1 Post-Submission and Post-Approval Requirements Monica S. Krieger, Ph.D, M.B.A. Vice President, Regulatory Affairs OncoGenex Technologies, Inc. 2 Outline Introduction to PDUFA/MDUFA Amendments to Unapproved Applications Approved Applications Scheduled – Annual Reports – Distribution Reports – Safety Reports – User Fees Unscheduled – Changes to Approved Applications – CMC Supplements – Efficacy Supplements – Reports of Problems and Errors – Promotional Literature – Individual Safety Reports 3 What I have tried to provide Reference in the CFR and/or FDC Act Available guidance documents on FDA website Brief summary of requirements Impact on the sponsor/company and various departments 4 PDUFA/MDUFA-What are they? What is PDUFA? Prescription Drug User Fee Act What are PDUFA I, II, III? – 5-year phases – Approved by Congress What is MDUFA Medical Device User Fee and Modernization Act

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

1
Post-Submission and Post-Approval Requirements
Monica S. Krieger, Ph.D, M.B.A.Vice President, Regulatory Affairs
OncoGenex Technologies, Inc.
2
Outline
Introduction to PDUFA/MDUFAAmendments to Unapproved ApplicationsApproved Applications
Scheduled– Annual Reports– Distribution Reports– Safety Reports– User FeesUnscheduled – Changes to Approved Applications
– CMC Supplements– Efficacy Supplements
– Reports of Problems and Errors– Promotional Literature– Individual Safety Reports
3
What I have tried to provide
Reference in the CFR and/or FDC ActAvailable guidance documents on FDA websiteBrief summary of requirements Impact on the sponsor/company and various departments
4
PDUFA/MDUFA-What are they?
What is PDUFA?Prescription Drug User Fee ActWhat are PDUFA I, II, III?– 5-year phases– Approved by Congress
What is MDUFAMedical Device User Fee and Modernization Act

5
PDUFA
Congress passed PDUFA in 1992 Goal was to decrease time to approval
FDA and the drug industry struck a dealIndustry would provide funding for the drug review program FDA would agree to meet drug-review time performance goals.
Authorized the agency to collect fees from companies that produce certain human drug and biological products
Application FeeManufacturing Facility FeeProduct Fee
6
PDUFA Goals
90% in 6 months * No Goal Discipline review letters for pre-submitted “Reviewable Units” of new drug and biologic applications
90% of class 1 in 2 months and 90% of class 2 in 6 months *
90% in 6 months No Goal Complete review of resubmitted efficacy supplements
90% of class 1 in 2 months and 90% of class 2 in 6 months 90% in 6 months Complete review of resubmitted new drug
and biologic applications
90% in 4 months if prior approval needed, 6 months otherwise 90% in 6 months Complete review of manufacturing
supplements
90% in 10 months 90% in 12 months Complete review of standard original new drug and biologic applications and efficacy supplements
90% in 6 monthsComplete review of priority original new drug and biologic applications and efficacy supplements
PDUFA IIIPDUFA IIPDUFA IGoal
7
PDUFA IIIPDUFA IIPDUFA IGoal
Enhanced by the end of FY 2007 In place by the end of FY 2002 No Goal Electronic application receipt
and review
90% within 45 days No Goal Complete review of special protocols
90% within 30 days No Goal Resolve major disputes appealed by industry
90% within 30 days No Goal
Communicate results of review of complete industry responses to FDA clinical holds
90% within 30 days No Goal Provide industry with meeting minutes
90% within 30, 60, or 75 days, depending on type of meeting No Goal Meet with industry within set times
90% within 14 days No Goal Respond to industry requests for meetings
90% within 14 days of filing date * No Goal
Report of substantive deficiencies (or lack thereof)
PDUFA Goals-Drugs and Biologics
8
Where can I find the PDUFA Goals?
Current PDUFA performance goals can be found at www.fda.gov/oc/pdufa/PDUFAIIIGoals.html

9
MDUFA
MDUFA is the Medical Device User Fee and Modernization Act Success with drug/biologic user fees prompted MUDFAPassed by Congress in October 2002 Goal was to provide three significant benefits:
Safe and effective medical treatments will reach patients more rapidlyAssure timely, high-quality reviewsMaintain high standards for safety and effectiveness
10
MDUFA
Current MDUFMA performance goals can be found at www.fda.gov/cdrh/mdufma/presentations/mdufma.html
11
USER FEES-2006
Application fees-Drugs /Biologics$767,400 for an application requiring clinical data $383,700 for an application not requiring clinical data$383,700 for a supplement requiring clinical data
Annual Fees$264,200 for establishment fees$42,130 for product fees
12
User Fees-Exceptions and Waivers
Designated Orphan Drug or Indication An application for product designated as an Orphan Drug A supplement for a new indication for an Orphan Drug
Small business or its affiliate (< 500 employees)Agency will waive the application fee for the first human drug application
13
MDUFA
Reviews Subject to User FeesEffective October 1, 2002, a user fee is assessed for FDA review of a —
510(k) Premarket application — PMA, PDP, Premarket Report (reprocessed device), or BLA. Panel-track supplement 180-day supplement Real-time supplement BLA efficacy supplement
14
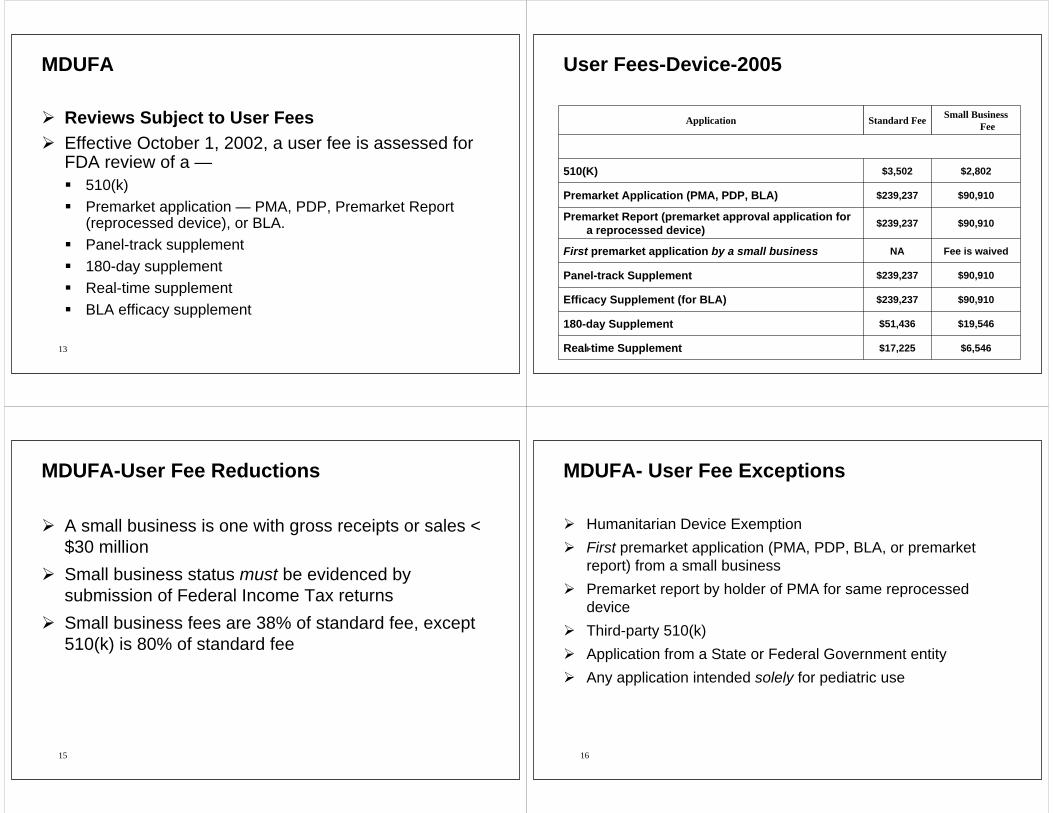
User Fees-Device-2005
Small Business FeeStandard FeeApplication
$6,546$17,225Real-time Supplement
$19,546$51,436180-day Supplement
$90,910$239,237Efficacy Supplement (for BLA)
$90,910$239,237Panel-track Supplement
Fee is waivedNAFirst premarket application by a small business
$90,910$239,237Premarket Report (premarket approval application for a reprocessed device)
$90,910$239,237Premarket Application (PMA, PDP, BLA)
$2,802$3,502510(K)
15
MDUFA-User Fee Reductions
A small business is one with gross receipts or sales < $30 millionSmall business status must be evidenced by submission of Federal Income Tax returnsSmall business fees are 38% of standard fee, except 510(k) is 80% of standard fee
16
MDUFA- User Fee Exceptions
Humanitarian Device ExemptionFirst premarket application (PMA, PDP, BLA, or premarketreport) from a small businessPremarket report by holder of PMA for same reprocessed deviceThird-party 510(k) Application from a State or Federal Government entityAny application intended solely for pediatric use

17
Post Submission RequirementsAmendments to Unapproved Applications
21CFR 314.60 Amendments 21 CFR 814.37-PMA Amendments
18
Amendments to Unapproved Applications
Guidances/SOPPs–CBER/CDERGood Review Management Principles and Practices for PDUFA Products (CBER/CDER)Information Request and Discipline Review Letters Under the Prescription Drug User Fee ActClassifying Resubmissions in Response to Action LettersSOPP 8401.1: Issuance and Review of Responses to Information Requests and Discipline Review Letters to Pending Applications
Guidances-CDRHFDA and Industry Actions on Premarket Approval Applications (PMAs): Effect on FDA Review Clock and Performance AssessmentPremarket Approval Application Filing Review
19
Amendments to Unapproved Applications
The BLA, NDA or PMA is rarely the last submission prior to an Approval LetterFDA will formally and informally communicate during the review processResponses need to be submitted to the application
20
FDA Review of Unapproved Applications
Designate whether priority or standard application (6-month vs. 12-month review clock)Check if fees paid Document presence of all of the required filing elementsForm the review committee and meet Make a refusal-to-file decision by the 45-day meetingMake Information Requests Complete review - issue an action letter (complete response or approval)Other actions - withdrawal or denial

21
Types of FDA Letters for Drugs/Biologics
Information request letters Requests for additional information
Discipline review letters Not a complete review Early thoughts from members of review teamIdentify deficiencies that need to be correctedMay describe actions that need to be taken
Action letterIssued after complete review of the filed application
Preliminary comments on draft labeling
22
Effect of Letters on Review Clock
The IR/DR letters will not affect the user fee review clockYour response affects the review clockReview team will review responses at their discretion
They may or may not consider a response to a DR a major amendmentMajor amendments extend the review time (3 months)Only one extension allowed
The Agency is not obligated to review the response before the issuance of an action letterThe action letter will include all deficiencies that must be answered to place the application in condition for approval
23
Resubmissions in Response to Action Letters
Class 1 Resubmission (2-month review)Final labelingSafety updatesCommitments of phase 4 studiesAssay validationLot release of last 1-2 lots used to support approval
Class 2 Resubmission (6-month review)Everything else
24
Types of Amendments
Answers to questionsAdditional information (i.e.)
Adverse eventsManufacturing tests
New analysesUpdated ISS and ISEData from studies in progressProtocols for Post-approval Commitments
25
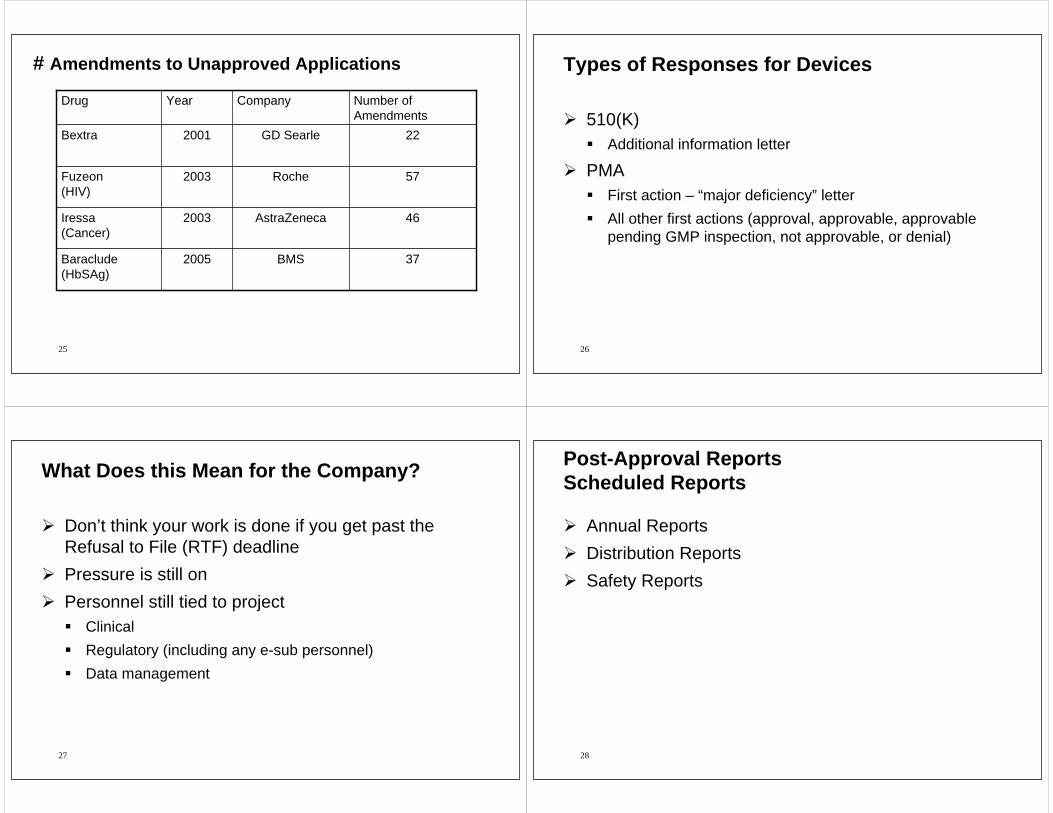
# Amendments to Unapproved Applications
22GD Searle2001Bextra
57Roche2003Fuzeon(HIV)
37BMS2005Baraclude(HbSAg)
46AstraZeneca2003Iressa(Cancer)
Number of Amendments
CompanyYearDrug
26
Types of Responses for Devices
510(K)Additional information letter
PMAFirst action – “major deficiency” letter All other first actions (approval, approvable, approvable pending GMP inspection, not approvable, or denial)
27
What Does this Mean for the Company?
Don’t think your work is done if you get past the Refusal to File (RTF) deadlinePressure is still onPersonnel still tied to project
ClinicalRegulatory (including any e-sub personnel)Data management
28
Post-Approval ReportsScheduled Reports
Annual ReportsDistribution ReportsSafety Reports

29
Annual Reports-Regulation
CDER -314.81 b (2)CBER
21 CFR 600.12 –Changes in Application21 CFR 600.70 –Post-marketing Commitments21 CFR 600.80 –Adverse Events21 CFR 600.81 –Distribution Report
CDRH21 CFR 814.84
30
Annual Reports-Guidance
Draft Guidance for Industry: Providing Regulatory Submissions in Electronic Format--Annual Reports for New Drug Applications and Abbreviated New Drug Applications - 8/27/2003
31
Annual Report-Drugs and Biologics
When is it due?within 60 days of the anniversary date of U.S. approval of the application
What time frame should it cover?must include all the information obtained during the interval that ends on the U.S. anniversary date
32
Annual Report-What is included (314.81)
SummaryNew information that might affect the safety, effectiveness, or labeling Actions the applicant has taken or planned based on this information
Distribution data Information about the quantity of the drug product distributed Total number of dosage units of each strength or potency Disclosure of financial or pricing data is not required.
LabelingCurrently used professional labeling, patient brochures or package inserts (if any), and a representative sample of the package labelsA summary of any changes in labeling since the last report

33
Annual Report-What is included (314.81)
Chemistry, manufacturing, and controls changesReports of experiences, investigations, studies, or tests that may affect FDA`sprevious conclusions about the safety or effectiveness of the drug product.A full description of the manufacturing and controls changes not requiring a supplemental application
Nonclinical laboratory studiesCopies of unpublished reports and summaries of published reports of new toxicological findings
Clinical dataPublished clinical trials of the drug (or abstracts ) including clinical trials on new uses; Review articles, papers describing the use in medical practice, Summaries of completed unpublished clinical trials or prepublication manuscripts
34
Annual Report-What is included (314.81)
Status reports of post-marketing study commitmentsA status report of each post-marketing study concerning clinical safety, clinical efficacy, clinical pharmacology, and nonclinical toxicology that is required by FDAMust be reported annually until FDA agrees the commitment has been fulfilled or the study is no longer feasible or would no longer provide useful information
Status of other post-marketing studiesChemistry, manufacturing, and controls studies that the applicant has agreed to perform Product stability studies
Log of outstanding regulatory businessTo facilitate communications between FDA and the applicant
35
Post-Marketing Commitments
RegulationFDA Modernization Act of 1997 added a new provision (section 506B) on post-marketing studies to the Federal Food, Drug, and CosmeticApplicable to drugs and biologics –NOT devices
GuidanceReports on the Status of Post-marketing Studies -Implementation of Section 130 of the Food and Drug Administration Modernization Act of 1997
36
Post-Marketing Commitments
Clinical StudiesCalled phase 4 commitmentsStudies — required of or agreed to by a sponsor – conducted after FDA has approved a product FDA may require commitment prior to approval– May even require study to have startedCommitments are published on FDA website with yearly progress results

37
Post-Marketing Commitments-When likely?
Commitment Required Under: Accelerated ApprovalApproved based on surrogate endpoints that reasonably predict clinical benefitClinical benefit must be confirmed through additional human studies
Commitment Required Under: Animal Efficacy RuleNew drug or biological used to reduce or prevent the toxicity of chemical, biological, radiological, or nuclear substancesApproved for marketing based on evidence of effectiveness in animalsSponsor must verify clinical benefit when such studies are feasible and ethical
Commitment Required Under: Pediatric Research Equity ActPediatric Research Equity Act of 2003 Designed to improve the quality of pediatric information in drug labelingFDA has explicit authority to require applications to include pediatric studies
Studies to gather additional information about a product's safety, efficacy, or optimal use
38
Post-Marketing Commitments
CMC RelatedMay request additional validationsStability dataCommitments are NOT published on FDA website– Considered confidential
39
Post-Marketing Commitments-Examples
*Accelerated Approval CommitmentOccurrence of MDSLong-term effects of HAMA-effect on IVD tests
Long-term effects of HAMA-effect on patients
Use of Prophylactic VaccinesRetreatment with BexxarBexxar vs ZevalinBexxar vs Rituximab*
DESCRIPTION OF STUDY
40
Post-Marketing Commitments-Examples
1,090 NDAs were approvedThese NDAs plus supplemental applications generated a total of 2,328 postmarketing commitments1,737 reporting under FDAMA 130

41
Post-Marketing Commitments
Failure of PMC Clinical TrialFDA can pull approvalConsidered with Astrazeneca’s IRESSA after PMC trial failedTo date –has not happened
Concern with companies not meeting the commitments
42
Post-Marketing Commitments-Impact on Company
Initial approval may not be the last clinical trialAccelerated approval=Confirmatory TrialComplicated products =follow-up trials$$$$Resources may be requiredRecent drug problems mean this is only going to increase
43
Annual Report-Devices (PMA)
Due annually at intervals of 1 year from the date of approval of the original PMAPost-approval reports for supplements approved under the original PMA are included in the next and subsequent annual reports for the original PMA
44
Annual Report-PMAs
Identification of changes described in 21CFR814.39(a) and changes required to be reported to FDA under 21CFR814.39(b) Bibliography and summary of the following information:
Unpublished reports of data from any clinical investigations or nonclinical laboratory studies involving the device or related devices Reports in the scientific literature concerning the device
45
Scheduled Reports
Annual ReportsDistribution ReportsSafety Reports
46
Distribution Reports-Biologics
21 CFR 600.81- Distribution reports. Must be submitted every 6 monthsFDA can require other time framesMust contain:
bulk lot number fill lot numbers label lot number (if different from fill lot number)labeled date of expirationnumber of doses in fill lot/label lotdate of release of fill lot/label lot for distribution
47
Distribution Reports-Devices
Form 3417-Medical Device Reporting-Baseline Report (for first report)Annual update requiredIncludes a line for # devices distributed
48
Medical Device Tracking
21 CFR Part 821Section 519(e) of the FDC Act Guidance for Industry and FDA Staff: Medical Device Tracking May 5, 2003

49
Medical Device Tracking
Devices that are life sustaining or life supporting and used; are permanently implanted; serious consequences if they fail Company must ensure that devices can be traced from the manufacturing facility to the patientRequires effective tracking of through the distributor network to the patient Must be capable of patient notification or device recall Can use a contractor but manufacturer is responsible
50
Examples of Products that must be Tracked
· Mandibular condyle prosthesis· Temporomandibular Joint (TMJ) prosthesis· Abdominal Aortic Aneurysm Stent Grafts· Automatic implantable cardioverter/defibrillator· Cardiovascular permanent implantable pacemaker electrode· Implantable pacemaker pulse generator· Replacement heart valve (mechanical only)· Implanted cerebellar stimulator· Implanted diaphragmatic/phrenic nerve stimulator· Implantable infusion pumps· Dura mater· Breathing frequency monitors· Continuous Ventilators· DC-defibrillators and paddles· Ventricular Bypass (assist) Device
51
Scheduled Reports
Annual ReportsDistribution ReportsSafety Reports
52
Safety Reports -
21 CFR 600.80Periodic adverse experience reports are due
at quarterly intervals, for 3 years from the date of licensing annual intervals from then onwithin 30 days of the close of the quarter
Annual report within 60 days of the anniversary date of the license
53
Contents of Annual Safety Report (example)
Introduction Initial Reports List of Serious Listed Initial Reports List of Non-Serious Unlisted Initial ReportsList of Non-Serious Listed Initial ReportsList of Serious Listed Follow-up Reports List of Non-Serious Unlisted Follow-up Reports List of Non-Serious Listed Follow-up ReportsList of 15-Day Reports by Body System Submitted During Reporting PeriodTabulation by Body System of AII Events Reported Analysis and Discussion of Serious Listed Initial Reports Analysis and Discussion of Non-Serious Unlisted Initial ReportsAnalysis and Discussion of Non-Serious Listed Initial ReportsSummary and Analysis of the 15-Day Reports Submitted During Reporting PeriodSummary of Actions Taken During Reporting PeriodAppendix A: Current United States Prescribing Information for the Product
54
Device-Annual Report -Safety
21 CFR 803If you are a manufacturer or importer, you must report – deaths and serious injuries that your device has or may have caused
or contributed to, – certain device malfunctions, and– specified follow-up and baseline reports. MDR regulation requires submission of a baseline report on a device model the first time it is the subject of a device report (Form 3417)Annual updates required– Summary report of all reportable adverse events submitted to
manufacturers or the FDA
55
Scheduled Reports
Annual ReportsDistribution ReportsSafety Reports
56
Unscheduled Reports
Changes to Approved ApplicationsCMC SupplementsEfficacy Supplements
Reports of Problems and ErrorsPromotional LiteratureIndividual Safety Reports
57
Changes in Applications-Regulation and Guidance
Regulation21 CFR 314.70-Drugs21 CFR 601.12-Biologics21 CFR 807.81 -510(k)21 CFR 814.39 -PMA
58
Changes in Applications-Regulation and Guidance
GuidanceChanges to an Approved NDA or ANDAGuidance for Industry: Changes to an Approved Application for Specified Biotechnology and Specified Synthetic Biological ProductsGuidance for Industry: Changes to an Approved Application: Biological Products30-Day Notices and 135-Day PMA Supplements for Manufacturing Method or Process
59
Changes in Applications-CBER/CDER
CBER and CDER system are similar A change to a product, production process, quality controls, equipment, facilities, or responsible personnel must be reported in one of the following:
1) supplement requiring approval prior to distribution, 2) supplement submitted 30 days prior to distribution3) an annual report
Which one depends on its potential to have an adverse effect on the identity, strength, quality, purity, or potency of the biological product as they may relate to the safety or effectiveness of the product.
60
Changes to Applications-Summary of Requirement
Minimal potentialAnnual Report
Moderate potential30-Day Prior
Substantial potentialPrior Approval
Risk of an Adverse Effect on the Identity, Strength, Quality, Purity, or Potency
Type of Submission

61
Changes to Applications-Specific Examples with Substantial Potential (Biologics/Drugs)
Changes in labeling, except those that increase safetyChanges in indicationChanges in route of administration
Change in drug formulation/quantity Change in test methods
Deletion of a specification or an analytical methodElimination of a test Change in acceptance criteria
Scale-up requiring larger equipment Extension of the expiration dating period / change in storage temperature w/o stability protocol
62
CMC Changes-Examples of Changes with Moderate Potential (Biologics/Drugs)
Addition or reduction in number of pieces of equipment to achieve a change in purification scaleChange in the fill volume (per vial) Changes in responsible individuals specified in the approved application, Modification of an approved manufacturing facility or room(s) that is not likely to have an adverse effectChange in the site of testing from one facility to another Change in the structure of a legal entity that would require issuance of new license(s),
63
CMC Changes-Examples of Changes with Minimal Potential (Biologics/Drugs)
Addition of equipment identical to the primary system Upgrade or minor corrective change to production air handling, water, or steam supply systems Relocation of testing laboratories in areas in the licenseRoom upgrades, (i.e. improved finishes on floors/walls)Installation of non-process equipment (ie.refrigerators) freezersModifications in analytical procedures with no change in the basic test methodology or existing release specifications Change in harvesting and/or pooling procedures which does not affect the method of manufacture, recovery, storage conditions, etc.Replacement of an in-house reference standard or reference panel according to SOPs and specifications in approved license application.
64
Changes to existing PMA
Must submit a supplement if the change affects the safety or effectiveness of the device
New indicationsLabeling changesDifferent facility to manufacture, process, or package the deviceChanges in sterilization proceduresChanges in the performance or design specifications, circuits, components, ingredients, principle of operation, or physical layout of the device Extension of the expiration date w/o approved protocol

65
Changes to existing PMA
Changes that may be made without submitting a supplement Labeling changes that– add or strengthen a contraindication, warning, precaution, or
information about an adverse reaction. – add or strengthen an instruction that is intended to enhance the safe
use of the device– delete misleading, false, or unsupported indicationsChanges in QC or manufacturing process that add a new specification or test method, or provide added assurance of purity, identity, strength, or reliability
These changes are reported in periodic reports
66
Changes to existing PMA
Exception to PMA supplementsSection 515(d) (6) of the act added by the FDA Modernization Act of 1998 requires a 30-day Notice for changes that that involve modifications to manufacturing procedures or method of manufactureIf the notice is inadequate, a 135-day PMA supplement will be required
67
Changes to an Existing 510(k)
New intended use for a device Requires a premarket notification submission for major changes in intended use. Most, if not all changes in intended use will require a 510(k)
Change or modification of a deviceNew submission required if that change could significantly affect safety or effectivenessThe burden is on the company to decideJustification should be reflected in your device master record and change control records
68
Efficacy Supplements
Required to submit for change in labelUsually requires clinical dataUser fee must be paid prior to submission if clinical data included
If not Orphan Drug
Consider meeting with FDA to present outline of submission and results of clinical data prior to submission
69
CMC or Label Changes-Impact on Company
Everyone needs to understand the requirementsNeed document control system
Record of product changes made during year Procedure to evaluate importance of change
Regulatory should review changes for importanceMake determination whether submission is required prior to implementation Documentation of decisions
Personnel to test and validate changes
70
Unscheduled Reports
Changes to Approved ApplicationsCMC SupplementsEfficacy Supplements
Reports of Problems and ErrorsPromotional LiteratureIndividual Safety Reports
71
Product Problems-Regulations-FD&C Act
Sec. 301 Prohibited ActsItems that are adulterated/misbrandedCounterfeit drugs
Sec. 501 Definition of AdulteratedFilthy, putrid or decomposedDifference (reduced) in strength/purity
Sec. 502 Definition of MisbrandedLabeling is false or misleadingAdequate directions for use or warnings
72
Reporting Product Problems
BiologicsProduct Deviations
DrugsField Alerts
DevicesProduct Malfunctions

73
Biologics-Product Deviation- Guidances
BiologicsRegulation-Biologic Products– 21 CFR 600.14– Guidance for Industry-Biological Product Deviation
Reporting for Licensed Manufacturers of Biological Products -Other than Blood and Blood Components
Regulation-Blood Products
– 21 CFR 606.171– Guidance for Industry-Biological Product Deviation Reporting for
Blood and Plasma Establishments
74
Product Deviations-Biologics or Blood
What is reported?Any event, associated with the manufacturing, if that event meets any of the following criteria:
(i) Represents a deviation that affects the safety, purity, or potency of that product; or
(ii) Represents an unexpected or unforeseeable event that may affect the safety, purity, or potency of that product; and
Occurs in your facility or another facility under contract with you; and Involves a distributed biological product or blood.
75
Product Deviations- When is a Report Required?
If problem is discovered AFTER PRODUCT DISTRIBUTION
76
Product Deviations-When is NO Report Required
When is a Report NOT REQUIRED: When no affected products are distributed, When it is determined prior to distribution that the safety, purity, or potency of a product is not affected. When a product is appropriately reprocessed or reworked by an FDA approved method, prior to distribution. When there is a minor record keeping omission, that has no potential to affect the safety, purity, or potency of the product.

77
Product Deviations-Examples
Examples:Raw material contaminated or does not meet spec
Manufacturing/processing performed using incorrect parametersBulk or intermediate product stored improperly Required testing was not performed, was performed incorrectlyPackage insert is incorrect, not the current approved version, or not included with productBlood establishment performed compatibility testing using a patient sample from the wrong patient
78
Drugs-Field Alerts
Regulations21 CFR 314.81 Other postmarketing reports
Guidance NDA Field Alert Form and InstructionsProduct Recalls, Including Removals and Corrections
SOPP Standing Operating Procedures for Handling NDA/ANDA Field Alert Reports
79
Field Alerts -Drugs
NDA—Field alert report Must submit information of the following kinds of incidentsLabeling errorBacteriological contaminationSignificant change or deteriorationFailure of to meet the specification in the application
Report to the FDA district office that is responsible for the facility involved within 3 working days of receipt by the applicant. The information may be provided by telephone with prompt written followup.
80
Field Alert-FDA Response
District OfficesPost-market Surveillance Team (PST) Coordinator evaluates the Field Alert Report and conducts follow-up, if necessaryThe District Recall and Emergency Coordinator reviewsMay follow up with company
Office of CompliancePostmarket Surveillance Team will evaluate the initial report and the
district's follow-up informationIf HFD-336 determines there is a potentially significant problem, they contact the Office of Compliance Drug Recall Staff (and the Division of Manufacturing and Product Quality
81
Malfunctions –DevicesRegulations and Guidance
21 CFR 803-Medical Device Reporting21 CFR 806-Reports of Corrections and Removals
82
Medical Device Reporting -Devices
Malfunction [§803.3(m)] Failure of the device to meet its performance specificationsReportable if:– chance of a death or serious injury is not remote;– device is considered life-supporting/life-sustaining– manufacturer takes/would be required to take action – reported even if it can be corrected in routine service
Who must reportManufacturerDistributorUser facility
83
Deviation Reports/Field Alerts/Malfunctions-Impact on Company
Need systems to collect incoming data with SOP on documenting problems and decisionsNeed written procedure for reporting product deviations, field alerts or malfunctionsAlso need a Recall ProcedureNeed a tracking system for lot releaseNeed a good lawyer
84
Promotional Literature
GuidancesFDA Form 2253 - Transmittal of Advertisements and Promotional Labeling for Drugs and Biologics for Human UseAccelerated Approval Products: Submission of Promotional Materials (Posted 3/26/1999)
85
Promotional Literature
Code of Federal Regulations21 CFR 99 - Dissemination of Information on Unapproved/New Uses
for Marketed Drugs, Biologics, and Devices21 CFR 200– General21 CFR 201- Labeling21 CFR 202- Prescription Drug Advertising21 CFR 312- Investigational New Drug Applications21 CFR 314- Applications to Market New Drug or Antibiotic
86
Promotional Literature
Labeling reviewed during approval processPackage insertPackage labelsOther (based on BEXXAR experience)– Training Manual– Software Manual
Reviewers are the individuals who reviewed the application
87
Promotional Literature
Review of all promotional material Must submit w/ Form 2253 at the time of use– DDMAC/APLS– Failure to submit can result in a Warning Letter
Except new product approved under Accelerated Approval Regulations
All promotional material pre-reviewed
88
Safety Reports- Relevant Regulations
DRUGS
21 CFR 314.80 – Post-marketing reporting of adverse drug experiences
BIOLOGICS
21 CFR 600.80 – Post-marketing reporting of adverse experiences
DEVICES
• 21 CFR 803 – Medical Device Reporting

89
Safety Reports-Guidance Documents
Post-marketing Reporting of AEsDrugs (March 1992)
Clarification of what to Report for Human Drug and Licensed Biological Products (August 1997)
Drugs and Biologics (draft guidance - March 2001)
Devices –Medical Device Reporting for Manufacturers (March 1997)
90
Safety Reports-ICH Documents
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH)
MedDRA Term Selection: Points to Consider - Release 3.4, based on MedDRA version 7.1, an ICH-Endorsed Guide for MedDRA UsersM1 MedDRA (Medical Dictionary for Regulatory Activities) M2 ESTRI (Electronic Standards for the Transfer of Regulatory Information) E2A (Clinical Safety Data Management) E2B (Guidance On Data Elements For Transmission of Individual Case Safety Reports) E2BM (Guidance on Data Elements for Transmission of Individual Case Safety Reports) E2C PSUR (Periodic Safety Update Report)
91
Safety Reports-Requirements
Obligated to Review Adverse Drug ExperiencesFrom foreign or domestic sources including– Commercial marketing experience– Post-marketing clinical investigations,– Post-marketing epidemiological/surveillance studies– Reports in the scientific literature, and – Unpublished scientific papers
Must Develop Written Procedures for SurveillanceReceipt and evaluation, as well as Reporting of post-marketing AEs to FDA
92
Safety Reports –Definitions-Drugs/Biologics
Serious adverse drug experienceDeathLife-threatening adverse drug experienceInpatient hospitalization or prolongation of existing hospitalizationPersistent or significant disability/incapacityCongenital anomaly/birth defectImportant medical events that may jeopardize the patient or subject and may require medical or surgical intervention to prevent one of the outcomes listed in this definition.
93
Safety Reports –Definitions-Drugs/Biologics
Unexpected adverse drug experienceAny adverse drug experience that is not listed in the current labeling for the drug product. Basically –Anything Not Previously Observed
94
Safety Reports-Drugs and Biologics
Drugs and BiologicsReporting requirements– Submit two copies of each report –Form 3500A
Postmarketing 15-day “Alert reports”– Must report each adverse drug experience that is
– both serious and unexpected– No later than 15 calendar days of initial receipt of the information
15-day “Alert reports”—follow-up– Must investigate all adverse drug experiences that are reported– Submit followup reports within 15 calendar days of receipt of new information
or as requested by FDA
95
Safety Reports-Devices
Reportable events
Serious injury/(Serious illness) [§803.3(aa)(1)]– life threatening, even if temporary– results in permanent impairment of a body function – requires medical or surgical intervention to preclude
permanent impairment
96
Safety Reports-Devices
Coincide with registration
FDA 3381Annual Certification
When device or family reported 1st
time
FDA 3417Baseline report-basic data on device
Within 30 daysof becoming aware
FDA 3500ADeath, serious injury, malfunctions
WhenFormWhat to Report

97
Safety Reports-Impact on Company
Usually Requires Development of a GroupExperienced health-care professionalsNurses and Pharm.D. typical members of teamCan be resource intensive
Requires written proceduresMay require specialized softwareMay require submissions to IND for RegulatoryMay require European Submissions
Related Documents











