Perylenediimide-Based Donor−Acceptor Dyads and Triads: Impact of Molecular Architecture on Self-Assembling Properties Pierre-Olivier Schwartz, † Laure Biniek, ‡ Elena Zaborova, § Benoît Heinrich, † Martin Brinkmann, ‡ Nicolas Leclerc, § and Ste ́ phane Me ́ ry* ,† † Institut de Physique et de Chimie des Mate ́ riaux de Strasbourg, UMR 7504, CNRS, Universite ́ de Strasbourg, 23 rue du Loess, BP 43, 67034 Strasbourg, Cedex 2, France ‡ Institut Charles Sadron, UPR22 CNRS, 23 rue du Loess, BP 84047, 67034 Strasbourg Cedex 2, France § Institut de Chimie et Proce ́ de ́ s pour l'Energie, l'Environnement et la Sante ́ , UMR 7515, CNRS, Universite ́ de Strasbourg, ECPM, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France * S Supporting Information ABSTRACT: Perylenediimide-based donor −acceptor co- oligomers are particularly attractive in plastic electronics because of their unique electro-active properties that can be tuned by proper chemical engineering. Herein, a new class of co-oligomers has been synthesized with a dyad structure (AD) or a triad structure (DAD and ADA) in order to understand the correlations between the co-oligomer molecular architecture and the structures formed by self-assembly in thin films. The acceptor block A is a perylene tetracarboxyl diimide (PDI), whereas the donor block D is made of a combination of thiophene, fluorene, and 2,1,3- benzothiadiazole derivatives. D and A blocks are linked by a short and flexible ethylene spacer to ease self-assembling in thin films. Structural studies using small and wide X-ray diffraction and transmission electron microscopy demonstrate that AD and ADA lamellae are made of a double layer of co-oligomers with overlapping and strongly π-stacked PDI units because the sectional area of the PDI is about half that of the donor block. These structural models allow rationalizing the absence of organization for the DAD co-oligomer and therefore to draw general rules for the design of PDI-based dyads and triads with proper self-assembling properties of use in organic electronics. 1. INTRODUCTION Nanostructured 3D morphologies composed of electronically active constituents have gained interest in the field of organic electronic. 1−3 In particular organic solar cells with a well- ordered phase-separated donor and acceptor morphology, i.e. a so-called nanostructured D−A bulk heterojunction has been proposed as an “ideal structure”, leading to OPV devices with enhanced efficiency and stability. 4,5 Generating sub-10 nm- sized donor and acceptor domains with a large interfacial area is expected to ease exciton separation, whereas a bicontinuous “percolation” network of the two components may improve charge-carrier conduction and extraction from the active layer to the electrodes. 6 Nanometer-scaled domains are also required because of the limited exciton diffusion length in organic thin films. 7 Ordered D and A arrays are also promising candidates for organic field effect transistors (OFETs) devices, since they could intrinsically exhibit an ambipolar charge transport through a bicontinuous p-type and n-type network. Various strategies were proposed to prepare thin films that fulfill all these requirements. They can be classified into two major categories: (i) methods based on chemical engineering that exploit the self-assembling properties of donor−acceptor (macro)molecular systems e.g., block copolymers 8 or co- oligomers 9,10 (including low-molecular weight com- pounds) 11−13 and (ii) physicochemical methods that use specific thin film processing methods e.g. nanoimprint lithography 14 or template-based methods. 15 While the latter methods seem very promising in terms of cost, reproducibility and large-scale integration, exploiting the self-assembling of precisely designed donor−acceptor block co-oligomers is of particular interest as the preparation methods may be facilitated by the intrinsic self-assembling properties of the co-oligomers. Several teams have investigated the donor−acceptor block (D−A) copolymer approach implying the synthesis of a macromolecule where an electron donor and an electron acceptor block are chemically bound. 16 For many copolymers, the acceptor block was made of pendant fullerene C 60 . However, in the past decade, perylenediimide (PDI) appeared as an interesting alternative acceptor unit to C 60 in OPV devices. 8d−h Many macromolecular architectures using PDI were synthesized and interesting properties such as n-type Received: January 7, 2014 Published: March 27, 2014 Article pubs.acs.org/JACS © 2014 American Chemical Society 5981 dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−5992

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Perylenediimide-Based Donor−Acceptor Dyads and Triads: Impact ofMolecular Architecture on Self-Assembling PropertiesPierre-Olivier Schwartz,† Laure Biniek,‡ Elena Zaborova,§ Benoît Heinrich,† Martin Brinkmann,‡
Nicolas Leclerc,§ and Stephane Mery*,†
†Institut de Physique et de Chimie des Materiaux de Strasbourg, UMR 7504, CNRS, Universite de Strasbourg, 23 rue du Loess, BP43, 67034 Strasbourg, Cedex 2, France‡Institut Charles Sadron, UPR22 CNRS, 23 rue du Loess, BP 84047, 67034 Strasbourg Cedex 2, France§Institut de Chimie et Procedes pour l'Energie, l'Environnement et la Sante, UMR 7515, CNRS, Universite de Strasbourg, ECPM, 25rue Becquerel, 67087 Strasbourg Cedex 2, France
*S Supporting Information
ABSTRACT: Perylenediimide-based donor−acceptor co-oligomers are particularly attractive in plastic electronics becauseof their unique electro-active properties that can be tuned byproper chemical engineering. Herein, a new class of co-oligomershas been synthesized with a dyad structure (AD) or a triadstructure (DAD and ADA) in order to understand the correlationsbetween the co-oligomer molecular architecture and the structuresformed by self-assembly in thin films. The acceptor block A is aperylene tetracarboxyl diimide (PDI), whereas the donor block Dis made of a combination of thiophene, fluorene, and 2,1,3-benzothiadiazole derivatives. D and A blocks are linked by a shortand flexible ethylene spacer to ease self-assembling in thin films.Structural studies using small and wide X-ray diffraction andtransmission electron microscopy demonstrate that AD and ADA lamellae are made of a double layer of co-oligomers withoverlapping and strongly π-stacked PDI units because the sectional area of the PDI is about half that of the donor block. Thesestructural models allow rationalizing the absence of organization for the DAD co-oligomer and therefore to draw general rules forthe design of PDI-based dyads and triads with proper self-assembling properties of use in organic electronics.
1. INTRODUCTION
Nanostructured 3D morphologies composed of electronicallyactive constituents have gained interest in the field of organicelectronic.1−3 In particular organic solar cells with a well-ordered phase-separated donor and acceptor morphology, i.e. aso-called nanostructured D−A bulk heterojunction has beenproposed as an “ideal structure”, leading to OPV devices withenhanced efficiency and stability.4,5 Generating sub-10 nm-sized donor and acceptor domains with a large interfacial area isexpected to ease exciton separation, whereas a bicontinuous“percolation” network of the two components may improvecharge-carrier conduction and extraction from the active layerto the electrodes.6 Nanometer-scaled domains are also requiredbecause of the limited exciton diffusion length in organic thinfilms.7 Ordered D and A arrays are also promising candidatesfor organic field effect transistors (OFETs) devices, since theycould intrinsically exhibit an ambipolar charge transportthrough a bicontinuous p-type and n-type network. Variousstrategies were proposed to prepare thin films that fulfill allthese requirements. They can be classified into two majorcategories: (i) methods based on chemical engineering thatexploit the self-assembling properties of donor−acceptor
(macro)molecular systems e.g., block copolymers8 or co-oligomers9,10 (including low-molecular weight com-pounds)11−13 and (ii) physicochemical methods that usespecific thin film processing methods e.g. nanoimprintlithography14 or template-based methods.15 While the lattermethods seem very promising in terms of cost, reproducibilityand large-scale integration, exploiting the self-assembling ofprecisely designed donor−acceptor block co-oligomers is ofparticular interest as the preparation methods may be facilitatedby the intrinsic self-assembling properties of the co-oligomers.Several teams have investigated the donor−acceptor block
(D−A) copolymer approach implying the synthesis of amacromolecule where an electron donor and an electronacceptor block are chemically bound.16 For many copolymers,the acceptor block was made of pendant fullerene C60.However, in the past decade, perylenediimide (PDI) appearedas an interesting alternative acceptor unit to C60 in OPVdevices.8d−h Many macromolecular architectures using PDIwere synthesized and interesting properties such as n-type
Received: January 7, 2014Published: March 27, 2014
Article
pubs.acs.org/JACS
© 2014 American Chemical Society 5981 dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−5992

transport and OPV activity were demonstrated.17 In particular,D−A copolymers based on perylenediimides have recentlygained high interest because of the combination of remarkableself-assembling properties along with a strong n-type chargetransport.8g,h More generally, D−A copolymers have been usedas active layers as well as stabilizers of donor/acceptor blends,but lead to rather limited charge transport and photovoltaicproperties in thin films.18
One possible way to improve the block copolymer approachconsists in the design of co-oligomers with a precisecomposition and molecular architecture avoiding, in particular,the issues of polydispersity and chemical purity inherent tocopolymer. Recently, Y. Geng et al. demonstrated interestingOPV properties in thin film devices prepared from PDI-basedoligomers forming lamellar mesophases with a perfect phaseseparation between donor and acceptor groups.10c,d The qualityof the organization was found to be significantly improved byincreasing the donor block length and by using solvent-vaporannealing to improve the film morphology. In these reports,however, only a series of oligomers of dyad architecture (AD)was investigated. Derived from the block co-oligomer approach,one has to mention the large number of low-molecular weightsystems reported in the literature which are made of a “small”donor conjugated unit (<800 Da) covalently linked to a C60,
11
peryleneimide12,13 or other acceptor unit,19 into dyad or triadarchitectures. Only a few systems, however, could successfullylead to microphase separation of the blocks clearly leading toself-assembling into 3-D supramolecular D/A arrays.11b−d,13,18
Liquid crystals seem to constitute a promising approach in thatdirection.10c,d,11b,c,13a,g With low-molecular weight systems,however, the small size of the electro-active species doesrepresent a challenge to build up well-defined D and A channelsover large-length scales to ease ambipolar charge trans-port.11b,c,12f,13
Herein we report the syntheses and the physicochemicalproperties of a series of donor−acceptor block co-oligomers ofdifferent molecular architectures within dyad (AD) and triad(ADA and DAD) systems. The donor block length has beenonly slightly varied to remain in the range of moderate
molecular weight systems. The aim of this contribution is toinvestigate the structure−property relationship of thesematerials with particular emphasis on spectroscopic, thermal,and structural properties. A careful look will be brought to themolecular architecture dependence of the material organization,by means of X-ray diffraction studies (powder and GIWAXS)and electron microscopy investigation (TEM).
2. RESULTS AND DISCUSSION
Molecular Design and Synthesis. We have designed newmolecular structures based on the approach of monodisperseblock co-oligomers including both electron-acceptor (A) andelectron donor blocks (D). Various molecular architectures e.g.AD, DAD and ADA (Figure 1) are prepared by assemblingproperly these two blocks.The studied co-oligomers are made of the association of
several chemical units: perylene tetracarboxyl diimides (PDI),thiophene, fluorene, and 2,1,3-benzothiadiazole derivatives(Figure 1). PDI presents an n-type character (A), while theother units compose the p-type block (D). PDI is made of alarge aromatic macrocycle providing strong π−π interactionsthat is the primary force to drive the formation of stackedstructures.20 The thienofluorene building block can be seen as arigid conjugated rod, which incidentally showed its ability toproduce mesophases when substituted by appropriate alkylchains.21 Its association with PDI was therefore expected topromote the formation of a lamellar organization withsegregated D and A domains. This assumption was recentlydemonstrated in the case of the AD molecular architecture.10c,d
The electron-deficient thiophene−benzothiadiazole−thiophene(TBzT) moiety has been included in the donor block with theintent of lowering the energy bandgap of the donor block bydecreasing its LUMO level and red-shifting its absorptionspectrum.22 Finally, the covalent bonding of the donor block byN-substitution of the PDI core ensures an electronicdisconnection between both blocks. In our systems, anonconjugated ethylene linker is inserted between the D andA blocks to impede the direct charge recombination.13c It isalso expected to bring some flexibility between both blocks to
Figure 1. Chemical structure of the donor−acceptor block co-oligomers categorized into the three molecular architectures: ADn, DnADn and ADA.
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925982
favor their respective packing. Moreover, this linker is expectedto be short enough to prevent the D and A backfolding, alreadynoticed for other systems with longer linkers,23 which indeed isdetrimental for the formation of nanostructures with segregatedD and A domains.Finally the choice of the solubilizing side chains, necessary to
process these materials from solution, is also extremelyinfluential for the material organization and consequently theoptoelectronic properties.24 In our case, we decided to uselinear octyl chains on the fluorene moieties and 2-ethylhexyl-ramified chains on the PDI cores (when asymmetrized) and onthiophene units adjacent to the benzothiadiazole. Ethylhexylside chains were chosen because of their higher solubilizingcapability with respect to that of their linear counterparts.All oligomers were synthesized using the same two-step
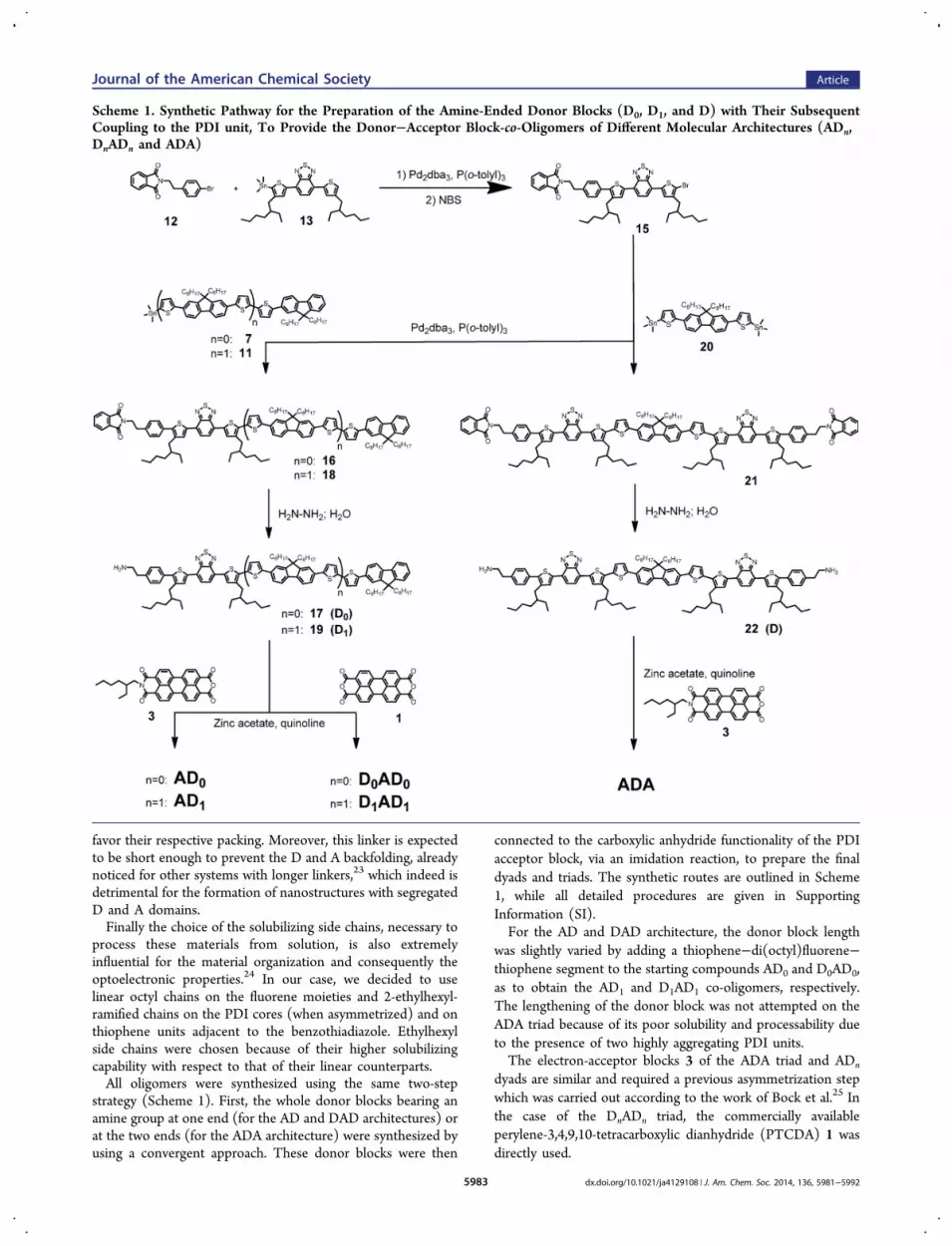
strategy (Scheme 1). First, the whole donor blocks bearing anamine group at one end (for the AD and DAD architectures) orat the two ends (for the ADA architecture) were synthesized byusing a convergent approach. These donor blocks were then
connected to the carboxylic anhydride functionality of the PDIacceptor block, via an imidation reaction, to prepare the finaldyads and triads. The synthetic routes are outlined in Scheme1, while all detailed procedures are given in SupportingInformation (SI).For the AD and DAD architecture, the donor block length
was slightly varied by adding a thiophene−di(octyl)fluorene−thiophene segment to the starting compounds AD0 and D0AD0,as to obtain the AD1 and D1AD1 co-oligomers, respectively.The lengthening of the donor block was not attempted on theADA triad because of its poor solubility and processability dueto the presence of two highly aggregating PDI units.The electron-acceptor blocks 3 of the ADA triad and ADn
dyads are similar and required a previous asymmetrization stepwhich was carried out according to the work of Bock et al.25 Inthe case of the DnADn triad, the commercially availableperylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA) 1 wasdirectly used.
Scheme 1. Synthetic Pathway for the Preparation of the Amine-Ended Donor Blocks (D0, D1, and D) with Their SubsequentCoupling to the PDI unit, To Provide the Donor−Acceptor Block-co-Oligomers of Different Molecular Architectures (ADn,DnADn and ADA)
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925983
The synthesis of electron-donor blocks is identical for that ofthe DnADn and ADn series (Scheme 1). It starts with thesynthesis of the electron-deficient TBzT derivative 15 bearingat one end a phthalimide moiety, precursor to the aminefunctionality and at the other end, a bromine atom forsubsequent adjunction of a thienofluorene moiety. Thisintermediate compound (15) was obtained from successivereaction steps, involving a Stille cross-coupling reactionbetween N-bromophenethyl(phthalimide) 12 and the stanny-lated TBzT derivative 13,23 and then a bromination using NBS.All reaction steps were achieved in rather high yield except theStille cross-coupling reaction (∼60% yield), most likely due tothe steric hindrance caused by the ethylhexyl chain adjacent tothe stannylated position in 13. In a next step, 15 was cross-coupled with the thienofluorene derivative 7 or 11 to producethe phthalimide-ended donor blocks 16 or 18 of differentlength (n = 0 or 1, respectively) in 78−90% yield. The latterwere obtained from multiple reaction steps (85−95% yieldeach), involving trimethylstannylation (via either a H/Li or Br/Li exchange), bromination (NBS), and Stille cross-couplingreactions. The whole amino-ended donor blocks 17 (D0) and19 (D1) were finally obtained after cleavage of the phthalimideprotecting group by hydrazinolysis in quantitative yield. These
donor blocks were used without further purification for the lastimidation step with the perylene carboxylic anhydride 1 ordianhydride 3 to provide the targeted dyads (AD0 and AD1) ortriads (D0AD0 and D1AD1), respectively. These final reactionswere carried out in quinoline at 200 °C in the presence of zincacetate. Several purification steps by column chromatographyon silica gel (petroleum ether/CH2Cl2) were required to affordpure oligomers. They were obtained in good yields (around80%) as purple solids and characterized by NMR and Maldi-TOF analyses.In the case of the ADA triad, we have synthesized a specific
donor block composed of two amino-functionalized TBzTmoieties 22 (D). This donor block synthesis was performed byusing a Stille cross-coupling reaction between the TBzTderivative 15 and the bis-stannylated thienofluorene 20,26
followed by a cleavage (hydrazinolysis) of the phthalimideprotecting groups. The final ADA triad was obtained from adouble imidation reaction between the bis-amino derivative 22and the perylene dicarboxylic anhydride 1 with a limited yieldof 26%. This low yield is ascribed to the increased amount ofbyproducts and the poor solubility of the final compound.Proper purification by several column chromatographies on
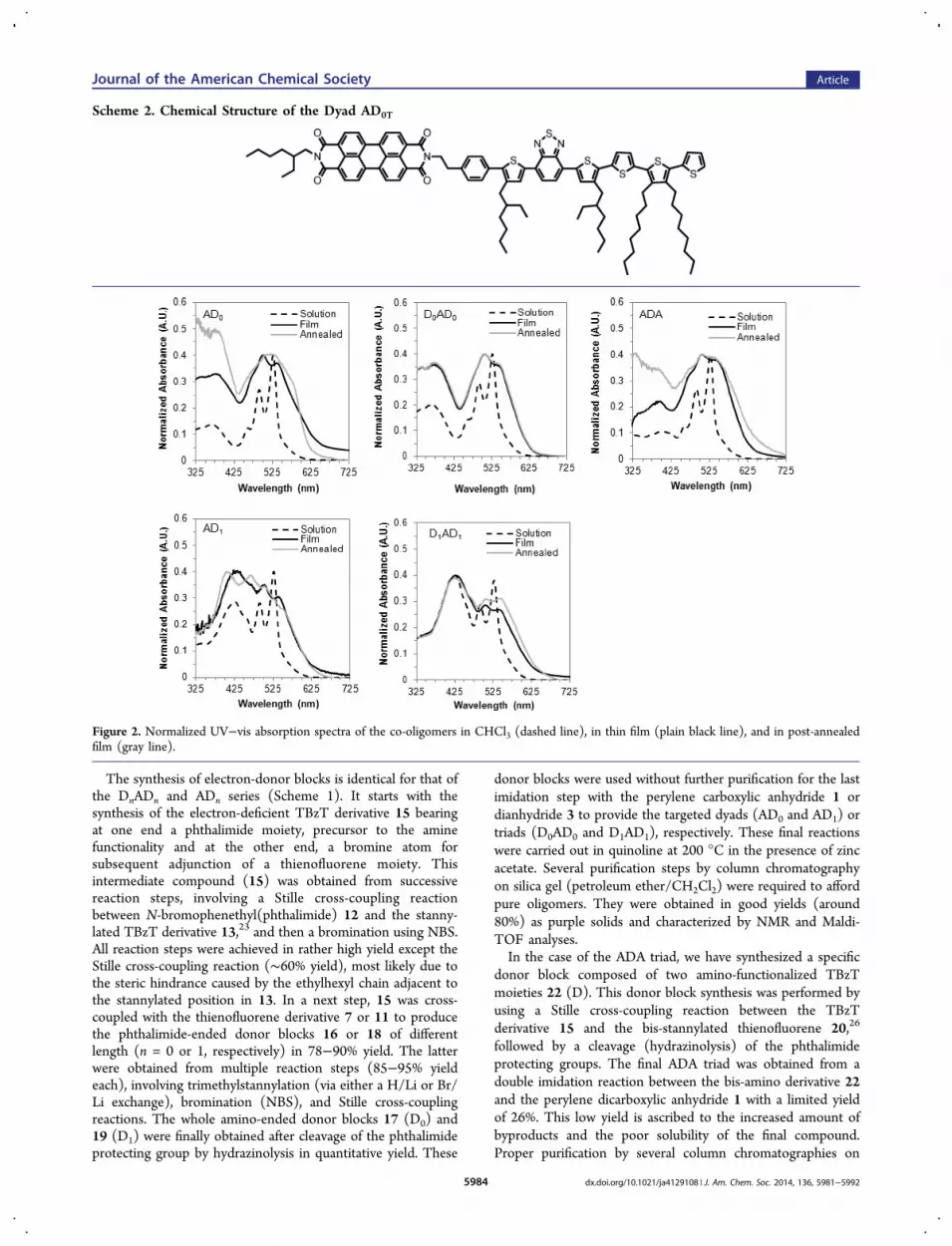
Scheme 2. Chemical Structure of the Dyad AD0T
Figure 2. Normalized UV−vis absorption spectra of the co-oligomers in CHCl3 (dashed line), in thin film (plain black line), and in post-annealedfilm (gray line).
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925984
silica gel (chloroform/methanol) provided the purple solid ofADA of good purity.In order to evaluate the role of the thienofluorene unit on the
molecular organization, another dyad (AD0T) was alsosynthesized in which the thienofluorene was replaced by aterthiophene unit (Scheme 2). Both units similarly contain twolinear octyl side chains. The synthetic pathway is reported in SI(Schemes S6 and S7). On the basis of the thin film morphologyanalysis performed by TEM, the structural organization ofAD0T will be compared with the one of AD0.Spectroscopic and Redox Properties. The UV−visible
absorption spectra of the co-oligomers recorded in solution (inchloroform) and in thin films (doctor-bladed from solution at45 °C) before and after post-annealing treatment (close to themelting) are presented in Figure 2. In solution, the absorptionspectra of all co-oligomers are dominated by the characteristicvibronic structure of the PDI core with components at 465,495, and 530 nm. The donor contribution is represented bytwo bands, one in the range 325−425 nm, whose position iscorrelated to the donor length conjugation, and the second one,centered at about 450 nm which extends beyond the absorptionrange of PDI, to 600 nm. This latter contribution can becorrelated to the electron-deficient benzothiadiazole-based partof the donor group (see Figure S1 in SI). In thin films, thecontribution of the PDI block broadens as a consequence of theexcitonic coupling in the solid state with an additional red-shifted contribution seen as a shoulder at λ ≈ 600 nm for AD0.After annealing of the films at a temperature close to theisotropization temperature, a clear variation of the fine structureof the absorption of the PDI block is observed for all theoligomers, except for D0AD0 which remains unchangedwhatever the thermal post-treatment applied. This is inagreement with the DSC results showing a clear first-orderphase transition for AD0, AD1, and ADA upon cooling from themelt, whereas such transition is observed neither for D0AD0 norfor D1AD1 (vide infra).As a first investigation of the excited states, the emission
spectra in solution (10−6 mol L−1) were recorded for onerepresentative material, the dyad AD1 (Figure S2 in SI). Uponexcitation of AD1 at 400 nm, a quasi-quantitative quenching offluorescence is observed. This implies the exciton dissociationvia a charge transfer from the donor to the acceptor block andno radiative recombination of separated charges. This impliesalso that the (−(CH2)2−) linker does not prevent theintramolecular charge transfer, contrary to previous observa-tions in another series of block co-oligomers made of hexa-peri-hexabenzocoronene and PDI units.13c Similar behavior wasobserved for all our synthesized co-oligomers (Figures S1−S4in SI). By comparison, excitation of the isolated donor block(not connected to A) at the same wavelength (400 nm), gives
rise to a typical emission band centered at about 650 nm. Moreremarkable is the almost complete fluorescence quenchingobserved whatever the excitation wavelength selected. Thus,upon excitation at 530 and 570 nm, an efficient quenching offluorescence was observed (Figures S1−S4 in SI). At suchwavelengths, only the electron-deficient benzothiadiazole-basedpart of the donor block and the PDI acceptor block isabsorbing. These results point at an efficient excitondissociation in those block co-oligomers, even in dilutedsolution.The electrochemical properties of the co-oligomers were
probed by cyclic voltammetry. Ferrocene was used as internalstandard to convert the values obtained with Ag/Ag+ referenceto the saturated calomel electrode scale (SCE). In order to havean insight into the intrinsic electrochemical properties of ourD−A block co-oligomers, the analyses were carried out insolution (methylene chloride) for all materials, except for ADAmolecule. Due to its poor solubility in most organic solvents,ADA was only analyzed in the solid state, using acetonitrile aselectrolytic support. Cyclic voltammograms are presented inthe SI (Figures S5, S6, and S7), while the values are reported inTable 1.All materials exhibited well-reversible oxidative and reductive
processes. In solution, they displayed two successive reversiblemonoelectronic reduction waves around −0.50 V and −0.70 V,respectively, that are classically found for PDI (Figures S5 andS6 in SI). In the case of ADA in the solid state, the two-electronreduction occurs in a single wave at −0.85 V (Figure S7 in SI).It is not a particular feature since all our block co-oligomersshow similar behaviors in the solid state. By using the NHEformal potential of −4.75 eV, LUMO levels of PDI blocks werecalculated at −4.25 eV for ADn dyads and DnADn triads insolution, and at −3.90 eV for ADA triad in solid state. Nooxidation of the PDI core could be observed in the range ofmeasured voltages. Regarding the electron-donor blocks, wewere able to determine the oxidative and reductive processesfor all molecules. Similarly to the LUMO level of PDI, minordifferences were observed in the HOMO levels of all moleculesin solution. They all exhibited two oxidation waves in the range0.7−1.1 V, corresponding to extracted HOMO levels rangingfrom −5.48 to −5.54 eV. The cathodic current increaseobserved for DnADn triads in comparison with ADn dyadsconfirms the bielectronic nature of those oxidative waves. Thisfeature is the result of the presence of two decoupled electrondonor blocks in one single DnADn molecule. For all molecules,we were also able to measure a reduction process around −1.2V, corresponding to the electron-donor blocks D0 and D1.Electrochemical band gaps of 1.9−2.0 eV were thus obtainedfor both donor blocks in solution, in good agreement with theband gaps measured in solid state for the pure electron-donor
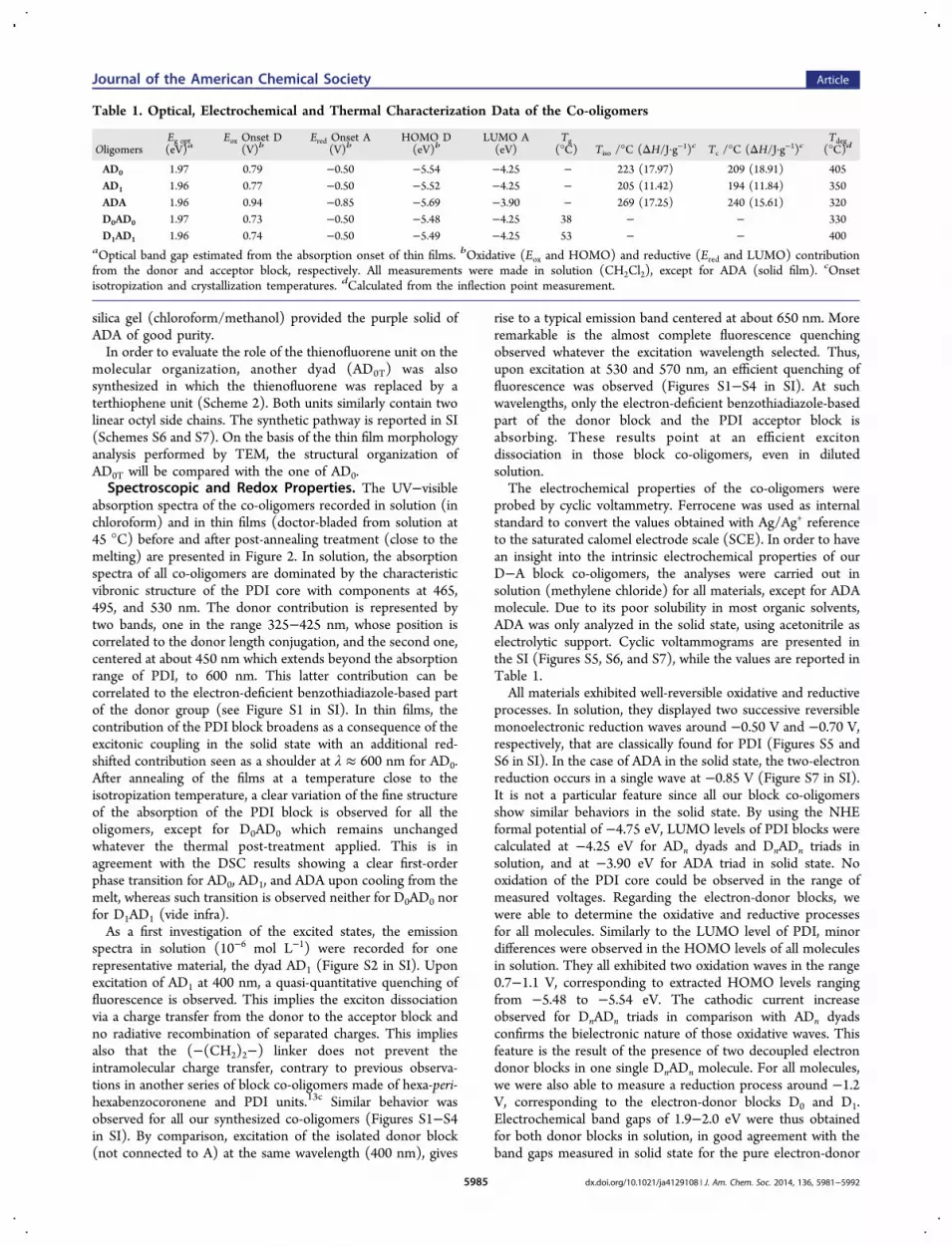
Table 1. Optical, Electrochemical and Thermal Characterization Data of the Co-oligomers
OligomersEg opt(eV)a
Eox Onset D(V)b
Ered Onset A(V)b
HOMO D(eV)b
LUMO A(eV)
Tg(°C) Tiso /°C (ΔH/J·g−1)c Tc /°C (ΔH/J·g−1)c
Tdeg(°C)d
AD0 1.97 0.79 −0.50 −5.54 −4.25 − 223 (17.97) 209 (18.91) 405AD1 1.96 0.77 −0.50 −5.52 −4.25 − 205 (11.42) 194 (11.84) 350ADA 1.96 0.94 −0.85 −5.69 −3.90 − 269 (17.25) 240 (15.61) 320D0AD0 1.97 0.73 −0.50 −5.48 −4.25 38 − − 330D1AD1 1.96 0.74 −0.50 −5.49 −4.25 53 − − 400
aOptical band gap estimated from the absorption onset of thin films. bOxidative (Eox and HOMO) and reductive (Ered and LUMO) contributionfrom the donor and acceptor block, respectively. All measurements were made in solution (CH2Cl2), except for ADA (solid film). cOnsetisotropization and crystallization temperatures. dCalculated from the inflection point measurement.
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925985
blocks. Regarding the electron-donor block of the ADA triad insolid state, similar voltammograms could be recorded with twooxidation waves and one reduction wave (Figure S7 in SI). Dueto a shift of the first oxidation wave toward the higherpotentials, a higher electrochemical band gap of 2.1 eV hasbeen calculated. Although the block co-oligomers presentedstructural variations, the HOMO and LUMO levels were notsignificantly changed which implies that the imide grouptogether with the flexible (−(CH2)2−) linker between donorand acceptor blocks ensures adequate decoupling of electronicstates of the blocks. This is in agreement with the absence ofcharge transfer (CT) band for co-oligomers in UV−vis spectra(diluted solution and thin films).Thermal and Structural Properties. The thermal proper-
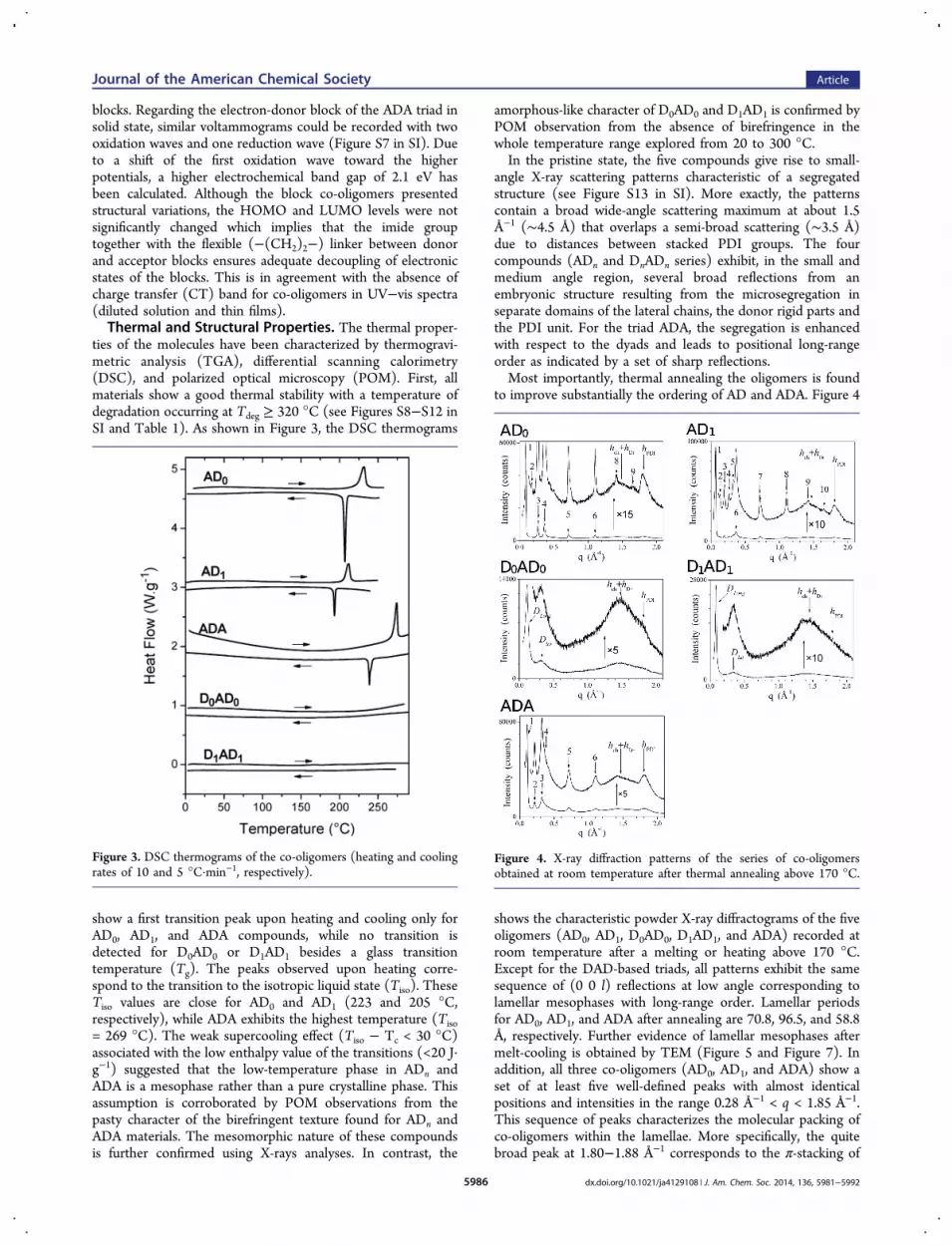
ties of the molecules have been characterized by thermogravi-metric analysis (TGA), differential scanning calorimetry(DSC), and polarized optical microscopy (POM). First, allmaterials show a good thermal stability with a temperature ofdegradation occurring at Tdeg ≥ 320 °C (see Figures S8−S12 inSI and Table 1). As shown in Figure 3, the DSC thermograms
show a first transition peak upon heating and cooling only forAD0, AD1, and ADA compounds, while no transition isdetected for D0AD0 or D1AD1 besides a glass transitiontemperature (Tg). The peaks observed upon heating corre-spond to the transition to the isotropic liquid state (Tiso). TheseTiso values are close for AD0 and AD1 (223 and 205 °C,respectively), while ADA exhibits the highest temperature (Tiso= 269 °C). The weak supercooling effect (Tiso − Tc < 30 °C)associated with the low enthalpy value of the transitions (<20 J·g−1) suggested that the low-temperature phase in ADn andADA is a mesophase rather than a pure crystalline phase. Thisassumption is corroborated by POM observations from thepasty character of the birefringent texture found for ADn andADA materials. The mesomorphic nature of these compoundsis further confirmed using X-rays analyses. In contrast, the
amorphous-like character of D0AD0 and D1AD1 is confirmed byPOM observation from the absence of birefringence in thewhole temperature range explored from 20 to 300 °C.In the pristine state, the five compounds give rise to small-
angle X-ray scattering patterns characteristic of a segregatedstructure (see Figure S13 in SI). More exactly, the patternscontain a broad wide-angle scattering maximum at about 1.5Å−1 (∼4.5 Å) that overlaps a semi-broad scattering (∼3.5 Å)due to distances between stacked PDI groups. The fourcompounds (ADn and DnADn series) exhibit, in the small andmedium angle region, several broad reflections from anembryonic structure resulting from the microsegregation inseparate domains of the lateral chains, the donor rigid parts andthe PDI unit. For the triad ADA, the segregation is enhancedwith respect to the dyads and leads to positional long-rangeorder as indicated by a set of sharp reflections.Most importantly, thermal annealing the oligomers is found
to improve substantially the ordering of AD and ADA. Figure 4
shows the characteristic powder X-ray diffractograms of the fiveoligomers (AD0, AD1, D0AD0, D1AD1, and ADA) recorded atroom temperature after a melting or heating above 170 °C.Except for the DAD-based triads, all patterns exhibit the samesequence of (0 0 l) reflections at low angle corresponding tolamellar mesophases with long-range order. Lamellar periodsfor AD0, AD1, and ADA after annealing are 70.8, 96.5, and 58.8Å, respectively. Further evidence of lamellar mesophases aftermelt-cooling is obtained by TEM (Figure 5 and Figure 7). Inaddition, all three co-oligomers (AD0, AD1, and ADA) show aset of at least five well-defined peaks with almost identicalpositions and intensities in the range 0.28 Å−1 < q < 1.85 Å−1.This sequence of peaks characterizes the molecular packing ofco-oligomers within the lamellae. More specifically, the quitebroad peak at 1.80−1.88 Å−1 corresponds to the π-stacking of
Figure 3. DSC thermograms of the co-oligomers (heating and coolingrates of 10 and 5 °C·min−1, respectively).
Figure 4. X-ray diffraction patterns of the series of co-oligomersobtained at room temperature after thermal annealing above 170 °C.
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925986

the PDI blocks, hPDI (maximum at 3.5 Å).27 The broadscattering maximum at 1.5 Å−1 comes from lateral distancesbetween molten alkyl side chains (hch) and likely between rigidmoieties of donor blocks segments (hDr). Additional peakscorrespond to the reflections of the three-dimensional structureemerging from the segregation of alkyl chains and donorblocks.Contrarily to ADn and ADA compounds, both DAD
derivatives exhibit, even after annealing, a poorly definedpattern (Figure 4). The latter is dominated by a slightlybroadened peak corresponding to lamellar periods of 56 and 76Å for D0AD0 and D1AD1, respectively, besides two broaderpeaks at 0.31 and 1.5 Å−1 coming from lateral distancesbetween chains and rigid moieties. The absence of a clear hPDIpeak combined to the persistence of a quite sharp lamellarperiod (DLong) shows that the layers of parallel packedmolecules are preserved, but without the regular PDI stacks
and only over intermediate correlation ranges (see Table 2).Finally, the lack of long-range order in D0AD0 and D1AD1
results in POM observation of textures, typical of an isotropicliquid, i.e. without birefringence in the liquefied-cooled thinfilms. In contrast to DAD-based triads, the ADA triad forms alamellar mesophase already in the pristine state. However, thispristine lamellar structure differs from that obtained after melt-cooling (see Figure S14 in SI and Table 2). In particular, thelamellar period is 30% smaller in the pristine state. Above 100°C the pristine structure gradually rearranges toward the stablelamellar mesophase, which is also the structure obtained inmelt-cooled thin films. The sequence of peaks in the range 0.28Å−1 < q < 1.85 Å−1 observed for ADA after annealing is almostidentical to that observed for the AD0 and AD1 dyads,indicating a strong similarity in the molecular packing of theADA triad and the ADn compound series.
Figure 5. ED patterns (a and d), two-dimensional grazing incidence X-ray diffraction (GIWAXS) intensity maps (b and e), and TEM-BF images (cand f) of AD0 (a,b,c) and AD1 (d,e,f) thin films after annealing above the melting temperature. The insets represent the dominant orientation of thelamellae on the SiO2 substrate. The inset in (c) highlights a set of standing lamellae also shown in an enlarged view in the lower left corner.
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925987

Thin Films Morphology and Structure. TEM inves-tigation of AD0 and AD1 dyads as-cast films do not give anyindication for long-range order or structure. The situationchanges upon melt-cooling of the samples. Figure 5 depicts themorphology and the ED and GIWAXS for thin films of AD0and AD1 dyads after melt-cooling. The films of AD0 consist of amajority of flat-lying lamellae with a typical terracedmorphology. However, a small fraction of standing lamellaeare also observed, and these areas give rise to a periodic fringedpattern characteristic of the lamellar mesophase with a period l= 70.6 Å. For the analogous AD0T dyad, shown in Scheme 2, asimilar terraced morphology was also evidenced by TEM andED after melt-cooling (Figure 6). In strong contrast, the films
of AD1 consist essentially of standing lamellae as indicated bythe observation of a periodic fringed pattern. The contrast inthe TEM-BF images (Figure 5f) is attributed to the differencebetween the packing density of PDI and of the donor blocks.The sharp and dark lines are attributed to dense layers of PDIblocks, whereas the brighter zones correspond to the layers ofdonor blocks. The difference in lamellar orientation for AD0
(and AD0T) and AD1 indicates that, for short donor blocks, thedyad molecules can stand up on the substrate upon annealing,whereas for AD1 the molecular long axis remains in the plane ofthe substrate. These differences in morphology are confirmedby ED. Indeed, the characteristic (0 0 l) reflections are onlyseen in the case of AD1, whereas for AD0 (and AD0T) the EDpattern shows exclusively the (h 0 0) reflections. The sameanalysis can be conducted for the GIWAXS patterns. If weconsider the in-plane reflections (along qx), one observes the (h
0 0) reflections with a weak (0 0 3) contribution for AD0.Instead, for AD1, the (0 0 l) reflections are dominant. Along qz,the situation is reversed: for AD0, the presence of both (h 0 0)and (0 0 l) reflections along qz confirm the coexistence of bothflat-on and edge-on oriented lamellae. For AD1, the (h 0 0)reflections are very strong without any contribution from the (00 l) reflections along qz.The difference in molecular orientation in dyad films
observed as a function of the donor block length has importantimplications regarding potential devices properties. Accord-ingly, in melt-cooled films, one can expect edge-on orientedlamellae with lying molecules of AD1 to be more favorable forOPV applications with respect to AD0 and AD0T filmsconsisting of terraced domains with standing molecules.As previously demonstrated by XRD on pristine powders
(Figure S14 in SI), the ADA triad presents enhanced orderingwith a well-defined lamellar mesophase. This is confirmed byTEM observations on as-cast films prepared by doctor-blading.It is worth mentioning that for ADA triad, doctor-blading hasbeen realized at 180 °C for solubility reasons. In this condition,a clear segregation of donor and acceptor blocks leads to alamellar mesophase (Figure 7a). However, as seen in Figure 7c,after melt-cooling, the dimension of the domains is substantiallyincreased and the contrast/sharpness of the lamellar morphol-ogy is strongly enhanced. The extension of long-range lamellarorder is also evidenced by a strong increase of the intensity ofthe (0 0 l) reflections in the corresponding ED pattern (seeFigure 7b and d).In strong contrast to ADn and ADA compounds, TEM
investigations on D0AD0 and D1AD1 films did not show anyevidence of long-range-ordering independently of the thermalannealing (see Figure S15 in SI).
Correlation between Molecular Packing and Molec-ular Architecture. The strong similarity in the XRD patternsof dyads based on different donor blocks (different lengths andchemical compositions) indicates that the self-organization ofdyads into lamellar mesophases is mainly determined by thepacking of the PDI blocks. Let us consider first the case of AD0.For AD0, the lamellar period lies between 1 and 2 times thelength of the extended molecule. This suggests strongly that thelamellae involve two overlapping dyad molecules. This overlapmay concern either the PDI block or the donor block. Todiscriminate between these two possibilities, we have to
Table 2. Mesomorphism and Structural Parameters of the Compound Series Annealed above 170 °C for about 1 ha
molec areas
cmpd name molecular length (Å) phase and lattice parameters ρ (g·cm−3) ADd APDI
e
orthorhombic phaseb
a ; b ; c (Å) V (Å)3 (Z)AD0 50 35.5 ; 7.43 ; 70.6 18600 (Z = 8) 1.14 65.9 33.0AD1 66 35.4 ; 7.44 ; 96.3 25400 (Z = 8) 1.13 65.8 32.9ADA 86 35.2 ; 7.45 ; 117.8 30800 (Z = 8) 1.21 65.4 32.7ADA (prist) 86 29.4 ; 12.7 ; 81.2 30400 (Z = 8) 1.23 94 47
isotropic-like phasec
D1at (ξ) (Å) DLong(ξ) (Å)D0AD0 84 21 (30) 56 (160) 1.17 66 66D1AD1 116 18 (50) 76 (400) 1.14 71 71
aThe structural parameters for both dyads and ADA triad have been determined considering the model proposed in Figure 8b and d. ba, b, c: latticeparameters; V: lattice volume; Z: number of molecules per lattice; ρ: density extracted from the unit cell parameters or calculated from referencedensity measurements (in italics). cDLong: lamellar period; D1at: periodicity from lateral packing; ξ: correlation length in Å extracted from the Scherrerformula. dAD: molecular area per donor group (i.e., the area of the layer portion covered by a single group), as obtained from the ratio of molecularvolume and either the parameter c or DLong.
eAPDI: molecular area per PDI group.
Figure 6. (a) ED pattern and (b) terrace-like morphology observed byBF-TEM of AD0T thin film after annealing above the melting state.
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925988
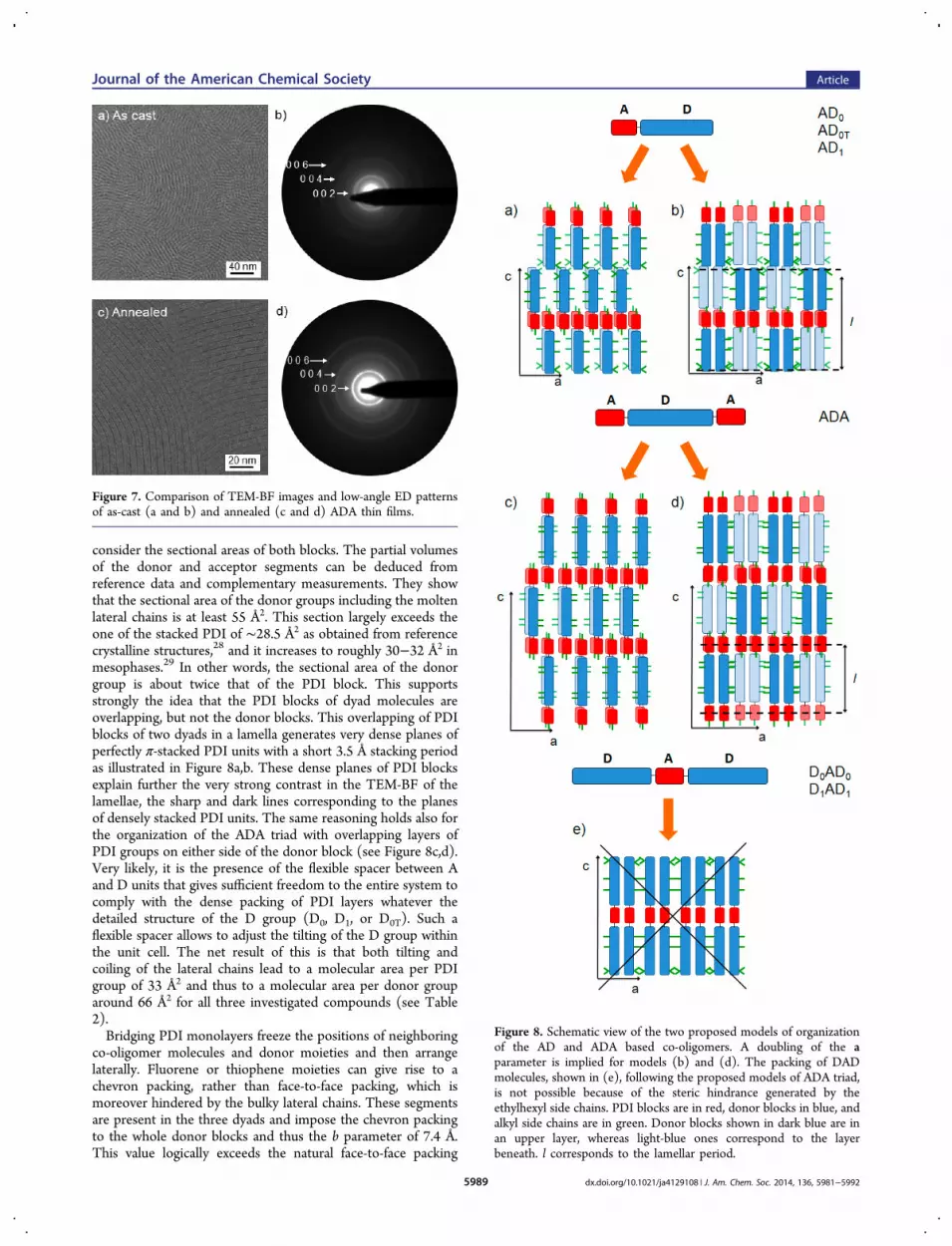
consider the sectional areas of both blocks. The partial volumesof the donor and acceptor segments can be deduced fromreference data and complementary measurements. They showthat the sectional area of the donor groups including the moltenlateral chains is at least 55 Å2. This section largely exceeds theone of the stacked PDI of ∼28.5 Å2 as obtained from referencecrystalline structures,28 and it increases to roughly 30−32 Å2 inmesophases.29 In other words, the sectional area of the donorgroup is about twice that of the PDI block. This supportsstrongly the idea that the PDI blocks of dyad molecules areoverlapping, but not the donor blocks. This overlapping of PDIblocks of two dyads in a lamella generates very dense planes ofperfectly π-stacked PDI units with a short 3.5 Å stacking periodas illustrated in Figure 8a,b. These dense planes of PDI blocksexplain further the very strong contrast in the TEM-BF of thelamellae, the sharp and dark lines corresponding to the planesof densely stacked PDI units. The same reasoning holds also forthe organization of the ADA triad with overlapping layers ofPDI groups on either side of the donor block (see Figure 8c,d).Very likely, it is the presence of the flexible spacer between Aand D units that gives sufficient freedom to the entire system tocomply with the dense packing of PDI layers whatever thedetailed structure of the D group (D0, D1, or D0T). Such aflexible spacer allows to adjust the tilting of the D group withinthe unit cell. The net result of this is that both tilting andcoiling of the lateral chains lead to a molecular area per PDIgroup of 33 Å2 and thus to a molecular area per donor grouparound 66 Å2 for all three investigated compounds (see Table2).Bridging PDI monolayers freeze the positions of neighboring
co-oligomer molecules and donor moieties and then arrangelaterally. Fluorene or thiophene moieties can give rise to achevron packing, rather than face-to-face packing, which ismoreover hindered by the bulky lateral chains. These segmentsare present in the three dyads and impose the chevron packingto the whole donor blocks and thus the b parameter of 7.4 Å.This value logically exceeds the natural face-to-face packing
Figure 7. Comparison of TEM-BF images and low-angle ED patternsof as-cast (a and b) and annealed (c and d) ADA thin films.
Figure 8. Schematic view of the two proposed models of organizationof the AD and ADA based co-oligomers. A doubling of the aparameter is implied for models (b) and (d). The packing of DADmolecules, shown in (e), following the proposed models of ADA triad,is not possible because of the steric hindrance generated by theethylhexyl side chains. PDI blocks are in red, donor blocks in blue, andalkyl side chains are in green. Donor blocks shown in dark blue are inan upper layer, whereas light-blue ones correspond to the layerbeneath. l corresponds to the lamellar period.
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925989

distance of PDI groups (∼3.5 Å deduced from the hPDIscattering), and the discrepancy needs to be compensated bysmall tilts of the stacked PDI groups. GIWAXS patternsconfirm these tilts, since the π-stacking scattering maximaappear tilted about 15−20° from the b vector direction (seeFigures 5b and e). Furthermore, the lamellae of donor moietiesare interrupted by perpendicular layers of molten aliphaticchains (see Figure 8). The lateral periodicity coinciding withthe parameter a = 35−35.5 Å involves the chains and rigidmoieties of two donor blocks and therefore four PDI groupsshared by two lamellae. The side-by-side packing of the PDIstacks then imposes a spacing of 8.8 Å per stack, close to valuesin crystalline phases (8.3 Å28). Except for the different cparameters related to the donor block lengths, the three dyadsand the ADA triad just show the same annealed structures andsimilar geometrical parameters. As mentioned above, the ADAtriad moreover shows a mesophase and long-range orderalready in the pristine state. The main specificities of thepristine geometry consist in a 40% reduced lamellar spacing anda 70% expansion along b, revealing an average 45° tilt ofmolecules from the b direction. As confirmed by the absence ofall crossed reflections, the regular microsegregated layeralternation along a of the orthorhombic phase does not formnatively, forcing rigid moieties to tilt. With the softening of thesample on first heat, this segregated structure develops,bringing rigid moieties back to the a × c plane and expandingthe parameter a to the size of a regular PDI monolayer, inconsistency with the gradual geometry change observed (seeFigure S14 in SI). For all cases, the few visible reflections andmolecular volumes are compatible with an orthorhombic phaseof similar nature. The structural parameters have been reportedin Table 2.Putting together all structural information gathered in this
study, one can propose two different packing schemes of ADdyads and the ADA triad, illustrated in Figure 8. As aconvention, the a, b, and c axes of the unit cell are orientedalong the alkyl side chains of the donor group, the π-stacking ofthe PDI, and the long molecular axis of the AD molecules,respectively. Considering the first model in Figure 8a, the ADmolecules are π-stacked into pairs with a strong π-overlap ofboth the PDI and the donor groups. Donor blocks in successivelayers along the a axis are pointing alternatively along c and −c.Within such AD pairs, the solubilizing chains of the two donorblocks are rejected on either side of the conjugated skeleton,and all alkyl chains are grouped together. Figure 8b shows analternative packing for which the AD molecules are π-stackedinto pairs of two molecules with a strong overlap of the PDIunits only, whereas donor blocks are arranged side-by-side withthe alkyl side chains rejected on each side of the donor pair.However, as for the first model, dense planes of π-stacked PDIblocks are formed also for this molecular arrangement. It islikely that the stacking of PDI units involves a slight lateraloffset (arrangement already observed in other studies27b,30).However, the PDI long axes are still colinear. These twopacking schemes can apply for AD0, AD0T, and AD1 dyads. Thetwo different packing schemes in Figure 8a,b can also apply forADA as illustrated in Figure 8c,d. The two terminal PDI groupson each side of the donor block in ADA can π-stack efficientlywithout perturbing the packing of donor blocks and can respectthe different sectional areas of the two blocks. However, themajor difference between ADn dyads and the ADA triad is thestrong interdigitation of the layers of ADA molecules. It can beanticipated that this strong interdigitation is responsible for the
enhanced long-range lamellar ordering in ADA films even inthe absence of melt-cooling. As a matter of fact, the stronginterdigitation of PDI groups on both sides of the donor couldpartly explain the higher density (1.21 g·cm−3) for the ADAtriad compared to the ADn analogues (1.13−1.14 g·cm−3).Regarding the DnADn triads, the large sectional area of donor
blocks imposes a high molecular area per PDI group which isincompatible with the generation of dense PDI layers. In somesense, the DAD self-assembling into a lamellar structure atlong-range order is prevented by the presence of solubilizingethylhexyl side chains on the donor block that are responsiblefor the high sectional area of this block as compared to PDI(Figure 8e). The marked discrepancy between molecular areasof D and A segments could similarly be held as responsible forthe lack of formation of well-defined D/A segregated structurein other peryleneimide-donor systems.13c,31,32 Note that thefew published examples of well-defined lamellar mesophases inDAD triads (with A = PDI) should be attributed to thepresence of a high density of chains (aliphatic13a or siloxane13g)at the triad extremities, which generates microphase separation.To conclude, a packing of DAD molecules following a
scheme similar to that given in Figure 8 for the AD dyads or theADA triad is virtually impossible. A more precise structuredetermination is needed for discriminating one or the otherpacking behavior of the molecules. Further studies are beingundertaken in order to provide a more complete assessment ofthe molecular packing of AD and ADA molecules.
3. CONCLUSIONSWe have successfully synthesized and characterized a new seriesof donor−acceptor block co-oligomers with different moleculararchitectures (AD, DAD, and ADA) with the aim of correlatingmolecular architecture and self-assembling properties, which isessential to improve optoelectronic devices such as OPV cellsor OFET. First, short ethylene linkers have been systematicallyinserted between the A and D blocks to favor their respectivepacking. Second, we have verified that these co-oligomerscomply with favorable optoelectronic properties, e.g. broadrange of absorption, appropriate HOMO and LUMO levels,decoupling of A and D electronic states. This was possible by acareful chemical design of the donor block made of acombination of thienofluorene segments of two differentlengths (D0 or D1) with the electron-deficient benzothiadiazoleunit. Efficient optoelectronic devices require ordered structureswith well-segregated acceptor and donor domains that areindeed determined by the molecular architecture. Intensivecharacterization of the co-oligomers by X-ray diffraction andTEM gave evidence for lamellar mesophases with long-rangeorder after melt-cooling for all systems except for DAD triads.Regardless of the length and chemical nature of the donorblock (D0, D0T, and D1), all the AD dyads and the ADA triadshow very similar packing of the co-oligomers within lamellae.The major difference between these two systems is the
interdigitated character of the lamellae for ADA, which is absentfor AD, and results in enhanced long-range ordering in bothpristine and melt-cooled ADA films. In the end, AD and ADAsuperstructures contain extended rows of PDI-stacks withinlamellae, leading to three-dimensional mesophases, while DADsystems show short-range lamellar correlations without markedPDI-stacks. The difference in this behavior is explained by thediscrepancy of the molecular cross sections between the PDIand the donor blocks. Placing the PDI unit at the end of themolecules allows to compensate its smaller cross-section by the
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925990

formation of interdigitated rows of PDI-stacks, ultimatelyleading to the cohesion of the lamellae. In contrast, with theDAD architecture, the large cross-section of the donor blocksprevents the central PDI unit from stacking.Another important aspect of the preparation of optoelec-
tronic devices is the control of the orientation of thenanostructure. In this work, we have evidenced the influenceof the donor block length on the orientation of the lamellardomains on a SiO2 substrate after melt-cooling. A majority oflying lamellae has been obtained for the short donor blocks(AD0 and AD0T), whereas AD1 molecules led to edge-onlamellae. In consequence, the modulation of the block lengthmight be an efficient tool to control the orientation of theoligomers on a substrate, depending on the optoelectronicapplication (OPV vs OFETs).Taken all together, these results are important as they allow
us to set some basic principles for the molecular design ofefficient self-assembling PDI-based donor−acceptor co-oligomers to be used in monocomponent optoelectronicdevices.
■ ASSOCIATED CONTENT*S Supporting InformationDetailed synthetic procedures, 1H NMR, 13C NMR, TGA, andMS spectra of final co-oligomers, UV−vis absorption andfluorescence spectra in solution, cyclic voltammetry traces,XRD patterns of ADA as a function of the temperature, ED-patterns and BF-TEM images of D0AD0. This material isavailable free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work has been supported by the French National ResearchAgency (ANR PICASSO Project) and by the EuropeanCommunity via the Interreg IV-A program (C25, Rhin-Solar). We thank Pohang Accelerator Laboratory (PAL) forgiving us the opportunity to perform the GIWAXS measure-ments, MEST and POSTECH for supporting these experi-ments, Dr. Tae Joo Shin for adjustments and help, and otherpeople from 9A U-SAXS beamline for assistance.
■ REFERENCES(1) Chen, J. T.; Hsu, C.-S. Polym. Chem. 2011, 2, 2707−2722.(2) Liu, H.; Xu, J.; Li, Y.; Li, Y. Acc. Chem. Res. 2010, 43, 1496−1508.(3) Hoeben, F. J. M.; Jonkhejim; Meijer, E. W.; Schenning, A. P. H. J.Chem. Rev. 2005, 105, 1491−1546.(4) Coakley, K. M.; McGehee, M. D. Chem. Mater. 2004, 16, 4533−4542.(5) Hoppe, H.; Sariciftci, N. S. Adv. Polym. Sci. 2008, 214, 1−86.(6) Yang, X.; Loos, J. Macromolecules 2007, 40, 1353−1362.(7) Thompson, B. C.; Frechet, J. M. J. Angew. Chem., Int. Ed. 2008,47, 58−77.(8) (a) De Boer, B.; Stalmach, U.; Melzer, C.; Krasnikov, V. V.;Hadziioannou, G. Polymer 2001, 43, 9097−9109. (b) Zhang, F.;Svensson, M.; Andersson, M. R.; Maggini, M.; Bucella, S.; Menna, E.;Inganas, O. Adv. Mater. 2001, 13, 1871−1874. (c) Miyanishi, S.;Zhang, Y.; Tajima, K.; Hashimoto, K. Chem. Commun. 2010, 46,6723−6725. (d) Lindner, S. M.; Thelakkat, M. Macromolecules 2004,37, 8832−8835. (e) Sommer, M.; Huettner, S.; Thelakkat, M. J. Mater.
Chem. 2010, 20, 10788−10797. (f) Zhang, Q.; Cirpan, A.; Russel, T.P.; Emrick, T. Macromolecules 2009, 42, 1079−1082. (g) Sommer, M.;Lindner, S. M.; Thelakkat, M. Adv. Funct. Mater. 2007, 17, 1493−1500.(h) Lohwasser, R. H.; Gupta, G.; Kohn, P.; Sommer, M.; Lang, A. S.;Thurn-Albrecht, T.; Thelakkat, M. Macromolecules 2013, 46, 4403−4410.(9) (a) Nierengarten, J.-F. Sol. Energy Mater. Sol. Cells 2004, 83, 187−199. (b) Roncali, J. Chem. Soc. Rev. 2005, 34, 483−495. (c) Segura, J.L.; Martin, N.; Guldi, D. M. Chem. Soc. Rev. 2005, 34, 31−47.(10) (a) Wurthner, F.; Chen, Z.; Hoeben, F. J. M.; Osswald, P.; You,C.-C.; Jonkheijm, P.; van Herrikhuyzen, J.; Schenning, A. P. H. J.; vander Schoot, P. P. A. M.; Meijer, E. W.; Beckers, E. H. A.; Meskers, S. C.J.; Janssen, R. A. J. J. Am. Chem. Soc. 2004, 126, 10611−10618.(b) Ramos, A. M.; Meskers, S. C. J.; Beckers, E. H. A.; Prince, R. B.;Brunsveld, L.; Janssen, R. A. J. J. Am. Chem. Soc. 2004, 126, 9630−9644. (c) Bu, L.; Guo, X.; Yu, B.; Qu, Y.; Xie, Z.; Yan, D.; Geng, Y.;Wang, F. J. Am. Chem. Soc. 2009, 131, 13242−13243. (d) Bu, l.; Guo,X.; Yu, B.; Fu, Y.; Qu, Y.; Xie, Z.; Yan, D.; Geng, Y.; Wang, F. Polymer2011, 52, 4253−4260.(11) (a) Nierengarten, J.-F.; Eckert, J.-F.; Nicoud, J.-F.; Ouali, L.;Krasnikov, V.; Hadziioannou, G. Chem. Commun. 1999, 617−618.(b) Li, W. S.; Yamamoto, Y.; Fukushima, T.; Saeki, A.; Seki, S.;Tagawa, S.; Masunaga, H.; Sasaki, S.; Takata, M.; Aida, T. J. Am. Chem.Soc. 2008, 130, 8886−8887. (c) Nishizawa, T.; Tajima, K.; Hashimoto,K. J. Mater. Chem. 2007, 17, 2440−2445. (d) Nishizawa, T.; Tajima,K.; Hashimoto, K. Nanotechnology 2008, 19, 424017.(12) (a) Chen, L. X.; Xiao, S.; Yu, L. J. Phys. Chem. B 2006, 110,111730−11738. (b) Cremer, J.; Mena-Osteritz, E.; Pschirere, N. G.;Mullen, K.; Bauerle, P. Org. Biomol. Chem. 2005, 3, 985−995.(c) Cremer, J.; Bauerle, P. Eur. J. Org. Chem. 2005, 3715−3723.(d) Cremer, J.; Bauerle, P. J. Mater. Chem. 2006, 16, 874−884.(e) Bullock, J. E.; Carmieli, R.; Mickley, S. M.; Vura-Weis, J.;Wasielewski, M. J. Am. Chem. Soc. 2009, 131, 11919−11929. (f) Balaji,G.; Kale, T. S.; Keerty, A.; Della Pelle, A. M.; Thayumanavan, S.;Valliyaveettil, S. Org. Lett. 2011, 13, 18−21.(13) (a) Peeters, E.; van Hal, P. A.; Meskers, S. C. J.; Janssen, R. J. A.;Meijer, E. W. Chem.Eur. J. 2002, 8, 4470−4474. (b) van der Boom,T.; Hayes, R. T.; Zhao, Y.; Bushard, P. J.; Weiss, E. A.; Wasielewski, M.R. J. Am. Chem. Soc. 2002, 124, 9582−9590. (c) Dossel, L. F.; Kamm,V.; Hoiward, I. A.; Laquai, F.; Pisula, W.; Feng, X.; Li, C.; Takase, M.;Kudernac, T.; de Feyter, S.; Mullen, K. J. Am. Chem. Soc. 2012, 134,5876−5886. (d) Kim, M. H.; Cho, M. J.; Kim, K. H.; Hoang, M. H.;Lee, T. W.; Jin, J.-I.; Kang, N. S.; Yu, J.-W.; Choi, D. H. Org. Electron.2009, 10, 1429−1441. (e) Jonkheijm, P.; Stutzmann, N.; Chen, Z.; deLeeuw, D. M.; Meijer, E. W.; Schenning, A. P. H. J.; Wurthner, F. J.Am. Chem. Soc. 2006, 128, 9535−9540. (f) Mativestsky, J. M.; Kastler,M.; Savage, R. C.; Gentilini, D.; Palma, M.; Pisula, W.; Mullen, K.;Samori, P. Adv. Funct. Mater. 2009, 19, 2486−2494. (g) Roland, T.;Leonard, J.; Hernandez Ramirez, G.; Mery, S.; Yurchenko, O.;Ludwigs, S.; Haacke, S. Phys. Chem. Chem. Phys. 2012, 14, 273−279.(14) Kim, M. S.; Kim, J. S.; Cho, J. C.; Shtein, M.; Guo, L. J.; Kim, J.Appl. Phys. Lett. 2007, 90, 123113.(15) (a) Fisslthaler, E.; Blumel, A.; Landfester, K.; Scherf, U.; List, E JW. Soft Matter 2008, 4, 2448−2453. (b) Kim, H.-C.; Park, S.-M.;Hinsberg, W. D. Chem. Rev. 2010, 110, 146−177. (c) He, X.; Gao, F.;Tu, G.; Hasko, D. G.; Huttner, S.; Greenham, N. C.; Steiner, U.;Friend, R. H.; Huck, W. T. S. Adv. Funct. Mater. 2011, 21, 139−146.(16) (a) Mishra, A.; MA, C.-Q.; Bauerle, P. Chem. Rev. 2009, 109,1141−1276. (b) Yassar, A.; Miozzo, L.; Gironda, R.; Horowitz, G.Prog. Polym. Sci. 2013, 38, 791−844. (c) Kozma, E.; Catellani, M. DyesPigments 2013, 98, 160−179. (d) Sommer, M.; Huettner, S.;Thelakkat, M. Adv. Polym. Sci. 2010, 228, 123−153.(17) (a) Wurthner, F. Chem. Commun. 2004, 1564−1579. (b) Zhan,X.; Facchetti, A.; Barlow, S.; Marks, T. J.; Ratner, M. A.; Wasielewski,M. R.; Marder, S. R. Adv. Mater. 2011, 23, 268−284. (c) Li, C.;Wonneberger, H. Adv. Mater. 2012, 24, 613−636. (d) Kozma, E.;Catellani, M. Dyes Pigments 2013, 98, 160−179.(18) (a) Richard, F.; Brochon, C.; Leclerc, N.; Eckhardt, D.; Heiser,T.; Hadziioannou, G. Macromol. Rapid Commun. 2008, 29, 885−891.
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925991

(b) Yassar, A.; Miozzo, L.; Gironda, R.; Horowitz, G. Prog. Polym. Sci.2013, 38, 791−844.(19) (a) Samori, P.; Yin, X.; Tchebotareva, N.; Wang, Z.; Pakula, T.;Jackel, F.; Watson, M. D.; Venturini, A.; Mullen, K.; Rabe, J. R. J. Am.Chem. Soc. 2004, 126, 3567−3575. (b) Yamamoto, Y.; Fukushima, T.;Suna, Y.; Ishii, N.; Saeki, A.; Seki, S.; Tagawa, S.; Taniguchi, M.; Kawai,T.; Aida, T. Science 2006, 314, 1761−1764.(20) (a) Langhals, H. Helv. Chim. Acta 2005, 88, 1309−1343.(b) Vura-Weis, J.; Ratner, M. A.; Wasielewski, M. R. J. Am. Chem. Soc.2010, 132, 1738−1739.(21) (a) Grell, M.; Bradley, D. D. C.; Inbasekaran, M.; Woo, E. P.Adv. Mater. 1997, 9, 798. (b) Sherf, U.; List, E. J. W. Adv. Mater. 2002,14, 477−487. Zhang, X.; Qu, Y.; Bu, L.; Tian, H.; Zhang, J.; Wang, L.;Geng, Y.; Wang, F. Chem.Eur. J. 2007, 13, 5238−6248.(22) (a) Biniek, L.; Fall, S.; Chochos, C. L.; Leclerc, N.; Leveque, P.;Heiser, T. Org. Electron. 2012, 13, 114−120. (b) Schwartz, P. O.;Zaborova, E.; Bechara, R.; Leveque, P.; Heiser, T.; Mery, S.; Leclerc,N. New J. Chem. 2013, 37, 2317−2323.(23) Ramos, A. M.; Beckers, E. H. A.; Offermans, T.; Meskers, S. C.J.; Janssen, R. A. J. J. Phys. Chem. A 2004, 108, 8201−8211.(24) (a) Biniek, L.; Fall, S.; Chochos, C. L.; Anokhin, D. V.; Ivanov,D.; Leclerc, N.; Leveque, P.; Heiser, T. Macromolecules 2010, 43,9779−9786. (b) Chu, T.-Y.; Lu, J.; Beaupre, S.; Zhang, Y.; Pouliot, P.-R.; Zhou, J.; Najari, A.; Leclerc, M.; Tao, Y. Adv. Funct. Mater. 2012,22, 2345−2351.(25) Kelber, J.; Bock, H.; Thiebaut, O.; Grelet, E.; Langhals, H. Eur. J.Org. Chem. 2011, 707−712.(26) Baek, M.-J.; Jang, W.; Lee, S.-H.; Lee, Y.-S. Synth. Met. 2012,161, 2785−2791.(27) (a) Hansen, M. R.; Graf, R.; Sekharan, S.; Sebastiani, D. J. Am.Chem. Soc. 2009, 131, 5251−5256. (b) Struijk, C. W.; Sieval, A. B.;Dackorst, J. E. J.; van Dijk, M.; Kimbes, P.; Koehorst, R. B. M.;Donker, H.; Schaafsma, T. J.; Picken, S. J.; van de Craats, A. M.;Warman, J. M.; Zuilhof, H.; Sudholter, E. J. R. J. Am. Chem. Soc. 2000,122, 11057−11066.(28) For instance in the structure DICLIK (N,N′-diethylperylene-3,4:9,10-bis(dicarboximide)) from the Cambridge Structural Database.(29) From the estimated partial volume expansion with respect tocrystal.(30) Kazmaier, P. M.; Hoffmann, R. J. Am. Chem. Soc. 1994, 116,9684−9691.(31) Neuteboom, E. E.; Meskers, S. C. J.; van Hal, P. A.; van Duren,J. K. J.; Meijer, E. W.; Janssen, R. A. J.; Dupin, H.; Pourois, G.; Cornil,J.; Lazzaroni, R.; Bredas, J.-L.; Beljonne, D. J. Am. Chem. Soc. 2003,125, 8625−8638.(32) Pron, A.; Reghu, R. R.; Rybakiewicz, R.; Cybulski, H.; Djurado,D.; Grazulevicius, J. V.; Zagorska, M.; Kulszewicz-Bajer, I.; Verilhac, J.-M. J. Phys. Chem. C 2011, 115, 15008−15017.
Journal of the American Chemical Society Article
dx.doi.org/10.1021/ja4129108 | J. Am. Chem. Soc. 2014, 136, 5981−59925992
Related Documents











