HEART FAILURE Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction Sarah M. Schumacher, 1,2 Erhe Gao, 1 Weizhong Zhu, 3 Xiongwen Chen, 3 J. Kurt Chuprun, 1,2 Arthur M. Feldman, 3 John J. G. Tesmer, 4 Walter J. Koch 1,2 * Heart failure (HF) is a disease of epidemic proportion and is associated with exceedingly high health care costs. G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor (GPCR) kinase 2 (GRK2), which is up-regulated in the failing human heart, appears to play a critical role in HF progression in part because enhanced GRK2 activity promotes dysfunctional adrenergic signaling and myocyte death. Recently, we found that the selec- tive serotonin reuptake inhibitor (SSRI) paroxetine could inhibit GRK2 with selectivity over other GRKs. Wild-type mice were treated for 4 weeks with paroxetine starting at 2 weeks after myocardial infarction (MI). These mice were compared with mice treated with fluoxetine, which does not inhibit GRK2, to control for the SSRI effects of paroxetine. All mice exhibited similar left ventricular (LV) dysfunction before treatment; however, although the con- trol and fluoxetine groups had continued degradation of function, the paroxetine group had considerably improved LV function and structure, and several hallmarks of HF were either inhibited or reversed. Use of genetically engi- neered mice indicated that paroxetine was working through GRK2 inhibition. The beneficial effects of paroxetine were markedly greater than those of b-blocker therapy, a current standard of care in human HF. These data dem- onstrate that paroxetine-mediated inhibition of GRK2 improves cardiac function after MI and represents a potential repurposing of this drug, as well as a starting point for innovative small-molecule GRK2 inhibitor development. INTRODUCTION With about 550,000 new cases of heart failure (HF) diagnosed in the United States alone each year, this disease represents a growing health care concern (1). Despite substantial improvements in its manage- ment, including improved mechanical and pharmacological therapy, outcomes in HF remain poor (1). Thus, there is an urgent need to de- velop new therapeutic strategies, including cell- and gene-based thera- pies, and recent research has been aimed at the underlying mechanisms of HF progression. A main driving force in the pathophysiology of HF is an increased sympathetic drive, which occurs to stimulate failing pump function. Although elevated norepinephrine causes an initial compensatory in- crease in heart rate (HR) and cardiac output, prolonged sympathetic nervous system (SNS) activation participates in the progressive, mal- adaptive changes characteristic of HF (2, 3). One mechanism by which increased circulating catecholamine levels contribute to HF progression is through dysregulation of GPCR [G protein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor] function in the heart. Enhanced stimulation of b-adrenergic receptors (bARs) followed by increased activation of GPCR kinases (GRKs) leads to enhanced phos- phorylation, internalization, and down-regulation of bAR density and signaling, leading to a loss in inotropic reserve (4, 5). In particular, in failing human hearts, the levels and activity of GRK2 are elevated (6, 7). Increased GRK2 has been shown to participate in adverse remodeling and contractile dysfunction during HF, whereas GRK2 inhibition through a C-terminal peptide that competes with GRK2 binding to Gbg (bARKct) enhances heart function and can prevent and reverse HF (8–14). Fur- ther, GRK2 is a pro-death kinase in the heart, inhibiting vital cell sur- vival pathways and promoting apoptosis after cardiac injury (15–17). These data present compelling evidence of a causal role for GRK2 in the maladaptive progression of cardiac remodeling and dysfunction leading to HF, especially after ischemic injury. Therefore, the develop- ment of small-molecule inhibitors of GRK2 appears warranted for pharmacologic treatment of HF. Recently, we discovered that the selective serotonin reuptake inhib- itor (SSRI) antidepressant drug paroxetine specifically bound to the catalytic domain of GRK2 as an off-target and inhibited kinase activity in the micromolar range of affinity (18). Further, paroxetine could in- hibit GRK2 with selectivity over other GRK subfamilies (18). Moderate concentrations of paroxetine inhibited GRK2 target phosphorylation in vitro and significantly potentiated the bAR-mediated increase in myocardial contractility in vitro and in vivo after isoproterenol (ISO) administration (18). Here, we directly investigated whether paroxetine- mediated inhibition of GRK2 could improve cardiovascular signaling and function in a mouse model of HF. RESULTS Chronic paroxetine treatment improves cardiac function after myocardial infarction To determine whether pharmacologic inhibition of GRK2 by paroxetine could provide improvement in cardiac function in an animal model of HF, wild-type C57BL/6 mice underwent myocardial infarction (MI) or sham surgery (19) and were allowed 2 weeks for infarct develop- ment and HF progression before 4 weeks of treatment with vehicle [dimethyl sulfoxide (DMSO) in water], paroxetine, or fluoxetine (both at 5 mg/kg per day) through subcutaneous miniosmotic pumps. These doses were determined on the basis of a literature search of murine studies investigating their SSRI effects, in which doses ranged from 1 to 10 mg/kg. Using this treatment protocol, we found that the serum paroxetine levels ranged between 27 and 192 ng/ml after 4 weeks of 1 Center for Translational Medicine, Temple University School of Medicine, Philadelphia, PA 19140, USA. 2 Department of Pharmacology, Temple University School of Medicine, Philadelphia, PA 19140, USA. 3 Cardiovascular Research Center, Temple University School of Medicine, Philadelphia, PA 19140, USA. 4 Life Sciences Institute, University of Michigan, Ann Arbor, MI 48109, USA. *Corresponding author. E-mail: [email protected] RESEARCH ARTICLE www.ScienceTranslationalMedicine.org 4 March 2015 Vol 7 Issue 277 277ra31 1 on July 24, 2015 Downloaded from

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

R E S EARCH ART I C L E
HEART FA I LURE
Paroxetine-mediated GRK2 inhibition reverses cardiacdysfunction and remodeling after myocardial infarctionSarah M. Schumacher,1,2 Erhe Gao,1 Weizhong Zhu,3 Xiongwen Chen,3 J. Kurt Chuprun,1,2
Arthur M. Feldman,3 John J. G. Tesmer,4 Walter J. Koch1,2*
15
Heart failure (HF) is a disease of epidemic proportion and is associated with exceedingly high health care costs. Gprotein (heterotrimeric guanine nucleotide–binding protein)–coupled receptor (GPCR) kinase 2 (GRK2), which isup-regulated in the failing human heart, appears to play a critical role in HF progression in part because enhancedGRK2 activity promotes dysfunctional adrenergic signaling andmyocyte death. Recently, we found that the selec-tive serotonin reuptake inhibitor (SSRI) paroxetine could inhibit GRK2 with selectivity over other GRKs. Wild-typemice were treated for 4 weeks with paroxetine starting at 2 weeks after myocardial infarction (MI). These micewere compared with mice treated with fluoxetine, which does not inhibit GRK2, to control for the SSRI effects ofparoxetine. All mice exhibited similar left ventricular (LV) dysfunction before treatment; however, although the con-trol and fluoxetine groups had continued degradation of function, the paroxetine group had considerably improvedLV function and structure, and several hallmarks of HF were either inhibited or reversed. Use of genetically engi-neered mice indicated that paroxetine was working through GRK2 inhibition. The beneficial effects of paroxetinewere markedly greater than those of b-blocker therapy, a current standard of care in human HF. These data dem-onstrate that paroxetine-mediated inhibition of GRK2 improves cardiac function after MI and represents a potentialrepurposing of this drug, as well as a starting point for innovative small-molecule GRK2 inhibitor development.
20
onJul
y 24
, D
ownl
oade
d fr
om
INTRODUCTION
With about 550,000 new cases of heart failure (HF) diagnosed in theUnited States alone each year, this disease represents a growing healthcare concern (1). Despite substantial improvements in its manage-ment, including improved mechanical and pharmacological therapy,outcomes in HF remain poor (1). Thus, there is an urgent need to de-velop new therapeutic strategies, including cell- and gene-based thera-pies, and recent research has been aimed at the underlying mechanismsof HF progression.
A main driving force in the pathophysiology of HF is an increasedsympathetic drive, which occurs to stimulate failing pump function.Although elevated norepinephrine causes an initial compensatory in-crease in heart rate (HR) and cardiac output, prolonged sympatheticnervous system (SNS) activation participates in the progressive, mal-adaptive changes characteristic ofHF (2, 3). Onemechanism bywhichincreased circulating catecholamine levels contribute toHF progressionis through dysregulation of GPCR [G protein (heterotrimeric guaninenucleotide–binding protein)–coupled receptor] function in the heart.Enhanced stimulation of b-adrenergic receptors (bARs) followed byincreased activation of GPCR kinases (GRKs) leads to enhanced phos-phorylation, internalization, and down-regulation of bAR density andsignaling, leading to a loss in inotropic reserve (4, 5). In particular, infailing human hearts, the levels and activity of GRK2 are elevated (6, 7).Increased GRK2 has been shown to participate in adverse remodelingand contractile dysfunction duringHF, whereasGRK2 inhibition throughaC-terminal peptide that competes withGRK2 binding toGbg (bARKct)enhances heart function and can prevent and reverse HF (8–14). Fur-ther, GRK2 is a pro-death kinase in the heart, inhibiting vital cell sur-
1Center for Translational Medicine, Temple University School of Medicine, Philadelphia, PA19140, USA. 2Department of Pharmacology, Temple University School of Medicine,Philadelphia, PA 19140, USA. 3Cardiovascular Research Center, Temple University School ofMedicine, Philadelphia, PA 19140, USA. 4Life Sciences Institute, University of Michigan, AnnArbor, MI 48109, USA.*Corresponding author. E-mail: [email protected]
www.Scie
vival pathways and promoting apoptosis after cardiac injury (15–17).These data present compelling evidence of a causal role for GRK2 inthe maladaptive progression of cardiac remodeling and dysfunctionleading to HF, especially after ischemic injury. Therefore, the develop-ment of small-molecule inhibitors of GRK2 appears warranted forpharmacologic treatment of HF.
Recently, we discovered that the selective serotonin reuptake inhib-itor (SSRI) antidepressant drug paroxetine specifically bound to thecatalytic domain of GRK2 as an off-target and inhibited kinase activityin the micromolar range of affinity (18). Further, paroxetine could in-hibit GRK2 with selectivity over other GRK subfamilies (18). Moderateconcentrations of paroxetine inhibited GRK2 target phosphorylationin vitro and significantly potentiated the bAR-mediated increase inmyocardial contractility in vitro and in vivo after isoproterenol (ISO)administration (18). Here, we directly investigated whether paroxetine-mediated inhibition of GRK2 could improve cardiovascular signalingand function in a mouse model of HF.
RESULTS
Chronic paroxetine treatment improves cardiac functionafter myocardial infarctionTo determine whether pharmacologic inhibition of GRK2 by paroxetinecould provide improvement in cardiac function in an animal model ofHF, wild-type C57BL/6 mice underwent myocardial infarction (MI)or sham surgery (19) and were allowed 2 weeks for infarct develop-ment and HF progression before 4 weeks of treatment with vehicle[dimethyl sulfoxide (DMSO) in water], paroxetine, or fluoxetine (bothat 5 mg/kg per day) through subcutaneous miniosmotic pumps. Thesedoses were determined on the basis of a literature search of murinestudies investigating their SSRI effects, in which doses ranged from1 to 10 mg/kg. Using this treatment protocol, we found that the serumparoxetine levels ranged between 27 and 192 ng/ml after 4 weeks of
nceTranslationalMedicine.org 4 March 2015 Vol 7 Issue 277 277ra31 1
R E S EARCH ART I C L E
treatment in both sham and post-MI animals (n = 6 per group, controlserum from vehicle-treated animals was below the low limit of detectionof 1 ng/ml). Quantitative determinations of paroxetine concentration inhuman serum have revealed that over clinically relevant doses used fordepression (10 to 60 mg/day), levels range between 5 and 190 ng/ml(20–22), indicating that levels in our HF mice are equivalent.
All post-MI mice exhibited similar cardiac function at baseline and2 weeks after MI (Fig. 1). For example, left ventricular (LV) ejectionfraction (EF) decreased from ~70 to ~35% after 2 weeks, which ischaracteristic of post-MI hearts (Fig. 1, A and B). In contrast to thecontinuing degradation of function and structure in the vehicle- andfluoxetine-treated groups, paroxetine treatment led to robust improve-ment (Fig. 1). Paroxetine produced an ~30% absolute increase in EFand an ~20% absolute increase in fractional shortening (FS) above ve-hicle at 6 weeks after MI (Fig. 1, B and C). Analysis of LV internaldiameter during diastole (LVIDd) and systole (LVIDs) revealed a sig-
www.Scie
nificant restoration of LV dimension with paroxetine treatment (Fig. 1,D and E). In addition, LV end-diastolic dimension (LVEDd) was sig-nificantly preserved with paroxetine (Fig. 1F).
At 6 weeks after MI, cardiovascular function and adrenergic re-sponsiveness were analyzed through terminal hemodynamics at base-line and upon challenge with increasing doses of the bAR agonist ISO(Fig. 2). No difference was observed inmean systemic pressure (Fig. 2B)orHR responses to ISO (Fig. 2C), consistent with previous studies thatobserved no changes in pressure or HR with GRK2 lowering (8, 23).Although there was no statistical difference at baseline, paroxetine en-hanced dP/dt maximum and minimum at every dose of ISO in treatedsham animals (Fig. 2, D and E). Paroxetine enhanced contractility afterMI (Fig. 2, D and E). Furthermore, the nearly 2.5-fold increase in LVend-diastolic pressure (LVEDP) after MI, as a physiological index ofHF, in the vehicle and fluoxetine groups was completely and selec-tively blocked with paroxetine (Fig. 2F).
nceTranslationalMedicine.org
on J
uly
24, 2
015
Dow
nloa
ded
from
Paroxetine treatmentlimits adverse ventricularremodeling after MIWe next investigated whether the en-hanced cardiac function seen with chron-ic paroxetine treatment was due in partto reduced or reversed maladaptive remod-eling of the LV, consistent with GRK2 low-ering (13, 14). Heart weight (HW) andlength (HL) normalized to tibia length (TL)were significantly reduced in paroxetine-treated post-MI mice compared to vehicle-and fluoxetine-treated controls (Fig. 3, Aand B). Further, the about twofold in-crease in lung weight (LW) in the vehicleand fluoxetine groups, showing evidenceof HF development, was totally normal-ized by paroxetine treatment (Fig. 3C). Tovisualize scar formation and fibrosis, weharvested hearts 6 weeks after MI, andparaffin-embedded sections were stainedwithMasson’s trichrome. Although no dif-ference was observed in sham hearts, pres-ervation of LV architecture was evident intrichrome-stained, four-chamber sectionsof post-MI hearts treated with paroxetine(Fig. 3D). Four-chamber images were ta-ken at the level of the aortic outflow tractwhere the long and short axes of the LVare maximum, for quantitative comparison.Only paroxetine treatment significantlyreduced luminal area after MI (Fig. 3F).
To investigate the integrity of the myo-cardium, we collected higher-magnificationimages of the border zone (×10), transi-tional zone (×20), and infarct area (×10)(Fig. 3G). These images revealed a sub-stantial reduction in fibrosis in the borderzone of paroxetine-treated post-MI hearts,as well as a potential preservation of car-diomyocyte microstructure in the border
Fig. 1. Paroxetine treatment reverses LV dysfunction after MI. (A) Representative short-axis M-modeechocardiography recordings from C57BL/6 mice treated with vehicle (DMSO and water), fluoxetine
(fluox), or paroxetine (parox) at baseline, 2 weeks (pretreatment) and 4 and 6 weeks after MI comparedto noninfarcted (sham) mice treated the same way. (B and C) Serial measures of noted experimentalgroups for (B) LVEF and (C) FS. (D and E) Serial measures of (D) LVIDd and (E) LVIDs in these mice. (F)Serial measures of LVEDd in these mice. *P = 0.004, **P = 0.001, ***P < 0.0001 as determined by one-wayanalysis of variance (ANOVA) relative to corresponding MI vehicle. n = 9 to 14 per group.4 March 2015 Vol 7 Issue 277 277ra31 2
R E S EARCH ART I C L E
on J
uly
24, 2
015
Dow
nloa
ded
from
zone and myocyte content in the infarct zone. Together, these data sug-gest that paroxetine inhibition of GRK2 preserves the structural integ-rity of the myocardium.Paroxetine treatment reverses SNS overdrive andnormalizes the myocardial b-adrenergic system after MISympathetic overdrive in injured and compromised myocardium canincrease GRK2 levels, which enhances bAR down-regulation in chron-ic HF through increased phosphorylation and internalization (2, 4).To determine whether paroxetine therapy can prevent or reverse sym-pathetic overdrive in the post-MI mouse, we measured serum cate-cholamine levels from sham and 6-week post-MI animals (Fig. 4, Aand B). Although serum epinephrine and norepinephrine levels weresignificantly enhanced 6 weeks after MI in animals treated with vehicle
www.Scie
or fluoxetine, they were restored to near sham levels with paroxetinetreatment (Fig. 4, A and B). To assess whether paroxetine therapy andits catecholamine-lowering properties translate to restoration of bARdensity, we used radioligand binding and found that at 6 weeks afterMI, there is significant bAR down-regulation as expected (Fig. 4C).Four weeks of paroxetine treatment was found to restore bAR densityto near sham levels (Fig. 4C). We also found similar restoration of bARmRNA in these samples (fig. S1). These data agree with the restorationof bAR sensitivity observed during hemodynamic analysis in theseanimals in Fig. 2 and are consistent with paroxetine inhibiting the nodalregulator of cardiac bAR dysfunction—GRK2 (13). Finally, to completethe mechanistic circuit of GRK2 inhibition in the failing heart, we foundthat paroxetine treatment, but not fluoxetine treatment, reduced myo-cardial GRK2 protein up-regulation seen 6 weeks after MI (Fig. 4D).
Fig. 2. Paroxetine treatment after MI enhances in vivo LV hemo-dynamic function and restores bAR inotropic reserve. Hemodynamics
maximum (*P = 0.0234 and 0.0130, **P = 0.0008, ***P ≤ 0.0001), and (E)LV −dP/dt average minimum at baseline and with increasing doses of ISO
were recorded from wild-type (WT) mice treated with vehicle, fluoxetine(fluox), or paroxetine (parox) at 6 weeks after MI (after 4 weeks of treat-ment) compared to sham. (A) Representative LV dP/dt hemodynamic re-cordings. Arrows mark administration of ISO. (B) Quantification of meansystemic pressure. (C to E) Quantification of (C) HR, (D) LV +dP/dt average
(0.1 to 10 ng) (*P = 0.0105 and 0.0134 for sham, and 0.0280 for MI; **P =0.0058, 0.0040, and 0.0087, respectively). bpm, beats per minute. (F) Quan-tification of LVEDP at baseline (no ISO) 6 weeks after sham and MI (**P =0.005). Statistics are relative to corresponding sham or MI vehicle by two-or one-way ANOVA as appropriate. n = 12 to 15 per group.
nceTranslationalMedicine.org 4 March 2015 Vol 7 Issue 277 277ra31 3
R E S EARCH ART I C L E
on J
uly
24, 2
015
Dow
nloa
ded
from
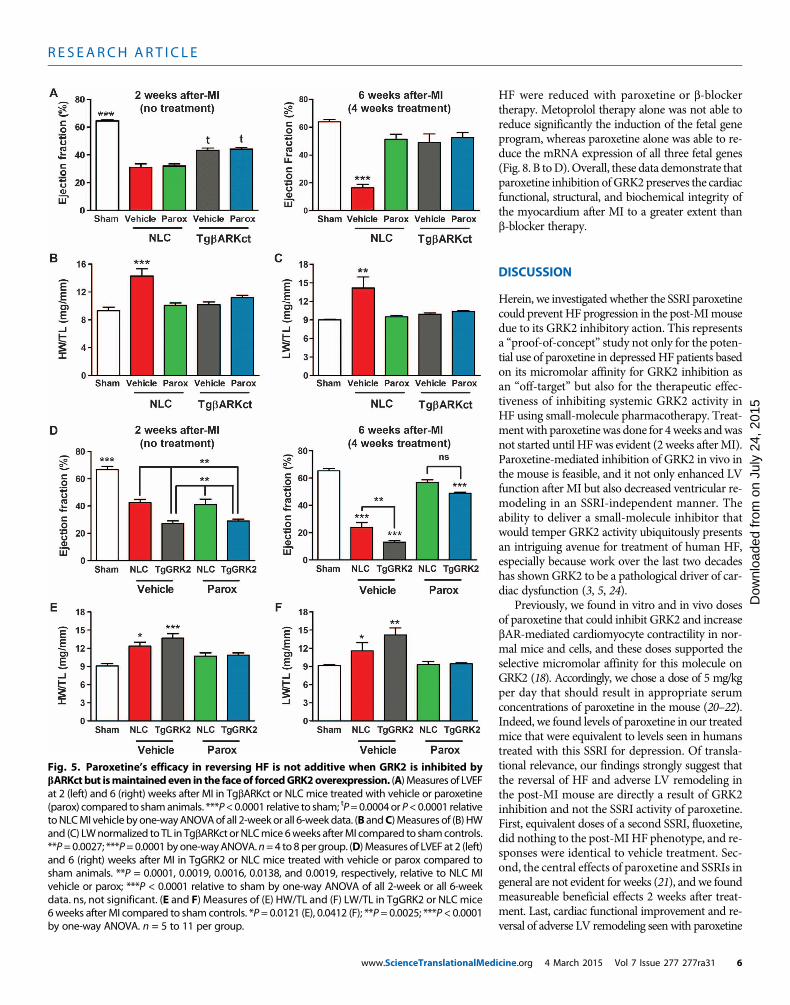
Cardiac-specific GRK2 gain- and loss-of-function mice supportparoxetine’s therapeutic mechanism after MI as GRK2 inhibitionOur laboratory has previously shown that myocardial expression of aC-terminal peptide ofGRK2 (bARKct), which competes forGbg bind-ing and subsequent membrane translocation and activation, can en-hance heart function and prevent as well as reverse HF in both smalland large animals (9, 12). To prove that paroxetine is indeed exertingbeneficial effects after MI through inhibition of this pathological ki-nase, we used cardiac-targeted bARKct transgenic mice (TgbARKct)
www.ScienceTranslationalMedicine.org
as well as cardiac GRK2–overexpressingmice (TgGRK2), where levels of GRK2are similar to those observed in humanHF (8). First, TgbARKct and their non-transgenic littermate control (NLC) micewere treated with MI or sham surgery asabove and then treated for 4 weeks withparoxetine (5 mg/kg per day) beginning2weeks afterMI. Consistentwith previousstudies, cardiac function (LVEF) was sig-nificantly improved in TgbARKct mice 2weeks after MI compared to NLC mice(Fig. 5A). As seen above, paroxetine en-hanced function in post-MI NLCmice af-ter 4 weeks of treatment (6 weeks afterMI); however, it had no additional effectsin post-MI TgbARKct mice over existingGRK2 inhibition (Fig. 5A). Consistentwiththese results and the in vivo GRK2 tar-geting of paroxetine, when isolated myo-cytes from NLC and TgbARKct mice werestudied, the enhanced ISO-mediated con-tractile actions of paroxetine were absentwhen GRK2 was already inhibited bybARKct expression (fig. S2).
Similar to the above in vivo cardiacfunctional effects, LVIDd andLVIDswereconsiderably improved in vehicle-treatedTgbARKct post-MI mice; however, thepositive effects of paroxetine were onlyfound in post-MI NLC mice with no ad-ditional therapeutic benefit in TgbARKctmice (table S1). In agreement, HW/TL andLW/TL were increased only in vehicle-treated NLC mice 6 weeks after MI, butpreserved in TgbARKct animals indepen-dent of treatment and restored in NLCmice with paroxetine (Fig. 5, B and C).Thus, the GRK2 inhibitory actions ofparoxetine in post-MI mice are equivalentto bARKct-mediated inhibition of thiskinase.
To investigate whether the therapeu-tic effects of paroxetine are still presentwhen GRK2 is up-regulated before MI,which would model the clinical scenarioof treating existing HF patients, we studiedTgGRK2 mice and their correspondingNLCs. Under the identical experimental
protocol as above, post-MI TgGRK2 mice showed considerably morecardiac dysfunction after 2 weeks compared to NLC mice (Fig. 5D),consistent with the pathological nature of this kinase in injured myo-cardium. Despite an overall greater degree of dysfunction in vehicle-treated TgGRK2 hearts 6 weeks after MI compared to NLC, paroxetinetreatment restored LVEF in TgGRK2 hearts to that of NLC (Fig. 5D).LV dilation was also greater in vehicle-treated TgGRK2 versus NLCmice at 2 and 6 weeks after MI as measured by LVIDs, but LV dimen-sionwas restored in both upon paroxetine treatment (fig. S3, A and B).
Fig. 3. Paroxetine reduces LV dilation and fibrosis after MI. (A to C) Measures of (A) HW normalizedto TL, (B) HL normalized to TL, and (C) LW to TL in WT C57BL/6 mice treated with vehicle, fluoxetine (fluox),
or paroxetine (parox) at 6 weeks after MI (4 weeks of treatment) compared to noninfarcted (sham) micetreated the same way. ***P ≤ 0.0001 relative to sham and tttP = 0.0075 relative to MI vehicle by one-wayANOVA. n = 12 to 19 per group. (D) Representative images of Masson’s trichrome–stained murine heartsections from WT mice treated with vehicle, fluox, or parox at 6 weeks after sham or MI surgery (4 weeksafter treatment). (E and F) Graphic representation of (E) infarct length and (F) lumen area in vehicle-, fluox-,or parox-treated mice 6 weeks after MI. *P = 0.004 by one-way ANOVA. n = 7, 6, and 8, respectively, for (D)to (G). (G) Representative images of Masson’s trichrome–stained murine heart sections focusing on theborder zone and the infarct area in vehicle-, fluox-, and parox-treated mice 6 weeks after MI.4 March 2015 Vol 7 Issue 277 277ra31 4
R E S EARCH ART I C L E
on J
uly
24, 2
015
Dow
nloa
ded
from
In agreement, althoughHW/TLwas greater in vehicle-treated TgGRK2mice compared to NLC mice, cardiac hypertrophy was completelyrestored in paroxetine-treated mice 6 weeks after MI (Fig. 5E). In-creased LWwas also normalized in these post-MI mice after 4 weeksof paroxetine treatment (Fig. 5F). Overall, paroxetine treatment wasno less effective at reducing LV dysfunction in TgGRK2 animals thanin wild-type mice, validating its therapeutic potential despite a lessfavorable biochemical cardiac milieu when this pathological enzymeis enhanced.
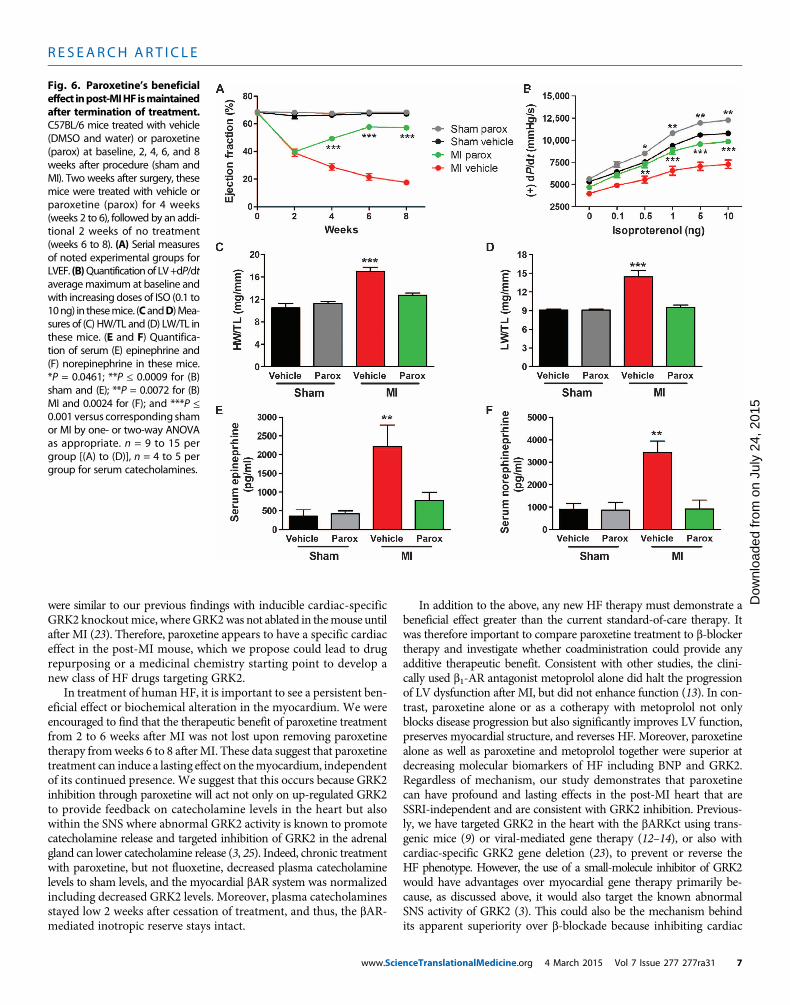
Inhibition of GRK2 activity by paroxetine signalingdemonstrates a persistent beneficial effect after MITo investigate whether the improved cardiac function, reverse remod-eling, and resetting of SNS overdrive and myocardial b-adrenergicsignaling seen after 4 weeks of paroxetine treatment in post-MI micewere persistent, we studied a cohort of animals 2 weeks after ter-mination of vehicle or paroxetine treatment (8 weeks after MI). Theparoxetine-mediated increase in EF and LV +/−dP/dtmaximum andminimum post-MI was maintained 2 weeks after cessation of treat-ment (Fig. 6, A and B, and fig. S4). Furthermore, the 2.5-fold increasein LVEDP in the vehicle-treated post-MI group was completely re-versed with paroxetine (fig. S4). The improvement in HW and LWwas also preserved after 2 weeks of no treatment in the post-MI micepreviously receiving paroxetine (Fig. 6, C and D), and no increase wasobserved in induction of the fetal gene program (fig. S4). Analysisof serum catecholamine levels revealed a maintenance near sham
www.ScienceTranslationalMedicine.org
values in post–paroxetine treatment micedespite a more pronounced elevation inthe vehicle group (Fig. 6, E and F). Thesedata are consistent with paroxetine nor-malizing the SNS/b-adrenergic axis after4 weeks of treatment in these mice (Fig.4), which apparently can preserve the im-provement in cardiac function and struc-ture even after acute GRK2 inhibition isstopped.
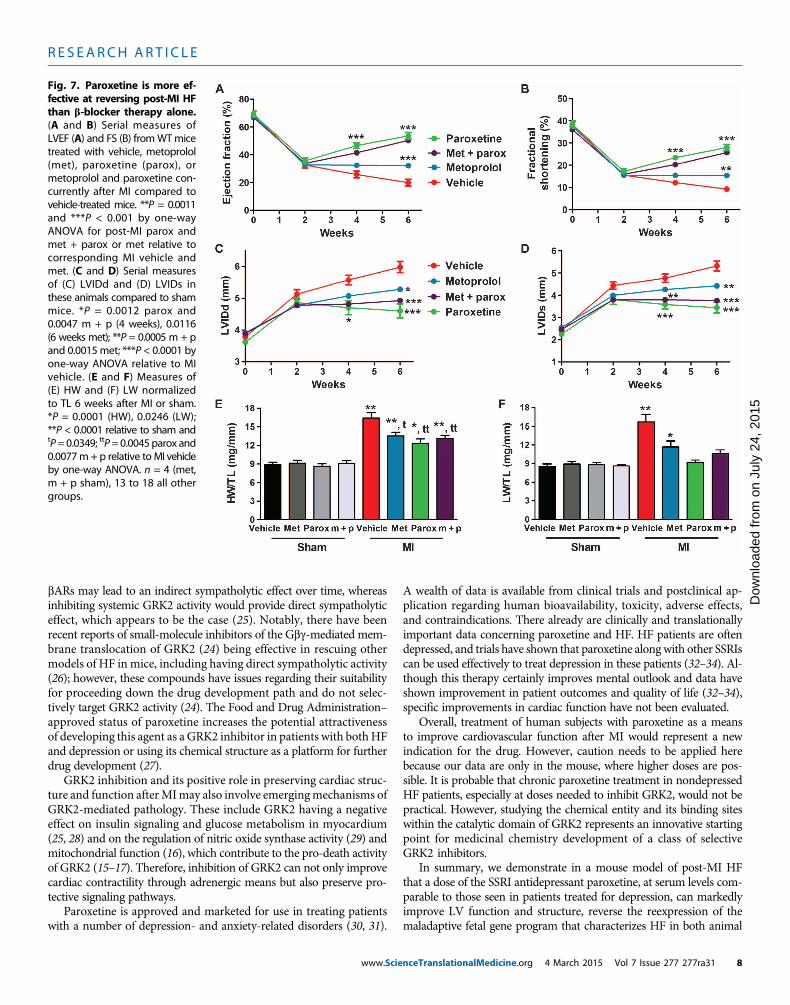
Paroxetine treatmentdemonstrates therapeutictranslational potential above andbeyond current standard of careTo increase the translational relevance ofa potential repurposing of paroxetine forHF therapy, at least in a depressed pop-ulation, we carried out an experiment toevaluate its post-MI effectiveness com-pared to a current human HF standardof care, bAR blockade. To do this, miceat 2 weeks afterMI were treated for 4 weekswith vehicle, the clinically used b1-AR an-tagonist metoprolol at 250 mg/kg per day,paroxetine at 5mg/kg per day, or both drugsconcurrently. In contrast to the progressivecontractile dysfunction in vehicle-treatedmice, metoprolol treatment stabilized LVfunction, maintaining EF and FS with nofurther deterioration from 2 to 6 weeksafter MI (Fig. 7, A and B). Metoprolol did
not improve function, whereas paroxetine treatment alone or its co-administrationwithmetoprolol demonstrated a reversal of LVdysfunc-tion, significantly improving EF and FS after MI (Fig. 7, A and B).Finally, metoprolol treatment alone reduced but did not prevent thecontinued expansion of LVIDd and LVIDs (Fig. 7, C and D). In con-trast, paroxetine alone or cotherapy with paroxetine and metoprololsignificantly improved LV dimension (Fig. 7, C andD).When lookingat the structure of the dissected heart, HW/TL ratios were reduced in alltreatment groups (Fig. 7E), and LWas ameasure of HFwas also reducedby all three treatments with the trend for paroxetine to be most effec-tive (Fig. 7F).
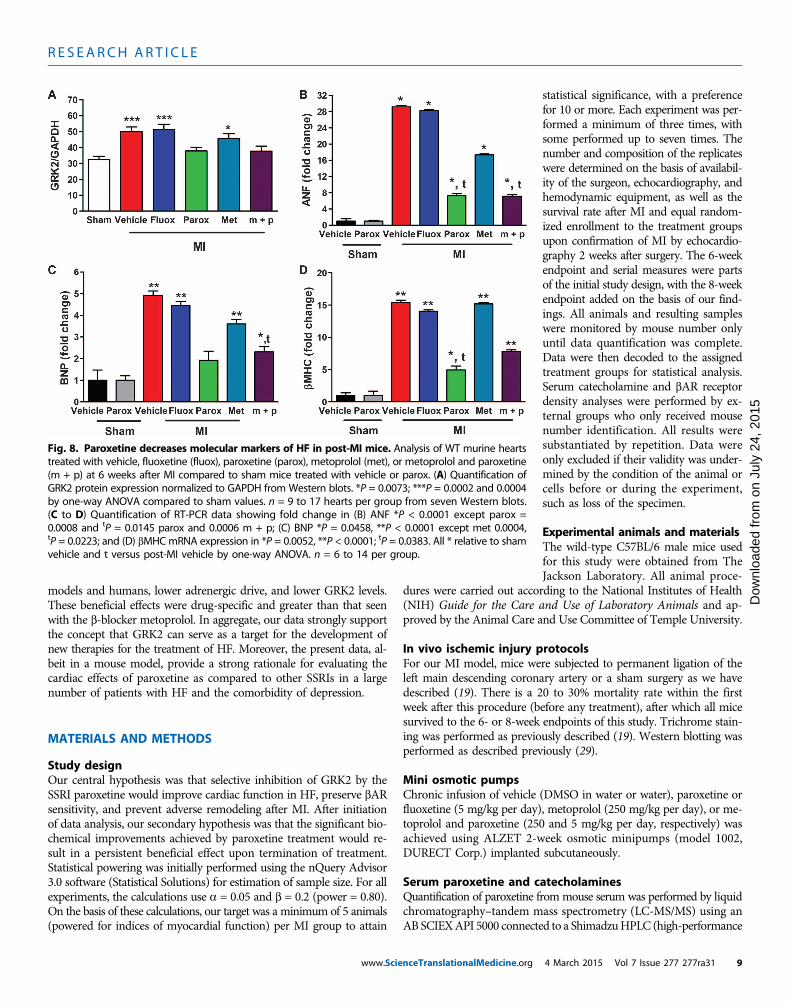
Finally, the molecular characteristics of HF were assessed in 6-weekpost-MI hearts to determine whether biomarkers of HF were reducedwith 4 weeks of paroxetine treatment compared to b-blocker therapy.In Fig. 4, paroxetine can have a positive effect on SNS and b-adrenergicsignaling, including a decrease in GRK2 levels in the heart, leadingto improved cardiac function. Under our experimental conditions,metoprolol alone was less effective at lowering GRK2 compared topost-MI treatment when paroxetine was given (Fig. 8A). One char-acteristic of HF is the myocardial reactivation of fetal gene expres-sion, and some of these, such as atrial natriuretic factor (ANF) andbrain natriuretic peptide (BNP), have been used as HF biomarkers.Therefore, we assessed the gene expression of ANF and BNP, as wellas b-myosin heavy chain (bMHC), which is also an HF marker, throughreverse transcription polymerase chain reaction (RT-PCR) in 6-weekpost-MI hearts to determine whether these molecular markers of
Fig. 4. Paroxetine reduces SNS overdrive and restores themyocardial bAR system afterMI. (A and B)Quantification of serum (A) epinephrine and (B) norepinephrine from WT mice, either sham or MI at
6weeks, treatedwith vehicle, fluoxetine (fluox), or paroxetine (parox). (C) bARdensity in shamversus 6-weekpost-MI hearts (Bmax values shown as femtomoles of receptor per milligram of sarcolemmal protein) withvehicle or parox treatment. (D) Representative Western blot image of GRK2 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) protein expression 6 weeks after sham or MI (after 4 weeks of treatmentwith vehicle, fluox, or parox). *P = 0.0167, 0.0047, 0.0427, and 0.0244, respectively (A and B), and *P =0.0003 (C) versus sham vehicle and *P= 0.0119 versusMI parox by one-way ANOVA. n= 6 to 9 per group.4 March 2015 Vol 7 Issue 277 277ra31 5
R E S EARCH ART I C L E
www.ScienceTranslationalMed
HF were reduced with paroxetine or b-blockertherapy. Metoprolol therapy alone was not able toreduce significantly the induction of the fetal geneprogram, whereas paroxetine alone was able to re-duce the mRNA expression of all three fetal genes(Fig. 8. B toD).Overall, these data demonstrate thatparoxetine inhibition of GRK2 preserves the cardiacfunctional, structural, and biochemical integrity ofthe myocardium after MI to a greater extent thanb-blocker therapy.
on J
uly
24, 2
015
Dow
nloa
ded
from
DISCUSSION
Herein, we investigated whether the SSRI paroxetinecould prevent HF progression in the post-MImousedue to its GRK2 inhibitory action. This representsa “proof-of-concept” study not only for the poten-tial use of paroxetine in depressedHF patients basedon its micromolar affinity for GRK2 inhibition asan “off-target” but also for the therapeutic effec-tiveness of inhibiting systemic GRK2 activity inHF using small-molecule pharmacotherapy. Treat-mentwith paroxetinewas done for 4weeks andwasnot started until HFwas evident (2 weeks afterMI).Paroxetine-mediated inhibition of GRK2 in vivo inthe mouse is feasible, and it not only enhanced LVfunction after MI but also decreased ventricular re-modeling in an SSRI-independent manner. Theability to deliver a small-molecule inhibitor thatwould temper GRK2 activity ubiquitously presentsan intriguing avenue for treatment of human HF,especially because work over the last two decadeshas shown GRK2 to be a pathological driver of car-diac dysfunction (3, 5, 24).
Previously, we found in vitro and in vivo dosesof paroxetine that could inhibit GRK2 and increasebAR-mediated cardiomyocyte contractility in nor-mal mice and cells, and these doses supported theselective micromolar affinity for this molecule onGRK2 (18). Accordingly, we chose a dose of 5 mg/kgper day that should result in appropriate serumconcentrations of paroxetine in the mouse (20–22).Indeed, we found levels of paroxetine in our treatedmice that were equivalent to levels seen in humanstreated with this SSRI for depression. Of transla-tional relevance, our findings strongly suggest thatthe reversal of HF and adverse LV remodeling inthe post-MI mouse are directly a result of GRK2inhibition and not the SSRI activity of paroxetine.First, equivalent doses of a second SSRI, fluoxetine,did nothing to the post-MI HF phenotype, and re-sponses were identical to vehicle treatment. Sec-ond, the central effects of paroxetine and SSRIs ingeneral are not evident for weeks (21), and we foundmeasureable beneficial effects 2 weeks after treat-ment. Last, cardiac functional improvement and re-versal of adverse LV remodeling seen with paroxetine
Fig. 5. Paroxetine’s efficacy in reversing HF is not additive when GRK2 is inhibited bybARKct but ismaintainedeven in the faceof forcedGRK2overexpression. (A)Measures of LVEF
at 2 (left) and 6 (right) weeks after MI in TgbARKct or NLC mice treated with vehicle or paroxetine(parox) compared to shamanimals. ***P<0.0001 relative to sham; tP=0.0004or P<0.0001 relativetoNLCMI vehicle byone-wayANOVAof all 2-weekor all 6-weekdata. (B andC)Measures of (B) HWand (C) LWnormalized to TL in TgbARKct orNLCmice 6weeks afterMI compared to shamcontrols.**P=0.0027; ***P=0.0001byone-wayANOVA.n=4 to 8per group. (D)Measures of LVEF at 2 (left)and 6 (right) weeks after MI in TgGRK2 or NLC mice treated with vehicle or parox compared tosham animals. **P = 0.0001, 0.0019, 0.0016, 0.0138, and 0.0019, respectively, relative to NLC MIvehicle or parox; ***P < 0.0001 relative to sham by one-way ANOVA of all 2-week or all 6-weekdata. ns, not significant. (E and F) Measures of (E) HW/TL and (F) LW/TL in TgGRK2 or NLC mice6weeks after MI compared to sham controls. *P= 0.0121 (E), 0.0412 (F); **P= 0.0025; ***P< 0.0001by one-way ANOVA. n = 5 to 11 per group.icine.org 4 March 2015 Vol 7 Issue 277 277ra31 6
R E S EARCH ART I C L E
on J
uly
24, 2
015
Dow
nloa
ded
from
were similar to our previous findings with inducible cardiac-specificGRK2 knockoutmice, where GRK2was not ablated in themouse untilafter MI (23). Therefore, paroxetine appears to have a specific cardiaceffect in the post-MI mouse, which we propose could lead to drugrepurposing or a medicinal chemistry starting point to develop anew class of HF drugs targeting GRK2.
In treatment of human HF, it is important to see a persistent ben-eficial effect or biochemical alteration in the myocardium. We wereencouraged to find that the therapeutic benefit of paroxetine treatmentfrom 2 to 6 weeks after MI was not lost upon removing paroxetinetherapy fromweeks 6 to 8 afterMI. These data suggest that paroxetinetreatment can induce a lasting effect on themyocardium, independentof its continued presence. We suggest that this occurs because GRK2inhibition through paroxetine will act not only on up-regulated GRK2to provide feedback on catecholamine levels in the heart but alsowithin the SNS where abnormal GRK2 activity is known to promotecatecholamine release and targeted inhibition of GRK2 in the adrenalgland can lower catecholamine release (3, 25). Indeed, chronic treatmentwith paroxetine, but not fluoxetine, decreased plasma catecholaminelevels to sham levels, and the myocardial bAR system was normalizedincluding decreased GRK2 levels. Moreover, plasma catecholaminesstayed low 2 weeks after cessation of treatment, and thus, the bAR-mediated inotropic reserve stays intact.
www.Scie
In addition to the above, any new HF therapy must demonstrate abeneficial effect greater than the current standard-of-care therapy. Itwas therefore important to compare paroxetine treatment to b-blockertherapy and investigate whether coadministration could provide anyadditive therapeutic benefit. Consistent with other studies, the clini-cally used b1-AR antagonist metoprolol alone did halt the progressionof LV dysfunction after MI, but did not enhance function (13). In con-trast, paroxetine alone or as a cotherapy with metoprolol not onlyblocks disease progression but also significantly improves LV function,preserves myocardial structure, and reverses HF. Moreover, paroxetinealone as well as paroxetine and metoprolol together were superior atdecreasing molecular biomarkers of HF including BNP and GRK2.Regardless of mechanism, our study demonstrates that paroxetinecan have profound and lasting effects in the post-MI heart that areSSRI-independent and are consistent with GRK2 inhibition. Previous-ly, we have targeted GRK2 in the heart with the bARKct using trans-genic mice (9) or viral-mediated gene therapy (12–14), or also withcardiac-specific GRK2 gene deletion (23), to prevent or reverse theHF phenotype. However, the use of a small-molecule inhibitor of GRK2would have advantages over myocardial gene therapy primarily be-cause, as discussed above, it would also target the known abnormalSNS activity of GRK2 (3). This could also be the mechanism behindits apparent superiority over b-blockade because inhibiting cardiac
Fig. 6. Paroxetine’s beneficialeffect inpost-MIHF ismaintained
after termination of treatment.C57BL/6 mice treated with vehicle(DMSO and water) or paroxetine(parox) at baseline, 2, 4, 6, and 8weeks after procedure (sham andMI). Two weeks after surgery, thesemice were treated with vehicle orparoxetine (parox) for 4 weeks(weeks 2 to 6), followed by an addi-tional 2 weeks of no treatment(weeks 6 to 8). (A) Serial measuresof noted experimental groups forLVEF. (B)Quantificationof LV+dP/dtaveragemaximum at baseline andwith increasing doses of ISO (0.1 to10ng) in thesemice. (CandD)Mea-sures of (C) HW/TL and (D) LW/TL inthese mice. (E and F) Quantifica-tion of serum (E) epinephrine and(F) norepinephrine in these mice.*P = 0.0461; **P ≤ 0.0009 for (B)sham and (E); **P = 0.0072 for (B)MI and 0.0024 for (F); and ***P ≤0.001 versus corresponding shamor MI by one- or two-way ANOVAas appropriate. n = 9 to 15 pergroup [(A) to (D)], n = 4 to 5 pergroup for serum catecholamines.nceTranslationalMedicine.org 4 March 2015 Vol 7 Issue 277 277ra31 7
R E S EARCH ART I C L E
on J
uly
24, 2
015
Dow
nloa
ded
from
bARs may lead to an indirect sympatholytic effect over time, whereasinhibiting systemic GRK2 activity would provide direct sympatholyticeffect, which appears to be the case (25). Notably, there have beenrecent reports of small-molecule inhibitors of the Gbg-mediated mem-brane translocation of GRK2 (24) being effective in rescuing othermodels of HF in mice, including having direct sympatholytic activity(26); however, these compounds have issues regarding their suitabilityfor proceeding down the drug development path and do not selec-tively target GRK2 activity (24). The Food and Drug Administration–approved status of paroxetine increases the potential attractivenessof developing this agent as a GRK2 inhibitor in patients with bothHFand depression or using its chemical structure as a platform for furtherdrug development (27).
GRK2 inhibition and its positive role in preserving cardiac struc-ture and function afterMImay also involve emergingmechanisms ofGRK2-mediated pathology. These include GRK2 having a negativeeffect on insulin signaling and glucose metabolism in myocardium(25, 28) and on the regulation of nitric oxide synthase activity (29) andmitochondrial function (16), which contribute to the pro-death activityof GRK2 (15–17). Therefore, inhibition of GRK2 can not only improvecardiac contractility through adrenergic means but also preserve pro-tective signaling pathways.
Paroxetine is approved and marketed for use in treating patientswith a number of depression- and anxiety-related disorders (30, 31).
www.Scie
A wealth of data is available from clinical trials and postclinical ap-plication regarding human bioavailability, toxicity, adverse effects,and contraindications. There already are clinically and translationallyimportant data concerning paroxetine and HF. HF patients are oftendepressed, and trials have shown that paroxetine alongwith other SSRIscan be used effectively to treat depression in these patients (32–34). Al-though this therapy certainly improves mental outlook and data haveshown improvement in patient outcomes and quality of life (32–34),specific improvements in cardiac function have not been evaluated.
Overall, treatment of human subjects with paroxetine as a meansto improve cardiovascular function after MI would represent a newindication for the drug. However, caution needs to be applied herebecause our data are only in the mouse, where higher doses are pos-sible. It is probable that chronic paroxetine treatment in nondepressedHF patients, especially at doses needed to inhibit GRK2, would not bepractical. However, studying the chemical entity and its binding siteswithin the catalytic domain of GRK2 represents an innovative startingpoint for medicinal chemistry development of a class of selectiveGRK2 inhibitors.
In summary, we demonstrate in a mouse model of post-MI HFthat a dose of the SSRI antidepressant paroxetine, at serum levels com-parable to those seen in patients treated for depression, can markedlyimprove LV function and structure, reverse the reexpression of themaladaptive fetal gene program that characterizes HF in both animal
Fig. 7. Paroxetine is more ef-fective at reversing post-MI HF
than b-blocker therapy alone.(A and B) Serial measures ofLVEF (A) and FS (B) fromWTmicetreated with vehicle, metoprolol(met), paroxetine (parox), ormetoprolol and paroxetine con-currently after MI compared tovehicle-treated mice. **P = 0.0011and ***P < 0.001 by one-wayANOVA for post-MI parox andmet + parox or met relative tocorresponding MI vehicle andmet. (C and D) Serial measuresof (C) LVIDd and (D) LVIDs inthese animals compared to shammice. *P = 0.0012 parox and0.0047 m + p (4 weeks), 0.0116(6 weeks met); **P = 0.0005 m + pand 0.0015met; ***P < 0.0001 byone-way ANOVA relative to MIvehicle. (E and F) Measures of(E) HW and (F) LW normalizedto TL 6 weeks after MI or sham.*P = 0.0001 (HW), 0.0246 (LW);**P < 0.0001 relative to sham andtP= 0.0349; ttP=0.0045 parox and0.0077m+ p relative toMI vehicleby one-way ANOVA. n = 4 (met,m + p sham), 13 to 18 all othergroups.nceTranslationalMedicine.org 4 March 2015 Vol 7 Issue 277 277ra31 8
R E S EARCH ART I C L E
on J
uly
24, 2
015
Dow
nloa
ded
from
models and humans, lower adrenergic drive, and lower GRK2 levels.These beneficial effects were drug-specific and greater than that seenwith the b-blocker metoprolol. In aggregate, our data strongly supportthe concept that GRK2 can serve as a target for the development ofnew therapies for the treatment of HF. Moreover, the present data, al-beit in a mouse model, provide a strong rationale for evaluating thecardiac effects of paroxetine as compared to other SSRIs in a largenumber of patients with HF and the comorbidity of depression.
MATERIALS AND METHODS
Study designOur central hypothesis was that selective inhibition of GRK2 by theSSRI paroxetine would improve cardiac function in HF, preserve bARsensitivity, and prevent adverse remodeling after MI. After initiationof data analysis, our secondary hypothesis was that the significant bio-chemical improvements achieved by paroxetine treatment would re-sult in a persistent beneficial effect upon termination of treatment.Statistical powering was initially performed using the nQuery Advisor3.0 software (Statistical Solutions) for estimation of sample size. For allexperiments, the calculations use a = 0.05 and b = 0.2 (power = 0.80).On the basis of these calculations, our target was a minimum of 5 animals(powered for indices of myocardial function) per MI group to attain
www.ScienceTranslationalMedicine.org
statistical significance, with a preferencefor 10 or more. Each experiment was per-formed a minimum of three times, withsome performed up to seven times. Thenumber and composition of the replicateswere determined on the basis of availabil-ity of the surgeon, echocardiography, andhemodynamic equipment, as well as thesurvival rate after MI and equal random-ized enrollment to the treatment groupsupon confirmation of MI by echocardio-graphy 2 weeks after surgery. The 6-weekendpoint and serial measures were partsof the initial study design, with the 8-weekendpoint added on the basis of our find-ings. All animals and resulting sampleswere monitored by mouse number onlyuntil data quantification was complete.Data were then decoded to the assignedtreatment groups for statistical analysis.Serum catecholamine and bAR receptordensity analyses were performed by ex-ternal groups who only received mousenumber identification. All results weresubstantiated by repetition. Data wereonly excluded if their validity was under-mined by the condition of the animal orcells before or during the experiment,such as loss of the specimen.
Experimental animals and materialsThe wild-type C57BL/6 male mice usedfor this study were obtained from TheJackson Laboratory. All animal proce-
dures were carried out according to the National Institutes of Health(NIH) Guide for the Care and Use of Laboratory Animals and ap-proved by the Animal Care and Use Committee of Temple University.
In vivo ischemic injury protocolsFor our MI model, mice were subjected to permanent ligation of theleft main descending coronary artery or a sham surgery as we havedescribed (19). There is a 20 to 30% mortality rate within the firstweek after this procedure (before any treatment), after which all micesurvived to the 6- or 8-week endpoints of this study. Trichrome stain-ing was performed as previously described (19). Western blotting wasperformed as described previously (29).
Mini osmotic pumpsChronic infusion of vehicle (DMSO in water or water), paroxetine orfluoxetine (5 mg/kg per day), metoprolol (250 mg/kg per day), or me-toprolol and paroxetine (250 and 5 mg/kg per day, respectively) wasachieved using ALZET 2-week osmotic minipumps (model 1002,DURECT Corp.) implanted subcutaneously.
Serum paroxetine and catecholaminesQuantification of paroxetine frommouse serum was performed by liquidchromatography–tandem mass spectrometry (LC-MS/MS) using anABSCIEXAPI 5000 connected to a ShimadzuHPLC (high-performance
Fig. 8. Paroxetine decreases molecular markers of HF in post-MI mice. Analysis of WT murine heartstreated with vehicle, fluoxetine (fluox), paroxetine (parox), metoprolol (met), or metoprolol and paroxetine
(m + p) at 6 weeks after MI compared to sham mice treated with vehicle or parox. (A) Quantification ofGRK2 protein expression normalized to GAPDH from Western blots. *P = 0.0073; ***P = 0.0002 and 0.0004by one-way ANOVA compared to sham values. n = 9 to 17 hearts per group from seven Western blots.(C to D) Quantification of RT-PCR data showing fold change in (B) ANF *P < 0.0001 except parox =0.0008 and tP = 0.0145 parox and 0.0006 m + p; (C) BNP *P = 0.0458, **P < 0.0001 except met 0.0004,tP = 0.0223; and (D) bMHCmRNA expression in *P = 0.0052, **P < 0.0001; tP = 0.0383. All * relative to shamvehicle and t versus post-MI vehicle by one-way ANOVA. n = 6 to 14 per group.4 March 2015 Vol 7 Issue 277 277ra31 9

R E S EARCH ART I C L E
on J
uly
24, 2
015
ded
from
liquid chromatography) 20AD with a C18 column (50 mm × 3 mm,3 mm). The calibration range was 1 to 500 ng/ml using 0.25 ml ofserum. Quantification of epinephrine and norepinephrine from mouseserum was performed through LC-MS/MS using an AB SCIEX API6500 connected to a Shimadzu HPLC 30ADwith a C18 HPLC column(50mm×2.1mm, 3 mm). The calibration range was 10 to 2000 pg/mlusing 0.25 ml of serum.
Membrane preparation and radioligand bindingassay for bARsAfter membrane preparation, radioligand [125I]CYP (PerkinElmer,iodo-(–)-cyanopindolol,[125I], NEX189100UC and NEX310010UC)binding to bARs was measured, and Kd (dissociation constant) andthe maximal number of binding sites (Bmax) for [
125I]CYP were deter-mined by Scatchard analysis of saturation binding isotherms withGraphPad Prism as described (35).
RNA isolation and semiquantitative PCRRNA isolation and analysis were performed as previously described(29). Semiquantitative PCR was carried out on complementary DNAusing SYBR Green (Bio-Rad) and 100 nM gene-specific oligonucleo-tides for 18S and ANF on a CFX96 real-time system with Bio-RadCFX Manager 2.1 software. Quantitation was established by comparing18S ribosomal RNA, which was similar between groups, for normal-ization and compared using the DDCt method.
Statistical analysisAll values in the text and figures are presented as means ± SEM ofindependent experiments for given n sizes. Statistical significance wasdetermined by one-way ANOVA with Tukey or two-way ANOVA withBonferroni post hoc as appropriate. Probabilities of 0.05 or less wereconsidered to be statistically significant.
Dow
nloa
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/7/277/277ra31/DC1Detailed MethodsFig. S1. Paroxetine restores bAR mRNA expression.Fig. S2. Paroxetine enhances myocyte contraction in response to ISO in control, but not inbARKct transgenic myocytes.Fig. S3. Paroxetine reduces LV dimension despite elevated GRK2 levels.Fig. S4. Paroxetine’s beneficial effect in post-MI HF is maintained after termination oftreatment.Table S1. Reduction of LV dimension by paroxetine is not additive with GRK2 inhibition bybARKct.Source Data (Excel file)
REFERENCES AND NOTES
1. S. M. Dunlay, N. L. Pereira, S. S. Kushwaha, Contemporary strategies in the diagnosis andmanagement of heart failure. Mayo Clin. Proc. 89, 662–676 (2014).
2. M. R. Bristow, R. Ginsburg, W. Minobe, R. S. Cubicciotti, W. S. Sageman, K. Lurie, M. E. Billingham,D. C. Harrison, E. B. Stinson, Decreased catecholamine sensitivity and b-adrenergic-receptordensity in failing human hearts. N. Engl. J. Med. 307, 205–211 (1982).
3. A. Lymperopoulos, G. Rengo, W. J. Koch, Adrenergic nervous system in heart failure: Patho-physiology and therapy. Circ. Res. 113, 739–753 (2013).
4. A. Claing, S. A. Laporte, M. G. Caron, R. J. Lefkowitz, Endocytosis of G protein-coupled recep-tors: Roles of G protein-coupled receptor kinases and b-arrestin proteins. Prog. Neurobiol. 66,61–79 (2002).
www.Scien
5. H. A. Rockman, W. J. Koch, R. J. Lefkowitz, Seven-transmembrane-spanning receptors andheart function. Nature 415, 206–212 (2002).
6. M. Ungerer, M. Böhm, J. S. Elce, E. Erdmann, M. J. Lohse, Altered expression of beta-adrenergicreceptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation 87,454–463 (1993).
7. G. Iaccarino, E. Barbato, E. Cipolletta, V. De Amicis, K. B. Margulies, D. Leosco, B. Trimarco,W. J. Koch, Elevated myocardial and lymphocyte GRK2 expression and activity in humanheart failure. Eur. Heart J. 26, 1752–1758 (2005).
8. W. J. Koch, H. A. Rockman, P. Samama, R. A. Hamilton, R. A. Bond, C. A. Milano, R. J. Lefkowitz,Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARKinhibitor. Science 268, 1350–1353 (1995).
9. H. A. Rockman, K. R. Chien, D. J. Choi, G. Iaccarino, J. J. Hunter, J. Ross Jr., R. J. Lefkowitz, W. J. Koch,Expression of a b-adrenergic receptor kinase 1 inhibitor prevents the development of myo-cardial failure in gene-targeted mice. Proc. Natl. Acad. Sci. U.S.A. 95, 7000–7005 (1998).
10. S. A. Akhter, A. D. Eckhart, H. A. Rockman, K. Shotwell, R. J. Lefkowitz, W. J. Koch, In vivoinhibition of elevated myocardial b-adrenergic receptor kinase activity in hybrid trans-genic mice restores normal b-adrenergic signaling and function. Circulation 100, 648–653(1999).
11. V. B. Harding, L. R. Jones, R. J. Lefkowitz, W. J. Koch, H. A. Rockman, Cardiac bARK1 inhi-bition prolongs survival and augments b blocker therapy in a mouse model of severeheart failure. Proc. Natl. Acad. Sci. U.S.A. 98, 5809–5814 (2001).
12. A. S. Shah, D. C. White, S. Emani, A. P. Kypson, R. E. Lilly, K. Wilson, D. D. Glower, R. J. Lefkowitz,W. J. Koch, In vivo ventricular gene delivery of a b-adrenergic receptor kinase inhibitor to thefailing heart reverses cardiac dysfunction. Circulation 103, 1311–1316 (2001).
13. G. Rengo, A. Lymperopoulos, C. Zincarelli, M. Donniacuo, S. Soltys, J. E. Rabinowitz, W. J. Koch,Myocardial adeno-associated virus serotype 6–bARKct gene therapy improves cardiac functionand normalizes the neurohormonal axis in chronic heart failure. Circulation 119, 89–98 (2009).
14. P. W. Raake, P. Schlegel, J. Ksienzyk, J. Reinkober, J. Barthelmes, S. Schinkel, S. Pleger, W. Mier,U. Haberkorn, W. J. Koch, H. A. Katus, P. Most, O. J. Müller, AAV6.bARKct cardiac gene therapyameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevantlarge animal heart failure model. Eur. Heart J. 34, 1437–1447 (2013).
15. H. Brinks, M. Boucher, E. Gao, J. K. Chuprun, S. Pesant, P. W. Raake, Z. M. Huang, X. Wang, G. Qiu,A. Gumpert, D. M. Harris, A. D. Eckhart, P. Most, W. J. Koch, Level of G protein–coupled receptorkinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mecha-nisms. Circ. Res. 107, 1140–1149 (2010).
16. M. Chen, P. Y. Sato, J. K. Chuprun, R. J. Peroutka, N. J. Otis, J. Ibetti, S. Pan, S. S. Sheu, E. Gao,W. J. Koch, Prodeath signaling of G protein–coupled receptor kinase 2 in cardiac myocytesafter ischemic stress occurs via extracellular signal–regulated kinase-dependent heatshock protein 90–mediated mitochondrial targeting. Circ. Res. 112, 1121–1134 (2013).
17. Q. Fan, M. Chen, L. Zuo, X. Shang, M. Z. Huang, M. Ciccarelli, P. Raake, H. Brinks, K. J. Chuprun,G. W. Dorn II, W. J. Koch, E. Gao, Myocardial ablation of G protein–coupled receptor kinase 2(GRK2) decreases ischemia/reperfusion injury through an anti-intrinsic apoptotic pathway.PLOS One 8, e66234 (2013).
18. D. M. Thal, K. T. Homan, J. Chen, E. K. Wu, P. M. Hinkle, Z. M. Huang, J. K. Chuprun, J. Song,E. Gao, J. Y. Cheung, L. A. Sklar, W. J. Koch, J. J. Tesmer, Paroxetine is a direct inhibitor of Gprotein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem. Biol.7, 1830–1839 (2012).
19. E. Gao, Y. H. Lei, X. Shang, Z. M. Huang, L. Zuo, M. Boucher, Q. Fan, J. K. Chuprun, X. L. Ma,W. J. Koch, A novel and efficient model of coronary artery ligation and myocardial infarction inthe mouse. Circ. Res. 107, 1445–1453 (2010).
20. H. Kirchherr, W. N. Kühn-Velten, Quantitative determination of forty-eight antidepressantsand antipsychotics in human serum by HPLC tandem mass spectrometry: A multi-level,single-sample approach. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 843, 100–113(2006).
21. J. H. Meyer, A. A. Wilson, N. Ginovart, V. Goulding, D. Hussey, K. Hood, S. Houle, Occupancyof serotonin transporters by paroxetine and citalopram during treatment of depression: A[11C]DASB PET imaging study. Am. J. Psychiatry 158, 1843–1849 (2001).
22. T. C. Tasker, C. M. Kaye, B. D. Zussman, C. G. Link, Paroxetine plasma levels: Lack of correlationwith efficacy or adverse events. Acta Psychiatr. Scand. Suppl. 350, 152–155 (1989).
23. P. W. Raake, L. E. Vinge, E. Gao, M. Boucher, G. Rengo, X. Chen, B. R. DeGeorge Jr., S. Matkovich,S. R. Houser, P. Most, A. D. Eckhart, G. W. Dorn II, W. J. Koch, G protein–coupled receptor kinase2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure.Circ. Res. 103, 413–422 (2008).
24. L. M. Casey, A. R. Pistner, S. L. Belmonte, D. Migdalovich, O. Stolpnik, F. E. Nwakanma, G. Vorobiof,O. Dunaevsky, A. Matavel, C. M. Lopes, A. V. Smrcka, B. C. Blaxall, Small molecule disruption ofGbg signaling inhibits the progression of heart failure. Circ. Res. 107, 532–539 (2010).
25. A. Lymperopoulos, G. Rengo, H. Funakoshi, A. D. Eckhart, W. J. Koch, Adrenal GRK2 upreg-ulation mediates sympathetic overdrive in heart failure. Nat. Med. 13, 315–323 (2007).
26. F. A. Kamal, D. M. Mickelsen, K. M. Wegman, J. G. Travers, J. Moalem, S. R. Hammes, A. V. Smrcka,B. C. Blaxall, Simultaneous adrenal and cardiac G-protein–coupled receptor-Gbg inhibitionhalts heart failure progression. J. Am. Coll. Cardiol. 63, 2549–2557 (2014).
ceTranslationalMedicine.org 4 March 2015 Vol 7 Issue 277 277ra31 10

R E S EARCH ART I C L E
27. K. T. Homan, E. Wu, M. W. Wilson, P. Singh, S. D. Larsen, J. J. Tesmer, Structural and func-tional analysis of G protein–coupled receptor kinase inhibition by paroxetine and a ration-ally designed analog. Mol. Pharmacol. 85, 237–248 (2014).
28. M. Ciccarelli, J. K. Chuprun, G. Rengo, E. Gao, Z. Wei, R. J. Peroutka, J. I. Gold, A. Gumpert,M. Chen, N. J. Otis, G. W. Dorn II, B. Trimarco, G. Iaccarino, W. J. Koch, G protein–coupledreceptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance aftermyocardial ischemia. Circulation 123, 1953–1962 (2011).
29. Z. M. Huang, E. Gao, F. V. Fonseca, H. Hayashi, X. Shang, N. E. Hoffman, J. K. Chuprun, X. Tian,D. G. Tilley, M. Madesh, D. J. Lefer, J. S. Stamler, W. J. Koch, Convergence of G protein–coupled receptor and S-nitrosylation signaling determines the outcome to cardiac ischemicinjury. Sci. Signal. 6, ra95 (2013).
30. R. Mandrioli, L. Mercolini, M. A. Saracino, M. A. Raggi, Selective serotonin reuptake inhibitors(SSRIs): Therapeutic drug monitoring and pharmacological interactions. Curr. Med. Chem. 19,1846–1863 (2012).
31. S. Gibiino, A. Serretti, Paroxetine for the treatment of depression: A critical update. ExpertOpin. Pharmacother. 13, 421–431 (2012).
32. S. S. Gottlieb, W. J. Kop, S. A. Thomas, S. Katzen, M. R. Vesely, N. Greenberg, J. Marshall, M. Cines,S. Minshall, A double-blind placebo-controlled pilot study of controlled-release paroxetine ondepression and quality of life in chronic heart failure. Am. Heart J. 153, 868–873 (2007).
33. A. Chittaranjan, K. B. Chethan, S. Sandarsh, Cardiovascular mechanisms of SSRI drugs andtheir benefits and risks in ischemic heart disease and heart failure. Int. Clin. Psychopharmacol.28, 145–155 (2013).
34. M. A. Silver, Depression and heart failure: An overview of what we know and don’t know.Cleve. Clin. J. Med. 77 (Suppl. 3), S7–S11 (2010).
www.Scien
35. Y. Kitagawa, S. Adachi-Akahane, T. Nagao, Determination of beta-adrenoceptor subtype onrat isolated ventricular myocytes by use of highly selective beta-antagonists. Br. J. Pharmacol.116, 1635–1643 (1995).
Acknowledgments: We thank Z. Qu and S. Baxter for technical assistance. Funding: This workwas supported by a Brody Family Medical Trust Fund Fellowship (S.M.S.) and NIH grantsHL086865 (J.J.G.T.), R37 HL061690 (W.J.K.), P01 HL08806 (W.J.K.), P01 HL075443 (W.J.K.), andP01 HL091799 (W.J.K, W.Z., and A.M.F). Author contributions: S.M.S. and W.J.K. wrote themanuscript and designed the experiments. S.M.S. conducted most of the experiments andanalyzed the data. E.G. performed the sham and MI surgeries as well as in vivo hemodynamics.S.M.S. and E.G. performed statistical analysis. W.Z. prepared membrane fractions and per-formed the radioligand binding assay and measurements. X.C. isolated adult mouse cardio-myocytes and performed and analyzed the single myocyte contraction assay. W.Z., X.C., J.K.C.,A.M.F., J.J.G.T., and W.J.K. provided intellectual guidance and manuscript revision. Competinginterests: The authors declare that they have no competing interests.
Submitted 7 October 2014Accepted 16 January 2015Published 4 March 201510.1126/scitranslmed.aaa0154
Citation: S. M. Schumacher, E. Gao, W. Zhu, X. Chen, J. K. Chuprun, A. M. Feldman, J. J. G. Tesmer,W. J. Koch, Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodelingafter myocardial infarction. Sci. Transl. Med. 7, 277ra31 (2015).
ceTranslationalMedicine.org 4 March 2015 Vol 7 Issue 277 277ra31 11
on J
uly
24, 2
015
Dow
nloa
ded
from

DOI: 10.1126/scitranslmed.aaa0154, 277ra31 (2015);7 Sci Transl Med
et al.Sarah M. Schumacherremodeling after myocardial infarctionParoxetine-mediated GRK2 inhibition reverses cardiac dysfunction and
Editor's Summary
therapy could be an additional or even additive strategy for treating heart failure.-blockers. If these data hold true in humans, paroxetineβenhanced in the presence of current standard-of-care
after myocardial infarction in a mouse model. These affects are separate from its SSRI functions and are further report that paroxetine can block or even reverse heart damageet al.to heart failure progression. Now Schumacher coupled receptor kinase 2 (GRK2), which is thought to contribute−binding protein)−(heterotrimeric guanine nucleotide
inhibitor (SSRI) paroxetine, which is used as an antidepressant, has been shown to selectively inhibit G protein has both economic and safety advantages over new drug development. The selective serotonin reuptake−−diseases approved drugs to treat additional−extending currently Food and Drug Administration (FDA)−−Drug repurposing
Taking antidepressants to heart
/content/7/277/277ra31.full.htmlcan be found at:
and other services, including high-resolution figures,A complete electronic version of this article
/content/suppl/2015/03/02/7.277.277ra31.DC1.html can be found in the online version of this article at: Supplementary Material
http://stm.sciencemag.org/content/scitransmed/6/224/224ra27.full.html http://stm.sciencemag.org/content/scitransmed/6/239/239rv1.full.html http://stm.sciencemag.org/content/scitransmed/7/270/270ra6.full.html
can be found online at:Related Resources for this article
http://www.sciencemag.org/about/permissions.dtl in whole or in part can be found at: article
permission to reproduce this of this article or about obtaining reprintsInformation about obtaining
is a registered trademark of AAAS. Science Translational Medicinerights reserved. The title NW, Washington, DC 20005. Copyright 2015 by the American Association for the Advancement of Science; alllast week in December, by the American Association for the Advancement of Science, 1200 New York Avenue
(print ISSN 1946-6234; online ISSN 1946-6242) is published weekly, except theScience Translational Medicine
on J
uly
24, 2
015
Dow
nloa
ded
from
Related Documents











