Ó 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Platinum Group Metals and Compounds HERMANN RENNER, Degussa AG, (retired), Hanau, Federal Republic of Germany G€ uNTHER SCHLAMP, Demetron GmbH, (retired), Hanau, Federal Republic of Germany INGO KLEINW € ACHTER, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic of Germany ERNST DROST, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic of Germany HANS MARTIN L€ uSCHOW, Degussa-H€ uls AG, (retired), Hanau, Federal Republic of Germany PETER TEWS, Allgem. Gold und Silberscheideanstalt (AGOSI), Pforzheim, Federal Republic of Germany PETER PANSTER, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic of Germany MANFRED DIEHL, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic of Germany JUTTA LANG, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic of Germany THOMAS KREUZER, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic of Germany ALFONS KN€ oDLER, Forschungsinstitut f€ ur Edelmetalle und Metallchemie, (retired), Schw€ abisch Gm€ und, Federal Republic of Germany KARL ANTON STARZ, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic of Germany KLAUS DERMANN, Ducera Dental GmbH und Co. KG, Hanau, Federal Republic of Germany JOSEF ROTHAUT, Ducera Dental GmbH und Co. KG, Hanau, Federal Republic of Germany RALF DRIESELMANN, dmc2 Degussa Metals Catalysts Cerdec AG, Frankfurt/M., Federal Republic of Germany CATRIN PETER, Klinikum der Friedrich Schiller Universit€ at, Jena, Federal Republic of Germany RAINER SCHIELE, Klinikum der Friedrich Schiller Universit€ at, Jena, Federal Republic of Germany 1. History ......................... 318 2. Properties ....................... 321 3. Occurrence ...................... 323 3.1. Abundance ...................... 323 3.2. Ores and Their Origin ............. 325 3.3. Primary Deposits ................. 326 3.4. Secondary Deposits ................ 328 3.5. Recovery of Secondary Platinum Group Metals .......................... 328 3.6. Reserves and Resources ............. 330 4. Mineral Dressing and Beneficiation .... 331 4.1. Treatment of Alluvial Platinum Deposits 331 4.2. Treatment of Primary Deposits ...... 331 4.3. Treatment of Nickel Ores ........... 332 4.4. Treatment of Metal Scrap .......... 333 4.5. Treatment of Dross ................ 333 4.6. Treatment of Supported Catalysts .... 334 4.7. Treatment of Solutions ............. 334 DOI: 10.1002/14356007.a21_075

Platinum Group Metals and Compounds
Oct 26, 2014
Platinum Group Metals and Compounds
Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

� 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Article No : a21_075
Platinum Group Metals and Compounds
HERMANN RENNER, Degussa AG, (retired), Hanau, Federal Republic of Germany
G€uNTHER SCHLAMP, Demetron GmbH, (retired), Hanau, Federal Republic of Germany
INGO KLEINW€ACHTER, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal
Republic of Germany
ERNST DROST, dmc2DegussaMetals Catalysts Cerdec AG, Hanau, Federal Republic of
Germany
HANS MARTIN L€uSCHOW, Degussa-H€uls AG, (retired), Hanau, Federal Republic of
Germany
PETER TEWS, Allgem. Gold und Silberscheideanstalt (AGOSI), Pforzheim, Federal
Republic of Germany
PETER PANSTER, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic
of Germany
MANFREDDIEHL, dmc2DegussaMetals Catalysts CerdecAG,Hanau, Federal Republic
of Germany
JUTTA LANG, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal Republic of
Germany
THOMAS KREUZER, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal
Republic of Germany
ALFONS KN€oDLER, Forschungsinstitut f€ur Edelmetalle und Metallchemie, (retired),
Schw€abisch Gm€und, Federal Republic of Germany
KARL ANTON STARZ, dmc2 Degussa Metals Catalysts Cerdec AG, Hanau, Federal
Republic of Germany
KLAUS DERMANN, Ducera Dental GmbH und Co. KG, Hanau, Federal Republic of
Germany
JOSEF ROTHAUT, Ducera Dental GmbH und Co. KG, Hanau, Federal Republic of
Germany
RALFDRIESELMANN, dmc2DegussaMetals Catalysts CerdecAG, Frankfurt/M., Federal
Republic of Germany
CATRIN PETER, Klinikum der Friedrich Schiller Universit€at, Jena, Federal Republic of
Germany
RAINER SCHIELE,Klinikumder Friedrich Schiller Universit€at, Jena, Federal Republic of
Germany
1. History . . . . . . . . . . . . . . . . . . . . . . . . . 318
2. Properties . . . . . . . . . . . . . . . . . . . . . . . 321
3. Occurrence . . . . . . . . . . . . . . . . . . . . . . 323
3.1. Abundance . . . . . . . . . . . . . . . . . . . . . . 323
3.2. Ores and Their Origin . . . . . . . . . . . . . 325
3.3. Primary Deposits . . . . . . . . . . . . . . . . . 326
3.4. Secondary Deposits . . . . . . . . . . . . . . . . 328
3.5. Recovery of Secondary Platinum Group
Metals . . . . . . . . . . . . . . . . . . . . . . . . . . 328
3.6. Reserves and Resources . . . . . . . . . . . . . 330
4. Mineral Dressing and Beneficiation . . . . 331
4.1. Treatment of Alluvial Platinum Deposits 331
4.2. Treatment of Primary Deposits . . . . . . 331
4.3. Treatment of Nickel Ores . . . . . . . . . . . 332
4.4. Treatment of Metal Scrap . . . . . . . . . . 333
4.5. Treatment of Dross . . . . . . . . . . . . . . . . 333
4.6. Treatment of Supported Catalysts . . . . 334
4.7. Treatment of Solutions . . . . . . . . . . . . . 334
DOI: 10.1002/14356007.a21_075

5. Dissolution Methods . . . . . . . . . . . . . . . 335
5.1. Dissolution in Aqua Regia . . . . . . . . . . . 335
5.2. Dissolution in Hydrochloric Acid –
Chlorine. . . . . . . . . . . . . . . . . . . . . . . . . 335
5.3. Dissolution in Hydrochloric Acid –
Bromine . . . . . . . . . . . . . . . . . . . . . . . . . 336
5.4. Other Dissolution Processes . . . . . . . . . . 336
5.5. Dissolution by Salt Fusion . . . . . . . . . . 336
6. Separation of Platinum Group Metals . 337
6.1. Chemistry of Platinum Group Metal
Separation . . . . . . . . . . . . . . . . . . . . . . 337
6.2. Older Separation Processes . . . . . . . . . 340
6.3. Current Separation Processes . . . . . . . . 341
6.4. Processes Used in Coarse Separation . . 343
6.5. Purification . . . . . . . . . . . . . . . . . . . . . . 346
6.6. Conversion of Salts into Metals . . . . . . 348
6.7. Partial Purification . . . . . . . . . . . . . . . . 350
6.8. Treatment of Internally Recycled Material 350
6.9. Construction Materials . . . . . . . . . . . . . 350
7. Platinum Group Metal Compounds . . . 351
7.1. Inorganic Compounds . . . . . . . . . . . . . . 351
7.1.1. Platinum Compounds. . . . . . . . . . . . . . . . 351
7.1.2. Palladium Compounds . . . . . . . . . . . . . . 352
7.1.3. Rhodium Compounds . . . . . . . . . . . . . . . 353
7.1.4. Iridium Compounds . . . . . . . . . . . . . . . . . 353
7.1.5. Ruthenium Compounds . . . . . . . . . . . . . . 354
7.1.6. Osmium Compounds . . . . . . . . . . . . . . . . 354
7.2. Organic Compounds . . . . . . . . . . . . . . . 354
8. Alloys . . . . . . . . . . . . . . . . . . . . . . . . . . 355
8.1. Alloy Systems. . . . . . . . . . . . . . . . . . . . . 355
8.2. Special Alloys. . . . . . . . . . . . . . . . . . . . . 356
8.3. Methods of Treatment . . . . . . . . . . . . . 361
9. Quality Specifications and Analysis . . . 361
9.1. Quality Specifications . . . . . . . . . . . . . . 361
9.2. Qualitative Analysis . . . . . . . . . . . . . . . . 362
9.3. Quantitative Analysis . . . . . . . . . . . . . . 362
9.4. Purity Analysis . . . . . . . . . . . . . . . . . . . 363
9.5. Trace Analysis . . . . . . . . . . . . . . . . . . . . 364
10. Uses . . . . . . . . . . . . . . . . . . . . . . . . . . . 364
10.1. Jewelry, Coinage, Investment . . . . . . . . 364
10.2. Apparatus . . . . . . . . . . . . . . . . . . . . . . . 364
10.3. Heterogeneous Catalysts . . . . . . . . . . . . 365
10.4. Fuel Cells . . . . . . . . . . . . . . . . . . . . . . . . 368
10.5. Homogeneous Catalysts . . . . . . . . . . . . 368
10.6. Automotive Emission Control Catalysts 369
10.7. Sensors . . . . . . . . . . . . . . . . . . . . . . . . . 369
10.8. Electrical Technology . . . . . . . . . . . . . . 371
10.9. Electronics . . . . . . . . . . . . . . . . . . . . . . . 372
10.10. Coatings . . . . . . . . . . . . . . . . . . . . . . . . 374
10.10.1. Coatings Produced by Electrolysis . . . . . . 375
10.10.2. Coatings Produced by Chemical Reaction 376
10.10.3. Coatings Produced by Physical Methods . 376
10.11. Dental Materials . . . . . . . . . . . . . . . . . . 376
11. Economic Aspects . . . . . . . . . . . . . . . . . 377
11.1. Supply . . . . . . . . . . . . . . . . . . . . . . . . . . 377
11.2. Demand . . . . . . . . . . . . . . . . . . . . . . . . . 378
11.3. Prices . . . . . . . . . . . . . . . . . . . . . . . . . . . 378
11.4. Commercial Aspects . . . . . . . . . . . . . . . 378
12. Toxicology . . . . . . . . . . . . . . . . . . . . . . . 379
References . . . . . . . . . . . . . . . . . . . . . . . 380
1. History [1–10], [166], [167]
Figure 1 gives an historical survey of importantplatinum group metal discoveries and platinumgroup metal technology.
Early Times. The earliest evidence of plat-inum is provided by a gold etui covered withhieroglyphic inscriptions, dating from the 7thcentury B.C. Around 1900, BERTHELOT (1827 –1907) investigated the etui, which was kept in theLouvre, and found that some of the inlays hith-erto thought to be silver were in fact platinum.
In ca. 1900, jewelry made of native platinumwas discovered in Ecuador, which was part of theInca empire in pre-Columbian times. This prob-ably dates from the first five centuries A.D.
In ancient times, the technique of washingriver sands and fusing together the grains ofplatinum was undoubtedly known, as was themaking of alloys by heating alluvial platinumand
gold with a blowpipe, these methods being simi-lar to those used in gold extraction and working.Pre-Columbian Indians were familiar with apowder metallurgy technique.
16th–18th Century [11], [12]. In theirsearch for gold in the New World, particularlyin the area of present-day Colombia, the Spanishoften found alluvial (‘‘placer’’) platinum. Theearliest written report of the metal was made in1557 by J. C. SCALIGER (1484 – 1558). Howev-er, platinum was worked to produce jewelry andutensils only afterman had learned that themetal,initially believed to be infusible, could be meltedand cast by first adding other metals to lower itsmelting point. Platinum was essentially regardedas a troublesome material, accompanying goldand silver, which lowered their workability. TheSpanish named the metal platina, the diminutiveform of plata (silver), as a derogatory termbecause it was found only in small quantities or
318 Platinum Group Metals and Compounds Vol. 28

Figure 1. Historical survey of important platinum group metal discoveries and platinum group metal technology
Vol. 28 Platinum Group Metals and Compounds 319

as small granules. The terms ‘‘white gold’’ and‘‘heavy silver’’ were also sometimes used. Whenthe metal became well known in Europe in 1748,thanks to the Spanish mathematician A. DE
ULLOA (1716 – 1795), the demand for platinumincreased. Its high density enabled it to be used toadulterate gold, and its introduction into Europewas therefore prohibited. Until 1908, the price ofplatinum was lower than that of gold. Around1750, the scientific investigation of platinumbegan, initiated largely by C. WOOD (1702 –1774), who learned of the metal in Jamaica in1741.He can be regarded as the true discoverer ofplatinum, having presented a paper in 1750 to theRoyal Society entitled ‘‘The New Semi-MetalCalled Platina.’’ In this initial period, fundamen-tal investigations into the chemistry of platinum(solubility in aqua regia, precipitation by addi-tion of ammonium chloride, fusion by addition ofarsenic, lead cupellation, etc.) were carried outby W. WATSON (1715 – 1787), W. LEWIS
(1708 – 1781), and others. These workers, in-cluding WOOD, often collaborated. Research atthis time had as its primary aim, distinguishingand separating platinum and gold, although plat-inum was at first widely believed to consist ofgold contaminated with other elements.
An important development in platinum tech-nology was the technique of converting the allu-vial (placer) deposits into platinum sponge,which could be satisfactorily formed by heat intocompact platinum or platinum artifacts. F. C.ACHARD (1753 – 1821) discovered the methodof oxidizing an easily fusible platinum – arsenicalloy to remove arsenic. As early as 1784, heproduced the first platinum crucible from plati-num sponge made in this way.
19th Century. Around 1800, the accompa-nying metals in native platinum, which generallycontains up to 80% Pt, were discovered. W. H.WOLLASTON (1766 – 1828) discovered palladium(initially also known as ‘‘new silver’’) and rho-dium; S. TENNANT (1761 – 1815) discoverediridium and osmium. At the same time, theexistence of platinum as a true element wasestablished. C. CLAUS (1796 – 1864) discoveredruthenium in 1844.
In 1823, J. W. DOEBREREINER (1780 – 1849)first used the catalytic action of platinum in thegas lighter named after him. In about 1810, theprocess invented by W. H. WOLLASTON became
established: dissolving the raw material in aquaregia, purifying it by precipitation of ammoniumhexachloroplatinate, (NH4)2[PtCl6], and heatingthis strongly to form platinum sponge. In 1817,this led to the formation of Johnson, Matthey &Co., London [13], which laid the foundation ofmodern platinum technology.
Until the beginning of the 19th century, al-most all platinum was obtained from the areanow known as Colombia, which from 1739 to1819 belonged to the Spanish Crown Dependen-cy of New Granada. At this time, ca. 1 t/aof platinum was extracted. ALEXANDER VON
HUMBOLDT was the most accomplished prospec-tor and developer of noble-metal deposits inNewGranada (1819) and of the newly discoveredplatinum deposit in the Urals. In 1819, platinumwas extracted from alluvial gold, but soon afterthis, the main production was switched to plati-num placers with low gold content. In 1825,Russia became the primary producer of platinum.The minting of platinum coins in Russia in1828 – 1845 necessitated an increase in produc-tion, which reached 3.5 t in 1843 [14–16].Almost all Russian platinum ore was used forcoinage (15 t total). Technology for the extrac-tion and treatment of placer deposits had reacheda high standard by this time.
After the closing of the refinery in St. Peters-burg, which was associated with the local mintand produced ametal of ca. 97% platinum, 1.2%iridium, 0.5% ruthenium, 0.25% palladium,1.5% iron, and 0.4% copper, the refining andworking ofRussian platinumorewere carried outalmost exclusively in Western Europe. Newcompanies for the refining and working of plati-num were founded, including Desmoutis, Paris(1822); Baker & Co., New York (from 1904:Engelhard, Newark, New Jersey); Heraeus,Hanau (1851); Siebert, Hanau (1881; from1930 part of Degussa, Frankfurt); J. Bishop &Co., Malverne, Pennsylvania and others.
20th Century [17–20]. The platinum indus-try grew vigorously after 1880 due to increasingdemands of the electrical industry, dentistry, andchemical technology. The primary consumerwasthe United States. In 1913, annual production ofcrude platinum reached 7 t. At this time, Russiabegan to make itself less dependent on othercountries by constructing a modern platinumrefinery and smelting point at Ekaterinburg (now
320 Platinum Group Metals and Compounds Vol. 28

Sverdlovsk). However, at the result of an ideo-logically negative attitude toward noble metalsand a false estimate of their economic impor-tance, platinum production was abandoned afterthe Russian Revolution.
The result was that, after World War I, Co-lombia once again became the largest platinumproducer. The raw materials were exclusivelyalluvial deposits, which were extracted by pan-ning, as they are even today. Almost all of theColombian crude platinum was refined in theUnited States.
In 1925, production in theUrals was restarted.New alluvial deposits were discovered in Siber-ia. Also, mining of primary platinum becameimportant. Today, a large proportion of the plati-num group metals supplied by the CIS comesfrom sulfidic nickel deposits. For some decades,the Soviet Union has been one of the largestproducers of platinum and especially palladium.Production figures are not available thus far.Estimates are approximate and are based onexport figures.
During World War I, the demand for nickelincreased, and Canada became an importantproducer of palladium and platinum, becausenickel ores also contained platinum group me-tals. Since 1890, these ores have been extractedby the Mond Nickel Co. (since 1961, INCO,London), later allied with the International Nick-el Co. of Canada. From 1925, platinum metalswere produced in their own refinery in Acton inLondon.Another important producer of platinumis Falconbridge Nickel Mines in Toronto.
After World War I, worldwide demand couldno longer be satisfied by Russia and other pro-ducers, and new sources were developed. InSouth Africa, the search was particularly wellrewarded. Platinum and osmiridium were dis-covered at Black Reef (1888), Witwatersrand(1892), Great Dyk (Rhodesia, 1918), and laterat Waterberg. The award of prospecting rights toprivate persons led to prospecting on a widescale. A systematic geological survey of theBushveld by J. MERENSKY was highly successful,leading to the discovery of platinum in the dunitepipes (1924) and subsequently in the stratiformplatinum deposits known as the Merensky Reef(1925), the largest platinum deposit in the world.
This discovery brought about the greatestupheaval yet seen in the platinum market. Itoccurred during a period of both high demand
(mainly for catalysts for ammonia oxidation) andhigh prices, the result being the foundation ofabout 50 producing companies. The primaryplatinum-bearing rock typical of South Africandeposits presented the ore treatment and smeltingtechnologies with completely new problems.Technical difficulties combined with a fall in theprice of platinum led to a slump. The largest ofthe surviving companies formed RustenburgPlatinum Mines Ltd. in 1931, whose outputreached 3 t/a during World War II.
AfterWorldWar II, theUnion of SouthAfrica(Republic of South Africa) became the mainproducer of platinum. From 1969, all of the crudeplatinum produced by South Africa was refinedby Johnson Matthey in England, althoughMatthey Rustenburg Refiners now refines con-siderable quantities of material produced inRustenburg. Recently, some new producers haveappeared in South Africa [e.g., Impala Platinum,Western Platinum (a subsidiary of Lonrho andFalconbridge), andAnglo-Transvaal Consolidat-ed Investment (Anglovaal) and its subsidiaryAtok Platinum Mines].
Figure 2 shows the history of world platinumoutput, andTable 1 lists the development of PGMproduction according to region.
2. Properties [166], [167]
Atomic Properties [21], [22]. The electron-ic structures of platinum group metals start fromthe inert gas structures of krypton and xenon:
Figure 2. History of world platinum output (including GUSsales to Western World)
Vol. 28 Platinum Group Metals and Compounds 321

Ru [Kr] 4 d 7 5 s1
Os [Xe] 4 f 14 5 d6 6 s2
Rh [Kr] 4 d 8 5 s1
Ir [Xe] 4 f 14 5 d7 6 s2
Pd [Kr] 4 d 10
Pt [Xe] 4 f 14 5 d9 6 s1
Two valence orbitals exist: an s shell and aninner d shell. The small energy difference be-tween them means that the electron shells arefilled in inconsistently, so that with some ele-ments, one or both electrons from the outer 5 s or6 s shells are taken up by the 4 d or 5 d shells.
The Platinum Group Metals show close simi-larities in general physical and chemical proper-ties. The very small differences in the atomicsizes of the elements of the second and the thirdrow of the periodic system are explained by thefull occupation of the 4 f14 electron orbitals of the‘‘heavy’’ elements Os, Ir, Pt (see ‘‘Lanthanidecontraction’’). Due to the electronic structure ofthese elements, which is also determined byrelativistic phenomena, causing splitting of p-and d-orbitals to altered energy levels, they show,compared to the ‘‘light’’ platinum group ele-ments (Ru, Rh, Pd), marked differences in theircatalytic activities both in homogeneous andheterogeneous catalysis [23] (see also Sec-tions 10.3 and 10.5).
Because of the small energy differences be-tween the valence shells, a number of oxidationstates occur. The following oxidation states areknown in the compounds of platinum groupmetals (principal oxidation states in bold print):
Ru: �2, 0, þ2, þ3, þ4, þ5, þ6, þ7, þ8
Rh: �1, 0, þ1, þ2, þ3, þ4, þ5, þ6
Pd: 0, þ2, þ3, þ4
Os: �2, 0, þ1, þ2, þ3, þ4, þ5, þ6, þ8
Ir: �1, 0, þ1, þ2, þ3, þ4, þ5, þ6
Pt: 0, þ2, þ4, þ5
The other properties typical of transitionmetals arevery marked; for example, catalytic activity due totheir readiness to change valence, formation ofintermediate compounds with different reagents,color, paramagnetism due to unpaired electrons,and strong tendency to form complexes.
Comparisons within the group of platinummetals, also including neighboring elements,often give an insight into the relationship be-tween electronic configuration and chemicalproperties.
Many properties show marked similaritiesalong the two horizontal rows:
Also, the tendency toward complex formationand higher oxidation states is more marked in therow of heavy elements than in the row of lightelements.
Vertical similarities between the elementsalso occur (e.g., in their behavior toward acids;(see Table 3):
Similarities also exist in the following diago-nal sequence, sometimes with ruthenium andiridium changing places:
The chemical properties of the platinumgroupmetals and the chemistry of their com-pounds fit
Table 1. Historical development of the production of platinum group metals according to region* (in t/a)
1800 1850 1900 1910 1920 1930 1940 1950 1960 1970 1975 1980 1985 1990 1995 1997
Colombia 1 < 1 < 1 < 1 2 2 2 1 1 < 1 < 1 < 1 < 1 < 1 < 1 < 1
Russia** 1 2 12 11 1 3 10 6 12 30 70 70 70 70 191
Canada � 1 � 1 1 2 6 7 12 12 12 12 12 12
South Africa 2 2 4 20 50 90 120 120 130 189
United States � 1 � 1 � 1 < 1 2 2 1 1 1 1 1 10 33
World 1 2 12 12 4 10 22 20 45 93 173 203 203 222 378 413
*Other countries account for only 1 – 2% of world production.**Estimated.
322 Platinum Group Metals and Compounds Vol. 28

less well into such a scheme than their physicalproperties.
All platinum group metals have several natu-rally occurring stable isotopes, with the excep-tion of rhodium, which has only one.
Physical Properties [22, 24–28], [167].The outstanding physical properties of the plati-num group metals are of great importance fortheir industrial use. These include high meltingpoint, low vapor pressure, high temperature co-efficient of electrical resistivity, and low coeffi-cient of thermal expansion. Table 2 lists atomicand crystal data and physical properties of theplatinum group metals.
Osmium is the densest metal known. Thedensity has been recalculated on the basis ofexperimental data and the atomic weight of190.23 þ/� 0.03 to be 22.587 þ/� 0.009 g/cm3, while for iridium the corresponding valuesare 192.217 þ/� 0.003 for the atomic weightand a density of 22.562 þ/� 0.009 g/cm.
Chemical Properties [29–34]. Table 3shows the chemical behavior of platinum groupmetals toward various reagents. Resemblancesare most clear within the two groups of elements.The aim is to quantify the reactions, so as toprovide useful information when choosing con-struction materials or dissolving platinum groupmetals. Some combinations of reagents that canbe used to dissolve these metals are also detailedin Chapter 5.
3. Occurrence
3.1. Abundance [10], [31], [36–39],[166], [167]
The abundance of platinum group metals (PGMs),which occupy an intermediate position based ontheir atomic number and atomic weight, would beexpected to be 10�4 ppm, based on the mode offormation of atomic nuclei [40]. They are concen-trated in planetary regions, reaching ca. 30 ppm in
Table 2. Atomic and physical properties of the platinum group metals [35, p. 267]
Ru Rh Pd Os Ir Pt
Atomic properties
Atomic number 44 45 46 76 77 78
Atomic weight 101.07 102.905 106.42 190.2 192.22 195.08
Crystal structure hcp fcc fcc hcp fcc fcc
Lattice constant, A�
a ¼ 2.701 3.803 3.887 a ¼ 2.735 3.839 3.924
c ¼ 4.275 c ¼ 4.319
c/a ¼ 1.583 c/a ¼ 1.579
Atomic radius, A�
1.325 1.345 1.376 1.377 1.357 1.377
Mechanical properties
Density (20 �C), g/cm3 12.45 12.41 12.02 22.59 22.56 21.45
Density liquid at melting point, g/cm3 10.90 10.70 10.49 20.1 19.39 18.91
Hardness (HV), Kg/mm2 250 – 500a 130 50 300 – 680a 200 48
Young’s modulus, GPa 485 386 124 570 538 173
Modulus of rigidity, MPa 172 153 51 220 214 67
Poisson’s ratio 0.29 0.26 0.39 0.25 0.26 0.39
Coefficient of compressibility 0.31 0.36 0.52 0.28 0.28 0.36
Thermal properties
Melting point, �C 2310 1966 1554 3045 2410 1772
Vapor pressure at mp, mbar 5.5 3.5 20 20 4 0.1
Boiling point, �C 3900 3700 2970 5000 4130 3827
Thermal conductivity (20 �C), W m�1 K�1 117 150 75 87 147 73
Coeff. of linear expansion (0 – 100 �C), 10�6 K 9.5 8.5 11.1 6.5 6.8 9.0
Electrical properties
Specific electrical resistance (0 �C), mW cm 6.7 4.34 9.725 8.2 4.7 9.825
Temperature coefficient of electrical
resistance (0 – 100 �C), 10�3 K
4.0 4.6 3.8 4.2 4.3 3.92
Transition temperature of superconductivity 0.47 0.9 0.71 0.11
Thermoelectric voltage against Pt (0 – 100 �C), mV 0.684 0.70 � 0.570 0.660 0
Work function 4.52 4.8 4.97 4.55 5.40 5.27
aDepending on crystal orientation
Vol. 28 Platinum Group Metals and Compounds 323

Table 3. Chemical resistance of the platinum group metals
Reagent Conditions Temperature, �C Pd Pt Rh Ir Ru Os
Hydrochloric acid 36% 20 – – – – – –
Hydrochloric acid 36% 100.
– – – – .
Nitric acid 65% 20 – – – – .
Nitric acid 65% 100 – – – –
Sulfuric acid 96% 20 – – – – – –
Sulfuric acid 96% 100 . –.
– – –
Sulfuric acid 96% 300. .
Hydrobromic acid 60% 20. .
– – –
Hydrobromic acid 60% 100 . – – .
Hydroiodic acid 57% 20 – – – –.
Hydroiodic acid 57% 100 – – – .
Hydrofluoric acid 40% 20 – – – – – –
Phosphoric acid 100. .
– – –
Acetic acid 99% 100 – – – – –
Hydrochloric acid/chlorine 20%/saturated 20 . .
Hydrochloric acid/chlorine 20%/saturated 80
Hydrochloric acid/chlorine 20 %/saturated 100 . . . .
Hydrochloric acid/bromine 20. .
Hydrochloric acid/bromine 100.
Aqua regia 20 – –
Aqua regia 100 – –
Aqua regia 150 .
Hydrochloric acid/H2O2 20
Hydrochloric acid/H2O2 100
Hydrobromic acid/bromine 60% 100
Water/bromine 20.
– – – – –
Ethanol/iodine 20.
–.
– –
Sodium hypochlorite solution 20 . –.
Sodium hypochlorite solution 100 –. .
Sodium cyanide solution 20 . –
Sodium cyanide solution 100 .
Copper(II) chloride solution 100.
–
324 Platinum Group Metals and Compounds Vol. 28

the earth. Considerable fractionation has takenplace in the earth’s interior, due mainly to thesiderophilic chemical character of PGMs, so thatvirtually the entire mass of PGMs is in the earth’smetallic core. The siliceous lithosphere is estimat-ed to contain 0.05 – 0.5 ppm.
On theoretical grounds, the PGMs in the earthare assumed to contain ca. 20%each of plat inum,palladium, ruthenium, and osmium, and ca. 6%each of rhodium, and iridium. In the case ofruthenium and especially osmium, these valuesare not reflected in the deposits mined to date.
3.2. Ores and Their Origin [10], [31],
[32], [36], [41–48], [166], [167]
The PGMs in the lithosphere have been trans-ferred from the earth’s interior. Tectonic move-
ments of the earth’s crust, followed by the erup-tion of magma, have led to their presence inregions close to the surface. Solidification pro-cesses, differences in melting point and density,gas emissions, convection due to heat, and theflow and eruption of magma, have all producedconcentration and separation effects, mainly inmarginal zones. Chemical interaction with high-temperature silicate layers, especially their sul-fide, arsenide, antimonide, selenide, and telluridecomponents, has also played a major role. Al-most invariably, the platinum group elementshave separated from ultrabasic magmas. Norite(Mg – Fe – Ca – Al silicate) contains mainlysulfidic intrusions, whereas those in dunite (Mg –Fe silicate) are mainly sulfide free. Platinum andpalladium (often with nickel, copper, chromium,etc.) sometimes undergo hydrothermal reactionswith chlorides in the earth’s interior. All of these
Table 3 (Continued)
Reagent Conditions Temperature, �C Pd Pt Rh Ir Ru Os
NaOH melt þair 500. . .
KOH melt þair 500. . .
NaOH melt þair 800 . .–
KOH melt þair 800 . .
KHSO4 melt þair 440.
– . –
NaCN melt þair 700 . . . .
KCN melt þair 700 . . .
NaCN/KCN melt (2 : 1) þair 550 . . . .
Chlorine, gaseous dry 20 . .– – – –
Chlorine, gaseous moist 20.
– – – .
Bromine liquid dry 20 . – – –
Bromine liquid moist 20 . – – – .
Iodine, solid dry 20 – – – – –.
Iodine, solid moist 20.
–.
– – –
Fluorine, gaseous 20.
Hydrogen sulfide, gaseous moist 20 – – – – – –
– Mass loss <0.01mg cm�2 h�1; ideal as construction material.Mass loss ca. 0.1mg cm�2 h�1; limited use as construction material
. Mass loss ca. 1mg cm�2 h�1; limited use for dissolution processes
Mass loss 10mg cm�2 h�1; suitable for dissolution processes.
Vol. 28 Platinum Group Metals and Compounds 325

processes have led to the formation of the prima-ry deposits of platinum-bearing rock. Workabil-ity depends on many factors–concentration ofplatinum metals, accessibility, size of deposit,value and potential uses of accompanying mate-rials–and is economical in only a few cases.
When primary deposits are altered and trans-ported by the natural action of the hydrosphereand atmosphere, secondary deposits, also knownas placers or alluvial deposits, are formed. Me-chanical concentration of the heavy constituentsby flowing water takes place, together withchemical dissolution and reprecipitation of theplatinum metals. Recently, hydrothermal pro-cesses have been shown to be considerably moreimportant than was at first thought. Alluvialdeposits usually originate from dunite.
The platinum metals occur in a large numberofminerals.Workable ore deposits containmain-ly sperrylite (PtAs2), cooperite (PtS), stibiopal-ladinite (Pd3Sb), laurite (RuS2), ferroplatinum(Fe – Pt), polyxene (Fe – Pt – other platinummetals), osmiridium (Os – Ir), and iridium plati-num (Ir – Pt). Theseminerals are associatedwithparticular carrier materials, which are often valu-able themselves (e.g., iron pyrites, nickel ironpyrites, or chrome iron ore).
The minerals are seldom present in an exactstoichiometric ratio. This is true of the platinumgroupmetals themselves,which are nearly alwayspresent in varying ratios, and of the accompa-nying elements with which they form compoundsor alloys. Isomorphism opens up the possibility offurther variations. Isomorphic intercalationsmake up a larger part of the economically work-able reserves. Electron probe microanalysis(EPM) has enabled a large number of definitecompounds and intermetallic phases to be identi-fied where mixtures or homogeneous solid solu-tions had formerly been assumed to be present.
Outside of true deposits, platinum group me-tals are widely distributed in very high dilution asisomorphous combinations with various metalssuch as nickel, cobalt, and copper, mainly in theirsulfides.
3.3. Primary Deposits [49–65], [166],[167]
The dunite bodies in the Urals are the mostimportant of the old Russian platinum deposits,
either as primary deposits or as material for theformation of secondary deposits. The only im-portant primary deposit is at Nishnij-Tagil,which is mined in many locations. It is a projec-tile-like intrusion of dunite through the earth’scrust, which appears to reach a depth of morethan 100 km and has an area on the earth’ssurface of 25 km2. Platinum metals are concen-trated in dunite in the form of striae, lenses, nests,and pillars. They are present mainly as polyxene(Fe – Pt), iridium-rich platinum, and osmiri-dium, and are often associated with serpentineor chromite. Most of the platinum has beenobtained from ores with a platinummetal contentof 10 – 20 ppm. However, ores containing400 ppm are found in some places. In the totaldunite body, the average platinum content is ca.0.1 ppm. Other dunite bodies in this region arenot economically important either for their metalcontent or for the extent of the deposit.
The dunite pipes at the eastern border of theSouth African Bushveld have a similar origin tothe Russian dunite bodies and are of the samebase material. The platinum-bearing core of thisnarrow intrusion often has a diameter of 20 m orless. Veins are rarely present. The excavations atDriekop and Onverwacht are the most wellknown.Mining is difficult. Since the exploitationof the Merensky Reef, they can be mined eco-nomically only in certain cases. Platinum groupmetals are sometimes present in themetallic stateand sometimes as sperrylite in association withchromite. Platinum concentrations are 1 –200 ppm, and locally higher.
The richest and scientifically most interestingdeposits, although small in extent, are the quartzlodes of Rietfontein on the Waterberg in theWestern Cape Province. Owing to their hydro-thermal origin, the platinummetals are present innative form, accompanied by hematite. ThePGMcontent of the ore often reaches almost5000 ppm. Deposits of similar structure have notbeen found elsewhere.
The largest known primary deposit of PGMsis the South African Bushveld Complex, with atotal extent of ca. 250 km from north to south and480 km from east to west. This oval-shaped zonewas apparently produced by an outflow of PGM-bearing norite magma into a flat basin. At thebottom of this, the minerals sperrylite (PtAs2)and cooperite (PtS) have separated along withiron pyrites, nickel pyrites, copper pyrites, and
326 Platinum Group Metals and Compounds Vol. 28

chromite. The relatively thin PGM-bearinglayers at the edges of the outflow near the earth’ssurface aremined. These are in both the oxidationand the sulfidic zones, and begin at a depth of< 100 m. The working depth extends to almost1000 m. The platinum content of themined ore is3 – 20 ppm. Palladium is less important thanplatinum; all the platinum group metals are pres-ent. The deposit has great economic importanceowing to its consistent quality, ease of extraction,and large extent. The Bushveld is mined at thewestern and southwestern edges (Rustenburgregion) and the eastern and northeastern edges(Lydenburg region) (Fig. 3) in two wide areas.The Bushveld is also the world’s largest chromi-um and vanadium deposit, and has large depositsof nickel, cobalt, copper, iron, tin, fluorspar, andalusite, magnesite, and asbestos.
These deposits were discovered in 1925 by thegeologist HANS MERENSKY (1871 – 1952) [17].He laid the foundations for the most importantphase in the development of platinum extractionby his wide-ranging prospecting work and fun-
damental observations on the geology of theBushveld. The most important areas of PGMdeposits in the western and eastern part of theBushveld are known as the Merensky Reef. Notfar from these deposits lie the less importantPlatreef and the UG 2 Reef (Upper Group Reef),which has recently increased in importance dueto its very high rhodium content in many places.This metal is in great demand and therefore veryexpensive.
The UG 2 reef is a chromite layer runningparallel to the Merensky Reef, but somewhatdeeper [66], [67].
Some examples of small deposits include theBlack Reef in the Bushveld and, a few hundredkilometers north, the Great Dyk Mine inZimbabwe.
The Stillwater deposit in Montana, UnitedStates is similar in origin and structure to theBushveld deposit. However, PGM separationoccurred at the boundary zone of a subterraneanmagma intrusion resembling a sloping lens. Thisdeposit extends for a length of ca. 80 km. The
Figure 3. Geology and platinum mines of the Bushveld Complex
Vol. 28 Platinum Group Metals and Compounds 327

total PGM content, with high levels of palladiumand rhodium, is higher than that of the MerenskyReef deposit.
The nickel sulfide deposits of the Sudbury,Ontario district of Canada, yielding iron pyrites(FeS), pentlandite (F – NiS), and copper pyrites(CuFeS2), are today important sources of plati-num, and more especially of palladium. Thesedeposits are associated with noritic magma. Theaverage platinum metal content of the untreatedore is only ca. 0.3 ppm, with a nickel content ofca. 2%. However, the treatment process for thenickel ore concentrates the platinum metals to> 50 ppm without extra cost.
Palladium-containing sperrylite (Pt – PdAs2)and stibiopalladinite (Pd3Sb) are present in theores, and the platinum group metals also formisomorphic mixtures with the heavy-metal sul-fides. During formation of these deposits, thesulfides of nickel and copper acted as collectors.The large reserves in Noril’sk in Central Siberia,which have a high palladium content, are similarto the Canadian deposits with regard to compo-sition and ore treatment methods. Some lead –zinc ores [e.g., in the Rammelsberg (Harz)moun-tains], contain small amounts of platinum andpalladium.
Comparisons with other metals indicate thatPGM reserves may exist in the Antarctic [68].The magma flow in the South African Bushveldoccurred in an early geological epoch (Cambri-an –more than 2�109 years ago), when the Afri-can and Antarctic continents were still part ofGondwanaland.
3.4. Secondary Deposits [166], [167]
Alluvial deposits are typical reserves of this type.They were produced in recent geological epochs(Quaternary – Holocene) by weathering andwashing of primary deposits, mainly dunitic,which resulted in concentration of the moreresistant and heavier components (i.e., the me-tallic and arsenidic platinum minerals, gold,magnetite, chromite, cassiterite, zircon, andgranite) in clay and sand. These oxidation zonesare often located above the primary deposits.River placers are formed from the alluvial de-posits by the erosive action of water, to yield so-called black sands. However, typical nuggets(mainly small granules but also sizable metallic
lumps weighing up to several kilograms) seem tohave been formedmainly bymechanical agglom-eration, usually involving chloridic dissolutionand reprecipitation. Very old deposits thatformed conglomerates by adhesive action aretermed fossil placers. The platinummetal contentof secondary deposits varies over a wide range.Deep alluvium and river headwater placers arethe most productive.
For easily washable sand, a PGM content of0.05 ppm is economic. Before World War I, thePGM content of economic deposits was ca.2 ppm, but today it ismuch lower.More platinummetals are recovered from river placers than fromalluvial deposits.
Secondary deposits occur mainly in the Urals,Siberia, Colombia, and Ethiopia. Often, the re-covery of gold and platinum metals from suchdeposits is so interdependent that sometimes onemetal, and sometimes the other, is the mainproduct. Osmiridium is obtained mainly in Alas-ka and in the Witwatersrand of South Africa,along with secondary deposits of gold.
3.5. Recovery of Secondary PlatinumGroup Metals [69–72]
Platinum metals not only are extracted from orebut,due to their high value, are also recoveredfrom a wide range of industrial residues. Theseresidues are of variable composition and quality,and recovery plants must be very flexible.
Often, the recovery operation is included inthe sales contract for semifinished and finishedgoods, so materials are sent directly for recoveryand do not appear on the raw materials market.For regular customers, accounts are kept of theweights of noble metals involved. The supply ofmaterial is facilitated, and risks due to pricevariation are minimized.
MetallicMaterials. Large quantities of me-tallic materials in the form of used platinum –rhodiumgauze catalysts result from the oxidationof ammonia (! Nitric Acid, Nitrous Acid, andNitrogen Oxides). These catalysts must be re-processed chemically after 3 – 18months of use.At present, ca. 50 t of Pt –Rh is bound up in thesegauzes. The Pd – Au gauzes used to recoverPt – Rh vaporized in ammonia oxidation plantsare also recycled.
328 Platinum Group Metals and Compounds Vol. 28

The glass industry generates large quantitiesof defective components for chemical recovery(e.g., frommelting vessels and other equipment).
Spinnerets from textile fiber manufacturemust normally be replaced after about one yearof operation owing to erosion of the holes.
Defective laboratory equipment, mainly cru-cibles and dishes, makes a considerable contri-bution, but chemical apparatus components areof little significance.
Considerable quantities of platinum – rhodi-um scrap are provided by the electrical measure-ment industry, mainly in thermocouple compo-nents. Other residues from the electrical andelectronic sectors include electrical contacts,heater elements, and electronic components.
Manufacture of fountain pen nibs yields re-sidues in the form of small spheres or dust thatcan contain ruthenium, osmium, iridium, rheni-um, tungsten, molybdenum, tantalum, nickel,and cobalt as alloy components. Treatment ofthese materials is among the most difficult of allseparation techniques.
Dross. Waste materials include slag, ash,furnace residue, corrosion residue from equip-ment, and precipitation residue. Catalyst residuesof poorly defined composition are also included.The PGM content of these materials is usuallylow; they are extremely variable and usuallynonmetallic.
Supported Catalysts [70]. Large quantitiesof platinum groupmetalsmust be recovered fromspent catalysts. Themost important of these, bothin quantity and in value, are the heterogeneouscatalysts used in the petroleum industry, espe-cially in reforming processes, where >50 t ofplatinum is bound up worldwide. The lifetime ofthese catalysts is 4 – 8 years. Residues usuallycontain 0.3 – 0.7% platinum on g-Al2O3, andusually also include palladium, rhodium, iridi-um, and rhenium, which must also be recovered.The other large-scale processes in the petroleumindustry–hydrofining and hydrocracking–yieldpalladium and platinum catalysts on aluminumsilicate carriers.
Another source of material for recovery ofplatinum metals is spent automobile catalyticconverters [69–71]. However, the PGM contentis low (2 g per unit), and the units are enclosed insteel sheet and widely scattered. Collection and
treatment are therefore difficult. A satisfactoryand economical solution to this problem has yetto be found. In Germany, collection logistics arecoupled to the recycling of automobiles.
The chemical industry produces considerableamounts of palladium catalysts on carbon car-riers, often in a moist state that presents samplingproblems. Similar residues come from plati-num – carbon, rhodium – carbon, and PtO2 cat-alysts, although these are sometimes unsupport-ed. Platinum asbestos, which was formerly usedwidely in sulfuric acid production, is now of verylittle importance.
Solutions [69]. Amounts of liquid residuesfrom homogeneous catalysts used in the oxoprocess (hydroformylation) have increased rela-tively rapidly. The rhodium content of the organ-ic solvents or oily process residues is between 50and 1000 ppm. Sometimes, these organic solu-tions contain iridium, ruthenium, or p alladium.Aqueous residues, especially homogeneous cat-alysts containing rhodium, are currently beingproduced in increasing quantities.
The electroplating industry yields exhaustedelectrolytes that cannot be regenerated. The mostimportant of these contain tetranitroplatinate(II),rhodium(III) sulfate, and rhodium(III) phosphate.
Finally, the processes used for separating theplatinum group metals also produce waste solu-tions that must be reclaimed (e.g., mother liquorfrom crystallization). These operations form partof the separation process.
Radioactive Residues. An as-yet unsolvedproblem is the treatment of radioactively con-taminated platinum equipment from chemicallaboratories and processes.
Fission of 235U in nuclear power stationsproduces considerable amounts of platinumgroup metals; one tonne of spent reactor fuelcontains 1.2 kg of palladium, 0.5 kg of rhodium,and 2.3 kg of ruthenium [72].
However, the radioactivity of the material hasnot permitted the commercial use of this PGMsource until now [73]. The most important PGMisotopes in spent reactor fuel are: 107Pd (t1/27 � 106 a), 102Rh (t1/2 3 a), and 106Ru (t1/21 a). 107Pd is a very low-energy b-emitter,which would not exclude its use in many majorapplications; alternatively, it may be removedby isotope separation. The active isotopes of
Vol. 28 Platinum Group Metals and Compounds 329

rhodium and ruthenium will have decayed tobackground levels after intermediate storage ofca. 30 years.
3.6. Reserves and Resources
Natural Reserves. Data concerning re-serves (discovered by prospecting and havingassessable economic value) and resources(which include additional supposed depositsand those with no current economic value) arevery dependent on the time prospecting wascarried out, and also on technical and economicparameters.
For platinum group metals, the current esti-mate of workable deposits is 70 000 t [46], [75],[76], 20 years ago, a figure of about one-third ofthis was assumed [77].
World reserves of the individual metals canbest be estimated from the observed composi-tions of the deposits (Table 4) and the totalamounts of PGMs that they contain (Fig. 4). Thisdoes not include osmiridium, which usually oc-curs with gold.
Industrial Residues. Industrial residueshave considerable potential for the supply ofplatinum group metals, in addition to their ex-traction from ore. In most sectors, the possibili-ties are now fully exploited. In others, recovery isdifficult because of the low PGM contents of thewaste materials (e.g., certain catalysts). Econom-ic recovery is also difficult when small PGM-containing components are widely distributed(e.g., in electronics).
In particular, no satisfactory solution hasbeen found to the problem of collecting used
Table 4. Relative proportions of platinum group metals in selected deposits, and their grades
Bushveld complex
Merensky UG 2 South Africa, Sudbury, Noril’sk, Stillwater,
Reef Reef Plat Reef Canada CIS Colombia United States Average
Platinum, % 59 42 42 38 25 93 19 45
Palladium, % 25 35 46 40 71 1 66.5 30
Ruthenium, % 8 12 4 2.9 1 4.0 5
Rhodium, % 3 8 3 3.3 3 2 7.6 4
Iridium, % 1 2.3 0.8 1.2 3 2.4 1
Osmium, % 0.8 0.6 1.2 1 <1
Gold, % 3.2 0.7 3.4 13.5 0.5
Grade, g/t 8.1 8.71 7 – 27 0.9 3.8 22.3
Figure 4. World platinum group metal reserves (total 70 000 t)
330 Platinum Group Metals and Compounds Vol. 28

automobile exhaust catalysts. In 1991, morethan 7 t of platinum was recovered from thissource in the United States and Europe and about0.5 t of rhodium in the United States [248].
A total of ca. 1 t/a of rhodium is currentlyproduced worldwide in nuclear power stations.Some of this is placed in intermediate storage,and some in final repositories.
4. Mineral Dressing andBeneficiation
4.1. Treatment of Alluvial PlatinumDeposits [5], [10], [19], [30–32], [36],[41], [43–48], [78–86], [166], [167]
Gangue materials must normally be removedfrom the platinum-bearing placer deposits. Somedeposits are extracted by subsurface mining.
Hydraulic classification can be carried out byhand washing with simple equipment, such asshovels, sieves, and troughs. This was the mostcommon method of treating the Colombian de-posits and is still used today. More modernmethods involve rotary sieves, troughs, and per-forated boxes with running water or water jets.
The most economical process is dredgingriver placers. This is a simple earth-movingtechnique linked with a natural inexhaustiblewater supply. Large installations have capacitiesof several thousand tonnes of sedimentary mate-rial per day, with a water consumption of ca. 10times this figure.Aboard the dredger, deposits aretreated mainly by gravity concentration withsieves and sedimentation equipment, using stir-rers, thickeners, washing boxes, etc.
The last stage of concentration is oftenmagnet-ic separation, with various field strengths used toseparate magnetite, ferroplatinum-containingminerals such as chromite, and nonmagnetic com-ponents into fractions. Often, a final hand washingis carried out. In some types of deposit, goldparticles or gold-containing platinum particles canbe separated as amalgam from the platinum con-centrate. Another process involves concentratingthe platinum metals chemically by dissolving theother components in nitric acid. The concentratesso produced can contain up to 90%platinumgroupmetals. These can be used directly by refineries.
A problem may occur with high losses ofplatinum carried out as very fine metal from thegravity separation process, often exceeding the
amount of the product itself. Losses can bereduced to some extent by recycling the lighterfraction during hydraulic classification of theplatinum deposits.
4.2. Treatment of Primary Deposits[46], [56], [66], [67], [86], [87], [166],
[167]
The treatment of platinum-bearing rock, which isalways supplied in lump form from primarydeposits, consists of an initial size reduction bycrushing and grinding, usually wet grinding.
After discovery of the South African primarydeposits in the Merensky Reef, attempts wereinitially made to treat this platinum-bearing rockby the methods used for alluvial deposits. Manyprocesses were investigated including gravityconcentration, flotation, and metallurgical andchemical processes, such as chloride formationby calcining the powdered ore at 500 – 600 �C inthe presence of sodiumchloride. The results wereunsatisfactory.
The modern process for the winning of PGMsfrom sulfide ores is shown in Figure 5. Groundores from the workable oxidation zone, whichcontain the platinum metals in native form, arefirst subjected to gravity concentration on cor-dyroy and James tables or by hydrocyclones, toseparate the metallic particles from the platinum-bearing minerals and give a concentrate with ahigh PGM content that can be processed quicklywith low losses.
Flotation is then carried out to remove thegangue from the sulfidic minerals, which are alsoassociated with arsenidic and sulfidic platinummetal compounds and very finely divided ele-mental platinum metals. This concentrates theplatinum group elements by a factor of 10 – 50.After filtration with a rotary filter, the platinummetals in the flotation concentrate are present at atotal concentration of several hundred parts permillion, along with a small percentage of sulfur,copper, nickel, and iron.
This sequence of process steps is not suitablefor all types of deposits and production equip-ment. Magnetic or electrostatic separation cansometimes be carried out before the smeltingoperations.
The pelletized material is smelted in a shaftfurnace to form a copper – nickel matte. Oxygen
Vol. 28 Platinum Group Metals and Compounds 331

is then blown into the converter to oxidize theiron sulfide selectively to iron oxide,which formsa slag. These two processes concentrate theplatinum metals in the copper – nickel matte to> 0.1%. The blowing operation is controlled soas to give the correct sulfur content for the nextconcentration stage. In this recently developed,
slow-cooling matte separation process, a finelycrystalline, homogeneously distributed Ni –Cu –Fe phase, in which the PGMs are concen-trated in high yield, is formed in the almost iron-free copper sulfide matte phase (see ! Nickel,Section 4.4.1.). The product is ground, and thePGM-containing magnetic Ni – Cu – Fe phaseis recovered from the PGM-free matte by mag-netic separation. Base metals are then removedby treatment with sulfuric acid and oxygen. Theconcentrate obtained contains 50 – 90% PGMs.The next stage is to separate the platinum groupmetals from one another.
The older pyrometallurgical process is timeconsuming and does not give such good separa-tion. After air blowing the iron, the convertermatte produced is smelted with sodium sulfideand separated into copper-containing and nickel-containing layers (tops and bottoms process).The nickel-containing material is roasted andthen reduced in a reverberatory furnace to givePGM-containing impure nickel. The platinumgroup metals are recovered from the anode slimeproduced during electrorefining. A method ofthis kind is apparently still used to treat sulfidicPGM deposits in the Urals.
Problems arise during treatment of the high-chromite ores of the UG 2 reef by the sulfidicroute; these are therefore often mixed with orefrom theMerensky deposit. A process that showsgreat economic promise for the future is to smelta metallic concentrate directly from the ore in aplasma furnace [66]. Both types of ore are suit-able, although the chromite-containing ore isbetter.
4.3. Treatment of NickelOres [63], [64]
In the processing of sulfide nickel ores, whichalways contain copper, the platinum group me-tals follow the nickel in the smelting process.When the crude copper is electrorefined, theamount of PGMs obtained in the anode slime isvery small.
When nickel is electrorefined, the platinumgroup metals remain behind in the slime formedat the nickel anode. The basemetals and the silverare dissolved by acid treatment, giving a concen-trate that contains ca. 70% platinum group me-tals and differs from the nickel anode slimeobtained from platinum ores of the Merensky
Figure 5. Winning of platinum group metals from sulfideores
332 Platinum Group Metals and Compounds Vol. 28

reef in having an appreciably higher palladiumcontent. Also, the concentration of platinumgroup elements in the unrefined nickel is about100 times lower than that in the nickel obtainedfrom platinum ore from the Merensky or UG 2reef (see Section 4.2).
When crude nickel is refined by the carbonylprocess (see ! Nickel, Section 6.3.), residueswith a lower PGM content are obtained. Theseare suitable for concentration by smelting underreducing conditions with lead(II) oxide and so-dium carbonate. The lead is driven off, and thesilver is dissolved and removed. These concen-trates are of high enough quality for separationinto individual metals.
The treatment of PGM-containing copper an-ode slime obtained from nickeliferous pyrrhotite(! Silver, Silver Compounds, and Silver Al-loys) is a long and costly process. The metalscopper, selenium, tellurium, arsenic and antimo-ny must first be removed (e.g., by forming theirsulfates in a high-temperature process), followedby dissolution or by producing slags via smeltingin the presence of potassium nitrate with an airblast. This yields so-called Dor�e metal. Theplatinum group metals are then concentrated inthe anode slime produced in the subsequent silverelectrorefining process (! Silver, Silver Com-pounds, and Silver Alloys).
Copper ores can also contain very smallamounts of platinum group metals. These appearin the copper anode slime during copperelectrorefining.
4.4. Treatment of Metal Scrap [166],
[167]
Ahigh proportion of themetallicwaste fromusedequipment and from semifinished products cansimply by dissolved without any prior treatment(see Chap. 5). These types of material in-cludecrucibles, dishes, thermocouple elements, gauzecatalysts, and fiber spinneret nozzles.
Massivematerials such as heavy-gauge sheetsor bars should be size reduced by crushing ormachining to produce swarf. For the highlyrefractory metals of the platinum group (Rh, Ir,Ru, andOs) and their alloys,mechanicalmethodsare usually not sufficient to produce a surfacesusceptible to dissolution. This is also true ofplatinum alloys with > 30% rhodium or 20%
iridium. In these cases, alloys of platinum orpalladium, which can be dissolved more easily,are preferable.
Very highly dispersed noble-metal black,which is often more soluble, is obtained byalloying the highly refractory platinum metalswith base metals and then dissolving the latterout. The noble-metal blackmust not be heated, orthe optimum surface properties for the solutionprocess would be adversely affected. Suitablealloying elements include copper, lead, nickel,zinc, aluminum, bismuth, and silver.
Commercial powdered rhodium and iridiumalso cannot be dissolved by direct chemicalmeans, but they can be treated with chlorine atca. 500 – 600 �C to form chlorides (also insolu-ble), and these can be reduced at low temperature(e.g., by hydrogen or by hydrazine in aqueoussuspension) to produce finely divided blacks thatcan be dissolved in hydrochloric acid – chlorine.
Concentrates of platinummetals often containoxides that are less soluble in oxidizing acidmixtures than the metals. In these cases, thematerial must first be reduced by heating in ahydrogen atmosphere or by treatingwith aqueoushydrazine hydrate at ca. 80 �C. For rhodiumoxides, solutions must be highly alkaline and attheir boiling point.
4.5. Treatment of Dross [88, 93–96]
If platinum metals cannot be separated by chem-ical or mechanical methods from accompanyingnonmetallic materials, as is usually the case withlow-grade waste, pyrometallurgical processesmust be used (as in ore treatment).
The most convenient and long-establishedpyrometallurgical process for low-grade wasteis smeltingwith lead in a shaft furnace to produceslag. Materials containing silver and gold (!Silver, Silver Compounds, and Silver Alloys;!Gold, Gold Alloys, and Gold Compounds, Sec-tion 6.2.) are treated in thisway. The lead acts as acollector for the platinum group metals. Thepresence of gold and silver also considerablyaffects the distribution equilibrium of the plati-num group metals in the melt. In the lead shaftfurnace, some of the high-melting noble metalsrhodium, iridium, and ruthenium separate as so-called furnace shows. When the lead is oxidizedand removed as litharge, most of the Rh, Ir, and
Vol. 28 Platinum Group Metals and Compounds 333

Ru (so-called bottom metals) precipitates fromthe increasingly silver-rich alloys. Considerableamounts of rhodium and iridium pass into theshaft furnace slag and are lost. Ruthenium andparticularly osmium are lost in large amounts influe dust and waste gases.
When the gold – silver alloy from the aboveprocess is electrorefined to obtain silver, gold andthe platinum group metals remain in the anodeslime. If this consists mainly of gold, it is con-verted to pure gold by Wohlwill electrolysis(! Gold, Gold Alloys, and Gold Compounds,Section 5.3.), in which platinum and palladiumare concentrated in the electrolyte, and silverchloride and the remaining platinum group me-tals in the anode slime. Alternatively, the silvercan be dissolved from the gold – silver alloy bynitric acid to form silver nitrate, which is purifiedby thermal decomposition of the accompanyingnitrates; the platinum group metals remain in thewater-insoluble oxide residue.
All the concentrates mentioned above can bedissolved, and the individual platinum groupmetals obtained from these solutions.
4.6. Treatment of Supported Catalysts[70], [71], [97–100]
Spent, inactive catalysts consisting of platinummetals supported on active carbon, or carrier-freenoble-metal catalysts that have become coatedwith organic residues, are concentrated by com-bustion. These materials are sometimes sponta-neously flammable.When they are being burned,strong air currents are suppressed to prevent dustlosses. If the resulting ash contains platinummetal oxides, these are reduced to the metal.
Catalysts with incombustible carriers insolu-ble in acid and alkali (e.g., g-Al2O3, silica gel,asbestos, and zeolites) can often be treated withoxidizing acid, but the noble metals dissolvecompletely only in the absence of organic resi-dues (especially tarry matter) and if the carriersare very porous. Otherwise, the platinum metalsmust be concentrated by the lead shaft furnaceprocess (see Section 4.5).
Reforming catalysts consist of g-Al2O3 impreg-nated with platinum, platinum – rhodium, plati-num – iridium, or platinum – rhenium. This carri-er material is soluble in acid and alkali, and isdissolved (e.g., in hot sulfuric acid or hot caustic
soda solution) leaving the noble metal as an insol-uble residue. However, small, but not negligible,amounts of noble metal also go into solution.Moreover, sulfidic impurities in the catalyst canlead to the release of toxic hydrogen sulfide. Otherpractical processes are dissolving the catalyst insodium carbonate solution (usually at 220 �C in apressurized reactor), or sintering with sodium hy-droxide or sodium carbonate. Carbon and otherproducts of the breakdown of mineral oil in thespent catalyst must be burned off before treatmentwith acidor alkali, because thesematerials interferewith the filtering of the platinummetal concentrate.
Spent automobile exhaust catalysts can beprocessed at high temperature in a plasma or asubmerged arc furnace. In the former, a plasma isproduced between an electrode and the moltenfeedmaterial. The energy of recombination of theplasma is released into the melt. In the resistancefurnace, slag serves as the electrical resistance inwhich heat is produced. In both processes, theoxide carrier is melted with or without addition ofa flux of lower the melting point. Iron or copper isadded as a collector, forming a metallic melt thattakes up the platinum group metals. The concen-tration of PGMs can reach 20%.
4.7. Treatment of Solutions [69], [101],[102]
In homogeneous catalysis, high-boiling distilla-tion residues are usually produced that contain novaluablematerials apart from rhodium and some-times ruthenium. These residues can be carefullyburned, and the ash treated by wet chemicalprocesses. Other techniques for recovering rho-dium have been suggested (i.e., liquid – liquidextraction, reductive precipitation of the metal,and pyrolytic hydrogenation). A process used inindustry, especially for the treatment of rhodium-containing oily residues from oxo synthesis, isprecipitation of acid-soluble rhodium tellurideby reacting the organically bound rhodium withtellurium. This process is notable for the highefficiency of rhodium recovery.
The methods used to produce concentratesfrom a variety of aqueous wastes are to a largeextent the same as those used for the internalrecycling of platinum metals in the solutionsproduced in metal winning processes (see Sec-tion 6.8).
334 Platinum Group Metals and Compounds Vol. 28
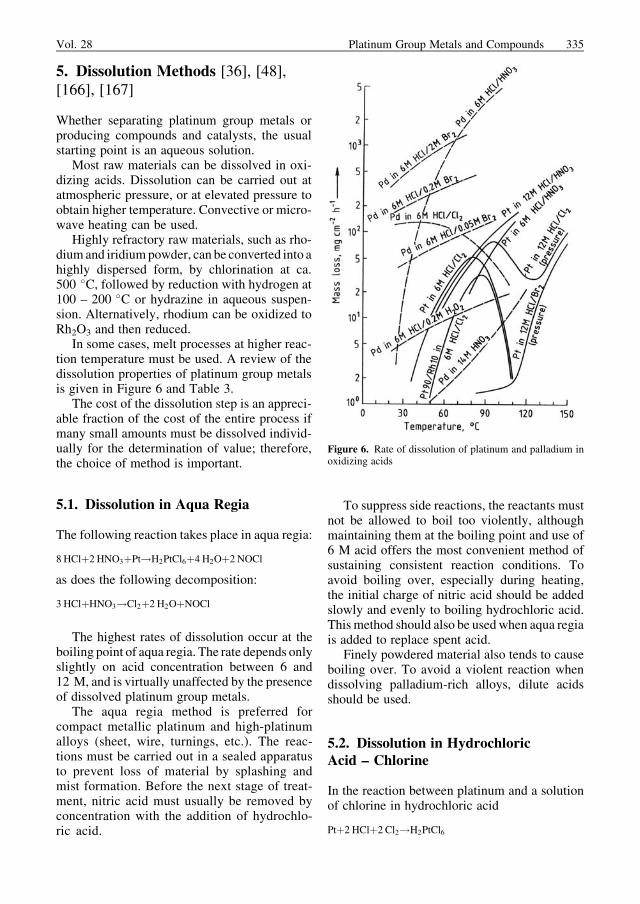
5. Dissolution Methods [36], [48],[166], [167]
Whether separating platinum group metals orproducing compounds and catalysts, the usualstarting point is an aqueous solution.
Most raw materials can be dissolved in oxi-dizing acids. Dissolution can be carried out atatmospheric pressure, or at elevated pressure toobtain higher temperature. Convective or micro-wave heating can be used.
Highly refractory raw materials, such as rho-diumand iridiumpowder, can be converted into ahighly dispersed form, by chlorination at ca.500 �C, followed by reduction with hydrogen at100 – 200 �C or hydrazine in aqueous suspen-sion. Alternatively, rhodium can be oxidized toRh2O3 and then reduced.
In some cases, melt processes at higher reac-tion temperature must be used. A review of thedissolution properties of platinum group metalsis given in Figure 6 and Table 3.
The cost of the dissolution step is an appreci-able fraction of the cost of the entire process ifmany small amounts must be dissolved individ-ually for the determination of value; therefore,the choice of method is important.
5.1. Dissolution in Aqua Regia
The following reaction takes place in aqua regia:
8 HClþ2 HNO3þPt!H2PtCl6þ4 H2Oþ2 NOCl
as does the following decomposition:
3 HClþHNO3!Cl2þ2 H2OþNOCl
The highest rates of dissolution occur at theboiling point of aqua regia. The rate depends onlyslightly on acid concentration between 6 and12 M, and is virtually unaffected by the presenceof dissolved platinum group metals.
The aqua regia method is preferred forcompact metallic platinum and high-platinumalloys (sheet, wire, turnings, etc.). The reac-tions must be carried out in a sealed apparatusto prevent loss of material by splashing andmist formation. Before the next stage of treat-ment, nitric acid must usually be removed byconcentration with the addition of hydrochlo-ric acid.
To suppress side reactions, the reactants mustnot be allowed to boil too violently, althoughmaintaining them at the boiling point and use of6 M acid offers the most convenient method ofsustaining consistent reaction conditions. Toavoid boiling over, especially during heating,the initial charge of nitric acid should be addedslowly and evenly to boiling hydrochloric acid.This method should also be usedwhen aqua regiais added to replace spent acid.
Finely powdered material also tends to causeboiling over. To avoid a violent reaction whendissolving palladium-rich alloys, dilute acidsshould be used.
5.2. Dissolution in HydrochloricAcid – Chlorine
In the reaction between platinum and a solutionof chlorine in hydrochloric acid
Ptþ2 HClþ2 Cl2!H2PtCl6
Figure 6. Rate of dissolution of platinum and palladium inoxidizing acids
Vol. 28 Platinum Group Metals and Compounds 335

the dissolution rates for platinum and platinum-rich alloys have a maximum in the range 80 –90 �C (see Fig. 6). As the boiling point of hydro-chloric acid (110 �C) is approached, the rate ofreaction decreases rapidly, increasing again athigher temperature. For palladium, the rate ofdissolution is considerably higher.
For these dissolution reactions, 6 – 8 M hy-drochloric acid is preferred since, in this con-centration range, both the amount of acid con-sumed and the amount of hydrogen chloride inthe waste gas are lower than if more concentrat-ed hydrochloric acid is used. The presence ofdissolved platinummetals does not decrease therate of dissolution. By careful control of addi-tion rates, a very slight excess of chlorine can beused, so that this method leads to lower levels ofwaste gas contamination than the aqua regiamethod. However, the rates of dissolution de-crease considerably in the absence of agitation,and the process is most suitable for easily stirredpowders, slurries, or concentrates and unsuit-able for bulk material.
Metal dissolves more slowly in the vaporphase or in refluxing hydrochloric acid contain-ing dissolved chlorine (see Fig. 6).
Hydrogen peroxide can be used in place ofchlorine as oxidizing agent. Optimum concen-trations are difficult to maintain owing to thedecomposition of hydrogen peroxide into oxygenand water; therefore, reaction rates are very lowcompared with those for HCl – Cl2. The HCl –H2O2 system has some limited use for palladium.
5.3. Dissolution in HydrochloricAcid – Bromine
Palladiumdissolvesmore rapidly in hydrochloricacid – bromine mixtures than in hydrochloricacid – chlorine (see Fig. 6). Bromine is moresoluble than chlorine in hydrochloric acid, en-abling high halogen concentrations to be used. Ifthe temperature is kept well below the boilingpoint of bromine (59 �C), losses in the wastegases become very small. Bromide formed in thedissolution reaction can be oxidized to bromineby chlorine in an easily controllable reaction,recovered by distillation, and recycled. Since theconversion of bromide to bromine
2 Br�þCl2!Br2þ2 Cl�
or
H2PtBr6þ3 Cl2!H2PtCl6þ3 Br2
can take place in parallel with the dissolutionprocess, bromine can be present in substoichio-metric amounts.
Platinum and its alloys with rhodium or iridi-um are more resistant to hydrochloric acid –bromine than to hydrochloric acid – chlorine.
5.4. Other Dissolution Processes
Concentrated nitric acid is suitable for dissolvingpalladium (see Fig. 6).
Concentrated sulfuric acid dissolves finelydivided rhodium at ca. 300 �C, but stronglyheated rhodium powder is largely insoluble insulfuric acid.
Concentrated hydrobromic acid is the onlyreagent that can directly dissolve oxides of theplatinum group metals such as PdO, PtO2,Rh2O3, and IrO2. Platinum can be dissolved ina mixture of hydrobromic acid and bromineabove 100 �C in a pressurized reactor. Metallicrhodium is quite soluble in concentrated hydro-bromic acid near its boiling point.
Powdered ruthenium or osmium can be trea-ted with an alkaline solution of potassium per-oxodisulfate (K2S2O8) to form solutions of ruthe-nate or osmate. In acidic media, osmium is oxi-dized by peroxodisulfate or chromic acid to formOsO4.
Perchloric acid has been reported to be asolvent for platinum and its alloys, ruthenium,and osmium.
5.5. Dissolution by Salt Fusion [34]
Aqueous dissolution processes are not successfulfor all types of concentrates and raw materials.Often, reactions in molten salts are more effec-tive. For many raw materials, molten salt meth-ods are selective and therefore also suitable forseparation.
Rhodium can be converted into water-solublerhodium(III) sulfate at ca. 600 �C by meltingwith potassium or sodium hydrogensulfate,which is converted to the pyrosulfate with lossof water. Platinum, iridium, and ruthenium arenot attacked.
336 Platinum Group Metals and Compounds Vol. 28

Ruthenium is converted into water-solubleK2[RuO4] when reacted with KOH – KNO3.Osmium reacts similarly. This reaction can beused to treat finely divided osmiridium, especial-ly after preliminary treatment with molten zinc,in which the iridium remains undissolved (seealso Section 4.4). Potassium salts are more ef-fective than sodium salts. Alkali-metal carbo-nates can be used instead of alkali-metalhydroxides.
Ruthenium and osmium are very readily at-tacked by molten Na2O2. The reaction can beprevented from becoming too violent by additionof NaOH or Na2CO3 to reduce the melt tempera-ture. Iridium can be oxidized to acid-solubleiridate by fusion with Na2O2.
Iridium can also be converted to acid-solubleiridate by treatment with fused BaO2 or BaO2 –Ba(NO3)2. This has the advantage of enabling thecation to subsequently be removed simply byprecipitation as BaSO4. The process is also usedto treat osmiridium, although osmium is con-verted to OsO4 and must be recovered from thewaste gas.
When mixtures of powdered platinum groupmetals are heated at 700 �C in a chlorine atmo-sphere, acid-soluble chlorides of palladium andplatinum, and acid-insoluble RhCl3, are formed.Chlorination of iridium and ruthenium producessubstoichiometric acid-insoluble products.
All of the platinum group metals form water-soluble complex chlorides when treated withsodium chloride in a chlorine atmosphere.
6. Separation of Platinum GroupMetals [31], [103], [263]
After the platinum group metals have been dis-solved, the individual metals must be recovered.Depending on the raw material, the solution maycontain all or some of the PGMs, together withgold, silver, and base metals.
Over the past 200 years, numerous separationprocesses have been developed and used [31],[32], [36], [41]. Many of the individual steps arestill very important in modern processes. Othersare of historical interest, but may be used inspecial cases to treat very small quantities. Inaddition to the large-scale separations carried outby mining companies and refineries a number ofsmall companies exist.
In general, coarse separation is followed by apurification stage. The process used for coarseseparation is determined largely by the compo-sition of the starting solution, and the purificationprocess depends on the particular PGM. Thepurification stage is necessary because, with fewexceptions, an individual platinum group metalof commercially acceptable purity cannot beisolated from complex solutions in a single step.
In practice, the composition of the raw mate-rial changes frequently and profoundly, especial-ly in the recovery of secondary metal. Here, thecrude separation process must be very flexible. Inseparation plants that are parts of mining opera-tions, the rawmaterial is usually fairly consistentover a long period of time.
The most important separation processes to-day make use of various combinations of precip-itation, crystallization, solvent extraction, anddistillation.
To achieve the necessary performance eco-nomically, a high selectivity, a good yield ofnoblemetal, and (tominimize loss of the interest-earning potential of high-value materials) a shorttreatment time are required.
6.1. Chemistry of Platinum GroupMetal Separation [104–115]
Of the many known reactions in PGM chemistry,certain ones have been developed to separate themetals or have been shown to be especially usefulfor this purpose. This is true both for crudeseparation and for purification; in many casesthe same reaction is equally important in bothstages.
Dissolution Properties. Differences in re-activity toward chemical reagents, especiallyoxidizing acids and molten salts, can often beutilized for coarse separation of platinum groupmetals. Good separation is possible only if plati-num and palladium do not form mixed crystalswith the refractory PGMs.
Solution Equilibria. In the crystallizationprocess (by either evaporation or precipitation),the solubilities of chloro complexes of the plati-num group metals are very important, as is thepossibility of altering these solubilities. Thetemperature dependence of these solubilities is
Vol. 28 Platinum Group Metals and Compounds 337
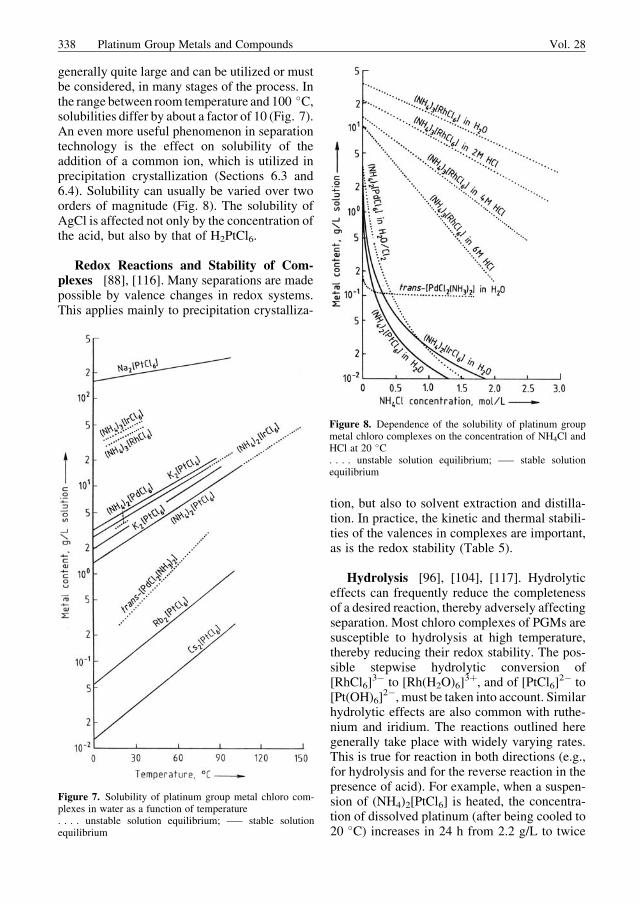
generally quite large and can be utilized or mustbe considered, in many stages of the process. Inthe range between room temperature and 100 �C,solubilities differ by about a factor of 10 (Fig. 7).An even more useful phenomenon in separationtechnology is the effect on solubility of theaddition of a common ion, which is utilized inprecipitation crystallization (Sections 6.3 and6.4). Solubility can usually be varied over twoorders of magnitude (Fig. 8). The solubility ofAgCl is affected not only by the concentration ofthe acid, but also by that of H2PtCl6.
Redox Reactions and Stability of Com-plexes [88], [116]. Many separations are madepossible by valence changes in redox systems.This applies mainly to precipitation crystalliza-
tion, but also to solvent extraction and distilla-tion. In practice, the kinetic and thermal stabili-ties of the valences in complexes are important,as is the redox stability (Table 5).
Hydrolysis [96], [104], [117]. Hydrolyticeffects can frequently reduce the completenessof a desired reaction, thereby adversely affectingseparation. Most chloro complexes of PGMs aresusceptible to hydrolysis at high temperature,thereby reducing their redox stability. The pos-sible stepwise hydrolytic conversion of[RhCl6]
3� to [Rh(H2O)6]3þ, and of [PtCl6]
2� to[Pt(OH)6]
2�, must be taken into account. Similarhydrolytic effects are also common with ruthe-nium and iridium. The reactions outlined heregenerally take place with widely varying rates.This is true for reaction in both directions (e.g.,for hydrolysis and for the reverse reaction in thepresence of acid). For example, when a suspen-sion of (NH4)2[PtCl6] is heated, the concentra-tion of dissolved platinum (after being cooled to20 �C) increases in 24 h from 2.2 g/L to twice
Figure 7. Solubility of platinum group metal chloro com-plexes in water as a function of temperature. . . . unstable solution equilibrium; —– stable solutionequilibrium
Figure 8. Dependence of the solubility of platinum groupmetal chloro complexes on the concentration of NH4Cl andHCl at 20 �C. . . . unstable solution equilibrium; —– stable solutionequilibrium
338 Platinum Group Metals and Compounds Vol. 28

this amount, and after 96 h it increases fourfold.This is due to partial hydrolysis to give solublecompounds. The reverse reaction, brought aboutby heating with hydrochloric acid at its azeotro-pic concentration, occurs at similar rates.
Ion Exchange Chromatography. The useof ion exchange is limited mainly to the separa-tion of PGMs from base metals [118]. For exam-ple, selective adsorption of [PtCl6]
2� on stronglybasic anion-exchange resins is possible, followedby elutionwith sodiumhydroxide solution, yield-ing [Pt(OH)6]
2�. The separation of a PGM mix-ture into individual PGMs by chromatography isdescribed in [119], [120].
Organic Precipitation Agents [121], [122].Palladium can be selectively precipitated by di-methylglyoxime, but the precipitate is difficult tofilter. Separation systems have also been reportedthat depend on the formation of complexes of thePGMwith substituted formazanes and substitutedbenzoylthiourea. Precipitable organometalliccompounds can often be recovered by solventextraction instead of precipitation.
Distillation and Sublimation. The only in-dustrial use of distillation or sublimation at pres-ent is the separation of RuO4 and OsO4, usuallyby steam distillation. These substances have highvapor pressures even at room temperature, so anoncondensable carrier gas cannot be used be-cause of the high carryover of material. A possi-ble future process is the separation of PGM byusing sublimation of PGM fluorides [123].
Reduction toMetal. Since the PGMs are allelectrochemically noble metals, selective reduc-tion and cementation by base metals are notpossible. In the past, collective cementation from
aqueous solution by zinc was often an importantstep, both for separation and for the recovery ofPGMs from recycled solutions. However, the useof zinc as a cementation agent is now ruled out forenvironmental reasons. Where cementation can-not be avoided, iron, aluminum, or Fe – Al alloyscan be used instead. In aqueous media, hydra-zine, formate, or boranate can be used to reduc-tively precipitate elemental PGMs.
Solvent Extraction [124–133]. Many liq-uid – liquid extraction systems have been de-scribed that can be used for solvent extractionof metals. Much research into the platinum groupmetals has been carried out, mainly aimed at theirseparation. The extraction processes do not usu-ally involve a true Nernst distribution of theextractable compounds between the organic andthe aqueous phases. More often, the extractablecompounds (generally organometallic) areformed in the extraction system itself. The re-actants are dissolved in an inert organic phase or,if they are themselves liquids, can be used assuch. The system is generally diluted with aninert solvent to lower the viscosity.
The extractants and extraction mechanismsare classified in the following groups:
1. Compound formation2. Anion exchange3. Cation exchange4. Solvation5. Solvent extraction without reaction
Of the many systems investigated, very feware of practical use.
The organic phase often increases significant-ly in viscosity as metal content increases and isusually diluted by a hydrocarbon mixture in the150 – 200 �C bp range.
Table 5. Stability of platinum group metal chloro complexes
Metal Oxidation state Complex Redox stability Kinetic stability Thermal stability
Pt IV [PtCl6]2� stable very stable very stable
Pt II [PtCl4]2� unstable unstable
Pd IV [PdCl6]2� unstable stable unstable
Pd II [PdCl4]2� stable very stable
Ir IV [IrCl6]2� stable stable
Ir III [IrCl6]3� stable stable
Rh III [RhCl6]3� stable stable unstable
Ru IV [RuCl6]2� unstable unstable
Vol. 28 Platinum Group Metals and Compounds 339

The extracted metal is stripped from the or-ganic phase by an aqueous phase.
6.2. Older Separation Processes [31]
Some separation methods, shown in Figures 9,10, 11, were commonly used for separatingPGMs in earlier times. Modifications of these
may still be used today or included with moremodern individual stages in an integrated pro-cess. In special cases, they can contribute to thesolution of separation problems.
Separation Process A (Fig. 9) [36], [43].The process used by INCO in the Acton refineryin England has been known for a long time. It isespecially suitable for treatment of the anodeslime from the electrolysis of nickel. Here, se-lective dissolution of Pt – Pd can be achievedFigure 9. Older separation process A
Figure 10. Older separation process B (Siebert)
340 Platinum Group Metals and Compounds Vol. 28

since the refractory PGMs are concentrated inseparate particles. Some special process steps areused, including alloying with lead, and selectivedissolution of rhodium, rutheniumand iridiumbysalt fusion.
Separation Process B (Fig. 10). A processformerly used by the platinum smelting companySiebert/Degussa is characterized by the recyclingof unacceptably large amounts of platinumdue tothe relatively high solubility of sodium chloro-
platinate. Recovery of the rarer platinum groupmetals by cementation led to serious pollution ofthe wastewater. The cemented metals were usu-ally dissolved with NaCl – Cl2. An advantage ofthe process was the efficient separation ofiridium.
Separation ProcessC (Fig. 11) [36], [134],[135] is suitable only for raw materials with lowpalladium, rhodium, and iridium content, be-cause of the difficulty of handling their hydrox-ide precipitates, which have adsorptive proper-ties. It has been used only in small separationplants for secondary metal and yields very pureplatinum.
6.3. Current Separation Processes
Modern separation processes (see Figs. 12, 13,14) are designed for high separation efficiency,minimum recycle, reduced holdup times of ex-pensive PGMs, and minimization of losses ofnoblemetals. Ecological considerations are oftencrucial, even when costs must be kept low. Thus,cementation on zinc, an extremely expensiveprocess even if the cost of necessary wastewatertreatment is excluded, has to a very large extentbeen eliminated from PGM separation processesfor environmental reasons.
Separation Process D (Fig. 12) [31], [88].All process steps can be carried out on the samesolution by using simple, similar process opera-tions, which is economically beneficial. Theprocess is very flexible, which is an advantage,particularly for the recovery of secondary metalwhen the ratio of PGMs changes continuously. Ifthe ruthenium content is high, RuO4 should beremoved by distillation as the first step of theseparation process.
Separation Process E (Fig. 13). Solventextraction with dialkyl sulfide is used. Unlikeprocess D, this provides a practically quantitativeand sharp separation of palladium, which yieldsfavorable conditions for the purification of pal-ladium and platinum. Solvent extraction of iridi-um can be carried out almost quantitatively and isvery useful when iridium content is low. Underthese conditions, use of precipitation crystalliza-tion would result in excessive amounts of iridium
Figure 11. Older separation process C (Gilchrist)
Vol. 28 Platinum Group Metals and Compounds 341

remaining as soluble (NH4)2[IrCl6] in the motherliquor.
Separation Process F (Fig. 14) [136]. Aprocess used mainly in mining operations (i.e.,with primary raw materials) has, as its mostnotable feature, solvent extraction of platinum,which is the metal present in highest concentra-tion. Oximes are better than thioethers for solvent
extraction; the higher rate of complex formationenables column technology to be used. Also, theseparation process can be set up as an integratedunit. Ion exchange, which is also carried out oncolumns, results only in separation of basemetals.
Similar separation processes based on solventextraction are in industrial use or development[129–133], [137–143].
Figure 12. Modern separation process DFigure 13. Modern separation process E
342 Platinum Group Metals and Compounds Vol. 28

6.4. Processes Used in CoarseSeparation [31]
Separation of Silver. Insoluble silver chlo-ride is formed in the dissolution process and isremoved by careful filtration from the startingsolution in concentrated hydrochloric acid. How-ever, this acid at its usual concentration leads tounacceptably high levels of silver remaining in
solution due to H[AgCl2] formation. Loweringthe free hydrochloric acid concentration, prefer-ably by evaporation and dilution, considerablyreduces the solubility of silver chloride. Thesettling rate and ease of filtration are improvedby flocculating agents.
Traces of silver (usually together with Cu andPd) can be extracted from chloride solutions ofplatinum group metals at pH 5.5 by dithizone inchloroform.
Separation of Gold. The usual process forgold separation involves reducing Au (III) toelemental gold in acid solution. Reducing agentsthat can be used include iron(II) salts, oxalic acid,sulfur dioxide, and ascorbic acid. Sodium nitrite,hydrogen peroxide, sodium formate, and ethylalcohol are also used [133]. The separation pro-cess is improved if reducing agents and reactionconditions are chosen such that Pt (IV) is notreduced to Pt (II). Otherwise, Pt (II) must bereoxidized to Pt (IV). The separation of goldfrom PGMs is becoming increasingly important[143].
Selective Dissolution Reactions and MeltReactions. Dissolution processes usually donot give very sharp separation of platinum groupmetals.
Problems with salt fusion lead to poor diffu-sion conditions. In dry chlorination of platinumgroup metals, the chlorides are thermodynami-cally stable over only a small range. Also, inreaction of the metals with sodium chloride andchlorine, temperatures must be chosen such thatunreacted metal particles are not blocked bymolten salt. In practice, achieving complete re-action in a single stage is usually impossible.
Separation of Platinum, Iridium, and Pal-ladium by Precipitation Crystallization[144–146]. The same basic process has been usedfor the separation of the largest quantities of plati-numgroupmetals since thebeginningof separationtechnology–precipitation crystallization of ammo-nium hexachloro complexes (NH4)2[MCl6]. Thisprocess can be optimized in many ways by influ-encing solubilities (e.g., by valence changes, addi-tion of a common ion, or changing the temperatureand rate of precipitation).
The first step in separation is usually to pre-cipitate platinum as (NH4)2[PtCl6]. If the
Figure 14. Modern separation process F (Matthey Rusten-burg refiners)
Vol. 28 Platinum Group Metals and Compounds 343

dissolved platinum is present as PtII, it must firstbe oxidized to PtIV (e.g., by chlorine). Excessdissolved chlorine is driven off by boiling,which also causes the palladium that had beenoxidized to palladium(IV) to be reduced topalladium(II). Any iridium(IV) present must beselectively reduced to iridium(III), for example,with iron(II) salts or ascorbic acid. The ammo-nium hexachloroplatinate(IV) is precipitatedpreferably by slow addition of concentratedammonium chloride solution at room tempera-ture with stirring. The hydrochloric acid con-centration should be 1 M, and the platinumcontent should be 50 – 200 g/L. After precipi-tation, the excess of NH4Cl in the solutionshould be at least 0.5 mol/L. The mother liquoris removed from the salt by washing with semi-saturated cold ammonium chloride solution.The deep-yellow ammonium hexachloroplati-nate(IV) is recovered with a purity of 99.0 –99.5% in > 99% yield.
Attempts have been made to precipitate the(NH4)2[PtCl6] slowly by gradual formation ofNHþ
4 ions in the reaction medium (e.g., byhydrolysis of urea or hexamethylenetetramine).In this way, an almost ideally homogeneousprecipitation medium is produced. However,the purities and yields achieved do not justifythe cost.
To precipitate iridium as (NH4)2[IrCl6], iridi-um(III) is converted to iridium(IV) by treatmentwith an oxidizing agent at ca. 100 �C, whilepalladium remains in the divalent state at thistemperature. Black (NH4)2[IrCl6] is filteredoff from the cold solution. It is more readilysoluble than the corresponding platinum salt, andtherefore sufficient excess of NH4Cl (0.5 –1.0 mol/L) and good cooling (20 �C) must beused, and the minimum amount of concentratedNH4Cl solution must be used for washing.Chlorine is then passed into the solution at roomtemperature, the excess NH4Cl in solution being> 1 mol/L, and palladium is precipitated in thepure state in the form of the sparingly soluble,brick-red ammonium hexachloropalladate(IV),(NH4)2[PdCl6]. The separation from rhodium(III) is, however, not sharp.
The main problem encountered in this precip-itation is the difficulty of producing sufficientlylarge crystals for filtration, which can be favoredby adding the palladium solution to the NH4Clsolution. High temperature favors crystal growth
but must be avoided to prevent the followingreaction from occurring:
A virtually quantitative yield is obtained if thecrystals are isolated quickly and washed with acold concentration NH4Cl solution saturatedwith Cl2.
The same basic separation process can becarried out by using potassium chloride insteadof ammonium chloride. Using potassium chlo-ride has advantages and disadvantages:
. As the solubilities of potassium hexa chlorosalts are lower than those of correspondingammonium salts, separation yields are better
. Precipitation of (NH4)2 [PdCl6] by passingchlorine through an NHþ
4 -containing solutionis dangerous, because explosive nitrogentrichloride is formed. This can be oxided byusing potassium chloride [147]
. Potassium ions are introduced into the purifi-cation processes (see below) and are moredifficult to remove than immonium ions
Separation of Rhodium. Ammonium hex-achlororhodate, (NH4)3[RhCl6], can be isolatedfrom rhodium solutions in hydrochloric acid suchas those obtained after removal of the otherplatinum group metals. Ammonium chloride isadded, followed by concentration and crystalli-zation.Much of the remaining (NH4)2[PtCl6] and(NH4)2[IrCl6] is precipitated at the same time.Chlorides of the base metals mostly remain insolution. The (NH4)3[RhCl6] can be selectivelyredissolved in water at room temperature to forman almost saturated solution. This gives a purifiedsolution of rhodium. Both (NH4)2[PtCl6] and(NH4)2[IrCl6] are recovered by filtration.
The rhodium can also be isolated as chloro-pentamminerhodium(III) chloride, [RhCl(NH3)5]Cl2, or potassium hexanitrorhodate(III),K3 [Rh(NO2)6], but the precipitations must becarried out in weakly alkaline solution. Thesemethods are unsuitable for solutions that containlarge quantities of base metals because, underreaction conditions, these form insoluble hydro-xides that are difficult to filter.
Cementation of rhodium by other metals (seeSection 6.3) can also be carried out. However, allof the noble metals and the copper remain with
344 Platinum Group Metals and Compounds Vol. 28

the rhodium.An advantage is that precipitation ofrhodium is complete in this process.
Distillation of Ruthenium(VIII) Oxide.The most important industrial process for theisolation of ruthenium is distillation of the veryvolatile ruthenium(VIII) oxide, RuO4, fromaqueous solution. This compound is formed insolutions of potassium ruthenate(VI), K2[RuO4],and hexachlororuthenate(III), K3 [RuCl6], byoxidation with chlorine at around neutral pH orwith sodium chlorate at pH 0 – 3. Oxidation canalso be carried out in dilute sulfuric acid solutionwith potassium permanganate, or with potassiumchlorate in sulfuric acid solution, but these meth-ods are less satisfactory for safety reasons. Thevolatile RuO4 is absorbed by dilute hydrochloricacid, and is converted into water-soluble chlor-oruthenate complexes.
Even in the absence of potassium permanga-nate, chlorate, or their reaction products, thedistillation of RuO4 is hazardous. The presenceof NHþ
4 salts can lead to the formation ofexplosive chlorides of nitrogen. Therefore, re-moving ruthenium at the start of a separationprocess is often preferable. The formation ofchlorine dioxide, chlorate, and perchlorate isalso possible. Moreover, RuO4 can explode byspontaneous decomposition, especially at> 100 �C and on contact with organic sub-stances. To improve safety, work should becarried out under an inert gas and by avoidingthe presence of large quantities (e.g., using acontinuous process).
Distillation of Osmium(VIII) Oxide. As arule, osmium is present in only a few primary andsecondary raw materials. Osmium should, ifpossible, be isolated as the first step of a separa-tion process, so that later operations will not leadto loss of volatile OsO4.
Solutions obtained from digestion of oresnormally contain osmates. Treatment of solu-tions in dilute sulfuric acid with oxidizing agentssuch as CrO3, K2S2O8, or HNO3 causes osmium(VIII) oxide to be formed and to distill off. Fromthis distillate, OsO4 can be isolated directly as awater-insoluble oil (mp 40 �C). Alternatively, itcan be absorbed in potassiumhydroxide solution,forming K2OsO4, or can be converted to metallicosmium by reaction with formaldehyde solution.Owing to the toxicity of OsO4, its escape into the
atmosphere must be prevented by use of the bestpossible absorption equipment. No danger ofexplosion exists.
Also, OsO4 can be formed and distilled off byheating fine osmium powder in a stream ofoxygen.
SolventExtraction. Liquid – liquid extrac-tion is often characterized by distribution coeffi-cients that vary greatly from element to element.These can usually be modified by chemical meth-ods to give conditions favorable for the separationof PGMs. However, the separation factors result-ing from the ratios of the distribution coefficientsrarely enable a single separation stage to providepurities that fulfill modern requirements formetalquality. Although separations are generally betterthan those effected by precipitation crystalliza-tion, coarse separation by solvent extraction mustbe followed by purification.
In practical industrial separation processes,mainly four extractants are used [109],[130–133], [143–146]. The most long-standingprocess is the separation of Pt (IV) andIr (IV) from Rh (III) with tributyl phosphate(TBP) [148], [149]. The extraction of H2[PtCl6]andH2[IrCl6]must be carried out in 4 – 6 MHCl(Fig. 15). If the usual mixture of 1 part TBP with
Figure 15. Extraction of platinum group metal chloro com-plexes by tributyl phosphate
Vol. 28 Platinum Group Metals and Compounds 345
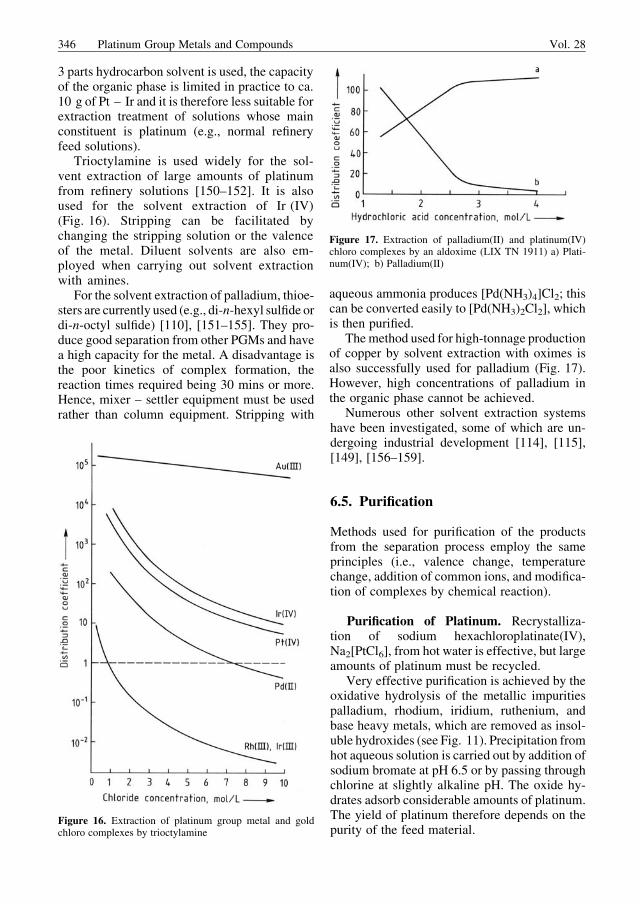
3 parts hydrocarbon solvent is used, the capacityof the organic phase is limited in practice to ca.10 g of Pt – Ir and it is therefore less suitable forextraction treatment of solutions whose mainconstituent is platinum (e.g., normal refineryfeed solutions).
Trioctylamine is used widely for the sol-vent extraction of large amounts of platinumfrom refinery solutions [150–152]. It is alsoused for the solvent extraction of Ir (IV)(Fig. 16). Stripping can be facilitated bychanging the stripping solution or the valenceof the metal. Diluent solvents are also em-ployed when carrying out solvent extractionwith amines.
For the solvent extraction of palladium, thioe-sters are currently used (e.g., di-n-hexyl sulfide ordi-n-octyl sulfide) [110], [151–155]. They pro-duce good separation from other PGMs and havea high capacity for the metal. A disadvantage isthe poor kinetics of complex formation, thereaction times required being 30 mins or more.Hence, mixer – settler equipment must be usedrather than column equipment. Stripping with
aqueous ammonia produces [Pd(NH3)4]Cl2; thiscan be converted easily to [Pd(NH3)2Cl2], whichis then purified.
Themethod used for high-tonnage productionof copper by solvent extraction with oximes isalso successfully used for palladium (Fig. 17).However, high concentrations of palladium inthe organic phase cannot be achieved.
Numerous other solvent extraction systemshave been investigated, some of which are un-dergoing industrial development [114], [115],[149], [156–159].
6.5. Purification
Methods used for purification of the productsfrom the separation process employ the sameprinciples (i.e., valence change, temperaturechange, addition of common ions, and modifica-tion of complexes by chemical reaction).
Purification of Platinum. Recrystalliza-tion of sodium hexachloroplatinate(IV),Na2[PtCl6], from hot water is effective, but largeamounts of platinum must be recycled.
Very effective purification is achieved by theoxidative hydrolysis of the metallic impuritiespalladium, rhodium, iridium, ruthenium, andbase heavy metals, which are removed as insol-uble hydroxides (see Fig. 11). Precipitation fromhot aqueous solution is carried out by addition ofsodium bromate at pH 6.5 or by passing throughchlorine at slightly alkaline pH. The oxide hy-drates adsorb considerable amounts of platinum.The yield of platinum therefore depends on thepurity of the feed material.
Figure 16. Extraction of platinum group metal and goldchloro complexes by trioctylamine
Figure 17. Extraction of palladium(II) and platinum(IV)chloro complexes by an aldoxime (LIX TN 1911) a) Plati-num(IV); b) Palladium(II)
346 Platinum Group Metals and Compounds Vol. 28

The classical method of purification is byrepeated precipitation of (NH4)2[PtCl6] orK2[PtCl6]. The crude salt is converted into me-tallic platinum, dissolved to form H2[PtCl6] (seeChap. 5), and reprecipitated. By repeating thismany times, very high purities are obtained,although the cost is also high. Also,(NH4)2[PtCl6] can be converted directly toH2PtCl6 by oxidation of the NHþ
4 or by cationexchange. In the oxidative decomposition ofNHþ
4 (e.g., by heating the aqueous solution withaddition of Cl2 or HNO3), care must be taken touse a carrier gas or distillative conditions toprevent accumulation of explosive nitrogen –halogen compounds.
Conventional crystallizations from water arepossible, but the solubility of (NH4)2[PtCl6] isrelatively low, so that despite favorable temper-ature coefficients (see Fig. 7), large volumesmust be used. Modified crystallization methodsare more economic (e.g., utilization of the con-siderably higher solubilities in water above100 �C at higher pressure). The purification ef-fected by crystallization is enhanced by hydro-lysis. In neutral or very slightly acidic mediaabove 90 �C, the conversion of the chloro com-plexes of the platinum group metals (especiallymetals other than platinum) into hydroxo com-plexes and oxide hydrates begins. These cannotform mixed crystals with (NH4)2[PtCl6]. Theextent of hydrolysis of hexachloroplatinate(IV)increases with increasing temperature and reac-tion time. Therefore, high temperature and longreaction time lead to a lower yield of crystallineproduct. The reaction is reversible in the presenceof hydrochloric acid.
When the products of solvent extraction bysubstituted amines are stripped, solutions of plat-inum compounds in hydrochloric acid are ob-tained. From these, pure (NH4)2[PtCl6] is pre-cipitated by addition of NH4Cl.
Purification ofPalladium [160]. Impure am-monium hexachloropalladate(IV), (NH4)2[PdCl6],can be dissolved directly for purification. This isachieved by the following reaction:
3 ðNH4Þ2½PdCl6�þ20 NH3!3½PdðNH3Þ4�Cl2þ12 NH4ClþN2
Acidification of a solution of [Pd(NH3)4]Cl2precipitates the sparingly soluble, pale yellowtrans-diamminedichloropalladium(II):
½PdðNH3Þ4�Cl2þ2 HCl!½PdCl2ðNH3Þ2�þ2 NH4Cl
The impure salt should be dissolved quickly atroom temperature with agitation to prevent ap-preciable quantities of the (NH4)2[PtCl6] presentas an impurity from dissolving. Undissolved ma-terial, mainly hydroxides and (NH4)2[PdCl6], isfiltered off. Hydrochloric acid is added to thesolution until a pH of 1 is reached, causing[PdCl2(NH3)2] to precipitate as easily filterablecrystals. Possible causes of poor purification aretoo high a concentration of (NH4)2[PdCl6] in theimpure material, too prolonged a reaction withammonia, too high a concentration of ammonia,or problems with filtration of the hydroxidicprecipitate before precipitating [PdCl2(NH3)2].High temperature is also detrimental. If the acidconcentration is too high, the yield is reduced dueto further reaction to form soluble (NH4)2[PdCl4].
Other possible methods for dissolving(NH4)2[PdCl6] are
2 ðNH4Þ2½PdCl6�þN2H4!2 ðNH4Þ2½PdCl4�þ4 HClþN2
or
ðNH4Þ2½PdCl6�!ðNH4Þ2½PdCl4�þCl2
Pure (NH4)2[PdCl6] can be obtained in almostquantitative yield by passing chlorine through thesolution and adding NH4Cl.
The purification of (NH4)2[PdCl6] by repre-cipitation is useful in the presence of silver andmost base metals. In the presence of rhodium,platinum, iridium, and copper, however, betterpurification is achieved by precipitating[PdCl2(NH3)2], although higher losses of palla-dium to the mother liquor must be tolerated.
Dichlorodiamminepalladium(II),[PdCl2(NH3)2], can be produced by directprecipitation from an impure solution of palladi-um resulting from crude separation or inadequatepurification. It is dissolved in aqueous ammoniasolution to form [Pd(NH3)4]Cl2 and reprecipi-tated by hydrochloric acid.
An ammoniacal stripping solution is usedafter solvent extraction of palladium by dialkylsulfide. Palladium can be precipitated from thisby acidifying to pH 1, thus producing [Pd(NH3)2Cl2] directly in a highly pure form.
Purification of Iridium. The purificationof crude ammonium hexachloroiridate(IV),(NH4)2[IrCl6], is carried out by precipitation of
Vol. 28 Platinum Group Metals and Compounds 347
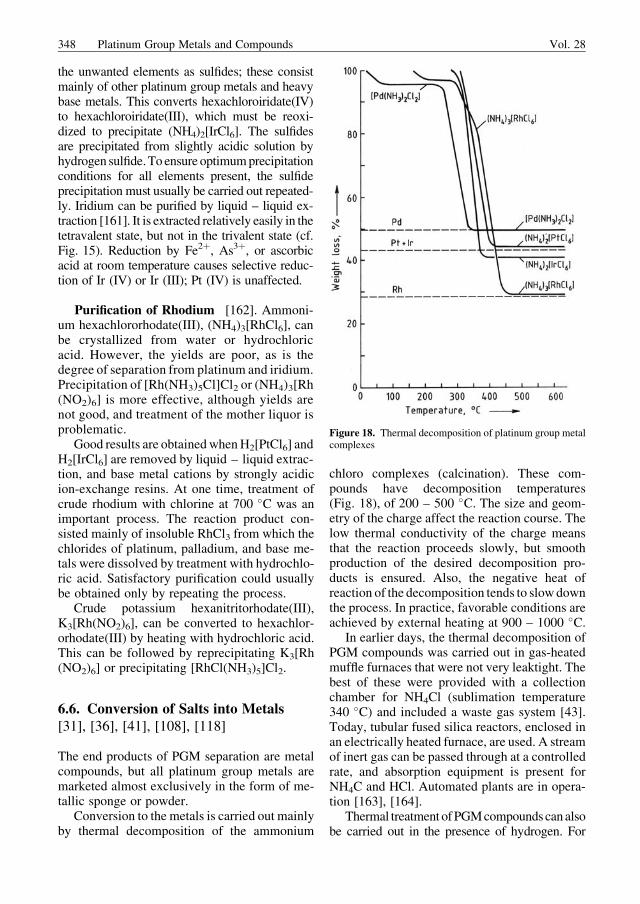
the unwanted elements as sulfides; these consistmainly of other platinum group metals and heavybase metals. This converts hexachloroiridate(IV)to hexachloroiridate(III), which must be reoxi-dized to precipitate (NH4)2[IrCl6]. The sulfidesare precipitated from slightly acidic solution byhydrogen sulfide.To ensure optimumprecipitationconditions for all elements present, the sulfideprecipitation must usually be carried out repeated-ly. Iridium can be purified by liquid – liquid ex-traction [161]. It is extracted relatively easily in thetetravalent state, but not in the trivalent state (cf.Fig. 15). Reduction by Fe2þ, As3þ, or ascorbicacid at room temperature causes selective reduc-tion of Ir (IV) or Ir (III); Pt (IV) is unaffected.
Purification of Rhodium [162]. Ammoni-um hexachlororhodate(III), (NH4)3[RhCl6], canbe crystallized from water or hydrochloricacid. However, the yields are poor, as is thedegree of separation from platinum and iridium.Precipitation of [Rh(NH3)5Cl]Cl2 or (NH4)3[Rh(NO2)6] is more effective, although yields arenot good, and treatment of the mother liquor isproblematic.
Good results are obtainedwhenH2[PtCl6] andH2[IrCl6] are removed by liquid – liquid extrac-tion, and base metal cations by strongly acidicion-exchange resins. At one time, treatment ofcrude rhodium with chlorine at 700 �C was animportant process. The reaction product con-sisted mainly of insoluble RhCl3 from which thechlorides of platinum, palladium, and base me-tals were dissolved by treatment with hydrochlo-ric acid. Satisfactory purification could usuallybe obtained only by repeating the process.
Crude potassium hexanitritorhodate(III),K3[Rh(NO2)6], can be converted to hexachlor-orhodate(III) by heating with hydrochloric acid.This can be followed by reprecipitating K3[Rh(NO2)6] or precipitating [RhCl(NH3)5]Cl2.
6.6. Conversion of Salts into Metals[31], [36], [41], [108], [118]
The end products of PGM separation are metalcompounds, but all platinum group metals aremarketed almost exclusively in the form of me-tallic sponge or powder.
Conversion to the metals is carried out mainlyby thermal decomposition of the ammonium
chloro complexes (calcination). These com-pounds have decomposition temperatures(Fig. 18), of 200 – 500 �C. The size and geom-etry of the charge affect the reaction course. Thelow thermal conductivity of the charge meansthat the reaction proceeds slowly, but smoothproduction of the desired decomposition pro-ducts is ensured. Also, the negative heat ofreaction of the decomposition tends to slowdownthe process. In practice, favorable conditions areachieved by external heating at 900 – 1000 �C.
In earlier days, the thermal decomposition ofPGM compounds was carried out in gas-heatedmuffle furnaces that were not very leaktight. Thebest of these were provided with a collectionchamber for NH4Cl (sublimation temperature340 �C) and included a waste gas system [43].Today, tubular fused silica reactors, enclosed inan electrically heated furnace, are used. A streamof inert gas can be passed through at a controlledrate, and absorption equipment is present forNH4C and HCl. Automated plants are in opera-tion [163], [164].
Thermal treatment ofPGMcompoundscanalsobe carried out in the presence of hydrogen. For
Figure 18. Thermal decomposition of platinum group metalcomplexes
348 Platinum Group Metals and Compounds Vol. 28

safety reasons, a mixture of hydrogen and nitrogenbelow the ignition temperature is preferable.
Another possibility to convert PGMcompoundsinto metals is chemical reduction with, e.g., hydra-zine in aqueousmedia. This processmust be used ifthe final products of purification are salts other thanthe ammonium salts (e.g., K2[PtCl6]).
Platinum. The compound (NH4)2PtCl6 canbe converted to platinum sponge by thermaldecomposition at 800 �C, according to
3 ðNH4Þ2½PtCl6�!3 Ptþ2 NH4Clþ16 HClþ2 N2
Excessive carryover of platinum can beavoided by introducing ammonium hexachloro-platinate(IV) into the reaction zone at ca. 300 �Cand heating it rapidly. Waste gas is washed withwater to extract ammonium chloride, both forenvironmental reasons and to prevent the loss oftraces of platinum.
If the reaction is carried out in a hydrogenatmosphere, elemental platinum is formed belowthe sublimation temperature of NH4Cl (ca.350 �C). Platinum carryover is then appreciablyless, and more ammonium chloride is formed:
ðNH4Þ2½PtCl6�þ2 H2!Ptþ2 NH4Clþ4 HCl
If NH4Cl is not sublimed but is removed bydissolving in water, finely divided platinum pow-der can be produced by this process. Calcinationof the hexachloroplatinate(IV) of an alkali metalis not recommended, because the alkali metalchloride produced is difficult to remove by aque-ous dissolution from the platinum sponge.
Hexachloroplatinates can usually be reducedin aqueous solution or suspension according to
ðNH4Þ2½PtCl6�þN2H4þ6 NaOH!Ptþ6 NaClþ2 NH3
þN2þ6 H2O
The reaction proceeds best above 80 �C, withexcess NaOHmaintained at 2 M and the reactioncontrolled by adjusting the rate of addition ofN2H4. The reaction must be flushed with nitro-gen, because hydrogen can be formed in a sidereaction. The platinum black formed is washed toremove salts, and either dried to produce powderor heated strongly to produce sponge. Overall,wet chemical reduction is more expensive thancalcination but should be used if alkali metal
hexachlorometallates are present, because theirdecomposition products cannot be sublimed.
Palladium. The thermal decomposition ofdichlorodiaminepalladium(II), [Pd(NH3)2Cl2],begins at ca. 290 �C in an inert atmosphere.Process conditions are the same as for(NH4)2[PtCl6]. The reaction is
3½PdðNH3Þ2Cl2�!3 Pdþ4 NH4Clþ2 HClþN2
For the calcination of ammonium hexachlor-opalladate(IV), which loses chlorine at ca.280 �C, the temperature should be increasedextremely slowly over the range 100 – 300 �C.If calcination is carried out in air, the palladiumsponge formed contains large amounts of PdO,which must be reduced by strong heating in ahydrogen atmosphere.
The wet chemical reduction
2½PdðNH3Þ2Cl2�þN2H4!2 Pdþ4 NH4ClþN2
and the reduction of (NH4)2[PdCl6] proceedquantitatively in strongly ammoniacal solutionabove 50 �C. Sodium formate in alkaline solu-tion can also be used as reducing agent. A nitro-gen atmosphere must be used in both cases toprevent formation of explosive gas mixtures.Chemical reduction often produces pyrophoricpalladium black, which must be dried in an inertgas atmosphere.
Iridium. The best method of producing irid-ium is to reduce ammonium hexachloroiridate(IV), (NH4)2[IrCl6], in a hydrogen atmosphereca. 800 �C, yielding the metal powder. Calcina-tion in air gives an oxide-containing material.Wet chemical reduction with hydrazine is possi-ble only in strongly alkaline sodium hydroxidesolution and does not always go to completion.
Rhodium. Ammonium hexachlororhodate(III) is subjected to the same process of thermalreduction as used for (NH4)2[IrCl6].
Wet chemical reduction with hydrazine ac-cording to
4 ðNH4Þ3RhCl6þ3 N2H4þ24 NaOH!4 Rhþ3 N2þ12 NH3
þ24 NaClþ24 H2O
must be carried out with a large excess of hydra-zine and NaOH at ca. 100 �C to ensure completereaction.
Vol. 28 Platinum Group Metals and Compounds 349

Ruthenium. Ammonium hexachlororuthe-nate(III – IV) is reduced, preferably in a hydro-gen atmosphere, at ca. 800 �C.Aqueous solutionsof ruthenates(IV) (e.g., produced by adding RuO4
to an alkali-metal hydroxide solution) are treatedwith ethanol to precipitate hydrated RuO2, whichis reduced in a hydrogen atmosphere at 800 �C.
Osmium. Osmium tetroxide that has beenabsorbed in potassium hydroxide solution isprecipitated as potassium osmate(IV) by addingethanol. This is reduced by strong heating in ahydrogen atmosphere or reacted with NaBH4 inaqueous solution [165]. The product must bewashed with water in both cases.
6.7. Partial Purification
Platinum – Rhodium Mixtures. For in-dustrial use, platinum and rhodium are oftenalloyed together, and separating these elementsduring the recovery process may be unnecessary.The solutions obtained by dissolving metallicplatinum – rhodium scrap can be used. The neu-tral solutions are treated with a sodium-loadedion exchanger to replace all unwanted cationswith sodium. Sometimes, Naþ is replaced by Hþ,giving a solution of H2[PtCl6] and H3[RhCl6]suitable for further treatment (e.g., reduction byhydrazine to give Pt – Rh powder).
Similar processes can be used for treatingsome single metal catalysts (e.g., material fromthe calcination of carbon – palladium).
Surface Cleaning. Some returned metallicmaterials with only surface contamination re-quire merely surface cleaning, thereby avoidinga wet separation process. They can be treatedwith acids, such as hydrofluoric acid, or withfused salt (e.g., NaHSO4), although some of theouter layer of platinum dissolves.
6.8. Treatment of Internally RecycledMaterial
A buildup of base metals occurs in recycled mate-rial such as mother liquor from precipitation andcrystallization. These base metals originate fromthe raw materials and from salts formed in thereactions, and also include small quantities ofplatinumgroupmetals from theseparationprocess.
The quantity or recycled noble metals shouldnormally not exceed1%of themetal in the startingmaterial. The extent of recycle must be limited,because losses are relatively high when noblemetals are recovered from this type of material.
Solutions containing platinum metals withonly small amounts of other materials should beconcentrated by evaporation in the presence ofhydrochloric acid. They can be combined withfresh raw material for separation.
If the solutions contain only ammonium chlo-ride and platinum groupmetals, they can often bereused directly as a precipitating agent afterconcentration in acid conditions.
If the solutions contain large amounts of saltsof alkali metals, alkaline earths, or other metalsalong with platinum and palladium residues, it isadvantageous to separate the ions [PtCl6]
2� and[PdCl4]
2� from the other materials by usingstrongly basic ion exchangers. Good separationis achieved if only the two chloro complexes arepresent. For rhodium and iridium this condition isoften not achieved, because of hydrolysis to givehydroxo complexes, so this method cannot beused unless special precautions are taken.
In earlier times, noble metals were recoveredfrom such solutions by cementationwith zinc in ahydrochloric acid solution. Aluminum is now thepreferred reducing agent because of the high costof zinc, which must be present in large excess,and the resultant environmental problems. Noblemetals can also be recovered with sodium boro-hydride. Alternatively, they can be reduced byhydrazine in alkaline solution. If no precautionsare taken, hydroxides of base metals are alsoprecipitated, but these can easily be redissolved.Copper is also precipitated in this reductionprocess. After redissolving the metals, the lattercan be reprecipitated as copper(II) oxalate oranother copper(I) or copper(II) salt that is spar-ingly soluble in slightly acidic solution.
All materials from which noble metals havebeen recovered must be monitored carefully forpossible noble-metal content before disposal, sothat no irrevocable losses occur.
6.9. Construction Materials
Many of the operations described above posespecial problems in the choice of materials forequipment and vessels.
350 Platinum Group Metals and Compounds Vol. 28

Plastics resistant to mineral acids can often beused. Vessels and reactors lined with hard rubberare resistant to acids and saturated chlorine solu-tions at 90 �C. Unsuitable metals for commonlyoccurring solutions of platinum and other plati-num group metals include stainless steel, titani-um, zirconium, the Hastelloys, and silver,because these materials cause cementation ofplatinumgroupmetals. Tantalum is an exception.It is resistant to dissolved halogens and does notcement platinumgroupmetals from solution. It isa suitable material for steam-heated heat exchan-gers. Equipment constructed of borosilicate glassand glass-lined steel reactors are chemicallyresistant to all of these materials. For solutionscontaining large amounts of ruthenium or osmi-um, other construction materials are recom-mended, including graphite for some items.
For treating raw materials with acidic moltensalts or salts that have a chlorinating action, andfor calcination, the use of fused silica equipmentis best. Treatment with molten oxidizing alkalinesalts is carried out in silver or nickel crucibles.
7. Platinum Group MetalCompounds [5], [6], [30], [32], [36],[41], [88], [105–107], [166], [263–270]
Because of the many possible oxidation states ofplatinumgroupmetals and their tendency to formcomplexes, the range of compounds is extremelywide.
7.1. Inorganic Compounds
7.1.1. Platinum Compounds
Hexachloroplatinic(IV) acid, H2[PtCl6]� 6 H2O,Mr 517.92, theoretical platinum content37.68%, platinum content of commercial prod-uct ca. 40% [commercial names: chloroplatinicacid, hydrogen hexachloroplatinate(IV)], formsreddish brown crystals (mp ca. 150 �C), whichdeliquesce in moist air and are readily soluble inwater or alcohol. The usual commercial productis an aqueous solution with a platinum content of25%.
Production (see Chap. 5). Platinum spongeis treated with moderately concentrated hydro-
chloric acid saturated with chlorine in a stirredvessel at ca. 80 �C. The solution obtained isevaporated until it reaches 150 �C.When cooled,it changes into solid H2PtCl6 � 4.5 H2O, whichcontains ca. 40% platinum, no mother liquor isproduced. If the platinum is dissolved in aquaregia instead of hydrochloric acid – chlorine, thenitric acid and NOCl formed must be removedcompletely, preferably by repeated evaporationto a syrupy consistency and redissolution inhydrochloric acid.
Uses. Hexachloroplatinic(IV) acid is themost industrially important platinum compound.It is used in the production ofmost other platinumcompounds and preparations. It is used primarilyto make catalysts by impregnating support ma-terials and as a precursor hydrosilylationcatalysts.
Hexachloroplatinates(IV). Ammoniumhexachloroplatinate(IV), (NH4)2[PtCl6], and, toa limited extent, the orange-red salt sodiumhexachloroplatinate(IV), Na2[PtCl6], are impor-tant in platinum separation processes (seeChap. 6).
Platinum Chlorides. Platinum(IV) chlo-ride, PtCl4, is produced by careful dehydrationof H2PtCl6 � 6 H2O at ca. 300 �C in a stream ofchlorine. It is a red-brown, crystalline, hygro-scopic powder with a relatively narrow range ofthermal stability.
Above 380 �C, PtCl4 liberates chlorine andforms platinum(II) chloride, PtCl2, which is sta-ble between 435 and 580 �C. Platinum(III) chlo-ride, PtCl3, is probably formed as an intermediatein this reaction. Above 580 �C, further decom-position occurs to yield metallic platinum. Plati-num(IV) chloride is the only chloride of platinumthat is soluble in water.
Platinum(II) chloride can also be obtainedfrom a solution of H2[PtCl4] by careful evap-oration under vacuum. Chlorides of platinumcan be produced by reacting chlorine withfinely divided platinum. The chloride obtainedwill be the one that is stable at the reactiontemperature.
Oxides of Platinum. The most industriallyimportant oxide of platinum is platinum(IV)oxide hydrate, PtO2 � H2O, a hydrogenation
Vol. 28 Platinum Group Metals and Compounds 351

catalyst. To prepare this substance, a solutionof 9 parts sodium nitrate and 1 part platinumas H2[PtCl6] is evaporated to dryness. Theproduct mixture is finely powdered and addedto molten NaNO3 (10 parts) at 520 �C withagitation. The product is dissolved in water,washed, and carefully dried to give a brownpowder, insoluble in aqua regia, with poorlydefined stoichiometry.
Platinum(II) oxide, PtO, and platinum(II) ox-ide hydrate, PtO � H2O, are also known, but havelittle or no industrial importance.
Other Platinum Compounds. The follow-ing compounds are used in aqueous electrochem-istry: potassium tetranitroplatinate(II), K2[Pt(NO2)4]; dinitrodiammineplatinum(II), [Pt(NH3)2(NO2)2]; and sodium hexahydroxoplati-nate(IV), Na2[Pt(OH)6].
Potassium tetracyanoplatinate(II),K2[Pt(CN)4], can be used in electroplating by moltensalt electrolysis. Barium tetracyanoplatinate(II),Ba[Pt(CN)4], is used in the manufacture of fluo-rescent screens.Many other cyano complexes areknown. Simple cyanides such as Pt(CN)2 and Pd(CN)2 also exist.
Potassium tetrachloroplatinate(II),K2[PtCl4],is a starting material for the synthesis of mostPt (II) compounds. Platinum(II) acetylacetonateis used in the pyrolytic production of platinumsurface coatings (see Section 10.10.2) and issuperior to other platinum compounds in thisapplication.
Dichlorodiammineplatinum(II), [PtCl2(NH3)2], is of historical interest and wasknown in two forms: ‘‘Peyrone’s chloride’’and ‘‘Reiset’s second chloride.’’ In 1893,WERNER recognized that these were cis andtrans isomers. cis-Diammine dichloroplati-num, also known as cisplatin or cis-platinum,is used in cancer therapy. Carboplatin (cis-diammine-1,1-cyclobutanedicarboxylatoplati-num), a second-generation cancer drug showsfewer side effects than cis-Platinum. Drugs fororal application like JM 216 are still in clinicaltrials [271].
7.1.2. Palladium Compounds [168]
Tetrachloropalladic(II) acid, H2[PdCl4],is stable only in solution. Commercial solutions
in hydrochloric acid contain 20% palladium andare dark brown.
Production. The method of producingH2[PdCl4] solution is similar to that forH2[PtCl4]solution. The metal is dissolved in HCl – Cl2 orHCl – HNO3, the rate of dissolution being higherthan with platinum (see Fig. 6). If dissolutionoccurs below ca. 50 �C, hexachloropalladic(IV)acid is formed first.
Uses. The solution of tetrachloropalladic(II)acid is the most industrially important palladiumpreparation. It is the starting material for almostall other palladium compounds, particularlycatalysts.
Palladium(II) Chloride, PdCl2,Mr 177.31,theoretical palladium content 60.0%, palladiumcontent of commercial product 59.9%, is abrown to brownish violet powder, insoluble inwater, but readily soluble in hydrochloric acidand solutions of alkali-metal chlorides. It sub-limes at 590 �C. Decomposition begins at600 �C and is complete at 740 �C.
Production. The best method of productionof PdCl2 is careful evaporation of a solution ofH2[PdCl4] in hydrochloric acid, preferably in arotary evaporator.
Uses. Anhydrous PdCl2 is the starting mate-rial for a number of palladium compounds.
Palladium Oxides. Poorly defined oxidehydrates are obtained by adding alkali to aqueoussolutions of Pd (II) compounds. The Pd (IV)oxide hydrates obtained from Pd (IV) solutionsrelease oxygen. A catalytically active palladiumpreparation analogous to PtO2 � x H2O can beobtained by evaporating a solution of H2[PdCl4]and NaNO3, and fusing the product.
Stoichiometric palladium(II) oxide, PdO,which is crystallographically well defined, isobtained by reaction of palladium black withoxygen or air at 750 �C. Decomposition occursat 850 �C.
Ammine Complexes of Palladium. Theaddition of ammonia to solutions of palladiumchloride first causes the formation of a pinkprecipitate of the binuclear complex
352 Platinum Group Metals and Compounds Vol. 28

[Pd(NH3)4PdCl4], known as Vauquelin’s salt,which is converted to soluble tetramminepalla-dium(II) chloride, [Pd(NH3)4]Cl2, by further ad-dition of NH3. Acidification of this solutionyields the sparingly soluble light-yellow trans-diamminedichloropalladium(II), [PdCl2(NH3)2].These compounds and reactions are important inthe industrial separation of palladium (seeChap. 6) and in electroplating processes.
Other Palladium Compounds. Ammoni-um hexachloropalladate(IV), (NH4)2[PdCl6], isimportant in separation technology. It is an oxi-dation product of tetrachloropalladate(II) (seeChap. 6). Palladium(II) sulfate, palladium(II)nitrate, and palladium(II) acetate are of someimportance in preparative chemistry. The readyformation of palladium hydride is exploited in-dustrially for example for the purification ofhydrogen.
7.1.3. Rhodium Compounds [169]
Hexachlororhodic(III) Acid, H3RhCl6.Wet separation processes and molten salt disso-lution usually produce (NH4)3[RhCl6] orNa3[RhCl6] � 12 H2O in crystalline form. Bothcompounds form deep-red crystals, which arevery soluble in water. Free hexachlororhodic(III) acid, H3[RhCl6], can be obtained by cationexchange, by oxidative decomposition of thecation in the case of the NHþ
4 salt (see Section6.2) or by dissolution of finely divided rhodiumpowder in hydrochlorine acid and chlorine gas.The composition of the chloro complexes ofrhodium depends strongly on the concentrationof hydrochloric acid, the temperature, the dura-tion of reaction, and the previous treatmentof the solution. By starting with a stronglyacidic medium, the following transformationsare possible:
½RhCl6�3���½RhCl5ðH2OÞ2��½RhCl4ðH2OÞ2���½RhCl3ðH2OÞ3��
½RhCl2ðH2O4Þ�þ��½RhClðH2OÞ5�2þ��½RhðH2OÞ6�3þ
The color varies from raspberry red to brown.
Rhodium Chlorides. Anhydrous, brownishred rhodium(III) chloride, RhCl3, which is insol-
uble in water and mineral acids, is obtained byheating rhodium powder to 700 �C in a chlorineatmosphere.
So-called soluble rhodium chloride, with theapproximate composition RhCl3 � 2.5 H2O, isobtained by evaporating a solution of H3[RhCl6]in hydrochloric acid, preferably in a rotary evap-orator. This compound is the most importantcommercial rhodium product, and is used as thestarting material for other rhodium compoundsand catalysts.
Rhodium Sulfate. Rhodium(III) sulfate,Rh2(SO4)3, can crystallize with varyingamounts of water; it can be obtained by dissol-ving Rh(OH)3 � x H2O in sulfuric acid or by theaction of hot sulfuric acid on freshly precipitat-ed rhodium black. It is used in rhodium platingbaths.
Chlorotris(triphenylphosphine)Rhodium(I),RhCl[P(C6H5)3]3, is formed by the reaction oftriphenylphosphinewith soluble rhodium(III) chlo-ride in ethanol solution under reflux. It separates asdeep-red crystals.
This complex, and rhodium compounds withsimilar structures, are important homogeneouscatalysts (see Section 8.1).
7.1.4. Iridium Compounds
Hexachloroiridic(IV) Acid, H2[IrCl6], isproduced in aqueous solution by the methodsused to obtain H3[RhCl6] (i.e., from the ammo-nium or sodium salts). It is by far the mostimportant iridium compound. The oxidation stateis relatively easily changed (in either direction),the very intense dark brown [IrIVCl6]
2� changingto the lighter colored [IrIIICl6]
3� or vice versa.The compounds are used as precursors in thepreparation of coated anodes for the productionof chlorine and sodium hydoxide. The ammoni-um salts of both anions are important in separa-tion technology (see Chap. 6).
Iridium(III) Chloride, IrCl3. A productstoichiometrically deficient in chlorine is ob-tained by heating iridium powder in a streamof chlorine at 650 �C. The chlorination reac-tion is assisted by the presence of carbonmonoxide.
Vol. 28 Platinum Group Metals and Compounds 353

7.1.5. Ruthenium Compounds
Ruthenium(VIII) Oxide, RuO4 is formedby the oxidation of aqueous solutions of ruthe-nates. It has a melting point of 25.5 �C and istherefore a liquid at room temperature. It is bothtoxic and explosive (see Section 10.1).
Soluble Ruthenium(IV) Chloride. So-called soluble ruthenium chloride consists main-ly of Ru(OH)Cl3 with some RuCl3 � H2O, and isprepared by reacting RuO4 with hydrochloricacid and evaporating the solution. The compound(NH4)2[RuCl6] can be precipitated by addingNH4Cl. Solutions of ruthenium chloride in hy-drochloric acid are used in the production ofcoated anodes.
Potassium Ruthenate(VI), K2[RuO4], isformed by fusing ruthenium with KOH – KNO3
or by dissolving RuO4 in potassium hydroxidesolution.
7.1.6. Osmium Compounds
Osmium(VIII) Oxide, OsO4, is the mostimportant osmium compound. It is formedeven at room temperature by the oxidation ofosmium powder by air. It is prepared bystrongly heating osmium in a stream of oxygenat 500 – 800 �C. The melting point is 40.6 �C,and the boiling point 131.2 �C. Sublimationtakes place even at room temperature. OsO4
used as an oxidation catalyst or as an aqueoussolution to stain organic tissue for microscopicanalysis.
Potassium osmate(VI), K2[OsO2(OH)4], isformed by the reaction of OsO4 with potassiumhydroxide solution and alcohol.
7.2. Organic Compounds [170], [171]
The PGMs form numerous compounds withdirect metal-carbon links both as carbonyls andas organometallic compounds. Many of these areof industrial importance in homogeneous cataly-sis (see Section 10.5). Complex organic com-pounds of ruthenium and osmium can facilitate
electron-transfermechanisms inmolecular struc-tures and also serve as sensitizer materials forphotoinduced hydrogen production (seeSection 10.9).
Osmium bipyridyl redox polymers com-bined with a redox enzyme can be used asminiature in vivo sensors for implantation intodiabetes patients for continuous monitoring oftheir blood glucose levels (wired enzyme bio-sensor, [271]). The divalent and trivalent com-plexed osmium ions can exchange electrons athigh rates. The osmium polymer connectselectrically the redox centres of the enzymesby introducing electron relays close to theenzyme active site. A sol – gel based ampero-metric biosensor incorporates an osmium re-dox polymer as mediator for detection oflactate [272].
Ruthenium phthalocyanine complex com-pounds have proven effective as drugs for pho-todynamic cancer therapy. It is administeredintraperitoneally, followed by laser irradiationof the tumor tissue. The compound acts in thepresence of light as sensitizer to induce thephotochemical destruction of the unwantedtissue [273].
Ruthenium complexes with polyaminocar-boxylate ligands act as effective nitric oxidescavengers. The binding property for nitric oxideenables macrophage cells to kill tumor cells andmay also form the base for drugs against othernitric oxide influenced diseases including hyper-tension, epilepsy, diabetes, arthritis, and septicshock [274].
Giant, dendritic organic coordination com-plexes of ruthenium and osmium (dendrimers,[276–279]) with up to 22 ruthenium atoms havebeen synthesized, which may be used for theproduction of thin films.
354 Platinum Group Metals and Compounds Vol. 28
Ruthenium forms catalytic active complexcompounds with C60 molecules [280–282].
8. Alloys [28], [30–32], [88–91], [166],[167], [172–184]
8.1. Alloy Systems
All the platinum group metals are infinitelymiscible with one another in the liquid state.When the molten alloys solidify, or when amixture of the powdered metals is sintered, plat-inum, iridium, palladium, and rhodium, whichcrystallize in the face-centered cubic system,form mixed crystals with each other. At lowertemperature, miscibility gaps are observed inmany cases.
Vol. 28 Platinum Group Metals and Compounds 355

Some phase diagrams of the binary alloys ofplatinum according to Massalski [90] are shownin Figures 19–33; of industrial importance aremainly the binary alloys Pt – Rh, Pt – Ir, Pt –Pd, Pt – Au, but Pt alloyed with base metals (Ni,Co,W) and a few ternary or higher alloys are alsoused.
8.2. Special Alloys
Platinum and palladium are the most importantbase components of industrially utilized PGMalloys. Special properties of interest concerncorrosion resistance, mechanical strength, high
temperature resistance, electrical conductivity,thermoelectric power, and magnetic behavior.
Platinum – palladium alloys are used injewelry, for electrical contacts, and for equip-ment parts for glass production and constructionelements in highly corrosive environments.
Platinum and palladium alloys with W, Re,Ni and other metals of Groups 6 and 8 – 10 aresuited for use in high-temperature strain gauges[283]. Pt alloys are used in spark plugs forautomotive engines.
Dispersion-strengthened platinum and plat-inumalloys are produced by coprecipitation of themetals and refractory oxides, for instance, withZrO2 (zirconium oxide grain-stabilized platinum,
Figure 19. Phase diagram of binary platinum group metalalloys: Ag – Pt
Figure 20. Phase diagram of binary platinum group metalalloys: Au – Pt
Figure 21. Phase diagram of binary platinum group metalalloys: Fe – Pt
Figure 22. Phase diagram of binary platinum group metalalloys: Co – Pt
356 Platinum Group Metals and Compounds Vol. 28

Figure 23. Phase diagram of binary platinum group metalalloys: Pt – Rh
Figure 24. Phase diagram of binary platinum group metalalloys: Ag – Pd
Figure 25. Phase diagram of binary platinum group metalalloys: Au – Pd
Figure 26. Phase diagram of binary platinum group metalalloys: Fe – Pd
Figure 27. Phase diagram of binary platinum group metalalloys: Ni – Pd
Figure 28. Phase diagram of binary platinum group metalalloys: Cu – Pd
Vol. 28 Platinum Group Metals and Compounds 357
ZGS, Johnson Matthey). Dispersion-strengthenedplatinum alloys with Zr and additional Y aresuperior in temperature stability and have higherrecrystallization temperatures [285].
Platinum can also be effectively dispersionhardened by incorporating very small amounts(0.04 – 0.08 wt%) of refractory carbides (TiC,ZrC). The strengthening is due to a partial de-composition and reaction of the carbide with thematrix. The low carbide concentration offers theadvantage that ductility, electrical conductivity,and the recrystallization behavior are not seri-ously impaired.
Platinum – gold and platinum – gold –rhodium alloys have higher strength than pure
Figure 29. Phase diagram of binary platinum group metalalloys: Pd – H
Figure 30. Phase diagram of binary platinum group metalalloys: Ag – Rh
Figure 31. Phase diagram of binary platinum group metalalloys: Fe – Rh
Figure 33. Phase diagram of binary platinum group metalalloys: Ir – Pt
Figure 32. Phase diagram of binary platinum group metalalloys: Ir – Rh
358 Platinum Group Metals and Compounds Vol. 28

platinum at 1000 �C, finer grain structure, andbetter resistance to wetting by molten glass.
Platinum – iridium alloys find different ap-plications in electrochemistry, chemical technol-ogy, medicine, and jewelry. Iridium contents (inwt%) are for laboratory ware 0.4 – 0.6%, jew-elry 5 – 15%, medical applications 10%, elec-trical contacts 10 – 25%, electrodes 10%, pens,needles, springs 25 – 30%. Maximum hardness(ca. 360 HB) is reached with 30% iridium.
W – Th – Al-doped platinum – 26 wt%iridium and platinum – 30 wt% rhodium areused to encapsulate plutonia fuel pellets forspacecraft heat sources [290].
Platinum – rhodium alloys with 10% rho-dium are used as catalyst for the oxidation ofNH3, as the standard element in thermocouples(Pt – Rh of different compositions or Pt), and asheater elements in electrically heated high-tem-perature furnace. Pt – Rh alloyswith additions ofAu are especially resistant to wetting by moltenglass. They are used in machine parts for theproduction of high-tech glasses for computermonitors, cathode ray tubes, television screens,liquid crystal displays, and plasma panels; glassfibers for telecommunication; and for use inglass-reinforced construction materials.
Platinum – ruthenium alloys. Rutheniumstrengthens platinum very effectively; 10% ru-thenium increases the hardness to 168 HV. Thealloy PtRu5 has the same strength, hardness, andcorrosion resistance as the alloy PtIr10 (‘‘hardplatinum’’). Applications are jewelry items, elec-trodes for electrochemistry, spark plug electro-des, electrical contacts, and wear-resistant wiresin potentiometers. Higher alloy compositions upto 14% ruthenium and ternary types with 15%rhodium and 6% ruthenium are used for electri-cal contacts and potentiometer wires for heavy-duty applications.
Platinum – cobalt multilayers on varioussubstrates are used for ultrahigh-capacity harddisks, for magneto-optical storage, and perpen-dicular recording applications. Special proces-sing enhances domain wall pinning effects andleads to higher coercivity [286]. Pt – Fe alloyshave maximum coercivity at 39.5 wt% platinum[287].
A new class of magnetic behavior, randomquantum spin chain paramagnetism, is exhibitedin one-dimensional compound Sr3CuPt1�xIrxO6
prepared as solid solution of the antiferromag-
netic Sr3CuPtO6 and the ferromagneticSr3CuIrO6. The platinum : iridium ratio deter-mines the magnetic behavior of the solid solution[288]. Alloys of the type Cr1�xIrxO2 (0 < x< 0.3) are ferromagnetic materials with highcoercivity [289].
Platinum – nickel alloys have been provenin electrical switches and as high-strength andhighly temperature resistant construction mate-rials in the chemical industry. Compositionsrange from trace amounts up to 20% nickel withmaximum tensile strength of about 1725 MPaafter 90% cold reduction.
Platinum – tungsten alloys, commonly usedin compositions with 5 – 8 wt% Co, have ex-cellent wear resistance. They are used for poten-tiometers, glow wires in copiers, diode switches,heater elements and cladding on rods for fuelelements in nuclear reactors.
Platinum electrodes are hardened with iridi-um for use in spark plugs and as corrosionresistant electrodes.
In equiatomic Ti – Ni alloys, (known for theshape-memory effect), substitution of nickel by2 atom% ruthenium stabilizes the high temper-ature B2-type phase at room temperature [292].Partial replacement of Ni by palladium in theequiatomic TiNi alloy improves the shape mem-ory properties [293].
Platinum alloys are components in cathetersfor pacemakers, in dental gold alloys, in markerbands, in guidewires and in temporary and per-manent implants.
Palladium – gold alloys are the basis of pre-cision resistance alloys. Addition of iron accent-uates the formation of long range ordering. Thealloy Au50Pd45Mo5 has a resistivity of100 mWcm and a temperature coefficient of re-sistance (TCR) of 0.00012 K�1 between 0 and100 �C.
The tensile strength reaches values of about1060 N/mm2.
Palladium – silver alloys are used as capac-itor electrode layers, as silver conductor paths,and as resistor elements in thick-film circuits.The palladium – silver alloy with 40 atom%Aghas the highest electrical resistivity at roomtemperature. It finds use for precision resistorsin electrical technology. The resistivity between20 and 100 �C is nearly constant and amounts to42.5 mW � cm; between 20 �C and 800 �C theresistivity changes from 42.5 to 44 mW � cm.
Vol. 28 Platinum Group Metals and Compounds 359

Ternary alloys containing silver and copperwith up to 10 to 25% palladium are hard soldersfor high-strength.
Palladium – ruthenium with 4.5 wt% ru-thenium is a standard jewelry alloy in the UnitedStates. Alloys with ruthenium contents up to12% serve as material for electrical contactsand as catalysts for the reduction of nitric oxide.
Palladium alloys with 60 – 80 wt% Pd foruse in dentistry contain copper and gallium ashardening elements and tin to lower the meltingtemperature. Best corrosion resistance is attainedwith a single-phase structure in the compositionrange of Cu 4 – 6 wt%, Ga 5 – 7 wt%, and Sn6 wt%. Addition of 1 wt% palladium to silver –tin – copper amalgam alloys improves hetero-geneous phase formation and the thermal stabil-ity [294].
Palladium alloys with iron and with cobaltshow ferromagnetism.
By alloying palladium with 0.1 wt% cobalt,the palladium atoms surrounding a cobalt atom,are strongly magnetized to magnetic moments inthe order of 10 mB, which is about six timeslarger than that of a single cobalt atom.
Palladium and certain palladium alloys areunique in their capability to dissolve largeamounts of hydrogen up to about 2800 timestheir own volume [295–297]. Hydrogen enterspalladium interstitially. The hydrogen atoms arelocated in the octahedral holes of the fcc lattice.Above 295 �C, palladium and hydrogen form acontinuous serious of solid solutions with fccstructure. Below that temperature, the phasesplits into a fcc palladium-rich phase and an fcchydrogen-rich phase, and the result is a miscibil-ity gap which broadens with decreasing temper-ature. The occupation of interstitial sites byhydrogen atoms causes expansion of the palladi-um lattice. The lattice parameters increase withincreasing hydrogen content from 3.891 A
�for
pure palladium up to 4.06 A�at 75 atom% hy-
drogen [298]. Thermal cycling of Pd – H alloysin the duplex phase region causes high mechani-cal stresses due to changes of the lattice dimen-sions with changing amounts of dissolved hydro-gen. Pure palladium, when repeatedly heated andcooled in hydrogen-containing atmosphere, be-comes brittle and cracks.
Palladium – silver alloys with 20 – 25 wt%silver dissolve larger amounts of hydrogen than
pure palladium and are resistant to thermal cy-cling [299]. Pd – Ag23 exhibits extremely highhydrogen permeability and is used for semiper-meable membranes for the extraction and purifi-cation of hydrogen up to 99.99999% purity.Palladium – silver permeation membranes areprovided in the form of bundled tubes withwall thickness of ca. 75 mm, sealed at oneend. Hydrogen is pressed from outside throughthe tubes, dissociating and diffusing throughthe alloy to the inner side of the tubes, leavingmost impurities outside the membrane [300],[301].
The solubilities of the hydrogen isotopes inpalladium and palladium – silver alloys differto an appreciable degree. Diffusion membranesof palladium or palladium – silver alloys cantherefore be used for separation and concentra-tion processes of different hydrogen isotopes[302]. The equilibrium hydrogen/deuteriumseparation factors vary from ca. 2.4 at 25 �C to3.7 at � 80 �C [303]. For the hydrogen/tritiumratio, a value of 2.8 was measured at 25 �C[304].
The ternary alloy Pd – Ti – Al is ferromag-netic, in contrast to the antiferromagnetic alloyPd – Ti [25].
Pt and Pt alloys are electrocatalysts in low-temperature fuel cells. The phosphoric acid fuelcell (PAFC) contains a Pt – Co – Cr alloy onthe cathode side and straight platinum on carbonas the anode. The higher activity of alloyscompared to pure platinum on the cathode sideis due to the fact that the non-noble metalcomponents are leached from the alloy crystal-lite surfaces, leaving a highly reticulated andactive surface [306], [307]. The catalytic activi-ty is effected by the preparation method and thecrystal structure of the three-component alloy[308]. The proton exchange membrane fuel cell(PEMFC) contains a platinum – ruthenium-loaded graphite anode and pure-platinum-load-ed graphite cathode [309]. The ruthenium alloyprotects the catalyst against poisoning by thecarbon monoxide which may be present in thehydrogen supply. Platinum – ruthenium alloysare also used as the anode catalyst in the directmethanol fuel cell (DMFC [310]). They preventthe formation of linearly bonded carbon mon-oxide (Pt–C�O) during the electrosorption ofmethanol on the platinum.
360 Platinum Group Metals and Compounds Vol. 28

Iridiumalloys are used inmicroelectrodes fortesting water quality by square-wave anodicstripping voltammetry [311].
Alloying of titanium and high-strength titani-um alloys (e.g., Ti – 6Al – 4V) with 0.1% ru-thenium effects improvements in the corrosionresistance against hydrochloric acid solutionsand can supersede corresponding Ti-Pd alloys.Selective dissolution generates on the surfacecathodic sites of low hydrogen overvoltage. Thisaccelerates hydrogen ion reduction and thuscauses a substantial shift in the corrosion poten-tial towards higher nobity, where the protectivesurface oxide film is stable. Applications areapparatus parts for processing corrosive chemi-cals [291].
Platinum metals based intermetallics in al-loyed or composite microstructures show inter-esting properties in high temperature strengthand oxidation resistance as needed for materialsin high temperature service. Prototypes are Ir –IrN-composites with refractorymetals. Pt – Al –Ru-alloys, ferritic alloys with Ru, and eutecticRu – RuAl and Ir – IrAl alloys [312].
8.3. Methods of Treatment [88], [107],[166], [167]
The starting material for the manufacture ofPGM alloys from platinum and palladium isgenerally metal sponge. The remaining ele-ments – rhodium, iridium, ruthenium, and osmi-um – are supplied as powders. Platinum and pal-ladium are also available in ingot form.
Owing to their high melting points, the plati-numgroupmetals are almost invariablymelted inan induction furnace, either under vacuum orwith argon as a protective gas. The vigorousmixing action produced by inductive heating isan additional advantage in producing alloys.Crucible materials can be aluminum oxide; zir-conium oxide; or magnesium oxide. Pure cal-cined calcium oxide was also used in the past.The molten metal is poured into water-cooledcopper molds. The refractory platinum groupmetals, ruthenium and osmium, are seldom need-ed as pure, solidmetals, butmay be compacted bysintering. Electric-arc melting and electron-beam melting are sometimes used. Electron-beam purification has recently become an impor-
tant method of treating iridium. This meltingprocess causes selective evaporation of traceelements and impurities at ca. 2700 �C.
Platinum, palladium, and most of their alloyscan be mechanically shaped by forging, rolling,drawing, and by deep drawing, etc., at roomtemperature, with intermittent heating. Form-ability decreases with increasing proportions ofthe other platinum group metals in the ascendingseries palladium, rhodium, iridium, ruthenium,and osmium. Special techniques are used forweaving and knitting Pt – Rh catalyst gauzesand for drilling extremely small holes in theplatinum alloy spinneret nozzles used in theproduction of fibers.
Platinum and the other metals can also beprocessed by powder metallurgy. The process ofinert gas spraying is used to produce powders.Particle sizes are around 50 mm.
Platinum and palladium can be fusionweldedwithout special precautions by TIG, plasma,and laser welding. Platinum metal can also bediffusion welded by hammering at red heat. Aprotective gas must be used with rhodium andiridium.
9. Quality Specifications and Analysis[5], [36], [185–198]
9.1. Quality Specifications [185], [200],[201]
In industry, the purity and fineness of platinumand platinum metals are even today often de-scribed by the classifications ‘‘technically pure’’(99%), ‘‘chemically pure’’ (99.9%), ‘‘physicallypure’’ (99.99%), and ‘‘spectroscopically pure’’(99.999%). These are taken from the recommen-dations of the Physikalisch-Technische Reich-sanstalt [29].
Present practice is to use the followingqualities:
99% (now hardly used)
99.9%
99.95%
99.98%
99.99%
99.999%
99.9999% (only in exceptional cases)
Vol. 28 Platinum Group Metals and Compounds 361

Other specifications are used for particularapplications (e.g., physical properties if used insensors or microelectronics [202], [203]).
Apart from osmium ASTM standard specifi-cations are available for the chemical composi-tion of some grades of the other refined PGMs(Table 6).
Most commercially available platinum andpalladium have purities of 99.95 – 99.98%.Higher purities usually sell at higher prices. Plati-num and palladium are generally supplied asingots or bares (stamped with the purity). Otherplatinum metals are supplied mainly as powder.
Platinum and palladium ingots of 500 –1000 g are preferred for investment purposes inJapan [1].
9.2. Qualitative Analysis
The older wet chemical qualitative reactionshave not entirely lost their usefulness.
Platinum can be identified by the precipitationof PtIV as yellow crystalline K2[PtCl6] or(NH4)2[PtCl6] from dilute hydrochloric acid so-lution by the addition of aKCl orNH4Cl solution.Other quadrivalent platinumgroup ions interfere.Analogous to Cassius gold purple, platinumforms platinum purple in weakly acid solutionsby reduction with SnCl2, Zn, or Al. The detectionlimit is ca. 1 mg/L, but Pd, Rh and Au caninterfere.
The chemical detection of palladium usesprecipitation of PdII as the yellow, ammonia-soluble dimethyldioxime complex. In contrastto the corresponding PtII compound, the Pd com-plex precipitates at low temperatures.
There is no simple specific detection reac-tion for the mainly trivalent rhodium. If othercolored ions are absent, it can be recognizedby its raspberry pink color in solutions ofrhodium(III) chloride or hexachlororhodate(III) complexes. If KOH is added to a RhIII
chloride solution, no precipitation occurs atfirst; the brown hydroxide precipitates onlywhen ethanol is added. This distinguishesbetween Rh and other PGMs.
Iridium can be detected by heating a solutionin concentrated sulfuric acid to fuming and, aftercooling, adding ammonium nitrate. Renewedwarming of the solution produces an intense bluecolor by formation of anionic complexes of IrIV.Chemically, even microgram quantities of ruthe-nium can be detected in hydrochloric acid solu-tion by the formation of a blue complex of RuIII
with thiourea.Osmium interferes with Ru detection with
thiourea by formation of a red complex of OsIII,which can be used for identification of Os. It canbe separated from Ru by distillation from nitricacid solution. The stinging odor of (the toxic)osmium tetroxide is also characteristic.
In the PGM industry spectrochemical meth-ods are used almost exclusively for qualitativeanalysis. The detection sensitivity for platinum insolutions by using the 265.85 nm emission line isonly slightly higher with inductively coupledplasma optical emission spectroscopy (ICP-OES) than with flame atomic absorption spec-troscopy (FAAS). X-ray fluorescence (XRF) of-fers quick detection or identification facilities forPGMs in metallic or nonmetallic materials. The244.8 nm, 340.46 nm, and 363.47 nm lines areused amongst others for the spectro-analyticaldetection of palladium by classical spectrogra-phy, FAAS, or ICP-OES. The surest way ofdetecting rhodium spectroanalytically is by usingthe 343.49 nm line with the above-mentionedmethods. Frequently used lines of Ir are at212.68 nm, at 215.27 nm, and at 224.27 nm. ForIr, the detection limit of ICP-OES is two orders ofmagnitude better than with FAAS. The spectro-analytical detection or identification of Ru bymeans of FAAS or ICP-OES offers no problems,as a series of lines, e.g., at 240.27 nm or349.89 nm, are available. Among all PGMs, Oshas the lowest detection limit when ICP is used.
9.3. Quantitative Analysis [313]
The methods of analysis used for economiccontrol reasons when buying and selling crudeproducts, alloys, compounds, etc., must reflectthe high value of PGMs.
Table 6. ASTM standard specifications for platinum group metals
Metal Standard Grades
Pt B 561 - 94 99.95, 99.99
Pd B 589 - 94 99.95
Rh B 616 - 96 99.80, 99.90, 99.95
Ir B 671 - 81 (Reappr. 1993) 99.80, 99.90
Ru B 717 - 96 99.80, 99.90
362 Platinum Group Metals and Compounds Vol. 28

Chemical Assay Methods. For commonlyoccurring combinations of elements, mandatorymethods of analysis and sampling have beendeveloped. The classical methods of separationand gravimetrical determination outlined brieflybelow are today of minor importance. PGMspresent in concentrations from mg/g to a fewpercent are often subjected to preliminary con-centration by fire assay with lead (see ! Gold,Gold Alloys, and Gold Compounds, Section9.3.), nickel sulfide, or copper as collector priorto the determination.
Platinum – Base Metal Alloys. Platinum isprecipitated as (NH4)2[PtCl6] from a nitrate-freesolution of H2[PtCl6] by addition of NH4Cl, theprecipitate is heated strongly,which converts it toplatinum, and then weighed [314]. Another pos-sibility to determine platinum gravimetrically isthe reduction with mercury(I) chloride [315]. ForPt on a-Al2O3 catalysts used in the petroleumindustry, the possible method of determiningplatinum is by the coloration produced with tin(II) chloride, which is determined photometrical-ly after extraction with ethyl acetate or afterpreliminary separation of platinum by reductionwith formic acid.
Platinum – Rhodium Alloys. A finely divid-ed black metal is obtained by fusion with zincfollowed by treatment with hydrochloric acid.Platinum is dissolved in aqua regia,(NH4)2[PtCl4] is precipitated and heated stronglyto yield platinum. The remaining noble metal isreductively precipitated from the mother liquor,combined with the residue remaining after aquaregia treatment, and treated with chlorine at650 – 700 �C. The RhCl3 obtained, which isinsoluble in aqua regia, is heated strongly in ahydrogen atmosphere to produce the metal,which is weighed.
Platinum – Palladium Alloys. Palladium isquantitatively precipitated with dimethylglyox-ime from a dilute solution in aqua regia. It isfiltered off, washed thoroughly, calcined, re-duced to the metal by hydrogen, and weighed.The dimethylglyoxime in solution is decom-posed by aqua regia, and platinum is convertedto (NH4)2[PtCl6], precipitated as sulfide, isolatedas the metal by reduction with hydrogen, andweighed.
Platinum – Iridium Alloys. Platinum – irid-ium alloys are converted into a finely dividedstate by fusion with zinc, and removing the zincwithHCl, and the platinum is dissolvedwith aquaregia. Iridium remains undissolved and, afterreduction with hydrogen, is weighed.
The classical dissolution for alloys like Pt – Irand Pt – Rh beginning with a fusion with zinccan be replaced advantageously by dissolving thealloys in a mixture of HCl and HNO3 underpressure in a sealed glass tube which is heatedin a drying furnace, or by dissolution in a PFA orquartz vessel by using a microwave furnace.
The PGM content of commercial compoundsand preparations is usually determined gravi-metrically by reduction in solution with hydra-zine or by reduction of the solid sample withhydrogen. Reductive precipitation from solu-tion in strong hydrochloric acid by treatmentwith zinc turnings is an exactmethod if carefullycarried out.
Physical Methods. Physical methods, espe-cially spectroscopic ones, are increasingly beingused, because they are inexpensive and, withimprovements in the equipment, have sufficientaccuracy for economic purposes. If brought intosolution by suitable acid treatment and/or fusion,such as Na2O2 fusion, and after separation frominterfering base metals, the PGMs can be deter-mined simultaneously by X-ray fluorescence, orICP-OES. Application of the principle of internalstandardization (! Gold, Gold Alloys, and GoldCompounds, Chap. 9.) is mandatory for precisework. The measurement procedures outlined in[316] for Pt and in [317] for Pd can be applied alsoto the ICP-OESofRh, Ir, andRu.Thedifficult andtime-consuming separationof thePGMs fromoneanother is no longer necessary. Therefore, analy-sis of complex materials containing preciousmetals, such as PGM ore concentrates containinggold, offers no problems. XRF can also be usedfor direct anlysis of metallic materials with suffi-cient accuracy for production control in themanufacturing of defined alloys. Calibration ofthis method may however be difficult.
9.4. Purity Analysis
The precious metal content of commercial gradesof PGMs can be determined more precisely by
Vol. 28 Platinum Group Metals and Compounds 363

measuring the total content of impurities by physi-cal methods than by direct determination of themain constituent. The most important of thesemethods are emission spectroscopy, e.g., withspark or glow discharge excitation. Recently, DCarc spectrometry became important again, becausethe application of a solid-state detector [318] en-ables simultaneous background corrections. Sparkablation ICP-OES offers an alternative method.When PGMs have been brought into solution (ifnecessary by dissolution in a mixture of HCl andHNO3 under pressure by conventional heating in asealedglass tubeorbyusing amicrowave furnace),the metallic impurities can be determined simulta-neously with a ICP-OE spectrometer with a solid-state detector [charge injection device (CID) orsegmented-array charge-coupled device (SCD)].Another solution technique of interest here is ICP-MS. Atomic absorption spectroscopy is of minorimportance. Mass spectrometry with glow dis-charge ablation (GD-MS) is used in special cases,e.g., for 99.995% or purer metals.
9.5. Trace Analysis
The determination of traces of precious metals isof great importance in mineral prospecting. Heremetal contents of only a few mg/g are of interest.Methods such as graphite tube AAS, neutronactivation analysis, total reflection X-ray fluores-cence analysis, and ICPmass spectrometry (ICP-MS) are used after appropriate enrichment pro-cedures (e.g., by fire assay with nickel sulfide ascollector).
The analysis of traces of precious metals inwaste materials from the precious metal proces-sing industry, especially refineries, has alwaysbeen very important inmonitoring the economicsof the process.
Trace analysis of platinum, palladium, andrhodium has become important in recent years.Whereas both the occurrence and theuse of PGMshave hitherto been located, automobile exhaust-gas catalysts now constitutes a widespread use.Although no toxic effects on the environmenthave thus been reported,methods of analysismustbe continually improved as permitted levels in thesoil and the atmosphere become increasinglystrict. The collection of the PGMs in a nickelsulfide phase by fire assay followed byGFAAS orICP-MS is suitable for analysis in the ng/g range.
10. Uses [3], [5], [30], [31], [204], [167]
10.1. Jewelry, Coinage, Investment[166]
In recent decades, platinum has been in suchdemand for jewelry and high-quality watchmak-ing that more than one-third of all the platinumconsumed has been used for this purpose. Of thisamount, Japan uses about 90%, with the UnitedStates taking the next largest share. Most of thealloys employed contain 95% platinum and arestamped PLAT 950. The mechanically morestable alloys Pt 95 – Cu 5 (Pt96/Cu 4), the hard-er alloys PtW 95/5, Pt 96 – Pd 4, and Pt 90 –Ir 10 are used for mounting precious stones. Pt/Co is used for jewelry casting. A relatively newalloy has a Pt content 99.9%; the hardness forjewelry is obtained with a small amount of sili-con. Forming costs for platinum are considerablyhigher than those for gold because the metal ismore difficult to work, and recycling costs arehigh. These factors markedly affect total cost.
Very good resistance to corrosion and erosionis required of alloys used for the nib tips of high-quality fountain pens [166]. These alloys arebased on ruthenium, iridium, and osmium.
In the Russian Empire in 1828 – 1845, a totalof 15 t of platinumwas formed into 3-, 6-, and 12-rouble pieces by sintering, minted, and broughtinto circulation.
Thefinenesswas99%platinumor99.2%PGM[15]. Between 1977 and 1980, platinum coinswereagain minted for the Moscow Olympic Games.These were 150-rouble pieces, fineness: 9999, 1/2ounce, in five designs. Since then, platinum coinshave been officially minted in other states (e.g., aPlatinum Noble in the UK, a PlatinumMaple Leafin Canada, and a PlatinumKoala in Australia). Thestandard issue of all these coins contained one troyounce (31.1035 g) of fine platinum. Small ingots,usually containing 99.995% platinum, are alsominted, intended mainly for small investors. Theuse of palladium for this purpose has recentlybegun on a small scale.
10.2. Apparatus [205–208]
Laboratory Technology. Platinum cruci-bles, dishes, boats, and electrodes (for electrogra-vimetry) have long been basic items of equipment
364 Platinum Group Metals and Compounds Vol. 28

in chemical laboratories. In earlier times, the purityof platinum was variable. Moreover, equipmentmade of pure platinum is not dimensionally stable,so today the more stable alloys Pt 97 – Ir 3, Pt 95– Au 5, and occasionally Pt 99.7 – Ir 0.3, aremainly used [209].
Another type of dimensionally stable plati-num is dispersion-hardened platinum, whichcontains 0.1 to 0.2 wt% of high-melting oxide(e.g., ZrO2, Y2O3) in finely divided form [88].The presence of this material reduces the mobili-ty of crystal imperfections. It is produced byprecipitation, internal oxidation, or mechanicalmixing followed by sintering of the powder. Themetal can be unalloyed platinum, or alloys ofplatinum with rhodium or gold. Rhodium andiridium crucibles can resist extremely aggressiveconditions in nonoxidizing atmospheres.
Chemical and Electrochemical Appara-tus [210], [319]. The platinum metals and theircompounds are of limited use in chemical pro-cess technology owing to their high cost. How-ever, platinum components are essential in fluo-rine chemistry. Bursting disks for protectionagainst excessive pressure are occasionally madeof platinum or palladium. Single crystals for theoptical industry and for laser technology aregrown in crucibles of platinum, rhodium, or(most commonly) iridium [210]. For the electro-chemical production of peroxo compounds, suchas hydrogen peroxide and peroxosulfate, massiveplatinum anodes or hollow platinum wires areused, although such use has decreased [319].
Glass Technology [210–213]. The corro-sion and erosion resistance of platinum metalstoward molten glass is not matched by any othermaterial [88]. High-purity optical glass is bestmelted in crucibles of unalloyed platinum. Spe-cial iron-free glass for sodium vapor lamps, X-ray windows, and cathode-ray tubes is melted inPt – Rh vessels.
In the automated production of bottles bylarge holding furnaces, the use of platinum com-ponents ensures problem-free operation overlong periods. These components include com-plete feeder systems, stirrers, plungers, skim-mers, pouring funnels, thermocouple protectiontubes, and level-measuring devices. Nowadaysdispersion-hardened platinum in increasingquantities is used in glassmaking technology.
In the manufacture of glass fibers and glasswool, the spinnerets are made of Pt – Rh. Rockwool and slag wool are made by a centrifugalspinning process with platinum centrifuges.
Textile Technology. Nozzles made of plat-inum alloys such as Au 70 – Pt 30 and Pt 92 –Ir 8 are used for spinning viscose fibers [214].
Soldering Materials. Gold and palladiumhave been used in some special solders.
10.3. Heterogeneous Catalysts [92],[215–220]
All platinum group metals have strong catalyticactivity, especially for hydrogenation reactions.The catalytic properties of the platinum metalsgroup elements can be regarded in relation tothose of the elements of group 8 – 10 of the firstrow of the periodic system. A clear correspon-dence to the differences in their catalytic behav-ior has been found in their behavior in coordi-nation formation, in complex formation, and inchemisorption behavior. The bond dissociationenergies of the related compounds, complexes,and adsorbed atoms or molecules are weaker forthe elements of the first two rows than for thoseof the third row. Insofar, closer relation existsbetween the catalytic characteristics of the ele-ments Fe, Co, Ni and Ru, Rh, Pd compared withthe elements of the third row. This specialenergetic situation allows easier intermolecularexchange of p- and ps electrons, which takesplace in numerous heterocyclic and homoge-neous catalytic reaction. Examples are the reac-tion of carbon monoxide with hydrogen, hydro-formylation, reactions with methanol, and thecatalysis of reactions with unsaturated hydro-carbons. The heterogeneous catalysts, includingsupported catalysts, are of major importance inchemical technology. As of 1998, 220 – 240 tof platinum group metals are in use worldwidein the form of supported catalysts. Next in orderof importance are bulk metallic catalysts in theform of wire gauzes, highly dispersed carrier-free metals, and oxides. Platinum and palladiumare technically much more important than rho-dium, ruthenium, iridium, and osmium. Often,two or more platinum group metals are com-bined (Table 7).
Vol. 28 Platinum Group Metals and Compounds 365

Supported Catalysts. The properties ofsupported catalysts are very dependent on theinteraction between noble-metal catalysts andsupport material. Classical support materialswere asbestos and pumice stone. Important sup-port materials today include aluminum oxides,activated carbon, silicates (e.g., zeolites), andsilica, also in combinations and as washcoats.Powdered support materials are used as suspen-sion catalysts; porous pressed shapes, pellets, andmonolithic structures in newer applications areused as fixed-bed catalysts. Reactions catalyzedby platinum group metals are listed in Table 8[319].
The largest demand for platinum catalystscomes from the petroleum industry [221],[222]. These are used in reforming high-boil-ing fractions from the distillation of crude oilat atmospheric pressure. The most importantreactions are the dehydrogenation of alkylcy-clohexanes, the isomerization and dehydroge-nation of alkylcyclopentanes, and the dehydro-genation and cyclization of alkanes, all ofwhich form aromatics, as well as hydrogena-tive cleavage of alkanes and naphthenes, anddealkylation of alkylaromatics. The resultingproduct mixtures are gasolines with high oc-tane numbers. The most important catalystsare Pt – g-Al2O3 Pt, Re/g-Al2O3, and Pt, Sn/g-Al2O3. Other catalysts such as Pt, Ir/g-Al2O3, Pt, Pd/g-Al2O3 or Pt, Ge/g-Al2O3
may also be used.Another use for PGM catalysts in the petro-
leum industry is in hydrotreating processes. Inaddition to base-metal catalysts, platinum andpalladium catalysts supported on g-Al2O3 or
aluminum silicates (zeolites) are also common.Supported palladium catalysts are used in hydro-gen peroxide production.
In the pharmaceutical industry, palladium/active carbon hydrogenation catalysts are widelyused, mainly with a palladium content of 5%.Other catalysts used include rhodium/activatedcarbon, platinum/activated carbon, palladium –platinum/activated carbon, and iridium/activatedcarbon, sometimes promoted by other elements,e.g., vanadium, iron, or bismuth. Lindlar cata-lysts (Pd, Pd/CaCO3) play an important rolein pharmaceutical industry. Platinum on electri-cally conducting carbon black is used as catalystin fuel cells. Furthermore precious metal formu-lations are used for exhaust gas catalysts, e.g., tocontrol automotive emissions (! AutomobileExhaust Control).
Solid Metal Catalysts [223–226]. Plati-num – rhodium gauzes are used as catalysts forthe large-scale oxidation of ammonia to nitricoxide (Ostwald and Brauer process) in the man-ufacture of nitric acid (! Nitric Acid, NitrousAcid, and Nitrogen Oxides).
The oxidation reaction
takes place in reactors in which many fine-mesh gauzes are stacked above one another.The rate of loss of noble metal varies between0.2 and 2.0 g per tonne of nitrogen reacted,depending on process conditions. If a rhodi-um – platinum alloy is used, the rate of loss of
Table 7. Modification of the catalytic activity of the platinum group metals by additional metals
Base metal Additional metal Reaction Effect of additive
Pt 5 – 20% Rh ammonia oxidation increased NO yield
lower Pt losses
Pt Ge, Sn, In, Ga dehydrogenation and hydrocracking of alkanes longer catalyst life due to fewer deposits
Pt Sn þ Re dehydrocyclization and aromatization of alkanes increased catalyst activity and stability
Pt Pb, Cu dehydrocyclization and aromatization of alkanes effective aromatization
Pt, Pd, Ir Au oxidative dehydrogenation of alkanes, n-butene
to butadiene, methanol to formaldehyde
better selectivity
Pd Sn, Pb selective hydrogenation of alkynes to alkenes
Pd Ni, Rh, Ag alkane deyhdrogenation and dehydrocyclization hinders coking
Pd Au, Cd ethylene and acetic acid to vinyl acetate
366 Platinum Group Metals and Compounds Vol. 28

noble metal is reduced; powdering of thegauze takes place more slowly, and its lifetimeis increased.
In the production of hydrogen cyanide(Andrussow process),
CH4þNH3þ3=2 O2!HCNþ3 H2O
platinum – rhodium or platinum – iridiumgauzes are used as catalysts (! Cyano Com-pounds, Inorganic, Section 1.2.1.).
Table 8. Catalysis by platinum group metals
Noble metal catalyst
Reaction Powder Compound Support
a) Oxidation and other reactions on noble metal catalysts
Methanol ! formaldehyde Ag - -
Ethylene ! ethylene oxide - - Ag/Al2O3
Ethylene ! acetaldehyde - PdCl2 in solution -
Amines ! nitriles Ag - -
Complete oxidation of organic compounds - - Pd/Al2O3
Disulfides to sulfoxides - RuO4 in CCl4 -
Alcohols to aldehydes, ketones - RuO4 in CCl4 -
Olefins, oxidative cleavage - RuO4 in CCl4 -
Synthesis or decomposition of organic compounds
CO þ H2 to methane - - Ru/Al2O3
to paraffin hydrocarbons - RuO2 Ru þ K2CO3
Addition of CO and H2 to
olefins to give alcohols - RhO3 -
olefins to give ketones - RhCl3 -
olefins to give esters - Rh oxide -
Addition of silane to olefins - - Pt/carbon Al2O3
Cleavage of formic acid Rh, Os Ru oxide Pt/asbestos
Leavage of formaldehyde Ru, Os OsO4 Pd/BaSO4
Inorganic catalysis
H2O2 ! H2O þ 1/2 O2 Ru, Pd - -
NO ! hydroxylamine - - Pt/C
PtAu5/C
PtIr1/C
Rh/C
NH3 ! NO PtRh10
b) Reduction and isomerization on noble metal catalysis
H2 addition to
acetylene and homologs Rh, Pd, Pt - Rh, Pd/Al2O3
Ag/Pd70/30/Al2O3
Pd and Pt/BaSO4
ethylene and homologs - - Ru/C
aromatic hydrocarbons - - Rh/Al2O3
aromatic compounds, hydrogenation of the aromatic ring - RuO2 Rh/Al2O3
heterocycles, hydrogenation of the aromatic ring - - Rh/Al2O3
Reforming of crude oil
dehydration (cracking) to aromatics Pd/Al2O3
isomerization/Cyclization - - Pt/Al2O3
PtIr/Al2O3
cleavage to alkanes - - Ru/SiO2
Reduction
of ketones to alcohols - - Ru/C
Pd/C
Pt/C
of carboxylic acids to alcohols - RuO2 Ru/C
Rh/C
nitrogen compounds to amines - PtO2 Pd/C
Pt/C
of disulfides to mercaptans - RuO2 -
of mercaptans to alcohols þ H2S - RuO2 -
Vol. 28 Platinum Group Metals and Compounds 367

The Degussa process
CH4þNH3!HCNþ3 H2
works with a-Al2O3 tubes coated on the insidewith a platinum film, forming a platinum – alu-minum alloy.
Finely DividedMetallic Catalysts. The so-called platinum metal blacks are well-estab-lished catalysts for hydrogenation, dehydroge-nation, and oxidation, mainly on a laboratoryscale. Their high degree of dispersion is shownby their large specific surface area (> 20 m2/g).These catalysts are very active even at low tem-perature, so reactions with labile molecules arepossible. In the production of hydrogen peroxideby the anthraquinone process (! Hydrogen Per-oxide, Section 4.1.2.2.), palladium black is usedto a considerable extent in suspension in a non-aqueous medium or as a fixed bed to catalyze thereduction of alkylanthraquinone to the corre-sponding hydroquinone. TheplatinumoxidePtO2
� H2O (Adam’s catalyst) is used widely as anunsupported catalyst in the pharmaceutical indus-try for the hydrogenation of alkenes and carbonylgroups. Nishimura’s catalyst contains rhodiumoxide in addition to platinum oxide.
Catalyst Poisoning and Regeneration[227]. All catalysts are liable to be deactivated(poisoned) by chemical,mechanical, and thermalmeans. Typical contact poisons for platinumgroup metals include sulfur, phosphorus, carbonmonoxide, hydrogen cyanide, lead, andmercury.
Reactivation is often possible, especially withsupported catalysts, by heating, steam treatment,reaction with oxidizing or reducing gases, orwashing.
10.4. Fuel Cells
Precious metals are used as anode and cathodecatalysts in low-temperature fuel cells such as theAFC (Alkaline Fuel Cell), PAFC (PhosphoricAcid Fuel Cell), PEMFC (Protone ExchangeMembrane Fuel Cell) and DMFC (Direct Meth-anol Fuel Cell). In the interesting PEMFC, whichhas a significant potential for replacing automo-bile internal combustion engines, for the cathodeside platinum catalysts supported on carbonblack are used. For the anode side, mainly be-
cause of the requiredCO tolerancewhenworkingwith reformed gas platinum/ruthenium catalystsare preferred. For AFC platinum/gold associatedwith nickel is applied. For PAFC quaternarycatalysts with platinum are used; platinum/rho-dium/iron was reported as cathode catalysts; onthe anode side normally platinum is used. TheDMFC, similar to the PEM,workswith platinum/ruthenium on the anode side, sometimes addi-tionally containing tungsten, and with platinumon the cathode side.
10.5. Homogeneous Catalysts [30],[88], [228–230], [322]
Homogeneous catalysis by platinum group me-tals, especially rhodium, has made a major im-pact in industrial chemistry. Important charac-teristics of these catalysts include high activityand, therefore, low concentration; high selectiv-ity leading to absence of side products; and mildreaction conditions, enabling the use of lowtemperature and pressure and facilitating thecontrol of reaction conditions. These factors areof great importance and allow expensive PGMsto be used economically. The discovery of chlor-otris(triphenylphosphine)rhodium(I), [RhCl{P(C6H5)3}3], a highly effective hydrogenation cat-alyst, byWILKINSON [231] opened a newchapter inhomogeneous catalysis and in the complex chem-istry of rhodium. Today, a number of modifiedcomplexes of this and similar types are known.
Homogeneous catalysis by rhodium com-plexes in oxo synthesis (hydroformylation) hasattained great importance and made possible thesynthesis of industrially important aldehydesfrom alkenes (see ! Oxo Synthesis). This pro-cess was formerly carried out with homogeneouscobalt catalysts.
By using [RhH(CO){P(C6H5)3}3] as the cata-lyst, reaction temperatures between 60 and120 �C and pressures of less than 50 bar couldbe realized, in contrast to the cobalt catalysts, forwhich temperatures up to 200 �C and pressuresas high as 300 bar are necessary. Often othercompounds are used as precursors from whichthe actual catalyst is formed during the reaction.These include [Rh(CO)2(acac)] and [Rh(CO){P(C6H5}3(acac)] (acac ¼ acetylacetonate).
Another approach to oxo synthesis is to workin a biphasic system. In this case a watersoluble
368 Platinum Group Metals and Compounds Vol. 28

ligand like P(C6H4SO3Na)3 and a rhodium salt(e.g., nitrate, acetate, or sulfate) is used as cata-lyst precursor [325].
The Monsanto acetic acid process uses RhIII
iodide as the catalyst precursor. In the cativaprocess [323] BP uses Ir acetate as complexprecursor.
Themost important application of platinum inhomogeneous catalysis is the vulcanization ofsilicon rubber. In addition, the synthesis of se-lected organofunctional silanes can be realizedby homogeneous precious metal catalysis [326].
A new era in homogeneous catalysis startedwith the discovery of asymmetric hydrogenationto produce enantiomerically pure compounds byusing metal complexes with chiral ligands [225],[328]. Many other known reactions [322], [324]are now also performed under asymmetricconditions like the isomerization of allylamines[327], [328], allylsubstitution [329] and Heckreactions [327–329]. The products are mainlyintermediates like amino acids, amines, and ster-oids for production of pharmaceutical and agro-chemicals. Examples of such ligands are DiAMP[331], BINAP [332], [333], Deguphos [334],Josiphos [335], DuPhos [336], [337]; productsinclude L-Dopa (drug for the treatment ofParkinson’s disease), L-Menthol (aroma), andMetolachlor (herbizide). The catalysts areformed in situ by reaction a precursor like [Rh(cod)2]BFa or [Rh(cod)acac] (cod ¼ 1,5-cy-clooctadiene) with the chiral ligand.
10.6. Automotive Emission ControlCatalysts [32], [233–236], [338–345]
The use of supported noble-metal catalysts for thetreatment of exhaust gases from automobiles hasbeen introduced beginning in the 1970s, aroundthe world (see also! Automobile Exhaust Con-trol). Regulations controlling the quality of ex-haust gases can be complied with only by the useof such catalysts. In particular, carbon monoxide(CO), hydrocarbons (CHx), and nitrogen oxides(NOx) must be reduced to a small percentage oftheir initial concentrations. Metals that have acatalytic action on exhaust gases include Pt, Pd,and Rh and all combinations thereof.
Formulations that can reduce all three harmfulcomponents are known as three-way catalysts.The three-way technique also includes control of
the air – fuel ratio by means of a l-sensor (seeSection 10.7). Exhaust control catalysts havetheir optimum activity only if the engine and thecatalytic unit are matched. The initially usedpellet catalysts have since been replaced bymonolith-type catalysts. The exhaust gas passesthrough numerous parallel channels of extrudedbodies (monoliths) that are coated with a catalyt-ically active layer, the so-called wash-coat. Thewash-coat consists of the PGM on g-Al2O3, rareearth oxides, and other metal oxides. The oxidemixture is designed to provide high thermalstability and the ability to compensate fluctua-tions of the air fuel ratio through oxygen storage.In some applications monoliths made from foilsare also used.
Each catalytic converter contains approxi-mately 2 g of PGM. The demand for thesematerials worldwide amounts to more thanone-third of the annual production of platinumand more than four-fifths of the annual pro-duction of rhodium. In the early 1990s the useof Pd has increased significantly. The PGMcatalysts are the only practicable materials forthis purpose. The required lifetime of an ex-haust-gas catalyst, as laid down by the widelyaccepted U.S. standard, corresponds to 80 000or 160 000 km.
Tighter standards in the future and the devel-opment of engines with higher fuel efficiency(lean-burn engines, new diesel engines) havespurred the further development of these tech-nologies, e.g., catalysts with the ability to tem-porarely store hydrocarbons or nitrogen oxides,electrically heated catalysts, and catalysts thatalso reduce soot emissions from diesel engines.All these catalyst types use PGMs.
Catalysts similar to those used for the treat-ment of automobile exhaust gases are also used totreat emissions from thermal power plants andindustrial waste gases contaminated with harm-ful organic materials.
10.7. Sensors [30], [88], [166], [167]
Thermocouples. Platinum metals and theiralloys are widely used in thermocouples (Table 9),because these have longer lifetimes than thosemade of base metals and can be used at highertemperature. The most important thermocoupleelement pair used in industry is Pt 90/Rh 10 – Pt
Vol. 28 Platinum Group Metals and Compounds 369

(wire thickness: ca. 0.5 mm),which gives accuratetemperature measurements over the range 800 –1600 �C.
Pt-Pt/Rh thermocouples have improved ther-mal stability and mechanical strength due todispersion hardening by alloying the platinumwith yttrium [346].
Resistance Thermometry. Platinum resis-tance thermometers are based on the temperaturedependence of the electrical resistivity. Thetemperature-coefficient of electrical resistance(TCR) of high-purity platinum is3.927 � 10�3/K. Basic values of the changes ofresistance over temperature ranges (e.g., � 200�C and þ 850 �C) of platinum resistance ther-mometers are specified in international standards(DIN IEC 751).
At temperatures below about 20 �K the TCRof pure metals becomes too low in comparisonto the absolute value of the resistance andcannot be measured. Alloys of rhodium with0.5 atom% iron or alloys of gold with 1.15 at-om% manganese show due to magnetic effectsan increasing TCR with decreasing temperature(Kondo effect [347]) to sufficient high, measur-able values.
Platinum wire-wound resistors, whose resis-tance is determined by the length and crosssection of the wire, have been almost completelydisplaced by so-called thin-film resistors, man-ufactured by thin-film technology. A film ofplatinum ca. 1 mm thick is deposited on aninsulating ceramic substrate by vacuum deposi-tion or cathodic sputtering. The required resis-tance value is adjusted by laser cutting. A mean-drous resistance path is formed in the film ofnoble metal by cutting with a laser beam or byionic etching through a masking coating pro-duced by photolithography (Fig. 34). The thick-ness of the film and the total path length deter-mine the resistance at 0 �C. Pathwidths are in theregion of 20 mm. The connecting leads, which
are joined to the thin film by special bondingprocesses, also consist of noble metals or theircompounds. Modern resistance thermometersbased on thin-film technology have the advan-tage over wire-wound resistance thermometersbecause they can be manufactured by highlyautomated methods and require only smallamounts of PGM (<1 mg per device).
Iridium-layers have values of thermal expan-sion which are comparable to those of the ce-ramic. They are therefore applied on devices fortemperaturemeasurements at temperatures downto about 200 �C.
Resistance thermometers are used today in awide range of technologies, from householdappliances to chemical equipment, and from roadtransportation to air and space travel.
Table 9. Commonly used noble metal thermocouple pairs
Positive leg Negative leg Operating temperature, �C
Cu Au 99.4 Co 0.6 �240 to 0
Pt 90 Ir 10 Au 60 Pd 40 0 to 700
Pt 90 Rh 10 Pt 850 to 1600
Pt 70 Rh 30 Pt 94 Rh 6 1000 to 1800
Rh 40 Ir 60 Ir 1000 to 2200
Figure 34. a) Platinum layer resistor element (dim 2 � 2.3mm) with welded connecting wires, layer resistance adjustedby laser cuts. b) enlarged view of platinum layer paths on theceramic substrate. Path width ca. 15 mm (Diehl et al. [167])
370 Platinum Group Metals and Compounds Vol. 28

Special Sensor Devices [348]. Oxygen se-lective sensors (‘‘lambda-probes’’), use the highelectrical conductivity imparted to solid electro-lytes (e.g., ZrO2) by the presence of oxygen ions,compared with their normal conductivity due toelectrons. The ionic conductor is provided onboth sides with platinum electrodes and it acts asa separator between two gas spaces. Any differ-ence in the oxygen partial pressure can be deter-mined by measuring the electromotive force(Nernst electromotive force) between the elec-trodes. Lambda probes are used in automotivecars to maintain an accurate air/fuel ratio and tomonitor the performance of the emissions controlsystem (automobile exhaust-gas catalysts inOtto-engines).
Noble-metal catalysts are also used in sensorsfor detecting combustible gases in air or explo-sive gas mixtures. Heat evolution is normallydetected by sensors known as Pellistors, whichconsist of platinum wire in a small sphere ofAl2O3 (diameter: 1.5 mm). The sphere is satu-rated with a noble-metal catalyst. If a chemicalreaction takes place on the catalyst, a temperatureincrease occurs that can be measured by thechange in resistance of the platinum wire. Theseheat evolution sensors are used in explosionprotection equipment.
Platinum and Palladium porphyrin com-plexes and ruthenium(II) diimines are used inoptical oxygen sensors. These sensors respondspecifically to molecular oxygen by a change inthe intensity of luminescence which is emittedfrom a probe molecule. The luminescence is re-versibly quenched by molecular oxygen. Opticalsensors have been developed for oxygen detectionin hydrogen, carbon dioxide, and as biosensor fordetermine the level of glucose [349].
Thin palladium layers act as hydrogen filtersin optical switching devices. These systems arebased on the hydrogen-dependent transition fromthe metallic state of yttrium hydride YH1.8 to thetransparent state YH2.9 (Fig. 35). Thin yttriumlayers are deposited by molecular-beam epitaxyon transparent glass plates covered with a pro-tective niobium layer and an upper palladiumlayer. The palladium layer impedes oxidation ofthe yttrium hydride and provides a catalyticsurface hydrogen dissociation and diffusion intoor out of the yttrium hydride layer. Depending onthe hydrogen concentration, the cell switches
from mirror reflection (YH1.8) to transparency(YH2.9). Switching occurs in less than 0.1 s.Applications are expected for automotive andconstruction parts [351], [352]. An other hydro-gen-concentration-dependent device consists ofan inner NbH layer, covered on both sides by ironplates. The system behaves ferromagnetically atlow H concentration; higher H concentrationeffect the transition to the antiferromagneticstate. The system is suited as magnetic-fieldsensor, for instance, in read and write heads ofcomputer hard disks.
Osmium -bipyridil-redox polymers com-bined with a redox enzyme can be used as minia-ture in vivo sensor for implantation into diabetespatients for continuous monitoring of their bloodglucose levels (‘‘Wired enzyme bio-sensor’’,[353]). The divalent and the trivalent complexedosmium ions can exchange electrons at high rates.The osmium polymer electrically connects theredox centres of the enzymes by introducingelectron relays close to the enzyme active site.
A sol-gel based amperometric biosensor in-corporates an osmium redox polymer asmediatorfor detection of lactate [354].
10.8. Electrical Technology [205], [237]
In low-current technology, electrical contacts ofgold, silver, palladium, rhodium, platinum, andtheir alloys are used. Contactsmade of bulk noblemetals are becoming less common than thosecoated by electrolytic and mechanical processes.
For a long time, large numbers of so-calledreed contacts have been used in telephone relays.These are Ag – Pd-, rhodium-, or ruthenium-coated contacts operating under a protective gas.Such contacts must be resistant to sparking,corrosion, and erosion, and must not become
Figure 35. Optical switching device with a palladium thinlayer [350]
Vol. 28 Platinum Group Metals and Compounds 371

welded together. During a lifetime of 30 years,they must carry out millions of switching opera-tions without damage. Furthermore, their contactresistance must be low, and their nominal resis-tance must be constant.
Platinum sealing wires are also now used onlyfor specialty light bulbs and transmitter tubes.Heating elements made of platinum metals ortheir compounds are used in resistance-heatedfurnaces above 1200 �C. Windings of Pt 70 –Rh 30 can be used up to 1500 �C, and ribbonwindings of rhodium up to 1800 �C. Furnaceelements made of platinum metals can be usedwithout a protective gas. Platinum, rhodium,iridium, and ruthenium are used as susceptormaterials in induction furnaces.
Platinum – iridium alloys are useful for sparkplug contacts for high-power aeroplane and au-tomobile engines.
10.9. Electronics
The platinum group metals and their alloys arekey materials in electronic devices and compo-nents (Table 10). Those include ‘‘active’’ discretesemiconductor devices (e.g., diodes, transistors,semiconductor memories), single-crystal inte-grated circuits (ICs), automotive electronic de-vices for engine-management systems, automo-tive oxygen sensors (lambda sensors), and safety
devices (ABS, airbag), and so-called passivecomponents (e.g., resistors, capacitors). The me-tals are mainly applied as surface layers, but alsoin formof bulkmaterial (sintered frompowder) indiscrete capacitor and resistor devices.
Important applications of platinum metals inelectronics are layer arrays of conductor andresistor paths deposited on substrate plates toform electronic circuits. The main types of elec-tronic circuits are printed circuits (PC) and thick-film and thin-film circuits. They are usuallyadditionally loaded with discrete active and pas-sive electronic components (hybrid technology).
On printed circuits boards, metallizing is pri-marily carried out by electroplating. In thick-filmcircuity, the noble metal layers are produced bythe use of paste preparations, which consist ofnoble metal powders, glass frits, and organicbinders. They are applied by screen printing ontoceramic substrates, followed either by drying orby firing at elevated temperatures. The glass fritsact as adhesives to the ceramic substrates.
In thin-film circuits the noble metal layers areprecipitated on glass or special ceramic sub-strates by sputtering, by evaporation, or by ther-mal decomposition of noble metal resinates.
The powders used in the pastes are generallyproduced by reduction of noble metal salts[355]. The reaction conditions determine shape,size, and specific surface area of the powder.Bimetallic (alloyed) powders are produced by
Table 10. Main applications of platinum group metals in electronics [92, p. 557]
Form
Surface coat
Element Bulk Bimetal electroplated sputtered or evaporated pastes, lacquers solder connection
Ru reed contacts Ru – O for cathodes in
special tubes
thick-film circuits
(resistor pastes for
hybrid circuits)
Mo – Ru
high-temperature
solders for electron
tubes
Rh slide contacts,
coating of Cu
disks for thyristors,
surface hardening
of Ag coatings on
pressure contacts
hard contact layers for
pressure contacts on Ag
surfaces, corrosion
protection
metallizing pastes
for sensor surfaces
Pd bonding wires
for transistors,
diodes
plug-in
contacts
plug-in contacts multilayer capacitors conductor and electrode
paste for thick-film
circuits, multilayer
capacitors
Ag – Cu – Pd solders
for electron tubes
package joining
Pt test resistors test resistors, diffusion
barriers, IC technology
372 Platinum Group Metals and Compounds Vol. 28

mechanical mixing to give composite blends orby coprecipitation, followed by thermal treat-ment of the dried powders. Their specific sur-face area is usually between 1.5 and 10 m2/g.The particle shapes can be spheroidal, micro-crystalline, or flake.
Platinum finds application as barrier layersand as contact pads on semiconductor chips.Platinum interlayers on RuO2 electrodes offerroelectric capacitors (lead zirconate titanatePZT) improve their stability and reduce leak-age losses [356]. Platinum additions in silver-based metallizing preparations and thick-filmconductor pastes for screen printing of hybridcircuits serve as stabilizing component againstsilver migration. In Ag-, AgPd-, or Au-con-taining thick-film pastes, Pt powder increasesthe resistance to scavenging during solderingoperations with Sn and Sn – Pb solder alloys.Palladium, pure or in combination with silver,is the most important component in pastesused for the production of the inner electrodesof multilayer capacitors. Special fine-grainpowders are needed to obtain sufficiently thinand homogeneous layer structures (Fig. 36). Itis produced by chemical reduction of aqueousPd(NO3)2 or PdCl2 solution. It is used in pureform or alloyed or mixed with silver powder(mainly of composition Ag – Pd30 wt%).Corresponding electrode pastes are applied toBaTiO3 substrates and fired at temperaturesdepending between 1100 and 1300 �C, de-
pending on grain size and substrate material.Palladium is also used as component in silver-based conductor pastes and for resistor ele-ments in thick-film circuity. As an additive insilver metallizing pastes, Pd improves theresistance to silver ion migration.
Electroless (by chemical reduction) deposit-ed, thin palladium coatings form the first met-allization step for metallizing bare polyimidesurfaces, followed by chemical and galvanicdeposition of thicker copper layers. This proce-dure is especially applied for the plating of holesin the polyimide substrates to connect electricalfunctions in multilayer PCs.
Ruthenium is the most important base ele-ment of screen-printed resistor layers in thick-film circuits. These layers are printed withpaste preparations consisting of RuO2 withadditions of PbO and Bi2O3. They form byfiring complex ruthenium compounds of highthermal stability and with very low tempera-ture coefficients of electrical resistivity. Exactresistance values are adjusted by applyingsmall cuts by means of laser beam into theresistor layer (‘‘laser trimming’’).
Platinum resinates are used for the productionof conductive coatings on ceramic substratesonto which further layers can be deposited elec-trolytically. Thin-film cobalt – platinum alloysand multilayers play an important role as mag-netic media on hard disks for magnetic andmagneto-optic storage and recording devices.Various organo platinum compounds, combinedwith special organic dyes, act as sensitizers toeffect the transformation of light energy intocharge separation in molecular systems. Appli-cations are the light-induced charge transfer inphotovoltaic equipment to produce electric cur-rent, also for the photochemical production ofhydrogen fromwater, fromhydrogen sulfide, or anonaqueous source. Organoruthenium com-pounds, deposited on nanocrystalline titaniumdioxide particles, serve as charge-transfer sensi-tizers in photovoltaic devices [357]. The parti-cles are placed as a thin transparent film betweentwo glass plates which have been coated withtransparent conducting oxide layers (Fig. 37).The sensitizer part is separated from the counter-electrode by an triiodide/iodide electrolyte. Aplatinum coat on the transparent conductingoxide layer catalyzes the cathodic reduction oftriiodide to iodide. The light-induced charge
Figure 36. Fine-grain palladium powder for the productionof electrodes in multilayer ceramic capacitors [167]
Vol. 28 Platinum Group Metals and Compounds 373

transfer from the excited sensitizer to the semi-conductor takes the route via interlocking organ-ic groups. The charge separation is accomplishedby the fact that the forward electron transfer fromthe metal via the ligand into the TiO2 semicon-ductor membrane is orders of magnitude faster(< 1 ps) than the back diffusion because of theless favorable electronic coupling between thesemiconductor and the oxidized ruthenium(III)ion. The overall efficiency for solar light conver-sion to electricity in this arrangement reaches10 – 11%. Higher efficiency is expected to beachieved by replacing the liquid electrolyte by a
solid electron-hole conductor in which conduc-tion does not involve mass transfer and whichallows the redox level to be matched to theground state oxidation potential of the sensitizer[358]. Applications are expected in the low-powder range (e.g., solar watches with the pho-tovoltaic cell incorporated in the cover glass),followed by development of interconnectedmodules, and production of solar tiles.
Organometallic platinum and ruthenium(II)compounds, anchored to a wide-band semicon-ductor, are promising materials for performingphotoinduced hydrogen production [359–363].A tandem device that achieves the direct cleav-age of water into hydrogen and oxygen by visiblelightworks by connection of two photosystems: afirst film of nanocrystalline WO3 and a second,dye-sensitized nanocrystalline TiO2 film. Theblue part of the spectrum is absorbed by the firstlayer, creating by band gap excitation valenceband holes which serve to oxidize water to oxy-gen. The second film captures the green and redpart, transmitted through the top electrode, there-by producing photovoltage, which enables theformation of hydrogen by the conducting elec-trons [359], [360].
Organic polypyridyl complexes of rutheniumand osmium are able to effect photoinducedelectron transfer in molecular structures (‘‘mo-lecular wires’’ [365]). Such components areneeded to link molecular-sized electronic de-vices (e.g., optical switches, fast recording de-vices, miniaturized sensors) electronically to thesurrounding assembly. Their function is to pro-vide electronic connection between spatiallyremote redox-active subunits. They must main-tain strict stereochemical integrity of the finalassembly. Unidirectional electron flow is pro-moted by external stimulation (e.g., absorptionof a photon, application of a potential at anattached electrode, or by chemical means).
RuO2/TiN storage electrodes serve for theproduction of dynamic random access memorycapacitors [366]. Surface layers of osmium andruthenium on dispenser cathodes lower the elec-tronic work function [367].
10.10. Coatings [30], [31], [93], [166]
Coatings of platinum metals may be applied to asubstrate by various methods.
Figure 37. Layout of a nanocrystalline TiO2 solar cell. Thetransparent dye derivatized titanium dioxide film is sand-wiched between two conducting glass electrodes. It harvestlight from both front and back sides. TCO is a transparentconducting oxide coating layer with a highly active, stableplatinum electrocatalyst [357](courtesy Platinum Metals Review)
374 Platinum Group Metals and Compounds Vol. 28

10.10.1. Coatings Produced byElectrolysis[338], [368], [369]
Platinum. Lustrous thin films for decorativepurposes are deposited from an electrolyte ob-tained by treating potassium tetranitritoplatinate(II), K2[Pt(NO2)4], with sulfuric acid. The activecomplex is H2[Pt(NO2)2SO4]. This ismore stablethan other platinum plating baths.
Relatively thick coatings can be depositedfrom alkaline solutions of sodium hexahydroxyplatinate(IV), Na2[Pt(OH)6].
A typical bath is sodium hexahydroxoplati-nate 20 g/L, sodium hydroxide 10 g/L, pH 13,75 �C, current density 0.8 A/dm2. A disadvan-tage is the low stability due to decomposition ofthe hydroxy salt. The use of the correspondingpotassium salts eliminates this difficulty [370],[371].
Platinum(II) diamminedinitrite, [Pt(NH3)2(NO2)2] (‘‘P salt’’), was formerly verywidely used and today along with Na2[Pt(OH)6],is used for coating titanium by aqueous electrol-ysis. These coatings are of lower quality thanthose produced by high-temperature electrolysis.Also, extensive pretreatment of the substratemetal is necessary, mainly to prevent cathodicabsorption of hydrogen.
The Q bath is based on a solution of [Pt(NH3)4]
2þ (Pt concentration 5 – 20 g/L) in adilute phosphate buffer, pH 10.3 – 10.5. At op-erating temperatures above 90 �C, high-quality,thick Pt deposites are obtained which are exten-sively used in the engineering industry whencomponents are subjected to high-temperaturecorrosion [372], [373].
Rhodium [169], [374–376]. Rhodium coat-ings are superior to all other PGM coatings,having better hardness, mechanical and chemi-cal stability, and reflectivity. For these reasons,rhodium is more widely used than the otherplatinum group metals (see also ! Electro-chemical and Chemical Deposition, Section8.16.).
Electrolytes consisting of rhodium sulfate(10 – 20 g/L Rh) in sulfuric acid can givecomparatively thick coatings (10 – 12 mm).Crack formation can be countered by additionof sulfite, selenic acid, magnesium sulfate, ormagnesium sulfamate. These baths are used toproduce rhodium coatings for heavy-duty slip
ring contacts and optical reflectors, and wereat one time used for reed contacts in telephonetechnology.
Rhodium sulfate or phosphate baths contain-ing 2 g/L of rhodium are used for decorativepurposes and give strongly reflective thin films(0.1 – 0.5 mm) that prevent silver or white goldfrom tarnishing. They are also used for platingeyeglass frames.
Palladium [168]. Palladium can sometimesreplace gold in coatings for electronics. Theplating baths contain palladium(II) diamminedi-nitrite, [Pd(NH3)2(NO2)2] (palladium P-salt),with additions of sodium and ammonium ni-trates, or sulfamates [377], [378].
Successful results have recently been ob-tained with plating baths based on organopalla-dium complexes. Concentrated hydrochloric ac-id plating bathswith dissolving palladiumanodesare rarely used now.
Palladium – nickel alloys with 70 – 75% Pdcan be electrodeposited from ammoniacal solu-tions. Such deposits, overflashed with gold areused as a replacement for gold in the electronicsindustry [379], [380].
Ruthenium [381–383] is deposited electro-lytically from solutions of ruthenium complexeswith a bridging nitrido ligand, for example,nitridooctachlorodiaquodiruthenate(IV),(NH4)3[H2O(Cl4Ru)–N–(RuCl4)H2O], the basisof the so-called RuNC baths. The correspondingcomplex with hydroxo ligands instead of chloroligands can also be used.
Ruthenium baths based on ruthenium nitro-sylsulfamate are also used.
High-Temperature Electrolysis (HTE)[30], [88], [239–241], [384], [385]. Platinumgroup metals can be deposited electrolyticallyfrom cyanide-containingmelts inwhich a solubleanode provides the noble metal that is depositedon the cathode. The metal most widely used inthis process is platinum, although iridium issometimes used.
The molten salt, an eutectic mixture ofKCN and NaCN, is maintained at 500 – 600 �Cin an electrically heated crucible. The platinumis deposited from the melt onto the substrate,which forms the cathode in the electriccircuit.
Vol. 28 Platinum Group Metals and Compounds 375

Process parameters are as follows:
Electrolyte K2[Pt(CN)4] in KCN – NaCN
Operating temperature 500 – 600 �CDeposition voltage 0.1 – 2 V
Cathodic current density 1 – 5 A/dm2
Cathodic current yield > 60%
Mean rate of deposition 10 – 20 mm/h
Oxidation of the melt and of the metal is pre-vented by a protective atmosphere of high-purityargon. Suitable substrate materials for HTE plat-inum deposition include refractory metals suchas titanium, niobium, tantalum, molybdenum,tungsten, zirconium; steels; and other metalssuch as copper. They must generally be pre-treated by sandblasting and degreasing.
Advantages of HTE platinum coatings, com-pared with those produced by aqueous electroly-sis, include low porosity; a wide range of filmthicknesses (1 – 400 mm), excellent adhesion;good resistance to mechanical impact, thermalshock, and sliding wear; high thermal conductiv-ity; and high ductility.
The largest use is in producing anodes forcathodic corrosion protection [242] (often in theform of expanded metal) or insoluble anodes forelectrolytic technology. Platinum-coatedmolybde-num wire is used in the construction of thermionictubes and for fusion with glass. High-temperatureelectrolysis is also used for electroforming.
10.10.2. Coatings Produced by ChemicalReaction [31]
Surface coatings of platinum metals can be de-posited onto metallic or nonmetallic substratesby chemical reduction or thermal decompositionof PGM compounds.
Platinum acetylacetonate is used for platini-zation of titanium. The titanium articles arepainted with a solution of this compound, dried,and heated. Repeated application is necessary.The process is not widely used due to allergicreaction and the low quality of the coatings.
Platinum coatings are produced on ceramictubes by a new technique using a lacquer [243],[386]. Such tubes are used as catalysts for thesynthesis of hydrogen cyanide.
Platinum metal resinates are used as ceramiccolorants [244], [245]. These can be employed to
produce conductive platinum coatings on ceram-ic substrates, onto which further layers can bedeposited electrolytically. Platinum coatings arealso used for decorative purposes.
Hexachlororuthenic(III) acid, H3[RuCl6],dissolved in alcohol, is painted onto the sub-strate, dried, and heated strongly in air toproduce an extremely adherent coating ofRuO2 [246], [247]. Titanium sheet, coated inthis way, has outstanding electrochemical prop-erties and is being used in chloralkali electroly-sis as dimensionally stable anodes. The RuO2
coatings are also used in the production ofcircuit boards by screen printing.
10.10.3. Coatings Produced by PhysicalMethods [88], [167]
Platinum can be deposited as a thin film on sub-strates by evaporating the metal in a high vacuum.Modern thin-film technology is based mainly onthe generation of fine particles from a cathode(cathodic sputtering). Due to the high kineticenergy of these metal particles, extremely goodadhesion is obtained. In the case of platinum groupmetals, sputtering targets are used for microelec-tronics and for resistance thermometers. Ion plat-ing is also used to produce high-quality coatings.
Roll-bonded PGM coatings are no longerimportant.
10.11. Dental Materials [88]
Platinum and palladium have major importancein dentistry (! Dental Materials, Section 2.3.).
Up to ca. 1900, dental protheses such as goldfillings, crowns, and bridges were made mainlyof alloyswith the same composition as those usedin coinage and jewelry, and later of 22-carat gold(83.3%). Platinum was used only to make fixingpins.
Important quality requirements such as oralstability, biological compatibility, color, strength,and good functional properties are achieved onlyby modern dental alloys. These complex alloys,unlike the earlier gold-containingones, have a veryfine-grained, homogeneous crystalline structure,givingmuchbetter corrosion resistance.Also, theirhardness is controllable over a wide range and canbe adapted to particular requirements.
376 Platinum Group Metals and Compounds Vol. 28

StandardAlloys. Modern alloys formakingdentures or prostheses have compositions mainlyin the range 65 – 85% gold, 1 – 10% platinum,1 – 4% palladium, 10 – 15% silver, 0 – 10%copper, and 0 – 1% zinc.
In low-gold alloys, which are cheaper, the goldis replaced partially by silver or palladium. Thesealloys contain 40 – 60% gold, 0 – 2% platinum,3 – 10% palladium, 23 – 35% silver, 0 –12%copper, and 0 – 5% tin, indium, and zinc.
Silver-base alloys have very low gold content.Adequate oral stability can be achieved by in-cluding at least 25% platinum group metals orgold. Inexpensive alloys in this category containat least 20 – 25% palladium, 0 – 3% gold, ca.55 –70% silver, and copper and zinc, and aretherefore no longer yellow, but white.
Alloys with Ceramic Veneering have beendeveloped in the early 1960s.With the help of thesealloys ceramics can be fused on a cast metalframework. The main properties of the alloys nec-essary for this task are a thermal expansion coeffi-cient similar to that of the ceramic and a solidustemperature sufficiently above the fusing tempera-ture of the ceramic. With early ceramics firing wascarried out at 980 �C. Alloys for these ceramicsneeded a solidus temperature above 1050 �C. Toachieve this goal the content of platinum and/orpalladium in the alloy had to be increased, whichcaused whitening of the alloy beside an increase ofthe melting range. In recent years, low fusingceramics became available, which allow to usealloys with compositions closer to the standardalloys and alloys with a more yellow color.
Alloys for high fusing ceramics with a highgold content have compositions in the range75 – 90% gold, 5 – 10% platinum, 1 – 10%palladium, 0 – 3% silver, with small additionsof iron, indium, tin, zinc, and other elements.
Attempts have beenmade to reduce the cost ofthese alloys by replacing gold by palladium andsilver. Typical Au – Pd – Ag alloys have com-positions in the following range: 42 – 62%gold,25 – 43% palladium, 5 – 20% silver, and 4 –10% base metals such as copper, tin, indium,gallium, and zinc. Typical silver-free Au – Pdalloys have the following composition: 45 –60% gold, 32 –40% palladium, and 0 – 14%of the base metals tin, indium, zinc, or gallium.
Palladium-base dental alloys were introducedin Germany in the early 1980s. These were
developed to reduce cost. Silver-free and sil-ver-containing alloys are available. Both containvery small amounts, if any, of gold and platinum.The range of compositions for the silver-contain-ing palladium-based alloys is 53 – 61% palladi-um, 28 – 38% silver, 0 – 12% tin, indium, zinc,and gallium. For the silver-free palladium-basedalloys, the composition range is 73 – 88% pal-ladium, 0 – 2% gold, and 12 – 26% of the basemetals copper, cobalt, tin, indium, and gallium.
11. Economic Aspects [5], [10], [24],[30], [46], [48], [57–63],[76], [235],[248–255]
11.1. Supply
The most important PGM-producing mines arelocated in
. South Africa (Amplats, Lonmin Platinum, Im-pala, Northham, Kroondal Platinum, TrojanPlatinum)
. Russia (Noril’sk Nickel, Koryak and Kondyormines)
. Canada (Inco, Falconbridge, North AmericanPalladium)
. USA (Stillwater)
. Zimbabwe (Hartley PLatinum, ZimbabwePlatinum, Anglo-American Zimbabwe).
The world total output 1998 of platinum,palladium and rhodium is listed in Table 11 andshown in Fig. 38.
In addition to PGMs obtained from ores, aconsiderable amount of secondary material ex-ists. The extent of this is very variable andfluctuates with time.
Averaged over all the PGMs, it amounts atpresent to ca. 20% of primary production, withthe greatest emphasis on platinum and especiallypalladium. The present effort directed toward
Table 11.World supply of Pt, Pd, andRh in 1998 by region (in 106 oz)
Pt Pd Rh
South Africa 3680 1820 400
Russia 1300 5800 110
North America 285 660 16
Others 135 120 4
Total 5400 8400 530
Vol. 28 Platinum Group Metals and Compounds 377

recovery from spent automobile catalysts willconsiderably increase the proportional secondaryproduction of platinum and rhodium.
A high proportion of the PGMs is recycled bythe users or, in the case of contract suppliers ofsemifinished products (e.g., catalysts), by theproducers. These materials do not appear on themarket either on the supply or on the demandside, so supply figures essentially reflect onlymined products.
11.2. Demand
Table 12 and Figure 39 show the demand forplatinum, palladium, rhodium, iridium and ru-thenium by application.
11.3. Prices
The prices of the PGMs are strongly influencedby factors of demand for new technical applica-tions, by availability of supply, and by specula-
tive stock building, leading to considerable dif-ferences between monthly and yearly high andlow price notations (see Table 13). Rising de-mand for technical applications, especially in theautocatalyst sector has caused strongly risingprices for platinum, palladium, and rhodium.
11.4. Commercial Aspects
Since 1975, a so-called London freemarket pricefor platinum is calculated and published twicedaily. Since 1976, a London palladium quotationhas also been available. Both of these were onlyprice indications, not to be compared with thefixed quotations for gold and silver. The LondonPlatinum and Palladium Market (LPPM) hassince been established and publishes fixed quota-tions twice daily.
Platinum and palladium are also traded on theNew York Mercantile exchange (NYMEX) andon the TOCOM in Tokyo.
Trading in platinum on the New Yorkexchange is in the form of contracts for unitsof 50 troy ounces. The purity of platinumsupplied in fulfillment of such a contract mustbe at least 99.9%, and the platinum mustoriginate from a producer or assayer registeredwith the exchange. For palladium, the contractamount is 100 troy ounces, and the purity mustbe 99.9%.
Spot transactions (immediate delivery andpayment) are guided by the London quotationand the New York exchange quotation.
Palladium in recent years has found increas-ing utilization in the production of automotivecatalysts. Due to very unstable supply from the
Table 12. PGM demand in 1998 by application (in 106 oz)
Pt Pd Rh Ir Ru
Autocatalyst 1830 4470 490 36
Chemical 265 230 31
Dental 1230
Electrical 320 8
Electronics 2070 184
Electrochemical 30 74
Glass 220 34
Investment 315
Jewelry 2370 250
Petrochemistry 130
Process catalysts 3 87
Others 305 115 10 36 30
Total 5755 8365 573 105 375
Figure 38. World palladium, rhodium, and platinum supplyin 1998 by region (in 106 oz) a) palladium (total 8400);b) rhodium (total 530); c) platinum (total 5400)
378 Platinum Group Metals and Compounds Vol. 28

main palladium producer of the world, Russia,palladiumprices increasedmore than threefold inthe period from December 1996 to May 1998.
12. Toxicology
Platinum is used in therapy; cisplatin is currentlyused for cancer treatment. Known side-effects
are sickness, vomiting, hypersensitivity reac-tions, ototoxicity and nephrotoxicity [387].
The tetroxides of osmium and rutheniumstrongly irritate and corrode the conjunctivaand cornea of the eye and the mucous mem-branes of the respiratory tract. Temporary toxiceffects can occur in bone marrow, liver, andkidneys. The salts of hexachloroplatinic acidH2[PtCl6] [16941-12-1] have a strong allergenicpotential (see below); platinum metal is notallergenic.
The MAK value for platinum compounds hasbeen set as low as 2 mg/m3 (as Pt), not because oftheir toxicity but due to allergenic potential. Forosmium tetroxide [20816-12-0] the MAK valueis even lower because of its strong irritatingeffect: 0.2 mg/m3. In the United States, TLVsfor water-soluble rhodium salts are 1 – 10 mg/m3. For other platinum group metals toxicologi-cal limits have not yet been established.
Table 13. PGM price lows and highs (1996 – 1998, in $/oz)
1996 1997 1998
low high low high low high
Pt 367 433 342 497 334 429
Pd 114 143 117 245 201 417
Rh 320 345 200 370 370 780
Ir 60 10 110 290 270 575
Ru 29 55 38 41 39 65
Figure 39. World PGM demand in 1998 by application (in 106 oz). Total quantities include recycled material (PA405 � 106 oz, Pd 175 � 106 oz, Rh 57 � 106 oz) a) platinum (total 5755); b) palladium (total 8365); c) rhodium (total573); d) iridium (total 105); e) ruthenium (total 375)
Vol. 28 Platinum Group Metals and Compounds 379

The median concentrations of platinum inblood, plasma/serum, and urine are about 1 ng/L[388]. The limit for platinum and osmium inindustrial effluents is 3 mg/L [389]. In theUnitedStates, most platinum group metals are classifiedas ‘‘poisonous’’ or ‘‘very poisonous’’ in waste-water treatment plants [390].
Automotive Emission Control Catalysts.Toxicological effects of platinum-group metalshave been studied increasingly because of theirwidespread use as automotive emission controlcatalysts. According to a recent investigation, anengine that is equipped with a modern three-waycatalyst emits approximately 15 ng of platinumper cubic meter of exhaust gas in an automobiletravelling at a constant speed of 100 km/h; plati-num metal is emitted, as well as platinum com-pounds that are yet to be analyzed [391]. Thestudy could not detect any health risk arisingfrom the use of automotive emission controlcatalysts, but they undoubtedly have an environ-mental benefit because they decrease the emis-sion of nitrous oxides, hydrocarbons, and carbonmonoxide. Even the most sensitive methodscould not detect a correlation between the uptakeof platinum from the environment and trafficdensity [392].
Hexachloroplatinate Allergy. Salts of hex-achloroplatinic acid H2[PtCl6] are among thestrongest synthetic allergens. Symptoms rangefrom irritation of skin and mucous membranes tolife-threatening attacks of asthma. Cigarettesmokers run a higher risk of sensitization toplatinum salts. The symptoms of allergy usuallystart after several weeks of exposure.
Hypersensitivity to hexachloroplatinate is ir-reversible, and further exposure must be avoidedimmediately.
For prophylaxis, exposure to dust and aerosolsmust be avoided by use of appropriate equip-ment. Special workplace hygiene is requiredwhen hot solutions of hexachloroplatinates, hex-achloroplatinic acid, or the products of theirthermal decomposition are handled. Exposedpersons must undertake skin tests (prick test) atregular intervals. In this way, sensitization isdetected at an early phase and appropriate mea-sures can be taken.
Other platinum and platinum group metalcompounds are not known to have an allergenic
potential. Recently, skin allergies to the plati-num-base alloys used in dental materials havebeen reported but are still under discussion. TheGerman government has recommended to testthe corrosion resistance and biocompatibility ofpalladium – copper alloys prior to their use indental restoration [393].
References
1 D. McDonald: A History of Platinum, J. Matthey Co.,
London, 1960.
2 D. McDonald, L. B. Hunt: A History of Platinum and its
Allied Metals, J. Matthey Co., London 1982.
3 PlatinumMetals Review, vol. 1 (1957) – vol. 44 (2000),
J. Matthey Co., London.
4 A. Binz: Edelmetalle – Ihr Fluch und Segen, W. Lim-
pert-Verlag, Berlin 1943.
5 Fonds der Chemischen Industrie: ‘‘Edelmetalle – Ge-
winnung, Verarbeitung, Anwendung,’’ Folienserie,
no. 12, Frankfurt 1989.
6 R€ompp Lexikon Chemie, 10th ed., Thieme, Stuttgart
1996 – 1999.
7 H. R€ompp: Chemie der Metalle, Franckh’sche Verlag-
shandlung, Stuttgart 1941.
8 P. Walden: Chronologische €ubersichtstabellen zur
Geschichte der Chemie (Altertum bis 1950), Spring-
er-Verlag, Berlin 1952.
9 S. Neufeld: Chronologie Chemie 1800 – 1980, VCH,
Weinheim, Germany 1987.
10 H. Quiring: ‘‘Platinmetalle’’, Die metallischen Roh-
stoffe, vol. 16, Enke Verlag, Stuttgart 1962.
11 J. Morin: La Platine, l’Or Blanc ou le Huitieme M�etal,Paris 1758.
12 A. Gal�an, R. Moreno: ‘‘Platinum in Eighteenth Centu-
ry – A Further Spanish Contribution to an Understand-
ing of Its Discovery and early Metallurgy,’’ Platinum
Met. Rev. 36 (1992) 40 – 47.
13 D. McDonald: ‘‘One Hundred and Fifty Years – An
Anniversary Review of Johnson Matthey’s Role in the
Economic History of Platinum’’, Plat. Met. Rev. 11
(1967) 18 – 29.
14 H. M. Severin: Gold and Platinum Coinage of Imperial
Russia from 1701 – 1911, Crown and Taler Publ. Co.,
New York 1958.
15 H.-G. Bachmann, H. Renner: ‘‘Nineteenth Century Plat-
inum Coins,’’ Platinum Met. Rev. 28 (1984) 126 – 131.
16 A. L. von Erengreuth: Allgemeine M€unzkunde und
Geldgeschichte des Mittelalters und der neueren Zeit,
R. Oldenbourg, M€unchen 1971.
17 L. B. Hunt: ‘‘Dr. Hans Merensky – Centenary of the
Discoverer of thePlatinumReef,’’PlatinumMet. Rev.15
(1971) 102 – 107.
18 J. Levin: The Story of Mintek 1934 – 1984, Council for
Mineral Technology (Mintek), Randburg 1985.
380 Platinum Group Metals and Compounds Vol. 28

19 A. M. Edwards, M. H. Silk: Platinum in South Africa,
Council for Mineral Technology (Mintek), Randburg
1987.
20 H. Houben: Festschrift zum f€unfzigj€ahrigen Bestehen
der Platinschmelze G. Siebert GmbH, Hanau, Alberti’s
Hofbuchhandlung, Hanau 1931.
21 D. G. Cooper:Das Periodensystem der Elemente, VCH,
Weinheim, Germany 1972.
22 E. Fluck,K.G.Heumann,PeriodensystemderElemente,
IUPAC-Empfehlungen, VCH, Weinheim, Germany
1985.
23 G. C. Bond, Plat. Met. Rev. 44 (2000) 146 – 155.
24 H. P. M€unster, Taschenbuch des Metallhandels, 8th ed.,
Metallverlag, Berlin 1989.
25 G. H. Aylward, I. J Findlay: Datensammlung Chemie,
VCH, Weinheim, Germany 1975.
26 National Bureau of Standards Circulars.
27 R. W. Douglass, F. C. Holden, R. J. Jaffee: High-
Temperature Properties and Alloying Behavior of the
Refractory Platinum-group Metals, Battelle Memorial
Institute, Columbus 1959.
28 E. M. Savitskii, V. P. Polyakova, M. A. Tylkina: Palla-
dium Alloys, Primary Sources/Publ., New York 1968.
29 Doduco: Datenbuch – Handbuch f€ur Techniker und
Kaufleute, 2nd ed., Pforzheim 1977.
30 The Platinum Asociation: Platinum, Production, Prop-
erties and Applications, a Short Course, Hill & Knowl-
ten, Frankfurt 1989.
31 Ullmann 5th ed., A21, 75 ff.
32 Kirk-Othmer, 4th ed., 19, 349.
33 Degussa: Platinger€ate – Best€andigkeit und Handha-
bung, Hanau 1976.
34 R. Bock: Aufschlußmethoden der anorganischen und
organischen Chemie, VCH, Weinheim, Germany
1972.
35 M. Fieberg, H. Renner, G. Schlamp in G. L. Trigg, E. H.
Immergut (eds.): Encyclopedia of Applied Physics,
vol. 18, Wiley– VCH, Weinheim, Germany 1997,
pp. 235 – 279.
36 Gmelin 8th ed., System no. 68 (Platin), 65 (Palladium),
64 (Rhodium), 67 (Iridium), 63 (Ruthenium), 66
(Osmium).
37 E. Merian:Metals and their Compounds in the Environ-
ment – Occurrence, Analysis, and Biological Rele-
vance, 2nd ed., VCH, Weinheim, Germany 1991.
38 E. Merian (ed.): Metalle in der Umwelt, VCH, Wein-
heim, Germany 1984.
39 R. G. Schwab, ‘‘Was wissen wir €uber die tieferen
Schichten der Erde ?’’, Angew. Chem. 86 (1974)
612 – 624.
40 C. Bresch, Zwischenstufe Leben, Piper-Verlag,
M€unchen 1977.
41 W. Schreiter: Seltene Metalle, vol. 2, Dtsch. Verlag f.
Grundstoffind., Leipzig 1961.
42 H. Schneiderh€ohn: Erzlagerst€atten, Fischer Verlag,
Stuttgart 1962.
43 V. Tafel: Lehrbuch der Metallh€uttenkunde, vol. 1, Hir-
zel Vlg., Leipzig 1951.
44 A. Cissarz: ‘‘Verbreitung der Platinmetalle, ihre la-
gerst€attenkundliche Stellung und ihre Anteile an der
Weltproduktion, Erzmetall 25 (1972) 7 – 16.
45 Gesellschaft Deutscher Metallh€utten- und Bergleute
(ed.): ‘‘Edelmetalle – Exploration und Gewinnung,’’
Schriftreihe der GDMB, no. 44, VCH, Weinheim,
Germany 1986.
46 W. Gocht:Handbuch der Metallm€arkte-Erzvorkommen,
Metallgewinnung, Metallverwendung, Preisbildung,
Handelsregelungen, 2nd ed., Springer-Verlag, Berlin
1985.
47 F. Pawlek, Metallh€uttenkunde, Walter de Gruyter Ver-
lag, Berlin 1983.
48 Winnacker-K€uchler, 4th ed., 4, pp. 541 – 572.
49 R. L. Bates, J. A. Jackson (eds.): Glossary of Geology,
2nd ed., American Geological Institute, 1980.
50 J. R. Loebenstein: Mineral Commodity Profiles, 1984,
Bureau of Mines, US Department of the Interior, Wa-
shington, annually.
51 V. I. Smirnov (ed.): Ore Deposits of the USSR, vol. II,
Pitman Publishing, 1977.
52 C. B. Coetzee: Mineral Resources of the Republic of
South Africa, Department of Mines, Pretoria 1976.
53 J. Lurie: South African Geology for Mining, Metallur-
gical, Hydrological and Civil Engineering, McGraw-
Hill, Johannesburg 1981.
54 W. Herzberg: ‘‘Der Bergbau S€udafrikas,’’ Metall 37
(1984) 407 – 411.
55 R. C. Hochreiter, D. C. Kennedy, W. Muir, A. I.
Woods: ‘‘Platinum in South Africa’’, J. S. Afr. Inst. Min
& Metall. 85 (1985) no. 6, 165.
56 A.M. Edwards, M. H. Silk: ‘‘Platinum in South Africa,’’
Mintek Special Publication, no. 12, Randburg 1987.
57 H. Renner, U. Tr€obs: ‘‘Rohstoffprofil Ruthenium’’,Me-
tall 42 (1988) 714 – 716.
58 H. Renner, U. Tr€obs: ‘‘Rohstoffprofil Osmium’’,Metall
44 (1990) 687–690.
59 H. Renner, U. Tr€obs: ‘‘Rohstoffprofil Iridium’’, Metall
40 (1986) 726 – 730.
60 H. Renner, U. Tr€obs: ‘‘Rohstoffprofil Rhodium’’,Metall
38 (1984) 1002 – 1005.
61 O. Stoppinski: Platinum Group Metals, National Re-
search Council, Washington 1977.
62 Hargreaves, Williamson (eds.): The Platinum Industry,
Prospects in Recovery, Shearson/American Express,
London 1984.
63 L. B. Cabril: Platinum-Group Elements, The Canadian
Institute of Mining and Metallurgy, Ottawa 1981.
64 ‘‘Metalle aus dem Meteoritenkrater – Nickel, Kupfer,
und Platin in Sudburry’’, Neue Z€urcher Zeitung 1990,
no. 258, Nov. 7.
65 T. P. Mohide: Platinum Group Metals – Ontario and
the World, Ministry of Natural Resources, Ontario
1979.
66 Mintek: ‘‘The Sucessful Development of an Industrial
Process for the Recovery of Platinum-groupMetals from
the UG-2 Reef, Application Report, no. 1, Randburg,
1987.
Vol. 28 Platinum Group Metals and Compounds 381

67 Mintek: ‘‘Recovery of PGMs from the UG-2 Reef’’,
Research Digest, no. 29, Randburg, 1988.
68 G. Hempel: ‘‘Wozu Polarforschung ?’’, Erzmetall 37
(1984) 577 – 584.
69 H. Renner:Behandlung und Verwertung von verbrauch-
ten edelmetallhaltigen Katalysatoren, M€ull- und Ab-
fallbeseitigung (M€ullhandbuch), 2nd ed., no. 8595, E.
Schmidt-Verlag, Berlin 1987.
70 W. Hasenpusch: ‘‘Aufarbeitung von Autoabgaskataly-
satoren – Die oberirdische Platin-Mine’’, EFCE Publ.
Ser. 10 (1989) no. 79.
71 H.Giegerich,C.Hensel: ‘‘Autokat-Recycling – einBei-
trag zur k€unftigen Edelmetallversorgung’’, Metall 44
(1990) 684 – 687.
72 G. A. Jensen: ‘‘Platinum-group Metals as a Possible
Byproduct of Nuclear Reactions,’’ Precious Met. Proc.
Int. Precious Met. Inst. Conf., 9th Meeting, Allentown,
Pa. 1985, 235 – 252.
73 G. Buckow et al.: Verwertung von Reststoffen aus einer
Wiederaufbereitungsanlage, Gesellschaft f€ur Reaktor-
sicherheit (GRS), K€oln 1986.
74 H. R€othemeyer: Endlagerung radioaktiver Abf€alle,VCH-Verlagsges., Weinheim, Germany 1991.
75 R. Saager: Metallische Rohstoffe von Antimon bis Zir-
konium, Bank Vontobel, Z€urich 1984.
76 V. E. McKelvey, E. N. Cameron: The Mineral Potential
of United States 1975 – 2000, University Wisconsin
Press, Madison 1973.
77 D. Meadows, E. Zahn, P. Milling: Die Grenzen des
Wachstums, 15th ed., DeutscheVerlagsanstalt, Stuttgart
1990.
78 L. Trueb: ‘‘Das Platin und seine f€unf Geschwister einer
Geschichte der Platinmetalle’’, Neue Z€urcher Zeitung
1989, no. 298 (Dec. 21).
79 L. Trueb: ‘‘Platinmetalle – Vom Bergwerk zum Kata-
lysator’’, Neue Z€urcher Zeitung 1989, no. 194
(Jun. 14).
80 O. S. North: Mineral Exploration, Mining and Proces-
sing Patents, McGraw Hill, New York, annually.
81 E. Henglein: Lexikon Chemische Technik, VCH, Wein-
heim, Germany 1988.
82 G.Agricola:VomBerg- undH€uttenwesen, DTV-Verlag,
M€unchen 1980.
83 W. Truthe: Hundert Jahre Gold- und Silber-Scheidung
nebst Gewinnung der Platinmetalle, Degussa 1943,
unpublished.
84 E. Jackson: Hydrometallurgical Extraction and Recla-
mation, Ellis Horwood, Chichester 1986.
85 E. M. Savitskii: Handbook of precious metals, Hemi-
sphere Publishing Corp., New York 1989.
86 J. C. Mostert, P. N. Roberts: ‘‘Electric Smelting at
Rustenburg PlatinumMines Ltd. of Nickel-Copper Con-
centrates containing Platinum Group Metals’’, J. S. Afr.
Inst. Min. Metall. 73 (1973) no. 9, 290.
87 C. F. Brugman, D. G. Kerfoot, ‘‘Treatment of Nickel-
Copper Matte at Western Platinum by the Sherritt
Acid Leach Process,’’ International Nickel Conference,
Toronto, Canada, 1985.
88 Degussa: Metall – Forschung und Entwicklung im
Degussa-Forschungszentrum Wolfgang, Frankfurt
1991.
89 G. Schlamp: ‘‘Noble Metals and Their Alloys,’’ in R.
Cahn (ed.): Materials Science and Technology, vols.
8/9, ‘‘Structure and Properties of Nonferrous Metals’’,
VCH, Weinheim, Germany 1995, pp. 467 – 587.
90 T. B. Massalski (ed.): Binary Alloys Phase Diagrams,
vols. 1 and 2, ASM Metals Park, OH 1986.
91 T. B. Massalski (ed.): et al. Noble Metal Alloys Phase
Diagrams, TheMetallurgical Soc.USAWarrendale, PA
1986.
92 G. Petzow, G. Effenberg: Ternary Alloys, VCH, Wein-
heim, Germany 1988.
93 J. E. Hoffmann, J. Met. 40 (1988) no. 6, 40 – 44.
94 E. J. Kohlmeyer, K. von Sprenger, Z. Anorg. Chem. 257
(1948) 199 – 214.
95 V. Jung, J. Met. 33 (1981) no. 10, 42 – 44.
96 Degussa, DE 2 059 235, 1970 (V. Jung).
97 V. Jung: ‘‘The Treatment of Automotive Exhaust Cat-
alysts’’, TMS/GDMB-Conference, Productivity and
Technology in the Metallurgical Industries, K€oln, Sept.1989.
98 D. Meredith et al., Stahl þ Eisen 17 (1988) 796 –
800.
99 S. E. Stenkvist, Steel Times, 1985, no. 10, 480 – 483.
100 Iron and Steel Society, Plasma Technology in Metallur-
gical Processing, Grand Junction, CO/USA, 1987.
101 Degussa, DE 3 223 501, 1983 (H. Renner, K. Kleis,
R. Schlodder).
102 W. C. Heraeus GmbH, DE 2 911 193, 1979.
103 International Precious Metals Institute (ed.): Current
Publications and Proceedings of Conferences and Semi-
nars, Dave Schneller, Boulder 1979, pp. 92 ff.
104 F. A. Cotton, G. Wilkinson: Advanced Inorganic Chem-
istry, 4th ed., John Wiley & Sons, New York 1980,
pp. 950 – 966.
105 F. A. Cotton, G. Wilkinson: Anorganische Chemie, 4th
ed., VCH, Weinheim, Germany 1985.
106 G. Wilkinson, R. D. Gillard, J. A. McCleaverty: Com-
prehensive Coordination Chemistry, vol. 5, Pergamon
Press, Oxford 1987.
107 F. R.Hartley:TheChemistry of PlatinumandPalladium,
John Wiley & Sons, New York 1973.
108 S. E. Livingstone, in J. C. Bailar et al. (eds.): Compre-
hensive Inorganic Chemistry, vol. III. Pergamon Press,
New York 1973, pp. 163 – 1370.
109 N. N. Greenwood, A. Earnshaw: Chemie der Elemente,
VCH, Weinheim, Germany 1988.
110 S. Daamach, G. Cote, D. Bauer: ‘‘Separation of Plati-
num-group Metals in Hydrochloric media: Solvent Ex-
traction of Palladium(II) with Dialkyl Sulfides’’, IPMI
1977, No. 22.
111 D. W. Agers, E. R. de Ment: ‘‘The Evaluation of New
LIX Reagents for the Extraction of Copper and Sugges-
tions for the Design of Commercial Mixer-Settler
Plants,’’ The Metallurgical Society of AIME, Paper
no. A 72 – 82, 1972.
382 Platinum Group Metals and Compounds Vol. 28

112 Matthey Rustenburg Ref., GB 2 013 644, 1978 1979
(K. J. Shanton, R. A. Grant).
113 MattheyRustenburgRef., DE-OS2 459 099, 1973 1975
(J. B. Payne).
114 L. Manziek (ed.): ‘‘Precious Metals Recovery and Re-
fining’’, Proceedings of the Precious Metals Recovery
and Refining Seminar, Nov. 12 – 14, 1989, Scottsdale,
Arizona.
115 V. H. Apprahamian, G. P. Demopoulos, G. B. Harris:
‘‘The Behavior of Impurities during the Solvent Extrac-
tion of Platinum Metals with TN 1911 Extractants’’ in
Precious Metals 1991, IPMI, Allentown, Pa. 1991,
p. 143.
116 W. Hasenpusch: ‘‘Die Trennung der Platinmetalle’’,
CLB, Chem. Labor Betr. 38 (1987) 454.
117 Ch. Baes, R. E. Mesmer: The Hydrolysis of Cations, J.
Wiley and Sons, New York 1976.
118 M. Knothe: ‘‘Untersuchungen zum Verhalten des Rh,
Pd, Ir und Pt im Chloridmedium an Anionenaus-
tauschern’’, Z. Anorg. Allgem. Chem. 463 (1980)
204 – 212.
119 A. P. Evers, R. J. Edwards, M. Fieberg, DE
27 26 558 B 2, 1976/1981.
120 G. Schmuckler, US 4 885 143, 1989.
121 W. Prodinger: Organische F€allungsmittel in der quan-
titativen Analyse, Enke Verlag, Stuttgart 1954.
122 J. Fries, H. Getrost, Organische Reagentien f€ur die
Spurenanalyse, E. Merck, Darmstadt 1977.
123 D. A. Boyd: A Novel Approach to PGMRefining, IPMI,
1992.
124 R. Marr, H.-J. Bart: ‘‘Metallsalz-Extraktion’’, Chem.-
Ing.-Tech. 54 (1979) 119 – 129.
125 L. Alders: Liquid-Liquid Extraction – Theory and Lab-
oratory Practice, Elsevier Publishing, Amsterdam
1959.
126 G. M. Ritcey, A. W. Ashbrook: Solvent Extraction:
Principles and Application to ProcessMetallurgy, Else-
vier, New York 1984.
127 F. Habashi: ‘‘Hydrometallurgy’’, Principles of Extrac-
tive Metallurgy, vol. 2, Gordon and Breach, New York
1970.
128 G. H. Morrison, H. Freiser: Solvent Extraction in
Analytical Chemistry, J. Wiley & Sons, New York
1957.
129 E. J. Barnes, J. D. Edwards: ‘‘Solvent Extractions at
Inco’s Acton Precious Metal Refinery’’, Chem. & Ind.
(London) 1982 no. 3, 151 – 164.
130 R. J. Edwards: ‘‘Refining of the Platinum-group Me-
tals’’, J. Met. 28 (1976) no. 8, 4 – 9.
131 R. J. Edwards: ‘‘Selective Solvent Extractants for the
Refining of PlatinumMetals’’, Proceedings of the Inter-
national Solvent Extraction Conference ISEC 7, Vol. 1,
CIM, Special Vol. 21, 24 (1979).
132 D. S. Flett: ‘‘Solvent Extraction in Precious Metal Re-
fining’’, IMPT Seminar, London 1982.
133 E. M. Elkin, P. W. Bennett: ‘‘A new Technique for the
Recovery of Palladium and Platinum from Gold Elec-
trolyte’’, J. Met. 17 (1965) no. 3, 252 –254.
134 G. Brauer: Handbuch der pr€aparativen anorganischen
Chemie, vol. 3, Enke, Stuttgart 1980.
135 R. Gilchrist: ‘‘The Platinum Metals’’, Chem. Rev. 32
(1943) 277 – 372.
136 Matthey Rustenburg Ref., GB 2 065 092, 1980 (R. A.
Grant).
137 Matthey Rustenburg Refiners: ‘‘The Separation Chem-
istry of Rhodium and Iridium’’,Proceeding of a Seminar
of the International Precious Metals Institute, IMPI,
Scottsdale, Arizona, 1989.
138 P. Charlesworth: ‘‘Separating the Platinum Group Me-
tals by Liquid-Liquid Extraction’’, Platinum Met. Rev.
25 (1981) 106 – 112.
139 L. R. P. Reavill, P. Charlesworth: ‘‘The Application of
Solvent Extraction to PlatinumGroupMetals Refining’’,
Proceedings of the International Solvent Extraction
Conference, ISEC ’80 vol. 3, Li�ege 1980.
140 M. J. Clearne, P. Charlesworth, D. J. Bryson: ‘‘Solent
Extraction in Platinum Group Metal Processing’’, J.
Chem. Technol. Biotechnol. 29 (1979) 210 –214.
141 Matthey Rustenburg Ref., US 3 979 207, 1974 (J. J.
McGregor).
142 INCO, US 4 390 366, 1983 (R. K. Lea, J. D. Edwards,
D. F. Colton).
143 M. Fieberg, R. J. Edwards: ‘‘The Extraction of Gold
from Chloride Solutions’’, Rep. Natl. Inst. Metall. (S.
Afr.) 1978, no. 1996.
144 A. S. Myerson, K. Toyokura (eds.): ‘‘Crystallisation as a
Separations Process,’’ ACS Symp. Ser. 438 (1990)
145 W. G€osele et al.: ‘‘Feststoffbildung durch Kristallisa-
tion und F€allung’’, Chem.-Ing.-Tech. 62 (1990) 544 –
552.
146 W. Hasenpusch, K. Bonin: ‘‘Kristallisation von Ammo-
nium-Hexachloroplatinat(IV),’’Chem.-Ztg. 1991, no. 5,
129 – 133.
147 M. Knothe, W. Hosenpusch, Inorg. Chem. 35 (1996)
4529 – 4530.
148 R. B. Wilson, W. D. Jacobs: ‘‘Separation of Iridium
from Rhodium by Extraction with Tributyl Phosphate’’,
Anal. Chem. 33 (1961) 1650 – 1652.
149 S. J. Tanaka: ‘‘The Recovery and Purification of Rhodi-
um using SuperligTM Technology from a Platinum-
group Metals Stream,’’ IPMI 1992.
150 Matthey Rustenburg Ref., US 4 382 067, 1982 1983
(R. A. Grant).
151 Matthey Rustenburg Ref., DE-OS 2 457 672, GB
5 682 673, 1973 1979 (J. J. McGregor).
152 PGM Industries, DE-OS 27 26 390, 1977 1979 (J. Baltz,
E. Coltrinari).
153 National Institute for Metallurgy (NIM), GB 1 490 815,
1974 1977 (R. J. Edwards).
154 AT & TTechnologies, US 4 479 922, 1983 (R.Haynes,
A. Jackson).
155 H. Renner: ‘‘The Selective Solvent Extraction of Palla-
dium by the Use of Di-Normal-Hexylsulfide’’, Rep.-
MINTEK, 1985, no. 217.
156 G. P. Demopoulos: ‘‘Solvent Extraction in Precious
Metals Refining’’, J. Met. 1986 no. 6, 13 – 17.
Vol. 28 Platinum Group Metals and Compounds 383

157 G. P. Demopoulos: ‘‘A Radically New Solvent Extrac-
tion Process for Rhodium Recovery’’, in Precious Me-
tals 1992, IPMI, 1992.
158 M. Grote, U. H€uppe, A. Kettrop: ‘‘Solvent Extraction of
Noble Metals by Formazons(II),’’ Hydrometallurgy 19
(1987) 51 – 68.
159 K.-H. K€onig: ‘‘Zur Solventextraktion von Platinmetal-
len,’’ Achema, Frankfurt 1985.
160 M.Knothe: ‘‘Die F€allung des PdII als Pd(NH3)2Cl2 sowie
des Verhaltens verschiedener Begleitelemente,’’ Z. An-
org. Allg. Chem. 503 (1983) 213 – 223.
161 A. P. Evers, R. J. Edwards, M. M. Fieberg, SA
7 63 681, 1976.
162 R. J. Edwards, M. M. Fieberg, W. te Riele, B. C. Want,
US 4 155 750, 1979.
163 A. Prior, K. H. Ohrbach, A. Kettrup, G. Matuschek:
‘‘Calcination of Platinum Group Metal Complexes to
Form Pure Metals’’, IPMI Seminar, 1988.
164 A. Prior, B. Lerwill: ‘‘Cost Management Strategies in
Refineries in the 1990s’’, IPMI Seminar, Tempe, Ari-
zona, 1991.
165 V. S. Khain, E. V. Fomina: ‘‘Reduction of OsO4 by
Sodium Tetrahydroborate in Acidic Medium’’, Russ.
J. Inorg. Chem. (Engl. Transl.) 25 (1980) 307 – 308.
166 Degussa: Edelmetall-Taschenbuch, Frankfurt 1967.
167 Degussa: Edelmetall-Taschenbuch, 2. Aufl., H€uthig-
Verlag, Heidelberg 1995.
168 E. M. Wise: Palladium, Academic Press, New York
1968.
169 J. A. Federov: Rhodium, Nauka Vlg., Moskau 1966.
170 A. F. Hollemann, E. Wiberg, N. Wiberg: Lehrbuch der
Anorganischen Chemie, 101. Aufl., W. de Gruyter,
Berlin – New York 1995, pp. 1546 (Ru, Os), 1572 (Rh,
Ir), 1602 (Pd, Pt), 1628 – 1716 (Carbonyle).
171 J. Huheey, E. Keiter, R. Keiter (eds.): Anorganische
Chemie, 2. Aufl., W. de Gruyter, Berlin – New York
1995, pp. 735 – 864.
172 A. S. Darling: ‘‘Some Properties and Applications of the
Platinum Group Metals’’, Int. Met. Rev. 18 (1973) 91 –
122.
173 E.Raub:DieEdelmetalleundihreLegierungen, Springer-
Verlag, Berlin 1940.
174 E. J€aneke: Kurzgefaßtes Handbuch aller Legierungen,
C. Winter Verlag, Heidelberg 1949.
175 J. L. Haughton, A. Prince: The Constitution Diagrams of
Alloys,ABibliography, Institute ofMetals, London1956.
176 A. Prince:Multicomponent Alloy Constitution, Bibliog-
raphy 1955 – 1973, Metals Society, London 1978.
177 A. Prince:Multicomponent Alloy Constitution, Bibliog-
raphy 1974 – 1977, Metals Society, London 1981.
178 W. Home-Rothery, G. V. Raynor: The Structure of
Metals and Alloys, Institute of Metals, London 1954.
179 M. Hansen: Der Aufbau der Zweistoff-Legierungen,
Springer-Verlag, Berlin 1936.
180 M. Hansen, K. Anderko: Constitution of Binary Alloys,
2nd ed., McGraw-Hill, New York 1958.
181 R. P. Eliott: Constitution of Binary Alloys, 1st suppl.,
McGraw-Hill, New York 1965.
182 F. A. Shunk: Constitution of Binary Alloys, 2nd suppl.,
McGraw-Hill, New York 1969.
183 W. G. Moffatt: The Handbook of Binary Phase Dia-
grams, General Electric Company, Schenectady, N.Y.,
1976 1977.
184 G. Petzow, G. Effenberg: Ternary Alloys, VCH, Wein-
heim, Germany 1988 onwards.
185 J. C. Chaston: ‘‘The Purity of Platinum’’, PlatinumMet.
Rev. 15 (1971) 122 – 128.
186 A.Wogrinz:Analytische Chemie der Edelmetalle, Enke
Verlag, Stuttgart 1936.
187 F. Ensslin:Edelmetall-Analyse, Springer-Verlag, Berlin
1964.
188 Chemikerausschuß der GDMB (ed.): Analyse der Me-
talle, 2nd ed., Springer-Verlag, Berlin 1961.
189 F. E. Beamish, J. C. van Loon:Analysis of NobleMetals,
Academic Press, New York 1977.
190 S. Kallmann: ‘‘A Survey of the Determination of the
Platinum Group Elements’’, Talanta 34 (1987) no. 8,
677 – 698.
191 H. G. Sigel, A. Sigel (eds.): Handbook on Toxicity of
Inorganic Compounds, Marcel Dekker, New York
1988.
192 E. D. Goldberg et al.: ‘‘Determination of Platinum
Sediments,’’ Anal. Chem. 58 (1986) 616 – 620.
193 H.-M. L€uschow: ‘‘Probenahme, Theorie und Praxis’’,
Schriftenreihe der GDMB, VCH, Weinheim 1980.
194 H.-M. L€uschow: ‘‘Zur Probenahme von Edelmetallen,
Teil 1: Probenahme von metallischen Materialien’’,
Erzmetall 42 (1989) 153 – 159.
195 H.-G. Bachmann, E. Koberstein, R. Straub: Chemie-
Technik 7 (1978) 441 – 446.
196 J. Suchomel, Fresenius’ Z. Anal. Chem. 300 (1980)
257 – 266; Fresenius’ Z. Anal. Chem. 307 (1981)
14 – 18.
197 B.Welz:Atomic Absorption Spectrometry, VCH,Wein-
heim, Germany 1985.
198 G. J€ager: Uber die Platinbestimmung in Platforming-
Katalysatoren, Dissertation, Mainz 1957.
199 W. Diehl: ‘‘Die quantitative Bestimmung der Verunrei-
nigungen der AG, Au, Pt und Rh mit einem 3,4-m-
Gitterspektrographen’’, Metall (Berlin) 23 (1969)
587 – 589.
200 F. Mylius: ‘‘Reinheitsgrade von Metallen des Handels’’,
Z. Anorg. Chem. 74 (1912) 407.
201 F. Mylius, A. Mazzuchelli: ‘‘€uber die Platinanalyse,’’ Z.
Anorg. Allg. Chem. 89 (1914) 1 – 38.
202 DIN 43 760, 1968.
203 DIN 43 710, 1977.
204 Gmelin, Platin, suppl. vol. A 1, 8.
205 H.Wolf: ‘‘Die Edelmetalle in Forschung und Industrie’’,
Metall 12 (1958) no. 7, 3 – 16.
206 J. Sagoschen: ‘‘Platin-Laborger€ate’’, Chem. Ztg. 88
(1964) 420 – 429.
207 G. Reinacher: ‘‘Platin-Ger€ate im Labor,’’ Chem. Anla-
gen þ Verfahren 1970, no. 1/2, 61 – 63.
208 Degussa: Platin-Ger€ate f€ur Labor und Praxis, no. 1 ff.,
Frankfurt 1977 ff.
384 Platinum Group Metals and Compounds Vol. 28

209 G. Reinacher: ‘‘Uber die Gewichtskonstanz von Ger€aten
aus der Legierung Platin/3% Iridium bei analytischen
Operationen’’, Werkst. Korros. 1964, no. 1, 84 – 88.
210 G. Reinacher, H. Roters: ‘‘Neuere Anwendungen von
Platinwerkstoffen in Chemie und Glasindustrie’’,Metall
23 (1969), 570 – 575.
211 G. Reinacher: ‘‘Anwendung von Platin-Werkstoffen in
Glash€utten’’, Chem. Anlagenþ Verfahren 1975, no. 10,
27 – 34.
212 D.B€ottger: ‘‘TheUse of Platinum in theGlass Industry,’’
Glass 62 (1985) no. 5, 177 – 178; Metall 39 (1985)
748 – 750.
213 M. Rowe: ‘‘NobleMetals in the Glass Industry,’’Ceram.
Eng. Sci. Proc. 6 (1985) 256 – 268.
214 W. Funk, R. Schumm: ‘‘Spinnd€usen – Bauteile f€ur die
Chemiefaserindustrie’’, Chemiefasern þ Text. Anwen-
dungstech./Text.-Ind. 22 (1972) 518 – 521.
215 J. R. Anderson, M. Baudart: Catalysis, Science and
Technology, vols. 12, Springer-Verlag, Berlin 1981.
216 Fonds der Chemischen Industrie: ‘‘Katalyse,’’ Foliens-
erie, no. 19, Frankfurt 1986.
217 K. Weissermel, H. J. Arpe: Industrial Organic Chemis-
try, VCH, Weinheim, Germany 1978.
218 P. N. Rylander: Catalysis Hydrogenation over Platinum
Metals, Academic Press, New York, 1967.
219 W. H€olderich, M. Schearzmann, W. D. Mross: ‘‘Hetero-
gene Katalysatoren in der chemischen Industrie,’’ Erz-
metall 39 (1986) 292 – 298.
220 A. J. Bird, A. B. Stiles:Catalyst Supports and Supported
Catalysts, Butterworth, London 1987.
221 J. Falbe, U. Haserodt: Katalysatoren, Tenside und
Mineral€oladditive, Thieme Verlag, Stuttgart 1978.
222 D.M. Little:Catalytic Reforming, Pennwall Pub., Tulsa
1985.
223 W. Ostwald, E. Brauer, in H. Huben (ed.): Festschrift
zum f€unfzigj€ahrigen Bestehen der Platinschmelze G.
Siebert GmbH, Hanau, Hanau 1931, pp. 240 – 256.
224 H. Holzmann: ‘‘€uber die katalytische Oxidation von
Ammoniak bei der industriellen Salpeters€aure-Herstel-
lung’’, Chem.-Ing.-Tech. 39 (1967) 89 – 95.
225 F. Sperner, W. Hohmann: ‘‘Rhodium-Platinum Gauzes
for Ammonia Oxidation,’’ PlatinumMet. Rev. 20 (1976)
12 – 20.
226 H. Connor: ‘‘The Role of PlatinumAlloys Gauzes in the
Ammonia Oxidation Process,’’ Platinum Met. Rev. 11
(1967) 2 – 9, 60 – 69.
227 E. G. Schlosser: ‘‘Die Katalysator-Desaktivierung und
-Vergiftung in verfahrenstechnischer Sicht,’’ Chem.-
Ing.-Tech. 47 (1975) 997 – 1005.
228 R. Ugo: ‘‘Aspects of Homogeneous Catalysis,’’ D. Re-
ibel Publ, Dordrecht 1970.
229 F. J. Smith: ‘‘New Technology for Industrial Hydrofor-
mulation’’, Platinum Met. Rev. 19 (1975) 93 –95.
230 K.-H. Schmidt: ‘‘Neuentwicklungen in der homogenen
Katalyse’’, Chem. Ind. 37 (1985) 762 – 765.
231 J. Osborn, G. Wilkinson, Chem. Commun. 1965, 131.
232 H. Brunner: ‘‘Rhodium-Katalysatoren f€ur die enantio-
selektive Hydrosylierung – ein neues Konzept zur
Entwicklung asymmetrischer Katalysatoren’’, Angew.
Chem. 95 (1985) 921 – 931.
233 G. J. K. Acres, B. J. Cooper: ‘‘Automobile Emission
Control Systems’’, PlatinumMet. Rev. 16 (1972) 74 –86.
234 W. Weigert, E. Koberstein, E. Lakatos: ‘‘Katalysatoren
zur Reinigung vonAutoabgasen’’,Chem.-Ztg. 97 (1973)
469 – 478.
235 O. Stopinski: Platinum-group Metals, National Re-
search Council, Washington 1977.
236 E. Koberstein: ‘‘Katalysatoren zur Reinigung von Auto-
abgasen’’,Chemie in unserer Zeit 18 (1984) no. 2, 37 –
45.
237 A. Keil: Werkstoffe f€ur elektrische Kontakte, Springer,
Berlin 1960.
238 A. von Krusenstjern: Edelmetall-Galvanotechnik,
Leuze-Vlg., Saulgau 1970.
239 D. Schlain et al.: ‘‘Elektrodesposition of Platinum Me-
tals from Molten Cyanides,’’ Platinum Met. Rev. 21
(1977) 38 – 42.
240 J. H. Notton: ‘‘Fused Salt Platinum Plating for Industrial
Applications,’’ Platinum Met. Rev. 21 (1977) 122 – 128.
241 H.-H. Beyer, F. Simon, Metall 34 (1980) 1016 – 1018.
242 J. Wurm, S. Sari: ‘‘Verchromen mit platinierten Titana-
noden,’’ Oberfl€ache-Surf. 1989 no. 12, 19 – 21.
243 F. Endter: ‘‘Die technische Synthese von Cyanwasserst-
off ausMethan undAmmoniakohneZusatz vonSauerst-
off,’’ Chem.-Ing.-Tech. 30 (1958) 305 –310.
244 L. B. Hunt: ‘‘The Platinum in the Decoration of Porce-
lain and Pottery,’’ Platinum Met. Rev. 22 (1978) 138 –
148.
245 Degussa: Glanzgoldfibel, Frankfurt 1979.
246 V. de Nora, J.-W. K€uhn-von Burgsdorff: ‘‘Der Beitrag
der dimensionsstabilen Anoden (DSA) zur Chlortech-
nologie,’’ Chem.-Ing.-Tech. 47 (1975) 125 –128.
247 J.-W. K€uhn-von Burgsdorff: ‘‘Anwendung dimensions-
stabiler Elektroden in der Elektrochemie,’’ Chem. Ind.
(D€usseldorf) 28 (1976) 406 – 408.
248 Johnson Mattey: Platinum, London, anually.
249 IMPT (ed.): Precious Metals – New and Review, vol. 1
ff, Allentown, 1976 ff.
250 Degussa, Gesch€aftsbericht, Frankfurt, annually.
251 Knies: ‘‘Die Marktsituation der Edelmetalle’’, Metall,
annually.
252 J. H. Jolly: Platinum-group Metals, US Dep. of the
Interior, Bureau of Mines, Washington 1968 ff.
253 K. Bolz, U. Harms, P. Pissulla, H. Schmidt: Gold,
Platinmetalle und Diamanten in der sowjetischen Han-
delspolitik, Verlag Weltarchiv, Hamburg 1985.
254 H.Kutzer: ‘‘Metalle – zwischenMangel und Uberfluß’’,
Handelsblatt-Schriftreihe, D€usseldorf, 1981.
255 F. H. Kemper: ‘‘Metalle – Belastung f€ur den
Menschen?’’ Erzmetall 40 (1987) 541 – 549.
256 J. Pepys, C. A. C. Pickering, E. G. Hughes: ‘‘Asthma due
to Inhaled Chemical Agents – Complex Salts of Plati-
num,’’ Clin. Allergy 2 (1972) 391 – 396.
257 M. C. Cleare, E. Hughes, B. Jacoby, J. Pepys: ‘‘Imme-
diate (typ I) Allergic Responses to Platinum Com-
pounds,’’ Clin. Allergy 6 (1976) 183 – 195.
Vol. 28 Platinum Group Metals and Compounds 385

258 Bundesverband der Pharmazeutischen Industrie: Rote
Liste – Verzeichnis von Fertigarzneimitteln, Edition
Cantor, Aulendorf, annually.
259 A. I. G. McLaughlin et al.: ‘‘Toxic Manifestations of
Osmium Tetroxide,’’ Brit. J. Ind. Med. 2 (1946) 183.
260 G. Rosner, R. F. Hertel: ‘‘Gef€ahrdungspotential von
Platinemissionen aus Autoabgas-Katalysatoren’’,
Staub-Reinhalt. Luft 46 (1986) 281 – 285.
261 A. B€artsch, C. Schlatter: ‘‘Platinemissionen aus Auto-
mobil-Katalysatoren,’’ Schriftenreihe Umweltschutz,
no. 55, Bundesamt f€ur Umweltschutz, Bern 1988.
262 W. Hasenpusch: ‘‘Explosive Reactions with Platinum-
group metals,’’ Chem.-Ztg. 111 (1987) 57 – 60.
263 W. Beck, W. Weigand: ‘‘Chemistry of Platinum Me-
tals,’’ Prax. Naturwiss. Chem. 40 (1991) 9 – 14.
264 S. D. Robinson, Annu. Rep. Prog. Chem., Sect. A: Inorg.
Chem. 85 (1988) 209 – 44.
265 S. D. Robinson, Annu. Rep. Prog. Chem., Sect. A: Inorg.
Chem. 87 (1990) 105 – 29.
266 S. D. Robinson, Annu. Rep. Prog. Chem., Sect. A: Inorg.
Chem. 88 (1991) 183 – 209.
267 D. T. Thompson, Annu. Rep. Prog. Chem., Sect. A:
Inorg. Chem. 89 (1992) 209 – 42.
268 G. Reid, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem.
90 (1993) 227 – 49.
269 M. A. Halcrow, Annu. Rep. Prog. Chem., Sect. A: Inorg.
Chem. 93 (1996) 241 – 267.
270 G. Reid, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem.
91 (1994) 261 – 94.
271 T. J. Ohara, Plat. Met. Rev. 39 (1995) 54 – 62.
272 T.-M. Park et al., Talanta 44 (1197) no. 6, 973 – 978.
273 M. J. Abrams, Plat. Met. Rev. 39 (1995) 14 – 18.
274 S. P. Fricker (ed.):Metal compounds in Cancer Therapy;
Metals in Health and Diseases Series, vol. 1, Chapman
and Hall, London 1994.
275 S. P. Fricker, Ruthenium,NitricOxide andDisease,Plat.
Met. Rev. 39 (1995) no. 4, 150 – 154.
276 M. J. Moss, Plat. Met. Rev. 39 (1995) 33 – 36.
277 G. R. Newcome et al., J. Chem. Soc., Chem. Commun.
(1993) 925.
278 G. R. Newcome et al., Macromol. Symp. 98 (1995)
467.
279 G. R. Newcome et al., Angew. Chem. Int. Ed. Engl. 34
(1995) 2023.
280 D. T. Thompson, Plat. Met. Rev. 40 (1996) 23 – 25.
281 D. T. Thompson, Plat. Met. Rev. 39 (1995) 108 – 11.
282 V. I. Sokolov, V. Bashilov, Plat. Met. Rev. 42 (1998)
18 – 24.
283 T. Lizhen, G. Jinxing, Plat. Met. Rev. 38 (1994) 98 –
108.
284 G. L. Selman, J. G.Day,A. A. Bourne,Plat.Met. Rev. 18
(1974) no. 2, 46 – 75.
285 Q. Zhang, D. Zhang, S. Jia, Plat. Met. Rev. 39 (1995)
167 – 171.
286 J. H. Kim, S.-C. Shin, Jpn. J. Appl. Phys. 35 (1996)
342 – 345.
287 Y. Tanaka et al., J. Magn. Magn. Mater. 170 (1997)
289 – 297.
288 T. N. Nguyen, P. A. Lee, H.-C. zur Loye, Science 271
(1996) 489 – 491.
289 G. Demazeau et al., J. Magn. Magn. Mater. 140 – 144
(1995) 165 – 166.
290 E. A. Franco-Ferreira, Plat. Met. Rev. 41 (1997) no. 4,
154 – 163.
291 R. W. Schutz, Plat. Met. Rev. 40 (1996) 54 – 61.
292 E. L. Semenova, N. Yu. Rusetskaya, V. M. Petyukh, V.
Ye. Listovnichiy, Plat. Met. Rev. 39 (1995) 174 – 179.
293 D. Goldberg, Y. Xu et al.,Mater. Lett. 22 (1995) nos. 5,
6, 241 – 248.
294 E. Herda, R. Higuchi-Rusli, Parangtopo,Mater. Lett. 30
(1997) nos. 5, 6, pp. 347 – 350.
295 G. A.Moore, Trans. Electrochem. Soc. 75 (1939) 237 –
269.
296 F. A. Lewis: The Palladium – Hydrogen System, Aca-
demic Press, New York 1967.
297 F. A. Lewis, Plat. Met. Rev. 38 (1994) 112 – 118.
298 G. Nelin, Phys. Stat. Solidi 45 (1971) no. 2, 527 – 536.
299 F. A. Lewis, Plat. Met. Rev. 26 (1982) 20 – 27, 70 – 78,
121 – 128.
300 J. B. Hunter, Plat. Met. Rev. 4 (1960) no. 4, 130 –
131.
301 A. G. Knapton, Plat. Met. Rev. 21 (1977) 44 – 50.
302 J. C. S. Booth, 11th World Hydrogen Energy Confer-
ence, Stuttgart, Germany, June 23 – 28, 1996.
303 E. Wicke, G. H. Nernst, Ber. Bunsenges. (1964) 224 –
235.
304 G. Sicking, Dissertation, M€unster, Germany, 1970.
305 K. U. Neumann et al., J. Magn. Magn. Mat. 137 (1994)
no. 3, 264 – 268.
306 A. Freund et al., Catalysis Today 27 (1996) 279 – 283.
307 K.-A. Starz, Third European Precious Metals Confer-
ence, Florence, Sept. 17 – 19, 1997.
308 M. Watanabe, K. Tsurumi, T. Mizukami, T. Nakamura,
P. Stonehart, J. Electrochem. Soc. 141 (1994) no. 10,
pp. 2659 – 2668.
309 T. R. Ralph, Plat. Met. Rev. 41 (1997) 102 – 113.
310 M. P. Hoghart, G. A. Hards, Plat. Met. Rev. 40 (1996)
150 – 159.
311 M.-L. Tercier, N. Parthasarathy, J. Buffle, Electroanal-
ysis 7 (1995) no. 1, 55 – 63.
312 I. M. Wolff, P. J. Hill, Plat. Met. Rev. 44 (2000) 158 –
166.
313 H.-M. L€uschow: Edelmetallanalytik, Eine Ubersicht
€uber bew€ahrte klassische und moderne Methoden, Heft
69 der Schriftenreihe der GDMB, Clausthal-Zellerfeld,
1993.
314 ISO StandardNo. 11210, ‘‘Determination of Platinum in
Platinum Jewellery Alloys – Gravimetric method after
precipitation of diammonium hexachlorolatinate’’.
315 ISO StandardNo. 11489, ‘‘Determination of Platinum in
Platinum Jewellery Alloys – Gravimetric Method after
Reduction with Mercury(I) Chloride’’.
316 ISO Standard No. 11494, ‘‘Determination of Palladium
in Palladium Jewellery Alloys – ICP-Solution-Spectro-
metric Method using Yttrium as Internal Standard
Element’’.
386 Platinum Group Metals and Compounds Vol. 28

317 ISO StandardNo. 11495, ‘‘Determination of Platinum in
Platinum Jewellery Alloys – ICP-Solution-Spectromet-
ric Method using Yttrium as Internal Standard
Element’’.
318 J. Roberts et al.: ‘‘Detection limits for trace elements in
high purity gold using a DC arc spectrograph with solid
state CID detection,’’ Appl. Note No. 60, Thermo Jarrell
Ash Corp. Franklin, MA 1995.
319 G. Ertl, H. Kn€ozinger, J. Weitkamp (eds.): Handbook
of Heterogeneous Catalysis, VCH, Weinheim,
Germany 1997. E. Drost et al., ‘‘Platinwerkstoffe f€urHochtemperatur-Einsatz,’’ Metall 50 (1996) nos. 7/8,
492.
320 E. Auer, A. Freund, J. Pietsch, T. Tacke, Applied Catal-
ysis A 173 (1998) 259 – 271.
321 H. Lindlar, Helvetica Chimica Acta 35 (1952) 446 –
450.
322 B. Cornils, W. A. Hermann (eds.): Applied Homoge-
neous Catalysis with Organometallic Compounds,
VCH, Weinheim, Germany 1996.
323 J. H. Jones, Plat. Met. Rev. 44 (2000) 94 – 105.
324 G. W. Parshall, S. D. Ittel: Homogeneous Catalysis,
Wiley Interscience, New York 1992.
325 B. Cornils, W. A. Hermann (eds.): Aqueous-Phase Or-
ganometallic Catalysis, Wiley-VCH, Weinheim, Ger-
many 1998.
326 B. Marciniec (ed.): Comprehensive Handbook on Hy-
drosilylation, Pergamon Press, Oxford 1992.
327 I. Ojima (ed.): Catalytic Asymmetric Synthesis, Wiley,
New York 1993.
328 R. Noyori (ed.): Asymmetric Catalysis in Organic Syn-
thesis, Wiley, New York 1994.
329 B. M. Trost, D. L. van Vranken, Angew. Chem. Int. Ed.
Engl. 31 (1992) 228.
330 O. Loiseleur, M. Hayashi, N. Schmees, A. Pfaltz, Syn-
thesis (1997) 1338.
331 D. B. Vineyard, W. S. Knowles, M. J. Sabacky, G. L.
Bachmann, D. J.Weinkauf, J. Am. Chem. Soc. 99 (1977)
5946.
332 A. Miyashita et al., J. Am. Chem. Soc. 102 (1980) 7932.
333 H. Takaya et al., J. Org. Chem. 51 (1986) 629.
334 U. Nagel, E. Kinzle, J. Andrade, G. Prescher,Chem. Ber.
119 (1986) 3326.
335 A. Togni, Angew. Chem. 108 (1996) 1581.
336 M. Burk, J. E. Feaster, R. L. Harlow,Organometallics 9
(1990) 2653.
337 M. J. Burk, J. Am. Chem. Soc. 113 (1991) 8518.
338 E. Lox, K. Ostgathe, P. Zelenka: ‘‘Reduction of Diesel
Exhaust Emission by Using Oxidation Catalysis,’’ SAE
Tech. Pap. Ser. 1990, 902 111.
339 B. H. Engler, E. Lox, K. Ostgathe, W. Cartellieri, P.
Zelenka: ‘‘DieselOxidationwithLowSulfate Formation
for HD-Diesel Engine Application,’’ SAE Tech. Pap.
Ser. 1991, 910 607.
340 B. H. Engler, E. Lox, K. Osthgathe, T. Ohata, K.
Tsuchitani, S. Ichihara, G. Garr, D. Psaras: ‘‘Recent
Trends in theApplication of Tri-Metal EmissionControl
Catalysts,’’ SAE Tech. Pap. Ser. 1994, 940928.
341 J. Leyrer, E. Lox,W. Strehlau: ‘‘Design Aspects of Lean
NOx-Catalysts for gasoline and Diesel Engine Applica-
tions,’’ SAE Tech. Pap. Ser. 1995, 952 495.
342 D. Lindner, E. Lox, R. v. Yperen, K. Ostgathe, T.
Kreuzer: ‘‘Reduction for Exhaust Gas Emissions by
Using Pd-based Three-way Catalysts,’’ SAE Tech. Pap.
Ser. 1996, 960 802.
343 W. Strehlau, J. Leyrer, E. Lox, T. Kreuzer, M. Hori, M.
Hoffmann: ‘‘NewDevelopments in Lean-NOx Catalysis
for Gasoline Fueled Passenger Cars in Europe,’’ SAE
Tech. Pap. Ser. 1996, 962 047.
344 W. Strehlau, J. v. d. Tillaart, J. Leyrer, E. Lox, W.
M€uller: ‘‘Evaluation of NOx-Storage Catalysis for Lean
Burn Gasoline Fueled Passenger Cars,’’ SAE Tech Pap.
Ser. 1997, 970 465.
345 L. Mußmann, D. Lindner, E. Lox, R. v. Yperen, T.
Kreuzer, I. Mitsushima: ‘‘The Role of Zirconium in
Novel Three-way Catalysts,’’ SAE Tech. Pap. Ser.
1997, 970 746.
346 B. Wu, G. Liu, Plat. Met. Rev. 41 (1997) 81 – 85.
347 J. Kondo, Progr. Theor. Physics 34 (1965) 372 – 382.
348 W. Diehl, Degussa Edelmetalltaschenbuch, H€uthigVerlag, Heidelberg 1995, p. 468.
349 A. Mills, Plat. Met. Rev. 41 (1997) 115 – 127.
350 J. N. Huiberts et al., Nature 380 (1996) 231 – 234.
351 R. Giessen, Phys. Bl. 53 (1997) 1207 – 1212.
352 A. Remhof, Phys. Rev. 56 (1997) 2897 – 2899.
353 T. J. Ohara, Plat. Met. Rev. 39 (1995) 54 – 62.
354 T.-M. Park et al., Talanta 44 (1997) no. 6, 973 – 978.
355 K. Lutz, Hagener Symp. Pulvermet. November 19th/
20th Hagen, 1998.
356 H. N. Shareef, K. R. Bellur, A. I. Kingon, O. Auciello,
Appl. Phys. Lett. 66 (1995) 239 – 241.
357 M. Gr€atzel, Plat. Met. Rev. 38 (1994) 151 – 159.
358 M. Gr€atzel in R. D. McDonell (ed.): AIP Conf. Proc.
404: FutureGenerationPhotovoltaic Technologies: First
NREL Conference, Colorado October 1997, 1–56396–
704–9, pp. 119 – 126.
359 K. Sayama, H. Arakawa, J. Chem. Soc. Faraday Trans.
93 (1997) no. 8, 1647 – 1654.
360 K. Gurunathan, P.Maruthamuthu,M. V. C. Sastri, Int. J.
Hydrogen Energy 22 (1997) no. 1, 57 – 62.
361 M. Misara, R. N. Pandey, O. N. Srivastava, Int. J.
Hydrogen Energy 22 (1997) no. 5, 501 – 508.
362 L. Shivalingappa, J. Sheng, T. Fukami, Vacuum 48
(1997) no. 5, 413 – 416.
363 T. Maruyama, T. Yamamoto, J. Phys. Chem. B. 101
(1997) no. 19, 3806.
364 R. Argazzi et al., J. Phys. Chem. B 101 (1997) no. 14,
2591 – 2597.
365 V. Grosshenny, A. Harriman, M. Hissler, R. Ziessel,
Plat. Met. Rev. 40 (1996) 26 – 35, 72 – 77.
366 K. Takemura et al., Jpn. J. Appl. Phys. 34 (1995) 5224 –
5229.
367 P. Zalm, J. A. van Stratum,Philips techn. Rev. 27 (1966)
nos. 3/4, 69 – 75.
368 F. H. Reid: ‘‘Electrodeposition of the Platinum Group
Metals,’’ Metallurg. Rev. 8 (1963) 167.
Vol. 28 Platinum Group Metals and Compounds 387

369 F. H. Reid: ‘‘Platinum Metal Plating –A Process and
Application Survey,’’ Trans. Inst. Met. Finish. 48 (1970)
115.
370 Johnson Matthey & Co., GB 363 569, 1931 (A. R.
Powell, A. W. Scott).
371 Engelhard Ind. Inc., FR 1 273 663, 1960 (H. W.
Robinson).
372 JohnsonMatthey, EP 358 375, 1996 (J. M. Albon,W. J.
Davis, P. E. Skinner, S. G. Warren).
373 P. E. Skinner: Platinum Met. Rev. 33 (1989) 102.
374 F. H. Reid: ‘‘Some Experimental and Practical Aspects
of Heavy Rhodium PLating,’’ Trans. Inst. Met. Finish.
33 (1956) 105.
375 M. Danemark, Y. Rusconi: ‘‘Rhodium–Eigenschaften
und elektrolytische Abscheidung,’’ Galvanotechn.
Oberfl€achensch. 3 (1062) 287.
376 M. Branik, F. Kummer: ‘‘Die galvanische Rhodiu-
mabscheidung,’’ Galvanotechnik 72 (1981) 1175.
377 F. H. Reid: ‘‘Palladium Plating – Processes and Applica-
tions in the United Kingdom,’’ Plating 52 (1965) 531.
378 J. A. Abys: ‘‘Palladium Plating,’’ Plat. Surf. Finish. 82
(1995) no. 8, 67.
379 P.Wilkinson: ‘‘The Replacement of Gold by Palladi-
um – Nickel on Connectors,’’ Trans. Inst. Met. Finish.
65 (1987) 33.
380 K. J. Whitlaw: ‘‘Gold-Flashed Palladium – Nickel for
Electronic Contacts,’’ Trans. Inst.Met. Finish. 64 (1986)
62.
381 F. H. Reid, J. C. Blake: ‘‘Electrodeposition of Rutheni-
um,’’ Trans. Inst. Met. Finish. 38 (1961) 45.
382 G. S. Reddy, P. Taimsalu: ‘‘Electrodeposition of Ruthe-
nium,’’ Trans. Inst. Met. Finish. 47 (1969) 187; US 3
576 724, 1971.
383 A. F. Bogensch€utz, J. L. Jostan, W. Mussinger: ‘‘Galva-
nische Abscheidungen von Rutheniumschichten,’’ Gal-
vanotechnik 67 (1976) 98.
384 G.-B. Dick: ‘‘Galvanische Abscheidungen von Platin
durch Hochtemperaturelektrolyse,’’ Galvanotechnik 79
(1988) 4066.
385 G.-B. Dick: ‘‘Galvanische Abscheidungen von Platin,
Tantal und Iridium durch Hochtemperaturelektrolyse,’’
Galvanotechnik 86 (1995) 1427.
386 Degussa, DE 196 17 040, 1998 (L. van Hippel, C. Bus-
sek, J. Sauer, M. Sauer, D. Arntz).
387 H. Marquardt, G. Sch€afer (eds.): Lehrbuch der Toxiko-
logie, Wiss.-Verl., Mannheim 1994.
388 J. Messerschmidt et al., Fresenius J. Anal. Chem. 343
(1992) 391.
389 E. Merian (ed.): Metals and Their Compounds in the
Environment, VCH, Weinheim, Germany 1991.
390 U. F€orstner: Umweltschutztechnik, 4. Aufl., Springer
Verlag, Berlin 1993.
391 Gesellschaft f€ur Strahlen- und Umweltforschung (GSF)
und Forschungszentrum J€ulich (eds.): Edelmetallemis-
sionen, Zwischenbericht GSF, M€unchen 1990.
392 J. Angerer, J. Schaller: ‘‘Akute und chronische Toxizit€atvon Spurenelementen,’’ Schriftenreihe der Gesellschaft
f€ur Mineralstoffe und Spurenelemente e.V., XIV, Wis-
senschaftliche Verlagsgesellschaft, Stuttgart 1993,
p. 214.
393 Bundesgesundheitsamt (BGA; ed.): Legierungen in der
zahn€arztlichen Praxis, Informationsschrift des Bundes-
gesundheitsamtes, BGA Berlin 1993.
Further Reading
A. Bonetti, R. Leone, F. Muggia, S. B. Howell (eds.): Plati-
num and Other Heavy Metal Compounds in Cancer
Chemotherapy, Humana Press, New York, NY 2009.
C. M. Giandomenico: Platinum-Group Metals, Compounds,
‘‘Kirk Othmer Encyclopedia of Chemical Technology’’,
5th edition, vol. 19, p. 635–667, John Wiley & Sons,
Hoboken, NJ, 2006, online: DOI: 10.1002/
0471238961.1612012007090114.a01.
R. J. Seymour, J. I. O’Farrelly: Platinum-Group Metals,
‘‘Kirk Othmer Encyclopedia of Chemical Technology’’,
5th edition, vol. 19, p. 596–635, John Wiley & Sons,
Hoboken, NJ, 2006, online: DOI: 10.1002/
0471238961.1612012019052513.a01.pub2.
F. Zereini, F. Alt (eds.): Palladium Emissions in the Environ-
ment, Springer, Berlin 2006.
388 Platinum Group Metals and Compounds Vol. 28
Related Documents











