PKA microdomain organisation and cAMP handling in healthy and dystrophic muscle in vivo Ira Verena Röder a , Valentina Lissandron b , Jessica Martin a , Yvonne Petersen a , Giulietta Di Benedetto b , Manuela Zaccolo b,c , Rüdiger Rudolf a, a Institute of Toxicology and Genetics, Forschungszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany b Venetian Institute of Molecular Medicine, 35129 Padua, Italy c Neuroscience and Molecular Pharmacology, FBLS, Glasgow G12 8QQ, UK abstract article info Article history: Received 28 November 2008 Received in revised form 20 January 2009 Accepted 20 January 2009 Available online 29 January 2009 Keywords: Cyclic adenosine monophosphate Dystrophy Förster resonance energy transfer Microdomain Protein kinase A Signalling Skeletal muscle Signalling through protein kinase A (PKA) triggers a multitude of intracellular effects in response to a variety of extracellular stimuli. To guarantee signal specicity, different PKA isoforms are compartmentalised by A- kinase anchoring proteins (AKAPs) into functional microdomains. By using genetically encoded uorescent reporters of cAMP concentration that are targeted to the intracellular sites where PKA type I and PKA type II isoforms normally reside, we directly show for the rst time spatially and functionally separate PKA microdomains in mouse skeletal muscle in vivo. The reporters localised into clearly distinct patterns within sarcomers, from where they could be displaced by means of AKAP disruptor peptides indicating the presence of disparate PKA type I and PKA type II anchor sites within skeletal muscle bres. The functional relevance of such differential localisation was underscored by the nding of mutually exclusive and AKAP-dependent increases in [cAMP] in the PKA type I and PKA type II microdomains upon application of different cAMP agonists. Specically, the sensors targeted to the PKA type II compartment responded only to norepinephrine, whereas those targeted to the PKA type I compartment responded only to α-calcitonin gene-related peptide. Notably, in dystrophic mdx mice the localisation pattern of the reporters was altered and the functional separation of the cAMP microdomains was abolished. In summary, our data indicate that an efcient organisation in microdomains of the cAMP/PKA pathway exists in the healthy skeletal muscle and that such organisation is subverted in dystrophic skeletal muscle. © 2009 Elsevier Inc. All rights reserved. 1. Introduction Duchenne muscular dystrophy is a severe neuromuscular disorder originating from deletions or non-sense mutations in the giant X-linked dystrophin gene. The 426 kDa dystrophin protein is part of the dystrophin-associated protein complex (DAPC), which links the extra- cellular matrix to the intracellular F-actin cytoskeleton [for review, see 1]. As such, it was proposed to deliver additional strength to the muscle bre, in order to withstand the strong shearing forces occurring during muscle contraction. Further discussed functions are regulation of ion channels [2–6], and of cellular signalling via nNOS [for review, see 7] or protein kinase A (PKA). Interest into the latter hypothesis was fostered by recent studies, suggesting an altered PKA-dependent signalling in dystrophic muscles. In one study, the electrophysiological characteristics of the skeletal muscle L-type Ca 2+ channel, which is responsible for muscle contraction, was found to be strongly altered in mdx myocytes [8]. Interestingly, these alterations could be reverted by addition of PKA [8]. Another report showed reduced in vitro PKA activity in mdx versus wildtype muscles [9]. The second messenger, cyclic adenosine monophosphate (cAMP), mediates pleiotropic signalling via G-protein coupled receptors (GPCRs) and PKA in response to a series of hormones and other cues [for review, see 10]. Understanding the mechanisms by which cAMP is able to relay a plethora of incoming signals to the multitude of its known cellular effects remains an important challenge. It is now largely accepted that much of the specicity of cAMP signalling is accomplished by a spatio-temporal compartmentalisation of cAMP transients as well as by physically coupling the relevant downstream signalling molecules close to their sites of action [for review, see 10]. The key cAMP effector, PKA, is in its inactive state a heterotetramer composed of two regulatory (R) and two catalytic (C) subunits [for Cellular Signalling 21 (2009) 819–826 Abbreviations: AKAP, A-kinase anchoring protein; cAMP, 3′–5′-cyclic adenosine monophosphate; CFP, cyan uorescent protein; CGRP, α-calcitonin gene-related peptide; CREB, cAMP response element-binding protein; DAPC, dystrophin associated protein complex; DD domain, dimerisation/docking domain; EPAC-camps, Exchange protein directly activated by cAMP–cAMP sensor; GPCR, G-protein coupled receptor; i.p., intraperitoneal; NE, norepinephrine; PKA-C, catalytic subunit of PKA; PKA-R, regulatory subunit of PKA; TA, tibialis anterior muscle; YFP, yellow uorescent protein. Corresponding author. Institute of Toxicology and Genetics, Forschungszentrum Karlsruhe, PO Box 3640, 76021 Karlsruhe, Germany. Tel.: +49 7247 82 3423; fax: +49 7247 82 3354. E-mail address: [email protected] (R. Rudolf). 0898-6568/$ – see front matter © 2009 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2009.01.029 Contents lists available at ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

PKA microdomain organisation and cAMP handling in healthy and dystrophicmuscle in vivo
Ira Verena Röder a, Valentina Lissandron b, Jessica Martin a, Yvonne Petersen a, Giulietta Di Benedetto b,Manuela Zaccolo b,c, Rüdiger Rudolf a,!a Institute of Toxicology and Genetics, Forschungszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germanyb Venetian Institute of Molecular Medicine, 35129 Padua, Italyc Neuroscience and Molecular Pharmacology, FBLS, Glasgow G12 8QQ, UK
a b s t r a c ta r t i c l e i n f o
Article history:Received 28 November 2008Received in revised form 20 January 2009Accepted 20 January 2009Available online 29 January 2009
Keywords:Cyclic adenosine monophosphateDystrophyFörster resonance energy transferMicrodomainProtein kinase ASignallingSkeletal muscle
Signalling through protein kinase A (PKA) triggers a multitude of intracellular effects in response to a varietyof extracellular stimuli. To guarantee signal speci!city, different PKA isoforms are compartmentalised by A-kinase anchoring proteins (AKAPs) into functional microdomains. By using genetically encoded "uorescentreporters of cAMP concentration that are targeted to the intracellular sites where PKA type I and PKA type IIisoforms normally reside, we directly show for the !rst time spatially and functionally separate PKAmicrodomains in mouse skeletal muscle in vivo. The reporters localised into clearly distinct patterns withinsarcomers, fromwhere they could be displaced by means of AKAP disruptor peptides indicating the presenceof disparate PKA type I and PKA type II anchor sites within skeletal muscle !bres. The functional relevance ofsuch differential localisation was underscored by the !nding of mutually exclusive and AKAP-dependentincreases in [cAMP] in the PKA type I and PKA type II microdomains upon application of different cAMPagonists. Speci!cally, the sensors targeted to the PKA type II compartment responded only to norepinephrine,whereas those targeted to the PKA type I compartment responded only to !-calcitonin gene-related peptide.Notably, in dystrophic mdx mice the localisation pattern of the reporters was altered and the functionalseparation of the cAMP microdomains was abolished. In summary, our data indicate that an ef!cientorganisation in microdomains of the cAMP/PKA pathway exists in the healthy skeletal muscle and that suchorganisation is subverted in dystrophic skeletal muscle.
© 2009 Elsevier Inc. All rights reserved.
1. Introduction
Duchenne muscular dystrophy is a severe neuromuscular disorderoriginating from deletions or non-sensemutations in the giant X-linkeddystrophin gene. The 426 kDa dystrophin protein is part of thedystrophin-associated protein complex (DAPC), which links the extra-cellular matrix to the intracellular F-actin cytoskeleton [for review, see1]. As such, it was proposed to deliver additional strength to themuscle!bre, in order to withstand the strong shearing forces occurring duringmuscle contraction. Further discussed functions are regulation of ion
channels [2–6], and of cellular signalling via nNOS [for review, see 7] orprotein kinase A (PKA). Interest into the latter hypothesis was fosteredby recent studies, suggesting an altered PKA-dependent signalling indystrophicmuscles. Inonestudy, theelectrophysiological characteristicsof the skeletal muscle L-type Ca2+ channel, which is responsible formuscle contraction, was found to be strongly altered in mdx myocytes[8]. Interestingly, these alterations could be reverted by addition of PKA[8]. Another report showed reduced in vitro PKA activity in mdx versuswildtype muscles [9].
The second messenger, cyclic adenosine monophosphate (cAMP),mediates pleiotropic signalling via G-protein coupled receptors(GPCRs) and PKA in response to a series of hormones and other cues[for review, see 10]. Understanding the mechanisms by which cAMP isable to relay a plethora of incoming signals to the multitude of itsknown cellular effects remains an important challenge. It is nowlargely accepted that much of the speci!city of cAMP signalling isaccomplished by a spatio-temporal compartmentalisation of cAMPtransients as well as by physically coupling the relevant downstreamsignalling molecules close to their sites of action [for review, see 10].The key cAMP effector, PKA, is in its inactive state a heterotetramercomposed of two regulatory (R) and two catalytic (C) subunits [for
Cellular Signalling 21 (2009) 819–826
Abbreviations: AKAP, A-kinase anchoring protein; cAMP, 3"–5"-cyclic adenosinemonophosphate; CFP, cyan "uorescent protein; CGRP, !-calcitonin gene-relatedpeptide; CREB, cAMP response element-binding protein; DAPC, dystrophin associatedprotein complex; DD domain, dimerisation/docking domain; EPAC-camps, Exchangeprotein directly activated by cAMP–cAMP sensor; GPCR, G-protein coupled receptor; i.p.,intraperitoneal; NE, norepinephrine; PKA-C, catalytic subunit of PKA; PKA-R, regulatorysubunit of PKA; TA, tibialis anterior muscle; YFP, yellow "uorescent protein.! Corresponding author. Institute of Toxicology and Genetics, Forschungszentrum
Karlsruhe, PO Box 3640, 76021 Karlsruhe, Germany. Tel.: +49 7247 82 3423; fax: +49 724782 3354.
E-mail address: [email protected] (R. Rudolf).
0898-6568/$ – see front matter © 2009 Elsevier Inc. All rights reserved.doi:10.1016/j.cellsig.2009.01.029
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig

review, see 11]. In mammals, PKA-R exist in four isoforms, named RI!,RI", RII! and RII", which keep PKA-C inactive until binding of cAMP toPKA-R unleashes the PKA-R–C interaction to liberate active PKA-C [forreview, see 11]. Furthermore, interaction of PKA-R with so called A-kinase anchoring proteins (AKAPs) leads to the localisation of differentPKA isoforms to speci!c subcellular compartments as it is observed inmany cells types [for review, see 12]. AKAPs are a group of more than50 heterogeneous proteins, which exhibit speci!c targeting sequencesthat mediate their subcellular localisation; furthermore, they all sharean amphipathic !-helical motif consisting of 14–18 residues, which isessential for the binding of the AKAP to PKA-R. Most AKAPs are alsoable to bind to additional signallingmolecules such as Ca2+-dependentprotein kinases or protein phosphatases and can thereby act as multi-scaffolding units [for review, see 12]. PKA-R exhibit a dimerisation/docking (DD) domain at their N-terminus, which mediates homo-dimerisation of the regulatory subunits and their docking to theAKAPs [13,14]. The DD domain sequence is PKA-R isoform-speci!c andtherefore different PKA-R isoforms interact with some AKAPs betterthan with others. On the basis of these binding characteristics,disruptor peptides have been designed, which allow to interferespeci!cally with the binding of PKA-RI or PKA-RII in a given cellularcontext and thus to discern the functional activities of the differentPKA isoforms. A prototype of such peptides is called Ht31 [15]. It isderived from human AKAP-Lbc and inhibits AKAP binding to both,PKA-RI and PKA-RII, albeit it is more effective in disrupting PKA-RIIanchoring [16,17]. Mutational screens and in silico-derived improve-ments led to the development of new peptides with nearly exclusiveaf!nities to just one PKA-R isoform [18–21]. These include the RIAD[19] and AKAP-IS peptides [21], which are highly speci!c for PKA-RI!and PKA-RII!, respectively, exhibiting several hundred fold higheraf!nities for one isoform than for the other.
In mammalian skeletal muscle the !-type R subunits appear toconstitute the major isoforms and their differential spatio-temporaldistribution suggests a functional diversity of RI! and RII! in thistissue [22,23]. In both cardiac and skeletal muscle, activation of the"-adrenergic receptor with norepinephrine (NE) or isoproterenolleads to force potentiation [24,25], i.e. upon equal stimulationmyocytes exhibit stronger contraction in the presence than in theabsence of "-agonists. Although downstreammolecular mechanismsleading to this effect may be partially different, in both muscle types"-agonists mediate a rise in cAMP [24,26], activation of PKA [27], andsubsequent phosphorylation of effector molecules, including L-typeCa2+ channels [28,29] and ryanodine receptors [30,31]. Thesechannels are located en face on the t-tubule and sarcoplasmicreticulum membranes, respectively.
With the advent of green "uorescent protein sensors technology ithas become possible to monitor dynamic changes of importantsignalling molecules such as Ca2+ or cAMP in live cells with highspatio-temporal resolution [32]. Using a genetically encoded Epac-based cAMP sensor [Epac-camps, 33] we have recently shown the!rst online visualisation of rising [cAMP] in live skeletal muscle upon"-adrenergic stimulation [26]. These sensors are composed of thecAMP binding domain of Epac sandwiched between the spectralmutants of GFP, CFP and YFP [33]. Upon binding or unbinding of cAMPthe cAMP binding domain undergoes conformational changes whichlead to a decrease or increase, respectively, of Förster resonanceenergy transfer (FRET) between CFP and YFP [33].We have previouslyshown that by N-terminal fusion of the DD domains of PKA-RI! orPKA-RII" to EPAC-camps it is possible to selectively target the Epac-camps sensor to the PKA type I and PKA type II compartments,respectively [34]. In the present study, we utilised transientexpression of such novel microdomain-speci!c cAMP sensors inlive mouse skeletal muscle to address the microdomain organisationof cAMP in healthy and dystrophic skeletal muscle. First, we showthat RI!- and RII!-EPAC-camps distribute in an AKAP-dependentmanner into different locations within the !bre. By in vivo two-
photon microscopic monitoring of whole muscle !bres we furtherdemonstrate that the rises in [cAMP] upon stimulationwith NE or !-calcitonin gene-related peptide (CGRP) in the RII!- or RI!-micro-domains, respectively, are spatially con!ned and are detected by thetargeted sensors in an AKAP anchoring-dependent manner. Finally,we describe a severe alteration of this signalling footprint indystrophic mdx mice.
2. Materials and methods
2.1. Expression plasmids and chemicals
Transfection experiments used the following constructs, allexpressed in pcDNA3 (Invitrogen) or pEGFP (Clontech Laboratories).The sensor RI!-EPAC-camps has been described in [34]. For theconstruction of RII!-EPAC-camps a similar approach was used. Brie"y,a chimera between the N-terminus of Epac-camps and the dimerisa-tion/docking (DD) domain of RIIa (47 aa) was generated via insertionof the 27 aa linker A (EAAAK)5 between the DD domain and the Epac-camps sequence. Ht31, RIAD and AKAP-IS were gifts from Dr. Scott(Oregon Health and Science University, U.S.A.). NE, CGRP andphalloidin-TRITC were from Sigma. All chemicals used were of highestavailable grade.
2.2. Animals and transfection
Adult C57BL/10J and 10J Dmdmdx mice were used. Animals wereobtained from Charles River and then maintained in the local animalfacility. Use and care of animals was as approved by Germanauthorities and according to national law (TierSchG §§7). An i.p.injection of a combination of Rompun (Bayer) and Zoletil 100(Laboratoires Virbac) was used for anaesthesia. Transfection wascarried out using an electroporation-based method of the TA muscle,as previously described [35,36].
2.3. In vivo two-photon microscopy
2.3.1. MicroscopyThis was performed as previously described [36], with minor
modi!cations. In brief, ten days after transfection animals wereanaesthetised and transfected muscles exposed for microscopy. Acombined confocal/two-photon system (Leica Microsystems) employ-ing a DMRE TCS SP2, equipped with a 20!/0.7 NA HC PL APO CS IMM/CORR UV multi-immersion objective (immersion medium, Visc-Ophtal gel, Winzer-Pharma) was used. Fibres were imaged fromsuper!cial layers up to a depth of about 400 µm in z-steps of 3 µm/slice. Images were taken at 512!512 pixel resolutionwith 200 Hz scanfrequency and 2! line average at 12-bit image depth with LeicaConfocal Software 2.61. RI!- and RII!-EPAC-camps "uorescence wasexcited with a mode-locked pulsed Maitai laser (Spectraphysics)tuned to 810 nm. CFP and YFP "uorescence signals were simulta-neously detected by non-descanned detectors equipped with a RSP505 dichroic mirror and BP485/30 and BP560/50 emission !lters (allLeica). Photomultiplier gains and offsets were kept constant, laserintensity was adjusted according to the depth of observed !bres.
2.3.2. Injection of agonists50 #l of 10 #M NE or CGRP were injected locally into the observed
muscle with a 30! gauge syringe. Note, that the !nal agonistconcentration was much lower than 10 #M, due to distribution ofthe solution in the large muscle volume and drain off by musclecirculation. For muscle recovery from injection-induced swellingmicroscopy was interrupted for 2 min after injection.
820 I.V. Röder et al. / Cellular Signalling 21 (2009) 819–826
2.4. Cryopreservation and sectioning of muscles
TA muscles were removed, washed in PBS (in mM: 2.67 KCl, 1.47KH2PO4, 137.93 NaCl, 8.06 Na2HPO4!7H2O), incubated for 10 min in
relaxation buffer (in mM: 100 KCl, 5 EGTA, 5 MgCl2, 3 2,3-butanedionemonoxime, 0.25 dithiothreitol, 10 histidine, pH 7.8), and then !xedover night at 4 °C in 4% wt/vol paraformaldehyde, pH 7.4. Muscleswere then dehydrated in sucrose solution (10% wt/vol sucrose in
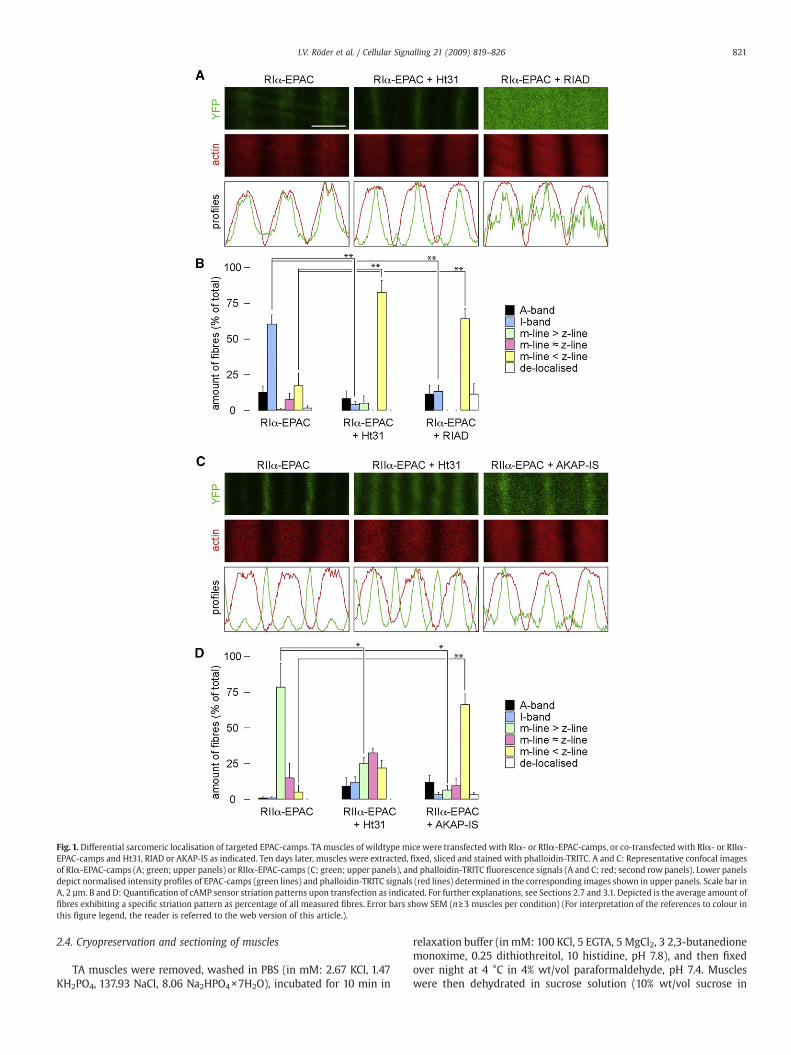
Fig. 1. Differential sarcomeric localisation of targeted EPAC-camps. TAmuscles of wildtype mice were transfected with RI!- or RII!-EPAC-camps, or co-transfected with RI!- or RII!-EPAC-camps and Ht31, RIAD or AKAP-IS as indicated. Ten days later, muscles were extracted, !xed, sliced and stained with phalloidin-TRITC. A and C: Representative confocal imagesof RI!-EPAC-camps (A; green; upper panels) or RII!-EPAC-camps (C; green; upper panels), and phalloidin-TRITC "uorescence signals (A and C; red; second rowpanels). Lower panelsdepict normalised intensity pro!les of EPAC-camps (green lines) and phalloidin-TRITC signals (red lines) determined in the corresponding images shown in upper panels. Scale bar inA, 2 µm. B and D: Quanti!cation of cAMP sensor striation patterns upon transfection as indicated. For further explanations, see Sections 2.7 and 3.1. Depicted is the average amount of!bres exhibiting a speci!c striation pattern as percentage of all measured !bres. Error bars show SEM (n!3 muscles per condition) (For interpretation of the references to colour inthis !gure legend, the reader is referred to the web version of this article.).
821I.V. Röder et al. / Cellular Signalling 21 (2009) 819–826

distilled water) for 6 h at 4 °C, embedded in tissue freezing medium(Labonord), frozen in liquid nitrogen and stored at "80 °C. Long-itudinal sections were made at "20 °C using a Leica CM1900 cryostatand then placed on electrostatic slides (Superfrost Plus, Labonord).
2.5. Staining of muscle slices
Sections were moistened with PBS, permeabilised with 0.1% TritonX-100 for 5 min, and then quenched in 50 mMNH4Cl for 10 min. Afterwashing with PBS, slices were blocked with 10% FBS/PBS for 20 minand incubated with 200 nM phalloidin-TRITC/10% FBS/PBS for!30 min. Slices were subsequently washed with 10% FBS/PBS, PBSand H2O, and then embedded in Mowiol (Calbiochem) for storage at4 °C for few days, or at "20 °C for long-term storage.
2.6. Confocal microscopy of slices
Images were taken with a DMRE TCS SP2 confocal microscopeequipped with an EL6000 lamp and a 63x/1.4 NA HCX PL APO CS oilimmersion objective (all Leica Microsystems). RI!- and RII!-EPAC-camps YFP "uorescence signals were excited with a KrAr laser at514 nm and emission was detected from 520–550 nm. Phalloidin-TRITC was monitored using a diode-pumped laser at 561 nm andemission detection from 580–630 nm. Images were taken at512!512 pixel and 8-bit resolution with Leica Confocal Software2.61. High-resolution images as shown in Figs. 1 and 3 were taken atthe same pixel resolution with an electronic zoom of about 10!.
2.7. Data analysis
Analysis of all image data employed ImageJ freeware program(http://rsb.info.nih.gov/ij/).
2.7.1. EPAC-camps sarcomeric localisationFirst, the phalloidin-TRITC striation patterns of sensor positive
!bres were oriented vertically. Then, sensor and phalloidin-TRITCsignal plot pro!les were determined, indicating the average"uorescence intensity of each pixel column as a function of distancealong the sarcomers, and plotted after normalisation in Figs. 1A, Cand 3A, A", C. As to Figs. 1B and D and 3 B and D, sensor striationswhich were clearly above !bre background and had a lateralhalfwidth extension of no more than one half that of thecorresponding phalloidin-TRITC pattern were considered as striated(either m-line or z-line). If they matched the phalloidin-TRITCpattern they were considered as I-band staining. If they had a similarextension, but were placed exactly in between the phalloidin-TRITCstaining they were considered as A-band staining. All other diffusesignals were considered as de-localised. Signi!cance tests employedStudent's t-test (! P#0.05; !! P#0.01).
2.7.2. Data-analysis of ratiometric videosCFP/YFP "uorescence ratios were calculated as follows. Upon
background subtraction, CFP- and YFP-image stacks were mean!ltered (1 pixel kernel) and a ratio-image stack was made. For eachsensor-positive !bre !ve corresponding regions of interest before andafter stimulus application were taken in subsequent optical sections.CFP/YFP ratio values were read out in these regions of interest in the
corresponding ratio-image stacks. Signi!cance tests employed Stu-dent's t-test (! P#0.05; !! P#0.01).
3. Results
3.1. Targeted cAMP sensors exhibit differential sarcomeric localisation
We !rst wanted to know whether RI!- and RII!-EPAC-campswould localise to distinct regions in skeletal muscle. To test this,
Fig. 2. cAMP responses to NE or CGRP are compartment speci!c in skeletal muscles invivo. TA muscles of wildtype mice were transfected with RI!- or RII!-EPAC-camps, orco-transfected with RI!- or RII!-EPAC-camps and Ht31, as indicated. Ten days later,"uorescence signals were monitored in situ with two-photon microscopy before andafter application of cAMP agonists, CGRP or NE. A: Representative two-photon images ofCFP (cyan) or YFP (yellow) signals or corresponding pseudo-coloured ratiometricimages (F(485 nm)/F(535 nm)), as indicated. In the ratiometric images, low and high[cAMP] are indicated by blue-green and yellow-red pseudo-colours, respectively. Thescale bar below the ratiometric images indicates the F(485 nm)/F(535 nm) valuescorresponding to speci!c pseudo-colours. The scale bar in the lower left panel depicts50 µm. B: Mean difference (in %) between CFP/YFP ratio values before and after agonistapplication and in the absence or presence of Ht31, as indicated. Mean±SEM (n!20!bres from at least 3 different experiments) (For interpretation of the references tocolour in this !gure legend, the reader is referred to the web version of this article.).
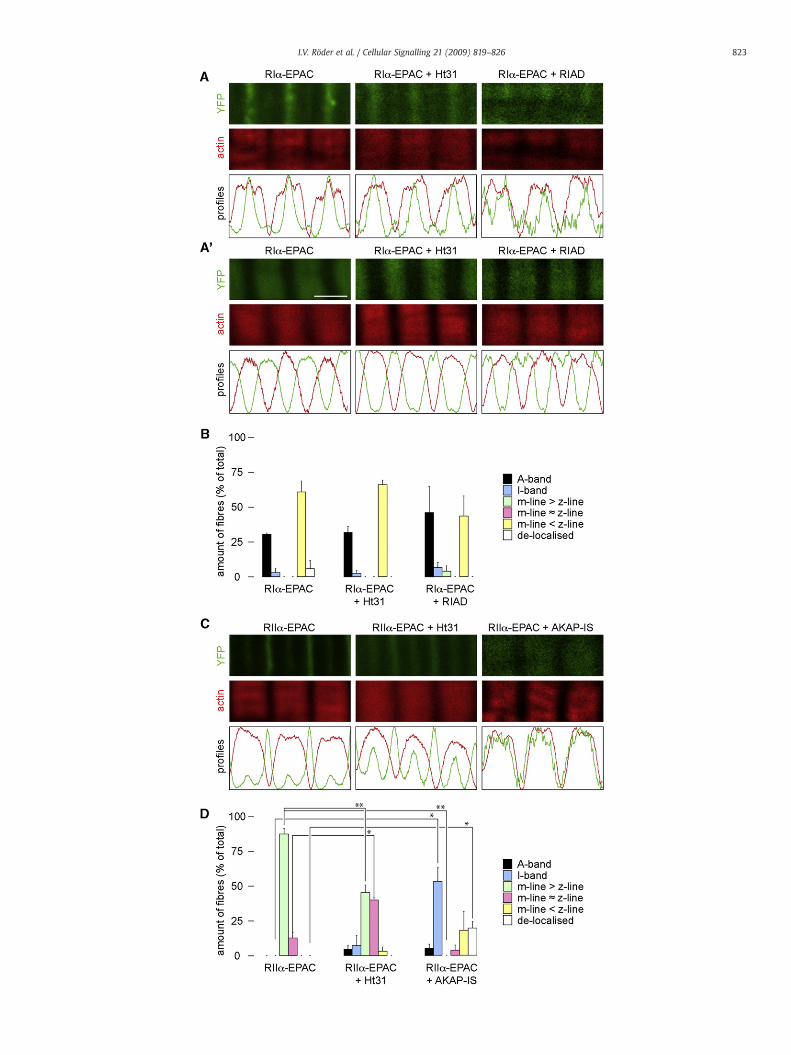
Fig. 3. Sarcomeric localisation of targeted EPAC-camps in dystrophic muscles. TAmuscles ofmdxmice were transfected with RI!- or RII!-EPAC-camps, or co-transfected with RI!- orRII!-EPAC-camps and Ht31, RIAD or AKAP-IS, as indicated. Ten days later, muscles were extracted, !xed, sliced and stainedwith phalloidin-TRITC. A, A" and C: Representative confocalimages of RI!-EPAC-camps (A and A"; green; upper panels) or RII!-EPAC-camps (C; green; upper panels), and phalloidin-TRITC "uorescence signals (A, A" and C; red; second rowpanels). Lower panels show normalised intensity pro!les of EPAC-camps (green lines) and phalloidin-TRITC signals (red lines) measured in the corresponding images shown in upperpanels. Scale bar in A", 2 µm. B and D: Quanti!cation of cAMP sensor striation patterns upon transfection as indicated. For further explanations, see Sections 2.7 and 3.3. Depicted isthe average amount of !bres exhibiting a speci!c striation pattern as percentage of all measured !bres. Error bars show SEM (n!3 muscles per condition) (For interpretation of thereferences to colour in this !gure legend, the reader is referred to the web version of this article.).
822 I.V. Röder et al. / Cellular Signalling 21 (2009) 819–826
823I.V. Röder et al. / Cellular Signalling 21 (2009) 819–826

muscles of wildtype mice were transfected with cDNA encoding foreither probe. Ten days later, muscles were taken, !xed, sliced andstained with phalloidin-TRITC, a marker for F-actin. Confocal analysisof the slices revealed a clearly different striation pattern of RI!- andRII!-EPAC-camps: RI!-EPAC-camps showed in most cases a localisa-tion in broad bands that largely co-localised with F-actin (I-bandregion, Fig. 1A, left panel; Fig. 1B), and only few !bres showedstriations in the A-band or z- or m-line regions (Fig. 1B). In contrast,RII!-EPAC-camps distributed in most !bres into two clear and slimstriations per sarcomer, corresponding to the localisation of them-lineand z-line, with the m-line signal being more pronounced (Fig. 1C, leftpanel). While in the case of RI!-EPAC-camps there was somevariability regarding the sharpness of the obtained striation patterns,more than 90% of all !bres expressing RII!-EPAC-camps exhibited aclear double striation pattern, similar to the one shown in Fig. 1C, leftpanel.
To investigate whether the different localisation patterns were dueto binding of the sensors to AKAPs, we co-transfected muscles withthe cAMP sensors and peptides disrupting the PKA-AKAP interaction,i.e. with Ht31, RIAD or AKAP-IS. For RI!-EPAC-camps the co-expression of Ht31 or RIAD led to a signi!cant re-distribution of thesensor from the I-band into the z-line region, although only a fractionof !bres was clearly affected (Fig. 1A, central panel; Fig. 1B). In thepresence of RIAD also a number of !breswith largely de-localised RI!-EPAC-camps "uorescence signals was found (Fig. 1A, right panel; Fig.1B). As to RII!-EPAC-camps, co-expression of Ht31 peptide signi!-cantly reduced the intensity difference between m-line and z-linelocalisation (Fig. 1C, central panel; Fig. 1D). Indeed, in a number of!bres the signal in the z-line was now more pronounced than in them-line (Fig. 1D). This effect was even stronger in the presence ofAKAP-IS where hardly any !bre with strong m-line staining wasdetected anymore (Fig. 1D). In summary, these data show that RI!-and RII!-EPAC-camps are distributed in different locations in thesarcomers of mouse skeletal muscle. Furthermore, they stronglysuggest that the striation patterns observed for the sensors are due tobinding to AKAPs.
3.2. Targeted cAMP sensors detect microdomain-speci!c rises in cAMPupon agonist application in vivo
Next we studied whether the microdomain-directed cAMP sensorswould also function to record changes in [cAMP] in live muscle in amicrodomain-speci!c manner. Therefore, muscles of wildtype micewere transfected with cDNAs encoding either RI!- or RII!-EPAC-camps or co-transfected with cDNAs encoding either RI!- or RII!-EPAC-camps and Ht31. Ten days later the specimens were monitoredwith in vivo two-photon microscopy as described previously [36]. Inbrief, during deep anaesthesia of the animal, transfectedmuscles wereexposed for monitoring, leaving muscle blood circulation andinnervation intact. The mouse was mounted onto a two-photonmicroscope and "uorescence emission signals of cyan (CFP, F(485 nm)) and yellow "uorescent protein (YFP, F(535 nm)) wererecorded simultaneously upon two-photon excitation at a wavelengthof 810 nm (Fig. 2A, left and central panels). For each !bre in the optic!eld the CFP/YFP emission ratio (F(485 nm)/F(535 nm), Fig. 2A, rightpanels) was determined prior to and after local injection of the cAMPagonists, !-calcitonin gene-related peptide or norepinephrine (CGRPor NE, respectively). Upon CGRP application we observed only withRI!- but not with RII!-EPAC-camps a signi!cant rise in the CFP/YFPemission ratio (Fig. 2B, compare !rst and !fth columns). Conversely,upon stimulation with NE only RII!-, but not RI!-EPAC-campsexhibited a signi!cant increase in CFP/YFP emission ratio (Fig. 2B,compare seventh and third columns).
To reveal whether the observed responses to NE and CGRP weredue to binding of the EPAC sensors to AKAPs, we performed in vivocAMP monitoring with RI!- and RII!-EPAC-camps in the presence of
Ht31. We found that the increases in CFP/YFP emission ratio obtainedin wildtype animals upon stimulation with CGRP and NE with RI!-and RII!-EPAC-camps, respectively, were strongly reduced in thepresence of Ht31 (Fig. 2B, second and eighth columns). This is a furtherindication that the responses to the cAMP agonists detected in theabsence of Ht31 were microdomain-speci!c and that RI!- and RII!-EPAC-camps resided in these disparate locales due to anchoring toAKAPs. In summary, by using the RI!- and RII!-EPAC-camps sensorswe were able to monitor microdomain-speci!c cAMP handling inskeletal muscle in vivo upon application of selected GPCR agonists.
3.3. Localisation of RI!-EPAC-camps is altered in dystrophic muscle
Recent reports about altered cAMP handling and AKAP expressionin dystrophic muscles prompted us to investigate the PKA micro-domain pro!le in mdx mice, which are a model for Duchenne/Beckermuscular dystrophy [37]. Therefore, muscles of mdx mice weretransfected with either RI!- or RII!-EPAC-camps or co-transfectedwith RI!- and RII!-EPAC-camps and Ht31, RIAD or AKAP-IS. Ten dayslater, muscles were taken, !xed, sliced and stained with phalloidin-TRITC. Confocal analysis showed that also in the dystrophic musclesthe striation patterns differed between RI!- and RII!-EPAC-camps.We observed that the "uorescence pattern of RII!-EPAC-camps wasvery similar in dystrophic as compared to wildtype muscles, i.e. inboth cases the sensor was concentrated in neatly con!ned strong m-lines and weaker z-line striations (compare Fig. 1C–D and Fig. 3C–D).In contrast, RI!-EPAC-camps in dystrophic muscles was almost neverfound in the I-band region as in thewildtype but either in the z-line orA-band regions (Fig. 3A–B). Furthermore, no signi!cant change in theRI!-EPAC-camps localisation was observed in the presence of Ht31 orRIAD (Fig. 3A–B). In contrast, the disruptor peptides Ht31 or AKAP-ISled to a similar, signi!cantly re-allocating effect of RII!-EPAC-campsas in wildtype animals (Fig. 3C–D). These results suggest that indystrophic muscles the PKA microdomain organisation is altered andthat, in particular, the occurrence of the RI!-microdomain shows littleor no dependence on AKAP binding.
3.4. Handling of cAMP is strongly altered in dystrophic muscles in vivo
By virtue of the observation of an altered microdomain organisa-tion in dystrophic mice, we asked if cAMP sensing by the PKA type I
Fig. 4. Aberrant agonist-dependent responses detected by targeted EPAC-camps uponstimulation with NE or CGRP in dystrophic skeletal muscles in vivo. TA muscles ofdystrophicmdxmice were transfected with RI!- or RII!-EPAC-camps, or co-transfectedwith RI!- or RII!-EPAC-camps and Ht31, as indicated. Ten days later, "uorescencesignals were monitored in situ with two-photon microscopy before and afterapplication of the cAMP agonists, CGRP or NE. Mean difference (%) between CFP/YFPratio values before and after agonist application and in the absence or presence of Ht31,as indicated. Mean±SEM (n!20 !bres from at least 3 different experiments).
824 I.V. Röder et al. / Cellular Signalling 21 (2009) 819–826

and PKA type II isozymes is also modi!ed in livemdx mice. Therefore,muscles of mdx mice were transfected with cDNAs encoding eitherRI!- or RII!-EPAC-camps or co-transfected with cDNAs encodingeither RI!- or RII!-EPAC-camps and Ht31. Ten days later thespecimens were monitored with in vivo two-photon microscopy asdescribed above, in the absence and presence of the cAMP agonistsCGRP or NE (Fig. 4). Notably, we observed major changes in theresponse patterns when compared with wildtype muscles. First, moststriking was that application of CGRP as well as NE resulted insigni!cant increases in [cAMP] in both, the PKA-RI! and PKA-RII!,microdomains (Fig. 4). Second, Ht31 did not reduce the responses ofCGRP or NE in the RI!- or the RII!-microdomains, respectively (Fig. 4).Third, Ht31 clearly ablated the NE response in the RI!-microdomainand slightly reduced the CGRP effect in the RII!-microdomain (Fig. 4).These results show that cAMP handling is severely altered indystrophic mouse skeletal muscle, in vivo.
4. Discussion
In the present study we have used genetically encoded "uor-escent cAMP sensors in combination with PKA-AKAP disruptorpeptides to study PKAmicrodomain localisation and cAMP dynamicsin skeletal muscles of healthy and dystrophic mice, in vivo. We showthat RI!- and RII!-EPAC-camps distribute into different locales in thesarcomeric region. Whereas RI!-EPAC-camps exhibited a ratheruniform distribution throughout the sarcomer or weak and broadstriations overlapping with F-actin signals (Fig. 1A–B), RII!-EPAC-camps showed a clear-cut double striation pattern on each m-lineand z-line (Fig. 1C–D). Both distribution patterns are thus in goodagreement with immunocytochemical studies on !xed samplesreporting sarcomeric localisations of endogenous PKA-RI! andPKA-RII! [22]. Furthermore, the effect of interfering peptides onsensor localisation strongly argues for an AKAP-speci!c binding ofPKA isoforms to these sites. In particular, RII!-EPAC-camps waspartially or completely displaced upon co-expression of Ht31 orAKAP-IS from both, m-line and z-line (Fig. 1C–D). Interestingly, them-line striations, which were strongest in the absence of anyinterfering peptide, were most affected in the presence of Ht31 andAKAP-IS. In general, the highly RII!-speci!c AKAP-IS peptide wasmore ef!cient in displacing RII!-EPAC-camps than Ht31. Together,these !ndings suggest that RII!-EPAC-camps binds to differentAKAPs in the m-line and z-line localisations and that the binding tothe m-line-speci!c AKAP(s) is of higher af!nity.
Our knowledge about favourite AKAPs, which could lead to theformation of the different microdomains in skeletal muscle, is stillrelatively scarce. However, three members of the AKAP protein superfamily might represent potential candidates. First, dystrobrevin, acomponent of the dystrophin-associated protein complex (DAPC) [forreview, see 1] was recently described to be an AKAP for the PKAisoforms RI!, RII!, and RII" [38]. Dystrobrevin localisation is strictlydependent on the presence of dystrophin and is almost entirelydetached from its normal localisation, i.e. the sarcolemma, in mice[37,39] and humans [40] lacking intact dystrophin. Furthermore,dystrophin itself normally exhibits a costameric organisation whichis partially lost in mdx muscles [41,42] and it seems reasonable tohypothesise, that the altered localisation of dystrobrevin may beresponsible at least partially for the observed changes in the RI!-EPAC-camps pattern in the dystrophin-lackingmdxmuscles. Second,the muscle-speci!c mAKAP was shown to be localised by immuno-electron microscopy in the z-line region and the triad, i.e. in closeproximity to t-tubules and terminal cisternae of the sarcoplasmicreticulum [43]. Furthermore, it was described to be necessary forproper phosphorylation of the ryanodine receptor [43]. Although thisphosphorylationwas normal also inmice lacking PKA-RII!, mAKAP ismost likely coupled to this PKA isoform [44]. While mAKAPexpression was apparently unaltered in dystrophic mdx skeletal
muscle [9], it was found to be reduced in dystrophic mdx hearts [45],suggesting that mAKAP may be a potential candidate for thelocalisation of PKA-RII! in the z-line region. Third, a series of recentreports have identi!ed a MEF2A-regulated and costamere-localisedprotein, myospryn [46], as an AKAP which is speci!c for theinteraction with PKA-RII! [47]. Expression of this AKAP was foundto be diminished, both at the mRNA as well as the protein level indystrophic mdx muscles [9].
Whatever AKAP(s) are mediating the PKA microdomain organisa-tion, which we observed using the targeted cAMP sensors, the [cAMP]responses upon agonist application showed that the handling of thissecond messenger is severely changed in dystrophic muscles in vivo.While in wildtype muscles the PKA-RI! and PKA-RII! microdomainsshowed a clear separation of signal speci!city towards either CGRP orNE (Fig. 2B), mdx muscles responded in both microdomains and toboth agonists in a similar manner (Fig. 4). On the one hand, this couldsuggest that the localisation of the G-protein coupled receptors, whichinduced the agonist-dependent rises in cAMP (i.e. the "-adrenergicreceptor and the CGRP receptor), was disrupted in these muscles. Onthe other hand, the microdomain organisation might have been socompromised in the case of dystrophic muscles, that mutual sensingof cAMP transients originating from the neighbouring microdomaincould occur. In particular, the apparent re-localisation of the PKA-RI!microdomain from the I-band to the A-band (i.e. close to the strongestPKA-RII! localisation in the m-line region; compare Figs. 1A and 3A"),could explain the reduced selectivity of the sensor targeted to the PKAtype I! microdomain. In addition, it should be noted that severelydystrophic muscle tissue presents areas within muscle !bres inwhichmost of the typically ordered striation pattern is lost. It is likely that insuch regions any microdomain organisation is subverted. Further-more, while in the wildtype upon agonist application all micro-domain-speci!c rises of [cAMP] were abolished by the addition of theAKAP disruptor peptide Ht31 (Fig. 2), the same treatment did notblock these responses in dystrophic muscles (Fig. 4). Conversely, inmdx muscles Ht31 completely blocked the rise in the PKA-RI!microdomain upon NE and reduced slightly (albeit not signi!cantly)the response in the PKA-RII! microdomain upon CGRP (Fig. 4). Ourlocalisation studies showing no changes of RI!-EPAC-camps localisa-tion in the presence of Ht31 but a clear effect on the PKA-RII!microdomain (Fig. 3), could argue for a release of PKA-RII! into thePKA-RI!microdomain where PKA-RII!might compete with PKA-RI!for cAMP. Finally, mis-regulation of the distributions and activities ofthe cAMP dissipating enzymes phosphodiesterases (PDEs) in thedystrophic muscles may also contribute to the alterations in cAMPhandling that we observed. The analysis of PDEs distribution andactivity in normal versus dystrophic muscles was beyond the scope ofthe present study and will represent the focus for furtherinvestigation.
5. Conclusions
In conclusion, we show for the !rst time microdomain-speci!ccAMP handling in mouse skeletal muscle in vivo. Speci!cally, wedemonstrate that PKA-RI! and PKA-RII! de!ne spatially distinctmicrodomains within which PKA is selectively activated by differentGPCR agonists. We furthermore show that this separation is absent indystrophic muscles frommdxmice, in vivo. This could lead to aberrantagonist-induced intracellular signalling and PKA-dependent activa-tion of effector molecules. Such mechanism may lead to both short-term dysregulations, such as the non-speci!c phosphorylation ofdihydropyridine or ryanodine receptors and to long-term effects, suchas faulty CREB-dependent gene expression. In fact, both short- andlong-term PKA-mediated processes were previously shown to bealtered in dystrophic muscles [8,9,48]. Disruption of the spatialorganisation of the cAMP/PKA signalling pathway may thus represent
825I.V. Röder et al. / Cellular Signalling 21 (2009) 819–826

an important component of Duchenne/Becker muscular dystrophypathogenesis.
Acknowledgements
We are grateful to Drs. J. Scott and K. Tasken for kindly providingRIAD and AKAP-IS cDNAs. RR was supported by the DeutscheForschungsgemeinschaft (grant RU923/3-1). RR and MZ were sup-ported by the Association Française contre les Myopathies (grant12056/13134). MZ was supported by the Fondation Leducq (O6 CVD02), the EC (LSHB-CT-2006-037189) and the British Heart Foundation(PG/07/091/23698).
References
[1] D.J. Blake, A. Weir, S.E. Newey, K.E. Davies, Physiol. Rev. 82 (2) (2002) 291.[2] A. Franco Jr., J.B. Lansman, Nature 344 (6267) (1990) 670.[3] P.R. Turner, P.Y. Fong, W.F. Denetclaw, R.A. Steinhardt, J. Cell. Biol. 115 (6) (1991)
1701.[4] C. Vandebrouck, D. Martin, M. Colson-Van Schoor, H. Debaix, P. Gailly, J. Cell. Biol.
158 (6) (2002) 1089.[5] F.X. Boittin, O. Petermann, C. Hirn, P. Mittaud, O.M. Dorchies, E. Roulet, U.T. Ruegg, J.
Cell. Sci. 119 (Pt 18) (2006) 3733.[6] N.P. Whitehead, M. Streamer, L.I. Lusambili, F. Sachs, D.G. Allen, Neuromuscul.
Disord. 16 (12) (2006) 845.[7] K.A. Lapidos, R. Kakkar, E.M. McNally, Circ. Res. 94 (8) (2004) 1023.[8] B.D. Johnson, T. Scheuer, W.A. Catterall, Proc. Natl. Acad. Sci. U. S. A. 102 (11) (2005)
4191.[9] J.G. Reynolds, S.A. McCalmon, J.A. Donaghey, F.J. Naya, J. Biol. Chem. 283 (13) (2008)
8070.[10] J.A. Beavo, L.L. Brunton, Nat. Rev. Mol. Cell. Biol. 3 (9) (2002) 710.[11] S.S. Taylor, D.R. Knighton, J. Zheng, L.F. Ten Eyck, J.M. Sowadski, Annu. Rev. Cell. Biol.
8 (1992) 429.[12] W. Wong, J.D. Scott, Nat. Rev. Mol. Cell. Biol. 5 (12) (2004) 959.[13] Z.E. Hausken, V.M. Coghlan, C.A. Hastings, E.M. Reimann, J.D. Scott, J. Biol. Chem.
269 (39) (1994) 24245.[14] Z.E. Hausken, M.L. Dell'Acqua, V.M. Coghlan, J.D. Scott, J. Biol. Chem. 271 (46)
(1996) 29016.[15] D.W. Carr, R.E. Stofko-Hahn, I.D. Fraser, S.M. Bishop, T.S. Acott, R.G. Brennan, J.D.
Scott, J. Biol. Chem. 266 (22) (1991) 14188.[16] K.A. Burton, B.D. Johnson, Z.E. Hausken, R.E.Westenbroek, R.L. Idzerda, T. Scheuer, J.D.
Scott, W.A. Catterall, G.S. McKnight, Proc. Natl. Acad. Sci. U. S. A. 94 (20) (1997) 11067.[17] F.W. Herberg, A. Maleszka, T. Eide, L. Vossebein, K. Tasken, J. Mol. Biol. 298 (2)
(2000) 329.[18] C. Hundsrucker, G. Krause, M. Beyermann, A. Prinz, B. Zimmermann, O. Diekmann, D.
Lorenz, E. Stefan, P. Nedvetsky, M. Dathe, F. Christian, T. McSorley, E. Krause, G.McConnachie, F.W.Herberg, J.D. Scott,W. Rosenthal, E. Klussmann, Biochem. J. 396 (2)(2006) 297.
[19] C.R. Carlson, B. Lygren, T. Berge, N. Hoshi, W. Wong, K. Tasken, J.D. Scott, J. Biol.Chem. 281 (30) (2006) 21535.
[20] L.L. Burns-Hamuro, Y. Ma, S. Kammerer, U. Reineke, C. Self, C. Cook, G.L. Olson, C.R.Cantor, A. Braun, S.S. Taylor, Proc. Natl. Acad. Sci. U. S. A. 100 (7) (2003) 4072.
[21] N.M. Alto, S.H. Soderling, N. Hoshi, L.K. Langeberg, R. Fayos, P.A. Jennings, J.D. Scott,Proc. Natl. Acad. Sci. U. S. A. 100 (8) (2003) 4445.
[22] G.A. Perkins, L.Wang, L.J. Huang,K.Humphries, V.J. Yao,M.Martone, T.J. Deerinck,D.M.Barraclough, J.D. Violin, D. Smith, A. Newton, J.D. Scott, S.S. Taylor,M.H. Ellisman, BMC.Neurosci. 2 (1) (2001) 17.
[23] T. Imaizumi-Scherrer, D.M. Faust, J.C. Benichou, R. Hellio, M.C. Weiss, J. Cell. Biol.134 (5) (1996) 1241.
[24] S.F. Steinberg, L.L. Brunton, Annu. Rev. Pharmacol. Toxicol. 41 (2001) 751.[25] S.P. Cairns, A.F. Dulhunty, Muscle. Nerve. 16 (12) (1993) 1317.[26] R. Rudolf, P.J. Magalhaes, T. Pozzan, J. Cell. Biol. 173 (2006) 187.[27] M. Zaccolo, T. Pozzan, Science 295 (5560) (2002) 1711.[28] B.P. Bean, M.C. Nowycky, R.W. Tsien, Nature 307 (5949) (1984) 371.[29] A. Sculptoreanu, T. Scheuer, W.A. Catterall, Nature 364 (6434) (1993) 240.[30] A. Yoshida, M. Takahashi, T. Imagawa, M. Shigekawa, H. Takisawa, T. Nakamura, J.
Biochem. (Tokyo). 111 (2) (1992) 186.[31] A. Sonnleitner, S. Fleischer, H. Schindler, Cell. Calcium. 21 (4) (1997) 283.[32] M. Zaccolo, F. De Giorgi, C.Y. Cho, L. Feng, T. Knapp, P.A. Negulescu, S.S. Taylor,
R.Y. Tsien, T. Pozzan, Nat. Cell. Biol. 2 (1) (2000) 25.[33] V.O. Nikolaev, M. Bunemann, L. Hein, A. Hannawacker, M.J. Lohse, J. Biol. Chem. 279
(36) (2004) 37215.[34] G. Di Benedetto, A. Zoccarato, V. Lissandron, A. Terrin, X. Li, M.D. Houslay, G.S. Baillie,
M. Zaccolo, Circ. Res. 103 (8) (2008) 836.[35] M. Dona, M. Sandri, K. Rossini, I. Dell'Aica, M. Podhorska-Okolow, U. Carraro,
Biochem. Biophys. Res. Commun. 312 (4) (2003) 1132.[36] R. Rudolf, M. Mongillo, P.J. Magalhaes, T. Pozzan, J. Cell. Biol. 166 (4) (2004) 527.[37] M. Durbeej, K.P. Campbell, Curr. Opin. Genet. Dev. 12 (3) (2002) 349.[38] M. Ceccarini, M. Grasso, C. Veroni, G. Gambara, B. Artegiani, G. Macchia, C. Ramoni,
P. Torreri, C. Mallozzi, T.C. Petrucci, P. Macioce, J. Mol. Biol. 371 (5) (2007) 1174.[39] B. Wang, J. Li, C. Qiao, C. Chen, P. Hu, X. Zhu, L. Zhou, J. Bogan, J. Kornegay, X. Xiao,
Gene. Ther. 15 (15) (2008) 1099.[40] L. Metzinger, D.J. Blake, M.V. Squier, L.V. Anderson, A.E. Deconinck, R. Nawrotzki,
D. Hilton-Jones, K.E. Davies, Hum. Mol. Genet. 6 (7) (1997) 1185.[41] M.W. Williams, R.J. Bloch, J. Cell. Biol. 144 (6) (1999) 1259.[42] R.J. Bloch, Y. Capetanaki, A. O'Neill, P. Reed, M.W. Williams, W.G. Resneck, N.C.
Porter, J.A. Ursitti, Clin. Orthop. Relat. Res. 403 (2002) S203 Suppl.[43] M.L. Ruehr,M.A.Russell,D.G. Ferguson,M. Bhat, J.Ma,D.S.Damron, J.D. Scott,M.Bond,
J. Biol. Chem. 278 (27) (2003) 24831.[44] M.S. Kapiloff, R.V. Schillace, A.M. Westphal, J.D. Scott, J. Cell. Sci. 112 (Pt 16) (1999)
2725.[45] M.S. Rohman, N. Emoto, Y. Takeshima, M. Yokoyama, M.Matsuo, Biochem. Biophys.
Res. Commun. 310 (1) (2003) 228.[46] J.T. Durham, O.M. Brand, M. Arnold, J.G. Reynolds, L. Muthukumar, H. Weiler,
J.A. Richardson, F.J. Naya, J. Biol. Chem. 281 (10) (2006) 6841.[47] J.G. Reynolds, S.A. McCalmon, T. Tomczyk, F.J. Naya, Biochim. Biophys. Acta 1773 (6)
(2007) 891.[48] B. Mayr, M. Montminy, Nat. Rev. Mol. Cell. Biol. 2 (8) (2001) 599.
826 I.V. Röder et al. / Cellular Signalling 21 (2009) 819–826
Related Documents











