ELSEVIER Biochimica et Biophysica Acta 1288 (1996) F37-F54 BB Biochi~ic~a et Biophysica A~ta P-glycoprotein multidrug resistance and cancer Irene Bosch, James Croop * Division of Pediatric Oncology, Dana-Farber Cancer Institute, Children's Hospital, Harvard Medical School, 44 Binney St., Boston MA 02115, USA Received 5 June 1996; accepted 7 June 1996 Contents 1. Introduction ................................................... F37 2. Isolation and characterization of the P-glycoprotein .............................. F38 3. P-glycoprotein functional activity ........................................ F39 4. P-glycoprotein expression in normal tissues .................................. F42 5. P-glycoprotein gene regulation ......................................... F43 6. P-glycoprotein expression in tumors ...................................... F44 7. Inhibition of multidrug resistance ........................................ F45 8. Clinical trials to circumvent multidrug resistance ............................... F47 9. Future directions ................................................. F48 10. Conclusion .................................................... F48 Acknowledgement .................................................. F49 References ...................................................... F49 1. Introduction The resistance of human malignancy to multiple chemotherapeutic agents remains a major obstacle in can- cer therapy. Resistance has been termed intrinsic when the tumor initially fails to respond to any of a wide range of anticancer agents. When tumors initially respond only to return refractory to many agents, the resistance is de- * Corresponding author. Fax: + 1 (617) 6322085; E-mail: j ames_croop @ dfci.harvard.edu scribed as acquired. Our understanding of the mechanisms responsible for drug resistance in tumors has improved dramatically over the past few years. Most organisms appear to have developed an array of strategies to protect themselves from noxious environmental compounds. A variety of these specific genetic changes have been identi- fied which are capable of protecting cancer cells from multiple chemotherapeutic agents. These include alteration in the cellular target as exemplified by decreased func- tional topoisomerase II activity due to either somatic muta- tions or decreased expression, increased inactivation of drugs by glutathione-S-transferase activity, aberrations in 0304-419X/96/$15.00 Copyright © 1996 Elsevier Science B.V. All rights reserved. PH S0304-419X(96)00022-4

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

ELSEVIER Biochimica et Biophysica Acta 1288 (1996) F37 -F54
BB Biochi~ic~a et Biophysica A~ta
P-glycoprotein multidrug resistance and cancer
Irene Bosch, James Croop * Division of Pediatric Oncology, Dana-Farber Cancer Institute, Children's Hospital, Harvard Medical School, 44 Binney St., Boston MA 02115, USA
Received 5 June 1996; accepted 7 June 1996
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F37
2. Isolation and characterization of the P-glycoprotein . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F38
3. P-glycoprotein functional activity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F39
4. P-glycoprotein expression in normal tissues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F42
5. P-glycoprotein gene regulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F43
6. P-glycoprotein expression in tumors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F44
7. Inhibit ion of mult idrug resistance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F45
8. Clinical trials to circumvent mult idrug resistance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F47
9. Future directions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F48
10. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F48
Acknowledgement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F49
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . F49
1. Introduction
The resistance of human malignancy to multiple chemotherapeutic agents remains a major obstacle in can- cer therapy. Resistance has been termed intrinsic when the tumor initially fails to respond to any of a wide range of anticancer agents. When tumors initially respond only to return refractory to many agents, the resistance is de-
* Cor responding author. Fax: + 1 (617) 6322085; E-mail :
j ames_croop @ dfci.harvard.edu
scribed as acquired. Our understanding of the mechanisms responsible for drug resistance in tumors has improved dramatically over the past few years. Most organisms appear to have developed an array of strategies to protect themselves from noxious environmental compounds. A variety of these specific genetic changes have been identi- fied which are capable of protecting cancer cells from multiple chemotherapeutic agents. These include alteration in the cellular target as exemplified by decreased func- tional topoisomerase II activity due to either somatic muta- tions or decreased expression, increased inactivation of drugs by glutathione-S-transferase activity, aberrations in
0 3 0 4 - 4 1 9 X / 9 6 / $ 1 5 . 0 0 Copyright © 1996 Elsevier Science B.V. All rights reserved. PH S 0 3 0 4 - 4 1 9 X ( 9 6 ) 0 0 0 2 2 - 4

F38 I. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
apoptosis pathways affecting the mechanisms of cell death, and increased efflux of cytotoxic compounds out of the cell due to increased expression of membrane transport proteins such as the P-glycoprotein (MDR1, PGY1) and the multidrug resistance associated protein (MRP). The role of each of these forms of drug resistance in the clinical outcome of cancer chemotherapy is the subject of intense research. P-glycoprotein overexpression is perhaps the best characterized of these mechanisms and strategies to circumvent P-glycoprotein functional activity have pro- gressed to the point where they are currently being evalu- ated in clinical practice [1-5]. An understanding of how P-glycoprotein has developed to one of the best currently available targets to improve cancer therapy highlights the challenges and problems which face the transition of bench research to its use at the bedside.
2. Isolation and characterization of the P-glycoprotein
Although drug resistance is intrinsically difficult to study in patients with cancer, cell lines developed in vitro have provided significant insights on the mechanisms of multidrug resistance. Cell lines which are highly resistant to a variety of anticancer agents have been generated by slowly increasing the concentration of a cytotoxic agent in a step-wise fashion [6]. This approach selects for sponta- neous or drug induced genetic alterations which provide a growth advantage in the selecting agent. A common phe- nomenon noted in cell lines selected with single, natural product cytotoxic agents was that these lines were often cross resistant to a well-defined spectrum of structurally diverse compounds with distinct cellular targets. These agents included many of the most active anticancer com- pounds - - the anthracyclines, the Vinca alkaloids, and actinomycin D (Table 1). Many, but not all, of the cyto- toxic compounds in the multidrug resistance phenotype are relatively small, lipophilic and cationic at physiological pH, however, the precise determinants for inclusion remain to be identified. Initial analysis of the cell lines indicated decreased accumulation of cytotoxic compounds [7,8], karyotypic abnormalities - - double minute chromosomes and homogeneous staining regions - - consistent with gene amplification [9-t2], and overexpression of a 170 kDa membrane glycoprotein, termed the P-glycoprotein [13]. Early observations also indicated that certain compounds appeared to be able to reverse multidrug resistance, restor- ing sensitivity of the resistant cells to cytotoxic compounds [14]. These cell lines represented key reagents for uncover- ing the basis of mutidrug resistance and could be manipu- lated to identify potentially useful agents with the promise of inhibiting clinical drug resistance. The suggestion that the gene was amplified and overexpressed directed the initial approaches to identify the molecular mechanism of multidrug resistance.
Table 1 P-glycoprotein substrates included in the multidrug resistance phenotype
Doxorubicin Daunorubicin Mitoxantrone Idarubicin Vinblastine Vincristine Colchicine Taxol Etoposide Tenoposide Actinomycin D Puromycin Valinomycin Mithramycin Gramacidin D Emetine Rhodamine 123 Ethidium bromide DiOC2 Hoeschst 33342 Lipophilic peptides Hydrophobic acetoxymethyl esters Ether lipids Cortisol [ 99 m Tc]Sestamibi
Molecular probes of the gene responsible for multidrug resistance were first isolated using a in-gel renaturation technique which detected amplified fragments of DNA [15]. Characterization of two independently derived mul- tidrug resistant Chinese hamster cell lines by in-gel renatu- ration identified approx. 150 kbp of amplified DNA not present in the parental cell lines [16]. A small amplified fragment was subcloned and used to isolate a contiguous 140 kbp region of amplified DNA [17]. The cloned, ampli- fied domain encoded sequences which recognized a 5 kbp mRNA overexpressed in the multidrug resistant cell lines. These genetic sequences were conserved across species and cross hybridized with human genomic DNA [18]. Probes from the amplified domain were used to isolate cDNAs from libraries derived from both murine and hu- man cell lines [19,20].
Similar cDNAs were isolated from multidrug resistance cell lines using a variety of techniques. Monoclonal anti- bodies generated against the P-glycoprotein [21] were used to screen an expression library generated from a Chinese hamster multidrug resistant cell line to successfully iden- tify a cDNA which hybridized to amplified genomic DNA and a 4.5 kbp mRNA overexpressed in multidrug resistant cell lines [22]. Likewise, cDNAs were identified using differential hybridization techniques for characterizing mRNAs overexpressed in multidrug resistant cell lines [23-25]. Comparative analysis indicated that the cDNAs represented species specific homologs of the P-glyco- protein [26].

L Bosch, J. Croop/Biochimica et Biophysica Acta 1288 (1996) F37-F54 F39
Transfection of the full length cDNAs isolated from libraries of either non-resistant [19,27,28] or drug resistant [29] cell lines into drug sensitive cells were both effective in conveying drug resistance, indicating that mutations were not required for the generation of multidrug resis- tance. Drug resistance correlated with overexpression of the recombinant RNA and a 170 kDa protein as well as decreased intracellular accumulation of cytotoxic com- pounds. Most transfection strategies included selection with a multidrug resistance substrate to achieve high levels of P-glycoprotein expression. However, even clones identi- fied without selection in cytotoxic agents displayed levels of drug resistance proportional to the amount of P-glyco- protein expressed, indicating that drug selection was not a prerequisite for induction of multidrug resistance [30,31].
The amino acid sequence derived from the cDNAs predicted an integral membrane glycoprotein with a tool wt. of approx. 140 kDa. This was consistent with the biochemical characterization of P-glycoprotein from mul- tidrug resistant cell lines. P-glycoprotein migration in poly- acrylamide gels is dependent on gel composition and sample preparation and is resolved at a mol wt. of 130-180 kDa which includes 10-15 kDa of N-linked glycosylation [32,33]. Hydrophobicity analysis suggested that the first half of the polypeptide was comprised of six transmem- brane domains followed by a large cytoplasmic domain which included the consensus for an ATP binding fold (Fig. 1). This nucleotide binding domain is comprised of two motifs separated by approximately 100 amino acids and is found in a wide range of ATPases [34]. The structure of the first half was repeated in the second half of the polypeptide with over 65% similar amino acid residues [35] suggesting duplication and /or fusion of ancestral moieties [36,37]. A short section which acts as a 'linker' region between the two halves of the polypeptide includes the consensus sequences for a variety of protein kinases. Although the topography based on the hydrophobicity analysis has recently been challenged based upon in vitro fusion proteins [38-41], functional analysis of recombi- nant, hemaglutinin tagged P-glycoprotein in drug resistant cells supports the original model [42].
Outside " ~ A
Fig. 1. Schematic representation of P-glycoprotein. Glycosylation sites in first external domain, phosphorylation sites in linker region O, and nucleotide binding folds identified. Adapted from Refs. [43] and [35].
Table 2 P-glycoprotein gene nomenclature
Species Class I Class II Class III
Human MDR 1 MDR2/3 Mouse mdr3 mdr l mdr2
mdr l a mdr l b mdr2 Hamster pgp l pgp2 pgp3
Nomenclature used by different groups. Class I and II convey multidrug resistance. Class tlI encodes a phosphatidylcholine flippase.
The molecular probes for the P-glycoprotein were ini- tially noted to cross hybridize with a series of genomic DNA fragments which were ultimately identified as encod- ing additional members of the P-glycoprotein multigene family. Three family members were identified in rodents [43-46] while only two were found in humans [20,47] and have been designated P-glycoprotein classes I, II, and lli based upon functional and sequence similarity [48] (Table 2). Rodent classes I and II and human class I convey multidrug resistance, while class III does not. All of the mammalian P-glycoproteins display a high level of amino acid identity (69-94%), with rodent classes I and II the highest [35]. A class II human P-glycoprotein does not appear to exist suggesting duplication of the rodent class I P-glycoprotein after divergence of primates and rodents [49].
A comparison of the predicted protein sequence of the P-glycoprotein with the genetic databases indicates that P-glycoprotein is included in a large superfamily of trans- port proteins termed ATP Binding Cassette (ABC) Trans- porters [50] or Traffic ATPases [51]. Members of this transport superfamily display high amino acid similarity in the 200 amino acids surrounding the ATP binding folds and utilize a functional transport unit comprised of approx- imately 12 membrane spanning domains and two cytoplas- mic domains each containing ATP binding consensus se- quences. These structural elements can be included in a single polypeptide as in the P-glycoprotein, as homo/het- erodimers or as 3-4 polypeptides encoded in multicompo- nent operons which assemble in the membrane to form the transport unit [35]. Proteins included in this superfamily transport a wide range of substrates including nutrients, ions, peptides, antibiotics and other toxins across a variety of membranes. Inclusion in this superfamily of transport proteins initially implicated the involvement of P-glyco- protein in the export of cytotoxic compounds out of the cell.
3. P-glycoprotein functional activity
Identification of the P-glycoprotein as the member of a superfamily of transport proteins and the decreased accu- mulation of cytotoxic agents in multidrug resistant cell lines suggested a direct role in the efflux of chemothera-

F40 I. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
peutic agents out of the cells. A large body of circumstan- tial and direct evidence continues to confirm this hypothe- sis, however, other possibilities have been entertained.
The vast majority of P-glycoprotein is located at the cell surface with smaller amounts associated with the Golgi apparatus [52,53]. Studies evaluating the binding of cyto- toxic drugs to plasma membranes from multidrug resistant cell lines have provided substantial evidence for a direct interaction between these compounds and the P-glyco- protein. The binding of [3H]vinblastine was enhanced in membrane vesicles from multidrug resistant cell lines when compared to preparations from drug sensitive cell lines [54]. The binding was saturable, temperature and trypsin sensitive, and could be inhibited by other cytotoxic com- pounds and multidrug resistance reversal agents [55]. The binding site in the multidrug resistant membranes was identified as a 150-180 kDa protein using a radioactive, photoactivatable vinblastine analog [56,57]. The identity of this protein as the P-glycoprotein was confirmed by im- munoprecipitation with specific antibodies. This direct in- teraction of the P-glycoprotein and cytotoxic agents has been extended with photoaffinity probes of transport sub- strates [58] and reversal agents such as [3H]azidopine [59,60], an analog of verapamil, [125I]iodoarylazidoprazo- sin [61], an analog of the transport substrate prazosin, iodoarylazidoforskolin [62] and progesterone [63]. These studies have shown a consistent rank order for the ability of cytotoxic compounds to displace the photoaffinity probe. The Vinca alkaloids are consistently the most potent, followed by actinomycin D, the anthracyclines, and colchicine. Reversal agents such as verapamil and cy- closporin A are quite effective inhibitors of photolabelling
and some compounds can enhance binding. Studies di- rected at determining the specific binding domains have indicated that both halves of the P-glycoprotein interact with the photoaffinity probes in the region of transmem- brane domains 6 and 12 based on peptide digests [64-68]. The common rank order and similar binding sites has suggested that there may be a single binding domain for many or even all of the compounds which interact with P-glycoprotein. However, verapamil, azidopine and quini- dine inhibit in a non-competitive manner while cy- closporine A is a competitive inhibitor of P-glycoprotein binding, suggesting that drug binding is complex and may involve multiple sites [69-72].
A variety of P-glycoprotein point mutations have been identified which affect drug binding and drug resistance profiles. The specificity acquired by these differences in primary structure lend further support for a direct interac- tion between P-glycoprotein and transport substrates. A common observation found with the point mutations is a differential effect on drug resistance with cells expressing the mutant protein being more resistant to some cytotoxic compounds, while being more sensitive to others (Table 3). The majority of mutations affecting drug specificity are located within or adjacent to a transmembrane domain. A glycine to valine mutation at position 185, in the first cytoplasmic loop just proximal to transmembrane domain three was identified in a highly resistant cell line selected in colchicine [73]. This somatic point mutation increased the relative resistance to colchicine but decreased the resistance to vinblastine. Cells overexpressing the mutant P-glycoprotein displayed an associated decrease in colchicine accumulation and a relative increase in vinblas-
Table 3 P-glycoprotein mutational analysis
Amino acid Pgp class Location Phenotype Reference
G ~ V Human I Cytoplasmic Loop 1 Vbl $, Col 1", Adr T, Act $ [73,74] 185
N --* S / G ~ V a Human I Cytoplasmic Loop 1 Vbl -, Col ]', Adr -, Act - [76] 183 185
G ~ V ~ Human I Cytoplasmic Loops 1 or 4 Vbl -, Col $, Adr T, Act $ [77] 141,187,188,812or830
S ~ F Mouse I T M l l Vbl ~,, Col $, Adr $, Act $ [78] 939
S ~ F Mouse II T M I I Vbl $, Col $, Adr $, Act $ [78] 941
G ~ A / A ~ P ~ Hamster I TM6 Vin $, Col $, Daun $, Act 1' [80,81] 338 339
P ~ A Human I TM4 Vbl -, Col ,1,, Adr $, Act $ [82] 223
P ~ A Human I TMI0 Vbl -, Col $, Adr $, Act ~ [82] 866
K ~ R Human I NBDI Vbl -, Col $, Adr '~ [83] 536
ERGA -~ DKGT Mouse I NBD1 Vbl $, Col $, Adr $, Act $ [84] (522 - 525)
T ~ C Mouse I NBD I Vbl -, Col $, Adr J,, Act $ [84] 578
Single letter amino acid codes are used to delineate residues. TM, transmembrane segment; NBD, nucleotide binding domain; Vbl, vinblastine; Col, colchicine; Adr, Adriamycin; Act, actinomycin D; Daun, daunorubicin; Pgp, P-glycoprotein. a Simulateneous mutation at both sites. b Mutation at any of these sites.

1. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54 F41
tine accumulation compared to cells expressing wild type P-glycoprotein. The mutant P-glycoprotein displayed in- creased binding of photoactive analogs of vinblastine and verapamil but decreased binding of a photoactive colchicine analog [74]. This has been interpreted to suggest that this mutation is associated with increased release of colchicine, but not vinblastine, from the cell to the extracellular space, implying discrete drug binding and release sites. In addi- tion to altering the specificity of drug resistance, this mutation also affects the ability of several reversal agents to differentially circumvent resistance to specific cytotoxic compounds, further supporting a direct interaction between P-glycoprotein and drug transport [75]. The importance of this region was further defined by replacing this first cytoplasmic loop and transmembrane domains 3 and 4 of human MDR1 P-glycoprotein with the analogous region from human MDR3 [76]. This created a chimeric P-glyco- protein with 17 amino acid differences that did not display drug resistance. The restoration of only four specific amino acids, all within the first cytoplasmic loop, back to the original MDR1 residues restored drug resistance. Further- more, the systematic substitution of each glycine to valine throughout the P-glycoprotein identified five sites which increased the relative resistance to colchicine and adriami- cyn without affecting resistance to vinblastine [77]. These sites were all located within cytoplasmic loops, three of which were adjacent to hydrophobic domains. These ob- servations suggest that the cytoplasmic loop/transmem- brane boundary and specifically, glycines in the cytoplas- mic loops contribute to P-glycoprotein drug specificity.
Similar observations have been made on mutations found within transmembrane domains. A serine to phenyl- alanine mutation, initially identified as reverse transcrip- tase error, in either mouse m d r l or m d r 3 within trans- membrane domain 11 produced an overall decrease in drug resistance that was very strong for colchicine and Adri- amycin but only modest for vinblastine [78]. There was also differential inhibition of drug resistance with a variety of reversal agents further confirming that their specificity is closely linked to cytotoxic drug binding sites [79]. Similarly, a tandem pair of substititions in transmembrane domain 6 of P-glycoprotein from highly resistant hamster cells selected with actinomycin D affects the cross resis- tance phenotype [80,81]. There was an overall decrease in vincristine, colchicine and daunorubicin resistance with an increase in resistance to actinomycin D. Similarly, replac- ing transmembrane domain 12, was the corresponding region of MDR3 impairs resistance to actinomycin D, vincristine, and doxorubicin but not colchicine [64]. Fi- nally, the systematic substitution of alanine for each of the prolines in the P-glycoprotein identified residues in trans- membrane domains 4 and 10 which drastically reduced resistance to colchicine, Adriamycin and actinomycin D, but retained resistance to vinblastine [82].
Although the large cytoplasmic domains which include the nucleotide binding folds have traditionally been consid-
ered the structural elements required for the energy pro- duction necessary for substrate translocation, recent evi- dence suggests that this region, too, may participate di- rectly or indirectly in substrate recognition. While analyz- ing mutations introduced in P-glycoprotein that are found in CFTR, a mutation of lysine to arginine at position 536 increased colchicine and Adriamycin resistance with little change in vinblastine sensitivity [83]. This site is a highly conserved domain of the ABC transporters, just proximal to the distal half of the nucleotide binding consensus in the amino half of protein. In addition, site directed mutagene- sis of the region surrounding the distal portion of the nucleotide binding fold in the amino half of the P-glyco- protein, identified two substitutions which decreased drug resistance to colchicine, Adriamycin, and actinomycin D, but had little effect on vinblastine [84]. These observations are consistent with the proposal that the regions between the motifs which define the nucleotide binding fold pro- vide the link between the ATPase activity and the specific functional activities of the polypeptide [85]. Taken to- gether, they indicate that P-glycoprotein primary structure is responsible for the recognition, specificity, and binding necessary for drug transport.
Early studies indicated that P-glycoprotein mediated multidrug resistance was energy dependent. The finding that two nucleotide binding folds associated with a wide range of ATPases were present in the primary sequence supported the idea that the P-glycoprotein encoded a mem- brane protein which binds [86] and utilizes ATP in the transport of substrates across the plasma membrane. P- glycoprotein immunoprecipitated from mammalian mul- tidrug resistant cell lines could be shown to display AT- Pase activity [87]. However, it was not until studies on insect cells expressing recombinant P-glycoprotein from baculoviral vectors that high levels of drug induced AT- Pase activity could be demonstrated [88]. A variety of cytotoxic substrates and reversal agents were shown to stimulate ATPase activity 2- to 5-fold. The ability to stimulate P-glycoprotein ATPase activity appears to be dependent on multiple parameters involved in substrate binding since both the specific substrate and the primary structure can influence stimulation. This is indicated by increased colchicine stimulated ATPase activity in mutant P-glycoprotein with the glycine to valine mutation at posi- tion 185 [89]. Purified P-glycoprotein reconstituted into proteoliposomes has similarly been shown to display drug stimulated ATPase activity [90-93]. ATPase activity was shown to be very sensitive to the detergents used during the extractions and the lipid composition of the bilayer suggesting that the tertiary structure is critical for optimal activity [94-96].
The movement of a variety of substances across plasma membranes has provided evidence that P-glycoprotein is directly responsible for the transport of cytotoxic com- pounds. Plasma membrane vesicles from multidrug resis- tant cell lines could be shown to accumulate [3H]vinblas-

F42 1. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
tine in an ATP dependent fashion [97]. This transport could be inhibited best by vinblastine followed by vera- pamil, actinomycin, daunorubicin, and colchicine. This rank order is similar to that observed with displacement of photoaffinity probes of P-glycoprotein (see above). Trans- port of cytotoxic agents against a concentration gradient has been demonstrated in multidrug resistant cell lines [98] and has been calculated with recombinant P-glycoprotein in erythrocytes [99] and yeast vesicles [100,101]. Finally, purified P-glycoprotein reconstituted in proteoliposomes has been demonstrated to transport substrates into vesicles across a concentration gradient, providing direct support for it's function as an active transporter of cytotoxic compounds [102,103].
Although these observations provide strong evidence for P-glycoprotein mediated transport, this hypothesis has been challenged based upon the broad substrate specificity, relatively small differences of cytotoxic agent accumula- tion found between multidrug resistant and sensitive cell lines and the low level of efflux predicted from the experi- mental observations [104-106]. An alternative hypothesis has been suggested where P-glycoprotein decreases the membrane electrical potential with an accompanied in- crease in internal pH. This is proposed to indirectly reduce the intracellular accumulation of cationic drugs due to electrophysiological parameters as opposed to direct P- glycoprotein mediated transport [107]. Analyses with puri- fied P-glycoprotein should provide the answers required to settle this controversy as well as better define the mecha- nism of drug translocation.
Compounds that activate protein kinase C such as phor- bol-12-myristate 13-acetate (TPA) and phorbol 12,13-di- butyrate increase P-glycoprotein phosphorylation and aug- ment drug resistance with decreased drug accumulation [108-110]. Furthermore, inhibitors of protein kinases de- crease P-glycoprotein phosphorylation, sensitize the cells to cytotoxic agents, and cause an increase in drug accumu- lation [111-114]. Although the changes in drug resistance due to differences in phosphorylation are modest, small differences in transport activity may be clinically impor- tant for drugs with steep dose response curves. Initial attempts to mutate the protein kinase consensus sites indi- cate that phosphorylation is not required for drug transport [115], however, phosphorylation does appear to increase drug binding and ATPase activity [116]. Post-translation modifications affecting drug resistance have also been identified in genetically engineered mutants lacking N- glycosylation [117]. Although non-glycosylated P-glyco- protein did not alter the level or pattern of drug resistance, the efficiency of obtaining drug resistant clones was drasti- cally reduced suggesting an effect on protein processing or stability. These observations suggest that subtle post-trans- lational modifications can affect drug resistance The physiological processes which influence these modifica- tions may provide new targets to inhibit P-glycoprotein function.
Recently, the concept of P-glycoprotein functioning solely as a drug transporter has been expanded with the observations that it is also responsible for ion movement. It is possible to demostrate that multidrug resistant cell lines excrete higher levels of extracellular ATP than drug sensi- tive cells [118]. This excretion is proportional to the level of P-glycoprotein expression. Electrophysiologic measure- ments on cells transfected with the mouse m d r l cDNA indicate that the increased extracellular ATP is due to ATP permeation across the plasma membrane as an ion flux. ATP channel activity in ABC transporters remains contro- versial, however, with several groups demonstrating simi- lar activity for CFFR after cAMP activation [119,120] while others have failed to reproduce the observations [121]. These discrepancies may in part be due to differ- ences in the methodologies used for CFFR channel stimu- lation. Additionally, chloride channel activity was origi- nally proposed for P-glycoprotein [122]. However, this chloride movement has been suggested more recently to be secondary to P-glycoprotein regulation of a distinct chlo- ride channel [123]. Until the precise mechanism of P- glycoprotein transport is clarified and the three dimen- sional structure is solved, it remains unclear whether these activities indicate multiple functional properties or a com- mon translocation mechanism for disparate substrates.
4. P-glycoprotein expression in normal tissues
P-glycoprotein expression has been characterized in normal tissues utilizing gene specific probes to determine mRNA levels [124-126] and combinations of antibodies [125,127-129] capable of delineating the different P- glycoprotein classes. These analyses indicate that the P- glycoprotein is differentially expressed in normal mam- malian tissues, usually at secretory surfaces. P-glyco- protein classes I and/or II are expressed in high levels in the biliary canaliculi, the proximal tubules of the kidney, intestinal and colonic epithelium, pancreatic ducts, bronchial mucosa, prostatic epithelia, ovarian follicles, and pregnant uterine epithelium [ 130,131 ]. The P-glycoprotein is also expressed in a number of specialized tissues, most notably the cortical regions of the adrenal gland. Interest- ingly, in hamsters, this expression is limited to males, suggesting P-glycoprotein involvement in the transport of sex specific hormones [132]. High levels of P-glycoprotein have been demonstrated in capillaries found in the central nervous system, testes, uterus and skin, suggesting a possi- ble role in the blood brain barrier [133]. Hematopoietic stem cells [134] and specific lymphocyte subclasses also express high levels of P-glycoprotein [135,136]. Class III P-glycoprotein is expressed at highest levels in the liver, spleen, and kidney, as well as the murine adrenal gland and heart [124,137-139].
The physiologic function of the P-glycoprotein has been difficult to identify. A role has been proposed both as a

L Bosch, J. Croop/Biochimica et Biophysica Acta 1288 (1996) F37-F54 F43
protective mechanism against noxious xenobiotics and as transporter of endogenous substrates. The analysis of mice with disruption of the murine P-glycoproteins has provided significant insight into its normal physiological functions. Mice with the class I P-glycoprotein gene disrupted appear normal and are fertile [140]. These mice, however, devel- oped paralytic symptoms and died after the administration of the antihelmintic ivermectin when the mouse colony in which they were housed became infected with mites. Char- acterization of ivermectin biodistribution indicated that the brains of mice deficient in class I P-glycoprotein accumu- lated almost 100-fold more ivermectin than control mice. This observation was consistent with P-glycoprotein hav- ing a role in the maintenance or generation of the blood brain barrier. Most other tissues accumulated approxi- mately 2- to 5-fold more ivermectin than found in tissues from wild type mice. Similar biodistributions, although not quite as extreme, were obtained with vinblastine. This analysis has raised the concern that certain organs, espe- cially the brain, are at risk for increased toxicity from chemotherapeutic agents when used in conjuction with agents aimed at interfering with P-glycoprotein function in the clinical setting. Mice lacking class III P-glycoprotein, which does not convey drug resistance, display an abnor- mal hepatic architecture characterized by lobule degenera- tion and expansion of the biliary ducts with a portal inflammatory reaction [141]. The mice appear to be unable to secrete phospholipids into the bile. Further analysis has confirmed that class III P-glycoprotein functions as flip- pase transporting phosphotidylcholine from the inner to outer leaflet of the plasma membrane [ 142,143].
5. P-glycoprotein gene regulation
Several lines of evidence suggests that P-glycoprotein is regulated at the transcriptional level. There is a dramatic rise in P-glycoprotein expression in the secretory, uterine epithelium during pregnancy [130,131]. This induction can be achieved with a combination of progesterone and estro- gen [144]. In addition, P-glycoprotein expression increases during the menstrual cycle reaching highest levels during the mid-secretory phase paralleling nuclear progesterone receptor expression and serum progesterone levels [131]. Estrogen has also been shown to increase P-glycoprotein expression in rat pituitary tumor cells within 24 h of administration [145]. P-glycoprotein expression increases during liver regeneration following partial hepatectomy and in chemically induced hyperplastic and neoplastic liver nodules [146,147]. Xenobiotics which increase cytochrome P-450 also induce liver P-glycoprotein expression [146,148]. Extracellular matrix [149], heat shock, arsenite [150] and differentiation agents such as butyrate [151,152] can induce P-glycoprotein transcription in certain cell lines. Cytotoxic compounds, including those not transported by P-glycoprotein, can increase expression, suggesting a gen-
eralized stress response to noxious stimuli [153,154]. Fi- nally, the high levels of P-glycoprotein mRNA in mul- tidrug resistant cell lines are due to both gene amplifica- tion and increases in RNA transcription [155].
Characterization of P-glycoprotein gene expression has identified a number of regulatory promoter sequences and transcription factors which may be responsible for P-glyco- protein transcriptional regulation. Two promoters have been identified which produce P-glycoprotein transcripts in hu- man cells [156]. A distal, upstream promoter appears to be active in certain drug resistant cell lines. Most drug resis- tant cell lines and normal tissues, however, utilize what has been designated the proximal or downstream promoter. Basal levels of transcription have been shown to be con- trolled by a negative regulatory, GC-rich region located at - 120 to - 100 which increases transcription when deleted, the consensus for a Y-box at - 7 3 to - 8 2 , the binding sites for SP1 and early growth response factors (EGR) in another GC rich region located at - 5 0 to - 7 0 , and a transcription initiation site (Inr) required for accurate initi- ation of transcription [154,157-159]. A TATA sequence is not present in the promoter and multiple transcription start sites have been identified [156,160]. A consensus, desig- nated multiple start site element downstream (MED-1), has been identified in the P-glycoprotein gene which is also present in several other TATA-Iess genes with multiple transcription start sites. This element is differentially over- expressed when stably transfected in multidrug resistant cell lines, but like other P-glycoprotein promoter regions only gives basal transcription levels in transient assays [ 161 ]. The latter observation has suggested that chromoso- mal integration may be required for characterization of multidrug resistant transcriptional elements.
Cellular stimuli and specific transcription factors have been identified which increase P-glycoprotein promoter activity. A progesterone responsive region is present in the first untranslated exon of murine class II P-glycoprotein [162]. This activity is mediated by the A form of the progesterone receptor and may in part be responsible for the induction of P-glycoprotein in the uterus during preg- nancy [130,144]. Several studies have indicated that P- glycoprotein promoter activity may be modulated via sig- nal transduction pathways. Stimulation of P-glycoprotein promoter activity has been demonstrated with c-Ha-Ras-1 and mutant p53 [163]. Induction of P-glycoprotein through the Ras signal transduction pathway may be mediated by the nuclear factor for interleukin-6 (NF-IL6) [ 164]. NF-IL6, which is activated by Ras, was identified from an expres- sion library using a conserved consensus from the mam- malian promoter located at position - 1 5 7 to -125 . Raf kinase, part of the Ras pathway, has similarly been impli- cated in serum-inducible stimulation of P-glycoprotein ex- pression [165]. Raf responsive elements appear to modu- late Spl signaling by enhancing the transactivation poten- tial of prebound Spl complexes. The ability of EGR factors to stimulate P-glycoprotein promoter activity can

F44 L Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
be separated from the overlapping Spl site by 12-O-tetra- decanoylphorbol-13-acetate (TPA) induction [166]. These observations suggest that P-glycoprotein expression is modulated at least in part by signal transduction pathways involved in growth regulation.
6. P-glycoprotein expression in tumors
The development of molecular and immunologic probes to detect P-glycoprotein expression provided a means to analyze human tumors for expression of the gene. The optimal means to quantitate P-glycoprotein expression re- mains controversial, however. Molecular methods relying on RNA expression may be confounded by a lack of correlation between RNA levels and protein expression as well as inclusion of contaminating normal tissues and stroma expressing high levels of the gene. Immunologic methods have been hampered by concerns of sensitivity with low levels of expression, cross reactivity of the antibodies to other proteins [167-169], and the specific protocols required to maintain epitope reactivity. Further- more, differences observed between individual laboratories with the same reagents have made comparisons difficult. Nevertheless, not only have specific tumors been consis- tently shown to overexpress high levels of the P-glyco- protein, overexpression has been demonstrated to have a negative prognostic value for certain tumors. Furthermore, numerous anecdotal cases have been documented where tumors initially had low levels of P-glycoprotein expressed when they were responsive to therapy, only to recur refrac- tory to therapy with high levels of expression.
The initial surveys of tumors found high levels of P-glycoprotein expression in most tumors derived from tissues expressing P-glycoprotein, although a discrepancy was sometimes found between the level in the tumor and adjacent normal tissue [125,128,129,170-173]. This in- cluded most tumors of the adrenal gland, colon, kidney, liver, pancreas and acute myelogenous leukemia. Occa- sionally sarcomas, breast cancer, lymphomas, neuroblas- tomas, acute lymphocytic leukemia, and chronic myeloge- nous leukemia expressed high levels. The systematic eval- uation of specific tumor types has consistently found sub- groups of tumors expressing high levels of P-glycoprotein. The challenge remains, however, in proving a relationship to clinical outcome which has been confounded by incon- sistencies observed between investigation groups.
Hematopoietic malignancies have been extensively studied for the expression of P-glycoprotein, due to the availability of tumor specimens and the ability to rapidly assay malignant cells on the fluorescent activated cell sorter with antibodies such as MRK-16 [174], 4E3.16 [175], HYB421 [176], MM4.17 [177] and UIC2 [178] directed against epitopes expressed on the exterior of the cell. In addition, the antibodies C219 [21] and JSB-1 [129] which detect intracellular epitopes have been used on
cytospins and smears. However, concerns over sensitivity with low levels of P-glycoprotein expression as well as different criteria for 'positivity' based on the relative intensity and the number of positive cells have made comparisons between different laboratories difficult. Most studies of acute myelogenous leukemia have demonstrated that approximately 30-50% of patients have high levels of P-glycoprotein detected on their leukemic blasts at diagno- sis and 50-75% will have high levels at the time of relapse. Whether this represents selection of blasts express- ing P-glycoprotein or selection of patients whose blasts had high levels of expression in the group which failed therapy is not clear. Multiple independent studies, how- ever, have demonstrated that high levels of P-glycoprotein expression have a negative impact on both induction of remission and long term survival [179-188]. Examination of acute lymphoblastic leukemia specimens indicates that approximately 10-20% of patients have blasts expressing high levels of P-glycoprotein [ 180,189-192]. Several stud- ies have shown a correlation between P-glycoprotein ex- pression and poor outcome [193-196], but this has not been a consistent finding [197,198]. Finally, malignant cells from patients with multiple myeloma have been demonstrated to express high levels of P-glycoprotein which specifically appears to be related to drug resistance [ 199-202]. This expression of P-glycoprotein and myeloma is frequently acquired after exposure to vincristine and doxorubicin [203].
The expression of P-glycoprotein has been evaluated in patients with breast cancer. The majority of studies evalu- ating P-glycoprotein expression have indicated that a sub- population of breast cancers express high levels of P- glycoprotein which may play a role in the response to chemotherapy. High levels of P-glycoprotein expression has been detected in 20-70% of patient samples using methods to detect RNA or protein expression [ 170,200,204-218]. Several important points can be made in addition to the detection of varying levels of P-glyco- protein expression in breast cancer specimens. First, previ- ously treated cancers were consistently more likely to e x p r e s s h i g h l e v e l s o f P - g l y c o p r o t e i n [170,200,204,208,209,211]. Next, several studies demon- strated that P-glycoprotein expression in patient samples correlated with resistance to doxorubicin using short term tumor cell assays in vitro [200,207,209,215]. Finally, five studies established an inverse correlation of P-glycoprotein e x p r e s s i o n and the r e s p o n s e to t h e r a p y [205,208,211,215,218]. In spite of these observations, a number of studies could not demonstrate high levels of e x p r e s s i o n by R N A or p ro t e in a n a l y s e s [212,216,217,219,220]. The initial conclusions from the latter studies were that P-glycoprotein was either not re- sponsible for resistance in breast cancer or that more sensitive methodogies were required to detect differences in P-glycoprotein expression. Indeed, the studies which could not find a correlation between clinical outcome and

L Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54 F45
P-glycoprotein expression could detect only very low lev- els of P-glycoprotein in small numbers of specimens. The analysis of P-glycoprotein expression in breast cancer, once again, highlights the need for consistent assessment to draw definitive conclusions.
Evaluation of renal cell carcinoma indicates that 50- 100% of specimens express high levels of P-glycoprotein [125,170,221-226]. Expression is usually found at the apical surface of the tubular epithelium and is highest in the more differentiated tumors. The expression is usually found to be patchy throughout the tumor and diffuse cellular staining can be found in tumors with the highest expression. This correlation between P-glycoprotein ex- pression and histologic subtype does not translate to a correlation with tumor stage or outcome. However, expres- sion does seem to correlate with chemoresistance in vitro [222,227,228]. Interestingly, well differentiated tubules found in Wilms tumor, which has a much better prognosis, also express high levels of P-glycoprotein, which is consis- tent with the lack of correlation with outcome in renal cell carcinoma [224].
Examination of colon carcinoma similarly reveals high levels of P-glycoprotein expression in 40-90% of tumors at the apical surface of differentiated tubular structures [ 125,128,129,170,173,229-232]. In addition, cells express- ing high levels of P-glycoprotein have been identified at the leading edge of invasive carcinoma [233]. These latter tumors appeared to have a higher incidence of metastases suggesting that P-glycoprotein may be a marker for aggres- sive tumors. The overall poor outcome for these tumors, however, along with heterogeneous expression of P-glyco- protein has suggested that if expression of this protein is responsible for chemoresistance, then other mechanisms must also be active.
Several studies have shown high levels of P-glyco- protein in a subset of neuroblastomas [234-239]. Although only 6-30% of tumors express high levels of P-glyco- protein at diagnosis, 30-40% of tumors are positive after therapy. This observation has suggested that resistance to chemotherapy in neuroblastoma may at least be partially mediated by the P-glycoprotein. Consistent with this hy- pothesis, P-glycoprotein expression has been shown to be predictor of neuroblastoma outcome [238]. In one study, all of the 13 P-glycoprotein positive patients relapsed compared to only 22% of the 31 P-glycoprotein negative patients. Although the highest percentage of positive tu- mors were in the most advanced stages, log-rank analysis for age, N-myc, tumor stage and histology indicated that P-glycoprotein expression significantly affected the rates of relapse and mortality. A separate analysis of neuroblas- toma initially identified a correlation between MRP, but not P-glycoprotein, gene expression and poor clinical out- come [240]. Further analysis, however, indicated that high levels of P-glycoprotein gene expression did, in fact, corre- late with a poor outcome in patients whose tumors did not have N-myc amplification or who were over one year of
age [241]. These studies are tempered by a report which found a better prognosis in patients whose tumors had high levels of P-glycoprotein RNA transcripts. High levels of P-glycoprotein gene expression were found in 91% of the tumors, with highest levels in low stage tumors. P-glyco- protein expression was inversely correlated with N-myc expression [237]. Interestingly, and to be contrary, an in vivo experimental model of neuroblastoma suggests that P-glycoprotein expression correlates with up regulation of N-myc expression [242]. These observations suggest that differences in study populations and methodologies for detection of P-glycoprotein may account for some of the observed discrepancies.
The expression of P-glycoprotein in both soft tissue and osteosarcoma has been evaluated. High expression levels have been found in 0-30% of soft tissue sarcomas [235,243-248]. The wide range of expression has been attributed to different techniques with varying sensitivities and the relatively small numbers of patients evaluated. Detection of P-glycoprotein in childhood sarcomas has been shown to be prognostic for final outcome [244]. All nine patients whose tumors were positive relapsed, while only one of 20 patients with P-glycoprotein negative tu- mors relapsed. Relapse-free and overall survival remained statistically different after adjustment for stages and sites. P-glycoprotein expression has consistently been found in 30-50% of osteosarcomas [249-252]. Two studies found RNA expression more sensitive than immunological tech- niques to detect P-glycoprotein [250,251]. P-glycoprotein expression correlated with outcome in two independent studies [250,252]. Interestingly, P-glycoprotein expression did not correlate with the extent of tumor necrosis after preoperative chemotherapy, the current best indicator of survival [252]. Although the initial response to chemo- therapy did not appear to be related to P-glycoprotein expression, it expression could be used to identify patients whose tumors progressed while on chemotherapy. Whether P-glycoprotein is responsible for this chemoresistance in osteosarcoma, or is only a marker of cell behavior, remains to be clarified [253].
P-glycoprotein expression has been found in a variety of brain tumors [254-257], retinoblastoma [258], certain gastric and esophageal adenocarcinomas [259], and a small number of ovarian cancers [260-262]. The systematic evaluation of specific tumors continues to show subgroups which express high levels of P-glycoprotein with as many as 50% of tumors expressing high levels [3].
7. Inhibition of multidrug resistance
Identification of the P-glycoprotein as a mechanism for drug resistance provided a theoretical target to improve anticancer therapy. Since the original observation that verapamil was capable of circumventing drug resistance in cells overexpressing P-glycoprotein [14], an increasing
F46 I. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
number of compounds capable of reversing multidrug re- sistance continues to be identified (Table 4) [263,264]. Just as there is significant heterogeneity in the cytotoxic com- pounds included in the multidrug resistance phenotype, there is great variability in the structures and cellular targets of these compounds. Included in this diverse group of chemosensitizers are: calcium channel blockers such as verapamil and nifedipine; calmodulin antagonists; steroids such as progesterone and tamoxifen; immunosuppressive agents such as cyclosporin A and FK506; as well as other compounds such as dipyridamole, quinidine, chloroquine, reserpine and amiodarone. Their previously identified ac- tivities do not appear to play a role in the mechanism in which they reverse multidrug resistance. Most of these agents reverse drug resistance in in vitro cytotoxicity assays in the range of 2 -10 /zM and increase intracellular accumulation of cytotoxic agents by 2- to 5-fold. Binding assays demostrate a direct interaction between the reversal agents and P-glycoprotein (see above).
Recently, [99mTc]Sestamibi, a lipophilic, cationic radio-
Table 4
Compounds which inhibit P-glycoprotein mediated multidrug resistance
Verapamil Nifedipine/AHC-52 Diltiazem Niguldpine Dihydropyridine analogs Thioridazine Trifluoperazine Chlorpromazine Benzquinamide Progesterone Tamoxifen Megestrol acetate RU-486 Cyclosporin A
SDZ PSC 833 FK506
Rapamycin Chloroquine Quinidine Quinine Dipyridamole/BIBW 22
Quinoline/MS-209 Reserpine/Yohimbine Amiodarone Acridonecarboxamides/GF 120918 Amido-keto pipecolinates/VX-710 Fluorocyclopropyldibenzosuberane/LY335979 Triazinoaminopiperidine/S9788 Thioxanthenes Erythromycin Valinomycin Ceftriaxone Itraconazole N-Acetyldaunorubicin Vindoline L-Histidinol Tween 80
Solutrol HS
120 .~ O
_~ 80
~ 60
g 4o
g N 2O E.-
r ~
b-
0 V79 A D 77A A LZ / /
0 0.1 1 10 100 1000
[Verapamil] (uM)
,~ 120
.~ 10o
~ 80
~ 6O
40
2o ~z ~< o
~5
o v79 B [] 77A t, LZ
/
0 .1 1 10 100
[Cyclosporin A] uM )
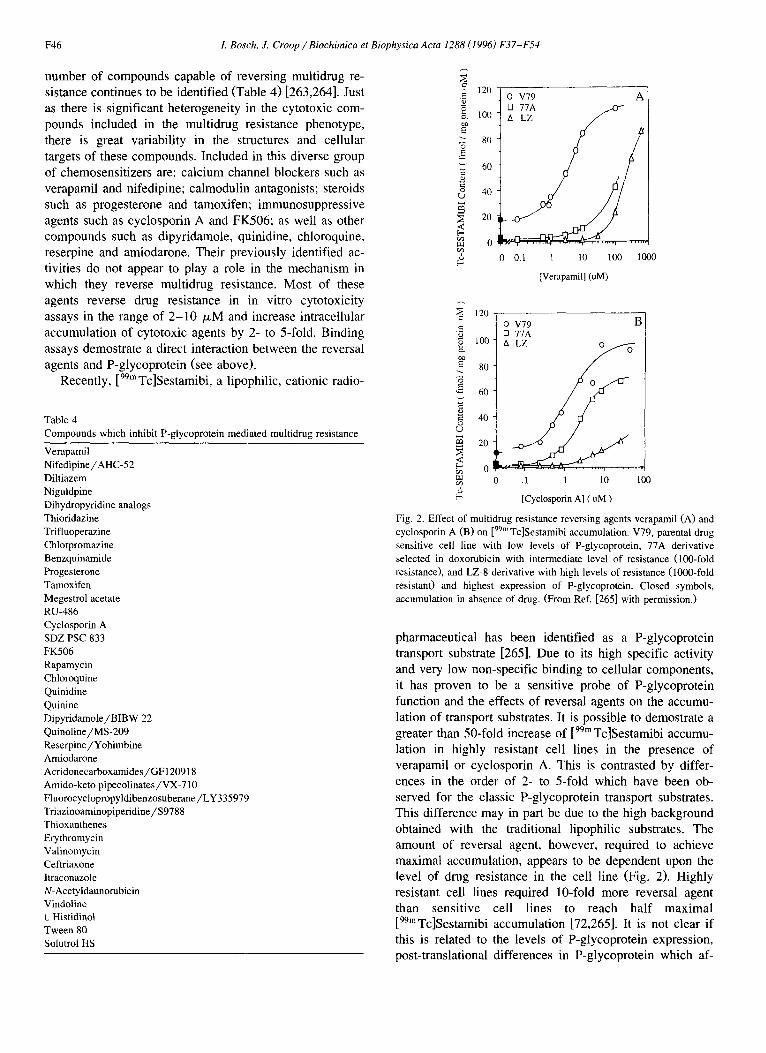
Fig. 2. Effect of multidrug resistance reversing agents verapamil (A) and cyclosporin A (B) on [99mTc]Sestamibi accumulation. V79, parental drug sensitive cell line with low levels of P-glycoprotein, 77A derivative selected in doxorubicin with intermediate level of resistance (100-fold resistance), and LZ-8 derivative with high levels of resistance (1000-fold resistant) and highest expression of P-glycoprotein. Closed symbols, accumulation in absence of drug. (From Ref. [265] with permission.)
pharmaceutical has been identified as a P-glycoprotein transport substrate [265]. Due to its high specific activity and very low non-specific binding to cellular components, it has proven to be a sensitive probe of P-glycoprotein function and the effects of reversal agents on the accumu- lation of transport substrates. It is possible to demostrate a greater than 50-fold increase of [99m Tc]Sestamibi accumu- lation in highly resistant cell lines in the presence of verapamil or cyclosporin A. This is contrasted by differ- ences in the order of 2- to 5-fold which have been ob- served for the classic P-glycoprotein transport substrates. This difference may in part be due to the high background obtained with the traditional lipophilic substrates. The amount of reversal agent, however, required to achieve maximal accumulation, appears to be dependent upon the level of drug resistance in the cell line (Fig. 2). Highly resistant cell lines required 10-fold more reversal agent than sensitive cell lines to reach half maximal [99mTc]Sestamibi accumulation [72,265]. It is not clear if this is related to the levels of P-glycoprotein expression, post-translational differences in P-glycoprotein which af-

1. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54 F47
feet transport activity or whether there exists differential compartmentation of the tracer in the highly drug resistant cells thereby altering its presentation to the transporter. Maximal inhibition of P-glycoprotein function, assayed by [99mTc]Sestamibi accumulation, appears to require 10- to 100-fold higher concentrations of the classic modulators verapamil or cyclosporin A than required to reverse drug resistance in cytotoxicity assays. Interestingly, the concen- trations of the current generation of high potency reversal agents required to achieve maximal [99mTc]Sestamibi ac- cumulation are closer to the concentration required for maximal cytotoxicity, implying improved efficacy in in- hibiting P-glycoprotein transport function [72]. These ob- servations highlight the difficulty in comparing results between different multidrug resistant cells lines, method- ologies and substrates and suggests that the levels required for maximal accumulation of cytotoxic substrates by some reversal agents may be much higher than previously esti- mated.
The continued interest in finding compounds with im- proved potency for reversing multidrug resistance has led to the systematic identification of a new generation of P-glycoprotein reversal agents. These compounds include SDZ PSC 833 [266], GF120918 [267], VX-170 [268], LY335979 [269], BIBW 22 [270], $9788 [271], dexniguldipine [272], MS-209 [273], and dexverapamil [274]. Many of these agents require less chemosensitizer to effectively inhibit drug resistance (in the 0.1 /~M range), achieve higher levels of reversal, or have fewer potential limiting side effects. Recent observations have indicated that at least some of the modulators are P-glycoprotein transport substrates and that efficiently transported com- pounds behave as poor cbemosensitizers [72,275]. Many of these compounds are currently undergoing preclinical as well as phase I / I I trials and hold the promise of an effective adjunct to improve the clinical efficacy of cancer chemotherapy.
8. Clinical trials to circumvent multidrug resistance
Numerous phase I and II trials with agents which reverse P-glycoprotein mediated multidrug resistance have been described in the literature [274,276-303]. The major- ity of these trials have had only a few patients whose tumors responded to a chemotherapeutic regimen which included the addition of a chemosensitizer such as vera- pamil, dexverapamit, quinine, quinidine, diltiazem, cy- closporin A, tamoxifen, nifedipine or trifluoperazine. This lack of response in the initial trials has prompted some investigators to believe that P-glycoprotein mediated mul- tidrug resistance is not an important factor in the therapeu- tic outcome in human malignancies [304]. A number of points must be considered before making such a conclu- sion, however. First, the number responders in phase I and II trials are expected to be low, since most patients who are entered on these trials have multiply relapsed or refrac-
tory disease. Second, as described above, the optimal methodologies for detecting P-glycoprotein have not been standardized. Until there is reproducible sensitivity and specificity in P-glycoprotein detection it is difficult to reliably understand its role in therapeutic outcomes. In- deed, most of the trials did not even attempt to categorize the level of expression in the patient's tumor. Third, achieving therapeutic levels of reversal agents in the mi- cromolar range has been problematic and often not deter- mined in many of the trials. Fourth, the amount of reversal agent required for complete inhibition of P-glycoprotein may have been underestimated based upon in vitro results, as described above. In addition, the pharmacokinetics and biodistribution of these compounds have not been totally defined for their use in cancer therapy and unacceptable side effects have been the limiting factor in administration of the reversal agent in a number of protocols. The results of clinical trials conducted early in the course of treatment with the second generation of P-glycoprotein reversal agents, which are approximately 10-fold more potent than the originally described compounds, will hopefully provide a clearer answer to the problem. Fifth, it is not clear if agents which inhibit P-glycoprotein would be most effec- tive in modifying intrinsic resistance or preventing ac- quired resistance. Finally, clinical resistance to chemother- apeutic agents is most certainly multifactorial. Agents which inhibit P-glycoprotein may ultimately be part of an armamentarium to circumvent or prevent the development of multiple mechanisms of drug resistance in human ma- lignancy.
Several important points have been identified from the trials to inhibit P-glycoprotein function. Most studies have been presented as feasibility studies and have not included controls for the administration of the chemotherapy alone. Although responses with the reversal agents have been observed in many trials, the highest number have been demonstrated in hematopoietic malignancies including leukemia, lymphoma, myeloma and Hodgkins disease [274,282,286,292,305]. Whether this relates to exposure of the malignant cells to the therapeutic agents or basic biological differences in tumor response is not clear [253]. Interestingly, a small number of patients with myeloma who were initially P-glycoprotein positive, were negative after therapy with cyclosporine A, suggesting successful elimination of the resistant cells [292]. Patients who re- spond to a reversal agent are often in the group whose tumor expressed P-glycoprotein [274,286,290,292] but this has not been a universal finding [302].
Several studies have demonstrated the critical finding that P-glycoprotein reversal agents can increase the area under the curve for the cytotoxic agent included in the therapeutic regimen by up to 200% [306-309]. Whether this is true for all reversal compounds or all chemothera- peutic agents is still being determined [287,306,3 I0]. This pharmacokinetic interaction may be due to a specific de- crease in P-glycoprotein mediated biliary effiux, nonspe-

F48 L Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
cific alterations in hepatobiliary metabolism, or a general- ized inhibition of P-glycoprotein in normal tissues which increases the serum concentration of the cytotoxic agent. This increased delivery of the chemotherapeutic agent to the tumor has the potential to confound the interpretation of P-glycoprotein reversal trials. Such trials need to be designed to demonstrate that a molecular interaction be- tween the P-glycoprotein and the reversal agent is respon- sible for improved efficacy and not simply an increase in the serum level of the cytotoxic agent. To approach this problem, many of the currently designed protocols use a maximal dose of reversal agent with approximately 50% of the routinely administered chemotherapeutic agent [311]. This strategy requires a new view on how to administer chemotherapy in an attempt to deliver equivalent systemic doses with similar anticancer agent toxicity.
These studies have also demonstrated that there is only a modest increase in the toxicity of the chemotherapeutic agents which has not limited administration of the thera- peutic regimens. Increased myelosuppression is the most common finding. There have not been any unusual toxici- ties, and any limitations to protocols have been by the known side effects of the reversal agent. Notably, there has not been an increase in central nervous system toxicity which might have been anticipated based on the expression of P-glycoprotein in the capillaries of the brain and the increased accumulation of cytotoxic agents in the brains of mice deficient in class I P-glycoprotein. Whether P-glyco- protein is simply not completely inhibited with the current regimens or whether levels which are toxic to the central nervous system have not yet been reached is not yet clear.
9. Future directions
Much of the current emphasis in P-glycoprotein re- search is focused on understanding drug translocation and demonstrating clinical efficacy of reversal agents. Several novel approaches have been attempted to further our abil- ity to exploit P-glycoprotein clinically at the molecular level.
A variety of specific and nonspecific approaches have been attempted to expand the options on how to overcome P-glycoprotein mediated multidrug resistance. A number of studies have demonstrated that monoclonal antibodies to exterior epitopes can modulate P-glycoprotein function [174,175,178], deliver toxins to kill cells expressing P- glycoprotein [312-314], and facilitate the immunological destruction of tumors expressing P-glycoprotein [176,315- 318]. In addition, the monoclonal antibody MRK-16 has been shown to increase the accumulation of cyclosporine A, although not verapamil, potentially increasing the po- tency of specific reversal agents and thus the efficacy of antitumor agents [319]. Both antisense oligonucleotides [320-329] and ribozymes [313,330-335] have been uti- lized to reverse drug resistance by decreasing the expres- sion of P-glycoprotein. Mechanisms to enhance cytotoxic-
ity in multidmg resistant cell lines have included using B4 blocked ricin [336] and antibodies to the Fas protein [337] to enhance anticancer drug cytotoxicity in multidrug resis- tant cell lines. Cytotoxic agents encapsulated in liposomes are consistently more effective in killing multidrug resis- tant cells and appear to increase the intracellular concentra- tion of the anticancer compounds [338-343]. Methodolo- gies including dye-mediated photolysis with benzopor- phyrin derivatives [344] and purging bone marrow of cells expressing P-glycoprotein with monoclonal antibodies and reversal agents [345,346] have been utilized to remove drug resistant cells in transplant models.
Expression of recombinant P-glycoprotein in bone mar- row progenitor cells has provided the means to protect hematopoietic cells from the toxicity of chemotherapeutic agents, potentially eliminating one of the limiting side effects of therapy. Retroviral vectors carrying human MDR1 cDNA have been successfully utilized to infect bone marrow progenitor cells for expression of P-glyco- protein both in vitro and in vivo [347-349]. A second approach has been the generation of a transgenic mouse which expresses recombinant human P-glycoprotein in hematopoietic precursors [350]. The recombinant P-glyco- protein is capable of protecting the murine bone marrow from the effects of anticancer agents and reversal agents have been able to inhibit the protective effect. This ap- proach may ultimately be utilized to deliver high doses of chemotherapy or even standard doses after bone marrow transplantation without myelosuppression as a dose limit- ing side effect. In addition, coupling recombinant P-glyco- protein with a second gene of interest provides a means to select and possibly maintain expression of the second gene with exposure to cytotoxic agents [351,352]. As methods to ensure delivery and sustain expression improve, this approach may find utility both in anticancer therapy and bone marrow transplantation for congenital deficiences.
I0. Conclusion
The P-glycoprotein is a fascinating protein which has captured the attention of both basic science researchers and clinicians. The analysis of its functional capacities has developed new insights on membrane transport and it undoubtedly remains one of the best targets to improve cancer chemotherapy. However, experimental analyses have been difficult due to the hydrophobic nature and large size of the polypeptide. This has been further confounded by inconsistencies with the available pharmacologic probes and immunologic reagents. In spite of these obstacles, sufficient evidence has accumulated to enable introduction of agents to inhibit P-glycoprotein function into clinical trials. P-glycoprotein reversal agents represent a clear ex- ample where molecular interactions studied in the labora- tory have moved to the bedside. The ability to manipulate P-glycoprotein function will most certainly enable clinical strategies to improve our therapies in a range of diseases.

L Bosch, J. Croop/Biochimica et Biophysica Acta 1288 (1996) F37-F54 F49
Acknowledgements
We would like to thank David Piwnica-Worms for helpful discussions. JMC is supported in part by a grant from Sandoz Pharmaceuticals and licensing agreements on patents for monoclonal antibody 4E3.16 and the mam- malian multidrug resistance gene.
References
[l] Croop. J.M., Gros, P. and Housman, D.E. (1988) J. Clin. Invest. 81, 1303-1309.
[2] Childs, S. and Ling, V. (1994) in Important Advances in Oncology (DeVita, V., Hellman, S. and Rosenberg, S,, eds.), pp. 21-36, Lippincott, Philadelphia.
[3] Gottesman, M.M. and Pastan, I. (1993) Ann. Rev. Biochem. 62, 385-427.
[4] Roninson, I.B. (1991) Plenum Press, New York. [5] Clynes, M. (1994), Kluwer Academic Publishers, Dordrecht/Bos-
ton/London. [6] Nielsen, D. and Skovsgaard. (1992) Biochim. Biophys. Acta 1139,
169-183. [7] Dano, K. (1973) Biochim. Biophys. Acta 323, 446-483. [8] Skovsgaard, T. (1978) Cancer Res. 38, 4722-4727. [9] Biedler, J.L. and Riehm, H. (1970) Cancer Res. 30, 1174-1184.
[I0] Baskin, F., Rosenberg, R.N. and Vaithinlingham, D. (198l) Proc. Natl. Acad. Sci. 78, 3654-3658.
[11] Meyers, M., Spengler, B. and Biedler, J. (1981) In Vitro 17, 221. [12] Grund, S.H., Patil, S.R., Shah, H.D., Panw, P.G. and Stadler, J.R.
(1983) Mol. Cell. Biol. 3, 1634-1641. [13] Juliano. R.L. and Ling, V. (1976) Biochim. Biophys. Acta 455,
152-162. [14] Tsuruo, T., Lida, H., Tsukagoshi, S. and Sakurai, Y. (1981) Cancer
Res. 41, 1967-1972. [15] Roninson, I.B. (1983)Nucl. Acids Res. 11, 5413-31. [16] Roninson, I.B., Abelson, H., Housman, D.E., Howell, N. and
Varshavsky, A. (1984) Nature 309, 626-628. [17] Gros, P., Croop, J., Roninson, I., Varshavsky, A. and Housman,
D.E. (1986) Proc. Natl. Acad. Sci. 83, 337-341. [18] Roninson, I.B., Chin, J.E., Choi, K.G., Gros, P., Housman, D.E.,
Fojo, A., Shen, D.W., Gottesman, M.M. and Pastan, 1. (1986) Proc. Natl. Acad. Sci. 83, 4538-42.
[19] Gros, P., Ben Neriah, Y., Croop, J.M. and Housman, D.E. (1986) Nature 323, 728-731.
[20] Chen, C.J., Chin, J.E., Ueda, K., Clark, D.P., Pastan, I., Gottesman, M.M. and Roninson, I.B. (1986) Cell 47, 381-389.
[21] Kartner, N., Evernden-Porelle, D., Bradley, G. and Ling, V. (1985) Nature 316, 820-823.
[22] Riordan, J.R., Deuchars, K., Kartner, N., Alon, N., Trent, J. and Ling, V. (1985) Nature 316, 817-819.
[23] de Bruijn, M.H., Van der Bliek, A.M., Biedler, J.L. and Borst, P. (1986) Mol. Cell. Biol. 6, 4717-22.
[24] Van der Bliek, A., Van der Velde-Koerts, T., Ling, V. and Borst, P. (1986) Mol. Cell. Biol. 6, 1671-1678.
[25] Scotto, K.W., Biedler, J.L. and Melera, P.W. (1986) Science 232, 751-755.
[26] Ueda, K., Cornwell, M.M., Gottesman, M.M., Pastan, I., Roninson, I.B., Ling, V. and Riordan, J.R. (1986) Biochem. Biophys. Res. Commun. 141,956-962.
[27] Croop, J., Guild, B., Gros, P. and Housman, D. (1987) Cancer Res. 47, 5982-5988.
[28] Lincke, C.R., van der Bliek, A.M., Schuurhuis, G.J., van der Velde Koerts, T., Smit, J.J. and Borst, P. (1990) Cancer Res. 50, 1779- 1785.
[29] Ueda, K., Cardarelli, C., Gottesman, M.M. and Pastam 1. (1987) Proc. Natl. Acad. Sci. 84, 3004-3008.
[30] Guild, B.C., Mulligan, R.C., Gros, P. and Housman, D.E. (1988) Proc. Natl. Acad. Sci. 85, 1595-1599.
[31] Choi, K., Frommel, T., Stem, R., Perex, C., Kriegler, M., Tsuruo, T. and Roninson, T. (1991) Proc. Natl. Acad. Sci. 88, 7386-7390.
[32] Greenberger, L.M., Lothstein, L., Williams, S.S. and Horwitz, S.B. (1988) Proc. Natl. Acad. Sci. 85, 3762-3766.
[33] Greenberger, L.M., Williams, S.S., Georges, E., Ling, V. and Horwitz, S.B. (1988) J. Natl. Cancer Inst. 80, 506-510.
[34] Walker, J.E., Saraste, M., Runswick, M.J. and Gay, N.J. (1982) EMBO J. 1,945-951.
[35] Croop, J. (1993) Cytotechnology 12, 1-32. [36] Raymond, M., Rose, E., Housman, D. and Gros, P. (1990) Mol.
Cell. Biol. 10, 1642-1651. [37] Chen, C.J., Clark, D., Ueda, K., Pastan, I., Gottesman. M.M. and
Roninson, I.B. (1990) J. Biol. Chem. 265, 506-514. [38] Zhang, J.T. and Ling, V. (1991) J. Biol. Chem. 266, 18224-18232. [39] Zhang, J.T., Duthie, M. and Ling, V. (1993) J. Biol. Chem. 268,
15101-15110. [40] Zhang, J.Y. and Ling, V. (1993) Biochim. Biophys. Acta 1153,
191-202. [41] Beja, O. and Bibi, E. (1995) J. Biol. Chem. 270, 12351-12354. [42] Kast, C., Canfield, V., Levenson, R. and Gros, P. (1995) Biochem-
istry 34, 4402-4411. [43] Gros, P., Croop, J. and Housman, D. (1986) Cell 47, 371-380. [44] Devault, A. and Gros, P. (1990) Mol. Cell. Biol. 10, 1652-1663. [45J Gros, P., Raymond, M., Bell, J. and Housman, D. (1988) Mol.
Cell. Biol. 8, 2770-2778. [46] Endicott, J.A., Sarangi, F. and Ling, V. (1991) DNA Seq. 2,
89-101. [47] Van der Bliek, A.M., Baas, F., Ten Houte de Lange, T., Kooiman,
P.M., Van der Velde Koerts, T. and Borst, P. (1987) EMBO J. 6, 3325-3331.
[48] Juranka, P.F., Zastawny, R.L. and Ling, V. (1989) FASEB J. 3, 2583-2592.
[49] Ng, W.F., Sarangi, F., Zastawny, R,L., Veinot, D.U and Ling, V. (1989) Mol. Cell. Biol. 9, 1224-1232.
[50] Higgins, C.F. (1992) Ann. Rev. Cell. Biol. 8, 67-113. [51] Ames, G.F., Mimura, C.S., Holbrook, S.R. and Shyamala, V.
(1992) Adv. Enzyme Rel. Areas Mol. Biol. 65, 1-47. [52] Willingham, M.C., Richert, N.D., Cornwell, M.M., Tsuruo, T.,
Hamada, H., Gottesman, M.M. and Pastan, I.H. (1987) J. Histol. Cytol. 35, 1451-1456.
[53] Molinari, A., Cianfriglia, M., Meschini, S., Calcabrini, A. and Arancia, G. (1994) Int. J. Cancer 59, 789-795.
[54] Cornwell, M.M., Gottesman, M.M. and Pastan, I.H. (1986) J. Biol. Chem. 261, 7921-7928.
[55] Cornwell, M.M., Pastan, I. and Gottesman, M.M. (1987) J. Biol. Chem. 262, 2166-2170.
[56] Cornwell, M.M., Sara, A.R., Felsted, R.L., Gottesman, M.M. and Pastan, I. (1986) Proc. Natl. Acad. Sci. 83, 3847-3850.
[57] Safa, A.R., Glover, C.J., Meyers, M.B., Biedler, J.L. and Felsted, R.L. (1986) J. Biol. Chem. 261, 6137-6140.
[58] Safa, A.R. (1993) Cancer Investig. 1 l, 46-56. [59] Safa, A.R. (1988) Proc. Natl. Acad. Sci. 85, 7187-7191. [60] Yang, C.P., Mellado, W. and Horwitz, S.B. (1988) Biochem.
Pharmacol. 37, 1417-1421. [61] Greenberger, L.M., Yang, C.P., Gindin, E. and Horwitz, S.B.
(1990) J. Biol. Chem. 265, 4394-4401. [62] Morris, D.I., Greenberger, L.M., Bruggemann, E.P., Cardarelli, C.,
Gottesman, M.M., Pastan, I. and Seamon, K.B. (1994) Mol. Phar- macol. 46, 329-337.
[63] Qian, X.D. and Beck, W.T. (1990) J. Biol. Chem. 265, 18753- 18756.
[64] Zhang, X., Collins, K.I. and Greenberger, L.M. (1995) J. Biol. Chem. 270, 5441-5448.

F50 L Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
[65] Greenberger, L.M., Lisanti, C.J., Silva, J.T. and Horwitz, S.B. (1991) J. Biol. Chem. 266, 20744-20751.
[66] Greenberger, L.M. (1993) J. Biol. Chem. 268, 11417-11425. [67] Yoshimura, A., Kuwazuru, Y., Sumizawa, T., Ichikawa, M., Ikeda,
S., Uda, T. and Akiyama, S. (1989) J. Biol. Chem. 264, 16282- 16291.
[68] Bruggemann, E.P., Germann, U.A., Gottesman, M.M. and Pastan, I. (1989) J. Biol. Chem. 264, 15483-15488.
[69] Tamai, I. and Safa, A.R. (1990) J. Biol. Chem. 265, 16509-16513. [70] Tamai, I. and Safa, A.R. (1991) J. Biol. Chem. 266, 16796-16800. [71] Pereira, E., Borrel, M.N., Fiallo, M. and Garnier-Suillerot, A.
(1994) Biochim. Biophys. Acta 1225, 209-216. [72] Piwnica-Worms, D., Rao, V.V., Kronauge, J.F. and Croop, J.M.
(1995) Biochemistry 34, 12210-12220. [73] Choi, K., Chen, C.J., Kriegler, M. and Roninson, I.B. (1988) Cell
53, 519-529. [74] Safa, A.R., Stern, R.K., Choi, K., Agresti, M., Tamai, I., Mehta,
N.D. and Roninson, I.B. (1990) Proc. Natl. Acad. Sci. 87, 7225- 7229.
[75] Cardarelli, C.O., Aksentijevich, I., Pastan, I. and Gottesman, M.M. (1995) Cancer Res. 55, 1086-1091.
[76] Currier, S.J., Kane, S.E., Willingham, M.C., Cardarelli, C.O., Pastan, I. and Gottesman, M.M. (1992) J. Biol. Chem. 267, 25153- 25159.
[77] Loo, T.W. and Clarke, D.M. (1994) J. Biol. Chem. 269, 7243-7248. [78] Gros, P., Dhir., R., Croop, J. and Talbot, F. (1991) Proc. Natl.
Acad. Sci. 88, 7289-7293~ [79] Kajiji, S., Dreslin, A., Grizzuti, K. and Gros, P. (1994) Biochem-
istry 33, 5041-5048. [80] Devine, S.E. and Melera, P.W. (1994) J. Biol. Chem. 269, 6133-
6139. [81] Devine, S.E., Ling, V. and Melera, P.W. (1992) Proc. Natl. Acad.
Sci. 89, 4564-4568. [82] Loo, T.W. and Clarke, D.M. (1993) J. Biol. Chem. 268, 3143-3149. [83] Hoof, T., Demmer, A., Hadam, M.R., Riordan, J.R. and Tummler,
B. (1994) J. Biol. Chem. 269, 20575-20583. [84] Beaudet, L. and Gros, P. (1995) J. Biol. Chem. 270, 17159-17170. [85] Starzyk, R.M., Webster, T. and Schimmel (1987) Science 237,
1614-1618. [86] Cornwell, M.M., Tsuruo, T., Gottesman, M.M. and Pastan, I.
(1987) FASEB J. 1, 51-54. [87] Hamada, H. and Tsuruo, T. (1988) Cancer Res. 48, 4926-4932. [88] Sarkadi, B., Price, E.M., Boucher, R.C., Germann, U.A. and
Scarborough, G.A. (1992) J. Biol. Chem. 267, 4854-4858. [89] Rao, U.S. (1995) J. Biol. Chem. 270, 6686-6690. [90] Doige, C.A., Yu, X. and Sharom, F.J. (1992) Biochim. Biophys.
Acta 1109, 149-160. [91] Ambudkar, S.V., Lelong, I.H., Zhang, J., Cardarelli, C.O., Gottes-
man, M.M. and Pastan, I. (1992) Proc. Natl. Acad. Sci. 89, 8472-8476.
[92] al-Shawi, M.K. and Senior, A~E. (1993) J. Biol. Chem. 268, 4197-4206.
[93] Shapiro, A.B. and Ling, V. (1994) J. Biol. Chem. 269, 3745-3754. [94] Doige, C.A., Yu, X. and Sharom, F.J. (1993) Biochim. Biophys.
Acta 1146, 65-72. [95] Shimabuku, A.M., Nishimoto, T., Ueda, K. and Komano, T. (1992)
J. Biol. Chem. 267, 4308-4311. [96] Urbatsch, I.L., al-Shawi, M.K. and Senior, A.E. (1994) Biochem-
istry 33, 7069-7076. [97] Horio, M., Gottesman, M.M. and Pastan, I. (1988) Proc. Natl.
Acad. Sci. 85, 3580-3584. [98] Lankelma, J., Spoelstra, E.C., Dekker, H. and Broxterman, H.J.
(1990) Biochim. Biophys. Acta 1055, 217-222. [99] Schlemmer, S.R. and Sirotnak, F.M. (1994) J. Biol. Chem. 269,
31059-31066. [100] Ruetz, S., Raymond, M. and Gros, P. (1993) Proc. Natl. Acad. Sci.
90, 11588-11592.
[101] Ruetz, S. and Gros, P. (1994) J. Biol. Chem. 269, 12277-12284. [102] Sharom, F.L, Yu, X. and Doige, C.A. (1993) J. Biol. Chem. 268,
24197-24202. [103] Shapiro, A.B. and Ling, V. (1995) J. Biol. Chem. 270, 16167-
16175. [104] Bornmann, W.G. and Roepe, P.D. (1994) Biochemistry 33, 12665-
12675. [105] Simon, S., Roy, D. and Schindler, M. (1994) Proc. Natl. Acad. Sci.
91, 1128-32. [106] Altenberg, G.A., Vanoye, C.G., Horton, J.K. and Reuss, L. (1994)
Proc, Natl. Acad. Sci. 91, 4654-4657. [107] Roepe, P.D. (1994) Trends Pharmacol. Sci. 15, 445-446. [108] Hamada, H., Hagiwara, K., Nakajima, T. and Tsuruo, T. (1987)
Cancer Res. 47, 2860-5. [109] Fine, R.L., Patel, J. and Chabner, B.A. (1988) Proc. Natl. Acad.
Sci. 85, 582-586. [110] Chambers, T.C., McAvoy, E.M., J.W., J. and Eilon, G. (1990) J.
Biol, Chem. 265, 7679-7686. [11l] Sato, W., Yusa, K., Naito, M. and Tsuruo, T. (1990) Biocbem.
Biopbys. Res. Commun. 173, 1252-1257. [112] Ma, L.D., Marquardt, D., Yakemoto, L. and Center, M.S. (1991) J.
Biol. Chem. 266, 5593-5599. [113] Chambers, T.C., Zheng, B. and Kuo, J.F. (1992) Mol. Pharmcol.
41, 1008-1015. [114] Bates, S.E., Lee, J.S., Dickstein, B., Spolyar, M. and Fojo, A.T.
(1993) Biochemistry 32, 9156-9164. [115] Germann, U., Chambers, T., Ambukar, S., Licht, T., Cardarelli, C.,
Pastan, I. and Gottesman, M. (1996) J. Biol. Chem. 271, 1708- 1716.
[116] Ahmad, S., Safa, A.R. and Glazer, R.I. (1994) Biochemistry 33, 10313-10318.
[117] Schinkel, A.H., Kemp, S., Dolle, M., Rudenko, G. and Wagenaar, E. (1993) J. Biol. Chem. 268, 7474-7481.
[118] Abraham, E.H., Prat, A.G., Gerweck, L., Seneveratne, T., Arceci, R., Kramer, R., Guidotti, G. and Cantiello, H.F. (1993) Proc. Natl. Acad. Sci. 90, 312-316.
[119] Reisin, I.L., Prat, A.G., Abraham, E.H., Amara, J.F., Gregory, R.J., Ausiello, D.A. and Cantiello, H.F. (1994) J. Biol. Chem. 269, 20584-20591.
[120] Schwiebert, E.M., Egan, M.E., Hwang, T.H., Fulmer, S.B., Allen, S.S., Cutting, G.R. and Guggino, W.B. (1995) Cell 81, 1063-1073.
[121] Reddy, M.M., Quinton, P.M., Haws, C., Wine, J.J., Grygorczyk, R., Tabcharani, J.A., Hanrahan, J.W., Gunderson, K.L. and Kopito, R.R. (1996) Science 271, 1876-1879.
[122] Valverde, M.A., Diaz, M., Sepulveda, F.V., Gill, D.R., Hyde, S.C. and Higgins, C.F. (1992) Nature 355, 830-833.
[123] Hardy, S.P., Goodfellow, H.R., Valverde, M.A., Gill, D.R., Sepul- veda, F.V. and Higgins, C.F. (1994) EMBO J. 14, 68-75.
[124] Croop, J.M., Raymond, M., Haber, D., Devault, A., Arceci, R.J., Gros, P. and Housman, D.E. (1989) Mol. Cell. Biol. 9, 1346-1350.
[125] Fojo, A.T., Ueda, K., Slamon, D.J., Poplack, D.G., Gottesman, M.M. and Pastan, I. (1987) Proc. Natl. Acad. Sci. 84, 265-269.
[126] Trezise, A.E.O., Romano, P.R., Gill, D.R., Hyde, S.C., Sepulveda, F.V., Buchwald, M. and Higgins, C.F. (1992) EMBO J. 11, 4291-4303.
[127] Georges, E., Bradley, G., Gariepy, J. and Ling, V. (1990) Proc. Natl. Acad. Sci. 87, 152-156.
[128] Cordon-Cardo, C., OBrien, J.P., Boccia, J., Casals, D., Bertino, J.R. and Melamed, M.R. (1990) J. Histochem. Cytochem. 38, 1277-1287.
[129] van der Valk, P., van Kalken, C.K., Ketelaars, H., Broxterman, H.J., Scheffer, G., Kuiper, C.M., Tsurno, T., Lankelma, J., Meijer, C.J., Pinedo, H. and Scheper, R. (1990) Ann. Oncol. 1, 56-64.
[130] Arceci, R.J., Croop, J.M., Horwitz, S.B. and Housman, D. (1988) Proc. Natl. Acad. Sci. 85, 4350-4354.
[131] Axiotis, C.A., Guarch, R., Merino, M.J., Laporte, N. and Neu- mann, R.D. (1991) Lab. Invest. 65,577-581.

1. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54 F51
[132] Bradley, G., Georges, E. and Ling, V. (1990) J. Cell. Physiol. 145, 398-408.
[133] Cordon-Cardo, C., O'Brien, J.P., Casals, D., Rittman-Grauer, L., Biedler, J.L., Melamed, M.R. and Bertino, J.R. (1989) Proc. Natl. Acad. Sci. 86, 695-698.
[134] Chaudhary, P.M. and Roninson, I.B. (1991) Cell 66, 85-94. [135] Chaudhary, P.M., Mechetner, E.B. and Roninson, I.B. (1992)
Blood 80, 2735-2739. [136] Drach, D., Zhao, S., Drach, J., Mahadevia, R.. Gattringer, C.,
Huber, H. and Andreeff, M. (1992) Blood 80, 2729-2734. [137] Chin, J.E., Sofflr, R., Noonan, K.E., Choi, K. and Roninson, I.B.
(1989) Mol. Cell. Biol. 9, 3808-3820. [138] Buschman, E., Arceci, R., Croop, J., Mingxin, C., Arias, I., Hous-
man, D. and Gros, P. (1992) J. Biol. Chem. 267, 18093-18099. [139] Smit, J.J., Schinkel, A.H., Mol, C.A., Majoor, D., Mooi, W.J.,
Jongsma, A.P., Lincke, C.R. and Borst, P. (1994) Lab. Invest. 71, 638-649.
[140] Schinkel, A.H., Smit, J.J., van Tellingen, O., Beijnen, J.H., Wage- naar, E., van Deemter, L., Mol, C.A., van der Valk, M.A., Robanus-Maandag, E.C., te Riele, H.P., Berns, A.J.M. and Borst, P. (1994) Cell 77, 491-502,
[141] Smit, J.J., Schinkel, A.H., Oude Elferink, R.P., Groen, A.K., Waganaaf, E., van Deemter, L., Mol, C.A., Ottenhofer, R., van der Lugt, N.M., van Room, M.A., van der Valk, M.A., Offerhous, G.L, Berns, A.J. and Borst, P. (1993) Cell 75, 451-462.
[142] Ruetz, S. and Gros, P. (1994) Cell 77, 1071-1081. [143] Smith, A.J., Timmermans-Hereijger, J., Roelofsen, B., Wirtz, K.,
van Blitterswijk, W.J., Smit, J.J.M., Schinkel, A.H. and Borst, P. (1994) FEBS Lett. 354, 263-266.
[144] Arceci. R.J., Baas, F., Raponi, R., Horwitz, S.B., Housman, D. and Croop, J. (1990) Mol. Reprod. Dev. 25, 101-109.
[145] Jancis, E.M., Chen, H.X., Carbone, R., Hochberg, R.B. and Dan- nies, P.S. (1993) Biochem. Pharm. 46, 1613-1619.
[146] Fairchild, C.R., Ivy, S.P., Rushmore, T., Lee, G., Koo, P., Gold- smith, M.E., Myers, C.E., Farber, E. and Cowan, K.H. (1987) Proc. Natl. Acad. Sci. 84, 7701-7705.
[147] Thorgeirsson, S.S., Huber, B.E., Sorrell, S., Fojo, A., Pastan, I. and Gottesman, M.M. (1987) Science 236, 1120-1122.
[148] Burt, R.K. and Thorgeirsson, S.S. (1988) J. Natl. Cancer Inst. 80, 1383-1386.
[149] Schuetz, J.D. and Schuetz, E.G. (1993) Cell Growth Diff. 4, 31-40.
[150] Chin, K.V., Tanaka, S., Darlington, G., Pastan, I. and Gottesman, M.M. (1990) J. Biol. Chem. 265, 221-226.
[151] Mickley, L.A., Bates, S.E., Richert, N.D., Currier, S., Tanaka, S., Foss, F., Rosen, N. and Fojo, A.T. (1989) J. Biol. Chem. 264, 18031-18040.
[152] Morrow, C.S., Nakagawa, M., Goldsmith, M.E., Madden, M.J. and Cowan, K.H. (1994) J. Biol. Chem. 269, 10739-10746.
[153] Chaudhary, P.M. and Roninson, I.B. (1993) J. Natl. Cancer Inst. 85, 632-639.
[154] Gottesman, M., Hrycyna, C., Schoelein, P., Gerrnann, U. and Pastan, I. (1995) Ann. Rev. Genet. 29, 607-649.
[155] Madden, M.J, Morrow, C.S., Nakagawa, M., Goldsmith, M.E., Fairchild, C.R. and Cowan, K.H. (1993) J. Biol. Chem. 268, 8290-8297.
[156] Ueda, K., Pastan, I. and Gottesman. M.M. (1987) J. Biol. Chem. 262, 17432-17436.
[157] Cornwell, M.M. and Smith, D.E. (1993) J. Biol. Chem. 268, 19505-19511.
[158] Goldsmith, M.E., Madden, M.J., Morrow, C.S. and Cowan, K.H. (1993) J. Biol. Chem. 268, 5856-5860.
[159] Cornwell, M.M. (1990) Cell Growth and Differ 1,607-615. [160] Ince, T.A. and Scotto, K.W. (1995) J. Biol. Chem. 270, 30249-
30252. [161] Ince, T.A. and Scotto, K.W. (1996) Cancer Res. 56, 2021-2024. [162] Piekarz, R.L., Cohen, D. and Horwitz, S.B. (1993) J. Biol. Chem.
268, 7613-7616.
[163] Chin, K., Kazumitsu, U., Pastan, I. and Gottesman, M. (1992) Science 255, 459-462.
[164] Comhates, N.J., Rzepka, R.W., Chen, Y.N. and Cohen, D. (1994) J. Biol. Chem. 269, 29715-29719.
[165] Miltenberger, R.J., Farnham, P.J., Smith, D.E., Stommel, J.M. and Cornwell, M.M. (1995) Cell Growth Diff. 6, 549-556.
[166] McCoy, C., Smith, D.E. and Cornwell, M.M. (1995) Mol. Cell. Biol. 15, 6100-6108.
[167] Thiebaut, F., Tsuruo, T., Hamada, H., Gottesman, M.M., Pastan, I. and Willingham, M.C. (1989) J. Histochem. Cytochem. 37, 159- 164.
[168] Rao, V.V., Anthony, D.C. and Piwnica-Worms, D. (1994) Cancer Res. 54, 1536-1541.
[169] Rao, V.V., Anthony, D.C. and Piwnica-Worms, D. (1995) J. Histochem. Cytochem. 43, 1187-1192.
[170] Goldstein, L.J., Galski, H., Fojo, A., Willingham, M., Lai, S.L., Gazdar, A., Pirker, R., Green, A., Crist, W., Brodeur, G.M., Lieber, M., Cossman, J., Gottesman, M. and Pastan, I. (1989) J. Natl. Cancer Inst. 81, 116-124.
[171] Sugawara, I. (1990) Acta Pathol. Jpn. 40, 545-553. [172] Kim, R. (1990) J. Histochem. Cytochem. 38, 1277-1287. [173] Noonan, K.E., Beck, C., Holzmayer, T.A., Chin, J.E., Wunder,
J.S., Andrulis, I.L., Gazdar, A.F., Willman, C.L., Griffith, B., Von Hoff, D.D. and Roninson, I.B. (1990) Proc. Natl. Acad. Sci. 87, 7160-7164.
[174] Hamada, H. and Tsuruo, T. (1986) Proc. Natl. Acad. Sci. 83, 7785-7789.
[175] Arceci, R., Stieglitz, K., Bras, J., Schinkel, A., Baas, F. and Croop, J. (1993) Cancer Res. 53, 310-317.
[176] Rittmann-Grauer, L.S., Yong, M., Sanders, V. and Mackensen, D. (1992) Cancer Res. 52, 1810-1816.
[177] Cianfriglia, M., Willingham, M.C., Tombesi, M., Scagliotti, G.V., Frasca, G. and Chersi, A. (1994) Int. J. Cancer 56, 153-160.
[178] Mechetner, E.B. and Roninson, I.B. (1992) Proc. Natl. Acad. Sci. 89, 5824-5828.
[179] Pirker, R., Wallner, J., Geissler, K., Linkesch, W., Haas, O.A., Bettelheim, P., Hopfner, M., Scherrer, R., Valent, P., Havelec, L., Ludwig, H. and Lechner, K. (1991) J. Natl. Cancer Inst. 83, 708-712.
[180] Marie, J.P., Zittoun, R. and Sikic, B.I. (1991) Blood 78, 586-592. [181] Campos, L., Guyotat, D., Archimbaud, E., Calmard, O.P., Tsuruo,
T., Troncy, J., Treille, D. and Fiere, D. (1992) Blood 79, 473-476. [182] te Boekhorst, P.A., de Leeuw, K., Schoester, M., Wittebol, S.,
Nooter, K., Hagemeijer, A., Lowenberg, B. and Sonneveld, P. (1993) Blood 82, 3157-3162.
[183] Zochbauer, S., Gsur, A., Brunner, R., Kyrle, P.A., Lechner, K. and Pirker, R. (1994) Leukemia 8, 974-977.
[184] Wood, P., Burgess, R., MacGregor, A. and Yin, J.A. (1994) Br. J. Haematol. 87, 509-514.
[185] Lamy, T., Goasguen, J.E., Mordelet, E., Grulois, I., Dauriac, C., Drenou, B., Chaperon, J., Fauchet, R, and le Prise, P.Y. (1994) Leukemia 8, 1879-1883.
[186] Nussler, V., Pelka-Fleischer, R., Zwierzina, H., Nerl, C., Beckert, B., Gullis, E., Gieseler, F., Bock, S., Bartl, R., Petrides, P.E. and Wilmanns, W. (1994) Ann. Hematol. 69, $25-29.
[187] Ino, T., Miyazaki, H., Isogai, M., Nomura, T., Tsuzuki, M., Tsuruo, T., Ezaki, K. and Hirano, M. (1994) Leukemia 8, 1492- 1497.
[188] Leith, C.P., Chen, I.M., Kopecky, K.J., Appelbaum, F.R., Head, D.R., Godwin, J.E., Weick, J.K. and Willman, C.L. (1995) Blood 86, 2329-2342.
[189] Rothenberg, M.L., Mickley, L.A., Cole, D.E., Balls, F.M., Tsnruo, T., Poplack, D.G. and Fojo, A.T. (1989) Blood 74, 1388-1395.
[190] Herweijer, H., Sonneveld, P., Baas, F. and Nooter, K. (1990) J. Natl. Cancer Inst. 82, 1133-1140.
[191] Kuwazuru, Y., Yoshimura, A., Hanada, S., Utsunomiya, A., Makino, T., Ishibashi, K., Kodama, M., lwahashi, M., Arima, T. and Akiyama, S. (1990) Cancer 66, 868-873.

F52 I. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
[192] Kuwazuru, Y., Hanada, S., Furukawa, T., Yoshimura, A., Sum- izawa, T., Utsunomiya, A., Ishibashi, K., Saito, T., Uozumi, K., Maruyama, M., Ishizawa, M., Arima, T. and Akiyama, S, (1990) Blood 76, 2065-2071.
[193] Goasguen, J.E., Dossot, J.M., Fardel, O., Le Mee, F., Le Gall, E., Leblay, R., LePrise, P.Y., Chaperon, J. and Fauchet, R. (1993) Blood 81, 2394-2398.
[194] Zhou, D.C., Hoang-Ngoc, L., Delmer, A., Faussat, A.M., Russo, D., Zittoun, R. and Marie, J.P. (1994) Leukem. Lymph. 13, 27-30.
[195] Sauerbrey, A., Zintl, F. and Volm, M. (1994) Br. J. Cancer 70, 1144-1149.
[196] Brophy, N.A., Marie, J.P., Rojas, V.A., Warnke, R.A., McFall, P.J., Smith, S.D. and Sikic, B.I. (1994) Leukemia 8, 327-335.
[197] Wattel, E., Lepelley, P., Merlat, A., Sartiaux, C., Bauters, F., Jouet, J.P. and Fenaux, P. (1995) Leukemia 9, 1870-1874.
[198] Pieters, R., Kaspers, G.J., Klumper, E. and Veerman, A.J. (1994) Med. Ped. Oncol. 22, 299-308.
[199] Dalton, W.S., Grogan, T.M., Rybski, J.A., Scheper, R.J., Richter, L., Kailey, J., Broxterman, H.J., Pinedo, H.M. and Salmon, S.E. (1989) Blood 73, 747-52.
[200] Salmon, S.E., Grogan, T.M., Miller, T., Scheper, R. and Dalton, W.S. (1989) J. Natl. Cancer Inst, 81,696-701.
[201] Epstein, J., Xiao, H.Q and Oba, B.K. (1989) Blood 74, 913-917. [202] Sonneveld, P., Durie, B.G., Lokhorst, H.M., Frutiger, Y., Schoester,
M. and Vela, E.E. (1993) Br. J. Haematol. 83, 63-67. [203] Grogan, T.M., Spier, C.M., Salmon, S.E., Matzner, M., Rybski, J.,
Weinstein, R.S., Scheper, R.J. and Dalton, W.S. (1993) Blood 81, 490-495.
[204] Schneider, J., Bak, M., Efferth, T., Kaufmann, M., Mattern, J. and Volm, M. (1989) Br. J. Cancer 60, 815-818.
[205] Kacinski, B.M., Yee, L.D., Carter, D., Li, D. and Kuo, M.T. (1989) Br. J. Cancer 60, 815-818.
[206] Wishart, G.C., Plumb, J.A., Going, J.J., McNicol, ARM., McArdle, C.S., Tsuruo, T. and Kaye, S.B. (1990) Br. J. Cancer 62, 758-761.
[207] Keith, W.N., Stallard, S. and Brown, R. (1990) Br. J. Cancer 61, 712-716.
[208] Ro, J., Sahiu, A., Ro, J.Y., Fritsche, H., Hortobagyi, G. and Blick, M. (1990) Human Pathology 21,787-791.
[209] Sanfilippo, O., Ronchi, E., De Marco, C., Di Fronzo, G. and Silvestrini, R. (1991) Eur J Cancer 27, 155-158.
[210] Wallner, J., Depisch, D., Hopfner, M., Haider, K., Spona, J., Ludwig, H. and Pirker, R. (1991) Eur J Cancer 27, 1352-1355.
[211] Verrelle, P., Meissonnier, F., Fonck, Y., Feillel, V., Dionet, C., Kwiatkowski, F., Plagne, R. and Chassagne, J. (1991) J Natl Cancer Inst 83, 111-116.
[212] Dixon, A., Bell, J., Ellis, I., Elston, C. and Blarney, R. (1992) Br. J. Cancer 66, 537-541.
[213] De La Torre, M., Larsson, R., Nygren, P., Lindgren, A. and Bergh, J. (1994) Acta Oncologica 33, 773-777.
[214] Charpin, C., Vielh, P., Duffaud, F., Devictor, B., Andrac, L., Lavant, M.N., Allasia, C., Horschowski, N. and Piana, L, (1994) J Natl Cancer Inst 86, 1539-1545.
[215] Veneroni, S., Zaffaroni, N., Daidone, M.G., Benini, E., Villa, R. and Silvestrini, R. (1994) Eur J Cancer 30A, 1002-1007.
[216] Decker, D.A., Morris, L.W., Levine, A.J., Pettinga, J.E., Grudzien, J.L. and Farkas, D.H. (1995) Ann Clin Lab Sci 25, 52-59.
[217] Schneider, J. and Romero, H. (1995) Anticancer Res 15, 1117- 1121.
[218] Bates, S.E., Meadows, B., Goldspiel, B.R., Denicoff, A., Le, T.B., Tucker, E., Steinberg, S.M. and Elwood, L.J. (1995) Cancer Chemotherapy and Pharmacology 35, 457-463.
[219] Merkel, D., Fuqua, S., Tandon, A., Hill, S., Buzdar, A. and Mcguire, W. (1989) J Clin Onc 7, 1129-1136.
[220] Moscow, J.A., Fairchild, C.R., Madden, M.J., Ransom, D.T., Wie- and, H.S., OBrien, E.E., Poplack, D.G., Cossman, J., Myers, C.E. and Cowan, K.H. (1989) Cancer Res. 49, 1422-1428.
[221] Fojo, A.T., Shen, D.W., Mickley, L.A., Pastan, I. and Gottesman, M.M. (1987) J Clin Oncol 5, 1922-1927.
[222] Kakehi, Y., Kanamaru, H., Yoshida, O., Ohkubo, H., Nakanishi, S., Gottesman, M.M. and Pastan, I. (1988) J Urol 139, 862-865.
[223] Bak, M.J., Efferth, T., Mickisch, G., Mattern, J. and Volm, M. (1990) Eur Urol 17, 72-75.
[224] van Kalken, C.K., van der Valk, P., Hadisaputro, M.M., Pieters, R., Broxterman, H.J., Kuiper, C.M., Scheffer, G.L., Veerman, A.J., Meyer, C.J., Scheper, R.J., Veerman, A., Meyer, C., Scheper, R. and Pinedo, H. (1991) Annals of Oncology 2, 55-62.
[225] Nishiyama, K., Shirahama, T., Yoshimura, A., Sumizawa, T., Furukawa, T., Ichikawa-Haraguchi, M., Akiyama, S. and Ohi, Y. (1993) Cancer 71,3611-3619.
[226] Rochlitz, C.F., Lobeck, H., Peter, S., Reuter, J., Mohr, B., de Kant, E., Huhn, D. and Herrmann, R. (1992) Cancer 69, 2993-2998.
[227] Kanamaru, H., Kakehi, Y., Yoshida, O., Nakanishi, S., Pastan, I. and Gottesman, M. (1989) J Natl Cancer Inst 81,844-849.
[228] Mickisch, G.H., Kossig, J., Keilhauer, G., Schlick, E., Tschada, R.K. and Alken, P.M. (1990) Cancer Res. 50, 3670-3674.
[229] Danova, M., Giordano, M., Erba, E., Palmeri, S., Candiloro, V., Riccardi, A., Ucci, G., Mazzini, G., D'Incalci, M. and Ascari, E. (1992) Journal of Cancer Res.earch and Clinical Oncology 118, 575-580.
[230] Fujii, H., Tanigawa, N., Muraoka, R., Shimomatsuya, T. and Tanaka, T. (1993) Anticancer Res 13, 2171-2175.
[231] Pirker, R., Wallner, J., Gsur, A., Gotzl, M., Zochbauer, S., Schei- thauer, W. and Depisch, D. (1993) Br. J. Cancer 68, 691-694.
[232] Sinicrope, F.A., Hart, J., Brasitus, T.A., Michelassi, F., Lee, J.J. and Safa, A.R. (1994) Cancer 74, 2908-2917.
[233] Weinstein, R.S., Jakate, S.M., Dominguez, J.M., Lebovitz, M.D., Koukoulis, G.K., Kuszak, J.R., Klusens, L.F., Grogan, T.M., Saclarides, T.J., Roninson, I.B. and Coon, J.S. (1991) Cancer Res. 51, 2720-2726.
[234] Bourhis, J., Benard, J., Hartmann, O., Boccon, G.L., Lemerle, J. and Riou, G. (1989) J Natl Cancer Inst 81, 1401-1405.
[235] Goldstein, L.J., Fojo, A.T., Ueda, K., Crist, W., Green, A., Brodeur, G., Pastan, I. and Gottesman, M.M. (1990) J Clin Oncol 8, 128-136.
[236] Corrias, M.V., Cornaglia, F.P., Di, M.D., Stenger, A.M., Lanino, E., Boni, L. and Tonini, G.P. (1990) Anticancer Res 10, 897-902.
[237] Nakagawara, A., Kadomatsu, K., Sato, S., Kohno, K., Takano, H., Nose, Y. and Kuwano, M. (1990) Cancer Res. 50, 3043-3047.
[238] Chan, H.S., Haddad, G., Thorner, P.S., DeBoer, G., Lin, Y.P., Ondrusek, N,, Yeger, H. and Ling, V. (1991) N Engl J Med 325, 1608-1614.
[239] Ramani, P. and Dewchand, H. (1995) J Path 175, 13-22. [240] Norris, M.D., Bordow, S.B., Marshall, G.M., Haber, P.S., Cohn,
S.L. and Haber, M. (1996) New Engl J Med 334, 231-8. [241] Norris, M.D., Bordow, S.B., Haber, P.S., Marshall, G.M., Stewart,
B.W. and Haber, M. (1996) AACR Proc 37, 313. [242] Da Silva, J., Riou, G. and Benard, I. (1994) Cancer Res. 54,
2256-2261. [243] Gerlach, J.H., Bell, D.R., Karakousis, C., Slocum, H.K., Kartner,
N., Rustum, Y.M., Ling, V. and Baker, R.M. (1987) J Clin Oncol 5, 1452-1460.
[244] Chan, H., Thorner, P., Haddad, G. and Ling, V. (1990) J Clin Oncol 8, 689-704.
[245] Schlaifer, D., Laurent, G., Chittal, S., Tsuruo, T., Soues, S., Muller, C., Charcosset, J.Y, Alard, C., Brousset, P., Mazerrolles, C. and Delsol, G. (1990) Br. J. Cancer 62, 177-182.
[246] Tawa, A., Inoue, M., Ishihara, S., Hara, J., Yumura, Y.K., Okada, A., Nihei, A., Taguchi, J., Kanai, N. and et, a.1. (1990) Cancer 66, 1980-1983.
[247] Toffoli, G., Frustaci, S., Tumiotto, L., Talamini, R., Gherlinzoni, F., Picci, P. and Boiocchi, M. (1992) Annals of Oncology 3, 63-69.

L Bosch, J. Croop/Biochimica et Biophysica Acta 1288 (1996) F37-F54 F53
[248] Hijazi, Y.M., Axiotis, C.A., Navarro, S., Steinberg, S.M., Horowitz, M.E. and Tsokos, M. (1994) Am J Clin Path 102, 61-67.
[249] Stein, U., Wunderlich, V,, Haensch, W. and Schmidt-Peter, P. (1993) Eur J Can 14, 1979-81.
[250] Imanishi, T., Abe, Y., Suto, R., Higaki, S., Ueyama, Y., Nakamura, M., Tamaoki, N., Fukuda, H. and Imai, N. (1994) Tokai J Exper and Clin Med 19, 39-46.
[251] Kandel, R.A., Campbell, S., Noble-Topham, S., Bell, R. and Andrulis, I.L. (1995) Diagnostic Molecular Pathology 4, 59-65.
[252] Baldini, N., Scotlandi, K., Barbanti-Brodano, G., Manara, M.C., Maurici, D., Bacci, G., Bertoni, F., Picci, P., Sottili, S., Cam- panacci, M. and et al. (1995) New Eng J Med 333, 1380-1385.
[253] Pinedo, H.M. and Giaccone, G. (1995) New Eng J Med 333, 1417-1419.
[254] Nabors, M.W., Griffin, C.A., Zehnbauer, B.A., Hruban, R.H., Phillips, P.C., Grossman, S.A., Brem, H. and Colvin, O.M. (1991) J Neurosurg 75,941-946.
[255] Tishler, D.M., Weinberg, K.I., Sender, L.S., Nolta, J.A. and Raffel, C. (1992) J Neurosurg 76, 507-512.
[256] Henson, J.W., Cordon-Cardo, C. and Posner, J.B. (1992) Journal of Neuro Oncology 14, 37-43.
[257] Tachibana, O., Yamashima, T., Yamashita, J. and Takabatake, Y. (1994) J Neurosurg 80, 79-84.
[258] Chan, H.S., Thorner, P.S., Haddad, G. and Gallie, B.L. (1991) Ophthal 98, 1425-1431.
[259] Robey-Cafferty, S.S., Rutledge, M.L. and Bruner, J.M. (1990) Am J Clin Pathol 93, 1-7.
[260] Bell, D.R., Gerlach, J.H., Kartner, N., Buick, R.N. and Ling, V. (19851J Clin Oncol 3, 311-315.
[26l] Bourhis, J., Goldstein, L.J., Riou, G., Pastan, I., Gottesman, M.M. and Benard, J. (1989) Cancer Res. 49, 5062-5065.
[262] van der Zee, A.G., Hollema, H., de Jong, S., Boonstra, H., Gouw, A., Willemse, P.H., Zijlstra, J.G. and de Vries, E.G. (1991) Cancer Res. 51, 5915-5920.
[263] Ford, J.M. and Hait, W.N. (1990) Pharmacol Rev 42, 155-199. [264] Ford, J.M. and Hait, W.N. (1993) Cytotechnology 12, 171-212. [265] Piwnica-Worms, D., Chiu, M.L., Budding, M., Kronauge, J.F.,
Kramer, R.A. and Croop, J,M. (1993) Cancer Res. 53, 977-984. [266] Boesch, D., Gaveriaux, C., Jachez, B., Pourtier-Manzanedo, A.,
Bollinger, P. and Loor, F. (1991) Cancer Res. 51, 4226-4233. [267] Hyafit, F., Vergely, C., Du Vignaud, P. and Grand-Perret, T.
(1993) Cancer Res. 53, 4595-4602. [268] Germann, U.A., Zelle, R.E., Shlyakhter, D., Duffy, J., Mason,
V.S., Galullo, V., Armistead, D.M., Saunders, J.O., Boger, J. and Harding, M.W. (1995) Cancer Res. 36, 346.
[269] Slate, D.L., Bruno, N.A., Casey, S.M., Zutshi, N., Garvin, L.J., Wu, H. and Pfister, J.R. (1995) Anticancer Res 15, 811-814.
[270] Chen, H.X., Bamberger, U., Heckel, A., Guo, X. and Cheng, Y.C. (1993) Cancer Res. 53, 1974-1977.
[271] Merlin, J.L., Guerci, A., Marchal, S., Missoum, N., Ramacci, C., Humbert, J.C., Tsuruo, T. and Guerci, O. (1994) Blood 84, 262- 269.
[272] Hofmann, J., Gekeler, V., Ise, W., Noller, A., Mitterdorfer, J., Hofer, S., Utz, I., Gotwald, M., Boer, R., Glossmann, H. and et, a. (1995) Biochem Pharm 49, 603-9.
[273] Sato, W., Fukazawa, N, Nakanishi, O., Baba, M., Suzuki, T., Yano, O., Naito, M. and Tsuruo, T. (1995) Cancer Chem Pharm 35, 271-277.
[274] Wilson, W.H., Bates, S.E., Fojo, A., Bryant, G., Zhan, Z., Regis, J., Wittes, R.E., Jaffe, E.S., Steinberg, S.M., Herdt, J. and Chabner, B. (1995) J Clin Onc 13, 1995-2004.
[275] Lee, J.S., Panll, K., Alvarez, M., Hose, C., Monks, A., Grever, M., Fojo, A.T. and Bates, S.E. (1994) Mol Pharm 46, 627-638.
[276] Bessho, F., Kinumaki, H., Kobayashi, M., Habu, H., Nakamura, K., Yokota, S., Tsuruo, T. and Kobayashi, N. (1985) Med Pediatr Oncol 13, 199-202.
[277] Ozols, R., Cunnion, R., Klecker, R., Hamilton, T., Ostchega, Y., Parrillo, J. and Young, R. (1987) J Clin Oncol 5, 641-647.
[278] Cairo, M., Siegel, S., Anas, N. and Sender, L. (1989) Cancer Res. 49, 1063-1066.
[279] Bartlett, N.L., Lum, B.L., Fisher, G.A., Brophy, N.A., Ehsan, M.N., Halsey, J. and Sikic, B.I. (1994) J Clin Onc 12, 835-842.
[280] Budd, G.T., Bukowski, R.M., Lichtin, A., Bauer, L,, Van Kirk, P. and Ganapathi, R. (1993) Investigational New Drugs 11, 75-79.
[281] Isonishi, S., Kirmani, S., Kim, S., Plaxe, S.C., Braly, P.S., McClay, E.F. and Howell, S.B. (1991) J Natl Cancer Inst 83, 621-626.
[282] List, A.F., Spier, C., Greer, J., Wolff', S., Hutter, J., Dorr, R., Salmon, S., Futscher, B, Baier, M. and Dalton, W. (1993) J Clin Oncol 11, 1652-1660.
[283] Miller, R.L., Bukowski, R.M., Budd, G.T., Purvis, J., Weick, J.K., Shepard, K., Midha, K.K. and Ganapathi, R. (1988) J Clin Oncol 6, 880-888.
[284] Jones, R., Kerr, D., Harnett, A., Rankin, E., Ray, S. and Kaye, S. (1990) Br. J. Cancer 62, 133-135.
[285] Mross, K., Bohn, C., Edler, L., Jonat, W., Queisser, W., Heide- mann, E., Goebel, M. and Hossfeld, D. (1993) Annals of Oncol 4, 45-50.
[286] Solary, E., Caillot, D., Chauffert, B., Casasnovas, R.O., Dumas, M., Maynadie, M. and Guy, H. (1992) J Clin Oncol 10, 1730-1736.
[287] Philip, P.A., Joel, S., Monkman, S.C., Dolega, O.E., Tonkin, K., Carmichael, J., Idle, J.R. and Harris, A.L. (1992) Br, J. Cancer 65, 267-270.
[288] Stuart, N.S., Philip, P., Harris, A.L., Tonkin, K., Houlbrook, S., Kirk, J., Lien, E.A. and Carmichael, J. (1992) Br. J. Cancer 66, 833-839.
[289] Workman, P., Kaye, S.B. and Schwartsmann, G. (1992) Current Opinion in Oncology 4, 1065-1072.
[290] Yahanda, A.M., Alder, K.M., Fisher, G.A., Brophy, N.A., Halsey, J., Hardy, R.I., Gosland, M.P., Lum, B.L. and Sikic, B.I. (1992) J Clin Onc 10, 1624-1634.
[291] Millward, M.J,, Cantwell, B.M., Lien, E.A,, Carmichael, J. and Harris, A.L. (1992) Ear J Cancer, 805-810.
[292] Sonneveld, P., Durie, B.G., Lokhorst, H.M., Marie, J.P., Solbu, G., Suciu, S., Zittoun, R., Lowenberg, B. and Nooter, K. (1992) Lancet 340, 255-259.
[293] Trump, D.L., Smith, D.C., Ellis, P.G., Rogers, M.P., Schold, S.C., Winer, E.P., Panella, T.J., Jordan, V.C. and Fine, R.L. (1992) J Natl Cancer Inst 84, 1811-1816.
[294] Millward, M.J., Cantwell, B.M., Munro, N.C., Robinson, A., Cor- ris, P.A. and Harris, A.L. (1993) Br. J. Cancer 67, 1031-1035.
[295] Milroy, R. (1993)Br. J. Cancer 68, 813-818. [296] Samuels, B.L., Mick, R., Vogelzang, N.J., Williams, S.F., Schilsky,
R.L., Safa, A.R., O'Brien, S.M. and Ratain, M.J, (1993) Clin Pharm Ther 54, 421-429.
[297] Christen, R.D., McClay, E.F., Plaxe, S.C., Yen, S.S., Kim, S., Kirmani, S., Wilgus, L.L., Heath, D.D., Shalinsky, D.R., Freddo, J.L. and et, a. (1993) J Clin Onc 11, 2417-2426.
[298] Wishart, G.C., Bissett, D., Paul, J., Jodrell, D., Harnett, A., Habe- shaw, T., Kerr, D.J., Macham, M.A., Soukop, M., Leonard, R.C. and et, a. (1994) J Clin Oncol 12, 1771-1777.
[299] Motzer, R.J., Lyn, P., Fischer, P., Lianes, P., Ngo, R.L., Cordon- Cardo, C. and O'Brien, J.P. (1995) J Clin Onc 13, 1958-1965.
[300] Warner, E., Tobe, S.W., Andrulis, I.L., Pei, Y., Trachtenberg, J. and Skorecki, K.L. (1995) Am J Clin Onc 18, 251-256.
[301] Agarwala, S.S., Bahnson, R.R., Wilson, J.W., Szumowski. J. and Ernstoff, M.S. (1995) Amer J Clin Oncol 18, 211-215.
[302] Dalton, W.S., Crowley, J.J., Salmon, S.S., Grogan, T.M., Laufman, L.R., Weiss, G.R. and Bonnet, J.D. (1995) Cancer 75, 815-820.
[303] Fisher, G.A. and Sikic, B.I. (1995) Hem/Onc Clinic NA 9, 363-382.
[304] Houghton, P.J. and Kaye, S.B. (1994) J NIH Research 6, 55-81. [305] Miller, T.P., Grogan, T.M., Dalton, W.S., Spier, C.M., Scheper,
R.J. and Salmon, S.E. (1991) J Clin Oncol 9, 17-24.- [306] Lum, B.L., Kaubisch, S., Yahanda, A.M., Adler, K.M., Jew, L.,
Ehsan, M.N., Brophy, N.A., Halsey, J., Gosland, M.P. and Sikic, B.1. (1992) J Clin Onc 10, 1635-1642.

F54 1. Bosch, J. Croop / Biochimica et Biophysica Acta 1288 (1996) F37-F54
[307] Mross, K., Hamm, K. and Hossfeld, D.K. (1993) Cancer Chem Pharm 31,369-375.
[308] Wilson, W.H., Jamis-Dow, C., Bryant, G., Balis, F.M., Klecker, R.W., Bates, S.E., Chabner, B.A., Steinberg, S.M., Kohler, D.R. and Wittes, R.E. (1995) J Clin Oncol 13, 1985-1994.
[309] Berg, S.L., Tolcher, A., O'Shaughnessy, J.A., Denicoff, A.M., Noone, M., Ognibene, F.P., Cowan, K.H. and Balis, F.M. (1995) J Clin Onc 13, 2039-2042.
[310] Gigante, M., Toffoli, G. and Boiocchi, M. (1995) Br. J. Cancer 71, 134-136.
[311 ] Sikic, B.I. (1993) Blood 11, 1629-1635. [312] Fitzgerald, D.J., Willingham, M.C., Cardarelli, C.O., Hamada, H.,
Tsuruo, T., Gottesman, M.M. and Pastan, I. (1987) Proc. Natl. Acad. Sci. 84, 4288-4292.
[313] Efferth, T. and Volm, M. (1993) Oncology 50, 303-308. [314] Hilselberger Kanitz, M.H., Mastro, J.M., Moore, R.E. and Starling,
J.J. (1994) Anticancer Res 14, 857-868. [315] Tsuruo, T., Hamada, H., Sato, S. and Heike, Y. (1989) Jpn J
Cancer Res. 80, 627-631. [316] Kulkarni, S.S., Wang, Z.M., Spitzer, G., Taha, M., Hamada, H.,
Tsuruo, T. and Dicke, K.A. (1989) Blood 74, 2244-2251. [317] Pearson, J.W., Fogler, W.E., Volker, K., Usui, N., Goldenberg,
S.K., Gruys, E., Riggs, C.W., Komschlies, K., Wiltrout, R.H., Tsurno, T., Pastan, I., Gottesman, M.M. and Longo, D. (1991) J Natl Cancer Inst 83, 1386-1391.
[318] Iwahashi, T., Okochi, E., Ariyoshi, K., Watabe, H., Amann, E., Mori, S., Tsuruo, T. and Ono, K. (1993) Cancer Res. 53, 5475- 5482.
[319] Naito, M., Tsuge, H., Kuroko, C., Koyama, T., Tomida, A., Tatsuta, T., Heike, Y. and Tsuruo, T. (1993) J Nat Cancer Inst 85, 311-316.
[320] Vasanthakumar, G. and Ahmed, N.K. (1989) Cancer Comm 1, 225-232.
[321] Clynes, M., Redmond, A., Moran, E. and Gilvarry, U. (1992) Cytotechnology 10, 75-89.
[322] Corrias, M.V. and Tonini, G.P. (1992) Anticancer Res 12, 1431- 1438.
[323] Redmond, A., Moran, E. and Clynes, M. (1993) Eur J Cancer 29A, 1078-1081.
[324] Schlaifer, D., Meyer, K., Muller, C., Attal, M., Smith, M.T., Tamaki, S., Weimels, J., Pris, J., Jaffrezou, J.P., Laurent, G. and et al. (1994) Leukemia 8, 289-291.
[325] Hanchett, L.A., Baker, R.M. and Dolnick, B.J. (1994) Somatic Cell and Molecular Genetics 20, 463-480.
[326] Thierry, A.R., Rahman, A. and Dritschilo, A. (1993) Biochem Biophys Res Com 190, 952-960.
[327] Bello-Reuss, E. and Ernest, S. (1994) American Journal of Physiol- ogy 267, C1351-1358.
[328] Quattrone, A., Papucci, L., Morganti, M., Coronnello, M., Mini, E., Mazzei, T., Colonna, F.P., Garbesi, A. and Capaccioli, S. (1994) Onc Res 6, 311-320.
[329] Tominaga, M., Tominaga, T., Miwa, A. and Okada, Y. (1995) J. Biol. Chem. 270, 27887-27893.
[330] Bertram, J., Palfner, K., Killian, M., Brysch, W., Schlingensiepen, K.H., Hiddemann, W. and Kneba, M. (1995) Anti-Cancer Drugs 6, 124-134.
[331] Kiehntopf, M., Brach, M.A., Licht, T., Petschauer, S., Karawajew, L., Kirschning, C. and Herrmann, F. (1994) EMBO Journal 13, 4645 -4652.
[332] Scanlon, K.J., Ishida, H. and Kashani-Sabet, M. (1994) Proc. Natl. Acad. Sci. 9l, 11123-11127.
[333] Kobayashi, H., Dorai, T., Holland, J.F. and Ohnuma, T. (1994) Cancer Res. 54, 1271-1275.
[334] Holm, P.S., Scanlon, K.J. and Dietel, M. (1994) Br. J. Cancer 70, 239-243.
[335] Kobayashi, H., Dorai, T,, Holland, J.F. and Ohnuma, T. (1993) FEBS Lett. 319, 71-74.
[336] O'Connor, R., Liu, C., Ferris, C.A., Guild, B.C., Teicher, B.A., Corvi, C., Liu, Y., Arceci, R.J., Goldmacher, V.S., Lambert, J.M. and et al. (1995) Blood 86, 4286-4294.
[337] Morimoto, H., Yonehara, S. and to, B. (1993) Cancer Res. 53, 2591-2596.
[338] Mickisch, G.H., Rahman, A., Pastan, I. and Gottesman, M.M. (1992) J. Natl. Cancer Inst. 84, 804-805.
[339] Rahman, A., Husain, S.R., Siddiqui, J., Verma, M., Agresti, M., Center, M., Safa, A.R. and Glazer, R.I. (1992) J. Natl. Cancer Inst. 84, 1909-1915.
[340] Sadasivan, R., Morgan, R., Fabian, C. and Stephens, R. (1991) Cancer Lett. 57, 165-171.
[341] Seid, C.A., Fidler, I.J., Clyne, R.K., Earnest, L.E. and Fan, D. (1991) Sel. Cancer Ther. 7, 103-112.
[342] Thierry, A.R., Jorgensen, T.J., Forst, D., Belli, J.A., Dritschilo, A. and Rahman, A. (1989) Cancer Commun. 1,311-316.
[343] Warren, L., Jardillier, J.C., Malarska, A. and Akeli, M.G. (1992) Cancer Res. 52, 3241-3245.
[344] Lemoli, R.M., Igarashi, T., Knizewski, M., Acaba, L., Richter, A., Jain, A., Mitchell, D., Levy, J. and Gulati, S.C. (1993) Blood 81, 793-800.
[345] Kuhl, J.S., Sikic, B.I., Blume, K.G. and Chao, N.J. (1992) Exp. Haematol. 20, 1048-1054.
[346] Aihara, M., Aihara, Y., Schmidt-Wolf, G., Schmidt-Wolf, I., Sikic, B.I., Blume, K.G. and Chao, N.J. (1991) Blood 77, 2079-2084.
[347] Sorrentino, B., Brandt, S., Bodine, D., Gottesman, M., Pastan, I., Cline, A. and Nienhuis, A. (1992) Science 257, 99-103.
[348] Podda, S., Ward, M., Himelstein, A., Richardson, C., de, 1.F., Weiss, E., Smith, L., Gottesman, M., Pastan, I. and Bank, A. (1992) Proc. Natl. Acad. Sci. 89, 9676-9680.
[349] Hegewisch-Becker, S., Hanania, E.G., Fu, S., Korbling, M., Deis- seroth, A.B. and Andreeff, M. (1995) Br. J. Haematol. 90, 876-883.
[350] Mickisch, G.H., Merlino, G.T., Galski, H., Gottesman, M.M. and Pastan, I. (1991) Proc. Natl. Acad. Sci. 88, 547-551.
[351] Germann, U.A., Chin, K.V., Pastan, I. and Gottesman, M.M. (1990) FASEB J. 4, 1501-1507.
[352] Aran, J.M., Gottesman, M.M. and Pastan, I. (1994) Proc. Natl. Acad. Sci. 91, 3176-3180.
Related Documents











