Research article The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005 1221 Oxidant stress from nitric oxide synthase–3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load Eiki Takimoto, 1 Hunter C. Champion, 1 Manxiang Li, 1 Shuxun Ren, 2 E. Rene Rodriguez, 3 Barbara Tavazzi, 4 Giuseppe Lazzarino, 5 Nazareno Paolocci, 1 Kathleen L. Gabrielson, 6 Yibin Wang, 2 and David A. Kass 1 1 Division of Cardiology, Department of Medicine, The Johns Hopkins Medical Institutions, Baltimore, Maryland, USA. 2 Division of Molecular Medicine, Departments of Anesthesiology and Medicine, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, California, USA. 3 Department of Pathology, Johns Hopkins University, Baltimore, Maryland, USA. 4 Institute of Biochemistry and Clinical Biochemistry, Catholic University of Rome “Sacro Cuore”, Rome, Italy. 5 Department of Chemical Sciences, Laboratory of Biochemistry University of Catania, Catania, Italy. 6 Department of Comparative Medicine, Johns Hopkins University, Baltimore, Maryland, USA. Cardiac pressure load stimulates hypertrophy, often leading to chamber dilation and dysfunction. ROS contribute to this process. Here we show that uncoupling of nitric oxide synthase–3 (NOS3) plays a major role in pressure load–induced myocardial ROS and consequent chamber remodeling/hypertrophy. Chronic transverse aortic constriction (TAC; for 3 and 9 weeks) in control mice induced marked cardiac hypertrophy, dilation, and dysfunction. Mice lacking NOS3 displayed modest and concentric hypertrophy to TAC with pre- served function. NOS3 –/– TAC hearts developed less fibrosis, myocyte hypertrophy, and fetal gene re-expres- sion (B-natriuretic peptide and α–skeletal actin). ROS, nitrotyrosine, and gelatinase (MMP-2 and MMP-9) zymogen activity markedly increased in control TAC, but not in NOS3 –/– TAC, hearts. TAC induced NOS3 uncoupling in the heart, reflected by reduced NOS3 dimer and tetrahydrobiopterin (BH4), increased NOS3- dependent generation of ROS, and lowered Ca 2+ -dependent NOS activity. Cotreatment with BH4 prevented NOS3 uncoupling and inhibited ROS, resulting in concentric nondilated hypertrophy. Mice given the anti- oxidant tetrahydroneopterin as a control did not display changes in TAC response. Thus, pressure overload triggers NOS3 uncoupling as a prominent source of myocardial ROS that contribute to dilatory remodeling and cardiac dysfunction. Reversal of this process by BH4 suggests a potential treatment to ameliorate the pathophysiology of chronic pressure-induced hypertrophy. Introduction Chronic cardiac ventricular pressure overload stimulates hypertro- phy that can progress to heart failure (1, 2). A major feature of this transition is pathologic remodeling with chamber dilation and pump dysfunction, and evidence increasingly supports an impor- tant role for ROS in this process (3–7). ROS generation is linked to hypertrophy stimulators such as G αq /G 11 -coupled agonists (e.g., phenylephrine, angiotensin) (8, 9), signaling kinases and phosphatases (4, 10), and mechanotransduction (11). ROS them- selves stimulate hypertrophy-associated kinases (4, 12), induce fetal gene re-expression (13), and contribute to chamber remodel- ing by activating MMPs (3, 14). ROS can be generated by mitochondrial electron transport leakage (10, 15), NADPH oxidases (3, 16, 17), xanthine oxidase (18), and nitric oxide synthase (NOS) (19, 20). Among these, NOS is intriguing because NO and its downstream target, pro- tein kinase G, are generally considered to blunt hypertrophy (21–24). However, NOS can be converted to a ROS generator, as demonstrated in vascular endothelium exposed to increased oxidant or hemodynamic stress (19, 20, 25). NOS3 is the domi- nant isoform in the endothelium, as well as in cardiac myocytes, where it is an important regulator of adrenergic, muscarinic, and rate-mediated reserve (26, 27). Active NOS3 is a homodimer that generates NO and L-citrulline from L-arginine. When exposed to oxidant stress, including peroxynitrite (ONOO – ), or deprived of its reducing cofactor tetrahydrobiopterin (BH4) or substrate L-arginine, NOS3 uncouples to the monomeric form that gener- ates O 2 – rather than NO (25, 28, 29). Uncoupled NOS3 is thought to be a prominent source of endothelial ROS in hypertension (19), neurohormonal stimulation, and hyperglycemia (8), and upon exposure to ONOO – (20). The present study tested the role of NOS3 in cardiac hyper- trophic remodeling in response to sustained pressure overload in mice with or without the NOS3 gene. We provide the first evi- dence to our knowledge that NOS3 uncoupling occurs in myo- cardium exposed to chronic pressure load, and that this serves as a major source for myocardial ROS, which is linked to dila- tive hypertrophy remodeling. We further show that oral supple- mentation with BH4 prevents NOS3 uncoupling and markedly blunts ROS generation and chamber dilation despite similar lev- els of chronic chamber loading. Nonstandard abbreviations used: BH4, tetrahydrobiopterin; DCF, dichlorofluo- rescein; DHE, dihydroethidium; Ea, arterial elastance; FS, fractional shortening; GSH/GSSH, ratio of reduced glutathione to oxidized glutathione; GTPCH-1, GTP- cyclohydrolase–1; H4N, tetrahydroneopterin; HW, heart weight; LVEDD, LV end- diastolic dimension; LVEF, LV ejection fraction; LVESD, left ventricular end-systolic dimension; β-MHC, β-myosin heavy chain; NOS, nitric oxide synthase; NP, natriuretic peptide; NT, nitrotyrosine; p-, phosphorylated; PLB, phospholamban; PV, pressure- volume; α-SA, α–skeletal actin; SERCA2, sarcoplasmic reticulum Ca 2+ ATPase 2; TAC, transverse aortic constriction; TL, tibia length. Conflict of interest: The authors have declared that no conflict of interest exists. Citation for this article: J. Clin. Invest. 115:1221–1231 (2005). doi:10.1172/JCI200521968.

Welcome message from author
This document is posted to help you gain knowledge. Please leave a comment to let me know what you think about it! Share it to your friends and learn new things together.
Transcript

Research article
The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005 1221
Oxidant stress from nitric oxide synthase–3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load
Eiki Takimoto,1 Hunter C. Champion,1 Manxiang Li,1 Shuxun Ren,2 E. Rene Rodriguez,3 Barbara Tavazzi,4 Giuseppe Lazzarino,5 Nazareno Paolocci,1 Kathleen L. Gabrielson,6
Yibin Wang,2 and David A. Kass1
1Division of Cardiology, Department of Medicine, The Johns Hopkins Medical Institutions, Baltimore, Maryland, USA. 2Division of Molecular Medicine, Departments of Anesthesiology and Medicine, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, California, USA.
3Department of Pathology, Johns Hopkins University, Baltimore, Maryland, USA. 4Institute of Biochemistry and Clinical Biochemistry, Catholic University of Rome “Sacro Cuore”, Rome, Italy. 5Department of Chemical Sciences, Laboratory of Biochemistry University of Catania,
Catania, Italy. 6Department of Comparative Medicine, Johns Hopkins University, Baltimore, Maryland, USA.
Cardiac pressure load stimulates hypertrophy, often leading to chamber dilation and dysfunction. ROS contribute to this process. Here we show that uncoupling of nitric oxide synthase–3 (NOS3) plays a major role in pressure load–induced myocardial ROS and consequent chamber remodeling/hypertrophy. Chronic transverse aortic constriction (TAC; for 3 and 9 weeks) in control mice induced marked cardiac hypertrophy, dilation, and dysfunction. Mice lacking NOS3 displayed modest and concentric hypertrophy to TAC with pre-served function. NOS3–/– TAC hearts developed less fibrosis, myocyte hypertrophy, and fetal gene re-expres-sion (B-natriuretic peptide and α–skeletal actin). ROS, nitrotyrosine, and gelatinase (MMP-2 and MMP-9) zymogen activity markedly increased in control TAC, but not in NOS3–/– TAC, hearts. TAC induced NOS3 uncoupling in the heart, reflected by reduced NOS3 dimer and tetrahydrobiopterin (BH4), increased NOS3-dependent generation of ROS, and lowered Ca2+-dependent NOS activity. Cotreatment with BH4 prevented NOS3 uncoupling and inhibited ROS, resulting in concentric nondilated hypertrophy. Mice given the anti-oxidant tetrahydroneopterin as a control did not display changes in TAC response. Thus, pressure overload triggers NOS3 uncoupling as a prominent source of myocardial ROS that contribute to dilatory remodeling and cardiac dysfunction. Reversal of this process by BH4 suggests a potential treatment to ameliorate the pathophysiology of chronic pressure-induced hypertrophy.
IntroductionChronic cardiac ventricular pressure overload stimulates hypertro-phy that can progress to heart failure (1, 2). A major feature of this transition is pathologic remodeling with chamber dilation and pump dysfunction, and evidence increasingly supports an impor-tant role for ROS in this process (3–7). ROS generation is linked to hypertrophy stimulators such as Gαq/G11-coupled agonists (e.g., phenylephrine, angiotensin) (8, 9), signaling kinases and phosphatases (4, 10), and mechanotransduction (11). ROS them-selves stimulate hypertrophy-associated kinases (4, 12), induce fetal gene re-expression (13), and contribute to chamber remodel-ing by activating MMPs (3, 14).
ROS can be generated by mitochondrial electron transport leakage (10, 15), NADPH oxidases (3, 16, 17), xanthine oxidase (18), and nitric oxide synthase (NOS) (19, 20). Among these,
NOS is intriguing because NO and its downstream target, pro-tein kinase G, are generally considered to blunt hypertrophy (21–24). However, NOS can be converted to a ROS generator, as demonstrated in vascular endothelium exposed to increased oxidant or hemodynamic stress (19, 20, 25). NOS3 is the domi-nant isoform in the endothelium, as well as in cardiac myocytes, where it is an important regulator of adrenergic, muscarinic, and rate-mediated reserve (26, 27). Active NOS3 is a homodimer that generates NO and L-citrulline from L-arginine. When exposed to oxidant stress, including peroxynitrite (ONOO–), or deprived of its reducing cofactor tetrahydrobiopterin (BH4) or substrate L-arginine, NOS3 uncouples to the monomeric form that gener-ates O2
– rather than NO (25, 28, 29). Uncoupled NOS3 is thought to be a prominent source of endothelial ROS in hypertension (19), neurohormonal stimulation, and hyperglycemia (8), and upon exposure to ONOO– (20).
The present study tested the role of NOS3 in cardiac hyper-trophic remodeling in response to sustained pressure overload in mice with or without the NOS3 gene. We provide the first evi-dence to our knowledge that NOS3 uncoupling occurs in myo-cardium exposed to chronic pressure load, and that this serves as a major source for myocardial ROS, which is linked to dila-tive hypertrophy remodeling. We further show that oral supple-mentation with BH4 prevents NOS3 uncoupling and markedly blunts ROS generation and chamber dilation despite similar lev-els of chronic chamber loading.
Nonstandard abbreviations used: BH4, tetrahydrobiopterin; DCF, dichlorofluo-rescein; DHE, dihydroethidium; Ea, arterial elastance; FS, fractional shortening; GSH/GSSH, ratio of reduced glutathione to oxidized glutathione; GTPCH-1, GTP-cyclohydrolase–1; H4N, tetrahydroneopterin; HW, heart weight; LVEDD, LV end-diastolic dimension; LVEF, LV ejection fraction; LVESD, left ventricular end-systolic dimension; β-MHC, β-myosin heavy chain; NOS, nitric oxide synthase; NP, natriuretic peptide; NT, nitrotyrosine; p-, phosphorylated; PLB, phospholamban; PV, pressure-volume; α-SA, α–skeletal actin; SERCA2, sarcoplasmic reticulum Ca2+ ATPase 2; TAC, transverse aortic constriction; TL, tibia length.
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 115:1221–1231 (2005). doi:10.1172/JCI200521968.
research article
1222 The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005
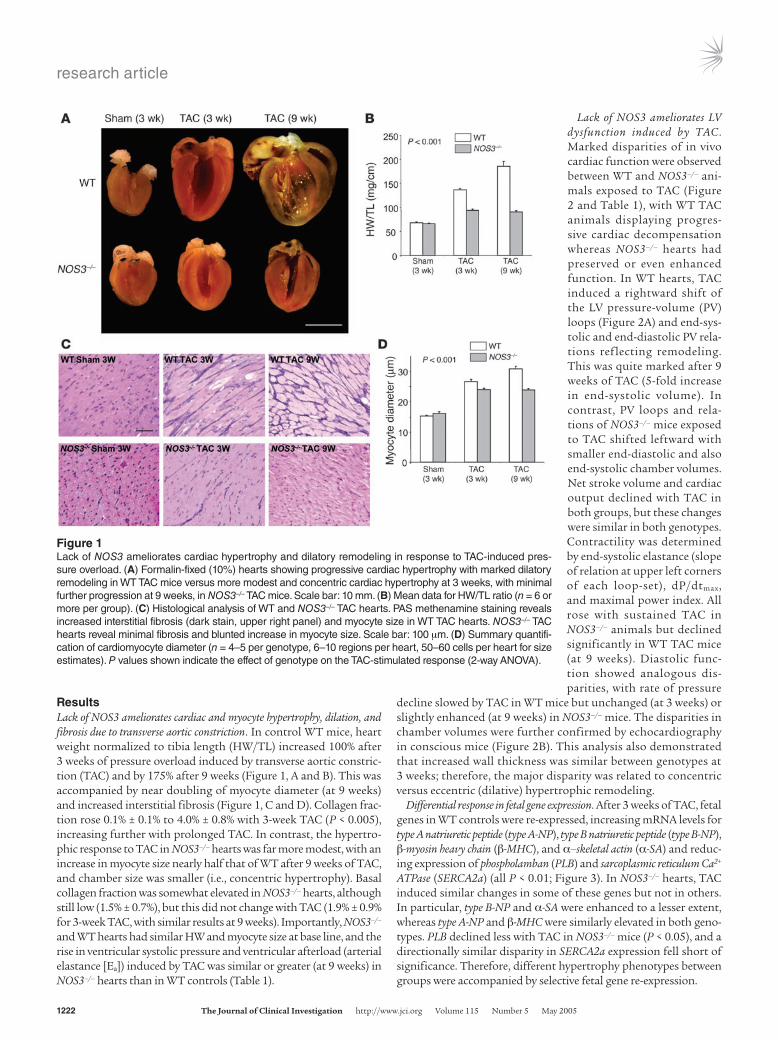
ResultsLack of NOS3 ameliorates cardiac and myocyte hypertrophy, dilation, and fibrosis due to transverse aortic constriction. In control WT mice, heart weight normalized to tibia length (HW/TL) increased 100% after 3 weeks of pressure overload induced by transverse aortic constric-tion (TAC) and by 175% after 9 weeks (Figure 1, A and B). This was accompanied by near doubling of myocyte diameter (at 9 weeks) and increased interstitial fibrosis (Figure 1, C and D). Collagen frac-tion rose 0.1% ± 0.1% to 4.0% ± 0.8% with 3-week TAC (P < 0.005), increasing further with prolonged TAC. In contrast, the hypertro-phic response to TAC in NOS3–/– hearts was far more modest, with an increase in myocyte size nearly half that of WT after 9 weeks of TAC, and chamber size was smaller (i.e., concentric hypertrophy). Basal collagen fraction was somewhat elevated in NOS3–/– hearts, although still low (1.5% ± 0.7%), but this did not change with TAC (1.9% ± 0.9% for 3-week TAC, with similar results at 9 weeks). Importantly, NOS3–/– and WT hearts had similar HW and myocyte size at base line, and the rise in ventricular systolic pressure and ventricular afterload (arterial elastance [Ea]) induced by TAC was similar or greater (at 9 weeks) in NOS3–/– hearts than in WT controls (Table 1).
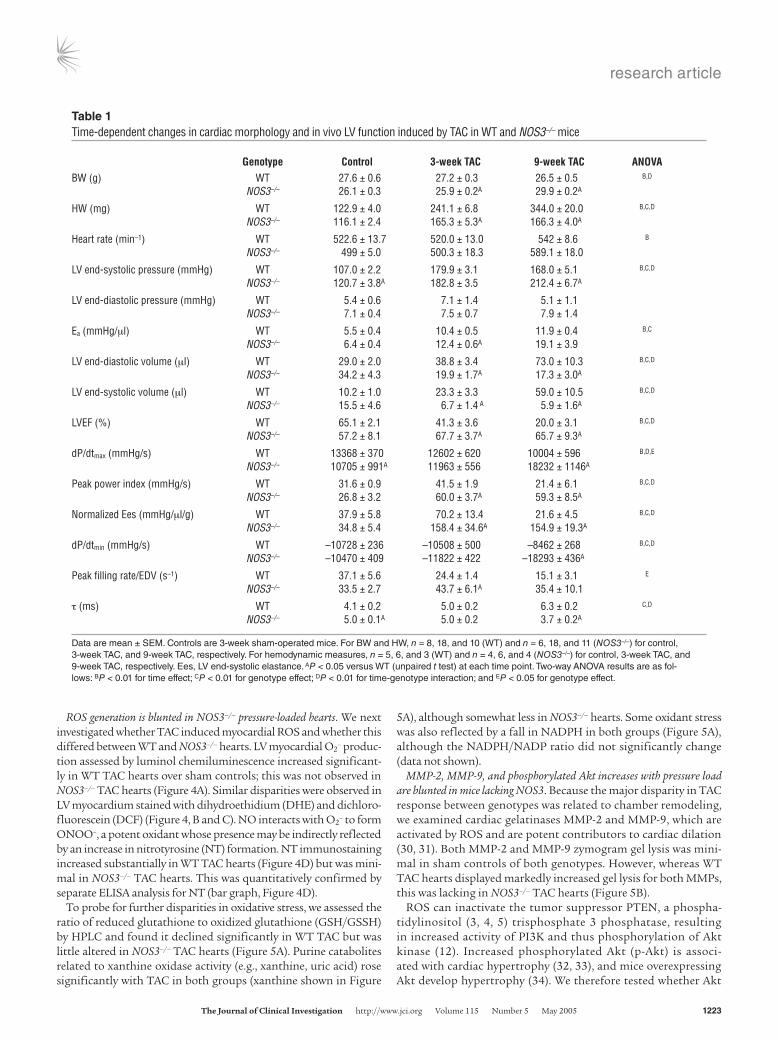
Lack of NOS3 ameliorates LV dysfunction induced by TAC. Marked disparities of in vivo cardiac function were observed between WT and NOS3–/– ani-mals exposed to TAC (Figure 2 and Table 1), with WT TAC animals displaying progres-sive cardiac decompensation whereas NOS3–/– hearts had preserved or even enhanced function. In WT hearts, TAC induced a rightward shift of the LV pressure-volume (PV) loops (Figure 2A) and end-sys-tolic and end-diastolic PV rela-tions reflecting remodeling. This was quite marked after 9 weeks of TAC (5-fold increase in end-systolic volume). In contrast, PV loops and rela-tions of NOS3–/– mice exposed to TAC shifted leftward with smaller end-diastolic and also end-systolic chamber volumes. Net stroke volume and cardiac output declined with TAC in both groups, but these changes were similar in both genotypes. Contractility was determined by end-systolic elastance (slope of relation at upper left corners of each loop-set), dP/dtmax, and maximal power index. All rose with sustained TAC in NOS3–/– animals but declined significantly in WT TAC mice (at 9 weeks). Diastolic func-tion showed analogous dis-parities, with rate of pressure
decline slowed by TAC in WT mice but unchanged (at 3 weeks) or slightly enhanced (at 9 weeks) in NOS3–/– mice. The disparities in chamber volumes were further confirmed by echocardiography in conscious mice (Figure 2B). This analysis also demonstrated that increased wall thickness was similar between genotypes at 3 weeks; therefore, the major disparity was related to concentric versus eccentric (dilative) hypertrophic remodeling.
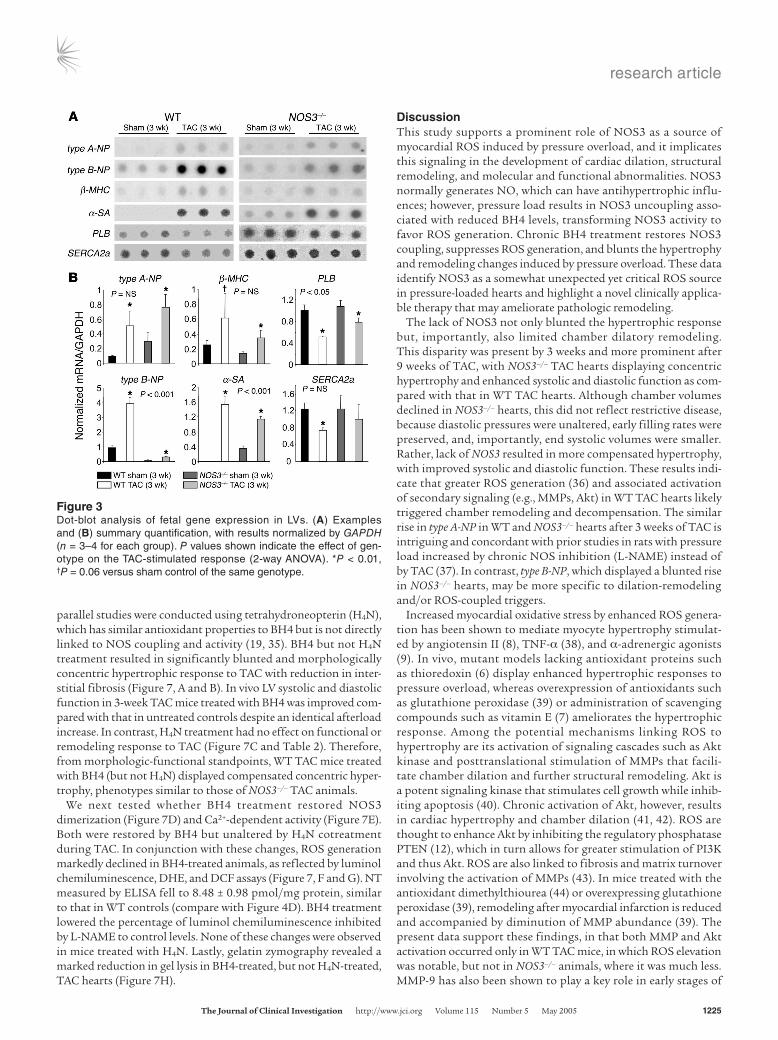
Differential response in fetal gene expression. After 3 weeks of TAC, fetal genes in WT controls were re-expressed, increasing mRNA levels for type A natriuretic peptide (type A-NP), type B natriuretic peptide (type B-NP), β-myosin heavy chain (β-MHC), and α–skeletal actin (α-SA) and reduc-ing expression of phospholamban (PLB) and sarcoplasmic reticulum Ca2+ ATPase (SERCA2a) (all P < 0.01; Figure 3). In NOS3–/– hearts, TAC induced similar changes in some of these genes but not in others. In particular, type B-NP and α-SA were enhanced to a lesser extent, whereas type A-NP and β-MHC were similarly elevated in both geno-types. PLB declined less with TAC in NOS3–/– mice (P < 0.05), and a directionally similar disparity in SERCA2a expression fell short of significance. Therefore, different hypertrophy phenotypes between groups were accompanied by selective fetal gene re-expression.
Figure 1Lack of NOS3 ameliorates cardiac hypertrophy and dilatory remodeling in response to TAC-induced pres-sure overload. (A) Formalin-fixed (10%) hearts showing progressive cardiac hypertrophy with marked dilatory remodeling in WT TAC mice versus more modest and concentric cardiac hypertrophy at 3 weeks, with minimal further progression at 9 weeks, in NOS3–/– TAC mice. Scale bar: 10 mm. (B) Mean data for HW/TL ratio (n = 6 or more per group). (C) Histological analysis of WT and NOS3–/– TAC hearts. PAS methenamine staining reveals increased interstitial fibrosis (dark stain, upper right panel) and myocyte size in WT TAC hearts. NOS3–/– TAC hearts reveal minimal fibrosis and blunted increase in myocyte size. Scale bar: 100 μm. (D) Summary quantifi-cation of cardiomyocyte diameter (n = 4–5 per genotype, 6–10 regions per heart, 50–60 cells per heart for size estimates). P values shown indicate the effect of genotype on the TAC-stimulated response (2-way ANOVA).
research article
The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005 1223
ROS generation is blunted in NOS3–/– pressure-loaded hearts. We next investigated whether TAC induced myocardial ROS and whether this differed between WT and NOS3–/– hearts. LV myocardial O2
– produc-tion assessed by luminol chemiluminescence increased significant-ly in WT TAC hearts over sham controls; this was not observed in NOS3–/– TAC hearts (Figure 4A). Similar disparities were observed in LV myocardium stained with dihydroethidium (DHE) and dichloro-fluorescein (DCF) (Figure 4, B and C). NO interacts with O2
– to form ONOO–, a potent oxidant whose presence may be indirectly reflected by an increase in nitrotyrosine (NT) formation. NT immunostaining increased substantially in WT TAC hearts (Figure 4D) but was mini-mal in NOS3–/– TAC hearts. This was quantitatively confirmed by separate ELISA analysis for NT (bar graph, Figure 4D).
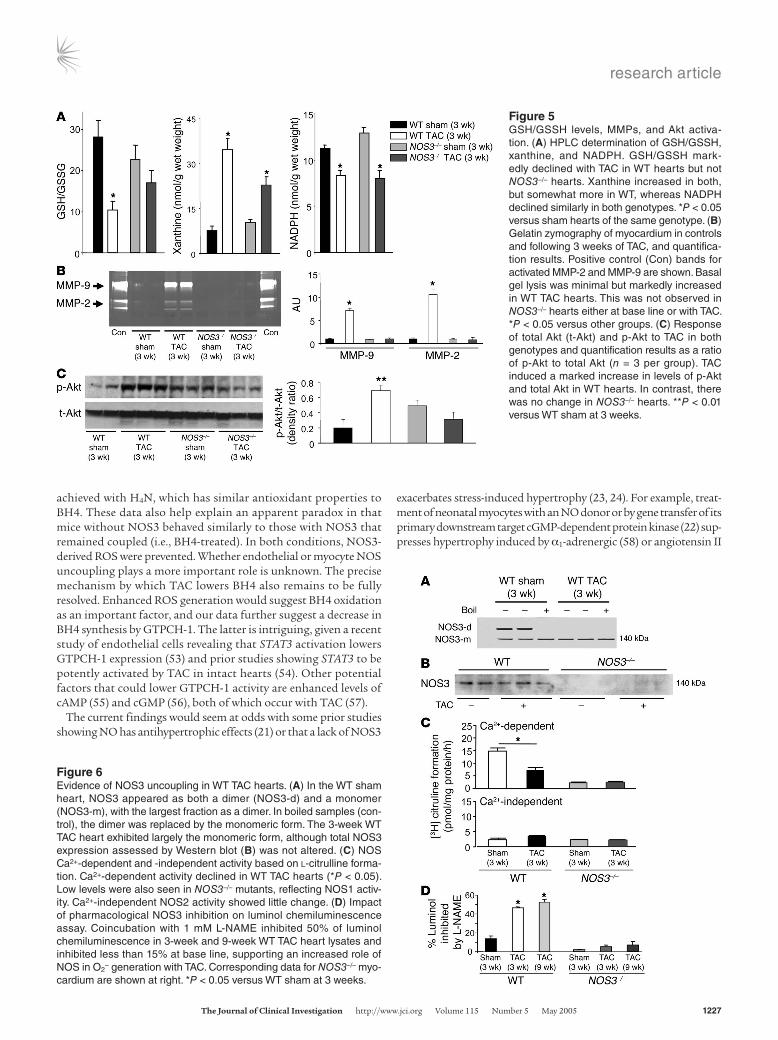
To probe for further disparities in oxidative stress, we assessed the ratio of reduced glutathione to oxidized glutathione (GSH/GSSH) by HPLC and found it declined significantly in WT TAC but was little altered in NOS3–/– TAC hearts (Figure 5A). Purine catabolites related to xanthine oxidase activity (e.g., xanthine, uric acid) rose significantly with TAC in both groups (xanthine shown in Figure
5A), although somewhat less in NOS3–/– hearts. Some oxidant stress was also reflected by a fall in NADPH in both groups (Figure 5A), although the NADPH/NADP ratio did not significantly change (data not shown).
MMP-2, MMP-9, and phosphorylated Akt increases with pressure load are blunted in mice lacking NOS3. Because the major disparity in TAC response between genotypes was related to chamber remodeling, we examined cardiac gelatinases MMP-2 and MMP-9, which are activated by ROS and are potent contributors to cardiac dilation (30, 31). Both MMP-2 and MMP-9 zymogram gel lysis was mini-mal in sham controls of both genotypes. However, whereas WT TAC hearts displayed markedly increased gel lysis for both MMPs, this was lacking in NOS3–/– TAC hearts (Figure 5B).
ROS can inactivate the tumor suppressor PTEN, a phospha-tidylinositol (3, 4, 5) trisphosphate 3 phosphatase, resulting in increased activity of PI3K and thus phosphorylation of Akt kinase (12). Increased phosphorylated Akt (p-Akt) is associ-ated with cardiac hypertrophy (32, 33), and mice overexpressing Akt develop hypertrophy (34). We therefore tested whether Akt
Table 1Time-dependent changes in cardiac morphology and in vivo LV function induced by TAC in WT and NOS3–/– mice
Genotype Control 3-week TAC 9-week TAC ANOVABW (g) WT 27.6 ± 0.6 27.2 ± 0.3 26.5 ± 0.5 B,D
NOS3–/– 26.1 ± 0.3 25.9 ± 0.2A 29.9 ± 0.2A
HW (mg) WT 122.9 ± 4.0 241.1 ± 6.8 344.0 ± 20.0 B,C,D
NOS3–/– 116.1 ± 2.4 165.3 ± 5.3A 166.3 ± 4.0A
Heart rate (min–1) WT 522.6 ± 13.7 520.0 ± 13.0 542 ± 8.6 B
NOS3–/– 499 ± 5.0 500.3 ± 18.3 589.1 ± 18.0
LV end-systolic pressure (mmHg) WT 107.0 ± 2.2 179.9 ± 3.1 168.0 ± 5.1 B,C,D
NOS3–/– 120.7 ± 3.8A 182.8 ± 3.5 212.4 ± 6.7A
LV end-diastolic pressure (mmHg) WT 5.4 ± 0.6 7.1 ± 1.4 5.1 ± 1.1 NOS3–/– 7.1 ± 0.4 7.5 ± 0.7 7.9 ± 1.4
Ea (mmHg/μl) WT 5.5 ± 0.4 10.4 ± 0.5 11.9 ± 0.4 B,C
NOS3–/– 6.4 ± 0.4 12.4 ± 0.6A 19.1 ± 3.9
LV end-diastolic volume (μl) WT 29.0 ± 2.0 38.8 ± 3.4 73.0 ± 10.3 B,C,D
NOS3–/– 34.2 ± 4.3 19.9 ± 1.7A 17.3 ± 3.0A
LV end-systolic volume (μl) WT 10.2 ± 1.0 23.3 ± 3.3 59.0 ± 10.5 B,C,D
NOS3–/– 15.5 ± 4.6 6.7 ± 1.4 A 5.9 ± 1.6A
LVEF (%) WT 65.1 ± 2.1 41.3 ± 3.6 20.0 ± 3.1 B,C,D
NOS3–/– 57.2 ± 8.1 67.7 ± 3.7A 65.7 ± 9.3A
dP/dtmax (mmHg/s) WT 13368 ± 370 12602 ± 620 10004 ± 596 B,D,E
NOS3–/– 10705 ± 991A 11963 ± 556 18232 ± 1146A
Peak power index (mmHg/s) WT 31.6 ± 0.9 41.5 ± 1.9 21.4 ± 6.1 B,C,D
NOS3–/– 26.8 ± 3.2 60.0 ± 3.7A 59.3 ± 8.5A
Normalized Ees (mmHg/μl/g) WT 37.9 ± 5.8 70.2 ± 13.4 21.6 ± 4.5 B,C,D
NOS3–/– 34.8 ± 5.4 158.4 ± 34.6A 154.9 ± 19.3A
dP/dtmin (mmHg/s) WT –10728 ± 236 –10508 ± 500 –8462 ± 268 B,C,D
NOS3–/– –10470 ± 409 –11822 ± 422 –18293 ± 436A
Peak filling rate/EDV (s–1) WT 37.1 ± 5.6 24.4 ± 1.4 15.1 ± 3.1 E
NOS3–/– 33.5 ± 2.7 43.7 ± 6.1A 35.4 ± 10.1
τ (ms) WT 4.1 ± 0.2 5.0 ± 0.2 6.3 ± 0.2 C,D
NOS3–/– 5.0 ± 0.1A 5.0 ± 0.2 3.7 ± 0.2A
Data are mean ± SEM. Controls are 3-week sham-operated mice. For BW and HW, n = 8, 18, and 10 (WT) and n = 6, 18, and 11 (NOS3–/–) for control, 3-week TAC, and 9-week TAC, respectively. For hemodynamic measures, n = 5, 6, and 3 (WT) and n = 4, 6, and 4 (NOS3–/–) for control, 3-week TAC, and 9-week TAC, respectively. Ees, LV end-systolic elastance. AP < 0.05 versus WT (unpaired t test) at each time point. Two-way ANOVA results are as fol-lows: BP < 0.01 for time effect; CP < 0.01 for genotype effect; DP < 0.01 for time-genotype interaction; and EP < 0.05 for genotype effect.
research article
1224 The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005
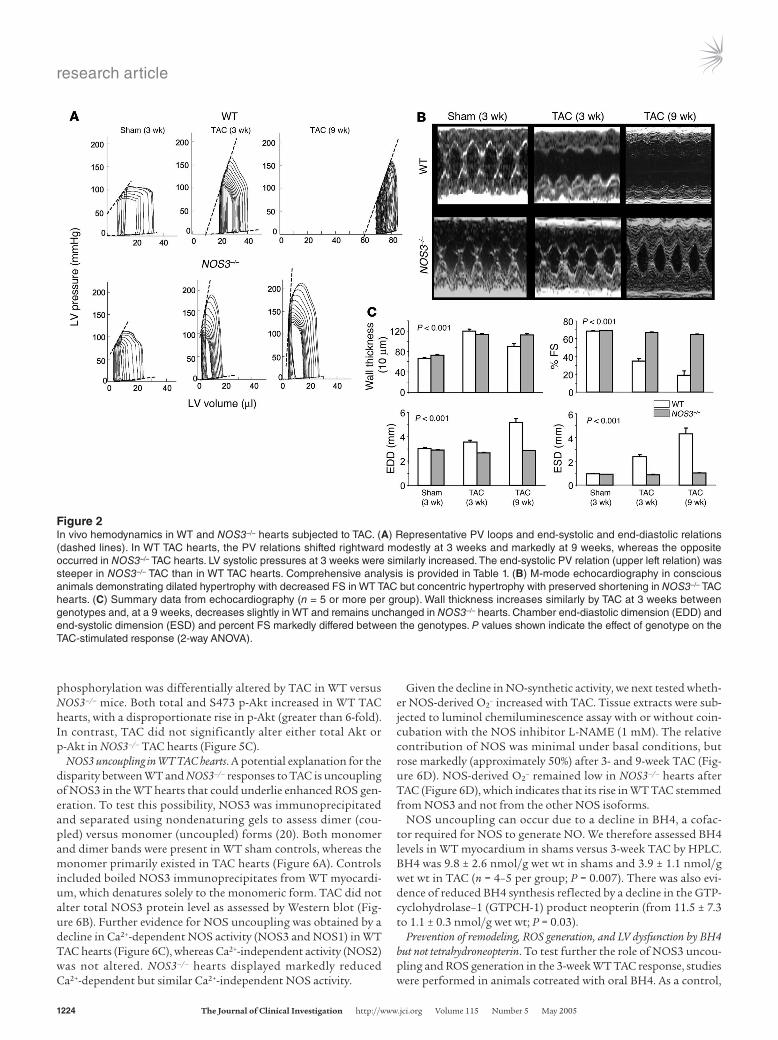
phosphorylation was differentially altered by TAC in WT versus NOS3–/– mice. Both total and S473 p-Akt increased in WT TAC hearts, with a disproportionate rise in p-Akt (greater than 6-fold). In contrast, TAC did not significantly alter either total Akt or p-Akt in NOS3–/– TAC hearts (Figure 5C).
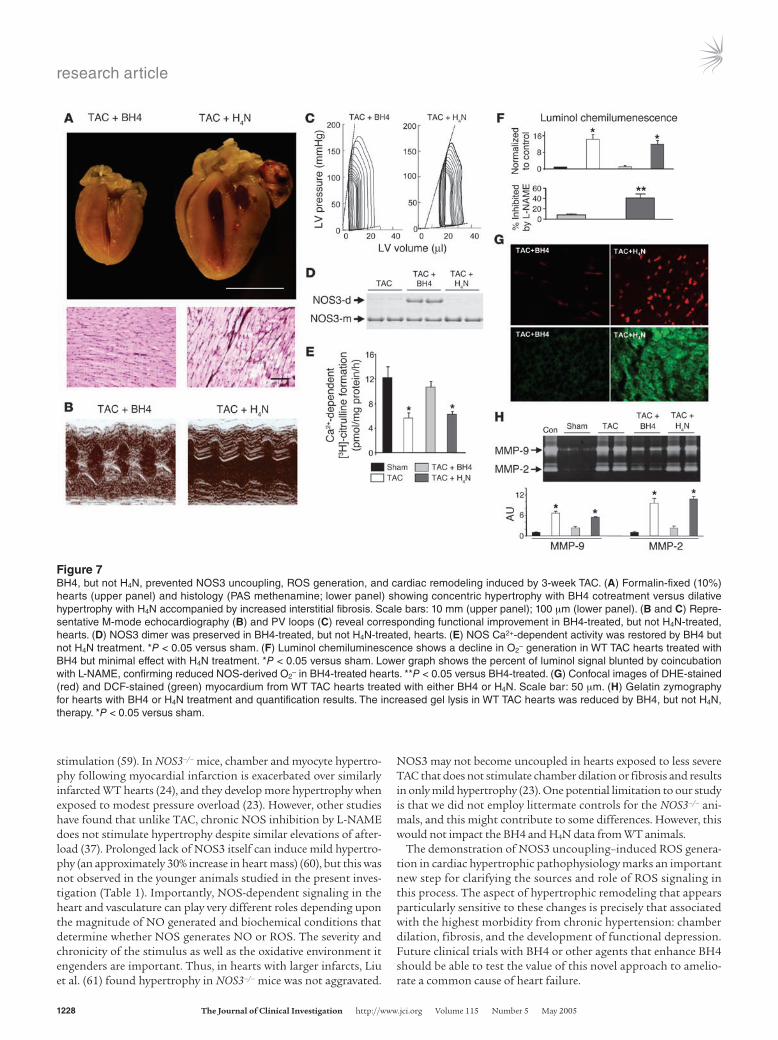
NOS3 uncoupling in WT TAC hearts. A potential explanation for the disparity between WT and NOS3–/– responses to TAC is uncoupling of NOS3 in the WT hearts that could underlie enhanced ROS gen-eration. To test this possibility, NOS3 was immunoprecipitated and separated using nondenaturing gels to assess dimer (cou-pled) versus monomer (uncoupled) forms (20). Both monomer and dimer bands were present in WT sham controls, whereas the monomer primarily existed in TAC hearts (Figure 6A). Controls included boiled NOS3 immunoprecipitates from WT myocardi-um, which denatures solely to the monomeric form. TAC did not alter total NOS3 protein level as assessed by Western blot (Fig-ure 6B). Further evidence for NOS uncoupling was obtained by a decline in Ca2+-dependent NOS activity (NOS3 and NOS1) in WT TAC hearts (Figure 6C), whereas Ca2+-independent activity (NOS2) was not altered. NOS3–/– hearts displayed markedly reduced Ca2+-dependent but similar Ca2+-independent NOS activity.
Given the decline in NO-synthetic activity, we next tested wheth-er NOS-derived O2
– increased with TAC. Tissue extracts were sub-jected to luminol chemiluminescence assay with or without coin-cubation with the NOS inhibitor L-NAME (1 mM). The relative contribution of NOS was minimal under basal conditions, but rose markedly (approximately 50%) after 3- and 9-week TAC (Fig-ure 6D). NOS-derived O2
– remained low in NOS3–/– hearts after TAC (Figure 6D), which indicates that its rise in WT TAC stemmed from NOS3 and not from the other NOS isoforms.
NOS uncoupling can occur due to a decline in BH4, a cofac-tor required for NOS to generate NO. We therefore assessed BH4 levels in WT myocardium in shams versus 3-week TAC by HPLC. BH4 was 9.8 ± 2.6 nmol/g wet wt in shams and 3.9 ± 1.1 nmol/g wet wt in TAC (n = 4–5 per group; P = 0.007). There was also evi-dence of reduced BH4 synthesis reflected by a decline in the GTP-cyclohydrolase–1 (GTPCH-1) product neopterin (from 11.5 ± 7.3 to 1.1 ± 0.3 nmol/g wet wt; P = 0.03).
Prevention of remodeling, ROS generation, and LV dysfunction by BH4 but not tetrahydroneopterin. To test further the role of NOS3 uncou-pling and ROS generation in the 3-week WT TAC response, studies were performed in animals cotreated with oral BH4. As a control,
Figure 2In vivo hemodynamics in WT and NOS3–/– hearts subjected to TAC. (A) Representative PV loops and end-systolic and end-diastolic relations (dashed lines). In WT TAC hearts, the PV relations shifted rightward modestly at 3 weeks and markedly at 9 weeks, whereas the opposite occurred in NOS3–/– TAC hearts. LV systolic pressures at 3 weeks were similarly increased. The end-systolic PV relation (upper left relation) was steeper in NOS3–/– TAC than in WT TAC hearts. Comprehensive analysis is provided in Table 1. (B) M-mode echocardiography in conscious animals demonstrating dilated hypertrophy with decreased FS in WT TAC but concentric hypertrophy with preserved shortening in NOS3–/– TAC hearts. (C) Summary data from echocardiography (n = 5 or more per group). Wall thickness increases similarly by TAC at 3 weeks between genotypes and, at a 9 weeks, decreases slightly in WT and remains unchanged in NOS3–/– hearts. Chamber end-diastolic dimension (EDD) and end-systolic dimension (ESD) and percent FS markedly differed between the genotypes. P values shown indicate the effect of genotype on the TAC-stimulated response (2-way ANOVA).
research article
The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005 1225
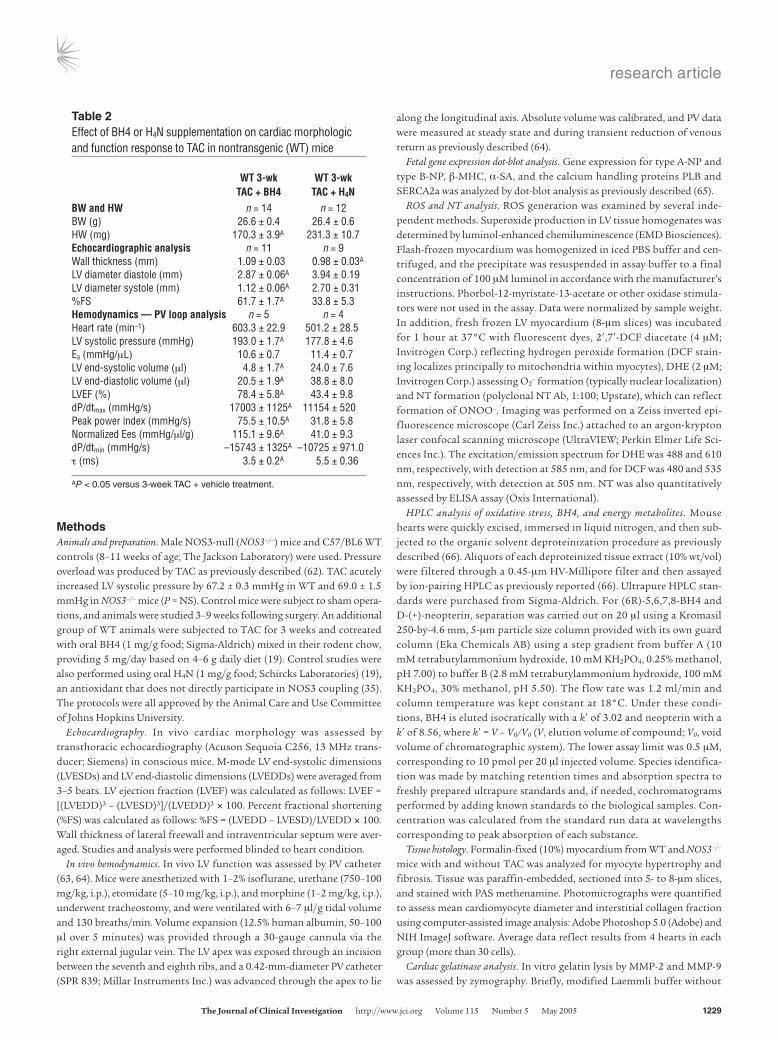
parallel studies were conducted using tetrahydroneopterin (H4N), which has similar antioxidant properties to BH4 but is not directly linked to NOS coupling and activity (19, 35). BH4 but not H4N treatment resulted in significantly blunted and morphologically concentric hypertrophic response to TAC with reduction in inter-stitial fibrosis (Figure 7, A and B). In vivo LV systolic and diastolic function in 3-week TAC mice treated with BH4 was improved com-pared with that in untreated controls despite an identical afterload increase. In contrast, H4N treatment had no effect on functional or remodeling response to TAC (Figure 7C and Table 2). Therefore, from morphologic-functional standpoints, WT TAC mice treated with BH4 (but not H4N) displayed compensated concentric hyper-trophy, phenotypes similar to those of NOS3–/– TAC animals.
We next tested whether BH4 treatment restored NOS3 dimerization (Figure 7D) and Ca2+-dependent activity (Figure 7E). Both were restored by BH4 but unaltered by H4N cotreatment during TAC. In conjunction with these changes, ROS generation markedly declined in BH4-treated animals, as reflected by luminol chemiluminescence, DHE, and DCF assays (Figure 7, F and G). NT measured by ELISA fell to 8.48 ± 0.98 pmol/mg protein, similar to that in WT controls (compare with Figure 4D). BH4 treatment lowered the percentage of luminol chemiluminescence inhibited by L-NAME to control levels. None of these changes were observed in mice treated with H4N. Lastly, gelatin zymography revealed a marked reduction in gel lysis in BH4-treated, but not H4N-treated, TAC hearts (Figure 7H).
DiscussionThis study supports a prominent role of NOS3 as a source of myocardial ROS induced by pressure overload, and it implicates this signaling in the development of cardiac dilation, structural remodeling, and molecular and functional abnormalities. NOS3 normally generates NO, which can have antihypertrophic influ-ences; however, pressure load results in NOS3 uncoupling asso-ciated with reduced BH4 levels, transforming NOS3 activity to favor ROS generation. Chronic BH4 treatment restores NOS3 coupling, suppresses ROS generation, and blunts the hypertrophy and remodeling changes induced by pressure overload. These data identify NOS3 as a somewhat unexpected yet critical ROS source in pressure-loaded hearts and highlight a novel clinically applica-ble therapy that may ameliorate pathologic remodeling.
The lack of NOS3 not only blunted the hypertrophic response but, importantly, also limited chamber dilatory remodeling. This disparity was present by 3 weeks and more prominent after 9 weeks of TAC, with NOS3–/– TAC hearts displaying concentric hypertrophy and enhanced systolic and diastolic function as com-pared with that in WT TAC hearts. Although chamber volumes declined in NOS3–/– hearts, this did not reflect restrictive disease, because diastolic pressures were unaltered, early filling rates were preserved, and, importantly, end systolic volumes were smaller. Rather, lack of NOS3 resulted in more compensated hypertrophy, with improved systolic and diastolic function. These results indi-cate that greater ROS generation (36) and associated activation of secondary signaling (e.g., MMPs, Akt) in WT TAC hearts likely triggered chamber remodeling and decompensation. The similar rise in type A-NP in WT and NOS3–/– hearts after 3 weeks of TAC is intriguing and concordant with prior studies in rats with pressure load increased by chronic NOS inhibition (L-NAME) instead of by TAC (37). In contrast, type B-NP, which displayed a blunted rise in NOS3–/– hearts, may be more specific to dilation-remodeling and/or ROS-coupled triggers.
Increased myocardial oxidative stress by enhanced ROS genera-tion has been shown to mediate myocyte hypertrophy stimulat-ed by angiotensin II (8), TNF-α (38), and α-adrenergic agonists (9). In vivo, mutant models lacking antioxidant proteins such as thioredoxin (6) display enhanced hypertrophic responses to pressure overload, whereas overexpression of antioxidants such as glutathione peroxidase (39) or administration of scavenging compounds such as vitamin E (7) ameliorates the hypertrophic response. Among the potential mechanisms linking ROS to hypertrophy are its activation of signaling cascades such as Akt kinase and posttranslational stimulation of MMPs that facili-tate chamber dilation and further structural remodeling. Akt is a potent signaling kinase that stimulates cell growth while inhib-iting apoptosis (40). Chronic activation of Akt, however, results in cardiac hypertrophy and chamber dilation (41, 42). ROS are thought to enhance Akt by inhibiting the regulatory phosphatase PTEN (12), which in turn allows for greater stimulation of PI3K and thus Akt. ROS are also linked to fibrosis and matrix turnover involving the activation of MMPs (43). In mice treated with the antioxidant dimethylthiourea (44) or overexpressing glutathione peroxidase (39), remodeling after myocardial infarction is reduced and accompanied by diminution of MMP abundance (39). The present data support these findings, in that both MMP and Akt activation occurred only in WT TAC mice, in which ROS elevation was notable, but not in NOS3–/– animals, where it was much less. MMP-9 has also been shown to play a key role in early stages of
Figure 3Dot-blot analysis of fetal gene expression in LVs. (A) Examples and (B) summary quantification, with results normalized by GAPDH (n = 3–4 for each group). P values shown indicate the effect of gen-otype on the TAC-stimulated response (2-way ANOVA). *P < 0.01, †P = 0.06 versus sham control of the same genotype.

research article
1226 The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005
vascular remodeling in response to hypertensive distending pres-sures (45). Although both NOS3–/– and WT hearts were exposed to the same pressure-load stimulus, only WT TAC hearts had increased gelatinolytic activity by zymography, which suggests that factors other than stress played an important role.
Although the role of ROS in hypertrophic signaling is increasing-ly accepted, its sources remain unclear. One candidate is NADPH oxidase, first shown to play a central role in ROS generation in vascular endothelium in response to mechanical (e.g., hyperten-sion; ref. 46) and humoral (e.g., angiotensin II; ref. 47) stimulation. Investigations showing that NADPH-dependent ROS generation rose in response to pressure overload and LV decompensation focused interest on these enzymes in the heart as well (16). How-ever, more recent studies found the hypertrophic response to TAC in mice lacking gp91phox was similar to controls (48, 49), suggest-ing that other Nox isoforms (48) or ROS generators played a role. One alternative source is xanthine oxidase, and although xanthine oxidase inhibition blunts after-infarction chamber remodeling (50), its role in pressure overload remains unclear (51). Further-more, we found that xanthine oxidase products, xanthine and uric acid, were elevated in both WT and NOS3–/– hearts exposed to TAC. Inducible NOS (NOS2) also can contribute to oxidant stress, but this did not appear important for the current studies, because Ca2+-independent NOS activity was low and unaltered.
The notion that NOS3 might serve as a dominant source for ROS that particularly played a role in chamber remodeling induced by pressure overload seems somewhat surprising, yet it is supported by prior work conducted in vascular tissue. NOS function is beneficial under normal conditions, but its exposure to mechanical/oxidant stress can shift it to a ROS generator to contribute to vascular disease. For example, deoxycorticosterone acetate-salt–induced hypertension stimulates endothelial dependent ROS generation that is markedly blunted by L-NAME and absent in vessels from mice lacking NOS3 (19). ROS generation in this model is associated with BH4 oxidation resulting in NOS uncoupling, and NADPH oxidases may provide an important initial trigger to this change. NOS uncoupling also occurs directly by oxidation of the zinc-thiolate complex, as shown by Zou et al. in studies employing ONOO– (20). Lastly, when BH4 synthesis is enhanced by endothelial-targeted expression of GTPCH-1, oxidant stress is suppressed and NO-mediated endothelial-dependent vaso-dilation is improved in a model of diabetes (52).
To our knowledge, the current study is the first to extend this mechanism to cardiac pathophysiology. Several lines of evi-dence supported NOS uncoupling due to TAC: the loss of NOS dimerization, the decline in BH4, the reduction in NOS NO-gen-erating activity, and the increase in NOS-dependent ROS gen-eration. Restoration of these changes by BH4 treatment further support the mechanism, particularly as similar effects were not
Figure 4ROS levels in WT and NOS3–/– hearts subjected to TAC. (A) Luminol chemiluminescence assay for superoxide in myocardial tissue extracts. TAC stimulated O2
– formation in WT hearts, but far less in NOS3–/– hearts. P value shown reflects the effect of genotype on the TAC-stimulated response (2-way ANOVA). (B and C) Intracellular ROS generation was also estimated by red DHE staining (B) and green DCF staining (C) in frozen sections imaged by confocal fluorescent microscopy. Both signals were increased in WT TAC and strongly attenuated in NOS3–/– TAC hearts. (D) NT measured by immunofluorescent staining and quantified by ELISA assay. Both methods revealed a marked increase in NT in WT TAC hearts but low levels in NOS3–/– TAC hearts, as shown in controls for both genotypes. *P < 0.05 versus other groups. Scale bars: 50 μm.
research article
The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005 1227
achieved with H4N, which has similar antioxidant properties to BH4. These data also help explain an apparent paradox in that mice without NOS3 behaved similarly to those with NOS3 that remained coupled (i.e., BH4-treated). In both conditions, NOS3-derived ROS were prevented. Whether endothelial or myocyte NOS uncoupling plays a more important role is unknown. The precise mechanism by which TAC lowers BH4 also remains to be fully resolved. Enhanced ROS generation would suggest BH4 oxidation as an important factor, and our data further suggest a decrease in BH4 synthesis by GTPCH-1. The latter is intriguing, given a recent study of endothelial cells revealing that STAT3 activation lowers GTPCH-1 expression (53) and prior studies showing STAT3 to be potently activated by TAC in intact hearts (54). Other potential factors that could lower GTPCH-1 activity are enhanced levels of cAMP (55) and cGMP (56), both of which occur with TAC (57).
The current findings would seem at odds with some prior studies showing NO has antihypertrophic effects (21) or that a lack of NOS3
exacerbates stress-induced hypertrophy (23, 24). For example, treat-ment of neonatal myocytes with an NO donor or by gene transfer of its primary downstream target cGMP-dependent protein kinase (22) sup-presses hypertrophy induced by α1-adrenergic (58) or angiotensin II
Figure 5GSH/GSSH levels, MMPs, and Akt activa-tion. (A) HPLC determination of GSH/GSSH, xanthine, and NADPH. GSH/GSSH mark-edly declined with TAC in WT hearts but not NOS3–/– hearts. Xanthine increased in both, but somewhat more in WT, whereas NADPH declined similarly in both genotypes. *P < 0.05 versus sham hearts of the same genotype. (B) Gelatin zymography of myocardium in controls and following 3 weeks of TAC, and quantifica-tion results. Positive control (Con) bands for activated MMP-2 and MMP-9 are shown. Basal gel lysis was minimal but markedly increased in WT TAC hearts. This was not observed in NOS3–/– hearts either at base line or with TAC. *P < 0.05 versus other groups. (C) Response of total Akt (t-Akt) and p-Akt to TAC in both genotypes and quantification results as a ratio of p-Akt to total Akt (n = 3 per group). TAC induced a marked increase in levels of p-Akt and total Akt in WT hearts. In contrast, there was no change in NOS3–/– hearts. **P < 0.01 versus WT sham at 3 weeks.
Figure 6Evidence of NOS3 uncoupling in WT TAC hearts. (A) In the WT sham heart, NOS3 appeared as both a dimer (NOS3-d) and a monomer (NOS3-m), with the largest fraction as a dimer. In boiled samples (con-trol), the dimer was replaced by the monomeric form. The 3-week WT TAC heart exhibited largely the monomeric form, although total NOS3 expression assessed by Western blot (B) was not altered. (C) NOS Ca2+-dependent and -independent activity based on L-citrulline forma-tion. Ca2+-dependent activity declined in WT TAC hearts (*P < 0.05). Low levels were also seen in NOS3–/– mutants, reflecting NOS1 activ-ity. Ca2+-independent NOS2 activity showed little change. (D) Impact of pharmacological NOS3 inhibition on luminol chemiluminescence assay. Coincubation with 1 mM L-NAME inhibited 50% of luminol chemiluminescence in 3-week and 9-week WT TAC heart lysates and inhibited less than 15% at base line, supporting an increased role of NOS in O2
– generation with TAC. Corresponding data for NOS3–/– myo-cardium are shown at right. *P < 0.05 versus WT sham at 3 weeks.
research article
1228 The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005
stimulation (59). In NOS3–/– mice, chamber and myocyte hypertro-phy following myocardial infarction is exacerbated over similarly infarcted WT hearts (24), and they develop more hypertrophy when exposed to modest pressure overload (23). However, other studies have found that unlike TAC, chronic NOS inhibition by L-NAME does not stimulate hypertrophy despite similar elevations of after-load (37). Prolonged lack of NOS3 itself can induce mild hypertro-phy (an approximately 30% increase in heart mass) (60), but this was not observed in the younger animals studied in the present inves-tigation (Table 1). Importantly, NOS-dependent signaling in the heart and vasculature can play very different roles depending upon the magnitude of NO generated and biochemical conditions that determine whether NOS generates NO or ROS. The severity and chronicity of the stimulus as well as the oxidative environment it engenders are important. Thus, in hearts with larger infarcts, Liu et al. (61) found hypertrophy in NOS3–/– mice was not aggravated.
NOS3 may not become uncoupled in hearts exposed to less severe TAC that does not stimulate chamber dilation or fibrosis and results in only mild hypertrophy (23). One potential limitation to our study is that we did not employ littermate controls for the NOS3–/– ani-mals, and this might contribute to some differences. However, this would not impact the BH4 and H4N data from WT animals.
The demonstration of NOS3 uncoupling–induced ROS genera-tion in cardiac hypertrophic pathophysiology marks an important new step for clarifying the sources and role of ROS signaling in this process. The aspect of hypertrophic remodeling that appears particularly sensitive to these changes is precisely that associated with the highest morbidity from chronic hypertension: chamber dilation, fibrosis, and the development of functional depression. Future clinical trials with BH4 or other agents that enhance BH4 should be able to test the value of this novel approach to amelio-rate a common cause of heart failure.
Figure 7BH4, but not H4N, prevented NOS3 uncoupling, ROS generation, and cardiac remodeling induced by 3-week TAC. (A) Formalin-fixed (10%) hearts (upper panel) and histology (PAS methenamine; lower panel) showing concentric hypertrophy with BH4 cotreatment versus dilative hypertrophy with H4N accompanied by increased interstitial fibrosis. Scale bars: 10 mm (upper panel); 100 μm (lower panel). (B and C) Repre-sentative M-mode echocardiography (B) and PV loops (C) reveal corresponding functional improvement in BH4-treated, but not H4N-treated, hearts. (D) NOS3 dimer was preserved in BH4-treated, but not H4N-treated, hearts. (E) NOS Ca2+-dependent activity was restored by BH4 but not H4N treatment. *P < 0.05 versus sham. (F) Luminol chemiluminescence shows a decline in O2
– generation in WT TAC hearts treated with BH4 but minimal effect with H4N treatment. *P < 0.05 versus sham. Lower graph shows the percent of luminol signal blunted by coincubation with L-NAME, confirming reduced NOS-derived O2
– in BH4-treated hearts. **P < 0.05 versus BH4-treated. (G) Confocal images of DHE-stained (red) and DCF-stained (green) myocardium from WT TAC hearts treated with either BH4 or H4N. Scale bar: 50 μm. (H) Gelatin zymography for hearts with BH4 or H4N treatment and quantification results. The increased gel lysis in WT TAC hearts was reduced by BH4, but not H4N, therapy. *P < 0.05 versus sham.
research article
The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005 1229
MethodsAnimals and preparation. Male NOS3-null (NOS3–/–) mice and C57/BL6 WT controls (8–11 weeks of age; The Jackson Laboratory) were used. Pressure overload was produced by TAC as previously described (62). TAC acutely increased LV systolic pressure by 67.2 ± 0.3 mmHg in WT and 69.0 ± 1.5 mmHg in NOS3–/– mice (P = NS). Control mice were subject to sham opera-tions, and animals were studied 3–9 weeks following surgery. An additional group of WT animals were subjected to TAC for 3 weeks and cotreated with oral BH4 (1 mg/g food; Sigma-Aldrich) mixed in their rodent chow, providing 5 mg/day based on 4–6 g daily diet (19). Control studies were also performed using oral H4N (1 mg/g food; Schircks Laboratories) (19), an antioxidant that does not directly participate in NOS3 coupling (35). The protocols were all approved by the Animal Care and Use Committee of Johns Hopkins University.
Echocardiography. In vivo cardiac morphology was assessed by transthoracic echocardiography (Acuson Sequoia C256, 13 MHz trans-ducer; Siemens) in conscious mice. M-mode LV end-systolic dimensions (LVESDs) and LV end-diastolic dimensions (LVEDDs) were averaged from 3–5 beats. LV ejection fraction (LVEF) was calculated as follows: LVEF = [(LVEDD)3 – (LVESD)3]/(LVEDD)3 × 100. Percent fractional shortening (%FS) was calculated as follows: %FS = (LVEDD – LVESD)/LVEDD × 100. Wall thickness of lateral freewall and intraventricular septum were aver-aged. Studies and analysis were performed blinded to heart condition.
In vivo hemodynamics. In vivo LV function was assessed by PV catheter (63, 64). Mice were anesthetized with 1–2% isoflurane, urethane (750–100 mg/kg, i.p.), etomidate (5–10 mg/kg, i.p.), and morphine (1–2 mg/kg, i.p.), underwent tracheostomy, and were ventilated with 6–7 μl/g tidal volume and 130 breaths/min. Volume expansion (12.5% human albumin, 50–100 μl over 5 minutes) was provided through a 30-gauge cannula via the right external jugular vein. The LV apex was exposed through an incision between the seventh and eighth ribs, and a 0.42-mm-diameter PV catheter (SPR 839; Millar Instruments Inc.) was advanced through the apex to lie
along the longitudinal axis. Absolute volume was calibrated, and PV data were measured at steady state and during transient reduction of venous return as previously described (64).
Fetal gene expression dot-blot analysis. Gene expression for type A-NP and type B-NP, β-MHC, α-SA, and the calcium handling proteins PLB and SERCA2a was analyzed by dot-blot analysis as previously described (65).
ROS and NT analysis. ROS generation was examined by several inde-pendent methods. Superoxide production in LV tissue homogenates was determined by luminol-enhanced chemiluminescence (EMD Biosciences). Flash-frozen myocardium was homogenized in iced PBS buffer and cen-trifuged, and the precipitate was resuspended in assay buffer to a final concentration of 100 μM luminol in accordance with the manufacturer’s instructions. Phorbol-12-myristate-13-acetate or other oxidase stimula-tors were not used in the assay. Data were normalized by sample weight. In addition, fresh frozen LV myocardium (8-μm slices) was incubated for 1 hour at 37°C with fluorescent dyes, 2′,7′-DCF diacetate (4 μM; Invitrogen Corp.) reflecting hydrogen peroxide formation (DCF stain-ing localizes principally to mitochondria within myocytes), DHE (2 μM; Invitrogen Corp.) assessing O2
– formation (typically nuclear localization) and NT formation (polyclonal NT Ab, 1:100; Upstate), which can reflect formation of ONOO–. Imaging was performed on a Zeiss inverted epi-fluorescence microscope (Carl Zeiss Inc.) attached to an argon-krypton laser confocal scanning microscope (UltraVIEW; Perkin Elmer Life Sci-ences Inc.). The excitation/emission spectrum for DHE was 488 and 610 nm, respectively, with detection at 585 nm, and for DCF was 480 and 535 nm, respectively, with detection at 505 nm. NT was also quantitatively assessed by ELISA assay (Oxis International).
HPLC analysis of oxidative stress, BH4, and energy metabolites. Mouse hearts were quickly excised, immersed in liquid nitrogen, and then sub-jected to the organic solvent deproteinization procedure as previously described (66). Aliquots of each deproteinized tissue extract (10% wt/vol) were filtered through a 0.45-μm HV-Millipore filter and then assayed by ion-pairing HPLC as previously reported (66). Ultrapure HPLC stan-dards were purchased from Sigma-Aldrich. For (6R)-5,6,7,8-BH4 and D-(+)-neopterin, separation was carried out on 20 μl using a Kromasil 250-by-4.6 mm, 5-μm particle size column provided with its own guard column (Eka Chemicals AB) using a step gradient from buffer A (10 mM tetrabutylammonium hydroxide, 10 mM KH2PO4, 0.25% methanol, pH 7.00) to buffer B (2.8 mM tetrabutylammonium hydroxide, 100 mM KH2PO4, 30% methanol, pH 5.50). The flow rate was 1.2 ml/min and column temperature was kept constant at 18°C. Under these condi-tions, BH4 is eluted isocratically with a k′ of 3.02 and neopterin with a k′ of 8.56, where k′ = V – V0/V0 (V, elution volume of compound; V0, void volume of chromatographic system). The lower assay limit was 0.5 μM, corresponding to 10 pmol per 20 μl injected volume. Species identifica-tion was made by matching retention times and absorption spectra to freshly prepared ultrapure standards and, if needed, cochromatograms performed by adding known standards to the biological samples. Con-centration was calculated from the standard run data at wavelengths corresponding to peak absorption of each substance.
Tissue histology. Formalin-fixed (10%) myocardium from WT and NOS3–/– mice with and without TAC was analyzed for myocyte hypertrophy and fibrosis. Tissue was paraffin-embedded, sectioned into 5- to 8-μm slices, and stained with PAS methenamine. Photomicrographs were quantified to assess mean cardiomyocyte diameter and interstitial collagen fraction using computer-assisted image analysis: Adobe Photoshop 5.0 (Adobe) and NIH ImageJ software. Average data reflect results from 4 hearts in each group (more than 30 cells).
Cardiac gelatinase analysis. In vitro gelatin lysis by MMP-2 and MMP-9 was assessed by zymography. Briefly, modified Laemmli buffer without
Table 2Effect of BH4 or H4N supplementation on cardiac morphologic and function response to TAC in nontransgenic (WT) mice
WT 3-wk WT 3-wk TAC + BH4 TAC + H4NBW and HW n = 14 n = 12 BW (g) 26.6 ± 0.4 26.4 ± 0.6HW (mg) 170.3 ± 3.9A 231.3 ± 10.7Echocardiographic analysis n = 11 n = 9Wall thickness (mm) 1.09 ± 0.03 0.98 ± 0.03A
LV diameter diastole (mm) 2.87 ± 0.06A 3.94 ± 0.19LV diameter systole (mm) 1.12 ± 0.06A 2.70 ± 0.31%FS 61.7 ± 1.7A 33.8 ± 5.3Hemodynamics — PV loop analysis n = 5 n = 4Heart rate (min–1) 603.3 ± 22.9 501.2 ± 28.5 LV systolic pressure (mmHg) 193.0 ± 1.7A 177.8 ± 4.6 Ea (mmHg/μL) 10.6 ± 0.7 11.4 ± 0.7LV end-systolic volume (μl) 4.8 ± 1.7A 24.0 ± 7.6 LV end-diastolic volume (μl) 20.5 ± 1.9A 38.8 ± 8.0 LVEF (%) 78.4 ± 5.8A 43.4 ± 9.8 dP/dtmax (mmHg/s) 17003 ± 1125A 11154 ± 520Peak power index (mmHg/s) 75.5 ± 10.5A 31.8 ± 5.8Normalized Ees (mmHg/μl/g) 115.1 ± 9.6A 41.0 ± 9.3 dP/dtmin (mmHg/s) –15743 ± 1325A –10725 ± 971.0τ (ms) 3.5 ± 0.2A 5.5 ± 0.36
AP < 0.05 versus 3-week TAC + vehicle treatment.

research article
1230 The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005
mercaptoethanol was added to lysed tissue samples and loaded on a 10% gelatin (Invitrogen Corp.). After electrophoresis, gels were washed twice with renaturing buffer at room temperature followed by developing buf-fer (Invitrogen Corp.) and then stained to visualize lytic bands (Simply-Blue; Invitrogen Corp.).
Determination of NOS dimerization and activity. SDS-resistant NOS3 dimers and monomers were assayed using low-temperature SDS-PAGE under reducing or nonreducing conditions, as previously described (20). NOS3 was immunoprecipitated as previously described (27), and the resulting samples were added to 5-fold Laemmli buffer (0.32 mol/l Tris-HCl, pH 6.8, 0.5 mol/l glycine, 10% SDS, 50% glycerol, and 0.03% bromophenol blue) in nonreducing gel (without 2-mercaptoethanol) to identify dimer dissociation due to reduced disulfide bridges. To pro-vide fully denatured control lanes, samples were boiled for 15 minutes prior to loading. Electrophoresis was performed using Tris glycine 6% gels (Invitrogen Corp.), and gels and buffers were maintained in an ice bath at 4°C and stained (SimplyBlue; Invitrogen Corp.). Ca2+-depen-dent and -independent NOS activity was determined from myocardial homogenates by [3H]-L-arginine to [3H]-L-citrulline conversion (Sigma-Aldrich) as previously described (27).
Akt activation. Akt activation was assessed by Western blotting for total Akt and S473 p-Akt (1:1,000 dilution; Cell Signaling Technology). Protein concentration was determined by bicinchoninic acid method and primary antibodies visualized by horseradish peroxidase–conjugated secondary antibodies and enhanced chemiluminescence (Pierce Biotechnology).
Statistical analysis. All values were expressed as mean ± SEM. Group data were compared using 1-way or 2-way ANOVA (with genotype and ± TAC as categories) and a Tukey’s post-hoc multiple comparisons test for between-
group differences. Unless specifically noted, analysis was performed with n = 4–6 per group in a given assay.
AcknowledgmentsThis study was supported by NIH grants PO1-HL59408, HL-47511, and AG18324 (to D.A. Kass); a Uehara Memorial Foun-dation grant and an American Heart Association (Mid-Atlantic Affiliate) Fellowship Grant (to E. Takimoto); a Shih-Chun Wang Young Investigator Award, a Giles F. Filley award of the American Physiological Society, and the Bernard Family Foundation (to H.C. Champion); and an American Heart Association Scientist Devel-opment Grant (to N. Paolocci). The authors thank Djahida Bedja for her technical support.
Received for publication April 26, 2004, and accepted in revised form February 22, 2005.
Address correspondence to: David A. Kass, Ross 835, Division of Cardiology, The Johns Hopkins Medical Institutions, 720 Rutland Avenue, Baltimore, Maryland 21205, USA. Phone: (410) 955-7153; Fax: (410) 502-2558; E-mail: [email protected].
E. Rene Rodriguez’s present address is: Division of Pathology and Laboratory Medicine, Department of Pathology, Cleveland Clinic Lerner College of Medicine, Cleveland, Ohio, USA.
Eiki Takimoto and Hunter C. Champion contributed equally to this work.
1. Frey, N., and Olson, E.N. 2003. Cardiac hypertro-phy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65:45–79.
2. Kenchaiah, S., and Pfeffer, M.A. 2004. Cardiac remodeling in systemic hypertension. Med. Clin. North Am. 88:115–130.
3. Sorescu, D., and Griendling, K.K. 2002. Reac-tive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest. Heart Fail. 8:132–140.
4. Takano, H., et al. 2003. Oxidative stress-induced signal transduction pathways in cardiac myocytes: involvement of ROS in heart diseases. Antioxid. Redox Signal. 5:789–794.
5. Sawyer, D.B., et al. 2002. Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell. Cardiol. 34:379–388.
6. Yamamoto, M., et al. 2003. Inhibition of endog-enous thioredoxin in the heart increases oxida-tive stress and cardiac hypertrophy. J. Clin. Invest. 112:1395–1406. doi:10.1172/JCI200317700.
7. Dhalla, A.K., Hill, M.F., and Singal, P.K. 1996. Role of oxidative stress in transition of hypertrophy to heart failure. J. Am. Coll. Cardiol. 28:506–514.
8. Mollnau, H., et al. 2002. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 90:E58–E65.
9. Amin, J.K., et al. 2001. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell. Cardiol. 33:131–139.
10. Sayen, M.R., Gustafsson, A.B., Sussman, M.A., Molkentin, J.D., and Gottlieb, R.A. 2003. Calcineurin transgenic mice have mitochondrial dysfunction and elevated superoxide production. Am. J. Physiol. Cell Physiol. 284:C562–C570.
11. Pimentel, D.R., et al. 2001. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in car-diac myocytes. Circ. Res. 89:453–460.
12. Leslie, N.R., et al. 2003. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 22:5501–5510.
13. Siwik, D.A., et al. 1999. Inhibition of copper-zinc superoxide dismutase induces cell growth, hyper-trophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro. Circ. Res. 85:147–153.
14. Spinale, F.G. 2002. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ. Res. 90:520–530.
15. Ide, T., et al. 1999. Mitochondrial electron trans-port complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 85:357–363.
16. Li, J.M., Gall, N.P., Grieve, D.J., Chen, M., and Shah, A.M. 2002. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 40:477–484.
17. Xiao, L., et al. 2002. Role of reactive oxygen spe-cies and NAD(P)H oxidase in alpha(1)-adrenocep-tor signaling in adult rat cardiac myocytes. Am. J. Physiol. Cell Physiol. 282:C926–C934.
18. Saavedra, W.F., et al. 2002. Imbalance between xanthine oxidase and nitric oxide synthase signal-ing pathways underlies mechanoenergetic uncou-pling in the failing heart. Circ. Res. 90:297–304.
19. Landmesser, U., et al. 2003. Oxidation of tetrahy-drobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111:1201–1209. doi:10.1172/JCI200314172.
20. Zou, M.H., Shi, C., and Cohen, R.A. 2002. Oxi-dation of the zinc-thiolate complex and uncou-pling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Invest. 109:817–826. doi:10.1172/JCI200214442.
21. Wollert, K.C., and Drexler, H. 2002. Regulation of cardiac remodeling by nitric oxide: focus on cardiac myocyte hypertrophy and apoptosis. Heart Fail. Rev. 7:317–325.
22. Wollert, K.C., et al. 2002. Gene transfer of cGMP-dependent protein kinase I enhances the antihyper-
trophic effects of nitric oxide in cardiomyocytes. Hypertension. 39:87–92.
23. Ichinose, F., et al. 2004. Pressure overload-induced LV hypertrophy and dysfunction in mice are exac-erbated by congenital NOS3 deficiency. Am. J. Physiol. Heart Circ. Physiol. 286:H1070–H1075.
24. Scherrer-Crosbie, M., et al. 2001. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation. 104:1286–1291.
25. Kuzkaya, N., Weissmann, N., Harrison, D.G., and Dikalov, S. 2003. Interactions of peroxynitrite, tet-rahydrobiopterin, ascorbic acid, and thiols: impli-cations for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 278:22546–22554.
26. Massion, P.B., and Balligand, J.L. 2003. Modula-tion of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J. Physiol. 546:63–75.
27. Champion, H.C., et al. 2004. Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circ. Res. 94:657–663.
28. Xia, Y., Dawson, V.L., Dawson, T.M., Snyder, S.H., and Zweier, J.L. 1996. Nitric oxide synthase gener-ates superoxide and nitric oxide in arginine-deplet-ed cells leading to peroxynitrite-mediated cellular injury. Proc. Natl. Acad. Sci. U. S. A. 93:6770–6774.
29. Rosen, G.M., et al. 2002. The role of tetrahydro-biopterin in the regulation of neuronal nitric-oxide synthase-generated superoxide. J. Biol. Chem. 277:40275–40280.
30. Iwanaga, Y., et al. 2002. Excessive activation of matrix metalloproteinases coincides with left ven-tricular remodeling during transition from hyper-trophy to heart failure in hypertensive rats. J. Am. Coll. Cardiol. 39:1384–1391.
31. Rajagopalan, S., Meng, X.P., Ramasamy, S., Har-rison, D.G., and Galis, Z.S. 1996. Reactive oxy-gen species produced by macrophage-derived foam cells regulate the activity of vascular matrix

research article
The Journal of Clinical Investigation http://www.jci.org Volume 115 Number 5 May 2005 1231
metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Invest. 98:2572–2579.
32. Matsui, T., et al. 2002. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 277:22896–22901.
33. Matsui, T., Nagoshi, T., and Rosenzweig, A. 2003. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2:220–223.
34. Matsui, T., et al. 2002. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 277:22896–22901.
35. Heitzer, T., et al. 2000. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ. Res. 86:E36–E41.
36. Ferdinandy, P., Danial, H., Ambrus, I., Rothery, R.A., and Schulz, R. 2000. Peroxynitrite is a major contributor to cytokine-induced myocardial con-tractile failure. Circ. Res. 87:241–247.
37. Bartunek, J., et al. 2000. Chronic N(G)-nitro-L-arginine methyl ester-induced hypertension: novel molecular adaptation to systolic load in absence of hypertrophy. Circulation. 101:423–429.
38. Suematsu, N., et al. 2003. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 107:1418–1423.
39. Shiomi, T., et al. 2004. Overexpression of gluta-thione peroxidase prevents left ventricular remod-eling and failure after myocardial infarction in mice. Circulation. 109:544–549.
40. Matsui, T., and Rosenzweig, A. 2005. Convergent signal transduction pathways controlling car-diomyocyte survival and function: the role of PI 3-kinase and Akt. J. Mol. Cell Cardiol. 38:63–71.
41. Matsui, T., et al. 2002. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J. Biol. Chem. 277:22896–22901.
42. Matsui, T., Nagoshi, T., and Rosenzweig, A. 2003. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2:220–223.
43. Siwik, D.A., Pagano, P.J., and Colucci, W.S. 2001. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibro-blasts. Am. J. Physiol. Cell Physiol. 280:C53–C60.
44. Kinugawa, S., et al. 2000. Treatment with dimeth-ylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ. Res. 87:392–398.
45. Lehoux, S., Lemarie, C.A., Esposito, B., Lijnen, H.R., and Tedgui, A. 2004. Pressure-induced matrix metalloproteinase-9 contributes to early hyperten-sive remodeling. Circulation. 109:1041–1047.
46. Griendling, K.K., Sorescu, D., and Ushio-Fukai, M. 2000. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ. Res. 86:494–501.
47. Griendling, K.K., Minieri, C.A., Ollerenshaw, J.D., and Alexander, R.W. 1994. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 74:1141–1148.
48. Byrne, J.A., et al. 2003. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 93:802–805.
49. Maytin, M., et al. 2004. Pressure overload-induced myocardial hypertrophy in mice does not require gp91phox. Circulation. 109:1168–1171.
50. Engberding, N., et al. 2004. Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation. 110:2175–2179.
51. de Jong, J.W., et al. 2000. Enhanced expression and activity of xanthine oxidoreductase in the failing heart. J. Mol. Cell. Cardiol. 32:2083–2089.
52. Alp, N.J., et al. 2003. Tetrahydrobiopterin-depen-dent preservation of nitric oxide-mediated endothelial function in diabetes by targeted trans-genic GTP-cyclohydrolase I overexpression. J. Clin. Invest. 112:725–735. doi:10.1172/JCI200317786.
53. Huang, A., Zhang, Y.Y., Chen, K., Hatakeyama, K., and Keaney, J.F., Jr. 2005. Cytokine-stimulated GTP cyclohydrolase i expression in endothelial cells requires coordinated activation of nuclear factor-kappaB and Stat1/Stat3. Circ. Res. 96:164–171.
54. Yasukawa, H., et al. 2001. Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J. Clin. Invest. 108:1459–1467. doi:10.1172/JCI200113939.
55. Ohtsuki, M., et al. 2002. cAMP inhibits cytokine-
induced biosynthesis of tetrahydrobiopterin in human umbilical vein endothelial cells. Life Sci. 70:2187–2198.
56. Shiraishi, H., et al. 2003. cGMP inhibits GTP cyclo-hydrolase I activity and biosynthesis of tetrahy-drobiopterin in human umbilical vein endothelial cells. J. Pharmacol. Sci. 93:265–271.
57. Takimoto, E., et al. 2005. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ. Res. 96:100–109.
58. Calderone, A., Thaik, C.M., Takahashi, N., Chang, D.L., and Colucci, W.S. 1998. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J. Clin. Invest. 101:812–818.
59. Ritchie, R.H., Schiebinger, R.J., LaPointe, M.C., and Marsh, J.D. 1998. Angiotensin II-induced hypertro-phy of adult rat cardiomyocytes is blocked by nitric oxide. Am. J. Physiol. 275:H1370–H1374.
60. Cappola, T.P., et al. 2003. Deficiency of different nitric oxide synthase isoforms activates divergent transcriptional programs in cardiac hypertrophy. Physiol. Genomics. 14:25–34.
61. Liu, Y.H., et al. 2002. Effect of ACE inhibitors and angiotensin II type 1 receptor antagonists on endothelial NO synthase knockout mice with heart failure. Hypertension. 39:375–381.
62. Takimoto, E., et al. 2002. Sodium calcium exchanger plays a key role in alteration of cardiac function in response to pressure overload. FASEB J. 16:373–378.
63. Georgakopoulos, D., et al. 1998. In vivo murine left ventricular pressure-volume relations by miniatur-ized conductance micromanometry. Am. J. Physiol. 274:H1416–H1422.
64. Isoda, T., et al. 2003. Novel regulation of cardiac force-frequency relation by CREM (cAMP response element modulator). FASEB J. 17:144–151.
65. Liao, P., et al. 2001. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 98:12283–12288.
66. Lazzarino, G., et al. 2003. Single sample prepara-tion for the simultaneous cellular and redox energy state determination. Anal. Biochem. 322:51–59.
Related Documents











