of Aktactivation. Phosphorylation of tuberin and 4EBP-1 was inhibited by PIAtreatment in both H1703 and H157 cells, but p70S6K phosphorylationwas inhibited to a much greater extent in H157 cells. The likely cumu-lative effect of modulating phosphorylation of these three proteins isdiminished protein synthesis. This may be relevant because the initiationof protein translation is associated with tumorigenesis (55). Interestingly,when we evaluated the effects of PIAs against two direct Akt substratesthat more directly control the apoptotic process, ASK-1 and MDM2, onlyASK-1 phosphorylation was slightly inhibited by PIAs 23, 24, and 25 inH157 cells. Collectively, these data suggest that decreased phosphoryla-
tion of downstream substrates of Akt may modulate many cellular pro-cesses including metabolism, transcription, protein translation, and/orapoptosis.
We also tested PIAs for inhibition of activation of other kinasesdownstream of Ras. Phosphorylation of c-Raf, a direct Akt substrate,was inhibited by PIA treatment in H1703 or H157 cells. Concomitantwith decreased c-Raf phosphorylation, ERK phosphorylation wasincreased in H1703 and H157 cells by PIAs 5 and 6 but not PIAs 23,24, and 25. Increased ERK phosphorylation after PIA treatment maybe a consequence of decreased c-Raf phosphorylation at Ser-259,because phosphorylation of c-Raf by Akt at Ser-259 has been shownto inhibit the mitogen-activated protein/ERK/ERK pathway (56). Thelack of correlation between inhibition of c-Raf phosphorylation andincreased ERK phosphorylation with PIAs 23, 24, and 25 may indi-cate that this group of PIAs has a different profile of “off-target”activities that affect the Raf/mitogen-activated protein/ERK/ERKpathway. Taken together, these data show that PIAs inhibit Akt andphosphorylation of downstream substrates without inhibiting up-stream kinases or ERK.
In addition to decreasing Akt phosphorylation and activity and inhib-iting the phosphorylation of several downstream targets, PIAs inducedapoptosis preferentially in cell lines with high levels of Akt activity.Based on historical comparisons of experiments performed in our labo-ratory using standard chemotherapy agents, PIAs induced more apoptosisat 24 h than did cisplatin (20 �M), paclitaxel (2.5 �M) etoposide (50 �M),trastuzumab (10.5 �M), or gemcitabine (100 �M) at 48 h (38). Moreover,the induction of apoptosis was related to Akt inhibition, because onlyPIAs that inhibited Akt activity effectively induced apoptosis, and aconstitutively active form of Akt that does not depend upon binding ofPIP3 to the PH domain for translocation bypassed the inhibitory effects ofPIAs.
The clinical implications of these apoptosis assays could be impor-tant, because Akt is becoming a highly validated therapeutic target incancer, a disease characterized by dysregulated apoptosis. Inhibitionof Akt might have great potential benefit for patients with cancer, andsupport for the concept of targeting Akt comes from many observa-tions. First, over 54% of human cancers have active Akt that isdetectable in situ.3 Thus, small molecule Akt inhibitors could havewide applicability as cancer drugs. Second, inhibition of the PI3k/Aktpathway by biochemical or genetic means increases the efficacy ofchemotherapy and/or radiation in vitro and in vivo (37, 38, 57, 58).Finally, several standard chemotherapeutic agents and chemopreven-tive agents inhibit the PI3k/Akt pathway as a consequence of admin-istration in vitro, and in some cases inhibition of Akt is directlyresponsible for cytotoxicity of these agents (59).
Despite the widely acknowledged need for Akt inhibitors, however,none are commercially available. Based on the properties we have de-scribed for PIAs 5, 6, 23, 24, and 25, we believe that PIAs are the bestcharacterized Akt inhibitors to date and that PIAs compare favorably withother inhibitors of the PI3k/Akt pathway such as wortmannin orLY294002, earlier generation PIAs, or nonspecific small molecule kinaseinhibitors engineered for greater specificity toward Akt. The PI3k inhib-itors wortmannin and LY294002 may have limited clinical utility due tospecificity concerns, adverse side effects, and solubility issues. Wortman-nin, in addition to inhibiting PI3k, inhibits myosin light chain kinase,phospholipase C, phospholipase D, phospholipase A2, and DNA-depend-ent protein kinase (59). LY294002 inhibits PI3k as well as the arylhydrocarbon receptor, which is a ligand-activated transcription factor(60). In vivo use of LY294002 in mice has been associated with manyadverse side effects, including death (61). Furthermore, wortmanninand LY294002 are only soluble in organic solvents, which mayadditionally limit their clinical application. Although DMSO was usedas a universal solvent for all PIAs in our studies because initial
2789
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from
evaluation revealed that PIAs with carbonate linkers were insoluble inwater (data not shown), PIAs 5, 6, 23, 24, and 25 are in fact solublein water. Therefore, although wortmannin and LY294002 inhibit thePI3k/Akt pathway, their lack of specificity, potential side effects, and
poor solubility raise doubts as to their suitability as lead compoundsfor additional drug development.
The biggest advantage of PIAs 5, 6, 23, 24, and 25 over early gener-ation PIAs such as DPIEL is greater efficacy. Although molecular mod-
Fig. 5. PIAs selectively increase apoptosis in cancer cells throughmodulation of Akt activity. A, cancer cell lines with high levels of Aktactivity (H1703, H157, H1155, and MB468 cells) or low levels of Aktactivity (A549, H1355, MCF-7, and MB231) were treated with PIA 5, 6,23, 24, 25, or 7 (10 �M) for 24 h. Apoptosis was assessed morphologicallyand by measuring the formation of sub2N DNA using flow cytometry.Con, control. B, PIAs inhibit Akt activation in cells that have a poorapoptotic response to PIA treatment. A549 or H1355 cells were treatedwith the indicated PIAs or DMSO (C) for 2 h, and lysates were harvestedfor immunoblotting with the indicated antibodies. Because levels of Aktphosphorylation were low in these cell lines, the immunoblots shown wereoverexposed to reveal inhibition of Akt phosphorylation induced by PIAs.C, time of exposure needed for apoptosis. H157 cells were exposed to theindicated PIAs for 2, 6, or 24 h. Apoptosis was assessed at 24 h using flowcytometry. Con, control. D, PIAs that do not inhibit Akt do not increaseapoptosis. H1703 cells were treated with PIA 5 or 8 (10 �M) for 24 h, andapoptosis was assessed as above. The structures of PIAs 5 and 8 aredepicted. E. constitutively active Akt protects against PIA-induced apo-ptosis. BEAS2B cells were infected with adenoviruses expressing b-galactosidase or Myr Akt. 24 h after infection, cells were treated withPIA24 or vehicle and samples were collected for assessment of Aktactivation and induction of apoptosis. Experiments were performed intriplicate at least three times.
2790
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

eling led to the synthesis of DPIEL, a first generation 3-deoxy PIA thatinhibited platelet derived growth factor-induced Akt activity and cellgrowth in vitro (40), DPIEL did not inhibit Akt in our cellular assays.This might reflect that fact that the inhibition of endogenous Akt activityin cancer cell lines that we used for screening is probably a more rigoroustest for Akt inhibition, because Akt is likely to be activated throughmultiple mechanisms in these cell lines, including activation of multiplegrowth factor receptors, activation of Ras, and inactivation of PTEN.Moreover, in xenograft studies, oral administration of DPIEL resulted inlow bioavailability due to acid lability of the compound, and i.v. admin-istration resulted in massive hemolysis and death (62). Structural simi-larities between DPIEL and PIAs raise the possibility that poor bioavail-ability and toxicity will also be characteristic of PIA administration invivo, which would preclude development of PIAs as drugs. However,preliminary acute toxicology experiments in mice suggest that PIAs maybe better tolerated than DPIEL (data not shown). Additional toxicologicand pharmacokinetic experiments will address these issues. Nevertheless,DPIEL lacks the efficacy and safety profile for an appropriate Aktinhibitor lead compound.
In a final comparison, PIAs 5, 6, 23, 24, and 25 are more fullycharacterized and have greater specificity than a recently described,nonspecific small molecule kinase inhibitor that was engineered to loseactivity against other kinases but retain activity against Akt. Chemicallymodifying H89, an inhibitor of protein kinase A, Reuveni et al. (63)synthesized a compound called NL-71-101 that had diminished activity
toward protein kinase A but retained modest inhibition of growth factor-induced Akt activity. NL-71-101 did induce apoptosis in one cell linewith high endogenous Akt activity (63). However, PIAs may be betterlead compounds than NL-71-101, because NL-71-101 was less specific,less potent, and slower acting (thus raising the possibility of indirectactivity) and did not inhibit endogenous Akt activity. Another potentialproblem with the design of NL-71-101 is that it functions as a competitiveinhibitor for ATP binding. Creating kinase inhibitors that target the ATPbinding site of a kinase can be fraught with specificity problems becauseall kinases possess ATP binding sites. This has been perhaps best ob-served with STI-571 (Gleevec), a competitive inhibitor of the ATP-binding site of many kinases (64). Interestingly, the wide clinical appli-cation of Gleevec is partially due to the fact that it inhibits many kinasesincluding bcr-abl, platelet-derived growth factor receptors, and c-Kit(65–67). Whether specific inhibition of Akt would be clinically valuableis unknown, but specific inhibition of Akt will be required to rigorouslyvalidate Akt as a molecular target.
The fact that rationally designed PIAs inhibit Akt and promoteapoptosis in cancer cells with constitutively high levels of Akt appearsto fulfill the need for development of small molecule Akt inhibitorsthat are specific and effective. Not only is this of potential importancefor cancer therapy and/or prevention, but also for other diseases suchas rheumatoid arthritis, AIDS, and other infectious diseases whosepathogenesis may depend upon Akt activation (68–73). These studiesraise many questions that will be addressed in ongoing and futureexperiments. Do other kinases contribute to the biological effects ofPIAs? Can PIAs be effectively combined with chemotherapy orradiation therapy? Are PIAs effective in vivo? Will this generation ofPIAs have better bioavailability and stability, with less toxicity invivo? Will one PIA emerge from the group of five as a lead com-pound? The answers to these questions will help to determine whetherPIAs can be advanced to realize the potential clinical benefit ofinhibiting Akt.
ACKNOWLEDGMENTS
We thank Dr. Dario Alessi for helpful discussions and the PH Akt-GFPconstruct. We also thank Amy Clark, Samantha Streicher, Mary Kunjappu,Erin Addis-Lieser, and Christina Dahlman for technical assistance and Dr. J. S.Gutkind for helpful discussions.
REFERENCES
1. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. GenesDev 1999;13:2905–27.
2. Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in humancancer. Nat Rev Cancer 2002;2:489–501.
3. Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding aserine-threonine kinase containing an SH2-like region. Science 1991;254:274–7.
4. Coffer PJ, Woodgett JR. Molecular cloning and characterisation of a novel putativeprotein-serine kinase related to the cAMP-dependent and protein kinase C families.Eur J Biochem 1991;201:475–81. Erratum in: Eur J Biochem 1992;205:1217
5. Jones PF, Jakubowicz T, Pitossi FJ, Maurer F, Hemmings BA. Molecular cloning andidentification of a serine/threonine protein kinase of the second-messenger subfamily.Proc Natl Acad Sci USA 1991;88:4171–5.
6. Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res 1999;253:210–29.
7. Leslie NR, Bennett D, Gray A, Pass I, Hoang-Xuan K, Downes CP. Targetingmutants of PTEN reveal distinct subsets of tumour suppressor functions. Biochem J2001;357:427–35.
8. Balendran A, Casamayor A, Deak M, et al. PDK1 acquires PDK2 activity in thepresence of a synthetic peptide derived from the carboxyl terminus of PRK2. CurrBiol 1999;9:393–404.
9. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinaseB/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA 1998;95:11211–6.
10. Lynch DK, Ellis CA, Edwards PA, Hiles ID. Integrin-linked kinase regulates phos-phorylation of serine 473 of protein kinase B by an indirect mechanism. Oncogene1999;18:8024–32.
11. Toker A, Newton AC. Akt/protein kinase B is regulated by autophosphorylation at thehypothetical PDK-2 site. J Biol Chem 2000;275:8271–4.
Fig.5. Continued
2791
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

12. Hill M, Feng J, Hemmings B. Identification of a plasma membrane raft-associatedPKB Ser473 kinase activity that is distinct from ILK and PDK1. Curr Biol 2002;12:1251.
13. Marmy-Conus N, Hannan KM, Cristiano BE, Hemmings BA, Pearson RB. Directidentification of tyrosine 474 as a regulatory phosphorylation site for the Akt proteinkinase. J Biol Chem 2002;277:38021–8.
14. Datta SR, Dudek H, Tao X, et al. Akt Phosphorylation of BAD couples survivalsignals to the cell-intrinsic death machinery. Cell 1997;91:231–41.
15. del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-inducedphosphorylation of BAD through the protein kinase Akt. Science 1997;278:687–9.
16. Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9by phosphorylation. Science 1998;282:1318–21.
17. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negativelyregulates apoptosis signal-regulating kinase 1. Mol Cell Biol 2001;21:893–901.
18. Ogawara Y, Kishishita S, Obata T, et al. Akt enhances Mdm2-mediated ubiquitinationand degradation of p53. J Biol Chem 2002;277:21843–50.
19. Zhou BP, Hung MC. Novel targets of Akt, p21(Cipl/WAF1), and MDM2. SeminOncol 2002;29:62–70.
20. Rossig L, Jadidi AS, Urbich C, Badorff C, Zeiher AM, Dimmeler S. Akt-dependentphosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothe-lial cells. Mol Cell Biol 2001;21:5644–57.
21. Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 pro-motes binding to 14-3-3 and cytoplasmic localization. J Biol Chem 2002;277:28706–13.
22. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature 2001;411:355–65.23. Biggs WH III, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase
B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helixtranscription factor FKHR1. Proc Natl Acad Sci USA 1999;96:7421–6.
24. Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-�B by the Akt/PKBkinase. Curr Biol 1999;9:601–4.
25. Cross D, Alessi D, Cohen P, Andjelkovich M, Hemmings B. Inhibition of glycogensynthase kinase-3 by insulin mediated by protein kinase B. Nature 1995;378:785–9.
26. Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target ofrapamycin is a direct target for protein kinase B: identification of a convergence pointfor opposing effects of insulin and amino-acid deficiency on protein translation.Biochem J 1999;344:427–31.
27. Sekulic A, Hudson CC, Homme JL, et al. A direct linkage between the phosphoi-nositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin inmitogen-stimulated and transformed cells. Cancer Res 2000;60:3504–13.
28. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Aktand suppresses mTOR signalling. Nat Cell Biol 2002;4:648–57.
29. Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2.Nat Cell Biol 2002;4:658–65.
30. Ackler S, Ahmad S, Tobias C, Johnson MD, Glazer RI. Delayed mammary glandinvolution in MMTV-AKT1 transgenic mice. Oncogene 2002;21:198–206.
31. Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combinedactivation of Ras and Akt in neural progenitors induces glioblastoma formation inmice. Nat Genet 2000;25:55–7.
32. Malstrom S, Tili E, Kappes D, Ceci JD, Tsichlis PN. Tumor induction by anLck-MyrAkt transgene is delayed by mechanisms controlling the size of the thymus.Proc Natl Acad Sci USA 2001;98:14967–72.
33. West KA, Brognard J, Clark AS, et al. Rapid Akt activation by nicotine and a tobaccocarcinogen modulates the phenotype of normal human airway epithelial cells. J ClinInvestig 2003;111:81–90.
34. Perez-Tenorio G, Stal O. Activation of AKT/PKB in breast cancer predicts a worseoutcome among endocrine treated patients. Br J Cancer 2002;86:540–5.
35. Lee JI, Soria JC, Hassan KA, et al. Loss of PTEN expression as a prognostic markerfor tongue cancer. Arch Otolaryngol Head Neck Surg 2001;127:1441–5.
36. Ermoian RP, Furniss CS, Lamborn KR, et al. Dysregulation of PTEN and proteinkinase B is associated with glioma histology and patient survival. Clin Cancer Res2002;8:1100–6.
37. Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activitypromotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancercells. Mol Cancer Ther 2002;1:707–17.
38. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase b is constitutively activein non-small cell lung cancer cells and promotes cellular survival and resistance tochemotherapy and radiation. Cancer Res 2001;61:3986–97.
39. Rong SB, Hu Y, Enyedy I, et al. Molecular modeling studies of the Akt PH domainand its interaction with phosphoinositides. J Med Chem 2001;44:898–908.
40. Hu Y, Qiao L, Wang S, et al. 3-(Hydroxymethyl)-bearing phosphatidylinositol etherlipid analogues and carbonate surrogates block PI3-K, Akt, and cancer cell growth.J Med Chem 2000;43:3045–51.
41. Kozikowski AP, Sun H, Brognard J, Dennis PA. Novel PI analogues selectively blockactivation of the Pro-survival serine/threonine kinase Akt. J Am Chem Soc 2003;125:1144–5.
42. Suhara T, Kim HS, Kirshenbaum LA, Walsh K. Suppression of Akt signaling inducesFas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol Cell Biol 2002;22:680–91.
43. Canman CE, Wolff AC, Chen CY, Fornace AJ Jr, Kastan MB. The p53-dependentG1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res 1994;54:5054–8.
44. Kastan MB, Zhan Q, el-Deiry WS, et al. A mammalian cell cycle checkpoint pathwayutilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992;71:587–97.
45. Gutkind JS, Lacal PM, Robbins KC. Thrombin-dependent association of phosphati-dylinositol-3 kinase with p60c-src and p59fyn in human platelets. Mol Cell Biol1990;10:3806–9.
46. Tripos Inc. Sybyl 6.9. St. Louis, MO: Tripos Inc; 2002.47. Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an
incremental construction algorithm. J Mol Biol 1996;261:470–89.48. Rarey M, Kramer B, Lengauer T. Docking of hydrophobic ligands with interaction-
based matching algorithms. Bioinformatics 1999;15:243–50.49. Meuillet EJ, Mahadevan D, Vankayalapati H, et al. Specific inhibition of the Akt1
pleckstrin homology domain by D-3-deoxy-phosphatidyl-myo-inositol analogues.Mol Cancer Ther 2003;2:389–99.
50. Thomas C, Deak M, Alessi D, van Aalten D. High-resolution structure of thepleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol(3,4,5)-trisphosphate. Curr Biol 2002;12:1256–62.
51. Thomas C, Deak M, Alessi D, van Aalten D. High-resolution structure of thepleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol(3,4,5)-trisphosphate. Curr Biol 2002;12:1256.
52. Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, Van Aalten DM. Binding ofphosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of pro-tein kinase B induces a conformational change. Biochem J 2003;375:531–8.
53. Hardt SE, Sadoshima J. Glycogen synthase kinase-3�: a novel regulator of cardiachypertrophy and development. Circ Res 2002;90:1055–63.
54. Burgering BM, Kops GJ. Cell cycle and death control: long live forkheads. TrendsBiochem Sci 2002;27:352–60.
55. Stoneley M, Willis AE. Aberrant regulation of translation initiation in tumorigenesis.Curr Mol Med 2003;3:597–603.
56. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (proteinkinase B). Science 1999;286:1741–4.
57. Bondar VM, Sweeney-Gotsch B, Andreeff M, Mills GB, McConkey DJ. Inhibition ofthe phosphatidylinositol 3�-kinase-AKT pathway induces apoptosis in pancreaticcarcinoma cells in vitro and in vivo. Mol Cancer Ther 2002;1:989–97.
58. Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol3�-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancermodels. Cancer Res 2002;62:1087–92.
59. West KA, Sianna Castillo S, Dennis PA. Activation of the PI3K/Akt pathway andchemotherapeutic resistance. Drug Resist Update 2002;5:234–48.
60. Guo M, Joiakim A, Reiners JJ. Suppression of 2,3,7,8-tetrachlorodibenzo-p-dioxin(TCDD)-mediated aryl hydrocarbon receptor transformation and CYP1A1 inductionby the phosphatidylinositol 3-kinase inhibitor 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). Biochem Pharmacol 2000;60:635–42.
61. Hu L, Zaloudek C, Mills GB, Gray J, Jaffe RB. In vivo and in vitro ovarian carcinomagrowth inhibition by a phosphatidylinositol 3-kinase inhibitor (LY294002). ClinCancer Res 2000;6:880–6.
62. Egorin MJ, Parise RA, Joseph E, et al. Plasma pharmacokinetics and bioavailablilityfor the phosphatidylinositide-3-kinase signalling inhibitor, OMDPI (NSC 710297) inCD2F1 mice. Proc Am Assoc Cancer Res 2002;43:604.
63. Reuveni H, Livnah N, Geiger T, et al. Toward a PKB inhibitor: modification of aselective PKA inhibitor by rational design. Biochemistry 2002;41:10304–14.
64. Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene 2002;21:5868–76.
65. McGary EC, Weber K, Mills L, et al. Inhibition of platelet-derived growth factor-mediated proliferation of osteosarcoma cells by the novel tyrosine kinase inhibitorSTI571. Clin Cancer Res 2002;8:3584–91.
66. von Bubnoff N, Schneller F, Peschel C, Duyster J. BCR-ABL gene mutations inrelation to clinical resistance of Philadelphia-chromosome-positive leukaemia toSTI571: a prospective study. Lancet 2002;359:487–91.
67. Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kitreceptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor.Blood 2000;96:925–32.
68. Zhang HG, Wang Y, Xie JF, et al. Regulation of tumor necrosis factor �-mediatedapoptosis of rheumatoid arthritis synovial fibroblasts by the protein kinase Akt.Arthritis Rheum 2001;44:1555–67.
69. Wolf D, Witte V, Laffert B, et al. HIV-1 Nef associated PAK and PI3-kinasesstimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. NatMed 2001;7:1217–24.
70. Francois F, Klotman ME. Phosphatidylinositol 3-kinase regulates human immunod-eficiency virus type 1 replication following viral entry in primary CD4(�) T lym-phocytes and macrophages. J Virol 2003;77:2539–49.
71. Chuenkova MV, Furnari FB, Cavenee WK, Pereira MA. Trypanosoma cruzi trans-sialidase: a potent and specific survival factor for human Schwann cells by means ofphosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci USA 2001;98:9936–41.
72. Celli J, Olivier M, Finlay BB. Enteropathogenic Escherichia coli mediates antiph-agocytosis through the inhibition of PI 3-kinase-dependent pathways. EMBO J2001;20:1245–58.
73. Avota E, Avots A, Niewiesk S, et al. Disruption of Akt kinase activation is important forimmunosuppression induced by measles virus. Nat Med 2001;7:725–31.
2792
PIAS INHIBIT Akt
Research. on August 28, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from
Related Documents











