Mini-review Pharmacogenetics of irinotecan metabolism and transport: An update Nicola F. Smith a , William D. Figg a,b, * , Alex Sparreboom b a Molecular Pharmacology Section, Medical Oncology Branch, National Cancer Institute, 9000 Rockville Pike, Building 10/Room 5A01, Bethesda, MD, USA b Clinical Pharmacology Research Core, Medical Oncology Branch, National Cancer Institute, 9000 Rockville Pike, Building 10/Room 5A01, Bethesda, MD, USA Received 1 November 2004; accepted 1 June 2005 Available online 3 November 2005 Abstract The anticancer agent irinotecan (CPT-11) is converted to SN-38, which is approximately 100 to 1000-fold more cytotoxic than the parent drug. The pharmacokinetics of irinotecan are extremely complex and have been the subject of intensive investigation in recent years. Irinotecan is subject to extensive metabolism by various polymorphic enzymes, including CES2 to form SN-38, members of the UGT1A subfamily, and CYP3A4 and CYP3A5, which form several pharmacologically inactive oxidation products. Elimination of irinotecan is also dependent on drug-transporting proteins, notably ABCB1 (P-glycoprotein), ABCC2 (cMOAT) and ABCG2 (BCRP), present on the bile canalicular membrane. The various processes mediating drug elimination, either through metabolic break- down or excretion, likely impact substantially on interindividual variability in drug handling. This report provides an update on current strategies to individualize irinotecan chemotherapy based on each patientÕs genetic constitution, which may ultimately lead to more selec- tive use of this agent. Published by Elsevier Ltd. Keywords: CPT-11; SN-38; CYP3A; CES; UGT1A; ABCB1; ABCC1; ABCC2; ABCG2 Contents 1. Introduction ............................................................................... 164 2. Irinotecan metabolism ........................................................................ 165 2.1. CYP3A-mediated metabolism .............................................................. 165 2.2. Carboxylesterase-mediated metabolism ........................................................ 168 2.3. UDP-glucuronosyltransferases .............................................................. 169 3. Irinotecan transport .......................................................................... 171 3.1. ABCB1-mediated transport ................................................................ 171 3.2. ABCC1-mediated transport ................................................................ 172 3.3. ABCC2-mediated transport ................................................................ 172 3.4. ABCG2-mediated transport ................................................................ 172 4. Conclusion ................................................................................ 172 References ................................................................................ 172 0887-2333/$ - see front matter Published by Elsevier Ltd. doi:10.1016/j.tiv.2005.06.045 * Corresponding author. Address: Clinical Pharmacology Research Core, Medical Oncology Branch, National Cancer Institute, 9000 Rockville Pike, Building 10/Room 5A01, Bethesda, MD, USA. Tel.: +1 301 402 3622; fax: +1 301 402 8606. E-mail address: wdfi[email protected] (W.D. Figg). www.elsevier.com/locate/toxinvit Toxicology in Vitro 20 (2006) 163–175

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

www.elsevier.com/locate/toxinvit
Toxicology in Vitro 20 (2006) 163–175
Mini-review
Pharmacogenetics of irinotecan metabolism and transport: An update
Nicola F. Smith a, William D. Figg a,b,*, Alex Sparreboom b
a Molecular Pharmacology Section, Medical Oncology Branch, National Cancer Institute, 9000 Rockville Pike, Building 10/Room 5A01, Bethesda, MD, USAb Clinical Pharmacology Research Core, Medical Oncology Branch, National Cancer Institute, 9000 Rockville Pike,
Building 10/Room 5A01, Bethesda, MD, USA
Received 1 November 2004; accepted 1 June 2005Available online 3 November 2005
Abstract
The anticancer agent irinotecan (CPT-11) is converted to SN-38, which is approximately 100 to 1000-fold more cytotoxic than theparent drug. The pharmacokinetics of irinotecan are extremely complex and have been the subject of intensive investigation in recentyears. Irinotecan is subject to extensive metabolism by various polymorphic enzymes, including CES2 to form SN-38, members ofthe UGT1A subfamily, and CYP3A4 and CYP3A5, which form several pharmacologically inactive oxidation products. Eliminationof irinotecan is also dependent on drug-transporting proteins, notably ABCB1 (P-glycoprotein), ABCC2 (cMOAT) and ABCG2(BCRP), present on the bile canalicular membrane. The various processes mediating drug elimination, either through metabolic break-down or excretion, likely impact substantially on interindividual variability in drug handling. This report provides an update on currentstrategies to individualize irinotecan chemotherapy based on each patient�s genetic constitution, which may ultimately lead to more selec-tive use of this agent.Published by Elsevier Ltd.
Keywords: CPT-11; SN-38; CYP3A; CES; UGT1A; ABCB1; ABCC1; ABCC2; ABCG2
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1642. Irinotecan metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
0887-2
doi:10.
* CoBuildin
E-m
2.1. CYP3A-mediated metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1652.2. Carboxylesterase-mediated metabolism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1682.3. UDP-glucuronosyltransferases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 169
3. Irinotecan transport . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
3.1. ABCB1-mediated transport . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1713.2. ABCC1-mediated transport . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1723.3. ABCC2-mediated transport . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1723.4. ABCG2-mediated transport . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1724. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
333/$ - see front matter Published by Elsevier Ltd.
1016/j.tiv.2005.06.045
rresponding author. Address: Clinical Pharmacology Research Core, Medical Oncology Branch, National Cancer Institute, 9000 Rockville Pike,g 10/Room 5A01, Bethesda, MD, USA. Tel.: +1 301 402 3622; fax: +1 301 402 8606.ail address: [email protected] (W.D. Figg).
164 N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175
1. Introduction
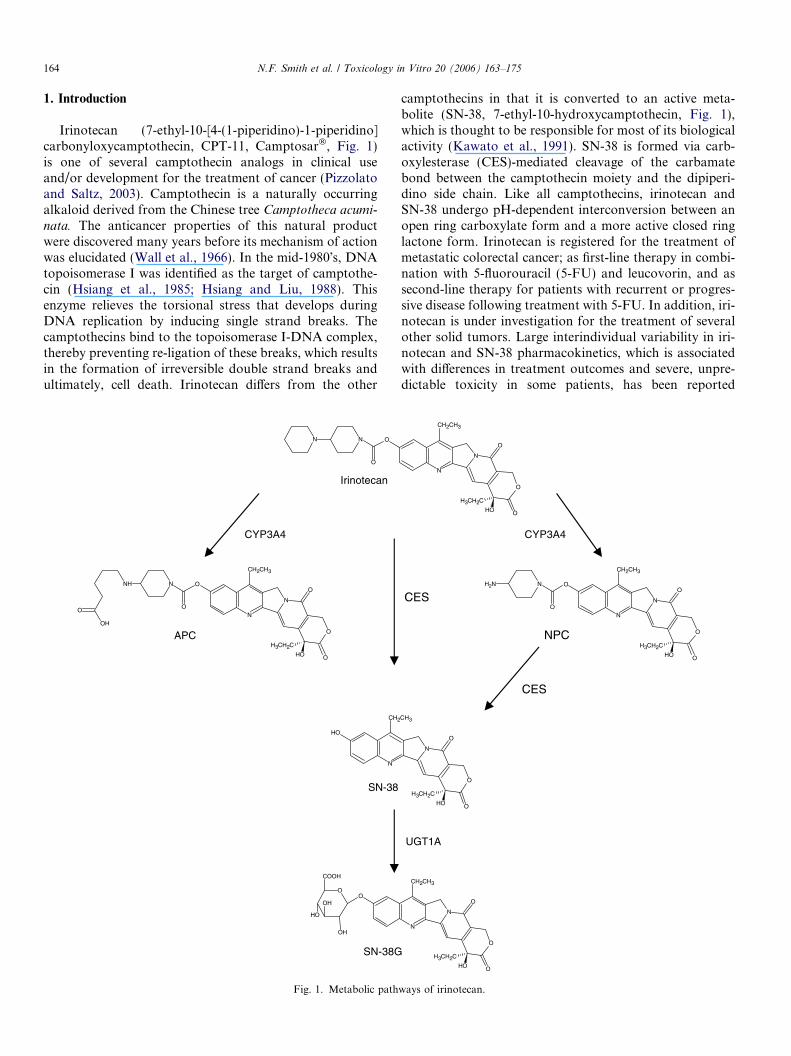
Irinotecan (7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin, CPT-11, Camptosar�, Fig. 1)is one of several camptothecin analogs in clinical useand/or development for the treatment of cancer (Pizzolatoand Saltz, 2003). Camptothecin is a naturally occurringalkaloid derived from the Chinese tree Camptotheca acumi-
nata. The anticancer properties of this natural productwere discovered many years before its mechanism of actionwas elucidated (Wall et al., 1966). In the mid-1980�s, DNAtopoisomerase I was identified as the target of camptothe-cin (Hsiang et al., 1985; Hsiang and Liu, 1988). Thisenzyme relieves the torsional stress that develops duringDNA replication by inducing single strand breaks. Thecamptothecins bind to the topoisomerase I-DNA complex,thereby preventing re-ligation of these breaks, which resultsin the formation of irreversible double strand breaks andultimately, cell death. Irinotecan differs from the other
N N O
O
Irinotecan
NH N O
ON
N
O
CH2CH3
O
O
H3CH2C
OH
OH
O
OH
N
CH2
OO
OH
COOH
OH
OH
APC
SN-38
SN-38G
CYP3A4
Fig. 1. Metabolic path
camptothecins in that it is converted to an active meta-bolite (SN-38, 7-ethyl-10-hydroxycamptothecin, Fig. 1),which is thought to be responsible for most of its biologicalactivity (Kawato et al., 1991). SN-38 is formed via carb-oxylesterase (CES)-mediated cleavage of the carbamatebond between the camptothecin moiety and the dipiperi-dino side chain. Like all camptothecins, irinotecan andSN-38 undergo pH-dependent interconversion between anopen ring carboxylate form and a more active closed ringlactone form. Irinotecan is registered for the treatment ofmetastatic colorectal cancer; as first-line therapy in combi-nation with 5-fluorouracil (5-FU) and leucovorin, and assecond-line therapy for patients with recurrent or progres-sive disease following treatment with 5-FU. In addition, iri-notecan is under investigation for the treatment of severalother solid tumors. Large interindividual variability in iri-notecan and SN-38 pharmacokinetics, which is associatedwith differences in treatment outcomes and severe, unpre-dictable toxicity in some patients, has been reported
N
N
O
CH2CH3
O
O
H3CH2C
OH
N O
ON
N
O
CH2CH3
O
O
H3CH2C
OH
NH2
N
O
CH3
O
O
H3CH2C
OH
N
N
O
CH2CH3
O
O
H3CH2C
OH
NPC
CES
CES
CYP3A4
UGT1A
ways of irinotecan.

Fig. 2. Schematic diagram of irinotecan metabolism and transport.
N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175 165
(Mathijssen et al., 2002; de Jong et al., 2004b). This vari-ability might be explained in part by genetic variationin the multiple metabolic enzymes and transporters in-volved in irinotecan disposition (Fig. 2). Pharmacogeneticapproaches are rapidly elucidating the inherited basis ofinterindividual differences in drug disposition and effectsin an attempt to individualize drug therapy (Evans andMcLeod, 2003; Smith et al., 2004). This review will describethe complex metabolism and transport of irinotecan,together with the current understanding of variability inthe genes encoding the enzymes and transporters involvedthese pathways (Tables 1 and 2).
2. Irinotecan metabolism
2.1. CYP3A-mediated metabolism
Several metabolites of irinotecan, which are much lessactive than the parent compound, have been identified inplasma, urine and bile from treated patients and followingincubation of irinotecan with human liver microsomes(Lokiec et al., 1996; Rivory et al., 1996; Dodds et al.,1998; Haaz et al., 1998a,b; Santos et al., 2000). The mostabundant of these is APC (7-ethyl-10-[4-N-(5-aminopenta-noic acid)-1-piperidino] carbonyloxycamptothecin; Fig. 1),which results from oxidation of the terminal piperidine ringof irinotecan. Whilst this metabolite does not appear to beresponsible for the efficacy or toxicity of irinotecan, its
formation may represent an important metabolic pathwayfor irinotecan clearance. In addition, NPC (7-ethyl-10-(4-amino-1-piperidino) carbonyloxycamptothecin; Fig. 1),resulting from cleavage of the distal piperidine ring, and sev-eral other oxidative metabolites have been detected in vivo(Lokiec et al., 1996; Santos et al., 2000). In vitro studiesusing cells transfected with cytochrome P450 (CYP)enzymes revealed that CYP3A4 is responsible for the for-mation of APC, NPC and a hydroxylated metabolite (Haazet al., 1998a,b; Santos et al., 2000), whereas incubation withCYP3A5 resulted in de-ethylation of the camptothecin moi-ety to form ametabolite (referred to asM4) that is undetect-able in vivo (Santos et al., 2000). The conversion of APC toNPC has not been reported. The role of the CYP3Aenzymes in irinotecan disposition was further exemplifiedin a recent study that investigated the relationship betweenCYP3A phenotype (assessed by the erythromycin breathtest and midazolam clearance) and the clearance of irino-tecan by 30 Caucasian patients. Whilst there was no associ-ation between irinotecan clearance and erythromycinmetabolism, the clearance of irinotecan and midazolamwere highly correlated (P < 0.001) (Mathijssen et al.,2004). The authors suggest that these discrepant findingsmay be due to the fact that the CYP3A-mediated metabo-lism of irinotecan more closely resembles the relatively slowand inefficient metabolism of midazolam, than the fastand extensive metabolism of erythromycin. Despite signifi-cant interindividual variation (Xie et al., 2004) and the
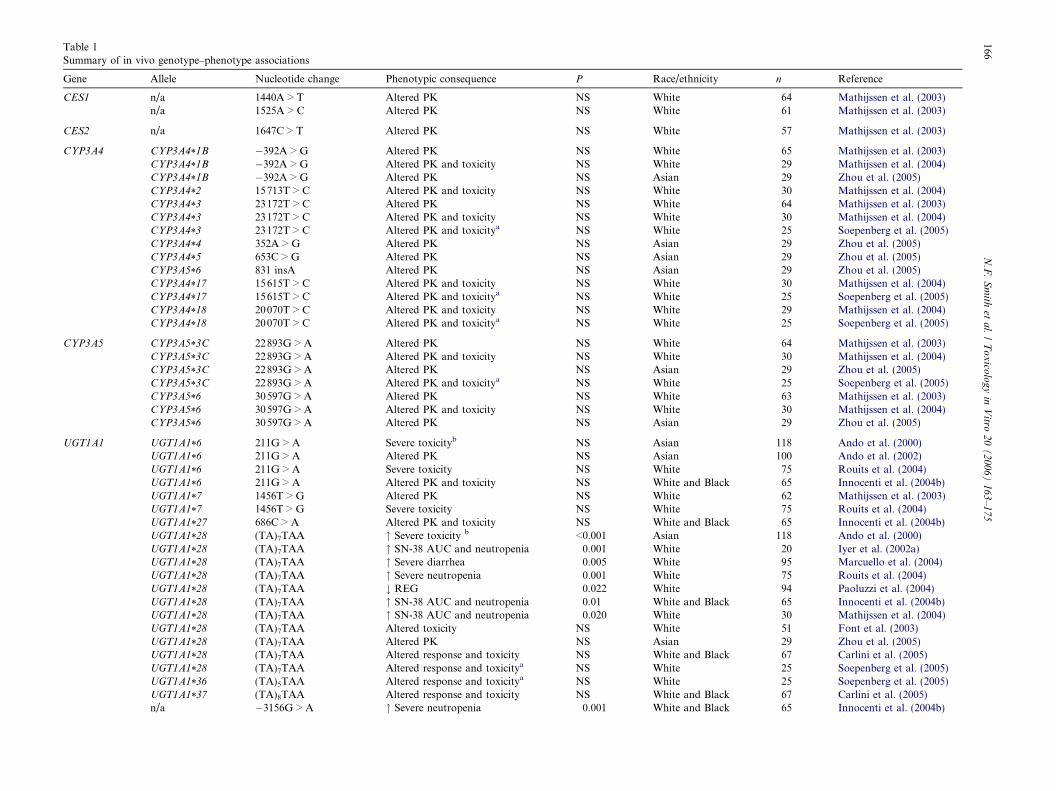
Table 1Summary of in vivo genotype–phenotype associations
Gene Allele Nucleotide change Phenotypic consequence P Race/ethnicity n Reference
CES1 n/a 1440A > T Altered PK NS White 64 Mathijssen et al. (2003)n/a 1525A > C Altered PK NS White 61 Mathijssen et al. (2003)
CES2 n/a 1647C > T Altered PK NS White 57 Mathijssen et al. (2003)
CYP3A4 CYP3A4*1B �392A > G Altered PK NS White 65 Mathijssen et al. (2003)CYP3A4*1B �392A > G Altered PK and toxicity NS White 29 Mathijssen et al. (2004)CYP3A4*1B �392A > G Altered PK NS Asian 29 Zhou et al. (2005)CYP3A4*2 15713T > C Altered PK and toxicity NS White 30 Mathijssen et al. (2004)CYP3A4*3 23172T > C Altered PK NS White 64 Mathijssen et al. (2003)CYP3A4*3 23172T > C Altered PK and toxicity NS White 30 Mathijssen et al. (2004)CYP3A4*3 23172T > C Altered PK and toxicitya NS White 25 Soepenberg et al. (2005)CYP3A4*4 352A > G Altered PK NS Asian 29 Zhou et al. (2005)CYP3A4*5 653C > G Altered PK NS Asian 29 Zhou et al. (2005)CYP3A5*6 831 insA Altered PK NS Asian 29 Zhou et al. (2005)CYP3A4*17 15615T > C Altered PK and toxicity NS White 30 Mathijssen et al. (2004)CYP3A4*17 15615T > C Altered PK and toxicitya NS White 25 Soepenberg et al. (2005)CYP3A4*18 20070T > C Altered PK and toxicity NS White 29 Mathijssen et al. (2004)CYP3A4*18 20070T > C Altered PK and toxicitya NS White 25 Soepenberg et al. (2005)
CYP3A5 CYP3A5*3C 22893G > A Altered PK NS White 64 Mathijssen et al. (2003)CYP3A5*3C 22893G > A Altered PK and toxicity NS White 30 Mathijssen et al. (2004)CYP3A5*3C 22893G > A Altered PK NS Asian 29 Zhou et al. (2005)CYP3A5*3C 22893G > A Altered PK and toxicitya NS White 25 Soepenberg et al. (2005)CYP3A5*6 30597G > A Altered PK NS White 63 Mathijssen et al. (2003)CYP3A5*6 30597G > A Altered PK and toxicity NS White 30 Mathijssen et al. (2004)CYP3A5*6 30597G > A Altered PK NS Asian 29 Zhou et al. (2005)
UGT1A1 UGT1A1*6 211G > A Severe toxicityb NS Asian 118 Ando et al. (2000)UGT1A1*6 211G > A Altered PK NS Asian 100 Ando et al. (2002)UGT1A1*6 211G > A Severe toxicity NS White 75 Rouits et al. (2004)UGT1A1*6 211G > A Altered PK and toxicity NS White and Black 65 Innocenti et al. (2004b)UGT1A1*7 1456T > G Altered PK NS White 62 Mathijssen et al. (2003)UGT1A1*7 1456T > G Severe toxicity NS White 75 Rouits et al. (2004)UGT1A1*27 686C > A Altered PK and toxicity NS White and Black 65 Innocenti et al. (2004b)UGT1A1*28 (TA)7TAA " Severe toxicity b <0.001 Asian 118 Ando et al. (2000)UGT1A1*28 (TA)7TAA " SN-38 AUC and neutropenia 0.001 White 20 Iyer et al. (2002a)UGT1A1*28 (TA)7TAA " Severe diarrhea 0.005 White 95 Marcuello et al. (2004)UGT1A1*28 (TA)7TAA " Severe neutropenia 0.001 White 75 Rouits et al. (2004)UGT1A1*28 (TA)7TAA # REG 0.022 White 94 Paoluzzi et al. (2004)UGT1A1*28 (TA)7TAA " SN-38 AUC and neutropenia 0.01 White and Black 65 Innocenti et al. (2004b)UGT1A1*28 (TA)7TAA " SN-38 AUC and neutropenia 0.020 White 30 Mathijssen et al. (2004)UGT1A1*28 (TA)7TAA Altered toxicity NS White 51 Font et al. (2003)UGT1A1*28 (TA)7TAA Altered PK NS Asian 29 Zhou et al. (2005)UGT1A1*28 (TA)7TAA Altered response and toxicity NS White and Black 67 Carlini et al. (2005)UGT1A1*28 (TA)7TAA Altered response and toxicitya NS White 25 Soepenberg et al. (2005)UGT1A1*36 (TA)5TAA Altered response and toxicitya NS White 25 Soepenberg et al. (2005)UGT1A1*37 (TA)8TAA Altered response and toxicity NS White and Black 67 Carlini et al. (2005)n/a �3156G > A " Severe neutropenia 0.001 White and Black 65 Innocenti et al. (2004b)
166N.F.Smith
etal./Toxico
logyin
Vitro
20(2006)163–175
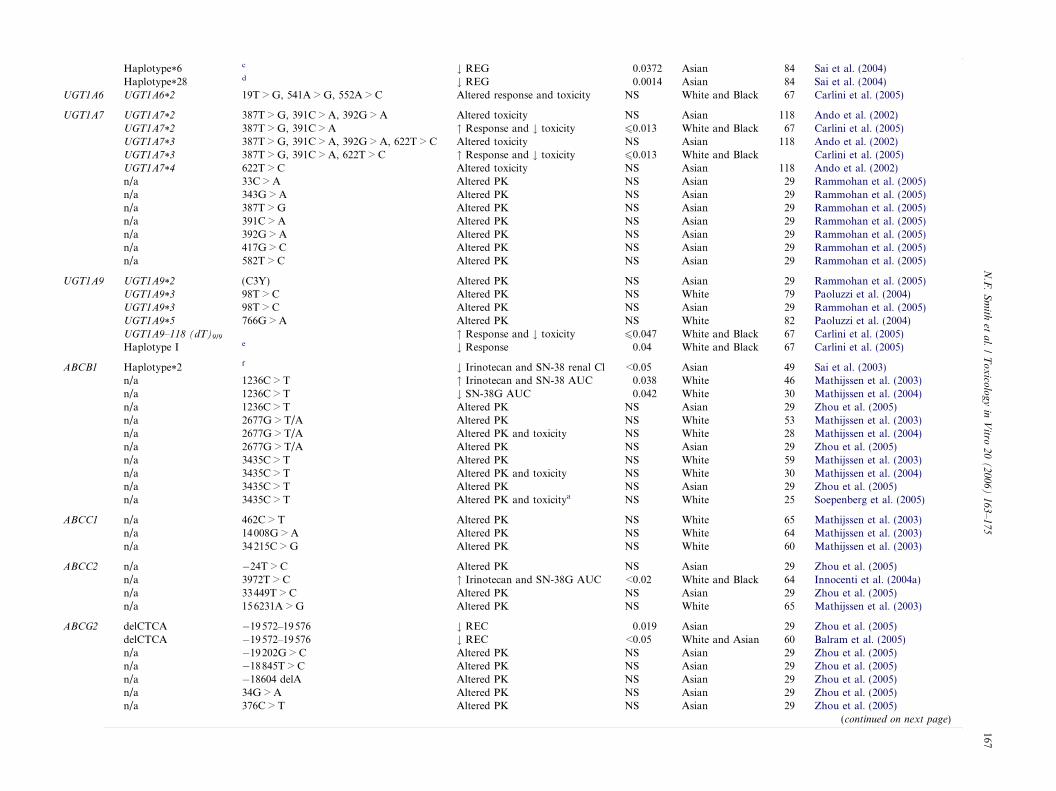
Haplotype*6c # REG 0.0372 Asian 84 Sai et al. (2004)
Haplotype*28d # REG 0.0014 Asian 84 Sai et al. (2004)
UGT1A6 UGT1A6*2 19T > G, 541A > G, 552A > C Altered response and toxicity NS White and Black 67 Carlini et al. (2005)
UGT1A7 UGT1A7*2 387T > G, 391C > A, 392G > A Altered toxicity NS Asian 118 Ando et al. (2002)UGT1A7*2 387T > G, 391C > A " Response and # toxicity 60.013 White and Black 67 Carlini et al. (2005)UGT1A7*3 387T > G, 391C > A, 392G > A, 622T > C Altered toxicity NS Asian 118 Ando et al. (2002)UGT1A7*3 387T > G, 391C > A, 622T > C " Response and # toxicity 60.013 White and Black Carlini et al. (2005)UGT1A7*4 622T > C Altered toxicity NS Asian 118 Ando et al. (2002)n/a 33C > A Altered PK NS Asian 29 Rammohan et al. (2005)n/a 343G > A Altered PK NS Asian 29 Rammohan et al. (2005)n/a 387T > G Altered PK NS Asian 29 Rammohan et al. (2005)n/a 391C > A Altered PK NS Asian 29 Rammohan et al. (2005)n/a 392G > A Altered PK NS Asian 29 Rammohan et al. (2005)n/a 417G > C Altered PK NS Asian 29 Rammohan et al. (2005)n/a 582T > C Altered PK NS Asian 29 Rammohan et al. (2005)
UGT1A9 UGT1A9*2 (C3Y) Altered PK NS Asian 29 Rammohan et al. (2005)UGT1A9*3 98T > C Altered PK NS White 79 Paoluzzi et al. (2004)UGT1A9*3 98T > C Altered PK NS Asian 29 Rammohan et al. (2005)UGT1A9*5 766G > A Altered PK NS White 82 Paoluzzi et al. (2004)UGT1A9–118 (dT)9/9 " Response and # toxicity 60.047 White and Black 67 Carlini et al. (2005)Haplotype I e # Response 0.04 White and Black 67 Carlini et al. (2005)
ABCB1 Haplotype*2f # Irinotecan and SN-38 renal Cl <0.05 Asian 49 Sai et al. (2003)
n/a 1236C > T " Irinotecan and SN-38 AUC 0.038 White 46 Mathijssen et al. (2003)n/a 1236C > T # SN-38G AUC 0.042 White 30 Mathijssen et al. (2004)n/a 1236C > T Altered PK NS Asian 29 Zhou et al. (2005)n/a 2677G > T/A Altered PK NS White 53 Mathijssen et al. (2003)n/a 2677G > T/A Altered PK and toxicity NS White 28 Mathijssen et al. (2004)n/a 2677G > T/A Altered PK NS Asian 29 Zhou et al. (2005)n/a 3435C > T Altered PK NS White 59 Mathijssen et al. (2003)n/a 3435C > T Altered PK and toxicity NS White 30 Mathijssen et al. (2004)n/a 3435C > T Altered PK NS Asian 29 Zhou et al. (2005)n/a 3435C > T Altered PK and toxicitya NS White 25 Soepenberg et al. (2005)
ABCC1 n/a 462C > T Altered PK NS White 65 Mathijssen et al. (2003)n/a 14008G > A Altered PK NS White 64 Mathijssen et al. (2003)n/a 34215C > G Altered PK NS White 60 Mathijssen et al. (2003)
ABCC2 n/a �24T > C Altered PK NS Asian 29 Zhou et al. (2005)n/a 3972T > C " Irinotecan and SN-38G AUC <0.02 White and Black 64 Innocenti et al. (2004a)n/a 33449T > C Altered PK NS Asian 29 Zhou et al. (2005)n/a 156231A > G Altered PK NS White 65 Mathijssen et al. (2003)
ABCG2 delCTCA �19572–19576 # REC 0.019 Asian 29 Zhou et al. (2005)delCTCA �19572–19576 # REC <0.05 White and Asian 60 Balram et al. (2005)n/a �19202G > C Altered PK NS Asian 29 Zhou et al. (2005)n/a �18845T > C Altered PK NS Asian 29 Zhou et al. (2005)n/a �18604 delA Altered PK NS Asian 29 Zhou et al. (2005)n/a 34G > A Altered PK NS Asian 29 Zhou et al. (2005)n/a 376C > T Altered PK NS Asian 29 Zhou et al. (2005)
(continued on next page)
N.F.Smith
etal./Toxico
logyin
Vitro
20(2006)163–175
167

Table
1(continued)
Gene
Allele
Nucleotidechan
gePhenotypic
consequence
PRace/ethnicity
nReference
n/a
421C
>A
Altered
PK
NS
White
84deJonget
al.(200
4a)
n/a
421C
>A
Altered
PK
NS
Asian
29Zhouet
al.(200
5)n/a
421C
>A
"REC
<0.05
Whitean
dAsian
60Balram
etal.(200
5)n/a
623T
>C
Altered
PK
NS
White
63Mathijssen
etal.(200
3)n/a
1444A
>G
Altered
PK
NS
Asian
29Zhouet
al.(200
5)n/a
1445G
>C
Altered
PK
NS
Asian
29Zhouet
al.(200
5)
n/a,notap
plicable;NS,notsign
ificant;REG,relative
extentofglucuronidation;REC,relative
extentofconversion.
aIrinotecanad
ministeredorally.
bLeukopenia
(grade4)
and/ordiarrhea
(grade3orworse).
cHap
lotypecontainsUGT1A1varian
tsat
positions211G
>A,378C
>T,an
d420G
>A.
dHap
lotypecontainsUGT1A1varian
tsat
positions�32
79T>G,�31
56G
>A,�36
4C>T,an
d(TA) 7TAA.
eHap
lotypecontainsUGT1A1*1
[(TA) 6TAA],UGT1A6*1
(19T
>G,54
1A>G,52
2A>C),UGT1A7*1
(342
G>A,38
7T>G,39
1C>A,62
2T>C),UGT1A9–118(dT)10/10.
fHap
lotypecontainsABCB1va
rian
tsat
positions12
36C>T,26
77G
>T,an
d34
35C>T.
168 N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175
occurrence of several polymorphisms within the CYP3A4
and CYP3A5 genes, no significant correlations betweenCYP3A4/5 genotype and irinotecan pharmacokinetics ortoxicity have been described to date (Mathijssen et al.,2003, 2004) (Table 1). A recent study by Mathijssen et al.(2003), investigated relationships between the pharmacoki-netics of irinotecan and its metabolites, and polymorphismsin CYP3A4 and CYP3A5, among other genes involvedin irinotecan disposition. Whilst there were no apparentpharmacokinetic associations with the CYP3A4*1B orCYP3A5*3C genotypes, the clearance of the lactone formof irinotecan was lower in patients heterozygous for theCYP3A4*3 polymorphism compared to wild-type individu-als. However, this difference was not significantly different(P = 0.059), possibly due to the small sample size. The lackof significant correlations between CYP3A4/5 genotype andirinotecan pharmacokinetics may be due to the low allelefrequency of most CYP3A variant genotypes in the Cauca-sian population (e.g., CYP3A4*17, CYP3A4*18, andCYP3A5*1), or may reflect the absence of a functionalchange in enzyme activity in vivo (e.g., CYP3A4*1B) (Xieet al., 2004). Thus, the role of CYP3A4/5 genotyping in iri-notecan therapy is currently unclear.
2.2. Carboxylesterase-mediated metabolism
Hydrolysis of the bulky dipiperidino moiety of irino-tecan produces the active metabolite SN-38 (Fig. 1). Theenzymes responsible for this reaction have been identifiedas the human carboxylesterases CES1, CES2 and, morerecently, CES3. However, the catalytic activity of theCES3 is so low, it is unlikely to be of importance for irino-tecan metabolism in vivo. The results of several studiespoint to CES2 as the major enzyme involved in irinotecanhydrolysis. Using purified enzymes, Humerickhouse et al.(2000) showed that CES2 has a much greater affinity for iri-notecan than CES1. In addition, HT29 cells overexpressingCES2 displayed a higher rate of irinotecan hydrolysis andwere more sensitive to irinotecan compared to those trans-fected with CES1 (Wu et al., 2002). Furthermore, Xu et al.(2002) found a significant correlation between liver micro-somal CES2 protein expression and SN-38 production.Studies using human liver microsomes and purifiedenzymes have shown that, in addition to irinotecan, CESis responsible for the conversion of NPC, but not APC,to SN-38 (Rivory et al., 1996; Dodds et al., 1998). How-ever, a more recent study demonstrated that APC is alsoa substrate for CES2, albeit a poor one (Sanghani et al.,2004). In addition to the liver, CES2 is expressed in multi-ple tissues including the small intestine, colon and plasma(Khanna et al., 2000; Guemei et al., 2001; Shingyojiet al., 2004). A recent study evaluated CES2 expressionin tumors and found that of the 18 different tumor typesanalyzed, only two did not express the protein. Large inter-individual variability in hepatic and intestinal CES2expression and irinotecan hydrolysis has been observed(Wu et al., 2002; Xu et al., 2002; Sanghani et al., 2003).

Table 2Summary of in vitro genotype–phenotype associations
Gene Allele Nucleotide change Phenotypic consequence P Reference
CES2 Haplotype a Altered irinotecan hydrolysis NS Wu et al. (2004)Haplotype b Altered irinotecan hydrolysis NS Wu et al. (2004)CES2*1 803C > G, 8721G > A, 9938G > A, 9943C > A Altered irinotecan hydrolysis NS Charasson et al. (2004)CES2*2 8721G > A Altered irinotecan hydrolysis NS Charasson et al. (2004)CES2*3 8721G > A, 9607C > T, 9624A > G, 9938G > A,
9943C > AAltered irinotecan hydrolysis NS Charasson et al. (2004)
CES2*4 8721G > A, 9938G > A, 9943C > A Altered irinotecan hydrolysis NS Charasson et al. (2004)CES2*7 4595T > C Altered irinotecan hydrolysis NS Charasson et al. (2004)CES2*8 7339G > A Altered irinotecan hydrolysis NS Charasson et al. (2004)CES2*10 1216T > C, 9938G > A, 9943C > A Altered irinotecan hydrolysis NS Charasson et al. (2004)
UGT1A1 UGT1A1*6 211G > A # SN-38 glucuronidationc <0.01 Jinno et al. (2003a)UGT1A1*6 211G > A # SN-38 glucuronidation <0.05 Gagne et al. (2002)UGT1A1*7 1456T > G # SN-38 glucuronidation <0.002 Gagne et al. (2002)UGT1A1*7 1456T > G # SN-38 glucuronidationc <0.01 Jinno et al. (2003a)UGT1A1*27 686C > A # SN-38 glucuronidation <0.002 Gagne et al. (2002)UGT1A1*27 686C > A # SN-38 glucuronidationc <0.01 Jinno et al. (2003a)n/a 686C > T # SN-38 glucuronidationc <0.05 Kaniwa et al. (2005)UGT1A1*28 (TA)7TAA # SN-38 glucuronidation <0.05 Iyer et al. (1999)UGT1A1*28 (TA)7TAA # SN-38 glucuronidation <0.001 Innocenti et al. (2002)UGT1A1*35 1291T > C # SN-38 glucuronidation <0.002 Gagne et al. (2002)
UGT1A7 UGT1A7*2 387T > G, 391C > A Altered SN-38 glucuronidation NS Gagne et al. (2002)UGT1A7*2 387T > G, 391C > A Altered SN-38 glucuronidation NS Villeneuve et al. (2003)UGT1A7*3 387T > G, 391C > A, 622T > C # SN-38 glucuronidation <0.05 Gagne et al. (2002)UGT1A7*3 387T > G, 391C > A, 622T > C # SN-38 glucuronidationd <0.05 Villeneuve et al. (2003)UGT1A7*4 622T > C # SN-38 glucuronidation <0.002 Gagne et al. (2002)UGT1A7*4 622T > C # SN-38 glucuronidationd <0.01 Villeneuve et al. (2003)UGT1A7*5 (G115S) # SN-38 glucuronidationd <0.05 Villeneuve et al. (2003)UGT1A7*6 (E139D) Altered SN-38 glucuronidation NS Villeneuve et al. (2003)UGT1A7*7 387T > G, 391C > A, (E139D) Altered SN-38 glucuronidation NS Villeneuve et al. (2003)UGT1A7*8 387T > G, 391C > A, (E139D), 622T > C # SN-38 glucuronidationd <0.05 Villeneuve et al. (2003)UGT1A7*9 (G115S), 387T > G, 391C > A # SN-38 glucuronidationd <0.05 Villeneuve et al. (2003)
UGT1A9 UGT1A9*2 8G > A Altered SN-38 glucuronidation NS Villeneuve et al. (2003)UGT1A9*3 98T > C # SN-38 glucuronidation <0.01 Villeneuve et al. (2003)UGT1A9*5 766G > A # SN-38 glucuronidationc <0.01 Jinno et al. (2003b)
a Haplotype between 50-UTR-363 and Intron1 + 1361.b Haplotypes formed on the four loci 50-UTR-363, Intron1 + 947, Intron1 + 1361, and Intron1 + 1643.c SN-38 glucuronidation efficiency ratio (normalized Vmax/Km).d Relative velocity (pmol/min/UGT protein levels).
N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175 169
The CES1 and/or CES2 genes were recently rese-quenced in an attempt to identify SNPs and determine hap-lotype structures (Charasson et al., 2004; Marsh et al.,2004; Wu et al., 2004). Multiple SNPs and haplotypes wereidentified and the frequencies of these were found to varybetween different ethnic populations. Marsh et al. (2004)examined the relationship between CES genotype andexpression in paired normal and tumor samples from 52patients with colorectal cancer. There was no associationbetween genetic polymorphisms and RNA expression innormal tissue. However, an intronic SNP in CES2 wasassociated with reduced CES2 expression in tumor tissue.
Charasson et al. (2004) identified 11 SNP�s that gave riseto 10 genotypes in addition to the wild-type. There was nosignificant difference in gene expression or carboxylesteraseactivity (measured using irinotecan as a substrate) betweenthe variant and the wild-type samples (Table 2). Wu et al.(2004) identified haplotype-expression correlations but didnot observe significant differences in the rate of irinotecan
hydrolysis by liver microsomes prepared from individualswith different haplotypes (Table 2). Jenabian et al. (2004)recently found a significant association between 2 SNP�s(�363C > G and 7555G > A) in CES2 and low RNAexpression levels. However, no significant difference inCES2 activity was observed among the different genotypes.Lastly, there was no association between the 1440A > Tand 1525A > C polymorphisms in CES1 or the 1647C >T polymorphism in CES2 and irinotecan pharmacoki-netics in 65 cancer patients (Mathijssen et al., 2003)(Table 1).
2.3. UDP-glucuronosyltransferases
UDP-glucuronosyltransferases (UGT�s) represent oneof the major classes of enzymes involved in phase II conju-gative metabolism. These enzymes catalyze the transfer of aglucuronic acid moiety from uridine diphosphoglucuronicacid to a wide range of structurally diverse endogenous

170 N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175
compounds and xenobiotics. The resulting glucuronideconjugates are more polar than the parent compoundsand are subsequently eliminated in the bile or urine.Knowledge of UGT�s, including their molecular genetics,substrate specificities and tissue distribution, has increasedconsiderably in recent years (Tukey and Strassburg, 2000).The liver represents one of the major sites of glucuronida-tion; however UGT�s are also expressed in extrahepatic tis-sues, including those of the gastrointestinal tract, thekidney and the brain. Seventeen UGT�s have been identi-fied in humans to date and these have been assigned totwo families, namely UGT1 and UGT2, which are furtherdivided on the basis of sequence homology into the sub-families UGT1A, UGT2A and UGT2B. Members of theUGT1 family are encoded by the UGT1A locus on chromo-some 2q37, which contains 13 first exons, each having itsown promoter and enhancer regions, which are spliced toidentical exons 2–5. In contrast, the UGT2 locus consistsof seven separate genes clustered on chromosome 4q13.A number of polymorphic variations have been describedfor both UGT1 and UGT2B gene families. However, onlya few of potential clinical relevance have been describedto date. The reader is referred to Guillemette (2003), fora recent review of UGT genetic polymorphisms and theirfunctional impact. Most efforts have focused on geneticalterations of the UGT1A1 gene; over 60 have beenreported so far and many of these influence its expressionand functional properties.
UGT1A1 is responsible for the glucuronidation of bili-rubin in humans and genetic variation of this isoform hasbeen extensively investigated in relation to hyperbilirubine-mic syndromes. These disorders, which include Gilbert�ssyndrome and Crigler–Najjar syndrome type I and II, arecharacterized by decreased or absent UGT1A1 activity asa result of UGT1A1 promoter or coding region polymor-phisms (Aono et al., 1993, 1994, 1995; Bosma et al.,1995). Of the known genetic variants, UGT1A1*28, whichresults from a dinucleotide (TA) insertion in the TATA ele-ment of the UGT1A1 promoter region [(TA)7TAA insteadof (TA)6TAA], is the most common and is associated withreduced gene expression together with a decrease in enzymeactivity, as measured by bilirubin and SN-38 glucuronida-tion (Iyer et al., 1999, 2002a). The frequency of these poly-morphic alleles varies considerably among different ethnicgroups, with the promoter variant being most prevalentamong Caucasians and African–Americans, whereas mis-sense mutations in the UGT1A1 coding region are morecommon in Asians (Desai et al., 2003). In vitro studieshave revealed that UGT1A1 is also involved in the glucu-ronidation of SN-38 to form the inactive SN-38-glucuro-nide (SN-38G), which undergoes subsequent biliaryexcretion (Iyer et al., 1998; Gagne et al., 2002). In addition,UGT1A9 and the extrahepatic UGT1A7 have beenreported to play a role in SN-38 inactivation. It followsthat in vitro and in vivo studies have been performed toinvestigate associations between UGT1A genotype and
phenotype. The results of these studies are summarized inTables 1 and 2.
Several groups have investigated the effects of polymor-phisms in UGT1A1, UGT1A7 and UGT1A9 enzymes onthe extent of SN-38 glucuronidation in vitro (Gagneet al., 2002; Iyer et al., 1999; Jinno et al., 2003a,b; Ville-neuve et al., 2003). This has been done using human livermicrosomes prepared from individuals with different geno-types or microsomal fractions prepared from cells (e.g.HEK293, COS-1) transiently or stably transfected withthe wild-type or variant cDNA�s. The majority of UGT1Apolymorphisms investigated to date are associated with adecrease in enzymatic activity in vitro, as evidenced by areduction in SN-38 glucuronide formation compared tothe wild-type enzyme (summarized in Table 2). The clinicalconsequences of these functional changes have been inves-tigated for many of these alleles. Several in vivo studieshave been performed to elucidate the effect of UGT1A
genotype on irinotecan pharmacokinetics and toxicity,which includes severe diarrhea and neutropenia in someindividuals (summarized in Table 1).
Ando et al. (2000) performed a retrospective study of118 Japanese cancer patients who had been treated with iri-notecan. In addition to the UGT1A1*28 promoter poly-morphism, the authors attempted to elucidate the clinicalsignificance of the coding region mutations that are morecommon in Asian populations, namely UGT1A1*6,UGT1A1*7, UGT1A1*27 andUGT1A1*29. The frequencyof UGT1A1*28 was 3.5-fold higher in those patients whoexperienced severe toxicity (grade 4 leukopenia and/orgrade 3 or 4 diarrhea) compared to those who did not.Of the 26 patients that experienced toxicity, theUGT1A1*28 variant alleles were homozygous in 15% andheterozygous in 31%. These frequencies were 3% and11%, respectively, among the 92 patients without toxicity.Multivariate analysis revealed that UGT1A1*28 was sig-nificantly associated with the development of irinotecantreatment-related toxicity (P < 0.001). The three individu-als that were heterozygous for UGT1A1*27 encounteredsevere toxicity. However, the frequency of UGT1A1*6was not significantly different between the two groups.None of the patients had the UGT1A1*7 or UGT1A1*29polymorphisms (Ando et al., 2000).
This was followed by a prospective study of 20 patientstreated with irinotecan, in which the relationship betweenUGT1A1 genotype and irinotecan pharmacokinetics wasevaluated in addition to toxicity (Iyer et al., 2002a). Therate of SN-38 glucuronidation was 3.9-fold lower inpatients with the UGT1A1*28 polymorphism comparedto those with the wild-type allele. In addition, it was foundthat more severe toxicity was observed in those patientswho were UGT1A1*28 carriers and that the presence ofthis allele was associated with a 2.5-fold lower absoluteneutrophil count at nadir (P = 0.04).
In 95 Caucasian colorectal cancer patients treated withirinotecan, the incidence of severe diarrhea was greater in

N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175 171
individuals homozygous (70%) and heterozygous (33%)for the variant allele compared to wild-type patients(17%) (P = 0.005). In addition, the occurrence of severehematological toxicity was higher, but this difference didnot reach statistical significance (Marcuello et al., 2004).Rouits et al. (2004) similarly reported increased toxicityin patients with the UGT1A1*28 allele receiving irinotecanand 5-FU.
Paoluzzi et al. (2004) investigated relationships betweenirinotecan pharmacokinetics and toxicity and UGT1A9
genotype, in addition to UGT1A1. The AUC ratio ofSN-38G to SN-38 was significantly reduced in patients withthe variant UGT1A1*28 allele (P = 0.022), but there wasno association with the occurrence of diarrhea (P = 0.74).One of 94 Caucasian patients had the variant UGT1A9*3(98T > C) allele and this did not affect irinotecan pharma-cokinetics. There were no UGT1A9*5 variants (766G > A)reported in this cohort. The authors concluded thatUGT1A9 functional variants are rare in Caucasians andare unlikely to be of clinical significance in irinotecan ther-apy (Paoluzzi et al., 2004).
Innocenti et al. (2004a,b) assessed genetic variants ofUGT1A1, including a novel promoter SNP �3156G > A,in 66 cancer patients receiving irinotecan therapy. Theseinvestigators found that grade 4 neutropenia was muchmore common in UGT1A1*28 homozygous patients(50%) than heterozygous patients (12.5%) and that noneof the patients with the normal allele experienced toxicity.Those patients with the 7/7 genotype had a 9.3-fold higherrisk of developing severe neutropenia versus the rest of thepatients (P = 0.01). In addition, there was an associationbetween pretreatment bilirubin levels and the risk of devel-oping toxicity following irinotecan therapy (P < 0.001)(Innocenti et al., 2004b).
Very recently, UGT1A haplotype-based studies havebeen performed (Liu et al., 2004; Sai et al., 2004). Saiet al. (2004), identified 6 UGT1A1 haplotype groups andinvestigated their association with irinotecan pharmacoki-netics and serum bilirubin levels in 85 Japanese cancerpatients. Two haplotype groups (*28 and *6) were associ-ated with a reduced SN-38G to SN-38 AUC ratio(P = 0.0014) and three (*28, *60 and *IB) were associatedwith increased bilirubin (P = 0.0007) (Sai et al., 2004).Haplotype analysis of UGT1A1 and UGT1A9 polymor-phisms in Asian and Caucasian populations was recentlyperformed and interethnic differences in haplotype frequen-cies observed. However, associations between these haplo-types and irinotecan have yet to be reported (Liu et al.,2004).
In summary, there is accumulating evidence to suggestthat screening for UGT1A1 polymorphisms might be clini-cally useful for predicting irinotecan toxicity in cancerpatients and a prospective trial is warranted to elucidatethis. UGT1A1 genotyping is not yet available in the clinicalsetting but may ultimately be used to determine whetherthe standard dose of irinotecan should be reduced or an
alternative treatment selected in order to avoid severe tox-icity in a susceptible individual.
3. Irinotecan transport
In addition to metabolism by multiple drug metaboliz-ing enzymes, irinotecan and its metabolites are subjectto transport by several members of the ATP-binding cas-sette (ABC) transporter superfamily ( Fig. 2). These trans-membrane proteins facilitate the cellular efflux of variousmolecules and are expressed in a wide variety of normal tis-sues (Sparreboom et al., 2003). The ABC transporters playan important role in the development of multi-drug resis-tance by cancer cells. In addition, they are known to influ-ence the disposition of drugs by way of their involvement inintestinal absorption and excretion by biliary and renalmechanisms. Moreover, expression of these transportersat the blood brain barrier limits the entry of many drugsinto the central nervous system. Several polymorphic vari-ants of the ABC transporter genes have recently beendescribed, which may alter the function and/or expressionof the corresponding proteins. Irinotecan, SN-38 andSN-38G are transported out of cells by ABCB1 (multi-drug resistance [MDR1]; P-glycoprotein [P-gp]), ABCC1(multi-drug resistance protein [MRP1]), ABCC2 (canalicu-lar multispecific organic anion transporter [C-MOAT];MRP2) and ABCG2 (breast cancer resistance protein[BCRP]).
3.1. ABCB1-mediated transport
The involvement of ABCB1 in irinotecan transport wasdemonstrated in recent studies using ABCB1 knockoutmice (Iyer et al., 2002b). The biliary recovery of irinotecanwas significantly lower in the knockout mice compared towild-type animals, implicating ABCB1 in irinotecan biliaryexcretion, whereas that of SN-38 and SN-38G was unaf-fected. Of all the ABC drug transporters involved inirinotecan disposition, evidence for a pharmacogeneticassociation is most compelling for ABCB1. Several poly-morphisms in the ABCB1 gene have been reported. Studiesevaluating the effect of these on irinotecan pharmacokinet-ics have focused on the commonly occurring polymor-phisms 1236C > T, 2677G > T/A, and 3435C > T, whichexist in strong linkage disequilibrium. In a study of 65 can-cer patients treated with irinotecan, a significant associa-tion between the 1236C > T polymorphism and exposureto irinotecan and SN-38 was observed (Mathijssen et al.,2004) (Table 1). The AUC of irinotecan (P = 0.038) andSN-38 (P = 0.031) was significantly higher in homozygouspatients compared to heterozygous and wild-type patients.In addition, the clearance of the lactone form of SN-38 dif-fered (P = 0.015). Sai et al. (2003) found a significant asso-ciation between the ABCB1 haplotype*2, which containsthe three SNP�s described, and reduced renal clearance ofirinotecan (P < 0.05), SN-38 (P < 0.01) and APC in a study

172 N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175
of 49 Japanese cancer patients, possibly due to reducedABCB1 expression in renal proximal tubules (Table 1).
3.2. ABCC1-mediated transport
A series of in vitro experiments identified ABCC1 as atransporter of irinotecan and SN-38. Transfection ofABCC1 cDNA into KB-3-1 human epidermoid carcinomacells resulted in decreased accumulation and increasedresistance to irinotecan and SN-38 compared to untrans-fected cells. Treatment with PAK-104P, a known ABCC1inhibitor, increased cellular accumulation and restored sen-sitivity (Chen et al., 1999). Polymorphisms in the ABCC1
gene have been identified, but no associations with irino-tecan pharmacokinetics or toxicity have been reported(Mathijssen et al., 2003) (Table 1).
3.3. ABCC2-mediated transport
Evidence for a role of ABCC2 in irinotecan transportcomes from studies comparing the biliary excretion of iri-notecan and its metabolites in Sprague–Dawley and Eisaihyperbilirubinemic (EHBR) rats, which are deficient inABCC2. The biliary excretion of irinotecan carboxylateand SN-38 carboxylate, and both the lactone and carboxyl-ate forms of SN-38G was lower in the ABCC2-deficientrats. In addition, uptake by hepatic canalicular membranevesicles isolated from the rats carrying the ABCC2 muta-tion was lower than those prepared from the other strain(Chu et al., 1997a,b).
Recently, polymorphisms in ABCC2 have been found toinfluence irinotecan disposition. Innocenti et al. (2004a,b)recently reported that the synonymous 3972T > C variantwas correlated with irinotecan AUC (P = 0.02), APCAUC (P < 0.0001) and APC:irinotecan AUC ratios(P < 0.0001) in a study of 64 patients (Innocenti et al.,2004a). Whilst there was no association with AUC itself,there was a correlation with SN-38G:SN-38 AUC ratio(P = <0.001) (Table 1). Kitigawa et al. (2004) investigatedrelationships between ABCC2 variants and irinotecan tox-icity in 120 Japanese patients. However, no significantassociation between the 1249G > A or �24C > T variantsand severe toxicity were observed (Table 1).
3.4. ABCG2-mediated transport
The results of multiple studies indicate that irinotecanand its metabolites are transported by ABCG2. Severalgroups have shown that overexpression of ABCG2 conferscancer cell resistance to irinotecan and SN-38 (Schellenset al., 2000; Nakatomi et al., 2001; Candeil et al., 2004).Furthermore, it has recently been demonstrated that theABCG2 mRNA content of liver metastatic tumor cellsfrom colorectal cancer patients treated with irinotecan ishigher than those from irinotecan-naı̈ve patients.
Several SNP�s in the ABCG2 gene have been identified(Iida et al., 2002; Itoda et al., 2003; Zamber et al., 2003).
Of these, the most extensively studied is the 421C > Atransversion, which results in an amino acid change of glu-tamine to lysine at codon 141 (Q141K). It has been demon-strated in vitro that this SNP results in markedly reducedprotein expression and low levels of irinotecan resistancecompared to wild-type transfected cells (Imai et al.,2002). de Jong et al. (2004a,b) recently evaluated the allelefrequency of this polymorphism in a large number of indi-viduals from different ethnic populations and its effect onirinotecan pharmacokinetics in 84 Caucasian patients. Sig-nificant interethnic variability in allele frequency wasobserved with the variant allele being most common inthe Han Chinese population (34%). There was no signifi-cant difference in irinotecan (P = 0.72) or SN-38 (P =0.67) AUC between wild-type patients and those carryingat least one variant allele (Table 1). However, one of thetwo homozygous patients showed extensive accumulationof SN-38 and SN-38G and suffered from severe toxicity.The authors noted that it is possible that other processesinvolved in irinotecan metabolism and elimination thatexhibit great interindividual variation might be over-shad-owing any effect of this ABCG2 polymorphism (de Jonget al., 2004a).
4. Conclusion
Irinotecan is one of the most important anticancer drugsdeveloped in the last few decades. The clinical pharmaco-kinetic profile of irinotecan has been explored extensivelyin recent years, and the generated information has beenof fundamental importance in our understanding of theclinical effects of this agent. In addition, a wealth of infor-mation has become available that has yielded valuableinsight into the role of polymorphic enzymes and trans-porters in irinotecan-mediated toxicities. Specifically, vari-ous investigators have now independently confirmed thatthe UGT1A1*28 genotype significantly affects the pharma-cokinetic profile of irinotecan as well as treatment-relatedmyelosuppression. Nonetheless, adequately powered pro-spective trials are required to evaluate the clinical utilityof UGT1A1*28 screening prior to chemotherapeutic treat-ment with irinotecan in order to identify patients that aregenetically predisposed to severe side effects.
References
Ando, Y., Saka, H., Ando, M., Sawa, T., Muro, K., Ueoka, H.,Yokoyama, A., Saitoh, S., Shimokata, K., Hasegawa, Y., 2000.Polymorphisms of UDP-glucuronosyltransferase gene and irinotecantoxicity: a pharmacogenetic analysis. Cancer Res. 60, 6921–6926.
Ando, Y., Ueoka, H., Sugiyama, T., Ichiki, M., Shimokata, K.,Hasegawa, Y., 2002. Polymorphisms of UDP-glucuronosyltransferaseand pharmacokinetics of irinotecan. Ther. Drug. Monit. 24, 111–116.
Aono, S., Yamada, Y., Keino, H., Hanada, N., Nakagawa, T., Sasaoka,Y., Yazawa, T., Sato, H., Koiwai, O., 1993. Identification of defect inthe genes for bilirubin UDP-glucuronosyltransferase in a patient withCrigler–Najjar syndrome type II. Biochem. Biophys. Res. Commun.197, 1239–1244.

N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175 173
Aono, S., Yamada, Y., Keino, H., Sasaoka, Y., Nakagawa, T., Onishi, S.,Mimura, S., Koiwai, O., Sato, H., 1994. A new type of defect in thegene for bilirubin uridine 5 0-diphosphate-glucuronosyltransferase in apatient with Crigler–Najjar syndrome type I. Pediatr. Res. 35, 629–632.
Aono, S., Adachi, Y., Uyama, E., Yamada, Y., Keino, H., Nanno, T.,Koiwai, O., Sato, H., 1995. Analysis of genes for bilirubin UDP-glucuronosyltransferase in Gilbert�s syndrome. Lancet 345, 958–959.
Balram, C., Li, J., Zhou, Q.-Y., Tan, E.-H., Mathijssen, R.H., van Schaik,R.H., Verweij, J., Sparreboom, A., Baker, S.D., 2005. Molecularmechanisms of interethnic differences in irinotecan disposition: impactof variants in ABCG2. J. Clin. Oncol. 23, 2018.
Bosma, P.J., Chowdhury, J.R., Bakker, C., Gantla, S., de Boer, A.,Oostra, B.A., Lindhout, D., Tytgat, G.N., Jansen, P.L., Oude Elferink,R.P., et al., 1995. The genetic basis of the reduced expression ofbilirubin UDP-glucuronosyltransferase 1 in Gilbert�s syndrome. NEngl. J. Med. 333, 1171–1175.
Candeil, L., Gourdier, I., Peyron, D., Vezzio, N., Copois, V., Bibeau, F.,Orsetti, B., Scheffer, G.L., Ychou, M., Khan, Q.A., Pommier, Y., Pau,B., Martineau, P., Del Rio, M., 2004. ABCG2 overexpression in coloncancer cells resistant to SN38 and in irinotecan-treated metastases. Int.J. Cancer 109, 848–854.
Carlini, L.E., Meropol, N.J., Bever, J., Andria, M.L., Hill, T., Gold, P.,Rogatko, A., Wang, H., Blanchard, R.L., 2005. UGT1A7 andUGT1A9 polymorphisms predict response and toxicity in colorectalcancer patients treated with capecitabine/irinotecan. Clin. Cancer Res.11, 1226–1236.
Charasson, V., Bellott, R., Meynard, D., Longy, M., Gorry, P., Robert, J.,2004. Pharmacogenetics of human carboxylesterase 2, an enzymeinvolved in the activation of irinotecan into SN-38. Clin. Pharmacol.Ther. 76, 528–535.
Chen, Z.S., Furukawa, T., Sumizawa, T., Ono, K., Ueda, K., Seto, K.,Akiyama, S.I., 1999. ATP-Dependent efflux of CPT-11 and SN-38 bythe multidrug resistance protein (MRP) and its inhibition by PAK-104P. Mol. Pharmacol. 55, 921–928.
Chu, X.Y., Kato, Y., Niinuma, K., Sudo, K.I., Hakusui, H., Sugiyama,Y., 1997a. Multispecific organic anion transporter is responsible forthe biliary excretion of the camptothecin derivative irinotecan and itsmetabolites in rats. J. Pharmacol. Exp. Ther. 281, 304–314.
Chu, X.Y., Kato, Y., Sugiyama, Y., 1997b. Multiplicity of biliaryexcretion mechanisms for irinotecan, CPT-11, and its metabolites inrats. Cancer Res. 57, 1934–1938.
de Jong, F.A., Marsh, S., Mathijssen, R.H., King, C., Verweij, J.,Sparreboom, A., McLeod, H.L., 2004a. ABCG2 pharmacogenetics:ethnic differences in allele frequency and assessment of influence onirinotecan disposition. Clin. Cancer Res. 10, 5889–5894.
de Jong, F.A., Mathijssen, R.H., Xie, R., Verweij, J., Sparreboom, A.,2004b. Flat-fixed dosing of irinotecan: influence on pharmacoki-netic and pharmacodynamic variability. Clin. Cancer Res. 10, 4068–4071.
Desai, A.A., Innocenti, F., Ratain, M.J., 2003. Pharmacogenomics: roadto anticancer therapeutics nirvana? Oncogene 22, 6621–6628.
Dodds, H.M., Haaz, M.C., Riou, J.F., Robert, J., Rivory, L.P., 1998.Identification of a new metabolite of CPT-11 (irinotecan): pharmaco-logical properties and activation to SN-38. J. Pharmacol. Exp. Ther.286, 578–583.
Evans, W.E., McLeod, H.L., 2003. Pharmacogenomics—drug disposition,drug targets, and side effects. N Engl. J. Med. 348, 538–549.
Font, A., Sanchez, J.M., Taron, M., Martinez-Balibrea, E., Sanchez, J.J.,Manzano, J.L., Margeli, M., Richardet, M., Barnadas, A., Abad, A.,Rosell, R., 2003. Weekly regimen of irinotecan/docetaxel in previouslytreated non-small cell lung cancer patients and correlation with uridinediphosphate glucuronosyltransferase 1A1 (UGT1A1) polymorphism.Invest. New Drugs 21, 435–443.
Gagne, J.F., Montminy, V., Belanger, P., Journault, K., Gaucher, G.,Guillemette, C., 2002. Common human UGT1A polymorphisms andthe altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38). Mol. Pharmacol. 62, 608–617.
Guemei, A.A., Cottrell, J., Band, R., Hehman, H., Prudhomme, M.,Pavlov, M.V., Grem, J.L., Ismail, A.S., Bowen, D., Taylor, R.E.,Takimoto, C.H., 2001. Human plasma carboxylesterase and butyr-ylcholinesterase enzyme activity: correlations with SN-38 pharmaco-kinetics during a prolonged infusion of irinotecan. Cancer Chemother.Pharmacol. 47, 283–290.
Guillemette, C., 2003. Pharmacogenomics of human UDP-glucuronosyl-transferase enzymes. Pharmacogenomics J. 3, 136–158.
Haaz, M.C., Riche, C., Rivory, L.P., Robert, J., 1998a. Biosynthesis of anaminopiperidino metabolite of irinotecan [7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecine] by human hepatic micro-somes. Drug Metab. Dispos. 26, 769–774.
Haaz, M.C., Rivory, L., Riche, C., Vernillet, L., Robert, J., 1998b.Metabolism of irinotecan (CPT-11) by human hepatic microsomes:participation of cytochrome P-450 3A and drug interactions. CancerRes. 58, 468–472.
Hsiang, Y.H., Liu, L.F., 1988. Identification of mammalian DNAtopoisomerase I as an intracellular target of the anticancer drugcamptothecin. Cancer Res. 48, 1722–1726.
Hsiang, Y.H., Hertzberg, R., Hecht, S., Liu, L.F., 1985. Camptothecininduces protein-linked DNA breaks via mammalian DNA topoiso-merase I. J. Biol. Chem. 260, 14873–14878.
Humerickhouse, R., Lohrbach, K., Li, L., Bosron, W.F., Dolan, M.E.,2000. Characterization of CPT-11 hydrolysis by human liver carboxy-lesterase isoforms hCE-1 and hCE-2. Cancer Res. 60, 1189–1192.
Iida, A., Saito, S., Sekine, A., Mishima, C., Kitamura, Y., Kondo, K.,Harigae, S., Osawa, S., Nakamura, Y., 2002. Catalog of 605 single-nucleotide polymorphisms (SNPs) among 13 genes encoding humanATP-binding cassette transporters: ABCA4, ABCA7, ABCA8,ABCD1, ABCD3, ABCD4, ABCE1, ABCF1, ABCG1, ABCG2,ABCG4, ABCG5, and ABCG8. J. Hum. Genet. 47, 285–310.
Imai, Y., Nakane, M., Kage, K., Tsukahara, S., Ishikawa, E., Tsuruo, T.,Miki, Y., Sugimoto, Y., 2002. C421A polymorphism in the humanbreast cancer resistance protein gene is associated with low expressionof Q141K protein and low-level drug resistance. Mol. Cancer Ther. 1,611–616.
Innocenti, F., Grimsley, C., Das, S., Ramirez, J., Cheng, C., Kuttab-Boulos, H., Ratain, M.J., Di Rienzo, A., 2002. Haplotype structure ifthe UDP-glucuronosyltransferase 1A1 promoter in different ethnicgroups. Pharmacogenetics 12, 725–733.
Innocenti, F., Undevia, S.D., Chen, P.X., Das, S., Ramirez, J., Dolan,M.E., Relling, M.V., Kroetz, D.L., Ratain, M.J., 2004a. Pharmaco-genetic analysis of interindividual irinotecan (CPT-11) pharmacoki-netic (PK) variability: Evidence for a functional variant of ABCC2.J. Clin. Oncol. 22, 2010.
Innocenti, F., Undevia, S.D., Iyer, L., Chen, P.X., Das, S., Kocherginsky,M., Karrison, T., Janisch, L., Ramirez, J., Rudin, C.M., Vokes, E.E.,Ratain, M.J., 2004b. Genetic variants in the UDP-glucuronosyltrans-ferase 1A1 gene predict the risk of severe neutropenia of irinotecan.J. Clin. Oncol. 22, 1382–1388.
Itoda, M., Saito, Y., Shirao, K., Minami, H., Ohtsu, A., Yoshida, T.,Saijo, N., Suzuki, H., Sugiyama, Y., Ozawa, S., Sawada, J., 2003.Eight novel single nucleotide polymorphisms in ABCG2/BCRP inJapanese cancer patients administered irinotecan. Drug Metab. Phar-macokinet. 18, 212–217.
Iyer, L., King, C.D., Whitington, P.F., Green, M.D., Roy, S.K., Tephly,T.R., Coffman, B.L., Ratain, M.J., 1998. Genetic predisposition to themetabolism of irinotecan (CPT-11). Role of uridine diphosphateglucuronosyltransferase isoform 1A1 in the glucuronidation of itsactive metabolite (SN-38) in human liver microsomes. J. Clin. Invest.101, 847–854.
Iyer, L., Hall, D., Das, S., Mortell, M.A., Ramirez, J., Kim, S., Di Rienzo,A., Ratain, M.J., 1999. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation inhuman liver tissue with UGT1A1 promoter polymorphism. Clin.Pharmacol. Ther. 65, 576–582.
Iyer, L., Das, S., Janisch, L., Wen, M., Ramirez, J., Karrison, T., Fleming,G.F., Vokes, E.E., Schilsky, R.L., Ratain, M.J., 2002a. UGT1A1*28

174 N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175
polymorphism as a determinant of irinotecan disposition and toxicity.Pharmacogenomics J. 2, 43–47.
Iyer, L., Ramirez, J., Shepard, D.R., Bingham, C.M., Hossfeld, D.K.,Ratain, M.J., Mayer, U., 2002b. Biliary transport of irinotecan andmetabolites in normal and P-glycoprotein-deficient mice. CancerChemother. Pharmacol. 49, 336–341.
Jenabian, A., Goasgen, N., Flinois, J.P., Gad, S., Beaune, P., 2004.Genetic polymorphisms of CES2 and their functional consequences.J. Clin. Oncol. 22, 2065.
Jinno, H., Tanaka-Kagawa, T., Hanioka, N., Saeki, M., Ishida, S.,Nishimura, T., Ando, M., Saito, Y., Ozawa, S., Sawada, J., 2003a.Glucuronidation of 7-ethyl-10-hydroxycamptothecin (SN-38), anactive metabolite of irinotecan (CPT-11), by human UGT1A1variants, G71R, P229Q, and Y486D. Drug Metab. Dispos. 31, 108–113.
Jinno, H., Saeki, M., Saito, Y., Tanaka-Kagawa, T., Hanioka, N., Sai, K.,Kaniwa, N., Ando, M., Shirao, K., Minami, H., Ohtsu, A., Yoshida,T., Saijo, N., Ozawa, S., Sawada, J., 2003b. Functional characteriza-tion of human UDP-glucuronosyltransferase 1A9 variant, D256N,found in Japanese cancer patients. J. Pharmacol. Exp. Ther. 306, 688–693.
Kaniwa, N., Kurose, K., Jinno, H., Tanaka-Kagawa, T., Saito, Y., Saeki,M., Sawada, J., Tohkin, M., Hasegawa, R., 2005. Racial variability inhaplotype frequencies of UGT1A1 and glucuronidation activity of anovel single nucleotide polymorphism 686C > T (P229L) found in anAfrican–American. Drug Metab. Dispos. 33, 458–465.
Kawato, Y., Aonuma, M., Hirota, Y., Kuga, H., Sato, K., 1991.Intracellular roles of SN-38, a metabolite of the camptothecinderivative CPT-11, in the antitumor effect of CPT-11. Cancer Res.51, 4187–4191.
Khanna, R., Morton, C.L., Danks, M.K., Potter, P.M., 2000. Proficientmetabolism of irinotecan by a human intestinal carboxylesterase.Cancer Res. 60, 4725–4728.
Kitigawa, C., Ando, M., Ando, Y., Sekido, Y., Usui, M., Takahashi, K.,Shimokata, Y., Hasegawa, Y., 2004. Genetic polymorphisms of themultidrug resistance-associated protein 2 gene (ABCC2) and Irino-tecan toxicity. J. Clin. Oncol. 22, 2009.
Liu, W., Innocenti, F., Chen, P.X., Desai, A.A., Grimsley, C., Di Rienzo,A., Das, S., Ratain, M.J., 2004. Haplotype analysis of UGT1A1 andUGT1A9 gene polymorphisms related to the glucuronidation of SN-38, the active metabolite of irinotecan. J. Clin. Oncol. 22, 2085.
Lokiec, F., du Sorbier, B.M., Sanderink, G.J., 1996. Irinotecan (CPT-11)metabolites in human bile and urine. Clin. Cancer Res. 2, 1943–1949.
Marcuello, E., Altes, A., Menoyo, A., Del Rio, E., Gomez-Pardo, M.,Baiget, M., 2004. UGT1A1 gene variations and irinotecan treatment inpatients with metastatic colorectal cancer. Br. J. Cancer 91, 678–682.
Marsh, S., Xiao, M., Yu, J., Ahluwalia, R., Minton, M., Freimuth, R.R.,Kwok, P.Y., McLeod, H.L., 2004. Pharmacogenomic assessment ofcarboxylesterases 1 and 2. Genomics 84, 661–668.
Mathijssen, R.H., Verweij, J., de Jonge, M.J., Nooter, K., Stoter, G.,Sparreboom, A., 2002. Impact of body-size measures on irinotecanclearance: alternative dosing recommendations. J. Clin. Oncol. 20, 81–87.
Mathijssen, R.H., Marsh, S., Karlsson, M.O., Xie, R., Baker, S.D.,Verweij, J., Sparreboom, A., McLeod, H.L., 2003. Irinotecan pathwaygenotype analysis to predict pharmacokinetics. Clin. Cancer Res. 9,3246–3253.
Mathijssen, R.H., de Jong, F.A., van Schaik, R.H., Lepper, E.R., Friberg,L.E., Rietveld, T., de Bruijn, P., Graveland, W.J., Figg, W.D., Verweij,J., Sparreboom, A., 2004. Prediction of irinotecan pharmacokineticsby use of cytochrome P450 3A4 phenotyping probes. J. Natl. CancerInst. 96, 1585–1592.
Nakatomi, K., Yoshikawa, M., Oka, M., Ikegami, Y., Hayasaka, S.,Sano, K., Shiozawa, K., Kawabata, S., Soda, H., Ishikawa, T.,Tanabe, S., Kohno, S., 2001. Transport of 7-ethyl-10-hydroxycam-ptothecin (SN-38) by breast cancer resistance protein ABCG2 inhuman lung cancer cells. Biochem. Biophys. Res. Commun. 288, 827–832.
Paoluzzi, L., Singh, A.S., Price, D.K., Danesi, R., Mathijssen, R.H.,Verweij, J., Figg, W.D., Sparreboom, A., 2004. Influence of geneticvariants in UGT1A1 and UGT1A9 on the in vivo glucuronidation ofSN-38. J. Clin. Pharmacol. 44, 854–860.
Pizzolato, J.F., Saltz, L.B., 2003. The camptothecins. Lancet 361, 2235–2242.
Rammohan, M., Jeevananthinee, J., Zhou, Q.-Y., Tan, E.-H., Sparre-boom, A., Verweij, J., Balram, C., 2005. The influence of functionalpolymorphisms in UGT1A7 and UGT1A9 on irinotecan pharmaco-kinetics in cancer patients. J. Clin. Oncol. 23, 2007.
Rivory, L.P., Riou, J.F., Haaz, M.C., Sable, S., Vuilhorgne, M.,Commercon, A., Pond, S.M., Robert, J., 1996. Identification andproperties of a major plasma metabolite of irinotecan (CPT-11)isolated from the plasma of patients. Cancer Res. 56, 3689–3694.
Rouits, E., Boisdron-Celle, M., Dumont, A., Guerin, O., Morel, A.,Gamelin, E., 2004. Relevance of different UGT1A1 polymorphisms inirinotecan-induced toxicity: a molecular and clinical study of 75patients. Clin. Cancer Res. 10, 5151–5159.
Sai, K., Kaniwa, N., Itoda, M., Saito, Y., Hasegawa, R., Komamura, K.,Ueno, K., Kamakura, S., Kitakaze, M., Shirao, K., Minami, H.,Ohtsu, A., Yoshida, T., Saijo, N., Kitamura, Y., Kamatani, N.,Ozawa, S., Sawada, J., 2003. Haplotype analysis of ABCB1/MDR1blocks in a Japanese population reveals genotype-dependent renalclearance of irinotecan. Pharmacogenetics 13, 741–757.
Sai, K., Saeki, M., Saito, Y., Ozawa, S., Katori, N., Jinno, H., Hasegawa,R., Kaniwa, N., Sawada, J., Komamura, K., Ueno, K., Kamakura, S.,Kitakaze, M., Kitamura, Y., Kamatani, N., Minami, H., Ohtsu, A.,Shirao, K., Yoshida, T., Saijo, N., 2004. UGT1A1 haplotypesassociated with reduced glucuronidation and increased serum bilirubinin irinotecan-administered Japanese patients with cancer. Clin. Phar-macol. Ther. 75, 501–515.
Sanghani, S.P., Quinney, S.K., Fredenburg, T.B., Sun, Z., Davis, W.I.,Murry, D.J., Cummings, O.W., Seitz, D.E., Bosron, W.F., 2003.Carboxylesterases expressed in human colon tumor tissue and theirrole in CPT-11 hydrolysis. Clin. Cancer Res. 9, 4983–4991.
Sanghani, S.P., Quinney, S.K., Fredenburg, T.B., Davis, W.I., Murry,D.J., Bosron, W.F., 2004. Hydrolysis of irinotecan and its oxidativemetabolites, 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino]-carbonyloxycamptothecin and 7-ethyl-10-[4-(1-piperidino)-1-amino]-carbonyloxycamptothecin, by human carboxylesterases CES1A1,CES2, and a newly expressed carboxylesterase isoenzyme, CES3.Drug Metab. Dispos. 32, 505–511.
Santos, A., Zanetta, S., Cresteil, T., Deroussent, A., Pein, F., Raymond,E., Vernillet, L., Risse, M.L., Boige, V., Gouyette, A., Vassal, G.,2000. Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 inhumans. Clin. Cancer Res. 6, 2012–2020.
Schellens, J.H., Maliepaard, M., Scheper, R.J., Scheffer, G.L., Jonker,J.W., Smit, J.W., Beijnen, J.H., Schinkel, A.H., 2000. Transport oftopoisomerase I inhibitors by the breast cancer resistance protein.Potential clinical implications. Ann. N.Y. Acad. Sci. 922, 188–194.
Shingyoji, M., Takiguchi, Y., Watanabe-Uruma, R., Asaka-Amano, Y.,Matsubara, H., Kurosu, K., Kasahara, Y., Tanabe, N., Tatsumi, K.,Kuriyama, T., 2004. In vitro conversion of irinotecan to SN-38 inhuman plasma. Cancer Sci. 95, 537–540.
Smith, N.F., Figg, W.D., Sparreboom, A., 2004. Recent advances inpharmacogenetic approaches to anticancer drug development. DrugDev. Res., 1–21.
Soepenberg, O., Dumez, H., Verweij, J., de Jong, F.A., de Jonge, M.J.,Thomas, J., Eskens, F.A., van Schaik, R.H., Selleslach, J., Ter Steeg,J., Lefebvre, P., Assadourian, S., Sanderlink, G.J., Sparreboom, A.,Oosterom, A.T., 2005. Phase I pharmacokinetic, food effect, andpharmacogenetic study of oral irinotecan given as semisolid matrixcapsules in patients with solid tumors. Clin. Cancer Res. 15, 1504–1511.
Sparreboom, A., Danesi, R., Ando, Y., Chan, J., Figg, W.D., 2003.Pharmacogenomics of ABC transporters and its role in cancerchemotherapy. Drug Resist. Updat. 6, 71–84.

N.F. Smith et al. / Toxicology in Vitro 20 (2006) 163–175 175
Tukey, R.H., Strassburg, C.P., 2000. Human UDP-glucuronosyltransfe-rases: metabolism, expression, and disease. Annu. Rev. Pharmacol.Toxicol. 40, 581–616.
Villeneuve, L., Girard, H., Fortier, L.C., Gagne, J.F., Guillemette, C.,2003. Novel functional polymorphisms in the UGT1A7 and UGT1A9glucuronidation enzymes in Caucasian and African–American subjectsand their impact on the metabolism of 7-ethyl-10-hydroxycamptothe-cin and flavopiridol anticancer drugs. J. Pharmacol. Exp. Ther. 307,117–128.
Wall, M.E., Wani, M.C., Cook, C.E., Palmer, K.H., 1966. Plantantitumor agents, I: the isolation and structure of camptothecin, anovel alkaloidal leukemia and tumor inhibitor from Camptotheca
acuminata. J. Am. Chem. Soc., 3888–3890.Wu, M.H., Yan, B., Humerickhouse, R., Dolan, M.E., 2002. Irinotecan
activation by human carboxylesterases in colorectal adenocarcinomacells. Clin. Cancer Res. 8, 2696–2700.
Wu, M.H., Chen, P., Wu, X., Liu, W., Strom, S., Das, S., Cook Jr., E.H.,Rosner, G.L., Dolan, M.E., 2004. Determination and analysis of single
nucleotide polymorphisms and haplotype structure of the humancarboxylesterase 2 gene. Pharmacogenetics 14, 595–605.
Xie, H.G., Wood, A.J., Kim, R.B., Stein, C.M., Wilkinson, G.R., 2004.Genetic variability in CYP3A5 and its possible consequences. Phar-macogenomics 5, 243–272.
Xu, G., Zhang, W., Ma, M.K., McLeod, H.L., 2002. Human carb-oxylesterase 2 is commonly expressed in tumor tissue and iscorrelated with activation of irinotecan. Clin. Cancer Res. 8, 2605–2611.
Zamber, C.P., Lamba, J.K., Yasuda, K., Farnum, J., Thummel, K.,Schuetz, J.D., Schuetz, E.G., 2003. Natural allelic variants of breastcancer resistance protein (BCRP) and their relationship to BCRPexpression in human intestine. Pharmacogenetics 13, 19–28.
Zhou, Q., Sparreboom, A., Tan, E.-H., Cheung, Y.-B., Lee, A., Poon, D.,Lee, E.D.J., Chowbay, B., 2005. Pharmacogenetic profiling across theirinotecan pathway in Asian patients with cancer. Br J. Clin.Pharmacol. 59, 415–424.
Related Documents











