Partial Atomic Charges and Their Impact on the Free Energy of Solvation Joakim P. M. J€ ambeck,* [a] Francesca Mocci, [a,b] Alexander P. Lyubartsev, [a] and Aatto Laaksonen [a,b] Free energies of solvation (DG) in water and n-octanol have been computed for common drug molecules by molecular dynamics simulations with an additive fixed-charge force field. The impact of the electrostatic interactions was investigated by computing the partial atomic charges with four methods that all fit the charges from the quantum mechanically determined electrostatic potential (ESP). Due to the redistribution of electron density that occurs when molecules are transferred from gas phase to condensed phase, the polarization impact was also investigated. By computing the partial atomic charges with the solutes placed in a conductor- like continuum, the charges were effectively polarized to take the polarization effects into account. No polarization correction term or similar was considered, only the partial atomic charges. Results show that free energies are very sensitive to the choice of atomic charges and that DG can differ by several k B T depending on the charge computing method. Inclusion of polarization effects makes the solutes too hydrophilic with most methods and in vacuo charges make the solutes too hydrophobic. The restrained-ESP methods together with effectively polarized charges perform well in our test set and also when applied to a larger set of molecules. The effect of water models is also highlighted and shows that the conclusions drawn are valid for different three-point models. Partitioning between an aqueous and a hydrophobic phase is also described better if the two environment’s polarization is taken into account, but again the results are sensitive to the charge calculation method. Overall, the results presented here show that effectively polarized charges can improve the description of solvating a drug-like molecule in a solvent and that the choice of partial atomic charges is crucial to ensure that molecular simulations produce reliable results. V C 2012 Wiley Periodicals, Inc. DOI: 10.1002/jcc.23117 Introduction Few would argue against that the free energy is one of the most important concepts in thermodynamics. Changes in the free energy control how small drug molecules are dissolved in water and how biologically active they are in terms of ligand binding and cell penetration. The knowledge of the free energy is therefore of great interest in rational drug design [1,2] and as a result of this, a great deal of effort has been put into developing methods and algorithms that can compute this property in an accurate and efficient fashion. With the increase in available computing power, the aspects of these calcula- tions that were once looked on as highly computationally in- tensive tasks, due to the prerequisite to sample, every ther- mally available part of phase space, are beginning to fade away. As this is true for the free energy of solvation of smaller molecules, the problem of predicting ligand binding affinities in a more efficient and systematic manner still remains. [1,3] In some thermodynamic cycles, the free energy of hydration (DG hyd ) has to be determined accurately to predict ligand binding affinities. [4] Further, DG hyd is important for drugs’ solu- bility [5] and their partitioning between between phases, usually water and n-octanol are used in experimental studies of drug cell permeation. The latter is often used as a model of the cell membrane in experimental studies with so-called log P val- ues. [6,7] Therefore, hydration free energies play a central role in our molecular understanding of several important processes. Computations of free energies of solvation also give impor- tance guidelines on which level of accuracy that should be expected in other applications as well act as a fundamental test of force fields (FFs). Usually, an empirical energy function, called FF, is used to describe the intermolecular and intramolecular interactions in a system composed of solvent and solute(s), that is, a model Hamiltonian. Molecular dynamics (MD) or Monte Carlo (MC) algorithms are then used to sample the different configura- tions of the system [8] and free energies can then be computed with the aid of statistical mechanics. Different methods can be applied to obtain free energies from these sampling proce- dures: thermodynamic integration [9] that has been used in a number of studies, [10–12] the Bennett acceptance ratio method, [13–16] free energy perturbation, [17–19] or the expanded ensemble (EE) method. [20–23] The mentioned studies are just a handful of the recent published work as it would be an over- whelming task to go through the whole field. The major [a] J. M. J € ambeck, F. Mocci, A. P. Lyubartsev, A. Laaksonen Division of Physical Chemistry, Arrhenius Laboratory, Stockholm University, Stockholm, SE-10691, Sweden E-mail: [email protected] or [email protected] [b] F. Mocci, A. Laaksonen Department of Chemical and Geological Sciences, University of Cagliari, Italy Contract/grant sponsor: Swedish Research Council (VR). V C 2012 Wiley Periodicals, Inc. Journal of Computational Chemistry 2013, 34, 187–197 187 FULL PAPER WWW.C-CHEM.ORG

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Partial Atomic Charges and Their Impact on the FreeEnergy of Solvation
Joakim P. M. J€ambeck,*[a] Francesca Mocci,[a,b] Alexander P. Lyubartsev,[a]
and Aatto Laaksonen[a,b]
Free energies of solvation (DG) in water and n-octanol have
been computed for common drug molecules by molecular
dynamics simulations with an additive fixed-charge force field.
The impact of the electrostatic interactions was investigated
by computing the partial atomic charges with four methods
that all fit the charges from the quantum mechanically
determined electrostatic potential (ESP). Due to the
redistribution of electron density that occurs when molecules
are transferred from gas phase to condensed phase, the
polarization impact was also investigated. By computing the
partial atomic charges with the solutes placed in a conductor-
like continuum, the charges were effectively polarized to take
the polarization effects into account. No polarization
correction term or similar was considered, only the partial
atomic charges. Results show that free energies are very
sensitive to the choice of atomic charges and that DG can
differ by several kBT depending on the charge computing
method. Inclusion of polarization effects makes the solutes too
hydrophilic with most methods and in vacuo charges make
the solutes too hydrophobic. The restrained-ESP methods
together with effectively polarized charges perform well in our
test set and also when applied to a larger set of molecules.
The effect of water models is also highlighted and shows that
the conclusions drawn are valid for different three-point
models. Partitioning between an aqueous and a hydrophobic
phase is also described better if the two environment’s
polarization is taken into account, but again the results are
sensitive to the charge calculation method. Overall, the results
presented here show that effectively polarized charges can
improve the description of solvating a drug-like molecule in a
solvent and that the choice of partial atomic charges is crucial
to ensure that molecular simulations produce reliable results.
VC 2012 Wiley Periodicals, Inc.
DOI: 10.1002/jcc.23117
Introduction
Few would argue against that the free energy is one of the
most important concepts in thermodynamics. Changes in the
free energy control how small drug molecules are dissolved in
water and how biologically active they are in terms of ligand
binding and cell penetration. The knowledge of the free
energy is therefore of great interest in rational drug design[1,2]
and as a result of this, a great deal of effort has been put into
developing methods and algorithms that can compute this
property in an accurate and efficient fashion. With the increase
in available computing power, the aspects of these calcula-
tions that were once looked on as highly computationally in-
tensive tasks, due to the prerequisite to sample, every ther-
mally available part of phase space, are beginning to fade
away. As this is true for the free energy of solvation of smaller
molecules, the problem of predicting ligand binding affinities
in a more efficient and systematic manner still remains.[1,3] In
some thermodynamic cycles, the free energy of hydration
(DGhyd) has to be determined accurately to predict ligand
binding affinities.[4] Further, DGhyd is important for drugs’ solu-
bility[5] and their partitioning between between phases, usually
water and n-octanol are used in experimental studies of drug
cell permeation. The latter is often used as a model of the cell
membrane in experimental studies with so-called log P val-
ues.[6,7] Therefore, hydration free energies play a central role in
our molecular understanding of several important processes.
Computations of free energies of solvation also give impor-
tance guidelines on which level of accuracy that should be
expected in other applications as well act as a fundamental
test of force fields (FFs).
Usually, an empirical energy function, called FF, is used to
describe the intermolecular and intramolecular interactions in
a system composed of solvent and solute(s), that is, a model
Hamiltonian. Molecular dynamics (MD) or Monte Carlo (MC)
algorithms are then used to sample the different configura-
tions of the system[8] and free energies can then be computed
with the aid of statistical mechanics. Different methods can be
applied to obtain free energies from these sampling proce-
dures: thermodynamic integration[9] that has been used in a
number of studies,[10–12] the Bennett acceptance ratio
method,[13–16] free energy perturbation,[17–19] or the expanded
ensemble (EE) method.[20–23] The mentioned studies are just a
handful of the recent published work as it would be an over-
whelming task to go through the whole field. The major
[a] J. M. J€ambeck, F. Mocci, A. P. Lyubartsev, A. Laaksonen
Division of Physical Chemistry, Arrhenius Laboratory, Stockholm University,
Stockholm, SE-10691, Sweden
E-mail: [email protected] or [email protected]
[b] F. Mocci, A. Laaksonen
Department of Chemical and Geological Sciences, University of Cagliari, Italy
Contract/grant sponsor: Swedish Research Council (VR).
VC 2012 Wiley Periodicals, Inc.
Journal of Computational Chemistry 2013, 34, 187–197 187
FULL PAPERWWW.C-CHEM.ORG

difference between the EE method and the other mentioned
methods is the way the simulations are set up. In order for
any free energy computation to be efficient, an ensemble of
intermediate states has to be assigned between the reference
state and the target state to secure a significant phase space
overlap. This results in a large number of simulations that have
to be performed and also requires a lot of postsimulation anal-
ysis to ensure proper overlap of states and so on. Therefore,
these procedures are very time-consuming and do not always
guarantee a precise value of the free energy.[10] Due to the
large throughput in drug design, it is desirable to have an effi-
cient methodology. With the EE method (described in more
detail later), the sampling of the phase space is governed by
random walk over the reference, a number of intermediate
and target states. As this is done in an automated fashion, it is
possible to obtain the free energy of solvation in one single
MD simulation which makes this procedure very efficient. The
EE method has been used in many studies to compute free
energies.[23–31]
It is important to point out that merely choosing an effec-
tive way of obtaining the free energy of solvation from simula-
tions is not enough. There are other vital choices one has to
make and perhaps the most crucial one is the FF. The set of
parameters in the FF that has the largest impact on the
computation of free energies are the partial atomic
charges[14,18,18,32,33] (if the discussion is limited to point charge,
additive FFs). Although atomic charges are determined by
quantum mechanical (QM) based calculations for FFs like
AMBER and to a lesser extent CHARMM, they have an inherent
problem—atomic charges are not QM observables, that is,
there is no strict physical definition of them. Electrostatic
moments such as dipole, quadrupole, and so forth can be
measured in laboratories and give (limited) information regard-
ing the charge distribution but for larger molecules this
becomes cumbersome. As there is no unambiguous way of
determining these partial atomic charges, a number of meth-
ods have been developed. One method of obtaining atomic
charges is by minimizing the difference between the electro-
static potential (ESP) determined from the wave function and
the ESP created by the point charges.[34] With this procedure,
the spherically symmetric point charges of each atom in the
molecule reproduce the ESP as well possible. As the ESP is a
QM observable, this is a straightforward procedure with a
more physical foundation than for instance Mulliken charges.
A problem that arises with this approach is how one should
sample the QM ESP. Several schemes have been suggested:
CHELP,[35] CHELPG,[36] and the Merz-Singh-Kollman (MK)
scheme.[34,37] All the mentioned methods sample the ESP at a
number of points outside the van der Waals (vdW) radii of the
constituting atoms of the molecule. The CHELP scheme uses
points at a distance of 2.5, 3.5, 4.5, 5.5, and 6.5 A from the
vdW surface, CHELPG samples the ESP at points evenly distri-
buted between 0 and 2.8 A from the vdW surface and the MK
scheme at shells at a distance of 1.4, 1.6, 1.8, and 2.0 times
the vdW radii. One inherent problem with all the mentioned
methods is the determination of the ESP ‘‘belonging’’ to the
more buried atoms such as sp3-hybridized carbons.[38,39] Fur-
ther, there is a pronounced conformational dependency of the
charges[40–42] which in turn will affects the computed free
energy of solvation.[43–45] One way of addressing the problem
with the charges of buried atoms and to temper the confor-
mational dependency is to use the restrained-ESP (RESP)[46]
approach. With this method, a hyperbolic penalty function is
used to restrain the charges to keep their magnitudes down,
and the ESP can be sampled with any of the three earlier
mentioned methods.
There are two problems with all the approaches described
above, namely the assumption that the charge density is sym-
metric around the atoms and the lack of polarization. Both
issues are inherent problems of additive FFs which requires a
lot of effort to correct. Still classical, point-charge FFs are used
due to their surprising accuracy and modest time cost com-
pared to, for example, polarizable FFs. The lack of polarization
is of great importance as for polar molecules, the charge distri-
bution can change considerably when transferring the mole-
cule from gas phase to condensed phase.[47,48] Water for
instance induces a strong electric field which polarizes the sol-
ute(s). Often charge calculations are performed with the solute
in gas phase where this redistribution of charges is ignored.
The effects of neglecting the polarization have been noted
previously in the literature.[23,49] One straightforward way to
include polarization effects would be to perform the QM calcu-
lations with a self-consistent reaction field. By doing so, the
ESP obtained would be polarized and therefore the partial
charges derived from this ESP would be effectively polarized
when used in simulations of condensed phase systems. This
solution has been proposed before[23] and has also been
applied.[14,49–51] Here, we further test if this method improves
the free energy calculations by systematically computing free
energies of solvation for a variety of molecules containing
functional groups of different nature. The aim is to identify the
scheme that results in the most reliable free energies of solva-
tion. Due to the freedom in how to sample the ESP, we here
systematically compare the results in terms of free energy of
solvation for 13 typical drug compounds for which the atomic
charges were computed with the four earlier mentioned
methods, with and without effectively polarized charges. This
resulted in 104 computed DGhyd values. To test the procedure
for a solvent with a smaller dielectric constant, the same types
of calculations were performed in n-octanol (from now on
referred to as octanol). This makes it possible to compute
log P values which are of great interest in drug design. In
total, 367 free energies of solvation and 104 log P values were
computed and compared to experiments when allowed.
Methodology
Modeling of solutes and solvents
Thirteen well-known drug molecules were chosen for compu-
tation of free energies of solvation (Figure 1). FF parameters
for the solute and octanol were taken from GAFF[52] as this FF
has proven to give reliable results in a number of free energy
studies.[14–16,30,31] Charges for octanol were taken from a
FULL PAPER WWW.C-CHEM.ORG
188 Journal of Computational Chemistry 2013, 34, 187–197 WWW.CHEMISTRYVIEWS.COM

previous investigation[23] and this set of charges was used
throughout the whole of this investigation. Partial charges for
the solutes were determined by first optimizing the geometry
of the solutes in vacuum at a DFT level of theory with the
B3LYP exchange-correlation functional[53–56] with the 6-
311þG(d,p) basis set. Frequency analysis was conducted to
verify the nature of the stationary points found. Polarization
effects were modeled by performing single-point calculations
on the optimized geometries with the conductor-like contin-
uum model[57,58] for two solvents, water (e ¼ 78.4) and octanol
(e ¼ 9.86). Once self-consistency was obtained, the polarized
ESPs were used for charge fitting. Four methods were used for
obtaining the partial charges: CHELP, CHELPG, MK, and RESP.
For the latter, the ESP was sampled with the MK scheme. Also,
charges from vacuum calculations were computed with all ESP
methods to see the impact of the effectively polarized charges.
All QM calculations were performed with the Gaussian09 pro-
gram suite[59] and the RESP fittings were done with the Red
program developed by Dupradeau et al.[60] Default parameters
for all calculations in Gaussian09 were used. The ESPs were
sampled at the same level of theory as the geometry optimiza-
tions were performed at, since it has been established that the
B3LYP functional gives reliable charges,[61,62] especially if used
with a basis set including polarized functions.[63] It should be
mentioned that the free energies reported here do not include
the energetic cost that comes with changing a molecule’s
in vacuo equilibrium electron distribution when transferring it
to condensed phase although there have been methods pur-
posed for this.[11,47–49] To simulate water, the flexible SPC
(fSPC) model by Toukan and Rahman[64] was chosen. The fSPC
water molecule is able to slightly change its geometry in
response to its environment and has proven to reproduce the
experimental dielectric constant for bulk water[65] which is of
great importance for solvation studies. Additional simulations
have been carried out with SPC/E[66] and TIP3P[67] water mod-
els as well.
Computation of free energies
In this work, we implemented some modifications of the origi-
nal EE method[20,21] which are briefly described below. The EE
method implies a gradual insertion/deletion of the studied sol-
ute molecule into/from the solvent. The insertion parameter ais introduced which describes the degree of insertion of the
solute, so that a ¼ 1 describes solute molecule fully interact-
ing with the solvent while a ¼ 0 represents the solute com-
pletely decoupled from the solvent (that is, case a ¼ 0
describes pure solvent and the solute molecule in a gas phase
of the same volume). The insertion parameter can accept a
number of (fixed) values {ai},i ¼ 0,…, M in the range between
0 and 1. During the simulation, which can be carried out by ei-
ther MC[20] or MD[21] algorithms, attempts are made to change
the insertion parameter to another (normally, neighboring)
value, with acceptance probability
Pði ! i61Þ ¼ min 1; exp � VSsðai61Þ � VSsðaiÞ � ðgi61 � giÞkBT
� �� �
ð1Þ
where VSs(ai) is the interaction energy of the solute particle
with the solvent corresponding to the given insertion parame-
ter ai and gi are so-called balancing (weighting) factors intro-
duced with the purpose to make distribution over subensem-
bles close enough to the uniform one. During the simulation,
Figure 1. Molecules for which free energies of solvation in water and n-octanol were calculated.
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 34, 187–197 189

probabilities of different subensembles qi are defined, and free
energy difference between states with fully inserted and fully
deleted solute (excess solvation free energy) is computed by
DG ¼ �kBT lnqMq0
þ gM � g0 (2)
In previous works on computation of the solvation free
energies,[23,68] a linear scaling of solute–solvent interaction
with a was used, namely VSs(a) ¼ aVSs(1). Such a scheme has a
shortage that the repulsive core of the Lennard-Jones (LJ)
potential decreases very slowly with decrease of a, which leads
to necessity to consider very small a values. Now, we scale
interaction VSs between the solute and solvent atoms (which is
supposed to be a sum of the LJ and electrostatic terms)
according to the following:
VSsðaÞ ¼ a4VSsLJ ð1Þ þ a2VSs
el ð1Þ (3)
The rationale between a4 scaling of the LJ interaction is that
the effective core radius of the LJ potential scales then as
(a4)1/12 ¼ a1/3, and thus the effective volume of the core
(approximately proportional to the free energy of cavity forma-
tion) scales linearly with a. Scaling a2 for the electrostatic inter-
actions is chosen to switch off them faster than the repulsive
LJ interaction, to minimize problems with hydrogen atoms of
water molecules which do not have LJ potential. With scaling
of the solute–solvent interaction, described by eq. 3, it is pos-
sible to choose ai points uniformly distributed in the range
[0:1], so that reasonable acceptance probability of transitions
is maintained through the whole range of subensembles. In
the Supporting Information, this approach is validated.
Another modification of the EE procedure implemented in
this work is the automatic choice of the balancing factors giby the Wang-Landau algorithm.[69] This algorithm is typically
used for equilibration of weights in the multicanonic[70] or
entropic sampling[71] simulations. In application to the EE
technique, the Wang-Landau algorithm can be reformulated as
described below. We start simulations with zero balancing fac-
tors. After visiting a subensemble, a small increment Dgi is
subtracted from the corresponding value of the balancing fac-
tor, which decreases the probability to go to already visited
states and favors to attaining a uniform distribution. After a
certain number of steps (a sweep), when the system visited all
subensembles and passed several times between the end
points, the value of the increment is decreased twice. After a
certain number of sweeps (usually 10-12), the value of the in-
crement becomes very small, and simultaneously the profile of
balancing factors is tuned in a way providing uniform walking
in the space of subensembles. After that the equilibration
stage ends, production run with fixed balancing factors is
made yielding the solvation free energies.
Simulation details
All MD simulations were performed in the isobaric–isothermal
ensemble, NPT, where the temperature was kept constant
at 310 K and the pressure at 1 atmosphere with the
Nos�e-Hoover[72,73] thermostat (with relaxation time 30 fs) and
barostat (relaxation time 1 ps), respectively. Periodic boundary
conditions were used in every dimension. Aqueous simulation
boxes contained 500 fSPC water molecules and one solute
that was gradually inserted. Octanol simulations were per-
formed with 125 octanol molecules and 46 fSPC water mole-
cules, which corresponds to the experimental molar ratio of a
saturated octanol/water solution. Note that saturation ratio of
the considered models may differ from the experimental one.
At start, the molecules were placed in the simulation box in a
regular fashion with the centers of mass at a FCC lattice, and
restriction for absolute value for forces between the atoms
was used to smoothly remove initial overlapping contacts. The
cutoff value for forces was set at the level which is never
reached in the equilibrated system. The systems were relaxed
in a 100 ps long simulation in the canonical ensemble (NVT)
followed by a 2 ns long equilibration in the NPT ensemble.
Production runs were run for 20 ns where for the first 10 ns,
the balancing factors used in the EE scheme were equilibrated
and the remaining 10 ns were used for actually computing the
free energy of solvation. In simulations with fSPC water as well
as in octanol simulations, the double time-step method of
Tuckerman et al.[74] was used with a 0.2 fs short time step for
faster molecular motion (internal motion) and a longer time
step of 2.0 fs for the slower molecular motions. For simulations
with rigid TIP3P and SPC/E water model, the bonds were con-
strained using SHAKE algorithm with tolerance 0.0001, and
leap-frog algorithm with time step 2 fs was used. LJ forces
were computed up to a distance of 0.5 nm for the short time
step and rcut ¼ 1.2 nm for the longer time step and the neigh-
bor list was updated every 10 steps. Electrostatic interactions
were treated by the Ewald summation method with the real-
space Ewald factor 2.8/rcut and cutoff in the reciprocal space
was set from the condition that the remaining terms do not
contribute by more than 0.0001 level of the total value. The
long-range dispersion corrections to the energy and pressure
were included. Between 25 and 40 subensembles, with uni-
formly distributed a values, were used in the calculations
depending on the solute size, which provided overall accep-
tance ratio of 30–35% for transitions between the subensem-
bles. All errors reported were computed via block averaging
over the production runs with blocks of 1 ns (excluding the
initial 10 ns were the balancing factors are equilibrated). The
EEMD calculations were performed by MDynaMix v. 5.2.4 pack-
age[75] (available from www.mmk.su.se/mdynamix).
Results and Discussion
Free energies of hydration
In Table 1, the free energies of hydration are shown together
with available experimental data. The spread of DGhyd is strik-
ing and shows how sensitive these types of calculations (and
in general molecular simulations with FFs) are. Previously, the
impact of partial atomic charges has been pointed out in free
energy calculations[14,18,23,33] but not to the same extent as
reported here. The present investigation includes molecules of
FULL PAPER WWW.C-CHEM.ORG
190 Journal of Computational Chemistry 2013, 34, 187–197 WWW.CHEMISTRYVIEWS.COM

very polar nature, which could be a reason for this very pro-
nounced dependency. When polarization effects are included,
all methods systematically underestimate DGhyd with the
exception of RESP. The partial atomic charges obtained in cal-
culations where polarization was taken into account appear to
be overpolarized. It is clear that the RESP charges do this to
the smallest extent and MK to the largest, and this is to be
expected as it has previously been shown that the MK scheme
gives charges of larger magnitude when compared with other
methods.[61,76] Actually, every free energy of hydration is
underestimated by the MK charges. In Figure 2, the correlation
between experiments and simulations are shown and as can
be seen, the underestimation is very evident. Further, in the
case of the polarized charges, it is clear that MK and CHELPG
give similar results. When in vacuo charges are used DGhyd is,
as expected, systematically overestimated, in agreement with
previously reported work.[15] This shows that the polarizing
effect of the solvent is a feature that should be considered for
molecules with several polar functional groups. It is, however,
worth noting that all charge methods overestimate DGhyd to a
Table 1. Free energies of hydration with the four different charge models with effectively polarized charges (Pol.) and gas phase charges (Gas).
Compound
CHELP CHELPG MK RESP
Pol. Gas Pol. Gas Pol. Gas Pol. Gas Exp.
4-Nitroaniline �65.4 6 0.4 �27.1 6 0.3 �70.9 6 0.3 �34.2 6 0.5 �78.3 6 0.4 �37.3 6 0.5 �54.2 6 0.4 �32.7 6 0.4 �41.4 6 1.8 [77]
Aspirin �54.1 6 0.6 �29.2 6 0.5 �57.3 6 0.5 �48.6 6 0.4 �61.9 6 0.5 �36.3 6 0.4 �51.3 6 0.6 �29.6 6 0.5 �51.6 6 0.8 [77]
Caffeine �71.8 6 0.3 �40.8 6 0.4 �89.1 6 0.4 �67.9 6 0.4 �90.3 6 0.4 �73.7 6 0.5 �69.1 6 0.4 �49.7 6 0.6 �53.7 6 0.6 [77]
D-glucose �73.7 6 0.5 �68.7 6 0.7 �126.8 6 0.7 �73.2 6 0.6 �128.3 6 0.7 �89.0 6 0.5 �95.9 6 0.9 �81.1 6 0.8 �106.5 6 0.9 [77]
Diflunisal �31.4 6 0.5 �37.7 6 0.7 �33.6 6 0.4 �32.4 6 0.5 �37.5 6 0.6 �34.9 6 0.6 �39.1 6 0.7 �11.6 6 0.6 �31.0 6 1.2 [77]
Ethyl paraben �40.6 6 0.4 �19.3 6 0.4 �56.0 6 0.6 �28.0 6 0.5 �58.6 6 0.5 �29.1 6 0.4 �35.9 6 0.5 �21.2 6 0.4 �38.5 6 0.4 [77]
Glycerol �64.8 6 0.3 �40.1 6 0.3 �74.2 6 0.5 �46.0 6 0.4 �76.8 6 0.3 �48.3 6 0.4 �57.9 6 0.5 �44.0 6 0.4 �58.3 6 0.2 [77]
Uracil �116.7 6 0.4 �65.1 6 0.3 �112.0 6 0.3 �61.5 6 0.3 �110.4 6 0.3 �59.3 6 0.3 �80.7 6 0.4 �54.0 6 0.4 �67.2 6 1.2 [77]
Amrinone �95.6 6 0.4 �44.6 6 0.4 �119.0 6 0.4 �57.5 6 0.5 �126.0 6 0.3 �60.4 6 0.4 �91.9 6 0.4 �56.9 6 0.5 –
Benzyl benzoate �20.8 6 0.6 �6.9 6 0.6 �25.4 6 0.5 �7.1 6 0.5 �39.7 6 0.5 �16.8 6 0.3 �27.5 6 0.5 �16.5 6 0.6 –
Metformin �111.8 6 0.8 �73.1 6 0.7 �102.1 6 0.6 �61.9 6 0.7 �108.0 6 0.6 �85.7 6 0.6 �71.0 6 0.7 �66.3 6 0.5 –
Mesalazine �76.5 6 0.3 �41.5 6 0.4 �97.6 6 0.4 �83.3 6 0.6 �105.3 6 0.6 �90.4 6 0.5 �63.0 6 0.8 �45.6 6 0.5 –
Oxazole �27.9 6 0.3 �11.8 6 0.3 �34.0 6 0.4 �15.6 6 0.3 �37.0 6 0.4 �17.1 6 0.4 �26.4 6 0.2 �16.3 6 0.3 –
RMS error 23.7 19.4 25.4 14.4 27.3 12.3 9.73 17.0 –
All free energies are shown in the unit kJ mol�1.
Figure 2. Correlation between experimental and computed free energies of hydration with effectively polarized charges a) and the relative errors b) and
for in vacuo charges, d) and e), respectively. In c) the molecular structure of the compounds are given.
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 34, 187–197 191

smaller amount when polarization is ignored than the meth-
ods underestimate the same property when charge polariza-
tion is taken into account (with the exception of RESP). There-
fore, it is evident that the polarization contribution is
important, but including it by merely placing the molecule in
polarizable continuum and using the CHELP, CHELPG, or MK
will not bring the simulated results closer to experimental find-
ings. The method that performs the best with the dataset pre-
sented in Figure 1 is RESP together with effectively polarized
charges. The root-mean-square error (RMSE) is around 3.8 kBT
which is relatively small. On the other end of the spectrum,
MK and CHELPG with effectively polarized charges are with a
RMSE of 11 kBT and 9.8 kBT, respectively. This is in agreement
with the findings of Shivakumar et al.[18] who concluded that
GAFF/RESP performs better than GAFF/CHELPG. As it is clear
that effectively polarized charges overestimate the molecule’s
hydrophilicity and the neglect of polarization overestimates
the molecule’s hydrophobicity, a middle way would be to pre-
fer. The reason for why the RESP methods work well could be
due to error cancellation. There is a nonuniqueness problem
whenever point charges are assigned to reproduce the quan-
tum mechanically determined ESP. The reason for the RESP
method to work so well here could be due the fact that the
ESP is well reproduced simultaneously as the charges are kept
as small as possible. The other methods lack this penalty func-
tion that tries to keep the magnitudes of the charges down
and therefore give too hydrophilic molecules although they
can still reproduce the ESP. One interesting observation is that
MK charges work relatively well if no polarization is taken into
account (a RMSE of 4.6 kBT).
Compounds that appear to be difficult contain functional
groups with nitrogen, for which the molecules’ hydrophilicity
is rather overestimated. It is also for these types of compounds
that the spread in free energy is the largest. In the SAMPL2
blind test, D-glucose, caffeine, and diflunisal were pointed out
as difficult compounds.[77] As can be seen in Table 1, RESP
with polarized charges performs quite well for these com-
pounds. The largest error is for caffeine and it is likely that
high density of electronegative species in these molecules
poses a problem for all charge determining methods.
The reason for why the applied charge computation meth-
ods give such a spread in free energies of hydration is difficult
to point out. A possible expla-
nation could be how the meth-
ods sample the ESP.[76] Most
methods only explore and try
to reproduce the QM ESP at
relative short distance from the
vdW surface of the molecule
despite the long-range nature
of Coulomb interactions. This
means that the point charges
are assured only to describe
the interactions between the
solute and the solvent at a
short distance. It is not certain
that the interactions at longer
distances are represented well by these partial atomic charges.
Another interesting point to make with charges is at which
QM level of theory the ESP was computed at. Often the Har-
tree-Fock method combined with the 6-31G(d) basis set is
used despite the fact that there are several available methods
for treating electron correlation that comes at a relatively low-
computational cost. It would be along one’s intuition that the
more accurate wave function is, the more accurate the ESP is
and hence the more accurate the partial atomic charge should
be. In a rather recent investigation, Mobley et al.[14] investigate
the impact of QM level of theory and draw the conclusions
that the dipole and multipole moments vary quite substan-
tially with the applied QM method. These effects together
with the LJ parameters were considered to be possible sources
of the discrepancy between simulations and experiments.
Extended simulations and molecular conformations
D-glucose and glycerol are difficult compounds due to their
flexibility and therefore one has to sample large parts of the
configurational space of these molecules to obtain a well-
defined free energy of hydration. To test the convergence of
the computed free energies, EEMD simulations were per-
formed with different lengths: 12, 14, 16, 18, 20, and 30 ns
where the first 10 ns were used to equilibrate the balancing
factors used in the EE scheme. Three molecules were consid-
ered, caffeine, D-glucose, and glycerol. The two latter were
chosen due to their flexibility and the former due to its rigid-
ity. This allows us to see if the simulation time is long enough
to sample a more complicated configurational space by compar-
ing the convergence of the free energy of solvation to that of a
molecule with less degrees of freedom. The results are pre-
sented in Table 2. As can be seen for DGhyd, the values do not
vary significantly which indicates that the free energies of hydra-
tion converge in simulations shorter than the 20 ns that were
first proposed here. The biggest impact is seen on the statistical
uncertainty as the free energy has converged. This is in agree-
ment with the findings of Paluch et al.[30] who used the EE
method to compute free energies of hydration for amino acid
analogs. This proves that the EE method is a robust method
that can provide accurate free energies within simulations of
shorter length than other previously mentioned methods.
Table 2. Convergence of computed free energies of solvation with increasing sampling time.
Caffeine D-glucose Glycerol
Sampling
time[a] DGhyd DGoct DGhyd DGoct DGhyd DGoct
12 �69.5 6 1.2 �66.7 6 4.9 �94.1 6 0.9 –[b] �58.7 6 0.7 �48.4 6 1.6
14 �69.4 6 1.4 �65.2 6 3.3 �93.7 6 1.5 –[b] �57.5 6 0.6 �47.2 6 1.0
16 �69.4 6 1.0 �63.2 6 2.0 �96.2 6 1.1 �58.6 6 3.0 �58.5 6 0.8 �47.0 6 1.0
18 �70.0 6 1.0 �68.2 6 3.0 �96.1 6 1.1 �58.6 6 3.0 �58.6 6 0.8 �47.8 6 0.9
20 �69.1 6 0.6 �68.7 6 2.7 �95.9 6 0.9 �73.4 6 1.4 �57.9 6 0.5 �49.1 6 1.0
30 �68.7 6 0.5 �69.0 6 1.0 �96.5 6 0.7 �80.1 6 1.2 �58.1 6 0.4 �50.6 6 0.8
Exp. �53.7 6 0.2[77] �53.3[c] �106.5 6 0.2[77] �88.0[c] �58.3 6 0.2[77] �43.7[c]
[a] Including a 10 ns equilibration of the EE balancing factors. [b] Insufficient sampling. [c] Calculated from
the experimental log P.
All energies are shown in kJ mol�1 and sampling time in ns.
FULL PAPER WWW.C-CHEM.ORG
192 Journal of Computational Chemistry 2013, 34, 187–197 WWW.CHEMISTRYVIEWS.COM

Other functional groups such as carboxylic acids have been
proven to be difficult to obtain converged results for due the
tendency of the hydroxyl hydrogen to be kinetically trapped
in either a cis or trans conformation with respect to the car-
bonyl oxygen.[16,31] To make sure that sampling of all thermally
relevant points in phase space was sufficient, a number of sim-
ulations of aspirin in water were performed. By choosing dif-
ferent starting conformations (either cis or trans) for the EEMD
simulations, the convergence of this degree of freedom can be
checked although there exist more sophisticated methods.[31]
As can be seen in Figures 3a and 3b, there is a difference in
the free energy of hydration for these starting conformations,
but as the simulations are longer, this difference more or less
disappears. The torsion angle changes repeatedly between a
cis or trans conformation and as a result, the starting confor-
mation is of less importance. Therefore, it is safe to conclude
that the problem concerning ergodicity is not present in our
simulations if they are run for a sufficient amount of time. This
is somewhat different from earlier finding in the literature
where the free energy barrier to cross between these confor-
mations was too high for any simulation on a ns-timescale to
pass.[16] The reason for this is probably the difference in stud-
ied systems. Klimovich and Mobley looked an isolated carbox-
ylic acid where in our investigations all the carboxylic groups
are in the vicinity of another polar group, making intramolecu-
lar hydrogen bonds possible. As a consequence of this, direct
comparisons are somewhat difficult.
The potential presence of intermolecular hydrogen bonds
further complicates the charge calculations. While computing
charges from molecular structures that have been optimized
in gas phase, it is very likely that the charges are derived from
a molecular conformation that is not representative of the
structure in condensed phase. In the case of aspirin, there is a
possibility of an intramolecular hydrogen bond, see Figure 3d.
In an aqueous solution, this hydrogen bond is very likely to be
broken in order for aspirin to form more hydrogen bonds with
the surrounding water. This poses a problem as it is not
always easy to beforehand choose the correct molecular ge-
ometry to use for computing the charges with. Multiple con-
formations can be used, but it is possible that conformations
that are not relevant for the condensed phase are included
then. As can be seen in Figures 3c and 3d, the charges
between the two geometries do not deviate significantly from
each other, yet the difference in DGhyd is around 12 kJ mol�1.
Our simulations dismiss the intramolecular hydrogen bond
when aspirin is dissolved in water (data not shown) and
indeed the charges obtained from the geometry without this
interaction provide free energies of hydration in excellent
agreement with experimental findings. An important conclu-
sion from this is that great caution not only has to be applied
when choosing the method to obtain the partial atomic
charges but also when choosing from which molecular geom-
etry to compute them from.
Sampling issues are more evident for solvation in octanol.
For caffeine, DGoct converges to a value of roughly �69 kJ
mol�1 but for the other two compounds, especially D-glucose,
the sampling time has an impact on the free energy of solva-
tion. D-glucose has many internal degrees of freedom and as
the dynamics in the octanol system is slower compared to the
aqueous system, the sampling of phase space is not very effi-
cient. When the production of EEMD simulations for D-glucose
Figure 3. Free energy of hydration for aspirin as a function of simulation
time with different starting conformations and charges. In a) the charges
are taken from c) and in b) the charges are taken from d). cis and trans
correspond to different initial positions of the hydrogen of the hydroxyl
group relative the carbonyl oxygen. For all calculations, an initial 10 ns pe-
riod of equilibration of the balancing factors is included.
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 34, 187–197 193

was prolonged even further than presented in Table 2, namely
to 40, 60, and 80 ns DGoct did not change, �79.2 6 1.2,
�79.17 6 1.0, and �79.8 6 0.9 kJ mol�1, respectively. Caf-
feine, on the other hand, is a rigid molecule with less internal
degrees of freedom and DGoct does not change drastically
between the different simulations. To ensure proper sampling
of the systems, dynamic properties were computed. The orien-
tational correlation time of octanol’s dipole was determined to
roughly 107 ps and the diffusion coefficient for the center of
mass of octanol in the octanol/water mixture was 0.081 �10�5cm2 s�1. From these results, we can conclude that sam-
pling in octanol is slow but not insufficient as the octanol mol-
ecules have time to decorrelate and diffuse. However, longer
simulations are to prefer as shown by the D-glucose simula-
tions for molecules with greater internal degrees of freedom.
Impact of solvent model
The impact of water model can affect the free energy of
hydration to large extents as noted by Shirts and Pande[78]
and Hess and van der Vegt.[11] To quantify this dependency in
the current manuscript, free energies of hydration were com-
puted for caffeine, glucose, glycerol, 4-nitroaniline, and ethyl
paraben using two popular water models, namely TIP3P and
SPC/E. For these simulations, all bonds were constrained and a
single time step of 2 fs was used. Other simulation parameters
were identical to those used for the fSPC calculations. The
results are presented in Table 3. As can be seen, the fSPC
water model results in a lower free energy of hydration in ev-
ery case except for ethyl paraben, which is the least hydro-
philic molecule of the this test. The possibility for the fPSC
water model to change its dipole moment is most likely the
reason for this. Due to fact that the fSPC water molecule can
alter its dipole moment, polarization effects are to a certain
extent taken into account.
In comparison with the values published by Klimovich and
Mobley[16] for caffeine, our values are consistently lower for the
polarized charge distributions and higher for the gas phase
charges. When comparing the values for glycerol in Table 3 to
the previously mentioned investigation, it is clear that the mole-
cule is more hydrophilic in the present investigation. The same
can be said for D-glucose. For the two latter compounds, the use
of effectively polarized charges brings the computed DGhyd val-
ues closer to the experiments and shows that inclusion of polar-
ization is important (as the charges used in ref. 16 were taken
from gas phase and the corresponding DGhyd were too high).
The differences in DGhyd are somewhat larger than observed
before by Shirts and Pande[78] and Hess and van der Vegt,[11]
but our drug molecules differ from the amino acid analogs
they studied. Furthermore, the strong dependency on the par-
tial atomic charges is shown by all water models which shows
that the conclusions previously drawn are valid, although the
trend is most evident with the fSPC model.
SAMPL2 blind test dataset
As the initial dataset was not of considerable size, we
extended our investigation by computing DGhyd for a larger
set of molecules, namely the one introduced in the SAMPL2
blind test.[77] It should be mentioned that one molecule,
5-iodouracil, was left out of the these simulations. Charges for
this set of molecules were computed with the methods
regarded to be the best from the previous section, that is, the
RESP method with effectively polarized charges. As the mole-
cules present in the SAMPL2 blind test are more heterogenous
and contain more functional groups this would provide more
insight to whether the conclusions drawn from the earlier,
Table 3. Impact of water model on the free energy of hydration.
Water model
Charges fSPC TIP3P SPC/E
Caffeine
CHELP �40.8 6 0.4 �41.1 6 0.5 �42.1 6 0.4
CHELPp �71.8 6 0.3 �65.3 6 0.4 �66.7 6 0.4
CHELPG �67.9 6 0.4 �67.7 6 0.5 �68.1 6 0.5
CHELPGp �89.1 6 0.4 �79.9 6 0.4 �81.9 6 0.5
MK �73.7 6 0.5 �66.1 6 0.4 �67.7 6 0.5
MKp �90.3 6 0.4 �81.5 6 0.5 �84.1 6 0.5
RESP �49.7 6 0.6 �47.6 6 0.5 �48.8 6 06
RESPp �69.1 6 0.4 �64.9 6 0.5 �65.6 6 0.3
Experiment �53.7 6 0.6[77]
D-glucose
CHELP �68.7 6 0.7 �64.2 6 0.6 �65.4 6 0.6
CHELPp �73.7 6 0.5 �69.7 6 0.6 �71.1 6 0.6
CHELPG �73.2 6 0.6 �69.0 6 0.6 �68.1 6 0.7
CHELPGp �126.8 6 0.7 �113.3 6 0.6 �119.4 6 0.8
MK �89.0 6 0.5 �82.4 6 0.7 �84.5 6 0.6
MKp �128.3 6 0.7 �115.8 6 0.8 �119.5 6 0.7
RESP �81.1 6 0.8 �76.7 6 0.6 �77.9 6 0.7
RESPp �95.9 6 0.9 �86.0 6 0.6 �88.1 6 0.7
Experiment �106.5 6 0.9[77]
Glycerol
CHELP �40.1 6 0.3 �39.1 6 0.4 �37.9 6 0.4
CHELPp �64.8 6 0.3 �58.8 6 0.3 �60.2 6 0.5
CHELPG �46.0 6 0.4 �44.1 6 0.5 �45.3 6 0.5
CHELPGp �74.2 6 0.5 �67.2 6 0.6 �68.4 6 0.4
MK �48.3 6 0.4 �45.0 6 0.3 �44.7 6 0.5
MKp �76.8 6 0.3 �58.8 6 0.5 �62.3 6 0.6
RESP �44.0 6 0.4 �42.3 6 0.6 �43.1 6 0.5
RESPp �57.9 6 0.5 �50.6 6 0.4 �52.2 6 0.5
Experiment �58.3 6 0.2[77]
4-nitroaniline
CHELP �27.1 6 0.3 �30.4 6 0.4 �28.1 6 0.3
CHELPp �65.4 6 0.4 �64.2 6 0.3 �62.1 6 0.5
CHELPG �34.2 6 0.5 �57.5 6 0.4 �54.8 6 0.3
CHELPGp �70.9 6 0.3 �72.6 6 0.4 �70.6 6 0.4
MK �37.3 6 0.5 �39.0 6 0.5 �36.4 6 0.4
MKp �78.3 6 0.4 �75.3 6 0.4 �73.0 6 0.5
RESP �32.7 6 0.4 �36.1 6 0.3 �33.0 6 0.5
RESPp �54.2 6 0.4 �57.5 6 0.5 �54.8 6 0.4
Experiment �41.4 6 1.8[77]
Ethyl paraben
CHELP �19.3 6 0.4 �21.8 6 0.5 �22.1 6 0.3
CHELPp �40.6 6 0.4 �41.0 6 0.4 �41.7 6 0.5
CHELPG �28.0 6 0.5 �29.4 6 0.4 �28.7 6 0.6
CHELPGp �56.0 6 0.6 �55.4 6 0.5 �57.0 6 0.4
MK �29.1 6 0.5 �32.1 6 0.7 �31.5 6 0.4
MKp �58.6 6 0.5 �61.1 6 0.4 �63.1 6 0.4
RESP �21.2 6 0.4 �24.2 6 0.5 �24.9 6 0.4
RESPp �35.9 6 0.5 �37.8 6 0.5 �36.8 6 0.6
Experiment �38.5 6 0.4[77]
All values are reported in kJ mol�1 and the p-superscript indicates that
the charges were effectively polarized as described in the Methods sec-
tion. Charges with no superscript are from gas phase.
FULL PAPER WWW.C-CHEM.ORG
194 Journal of Computational Chemistry 2013, 34, 187–197 WWW.CHEMISTRYVIEWS.COM
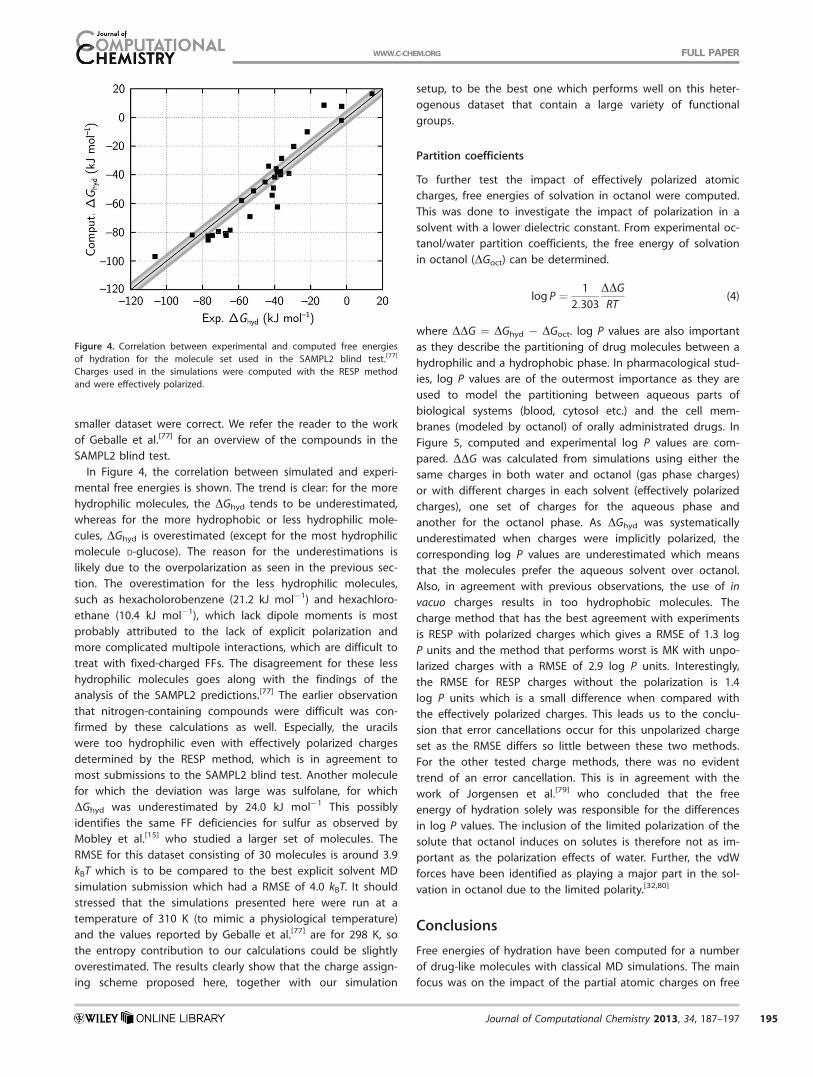
smaller dataset were correct. We refer the reader to the work
of Geballe et al.[77] for an overview of the compounds in the
SAMPL2 blind test.
In Figure 4, the correlation between simulated and experi-
mental free energies is shown. The trend is clear: for the more
hydrophilic molecules, the DGhyd tends to be underestimated,
whereas for the more hydrophobic or less hydrophilic mole-
cules, DGhyd is overestimated (except for the most hydrophilic
molecule D-glucose). The reason for the underestimations is
likely due to the overpolarization as seen in the previous sec-
tion. The overestimation for the less hydrophilic molecules,
such as hexacholorobenzene (21.2 kJ mol�1) and hexachloro-
ethane (10.4 kJ mol�1), which lack dipole moments is most
probably attributed to the lack of explicit polarization and
more complicated multipole interactions, which are difficult to
treat with fixed-charged FFs. The disagreement for these less
hydrophilic molecules goes along with the findings of the
analysis of the SAMPL2 predictions.[77] The earlier observation
that nitrogen-containing compounds were difficult was con-
firmed by these calculations as well. Especially, the uracils
were too hydrophilic even with effectively polarized charges
determined by the RESP method, which is in agreement to
most submissions to the SAMPL2 blind test. Another molecule
for which the deviation was large was sulfolane, for which
DGhyd was underestimated by 24.0 kJ mol�1 This possibly
identifies the same FF deficiencies for sulfur as observed by
Mobley et al.[15] who studied a larger set of molecules. The
RMSE for this dataset consisting of 30 molecules is around 3.9
kBT which is to be compared to the best explicit solvent MD
simulation submission which had a RMSE of 4.0 kBT. It should
stressed that the simulations presented here were run at a
temperature of 310 K (to mimic a physiological temperature)
and the values reported by Geballe et al.[77] are for 298 K, so
the entropy contribution to our calculations could be slightly
overestimated. The results clearly show that the charge assign-
ing scheme proposed here, together with our simulation
setup, to be the best one which performs well on this heter-
ogenous dataset that contain a large variety of functional
groups.
Partition coefficients
To further test the impact of effectively polarized atomic
charges, free energies of solvation in octanol were computed.
This was done to investigate the impact of polarization in a
solvent with a lower dielectric constant. From experimental oc-
tanol/water partition coefficients, the free energy of solvation
in octanol (DGoct) can be determined.
log P ¼ 1
2:303
DDGRT
(4)
where DDG ¼ DGhyd � DGoct. log P values are also important
as they describe the partitioning of drug molecules between a
hydrophilic and a hydrophobic phase. In pharmacological stud-
ies, log P values are of the outermost importance as they are
used to model the partitioning between aqueous parts of
biological systems (blood, cytosol etc.) and the cell mem-
branes (modeled by octanol) of orally administrated drugs. In
Figure 5, computed and experimental log P values are com-
pared. DDG was calculated from simulations using either the
same charges in both water and octanol (gas phase charges)
or with different charges in each solvent (effectively polarized
charges), one set of charges for the aqueous phase and
another for the octanol phase. As DGhyd was systematically
underestimated when charges were implicitly polarized, the
corresponding log P values are underestimated which means
that the molecules prefer the aqueous solvent over octanol.
Also, in agreement with previous observations, the use of in
vacuo charges results in too hydrophobic molecules. The
charge method that has the best agreement with experiments
is RESP with polarized charges which gives a RMSE of 1.3 log
P units and the method that performs worst is MK with unpo-
larized charges with a RMSE of 2.9 log P units. Interestingly,
the RMSE for RESP charges without the polarization is 1.4
log P units which is a small difference when compared with
the effectively polarized charges. This leads us to the conclu-
sion that error cancellations occur for this unpolarized charge
set as the RMSE differs so little between these two methods.
For the other tested charge methods, there was no evident
trend of an error cancellation. This is in agreement with the
work of Jorgensen et al.[79] who concluded that the free
energy of hydration solely was responsible for the differences
in log P values. The inclusion of the limited polarization of the
solute that octanol induces on solutes is therefore not as im-
portant as the polarization effects of water. Further, the vdW
forces have been identified as playing a major part in the sol-
vation in octanol due to the limited polarity.[32,80]
Conclusions
Free energies of hydration have been computed for a number
of drug-like molecules with classical MD simulations. The main
focus was on the impact of the partial atomic charges on free
Figure 4. Correlation between experimental and computed free energies
of hydration for the molecule set used in the SAMPL2 blind test.[77]
Charges used in the simulations were computed with the RESP method
and were effectively polarized.
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 34, 187–197 195

energy calculations. As it is well-known that solvent can have
a pronounced polarization effect on the electron distribution
of solutes, atomic charges were obtained by performing QM
calculations in polarizable continuum and then minimizing the
difference between the QM ESP and the one obtained from
point charges. These lead to effectively polarized charges that
performed better than in vacuo charges when the RESP
method was applied. To further test this conclusion, the free
energies of hydration for 30 molecules of the SAMPL2 dataset
were computed which showed in a small deviation when com-
pared with experimental data. The polarization effects of less
polar solvent like octanol were also tested by computing log P
values and again the RESP method with effectively polarized
charges performed the best.
Some important conclusions can be drawn based on the
data presented here. First, the results presented here suggest
that RESP charges at the B3LYP/6-311þG(d,p) level of theory
with polarization effects taken into account perform well for
computing free energies of solvation together with the simula-
tion setup used here and use of fSPC water model as a sol-
vent. It does not seem unlikely that this is due to the hyper-
bolic penalty function in RESP which assures that the charges
are kept as small as possible in magnitude while they simulta-
neously reproduce the ESP. Most importantly, the impact of
partial atomic charges on simulations based on classical point-
charge FFs has been verified. The enormous spread in DGhyd
illustrates that atomic charges are not a set of parameters to
take lightly. They can affect the results of molecular simula-
tions significantly as there is no ‘‘black-box’’ method available
to obtain reliable charges, great caution has to be applied.
Acknowledgments
The authors are grateful to PDC Center for High Performance Com-
puting, Stockholm and also High Performance Computing Center
North (HPC2N), Umea for kindly supplying allocated computer time.
A.L wants to thank the Sardinian Visiting Professors program at the
University of Cagliari, Italy. F.M. acknowledges the Wenner-Gren
Foundation for funding for Visiting Professorship. The authors
would also like to thank the referees for many helpful comments.
Keywords: molecular dynamics � free energy � computational
drug design � expanded ensemble � atomic charges
How to cite this article: J. P. M. J€ambeckmbeck, F. Mocci, A. P.
Lyubartsev, A. Laaksonen, J. Comput. Chem. 2013, 34, 187–197.
DOI: 10.1002/jcc.23117
Additional Supporting Information may be found in the
online version of this article
[1] J. D. Chodera, D. L. Mobley, M. R. Shirts, R. W. Dixon, K. Branson, V. S.
Pande, Curr. Opin. Struct. Biol. 2011, 21, 150.
[2] P. Kollman, Chem. Rev. 1993, 93, 2395.
[3] C. Chipot, D. A. Pearlman, Mol. Simul. 2002, 28, 1.
[4] M. R. Shirts, D. L. Mobley, J. D. Chodera, Annu. Rep. Comput. Chem.
2007, 3, 41.
[5] J. Westergren, L. Lindfors, T. H€oglund, K. Luder, S. Nordholm, R. Kjel-
lander, J. Phys. Chem. B 2007, 111, 1872.
[6] A. Leo, C. Hansch, D. Elkins, Chem. Rev. 1971, 71, 525.
[7] I. V. Tetko, V. Y. Tanchuk, A. E. Mark, J. Chem. Inform. Comput. Sci.
2001, 41, 1407.
[8] C. D. Christ, A. E. Mark, W. F. van Gunsteren, J. Comput. Chem. 2009,
31, 1569.
[9] J. G. Kirkwood, J. Chem. Phys. 1935, 3, 300.
[10] M. R. Shirts, J. W. Pitera, W. C. Swope, V. S. Pande, J. Chem. Phys. 2003,
119, 5740.
[11] B. Hess, N. F. A. van der Vegt, J. Phys. Chem. B 2006, 110, 17616.
[12] N. M. Garrido, A. J. Queimada, M. Jorge, E. A. Macedo, I. G. Economou,
J. Chem. Theory Comput. 2009, 5, 2346.
[13] C. H. Bennett, J. Comput. Phys. 1976, 22, 245.
[14] D. L. Mobley, E. Dumont, J. D. Chodera, K. A. Dill, J. Phys. Chem. B
2007, 111, 2242.
[15] D. L. Mobley, C. I. Bayly, M. D. Cooper, M. R. Shirts, K. A. Dill, J. Chem.
Theory Comput. 2009, 5, 350.
[16] P. V. Klimovich, D. L. Mobley, J. Comput. Aided Mol. Des. 2010, 24, 307.
[17] R. W. Zwanzig, J. Chem. Phys. 1954, 22, 1420.
[18] D. Shivakumar, Y. Deng, B. Roux, J. Chem. Theory Comput. 2009, 5, 919.
[19] D. Shivakumar, J. Williams, Y. Wu, W. Damm, J. Shelley, W. Sherman,
J. Chem. Theory Comput. 2010, 6, 1509.
[20] A. P. Lyubartsev, A. A. Martsinovski, S. Shekunov, P. N. Vorontsov-
Velyaimnov, J. Chem. Phys. 1992, 96, 1776.
[21] A. P. Lyubartsev, A. Laaksonen, P. N. Vorontsov-Velyaimnov, Mol. Phys.
1994, 82, 455.
Figure 5. Correlation between experimental and computed log p values.
Experimental log p values are taken from the PHYSPROP database.[81]
FULL PAPER WWW.C-CHEM.ORG
196 Journal of Computational Chemistry 2013, 34, 187–197 WWW.CHEMISTRYVIEWS.COM

[22] A. P. Lyubartsev, A. Laaksonen, P. N. Vorontsov-Velyaimnov, Mol. Simul.
1996, 18, 43.
[23] A. P. Lyubartsev, S. P. Jacobsson, G. Sundholm, A. Laaksonen, J. Phys.
Chem. B 2001, 105, 7775.
[24] K. M. Aberg, A. P. Lyubartsev, S. P. Jacobsson, A. Laaksonen, J. Chem.
Phys. 2004, 120, 3770.
[25] E. C. Cichowski, T. R. Schmidt, J. R. Errington, Fluid Phase Equilib. 2005,
236, 58.
[26] J. K. Shah, E. J. Maginn, J. Phys. Chem. B 2005, 109, 10395.
[27] F. J. Martinez-Veracoechea, F. A. Escobedo, J. Phys. Chem. B 2008, 112,
8120.
[28] J. Chang, J. Chem. Phys. 2009, 131, 074103.
[29] A. S. Paluch, S. Jayaraman, J. K. Shah, E. J. Maginn, J. Chem. Phys.
2010, 133, 124504.
[30] A. S. Paluch, J. K. Shah, E. J. Maginn, J. Chem. Theory Comput. 2011, 7,
1394.
[31] A. S. Paluch, D. L. Mobley, E. J. Maginn, J. Chem. Theory Comput. 2011,
7, 2910.
[32] E. M. Duffy, W. L. Jorgensen, J. Am. Chem. Soc. 2000, 122, 2878.
[33] Y. Deng, B. Roux, J. Phys. Chem. B 2004, 108, 16567.
[34] U. C. Singh, P. A. Kollman, J. Comput. Chem. 1984, 5, 129.
[35] L. E. Chirlian, M. M. Francl, J. Comput. Chem. 1987, 8, 894.
[36] C. M. Breneman, K. B. Wiberg, J. Comput. Chem. 1990, 11, 361.
[37] B. H. Besler, K. M. Merz, P. A. Kollman, J. Comput. Chem. 1990, 11, 431.
[38] M. M. Francl, C. Carey, L. E. Chirlian, D. M. Gange, J. Comput. Chem.
1996, 17, 367.
[39] M. M. Francl, L. E. Chirlian, Rev. Comput. Chem. 2000, 14, 1.
[40] C. A. Reynolds, J. W. Essex, W. G. Richards, J. Am. Chem. Soc. 1992,
114, 9075.
[41] W. D. Cornell, P. Cieplak, C. I. Bayly, P. A. Kollman, J. Am. Chem. Soc.
1993, 115, 9620.
[42] M. Basma, S. Sundara, D. Calgan, T. Vernalli, R. J. Woods, J. Comput.
Chem. 2001, 22, 1125.
[43] J. W. Essex, C. A. Reynolds, W. G. Richards, J. Am. Chem. Soc. 1992,
114, 3634.
[44] C. A. Reynolds, J. W. Essex, W. G. Richards, Chem. Phys. Lett. 1992, 199,
257.
[45] S. G. Lister, C. A. Reynolds, W. G. Richards, Int. J. Quantum Chem. 1992,
41, 293.
[46] C. I. Bayly, P. Cieplak, W. D. Cornell, P. A. Kollman, J. Phys. Chem. 1993,
97, 10269.
[47] W. C. Swope, H. W. Horn, J. E. Rice, J. Phys. Chem. B 2010, 114, 8621.
[48] W. C. Swope, H. W. Horn, J. E. Rice, J. Phys. Chem. B 2010, 114, 8631.
[49] C. Chipot, J. Comput. Chem. 2002, 24, 409.
[50] D. L. Mobley, E. Dumont, J. D. Chodera, K. A. Dill, J. Phys. Chem. B
2011, 115, 1329.
[51] D. L. Mobley, S. Liu, D. S. Cerutti, W. C. Swope, J. E. Rice, J. Comput.
Aided Mol. Des. 2012, 26, 551.
[52] J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman, D. A. Case, J. Comput.
Chem. 2004, 25, 1157.
[53] S. H. Vosko, R. G. Wilk, M. Nusair, Can. J. Phys. 1980, 58, 1200.
[54] C. Lee, W. Yang, R. G. Parr, Phys. Rev. B 1988, 37, 785.
[55] A. D. Becke, J. Chem. Phys. 1993, 98, 5648.
[56] P. J. Stephens, F. J. Devlin, C. F. Chabalowski, M. J. Frisch, J. Chem.
Phys. 1995, 98, 11623.
[57] V. Barone, M. Cossi, J. Tomasi, J. Comput. Chem. 1998, 19, 404.
[58] M. Cossi, N. Rega, G. Scalmani, V. Barone, J. Comput. Chem. 2003, 24,
669.
[59] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J.
R. Cheeseman, G. Scalmani, V. Barone, B. Mennuci, G. A. Peterson, H.
Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmayov, J. Bloino, G.
Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J.
Hasegawa, M. Ishilda, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T.
Montgomery Vreven, J. A. Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J.
Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Nor-
mand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi,
M. Cossi, N. Rega, J. M. Milliam, M. Klene, J. E. Knox, J. B. Cross, V.
Bakken, C. Adamo, J. Jaramillo, R. gomperts, R. E. Stratmann, O. Yazyev,
A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Moro-
kuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S.
Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslow-
ski, D. J. Fox, Gaussian09, Revision A.02, Gaussian Inc., Wallingford CT,
2009.
[60] F. Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel,
D. Lelong, W. Rosanski, P. Cieplak, Phys. Chem. Chem. Phys. 2010, 12,
7821.
[61] G. S. Maciel, E. Garcia, Chem. Phys. Lett. 2005, 409, 29.
[62] F. De Proft, J. M. L. Martin, P. Geerlings, Chem. Phys. Lett. 1996, 250,
393.
[63] S. Tsuzuki, T. Uchimaru, K. Tanabe, A. Yliniemela, J. Mol. Struct. (Theo-
chem) 1996, 365, 81.
[64] K. Toukan, A. Rahman, Phys. Rev. B 1985, 31, 2643.
[65] A. M. Nikitin, A. P. Lyubartsev, J. Comput. Chem. 2007, 28, 2020.
[66] H. J. C. Berendsen, J. R. Grigera, T. P. Straatsma, J. Chem. Phys. 1987,
91, 6269.
[67] W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey, M. L.
Klein, J. Chem. Phys. 1983, 79, 926.
[68] A. P. Lyubartsev, O. K. Forrisdahl, A. Laaksonen, J. Chem. Phys. 1998,
108, 227.
[69] F. Wang, D. P. Landau, Phys. Rev. Lett. 2001, 86, 2050.
[70] B. A. Berg, T. Neuhaus, Phys. Rev. Lett. 1992, 68, 9.
[71] J. Lee, Phys. Rev. Lett. 1993, 71, 211.
[72] S. Nos�e, Mol. Phys. 1984, 52, 255.
[73] W. G. Hoover, Phys. Rev. A 1985, 31, 1695.
[74] M. E. Tuckerman, B. J. Berne, G. J. Martyna, J. Chem. Phys. 1992, 97,
1990.
[75] A. P. Lyubartsev, A. Laaksonen, Comput. Phys. Commun. 2000, 128, 565.
[76] E. Sigfridsson, U. Ryde, J. Comput. Chem. 1998, 19, 377.
[77] M. T. Geballe, A. G. Skillman, A. Nicholls, J. P. Guthrie, P. J. Taylor,
J. Comput. Aided Mol. Des. 2010, 24, 259.
[78] M. R. Shirts, V. S. Pande, J. Chem. Phys. 2005, 122, 134508.
[79] W. L. Jorgensen, J. M. Briggs, M. L. Contreras, J. Phys. Chem. 1990, 94,
1683.
[80] P. Cieplak, J. Caldwell, P. Kollman, J. Comput. Chem. 2001, 22, 1048.
[81] PHYSPROP, Physical/Chemical Property Database; Syracuse Research
Corporation, SRC Environmental Research Centre: New York, 1994.
Received: 12 March 2012Revised: 23 August 2012Accepted: 24 August 2012Published online on 20 September 2012
FULL PAPERWWW.C-CHEM.ORG
Journal of Computational Chemistry 2013, 34, 187–197 197
Related Documents











