Available online at www.sciencedirect.com Biosensors and Bioelectronics 23 (2008) 1229–1235 Oxygen-reducing enzyme cathodes produced from SLAC, a small laccase from Streptomyces coelicolor Joshua Gallaway a , Ian Wheeldon a , Rosalba Rincon b , Plamen Atanassov b , Scott Banta a , Scott Calabrese Barton c,∗ a Columbia University, Department of Chemical Engineering, New York, NY 10027, USA b The University of New Mexico, Department of Chemical and Nuclear Engineering, Albuquerque, NM 87131, USA c Michigan State University, Department of Chemical Engineering and Material Science, East Lansing, MI 48824, USA Received 14 August 2007; received in revised form 2 November 2007; accepted 5 November 2007 Available online 13 November 2007 Abstract The bacterially-expressed laccase, small laccase (SLAC) of Streptomyces coelicolor, was incorporated into electrodes of both direct electron transfer (DET) and mediated electron transfer (MET) designs for application in biofuel cells. Using the DET design, enzyme redox kinetics were directly observable using cyclic voltammetry, and a redox potential of 0.43V (SHE) was observed. When mediated by an osmium redox polymer, the oxygen-reducing cathode retained maximum activity at pH 7, producing 1.5 mA/cm 2 in a planar configuration at 900 rpm and 40 ◦ C, thus outperforming enzyme electrodes produced using laccase from fungal Trametes versicolor (0.2 mA/cm 2 ) under similar conditions. This improvement is directly attributable to differences in the kinetics of SLAC and fungal laccases. Maximum stability of the mediated SLAC electrode was observed at pH above the enzyme’s relatively high isoelectric point, where the anionic enzyme molecules could form an electrostatic adduct with the cationic mediator. Porous composite SLAC electrodes with increased surface area produced a current density of 6.25mA/cm 2 at 0.3 V (SHE) under the above conditions. © 2007 Elsevier B.V. All rights reserved. Keywords: SLAC; Laccase; Biofuel cell; Oxygen reduction; Direct electron transfer; Mediated electron transfer 1. Introduction The inherent catalytic capacity of enzymes holds great promise for use in portable, flexible, and inexpensive micro- power sources able to refuel from ambient sources. Enzymatic biofuel cells could be designed to operate efficiently on any number of fuels, including ambient carbohydrates and macro- molecules (Bullen et al., 2006; Davis and Higson, 2007; Calabrese Barton et al., 2004). The high selectivity of enzymes allow for the elimination of many of the seals and balance of plant components of a traditional fuel cell, simplifying the design to essentially an anode and cathode (Heller, 2004). For applications demanding operation at low temperature and mild pH, with extremely small device volume and footprint ∗ Corresponding author. Tel.: +1 517 355 0222; fax: +1 517 432 1105. E-mail address: [email protected] (S.C. Barton). and at a cost low enough to be disposable, enzymatic biofuel cells have many advantages over batteries or traditional fuel cells. The cathode of any ambient biofuel cell will make use of the four-electron reduction of dissolved or atmospheric dioxygen to water. The study of enzymatic cathodes has focused chiefly on the copper-containing oxidoreductases laccase and bilirubin oxi- dase, with important considerations being high redox potential, complete four-electron reaction, desired pH of operation, and insensitivity to interfering anions. Results have been reported for the fungal laccases from Coriolus hirsutus, Trametes ver- sicolor, Coriolopsis gallica, and Pleurotus ostreatus, the tree laccase from Rhus vernicifera, and the fungal bilirubin oxidase from Myrothecium verrucaria (Mano et al., 2002; Calabrese Barton et al., 2002; Gupta et al., 2004; Tarasevich et al., 2001; Shleev et al., 2006). Enzymatic biofuel cells may be categorized in two types, based upon the method of electrical connection between the 0956-5663/$ – see front matter © 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.bios.2007.11.004

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

A
twptiww(©
K
1
ppbnmCaodFm
0d
Available online at www.sciencedirect.com
Biosensors and Bioelectronics 23 (2008) 1229–1235
Oxygen-reducing enzyme cathodes produced from SLAC,a small laccase from Streptomyces coelicolor
Joshua Gallaway a, Ian Wheeldon a, Rosalba Rincon b, Plamen Atanassov b,Scott Banta a, Scott Calabrese Barton c,∗
a Columbia University, Department of Chemical Engineering, New York, NY 10027, USAb The University of New Mexico, Department of Chemical and Nuclear Engineering, Albuquerque, NM 87131, USA
c Michigan State University, Department of Chemical Engineering and Material Science, East Lansing, MI 48824, USA
Received 14 August 2007; received in revised form 2 November 2007; accepted 5 November 2007Available online 13 November 2007
bstract
The bacterially-expressed laccase, small laccase (SLAC) of Streptomyces coelicolor, was incorporated into electrodes of both direct electronransfer (DET) and mediated electron transfer (MET) designs for application in biofuel cells. Using the DET design, enzyme redox kineticsere directly observable using cyclic voltammetry, and a redox potential of 0.43 V (SHE) was observed. When mediated by an osmium redoxolymer, the oxygen-reducing cathode retained maximum activity at pH 7, producing 1.5 mA/cm2 in a planar configuration at 900 rpm and 40 ◦C,hus outperforming enzyme electrodes produced using laccase from fungal Trametes versicolor (0.2 mA/cm2) under similar conditions. Thismprovement is directly attributable to differences in the kinetics of SLAC and fungal laccases. Maximum stability of the mediated SLAC electrode
as observed at pH above the enzyme’s relatively high isoelectric point, where the anionic enzyme molecules could form an electrostatic adductith the cationic mediator. Porous composite SLAC electrodes with increased surface area produced a current density of 6.25 mA/cm2 at 0.3 VSHE) under the above conditions.2007 Elsevier B.V. All rights reserved.
eywords: SLAC; Laccase; Biofuel cell; Oxygen reduction; Direct electron transfer; Mediated electron transfer
acc
fwtdcif
. Introduction
The inherent catalytic capacity of enzymes holds greatromise for use in portable, flexible, and inexpensive micro-ower sources able to refuel from ambient sources. Enzymaticiofuel cells could be designed to operate efficiently on anyumber of fuels, including ambient carbohydrates and macro-olecules (Bullen et al., 2006; Davis and Higson, 2007;alabrese Barton et al., 2004). The high selectivity of enzymesllow for the elimination of many of the seals and balancef plant components of a traditional fuel cell, simplifying the
esign to essentially an anode and cathode (Heller, 2004).or applications demanding operation at low temperature andild pH, with extremely small device volume and footprint∗ Corresponding author. Tel.: +1 517 355 0222; fax: +1 517 432 1105.E-mail address: [email protected] (S.C. Barton).
slfBS
b
956-5663/$ – see front matter © 2007 Elsevier B.V. All rights reserved.oi:10.1016/j.bios.2007.11.004
nd at a cost low enough to be disposable, enzymatic biofuelells have many advantages over batteries or traditional fuelells.
The cathode of any ambient biofuel cell will make use of theour-electron reduction of dissolved or atmospheric dioxygen toater. The study of enzymatic cathodes has focused chiefly on
he copper-containing oxidoreductases laccase and bilirubin oxi-ase, with important considerations being high redox potential,omplete four-electron reaction, desired pH of operation, andnsensitivity to interfering anions. Results have been reportedor the fungal laccases from Coriolus hirsutus, Trametes ver-icolor, Coriolopsis gallica, and Pleurotus ostreatus, the treeaccase from Rhus vernicifera, and the fungal bilirubin oxidaserom Myrothecium verrucaria (Mano et al., 2002; Calabrese
arton et al., 2002; Gupta et al., 2004; Tarasevich et al., 2001;hleev et al., 2006).Enzymatic biofuel cells may be categorized in two types,ased upon the method of electrical connection between the

1 d Bio
e1aftuoerasdadts
rb(u(1cpG1bcpe
GomLsiKp1ape7
itcrvSgte
e
cSmtrfiaeapf
2
2
Awi4tphpoftNTlC
2
SB(ipocgpcs7
a1sS
230 J. Gallaway et al. / Biosensors an
nzyme and the current collector (Ghindilis et al., 1997; Heller,992). In direct electron transfer (DET) systems, the enzymend current collector are in direct contact, and electrons passrom the electrode surface to the reducing substrate center ofhe enzyme. DET affords a simplicity of design and allows these of the full thermodynamic potential of the enzyme, with-ut introducing an overpotential for electron transfer from thenzyme to a mediator. In mediated electron transfer (MET), aedox-active molecule, whether organic or inorganic, is used toct as a reducing substrate and carry electrons from the electrodeurface to the enzyme. MET generally offers a higher currentensity than DET if the system has been well-optimized and theppropriate mediator has been employed. However, the METesign introduces an additional level of complexity, and elec-rode performance becomes a matter of mediator integrity andtability, as well as stability of the enzyme itself.
MET electrodes are often optimized on planar, glassy carbonotating disk electrodes and then scaled to composite electrodesased on a porous carbon cloth matrix. Calabrese Barton et al.2001) first reported carbon paper composite enzyme electrodes,sing laccase of C. hirsutus and producing 5.0 mA/cm2 at 0.62 VSHE) in 0.2 M pH 5 oxygen-saturated citrate buffer at 37 ◦C and000 rpm. Mano et al. (2002) constructed a bilirubin oxidaseathode producing 4.5 mA/cm2 at 0.57 V (SHE) in oxygenatedhosphate buffered saline (PBS) pH 7.4 at 37.5 ◦C at 1000 rpm.allaway (2007) report a T. versicolor electrode resulting in3 mA/cm2 at 0.53 V (SHE) in 0.1 M pH 4 oxygenated citrateuffer at 40 ◦C and 900 rpm. All of these MET examples use arosslinked osmium-based redox mediator, producing the appro-riate redox potential for mediation and resulting in a stablelectrode film construction.
Several cathodes are also reported using a DET construction.upta et al. (2004) reported oxygen-reducing cathodes basedn laccases from C. hirsutus and R. vernicifera on monolayer-odified gold electrodes operating in pH 7 phosphate buffer.im et al. (2007) reported bilirubin oxidase electrodes onilica sol–gel/carbon nanotube composite electrodes resultingn 140 �A/cm2 at 25 ◦C in quiescent, oxygen-saturated PBS.amitaka et al. (2007) constructed a cathode on highly orientedyrolytic graphite (HOPG) with a Trametes laccase, getting70 �A/cm2 under oxygen in pH 5 buffer at 25 ◦C. Tsujimura etl. (2005) have reported bilirubin oxidase-based electrodes in aoly-l-lysine matrix, characterized as DET-like with a diffusingnzyme, which result in 0.85 mA/cm2 in oxygen-saturated pHphosphate buffer, rotating at 1400 rpm.Electrodes based on bacterial enzymes appear only seldom
n the literature. Miura et al. (2007) constructed a DET elec-rode on HOPG using copper efflux oxidase (CueO) from E.oli, with the enzyme free in solution at pH 5 and 25 ◦C. Theesult is 3.7 mA/cm2 catalytic current, although the stability andersatility of construction for a non-diffusing version is unclear.uch use of a bacterial enzyme may be desirable, as heterolo-ous expression of recombinant proteins in bacteria is facile, and
he resulting proteins are non-glycosylated, which may facilitatelectron transfer.In the present work, we report the evaluation of a bacterially-xpressed laccase, small laccase (SLAC) of Streptomyces
tca2
electronics 23 (2008) 1229–1235
oelicolor, using both MET and DET-type electrode schemes.olution phase results are compared to enzyme electrode perfor-ance, and electrochemical polarization results are interpreted
o identify rate-limiting steps. This laccase was previouslyeported to be highly active at pH 7, displaying the same speci-city to oxygen and non-specificity to the reducing substrates other laccases, making it an ideal candidate for enzyme bio-lectrocatalysis (Machczynski et al., 2004). Here we show thatcurrent density approaching 7 mA/cm2 can be obtained in a
orous composite electrode, the highest reported current densityor enzymatic oxygen reduction at neutral pH.
. Experimental
.1. Materials
Ultrapure O2, air, N2, and argon were purchased from Techir (White Plains, NY). Laccase from Trametes versicoloras purchased from Sigma-Aldrich (St. Louis, MO). Vinylim-
dazole, potassium hexachloroosmiate, 2,2′-bipyridyl (bpy),,4′-dimethyl-2,2′-bipyridine (dm-bpy), sodium dithionite, glu-araldehyde, citric acid monohydrate, sodium citrate, sodiumhosphate, sodium phosphate monobasic, sodium hydroxide,ydrochloric acid, ethanol, dimethylformamide, and ether wereurchased from Fisher Chemical (Fair Lawn, NJ) and used with-ut purification. Azobisisobutyronitrile (AIBN) was purchasedrom Fisher Chemical and recrystallized from methanol prioro use. Single-walled carbon nanotube powder (CNT) and 5%afion solution in alcohol were purchased from Sigma-Aldrich.he plasmid pSLAC encoding the small laccase gene from iso-
ated from Streptomyces coelicolor was a kind gift from Gerardanters (Leiden University, Netherlands).
.2. Enzyme expression and purification
Laccase from Trametes versicolor was purified using a DEAEephacel resin as reported previously (Hudak and Calabresearton, 2005). SLAC was expressed as previously described
Machczynski et al., 2004). Briefly, 1 L of 2×YT media wasnoculated with mature E. coli (BL21) culture harboring theSLAC expression plasmid. The culture was grown to an OD600f 1.5 at 30 ◦C prior to induction with 0.4 mM IPTG. Expressionontinued for 20 h at 25 ◦C. Cells were harvested by centrifu-ation and soluble protein was released from re-suspended cellellets (10 mM phosphate, pH 7.3) by sonication. Prior to purifi-ation, the crude lysate was incubated with 1 mM of CuSO4 foreveral hours and dialyzed against 10 mM phosphate buffer, pH.3 containing 1 mM of EDTA.
SLAC was purified from the crude lysate by DEAE weaknion exchange chromatography (AKTA FPLC with HiPrep6/10 DEAE FF, GE Healthcare). Protein was eluted with ateep gradient of NaCl from 0 to 100 mM. Fractions containingLAC, blue in color, were pooled, concentrated by ultrafiltra-
ion, and purified to greater than 95% purity using size exclusionhromatography (HiLoad 16/60 Superdex 200, GE Healthcare)s judged by SDS-polyacrylamide gel electrophoresis. Yields of0–25 mg of purified SLAC were obtained.

d Bio
2
s(woticIarow
2
pbMabaioAs
2
rpwcoabdrbdfi0dirioocdite
mwpp0ts
tGd2pphda
2
jbptc3MrPiuto
cuehTitdos
fidwti
3
J. Gallaway et al. / Biosensors an
.3. Activity assay with [Os(dm-bpy)2(1-MeIm) Cl]Cl2
The low molecular weight osmium complex was synthe-ized with an analogous procedure to that previously reportedBuckingham et al., 1964). The activities of SLAC and TvLere determined by measuring the initial rate of oxidation of thesmium complex from a 1+ to a 2+ state in air-saturated solu-ion. The change in oxidation state was observed by the changen absorption at 520 nm (ε = 6700 M−1 cm−1). All assays werearried out in at least triplicate in clear 96 well plates (Corningnc., Corning NY) and the UV–vis measurements acquired withSpectraMax M2 microplate reader (Molecular Devices Corpo-
ation, Sunnyvale CA). Solutions were buffered with 100 mMf citrate, pH 4 and 5, or phosphate, pH 6, 7, 8 and 9. Buffer pHas adjusted with HCl or NaOH as required.
.4. DET electrode preparation
A carbon nanotube ink was prepared with 0.9 mL of pH 7hosphate buffer, 0.1 mL of 5% Nafion modified with tetra-utylammonium salt, and 5 mg of CNT powder (Topcagic andinteer, 2006). The ink was sonicated for 30 min. A 10 �L
liquot of the CNT ink was dropped on a screen-printed car-on electrode (Alderon Biosciences, Durham, NC) and left inhumid chamber for at least 24 h, until dry. The enzyme was
mmobilized on the CNT-modified electrode by adding 10 �Lf 5% glutaraldehyde and 10 �L of 10 mg/mL SLAC solution.
schematic of both the DET and MET electrodes is shown inupplementary material.
.5. MET electrode preparation
The redox polymer poly(n-VI12[Os(bpy)2Cl]+/2+), with aedox potential of 0.43 V (SHE), was synthesized as describedreviously (Forster and Vos, 1990). Briefly, polyvinylimidazoleas produced by free-radical polymerization in ethanol and pre-
ipitated in acetone (Dambatta and Ebdon, 1986). The precursorsmium complex was synthesized by reaction of bipyridine withmmonium hexachloroosmiate in dimethylformamide, followedy precipitation in ether. The complex was reduced by sodiumithionite and attached to the polyvinylimidazole backbone byefluxing in ethanol for three days, followed by purificationy ultrafiltration. Glassy carbon rotating disc electrodes (3 mmiameter, produced in-house) were sanded with 1200 grit ultrane sandpaper (Buehler, IL), and polished with 0.3 �m and.05 �m alumina slurries, followed by sonication for 10 min ineionized water. After the cleaning procedure, no electrochem-cal features were visible for a bare electrode over the potentialange of water stability between hydrogen and oxygen evolutionn pH 4 100 mM citrate buffer at 50 mV/s scan rate. A solutionf the redox polymer (10 mg/mL) was combined with a solutionf either SLAC or TvL (20 mg/mL), and a solution of PEGDGEross-linker (5 mg/mL) was added, in amounts necessary to pro-
uce a film with a total protein composition of 19–42% by weightn a matrix of cross-linked redox polymer. The redox polymero cross-linker mass ratio was held constant at 9:1. For bothnzymes, the optimum composition was found to be 61% poly-ts
electronics 23 (2008) 1229–1235 1231
er, 32% laccase, and 7% cross-linker by weight, in agreementith previous findings (Calabrese Barton et al., 2001). This com-osition was used for all experiments. An aliquot of 5 �L wasipetted onto the electrode surface to give a material loading of.69 mg/cm2. The electrode was allowed to cure for 5 h at roomemperature in a low-humidity environment, R.H. < 10%. Seeupplementary material for a schematic.
Composite carbon paper electrodes were constructed by cut-ing a 4 mm disk of GDL10AA (SGL Carbon AG, Weisbaden,ermany) and affixing to the glassy carbon RDE with con-uctive carbon paint (SPI, West Chester, PA). After drying forh, the carbon paper was made hydrophilic by treatment in airlasma for 5 min, and the film solution was added as in thelanar electrode case and allowed to cure overnight in ambient-umidity conditions. To minimize the effects of deactivationuring storage, all electrodes were characterized immediatelyfter the curing step.
.6. Electrochemical instrumentation and methods
All electrochemical experiments were carried out in a water-acketed cell containing 100 mL of the indicated 100 mMuffer with a jacket temperature of 40 ◦C. Measurements wereerformed with a �Autolab III potentiostat (Eco Chemie). Elec-rodes were rotated using a Pine Instruments Rotator. Glassyarbon rotating disk electrodes (RDEs) were constructed frommm diameter, type 1 glassy carbon rods (Alfa Aesar, Ward Hill,A). Potentials were measured relative to a silver-silver chlo-
ide (3 M NaCl) reference electrode (BAS, West Lafayette, IN).latinum gauze was used as the counter electrode. Electrochem-
cal measurements for the DET experiments were performedsing the screen-printed electrodes, with a carbon counter elec-rode and Ag|AgCl reference electrode, in pH 7 phosphate bufferr pH 4.8 acetate buffer.
Apparent electron diffusion of the MET mediator, Dm, wasalculated from the current response following a potential step,sing the Cottrell equation (Bard and Faulkner, 2001). In qui-scent, nitrogen-saturated solution, the working electrode waseld at a reducing potential until negligible current was recorded.he potential was stepped to an oxidizing potential, and after an
nitial nonlinear regime, current decayed linearly to zero withhe inverse square root of time, indicating an apparent Fickianiffusion of charge (Forster et al., 1989; Majda, 1992). The slopef the linear region in 10–30 ms period following the potentialtep was used to calculate Dm.
Stability of the operating electrodes was characterized bytting the initial 30 min of potentiostatic operation to a first-orderecay of the form i(t) = i0 exp(−t/τd). A selection of electrodesere run for longer intervals, up to 12 h, and it was found that
he time constant, τd, calculated from the initial 30 min wasndicative of electrode decay at longer times.
. Results
Direct connection of SLAC to a carbon electrode allows elec-ron transfer directly to the copper active sites of the enzyme,hown in Fig. 1a at pH 7. Oxidation and reduction peaks are
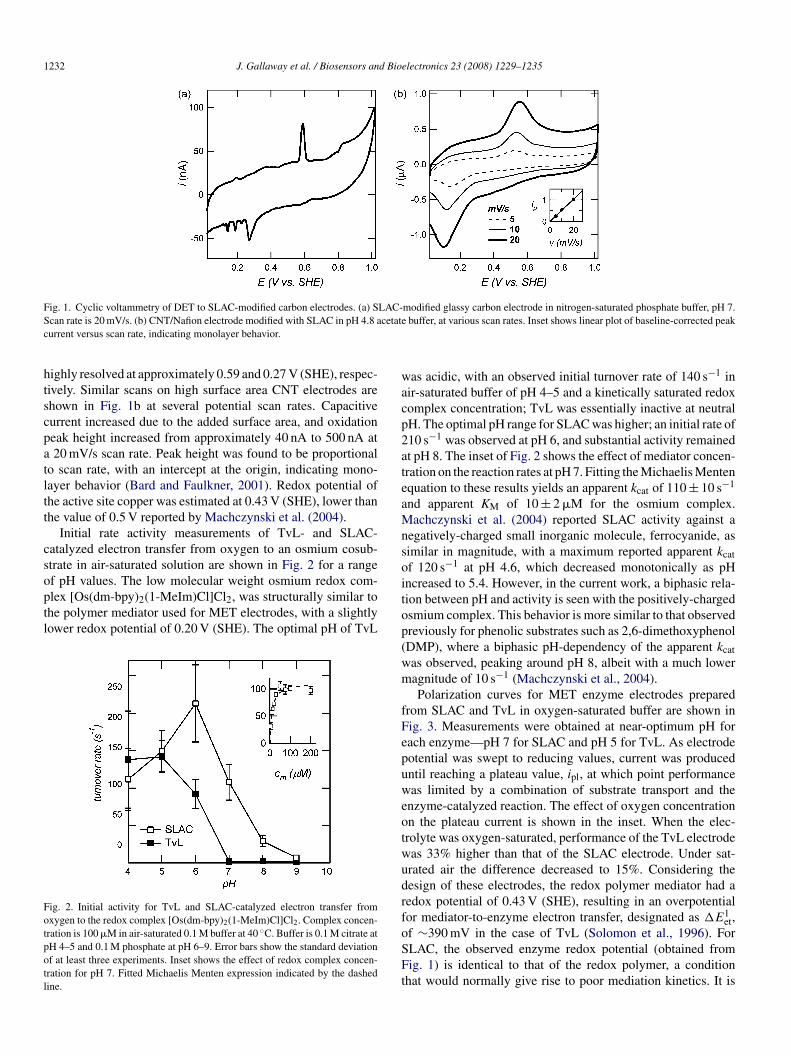
1232 J. Gallaway et al. / Biosensors and Bioelectronics 23 (2008) 1229–1235
F LAC-S cetatec
htscpatltt
csoptl
Fotpotl
wacp2ateaMnso
ig. 1. Cyclic voltammetry of DET to SLAC-modified carbon electrodes. (a) Scan rate is 20 mV/s. (b) CNT/Nafion electrode modified with SLAC in pH 4.8 aurrent versus scan rate, indicating monolayer behavior.
ighly resolved at approximately 0.59 and 0.27 V (SHE), respec-ively. Similar scans on high surface area CNT electrodes arehown in Fig. 1b at several potential scan rates. Capacitiveurrent increased due to the added surface area, and oxidationeak height increased from approximately 40 nA to 500 nA at20 mV/s scan rate. Peak height was found to be proportional
o scan rate, with an intercept at the origin, indicating mono-ayer behavior (Bard and Faulkner, 2001). Redox potential ofhe active site copper was estimated at 0.43 V (SHE), lower thanhe value of 0.5 V reported by Machczynski et al. (2004).
Initial rate activity measurements of TvL- and SLAC-atalyzed electron transfer from oxygen to an osmium cosub-trate in air-saturated solution are shown in Fig. 2 for a range
f pH values. The low molecular weight osmium redox com-lex [Os(dm-bpy)2(1-MeIm)Cl]Cl2, was structurally similar tohe polymer mediator used for MET electrodes, with a slightlyower redox potential of 0.20 V (SHE). The optimal pH of TvLig. 2. Initial activity for TvL and SLAC-catalyzed electron transfer fromxygen to the redox complex [Os(dm-bpy)2(1-MeIm)Cl]Cl2. Complex concen-ration is 100 �M in air-saturated 0.1 M buffer at 40 ◦C. Buffer is 0.1 M citrate atH 4–5 and 0.1 M phosphate at pH 6–9. Error bars show the standard deviationf at least three experiments. Inset shows the effect of redox complex concen-ration for pH 7. Fitted Michaelis Menten expression indicated by the dashedine.
itop(wm
fFepuweotwudrfoSFt
modified glassy carbon electrode in nitrogen-saturated phosphate buffer, pH 7.buffer, at various scan rates. Inset shows linear plot of baseline-corrected peak
as acidic, with an observed initial turnover rate of 140 s−1 inir-saturated buffer of pH 4–5 and a kinetically saturated redoxomplex concentration; TvL was essentially inactive at neutralH. The optimal pH range for SLAC was higher; an initial rate of10 s−1 was observed at pH 6, and substantial activity remainedt pH 8. The inset of Fig. 2 shows the effect of mediator concen-ration on the reaction rates at pH 7. Fitting the Michaelis Mentenquation to these results yields an apparent kcat of 110 ± 10 s−1
nd apparent KM of 10 ± 2 �M for the osmium complex.achczynski et al. (2004) reported SLAC activity against a
egatively-charged small inorganic molecule, ferrocyanide, asimilar in magnitude, with a maximum reported apparent kcatf 120 s−1 at pH 4.6, which decreased monotonically as pHncreased to 5.4. However, in the current work, a biphasic rela-ion between pH and activity is seen with the positively-chargedsmium complex. This behavior is more similar to that observedreviously for phenolic substrates such as 2,6-dimethoxyphenolDMP), where a biphasic pH-dependency of the apparent kcatas observed, peaking around pH 8, albeit with a much loweragnitude of 10 s−1 (Machczynski et al., 2004).Polarization curves for MET enzyme electrodes prepared
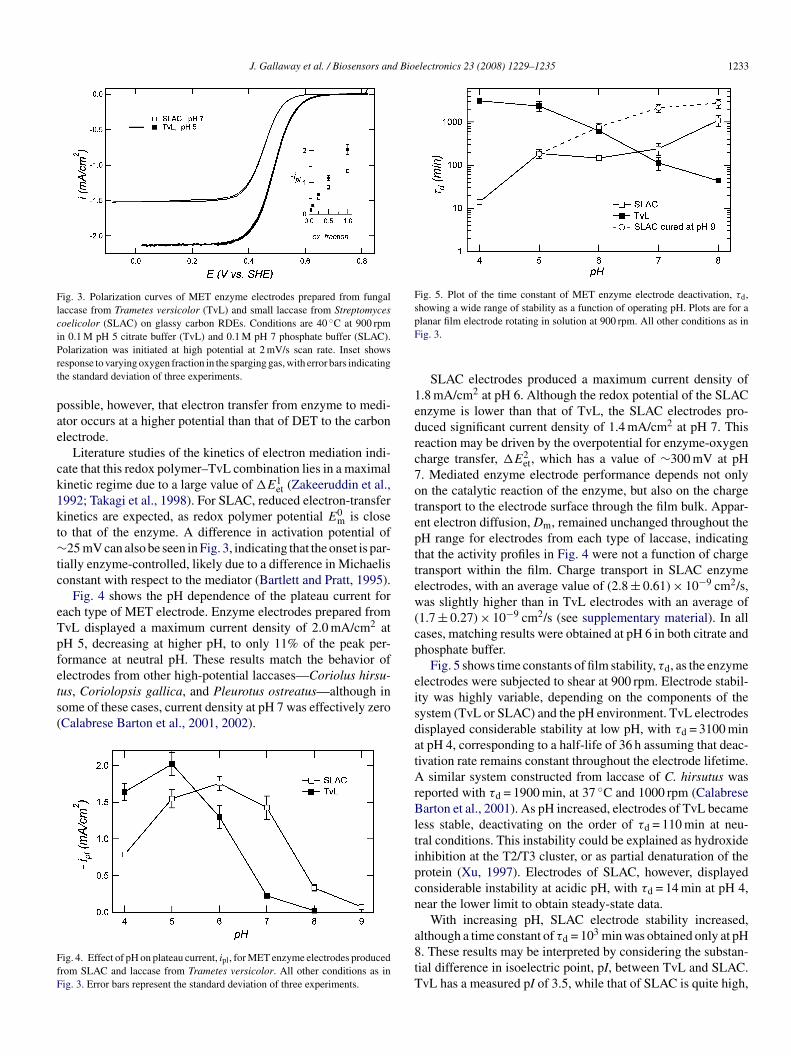
rom SLAC and TvL in oxygen-saturated buffer are shown inig. 3. Measurements were obtained at near-optimum pH forach enzyme—pH 7 for SLAC and pH 5 for TvL. As electrodeotential was swept to reducing values, current was producedntil reaching a plateau value, ipl, at which point performanceas limited by a combination of substrate transport and the
nzyme-catalyzed reaction. The effect of oxygen concentrationn the plateau current is shown in the inset. When the elec-rolyte was oxygen-saturated, performance of the TvL electrodeas 33% higher than that of the SLAC electrode. Under sat-rated air the difference decreased to 15%. Considering theesign of these electrodes, the redox polymer mediator had aedox potential of 0.43 V (SHE), resulting in an overpotentialor mediator-to-enzyme electron transfer, designated as �E1
et,
f ∼390 mV in the case of TvL (Solomon et al., 1996). ForLAC, the observed enzyme redox potential (obtained fromig. 1) is identical to that of the redox polymer, a conditionhat would normally give rise to poor mediation kinetics. It is
J. Gallaway et al. / Biosensors and Bioelectronics 23 (2008) 1229–1235 1233
Fig. 3. Polarization curves of MET enzyme electrodes prepared from fungallaccase from Trametes versicolor (TvL) and small laccase from Streptomycescoelicolor (SLAC) on glassy carbon RDEs. Conditions are 40 ◦C at 900 rpmin 0.1 M pH 5 citrate buffer (TvL) and 0.1 M pH 7 phosphate buffer (SLAC).Prt
pae
ck1kt∼tc
eTpfets(
FfF
Fig. 5. Plot of the time constant of MET enzyme electrode deactivation, τd,showing a wide range of stability as a function of operating pH. Plots are for apF
1edrc7otepttew(cp
olarization was initiated at high potential at 2 mV/s scan rate. Inset showsesponse to varying oxygen fraction in the sparging gas, with error bars indicatinghe standard deviation of three experiments.
ossible, however, that electron transfer from enzyme to medi-tor occurs at a higher potential than that of DET to the carbonlectrode.
Literature studies of the kinetics of electron mediation indi-ate that this redox polymer–TvL combination lies in a maximalinetic regime due to a large value of �E1
et (Zakeeruddin et al.,992; Takagi et al., 1998). For SLAC, reduced electron-transferinetics are expected, as redox polymer potential E0
m is closeo that of the enzyme. A difference in activation potential of
25 mV can also be seen in Fig. 3, indicating that the onset is par-ially enzyme-controlled, likely due to a difference in Michaelisonstant with respect to the mediator (Bartlett and Pratt, 1995).
Fig. 4 shows the pH dependence of the plateau current forach type of MET electrode. Enzyme electrodes prepared fromvL displayed a maximum current density of 2.0 mA/cm2 atH 5, decreasing at higher pH, to only 11% of the peak per-ormance at neutral pH. These results match the behavior of
lectrodes from other high-potential laccases—Coriolus hirsu-us, Coriolopsis gallica, and Pleurotus ostreatus—although inome of these cases, current density at pH 7 was effectively zeroCalabrese Barton et al., 2001, 2002).ig. 4. Effect of pH on plateau current, ipl, for MET enzyme electrodes producedrom SLAC and laccase from Trametes versicolor. All other conditions as inig. 3. Error bars represent the standard deviation of three experiments.
eisdatArBltipcn
a8tT
lanar film electrode rotating in solution at 900 rpm. All other conditions as inig. 3.
SLAC electrodes produced a maximum current density of.8 mA/cm2 at pH 6. Although the redox potential of the SLACnzyme is lower than that of TvL, the SLAC electrodes pro-uced significant current density of 1.4 mA/cm2 at pH 7. Thiseaction may be driven by the overpotential for enzyme-oxygenharge transfer, �E2
et, which has a value of ∼300 mV at pH. Mediated enzyme electrode performance depends not onlyn the catalytic reaction of the enzyme, but also on the chargeransport to the electrode surface through the film bulk. Appar-nt electron diffusion, Dm, remained unchanged throughout theH range for electrodes from each type of laccase, indicatinghat the activity profiles in Fig. 4 were not a function of chargeransport within the film. Charge transport in SLAC enzymelectrodes, with an average value of (2.8 ± 0.61) × 10−9 cm2/s,as slightly higher than in TvL electrodes with an average of
1.7 ± 0.27) × 10−9 cm2/s (see supplementary material). In allases, matching results were obtained at pH 6 in both citrate andhosphate buffer.
Fig. 5 shows time constants of film stability, τd, as the enzymelectrodes were subjected to shear at 900 rpm. Electrode stabil-ty was highly variable, depending on the components of theystem (TvL or SLAC) and the pH environment. TvL electrodesisplayed considerable stability at low pH, with τd = 3100 mint pH 4, corresponding to a half-life of 36 h assuming that deac-ivation rate remains constant throughout the electrode lifetime.
similar system constructed from laccase of C. hirsutus waseported with τd = 1900 min, at 37 ◦C and 1000 rpm (Calabresearton et al., 2001). As pH increased, electrodes of TvL became
ess stable, deactivating on the order of τd = 110 min at neu-ral conditions. This instability could be explained as hydroxidenhibition at the T2/T3 cluster, or as partial denaturation of therotein (Xu, 1997). Electrodes of SLAC, however, displayedonsiderable instability at acidic pH, with τd = 14 min at pH 4,ear the lower limit to obtain steady-state data.
With increasing pH, SLAC electrode stability increased,lthough a time constant of τd = 103 min was obtained only at pH
. These results may be interpreted by considering the substan-ial difference in isoelectric point, pI, between TvL and SLAC.vL has a measured pI of 3.5, while that of SLAC is quite high,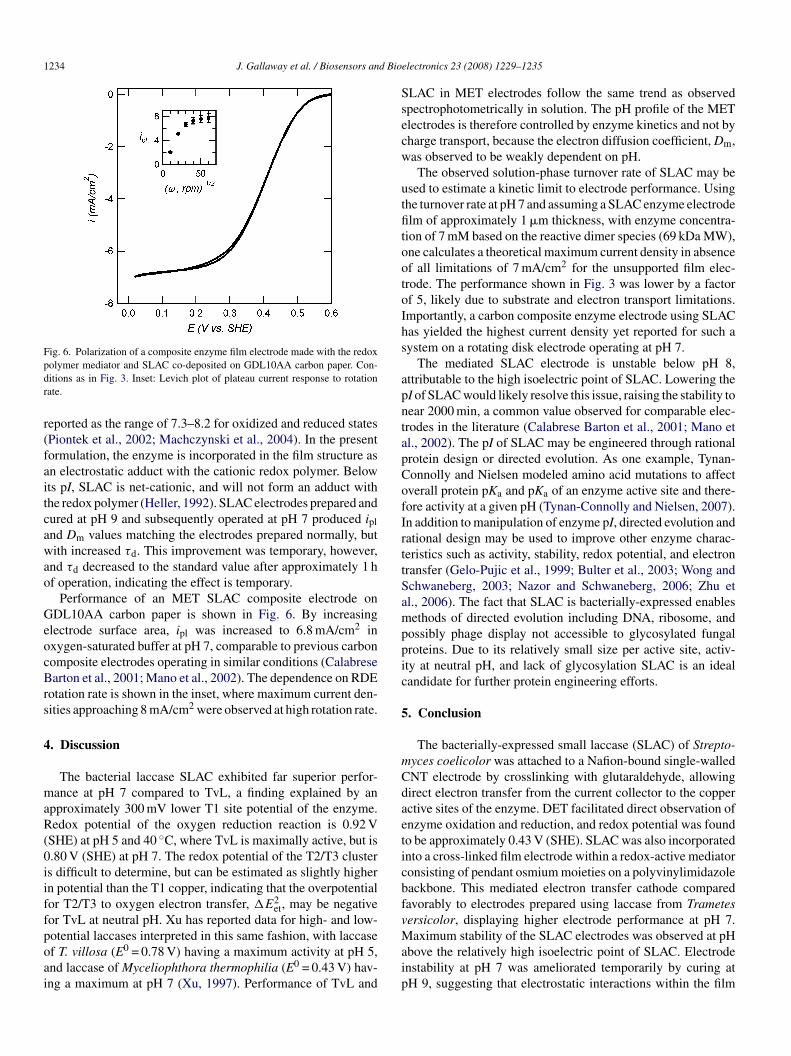
1234 J. Gallaway et al. / Biosensors and Bio
Fig. 6. Polarization of a composite enzyme film electrode made with the redoxpdr
r(faitcawao
GeocBrs
4
maR(0iiffpoai
Ssecw
utfitootoIhs
apntapCofIrttSamppic
5
mCdaeticbfv
olymer mediator and SLAC co-deposited on GDL10AA carbon paper. Con-itions as in Fig. 3. Inset: Levich plot of plateau current response to rotationate.
eported as the range of 7.3–8.2 for oxidized and reduced statesPiontek et al., 2002; Machczynski et al., 2004). In the presentormulation, the enzyme is incorporated in the film structure asn electrostatic adduct with the cationic redox polymer. Belowts pI, SLAC is net-cationic, and will not form an adduct withhe redox polymer (Heller, 1992). SLAC electrodes prepared andured at pH 9 and subsequently operated at pH 7 produced iplnd Dm values matching the electrodes prepared normally, butith increased τd. This improvement was temporary, however,
nd τd decreased to the standard value after approximately 1 hf operation, indicating the effect is temporary.
Performance of an MET SLAC composite electrode onDL10AA carbon paper is shown in Fig. 6. By increasing
lectrode surface area, ipl was increased to 6.8 mA/cm2 inxygen-saturated buffer at pH 7, comparable to previous carbonomposite electrodes operating in similar conditions (Calabresearton et al., 2001; Mano et al., 2002). The dependence on RDE
otation rate is shown in the inset, where maximum current den-ities approaching 8 mA/cm2 were observed at high rotation rate.
. Discussion
The bacterial laccase SLAC exhibited far superior perfor-ance at pH 7 compared to TvL, a finding explained by an
pproximately 300 mV lower T1 site potential of the enzyme.edox potential of the oxygen reduction reaction is 0.92 V
SHE) at pH 5 and 40 ◦C, where TvL is maximally active, but is.80 V (SHE) at pH 7. The redox potential of the T2/T3 clusters difficult to determine, but can be estimated as slightly highern potential than the T1 copper, indicating that the overpotentialor T2/T3 to oxygen electron transfer, �E2
et, may be negativeor TvL at neutral pH. Xu has reported data for high- and low-
otential laccases interpreted in this same fashion, with laccasef T. villosa (E0 = 0.78 V) having a maximum activity at pH 5,nd laccase of Myceliophthora thermophilia (E0 = 0.43 V) hav-ng a maximum at pH 7 (Xu, 1997). Performance of TvL andMaip
electronics 23 (2008) 1229–1235
LAC in MET electrodes follow the same trend as observedpectrophotometrically in solution. The pH profile of the METlectrodes is therefore controlled by enzyme kinetics and not byharge transport, because the electron diffusion coefficient, Dm,as observed to be weakly dependent on pH.The observed solution-phase turnover rate of SLAC may be
sed to estimate a kinetic limit to electrode performance. Usinghe turnover rate at pH 7 and assuming a SLAC enzyme electrodelm of approximately 1 �m thickness, with enzyme concentra-
ion of 7 mM based on the reactive dimer species (69 kDa MW),ne calculates a theoretical maximum current density in absencef all limitations of 7 mA/cm2 for the unsupported film elec-rode. The performance shown in Fig. 3 was lower by a factorf 5, likely due to substrate and electron transport limitations.mportantly, a carbon composite enzyme electrode using SLACas yielded the highest current density yet reported for such aystem on a rotating disk electrode operating at pH 7.
The mediated SLAC electrode is unstable below pH 8,ttributable to the high isoelectric point of SLAC. Lowering theI of SLAC would likely resolve this issue, raising the stability toear 2000 min, a common value observed for comparable elec-rodes in the literature (Calabrese Barton et al., 2001; Mano etl., 2002). The pI of SLAC may be engineered through rationalrotein design or directed evolution. As one example, Tynan-onnolly and Nielsen modeled amino acid mutations to affectverall protein pKa and pKa of an enzyme active site and there-ore activity at a given pH (Tynan-Connolly and Nielsen, 2007).n addition to manipulation of enzyme pI, directed evolution andational design may be used to improve other enzyme charac-eristics such as activity, stability, redox potential, and electronransfer (Gelo-Pujic et al., 1999; Bulter et al., 2003; Wong andchwaneberg, 2003; Nazor and Schwaneberg, 2006; Zhu etl., 2006). The fact that SLAC is bacterially-expressed enablesethods of directed evolution including DNA, ribosome, and
ossibly phage display not accessible to glycosylated fungalroteins. Due to its relatively small size per active site, activ-ty at neutral pH, and lack of glycosylation SLAC is an idealandidate for further protein engineering efforts.
. Conclusion
The bacterially-expressed small laccase (SLAC) of Strepto-yces coelicolor was attached to a Nafion-bound single-walledNT electrode by crosslinking with glutaraldehyde, allowingirect electron transfer from the current collector to the copperctive sites of the enzyme. DET facilitated direct observation ofnzyme oxidation and reduction, and redox potential was foundo be approximately 0.43 V (SHE). SLAC was also incorporatednto a cross-linked film electrode within a redox-active mediatoronsisting of pendant osmium moieties on a polyvinylimidazoleackbone. This mediated electron transfer cathode comparedavorably to electrodes prepared using laccase from Trametesersicolor, displaying higher electrode performance at pH 7.
aximum stability of the SLAC electrodes was observed at pHbove the relatively high isoelectric point of SLAC. Electrodenstability at pH 7 was ameliorated temporarily by curing atH 9, suggesting that electrostatic interactions within the film

d Bio
wtor
A
FR(mN
A
i
R
B
BB
B
B
C
C
C
DD
F
FGG
GGHHHK
L
M
M
M
M
NP
S
S
TT
TTTW
J. Gallaway et al. / Biosensors an
ere responsible for stability. High surface-area carbon elec-rodes further increased current density, producing 7 mA/cm2 inxygen-saturated buffer at pH 7, 900 rpm and 40 ◦C, the highesteported performance for such a system.
cknowledgements
The authors gratefully acknowledge support under contractA9550-06-1-0264 from the Air Force Office of Scientificesearch. The authors also wish to thank Shelley Minteer
St. Louis University) for providing the tetrabutylammonium-odified Nafion, and Gerard Canters (Leiden University, Theetherlands) for providing the pSLAC plasmid.
ppendix A. Supplementary data
Supplementary data associated with this article can be found,n the online version, at doi:10.1016/j.bios.2007.11.004.
eferences
ard, A.J., Faulkner, L.R., 2001. Electrochemical Methods, second ed. JohnWiley & Sons, Inc, New York.
artlett, P.N., Pratt, K.F.E., 1995. J. Electroanal. Chem. 397, 61–78.uckingham, D.A., Dwyer, F.P., Goodwin, H.A., Sargeson, A.M., 1964. Austr.
J. Chem. 17, 325–336.ullen, R.A., Arnot, T.C., Lakeman, J.B., Walsh, F.C., 2006. Biosens. Bioelec-
tron. 21, 2015–2045.ulter, T., Alcalde, M., Sieber, V., Meinhold, P., Schlachtbauer, C., Arnold, F.H.,
2003. Appl. Environ. Microbiol. 69, 987–995.alabrese Barton, S., Gallaway, J., Atanassov, P., 2004. Chem. Rev. 104,
4867–4886.alabrese Barton, S., Kim, H.H., Binyamin, G., Zhang, Y.C., Heller, A., 2001.
J. Phys. Chem. B 105, 11917–11921.alabrese Barton, S., Pickard, M., Vazquez-Duhalt, R., Heller, A., 2002.
Biosens. Bioelectron. 17, 1071–1074.ambatta, B.B., Ebdon, J.R., 1986. Eur. Polym. J. 22, 783–786.avis, F., Higson, S.P.J., 2007. Biosens. Bioelectron. 22, 1224–1235.
XZ
Z
electronics 23 (2008) 1229–1235 1235
orster, R.J., Kelly, A.J., Vos, J.G., Lyons, M.E.G., 1989. J. Electroanal. Chem.270, 365–379.
orster, R.J., Vos, J.G., 1990. Macromolecules 23, 4372–4377.allaway, J., 2007. PhD Dissertation, Columbia University, New York.elo-Pujic, M., Kim, H.H., Butlin, N.G., Palmore, G.T.R., 1999. Appl. Environ.
Microbiol. 65, 5515–5521.hindilis, A.L., Atanasov, P., Wilkins, E., 1997. Electroanalysis 9, 661–674.upta, G., Rajendran, V., Atanassov, P., 2004. Electroanalysis 16, 1182–1185.eller, A., 1992. J. Phys. Chem. 96, 3579–3587.eller, A., 2004. Phys. Chem. Chem. Phys. 6, 209–216.udak, N.S., Calabrese Barton, S., 2005. J. Electrochem. Soc. 152, A876–A881.amitaka, Y., Tsujimura, S., Setoyama, N., Kajino, T., Kano, K., 2007. Phys.
Chem. Chem. Phys. 9, 1793–1801.im, J., Cirigliano, N., Wang, J., Dunn, B., 2007. Phys. Chem. Chem. Phys. 9,
1809–1814.achczynski, M.C., Vijgenboom, E., Samyn, B., Canters, G.W., 2004. Protein
Sci. 13, 2388–2397.ajda, M., 1992. In: Murray, R.W. (Ed.), Molecular Design of Electrode Sur-
faces. John Wiley & Sons, Inc., New York.ano, N., Kim, H.H., Zhang, Y.C., Heller, A., 2002. J. Am. Chem. Soc. 124,
6480–6486.iura, Y., Tsujimura, S., Kamitaka, Y., Kurose, S., Kataoka, K., Sakurai, T.,
Kano, K., 2007. Chem. Lett. 36, 132–133.azor, J., Schwaneberg, U., 2006. Chembiochem 7, 638–644.iontek, K., Antorini, M., Choinowski, T., 2002. J. Biol. Chem. 277,
37663–37669.hleev, S., Pita, M., Yaropolov, A.I., Ruzgas, T., Gorton, L., 2006. Electroanal-
ysis 18, 1901–1908.olomon, E.I., Sundaram, U.M., Machonkin, T.E., 1996. Chem. Rev. 96,
2563–2605.akagi, K., Kano, K., Ikeda, T., 1998. J. Electroanal. Chem. 445, 211–219.arasevich, M.R., Bogdanovskaya, V.A., Kuznetsova, L.N., 2001. Russ. J. Elec-
trochem. 37, 833–837.opcagic, S., Minteer, S.D., 2006. Electrochim. Acta 51, 2168–2172.sujimura, S., Kano, K., Ikeda, T., 2005. J. Electroanal. Chem. 576, 113–120.ynan-Connolly, B.M., Nielsen, J.E., 2007. Protein Sci. 16, 239–249.ong, T.S., Schwaneberg, U., 2003. Curr. Opin. Biotechnol. 14, 590–596.
u, F., 1997. J. Biol. Chem. 272, 924–928.akeeruddin, S.M., Fraser, D.M., Nazeeruddin, M.K., Gratzel, M., 1992. J.Electroanal. Chem. 337, 253–283.hu, Z.W., Momeu, C., Zakhartsev, M., Schwaneberg, U., 2006. Biosens. Bio-
electron. 21, 2046–2051.
Related Documents











